
Identification of potential miRNAs and their targets in Vriesea carinata (Poales, Bromeliaceae)
11
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/authorsrights
Transcript of Identification of potential miRNAs and their targets in Vriesea carinata (Poales, Bromeliaceae)

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
Plant Science 210 (2013) 214– 223
Contents lists available at SciVerse ScienceDirect
Plant Science
jo u r n al homep age: www.elsev ier .com/ locate /p lantsc i
Identification of potential miRNAs and their targets in Vriesea carinata(Poales, Bromeliaceae)
Frank Guzmana, Mauricio Pereira Almerãob, Ana Paula Korbesa, Ana Paula Christoff a,Camila Martini Zanellaa, Fernanda Bereda, Rogério Margisa,b,∗
a PPGBM at Federal University of Rio Grande do Sul – UFRGS, Porto Alegre, RS, Brazilb Centre of Biotechnology and PPGBCM, Laboratory of Genomes and Plant Population, Federal University of Rio Grande do Sul – UFRGS, Porto Alegre, RS,Brazil
a r t i c l e i n f o
Article history:Received 8 September 2012Received in revised form 24 April 2013Accepted 23 May 2013Available online xxx
Keywords:Vriesea carinatamiRNAsHigh-throughput sequencingStress response
a b s t r a c t
The miRNAs play important roles in regulation of gene expression at the post-transcriptional level. A smallRNA and RNA-seq of libraries were constructed to identify miRNAs in Vriesea carinata, a native bromeliadspecies from Brazilian Atlantic Rainforest. Illumina technology was used to perform high throughputsequencing and data was analyzed using bioinformatics tools. We obtained 2,191,509 mature miRNAssequences representing 54 conserved families in plant species. Further analysis allowed the predictionof secondary structures for 19 conserved and 16 novel miRNAs. Potential targets were predicted frompre-miRNAs by sequence homology and validated using RTqPCR approach. This study provides the firstidentification of miRNAs and their potential targets of a bromeliad species.
© 2013 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
MicroRNAs (miRNAs) are small non-coding regulatory RNAswidely found in unicellular and multicellular organisms that actas regulators of gene expression at the post-transcriptional levelon genes containing miRNA binding sites [1]. Mature miRNAs aresingle-stranded RNA molecules of approximately 21 nucleotides(nt) in length processed from a precursor molecule (pre-miRNA)[2]. To regulate protein-coding genes the mature miRNA binds inthe mRNA target site leading to mRNA degradation or translationrepression [3]. In plants, miRNAs have diverse biological functionsand are involved in the regulation of optimal growth and devel-opment, as well as other physiological processes, including abioticand biotic stress responses [4]. Several studies showed that manymiRNAs are conserved across different plant families [5]. However,it was also reported species and family specific miRNAs that areexpressed in low levels and probably have evolved recently [6].
Bromeliaceae family, with 3248 species distributed in 58 genera[7], is an example of a large and well described adaptive radiation
Abbreviations: miRNA, microRNA; sRNA, small RNA; pre-miRNA, microRNA pre-cursor; MFEI, minimal folding energy index; nt, nucleotides; vca, Vriesea carinata.
∗ Corresponding author at: Centre of Biotechnology and PPGBCM, Laboratory ofGenomes and Plant Population, Building 43431, Federal University of Rio Grande doSul – UFRGS, P.O. Box 15005, CEP 91501-970, Porto Alegre, RS, Brazil.Tel.: +55 51 33087766; fax: +55 51 33087309.
E-mail addresses: [email protected], [email protected](R. Margis).
of plant families in the Neotropics. The family is composed of ter-restrial xerophytes and both facultative and obligatory epiphytesspecies which acquired, throughout their evolution, interesting anddifferent adaptive mechanisms such as central tanks, CAM photo-synthetic pathway, and bear absorptive trichomes, characteristicsthat allowed them to occupy a wide range of habitats [8,9]. Con-sequently, Bromeliaceae constitutes one of the most ecologicallydiverse and species-rich clades of flowering plants native to theNew World [10]. In Brazil, the Atlantic Rainforest is considered oneof the main centers of diversity and endemism of Bromeliaceae,showing 31 genera and 803 species of which 10 genera and 653species are endemic [11]. Vriesea carinata is an epiphytic or ter-restrial species distributed along the Brazilian Atlantic Rainforest.As a typical species of this biome [12], V. carinata is an interest-ing model for studying the expression of miRNAs in Bromeliaceae.The first step to study the expression of miRNAs is to identify miR-NAs and its targets in different natural conditions. For this purpose,we performed a high-throughput sequencing analysis (Solexa tech-nology) of small RNAs (sRNAs) from the endemic Brazilian AtlanticRainforest species V. carinata.
2. Materials and methods
2.1. Plant material and RNA isolation
Total RNA was isolated from V. carinata leaves using Trizolreagent (Invitrogen, CA, USA), according to manufacturer’s pro-tocol. The RNA quality was evaluated by electrophoresis on a 1%
0168-9452/$ – see front matter © 2013 Elsevier Ireland Ltd. All rights reserved.http://dx.doi.org/10.1016/j.plantsci.2013.05.013

Author's personal copy
F. Guzman et al. / Plant Science 210 (2013) 214– 223 215
agarose gel and quantification was determined using a Nanodrop(Nanodrop Technologies, Wilmington, DE, USA).
2.2. Deep sequencing
Total RNA (>10 �g) from leaves was sent to Fasteris SA (Plan-les-Ouates, Switzerland) for processing. One small RNA (sRNA) librarywas constructed and sequenced the Illumina HiSeq2000 platform.Briefly, the construction of the small RNA libraries consisted of thefollowing successive steps: acrylamide gel purification of the RNAfraction corresponding to the size range 20–30 nt, ligation of the 3pand 5p adapters to the RNA in two separate subsequent steps, eachfollowed by acrylamide gel purification, cDNA synthesis followedby acrylamide gel purification, and a final step of PCR amplificationto generate a cDNA colony template library for Illumina sequencing.
A polyadenylated transcript sequencing (mRNA-seq) was per-formed using the following successive steps: poly-A purification,cDNA synthesis using poly-T primer shotgun to generate inserts of500 nt, 3p and 5p adapters ligations, pre-amplification, colony gen-eration and sequencing. The Illumina output data correspondes tosequence tags of 100 bases.
2.3. Accession numbers
Sequencing data is available in Gene Expression Omnibus (GEO)under the series accession GSE38250 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38250). This accession contains theRNA-seq and sRNA libraries derived from V. carinata leaves.
2.4. Analysis of small RNA library
The overall procedure for analyzing Illumina small RNA libraryis shown in Fig. S1. All low quality reads (with FASTq value bel-low 13) were removed and 5′ and 3′ adapter sequences weretrimmed using Genome Analyzer Pipeline (Illumina) by Fasteris.The remaining low quality reads with ‘n’ were removed with Prin-Seq script [13]. Sequences shorter than 18 nt and larger than 25 ntwere excluded from further analysis. Small RNAs derived fromViridiplantae rRNAs, tRNAs, snRNAs and snoRNAs deposited at thetRNAdb [14], SILVA rRNA [15], and NONCODE v3.0 [16] databasesand from Poales mtRNA and cpRNA deposited at NCBI GenBankdatabase (http://ftp.ncbi.nlm.nih.gov) were identified by mappingwith Bowtie [17]. After data cleaning (low quality reads, adaptersequences), mRNA-seq data was assembled de novo in contigs usingCLC Genome Workbench version 4.0.2 (CLCbio, Aarhus, Denmark)with default parameters. In total, 41,171 contigs were assembledand used as reference for pre-miRNA and target sequence identifi-cation.
2.5. Identification of conserved and novel miRNAs
In order to determine plant conserved miRNAs, sRNA sequenceswere aligned with conserved non-redundant Viridiplantae miRNAsdeposited at miRBase (Release 18, November 2011) using Bowtie.Complete alignment of the sequences was required and no mis-matches allowed. To search for novel miRNAs, sRNA sequenceswere matched against a set of proprietary V. carinata mRNAtranscript database using SOAP2 [18]. The SOAP2 output was fil-tered with an in house filter precursor tool to separate candidatesequences as miRNA precursors with an anchoring pattern of ablock of aligned small RNAs with perfect matches. The candidateprecursors obtained were confirmed manually using Tablet soft-ware [19] to visualize the anchoring pattern. As miRNAs precursorshave a characteristic hairpin structure, the next step to select pre-cursor candidates was the secondary structure analysis by RNAfold
with RNAfold webserver and using annotation algorithm from theUEA sRNA toolkit [20].
We used the mfold web server (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) to identify the minimal foldingfree energy (MFE, �G kcal/mol) of each miRNA precursor. This valueestimates the stability of the miRNA candidate-target duplex. Then,the adjust minimal folding free energy (AMFE) and the minimalfolding free energy index (MFEI) were calculated according to theprevious report [21]. AMFE means the MFE of a RNA sequencewith 100 nt in length, which is equal to MFE/(length of a poten-tial pre-miRNA) × 100. MFEI is equal to MFE/(length of a potentialpre-miRNA)/(The percentage of nucleotides G and C). In addition,perfect stem-loop structures should have the sRNA sequence at onearm of the stem and a respective anti-sense sequence at the oppo-site arm. Finally, precursor candidate sequences were confirmed asnovel by BLASTn algorithm from the miRBase (www.mirbase.org)and NCBI databases.
2.6. Prediction of miRNA targets
The mRNA contigs previously assembled were clustered usingthe Gene Indices Clustering Tools (http://compbio.dfci.harvard.edu/tgi/software/) [22] to reduce any sequence redundancy. Theclustering output was passed to CAP3 assembler [23] for multi-ple alignment and consensus building. Contigs that cannot reachthe threshold set and fall into any assembly should remain as alist of singletons. The prediction of target genes of the most abun-dant mature miRNAs from the conserved and novel pre-miRNAswas performed by psRNAtarget [24] using V. carinata assembledunigenes longer than 600 bp (default parameters and expectationvalue of 3.5). Candidate RNA sequences were then annotated byassignment of putative gene descriptions based on sequence simi-larity with previously identified genes. These genes were annotatedwith those details deposited in the protein database of NR andSwiss Prot/Uniprot protein database using BLASTx implementedin blast2GO v2.3.5 software [25]. The annotation was improvedby analysis of conserved domains/families using InterProScan tooland Gene Ontology terms were determined by GOslim tool fromblast2GO software. At the same time the orientation of the trans-cripts were obtained from BLAST annotations.
2.7. miRNAs and target confirmation by RT-qPCR
In order to validate the in silico predicted V. carinata miRNAs andsome of their mRNAs targets, RT-qPCR reactions were performed asdescribed in previous works [26,27]. RNA samples were extractedfrom two different tissues, leaf and ovary in six biological repli-cates, with the Trizol reagent (Invitrogen, CA, USA). The RNA qualitywas accessed by electrophoresis on a 1% agarose gel. Thereafter thecDNA were obtained for 17 miRNAs based on the stem-loop method[28]. Also the cDNA for mRNA targets validation were obtainedbased on the poli-T amplification by reverse transcription of anM-MLV RNA Polymerase (Invitrogen, CA, USA), accordingly withthe manufacturer instructions. Primers used for Stem loop cDNAsynthesis, mature miRNA expression and mRNA target amplifica-tion were described in Supplementary Tables S1–S3, respectively.The RT-qPCR amplifications were performed in a CFX 384 Real-Time PCR System (Bio Rad), using SYBR Green (Invitrogen). PCRreactions were carried out in a final volume of 10 �L, containing5 �L of diluted cDNA (1:100) and 5 �L of reagents mix: 1X SYBRGreen, 0.025 mM dNTP, 1X PCR buffer, 3 mM MgCl2, 0.25 U Plati-nun Taq DNA Polimerase (Invitrogen) and 200 nM of each reverseand forward primer. The RT-qPCR conditions were set as follow:94 ◦C for 5 min, 40 cycles of 94 ◦C for 15 s, 60 ◦C for 10 s and 25 sat 72 ◦C. In the end of the PCR run, a melting curve were evalu-ated. Samples were analyzed in four technical replicates, and a no
Author's personal copy
216 F. Guzman et al. / Plant Science 210 (2013) 214– 223
Table 1Summary of data from sequencing of V. carinata small RNA libraries.
Type Number of reads Percentage (%)
Total readsa 15,986,233 10018–25 nt 13,834,378 86<18 nt 752,929 5>25 nt 1,398,926 9
a Reads with high quality.
template negative control was included. RNA input normalizationsfor miRNA were performed with the miR011 and miR397, selectedby geNorm [29] as the best combination of normalizers. For targetmRNAs the combination of the genes vca-miR011-5p-2-t (his-tone acetyl tranferase mbd9) and vca-miR396-5p-2-t (nuclear porecomplex protein nup98-nup96) were set as the best combinationfor RNA input normalization by geNorm. To calculate the relativeexpression of miRNAs and mRNA targets the 2−��ct method wereused [30]. To compare pairwise differences in expression, Student’st-test was performed considering p < 0.05.
3. Results
3.1. V. carinata RNA library sequencing
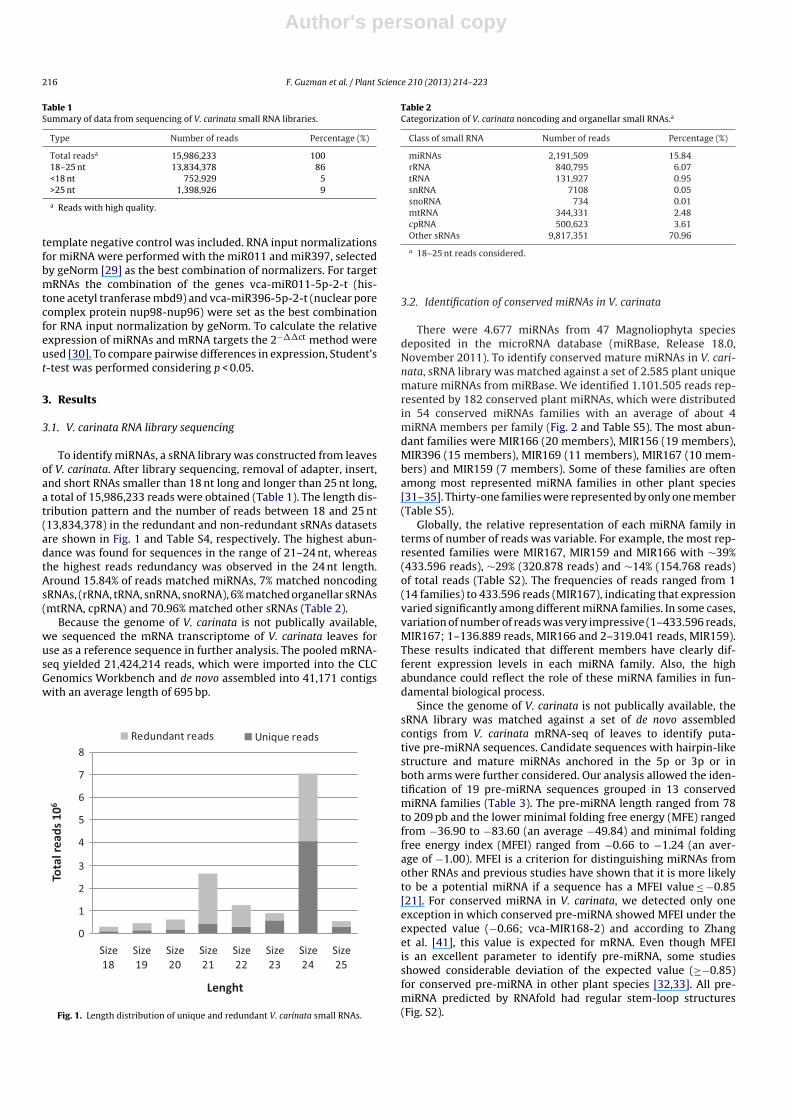
To identify miRNAs, a sRNA library was constructed from leavesof V. carinata. After library sequencing, removal of adapter, insert,and short RNAs smaller than 18 nt long and longer than 25 nt long,a total of 15,986,233 reads were obtained (Table 1). The length dis-tribution pattern and the number of reads between 18 and 25 nt(13,834,378) in the redundant and non-redundant sRNAs datasetsare shown in Fig. 1 and Table S4, respectively. The highest abun-dance was found for sequences in the range of 21–24 nt, whereasthe highest reads redundancy was observed in the 24 nt length.Around 15.84% of reads matched miRNAs, 7% matched noncodingsRNAs, (rRNA, tRNA, snRNA, snoRNA), 6% matched organellar sRNAs(mtRNA, cpRNA) and 70.96% matched other sRNAs (Table 2).
Because the genome of V. carinata is not publically available,we sequenced the mRNA transcriptome of V. carinata leaves foruse as a reference sequence in further analysis. The pooled mRNA-seq yielded 21,424,214 reads, which were imported into the CLCGenomics Workbench and de novo assembled into 41,171 contigswith an average length of 695 bp.
Size 18
Size 19
Size 20
Size 21
Size 22
Size 23
Size 24
Size 25
lenght
Redund ant reads Unique reads
Lenght
Tota
l rea
ds 1
06
0
1
2
3
4
5
6
7
8
Fig. 1. Length distribution of unique and redundant V. carinata small RNAs.
Table 2Categorization of V. carinata noncoding and organellar small RNAs.a
Class of small RNA Number of reads Percentage (%)
miRNAs 2,191,509 15.84rRNA 840,795 6.07tRNA 131,927 0.95snRNA 7108 0.05snoRNA 734 0.01mtRNA 344,331 2.48cpRNA 500,623 3.61Other sRNAs 9,817,351 70.96
a 18–25 nt reads considered.
3.2. Identification of conserved miRNAs in V. carinata
There were 4.677 miRNAs from 47 Magnoliophyta speciesdeposited in the microRNA database (miRBase, Release 18.0,November 2011). To identify conserved mature miRNAs in V. cari-nata, sRNA library was matched against a set of 2.585 plant uniquemature miRNAs from miRBase. We identified 1.101.505 reads rep-resented by 182 conserved plant miRNAs, which were distributedin 54 conserved miRNAs families with an average of about 4miRNA members per family (Fig. 2 and Table S5). The most abun-dant families were MIR166 (20 members), MIR156 (19 members),MIR396 (15 members), MIR169 (11 members), MIR167 (10 mem-bers) and MIR159 (7 members). Some of these families are oftenamong most represented miRNA families in other plant species[31–35]. Thirty-one families were represented by only one member(Table S5).
Globally, the relative representation of each miRNA family interms of number of reads was variable. For example, the most rep-resented families were MIR167, MIR159 and MIR166 with ∼39%(433.596 reads), ∼29% (320.878 reads) and ∼14% (154.768 reads)of total reads (Table S2). The frequencies of reads ranged from 1(14 families) to 433.596 reads (MIR167), indicating that expressionvaried significantly among different miRNA families. In some cases,variation of number of reads was very impressive (1–433.596 reads,MIR167; 1–136.889 reads, MIR166 and 2–319.041 reads, MIR159).These results indicated that different members have clearly dif-ferent expression levels in each miRNA family. Also, the highabundance could reflect the role of these miRNA families in fun-damental biological process.
Since the genome of V. carinata is not publically available, thesRNA library was matched against a set of de novo assembledcontigs from V. carinata mRNA-seq of leaves to identify puta-tive pre-miRNA sequences. Candidate sequences with hairpin-likestructure and mature miRNAs anchored in the 5p or 3p or inboth arms were further considered. Our analysis allowed the iden-tification of 19 pre-miRNA sequences grouped in 13 conservedmiRNA families (Table 3). The pre-miRNA length ranged from 78to 209 pb and the lower minimal folding free energy (MFE) rangedfrom −36.90 to −83.60 (an average −49.84) and minimal foldingfree energy index (MFEI) ranged from −0.66 to −1.24 (an aver-age of −1.00). MFEI is a criterion for distinguishing miRNAs fromother RNAs and previous studies have shown that it is more likelyto be a potential miRNA if a sequence has a MFEI value ≤ −0.85[21]. For conserved miRNA in V. carinata, we detected only oneexception in which conserved pre-miRNA showed MFEI under theexpected value (−0.66; vca-MIR168-2) and according to Zhanget al. [41], this value is expected for mRNA. Even though MFEIis an excellent parameter to identify pre-miRNA, some studiesshowed considerable deviation of the expected value (≥−0.85)for conserved pre-miRNA in other plant species [32,33]. All pre-miRNA predicted by RNAfold had regular stem-loop structures(Fig. S2).
Author's personal copy
F. Guzman et al. / Plant Science 210 (2013) 214– 223 217
Tab
le
3C
har
acte
rist
ics
of
pre
-miR
NA
s
iden
tifi
ed
in
V. c
arin
ata
mat
chin
g
con
serv
ed
miR
NA
fam
ilie
s
in
oth
er
pla
nt
spec
ies.
Pre-
miR
NA
Prec
urs
or
miR
NA
s
Mat
ure
miR
NA
s
Con
tig
nam
e
Len
gth
MFE
AM
FE
MFE
I
5p
mor
e
abu
nd
ant
3p
mor
e
abu
nd
ant
Tota
l rea
ds
Tota
l iso
miR
NA
s
Sequ
ence
mem
ber
Rea
ds
Sequ
ence
mem
ber
Rea
ds
vca-
MIR
156-
1
con
tig
1187
5210
3
−58.
70−5
6.99
−1.0
5U
GA
CA
GA
AG
AG
AG
UG
AG
CA
C
9473
UG
CU
CA
CU
UC
UC
UU
UC
UG
UC
AG
730
14,9
63
55vc
a-M
IR15
6-2
con
tig
1376
86
−47.
20
−54.
88
−1.2
4
UU
GA
CA
GA
AG
AU
AG
AG
AG
CA
C
8388
GC
UC
UC
UA
UG
CU
UC
UG
UC
AU
C
359
8921
22vc
a-M
IR15
9
con
tig
1404
10
209
−83.
60
−40.
00
−0.9
1
GA
GC
UU
CC
UU
GG
GU
CC
AA
AG
A
3500
UU
UG
GA
UU
GA
AG
GG
AG
CU
CU
A
319,
041
332,
989
49vc
a-M
IR16
0
con
tig
1460
36
103
−54.
40
−52.
82
−0.9
9
UG
CC
UG
GC
UC
CC
UG
AA
UG
CC
A
291
ND
–
307
4vc
a-M
IR16
6-1
con
tig
1327
16
96
−52.
60
−54.
79
−1.0
1
GG
AA
UG
UU
GU
CU
GG
UU
CG
AA
A
240
UC
GG
AC
CA
GG
CU
UC
AU
UC
CC
C
136,
889
148,
317
30vc
a-M
IR16
6-2
con
tig
1452
5512
9
−46.
31−3
5.89
−0.8
4
GG
AA
UG
UU
GU
CU
GG
CU
CG
AG
G
1564
UC
GG
AC
CA
GG
CU
UC
AU
UC
CC
C
136,
889
149,
916
34vc
a-M
IR16
6-3
con
tig
7568
4
89
−39.
80
−44.
72
−0.8
3
GG
AA
UG
UU
GU
CC
GG
CU
CG
AG
G
1842
UC
GG
AC
CA
GG
CU
UC
AU
UC
CC
C
136,
889
149,
967
35vc
a-M
IR16
7-1
con
tig
1269
3910
4
−56.
10−5
3.94
−1.2
2U
GA
AG
CU
GC
CA
GC
AU
GA
UC
UG
A
410,
104
CA
GA
UC
AU
GU
GG
CA
GU
UU
CA
U
17
411,
899
30vc
a-M
IR16
7-2
con
tig
8566
7
107
−38.
70
−36.
17
−0.7
6
UG
AA
GC
UG
CC
AG
CA
UG
AU
CU
GA
410,
104
AG
AU
CA
UG
UU
GC
AG
UU
UC
AU
C
40
411,
769
19vc
a-M
IR16
8-1
con
tig
8494
1
91
−54.
90
−60.
33
−0.9
6
UC
GC
UU
GG
UG
CA
GG
UC
GG
GA
A
80,4
71
UC
CC
GG
CU
UG
CA
CC
AA
CU
GA
A
357
87,3
36
61vc
a-M
IR16
8-2
con
tig
9753
4
90
−39.
70
−44.
11
−0.6
6
UC
GC
UU
GG
UG
CA
GG
UC
GG
GA
A
80,4
71
CC
CG
CC
UU
GC
AC
CA
AC
UG
AA
U
124
86,3
14
23vc
a-M
IR17
2-1
con
tig
1421
80
130
−50.
80
−39.
08
−1.0
8
GU
GG
CA
UC
AU
GA
AG
AU
UC
AC
A
484
AG
AA
UC
UU
GA
UG
AU
GC
UG
CA
U
1783
67,6
57
35vc
a-M
IR17
2-2
con
tig
6959
6
94
−46.
80
−49.
79
−1.1
7
CA
GC
AU
CA
UC
CA
GA
UU
CA
CA
U
697
AG
AA
UC
UU
GA
UG
AU
GC
UG
CA
U
1783
68,2
48
53vc
a-M
IR39
3
con
tig
1443
92
79
−40.
20
−50.
89
−1.2
2
UC
CA
AA
GG
GA
UC
GC
AU
UG
AU
C
1017
UC
AA
UG
CG
AU
UU
CU
UU
GG
AA
U
50,3
29
52,0
13
28vc
a-M
IR39
6
con
tig
1039
4290
−36.
90
−41.
00
−1.0
5
UU
CC
AC
AG
CU
UU
CU
UG
AA
CU
4329
GU
UC
AA
UA
AA
GC
UG
UG
GG
AA
A
1116
10,0
58
37vc
a-M
IR39
7
con
tig
2034
7
78
−37.
70
−48.
33
−0.9
9
UC
AU
UG
AG
UG
CA
GC
GU
UG
AU
G
1514
UC
AG
CG
CU
UC
AC
UC
AA
UC
AU
G
54
2423
16vc
a-M
IR40
8
con
tig
7432
7
104
−61.
50
−59.
13
−0.9
9
AC
GG
GG
AC
GA
GG
UC
GG
GC
AU
G
54
UG
CA
CU
GC
CU
CU
UC
CC
UG
GC
U
848
1953
12vc
a-M
IR52
8
con
tig
8393
83
−45.
50
−54.
82
−0.9
9
UG
GA
AG
GG
GC
AU
GC
AG
AG
GA
G
16,0
88
UU
UC
CU
GU
GC
UC
GC
CC
CU
UC
C
413
17,0
25
27vc
a-M
IR53
5
con
tig
1422
06
94
−55.
60
−59.
15
−1.1
1
UG
AC
GA
UG
AG
AG
AG
AG
CA
CG
C
46,8
06
GU
GC
UC
UC
UC
UC
GU
UG
UC
AC
C
806
49,5
98
78
ND
:
Not
det
ecte
d.
3.3. Identification of novel miRNAs in V. carinata
Following the criteria in the identification of conserved pre-miRNAs, we obtained 17 potential novel miRNAs grouped in 16miRNA families (Table 4). In addition to the hairpin structure, thedetection of complementary antisense miRNA (miRNA*) in all pre-miRNA was a strong indication to consider these sequences as truecandidates. Comparison between the mature sequences of candi-date miRNAs and sequences deposited in miRBase suggest thatthese candidates are novel miRNAs not identified in others speciesand probably specific to Bromeliacea. These sequences (miRNA*)are rarely found by cloning because of their quick degradation incells and its detection represented further evidence for the exist-ence of mature miRNAs [36].
The novel pre-miRNA sequences in V. carinata ranged from 79to 235 nt and the lower minimal folding free energy (MFE) rangedfrom −26.20 to −109.50 and minimal folding free energy index(MFEI) ranged from −0.87 to −1.48 (Table 4). All novel pre-miRNAidentified were in accordance with the expected value (≥−0.85)[21]. Likewise to conserved pre-miRNA, all novel pre-miRNA pre-dicted by RNAfold had regular stem-loop structures (Fig. S2).
3.4. IsomiRs sequences
We detected multiple mature miRNAs variants named isoformsor isomiRs in both arms (5p and 3p) of identified conserved andnovel pre-miRNAs (Tables 3 and 4). Because they vary widely inabundance, we considered all isomiRs in each pre-miRNA even ifthey showed only one read. For example, vca-nMIR016 showedsix isomiRs in 3p arm varying from 2 to 13 reads and six isomiRsin 5p arm with reads ranging from 1 to 17 (Fig. 3). The exist-ence of these multiple variants reflects the variability in miRNAbiogenesis, specifically in DCL cleavage sites or subsequent pro-cessing/degradation or later processing steps [37]. Although thebiological functions of isomiRs remain to be determined, isomiRsfrom a single pre-miRNA could be a way of broadening the regula-tory network [38,39].
3.5. Identification and classification of targets for pre-miRNAsidentified
To understand the biological function of miRNAs in V. carinata,the putative targets sites of the miRNA candidates were identifiedby aligning the most abundant mature miRNAs of each conservedand novel pre-miRNA identified to a set of V. carinata assembledunigenes using psRNAtarget. Despite the use of 4 as cut-off in pre-vious works to infer miRNA targets [40,41], we adopted the strictercut-off threshold of 3.5. Based on this criterion, we found 145 poten-tial targets in total (for 27 miRNA families), of which 79 weretargets of conserved miRNAs (13 miRNA families) and 66 were tar-gets of novel miRNAs (14 miRNA families). Only for three novelmiRNAs families (vca-nMIR002, vca-nMIR003 and vca-nMIR016)targets were not detected. Detailed annotation results are given inTables 5and S6.
All targets regulated by the identified conserved and novel miR-NAs in this study were subjected to GO analysis to evaluate theirpotential functions. The categorization of these genes according tobiological process, cellular component and molecular function aresummarized in Fig. 4. Based on biological process, these genes wereclassified into 12 categories and the four most overrepresentedGO terms were response to metabolic, cellular, developmental andmulticellular processes, suggesting that V. carinata miRNAs areinvolved in a broad range of physiological functions. Interestingly,we found 8 and 6 candidate genes in the category of responseto abiotic stimulus in conserved and novel miRNAs, respectively(Tables 5 and S6). Categories based on molecular function revealed

Author's personal copy
218 F. Guzman et al. / Plant Science 210 (2013) 214– 223Ta
ble
4C
har
acte
rist
ics
of
nov
el
pre
-miR
NA
s
iden
tifi
ed
in
V. c
arin
ata.
pre
-miR
NA
Prec
urs
or
miR
NA
s
Mat
ure
miR
NA
s
Con
tig
nam
e
Len
gth
MFE
AM
FE
MFE
I
5p
mor
e
abu
nd
ant
3p
mor
e
abu
nd
ant
Tota
l rea
ds
Tota
l iso
miR
NA
s
Sequ
ence
mem
ber
Rea
ds
Sequ
ence
mem
ber
Rea
ds
vca-
MIR
001
con
tig
1041
45
178
−60.
60
−34.
04
−0.8
7
AA
TAA
TAA
TTC
GTT
GG
GC
CC
AA
AC
12
TGA
AC
CC
AA
CG
AG
TTA
TTA
TTG
CT
48
120
24vc
a-M
IR00
2
con
tig
1041
45
180
−72.
80
−45.
50
−1.2
3
TAA
TAA
CTC
GTT
GG
GTT
CA
AA
CC
A
20
GG
TTTG
GG
CC
CA
AC
GA
ATT
1
42
9vc
a-M
IR00
3
con
tig
1093
07
90
−26.
20
−29.
11
−1.0
9
AA
ATA
ATG
ATG
TGTC
GTT
GA
TGC
G
78
ATA
TAG
CA
TTA
TTA
TGG
TTG
ATG
A
4
159
17vc
a-M
IR00
4
con
tig
1233
19
88
−55.
80
−63.
41
−1.4
0
TTG
TGC
TTTT
AG
AA
GC
ATT
CC
GG
C
18
GG
ATC
GG
AA
TGC
TTC
TAA
TAG
CA
C
1
29
8vc
a-M
IR00
5
con
tig
1245
61
222
−109
.50
−49.
32
−1.2
3
TTC
CG
GTC
TGG
CTT
AC
GA
CA
T
158
TTG
AC
AG
CTT
TCA
AA
GG
GTT
T
2444
3035
58vc
a-M
IR00
6
con
tig
1256
68
145
−58.
20
−40.
14
−1.1
4
TCC
TGG
ATA
ATA
CA
ATA
ATT
GC
GC
11
AA
TTA
TTG
TATC
ATT
TAG
GG
GG
GA
9
31
9vc
a-M
IR00
7
con
tig
7768
1
105
−32.
60
−31.
05
−1.2
5
AG
GA
CA
GA
TCA
GA
TTC
ATT
GA
ATC
4
ATT
CA
ATG
AA
TCTA
TCTG
TCA
AT
8
23
8vc
a-M
IR00
8
con
tig
7928
6
90
−37.
00
−40.
66
−1.4
8
AG
AC
AG
ATC
AG
GTA
CA
TTG
AA
TCT
144
AA
TGTA
TCTA
TCTG
TCA
ATA
GTA
A
7
174
21vc
a-M
IR00
9
con
tig
1470
42
230
−108
.00
−46.
96
−1.1
9
AG
TAA
AG
ATG
GG
ATG
GA
TGG
CA
GA
1112
AG
ATT
CA
CTA
GG
TGG
TCC
ATG
GC
T
25
1862
76vc
a-M
IR01
0
con
tig
1272
91
102
−48.
10
−47.
16
−1.1
2
TGC
GG
TTA
GG
ATT
ATA
GTG
GT
1027
AC
TATT
TCA
CC
AA
CA
AC
CC
AC
TAA
122
5183
95vc
a-M
IR01
1
con
tig
7253
910
9
−54.
30−4
9.82
−1.2
3
TTC
AG
AG
CA
TCTG
TTC
AG
TGC
717
TAG
GTG
CTC
CG
AA
TCA
TCTA
GG
23
1199
37vc
a-M
IR01
2
con
tig
1770
6
141
−84.
60
−60.
00
−1.0
2
TCC
GC
CA
CG
TCA
TCTG
TGTA
C
99
TCA
CG
GA
TGA
CG
TGG
CG
GA
CC
2399
2992
35vc
a-M
IR01
3
con
tig
6846
1
86
−50.
70
−58.
95
−1.2
4
TTC
GC
AG
GA
GA
GA
TGA
TGC
CG
457
GC
ATC
GTC
TCTC
CG
GC
GA
CG
G
16
562
19vc
a-M
IR01
4
con
tig
8322
8
235
−82.
80
−35.
23
−0.8
8
GTT
GTC
CTA
TGTT
CC
GG
CA
GA
9
TGC
AG
GA
GTG
TAG
GA
TAA
CG
C
506
527
10vc
a-M
IR01
5
con
tig
9188
9
188
−75.
00
−39.
89
−1.3
4
TCC
AC
AG
GC
TTTC
TTG
AA
CTA
310,
363
TTC
AA
GA
AA
GTT
CG
TGG
AA
A
4962
335,
732
56vc
a-M
IR01
6
con
tig
7074
1
79
−48.
00
−60.
76
−1.0
2
TCA
GG
AG
AG
ATG
AC
AC
CC
A
169
GG
TGTC
AC
CTC
TCC
TGG
AC
55
306
14
that the target genes were related to eight functions and thefour most frequent terms were hydrolase activity, protein binding,small molecule binding and nucleic acid binding. In the categoryof cellular component, the analysis revealed that the target genesare expressed on a high percentage in intracellular membrane-bounded organelle and plastid.
3.6. Biological confirmation of identified miRNAs and targets
The RT-qPCR method was used to validate the expression of themost abundant mature sequences from seventeen miRNAs (vca-miR156-1, vca-miR156-2, vca-miR159, vca-miR166, vca-miR168,vca-miR393, vca-miR396, vca-miR397, vca-miR528, vca-miR535,vca-miR009, vca-miR010, vca-miR011, vca-miR012, vca-miR013,vca-miR014 and vca-miR015) and some of their targets. We con-firmed that these miRNAs were expressed in leaf and ovary tissues(results not shown). For the miRNAs vca-miR166 and vca-miR393we found that their expressions were inversely correlated with theexpression of some of its targets (Fig. S3).
4. Discussion
Several miRNAs have been identified via computational orexperimental approaches in different plant families, but there is nosequence or functional information available about miRNAs in anyBromeliaceae species. V. carinata is a typical representative of theBromeliaceae in the Brazilian Atlantic Rainforest biome and there-fore emerges as interesting model for this botanical family. We usedIllumina technology for deep sequencing of sRNA library to identifymiRNAs in this species and results are discussed.
Size profile is an important feature to distinguish miRNA fromother sRNAs and most of the mature miRNAs are of 21–25 nt. Thelength distribution pattern indicated that the majority of the sRNAwas 24 nt, accounting for ∼51% of the total reads, followed by 21 nt(∼19%), 22 nt (∼9%) and 23 nt (∼6%). This distribution pattern ishighly consistent with previous studies in other plants [42,43] anddifferences in distribution pattern (e.g., for some species 21 nt wasthe most abundant) may reflect the composition of the sRNAs ofa given plant species according to tissue (or cell) and physiologi-cal conditions [44]. Another explanation is that molecules of 24 ntare the typical size of Dicer-digestion product and are often themost abundant endogenous plant sRNAs [45,46]. sRNAs of 24 nt areknown to be involved in heterochromatin modification, especiallyin a genome with a high content of repetitive sequences [47,48]. Thehigh percentage of 24 nt sRNAs found in V. carinata could reflect thecomplexity of the genome of bromeliads [49].
Computational methods have been successfully used to predicthundreds of miRNAs in a wide variety of plant species. Illuminasequencing is a powerful tool to estimate expression profiles ofmiRNA. This technology provides the resources to know the abun-dance of various miRNA families and even distinguish betweendifferent members of a given family. In our case, we found sig-nificant differences among the number and abundance of themembers identified in each family, similarity observed in otherstudies [50,51], suggesting that this wide variation is due to a func-tional divergence in the conserved miRNA families. In this study, weidentified 182 conserved plant miRNAs distributed in 54 miRNAsfamilies, very similar to found for barley, using the same approach(126 miRNA distributed in 58 miRNA families) [52].
Although conserved miRNAs have been identified by sequencingand by comparison again miRNAs identified in other species, mostof plant species-specific miRNAs remain unidentified due to the lev-els of expression of these are very low producing a small numberof reads sequenced in comparison to the conserved miRNAs [53].For this reason, we used a new approach to identify novel miRNAs

Author's personal copy
F. Guzman et al. / Plant Science 210 (2013) 214– 223 219
1
10
100
1.000
10.00 0
100.00 0
1.000 .00 0
miR
156
miR
157
miR
158
miR
159
miR
160
miR
161
miR
162
miR
164
miR
165
miR
166
miR
167
miR
168
miR
169
miR
170
miR
171
miR
172
miR
319
miR
390
miR
393
miR
394
miR
395
miR
396
miR
397
miR
398
miR
399
miR
400
miR
403
miR
408
miR
447
miR
528
miR
529
miR
530
miR
535
miR
824
miR
827
miR
828
miR
844
miR
845
miR
858
miR
859
miR
866
miR
1030
miR
1507
miR
1509
miR
1885
miR
2118
miR
2934
miR
3711
miR
4995
miR
5083
miR
5139
miR
5179
miR
5368
miR
5538
miRNA famili es
19
2
3
7
3
3
3
7
2
20
10
3
11
1
7 5
1
1
1
5
15
5 4
2
2
1
1
1
22
1
11
1
11
11
1
1 11 1 1 1
11
1
1 1 111
miRNA families
10
0
1
2
3
4
5
6
Tota
l rea
ds (l
og10
)
Fig. 2. Number of identified miRNAs in each conserved miRNA family in plants. The values above the bars indicate the number of members identified fot each conservedmiRNA family.
in species without availability of genomic resources using simul-taneously sequences from sRNA and RNAseq libraries. Using thismethodology, we identified 16 potential miRNAs candidates spe-cific for V. carinata. In all cases, complementary antisense miRNA
(miRNA*) was identified providing more evidence for their exist-ence as novel miRNAs [54,55].
To understand the function of the identified miRNAs, theirputative targets were predicted using a bioinformatics approach.
Fig. 3. Predicted secondary structures of conserved and novel miRNAs of V. carinata. Secondary structures of vca-nMIR013 and vca-nMIR016 precursors, their locations andthe expression of small RNAs mapped onto these precursors. The sequences corresponding to the most abundant mature miRNAs in the 5p and 3p arms are labeled in redand purple, respectively. Values on the left side of the miRNA sequence represent read counts in the leaf library.

Author's personal copy
220 F. Guzman et al. / Plant Science 210 (2013) 214– 223
Table 5Predicted putative targets of novel miRNAs in V. carinata.
Mature miRNA Contig Scoreb Inhibition Putative function
vca-miR001-3p contig 98870 2 Cleavage Rna binding proteinvca-miR004-5p contig 69835 rc 2.5 Cleavage Kinesin family member 6vca-miR005-3p contig 119481a 2 Cleavage Tetratricopeptide repeat-containing protein
contig 75544a 2.5 Cleavage Two-component response regulator arr3contig 66635 rca 3 Cleavage Heat shock 70 kda mitocondrialcontig 67788a 3 Cleavage Pyruvate kinase isozyme chloroplasticcontig 67948 rc 3.5 Cleavage E3 ubiquitin ligase big brothercontig 69145 3.5 Cleavage Fertility restorercontig 95670 3.5 Cleavage Probable lrr receptor-like serine threonine-protein kinasecontig 143421 3.5 Cleavage R3h domain-containing proteincontig 70604 rc 3.5 Cleavage Reticulon-like protein b17contig 67529 3.5 Cleavage Tho complex subunit 1
vca-miR006-5p contig 139947 rc 3 Cleavage Lysosomal alpha-mannosidasecontig 75839 3 Translation Probable beta- -galactosyltransferase 2contig 140146 3.5 Translation Chloroplastic group iia intron splicing facilitator chloroplasticcontig 81980 rc 3.5 Cleavage Omega-amidase nit2
vca-miR007-3p contig 140165 rc 0.5 Cleavage Calmodulin-like proteincontig 72901 rc 3 Cleavage Chlorophyllase- chloroplasticcontig 68350 3 Cleavage Soluble starch synthase chloroplastic amyloplasticcontig 140661 rc 3.5 Translation Tubulin-specific chaperone d
vca-miR008-5p contig 69155 rc 2 Translation Probable protein phosphatase 2c 18contig 140200 rc 3 Cleavage Abc transporter f family member 3contig 71460 rc 3 Translation Membrane proteincontig 79377 rc 3.5 Cleavage 1-phosphatidylinositol 3-phosphate 5-kinase fab1contig 67906 rc 3.5 Cleavage Aminopeptidase c
vca-miR009-5p contig 140774 rca 3 Cleavage Mitochondrial substrate carrier family proteincontig 118843 rc 3.5 Cleavage Cellulose synthase
vca-miR010-5p contig 71336a 1 Cleavage Abc transporter i family member 1vca-miR011-5p contig 89802a 3 Cleavage Dna binding protein
contig 140009a 3 Cleavage Methyl- -binding domain-containing protein 9contig 67880 rc 3.5 Cleavage Phenylalanyl-trna synthetase beta subunitcontig 122123 3.5 Cleavage Serine threonine-protein kinase nek1
vca-miR012-3p contig 71550 rca 3 Translation Ribonucleoside-diphosphate reductase small chaincontig 69862a 3 Cleavage Wrky transcription factor 11contig 34475 rc 3.5 Cleavage Oligopeptidase bcontig 140456 rca 3.5 Translation Protein resurrection 1
vca-miR013-5p contig 139979a 2.5 Cleavage Calcium-transporting atpase plasma membrane-typecontig 66522a 3 Cleavage Glucan -beta-glucosidasecontig 66366a 3 Cleavage Glutamyl-trna amidotransferase subunit acontig 68601a 3 Cleavage Probable protein phosphatase 2c 38contig 118464 rc 3.5 Cleavage Casein kinase ii subunit betacontig 705 3.5 Cleavage Retrotransposon ty3-gypsy subclasscontig 66929 rc 3.5 Translation Transcription factor gte4contig 141231 rc 3.5 Translation U3 small nucleolar ribonucleoprotein protein imp4
vca-miR014-3p contig 83228 rca 0 Cleavage Nucleoid dna-binding protein cnd41contig 66712a 3 Cleavage Uncharacterized protein ycf36contig 140254 rc 3.5 Cleavage Pumilio homolog 1
vca-miR015-5p contig 140456 rca 3 Translation Protein resurrection1contig 72376 rc 3.5 Translation Ethylene-responsive transcription factor 1contig 140188 rc 3.5 Cleavage Protein root hair defective 3contig 66270 rc 3.5 Cleavage Ubiquitin carboxyl-terminal hydrolase 7
In bold: putative targets with response to abiotic stimulus.a Targets evaluated in RTqPCR experiments.b psRNATarget value.
As expected, many putative targets of conserved miRNAs weretranscriptions factors, similar reported in other studies [56–58].Among the most important transcription factors identified, wefound squamosa promoter binding protein (SBP)-like (SPL) genes,which are targets of MIR156 and MIR535 families, affecting diversedevelopmental processes such as leaf development, shoot mat-uration, phase change and flowering in plants [59,60]. We alsoidentified the auxin response factor (ARF), a plant-specific family ofDNA binding proteins involved in hormone signal transduction thatare targets of MIR160 and MIR167 families [61,62]. Other importantgenes identified and targeted by MIR156 and MIR396 were penta-tricopeptide repeat genes (PPR) which show high importance inthe regulation of gene expression. This gene family is implicated inpost-transcriptional processes such as splicing, editing, processingand translation specifically in organelles such as mitochondria andchloroplasts [63].
In the case of the novel miRNAs, we also identified othertranscription factors as targets. WRKY is the putative target ofvca-nMIR012 and belongs to a large family of transcription factorsthat regulate various physiological processes, including pathogendefense, senescence, trichome development and signal transduc-tion [64]. The ethylene-responsive transcription factor 1 (ESR1)was targeted by vca-nMIR015 and is involved in regulating geneexpression patterns in meristems, modulating organ developmentand promoting shoot regeneration through the cytokinin signalingpathway [65,66]. Other important putative target identified for vca-nMIR013 was the transcription factor GTE4. It is involved in theactivation and maintenance of cell division in the meristems andby this controls cell numbers in differentiated organs [67].
Based in the GO analysis, we found 14 candidate genes withresponse to abiotic stimulus. These results indicate that the puta-tive targets from identified miRNAs are involved in a large number

Author's personal copy
F. Guzman et al. / Plant Science 210 (2013) 214– 223 221
Fig. 4. Gene categories and the distribution of target genes of the most abundant mature miRNAs in the conserved and novel pre-miRNA identified in V. carinata. (A) Molecularfunction, (B) biological process and (C) cellular component.
of primary physiological and metabolic processes and are likelyinvolved in the adaptation of V. carinata to different types of stressand environmental conditions observed in natura. Similar resultsin other plants confirmed the miRNA molecular regulation in dif-ferent plant abiotic stresses [68]. Future experimental validationwill determine how many of these predicted targets are genuinelytargeted by miRNAs in V. carinata in specific environmental andphysiological conditions.
This work provides a comprehensive investigation of the miRNApopulation in leaves of the bromeliad species V. carinata. The resultssupport the currently idea that miRNAs are conserved in plantsand play an important role in several physiological processes asshown by bioinformatics analysis of miRNA putative target genes.Although the exact function of these putative target genes remainsto be confirmed, the present study provides novel insights for thecomprehension of the molecular processes involved in the con-served miRNA function.
Acknowledgements
This work was sponsored by PDJ (509828/2010-8) and Pro-ductivity and Research Grant (307868/2011-7) from the National
Council for Scientific and Technological Development (CNPq,Brazil).
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at http://dx.doi.org/10.1016/j.plantsci.2013.05.013.
References
[1] A.C. Mallory, H. Vaucheret, Functions of microRNAs and related small RNAs inplants, Nat. Genet. 38 (Suppl.) (2006) S31–S36.
[2] A.M. Denli, B.B.J. Tops, R.H.A. Plasterk, R.F. Ketting, G.J. Hannon, Processingof primary microRNAs by the Microprocessor complex, Nature 432 (2004)231–235.
[3] M.W. Jones-Rhoades, D.P. Bartel, B. Bartel, MicroRNAS and their regulatory rolesin plants, Annu. Rev. Plant Biol. 57 (2006) 19–53.
[4] R. Sunkar, Y.-F. Li, G. Jagadeeswaran, Functions of microRNAs in plant stressresponses, Trends Plant Sci. 17 (2012) 196–203.
[5] T. Dezulian, J. Palatnik, D. Huson, D. Weigel, Conservation and divergence ofmicroRNA families in plants, Genome Biol. 6 (2005) P13.
[6] E. Allen, Z. Xie, A.M. Gustafson, G.-H. Sung, J.W. Spatafora, J.C. Carrington, Evo-lution of microRNA genes by inverted duplication of target gene sequences inArabidopsis thaliana, Nat. Genet. 36 (2004) 1282–1290.

Author's personal copy
222 F. Guzman et al. / Plant Science 210 (2013) 214– 223
[7] H.E. Luther, An alphabetical list of bromeliad binomials, in: Sarasota BromeliadSociety and Marie Selby Botanical Gardens, 2010.
[8] T.A. Ranker, D.E. Soltis, P.S. Soltis, A.J. Gilmartin, Subfamilial relationships of theBromeliaceae: evidence from chloroplast DNA restriction site variation, Syst.Bot. 15 (1990) 425–434.
[9] D.H. Benzing, Bromeliaceae, in: Profile of an Adaptive Radiation, CambridgeUniversity Press, Cambridge, 2000.
[10] T.J. Givnish, M.H.J. Barfuss, B. Van Ee, R. Riina, K. Schulte, R. Horres, P.A. Gonsiska,R.S. Jabaily, D.M. Crayn, J.A.C. Smith, K. Winter, G.K. Brown, T.M. Evans, B.K.Holst, H. Luther, W. Till, G. Zizka, P.E. Berry, K.J. Sytsma, Phylogeny, adaptiveradiation, and historical biogeography in Bromeliaceae: insights from an eight-locus plastid phylogeny, Am. J. Bot. 98 (2011) 872–895.
[11] S. Porembski, G. Martinelli, R. Ohlemuller, W. Barthlott, Diversity and ecologyof saxicolous vegetation mats on inselbergs in the Brazilian Atlantic rainforest,Divers. Distrib. 4 (1998) 107–119.
[12] L.B. Smith, R.J. Downs, Flora Neotropica: Monography 14, Part 2 – Tilland-sioideae, The New York Botanical Garden Hafner Press, New York, 1977.
[13] R. Schmieder, R. Edwards, Quality control and preprocessing of metagenomicdatasets, Bioinformatics (Oxford, Engl.) 27 (2011) 863–864.
[14] F. Jühling, M. Mörl, R.K. Hartmann, M. Sprinzl, P.F. Stadler, J. Pütz, tRNAdb 2009:compilation of tRNA sequences and tRNA genes, Nucleic Acids Res. 37 (2009)D159–D162.
[15] E. Pruesse, C. Quast, K. Knittel, B.M. Fuchs, W. Ludwig, J. Peplies, F.O. Glöckner,SILVA: a comprehensive online resource for quality checked and aligned ribo-somal RNA sequence data compatible with ARB, Nucleic Acids Res. 35 (2007)7188–7196.
[16] S. He, C. Liu, G. Skogerbø, H. Zhao, J. Wang, T. Liu, B. Bai, Y. Zhao, R. Chen, NON-CODE v2.0: decoding the non-coding, Nucleic Acids Res. 36 (2008) D170–D172.
[17] B. Langmead, C. Trapnell, M. Pop, S.L. Salzberg, Ultrafast and memory-efficientalignment of short DNA sequences to the human genome, Genome Biol. 10(2009) R25.
[18] R. Li, C. Yu, Y. Li, T.-W. Lam, S.-M. Yiu, K. Kristiansen, J. Wang, SOAP2: animproved ultrafast tool for short read alignment, Bioinformatics (Oxford, Engl.)25 (2009) 1966–1967.
[19] I. Milne, M. Bayer, L. Cardle, P. Shaw, G. Stephen, F. Wright, D. Marshall, Tablet– next generation sequence assembly visualization, Bioinformatics (Oxford,Engl.) 26 (2010) 401–402.
[20] S. Moxon, F. Schwach, T. Dalmay, D. Maclean, D.J. Studholme, V. Moulton,A toolkit for analysing large-scale plant small RNA datasets, Bioinformatics(Oxford, Engl.) 24 (2008) 2252–2253.
[21] B.H. Zhang, X.P. Pan, S.B. Cox, G.P. Cobb, T.A. Anderson, Evidence that miRNAsare different from other RNAs, CMLS 63 (2006) 246–254.
[22] G. Pertea, X. Huang, F. Liang, V. Antonescu, R. Sultana, S. Karamycheva, Y. Lee,J. White, F. Cheung, B. Parvizi, J. Tsai, J. Quackenbush, TIGR Gene Indices clus-tering tools (TGICL): a software system for fast clustering of large EST datasets,Bioinformatics (Oxford, Engl.) 19 (2003) 651–652.
[23] X. Huang, A. Madan, CAP3: A DNA sequence assembly program, Genome Res. 9(1999) 868–877.
[24] X. Dai, P.X. Zhao, psRNATarget: a plant small RNA target analysis server, NucleicAcids Res. 39 (2011) W155–W159.
[25] A. Conesa, S. Götz, Blast2GO: a comprehensive suite for functional analysis inplant genomics, Int. J. Plant Genom. 2008 (2008) 619832.
[26] F.R. Kulcheski, F.C. Marcelino-Guimaraes, A.L. Nepomuceno, R.V. Abdelnoor, R.Margis, The use of microRNAs as reference genes for quantitative polymerasechain reaction in soybean, Anal. Biochem. 406 (2010) 185–192.
[27] F.R. Kulcheski, L.F. de Oliveira, L.G. Molina, M.P. Almerao, F.A. Rodrigues, J.Marcolino, J.F. Barbosa, R. Stolf-Moreira, A.L. Nepomuceno, F.C. Marcelino-Guimaraes, R.V. Abdelnoor, L.C. Nascimento, M.F. Carazzolle, G.A. Pereira, R.Margis, Identification of novel soybean microRNAs involved in abiotic andbiotic stresses, BMC Genom. 12 (2011) 307.
[28] C. Chen, D.A. Ridzon, A.J. Broomer, Z. Zhou, D.H. Lee, J.T. Nguyen, M. Barbisin,N.L. Xu, V.R. Mahuvakar, M.R. Andersen, K.Q. Lao, K.J. Livak, K.J. Guegler, Real-time quantification of microRNAs by stem-loop RT-PCR, Nucleic Acids Res. 33(2005) e179.
[29] J. Vandesompele, K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe,F. Speleman, Accurate normalization of real-time quantitative RT-PCR data bygeometric averaging of multiple internal control genes, Genome Biol. 3 (2002),research0034.
[30] K.J. Livak, T.D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method, Methods 25 (2001)402–408.
[31] R. Sunkar, G. Jagadeeswaran, In silico identification of conserved microRNAs inlarge number of diverse plant species, BMC Plant Biol. 8 (2008) 37.
[32] T.P. Frazier, F. Xie, A. Freistaedter, C.E. Burklew, B. Zhang, Identification andcharacterization of microRNAs and their target genes in tobacco (Nicotianatabacum), Planta 232 (2010) 1289–1308.
[33] T. Unver, I. Parmaksiz, E. Dündar, Identification of conserved micro-RNAs andtheir target transcripts in opium poppy (Papaver somniferum L.), Plant Cell Rep.29 (2010) 757–769.
[34] F. Guzman, M.P. Almerao, A.P. Korbes, G. Loss-Morais, R. Margis, Identifica-tion of microRNAs from Eugenia uniflora by high-throughput sequencing andbioinformatics analysis, PLoS ONE 7 (2012) e49811.
[35] A.P. Korbes, R.D. Machado, F. Guzman, M.P. Almerao, L.F. de Oliveira, G. Loss-Morais, A.C. Turchetto-Zolet, A. Cagliari, F. dos Santos Maraschin, M. Margis-Pinheiro, R. Margis, Identifying conserved and novel microRNAs in developingseeds of Brassica napus using deep sequencing, PLoS ONE 7 (2012) e50663.
[36] Q.-X. Song, Y.-F. Liu, X.-Y. Hu, W.-K. Zhang, B. Ma, S.-Y. Chen, J.-S. Zhang, Identi-fication of miRNAs and their target genes in developing soybean seeds by deepsequencing, BMC Plant Biol. 11 (2011) 5.
[37] R.D. Morin, G. Aksay, E. Dolgosheina, H.A. Ebhardt, V. Magrini, E.R. Mardis, S.C.Sahinalp, P.J. Unrau, Comparative analysis of the small RNA transcriptomes ofPinus contorta and Oryza sativa, Genome Res. 18 (2008) 571–584.
[38] L. Guo, Z. Lu, Global expression analysis of miRNA gene cluster and familybased on isomiRs from deep sequencing data, Comput. Biol. Chem. 34 (2010)165–171.
[39] L. Guo, H. Li, T. Liang, J. Lu, Q. Yang, Q. Ge, Z. Lu, Consistent isomiR expressionpatterns and 3′ addition events in miRNA gene clusters and families implicatefunctional and evolutionary relationships, Mol. Biol. Rep. 39 (2012) 6699–6706.
[40] R. Schwab, J.F. Palatnik, M. Riester, C. Schommer, M. Schmid, D. Weigel, Specificeffects of microRNAs on the plant transcriptome, Dev. Cell 8 (2005) 517–527.
[41] B. Zhang, X. Pan, E.J. Stellwag, Identification of soybean microRNAs and theirtargets, Planta 229 (2008) 161–182.
[42] G. Szittya, S. Moxon, D.M. Santos, R. Jing, M.P.S. Fevereiro, V. Moulton, T. Dalmay,High-throughput sequencing of Medicago truncatula short RNAs identifies eightnew miRNA families, BMC Genom. 9 (2008) 593.
[43] C.-Z. Zhao, H. Xia, T.P. Frazier, Y.-Y. Yao, Y.-P. Bi, A.-Q. Li, M.-J. Li, C.-S. Li, B.-H.Zhang, X.-J. Wang, Deep sequencing identifies novel and conserved microRNAsin peanuts (Arachis hypogaea L.), BMC Plant Biol. 10 (2010) 3.
[44] Q.-H. Zhu, A. Spriggs, L. Matthew, L. Fan, G. Kennedy, F. Gubler, C. Helliwell,A diverse set of microRNAs and microRNA-like small RNAs in developing ricegrains, Genome Res. 18 (2008) 1456–1465.
[45] H. Vaucheret, Post-transcriptional small RNA pathways in plants: mechanismsand regulations, Genes Develop. 20 (2006) 759–771.
[46] R. Rajagopalan, H. Vaucheret, J. Trejo, D.P. Bartel, A diverse and evolutionar-ily fluid set of microRNAs in Arabidopsis thaliana, Genes Develop. 20 (2006)3407–3425.
[47] A.J. Herr, Pathways through the small RNA world of plants, FEBS Lett. 579 (2005)5879–5888.
[48] F. Vazquez, Arabidopsis endogenous small RNAs: highways and byways, TrendsPlant Sci. 11 (2006) 460–468.
[49] F.C. Favoreto, C.R. Carvalho, A.B.P. Lima, A. Ferreira, W.R. Clarindo, Genome sizeand base composition of Bromeliaceae species assessed by flow cytometry,Plant Syst. Evol. 298 (2012) 1185–1193.
[50] D. Gonzalez-Ibeas, J. Blanca, L. Donaire, M. Saladié, A. Mascarell-Creus, A. Cano-Delgado, J. Garcia-Mas, C. Llave, M.A. Aranda, Analysis of the melon (Cucumismelo) small RNAome by high-throughput pyrosequencing, BMC Genom. 12(2011) 393.
[51] J.R. Puzey, A. Karger, M. Axtell, E.M. Kramer, Deep annotation of Popu-lus trichocarpa microRNAs from diverse tissue sets, PLoS ONE 7 (2012)e33034.
[52] S. Lv, X. Nie, L. Wang, X. Du, S.S. Biradar, X. Jia, S. Weining, Identificationand Characterization of MicroRNAs from Barley (Hordeum vulgare L.) by High-Throughput Sequencing, Int. J. Mol. Sci. 13 (2012) 2973–2984.
[53] N. Fahlgren, M.D. Howell, K.D. Kasschau, E.J. Chapman, C.M. Sullivan, J.S.Cumbie, S.A. Givan, T.F. Law, S.R. Grant, J.L. Dangl, J.C. Carrington, High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birthand death of MIRNA genes, PLoS ONE 2 (2007) e219.
[54] G. Martínez, J. Forment, C. Llave, V. Pallás, G. Gómez, High-throughput sequenc-ing, characterization and detection of new and conserved cucumber miRNAs,PLoS ONE 6 (2011) e19523.
[55] C. Wang, X. Wang, N.K. Kibet, C. Song, C. Zhang, X. Li, J. Han, J. Fang, Deepsequencing of grapevine flower and berry short RNA library for discovery ofnovel microRNAs and validation of precise sequences of grapevine microRNAsdeposited in miRBase, Physiol. Plant. 143 (2011) 64–81.
[56] V. Pantaleo, G. Szittya, S. Moxon, L. Miozzi, V. Moulton, T. Dalmay, J. Burgyan,Identification of grapevine microRNAs and their targets using high-throughputsequencing and degradome analysis, Plant J. Cell. Mol. Biol. 62 (2010)960–976.
[57] D. De Paola, F. Cattonaro, D. Pignone, G. Sonnante, The miRNAome of globeartichoke: conserved and novel micro RNAs and target analysis, BMC Genom.13 (2012) 41.
[58] M. Colaiacovo, A. Subacchi, P. Bagnaresi, A. Lamontanara, L. Cattivelli, P. Fac-cioli, A computational-based update on microRNAs and their targets in barley(Hordeum vulgare L.), BMC Genom. 11 (2010) 595.
[59] G. Wu, M.Y. Park, S.R. Conway, J.-W. Wang, D. Weigel, R.S. Poethig, Thesequential action of miR156 and miR172 regulates developmental timing inArabidopsis, Cell 138 (2009) 750–759.
[60] M. Shikata, T. Koyama, N. Mitsuda, M. Ohme-Takagi, Arabidopsis SBP-box genesSPL10, SPL11 and SPL2 control morphological change in association with shootmaturation in the reproductive phase, Plant Cell Physiol. 50 (2009) 2133–2145.
[61] M.-F. Wu, Q. Tian, J.W. Reed, Arabidopsis microRNA167 controls patterns ofARF6 and ARF8 expression, and regulates both female and male reproduction,Development (Cambr., Engl.) 133 (2006) 4211–4218.
[62] J.H. Yang, S.J. Han, E.K. Yoon, W.S. Lee, Evidence of an auxin signal pathway,microRNA167-ARF8-GH3, and its response to exogenous auxin in cultured ricecells, Nucleic Acids Res. 34 (2006) 1892–1899.
[63] S. Fujii, I. Small, The evolution of RNA editing and pentatricopeptide repeatgenes, New Phytol. 191 (2011) 37–47.
[64] T. Eulgem, P.J. Rushton, S. Robatzek, I.E. Somssich, The WRKY superfamily ofplant transcription factors, Trends Plant Sci. 5 (2000) 199–206.
[65] H. Banno, Y. Ikeda, Q.W. Niu, N.H. Chua, Overexpression of Arabidopsis ESR1induces initiation of shoot regeneration, Plant Cell 13 (2001) 2609–2619.

Author's personal copy
F. Guzman et al. / Plant Science 210 (2013) 214– 223 223
[66] T. Kirch, R. Simon, M. Grunewald, W. Werr, The Dornroschen/Enhancer of shootregeneration1 gene of Arabidopsis acts in the control of meristem cell fate andlateral organ development, Plant Cell 15 (2003) 694–705.
[67] C.A. Airoldi, F.D. Rovere, G. Falasca, G. Marino, M. Kooiker, M.M. Altamura, S. Cit-terio, M.M. Kater, The Arabidopsis BET bromodomain factor GTE4 is involved in
maintenance of the mitotic cell cycle during plant development, Plant Physiol.152 (2010) 1320–1334.
[68] S. Zhang, Y. Yue, L. Sheng, Y. Wu, G. Fan, A. Li, X. Hu, M. ShangGuan, C. Wei,PASmiR: a literature-curated database for miRNA molecular regulation in plantresponse to abiotic stress, BMC Plant Biol. 13 (2013) 1–8.




















