
Highly Stable Binding Proteins Derived from the Hyperthermophilic Sso7d Scaffold
16
Highly Stable Binding Proteins Derived from the Hyperthermophilic Sso7d Scaffold Nimish Gera, Mahmud Hussain, Robert C. Wright and Balaji M. Rao⁎ Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, USA Received 11 March 2011; accepted 8 April 2011 Available online 16 April 2011 Edited by F. Schmid Keywords: hyperthermophilic protein scaffolds; yeast surface display; protein engineering; stability; alternate scaffold We have shown that highly stable binding proteins for a wide spectrum of targets can be generated through mutagenesis of the Sso7d protein from the hyperthermophilic archaeon Sulfolobus solfataricus. Sso7d is a small (∼ 7 kDa, 63 amino acids) DNA-binding protein that lacks cysteine residues and has a melting temperature of nearly 100 °C. We generated a library of 10 8 Sso7d mutants by randomizing 10 amino acid residues on the DNA-binding surface of Sso7d, using yeast surface display. Binding proteins for a diverse set of model targets could be isolated from this library; our chosen targets included a small organic molecule (fluorescein), a 12 amino acid peptide fragment from the C-terminus of β-catenin, the model proteins hen egg lysozyme and streptavidin, and immunoglobulins from chicken and mouse. Without the application of any affinity maturation strategy, the binding proteins isolated had equilibrium dissociation constants in the nanomolar to micromolar range. Further, Sso7d-derived binding proteins could discrim- inate between closely related immunoglobulins. Mutant proteins based on Sso7d were expressed at high yields in the Escherichia coli cytoplasm. Despite extensive mutagenesis, Sso7d mutants have high thermal stability; five of six mutants analyzed have melting temperatures N 89 °C. They are also resistant to chemical denaturation by guanidine hydrochloride and retain their secondary structure after extended incubation at extreme pH values. Because of their favorable properties, such as ease of recombinant expression, and high thermal, chemical and pH stability, Sso7d-derived binding proteins will have wide applicability in several areas of biotechnology and medicine. © 2011 Elsevier Ltd. All rights reserved. Introduction The generation of binding molecules that specifi- cally recognize target species is of great importance in clinical applications, biotechnology and funda- mental research in biology. Historically, antibodies or immunoglobulins have been the most commonly used reagents for molecular recognition. Antibodies that bind specifically to a wide variety of targets can be obtained through alteration of the amino acid composition of six hypervariable loop regions, called complementarity determining regions, on the im- munoglobulin framework. However, antibodies are limited by the low level of stability resulting from *Corresponding author. E-mail address: [email protected]. Abbreviations used: FACS, fluorescence-activated cell sorting; BSA, bovine serum albumin; mIgG, mouse immunoglobulin G; cIgY, chicken immunoglobulin G; rIgG, rabbit immunoglobulin G; gIgG, goat immunoglobulin G; hFc, Fc portion of human immunoglobulin G; PBS-BSA, phosphate-buffered saline with 0.1% (w/v) bovine serum albumin; Strep-PE, streptavidin-phycoerythrin; DSC, differential scanning calorimetry; Gdn-HCl, guanidine hydrochloride; SH3, src homology 3; IMAC, immobilized metal affinity chromatography; DARPin, designed ankyrin repeat protein. doi:10.1016/j.jmb.2011.04.020 J. Mol. Biol. (2011) 409, 601–616 Contents lists available at www.sciencedirect.com Journal of Molecular Biology journal homepage: http://ees.elsevier.com.jmb 0022-2836/$ - see front matter © 2011 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Highly Stable Binding Proteins Derived from the Hyperthermophilic Sso7d Scaffold

doi:10.1016/j.jmb.2011.04.020 J. Mol. Biol. (2011) 409, 601–616
Contents lists available at www.sciencedirect.com
Journal of Molecular Biologyj ourna l homepage: ht tp : / /ees .e lsev ie r.com. jmb
Highly Stable Binding Proteins Derived from theHyperthermophilic Sso7d Scaffold
Nimish Gera, Mahmud Hussain, Robert C. Wright and Balaji M. Rao⁎Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, NC 27695, USA
Received 11 March 2011;accepted 8 April 2011Available online16 April 2011
Edited by F. Schmid
Keywords:hyperthermophilic proteinscaffolds;yeast surface display;protein engineering;stability;alternate scaffold
*Corresponding author. E-mail addrAbbreviations used: FACS, fluore
sorting; BSA, bovine serum albuminimmunoglobulin G; cIgY, chicken imrIgG, rabbit immunoglobulin G; gIgimmunoglobulin G; hFc, Fc portionimmunoglobulin G; PBS-BSA, phospwith 0.1% (w/v) bovine serum albustreptavidin-phycoerythrin; DSC, dicalorimetry; Gdn-HCl, guanidine hyhomology 3; IMAC, immobilized mchromatography; DARPin, designedprotein.
0022-2836/$ - see front matter © 2011 E
We have shown that highly stable binding proteins for a wide spectrum oftargets can be generated through mutagenesis of the Sso7d protein from thehyperthermophilic archaeon Sulfolobus solfataricus. Sso7d is a small (∼7 kDa,63 amino acids) DNA-binding protein that lacks cysteine residues and has amelting temperature of nearly 100 °C. We generated a library of 108 Sso7dmutants by randomizing 10 amino acid residues on the DNA-bindingsurface of Sso7d, using yeast surface display. Binding proteins for a diverseset of model targets could be isolated from this library; our chosen targetsincluded a small organic molecule (fluorescein), a 12 amino acid peptidefragment from the C-terminus of β-catenin, the model proteins hen egglysozyme and streptavidin, and immunoglobulins from chicken andmouse.Without the application of any affinity maturation strategy, the bindingproteins isolated had equilibrium dissociation constants in the nanomolar tomicromolar range. Further, Sso7d-derived binding proteins could discrim-inate between closely related immunoglobulins. Mutant proteins based onSso7d were expressed at high yields in the Escherichia coli cytoplasm.Despite extensive mutagenesis, Sso7d mutants have high thermal stability;five of six mutants analyzed have melting temperatures N89 °C. They arealso resistant to chemical denaturation by guanidine hydrochloride andretain their secondary structure after extended incubation at extreme pHvalues. Because of their favorable properties, such as ease of recombinantexpression, and high thermal, chemical and pH stability, Sso7d-derivedbinding proteins will have wide applicability in several areas ofbiotechnology and medicine.
© 2011 Elsevier Ltd. All rights reserved.
ess: [email protected] cell; mIgG, mousemunoglobulin G;G, goatof humanhate-buffered salinemin; Strep-PE,fferential scanningdrochloride; SH3, srcetal affinityankyrin repeat
lsevier Ltd. All rights reserve
Introduction
The generation of binding molecules that specifi-cally recognize target species is of great importancein clinical applications, biotechnology and funda-mental research in biology. Historically, antibodiesor immunoglobulins have been the most commonlyused reagents for molecular recognition. Antibodiesthat bind specifically to a wide variety of targets canbe obtained through alteration of the amino acidcomposition of six hypervariable loop regions, calledcomplementarity determining regions, on the im-munoglobulin framework. However, antibodies arelimited by the low level of stability resulting from
d.

602 Sso7d Scaffold Stable Binding Proteins
their complex multi-subunit structure, and relativelyhigh cost of production.To overcome the limitations of antibodies, several
alternative protein frameworks have been used asscaffolds or “templates” for engineering molecularrecognition.1-3 A protein variant that binds aparticular target with high affinity and specificityis generated through a combination of amino acidsubstitutions and sequence insertions or deletions incertain regions of the scaffold protein. Typically,binding proteins are isolated from a combinatoriallibrary of scaffold protein mutants. Common exam-ples include the 10th domain of fibronectin,4-5
designed ankyrin repeat proteins (DARPins)7,8 andaffibodies.9,10 Proteins derived from alternative non-immunoglobulin scaffolds can provide severaladvantages over antibodies such as low molecularmass, higher stability and ease of recombinantexpression in bacterial systems.Since binding proteins are obtained through
mutagenesis of the scaffold protein, tolerance to adiverse set of amino acid changes while retaining itsnative structure is the hallmark of an ideal scaffold.Highly stable proteins are more tolerant of a widerange of mutations and therefore have enhancedevolvability.11 Consequently, highly stable scaffoldproteins are more likely to yield binding proteins toa wide variety of target species. In nature, hyper-thermophilic archaea and bacteria have evolved towithstand extreme conditions of temperature andpH. Not surprisingly, several proteins from theseorganisms have very high thermal stability andresistance to proteolysis.12,13 Further, many of theseproteins also have low molecular mass and lackdisulfide bonds, enabling their facile expression inthe reducing cytoplasm of Escherichia coli. As aresult, hyperthermophilic proteins are excellentcandidates for use as protein scaffolds to engineermolecular recognition, in addition to being attrac-tive model proteins for obtaining insight into proteinstability. Nevertheless, the generation of bindingproteins from proteins of hyperthermophilic originusing combinatorial library screening methods hasreceived scant attention. Here, we show that theultrastable Sso7d protein from the hyperthermo-philic archaeon Sulfolobus solfataricus can be used asa versatile scaffold to generate highly stable bindingproteins for a wide range of target species.The Sso7d protein is a small (∼7 kDa, 63 amino
acids) DNA-binding protein containing an SH3-likefold consisting of an incomplete β-barrel with fiveβ strands and a C-terminal α helix.14-17 Sso7d ishighly stable with a melting temperature of nearly100 °C and lacks cysteine.18 We investigated thepossibility of generating proteins that bind to awide spectrum of target species through mutagen-esis of the DNA-binding surface of Sso7d. Towardsthis end, we generated a modestly sized combina-torial library of ∼108 Sso7d mutants using yeast
surface display,19,20 through randomization of 10amino acid residues on the DNA-binding surface ofSso7d. Subsequently, we screened this library andisolated binding proteins for a diverse set of modeltargets, with equilibrium dissociation constants(KD) in the micromolar to nanomolar range. Ourset of chosen targets included a small organicmolecule, a peptide and model proteins of varioussizes, including immunoglobulin proteins fromdifferent species. Isolation of binding proteins thatcan discriminate between closely related immuno-globulins demonstrates the ability to engineerhighly specific molecular recognition through mu-tagenesis of the Sso7d scaffold. Further, we recom-binantly expressed several Sso7d-derived bindingproteins in E. coli and evaluated their stability underdenaturing conditions. Despite mutation of 10 outof 63 residues (15%) in Sso7d, the mutant bindingproteins show remarkable stability against thermaland chemical stress as well as extended exposure toa wide range of pH values (0.33 12.5). Previously,binding proteins to the membrane protein PulDhave been isolated using ribosome display, from acombinatorial library of 3×1012 mutants obtainedthrough randomization of 14 residues on the nearlyidentical Sac7d from Sulfolobus acidocaldarius.21
Taken together, our results show that stable bindingproteins for diverse target species can be generatedusing the Sso7d scaffold. Because of their favorableproperties such as low molecular mass, ease ofbacterial expression and high level of stability, weexpect that the Sso7d-derived binding proteinscan be used in several clinical and biotechnologyapplications.
Results
Construction of Sso7d mutant library usingyeast surface display
We constructed a library of Sso7d variantsthrough mutation of 10 surface-exposed residuesin the DNA-binding region of Sso7d, as shown inFig. 1. The wild type Sso7d protein is reported tohave RNase activity, which could limit the appli-cation of Sso7d-derived mutants in the context ofspecific intracellular inhibition of target proteins ordomains thereof. Interestingly, single point muta-tions K12L and E35L completely abolish the RNaseactivity of Sso7d without affecting its thermalstability.22 Therefore, an E35L mutant of Sso7dwas used as the starting point for construction ofthe library. The DNA sequences encoding the Sso7dmutant library were obtained through synthesis ofa single oligonucleotide with NNN degeneratecodons at positions encoding the residues K20,K21, W23, V25, M28, S30, T32, T40, R42 and A44.
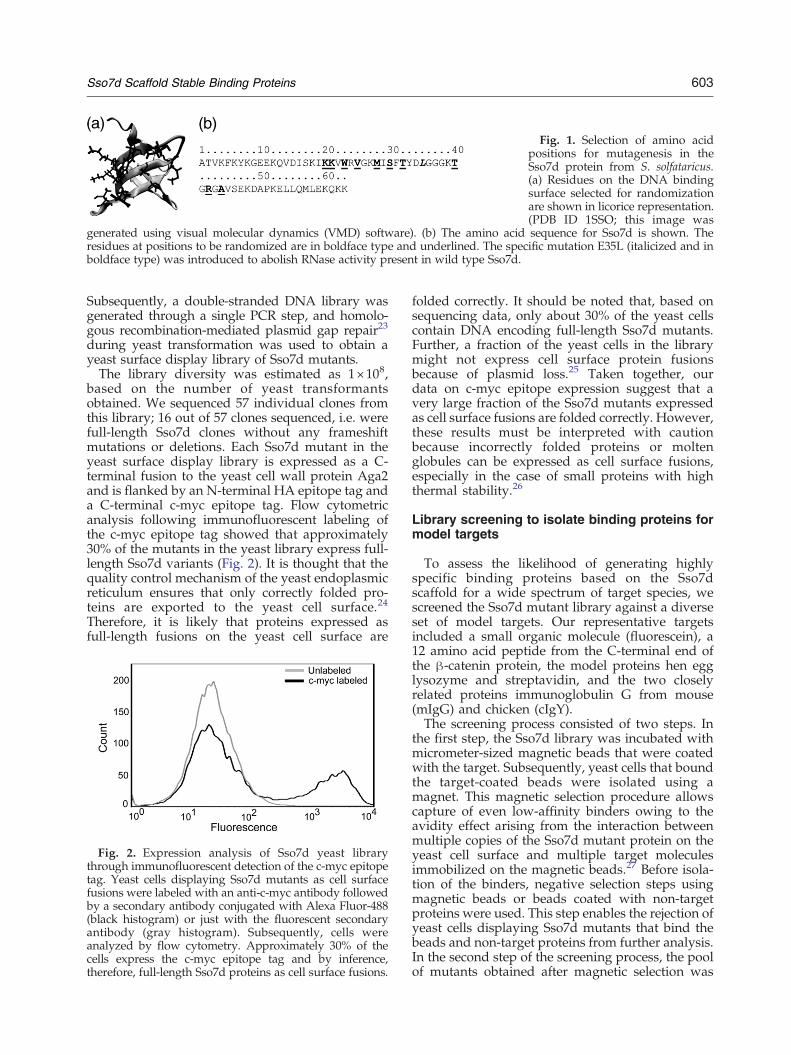
Fig. 1. Selection of amino acidpositions for mutagenesis in theSso7d protein from S. solfataricus.(a) Residues on the DNA bindingsurface selected for randomizationare shown in licorice representation.(PDB ID 1SSO; this image was
generated using visual molecular dynamics (VMD) software). (b) The amino acid sequence for Sso7d is shown. Theresidues at positions to be randomized are in boldface type and underlined. The specific mutation E35L (italicized and inboldface type) was introduced to abolish RNase activity present in wild type Sso7d.
603Sso7d Scaffold Stable Binding Proteins
Subsequently, a double-stranded DNA library wasgenerated through a single PCR step, and homolo-gous recombination-mediated plasmid gap repair23
during yeast transformation was used to obtain ayeast surface display library of Sso7d mutants.The library diversity was estimated as 1×108,
based on the number of yeast transformantsobtained. We sequenced 57 individual clones fromthis library; 16 out of 57 clones sequenced, i.e. werefull-length Sso7d clones without any frameshiftmutations or deletions. Each Sso7d mutant in theyeast surface display library is expressed as a C-terminal fusion to the yeast cell wall protein Aga2and is flanked by an N-terminal HA epitope tag anda C-terminal c-myc epitope tag. Flow cytometricanalysis following immunofluorescent labeling ofthe c-myc epitope tag showed that approximately30% of the mutants in the yeast library express full-length Sso7d variants (Fig. 2). It is thought that thequality control mechanism of the yeast endoplasmicreticulum ensures that only correctly folded pro-teins are exported to the yeast cell surface.24
Therefore, it is likely that proteins expressed asfull-length fusions on the yeast cell surface are
Fig. 2. Expression analysis of Sso7d yeast librarythrough immunofluorescent detection of the c-myc epitopetag. Yeast cells displaying Sso7d mutants as cell surfacefusions were labeled with an anti-c-myc antibody followedby a secondary antibody conjugated with Alexa Fluor-488(black histogram) or just with the fluorescent secondaryantibody (gray histogram). Subsequently, cells wereanalyzed by flow cytometry. Approximately 30% of thecells express the c-myc epitope tag and by inference,therefore, full-length Sso7d proteins as cell surface fusions.
folded correctly. It should be noted that, based onsequencing data, only about 30% of the yeast cellscontain DNA encoding full-length Sso7d mutants.Further, a fraction of the yeast cells in the librarymight not express cell surface protein fusionsbecause of plasmid loss.25 Taken together, ourdata on c-myc epitope expression suggest that avery large fraction of the Sso7d mutants expressedas cell surface fusions are folded correctly. However,these results must be interpreted with cautionbecause incorrectly folded proteins or moltenglobules can be expressed as cell surface fusions,especially in the case of small proteins with highthermal stability.26
Library screening to isolate binding proteins formodel targets
To assess the likelihood of generating highlyspecific binding proteins based on the Sso7dscaffold for a wide spectrum of target species, wescreened the Sso7d mutant library against a diverseset of model targets. Our representative targetsincluded a small organic molecule (fluorescein), a12 amino acid peptide from the C-terminal end ofthe β-catenin protein, the model proteins hen egglysozyme and streptavidin, and the two closelyrelated proteins immunoglobulin G from mouse(mIgG) and chicken (cIgY).The screening process consisted of two steps. In
the first step, the Sso7d library was incubated withmicrometer-sized magnetic beads that were coatedwith the target. Subsequently, yeast cells that boundthe target-coated beads were isolated using amagnet. This magnetic selection procedure allowscapture of even low-affinity binders owing to theavidity effect arising from the interaction betweenmultiple copies of the Sso7d mutant protein on theyeast cell surface and multiple target moleculesimmobilized on the magnetic beads.27 Before isola-tion of the binders, negative selection steps usingmagnetic beads or beads coated with non-targetproteins were used. This step enables the rejection ofyeast cells displaying Sso7d mutants that bind thebeads and non-target proteins from further analysis.In the second step of the screening process, the poolof mutants obtained after magnetic selection was

604 Sso7d Scaffold Stable Binding Proteins
subjected to fluorescence-activated cell sorting(FACS). Multiple rounds of FACS resulted in apool of mutants with the highest binding affinity fora given target. For each target, we sequencedplasmid DNA isolated from five individual yeastclones in the final pool of mutants after FACS andthe results are shown in Table 1.Five distinct Sso7d mutants were obtained from
the pool of binders for lysozyme and the peptidefragment from β-catenin, two distinct clones wereobtained in the case of binders to mIgG and a singledominant clone was found for binders to cIgY,streptavidin and fluorescein. On the basis of theanalysis of the 15 distinct Sso7d mutants obtainedfor the six targets using their GRAVY score,28 it isseen that the mutant Sso7d proteins are, in general,more hydrophobic than the wild type Sso7d protein.An overall increase in aromaticity of the bindinginterface, relative to that of the wild type protein, isobserved in the case of binders to mIgG, streptavi-din, lysozyme and fluorescein; three or morearomatic residues are present in these proteins.
Analysis of binding affinity and specificity
A single clone from the pool of mutants with thehighest affinity for each target was selected forfurther analysis of binding affinity and specificityand the selected clones are given in Table 1. Theequilibrium dissociation constants (KD) for thebinding interaction between the selected Sso7d
Table 1. Protein sequences of the Sso7d mutants
Wild type Sso7dTargetmIgG
Sso7d-mIgGa (4)b
Sso7d-mIgG-21Streptavidin
Sso7d-streptavidina (5)b
cIgYSso7d-cIgYa (5)b
FluoresceinSso7d-fluoresceina (5)b
β-Catenin peptideSso7d-β-catenin peptidea
Sso7d-β- catenin peptide -1Sso7d-β- catenin peptide -6Sso7d-β- catenin peptide -9Sso7d-β- catenin peptide -10
LysozymeSso7d-lysozymea
Sso7d-lysozyme-12Sso7d-lysozyme-13Sso7d-lysozyme-18Sso7d-lysozyme-20
The corresponding sequence for the wild type protein is shown as a ra These proteins were chosen for further characterization.b For each target, five individual clones from the pool of binders
parentheses is the number of identical DNA sequences obtained.
mutants and their respective targets were deter-mined through titration using proteins displayed onyeast cells. Briefly, yeast cells expressing the Sso7dmutants as cell surface fusions were incubated withvarious concentrations of the target species. At eachconcentration of target, the fraction of cell surfacefusions bound by the target was determined by flowcytometric analysis following indirect immunofluo-rescent labeling of the target. Subsequently, for eachmutant, data from at least three different experi-ments were fit to a simple one-step binding isothermand KD was estimated as described.29 It is importantto note that many studies have demonstrated theequivalence of KD values determined using yeastcell surface titrations to those obtained when solubleprotein is used for analysis.6 Estimated KD valuesand their associated 68% confidence intervals aregiven in Table 2. The Sso7dmutants analyzed have arange of binding affinities for their respectivetargets. Not surprisingly, the lowest binding affin-ities were observed in the case of Sso7d mutants thatbind the small molecule fluorescein and the peptidefragment from β-catenin. The estimates of KD forSso7d mutants binding immunoglobulins andstreptavidin might be influenced by the avidityeffect; the immunoglobulins and streptavidin havetwo and four identical subunits, respectively, anda single target molecule might interact with morethan one cell surface-displayed Sso7d protein.Nevertheless, assuming that steric considerationsallow the immunoglobulins and streptavidin to
20........30........40....
KKVWRVGKMISFTYDLGGGKTGRGAVSso7d mutant sequence
YLVLRLGKFIYFYYDLGGGKLGLGHVLYVKRAGKYITFWYDLGGGKLGSGFV
WIVARDGKWIDFSYDLGGGKSGIGYV
DGVSRMGKLIVFTYDLGGGKRGIGIV
FRVIRSGKAIRFLYDLGGGKFGYGVV
GVVGRCGKVIVFPYDLGGGKLGLGTVKSVLRTGKIIIFPYDLGGGKLGIGIVAIVARFGKQILFPYDLGGGKLGIGIVSWVSRYGKAILFLYDLGGGKMGRGVVKCVHRCGKAIFFLYDLGGGKFGAGLV
CFVFRWGKCICFDYDLGGGKQGSGCVFLVNRGGKWIFFLYDLGGGKEGEGNVFLVYRWGKSIEFEYDLGGGKLGAGLVSLVLRHGKLILFIYDLGGGKLGGGEVAYVNRSGKFILFSYDLGGGKFGTGEV
eference. Mutated positions are in boldface type and underlined.
after the final cytometric sort were sequenced. The number in
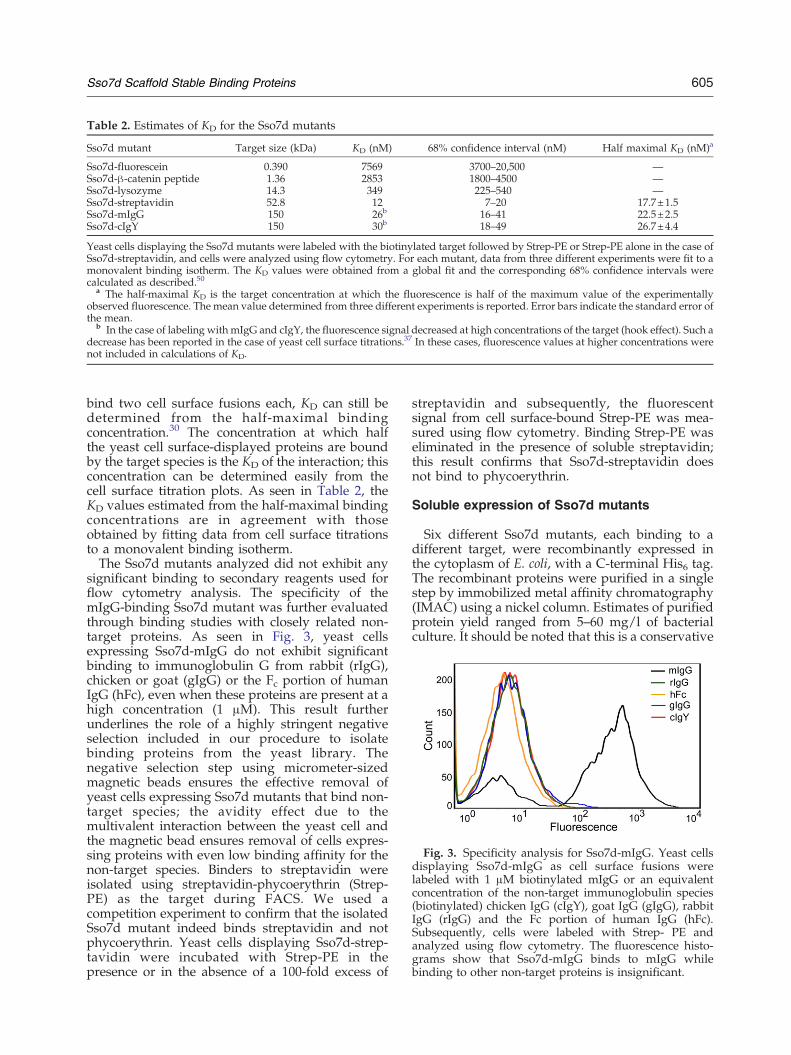
Fig. 3. Specificity analysis for Sso7d-mIgG. Yeast cellsdisplaying Sso7d-mIgG as cell surface fusions werelabeled with 1 μM biotinylated mIgG or an equivalentconcentration of the non-target immunoglobulin species(biotinylated) chicken IgG (cIgY), goat IgG (gIgG), rabbitIgG (rIgG) and the Fc portion of human IgG (hFc).Subsequently, cells were labeled with Strep- PE andanalyzed using flow cytometry. The fluorescence histo-grams show that Sso7d-mIgG binds to mIgG whilebinding to other non-target proteins is insignificant.
Table 2. Estimates of KD for the Sso7d mutants
Sso7d mutant Target size (kDa) KD (nM) 68% confidence interval (nM) Half maximal KD (nM)a
Sso7d-fluorescein 0.390 7569 3700–20,500 —Sso7d-β-catenin peptide 1.36 2853 1800–4500 —Sso7d-lysozyme 14.3 349 225–540 —Sso7d-streptavidin 52.8 12 7–20 17.7±1.5Sso7d-mIgG 150 26b 16–41 22.5±2.5Sso7d-cIgY 150 30b 18–49 26.7±4.4
Yeast cells displaying the Sso7d mutants were labeled with the biotinylated target followed by Strep-PE or Strep-PE alone in the case ofSso7d-streptavidin, and cells were analyzed using flow cytometry. For each mutant, data from three different experiments were fit to amonovalent binding isotherm. The KD values were obtained from a global fit and the corresponding 68% confidence intervals werecalculated as described.50
a The half-maximal KD is the target concentration at which the fluorescence is half of the maximum value of the experimentallyobserved fluorescence. The mean value determined from three different experiments is reported. Error bars indicate the standard error ofthe mean.
b In the case of labeling with mIgG and cIgY, the fluorescence signal decreased at high concentrations of the target (hook effect). Such adecrease has been reported in the case of yeast cell surface titrations.37 In these cases, fluorescence values at higher concentrations werenot included in calculations of KD.
605Sso7d Scaffold Stable Binding Proteins
bind two cell surface fusions each, KD can still bedetermined from the half-maximal bindingconcentration.30 The concentration at which halfthe yeast cell surface-displayed proteins are boundby the target species is the KD of the interaction; thisconcentration can be determined easily from thecell surface titration plots. As seen in Table 2, theKD values estimated from the half-maximal bindingconcentrations are in agreement with thoseobtained by fitting data from cell surface titrationsto a monovalent binding isotherm.The Sso7d mutants analyzed did not exhibit any
significant binding to secondary reagents used forflow cytometry analysis. The specificity of themIgG-binding Sso7d mutant was further evaluatedthrough binding studies with closely related non-target proteins. As seen in Fig. 3, yeast cellsexpressing Sso7d-mIgG do not exhibit significantbinding to immunoglobulin G from rabbit (rIgG),chicken or goat (gIgG) or the Fc portion of humanIgG (hFc), even when these proteins are present at ahigh concentration (1 μM). This result furtherunderlines the role of a highly stringent negativeselection included in our procedure to isolatebinding proteins from the yeast library. Thenegative selection step using micrometer-sizedmagnetic beads ensures the effective removal ofyeast cells expressing Sso7d mutants that bind non-target species; the avidity effect due to themultivalent interaction between the yeast cell andthe magnetic bead ensures removal of cells expres-sing proteins with even low binding affinity for thenon-target species. Binders to streptavidin wereisolated using streptavidin-phycoerythrin (Strep-PE) as the target during FACS. We used acompetition experiment to confirm that the isolatedSso7d mutant indeed binds streptavidin and notphycoerythrin. Yeast cells displaying Sso7d-strep-tavidin were incubated with Strep-PE in thepresence or in the absence of a 100-fold excess of
streptavidin and subsequently, the fluorescentsignal from cell surface-bound Strep-PE was mea-sured using flow cytometry. Binding Strep-PE waseliminated in the presence of soluble streptavidin;this result confirms that Sso7d-streptavidin doesnot bind to phycoerythrin.
Soluble expression of Sso7d mutants
Six different Sso7d mutants, each binding to adifferent target, were recombinantly expressed inthe cytoplasm of E. coli, with a C-terminal His6 tag.The recombinant proteins were purified in a singlestep by immobilized metal affinity chromatography(IMAC) using a nickel column. Estimates of purifiedprotein yield ranged from 5–60 mg/l of bacterialculture. It should be noted that this is a conservative
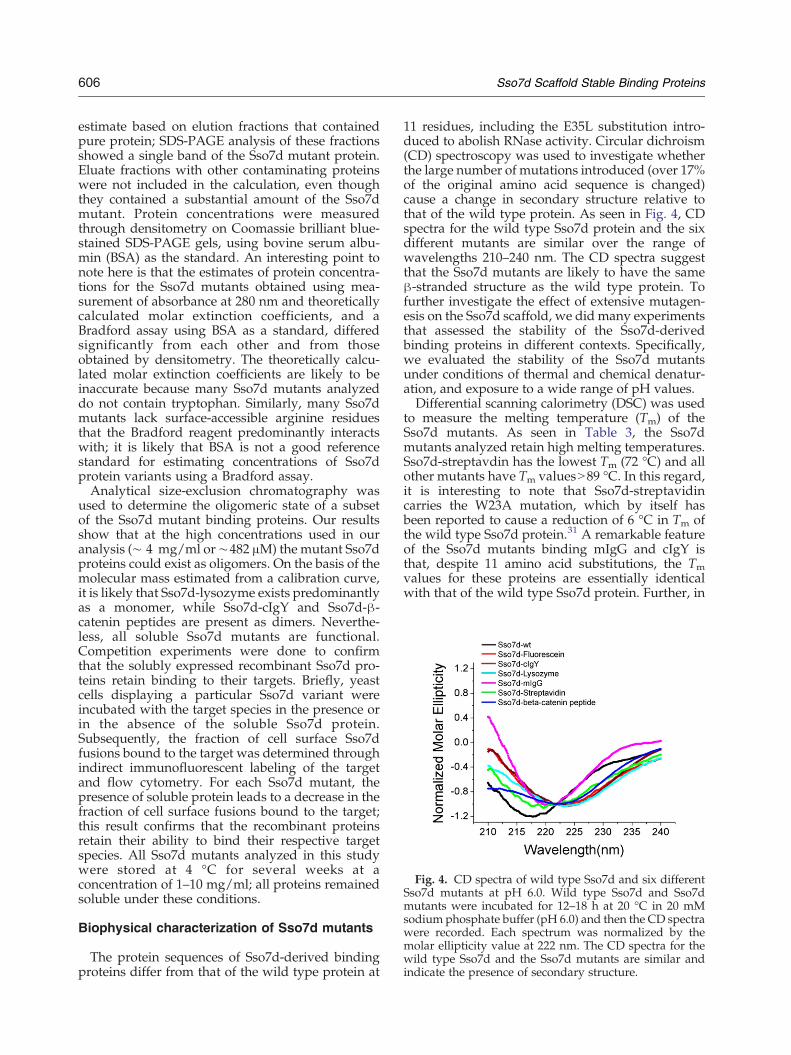
Fig. 4. CD spectra of wild type Sso7d and six differentSso7d mutants at pH 6.0. Wild type Sso7d and Sso7dmutants were incubated for 12–18 h at 20 °C in 20 mMsodium phosphate buffer (pH 6.0) and then the CD spectrawere recorded. Each spectrum was normalized by themolar ellipticity value at 222 nm. The CD spectra for thewild type Sso7d and the Sso7d mutants are similar andindicate the presence of secondary structure.
606 Sso7d Scaffold Stable Binding Proteins
estimate based on elution fractions that containedpure protein; SDS-PAGE analysis of these fractionsshowed a single band of the Sso7d mutant protein.Eluate fractions with other contaminating proteinswere not included in the calculation, even thoughthey contained a substantial amount of the Sso7dmutant. Protein concentrations were measuredthrough densitometry on Coomassie brilliant blue-stained SDS-PAGE gels, using bovine serum albu-min (BSA) as the standard. An interesting point tonote here is that the estimates of protein concentra-tions for the Sso7d mutants obtained using mea-surement of absorbance at 280 nm and theoreticallycalculated molar extinction coefficients, and aBradford assay using BSA as a standard, differedsignificantly from each other and from thoseobtained by densitometry. The theoretically calcu-lated molar extinction coefficients are likely to beinaccurate because many Sso7d mutants analyzeddo not contain tryptophan. Similarly, many Sso7dmutants lack surface-accessible arginine residuesthat the Bradford reagent predominantly interactswith; it is likely that BSA is not a good referencestandard for estimating concentrations of Sso7dprotein variants using a Bradford assay.Analytical size-exclusion chromatography was
used to determine the oligomeric state of a subsetof the Sso7d mutant binding proteins. Our resultsshow that at the high concentrations used in ouranalysis (∼ 4 mg/ml or∼482 μM) the mutant Sso7dproteins could exist as oligomers. On the basis of themolecular mass estimated from a calibration curve,it is likely that Sso7d-lysozyme exists predominantlyas a monomer, while Sso7d-cIgY and Sso7d-β-catenin peptides are present as dimers. Neverthe-less, all soluble Sso7d mutants are functional.Competition experiments were done to confirmthat the solubly expressed recombinant Sso7d pro-teins retain binding to their targets. Briefly, yeastcells displaying a particular Sso7d variant wereincubated with the target species in the presence orin the absence of the soluble Sso7d protein.Subsequently, the fraction of cell surface Sso7dfusions bound to the target was determined throughindirect immunofluorescent labeling of the targetand flow cytometry. For each Sso7d mutant, thepresence of soluble protein leads to a decrease in thefraction of cell surface fusions bound to the target;this result confirms that the recombinant proteinsretain their ability to bind their respective targetspecies. All Sso7d mutants analyzed in this studywere stored at 4 °C for several weeks at aconcentration of 1–10 mg/ml; all proteins remainedsoluble under these conditions.
Biophysical characterization of Sso7d mutants
The protein sequences of Sso7d-derived bindingproteins differ from that of the wild type protein at
11 residues, including the E35L substitution intro-duced to abolish RNase activity. Circular dichroism(CD) spectroscopy was used to investigate whetherthe large number of mutations introduced (over 17%of the original amino acid sequence is changed)cause a change in secondary structure relative tothat of the wild type protein. As seen in Fig. 4, CDspectra for the wild type Sso7d protein and the sixdifferent mutants are similar over the range ofwavelengths 210–240 nm. The CD spectra suggestthat the Sso7d mutants are likely to have the sameβ-stranded structure as the wild type protein. Tofurther investigate the effect of extensive mutagen-esis on the Sso7d scaffold, we did many experimentsthat assessed the stability of the Sso7d-derivedbinding proteins in different contexts. Specifically,we evaluated the stability of the Sso7d mutantsunder conditions of thermal and chemical denatur-ation, and exposure to a wide range of pH values.Differential scanning calorimetry (DSC) was used
to measure the melting temperature (Tm) of theSso7d mutants. As seen in Table 3, the Sso7dmutants analyzed retain high melting temperatures.Sso7d-streptavdin has the lowest Tm (72 °C) and allother mutants have Tm valuesN89 °C. In this regard,it is interesting to note that Sso7d-streptavidincarries the W23A mutation, which by itself hasbeen reported to cause a reduction of 6 °C in Tm ofthe wild type Sso7d protein.31 A remarkable featureof the Sso7d mutants binding mIgG and cIgY isthat, despite 11 amino acid substitutions, the Tmvalues for these proteins are essentially identicalwith that of the wild type Sso7d protein. Further, in

Table 3. Biophysical characterization of Sso7d mutants and comparison with wild type Sso7d
Sso7d variant Tm (°C)a [Gdn-HCl]1/2 (M)b pH stability rangec
Sso7d-wt 98d 4 0.33–12.5Sso7d-fluorescein 89.6 (89.5,89.6) 3.4 0.33–12.5Sso7d-β-catenin peptide 92.7 (92.5,92.8) 2.5 0.33–12.5Sso7d-lysozyme 92.7 (92.6,92.7) 5 0.33–12.5Sso7d-streptavidin 72.5 (72.4,72.5) 2.8 0.33–12.5Sso7d-mIgG 102.8 (102.8,102.8) 4 0.33–12.5Sso7d-cIgY 100 (100.3,99.7) 3.2 0.33–12.5
a Average value from duplicate experiments is reported. Numbers in parentheses show the experimentally determined Tm value ineach experiment.
b [Gdn-HCl]1/2 calculated from Fig. 5.c This is the range of pH over which the CD spectrum remains largely unchanged, which indicates the presence of secondary structure.d Tm for wild type Sso7d in the literature.18
607Sso7d Scaffold Stable Binding Proteins
the case of Sso7d-mIgG, Sso7d-fluorescein andSso7d-streptavidin, the melting curves obtained intwo successive heating and cooling cycles werenearly identical. These data suggests that a signif-icant fraction of these three mutant proteinsundergo a reversible folded-to-unfolded state tran-sition under thermal denaturation conditions.CD spectroscopy was used to monitor the unfold-
ing of the Sso7d mutants in the presence ofguanidine hydrochloride (Gdn-HCl). The Sso7dmutants were incubated with various concentrationsof Gdn-HCl at 4 °C and pH 7.3 for 12–18 h.Subsequently, molar ellipticity measurements at222 nm were used to estimate the fraction ofunfolded protein at a given concentration ofGdn-HCl. Figure 5 shows the Gdn-HCl denatur-ation curves for the six Sso7d mutants analyzed.Gdn-HCl concentrations corresponding to themid-point of the folded-to-unfolded state transi-tion for the Sso7d mutants ([Gdn-HCl]1/2) aregiven in Table 3. Most Sso7d mutants are lessresistant to Gdn-HCl denaturation than the wildtype protein ([Gdn-HCl]1/2=4 M). Of the mutantsanalyzed, Sso7d-β-catenin peptide has the lowest[Gdn-HCl]1/2 (2.5 M) and Sso7d-lysozyme has ahigher value of [Gdn-HCl]1/2 (5 M) than that of thewild-type protein. To put these data into context,the [Gdn-HCl]1/2 for a typical single-chain antibodyhas been reported as 1.5 M.32
To evaluate the pH stability of the Sso7d mutants,protein denaturation as a result of extended expo-sure to various pH conditions was measured usingCD spectroscopy. The Sso7d mutants and the wildtype protein were incubated at 20 °C for 12–16 h atvarious pH values. Figure 6 shows the CD spectrafor a range of pH conditions for the wild type Sso7dprotein and Sso7d-lysozyme. As with the wild typeprotein, the CD spectrum of Sso7d-lysozyme overthe range of wavelengths 210–240 nm was largelyunchanged under a wide range of pH conditions.CD spectra for the other mutants show similarbehavior. The pH range in which the CD spectrumremains unchanged for each Sso7d mutant is given
in Table 3. The CD spectra at pH 13.5 were verynoisy, likely because of the high ionic strength, andwere not included in this analysis.These results indicate that, despite extensive
mutagenesis, binding proteins derived from theSso7d scaffold are highly resistant to thermal andchemical denaturation and can withstand extendedexposure to pH extremes.
Mutational analysis of Sso7d-lysozyme
The Sso7d library was generated through muta-genesis of 10 residues on the DNA-binding surfaceof Sso7d. To assess the number and location ofmutations that contribute towards the generation ofbinding affinity for a specific target, we conducted adetailed mutational analysis on Sso7d-lysozyme.Specifically, the mutations in Sso7d-lysozyme werereverted to the wild type Sso7d residues, one by one.Subsequently, we quantified the lysozyme-bindingactivity of these single mutants. Briefly, yeastsurface-displayed mutants were labeled with vari-ous concentrations of soluble lysozyme, rangingfrom 25 nM to 1 μM. The fraction of yeast cellsurface fusions that bind lysozyme at each concen-tration was used to classify the mutants in thefollowing four categories: binding similar to Sso7d-lysozyme (+++), binding somewhat reduced (++),binding greatly reduced (+) and binding abolishedin the given range of concentrations (–). Thisanalysis allows us to assess the contribution of aparticular mutation to the overall binding todetermine the energetic hotspot in the binding siteand the results are summarized in Fig. 7.Out of the ten mutations analyzed, seven (F21K,
F23W, W25V, C30S, D32T, S42R and C44A) have aprofound effect on binding affinity, as shown by thecomplete loss of binding to lysozyme upon rever-sion to the wild type residue. The reversion mutantscorresponding to Q40 and M28 retain some bindingaffinity for lysozyme, albeit reduced, suggestingthat these positions contribute somewhat to thebinding interaction with lysozyme. On the other
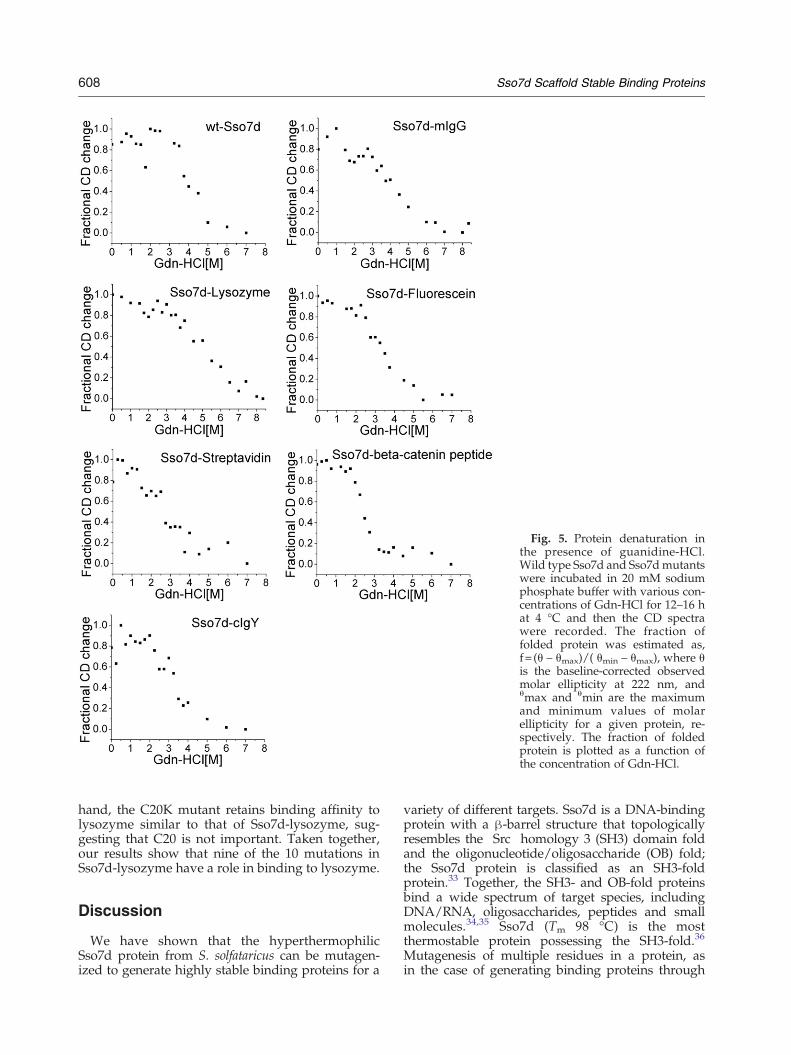
Fig. 5. Protein denaturation inthe presence of guanidine-HCl.Wild type Sso7d and Sso7dmutantswere incubated in 20 mM sodiumphosphate buffer with various con-centrations of Gdn-HCl for 12–16 hat 4 °C and then the CD spectrawere recorded. The fraction offolded protein was estimated as,f=(θ – θmax)/( θmin – θmax), where θis the baseline-corrected observedmolar ellipticity at 222 nm, andθmax and θmin are the maximumand minimum values of molarellipticity for a given protein, re-spectively. The fraction of foldedprotein is plotted as a function ofthe concentration of Gdn-HCl.
608 Sso7d Scaffold Stable Binding Proteins
hand, the C20K mutant retains binding affinity tolysozyme similar to that of Sso7d-lysozyme, sug-gesting that C20 is not important. Taken together,our results show that nine of the 10 mutations inSso7d-lysozyme have a role in binding to lysozyme.
Discussion
We have shown that the hyperthermophilicSso7d protein from S. solfataricus can be mutagen-ized to generate highly stable binding proteins for a
variety of different targets. Sso7d is a DNA-bindingprotein with a β-barrel structure that topologicallyresembles the Src homology 3 (SH3) domain foldand the oligonucleotide/oligosaccharide (OB) fold;the Sso7d protein is classified as an SH3-foldprotein.33 Together, the SH3- and OB-fold proteinsbind a wide spectrum of target species, includingDNA/RNA, oligosaccharides, peptides and smallmolecules.34,35 Sso7d (Tm 98 °C) is the mostthermostable protein possessing the SH3-fold.36
Mutagenesis of multiple residues in a protein, asin the case of generating binding proteins through

Fig. 6. pH stability analysis of (a) wild type Sso7d and (b) Sso7d-lysozyme. Protein samples were incubated in 20 mMsodium phosphate buffer at various pH values for 12–18 h at 20 °C and then the CD spectra in the 210–240 nmwavelengthrange were measured. Each CD spectrum was normalized by the molar ellipticity value at 222 nm. As with the wild typeSso7d, normalized CD spectra for Sso7d-lysozyme are essentially identical, suggesting no appreciable change insecondary structure over a wide range of pH values.
609Sso7d Scaffold Stable Binding Proteins
mutation of a scaffold protein, often results indestabilization of the protein and a concomitantdecrease in Tm of the mutant relative to the wildtype protein. It can therefore be naively reasonedthat a scaffold protein with high thermal stability ismore likely to yield mutant binding proteins thatare stable. Indeed, it has been shown that highlythermostable proteins are more tolerant to mutationand consequently have enhanced evolvability.11
Owing to the seemingly inherent adaptability ofthe SH3- and OB-fold and the highly thermostablenature of Sso7d, we hypothesized that the Sso7dprotein is likely to be a versatile scaffold forgenerating binding proteins. In this study, weinvestigated the ability of the Sso7d scaffold toyield proteins binding to a diverse set of targets.We mutagenized 10 surface-accessible residues on
β-strands in the DNA-binding interface of Sso7d(Fig. 1) to generate a library of 108 Sso7d mutants
Sso7d-lysozyme and each mutant was classified into one of fou(+++), binding somewhat reduced (++), binding greatly redconcentration range (-). Shades of gray are used to indicalysozyme; a darker shade of gray indicates a greater contribu
using yeast surface display. DNA sequencing sug-gests that this yeast library contains about 3×107
full-length Sso7d mutants. We were able to isolateproteins binding to a set of six diverse model targetsfrom this modest library; our targets included asmall organic molecule (fluorescein), a peptide(a 12 amino acid peptide from the C-terminusof the β-catenin protein), hen egg lysozyme, strepta-vidin and immunoglobulins. Further, the selectedSso7d mutants could discriminate between theclosely related immunoglobulin targets (Fig. 3).Earlier studies reported the isolation of de novobinding proteins from small yeast surface displaylibraries.5,37–39 In such cases, the number of correctlyfolded mutants in the library can be maximizedthrough careful choice of residues to be mutagen-ized. Due to the high thermal stability of the Sso7dprotein, and the choice of surface-accessible residuesfor mutagenesis based on data from previous studies
Fig. 7. Mutational analysis forSso7d-lysozyme. Each mutated res-idue in Sso7d-lysozyme wasreverted to the corresponding resi-due in wild type Sso7d. (a) TheSso7d scaffold with randomizedresidues is shown in licorice repre-sentation. (PDB ID 1SSO; this imagewas generated using visual molec-ular dynamics (VMD) software). (b)Mutational analysis grid showingthe effect of reversion mutations;lysozyme binding of each reversionmutant was compared to that of
r categories as follows: binding similar to Sso7d-lysozymeuced (+) and binding completely abolished in the givente the importance of residue for maintaining binding totion to binding.

Table 4.Comparison of the melting temperatures of Sso7dmutants with those of mutants obtained from other non-immunoglobulin scaffolds
ScaffoldNo. amino
acid residues
Tm(°C)
Wildtype Mutants
Affibody9 58 78 40–57Tenth domain
of fibronectin5,52,5394 84 56–74
DARPins7,54 130–190 —a 66–100Lipocalin (bilin-binding
protein)55,56b180 61 62–73
Sso7d (this study) 60 98 72–103a The melting temperature (Tm) depends on the number of
ankyrin repeats.
610 Sso7d Scaffold Stable Binding Proteins
with Sso7d, it is likely that a very large fraction of thefull-length Sso7d mutants are folded correctly.Indeed, the high thermal stability of the six differentmutants characterized in this study suggests stronglythat the Sso7d scaffold is highly tolerant to mutation.In this regard, it is interesting to note that indirectimmunofluorescent detection of the c-myc epitope tagshows 30% of the total library, i.e.∼3×107 mutants, iscomposed of full-length Sso7d mutants displayed asyeast cell surface fusions (Fig. 2). To put thisnumber into context, sequencing data shows thatthere are only ∼3×107 (30%) full-length Sso7dmutants in the library. It has been proposed thatthe quality control mechanism in the yeast endo-plasmic reticulum ensures cell surface display ofonly correctly folded proteins,24 although there issome evidence to suggest that incorrectly foldedproteins or molten globules might be displayed ascell surface fusions.26
The Sso7d mutants obtained have binding affin-ities in the nanomolar to micromolar range for theirrespective targets (Table 2). It is important to notethat no affinity maturation strategy was applied inthis study. Additional rounds of mutagenesis andscreening can be used to obtain binders with higheraffinities. Alternatively, it might be possible toobtain binders with higher affinity through screen-ing of an Sso7d library with higher diversity. In thisstudy, we used a yeast surface display library of∼108 Sso7d mutants, with ∼3×107 full-lengthclones. In a yeast surface display, library diversityis limited by the transformation efficiency of yeast;the largest reported diversity of a yeast displaylibrary is ∼109 mutants.40 A lysozyme-bindingprotein with a KD of 5 nM has been isolated froma library of ∼1012 mutants derived from the nearlyidentical Sac7d scaffold, using ribosome display.41
By contrast, the lysozyme-binding Sso7d mutantisolated in this study using yeast surface display hasa KD of ∼350 nM. Nevertheless, the yeast displaysystem provides certain key advantages, such as theuse of flow cytometric sorting, the ability todiscriminate between clones with small differencesin binding affinity and ease of quantitative determi-nation of binding affinities (KD) through cell surfacetitrations. Therefore, a screening strategy thatharnesses the large library diversity accessibleusing ribosome display or mRNA display in apreliminary screening step, followed by subsequentsteps using yeast surface display might be optimal.An interesting question that arises in the context of
generating de novo binders from scaffold proteins is:how many mutations play a role in the bindinginteraction with the target? To address this question,we conducted a mutational analysis on Sso7d-lysozyme where each mutation was reverted to thewild type residue and the binding affinity of thereversion mutant was compared to that of Sso7d-lysozyme. Strikingly, our analysis (Fig. 7) showed
that nine of the 10 mutations in Sso7d-lysozymecontribute to the binding interaction with lysozyme.This result cannot be extrapolated to any target, butour data show that the DNA-binding surface ofSso7d can be mutagenized in a manner such thatnearly all the mutated residues contribute to targetbinding.The mutant Sso7d proteins analyzed in this
study were easily expressed at reasonable yieldsin the reducing cytoplasm of E. coli and purifiedusing conventional IMAC protocols. Despite exten-sive mutagenesis relative to the wild type protein(11 mutations out of 63 residues, including E35L), theSso7d mutants exhibited remarkably high meltingtemperatures. Five of the six mutants analyzedhad Tm N89 °C. To the best of our knowledge, Sso7d-mIgG (Tm 102.8 °C) has the highest reported meltingtemperature amongst mutant proteins derivedfrom non-immunoglobulin scaffolds. Table 4 givesthe Tm ranges for mutants derived from commonlyused non-antibody scaffolds. Simplistically, mutantsderived from a protein scaffold will be thermallystable if the non-mutagenized regions of the scaffoldare key determinants of scaffold protein stability.For instance, mutation of 11 amino acid residues inthe non-binding surface results in an optimizedaffibody scaffold with greater thermal stability.42
This optimized scaffold, in turn, yields bindingproteins with improved thermal stability. In the caseof Sso7d, the hydrophobic core comprised of 11residues (V3, F5, V14, I19, I29, F31, Y33, G43, V45,A50 and L54) as well as the C-terminal α helix play amajor role in protein stability.31,43 Mutagenesis ofresidues in the hydrophobic core results in asignificant decrease in the melting temperature ofSso7d.44 In particular, the single point mutationF31A was shown to decrease the melting tempera-ture by 24 °C.45 Similarly, deletion of the C-terminalα helix causes a 46 °C decrease in meltingtemperature.43 The Sso7d mutants analyzed werealso resistant to chemical denaturation mediated by

611Sso7d Scaffold Stable Binding Proteins
Gdn-HCl and extreme conditions of pH. Of themutants analyzed, Sso7d-β-catenin peptide has thelowest [Gdn-HCl]1/2 of 2.5 M. By comparison, thecorresponding [Gdn-HCl]1/2 for a typical singlechain antibody was reported as 1.5 M.32 All Sso7dmutants analyzed could also withstand extendedincubation in a wide range of pH values (0.33–12.5),while retaining their secondary structure; the dataobtained at pH 13.5 were inconclusive. Takentogether, our results show that binding proteinsderived from the Sso7d scaffold exhibit severaldesirable properties, such as easy recombinantexpression, high thermal stability and resistance tochemical denaturation and pH extremes.To date, antibodies are the most commonly used
scaffolds for generating binding proteins. Apartfrom historical reasons, this is largely because thetopological diversity generated through altering thelength and amino acid compositions of six hyper-variable loops in the complementarity determiningregions of antibodies can yield binding proteinswith high affinity to a wide spectrum of targets. Inour work, the lowest binding affinities wereobtained for binders to fluorescein (KD ∼ 7 μM)and the β-catenin peptide fragment (KD ∼2.5 μM).It is possible that the flat surface topology of theDNA-binding surface of Sso7d is not optimal forgenerating binding proteins with high affinity tothese targets. Nevertheless, the Sso7d protein yieldsbinding proteins with several desirable properties,such as small size, easy expression in E. coli andhigh thermal stability. The tolerance to mutationthat makes Sso7d a versatile scaffold is arguably aresult of its high thermal stability and hyperther-mophilic origin. Hyperthermophilic archaea andbacteria could provide a rich source of small stableproteins that can be used as scaffolds for generatingbinding proteins. An ensemble of hyperthermophil-ic scaffolds where multiple different topologies aremutagenized might be well suited for generatingbinding proteins for a wide spectrum of targets.Further, a different topology on the Sso7d proteinmight be mutagenized to obtain binding proteins.Previously, binding proteins have been isolatedfrom a library generated through mutagenesis ofthe RT- and src-loops in an SH3-fold-containingprotein.46 Therefore, it is conceivable that the loopregions in Sso7d near the ATP-binding pocket(residues Y7 and K39)47 might be targeted formutagenesis. We are currently investigating theuse of an ensemble of hyperthermophilic proteinscaffolds as well as mutagenesis of loop regions ofSso7d to generate binding proteins.In summary, we have shown that the Sso7d
protein scaffold can be engineered to generatebinding proteins with high specificity for a widerange of targets. Sso7d variants are highly resistantto thermal and chemical denaturation as well as pHextremes. Consequently, Sso7d-derived binding
proteins are well suited for use in several applica-tions where antibodies cannot or need not be used.The lack of disulfide bonds and low molecular massmake them ideal for use as intracellular inhibitorsfor “protein interference”. Unlike RNA interference,mutant proteins based on the Sso7d scaffold can beused to bind and inhibit the function of a specificdomain or a post-translationally modified form of aprotein.21 Sso7d proteins are also well suited for useas affinity ligands in protein purification applica-tions. Due to their resistance to pH extremes, theSso7d mutants are likely to be compatible withclean-in-place sterilization cycles involving washsteps with sodium hydroxide. It is worthwhilenoting that a very high binding affinity of interactionis not required in these aforementioned applications.Finally, due to their high thermal stability and easeof recombinant expression, Sso7d mutant proteinscan be used as low-cost affinity reagents in imagingand diagnostics. Thus, the Sso7d protein is aversatile scaffold that is relevant to multiple appli-cations in biotechnology and medicine.
Materials and Methods
Sso7d library construction
Surface-exposed residues in the DNA binding surface ofthe wild type Sso7d protein were identified using visualmolecular dynamics software (VMD, University of Illi-nois, Urbana-Champaign, IL). Ten residues in the flatDNA-binding surface of Sso7d were chosen for random-ization. Subsequently, the Sso7d library was synthesizedas a single oligonucleotide, U1, containing degenerateNNN codons (Integrated DNA Technologies, Coralville,IA). The DNA sequence of U1 is 5′-ATG GCG ACC GTGAAA TTT AAA TAT AAA GGC GAA GAA AAA CAGGTG GAT ATT AGC AAA ATT NNN NNN GTG NNNCGC NNN GGC AAA NNN ATT NNN TTT NNN TATGAT CTG GGC GGC GGC AAA NNN GGC NNN GGCNNN GTG AGC GAA AAA GAT GCG CCG AAA GAACTGCTGCAGATGCTGGAAAAACAGAAAAAA-3′.The non-randomized regions of U1 contain codons thatare optimized for expression in E. coli.A double-stranded DNA library of Sso7d mutants was
generated by amplification of U1 in a single PCR stepusing oligonucleotide primers Pf1 and Pr1 that containyeast surface display consensus sequences as well as therestriction sites for the enzymes NheI (Pf1) and BamHI(Pr1). The primer sequences are:
Pf1 5′AGT GGT GGT GGT GGT TCT GGT GGT GGTGGT TCT GGT GGT GGT GGT TCT GCT AGC ATGGCG ACC GTG AAA TTT AAA TAT AAA G 3′Pr1 5′CTC GAG CTA TTA CAA GTC CTC TTC AGAAAT AAG CTT TTG TTC GGA TCC TTT TTT CTG TTTTTC CAG CAT CTG 3′
restriction sites are italicized. Each 50 μl PCR reaction hadthe following components: Phusion™ high-fidelity DNA

612 Sso7d Scaffold Stable Binding Proteins
polymerase (1 U/50 μl; New England Biolabs, Ipswich,MA) in high-fidelity Phusion™ buffer, 0.2 mM deoxynu-cleotide triphosphate (dNTPs), 0.5 μM primers, 1 Mbetaine and 3% (v/v) dimethyl sulfoxide (DMSO). Themixture was denatured at 98 °C for 2 min followed by 25cycles of 98 °C for 1 min, 68 °C for 1 min, 72 °C for 15 s anda final extension step at 72 °C for 10 min. PCR productswere concentrated using PelletPaint™ (Novagen, SanDiego, CA).A yeast surface display library of Sso7d mutants was
generated by homologous recombination-mediated plas-mid gap repair, using published protocols as a guide.23,29
The Saccharomyces cerevisiae strain EBY100 and the yeastsurface display plasmid vector (pCTCON) were kindgifts from Professor K. Dane Wittrup (MassachusettsInstitute of Technology, Cambridge, MA). Briefly, thepCTCON vector was linearized by digestion with therestriction enzymes NheI, BamHI and SalI. Linearizedvector was purified by PCR using a Qiagen PCRpurification kit (Qiagen, Valencia, CA) and then concen-trated using PelletPaint™. Subsequently, the linearizedvector was transformed into yeast along with the linearDNA library of Sso7d mutants. Electroporation was doneat 0.54 kV, 25 μF and 1000 Ω in a Bio-Rad Gene Pulsersystem (Bio-Rad, Hercules, CA) with 1 μg of cutpCTCON vector and 3 μg of concentrated Sso7d libraryPCR product. Electrocompetent EBY100 cells wereprepared as described.29 Transformed cells were grownin YPD medium (10 g/l yeast extract, 20 g/l peptone,20 g/l dextrose) for 1 h at 30 °C and 250 rpm. At thispoint, serial dilutions of a small fraction of the trans-formed cells were plated onto SDCAA plates (20 g/ldextrose, 5 g/l Casamino acids, 6.7 g/l yeast nitrogenbase, 182 g/l sorbitol, 5.40 g/l Na2HPO4, 7.45 g/lNaH2PO4, 15 g/l agar) to determine the library diversity.The remainder of the transformed cells were grown in250 ml of selective medium SDCAA (20 g/l dextrose,5 g/l Casamino acids, 6.7 g/l yeast nitrogen base, 5.40 g/lNa2HPO4, 7.45 g/l NaH2PO4) with 1:100 pen-strepsolution (Invitrogen, Carlsbad, CA) at 30 °C and250 rpm for 24–48 h. Subsequently, the yeast library waspassaged three times in SDCAA medium and frozen inaliquots of 2×109 cells per vial. The library diversity wasdetermined to be 1×108 transformants.
Magnetic selection
Magnetic selection was done as described.27 Yeast cellsgrown in SDCAA were pelleted and suspended at 1×107
cells/ml (A600 1) in SGCAA(20 g/l galactose, 5 g/lCasamino acids, 6.7 g/l yeast nitrogen base, 5.40 g/lNa2HPO4, 7.45 g/l NaH2PO4). Cells were incubated at20 °C and 250 rpm for 20–24 h to induce expression ofyeast cell surface protein fusions. Dynal™ biotin binderbeads (100 μl containing 4×108 beads/ml; Invitrogen,Carlsbad, CA) were washed in PBS-BSA (8 g/l NaCl,0.2 g/l KCl, 1.44 g/l Na2HPO4, 0.24 g/l KH2PO4, pH 7.4,0.1 % (w/v) BSA) and incubated with biotinylated target(mIgG, cIgY, hen egg lysozyme, β-catenin C-terminalpeptide, fluorescein) overnight at 4 °C with rotation, toobtain the target-coated magnetic beads. Biotinylatedversions of these targets were obtained from the followingsources: mIgG and cIgY from Jackson Immunoresearch(Westgrove, PA), hen egg lysozyme from Biomeda
Corporation (Foster City, CA) and Sigma-Aldrich (StLouis, MO), β-catenin C-terminal peptide from Genscript(Piscataway, NJ) and fluorescein from Thermo Scientific(Rockford, IL). Biotin binder beads were used as a targetfor isolating binders to streptavidin.Negative selection against the biotin binder beads was
performed for all targets (except streptavidin) byincubating ∼2×109 induced cells (20 times the librarydiversity) with the washed beads in a 1.5 ml tube for1–2 h at 4 °C with rotation. For mIgG and cIgY,additional negative selections were done against cIgY,rIgG and hFc, and mIgG, rIgG and hFc, respectively.The tube was then placed onto a magnetic particleconcentrator, and bead-bound cells were discarded.Unbound cells were added to target pre-coated beadswashed with PBS-BSA and incubated for 1–2 h at 4 °C.Finally, bead-bound cells were collected using a mag-netic particle concentrator, washed four times with 1 mlof PBS-BSA and grown in 5 ml of SDCAA for 24–48 h at30 °C and 250 rpm.
Fluorescence-activated cell sorting (FACS)
Magnetically sorted cells were expanded and inducedin SGCAA at 107 cells/ml at 20 °C and 250 rpm. FACSwas used to isolate binders with highest affinity for thetargets using published protocols.19,29,48,49 Briefly, 2×107
cells were labeled simultaneously with biotinylated target(mIgG, cIgY, hen egg lysozyme, β-catenin C-terminalpeptide, and fluorescein) and an anti-c-myc chickenantibody (Invitrogen, Carlsbad, CA) or an anti-HAmouse antibody (Roche, Indianapolis, IN). Indirect im-munofluorescent detection of the target was achievedusing secondary labeling with Strep-PE or neutravidin-FITC (Invitrogen, Carlsbad, CA). A goat anti-chickenantibody conjugated with Alexa Fluor-488 or Alexa Fluor-633 and a goat anti-mouse antibody conjugated withAlexa Fluor-488 or Alexa Fluor-633 (Invitrogen, Carlsbad,CA) were used for secondary detection of the anti-c-mycand the anti-HA antibodies, respectively. Strep-PE wasused as a target for isolation of streptavidin binders. Cellsamples were analyzed and sorted on a flow cytometer(FACS Aria, Becton Dickinson, San Jose, CA). Themagnetically sorted population was sorted four times byFACS for cIgY and mIgG, three times for fluorescein andstreptavidin and twice for hen egg lysozyme and β-catenin peptide, with successively lower target concentra-tions. Cells from the final sort were plated onto an SDCAAplate. For each target, DNA sequences were obtained forfive individual clones and the equilibrium dissociationconstant (KD) was determined for a single clone.
Measurement of Kd
The equilibrium dissociation constant (KD) was deter-mined using cell surface titrations as described.29 Briefly,yeast cells displaying the cell surface Sso7d mutant wereincubated with various concentrations of the biotinylatedtarget at room temperature and pH 7.4, followed by Strep-PE; in the case of Sso7d-streptavidin, cells were labeleddirectly with Strep-PE. Subsequently, flow cytometricanalysis was used to measure the mean fluorescenceintensity of the cells. The KD of the binding interaction

613Sso7d Scaffold Stable Binding Proteins
between the Sso7d mutant and its target was estimatedusing the following relationship:
F =c L½ �0
KD + L½ �0where F is the observed mean fluorescence intensity, [L]0is the corresponding concentration of target used forlabeling the cells, and c is a constant. The constants c andKD were determined through a global fit, using data fromat least three different experiments and the 68% confi-dence intervals, which correspond to the commonlyreported standard deviations associated with triplicateexperiments, were calculated as described.50
Sequence determination of Sso7d variants
Plasmid DNA from the Sso7d library and five Sso7dvariants for each target was isolated using the ZymoprepKit II (Zymoresearch Corporation, Orange, CA). Theisolated DNA was subsequently transformed into Nova-blue™ (E . coli) cells (EMD Biosciences, San Diego, CA).Plasmids were sequenced by Genewiz ( La Jolla, CA).Oligonucleotide P3 (5′ ACT ACG CTC TGC AGG CTAGT 3′) was used as the primer for sequencing.
†http://rsb.info.nih.gov/ij/.
Recombinant expression and purification ofSso7d mutants
Sso7d variants were cloned into the pET22b (+) vectorusing NdeI and XhoI restriction sites. Oligonucleotideprimers Pf2 and Pr2 (Integrated DNA Technologies,Coralville, IA) were used to introduce the restrictionsites. The primer sequences are as follows:
Pf2-5′ GAA TCC CAT ATGATG GCG ACC GTG AAATTT AAA 3′Pr2-5′ CCG CCG CTC GAG TTT TTT CTG TTT TTCCAG CAT C 3′
where restriction sites are in italics. Plasmid constructswere subsequently transformed into Rosetta™ (E . coli)cells (EMD Biosciences, San Diego, CA). A 5 ml overnightculture in LB medium (10 g/l Bacto-tryptone, 5 g/l yeastextract, 10 g/l NaCl) containing 1% (w/v) glucose and50 μg/ml ampicillin at 37 °C and 250 rpm was used toinoculate a 1 liter culture in 2xYT medium (16 g/l Bacto-tryptone, 10 g/l yeast extract, 5 g/l NaCl). The cells wereinduced with 0.5 mM IPTG at an A600 of 1.0 and grownfor 19–20 h at 37 °C and 250 rpm. Subsequently, cellswere harvested by centrifugation at 5000 rpm for 20 min,suspended in buffer A (50 mM Tris, pH 7.6, 300 mMNaCl) and lysed by sonication for 10 min. The cell lysatewas centrifuged and the final supernatant was passedthrough a 0.22 μm pore size filter then loaded onto a Hi-Trap 5 ml Ni column (GE Healthcare Bio-Sciences,Piscataway, NJ) on a Bio-Rad BioLogic Duoflow FPLC(Hercules, CA). His6-tagged Sso7d mutants were elutedwith a linear gradient using buffer A and buffer B (50 mMTris, pH 7.6, 300 mM NaCl, 500 mM imidazole). Theeluted fractions containing pure protein were pooled,dialyzed against PBS (pH 7–8) (Sso7d-β-catenin peptideand cIgY binding proteins) or 50 mM sodium acetate
buffer, pH 5.0 (Sso7d-mIgG, Sso7d-lysozyme, Sso7d-fluorescein and Sso7d-streptavidin) and concentrated.Protein concentrations were determined by densitometryin Coomassie brilliant blue-stained SDS-PAGE gels usingImageJ† software.51
Soluble competition experiments
Yeast cells expressing an Sso7d variant as cell surfacefusions were incubated with biotinylated target in thepresence or in the absence of 100–200 fold excess of thecorresponding soluble Sso7d mutant protein, recombi-nantly expressed in E. coli. Subsequently, yeast cells werelabeled with Strep-PE (Invitrogen, Carlsbad, CA) to detecttarget-bound cell surface fusions.
Differential scanning calorimetry (DSC)
The DSC experiments were done with a Nano DSC IIdifferential scanning calorimeter (TA Instruments, New-castle, DE). PBS was used as a buffer for Sso7d-β-cateninpeptide and Sso7d-cIgY; 50 mM sodium acetate bufferwas used for Sso7d-fluorescein, Sso7d-streptavidin,Sso7d-mIgG and Sso7d-lysozyme. Protein samples andthe corresponding buffers were degassed for 30–60 minbefore analysis. Four scans (1 °C/min) consisting of twoheating and two cooling cycles were done for each sample.In each case, a buffer baseline run was done, consisting ofthe same four scans in the same temperature range usedfor the sample. Data were recorded in DSC Run softwareand exported to CpCalc software for analysis. Each bufferand sample run was done twice.
CD spectroscopy
Gdn-HCl stability experiments
Protein samples were incubated overnight (12–16 h) at4 °C in 20 mM sodium phosphate buffer (pH 7.3) withvarious concentrations of Gdn-HCl (0–8 M). Sampleswere analyzed on a JASCO-815 spectropolarimeter andCD spectra over the range of wavelengths 210–240 nmwere recorded. A 50 nm/min scan rate, 0. 1 nm pitch,1 nm bandwidth and 2 s response time were used. Threeaccumulation scans were done for each sample. Bufferruns were done for baseline correction. The molarellipticity at 222 nm was used as a metric to trackprotein unfolding upon chemical denaturation. For eachprotein, the baseline-corrected molar ellipticity values (θ)were normalized to estimate the fraction of foldedprotein as:
f =u − umax
umin − umax
where θmin and θmax are the minimum and maximumvalues of baseline-corrected molar ellipticity. Normalizedellipticity values are plotted against the concentration ofGdn-HCl in Fig. 5.

614 Sso7d Scaffold Stable Binding Proteins
pH stability
Protein samples were incubated in 20 mM sodiumphosphate buffer from pH 0.33 to 13.5 at 20 °C for 12–18 h.Subsequently, samples were analyzed on a JASCO-815spectropolarimeter to record CD spectra over the range ofwavelengths 210–240 nm, at 50 nm/min, 0.1 nm pitch,1 nm bandwidth and 2 s response time. Three accumu-lation scans were done for each sample. Each spectrumwas normalized by the molar ellipticity value at 222 nm.
Analytical size-exclusion chromatography
Size-exclusion chromatography/gel filtration was usedto determine the oligomeric state of wild type Sso7d andSso7d-derived binding proteins (Sso7d-lysozyme, Sso7d-cIgY and Sso7d-β-catenin peptide) using a Superdex 75HR 10/300 column (GE Healthcare Biosciences, Piscat-away, NJ). The calibration standards used for molecularmass determination were BSA (66 kDa), carbonic anhy-drase (29 kDa), cytochrome c (12.4 kDa) and aprotinin(6.5 kDa). Blue dextran (2000 kDa) was used to measurethe void volume. All proteins were purified and dialyzedextensively against PBS before analysis. The column wasequilibrated with five column volumes of PBS and 500 μlof each sample (∼4 mg/ml) was loaded onto the column.Absorbance at 280 nm was monitored and molecularmass was calculated as the elution time of the samplecompared to that of the calibration standards.
Mutational analysis for Sso7d-lysozyme
Overlap extension PCR and homologous recombina-tion were used to generate Sso7d-lysozyme reversionmutants in which each mutation was reverted to the wildtype sequence, one at a time. Two different fragments(F1 and F2) were created from the Sso7d-lysozymemutant using internal primers, and primers Pf1 and Pr1as described in the library construction protocol.Fragment F1 was obtained by using primer Pf1 and areverse internal primer. Fragment F2 was generatedusing a forward internal primer and the Pr1 primer. Themutation sites were introduced into the internal primersand were incorporated during the PCR steps. The twofragments F1 and F2 had 30–40 bp homology with eachother and 50 bp homology with the pCTCON vector tofacilitate homologous recombination. Subsequently,yeast transformation was done as described above andthe identity of each reversion mutant was confirmed byDNA sequencing.Yeast-displayed proteins were used to compare the
binding affinities of the reversion mutants to that of Sso7d-lysozyme. Briefly, yeast cells expressing the mutants orSso7d-lysozyme were labeled with five different concen-trations of biotinlyated lysozyme at pH 7.4, followed byStrep-PE and flow cytometry was used to measure themean fluorescence intensity of the cells. In a parallelexperiment, mean fluorescence intensity of cells due toimmunofluorescent labeling of the c-myc tag was mea-sured. The ratio of mean fluorescence intensity due tolysozyme binding to that due to c-myc labeling was usedas a quantitative metric for the fraction of cell surfacefusions that bind lysozyme at a given concentration (f).Plots of f versus lysozyme concentration for each mutant
were compared with the corresponding plot for Sso7d-lysozyme and each mutant was classified into one of thefollowing four categories: binding similar to tat of Sso7d-lysozyme (+++), binding somewhat reduced (++), bindinggreatly reduced (+) and binding abolished in the givenconcentration range (-).
Acknowledgements
We thank Paige Luck from the Department ofFood Science, North Carolina State University(NCSU) for help with differential scanning calorim-etry experiments and Dr John van Zanten from theBiomanufacturing Training and Education Center(BTEC), NCSU for help with the CD experiments.This work was supported by the National ScienceFoundation ( grant CBET 0853771) and the US ArmyMedical Research and Materiel Command undercontract no. W81XWH-08-C-0019. The views, opin-ions and/or findings contained in this report arethose of the authors and should not be construed asan official Department of the Army position, policyor decision unless so designated by other documen-tation. In conducting work involving the use ofrecombinant DNA the investigators adhered to thecurrent version of the National Institutes of Health(NIH) Guidelines for Research Involving Recombi-nant DNA Molecules.
Supplementary Data
Supplementary data to this article can be foundonline at doi:10.1016/j.jmb.2011.04.020
References
1. Binz, H. K., Amstutz, P. & Pluckthun, A. (2005).Engineering novel binding proteins from nonimmu-noglobulin domains. Nat. Biotechnol. 23, 1257–1268.
2. Skerra, A. (2007). Alternative non-antibody scaffoldsfor molecular recognition. Curr. Opin. Biotechnol. 18,295–304.
3. Gebauer, M. & Skerra, A. (2009). Engineered proteinscaffolds as next-generation antibody therapeutics.Curr. Opin. Chem. Biol. 13, 245–255.
4. Koide, A. & Koide, S. (2007). Monobodies: antibodymimics based on the scaffold of the fibronectin type IIIdomain. Methods Mol. Biol. 352, 95–109.
5. Hackel, B. J., Kapila, A. & Wittrup, K. D. (2008).Picomolar affinity fibronectin domains engineeredutilizing loop length diversity, recursive mutagenesis,and loop shuffling. J. Mol. Biol. 381, 1238–1252.
6. Lipovsek, D., Lippow, S. M., Hackel, B. J., Gregson, M.W., Cheng, P., Kapila, A. & Wittrup, K. D. (2007).Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III

615Sso7d Scaffold Stable Binding Proteins
domain and selected by yeast surface display:molecular convergence with single-domain camelidand shark antibodies. J. Mol. Biol. 368, 1024–1041.
7. Stumpp, M. T., Binz, H. K. & Amstutz, P. (2008).DARPins: a new generation of protein therapeutics.Drug Discov. Today, 13, 695–701.
8. Zahnd, C., Wyler, E., Schwenk, J. M., Steiner, D.,Lawrence, M. C., McKern, N. M. et al. (2007). Adesigned ankyrin repeat protein evolved to picomolaraffinity to Her2. J. Mol. Biol. 369, 1015–1028.
9. Nygren, P. A. (2008). Alternative binding proteins:affibody binding proteins developed from a smallthree-helix bundle scaffold. FEBS J. 275, 2668–2676.
10. Lofblom, J., Feldwisch, J., Tolmachev, V., Carlsson, J.,Stahl, S. & Frejd, F. Y. (2010). Affibody molecules:engineered proteins for therapeutic, diagnostic andbiotechnological applications. FEBS Lett. 584, 2670–2680.
11. Bloom, J. D., Labthavikul, S. T., Otey, C. R. & Arnold,F. H. (2006). Protein stability promotes evolvability.Proc. Natl Acad. Sci. USA, 103, 5869–5874.
12. Karshikoff, A. & Ladenstein, R. (2001). Ion pairs andthe thermotolerance of proteins from hyperthermo-philes: a “traffic rule” for hot roads. Trends Biochem.Sci. 26, 550–556.
13. Ladenstein, R. & Antranikian, G. (1998). Proteins fromhyperthermophiles: stability and enzymatic catalysisclose to the boiling point of water. Adv. Biochem. Eng.Biotechnol. 61, 37–85.
14. Baumann, H., Knapp, S., Lundback, T., Ladenstein, R.&Hard, T. (1994). Solution structure andDNA-bindingproperties of a thermostable protein from the archaeonSulfolobus solfataricus. Nat. Struct. Biol. 1, 808–819.
15. Edmondson, S. P. & Shriver, J. W. (2001). DNAbinding proteins Sac7d and Sso7d from Sulfolobus.Methods Enzymol. 334, 129–145.
16. Knapp, S., Mattson, P. T., Christova, P., Berndt, K. D.,Karshikoff, A., Vihinen, M. et al. (1998). Thermalunfolding of small proteins with SH3 domainfolding pattern. Proteins: Struct. Funct. Bioinformatics,31, 309–319.
17. Baumann, H., Knapp, S., Karshikoff, A., Ladenstein,R. & Hard, T. (1995). DNA-binding surface of theSso7d protein from Sulfolobus solfataricus. J. Mol. Biol.247, 840–846.
18. Knapp, S., Karshikoff, A., Berndt, K. D., Christova, P.,Atanasov, B. & Ladenstein, R. (1996). Thermalunfolding of the DNA-binding protein Sso7d fromthe hyperthermophile Sulfolobus solfataricus. J. Mol.Biol. 264, 1132–1144.
19. Boder, E. T. & Wittrup, K. D. (1997). Yeast surfacedisplay for screening combinatorial polypeptidelibraries. Nat. Biotechnol. 15, 553–557.
20. Gai, S. A. & Wittrup, K. D. (2007). Yeast surfacedisplay for protein engineering and characterization.Curr. Opin. Struct. Biol. 17, 467–473.
21. Mouratou, B., Schaeffer, F., Guilvout, I., Tello-Manigne,D., Pugsley, A. P., Alzari, P. M. & Pecorari, F. (2007).Remodeling a DNA-binding protein as a specific invivo inhibitor of bacterial secretin PulD. Proc. Natl Acad.Sci. USA, 104, 17983–17988.
22. Shehi, E., Serina, S., Fumagalli, G., Vanoni, M.,Consonni, R., Zetta, L. et al. (2001). The Sso7d DNA-binding protein from Sulfolobus solfataricus has ribo-nuclease activity. FEBS Lett. 497, 131–136.
23. Swers, J. S., Kellogg, B. A. & Wittrup, K. D. (2004).Shuffled antibody libraries created by in vivo homol-ogous recombination and yeast surface display.NucleicAcids Res. 32, e36.
24. Shusta, E. V., Kieke, M. C., Parke, E., Kranz, D. M. &Wittrup, K. D. (1999). Yeast polypeptide fusionsurface display levels predict thermal stability andsoluble secretion efficiency. J. Mol. Biol. 292, 949–956.
25. Kieke, M. C., Cho, B. K., Boder, E. T., Kranz, D. M. &Wittrup, K. D. (1997). Isolation of anti-T cell receptorscFv mutants by yeast surface display. Protein Eng. 10,1303–1310.
26. Park, S., Xu, Y., Stowell, X. F., Gai, F., Saven, J. G. &Boder, E. T. (2006). Limitations of yeast surface displayin engineering proteins of high thermostability. ProteinEng. Des. Sel. 19, 211–217.
27. Ackerman, M., Levary, D., Tobon, G., Hackel, B.,Orcutt, K. D. & Wittrup, K. D. (2009). Highly avidmagnetic bead capture: an efficient selection methodfor de novo protein engineering utilizing yeast surfacedisplay. Biotechnol. Prog. 25, 774–783.
28. Kyte, J. & Doolittle, R. F. (1982). A simple methodfor displaying the hydropathic character of a protein.J. Mol. Biol. 157, 105–132.
29. Chao, G., Lau, W. L., Hackel, B. J., Sazinsky, S. L.,Lippow, S. M. & Wittrup, K. D. (2006). Isolating andengineering human antibodies using yeast surfacedisplay. Nat. Protoc. 1, 755–768.
30. Hackel, B. J. & Wittrup, K. D. (2010). The full aminoacid repertoire is superior to serine/tyrosine forselection of high affinity immunoglobulin G bindersfrom the fibronectin scaffold. Protein Eng. Des. Sel. 23,211–219.
31. Catanzano, F., Graziano, G., Fusi, P., Tortora, P. &Barone, G. (1998). Differential scanning calorimetrystudy of the thermodynamic stability of somemutantsof Sso7d from Sulfolobus solfataricus. Biochemistry, 37,10493–10498.
32. Worn, A. & Pluckthun, A. (1999). Different equilibriumstability behavior of ScFv fragments: identification,classification, and improvement by protein engineering.Biochemistry, 38, 8739–8750.
33. Agrawal, V. & Kishan, R. K. (2001). Functionalevolution of two subtly different (similar) folds. BMCStruct. Biol. 1, 5.
34. Kishan, K. V. & Agrawal, V. (2005). SH3-like foldproteins are structurally conserved and functionallydivergent. Curr. Protein Pept. Sci. 6, 143–150.
35. Arcus, V. (2002). OB-fold domains: a snapshot ofthe evolution of sequence, structure and function.Curr. Opin. Struct. Biol. 12, 794–801.
36. Granata, V., Vecchio, P. D., Barone, G., Shehi, E., Fusi,P., Tortora, P. & Graziano, G. (2004). Guanidine-induced unfolding of the Sso7d protein from thehyperthermophilic archaeon Sulfolobus solfataricus. Int.J. Biol. Macromol. 34, 195–201.
37. Pavoor, T. V., Cho, Y. K. & Shusta, E. V. (2009).Development of GFP-based biosensors possessing thebinding properties of antibodies. Proc. Natl Acad. Sci.USA, 106, 11895–11900.
38. Hackel, B. J., Ackerman, M., Howland, S. W. &Wittrup, K. D. (2010). Stability and CDR compositionbiases enrich binder functionality landscapes. J. Mol.Biol. 401, 84–96.

616 Sso7d Scaffold Stable Binding Proteins
39. Silverman, A. P., Levin, A. M., Lahti, J. L. & Cochran,J. R. (2009). Engineered cystine-knot peptides thatbind alpha(v)beta(3) integrin with antibody-likeaffinities. J. Mol. Biol. 385, 1064–1075.
40. Feldhaus, M. J., Siegel, R. W., Opresko, L. K.,Coleman, J. R., Feldhaus, J. M., Yeung, Y. A. et al.(2003). Flow-cytometric isolation of human antibodiesfrom a nonimmune Saccharomyces cerevisiae surfacedisplay library. Nat. Biotechnol. 21, 163–170.
41. Cinier, M., Petit, M., Williams, M. N., Fabre, R. M.,Pecorari, F., Talham, D. R. et al. (2009). Bispho-sphonate adaptors for specific protein binding onzirconium phosphonate-based microarrays. Bioconjug.Chem. 20, 2270–2277.
42. Feldwisch, J., Tolmachev, V., Lendel, C., Herne, N.,Sjoberg, A., Larsson, B. et al. (2010). Design of anoptimized scaffold for affibody molecules. J. Mol. Biol.398, 232–247.
43. Shehi, E., Granata, V., Del Vecchio, P., Barone, G.,Fusi, P., Tortora, P. & Graziano, G. (2003). Thermalstability and DNA binding activity of a variant formof the Sso7d protein from the archeon Sulfolobussolfataricus truncated at leucine 54. Biochemistry, 42,8362–8368.
44. Consonni, R., Arosio, I., Recca, T., Fusi, P. & Zetta, L.(2007). Structural determinants responsible for thethermostability of Sso7d and its single point mutants.Proteins: Struct. Funct. Bioinformatics, 67, 766–775.
45. Consonni, R., Santomo, L., Fusi, P., Tortora, P. & Zetta,L. (1999). A single-point mutation in the extreme heat-and pressure-resistant sso7d protein from Sulfolobussolfataricus leads to a major rearrangement of thehydrophobic core. Biochemistry, 38, 12709–12717.
46. Bertschinger, J., Grabulovski, D. & Neri, D. (2007).Selection of single domain binding proteins bycovalent DNA display. Protein Eng. Des. Sel. 20, 57–68.
47. Renzone, G., Vitale, R. M., Scaloni, A., Rossi, M.,Amodeo, P. & Guagliardi, A. (2007). Structural
characterization of the functional regions in thearchaeal protein Sso7d. Proteins: Struct. Funct. Bioinfor-matics, 67, 189–197.
48. VanAntwerp, J. J. &Wittrup, K. D. (2000). Fine affinitydiscrimination by yeast surface display and flowcytometry. Biotechnol. Prog. 16, 31–37.
49. Boder, E. T. & Wittrup, K. D. (1998). Optimalscreening of surface-displayed polypeptide libraries.Biotechnol. Prog. 14, 55–62.
50. Rao, B. M., Girvin, A. T., Ciardelli, T., Lauffenburger,D. A. & Wittrup, K. D. (2003). Interleukin-2 mutantswith enhanced alpha-receptor subunit binding affin-ity. Protein Eng. 16, 1081–1087.
51. Rasband, W. S. (1997-2009). ImageJ. U. S. NationalInstitutes of Health, Bethesda, Maryland, USA;http://rsb.info.nih.gov/ij/.
52. Bloom, L. & Calabro, V. (2009). FN3: a new proteinscaffold reaches the clinic. Drug Discov. Today, 14,949–955.
53. Parker, M. H., Chen, Y., Danehy, F., Dufu, K.,Ekstrom, J., Getmanova, E. et al. (2005). Antibodymimics based on human fibronectin type threedomain engineered for thermostability and high-affinity binding to vascular endothelial growth factorreceptor two. Protein Eng. Des. Sel. 18, 435–444.
54. Wetzel, S. K., Settanni, G., Kenig, M., Binz, H. K. &Pluckthun, A. (2008). Folding and unfolding mecha-nism of highly stable full-consensus ankyrin repeatproteins. J. Mol Biol. 376, 241–257.
55. Skerra, A. (2008). Alternative binding proteins:anticalins – harnessing the structural plasticity ofthe lipocalin ligand pocket to engineer novel bindingactivities. FEBS J. 275, 2677–2683.
56. Schlehuber, S. & Skerra, A. (2002). Tuning ligandaffinity, specificity, and folding stability of anengineered lipocalin variant – a so-called ‘anticalin’ –using a molecular random approach. Biophys. Chem.96, 213–228.




















