
High Pressure Phase Transition and Elastic Properties of Lutetium Monopnictides
6
High pressure phase transition and elastic properties of Lutetium chalcogenide T. Seddik a, , F. Semari b , R. Khenata a, , A. Bouhemadou c , B. Amrani a a Laboratoire de Physique Quantique et de Mod elisation Math ematique (LPQ3M), D epartement de Technologie, Universit e de Mascara, 29000 Mascara, Algeria b Physics Department, Faculty of Sciences, University of Sidi-Bel-Abb es, 22000, Algeria c Laboratory for Developing New Materials and their Characterisation, Department of Physics, Faculty of Sciences, University of Setif, Algeria article info Article history: Received 12 August 2009 Accepted 25 August 2009 Keywords: Lutetium chalcogenides Ab initio calculation FP APW+lo Structural properties Phase transition Elastic constants abstract Using first-principles density functional calculation, the pressure induced structural phase transforma- tion and mechanical properties of NaCl type (B1) structure in Lutetium chalcogenides (LuX: X =S, Se, Te) were studied by means of the full-potential augmented plane wave plus local orbitals (FP APW+lo) method. The calculations were performed within the generalized gradient approximation (GGA) for the exchange-correlation potential. The calculated ground state properties such us lattice constants agree quit well with the experimental findings. We have determined the full set of first-order elastic constants and their pressure dependence, which have not been calculated and measured yet. The Debye temperature is estimated from the average sound velocity. To our knowledge this is the first quantitative theoretical prediction of the structural phase transition and elastic properties for these compounds and still awaits experimental confirmations. & 2009 Elsevier B.V. All rights reserved. 1. Introduction The theoretical studies have been fundamental in the devel- opment of new materials and new devices for diverse industrial applications. With advanced ab initio method, it is now feasible to access a database of crystal structure and use computer software to obtain interesting properties in the case in which experimental measurements are absent. In the last years a great number of works on the structural phase transition and elastic properties in rare-earth chalcogenide and pnictides has been investigated [1–14]. Several of these compounds exhibit no integer valence at high pressure and display numerous allotropic structures and properties, which can be in general interpreted in terms of valence fluctuations, arising from instability of f-electrons of the heavy RE atoms. Like the lanthanides, actinides chalcogenides and pnictides, the Lutetium chalcogenides LuX (X=S,Se,Te) compounds crystallize in NaCl- type structure (B1) at ambient conditions with space group Fm3m (2 2 5). The rare-earth (Lu) atom is positioned at (0;0;0) and the X atom at (1/2,1/2,1/2). To the best of our knowledge, apart the crystal structure and the superconducting behavior in the NaCl-phase in these compounds [15], experimental information on LuX compound is scarce. There is no theoretical and experimental reports on the structural phase transformation on these compounds have been appeared in the literature. Moreover, the elastic constants and their pressure dependence of the investigated compounds have not been calculated and measured yet. The raisons mentioned above, motivate us to perform these calculations using the full- relativistic version of the full-potential augmented plane-wave plus local orbitals method (FP APW+lo), based on the density functional theory (DFT), in order to provide reference data for the experimentalists and to complete existing theoretical and experi- mental works on these compounds. This paper will proceed as follows: in Section 2, we make a brief review of the computational technique used in this study. The most relevant results obtained for the structural phase transitions as well as the elastic and their related properties for Lutetium chalcogenides LuX compounds are presented and discussed in Section 3. Finally, in Section 4, we summarize the main conclusions of our work. 2. Computational method The calculations presented in this work were performed using a full-relativistic version of the full-potential with the mixed basis APW+lo method [16,17] as incorporated in WIEN2K code [18]. This is an implementation of the density functional theory (DFT) [19] with different possible approximations for the exchange correlation (XC) potentials. The XC potential was taken to be the ARTICLE IN PRESS Contents lists available at ScienceDirect journal homepage: www.elsevier.com/locate/physb Physica B 0921-4526/$ - see front matter & 2009 Elsevier B.V. All rights reserved. doi:10.1016/j.physb.2009.08.113 Corresponding authors. Fax: +213 45802923. E-mail addresses: [email protected] (T. Seddik), [email protected] (R. Khenata). Physica B 405 (2010) 394–399
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of High Pressure Phase Transition and Elastic Properties of Lutetium Monopnictides

ARTICLE IN PRESS
Physica B 405 (2010) 394–399
Contents lists available at ScienceDirect
Physica B
0921-45
doi:10.1
� Corr
E-m
khenata
journal homepage: www.elsevier.com/locate/physb
High pressure phase transition and elastic propertiesof Lutetium chalcogenide
T. Seddik a,�, F. Semari b, R. Khenata a,�, A. Bouhemadou c, B. Amrani a
a Laboratoire de Physique Quantique et de Mod�elisation Math�ematique (LPQ3M), D�epartement de Technologie, Universit�e de Mascara, 29000 Mascara, Algeriab Physics Department, Faculty of Sciences, University of Sidi-Bel-Abb�es, 22000, Algeriac Laboratory for Developing New Materials and their Characterisation, Department of Physics, Faculty of Sciences, University of Setif, Algeria
a r t i c l e i n f o
Article history:
Received 12 August 2009
Accepted 25 August 2009
Keywords:
Lutetium chalcogenides
Ab initio calculation
FP�APW+lo
Structural properties
Phase transition
Elastic constants
26/$ - see front matter & 2009 Elsevier B.V. A
016/j.physb.2009.08.113
esponding authors. Fax: +213 45802923.
ail addresses: [email protected] (T. Seddik),
[email protected] (R. Khenata).
a b s t r a c t
Using first-principles density functional calculation, the pressure induced structural phase transforma-
tion and mechanical properties of NaCl type (B1) structure in Lutetium chalcogenides (LuX: X=S, Se, Te)
were studied by means of the full-potential augmented plane wave plus local orbitals (FP�APW+lo)
method. The calculations were performed within the generalized gradient approximation (GGA) for the
exchange-correlation potential. The calculated ground state properties such us lattice constants agree
quit well with the experimental findings. We have determined the full set of first-order elastic constants
and their pressure dependence, which have not been calculated and measured yet. The Debye
temperature is estimated from the average sound velocity. To our knowledge this is the first quantitative
theoretical prediction of the structural phase transition and elastic properties for these compounds and
still awaits experimental confirmations.
& 2009 Elsevier B.V. All rights reserved.
1. Introduction
The theoretical studies have been fundamental in the devel-opment of new materials and new devices for diverse industrialapplications. With advanced ab initio method, it is now feasible toaccess a database of crystal structure and use computer softwareto obtain interesting properties in the case in which experimentalmeasurements are absent.
In the last years a great number of works on the structuralphase transition and elastic properties in rare-earth chalcogenideand pnictides has been investigated [1–14]. Several of thesecompounds exhibit no integer valence at high pressure anddisplay numerous allotropic structures and properties, which canbe in general interpreted in terms of valence fluctuations, arisingfrom instability of f-electrons of the heavy RE atoms. Like thelanthanides, actinides chalcogenides and pnictides, the Lutetiumchalcogenides LuX (X=S,Se,Te) compounds crystallize in NaCl-type structure (B1) at ambient conditions with space group Fm3m(2 2 5). The rare-earth (Lu) atom is positioned at (0;0;0) and the Xatom at (1/2,1/2,1/2).
To the best of our knowledge, apart the crystal structure andthe superconducting behavior in the NaCl-phase in thesecompounds [15], experimental information on LuX compound is
ll rights reserved.
scarce. There is no theoretical and experimental reports on thestructural phase transformation on these compounds have beenappeared in the literature. Moreover, the elastic constants andtheir pressure dependence of the investigated compounds havenot been calculated and measured yet. The raisons mentionedabove, motivate us to perform these calculations using the full-relativistic version of the full-potential augmented plane-waveplus local orbitals method (FP�APW+lo), based on the densityfunctional theory (DFT), in order to provide reference data for theexperimentalists and to complete existing theoretical and experi-mental works on these compounds.
This paper will proceed as follows: in Section 2, we make abrief review of the computational technique used in this study.The most relevant results obtained for the structural phasetransitions as well as the elastic and their related properties forLutetium chalcogenides LuX compounds are presented anddiscussed in Section 3. Finally, in Section 4, we summarize themain conclusions of our work.
2. Computational method
The calculations presented in this work were performed usinga full-relativistic version of the full-potential with the mixed basisAPW+lo method [16,17] as incorporated in WIEN2K code [18].This is an implementation of the density functional theory (DFT)[19] with different possible approximations for the exchangecorrelation (XC) potentials. The XC potential was taken to be the

ARTICLE IN PRESS
T. Seddik et al. / Physica B 405 (2010) 394–399 395
generalized gradient approximation (GGA) of Perdew-Burke andErnzerhof (PBE) [20]. The convergence parameter RMTKmax, whichcontrols the size of the basis sets in these calculations, was set to12. The RMT muffin-tin radii were taken as 2.45, 2.03, 2.3 and 2.45atomic units (a.u) for Lu, S, Se and Te, respectively. The valencewave functions inside MT spheres were expanded up to lmax=10,while the charge density was Fourier expanded up to Gmax=14(a.u)�1. The self-consistent calculations are considered to beconverged when the total energy of the system is stable within10�5 Ry. The integrals over the Brillouin zone are performed up to11�11�11 mesh for both B1 and B2 phases in the irreducibleBrillouin zone, using the Monkhorst–Pack special k-pointsapproach [21]. Both the plane wave cut-off and the number ofk-point were varied to ensure total energy convergence.
3. Results and discussions
3.1. Structural properties
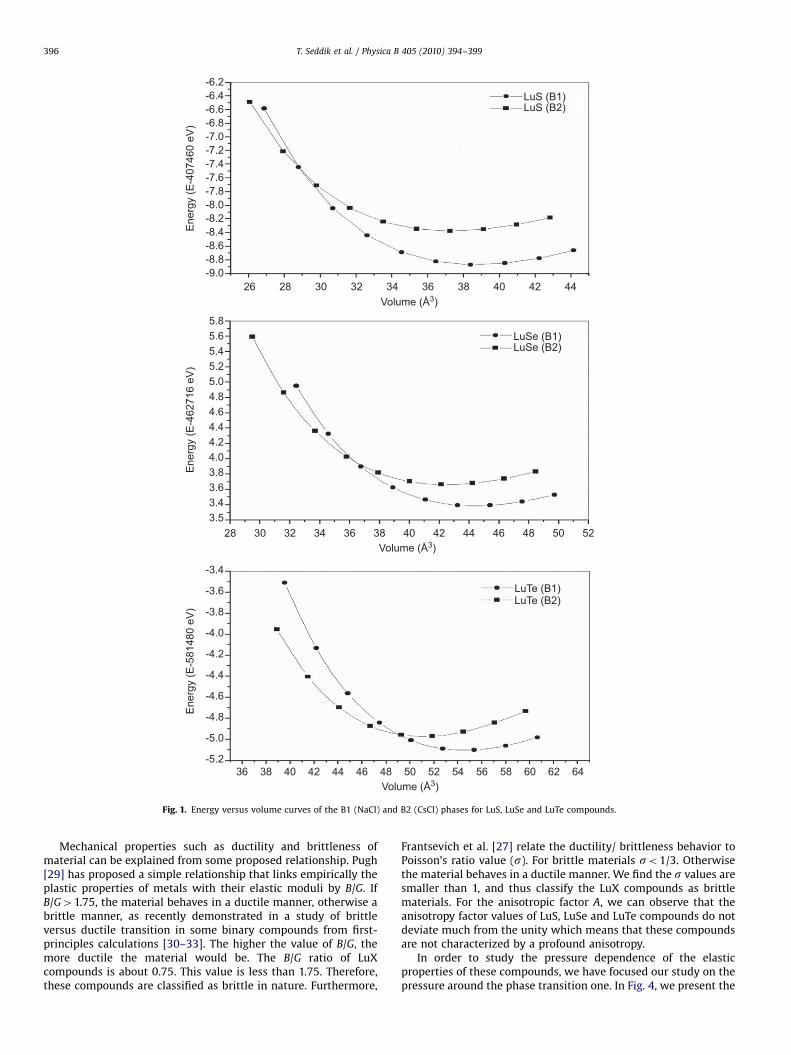
The total energy of the Lutetium chalcogenide LuS, LuSe andLuTe was calculated as a function of the volume in the B1-(NaCl)and B2-CsCl phases using the APW+lo method. The plots ofcalculated total energies versus reduced volume for thesecompounds are given in Fig. 1. It is seen from these E–V curvesthat the NaCl phase is stable than the CsCl phase at ambientconditions. The calculated total energies are fitted to Murnaghan’sequation of state [22] to determine the ground state propertiessuch as the equilibrium lattice constant a0, the bulk modulus B
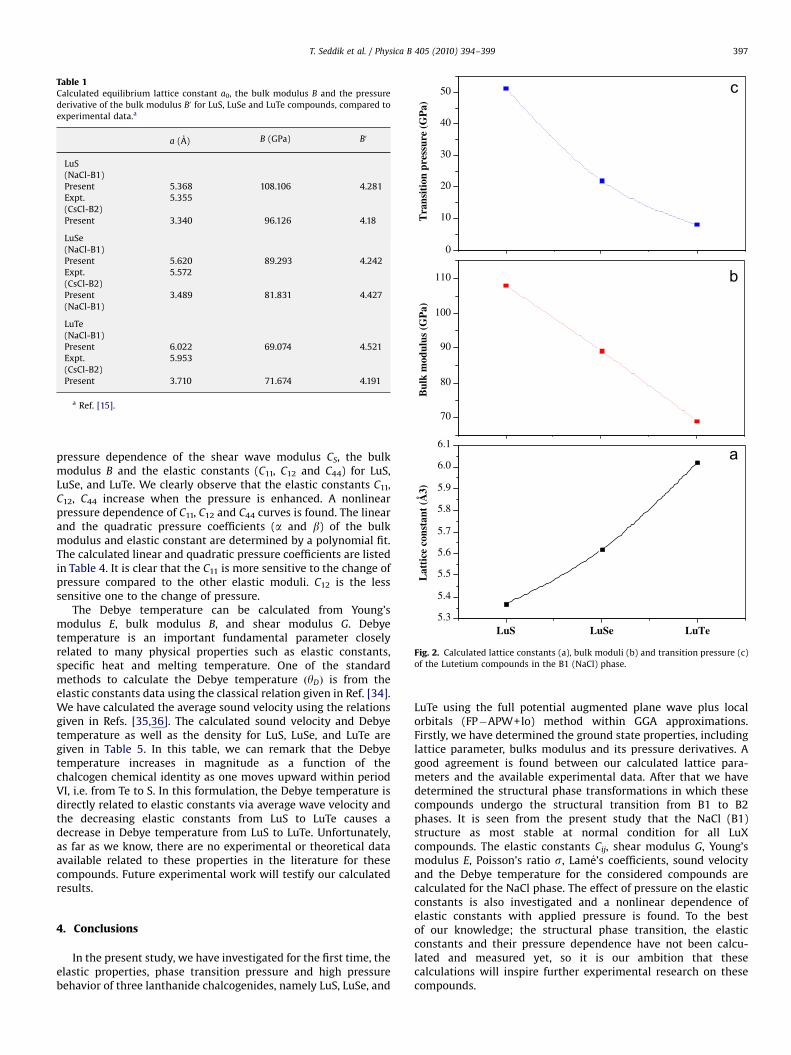
and its pressure derivative B0. The calculated equilibriumparameters (a0, B and B0) in both structures are given in Table 1,which also contains experimental data for comparison. Thecalculated lattice parameters are in reasonable agreement withexperimental data [15]. We not that the cation atoms are the samein the three compounds, the anion atoms size are different. Thisdifference could be the responsible for increasing of the latticeconstant from LuS to LuTe. The calculated bulk modulus decreasesfrom LuS to LuTe i.e. from the lower to the higher atomic numberFig. 2. This suggests that LuTe is more compressible than theother tow compounds, no experimental data for the bulk moduliof these compounds are available to be compared with ourtheoretical results.
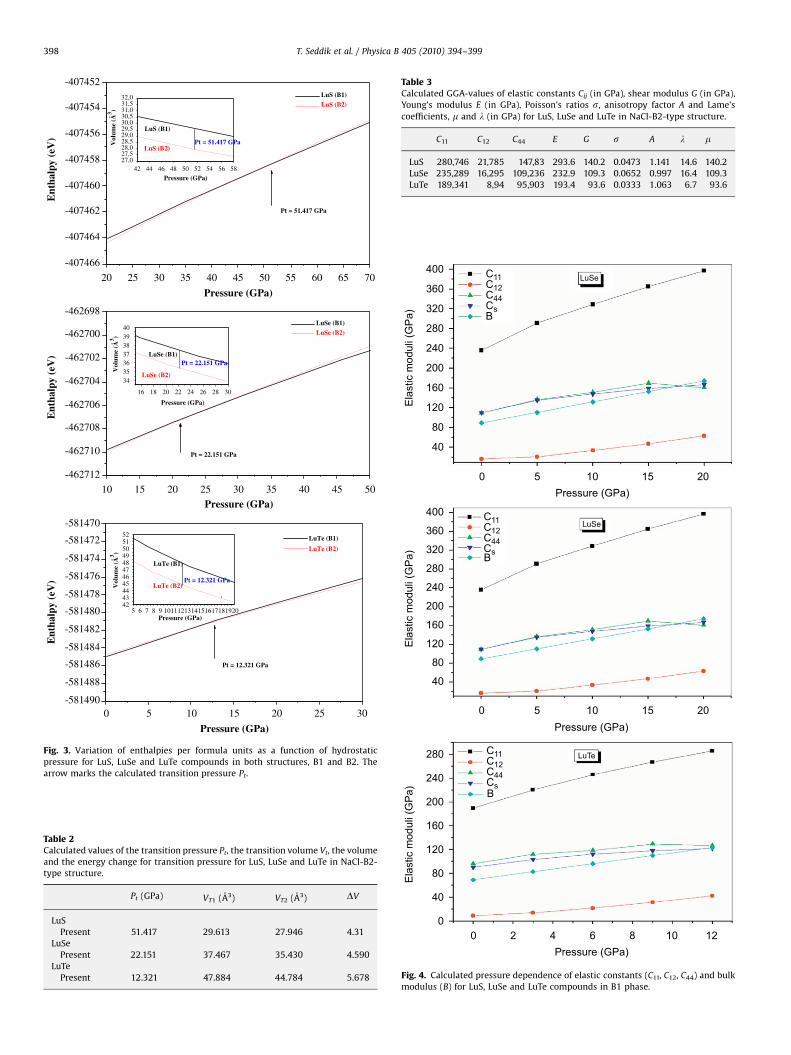
With application of high pressure, the calculation shows that anew crystal phases appear in LuX compounds, and the relativestability of two crystal structures requires an extremely accurateprediction. To discuss the stability of crystal structures and thepressure induced phase transition theoretically, it is essential toestimate the Gibbs free energy (G) [23] for the two phases, whichis given by G=E0+PV+TS. Since, the theoretical calculations areperformed at T=0 K, Gibbs free energy becomes equal to theenthalpy H=E0+PV, then the transition pressures are calculated atwhich the enthalpy for the two phases are equal. For a givenpressure, a stable structure is one for which the enthalpy has itslowest value. The enthalpy versus pressure curves for the bothstructures for LuS, LuSe and LuTe are displayed in Fig. 3. Thecalculated values of Pt and the transition volume are listed inTable 2. It is revealed from these results that present FP�APW+lomethod has correctly predicted the stable crystallographicstructure as B1 phase for all the LuX compounds. This can beunderstood from the obtained values of the enthalpy, which aresmaller in NaCl phase, as compared to those in CsCl phase.Following Fig. 2, it can be seen that the B1-B2 transition pressuredecrease systematically and consistently with the atomic numberof the chalcogen. To the best of our knowledge the values of thetransition pressure for these compounds have not been measured
or calculated, hence our results can serve as a prediction for futureinvestigation.
3.2. Elastic constants and their related properties
The knowledge of the elastic properties plays an importantpart in providing valuable information about the mechanical anddynamical properties, such as inter atomic potentials, equation ofstate, and phonon spectra. Elastic properties are also thermo-dynamically related to the specific heat, thermal expansion, Debyetemperature, melting point and Gruneisen parameter. The elasticconstants are believed to be related to the strength of materials.Indeed, the latter has been often related to the bulk modulus B,shear modulus G, Young’s modulus E, and Poisson’s ratio n. Whichare frequently measured for polycrystalline materials wheninvestigating their hardness. The elastic moduli can be alsopredict the structural stability of materials.
Wherefore we have calculated the Cij of these compounds inNaCl-B1 phase at ambient and under hydrostatic pressure byusing the method developed by Mehl [24]. In our calculations, weconsider only small lattice distortion in order to remain withinelastic domain of the crystal. A cubic structure is characterized bythree independent elastic constants namely C11, C12, and C44. Thecalculated values of elastic constants Cij, shear wave modulus CS
for LuS, LuSe and LuTe are listed in Table 3. In view of Table 3, it isclear that the elastic constants increase in magnitude as afunction of the anion chemical identity as one moves upwardswithin period VI, i.e. from Te to S. No elastic constants have beenreported experimentally and theoretically for LuX compounds.It is noticeable that for these compounds, all elastic constantssatisfy the following criterions: C11�C1240; C4440; C11+2C1240; and the bulk modulus also satisfy a criterionC11oBoC12 [25,26], implying that those compounds are stableagainst elastic deformation.
Elastic constants are correlated to numerous macroscopicparameters as the shear modulus G, Young’s modulus E, Poisson’sratio s and Lam�e’s coefficients m and l, which are the importantelastic moduli for mechanical applications. The latter can becalculated from the elastic constants Cij using the followingstandard relations:
G¼ ðC11 � C12þ3C44Þ=5 ð1Þ
E¼ 9BG=ð3BþGÞ ð2Þ
s¼ ð3B� EÞ=ð6BÞ ð3Þ
m¼ E=ð2ð1þsÞ ð4Þ
l¼ sE=fð1þsÞð1� 2sÞg ð5Þ
The calculated values of the mentioned elastic moduli forpolycrystalline LuS, LuSe and LuTe aggregates are also listed inTable 3. From this table it is observed that a decrease in the latticeconstant leads to an enhancement in the bulk modulus, Young’smodulus, and the shear modulus. It is known that the hardnessand strength of materials are related to their elastic moduli, suchas Young’s modulus E, bulk modulus B and the shear modulus G
[27]. Although the relationship between hardness and themodulus is not identical for different materials, the general trendis, the larger the modulus, the harder the material. Therefore, forLuS is expected to be the hardest due to its largest modulus.
The value of the Poisson ratio (s) for covalent materials issmall (s=0.1), whereas for ionic materials a typical value of s is0.25 [28]. In our case the value of n varies around 0.1, i.e. a strongcovalent contribution in intra-atomic bonding for these com-pounds should be assumed.
ARTICLE IN PRESS
-6.2-6.4-6.6-6.8-7.0-7.2-7.4-7.6-7.8-8.0-8.2-8.4-8.6-8.8-9.0
5.85.65.45.25.04.84.64.44.24.03.83.63.43.5
28
-3.4
-3.6
-3.8
-4.0
-4.2
-4.4
-4.6
-4.8
-5.0
-5.2
26 28 30 32 34 36 38 40 42 44Volume (Å3)
30 32 34 36 38Volume (Å3)
40 42 44 46 48 50 52
36 38 40 42 44 46 48 50 52 54 56 58 60 62 64Volume (Å3)
LuS (B1)LuS (B2)
LuSe (B1)LuSe (B2)
LuTe (B1)LuTe (B2)
Ene
rgy
(E-4
0746
0 eV
)E
nerg
y (E
-462
716
eV)
Ene
rgy
(E-5
8148
0 eV
)
Fig. 1. Energy versus volume curves of the B1 (NaCl) and B2 (CsCl) phases for LuS, LuSe and LuTe compounds.
T. Seddik et al. / Physica B 405 (2010) 394–399396
Mechanical properties such as ductility and brittleness ofmaterial can be explained from some proposed relationship. Pugh[29] has proposed a simple relationship that links empirically theplastic properties of metals with their elastic moduli by B/G. IfB/G41.75, the material behaves in a ductile manner, otherwise abrittle manner, as recently demonstrated in a study of brittleversus ductile transition in some binary compounds from first-principles calculations [30–33]. The higher the value of B/G, themore ductile the material would be. The B/G ratio of LuXcompounds is about 0.75. This value is less than 1.75. Therefore,these compounds are classified as brittle in nature. Furthermore,
Frantsevich et al. [27] relate the ductility/ brittleness behavior toPoisson’s ratio value (s). For brittle materials so1/3. Otherwisethe material behaves in a ductile manner. We find the s values aresmaller than 1, and thus classify the LuX compounds as brittlematerials. For the anisotropic factor A, we can observe that theanisotropy factor values of LuS, LuSe and LuTe compounds do notdeviate much from the unity which means that these compoundsare not characterized by a profound anisotropy.
In order to study the pressure dependence of the elasticproperties of these compounds, we have focused our study on thepressure around the phase transition one. In Fig. 4, we present the
ARTICLE IN PRESS
Table 1Calculated equilibrium lattice constant a0, the bulk modulus B and the pressure
derivative of the bulk modulus B0 for LuS, LuSe and LuTe compounds, compared to
experimental data.a
a (A) B (GPa) B0
LuS
(NaCl-B1)
Present 5.368 108.106 4.281
Expt. 5.355
(CsCl-B2)
Present 3.340 96.126 4.18
LuSe
(NaCl-B1)
Present 5.620 89.293 4.242
Expt. 5.572
(CsCl-B2)
Present 3.489 81.831 4.427
(NaCl-B1)
LuTe
(NaCl-B1)
Present 6.022 69.074 4.521
Expt. 5.953
(CsCl-B2)
Present 3.710 71.674 4.191
a Ref. [15].70
80
90
100
110
Bul
k m
odul
us (
GP
a)
LuS5.3
5.4
5.5
5.6
5.7
5.8
5.9
6.0
6.1
Lat
tice
con
stan
t (Å
3)
0
10
20
30
40
50
Tra
nsit
ion
pres
sure
(G
Pa)
LuSe LuTe
Fig. 2. Calculated lattice constants (a), bulk moduli (b) and transition pressure (c)
of the Lutetium compounds in the B1 (NaCl) phase.
T. Seddik et al. / Physica B 405 (2010) 394–399 397
pressure dependence of the shear wave modulus CS, the bulkmodulus B and the elastic constants (C11, C12 and C44) for LuS,LuSe, and LuTe. We clearly observe that the elastic constants C11,C12, C44 increase when the pressure is enhanced. A nonlinearpressure dependence of C11, C12 and C44 curves is found. The linearand the quadratic pressure coefficients (a and b) of the bulkmodulus and elastic constant are determined by a polynomial fit.The calculated linear and quadratic pressure coefficients are listedin Table 4. It is clear that the C11 is more sensitive to the change ofpressure compared to the other elastic moduli. C12 is the lesssensitive one to the change of pressure.
The Debye temperature can be calculated from Young’smodulus E, bulk modulus B, and shear modulus G. Debyetemperature is an important fundamental parameter closelyrelated to many physical properties such as elastic constants,specific heat and melting temperature. One of the standardmethods to calculate the Debye temperature ðyDÞ is from theelastic constants data using the classical relation given in Ref. [34].We have calculated the average sound velocity using the relationsgiven in Refs. [35,36]. The calculated sound velocity and Debyetemperature as well as the density for LuS, LuSe, and LuTe aregiven in Table 5. In this table, we can remark that the Debyetemperature increases in magnitude as a function of thechalcogen chemical identity as one moves upward within periodVI, i.e. from Te to S. In this formulation, the Debye temperature isdirectly related to elastic constants via average wave velocity andthe decreasing elastic constants from LuS to LuTe causes adecrease in Debye temperature from LuS to LuTe. Unfortunately,as far as we know, there are no experimental or theoretical dataavailable related to these properties in the literature for thesecompounds. Future experimental work will testify our calculatedresults.
4. Conclusions
In the present study, we have investigated for the first time, theelastic properties, phase transition pressure and high pressurebehavior of three lanthanide chalcogenides, namely LuS, LuSe, and
LuTe using the full potential augmented plane wave plus localorbitals (FP�APW+lo) method within GGA approximations.Firstly, we have determined the ground state properties, includinglattice parameter, bulks modulus and its pressure derivatives. Agood agreement is found between our calculated lattice para-meters and the available experimental data. After that we havedetermined the structural phase transformations in which thesecompounds undergo the structural transition from B1 to B2phases. It is seen from the present study that the NaCl (B1)structure as most stable at normal condition for all LuXcompounds. The elastic constants Cij, shear modulus G, Young’smodulus E, Poisson’s ratio s, Lam�e’s coefficients, sound velocityand the Debye temperature for the considered compounds arecalculated for the NaCl phase. The effect of pressure on the elasticconstants is also investigated and a nonlinear dependence ofelastic constants with applied pressure is found. To the bestof our knowledge; the structural phase transition, the elasticconstants and their pressure dependence have not been calcu-lated and measured yet, so it is our ambition that thesecalculations will inspire further experimental research on thesecompounds.
ARTICLE IN PRESS
20
-407466
-407464
-407462
-407460
-407458
-407456
-407454
-407452
Pt = 51.417 GPa
Ent
halp
y (e
V)
Pressure (GPa)
LuS (B1)
LuS (B2)
4227,027,528,028,529,029,530,030,531,031,532,0
Pt = 51.417 GPaLuS (B2)
LuS (B1)
Vol
ume
(Å3 )
Pressure (GPa)
10
-462712
-462710
-462708
-462706
-462704
-462702
-462700
-462698
Pt = 22.151 GPa
Ent
halp
y (e
V)
Pressure (GPa)
LuSe (B1)
LuSe (B2)
16
34353637383940
Pt = 22.151 GPa
LuSe (B2)
LuSe (B1)
Vol
ume
(Å3 )
Pressure (GPa)
0-581490
-581488
-581486
-581484
-581482
-581480
-581478
-581476
-581474
-581472
-581470
Pt = 12.321 GPa
Ent
halp
y (e
V)
Pressure (GPa)
LuTe (B1)
LuTe (B2)
54243444546474849505152
Pt = 12.321 GPaLuTe (B2)
LuTe (B1)
Vol
ume
(Å3 )
Pressure (GPa)
25 30 35 40 45 50 55 60 65 70
15 20 25 30 35 40 45 50
5 10 15 20 25 30
44 46 48 50 52 54 56 58
18 20 22 24 26 28 30
6 7 8 9 1011121314151617181920
Fig. 3. Variation of enthalpies per formula units as a function of hydrostatic
pressure for LuS, LuSe and LuTe compounds in both structures, B1 and B2. The
arrow marks the calculated transition pressure Pt.
Table 2Calculated values of the transition pressure Pt, the transition volume Vt, the volume
and the energy change for transition pressure for LuS, LuSe and LuTe in NaCl-B2-
type structure.
Pt (GPa) VT1 (A3) VT2 (A3) DV
LuS
Present 51.417 29.613 27.946 4.31
LuSe
Present 22.151 37.467 35.430 4.590
LuTe
Present 12.321 47.884 44.784 5.678
Table 3Calculated GGA-values of elastic constants Cij (in GPa), shear modulus G (in GPa),
Young’s modulus E (in GPa), Poisson’s ratios s, anisotropy factor A and Lame’s
coefficients, m and l (in GPa) for LuS, LuSe and LuTe in NaCl-B2-type structure.
C11 C12 C44 E G s A l m
LuS 280,746 21,785 147,83 293.6 140.2 0.0473 1.141 14.6 140.2
LuSe 235,289 16,295 109,236 232.9 109.3 0.0652 0.997 16.4 109.3
LuTe 189,341 8,94 95,903 193.4 93.6 0.0333 1.063 6.7 93.6
400
360
320
280
240
200
160
120
80
40
400
360
320
280
240
200
160
120
80
40
280
240
200
160
120
80
40
0
0 5 10 15 20Pressure (GPa)
Pressure (GPa)
Pressure (GPa)
0 5 10 15 20
0 2 4 6 8 10 12
Ela
stic
mod
uli (
GP
a)E
last
ic m
odul
i (G
Pa)
Ela
stic
mod
uli (
GP
a) B
B
C11C12C44Cs
C11C12C44Cs
C11C12C44Cs
B
LuSe
LuSe
LuTe
Fig. 4. Calculated pressure dependence of elastic constants (C11, C12, C44) and bulk
modulus (B) for LuS, LuSe and LuTe compounds in B1 phase.
T. Seddik et al. / Physica B 405 (2010) 394–399398

ARTICLE IN PRESS
Table 4Calculated pressure derivatives of the elastic constants and bulk modulus for LuS,
LuSe and LuTe in NaCl-B2-type structure.
LuS LuSe LuTe
a b a b a b
C11 8.76 �0.048 10.66 �0.136 10.64 �0.223
C12 2.042 0.0241 1.029 0.0685 1.455 0.111
C44 3.228 �0.041 6.61 �0.194 5.739 �0.259
B 4.281 �0.0 4.242 �0.0 4.521 �0.0
Table 5Calculated GGA-density (r, in g/cm), longitudinal, transverse and average sound
velocity (vl, vt and vm, in m/s) and Debye temperature (yD, in K) for LuS, LuSe and
LuTe in NaCl-B2-type structure.
P vl vt vm yD
LuS 8.89 5760.7 3971.1 4841.8 537.1
LuSe 9.50 4973.5 3391.6 4152.8 440.0
LuTe 9.2 4589.6 3188.9 3875.8 383.2
T. Seddik et al. / Physica B 405 (2010) 394–399 399
References
[1] G. Pagare, S.P. Sanyal, P.K. Jha, J. Alloys Compounds 398 (2005) 16.[2] G. Pagare, V. Srivastava, S.P. Sanyal, J. Phys. Chem. Solids 66 (2005) 1177.[3] M. Aynyas, N. Kaurav, S.P. Sanyal, J. Phys. Chem. Solids 63 (2002) 821.[4] M. Aynyas, S.P. Sanyal, P.K. Jha, Phys. Stat. Sol. (b) 229 (2002) 1459.[5] P.K. Jha, S.P. Sanyal, Phys. Stat. Sol. (b) 13 (1997) 200;
P.K. Jha, S.P. Sanyal, J. Phys. Chem. Solids 64 (2003) 1237;P.K. Jha, S.P. Sanyal, J. Phys. Chem. Solids 64 (2003) 127.
[6] V. Srivastava, A.K. Bandyopadhyay, P.K. Jha, S.P. Sanyal, J. Phys. Chem. Solids 64(2003) 907.
[7] D. Varshney, N. Kaurav, R. Kinge, S. Shah, R.K. Singh, High Pressure Res. 25 (2)(2005) 145.
[8] D. Varshney, N. Kaurav, R. Kinge, R.K. Singh, J. Phys. Condens. Matter 19 (2007)236204.
[9] K. Kholiya, B.R.K. Gupta, PRANAMA—J. Phys. 68 (2007) 649.[10] I.R. Shein, K.I. Shein, A.L. Ivanovskii, Pis’ma Zh. Tekhn. Fiz. 33 (2007) 72.[11] A. Bouhemadou, R. Khenata, M. Sahnoun, H. Baltache, M. Kharoubi, Physica B
363 (2005) 255.[12] A. Bouhemaou, R. Khenata, M. Maamache, J. Mol. Struct. THEOCHEM 777
(2006) 5.[13] D. Rached, M. Rabah, R. Khenata, B. Abidri, S. Benalia, Solid State Comm.
(2009), in press.[14] F. Soyalp, Comput. Mater. Sci. 44 (2009) 1371;
F. Soyalp, Comput. Mater. Sci. 43 (2008) 313.[15] F. Hulliger, G.W. Hull. Jr., Solid State Comm. 8 (1970) 1379.[16] G.K.H. Madsen, P. Blaha, K. Schwarz, E. Sjostedt, L. Nordstrom, Phys. Rev. B 64
(2001) 195134.[17] K. Schwarz, P. Blaha, G.K.H. Madsen, Comput. Phys. Comm. 147 (2002) 71.[18] P. Blaha, K. Schwarz, G.K.H. Madsen, D. Kvasnicka, J. Luitz, WIEN2k, an
augmented plane wave plus local orbitals program for calculating crystalproperties, Vienna University of Technology, Austria, 2001.
[19] P. Hohenberg, W. Kohn, Phys. Rev. 136 (1964) 86.[20] J.P. Perdew, S. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865.[21] H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188.[22] F.D. Murnaghan, Proc. Nat. Acad. Sci. USA 30 (1944) 244.[23] W. Wettling, J. Windscheif, Solid State Comm. 50 (1984) 33.[24] M.J. Mehl, Phys. Rev. B 47 (1993) 2493.[25] D.C. Wallace, Thermodynamics of Crystals, Wiley, New York, 1972 (Chapter 1).[26] O. Beckstein, J.E. Klepeis, G.L.W. Hart, O. Pankratov, Phys. Rev. B 63 (2001)
134112.[27] I.N. Frantsevich, F.F. Voronov, S.A. Bokuta, in: I.N. Frantsevich (Ed.), Elastic
Constants and Elastic Moduli of Metals and Insulators, Naukova Dumka, Kiev,1983, pp. 60–180.
[28] J. Haines, J.M. Leger, G. Bocquillon, Annu. Rev. Mater. Res. 31 (2001) 1.[29] S.F. Pugh, Philos. Mag. 45 (1954) 823.[30] D. Varshney, G. Joshi, N. Kaurav, R.K. Singh, J. Phys. Chem. Solids 70 (2009)
451.[31] Y. Wu, W. Hu, European Phys. J. B 60 (2007) 75.[32] Y. Wu, W. Hu, S. Han, Physica B 403 (2008) 3792.[33] J.R. Morris, Y. Ye, Y.-B. Lee, B.N. Harmon, K.A. Gschneidner Jr., A.M. Russell,
Issue Series Title: Acta Mater. 52 (2004) 4849.[34] P. Wachter, M. Filzmoser, J. Rebizant, Physica B 293 (2001) 199.[35] O.L. Anderson, J. Phys. Chem. Solids 24 (1963) 909.[36] E. Schreiber, O.L. Anderson, N. Soga, Elastic Constants and their Measure-
ments, McGraw-Hill, New York, 1996.




















