
Host Gene Regulation During Coxsackievirus B3 Infection in Mice
12
Host Gene Regulation During Coxsackievirus B3 Infection in Mice Assessment by Microarrays Lydia A. Taylor, Christopher M. Carthy, Decheng Yang, Kareem Saad, Donald Wong, George Schreiner, Lawrence W. Stanton, Bruce M. McManus Abstract—Host genetic responses that characterize enteroviral myocarditis have not yet been determined. The injurious and inflammatory process in heart muscle may reflect host responses of benefit to the virus and ultimately result in congestive heart failure and dilated cardiomyopathy. On the other hand, host responses within the myocardium may secure the host against acute or protracted damage. To investigate the nature of modified gene expression in comparison with normal tissue, mRNA species were assessed in myocardium using cDNA microarray technology at days 3, 9, and 30 after infection. Of 7000 clones initially screened, 169 known genes had a level of expression significantly different at 1 or more postinfection time points as compared with baseline. The known regulated genes were sorted according to their functional groups and normalized expression patterns and, subsequently, interpreted in the context of viremic, inflammatory, and healing phases of the myocarditic process. (Circ Res. 2000;87:328-334.) Key Words: coxsackieviruses n gene regulation n microarrays n myocardium N early 50% of North American clinical myocarditis cases are attributable to Picornaviradae infections, with the coxsackievirus B (CVB) serogroup making up the most significant proportion of such infections. 1 Coxsackieviruses generally have a high attack rate with a relatively low myocarditic potential, and a relatively small percentage of the human population develop clinically visible viral myocarditis in a lifetime. 2 The susceptible human population appears to be nonrandom, with genotypic factors related to age and sex, and environmental factors such as nutrition and pregnancy, modulating host susceptibility to viral myocarditis. Mouse models of CVB3-mediated myocarditis mimic the human disease process. Indeed, disease progression and pathology closely resemble the human disease, 3 and both age and genetic background affect susceptibility. 4–6 As such, CVB3- mediated murine myocarditis is a valuable model for studying the pathogenesis of virus-induced human myocarditis and has allowed partial dissection of the disease progression of myocarditis, which is otherwise impossible in humans. Three distinct phases of the infectious and injurious pro- cess are appreciated. 7 The first phase, encompassing peak viremia, occurs 2 to 4 days after infection wherein direct virus-induced tissue injury and death of infected cardiac myocytes are profound. 7,8 Impairment of left ventricular function in physiologically loaded working mouse hearts with histopathologically graded moderate injury after CVB3 infec- tion reinforces the important pathophysiological role of direct viral damage of myocardium. This myocardial injury be- comes progressively more noticeable by days 3 and 4 and is significantly advanced by day 5, after which direct injury is complicated by an inflammatory infiltrate, thus marking the beginning of the second phase. This inflammatory infiltrate includes both innate and specific immune participants largely localized to regions of dead, dying, and residually infected myocytes 9 and serves to clear both infectious virus particles and dying cells. This phase typically lasts through day 14 after infection and is followed by a healing phase character- ized by steady tissue reparation and ventricular remodeling wherein dead myocytes are removed and many remaining myocytes change their structural, ionic, metabolic, and con- tractile properties. 10 In addition, nonmyocytic cells, fibro- blasts for example, often increase in number in the cardiac parenchyma during the healing phase, participating in the secretion of cytokines, growth factors, and extracellular matrix (ECM). The host genetic elements responsible for the changes observed in the above 3 phases of CVB3-mediated myocar- ditis have not yet been determined. Although previous inves- tigations by our laboratory 11 and by others have yielded clues to gene expression involved in the pathology of myocarditis at certain time points in murine models, including those of virus and host, the majority of host genes involved in this Received March 10, 2000; revision received June 19, 2000; accepted June 20, 2000. From the Cardiovascular Research Laboratory (L.A.T., C.M.C., D.Y., K.S., D.W., B.M.M.), Department of Pathology and Laboratory Medicine, University of British Columbia, St. Paul’s Hospital, Vancouver, Canada, and SCIOS, Inc (G.S., L.W.S.), Sunnyvale, Calif. Correspondence to Bruce McManus, MD, PhD, Department of Pathology and Laboratory Medicine, University of British Columbia, St. Paul’s Hospital, 1081 Burrard St, Vancouver, BC, Canada V6Z 1Y6. E-mail [email protected] © 2000 American Heart Association, Inc. Circulation Research is available at http://www.circresaha.org 328 Molecular Medicine by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from by guest on January 19, 2016 http://circres.ahajournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Host Gene Regulation During Coxsackievirus B3 Infection in Mice

Host Gene Regulation During Coxsackievirus B3Infection in Mice
Assessment by Microarrays
Lydia A. Taylor, Christopher M. Carthy, Decheng Yang, Kareem Saad, Donald Wong,George Schreiner, Lawrence W. Stanton, Bruce M. McManus
Abstract—Host genetic responses that characterize enteroviral myocarditis have not yet been determined. The injurious andinflammatory process in heart muscle may reflect host responses of benefit to the virus and ultimately result incongestive heart failure and dilated cardiomyopathy. On the other hand, host responses within the myocardium maysecure the host against acute or protracted damage. To investigate the nature of modified gene expression in comparisonwith normal tissue, mRNA species were assessed in myocardium using cDNA microarray technology at days 3, 9, and30 after infection. Of 7000 clones initially screened, 169 known genes had a level of expression significantly differentat 1 or more postinfection time points as compared with baseline. The known regulated genes were sorted according totheir functional groups and normalized expression patterns and, subsequently, interpreted in the context of viremic,inflammatory, and healing phases of the myocarditic process.(Circ Res. 2000;87:328-334.)
Key Words: coxsackievirusesn gene regulationn microarraysn myocardium
Nearly 50% of North American clinical myocarditis casesare attributable toPicornaviradaeinfections, with the
coxsackievirus B (CVB) serogroup making up the mostsignificant proportion of such infections.1 Coxsackievirusesgenerally have a high attack rate with a relatively lowmyocarditic potential, and a relatively small percentage of thehuman population develop clinically visible viral myocarditisin a lifetime.2 The susceptible human population appears tobe nonrandom, with genotypic factors related to age and sex,and environmental factors such as nutrition and pregnancy,modulating host susceptibility to viral myocarditis. Mousemodels of CVB3-mediated myocarditis mimic the humandisease process. Indeed, disease progression and pathologyclosely resemble the human disease,3 and both age andgenetic background affect susceptibility.4–6 As such, CVB3-mediated murine myocarditis is a valuable model for studyingthe pathogenesis of virus-induced human myocarditis and hasallowed partial dissection of the disease progression ofmyocarditis, which is otherwise impossible in humans.
Three distinct phases of the infectious and injurious pro-cess are appreciated.7 The first phase, encompassing peakviremia, occurs 2 to 4 days after infection wherein directvirus-induced tissue injury and death of infected cardiacmyocytes are profound.7,8 Impairment of left ventricularfunction in physiologically loaded working mouse hearts withhistopathologically graded moderate injury after CVB3 infec-
tion reinforces the important pathophysiological role of directviral damage of myocardium. This myocardial injury be-comes progressively more noticeable by days 3 and 4 and issignificantly advanced by day 5, after which direct injury iscomplicated by an inflammatory infiltrate, thus marking thebeginning of the second phase. This inflammatory infiltrateincludes both innate and specific immune participants largelylocalized to regions of dead, dying, and residually infectedmyocytes9 and serves to clear both infectious virus particlesand dying cells. This phase typically lasts through day 14after infection and is followed by a healing phase character-ized by steady tissue reparation and ventricular remodelingwherein dead myocytes are removed and many remainingmyocytes change their structural, ionic, metabolic, and con-tractile properties.10 In addition, nonmyocytic cells, fibro-blasts for example, often increase in number in the cardiacparenchyma during the healing phase, participating in thesecretion of cytokines, growth factors, and extracellularmatrix (ECM).
The host genetic elements responsible for the changesobserved in the above 3 phases of CVB3-mediated myocar-ditis have not yet been determined. Although previous inves-tigations by our laboratory11 and by others have yielded cluesto gene expression involved in the pathology of myocarditisat certain time points in murine models, including those ofvirus and host, the majority of host genes involved in this
Received March 10, 2000; revision received June 19, 2000; accepted June 20, 2000.From the Cardiovascular Research Laboratory (L.A.T., C.M.C., D.Y., K.S., D.W., B.M.M.), Department of Pathology and Laboratory Medicine,
University of British Columbia, St. Paul’s Hospital, Vancouver, Canada, and SCIOS, Inc (G.S., L.W.S.), Sunnyvale, Calif.Correspondence to Bruce McManus, MD, PhD, Department of Pathology and Laboratory Medicine, University of British Columbia, St. Paul’s
Hospital, 1081 Burrard St, Vancouver, BC, Canada V6Z 1Y6. E-mail [email protected]© 2000 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
328
Molecular Medicine
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from

complex process remain elusive. Traditional molecular ap-proaches continue to add to our understanding of the patho-genesis and progression of viral myocarditis one gene at atime; however, new technologies in molecular biology,namely cDNA microarrays, have revolutionized the power ofunbiased expression searches and afford the opportunity toprofile the expression of thousands of genes in a singleexperiment.12
Thus, to rapidly expand the portrait of host gene expressioninvolved in the pathogenesis of viral myocarditis and partic-ularly to examine gene expression during all phases of viralmyocarditis, we used cDNA microarrays to evaluate therelative abundance of myocardial mRNA species in CVB3-infected male adolescent A/J mice at 3, 9, and 30 days afterinfection as compared with sham-infected myocardium. Froman initial screen of'7000 clones, 614 sequences including169 known genes were identified as having either a signifi-cant (1.8-fold) decrease or increase in expression at 1 or morepostinfection time points. The 169 known genes were sortedaccording to their functional groups including those involvedin cell division, structure/motility, signaling, host defense,gene/protein expression, metabolism, mitochondrial se-quences, and presently unknown function. These genes werethen interpreted in light of known contributors to the myo-carditic process.
Materials and MethodsAnimals and VirusA/J (H-2a) mice (The Jackson Laboratories) were 5 weeks old at theonset of the experiment. Myocarditic CVB3 was kindly provided byDr C.J. Gauntt (University of Texas, San Antonio, Tex) and stored at–80°C. Virus was propagated in HeLa cells (American Type CultureCollection). Stock virus was titered before the onset of experimentsusing the plaque assay method. Mice were infected with 13105 pfuof CVB3 (n515 per time point) or PBS sham (n515 per time point)and euthanized by CO2 narcosis on days 3, 9, and 30 after infection.Animals that died naturally after infection ('25%) were not includedin experimental analysis (because of degraded RNA).
Tissue HistopathologyHeart tissue was fixed in D-PBS–buffered 4% paraformaldehydeovernight, dehydrated in graded alcohols, cleared in xylene, embed-ded in paraffin, and sectioned (4mm) for in situ hybridization.
In Situ HybridizationIn situ hybridization was performed as previously described.13
Tissue sections were incubated overnight in hybridization mixturecontaining digoxigenin-labeled, CVB3-specific antisense ribo-probes. Blocking with 2% lamb serum followed posthybridizationwashing. Hybridized riboprobe was detected with a sheep antidigoxi-genin polyclonal antibody conjugated to alkaline phosphatase (Boeh-ringer Mannheim PQ). Slides were developed in Sigma-Fast ni-troblue tetrazolium-BCIP (5-bromo-4-chloro-3-indolylphosphatetuluidinium) (Sigma), counterstained in fresh carmalum, and exam-ined for reaction product by light microscopy.
cDNA ProbesMouse hearts from each experimental group were pooled, flash-frozen in liquid nitrogen, pooled, and stored at270°C. mRNA foreach group was extracted and twice selected by oligo(dT) chroma-tography. Fluorescently labeled cDNA probes were generated byreverse transcription of poly(A)1mRNA in the presence of Cy3 orCy5 dCTP (Amersham).
DNA MicroarrayMicroarrays contained'7000 cDNA clones randomly collectedfrom a normalized male Wistar rat heart cDNA library.14 Microarrayfabrication and hybridization were performed at Incyte Pharmaceu-ticals.15 cDNA inserts were generated by PCR amplification withprimers derived from flanking vector sequences.16 PCR productswere arrayed from 96-well microtiter plates onto silanated micro-scope slides in an area of 1.8 cm2 using print tips driven byhigh-speed robotics. Printed arrays were incubated for 4 hours in ahumid chamber and rinsed once in 0.2% SDS (1 minute), twice inH2O (1 minute), and once in sodium borohydride solution (1.9 g ofNaBH4 dissolved in 300 mL of PBS and 100 mL of 100% ethanol;5 minutes). The arrays were submerged in H2O (2 minutes) at 95°C,transferred quickly into 0.2% SDS (1 minute), rinsed twice in H2O,air dried, and stored in the dark at 25°C.17 Each pair of fluorescentlylabeled cDNA sample probes was applied to the microarray andallowed to hybridize competitively to the 7000 elements. Degree ofhybridization was quantified by sequential excitation of the 2fluorophores with a scanning laser. Differential expression valueswere represented as a ratio of intensities. Expression data wereomitted if the signal was,2.5-fold over local background or derivedfrom ,40% of the area of the printed spot.
ResultsViral Time Course in the HeartIn situ hybridization using antisense CVB3-specific ribo-probes was performed on myocardial sections from mice ateach time point to determine the viral load, infected cell type,and infection variability among animals. At day 3 afterinfection, viral RNA is detectable in the myocardium of allanimals. This amount of viral RNA is typically increased byday 9 after infection and completely absent by day 30 afterinfection, when no viral RNA is detectable in the myocardi-um of infected animals (Figure 1).
The characteristic localization of viral RNA within themyocardium at days 3 and 9 after infection exhibits a clearlack of preference for infection of endothelial cells, eitherthose lining the ventricular cavities or those in small arteries,veins, or lymphatics within the tissue. In addition, there is nodetectable infection of interstitial cells, such as fibroblasts.Indeed, the most visibly and clearly infected cell type is thecardiac myocyte. The range of cellular dynamics in thecardiac myocyte is reflected by the coexistence of cardiacmuscle cells with or without a high-titer viral replication, butwhich have undergone cytopathic effects and may be resist-ing death in a fashion distinct from that of neighboringcoagulated cells on the one hand and neighboring uninfectedor “healthy” cells on the other. Additionally, infectiousinvolvement of the ventricular septum and the left and right
Figure 1. CVB3 RNA localization in A/J mice by in situ hybridiza-tion. A/J mice were infected, and tissues were harvested at 3 (A), 9(B), and 30 (C) days after infection. Positive myocytes were presentat days 3 and 9 after infection. Positively stained myocytes weredetected at day 30 after infection. Magnifications 320.
Taylor et al Host Gene Regulation During CVB3 Infection 329
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from

ventricular free walls appears generally similar in pattern,whereas atrial tissue has a more limited presence of viralgenome in comparison.
Examination of contiguous sections stained with in situhybridization for viral RNA, hematoxylin and eosin, andMasson’s trichrome (Figure 2) illustrates that infected myo-cytes are undergoing virus-induced cell death without im-mune involvement.
Microarray Analysis of Gene ExpressionDNA microarrays were used to evaluate changes in geneexpression in the myocardium at 3, 9, and 30 days afterinfection. Approximately 7000 heart cDNA clones represent-ing '4200 distinct genes were printed as high-density arrays.Arrays were probed in duplicate with fluorescently labeledprobes generated from mRNA extracted from infected andcontrol myocardial tissue at each time point. Duplicate resultswere averaged, clones with discordant or absent results werediscarded entirely, and remaining clones increased or de-creased by 1.8-fold relative to control were considereddifferentially expressed. A total of 619 cDNA clones weredifferentially expressed in CVB3-infected myocardium.cDNA clones that had no significant match, or matched onlyto an expressed sequence tag in GenBank (http://www.ncbi.n-lm.nih.gov/), were not considered further. In total, differen-tially expressed cDNA sequences corresponded to 169 indi-vidual known genes Figure 3.
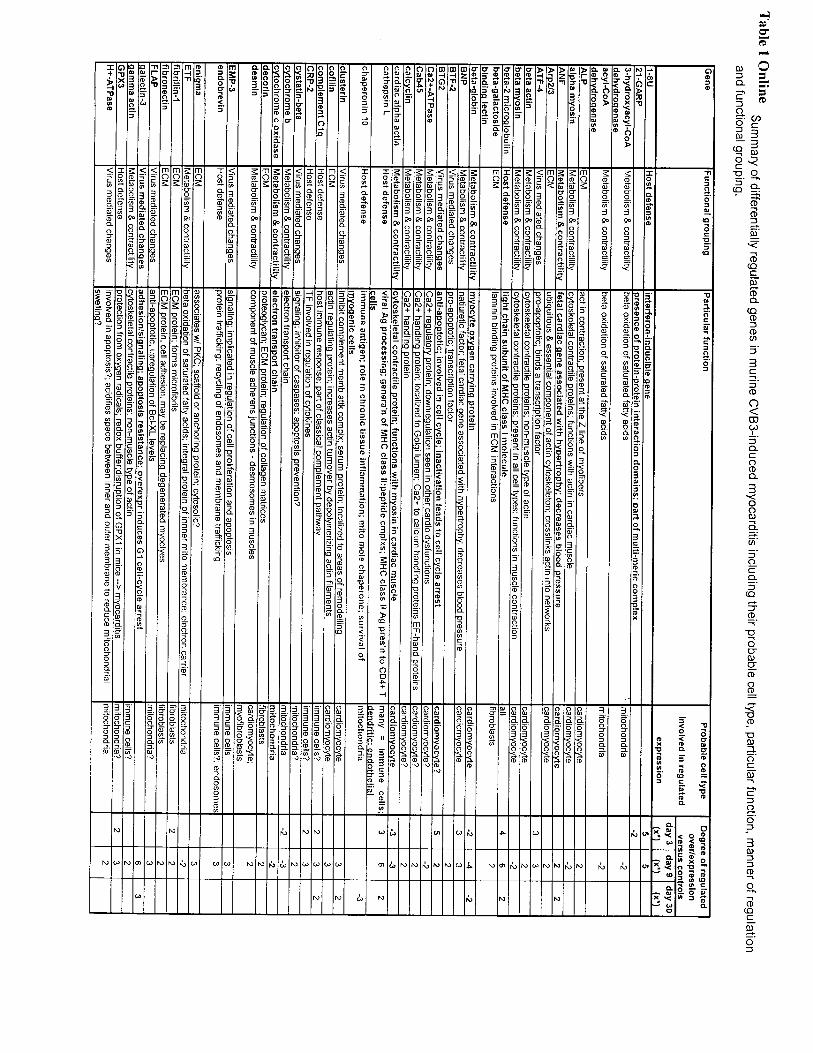
DiscussionIn a well-established model of viral infection, with conse-quent myocyte cell death, myocarditis, and reparative heartdisease, and with a microarray containing a random andsubstantial repertoire of known sequences, the temporalchanges in gene expression in a single target organ systemwere examined. Table 1 online (available at http://www.circresaha.org) summarizes 85 of the 169 known gene se-quences found to be differentially expressed in this analysis
in the context of their expression trends at each of the 3postinfection time points.
Virus-Mediated ChangesEarly events in CVB3 infection of susceptible mice have beenpostulated to include gene regulation directly related to viralreplication in the host cell. In this study, the upregulation ofpoly(A) binding protein (PABP), ubiquitin-specific proteaseUBP41, and inorganic pyrophosphatase genes appear tosupport this concept. PABP transcript upregulation is seem-ingly compensatory for viral degradation of the PABP, whichis cleaved by the CVB3 viral genome–encoded protease2Apro.18 PABP is thought to play an important role inmediating the translation of eukaryotic genes through facili-tating the binding poly(A) tails of different mRNA, therebycreating highly ordered protein-RNA structures.19 Survivingcells may require an elevated level of PABP to increasetranslation of other cellular proteins that facilitate myocyterepair and the restoration of normal cardiac muscle cellactivity and integrity.
In addition to PABP, the gene of another protein involvedin ribonucleoprotein complexes, heterogeneous nuclear ribo-nucleoprotein (hnRNP), was found to be upregulated at days3 and 9 in our model of viral myocarditis. hnRNP maysignificantly impact gene expression by modulating the ac-cessibility and/or interactions oftrans-acting factors withparticular DNA/RNA sequence elements and has been impli-cated in the translational inhibition of certain viral mRNA’s.20
The expression of other host genes modulated eitherthrough viral action or by host reaction involves a “struggle”between the expression of proapoptotic and antiapoptoticmolecules. There is normally a balance between apoptoticand antiapoptotic signals, and cell death occurs in response toa persistent shift in this balance. Although the expressions ofvarious apoptotic and antiapoptotic signaling molecules weredifferentially regulated in our study, their potential diverseand unknown functions make it impossible, in many in-stances, to make strong conclusions regarding the impact oftheir transcriptional regulation.
Of note, however, is the upregulation of the peripheral-typebenzodiazepine receptor (PBR) gene on days 3 and 9 afterinfection, which appears to contribute to cell survival. ThePBR localizes to the mitochondrial membrane, where it hasbeen found physically associated with a voltage-dependentanion channel and is a component of the mitochondrialpermeability transition pore complex (PT) formed at thecontact site between the mitochondrial inner and outer mem-branes.21 Because the PT is known to be involved in apoptosissignaling22 and opening of the PT and voltage-dependentanion channel pores are regulated by Bcl-2 and Bcl-XL,23 itis likely that PBR is a critical component in an antiapoptoticprocess. This is supported by the wealth of PBR agonists thatare potent antiapoptotic compounds.21 Thus, upregulation ofthe PBR transcript at days 3 and 9 may represent a host stressresponse aimed at cell survival.
Host Defense ResponseThere is increasing evidence that viral infection generatesreactive oxygen species24 and oxygen free radical scavengers
Figure 2. Direct viral injury of CVB3-infected myocytes. Contig-uous sections were in situ hybridized for viral RNA (A and D)and stained with hematoxylin and eosin (B and E) and Masson’strichrome (C and F) for histological examination. At day 3 afterinfection, cells that contained viral RNA were also injured asdetermined by the presence of cytopathic effect (A through C)and coagulation necrosis (D through F). Magnifications 340.
330 Circulation Research August 18, 2000
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from
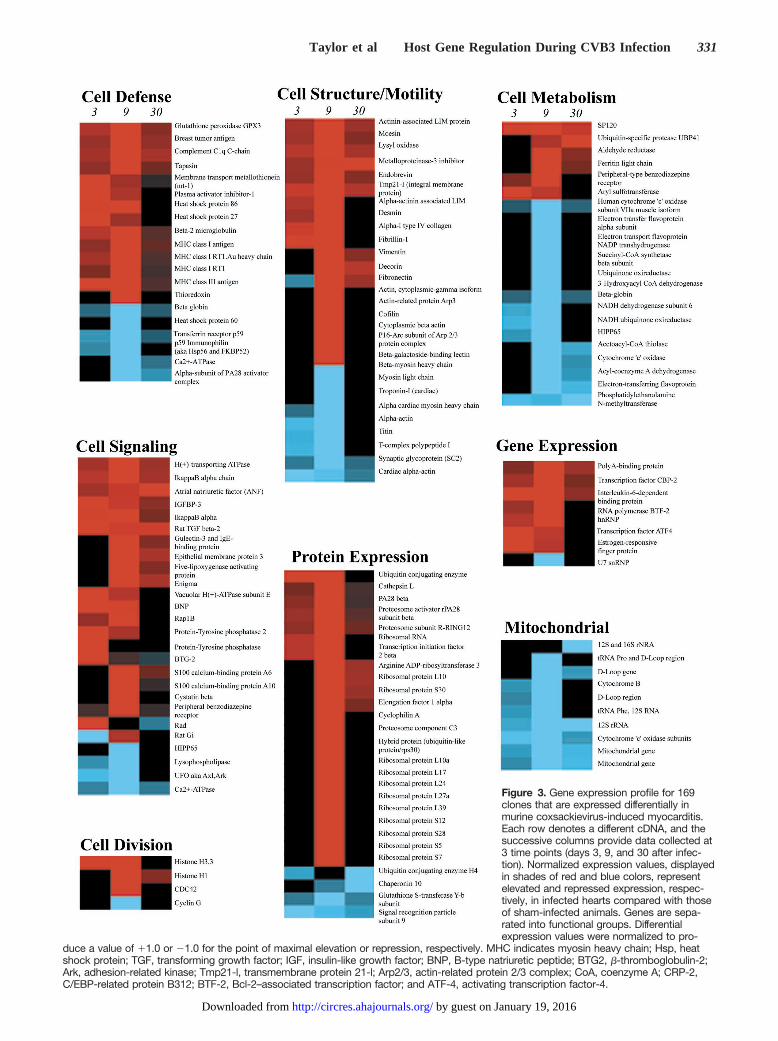
Figure 3. Gene expression profile for 169clones that are expressed differentially inmurine coxsackievirus-induced myocarditis.Each row denotes a different cDNA, and thesuccessive columns provide data collected at3 time points (days 3, 9, and 30 after infec-tion). Normalized expression values, displayedin shades of red and blue colors, representelevated and repressed expression, respec-tively, in infected hearts compared with thoseof sham-infected animals. Genes are sepa-rated into functional groups. Differentialexpression values were normalized to pro-
duce a value of 11.0 or 21.0 for the point of maximal elevation or repression, respectively. MHC indicates myosin heavy chain; Hsp, heatshock protein; TGF, transforming growth factor; IGF, insulin-like growth factor; BNP, B-type natriuretic peptide; BTG2, b-thromboglobulin-2;Ark, adhesion-related kinase; Tmp21-l, transmembrane protein 21-l; Arp2/3, actin-related protein 2/3 complex; CoA, coenzyme A; CRP-2,C/EBP-related protein B312; BTF-2, Bcl-2–associated transcription factor; and ATF-4, activating transcription factor-4.
Taylor et al Host Gene Regulation During CVB3 Infection 331
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from

protect against virus-induced myocarditis.25 Thus, it is ofinterest that host responses to CVB3 infection include in-creased expression of oxygen free radical scavenger genesconcordant with the timing of a strong, early reactive oxygenspecies presence and stress response that diminishes as theremodeling process evolves. These scavenger genes includeglutathione peroxidase, metallothionein, and thioredoxin, allof which convey protection to cardiac myocytes from reactiveoxygen species.25–27
Further response to reactive oxygen species in this modelwas seen with the day 30 downregulation of the phosphoti-dylethanolamineN-methyltransferase gene, the protein ofwhich is necessary for maintenance of membrane permeabil-ity integrity and is decreased in cardiac subcellular fractionsby excess oxygen free radicals. PhosphotidylethanolamineN-methyltransferase activity has been found to be decreasedin diseased hearts wherein oxygen free radicals are suspectedto play a role in the pathogenesis of cardiac dysfunction.28
Correspondingly, the gene expression of another enzymeinvolved in the recycling of phospholipids,29 lysophospho-lipase, was found to be downregulated at days 3 and 9. Thesefindings are intriguing, because mitochondrial membranephospholipid composition may impact electron transportchain enzyme membership and cellular respiration.30 Suchchanges are supported by our model and are described inother models of cardiac dysfunction.31,32
Changes in Host Metabolism and ContractilityTypically, a switch of the chief myocardial energy sourcefrom fatty acidb-oxidation to glycolysis is observed both inhypertrophy and in the failing heart. These changes werehighlighted by human patients with dilated cardiomyopathywherein enzymes in the mitochondrial fatty acid oxidationcycle are downregulated.33 Furthermore, downregulation ofthe expression of the medium-chain acyl–coenzyme A dehy-drogenase gene, a key fatty acidb-oxidation enzyme, hasbeen demonstrated during the progression from cardiac hy-pertrophy to ventricular dysfunction.33 In our model, theobserved downregulation of genes encoding enzymes appar-ently involved in theb-oxidation of saturated fatty acids andmetabolic mitochondrial enzymes involved in the citric acidcycle and electron transport chain agrees with other models ofcardiac dysfunction. In addition, transcripts encoded by themitochondrial genome, namely cytochromeb, the mutants ofwhich have been found to include cardiac conduction blockphenotypes,34 and 12S and 16S rRNA genes, the mutants ofwhich have been reported with hypertrophic cardiomyopa-thy,35 were found to be downregulated on days 3, 9, and 30,respectively. Because mitochondrial mutations have beenimplicated in both hypertrophic and dilated cardiomyopa-thies,35 it is tempting to speculate that downregulation ofthese and other mitochondrial transcripts may be responsiblefor the loss of function/cardiomyopathy observed in ourmodel.
In parallel to myocardial energy source switching fromb-oxidation of fatty acids to glycolysis, a reversion to thefetal energy substrate preference pattern and the regulation ofother “hypertrophy” genes in the myocarditis model may beimportant. Myocardial hypertrophy is often an early hallmark
of heart failure, occurring in response to a variety of stimuli,including pathologic factors. The hypertrophic response oc-curs as the heart attempts adaptation to increased demands onindividual myocytes for cardiac work. Our model wouldsatisfy such a setting, as noninjured myocytes compensate forinjured and dead myocytes. In most forms of cardiac hyper-trophy, there is an increase in the expression of embryonicgenes, including the genes for natriuretic peptides and fetalcontractile proteins.36 This re-expression of fetal isoformsmimics the mitogenic response of many terminally differen-tiated cell types, such as cardiomyocytes. Such responseswere seen in our model with the upregulation of genesencoding B-type natriuretic peptide, atrial natriuretic factor,b-actin, andg-actin.
Changes in ECM GenesGenetically modified mice have also implicated ECM pro-teins in the pathogenesis of heart disease. Both disruption ofdesmin, a component of muscle adherens junctions in mice,and an ECM protein defect in Syrian hamsters lead tocardiomyopathy, implicating a link between myocardial ECMand the pathogenesis of cardiomyopathy.36 In our model, alarge number of genes encoding ECM proteins and ECM/cytoskeletal linker proteins were upregulated during thecourse of disease, beginning as early as day 3. Overexpres-sion of these genes not only corresponds to an observedincrease in ECM seen in the day 30 postinfection myocarditicphenotype but also suggests an active reparative and remod-eling effort in response to myocardial damage and cell loss.
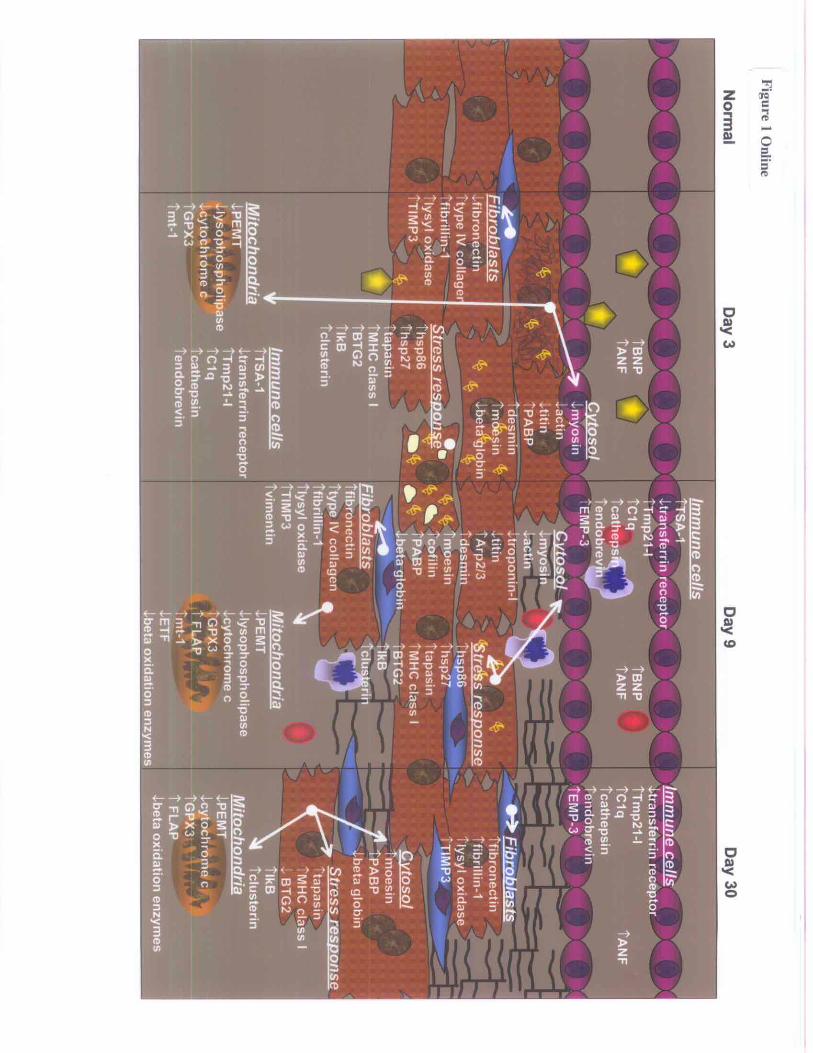
ConclusionsGene regulatory phenomena observed in our study provide anew framework for understanding the initiation and progres-sion of viral myocarditis, including several new and poten-tially important discoveries. Figure 1 online (available athttp://www.circresaha.org) summarizes several of the impor-tant regulatory events and places a microenvironmental andtemporal context for their expressive changes. Our approachto this study was based on a powerful molecular tool in anunbiased assessment of differential gene regulation. Theinter-rodent sequence similarities and homologies in manygenes (ftp://devftp.informatics.jax.org/pub/informatics/reports/index.html; http://www.latrobe.edu.au/www/genetics/reports.html) made it feasible to use the carefully prepared ratcDNA library. We used this library as our collaborators werestudying rat models of heart disease with preprepared ratchips, and no comparable ability to screen against a mouselibrary or develop a rat model of viral myocarditis wasavailable. It should be noted, however, that qualitatively thehybridization of the 2 rodent mRNAs were found to beequivalent, indicating no quantitative species-specific varia-tion, and the mouse-rat hybridization demonstrated higherquality than the rat-rat hybridization with respect to hybrid-ization specificity. Furthermore, the relative labeling intensityfor any given gene across these 2 species was identical,indicating that under our stringency conditions, no loss insensitivity occurred. Finally, and perhaps unexpectedly, aconsistently higher signal-to-noise ratio was observed withthe hybridization of mouse cDNA in comparison with rat
332 Circulation Research August 18, 2000
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from

cDNA against the rat chip. The authors have no explanationfor this except to surmise that perhaps background labeling ofthe cDNA spots includes hybridization to species-specificrepeats (the Alu repeats, for example) found not only in thegenome but also in the 39, untranslated end of the mRNAstrands. If so, cDNA made from mouse mRNA might showless complementary binding to the Alu-type repeats found inthe rat cDNA spots and demonstrate less background binding.Nevertheless, an additional species-specific approach couldpossibly yield additional insights regarding the regulation ofviral receptors, mouse-specific cytokines, and other species-specific gene expression, yet the richness of our observationsthus far has provided many potentially important new oppor-tunities to understand the pathogenesis of enteroviral heartdisease.
This initial portrait of an in vivo model of human diseaseis considered a first step, inasmuch as microarray results andtrends are limited to a portion of the total regulatory picturein cellular processes. In addition, the demonstration andexploration of transcriptionally regulated events using ex-pression microarray technologies, while qualitative, is onlyquantitative in a relative fashion.12 Furthermore, the processinvolves a mixed, interactive cell population for origin of theprobe transcripts in our model tissue. The mixed populationof cells used for probe construction not only contained manydifferent cell types including infected myocytes, but alsononinfected myocytes with infected neighbors in close prox-imity, releasing stimuli or signaling factors (autocrine orparacrine). Thus, gene regulation observed in these whole-heart gene expression assays was due to a composite portraitrather than sole regulation of myocyte gene expression inresponse to viral infection. Although not unicellular, ourapproach does capture the entire pathophysiological micro-environment associated with the myocarditic disease processand as such may provide directions of greater pertinence tothe pathogenesis and ultimate treatment of viral myocarditis.Our observations do not exclude the possible influence ofsystemic factors, originating from other organs and tissues,on the gene expression profile of the myocardium.
Future DirectionsThe value of additional studies that assess gene expressionprofiles of individual cells or cell populations are, of course,appreciated.37 However, the strengths and limitations ofeither approach necessitate gleaning insights from both. Lasercapture microdissection or laser pressure catapult will in partresolve the “whole-organ” issue, recognizing the limits interms of members of a given cell type (ie, myocytes) that canbe effectively evaluated in an experiment designed for pur-poses such as ours.38–40
Additionally, these studies on gene expression changes inmice after CVB3 infection will be expanded by our group andothers to include both more time points and additionalcomparisons such as myocarditis susceptible versus resistantand immunocompromised versus immunocompetent mousehearts. Together, these integrated approaches toward charac-terizing gene regulation in the murine model of CVB3-induced myocarditis will both further present understandingand generate numerous leads regarding investigations into
genetic governance of susceptibility, pathogenesis, and pro-gression. Furthermore, these studies will lead to heightenedunderstanding of myocardial viral infection in specific andheart dysfunction in general and may direct our attention topotential therapeutic targets for both.
AcknowledgmentsWe thank the British Columbia Health Research Foundation (20R-52234), Medical Research Council (20R-90214), and Heart andStroke Foundation of British Columbia and Yukon (20R-53837) forgrant-in-aid support. Dr Michael Allard provided helpful critique andadvice in the preparation of the manuscript. We also thank thefollowing people for their valuable contributions to the experimentsand manuscript preparation: Deborah Damm, Andrew Lam, LisaGarrard, Paul Cheung, David Granville, and Zongshu Luo.
References1. Huber SA, Gauntt CJ, Sakkinen P. Enteroviruses and myocarditis: viral
pathogenesis through replication, cytokine induction, and immunopatho-genicity.Adv Virus Res. 1998;51:35–80.
2. Grist NR, Reid D. Epidemiology of viral infections of the heart. In: SmithKM, Lauffer MA, eds. Viral Infections in the Heart.London, UK:Edward Arnold; 1993.
3. Woodruff JF. Viral myocarditis: a review.Am J Pathol. 1980;101:425–484.
4. Wolfgram LJ, Beisel KW, Herskowitz A, Rose NR. Variations in thesusceptibility to Coxsackievirus B3-induced myocarditis among differentstrains of mice.J Immunol. 1986;136:1846–1852.
5. McManus BM, Gauntt CJ, Cassling RS. Immunopathologic basis ofmyocardial injury.Cardiovasc Clin. 1988;18:163–184.
6. Huber SA. Coxsackievirus-induced myocarditis is dependent on distinctimmunopathogenic responses in different strains of mice.Lab Invest.1997;76:691–701.
7. McManus BM, Chow LH, Wilson JE, Anderson DR, Gulizia JM, GaunttCJ, Klingel KE, Beisel KW, Kandolf R. Direct myocardial injury byenterovirus: a central role in the evolution of murine myocarditis.ClinImmunol Immunopathol. 1993;68:159–169.
8. Chow LH, Gauntt CJ, McManus BM. Differential effects of myocarditicvariants of Coxsackievirus B3 in inbred mice: a pathologic character-ization of heart tissue damage.Lab Invest. 1991;64:55–64.
9. Chow LH, Gauntt CJ, McManus BM. Early cellular infiltrates in cox-sackievirus B3 murine myocarditis. In: Schultheiss H-P, ed.NewConcepts in Viral Heart Disease: Virology, Immunology, and ClinicalManagement. Berlin, Germany; 1988.
10. Gomez RM, Castagnino CG, Berria MI. Extracellular matrix remodellingafter coxsackievirus B3-induced murine myocarditis.Int J Exp Pathol.1992;73:643–653.
11. Yang D, Yu J, Luo Z, Carthy CM, Wilson JE, Liu Z, McManus BM. Viralmyocarditis: identification of five differentially expressed genes in cox-sackievirus B3-infected mouse heart.Circ Res. 1999;84:704–712.
12. Duggan DJ, Bittner M, Chen Y, Meltzer P, Trent JM. Expression pro-filing using cDNA microarrays.Nat Genet. 1999;21:10–14.
13. Anderson DR, Carthy CM, Wilson JE, Yang D, Devine DV, McManusBM. Complement component 3 interactions with coxsackievirus B3capsid proteins: innate immunity and the rapid formation of splenicantiviral germinal centers.J Virol. 1997;71:8841–8845.
14. Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM,Zheng Q, Protter AA, Schreiner GF, White RT. Altered patterns of geneexpression in response to myocardial infarction.Circ Res. 2000;86:939–945.
15. Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JCF, Trent JM,Staudt LM, Hudson J Jr, Boguski MS, Lashkari D, Shalon D, Botstein D,Brown PO. The transcriptional program in the response of human fibro-blasts to serum.Science. 1999;283:83–87.
16. Aitman TJ, Glazier AM, Wallace CA, Cooper LD, Norsworthy PJ, WahidFN, Al-Majali KM, Trembling PM, Mann CJ, Shoulders CC, Graf D, St.Lezin E, Kurtz TW, Kren V, Pravenec M, Ibrahimi A, Abumrad NA,Stanton LW, Scott J. Identification of Cd36 (Fat) as an insulin-resistancegene causing defective fatty acid and glucose metabolism in hypertensiverats.Nat Genet1999;21:76–83.
Taylor et al Host Gene Regulation During CVB3 Infection 333
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from

17. Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallelhuman genome analysis: microarray-based expression monitoring of1000 genes.Proc Natl Sci U S A.1996;93:10614–10619.
18. Kerekatte V, Keiper BD, Badorff C, Cai A, Knowlton KU, Rhoads RE.Cleavage of poly(A)-binding protein by Coxsackievirus 2A protease invitro and in vivo: another mechanism for host protein synthesis shutoff?J Virol. 1999;73:709–717.
19. Kuhn U, Pieler T.Xenopuspoly(A) binding protein: functional domainsin RNA binding and protein-protein interaction.J Mol Biol. 1996;256:20–30.
20. Krecic AM, Swanson MS. hnRNP complexes: composition, structure,and function.Curr Opin Cell Biol. 1999;11:363–371.
21. Bono F, Lamarche I, Prabonnaud V, Le Fur G, Herbert JM. Peripheralbenzodiazepine receptor agonists exhibit potent antiapoptotic activities.Biochem Biophys Res Commun. 1999;265:457–461.
22. Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B,Andrews D, Duclohier H, Reed JC, Kroemer G. Bcl-2 and Bax regulatethe channel activity of the mitochondrial adenine nucleotide translocator.Oncogene. 2000;19:329–336.
23. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate therelease of apoptogenic cytochromec by the mitochondrial channelVDAC. Nature. 1999;399:483–487.
24. Schwarz KB. Oxidative stress during viral infection: a review.Free RadicBiol Med. 1996;21:641–649.
25. Beck MA, Esworthy RS, Ho YS, Chu FF. Glutathione peroxidase protectsmice from viral-induced myocarditis.FASEB J. 1998;12:1143–1149.
26. Kang YJ, Li G, Saari JT. Metallothionein inhibits ischemia-reperfusioninjury in mouse heart.Am J Physiol. 1999;276:H993–H997.
27. Isowa N, Yoshimura T, Kosaka S, Liu M, Hitomi S, Yodoi J, Wada H.Human thioredoxin attenuates hypoxia-reoxygenation injury of murineendothelial cells in a thiol-free condition.J Cell Physiol. 2000;182:33–40.
28. Kaneko M, Panagia V, Paolillo G, Majumder S, Ou C, Dhalla NS.Inhibition of cardiac phosphatidylethanolamineN-methylation by oxygenfree radicals.Biochim Biophys Acta. 1990;1021:33–38.
29. Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. NewYork: Worth Publishers; 1993.
30. Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Alterations incarnitine-acylcarnitine translocase activity and in phospholipid compo-sition in heart mitochondria from hypothyroid rats.Biochim BiophysActa. 1997;1362:193–200.
31. Dhalla NS, Golfman L, Takeda S, Takeda N, Nagano M. Evidence for therole of oxidative stress in acute ischemic heart disease: a brief review.Can J Cardiol. 1999;15:587–593.
32. Vogt AM, Kubler W. Heart failure: is there an energy deficit contributingto contractile dysfunction?Basic Res Cardiol. 1998;93:1–10.
33. Sack MN, Kelly DP. The energy substrate switch during development ofheart failure: gene regulatory mechanisms.Int J Mol Med. 1998;1:17–24.
34. Zanssen S, Molnar M, Buse G, Schroder JM. Mitochondrial cytochromeb gene deletion in Kearns-Sayre syndrome associated with a subclinicaltype of peripheral neuropathy.Clin Neuropathol. 1998;17:291–296.
35. DiMauro S, Hirano M. Mitochondria and heart disease.Curr OpinCardiol. 1998;13:190–197.
36. Hunter JJ, Chien KR. Mechanisms of disease: signaling pathways forcardiac hypertrophy and failure.N Engl J Med. 1999;341:1276–1283.
37. Geiss GK, Bumgarner RE, An MC, Agy MB, van ’t Wout AB,Hammersmark E, Carter VS, Upchurch D, Mullins JI, Katze MG.Large-scale monitoring of host cell gene expression during HIV-1infection using cDNA microarrays.Virology. 2000;266:8–16.
38. Banks RE, Dunn MJ, Forbes MA, Stanley A, Pappin D, Naven T, GoughM, Harnden P, Selby PJ. The potential use of laser capture microdis-section to selectively obtain distinct populations of cells for proteomicanalysis: preliminary findings.Electrophoresis. 1999;20:689–700.
39. McCarthy RE 3rd, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK,Hare JM, Baughman KL. Long-term outcome of fulminant myocarditis ascompared with acute (nonfulminant) myocarditis.N Engl J Med. 2000;342:690–695.
40. Kohda Y, Murakami H, Moe OW, Star RA. Analysis of segmental renalgene expression by laser capture microdissection.Kidney Int. 2000;57:321–331.
334 Circulation Research August 18, 2000
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from


Schreiner, Lawrence W. Stanton and Bruce M. McManusLydia A. Taylor, Christopher M. Carthy, Decheng Yang, Kareem Saad, Donald Wong, George
MicroarraysHost Gene Regulation During Coxsackievirus B3 Infection in Mice: Assessment by
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2000 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.87.4.3282000;87:328-334Circ Res.
http://circres.ahajournals.org/content/87/4/328World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2000/08/11/87.4.328.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on January 19, 2016http://circres.ahajournals.org/Downloaded from




















