
Human dopamine transporter gene: coding region conservation among normal, Tourette's disorder,...
10
Molecular Psychiatry (2000) 5, 283–292 2000 Macmillan Publishers Ltd All rights reserved 1359-4184/00 $15.00 www.nature.com/mp ORIGINAL RESEARCH ARTICLE Human dopamine transporter gene: coding region conservation among normal, Tourette’s disorder, alcohol dependence and attention-deficit hyperactivity disorder populations DJ Vandenbergh 1,2 , MD Thompson 3 , EH Cook 4 , E Bendahhou 1 , T Nguyen 3 , MD Krasowski 4 , D Zarrabian 3 , D Comings 5 , EM Sellers 6,7 , RF Tyndale 6 , SR George 6 , BF O’Dowd 3 and GR Uhl 1 1 Molecular Neurobiology Branch, National Institute on Drug Abuse, NIH; 3 Department of Pharmacology, University of Toronto, Toronto, Ontario M5S 2S1, Canada, 4 Laboratory of Developmental Neuroscience, Department of Psychiatry, University of Chicago, Chicago; 5 Department of Medical Genetics, City of Hope Medical Center, Duarte, California; 6 Centre for Addiction and Mental Health, and Departments of Pharmacology, Psychiatry and Medicine; 7 Sunnybrook and Women’s College Health Science Centre, University of Toronto, Toronto, Ontario, Canada The dopamine transporter (DAT) provides major regulation of the synaptic levels of dopamine and is a principal target of psychostimulant drugs. Associations between DAT gene polymor- phisms and human disorders with possible links to dopaminergic neurotransmission, includ- ing attention-deficit/hyperactivity disorder (ADHD) and consequences of cocaine and alcohol administration, have been reported. We now report approximately 60 000 bp of genomic sequence containing the entire DAT gene. This sequence was used to amplify each of the 15 DAT gene exons and several introns and analyze these amplification products by single- stranded sequence conformation (SSCP) and/or direct sequencing. These results define silent allelic single nucleotide sequence variants in DAT gene exons 2, 6, 9 and 15. Rare conserva- tive mutations are identified in amino acids encoded by DAT exons 2 and 8. Analyses of the common nucleotide variants and the previously reported VNTR in the non-coding region of exon 15 define the pattern of linkage disequilibrium across the DAT locus. These comprehen- sive analyses, however, fail to identify any common protein coding DAT sequence variant in more than 150 unrelated individuals free of neuropsychiatric disease, 109 individuals meeting City of Hope criteria for Tourette’s syndrome, 64 individuals with DSM-IV diagnoses of ethanol dependence, or 15 individuals with ADHD. These data are consistent with substantial evol- utionary conservation of the DAT protein sequence. They suggest that gene variants that alter levels of DAT expression provide the best current candidate mechanism for reported associations between DAT gene markers, ADHD and other more tentatively associated neuro- psychiatric disorders. Molecular Psychiatry (2000) 5, 283–292. Keywords: polymorphism; chromosome 5p15.3; sequence; transmission disequilibrium test; psychi- atric disorder Introduction The human dopamine transporter (DAT) gene (SLC6A3) encodes the member of the neurotransmitter transporter gene family 1 that reaccumulates the neuro- transmitter dopamine into presynaptic terminals. This Correspondence: Dr GR Uhl, Molecular Neurobiology, NIDA-IRP, 5500 Nathan Shock Dr, Baltimore, MD 21224, USA. E-mail: guhlKintra.nida.nih.gov Current NIDA policy prevents Dr Uhl from listing his non- NIDA affiliations. 2 Present address: Center for Development and Health Genetics and Department of Biobehavioral Health, The Pennsylvania State University, 101 Amy Gardner House, University Park, PA 16802, USA Received 21 June 1999; revised 17 August 1999 and 3 September 1999; accepted 3 September 1999 transporter is the primary regulator of the time course and synaptic concentration of released dopamine, which is a key neurotransmitter for regulation of mood and movement. Several lines of evidence suggest that variants of the dopamine transporter gene could play important roles in a number of neuropsychiatric disorders. Cocaine and other psychostimulants block DAT function; DAT has been postulated to be a key site for psychostimul- ant reward. 2 The ability of virtually all abused sub- stances to enhance dopamine release makes DAT a candidate contributor to vulnerability to many classes of abused substances. Other disorders with docu- mented dopaminergic involvement in pathogenesis and/or treatment include Parkinson’s disease, schizo- phrenia, attention deficit hyperactivity disorder (ADHD) and Tourette’s syndrome. 3–5 Dopamine trans-
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Human dopamine transporter gene: coding region conservation among normal, Tourette's disorder,...

Molecular Psychiatry (2000) 5, 283–292 2000 Macmillan Publishers Ltd All rights reserved 1359-4184/00 $15.00
www.nature.com/mp
ORIGINAL RESEARCH ARTICLE
Human dopamine transporter gene: coding regionconservation among normal, Tourette’s disorder, alcoholdependence and attention-deficit hyperactivity disorderpopulationsDJ Vandenbergh1,2, MD Thompson3, EH Cook4, E Bendahhou1, T Nguyen3, MD Krasowski4,D Zarrabian3, D Comings5, EM Sellers6,7, RF Tyndale6, SR George6, BF O’Dowd3 and GR Uhl1
1Molecular Neurobiology Branch, National Institute on Drug Abuse, NIH; 3Department of Pharmacology, University ofToronto, Toronto, Ontario M5S 2S1, Canada, 4Laboratory of Developmental Neuroscience, Department of Psychiatry,University of Chicago, Chicago; 5Department of Medical Genetics, City of Hope Medical Center, Duarte, California;6Centre for Addiction and Mental Health, and Departments of Pharmacology, Psychiatry and Medicine; 7Sunnybrook andWomen’s College Health Science Centre, University of Toronto, Toronto, Ontario, Canada
The dopamine transporter (DAT) provides major regulation of the synaptic levels of dopamineand is a principal target of psychostimulant drugs. Associations between DAT gene polymor-phisms and human disorders with possible links to dopaminergic neurotransmission, includ-ing attention-deficit/hyperactivity disorder (ADHD) and consequences of cocaine and alcoholadministration, have been reported. We now report approximately 60000 bp of genomicsequence containing the entire DAT gene. This sequence was used to amplify each of the 15DAT gene exons and several introns and analyze these amplification products by single-stranded sequence conformation (SSCP) and/or direct sequencing. These results define silentallelic single nucleotide sequence variants in DAT gene exons 2, 6, 9 and 15. Rare conserva-tive mutations are identified in amino acids encoded by DAT exons 2 and 8. Analyses of thecommon nucleotide variants and the previously reported VNTR in the non-coding region ofexon 15 define the pattern of linkage disequilibrium across the DAT locus. These comprehen-sive analyses, however, fail to identify any common protein coding DAT sequence variant inmore than 150 unrelated individuals free of neuropsychiatric disease, 109 individuals meetingCity of Hope criteria for Tourette’s syndrome, 64 individuals with DSM-IV diagnoses of ethanoldependence, or 15 individuals with ADHD. These data are consistent with substantial evol-utionary conservation of the DAT protein sequence. They suggest that gene variants thatalter levels of DAT expression provide the best current candidate mechanism for reportedassociations between DAT gene markers, ADHD and other more tentatively associated neuro-psychiatric disorders. Molecular Psychiatry (2000) 5, 283–292.
Keywords: polymorphism; chromosome 5p15.3; sequence; transmission disequilibrium test; psychi-atric disorder
Introduction
The human dopamine transporter (DAT) gene(SLC6A3) encodes the member of the neurotransmittertransporter gene family1 that reaccumulates the neuro-transmitter dopamine into presynaptic terminals. This
Correspondence: Dr GR Uhl, Molecular Neurobiology, NIDA-IRP,5500 Nathan Shock Dr, Baltimore, MD 21224, USA. E-mail:guhlKintra.nida.nih.govCurrent NIDA policy prevents Dr Uhl from listing his non-NIDA affiliations.2Present address: Center for Development and Health Geneticsand Department of Biobehavioral Health, The Pennsylvania StateUniversity, 101 Amy Gardner House, University Park, PA16802, USAReceived 21 June 1999; revised 17 August 1999 and 3 September1999; accepted 3 September 1999
transporter is the primary regulator of the time courseand synaptic concentration of released dopamine,which is a key neurotransmitter for regulation of moodand movement.
Several lines of evidence suggest that variants of thedopamine transporter gene could play important rolesin a number of neuropsychiatric disorders. Cocaineand other psychostimulants block DAT function; DAThas been postulated to be a key site for psychostimul-ant reward.2 The ability of virtually all abused sub-stances to enhance dopamine release makes DAT acandidate contributor to vulnerability to many classesof abused substances. Other disorders with docu-mented dopaminergic involvement in pathogenesisand/or treatment include Parkinson’s disease, schizo-phrenia, attention deficit hyperactivity disorder(ADHD) and Tourette’s syndrome.3–5 Dopamine trans-

DAT polymorphismsDJ Vandenbergh et al
284
Molecular Psychiatry
porter inhibitors, including psychostimulants and bup-ropion, are currently among the most efficacious treat-ments in ADHD. In vitro studies of DAT site-directedmutants document that many single amino acid DATsubstitutions selectively alter aspects of DAT functionlikely to be important for its selective properties.6
Transgenic mice with altered DAT expression, bothknockouts and transgenic over-expressors, are differen-tially vulnerable to Parkinsonism-inducing neurotox-ins,7 display different locomotor habituation patterns,and reveal differing locomotor responses to psychosti-mulants.8,9 DAT gene sequence variants are thusattractive candidates for contributing to several humandisorders with significant morbidity and mortality.
Previously-described DAT sequence variants havebeen used to analyze the potential genetic contributionof this gene to several of the disorders mentionedabove. An allelic variant marked by a 10-copy variablenumber tandem repeat (VNTR) marker at the 39 end ofthe DAT gene locus has been associated with ADHD inthree independent family-based controlled studies,10–13
athough it could not be found in one associationstudy.14 The 9-copy repeat has been found more fre-quently in unrelated cocaine abusers who report para-noid ideation with cocaine use.15 Suggestive linkagehas been reported between DAT and bipolar disorder.16
More recently, alcohol withdrawal symptoms werefound to be more pronounced in chronically intoxi-cated patients who had the 9-copy DAT allele.17
Association and/or linkage studies of VNTR marker fre-quencies have also been reported in drug abuse,18 alco-hol abuse,19,20 schizophrenia,21,22 ADHD23 and Parkin-son’s disease.24–26 These marker studies, however, donot provide maximal sensitivity for detecting polymor-phisms across the entire DAT locus. More adequateevaluation of the possibility that DAT gene variantscould contribute differences in protein coding requiresadditional DAT genomic sequence information as wellas information about individual variants in theadditional sequences.
Initial work in elucidating human DAT genomicsequences revealed that the first 12 human DAT geneexons were distributed over at least 50 kb of genomicDNA located at chromosome 5p15.3.27 Alignment ofthese partial genomic clones with human DAT cDNAsequences indicated the presence of additional 39exons, including an exon containing the VNTR poly-morphism that lies in the DAT cDNAs 39 untranslated
Table 1 Research volunteers and DAT sequence variation detection
Institution na Study DNA variant detection
NIDA 20 Addiction Direct sequencing of PCR productsU. Chicago 47 ADHD SSCPb and direct sequencing of PCR productsU. Toronto 128 Alcoholism SSCP and sequencing of cloned PCR productsc
U. Toronto/ City of Hope 174 Tourette’s syndrome SSCP and sequencing of cloned PCR products
aNumber of individuals including controls for each group.bSSCP – Single Strand Conformation Polymorphism.cMultiple isolates of PCR products were sequenced independently to minimize cloning artifacts.
region. We now report complete elucidation of the pri-mary sequence of the entire DAT gene and more than8 kb of 59-flanking sequence. Variants in thesesequences have been identified by single-stranded con-formational polymorphism and direct sequencing ofDNA amplified from each DAT genomic exon and fromseveral introns. We have examined both disease popu-lations and individuals free of neuropsychiatric dis-orders.
Materials and methods
DAT exons 13, 14 and 15. Isolation and completesequenceGenomic clones were isolated by standard hybridiz-ation screening of phage libraries (Clontech, Palo Alto,CA, USA) using probes from the 39-end of the hDATcDNA.28 Phage DNA was isolated from plate lysatesusing a phage DNA isolation kit (Stratagene, La Jolla,CA, USA) and subcloned into pBluescript II for sub-sequent analysis. Regions of the gene that were notfound in these plasmids were cloned by long PCRusing the LA PCR kit (TaKaRa, Panvera, Madison, WI,USA). Clones from the 59-flank through exon 12,described in Donovan et al,27 have now beensequenced in their entirety. Automated sequencing ofintrons (single stranded) and exons (double stranded)was performed using ABI sequencers (Applied Biosys-tems, Foster City, CA, USA) at the Johns HopkinsCore Facility.
Research volunteersIndividuals volunteered for inclusion in study groupsunder appropriate informed consent provisions. Racialand ethnic classifications were determined by self-report. Table 1 provides a brief summary of the volun-teers from the three institutions that participated inthis work. No research volunteer from the NationalInstitute of Drug Abuse Intramural Research Programhad any prominent current psychiatric symptoma-tology. None had a Symptomatology Check List (SCL-90) t score more than 70, and high values in any singlecategory excluded potential subjects from partici-pation. Control individuals from Toronto were freefrom psychiatric disorders as determined by the Phar-maco Dependence Health Questionnaire.29
A total of 128 Caucasian controls of western andnorthern European descent were recruited in a study

DAT polymorphismsDJ Vandenbergh et al
285of drug abuse at the National Institute on Drug AbuseIntramural Research Program. Additional control vol-unteers, free from diagnoses of psychiatric disorders,were recruited from an adjacent hemodialysis unit anda public health facility studying HIV infection in Balti-more. For exon sequencing to detect new mutations,individuals were selected on the basis of DAT VNTRallele status (described below under direct sequencescreening for variants) and included some African-Americans who attended a Baltimore area sickle-cellanemia clinic.30
The families of 47 child or adolescent probands withDSM-III-R ADHD31 (Am. Psych. Assoc.) diagnosesmade at the University of Chicago Hyperactivity,Attention, and Learning Problems (HALP) clinicbetween April 1993 and October 1994 formed theADHD sample tested previously for linkage disequilib-rium with the 39-DAT VNTR.10 Fifteen of these individ-uals were chosen based on their having received the39-VNTR 10-copy allele from at least one heterozygousparent. These individuals were screened for polymor-phisms in the DAT gene by both SSCP and bidirec-tional sequencing.
Unrelated Caucasian individuals of non-Hispanic,northern or western European descent were sampledat the Centre for Addiction and Mental Health (CAMH),Toronto, Canada. Sixty-four individuals meeting DSM-III-R diagnoses of alcohol dependence were comparedto 64 matched controls free from alcohol dependenceselected from the CAMH.
Tourette’s clinic patients consisted of consecutive,unrelated individuals of non-Hispanic, northern orwestern European descent. There were 109 probandstreated at the TS clinic of the City of Hope NationalMedical Center (COH). Eighty-two percent met DSMdiagnosis of TS and 18% had other chronic motor orvocal tic disorders. Thirty-five of these volunteersresponded positively to DIS/DSM-III-R questions aboutADHD and 22 to questions about obsessive compulsivedisorder. These results were compared to those of acontrol group consisting of 67 individuals from theCAMH.
All comparisons of unrelated individuals by allelestatus and disorder phenotype were made by x2 analy-ses, with the exception of the ADHD families. ADHDfamilies, who were analyzed by the Transmission Dis-equilibrium Test (TDT)32 were not separated by racesince the TDT provides an internal control for racialstratification, all other subjects were of northern andwestern European descent.
Research volunteer DNABlood samples were obtained by venipuncture follow-ing informed consent. Genomic DNA was isolated byphenol extraction and ethanol precipitation of DNAfrom most blood samples as previously described.30
DNA samples from the University of Chicago were iso-lated without phenol, as previously described.10
Molecular Psychiatry
DAT VNTR and TaqA polymorphism detection byPCR and PCR-RFLPA previously described polymorphism initiallydetected by Southern analyses of TaqI digests28 wasdetected by PCR amplification using the oligonucleo-tide primers HDATTaqAFor (59-CCGTGCCAGCTCCTGCTG-39) and HDATTaqARev (59-GACCTCAGTGGTGTCTGTTGATACGG-39) for 35 cycles of 98°C for10 s, 67°C for 60 s, 72°C for 60 s followed by TaqIdigestion and polyacrylamide gel electrophoresis. Theexpected band sizes are 623 bp for the A1 allele, and423 plus 200 bp for the A2 allele.
The exon 15 VNTR was amplified using the oligo-nucleotide primers T3-5Long (59-TGTGGTGTAGGGAACGGCCTGAG-39) and T7-3aLong (59-CTTCCTGGAGGTCACGGCTCAAGG-39) under con-ditions previously described.33
Intron polymorphismsAlleles of an intron 8 VNTR were detected by PCRamplification of 40 ng of genomic DNA from 21 Cauca-sian and 24 African-American control individualsusing the primers Int8RepFor (59-GCACAAATGAGTGTTCGTGCATGTG-39) and Int8RepRev (59-AGCAGGAGGGGCTTCCAGGC-3) and 35 cycles of98°C for 10 s and 67°C for 60 s, which was initiated bythe addition of Taq polymerase in the first cycle at 82°C(Hot Start). Fragment sizes are 260 bp and 290 bp forthe five- and six-copy alleles respectively, determinedusing 5% polyacrylamide gel electrophoresis.
SSCP analysesSequences of the oligonucleotides used as PCR primersto amplify each DAT exon and intron 14 are listed inTable 2. PCR reactions used 200 ng genomic DNA,50 pmol of each oligonucleotide primer, 300 mM dNTP,1.5 mM MgCl2, 0.5 U Taq DNA polymerase (Life Tech-nologies, Gaithersburg, MD, USA) and 40 cycles of95°C for 30 s, 60–68°C for 40 s, and 72°C for 40 s in aPerkin Elmer GeneAmp 9600. For SSCP analyses con-ducted at the University of Toronto on samples fromthe COH and CAMH, PCR products were diluted 1:4 inSSCP loading buffer (0.04% SDS, 16 mM EDTA, 57%formamide, 0.03% bromophenol blue and 0.03%xylene cyanol) heated at 95°C for 5 min, and rapidlycooled to 0°C for 5 min before electrophoresis usingNovex Xcell II minicells.34,35 Five sets of conditionvariants were used: (a) 400 volts, 10 mA at 2.5 W; (b)100 V-h; (c) 6°C in loading buffer without glycerol; (d)15°C in loading buffer containing glycerol; and (e) 100volts for 18 h at 4°C on 4–20% TBE gradient gels. DNAwas visualized by silver staining. Samples in whichvariants were detected by SSCP were cloned(pBluescript), and multiple isolates were sequencedmanually [USB Sequenase PCR Product Sequencing(Cleveland, OH, USA), or Pharmacia-Biotech T7sequencing (Uppsala, Sweden)] to identify the nucleo-tides responsible for the variations.
SSCP electrophoresis at the University of Chicagowas performed using Phast System 20% polyacryla-mide gels at 6°C without glycerol in the loading buffer,
DAT polymorphismsDJ Vandenbergh et al
286
Molecular Psychiatry
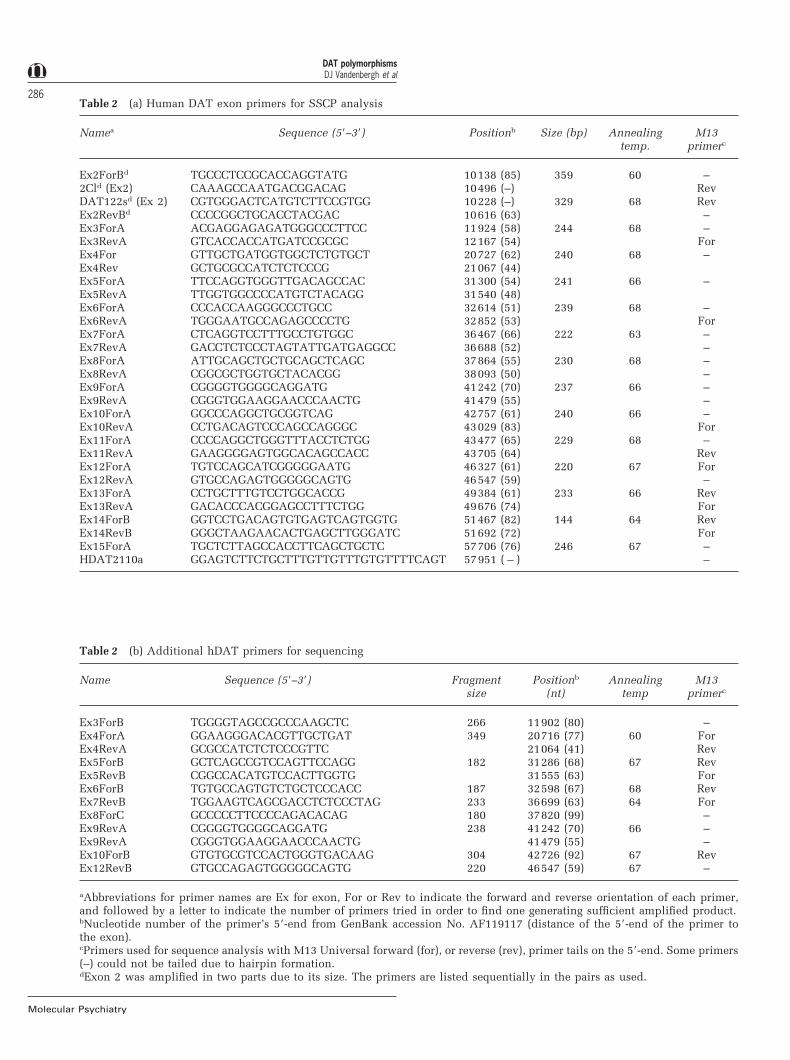
Table 2 (a) Human DAT exon primers for SSCP analysis
Namea Sequence (59–39) Positionb Size (bp) Annealing M13temp. primerc
Ex2ForBd TGCCCTCCGCACCAGGTATG 10138 (85) 359 60 –2Cld (Ex2) CAAAGCCAATGACGGACAG 10496 (–) RevDAT122sd (Ex 2) CGTGGGACTCATGTCTTCCGTGG 10228 (–) 329 68 RevEx2RevBd CCCCGGCTGCACCTACGAC 10616 (63) –Ex3ForA ACGAGGAGAGATGGGCCCTTCC 11924 (58) 244 68 –Ex3RevA GTCACCACCATGATCCGCGC 12167 (54) ForEx4For GTTGCTGATGGTGGCTCTGTGCT 20727 (62) 240 68 –Ex4Rev GCTGCGCCATCTCTCCCG 21067 (44)Ex5ForA TTCCAGGTGGGTTGACAGCCAC 31300 (54) 241 66 –Ex5RevA TTGGTGGCCCCATGTCTACAGG 31540 (48)Ex6ForA CCCACCAAGGGCCCTGCC 32614 (51) 239 68 –Ex6RevA TGGGAATGCCAGAGCCCCTG 32852 (53) ForEx7ForA CTCAGGTCCTTTGCCTGTGGC 36467 (66) 222 63 –Ex7RevA GACCTCTCCCTAGTATTGATGAGGCC 36688 (52) –Ex8ForA ATTGCAGCTGCTGCAGCTCAGC 37864 (55) 230 68 –Ex8RevA CGGCGCTGGTGCTACACGG 38093 (50) –Ex9ForA CGGGGTGGGGCAGGATG 41242 (70) 237 66 –Ex9RevA CGGGTGGAAGGAACCCAACTG 41479 (55) –Ex10ForA GGCCCAGGCTGCGGTCAG 42757 (61) 240 66 –Ex10RevA CCTGACAGTCCCAGCCAGGGC 43029 (83) ForEx11ForA CCCCAGGCTGGGTTTACCTCTGG 43477 (65) 229 68 –Ex11RevA GAAGGGGAGTGGCACAGCCACC 43705 (64) RevEx12ForA TGTCCAGCATCGGGGGAATG 46327 (61) 220 67 ForEx12RevA GTGCCAGAGTGGGGGCAGTG 46547 (59) –Ex13ForA CCTGCTTTGTCCTGGCACCG 49384 (61) 233 66 RevEx13RevA GACACCCACGGAGCCTTTCTGG 49676 (74) ForEx14ForB GGTCCTGACAGTGTGAGTCAGTGGTG 51467 (82) 144 64 RevEx14RevB GGGCTAAGAACACTGAGCTTGGGATC 51692 (72) ForEx15ForA TGCTCTTAGCCACCTTCAGCTGCTC 57706 (76) 246 67 –HDAT2110a GGAGTCTTCTGCTTTGTTGTTTGTGTTTTCAGT 57951 ( – ) –
Table 2 (b) Additional hDAT primers for sequencing
Name Sequence (59–39) Fragment Positionb Annealing M13size (nt) temp primerc
Ex3ForB TGGGGTAGCCGCCCAAGCTC 266 11902 (80) –Ex4ForA GGAAGGGACACGTTGCTGAT 349 20716 (77) 60 ForEx4RevA GCGCCATCTCTCCCGTTC 21064 (41) RevEx5ForB GCTCAGCCGTCCAGTTCCAGG 182 31286 (68) 67 RevEx5RevB CGGCCACATGTCCACTTGGTG 31555 (63) ForEx6ForB TGTGCCAGTGTCTGCTCCCACC 187 32598 (67) 68 RevEx7RevB TGGAAGTCAGCGACCTCTCCCTAG 233 36699 (63) 64 ForEx8ForC GCCCCCTTCCCCAGACACAG 180 37820 (99) –Ex9RevA CGGGGTGGGGCAGGATG 238 41242 (70) 66 –Ex9RevA CGGGTGGAAGGAACCCAACTG 41479 (55) –Ex10ForB GTGTGCGTCCACTGGGTGACAAG 304 42726 (92) 67 RevEx12RevB GTGCCAGAGTGGGGGCAGTG 220 46547 (59) 67 –
aAbbreviations for primer names are Ex for exon, For or Rev to indicate the forward and reverse orientation of each primer,and followed by a letter to indicate the number of primers tried in order to find one generating sufficient amplified product.bNucleotide number of the primer’s 59-end from GenBank accession No. AF119117 (distance of the 59-end of the primer tothe exon).cPrimers used for sequence analysis with M13 Universal forward (for), or reverse (rev), primer tails on the 59-end. Some primers(–) could not be tailed due to hairpin formation.dExon 2 was amplified in two parts due to its size. The primers are listed sequentially in the pairs as used.

DAT polymorphismsDJ Vandenbergh et al
287or at 15°C with glycerol. All samples from the Univer-sity of Chicago were sequenced bidirectionally fromPCR products, regardless of whether a variant wasdetected by SSCP or not. Direct sequencing failed toidentify any variants not detected by SSCP screeningunder at least one set of conditions (see below).
Direct sequence screening for variantsTen of the individuals from the NIDA samples wereselected for direct sequence analysis without precedingSSCP analysis, based on the occurrence of exon 15VNTR alleles containing at least one copy of the lesscommon 3, 5, 7, 8 or 11 repeat variants. Ten additionalindividuals were selected based on their expression ofthe more common 9- or 10-copy variants to maximizethe opportunities for finding novel alleles. Fifteen chil-dren from the University of Chicago, who had ADHDand evidence of transmission of the 10-copy allele froma heterozygous parent, were directly sequenced fromboth strands using primers that flanked each of theexons using an ABI 310 sequencer. DAT exons fromDNA from NIDA and the University of Chicago, wereamplified by PCR using the primers described in Table2 and sequenced using universal M13 forward orreverse sequences (footnote and Table 2b), which hadbeen included at the 39 and 59 ends of the primersequences. For those exon primers that formed hair-pins with an M13 tail, one of the two PCR primers wasused for sequencing. Sequences generated have beensubmitted to GenBank (Accession No. AF119117).
Figure 1 (a) Map of the relative positions of the 15 DAT exons. Below the line are the single nucleotide polymorphisms foundby direct sequencing and by SSCP. The two polymorphisms changing codons are shown by single letter code for the aminoacids (V/A under exon 2 and A/V under exon 8; see text for nucleotide changes). The frequency of the less common allele(the second in each pair) is included. The TaqI polymorphism in intron 4,28 the VNTR in exon 15,33 and the anonymous markerD5S678 are also shown. (b) Linkage disequilibrium with corresponding D9 values between polymorphic sites in exons 2 (nt215), 9 (nt 1316) and 15 (nt 1999, and VNTR). Significance values are shown by #P , 0.05; ##P , 0.001. (c) l clones and PCRfragments containing DAT gene sequences are shown in position relative to the gene. Clones containing the first 12 exons weredescribed in Donovan et al,27 and have been included here for a complete description of the gene.
Molecular Psychiatry
Results
Gene structureA total of 59747 bp of sequence comprising the com-plete DAT gene has been determined, including 8.6 kbof 59-flanking sequence (Figure 1a) (GenBank accessionNo. AF119117). The sequences substantially sup-plement the 5622 bp of sequence previously reported,27
which described the immediate 59 flank of the firstexon, the first 12 exons, and minimal intron sequence.Exons 13 and 14, which could not be detected in gen-omic DNA libraries, were cloned by long PCR amplifi-cation using primer pairs specific to exons 13 and 14,and 14 and 15, followed by subcloning the PCR pro-ducts (Figure 1c). Exon 15 of the DAT gene was ident-ified in genomic clones by hybridization to DAT cDNAsequences. One of these clones, cosmid M77-1, was agenerous gift from John Wasmuth and Deanna Church(University of California at Irvine) and also containsthe anonymous marker D5S678 approximately 25 kbdownstream of DAT exon 15 (data not shown). Thisindependently determined DAT gene structure andsequence also completes the structure recentlypresented by Kawarai et al.36
The positions of exon/intron boundaries, shown inTable 3, are completely conserved with those of thenorepinephrine,37 serotonin,38 and GABA trans-porters.39 Exon 13 encodes a small part of putativetransmembrane domain 11 and all of transmembranedomain 12 of the DAT protein. Exon 14 is the smallest
DAT polymorphismsDJ Vandenbergh et al
288
Molecular Psychiatry
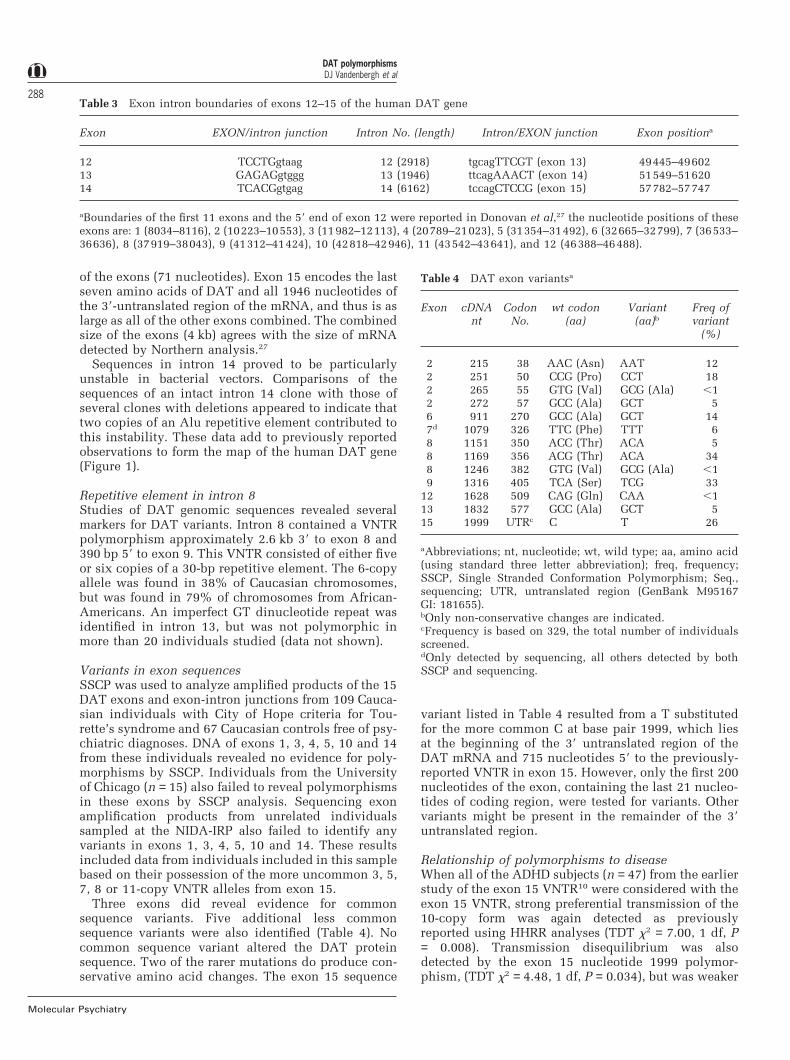
Table 3 Exon intron boundaries of exons 12–15 of the human DAT gene
Exon EXON/intron junction Intron No. (length) Intron/EXON junction Exon positiona
12 TCCTGgtaag 12 (2918) tgcagTTCGT (exon 13) 49445–4960213 GAGAGgtggg 13 (1946) ttcagAAACT (exon 14) 51549–5162014 TCACGgtgag 14 (6162) tccagCTCCG (exon 15) 57782–57747
aBoundaries of the first 11 exons and the 59 end of exon 12 were reported in Donovan et al,27 the nucleotide positions of theseexons are: 1 (8034–8116), 2 (10223–10553), 3 (11982–12113), 4 (20789–21023), 5 (31354–31492), 6 (32665–32799), 7 (36533–36636), 8 (37919–38043), 9 (41312–41424), 10 (42818–42946), 11 (43542–43641), and 12 (46388–46488).
of the exons (71 nucleotides). Exon 15 encodes the lastseven amino acids of DAT and all 1946 nucleotides ofthe 39-untranslated region of the mRNA, and thus is aslarge as all of the other exons combined. The combinedsize of the exons (4 kb) agrees with the size of mRNAdetected by Northern analysis.27
Sequences in intron 14 proved to be particularlyunstable in bacterial vectors. Comparisons of thesequences of an intact intron 14 clone with those ofseveral clones with deletions appeared to indicate thattwo copies of an Alu repetitive element contributed tothis instability. These data add to previously reportedobservations to form the map of the human DAT gene(Figure 1).
Repetitive element in intron 8Studies of DAT genomic sequences revealed severalmarkers for DAT variants. Intron 8 contained a VNTRpolymorphism approximately 2.6 kb 39 to exon 8 and390 bp 59 to exon 9. This VNTR consisted of either fiveor six copies of a 30-bp repetitive element. The 6-copyallele was found in 38% of Caucasian chromosomes,but was found in 79% of chromosomes from African-Americans. An imperfect GT dinucleotide repeat wasidentified in intron 13, but was not polymorphic inmore than 20 individuals studied (data not shown).
Variants in exon sequencesSSCP was used to analyze amplified products of the 15DAT exons and exon-intron junctions from 109 Cauca-sian individuals with City of Hope criteria for Tou-rette’s syndrome and 67 Caucasian controls free of psy-chiatric diagnoses. DNA of exons 1, 3, 4, 5, 10 and 14from these individuals revealed no evidence for poly-morphisms by SSCP. Individuals from the Universityof Chicago (n = 15) also failed to reveal polymorphismsin these exons by SSCP analysis. Sequencing exonamplification products from unrelated individualssampled at the NIDA-IRP also failed to identify anyvariants in exons 1, 3, 4, 5, 10 and 14. These resultsincluded data from individuals included in this samplebased on their possession of the more uncommon 3, 5,7, 8 or 11-copy VNTR alleles from exon 15.
Three exons did reveal evidence for commonsequence variants. Five additional less commonsequence variants were also identified (Table 4). Nocommon sequence variant altered the DAT proteinsequence. Two of the rarer mutations do produce con-servative amino acid changes. The exon 15 sequence
Table 4 DAT exon variantsa
Exon cDNA Codon wt codon Variant Freq ofnt No. (aa) (aa)b variant
(%)
2 215 38 AAC (Asn) AAT 122 251 50 CCG (Pro) CCT 182 265 55 GTG (Val) GCG (Ala) ,12 272 57 GCC (Ala) GCT 56 911 270 GCC (Ala) GCT 147d 1079 326 TTC (Phe) TTT 68 1151 350 ACC (Thr) ACA 58 1169 356 ACG (Thr) ACA 348 1246 382 GTG (Val) GCG (Ala) ,19 1316 405 TCA (Ser) TCG 33
12 1628 509 CAG (Gln) CAA ,113 1832 577 GCC (Ala) GCT 515 1999 UTRc C T 26
aAbbreviations; nt, nucleotide; wt, wild type; aa, amino acid(using standard three letter abbreviation); freq, frequency;SSCP, Single Stranded Conformation Polymorphism; Seq.,sequencing; UTR, untranslated region (GenBank M95167GI: 181655).bOnly non-conservative changes are indicated.cFrequency is based on 329, the total number of individualsscreened.dOnly detected by sequencing, all others detected by bothSSCP and sequencing.
variant listed in Table 4 resulted from a T substitutedfor the more common C at base pair 1999, which liesat the beginning of the 39 untranslated region of theDAT mRNA and 715 nucleotides 59 to the previously-reported VNTR in exon 15. However, only the first 200nucleotides of the exon, containing the last 21 nucleo-tides of coding region, were tested for variants. Othervariants might be present in the remainder of the 39untranslated region.
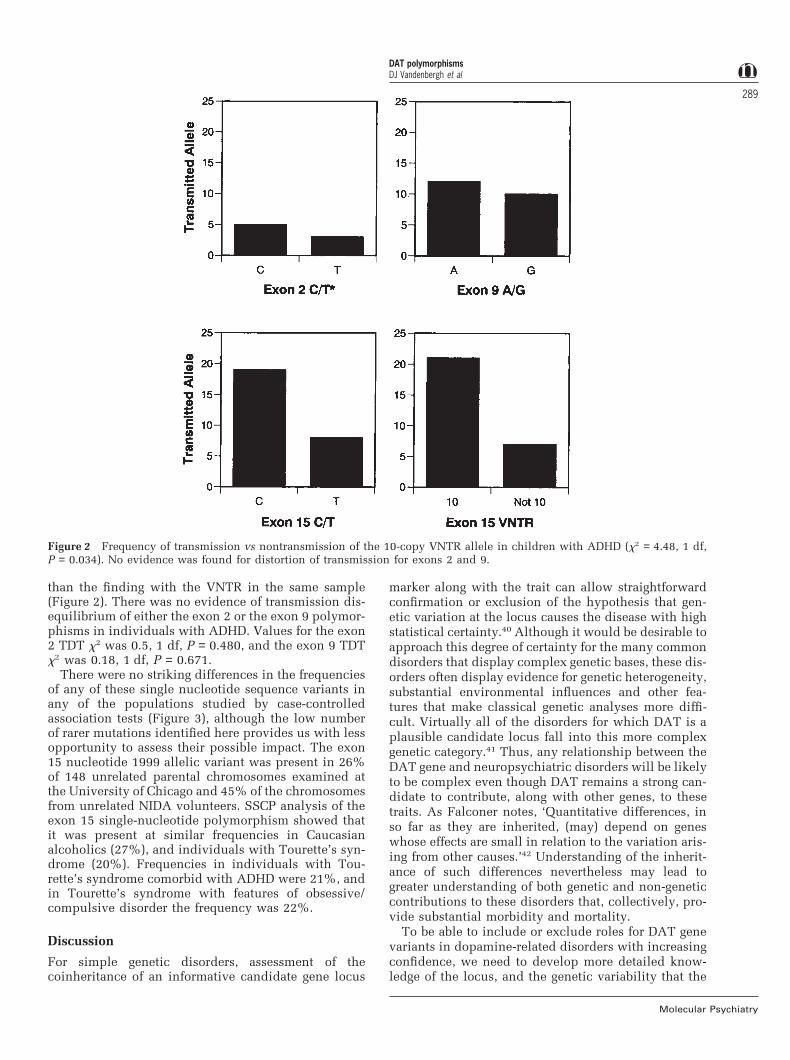
Relationship of polymorphisms to diseaseWhen all of the ADHD subjects (n = 47) from the earlierstudy of the exon 15 VNTR10 were considered with theexon 15 VNTR, strong preferential transmission of the10-copy form was again detected as previouslyreported using HHRR analyses (TDT x2 = 7.00, 1 df, P= 0.008). Transmission disequilibrium was alsodetected by the exon 15 nucleotide 1999 polymor-phism, (TDT x2 = 4.48, 1 df, P = 0.034), but was weaker
DAT polymorphismsDJ Vandenbergh et al
289
Figure 2 Frequency of transmission vs nontransmission of the 10-copy VNTR allele in children with ADHD (x2 = 4.48, 1 df,P = 0.034). No evidence was found for distortion of transmission for exons 2 and 9.
than the finding with the VNTR in the same sample(Figure 2). There was no evidence of transmission dis-equilibrium of either the exon 2 or the exon 9 polymor-phisms in individuals with ADHD. Values for the exon2 TDT x2 was 0.5, 1 df, P = 0.480, and the exon 9 TDTx2 was 0.18, 1 df, P = 0.671.
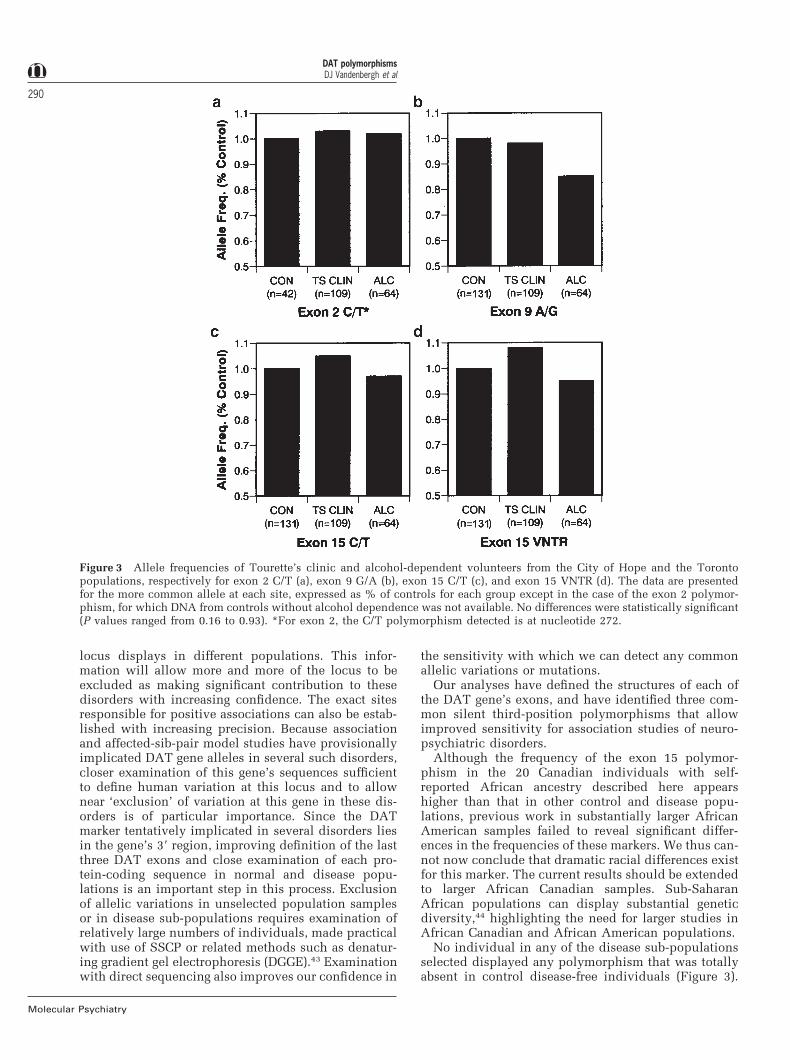
There were no striking differences in the frequenciesof any of these single nucleotide sequence variants inany of the populations studied by case-controlledassociation tests (Figure 3), although the low numberof rarer mutations identified here provides us with lessopportunity to assess their possible impact. The exon15 nucleotide 1999 allelic variant was present in 26%of 148 unrelated parental chromosomes examined atthe University of Chicago and 45% of the chromosomesfrom unrelated NIDA volunteers. SSCP analysis of theexon 15 single-nucleotide polymorphism showed thatit was present at similar frequencies in Caucasianalcoholics (27%), and individuals with Tourette’s syn-drome (20%). Frequencies in individuals with Tou-rette’s syndrome comorbid with ADHD were 21%, andin Tourette’s syndrome with features of obsessive/compulsive disorder the frequency was 22%.
Discussion
For simple genetic disorders, assessment of thecoinheritance of an informative candidate gene locus
Molecular Psychiatry
marker along with the trait can allow straightforwardconfirmation or exclusion of the hypothesis that gen-etic variation at the locus causes the disease with highstatistical certainty.40 Although it would be desirable toapproach this degree of certainty for the many commondisorders that display complex genetic bases, these dis-orders often display evidence for genetic heterogeneity,substantial environmental influences and other fea-tures that make classical genetic analyses more diffi-cult. Virtually all of the disorders for which DAT is aplausible candidate locus fall into this more complexgenetic category.41 Thus, any relationship between theDAT gene and neuropsychiatric disorders will be likelyto be complex even though DAT remains a strong can-didate to contribute, along with other genes, to thesetraits. As Falconer notes, ‘Quantitative differences, inso far as they are inherited, (may) depend on geneswhose effects are small in relation to the variation aris-ing from other causes.’42 Understanding of the inherit-ance of such differences nevertheless may lead togreater understanding of both genetic and non-geneticcontributions to these disorders that, collectively, pro-vide substantial morbidity and mortality.
To be able to include or exclude roles for DAT genevariants in dopamine-related disorders with increasingconfidence, we need to develop more detailed know-ledge of the locus, and the genetic variability that the
DAT polymorphismsDJ Vandenbergh et al
290
Molecular Psychiatry
Figure 3 Allele frequencies of Tourette’s clinic and alcohol-dependent volunteers from the City of Hope and the Torontopopulations, respectively for exon 2 C/T (a), exon 9 G/A (b), exon 15 C/T (c), and exon 15 VNTR (d). The data are presentedfor the more common allele at each site, expressed as % of controls for each group except in the case of the exon 2 polymor-phism, for which DNA from controls without alcohol dependence was not available. No differences were statistically significant(P values ranged from 0.16 to 0.93). *For exon 2, the C/T polymorphism detected is at nucleotide 272.
locus displays in different populations. This infor-mation will allow more and more of the locus to beexcluded as making significant contribution to thesedisorders with increasing confidence. The exact sitesresponsible for positive associations can also be estab-lished with increasing precision. Because associationand affected-sib-pair model studies have provisionallyimplicated DAT gene alleles in several such disorders,closer examination of this gene’s sequences sufficientto define human variation at this locus and to allownear ‘exclusion’ of variation at this gene in these dis-orders is of particular importance. Since the DATmarker tentatively implicated in several disorders liesin the gene’s 39 region, improving definition of the lastthree DAT exons and close examination of each pro-tein-coding sequence in normal and disease popu-lations is an important step in this process. Exclusionof allelic variations in unselected population samplesor in disease sub-populations requires examination ofrelatively large numbers of individuals, made practicalwith use of SSCP or related methods such as denatur-ing gradient gel electrophoresis (DGGE).43 Examinationwith direct sequencing also improves our confidence in
the sensitivity with which we can detect any commonallelic variations or mutations.
Our analyses have defined the structures of each ofthe DAT gene’s exons, and have identified three com-mon silent third-position polymorphisms that allowimproved sensitivity for association studies of neuro-psychiatric disorders.
Although the frequency of the exon 15 polymor-phism in the 20 Canadian individuals with self-reported African ancestry described here appearshigher than that in other control and disease popu-lations, previous work in substantially larger AfricanAmerican samples failed to reveal significant differ-ences in the frequencies of these markers. We thus can-not now conclude that dramatic racial differences existfor this marker. The current results should be extendedto larger African Canadian samples. Sub-SaharanAfrican populations can display substantial geneticdiversity,44 highlighting the need for larger studies inAfrican Canadian and African American populations.
No individual in any of the disease sub-populationsselected displayed any polymorphism that was totallyabsent in control disease-free individuals (Figure 3).

DAT polymorphismsDJ Vandenbergh et al
291The higher frequencies of the more common exon 2and exon 9 polymorphisms allow us to relatively con-fidently exclude any robust association between vari-ants at these portions of the DAT gene locus and thedisorders for which significant numbers of individualswere sampled. No contribution to either Tourette’s syn-drome spectrum or alcohol dependence is supportedfrom this work. These marker frequencies are also simi-lar in polysubstance abuser and control clinical popu-lations.18 Single studies have also suggested that 9-copy VNTR frequencies are elevated in the cocaineabuser subpopulation that experience paranoia withcocaine use,15 alcoholics who are habitually violent45
and alcoholics who display more pronounced alcoholwithdrawal.17
In the ADHD samples in which the exon 15 VNTRassociation was initially described,10,46 the singlenucleotide 1999 exon 15 C allelic variant was also pref-erentially transmitted to children with ADHD. Therewas no evidence of preferential transmission of eitherDAT exon 2 or 9 markers to the ADHD children, how-ever.
This comprehensive evaluation of 2053 bp of DATgene exons and about 700 bp of flanking sequence hasidentified 16 sequence variants, 12 in exon sequences.Only two of the rarer variants result in amino acid sub-stitution, none of the others change the DAT proteincoding sequence. This substantial conservation is con-sistent with the idea that normal DAT function is evol-utionarily important. These data are also consistentwith the conservation of this transporter gene’s struc-ture with those of other transporter gene family mem-bers, even those located on other chromosomes.37–39
Such data, as well as other work mentioned above, pro-vide increasing interest in the possibility that level-of-expression variation, rather than protein sequence vari-ants, may provide the common individual differencesat the human DAT gene locus. Mutations inpromoter/enhancer regions of this gene could even pro-vide a significant impact on the dopaminergic brainfunction and dysfunction.
Acknowledgements
The authors would like to thank Dr Carlo Contoreggi,Ms Judith Hess, Ms Brenda Campbell, and Dr MarkStein for assistance in clinical characterization samplecollection from NIDA-IRP subjects. We would also liketo thank Shuya Yan and Roxann Ingersoll for experttechnical assistance. This work was supported finan-cially by NIDA’s Intramural Research Program, andgrants from CAMH, the Medical Research Council ofCanada, and NIDA.
References1 Uhl GR, Johnson PS. Neurotransmitter transporter: three important
gene families for neuronal function. J Exp Biol 1994; 196: 229–236.2 Ritz MC, Lamb RJ, Goldberg SR, Kukor MJ. Cocaine receptors on
dopamine transporters are related to self-administration of cocaine.Science 1987; 237: 1219–1223.
3 Singer HS, Hahn I-H, Moran T. Abnormal dopamine uptake sites
Molecular Psychiatry
in postmortem striatum from patients with Tourette’s syndrome.Ann Neurol 1991; 30: 558–562.
4 Wong DF, Harris JC, Naidu S, Yokoi F, Marenco S, Dannals RF etal. Dopamine transporters are markedly reduced in Lesch-Nyhandisease in vivo. Proc Natl Acad Sci 1996; 93: 5539–5543.
5 Wong DF, Ricaurte G, Grunder G, Rothman R, Naidu S, Singer Het al. Dopamine transporter changes in neuropsychiatric disorders.Adv Pharm 1998; 42: 219–223.
6 Uhl GR, Lin Z, Metzger T, Dar D. Dopamine transporter mutants,small molecules, and approaches to cocaine antagonist/dopaminetransporter disinhibiter development. Met Enz 1998; 296: 456–465.
7 Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopaminetransporter is required for in vivo MPTP neurotoxicity: evidencefrom mice lacking the transporter. J Neurochem 1997; 69: 1322–1325.
8 Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperloco-motion and indifference to cocaine and amphetamine in mice lack-ing the dopamine transporter. Nature 1996; 379: 606–612.
9 Sora I, Wichems C, Takahashi N, Li X-F, Zeng Z, Revay R et al.Cocaine reward models: conditioned place preference can be estab-lished in dopamine- and in serotonin-transporter knockout mice.Proc Natl Acad Sci 1998; 95: 7699–7704.
10 Cook EHJ, Stein MA, Krasowski MD, Cox NJ, Olkon DM, Kieffer JEet al. Association of attention-defect disorder and the dopaminetransporter gene. Am J Hum Genet 1995; 56: 993–998.
11 Waldman ID, Rowe DC, Abramowitz A, Kozel S, Mohr J, ShermanSL et al. Association and linkage of the dopamine transporter geneand attention-deficit hyperactivity disorder in children: heterogen-eity owing to diagnostic subtype and severity. Am J Hum Genet1998; 63: 1767–1776.
12 Gill M, Daly G, Heron S, Hawi Z, Fitzgerald M. Confirmation ofassociation between attention deficit hyperactivity disorder and adopamine transporter polymorphism. Mol Psychiatry 1997; 2:311–313.
13 Daly G, Hawi Z, Fitzgerald M, Gill M. Mapping susceptibility lociin attention deficit hyperactivity disorder: preferential trans-mission of parental alleles at DAT1, DBH and DRD5 to affectedchildren. Mol Psychiatry 1999; 4: 192–196.
14 LaHoste GJ, Swanson JM, Wigal SB, Glabe C, Wigal T, King N etal. Dopamine D4 receptor gene polymorphism is associated withattention deficit-hyperactivity disorder. Mol Psychiatry 1996; 1:121–124.
15 Gelernter J, Kranzler HR, Satel SL, Rao PA. Genetic associationbetween dopamine transporter protein alleles and cocaine-inducedparanoia. Neuropsychoparmacology 1994; 11: 195–200.
16 Kelsoe J, Sadovnick AD, Kristbjarnarson H, Bergesch P, Mroczkow-ski-Parker Z, Drennan M et al. Possible locus for bipolar disordernear the dopamine transporter on chromosome 5. Am J Med Genet1996; 67: 533–540.
17 Schmidt LG, Harms H, Kuhn S, Rommelspacher H, Sander T. Modi-fication of alcohol withdrawal by the A9 allele of the dopaminetransporter gene. Am J Psychiatry 1998; 155: 474–478.
18 Persico AM, Vandenbergh DJ, Smith SS, Uhl GR. Dopamine trans-porter gene markers are not associated with polysubstance abuse.Biol Psychiatry 1993; 34: 265–267.
19 Muramatsu T, Higuchi S. Dopamine transporter gene polymor-phism and alcoholism. Biochem Biophys Res Com 1995; 211: 28–32.
20 Sander T, Harms H, Podschus J, Finckh U, Nickel B, Rolfs A et al.Allelic association of a dopamine transporter gene polymorphismin alcohol dependence with withdrawal seizures or delirium. BiolPsychiatry 1997; 41: 299–304.
21 Daniels J, Williams JD, Asherton P, McGuffin P, Owen M. Noassociation between schizophrenia and polymorphisms within thegenes for debrisoquine 4-hydroxylase (CYP2D6) and the dopaminetransporter (DAT). Am J Med Gen 1995; 60: 85–87.
22 Persico AM, Wang ZW, Black DW, Andreasen NC, Uhl GR, CroweRR. Exclusion of close linkage of the dopamine transporter genewith schizophrenia spectrum disorders. Am J Psychiatry 1995; 152:134–136.
23 Comings DE, Wu S, Chui C, Ring RH, Gade R, Ahn C et al. Polygen-etic inheritance of Tourette’s syndrome, stuttering, attention defecthyperactivity, conduct and oppositional defiant disorder: the addi-

DAT polymorphismsDJ Vandenbergh et al
292
Molecular Psychiatry
tive and subtractive effect of the three dopaminergic genes—DRD2,DBH, and DAT1. Am J Med Genet 1996; 67: 264–288.
24 Higuchi S, Muramatsu T, Arai H, Hayashida M, Sasaki H, Tro-janowski JQ. Polymorphisms of dopamine receptor and transportergenes and Parkinson’s disease. J Neural Transm 1995; 10: 107–113.
25 LeCouteur DG, Leighton PW, McCann SJ, Pond SM. Association ofa polymorphism in the dopamine transporter gene with Parkinson’sdisease. Mov Disord 1997; 12: 760–763.
26 Leighton PW, LeCouteur DG, Pang CC, McCann SJ, Chan D, LawLK et al. The dopamine transporter gene and Parkinson’s diseasein a Chinese population. Neurology 1997; 49: 1577–1579.
27 Donovan DM, Vandenbergh DJ, Perry MP, Bird GS, Ingersoll R,Nanthakumar E et al. Human and mouse dopamine transportergenes: conservation of 59-flanking sequence elements and genestructures. Am J Med Genet 1995; 3: 327–335.
28 Vandenbergh DJ, Persico AM, Uhl GR. A human dopamine trans-porter cDNA predicts reduced glycosylation, displays a novelrepetitive element and provides racially-dimorphic Taq I RFLPs.Mol Brain Res 1992; 15: 161–166.
29 Tyndale RM, Droll KP, Sellers EM. Genetically defined CYP2D6metabolism provides protection against oral opiate dependence.Pharmacogenetics 1997; 7: 375–379.
30 Smith SS, O’Hara BF, Persico AM, Gorelick DA, Newlin DB, VlahovD et al. Genetic vulnerability to drug abuse: the dopamine D2receptor TaqI B1 RFLP appears more frequently in polysubstanceabusers. Arch Gen Psychiatry 1992; 49: 723–727.
31 American Psychiatric Association. Diagnostic and Statistical Man-ual of Mental Disorders. American Psychiatric Association: Wash-ington DC, 1987.
32 Spielman RS, McGinnis RE, Ewens WJ. Transmission test for link-age disequilibrium: the insulin gene region and insulin-dependentdiabetes mellitus (IDDM). Am J Hum Genet 1993; 52: 506–516.
33 Vandenbergh DJ, Persico AM, Hawkins AL, Griffin CA, Li X, JabsEW et al. Human dopamine transporter gene (DAT1) maps to chro-mosome 5p15.3 and displays a VNTR. Genomics 1992; 14: 1104–1106.
34 Bardeesy N, Pelletier J. Mutational detection by single strandedconformational polymorphism. Meth Neurosci 1995; 26: 163–183.
This article is a ‘United States Government Work’ paper as defined by the US Copyright Act.
35 Thompson M, Comings DE, Feder L, George SR, O’Dowd BF.Mutation screening of the dopamine D1 receptor gene in Tourette’ssyndrome and alcohol dependent patients. Am J Med Genet 1998;81: 241–244.
36 Kawarai T, Kawakami H, Yamamura Y, Nakamura S. Structure andorganization of the gene encoding human dopamine transporter.Gene 1997; 195: 11–18.
37 Porzgen P, Bonisch H, Bruss M. Molecular cloning and organiza-tion of the coding region of the human norepinephrine transportergene. Biochem Biophys Res Com 1995; 215: 1145–1150.
38 Lesch KP, Balling U, Gross J, Strauss K, Wolozin BL, Murphy DL etal. Organization of the human serotonin transporter gene. J NeuralTransm 1994; 95: 157–162.
39 Liu QR, Mandiyan S, Nelson H, Nelson N. A family of genes enco-ding neurotransmitter transporters. Proc Natl Acad Sci 1992; 89:6639–6643.
40 Lander ES, Schork NJ. Genetic dissection of complex traits. Science1994; 265: 2037–2048.
41 Uhl GR, Gold LH, Risch N. Genetic analyses of complex behavioraldisorders. Proc Natl Acad Sci 1997; 94: 2785–2786.
42 Falconer DS. Introduction to Quantitative Genetics, 3rd edn. JohnWiley & Sons: New York, 1989, p 1.
43 Abrams ES, Murdaugh SE, Lerman LS. Comprehensive detectionof single base changes in human genomic DNA using denaturinggradient gel electrophoresis and a GC clamp. Genomics 1990; 7:463–475.
44 Tishkoff SA, Dietzsch E, Speed W, Pakstis AJ, Kidd JR, Cheung Ket al. Global patterns of linkage disequilibrium at the CD4 locusand modern human origins. Science 1996; 271: 1380–1387.
45 Tiihonen J, Kuikka J, Bergstrom K, Hakola P, Karhu J, RyynanenO-P et al. Altered striatal dopamine re-uptake site densities inhabitually violent and non-violent alcoholics. Nature Med 1995; 1:654–657.
46 Cook EJ, Stein MA, Leventhal BL. Family-based association of thedopamine transport gene in attention deficit disorder. In: Blum K,Nobel EP (eds). Handbook of Psychiatric Genetics. CRC Press: NewYork, 1997, pp 297–310.




















