
Identification of a premature stop codon mutation in the PHGDH gene in severe Neu-Laxova...
7
RESEARCH ARTICLE Identification of a Premature Stop Codon Mutation in the PHGDH Gene in Severe Neu-Laxova Syndrome—Evidence for Phenotypic Variability Eduardo P. Mattos, 1,4 Andre ´ Anjos da Silva, 1,4 Jose ´ Anto ˆnio A Magalha ˜es, 2,5 Ju ´lio Ce ´sar L. Leite, 1 Sandra Leistner-Segal, 1 Rejane Gus-Kessler, 1 Juliano Adams Perez, 3 Leonardo M. Vedolin, 3,5 Albertina Torreblanca-Zanca, 6,7 Pablo Lapunzina, 6,8 Victor L. Ruiz-Perez, 7,8 and Maria Teresa V. Sanseverino 1 * 1 Medical Genetics Service, Hospital de Clı ´nicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil 2 Fetal Medicine Group, Hospital de Clı ´nicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil 3 Radiology Service, Hospital de Clı ´nicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil 4 Department of Genetics, Federal University of Rio Grande do Sul, Porto Alegre, Rio Grande do Sul, Brazil 5 Internal Medicine Department, Federal University of Rio Grande do Sul, Porto Alegre, Rio Grande do Sul, Brazil 6 INGEMM, Instituto de Gene ´tica Me ´dica Y Molecular, Hospital Universitario La Paz, Madrid, Spain 7 Instituto de Investigaciones Biome ´dicas, Consejo Superior de Investigaciones Cientı ´ficas-Universidad Auto ´noma de Madrid, Madrid, Spain 8 Centro de Investigacio ´n Biome ´dica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III (ISCIII), Madrid, Spain Manuscript Received: 30 September 2014; Manuscript Accepted: 21 November 2014 In some cases Neu-Laxova syndrome (NLS) is linked to serine deficiency due to mutations in the phosphoglycerate dehydro- genase (PHGDH) gene. We describe the prenatal and postnatal findings in a fetus with one of the most severe NLS phenotypes described so far, caused by a homozygous nonsense mutation of PHGDH. Serial ultrasound (US) and pre- and postnatal magnetic resonance imaging (MRI) evaluations were performed. Prena- tally, serial US evaluations suggested symmetric growth restric- tion, microcephaly, hypoplasia of the cerebellar vermis, micrognathia, hydrops, shortened limbs, arthrogryposis, and talipes equinovarus. The prenatal MRI confirmed these findings prompting a diagnosis of NLS. After birth, radiological imaging did not detect any gross bone abnormalities. DNA was extracted from fetal and parental peripheral blood, all coding exons of PHGDH were PCR-amplified and subjected to Sanger sequenc- ing. Sequencing of PHGDH identified a homozygous premature stop codon mutation (c.1297C>T; p.Gln433*) in fetal DNA, both parents (first-cousins) being heterozygotes. Based on previous associations of mutations in this gene with a milder NLS pheno- type, as well as cases of serine deficiency, these observations lend further support to a genotype-phenotype correlation between the degree of PHGDH inactivation and disease severity. Ó 2015 Wiley Periodicals, Inc. Key words: Neu-Laxova syndrome; prenatal diagnosis; mag- netic resonance imaging; phosphoglycerate dehydrogenase; serine metabolism Conflict of interest: none Grant sponsor: Spanish Ministry of Science and Innovation; Grant number: SAF2013-43365-R; Grant sponsor: The CIBERER Program of Research on Pediatric diseases (ACCI 2012); Grant sponsor: The Brazilian National Council for Research and Development (CNPq); Grant sponsor: Hospital de Clı ´nicas de Porto Alegre (FIPE-HCPA). Correspondence to: Maria Teresa Vieira Sanseverino, Servic ¸o de Gene ´tica Me ´dica, Hospital de Clı´nicas de Porto Alegre, Rua Ramiro Barcelos, 2350, Porto Alegre, RS, Brazil CEP 90035-903. E-mail: [email protected] Article first published online in Wiley Online Library (wileyonlinelibrary.com): 00 Month 2014 DOI 10.1002/ajmg.a.36930 How to Cite this Article: Mattos EP, Silva AA, Magalha ˜es JAA, Leite JCL, Segal SL, Gus-Kessler R, Perez JA, Vedolin LM, Torreblanca-Zanca A, Lapunzina P, Ruiz-Perez VL, Sanseverino MTV. 2015. Identification of a premature stop codon mutation in the PHGDH gene in severe Neu-Laxova syndrome—Evidence for phenotypic variability. Am J Med Genet Part A. 9999A:1–7. Ó 2015 Wiley Periodicals, Inc. 1
Transcript of Identification of a premature stop codon mutation in the PHGDH gene in severe Neu-Laxova...

�
RESEARCH ARTICLE
Identification of a Premature Stop Codon Mutationin the PHGDH Gene in Severe Neu-LaxovaSyndrome—Evidence for Phenotypic Variability
Eduardo P. Mattos,1,4 Andre Anjos da Silva,1,4 Jose Antonio A Magalhaes,2,5 Julio Cesar L. Leite,1Sandra Leistner-Segal,1 Rejane Gus-Kessler,1 Juliano Adams Perez,3 Leonardo M. Vedolin,3,5
Albertina Torreblanca-Zanca,6,7 Pablo Lapunzina,6,8 Victor L. Ruiz-Perez,7,8
and Maria Teresa V. Sanseverino1*1Medical Genetics Service, Hospital de Clınicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil2Fetal Medicine Group, Hospital de Clınicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil3Radiology Service, Hospital de Clınicas de Porto Alegre, Porto Alegre, Rio Grande do Sul, Brazil4Department of Genetics, Federal University of Rio Grande do Sul, Porto Alegre, Rio Grande do Sul, Brazil5Internal Medicine Department, Federal University of Rio Grande do Sul, Porto Alegre, Rio Grande do Sul, Brazil6INGEMM, Instituto de Genetica Medica Y Molecular, Hospital Universitario La Paz, Madrid, Spain7Instituto de Investigaciones Biomedicas, Consejo Superior de Investigaciones Cientıficas-Universidad Autonoma de Madrid, Madrid, Spain8Centro de Investigacion Biomedica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III (ISCIII), Madrid, Spain
Manuscript Received: 30 September 2014; Manuscript Accepted: 21 Novemb
er 2014Conflict of interest: none
Grant sponsor: Spanish Ministry of Science and Innovation;
Grant number: SAF2013-43365-R; Grant sponsor: The CIBERER
Program of Research on Pediatric diseases (ACCI 2012);
Grant sponsor: The Brazilian National Council for Research and
Development (CNPq); Grant sponsor: Hospital de Clınicas de Porto
Alegre (FIPE-HCPA).�Correspondence to:
Maria Teresa Vieira Sanseverino, Servico de Genetica Medica, Hospital
de Clınicas de Porto Alegre, Rua Ramiro Barcelos, 2350, Porto Alegre,
RS, Brazil CEP 90035-903.
E-mail: [email protected]
Article first published online in Wiley Online Library
How to Cite this Article:Mattos EP, Silva AA, Magalhaes JAA, Leite
JCL, Segal SL, Gus-Kessler R, Perez JA,
Vedolin LM, Torreblanca-Zanca A,
Lapunzina P, Ruiz-Perez VL, Sanseverino
MTV. 2015. Identification of a premature
stop codon mutation in the PHGDH gene
in severe Neu-Laxova syndrome—Evidence
for phenotypic variability.
Am J Med Genet Part A. 9999A:1–7.
In some cases Neu-Laxova syndrome (NLS) is linked to serine
deficiency due to mutations in the phosphoglycerate dehydro-
genase (PHGDH) gene. We describe the prenatal and postnatal
findings in a fetus with one of the most severe NLS phenotypes
described so far, caused by a homozygous nonsense mutation of
PHGDH. Serial ultrasound (US) andpre- andpostnatalmagnetic
resonance imaging (MRI) evaluations were performed. Prena-
tally, serial US evaluations suggested symmetric growth restric-
tion, microcephaly, hypoplasia of the cerebellar vermis,
micrognathia, hydrops, shortened limbs, arthrogryposis, and
talipes equinovarus. The prenatal MRI confirmed these findings
prompting a diagnosis of NLS. After birth, radiological imaging
did not detect any gross bone abnormalities. DNAwas extracted
from fetal and parental peripheral blood, all coding exons of
PHGDH were PCR-amplified and subjected to Sanger sequenc-
ing. Sequencing of PHGDH identified a homozygous premature
stop codonmutation (c.1297C>T; p.Gln433*) in fetalDNA, both
parents (first-cousins) being heterozygotes. Based on previous
associations of mutations in this gene with a milder NLS pheno-
type, as well as cases of serine deficiency, these observations lend
further support to a genotype-phenotype correlation between
the degree of PHGDH inactivation and disease severity.
� 2015 Wiley Periodicals, Inc.
Key words: Neu-Laxova syndrome; prenatal diagnosis; mag-
netic resonance imaging; phosphoglycerate dehydrogenase; serine
metabolism
(wileyonlinelibrary.com): 00 Month 2014
DOI 10.1002/ajmg.a.36930
2015 Wiley Periodicals, Inc. 1
2 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
INTRODUCTION
Neu-Laxova syndrome (NLS) is a lethal genetic condition charac-
terized by multiple congenital anomalies and ectodermal abnor-
malities [Neu et al., 1971; Laxova et al., 1972]. The diagnostic
findings of NLS are characteristic and include intrauterine growth
restrictionwith reduced fetalmobility and arthrogryposis, ichthyo-
sis, microcephaly, short neck, and hypoplastic lungs [Manar &
Asma, 2010]. Other neurologic and facial findings are suggestive of
a NLS diagnosis, such as lissencephaly, cerebellar hypoplasia,
corpus callosum abnormalities, severe proptosis with ectropion
and hypertelorism, micrognathia, flat nose and malformed ears.
Most NLS patients die shortly after birth [Manning et al., 2004].
Cerebro-oculo-facio-skeletal, Miller-Dieker, Roberts, Refsum,
Pena-Shokeir and Smith-Lemli-Opitz syndromes, congenital ich-
thyosis and some chromosomal abnormalities (trisomy 12 and 4p
deletion) should be considered in the differential diagnosis.
As it is the case for most rare genetic disorders, there are no
reliable prevalence estimates for NLS; however, it appears to occur
more commonly among inbred populations, and at least a dozen
families with multiple gestations affected by NLS have been de-
scribed [Neu et al., 1971; Laxova et al., 1972; Shved et al., 1985;
Ejeckam et al., 1986; Karimi-Nejad et al., 1987; Tolmie et al., 1987;
Ostrovskaya & Lazjuk, 1988; Naveed et al., 1990; Abdel Meguid &
Temtamy, 1991; Rode et al., 2001; Acuna-Hidalgo et al., 2014;
Shaheen et al., 2014]. Accordingly, autosomal recessive inheritance
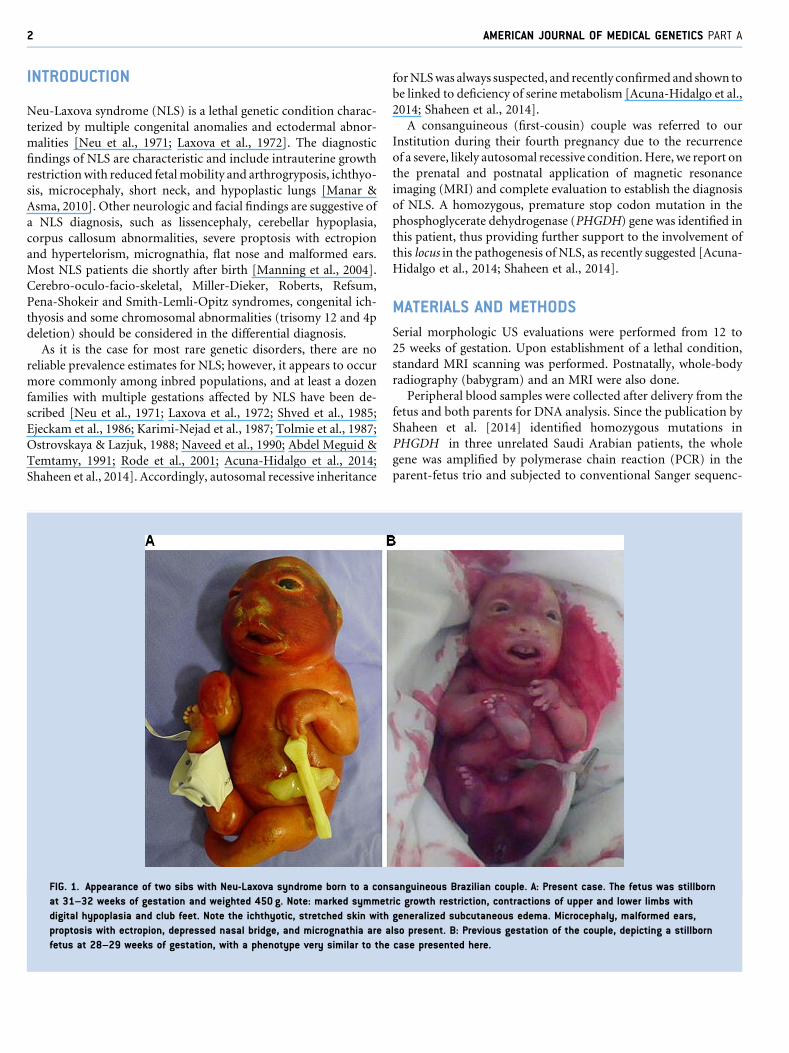
FIG. 1. Appearance of two sibs with Neu-Laxova syndrome born to a con
at 31–32 weeks of gestation and weighted 450 g. Note: marked symmet
digital hypoplasia and club feet. Note the ichthyotic, stretched skin with
proptosis with ectropion, depressed nasal bridge, and micrognathia are a
fetus at 28–29 weeks of gestation, with a phenotype very similar to the
forNLSwas always suspected, and recently confirmed and shown to
be linked to deficiency of serine metabolism [Acuna-Hidalgo et al.,
2014; Shaheen et al., 2014].
A consanguineous (first-cousin) couple was referred to our
Institution during their fourth pregnancy due to the recurrence
of a severe, likely autosomal recessive condition.Here, we report on
the prenatal and postnatal application of magnetic resonance
imaging (MRI) and complete evaluation to establish the diagnosis
of NLS. A homozygous, premature stop codon mutation in the
phosphoglycerate dehydrogenase (PHGDH) gene was identified in
this patient, thus providing further support to the involvement of
this locus in the pathogenesis of NLS, as recently suggested [Acuna-
Hidalgo et al., 2014; Shaheen et al., 2014].
MATERIALS AND METHODS
Serial morphologic US evaluations were performed from 12 to
25 weeks of gestation. Upon establishment of a lethal condition,
standard MRI scanning was performed. Postnatally, whole-body
radiography (babygram) and an MRI were also done.
Peripheral blood samples were collected after delivery from the
fetus and both parents for DNA analysis. Since the publication by
Shaheen et al. [2014] identified homozygous mutations in
PHGDH in three unrelated Saudi Arabian patients, the whole
gene was amplified by polymerase chain reaction (PCR) in the
parent-fetus trio and subjected to conventional Sanger sequenc-
sanguineous Brazilian couple. A: Present case. The fetus was stillborn
ric growth restriction, contractions of upper and lower limbs with
generalized subcutaneous edema. Microcephaly, malformed ears,
lso present. B: Previous gestation of the couple, depicting a stillborn
case presented here.

FIG. 2. Prenatal magnetic resonance imaging of the fetus at 25–26 weeks of gestation confirms the ultrasonographic diagnostic hypothesis
of Neu-Laxova syndrome. A: Fetal profile shows proportionate growth restriction and arthrogryposis with contracted upper limbs and club feet
(RA: right arm; RF: right foot; LF: left foot). B: Sagittal section of the fetus shows microcephaly, micrognathia, and a hypoplastic cerebellum,
but no neural tube defects. C and D: Coronal sections of the fetus depicting a hypersignal with thickening of the cranial subcutaneous
tissues, absence of the corpus callosum and lissencephaly.
MATTOS ET AL. 3
ing (primers and PCR conditions available upon request). The
nomenclature and position of the identified mutation is in
accordance to the PHGDH reference messenger RNA sequence
NM_006623.3, obtained from the National Center for Biotech-
nology Information. Functional prediction of the identified
mutation was performed using the MutationTaster algorithm
[Schwarz et al., 2010].
Informed consent for the anonymous disclosure of patient data
and pictures was obtained from the family reported here.
RESULTS
A consanguineous couple (first-degree cousins; 29-year-old moth-
er and 43-year-old father) was referred for evaluation during their
fourth pregnancy due to detection of a fetus with multiple anoma-
lies with familial recurrence. Their first pregnancy ended in spon-
taneous abortion at 8–9 weeks of gestation, while the second
pregnancy gave rise to a healthy daughter, who is now 8 years
old. In the third gestation, the fetus presented with severe growth
restriction and multiple abnormalities. Preterm labor occurred at
28 weeks of gestation and the baby died shortly after birth, but,
unfortunately, the patient was not further evaluated and remained
undiagnosed. Retrospectively, clinical pictures taken by the father
were reviewed and compared to the prospective case, fitting the
diagnosis of NLS (Fig. 1).
In the present case, an ultrasound (US) evaluation at 19 weeks of
gestation detected symmetric growth restriction, hypoplasia of the
cerebellar vermis, micrognathia, hydrops, possible spina bifida,
shortened limbs, articular rigidity, and club feet. These abnormali-
ties were confirmed in a subsequent US examination at 22 weeks of
gestation. Supplementary Table I shows the body measurements
obtained in the sequential US investigations, evidencing marked
growth retardation with several data points falling below the first
centile for gestational age.
Amniocentesis showed a normal female chromosome constitu-
tion (46, XX). Considering the severity of the case, magnetic
resonance imaging (MRI) was elected at 25–26 weeks of gestation
in an attempt to better characterize the abnormal findings evi-
denced by the US evaluations (Fig. 2). It showed marked fetal
growth restriction, microcephaly, a flat nose, micrognathia, talipes
equinovarus, and arthrogryposis of the upper limbs. There was also
a hypersignal with thickening of cranial subcutaneous tissues, but
no evidence of myelomeningocele. Collectively, these findings led
to the prenatal diagnostic hypothesis of NLS, confirmed
postnatally.
Preterm labor followed by spontaneous delivery occurred at 31–
32weeks of gestation. The stillborn infantweighted 450 g (below1st
centile), and had anthropometric measures compatible with 22–24
weeks of gestation, severe growth restriction, along with contrac-
tures of upper and lower limbswithhypoplasiaof digits, “stretched”
skin with mild ichthyosis and generalized subcutaneous edema,
short neck, scoliosis, club feet, and female external genitalia (Fig. 1).
Craniofacial findings included microcephaly, hypertelorism and
malformed ears, proptosis with ectropion, depressed nasal bridge,
micrognathia, and complete posterior cleft palate. An autopsy was
performed and internal organ malformations consisted mainly of
pulmonary, thymic, and left ventricle hypoplasia. Skeletal evalua-
tion by radiography showed fusion of the bones of the cranial vault
and scoliosis; no signs of other major malformation or dysplasia
were detected (Fig. 3).
Postnatal MRI (Fig. 4) confirmed the severe involvement of the
cerebellum and agenesis of the corpus callosum, as indicated by the
diagnostic procedures performed inutero. Furthermore, absence of
cerebral gyri and sulci reinforced the prenatal finding of
lissencephaly.
By the time this study was initiated, there were yet no genetic loci
consistently associated to NLS, which led us to consider exome
sequencing of the parent-offspring trio, in an attempt to identify
FIG. 3. Whole-body radiography of the patient in antero-posterior (A) and lateral (B) views, showing scoliosis but no additional gross
abnormalities.
4 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
potentially pathogenic coding variants in the fetus. However, the
data subsequently published by Shaheen et al. [2014] prompted us
to focus the analysis on the PHGDH gene [Shaheen et al., 2014].
Indeed, a potentially pathogenic, homozygous single-nucleotide
substitution (c.1297C>T) was identified in the fetal DNA, both
parents being heterozygous (Fig. 5A and B). Structurally, this
mutation changes glutamine residue 433 for a premature stop
codon (p.Gln433*) at the C-terminal regulatory domain of
PHGDG, anticipating the deletion of the last 101 amino acids
(Fig. 5C). Although no functional studies were performed, this
mutation was predicted to behave as a null allele, preventing the
expression of PHGDH from both alleles due to surveillance by the
mRNA nonsense-mediated decay system.
DISCUSSION
Although NLS has been recognized as a distinct genetic entity for
over 40 years, its molecular basis remained elusive until recently,
when Shaheen et al. performed autozygosity mapping and gene
sequencing in three NLS patients from distinct consanguineous
Saudi Arabian families [Shaheen et al., 2014]. Prior to this study,
some authors suggested that NLS might belong to the neuro-
ectodermal dysplasia disease group [Lazjuk et al., 1979; Ejeckam
et al., 1986; Naveed et al., 1990; Bronshtein et al., 1993]. Others
hypothesized defects in one or more genes on the 6q and/or 9p
regions, based onmousemodels of restrictive dermopathies, which
resemble the “cocoon”-like aspect of the fetus [Manning et al.,
2004].Hence, the associationof thePHGDHlocuswithNLScameas
a surprise [Shaheen et al., 2014], since coding variants in this gene
were already known to cause PHGDH deficiency, a less severe
condition characterized by delayed development, microcephaly,
seizures and growth retardation [Jaeken et al., 1996; Van der
Crabben et al., 2013]. Interestingly, the conceptual link between
phenotype and an inborn error of metabolism in NLS was estab-
lished more than two decades after its initial proposal by Shapiro
et al. [1992], who argued that the abnormal fat deposits, the

FIG. 4. Postnatal magnetic resonance imaging of the fetus after delivery (31–32 weeks of gestation) confirms the prenatal detection of central nervous
system abnormalities. Coronal (A), sagittal (B), and transverse (C) sections of the fetus illustrate cerebellar hypoplasia and generalized lissencephaly.
MATTOS ET AL. 5
ichthyotic skin, and the eventual development of cataract seen in
NLS should relate to an underlyingmetabolic defect [Shapiro et al.,
1992].
Classification of NLS phenotypes was proposed by Curry
[1982], who divided NLS into three subgroups according to
FIG. 5. Molecular analysis identifies a homozygous premature termination c
Pedigree of the reported family. B: Chromatograms illustrating the sequencin
parents, who are heterozygous carriers for the mutation (C; Individuals I-1 a
PHGDH structure with depicted mutations identified so far in both serine defi
identified in this study is depicted in bold. SB, substrate binding domain; NA
selected clinical findings. Patients of group I would be those
who present mainly with arthrogryposis, partial syndactyly,
squamous skin and bone hypomineralization. Group II would
include patients with severe swelling of hands and feet—also
known as “glove-shaped” limbs—secondary to oedema and fat
odon mutation in the PHGDH gene in a patient with NLS syndrome. A:
g profile at the PHGDH locus of a normal control individual (N), both
nd I-2), and the patient (P; Proband II-4). C: Schematic representation of
ciency (lower notations) and NLS (upper notations). The variation
D(P), NADþ/NADPþ binding domain; RD, regulatory domain.

6 AMERICAN JOURNAL OF MEDICAL GENETICS PART A
deposition, pronounced ichthyosis and poor bone mineraliza-
tion. Patients with the most typical clinical findings of NLS
would be placed in this group [Manning et al., 2004]. Finally,
group III would be reserved for patients with hypoplastic digits,
short limbs, microtia, and severe ichthyosis similar to that
observed in harlequin ichthyosis. It is not known whether these
phenotypic differences are due to variable expressivity of a
single mutated gene, or mutations in different genes.
Besides variable expression in NLS, mutations in the phospho-
serine aminotransferase (PSAT1) and phosphoserine phosphatase
(PSPH) genes have also been reported, supporting the hypothesis of
allelic heterogeneity for NLS. These results also show that NLS
might arise frommutations in at least three different genes involved
in the L-serine biosynthesis pathway, probably reflecting severe
forms ofmilder inborn errors ofmetabolism [Acuna-Hidalgo et al.,
2014; Shaheen et al., 2014].
In light of our results and those presented by Shaheen and
colleagues and Acuna-Hidalgo and colleagues [Acuna-Hidalgo
et al., 2014; Shaheen et al., 2014], we propose that at least part of
the variable clinical presentations of NLS might be explained by
different PHGDH mutations that give rise to a variable of
disease. Accordingly, the NLS-associated variants described by
Shaheen et al., who reported on patients that seem phenotypically
less affected then the one we describe here, are missense sub-
stitutions not entirely different from other PHGDH missense
mutations observed in individuals with the inborn error of serine
metabolism known as PHGDH deficiency [Jaeken et al., 1996;
Shaheen et al., 2014]. Despite missense mutations located in
conserved catalytic residues of the enzyme [Shaheen et al., 2014],
PHGDHmight still have some degree of residual activity in those
cases (although functional studies remain to be conducted to
experimentally confirm this hypothesis). On the other hand, the
patient reported here was shown to encode a homozygous
premature termination codon mutation, which is predicted to
result in complete PHGDH absence, due to the activity of the
cellular nonsense mRNA-mediated decay system [Chang et al.,
2007], which in turn might result in the more severe phenotype
observed here.
In conclusion, we present a clinical characterization of a NLS
patient using MRI evaluations. A homozygous premature stop
codon mutation in the PHGDH gene was identified in the patient
reported here, which corroborates a recent association of this locus
with NLS [Acuna-Hidalgo et al., 2014; Shaheen et al., 2014]. The
identification of additional patients will likely shed light on the
phenotypic and genetic diversity of NLS, leading to a better
understanding of serine-deficiency diseases and its genotype-phe-
notype correlations.
ACKNOWLEDGMENTS
The authors are indebted to the family reported here, for their
support for publication. Part of this work was supported by the
Spanish Ministry of Science and Innovation SAF2013-43365-R,
the CIBERER Program of Research on Pediatric diseases (ACCI
2012), the Brazilian National Council for Research and Devel-
opment (CNPq), and Hospital de Clınicas de Porto Alegre (FIPE-
HCPA).
REFERENCES
Abdel Meguid N, Temtamy SA. 1991. Neu Laxova syndrome in twoEgyptian families. Am J Med Genet 41:30–31.
Acuna-Hidalgo R, Schanze D, Kariminejad A, Nordgren A, KariminejadMH, Conner P, Grigelioniene G, Nilsson D, Nordenskjold M, WedellA, Freyer C, Wredenberg A, Wieczorek D, Gillessen-Kaesbach G,Kayserili H, Elcioglu N, Ghaderi-Sohi S, Goodarzi P, Setayesh H,van de Vorst M, Steehouwer M, Pfundt R, Krabichler B, Curry C,MacKenzie MG, Boycott KM, Gilissen C, Janecke AR, Hoischen A,Zenker M. 2014. Neu-Laxova syndrome is a heterogeneous metabolicdisorder caused by defects in enzymes of the L-serine biosynthesispathway. Am J Hum Genet 95:285–293.
Bronshtein M, Blumenfeld I, Cohen I, Blumenfeld Z. 1993. Fetal ultraso-nographic detection of hypodontia in the Neu-Laxova syndrome. J ClinUltrasound 21:648–650.
Chang Y-F, Imam JS, Wilkinson MF. 2007. The nonsense-mediated decayRNA surveillance pathway. Annu Rev Biochem 76:51–74.
Curry CJ. 1982. Further comments on the Neu-Laxova syndrome. Am JMed Genet 13:441–444.
Ejeckam GG, Wadhwa JK, Williams JP, Lacson AG. 1986. Neu-Laxovasyndrome: report of two cases. Pediatr Pathol 5:295–306.
Jaeken J, Detheux M, Van Maldergem L, Foulon M, Carchon H, VanSchaftingen E. 1996. 3-Phosphoglycerate dehydrogenase deficiency: Aninborn error of serine biosynthesis. Arch Dis Child 74:542–545.
Karimi-Nejad MH, Khajavi H, Gharavi MJ, Karimi-Nejad R. 1987. Neu-Laxova syndrome: Report of a case and comments. Am J Med Genet28:17–23.
Laxova R, Ohara PT, Timothy JA. 1972. A further example of a lethalautosomal recessive condition in sibs. J Ment Defic Res 16:139–143.
Lazjuk GI, Lurie IW, Ostrowskaja TI, Cherstvoy ED, Kirillova IA, NedzvedMK, Usoev SS. 1979. Brief clinical observations: The Neu-Laxova syn-drome—a distinct entity. Am J Med Genet 3:261–267.
Manar A-L, Asma B. 2010. Neu-Laxova syndrome: A new patient withdetailed antenatal and post-natal findings. Am J Med Genet A152A:3193–3196.
Manning MA, Cunniff CM, Colby CE, El-Sayed YY, Hoyme HE. 2004.Neu-Laxova syndrome: Detailed prenatal diagnostic and post-mor-tem findings and literature review. Am J Med Genet A 125A:240–249.
Naveed Manjunath CS, Sreenivas V. 1990. New manifestations of Neu-Laxova syndrome. Am J Med Genet 35:55–59.
Neu RL, Kajii T, Gardner LI, Nagyfy SF. 1971. A lethal syndrome ofmicrocephaly with multiple congenital anomalies in three siblings.Pediatrics 47:610–612.
OstrovskayaTI, LazjukGI. 1988.Cerebral abnormalities in theNeu-Laxovasyndrome. Am J Med Genet 30:747–756.
RodeME,MennutiMT,Giardine RM, Zackai EH,Driscoll DA. 2001. Earlyultrasound diagnosis of Neu-Laxova syndrome. Prenat Diagn 21:575–580.
Schwarz JM, Rodelsperger C, SchuelkeM, SeelowD. 2010.MutationTasterevaluates disease-causing potential of sequence alterations. NatMethods7:575–576.
Shaheen R, Rahbeeni Z, Alhashem A, Faqeih E, Zhao Q, Xiong Y,Almoisheer A, Al-Qattan SM, Almadani HA, Al-Onazi N, Al-BaqawiBS, SalehMA,Alkuraya FS. 2014.Neu-Laxova syndrome, an inborn errorof serine metabolism is caused by mutations in PHGDH. Am J HumGenet 94:898–904.

MATTOS ET AL. 7
Shapiro I, Borochowitz Z, Degani S, Dar H, Ibschitz I, Sharf M. 1992. Neu-Laxova syndrome: prenatal ultrasonographic diagnosis, clinical and patho-logical studies, and new manifestations. Am J Med Genet 43:602–605.
ShvedIA,LazjukGI,CherstvoyED.1985.Elaborationof thephenotypicchangesof the upper limbs in the Neu-Laxova syndrome. Am J Med Genet 20:1–11.
Tolmie JL, Mortimer G, Doyle D, McKenzie R, McLaurin J, Neilson JP. 1987.The Neu-Laxova syndrome in female sibs: Clinical and pathological featureswith prenatal diagnosis in the second sib. Am J Med Genet 27:175–182.
VanderCrabben SN,Verhoeven-DuifNM, Brilstra EH,VanMaldergemL,CoskunT, Rubio-Gozalbo E, Berger R, deKoning TJ. 2013. An update onserine deficiency disorders. J Inherit Metab Dis 36:613–619.
SUPPORTING INFORMATION
Additional supporting information may be found in the online
version of this article at the publisher’s web-site.




















