
STAT5 Outcompetes STAT3 To Regulate the Expression of the Oncogenic Transcriptional Modulator BCL6
Upload
independentCategory
view
0download
0
www.elsevier.com/locate/yviro
Virology 322 (2004) 51–60
Hepatitis C virus NS5A mediated STAT3 activation requires
co-operation of Jak1 kinase
Bhaswati Sarcar,a Asish K. Ghosh,a Robert Steele,a Ranjit Ray,b and Ratna B. Raya,b,*
aDepartment of Pathology, Saint Louis University, St. Louis, MO 63104, USAbDepartment of Internal Medicine, Saint Louis University, St. Louis, MO 63104, USA
Received 15 November 2003; returned to author for revision 9 December 2003; accepted 5 January 2004
Abstract
Hepatitis C virus (HCV) is a major etiologic agent for chronic hepatitis worldwide and often leads to cirrhosis and hepatocellular
carcinoma. However, the mechanism for development of chronic hepatitis or hepatocarcinogenesis by HCV remains unclear. Signal
transducers and activators of transcription (STATs) family proteins function as the downstream effectors of cytokine signaling and play a
critical role in cell growth regulation. In many cancers including liver, STAT3 is often constitutively activated, although the mechanism of
persistent activation of STAT3 is unknown. The nonstructural protein 5A (NS5A) encoded from the HCV genome has shown cell growth
regulatory properties. In this study, we have observed that HCV NS5A activates STAT3 phosphorylation, which in turn translocates into the
nucleus. In vivo activation of STAT3 was also observed in the liver of transgenic mice expressing HCV NS5A. Introduction of NS5A in
hepatoma cells modulated STAT3 downstream molecules Bcl-xL and p21 expression. To determine if STAT3 activation by NS5A could
induce STAT3 mediated gene expression, a luciferase reporter construct based on a synthetic promoter was used to transfect hepatoma cells.
Activation of endogenous cellular STAT3 by HCV NS5A induced luciferase gene expression through STAT3 specific binding elements. Our
subsequent studies suggested that NS5A forms a complex with Jak1 and recruits STAT3 for activation. Taken together, our results suggested
that NS5A activates STAT3 through co-operation of Jak1 kinase and activated STAT3 may contribute to HCV-mediated pathogenesis.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Hepatitis C virus; STAT3; Hepatoma cells
Introduction (Clarke, 1997; Grakoui et al., 1993; Hijikata et al., 1991).
Hepatitis C virus (HCV) is a major etiologic agent for
chronic hepatitis worldwide and may lead to the develop-
ment of hepatocellular carcinoma. However, the mechanism
for development of chronic hepatitis or hepatocarcinogene-
sis by HCV remains unclear. HCV genome contains a linear,
positive-strand RNA molecule of approximately 9500
nucleotides (Choo et al., 1989). Many HCV genomes have
been cloned and the sequence divergence indicates several
genotypes and a series of subtypes for this virus (Simmonds,
2001). The HCV genome encodes a single polyprotein
precursor of approximately 3000 amino acids which is
cleaved by both host and viral proteases to generate at least
10 individual proteins, Core, E1, E2, p7, nonstructural
protein (NS) 2, NS3, NS4A, NS4B, NS5A, and NS5B
0042-6822/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.virol.2004.01.008
* Corresponding address. Department of Pathology, Saint Louis
University, 4th Floor, 1402 South Grand Boulevard, St. Louis, MO
63104. Fax: +1-314-771-3816.
E-mail address: [email protected] (R.B. Ray).
HCV NS5A exists as two phosphoproteins p56 and p58 that
are phosphorylated at serine residues after the mature NS5A
protein and is released from the polyprotein (Kaneko et al.,
1994; Tanji et al., 1995). NS5A modulates cell cycle and
immunoregulatory genes (Ghosh et al., 1999a, 2000a;
Majumder et al., 2001; Polyak et al., 2001) and promotes
murine fibroblasts to transformed phenotype (Gale et al.,
1999; Ghosh et al., 1999a). Because NS5A does not bind to
the DNA directly, it probably exerts its cell growth regula-
tory effect through other protein(s). In fact, NS5A down-
regulates the p21 gene by sequestering p53 in the cytoplasm
(Lan et al., 2002; Majumder et al., 2001). NS5A also exerts
anti-apoptotic activity in mammalian cells (Gale et al., 1999;
Ghosh et al., 2000b). NS5A physically associates with PKR
(Gale et al., 1999), human vesicle associated protein
hVAP33 (Tu et al., 1999), growth factor receptor-binding
protein 2 (Tan et al., 1999), a novel transcription factor
SRCAP (Ghosh et al., 2000a), and a nuclear import ma-
chinery component karyopherin h3 protein (Chung et al.,
2000). These results together suggest that HCV NS5A
B. Sarcar et al. / Virology 322 (2004) 51–6052
protein is likely to play an important role in virus–host
interaction.
Signal transducers and activators of transcription (STATs)
are a family of latent cytoplasmic transcription factors that
are activated in response to various extracellular polypeptide
ligands, including cytokines and growth factors (Darnell et
al., 1994; Schindler and Darnell, 1995). STATs have an
amino-terminal protein–protein interaction domain, a DNA
interaction domain, a SH2 domain, and a single tyrosine
phosphorylation site. Phosphorylation of this tyrosine resi-
due stabilizes the association of two STAT monomers
through the interaction between the phosphotyrosine and
SH2 domain (Bowman et al., 2000; Bromberg and Darnell,
2000; Chatterjee-Kishore et al., 2000; Darnell, 1997; Schin-
dler and Darnell, 1995). These activated STAT dimers
translocate into the nucleus, where they bind to their cognate
DNA response elements. In addition to their normal roles in
cell signaling, a relationship between the activation of STATs
and oncogenesis has been observed (Bowman et al., 2000;
Darnell, 1997). Many oncoproteins can activate specific
STATs, and the activated STAT protein participates in
oncogenesis by stimulating cell proliferation and preventing
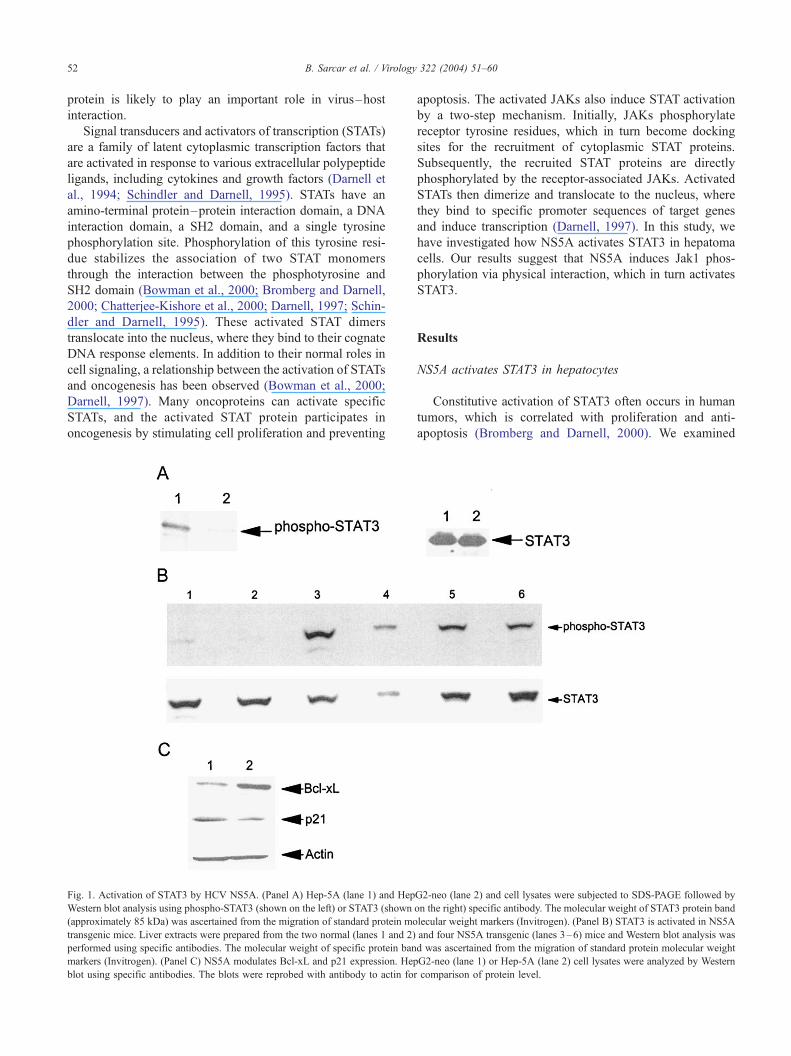
Fig. 1. Activation of STAT3 by HCV NS5A. (Panel A) Hep-5A (lane 1) and Hep
Western blot analysis using phospho-STAT3 (shown on the left) or STAT3 (shown
(approximately 85 kDa) was ascertained from the migration of standard protein mo
transgenic mice. Liver extracts were prepared from the two normal (lanes 1 and 2)
performed using specific antibodies. The molecular weight of specific protein ban
markers (Invitrogen). (Panel C) NS5A modulates Bcl-xL and p21 expression. Hep
blot using specific antibodies. The blots were reprobed with antibody to actin for
apoptosis. The activated JAKs also induce STAT activation
by a two-step mechanism. Initially, JAKs phosphorylate
receptor tyrosine residues, which in turn become docking
sites for the recruitment of cytoplasmic STAT proteins.
Subsequently, the recruited STAT proteins are directly
phosphorylated by the receptor-associated JAKs. Activated
STATs then dimerize and translocate to the nucleus, where
they bind to specific promoter sequences of target genes
and induce transcription (Darnell, 1997). In this study, we
have investigated how NS5A activates STAT3 in hepatoma
cells. Our results suggest that NS5A induces Jak1 phos-
phorylation via physical interaction, which in turn activates
STAT3.
Results
NS5A activates STAT3 in hepatocytes
Constitutive activation of STAT3 often occurs in human
tumors, which is correlated with proliferation and anti-
apoptosis (Bromberg and Darnell, 2000). We examined
G2-neo (lane 2) and cell lysates were subjected to SDS-PAGE followed by
on the right) specific antibody. The molecular weight of STAT3 protein band
lecular weight markers (Invitrogen). (Panel B) STAT3 is activated in NS5A
and four NS5A transgenic (lanes 3–6) mice and Western blot analysis was
d was ascertained from the migration of standard protein molecular weight
G2-neo (lane 1) or Hep-5A (lane 2) cell lysates were analyzed by Western
comparison of protein level.
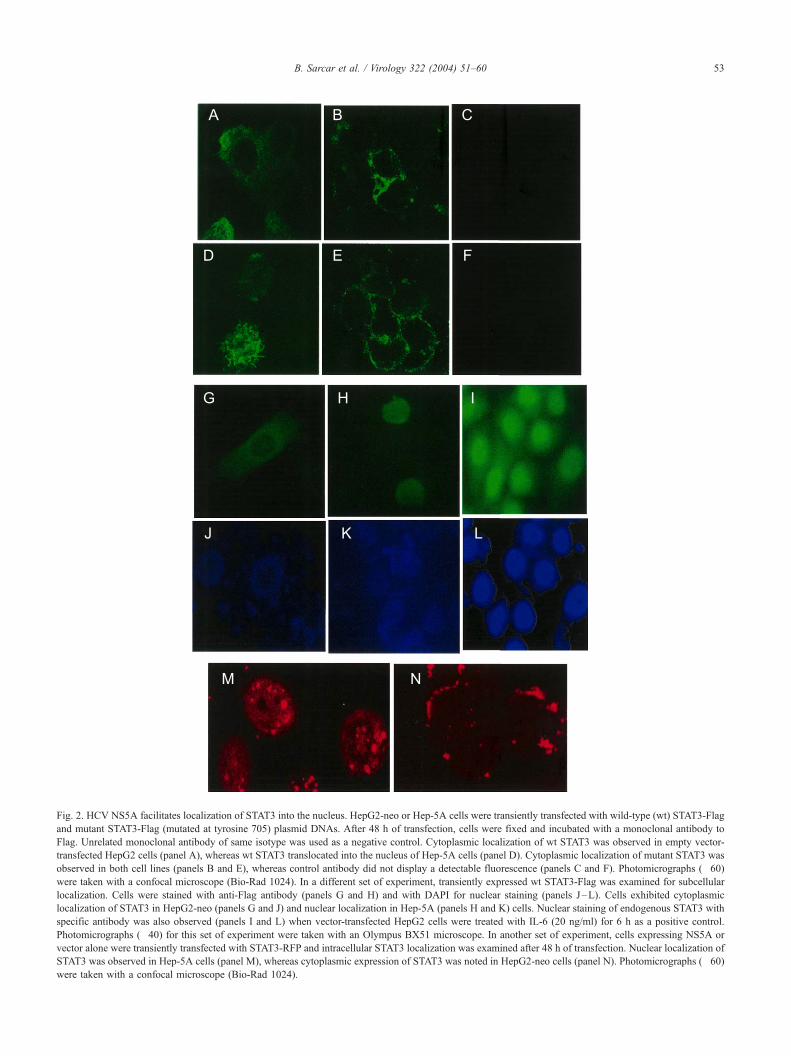
Fig. 2. HCV NS5A facilitates localization of STAT3 into the nucleus. HepG2-neo or Hep-5A cells were transiently transfected with wild-type (wt) STAT3-Flag
and mutant STAT3-Flag (mutated at tyrosine 705) plasmid DNAs. After 48 h of transfection, cells were fixed and incubated with a monoclonal antibody to
Flag. Unrelated monoclonal antibody of same isotype was used as a negative control. Cytoplasmic localization of wt STAT3 was observed in empty vector-
transfected HepG2 cells (panel A), whereas wt STAT3 translocated into the nucleus of Hep-5A cells (panel D). Cytoplasmic localization of mutant STAT3 was
observed in both cell lines (panels B and E), whereas control antibody did not display a detectable fluorescence (panels C and F). Photomicrographs (�60)
were taken with a confocal microscope (Bio-Rad 1024). In a different set of experiment, transiently expressed wt STAT3-Flag was examined for subcellular
localization. Cells were stained with anti-Flag antibody (panels G and H) and with DAPI for nuclear staining (panels J–L). Cells exhibited cytoplasmic
localization of STAT3 in HepG2-neo (panels G and J) and nuclear localization in Hep-5A (panels H and K) cells. Nuclear staining of endogenous STAT3 with
specific antibody was also observed (panels I and L) when vector-transfected HepG2 cells were treated with IL-6 (20 ng/ml) for 6 h as a positive control.
Photomicrographs (�40) for this set of experiment were taken with an Olympus BX51 microscope. In another set of experiment, cells expressing NS5A or
vector alone were transiently transfected with STAT3-RFP and intracellular STAT3 localization was examined after 48 h of transfection. Nuclear localization of
STAT3 was observed in Hep-5A cells (panel M), whereas cytoplasmic expression of STAT3 was noted in HepG2-neo cells (panel N). Photomicrographs (�60)
were taken with a confocal microscope (Bio-Rad 1024).
B. Sarcar et al. / Virology 322 (2004) 51–60 53

B. Sarcar et al. / Virology 322 (2004) 51–6054
whether NS5A activates the endogenous STAT3 in stable
HepG2 cells expressing NS5A (Hep-5A cells). For this
purpose, cell lysates were analyzed by Western blot using
specific antibodies. Hep-5A cells expressed a higher level of
phosphorylated STAT3 as compared to cells expressing
empty vector (HepG2-neo) (Fig. 1, panel A). However, total
STAT3 expression level was similar in both HepG2-neo and
Hep-5A cells. We have also investigated whether HCV
NS5A protein activates the phosphorylation of STAT3
expression in transgenic mouse liver. Mouse liver was
collected in cold perfusion buffer (50 mM phosphate buffer,
pH 7.4, 120 mM NaCl, and 10 mM EDTA) and a small
portion was homogenized in 2 ml ice-cold glycerol buffer
using a Dounce homogenizer as previously described
(Majumder et al., 2002). Liver homogenates were subjected
Fig. 3. NS5A enhances transcriptional activity of the STAT3-regulated promote
reporter (3 Ag) and increasing concentrations of the CMV NS5A expression vecto
plasmid DNA (6 ug) was used as a negative control. Plasmid DNA expressing v
constant in each transfection by adding empty vector DNA. Luciferase assay was p
each set of experiments, and relative luciferase activity is presented. (Panel B) Huh
reporter plasmid DNA (3 Ag) and luciferase activity was measured after 48 h of tran
and relative luciferase activity is presented with standard deviation from triplicate
to Western blot analysis using specific antibodies for phos-
pho-STAT3 and STAT3 expression. Phosphorylation of
STAT3 was observed from the liver extracts of four NS5A
transgenic mice (Fig. 1, panel B, lanes 3–6), but not from
two normal littermates (lanes 1–2). The blot was reprobed
with a rabbit antibody to STAT3, and comparable level of
specific protein was observed from all mice liver. We also
examined the expression of p21 and Bcl-xL (downstream
molecules of STAT3) in Hep-5A and HepG2-neo cells.
Densitometric scanning of the results from Western blot
analysis suggested an approximately 3.5-fold increase of
Bcl-xL and approximately 3-fold reduction of p21 expres-
sion (Fig. 1, panel C). Results from these set of experiments
suggest that HCV NS5A expression increases phosphoryla-
tion of STAT3 level and alters Bcl-xL and p21 expression.
r. (Panel A) HepG2 cells were transiently co-transfected with pLucTKS3
r (1, 3, and 6 Ag). Carboxy-terminal deletion mutant of NS5A (NS5A1– 332)
-Src (4 Ag) was used as a positive control. The amount of DNA was kept
erformed at 48 h post-transfection. Triplicate transfection was performed in
7, Huh7-NS5A, and Rep-1 cells were separately transfected with pLucTKS3
sfection. Triplicate transfections were performed in each set of experiments,
analysis. Basal value was arbitrarily set at 1.

Fig. 4. Jak1 kinase inhibitor, AG490, inhibits NS5A induced activation of
Jak1 kinase. Huh7 cells were transduced with Adh-gal (lanes 1 and 2) or
Ad-NS5A (lanes 3 and 4) and treated with 50 AM of AG490 (lanes 2 and 4).
After 48 h, cell lysates were analyzed by Western blot using specific
antibodies. Phosphorylation of Jak1 kinase was inhibited in presence of
AG490. When these samples were analyzed for phospho-STAT3, similar
results were observed (middle panel). Expression of total STAT3 remained
unaltered as seen from reprobed blot (bottom panel). The molecular weight
of specific protein band was ascertained from the migration of standard
protein molecular weight markers (Invitrogen).
B. Sarcar et al. / Virology 322 (2004) 51–60 55
NS5A facilitates translocation of STAT3 into the nucleus
To examine the localization of phospho-STAT3 in the
presence of NS5A, indirect immunofluorescence was per-
formed. HepG2 or Hep-5A cells were transiently trans-
fected with wild-type (wt) STAT3-Flag and mutant STAT3-
Flag (mutated at tyrosine 705) plasmid DNAs. After 48 h of
transfection, cells were fixed and incubated with a mono-
clonal antibody to Flag. Unrelated monoclonal antibody of
the same isotype was used as a negative control. STAT3
translocated into the nucleus of Hep-5A cells (Fig. 2, panel
D), whereas cytoplasmic localization of STAT3 was ob-
served in empty vector-transfected HepG2 cells (panel A).
Predominant perinuclear localization of mutant STAT3 was
observed in both the cell lines (panels B and E). Detectable
fluorescence was not observed from the control antibody of
same isotype (panels C and F). We further examined the
nuclear staining with DAPI (panels J–L) to confirm the
subcellular localization of STAT3 in Hep-5A cells. Our
results demonstrated cytoplasmic localization of STAT3 in
HepG2-neo cells (panels G and J) and nuclear localization
of STAT3 in Hep-5A cells (panels H and K). When we
transiently expressed the STAT3-RFP (Bild et al., 2002),
nuclear localization of STAT3 was observed in Hep-5A
cells (panel M); however, cytoplasmic localization of
STAT3 was observed in HepG2-neo cells (panel N). We
have also treated the HepG2-neo cells with IL-6 (20 ng/ml)
as a positive control for nuclear localization as described
earlier (Barre et al., 2003). STAT3 localization was ob-
served after 6 h of IL-6 treatment (panels I and L), as
expected. These results suggest that NS5A activates phos-
phorylation of STAT3, which in turn, accumulates in the
nucleus. To examine whether HCV NS5A physically asso-
ciates with STAT3 for activation, a co-immunoprecipitation
experiment was performed. Cells were co-transfected with
CMV Flag STAT3 plasmid DNA and different concentra-
tions of CMV NS5A to examine in vivo complex forma-
tion. However, after several attempts, we did not observe a
direct association between NS5A and STAT3 in HepG2 or
Huh7 cells. Thus, our results suggest that HCV NS5A
facilitates translocation of activated STAT3 into the nucleus,
although a direct interaction of NS5A and STAT3 could not
be determined.
NS5A enhances transcriptional activity of STAT3-regulated
promoter
We next performed an in vitro transient transfection
assay using STAT3-regulated reporter plasmid DNA. The
STAT3 reporter pLucTKS3 was constructed from pLucTK
by inserting seven copies of an annealed oligonucleotide
corresponding to a STAT3-specific binding site from the
human C-reactive protein (CRP) gene and was used as a
reporter plasmid for determining transcriptional activity of
the STAT3 protein (Turkson et al., 1998). Cells were co-
transfected with STAT3 reporter construct pLucTKS3 and
CMV NS5A. After 48 h of transfection, cells were harvested
using the reporter lysis buffer, and luciferase activity was
measured. HCV NS5A enhanced luciferase expression by 4-
to 5-fold in HepG2 cells (Fig. 3, panel A). However,
carboxy-terminal deletion mutant of NS5A (CMV
NS5A1–332) did not display an increase of luciferase activ-
ity. The v-src expression vector was used as a positive
control and enhancement of luciferase activity was ex-
hibited, as expected. Similar results were also obtained from
Huh7 cells in an in vitro reporter assay (data not shown). To
examine whether similar effects are observed in presence of
other HCV proteins, we used Huh7 cells expressing NS5A
(Huh-NS5A) or full-length replicon (Rep-1). Enhancement
of luciferase activity was observed when we used stable cell
lines expressing NS5A or Rep-1 (Fig. 3, panel B), sug-
gesting that NS5A activates STAT3 in the presence of
other HCV proteins. These results suggest that HCV
NS5A enhances transcriptional activity of STAT3 regulated
promoter.
Jak1 is essential for STAT3 activation induced by NS5A in
hepatoma cells
Our initial examination suggested that NS5A does not
physically associate with STAT3 in HepG2 or Huh7 cells.
Jak kinases have been shown to phosphorylate STATs for
translocation into the nucleus and modulate various genes
involved in cell growth (Bowman et al., 2000). We exam-
ined whether NS5A mediated STAT3 activation in HepG2
or Huh7 cells is inhibited in the presence of a JAK1 kinase
inhibitor, AG490, by Western blot analysis. AG490 is
selective for JAK family kinases and reported not to inhibit
Src, Lck, Lyn, Btk, and Syk kinases (Zhang et al., 2000).
Cells were infected with nonreplicating adenovirus express-
ing NS5A (Ad-NS5A) or h-galactosidase (Adh-gal). Trans-
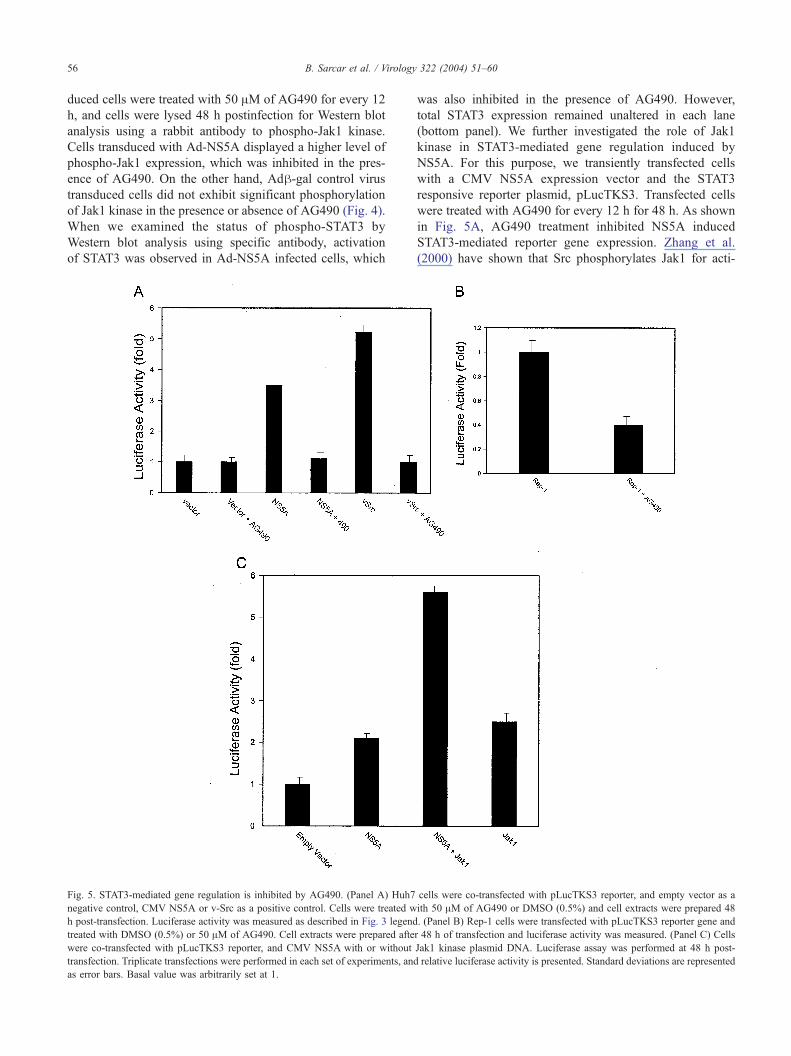
B. Sarcar et al. / Virology 322 (2004) 51–6056
duced cells were treated with 50 AM of AG490 for every 12
h, and cells were lysed 48 h postinfection for Western blot
analysis using a rabbit antibody to phospho-Jak1 kinase.
Cells transduced with Ad-NS5A displayed a higher level of
phospho-Jak1 expression, which was inhibited in the pres-
ence of AG490. On the other hand, Adh-gal control virustransduced cells did not exhibit significant phosphorylation
of Jak1 kinase in the presence or absence of AG490 (Fig. 4).
When we examined the status of phospho-STAT3 by
Western blot analysis using specific antibody, activation
of STAT3 was observed in Ad-NS5A infected cells, which
Fig. 5. STAT3-mediated gene regulation is inhibited by AG490. (Panel A) Huh7
negative control, CMV NS5A or v-Src as a positive control. Cells were treated w
h post-transfection. Luciferase activity was measured as described in Fig. 3 legend
treated with DMSO (0.5%) or 50 AM of AG490. Cell extracts were prepared afte
were co-transfected with pLucTKS3 reporter, and CMV NS5A with or without
transfection. Triplicate transfections were performed in each set of experiments, and
as error bars. Basal value was arbitrarily set at 1.
was also inhibited in the presence of AG490. However,
total STAT3 expression remained unaltered in each lane
(bottom panel). We further investigated the role of Jak1
kinase in STAT3-mediated gene regulation induced by
NS5A. For this purpose, we transiently transfected cells
with a CMV NS5A expression vector and the STAT3
responsive reporter plasmid, pLucTKS3. Transfected cells
were treated with AG490 for every 12 h for 48 h. As shown
in Fig. 5A, AG490 treatment inhibited NS5A induced
STAT3-mediated reporter gene expression. Zhang et al.
(2000) have shown that Src phosphorylates Jak1 for acti-
cells were co-transfected with pLucTKS3 reporter, and empty vector as a
ith 50 AM of AG490 or DMSO (0.5%) and cell extracts were prepared 48
. (Panel B) Rep-1 cells were transfected with pLucTKS3 reporter gene and
r 48 h of transfection and luciferase activity was measured. (Panel C) Cells
Jak1 kinase plasmid DNA. Luciferase assay was performed at 48 h post-
relative luciferase activity is presented. Standard deviations are represented

B. Sarcar et al. / Virology 322 (2004) 51–60 57
vation of STAT3 and Jak1 phosphorylation can be specif-
ically inhibited by treatment with AG490. We treated Src-
transfected cells with AG490 to use as a positive control for
inhibition of Jak1 phosphorylation. Inhibition of STAT3
activation was also observed in Huh7 cells expressing HCV
full-length replicon (Rep-1) in the presence of AG490 (Fig.
5, panel B). An in vitro reporter assay was performed using
pLucTKS3, NS5A, and Jak1 plasmid DNAs to further
examine whether NS5A-mediated STAT3 activation is al-
tered in the presence of Jak1 kinase. Our results suggested
that co-expression of Jak1 and NS5A further induces
STAT3 activation (at least 2-fold) as compared to NS5A
or Jak1 kinase alone (Fig. 5, panel C).
Does NS5A physically associate with Jak1?
To determine whether NS5A and Jak1 form a complex in
vivo, a co-immunoprecipitation assay was performed. Huh7
cells were co-transfected with myc-tagged Jak1 and CMV-
NS5A plasmid DNAs following vvT7 infection. Cell lysates
were immunoprecipitated with c-myc monoclonal antibody
for Jak1 kinase and subjected to Western blot analysis for
NS5A using specific antibody. Co-precipitation of Jak1 with
NS5A was evident from the specificity of the antibody and
Fig. 6. NS5A physically associates with Jak1 kinase. Huh7 cells were
infected with vvT7 and co-transfected with myc-tagged Jak1 and empty
vector (lane 1) or CMV NS5A (lane 2). After 24 h of transfection, cell
lysates were immunoprecipitated with a monoclonal antibody to c-myc and
immunoblotted with a monoclonal antibody to NS5A. Cells transfected
with myc-tagged Jak1 (lane 3) and NS5A (lane 4) were immunoblotted with
a monoclonal antibody to NS5A to examine the exogenous expression of
NS5A. The molecular weight of the NS5A protein was ascertained from the
migration of standard protein molecular weight markers (Invitrogen). The
blot was reprobed with a monoclonal antibody for detection of Jak1
(bottom panel). The positions of NS5A and immunoglobulin (Ig) heavy
chain (from experimental reagents) are shown.
the size of the NS5A protein (Fig. 6, lane 2). On the other
hand, cell lysates when similarly analyzed from myc-tagged
Jak1 and empty vector plasmid DNA as a negative control
did not exhibit precipitation of NS5A protein (Fig. 6, lane
1). The exogenous expression of NS5A from transfected cell
lysates was analyzed by Western blot (Fig. 6, lanes 3 and 4).
The blot when reprobed with a specific antibody to Jak1
displayed the Jak1 kinase protein band (bottom of panel).
However, endogenous Jak1 could not be detected as co-
precipitate with NS5A in Hep-5A cells, possibly for a low
level of Jak1 expression.
Discussion
STAT3 generally remains latent in the cytoplasm in an
unphosphorylated state (Schindler et al., 1992). Activation of
STAT3 requires tyrosine phosphorylation, which occurs
upon cytokine signaling or other stimuli (Wen et al.,
1995). Upon stimulation, STAT3 is phosphorylated at tyro-
sine residues and translocates into the nucleus, where it binds
cognate DNA sequences as dimers (Doria et al., 1995; Zhong
et al., 1994). Activation of STAT3 by NS5A in Huh7 cells
was observed earlier (Gong et al., 2001); however, the
mechanism of STAT3 activation is unknown. In this study,
we have shown that HCV NS5A activates STAT3 phosphor-
ylation, which in turn accumulates into the nucleus, although
a direct interaction between NS5A and STAT3 was not
observed. We have also shown that activation of STAT3
occurs in the presence of other HCV proteins using full-
length HCV replicon. Our results from transgenic mice
expressing HCV NS5A also suggested activation of STAT3
in mouse liver. Several tumor viruses are associated with
STAT activation. Epstein–Barr virus transformation leads to
uncontrolled activation of STATs (Weber-Nordt et al., 1996).
Kaposi’s sarcoma-associated herpesvirus ORF50 stimulates
transcriptional activity of STAT3 (Gwack et al., 2002). Rous
sarcoma virus v-src oncoprotein has been shown to phos-
phorylate and activate STAT3 (Yu et al., 1995; Cao et al.,
1996). The ability of these viruses to activate STATs is
closely related to viral oncogenic potential.
Jak kinases are large enzymes (120–140 kDa) that are
cytoplasmically pre-associated with signal-transducing cy-
tokine receptor subunits (Ihle, 1995). Upon cytokine-
induced receptor aggregation, Jaks are probably activated
by auto- and transphosphorylation. Tyrosine residues with-
in the cytoplasmic tail of the receptor are subsequently
phosphorylated by the kinases, providing docking sites for
SH2 domain containing signaling proteins including
STATs, tyrosine phosphatases, and suppressors of cytokine
signaling. Tyrosine-phosphorylated STATs homo- or heter-
odimerize and translocate to the nucleus where they bind
to specific DNA sequences in the promoter regions of their
respective target genes (Darnell et al., 1994; Heinrich et
al., 1998). We have demonstrated that NS5A interacts with
and activates Jak1 kinase, which in turn phosphorylates

B. Sarcar et al. / Virology 322 (2004) 51–6058
STAT3. Phosphorylation of STAT3 was also inhibited by a
specific Jak1 kinase inhibitor, further suggesting the in-
volvement of Jak1 kinase. NS5A does not possess tyrosine
kinase activity; therefore, the precise mechanism of NS5A-
mediated Jak1 phosphorylation remains to be elucidated.
Other transforming viruses have been shown to activate
STAT3 in cooperation with other kinases. Herpesvirus
saimiri Tip-484 activates STAT3 in cooperation with
p56lck (Lund et al., 1997). Human T-cell lymphotropic
virus type-1 transformed cells exhibit constitutive activa-
tion of the Jak–STAT pathway (Migone et al., 1995). Our
results suggest that NS5A forms complex with Jak1 kinase
and recruits STAT3 for activation. The activated STAT3
then translocates into the nucleus and modulates cell
growth regulatory genes Bcl-xL and p21, which may lead
to HCV mediated pathogenesis.
Many different cell lines and fresh isolates derived from
a variety of tumors express activated form of STAT3
(O’Shea et al., 2002). They are often activated upon
different stimuli and facilitate cellular expansion by trans-
activating genes encoding proteins to enhance cell surviv-
al. In fact, introduction of NS5A in hepatoma cells
enhances Bcl-xL expression and represses p21. We have
shown previously that NS5A associates and sequesters p53
into cytoplasm and downregulates p21 expression
(Majumder et al., 2001). Recently, Lin et al. (2002)
reported that transduction of p53 in human breast carcino-
ma cells inhibits phospho-STAT3 and enhances p21 ex-
pression. Therefore, it is possible that p53–NS5A complex
formation allows STAT3 to phosphorylate and translocate
into the nucleus for promotion of cell growth. HCV-
mediated promotion of cell growth may also involve
additional viral protein. Recently, STAT3 activation by
HCV core was reported in transgenic mice (Yoshida et
al., 2002). Co-expression of core and STAT3 was sug-
gested to decrease the dephosphorylation rate of phosphor-
ylated STAT3. Therefore, it is possible that expression of
core and NS5A together may have stronger oncogenic
potential than the individual proteins. Our results along
with others suggest that HCV evolves several mechanisms
to activate STAT3, which may contribute to virus-mediated
pathogenesis.
Materials and methods
Cell lines and reagents
Human hepatoma cell lines, HepG2 and Huh7, were
grown in DMEM containing 10% fetal bovine serum.
Expression vectors for STAT3-luc (pLucTKS3), STAT3,
STAT3-RFP, and v-src were generously provided by
Richard Jove (University of South Florida, FL); wild-type
and mutant STAT3-Flag was kindly provided by James
Darnell (Rockefeller University, NY); Jak1 cDNA was
kindly provided by James Ihle (St. Jude Children Hospital,
Memphis, TN); and Jak1-myc was kindly provided by
Robert Schriber (Washington University, St. Louis, MO).
Antibodies to Jak1, phospho-STAT3, and STAT3 were
obtained from Cell Signaling, MA. Monoclonal antibodies
to Flag and c-myc were obtained from Sigma (Missouri).
Jak1 kinase inhibitor, AG490, was obtained from Pharmin-
gen (California); protein A/G-agarose and enhanced chemi-
luminescence (ECL) detection kit were from Amersham
Pharmacia Biotech (Illinois). HCV full-length replicon from
Con1 strain was kindly provided by Ralf Bartenschlager,
University of Heidelberg, Germany.
Luciferase assay
Cells (5 � 105) were transfected with 3 Ag of reporter
plasmid pLucTSK3 and different doses of effector plasmid
DNAs using lipofectamine (Invitrogen, California). Empty
vector DNA was used as a negative control for the
luciferase assay. The total amount of DNA was kept
constant by addition of empty vector. Cells were lysed
using the reporter lysis buffer (Promega, Wisconsin), and
the luciferase activity was determined using a luminometer
(Optocomp II, MGM Instruments) as previously described
(Majumder et al., 2001).
Co-immunoprecipitation
Cells were co-transfected with 2 Ag of CMV Flag
STAT3, or myc-tagged Jak1 plasmid DNA and CMV
NS5A, and cell lysates were prepared after 48 h using 0.3
ml lysis buffer (150 mM NaCl, 10 mM HEPES, pH 7.6,
0.1–1% Nonidet P-40, 5 mM EDTA) containing a cocktail
of protease inhibitors (aprotinin, leupeptin, pepstatin, and
phenylmethylsulphonylfluoride). In some cases, plasmid
DNAs were transiently expressed using vvT7 infection–
transfection system (Sen et al., 2003) and cells were lysed
after 24 h. Cell lysates were incubated with a monoclonal
antibody to Flag (M2) or myc (9E-10) epitopes for 4 h at
4 jC, followed by an overnight incubation with protein
A-Sepharose beads (Pharmacia). The immunoprecipitates
were separated by SDS-polyacrylamide gel electrophoresis
and electroblotted onto a nitrocellulose membrane. Immu-
noblotting was performed by incubation of the membrane for
1 h with a rabbit antibody to STAT3 or Jak1 or monoclonal
antibody to NS5A (Biogenesis, New Hampshire). Proteins
were detected by enhanced chemiluminescence.
Immunofluorescence study
HepG2 or Hep-5A (HepG2 cells stably expressing NS5A)
cells (Ghosh et al., 2000b) were transfected with wild-type or
mutant STAT3 with Flag tag. After 48 h of transfection, cells
were washed and fixed with 3.7% formaldehyde followed by
blocking with 3% BSA. Cells were incubated with mono-
clonal antibody to Flag epitope or unrelated monoclonal
antibody with same isotype (IgG1) or STAT3 specific

B. Sarcar et al. / Virology 322 (2004) 51–60 59
antibody for 1 h at room temperature. Cells were washed and
incubated with anti-rabbit Ig conjugated with Alexa 488
(Molecular Probes, Oregon) for 30 min at room temperature.
Finally, cells were rinsed and mounted for immunofluores-
cence microscopy (confocal microscope, Bio-Rad model
1024 or Olympus BX51). A parallel set of experiments
was performed with wild-type STAT3 for nuclear staining
with DAPI. For this set of experiment, photomicrographs
were taken with Olympus microscope.
Generation of recombinant adenovirus expressing NS5A
Recombinant adenovirus expressing NS5A (Ad-NS5A)
was generated as previously described (Ghosh et al.,
1999b). Briefly, NS5A was cloned into the pAdTrack-
CMV shuttle vector (He et al., 2002) under the control of
CMV promoter (kindly provided by Bert Vogelstein, John
Hopkins University, Maryland). The resulting AdTrack
CMV-NS5A plasmid DNA was linearlized with PmeI re-
striction enzyme and co-transformed into E. coli BJ5183
cells with pAdEasy adenoviral backbone plasmid DNA.
Recombinants were selected and confirmed by restriction
enzyme endonuclease digestion and sequence analysis.
Finally, linearized recombinant plasmid DNA was trans-
fected into an adenovirus packaging cell line (293 cells) for
generation of recombinant adenovirus (Ad-NS5A). The
integration and expression of the NS5A in the recombinant
adenovirus was verified by PCR. Ad-NS5A was purified by
CsCl density gradient (Ghosh et al., 2002) and used for this
study. Adh-gal (He et al., 2002) was used as negative
control.
Establishment of Huh7 cells stably expressing HCV
full-length replicon
Huh7 cells expressing full-length HCV replicon were
established as described earlier (Pietschmann et al., 2002).
Briefly, HCV full-length RNA was generated by in vitro
transcription from plasmid DNA containing Con1 sequen-
ces using T7 polymerase (Promega). Huh7 cells were
electroporated with 1 Ag of purified in vitro transcripts
containing HCV full-length RNA. Cells were selected with
G418 at a concentration of 500 Ag/ml. Approximately 4
weeks post-electroporation, small colonies were visible.
These colonies were characterized for the presence of
HCV RNA and protein expression. One of the colonies
was named as Rep-1 and used in this study.
Acknowledgments
We thank Ralf Bartenschlager, James Darnell, James
Ihle, Richard Jove, and Robert Schriber for providing us
the reagents for our study, and Richard Jove for helpful
suggestions. We also thank David Lagunoff for helping
us in immunofluorescence study and Adrish Sen for
initial work on this project. This research was supported
by PHS Grant AI45144 from the National Institutes of
Health.
References
Barre, B., Avril, S., Coqueret, O., 2003. Opposite regulation of myc and
p21waf1 transcription by STAT3 proteins. J. Biol. Chem. 278,
2990–2996.
Bild, A.H., Turkson, J., Jove, R., 2002. Cytoplasmic transport of Stat3 by
receptor-mediated endocytosis. EMBO J. 21, 3255–3263.
Bowman, T., Garcia, R., Turkson, J., Jove, R., 2000. STATs in oncogenesis.
Oncogene 19, 2474–2488.
Bromberg, J., Darnell, J.E., 2000. The role of STATs in transcriptional con-
trol and their impact on cellular function. Oncogene 19, 2468–2473.
Cao, X., Tay, A., Guy, G.R., Tan, Y.H., 1996. Activation and association of
Stat3 with Src in v-Src-transformed cell lines. Mol. Cell. Biol. 16,
1595–1603.
Chatterjee-Kishore, M., van den Akker, F., Stark, G.R., 2000. Association
of STATs with relatives and friends. Trends Cell Biol. 10, 106–111.
Choo, Q.L., Kuo, G., Weiner, A.J., Overby, L.R., Bradley, D.W., Houghton,
M., 1989. Isolation of a cDNA clone derived from a blood-borne non-A,
non-B viral hepatitis genome. Science 244, 359–362.
Chung, K.M., Lee, J., Kim, J.E., Song, O.K., Cho, S., Lim, J., Seedorf, M.,
Hahm, B., Jang, S.K., 2000. Nonstructural protein 5A of hepatitis C virus
inhibits the function of karyopherin beta3. J. Virol. 74, 5233–5241.
Clarke, B., 1997. Molecular virology of hepatitis C virus. J. Gen. Virol. 78,
2397–2410.
Darnell, J.E., 1997. STATs and gene regulation. Science 277, 1630–1635.
Darnell Jr., J.E., Kerr, I.M., Stark, G.R., 1994. Jak–STAT pathways and
transcriptional activation in response to IFNs and other extracellular
signaling proteins. Science 264, 1415–1421.
Doria, M., Klein, N., Lucito, R., Schneider, R.J., 1995. The hepatitis B
virus HBx protein is a dual specificity cytoplasmic activator of Ras and
nuclear activator of transcription factors. EMBO J. 14, 4747–4757.
Gale Jr., M., Kwieciszewski, B., Dossett, M., Nakao, H., Katze, M.G.,
1999. Antiapoptotic and oncogenic potentials of hepatitis C virus are
linked to interferon resistance by viral repression of the PKR protein
kinase. J. Virol. 73, 6506–6516.
Ghosh, A.K., Steele, R., Meyer, K., Ray, R., Ray, R.B., 1999a. Hepatitis C
virus NS5A protein modulates cell cycle regulatory genes and promotes
cell growth. J. Gen. Virol. 80, 1179–1183.
Ghosh, A.K., Grigorieva, I., Steele, R., Hoover, R.G., Ray, R.B., 1999b.
PTEN transcriptionally modulates c-myc gene expression in human
breast carcinoma cells and is involved in cell growth regulation. Gene
235, 85–91.
Ghosh, A.K., Majumder, M., Steele, R., Yaciuk, P., Chrivia, J., Ray, R.,
Ray, R.B., 2000a. Hepatitis C virus NS5A protein modulates transcrip-
tion through a novel cellular transcription factor SRCAP. J. Biol. Chem.
275, 7184–7188.
Ghosh, A.K., Majumder, M., Steele, R., Meyer, K., Ray, R., Ray, R.B.,
2000b. Hepatitis C virus NS5A protein protects against TNF-alpha
mediated apoptotic cell death. Virus Res. 67, 173–178.
Ghosh, A.K., Majumder, M., Steele, R., Liu, T.J., Ray, R.B., 2002. MBP-1
mediated apoptosis involves cytochrome c release from mitochondria.
Oncogene 21, 2775–2784.
Gong, G., Waris, G., Tanveer, R., Siddiqui, A., 2001. Human hepatitis C
virus NS5A protein alters intracellular calcium levels, induces oxidative
stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci.
U.S.A. 98, 9599–9604.
Grakoui, A., Wychowski, C., Lin, C., Feinstone, S.M., Rice, C.M., 1993.
Expression and identification of hepatitis C virus polyprotein cleavage
products. J. Virol. 67, 1385–1395.
Gwack, Y., Hwang, S., Lim, C., Won, Y.S., Lee, C.H., Choe, J., 2002.

B. Sarcar et al. / Virology 322 (2004) 51–6060
Kaposi’s Sarcoma-associated herpesvirus open reading frame 50 stim-
ulates the transcriptional activity of STAT3. J. Biol. Chem. 277,
6438–6442.
He, Y., Nakao, H., Tan, S.L., Polyak, S.J., Neddermann, P., Vijaysri, S.,
Jacobs, B.L., Katze, M.G., 2002. Subversion of cell signaling pathways
by hepatitis C virus nonstructural 5A protein via interaction with Grb2
and P85 phosphatidylinositol 3-kinase. J. Virol. 76, 9207–9217.
Heinrich, P.C., Behrmann, I., Muller-Newen, G., Schaper, F., Graeve, L.,
1998. Interleukin-6-type cytokine signalling through the gp130/Jak/
STAT pathway. Biochem. J. 334, 297–314.
Hijikata, M., Kato, N., Ootsuyama, Y., Nakagawa, M., Shimotohno, K.,
1991. Gene mapping of the putative structural region of the hepatitis C
virus genome by in vitro processing analysis. Proc. Natl. Acad. Sci.
U.S.A. 88, 5547–5551.
Ihle, J.N., 1995. Cytokine receptor signalling. Nature 377, 591–594.
Kaneko, T., Tanji, Y., Satoh, S., Hijikata, M., Asabe, S., Kimura, K.,
Shimotohno, K., 1994. Production of two phosphoproteins from the
NS5A region of the hepatitis C viral genome. Biochem. Biophys.
Res. Commun. 205, 320–326.
Lan, K.H., Sheu, M.L., Hwang, S.J., Yen, S.H., Chen, S.Y., Wu, J.C.,
Wang, Y.J., Kato, N., Omata, M., Chang, F.Y., Lee, S.D., 2002. HCV
NS5A interacts with p53 and inhibits p53-mediated apoptosis. Onco-
gene 21, 4801–4811.
Lin, J., Jin, X., Rothman, K., Lin, H.J., Tang, H., Burke, W., 2002. Mod-
ulation of signal transducer and activator of transcription 3 activities by
p53 tumor suppressor in breast cancer cells. Cancer Res. 62, 376–380.
Lund, T.C., Garcia, R., Medveczky, M.M., Jove, R., Medveczky, P.G.,
1997. Activation of STAT transcription factors by herpesvirus Saimiri
Tip-484 requires p56lck. J. Virol. 71, 6677–6682.
Majumder, M., Ghosh, A.K., Steele, R., Ray, R., Ray, R.B., 2001. Hepatitis
C virus NS5A physically associates with p53 and regulates p21/waf1
gene expression in a p53-dependent manner. J. Virol. 75, 1401–1407.
Majumder, M., Ghosh, A.K., Steele, R., Zhou, X.Y., Phillips, N.J., Ray, R.,
Ray, R.B., 2002. Hepatitis C virus NS5A protein impairs TNF-mediated
hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice.
Virology 294, 94–105.
Migone, T.S., Lin, J.X., Cereseto, A., Mulloy, J.C., O’Shea, J.J., Franchini,
G., Leonard, W.J., 1995. Constitutively activated Jak-STAT pathway in
T cells transformed with HTLV-I. Science 269, 79–81.
O’Shea, J.J., Gadina, M., Schreiber, R.D., 2002. Cytokine signaling in
2002: new surprises in the Jak/Stat pathway. Cell 109, S121–S131.
Pietschmann, T., Lohmann, V., Kaul, A., Krieger, N., Rinck, G., Rutter, G.,
Strand, D., Bartenschlager, R., 2002. Persistent and transient replication
of full-length hepatitis C virus genomes in cell culture. J. Virol. 76,
4008–4021.
Polyak, S.J., Khabar, K.S., Paschal, D.M., Ezelle, H.J., Duverlie, G., Bar-
ber, G.N., Levy, D.E., Mukaida, N., Gretch, D.R., 2001. Hepatitis C
virus nonstructural 5A protein induces interleukin-8, leading to partial
inhibition of the interferon-induced antiviral response. J. Virol. 75,
6095–6106.
Schindler, C., Darnell, J.E., 1995. Transcriptional responses to polypeptide
ligands: the JAK–STAT pathway. Annu. Rev. Biochem. 64, 621–651.
Schindler, C., Shuai, K., Prezioso, V.R., Darnell Jr., J.E., 1992. Interferon-
dependent tyrosine phosphorylation of a latent cytoplasmic transcrip-
tion factor. Science 257, 809–813.
Sen, A., Steele, R., Ghosh, A.K., Basu, A., Ray, R., Ray, R.B., 2003.
Inhibition of hepatitis C virus protein expression by RNA interference.
Virus Res. 96, 27–35.
Simmonds, P., 2001. The origin and evolution of hepatitis viruses in
humans. J. Gen. Virol. 82, 693–712.
Tan, S.L., Nakao, H., He, Y., Vijaysri, S., Neddermann, P., Jacobs, B.L.,
Mayer, B.J., Katze, M.G., 1999. NS5A, a nonstructural protein of hep-
atitis C virus, binds growth factor receptor-bound protein 2 adaptor
protein in a Src homology 3 domain/ligand-dependent manner and
perturbs mitogenic signaling. Proc. Natl. Acad. Sci. U.S.A. 96,
5533–5538.
Tanji, Y., Kaneko, T., Satoh, S., Shimotohno, K., 1995. Phosphorylation of
hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 69,
3980–3986.
Tu, H., Gao, L., Shi, S.T., Taylor, D.R., Yang, T., Mircheff, A.K., Wen, Y.,
Gorbalenya, A.E., Hwang, S.B., Lai, M.M., 1999. Hepatitis C virus
RNA polymerase and NS5A complex with a SNARE-like protein. Vi-
rology 263, 30–41.
Turkson, J., Bowman, T., Garcia, R., Caldenhoven, E., De Groot, R.P.,
Jove, R., 1998. Stat3 activation by Src induces specific gene regula-
tion and is required for cell transformation. Mol. Cell. Biol. 18,
2545–2552.
Weber-Nordt, R.M., Egen, C., Wehinger, J., Ludwig, W., Gouilleux-Gruart,
V., Mertelsmann, R., Finke, J., 1996. Constitutive activation of STAT
proteins in primary lymphoid and myeloid leukemia cells and in
Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 88,
809–816.
Wen, Z., Zhong, Z., Darnell Jr., J.E., 1995. Maximal activation of tran-
scription by Stat1 and Stat3 requires both tyrosine and serine phosphory-
lation. Cell 82, 241–250.
Yoshida, T., Hanada, T., Tokuhisa, T., Kosai, K., Sata, M., Kohara, M.,
Yoshimura, A., 2002. Activation of STAT3 by the hepatitis C virus
core protein leads to cellular transformation. J. Exp. Med. 196,
641–653.
Yu, C.L., Meyer, D.J., Campbell, G.S., Larner, A.C., Carter-Su, C.,
Schwartz, J., Jove, R., 1995. Enhanced DNA-binding activity of a
Stat3-related protein in cells transformed by the Src oncoprotein. Sci-
ence 269, 81–83.
Zhang, Y., Turkson, J., Carter-Su, C., Smithgall, T., Levitzki, A., Kraker, A.,
Krolewski, J.J., Medveczky, P., Jove, R., 2000. Activation of Stat3 in v-
Src-transformed fibroblasts requires cooperation of Jak1 kinase activity.
J. Biol. Chem. 275, 24935–24944.
Zhong, Z., Wen, Z., Darnell Jr., J.E., 1994. Stat3: a STAT family member
activated by tyrosine phosphorylation in response to epidermal growth
factor and interleukin-6. Science 264, 95–98.
Copyright © 2022 FDOKUMEN













![Activation of HydA ΔEFG Requires a Preformed [4Fe4S] Cluster](https://static.fdokumen.com/doc/165x107/63164e0f0c69af6c1c0050c7/activation-of-hyda-defg-requires-a-preformed-4fe4s-cluster.jpg)






