
He thong quan ly diem sinh vien
12
The Prostate 65:287^298 (2005) Role of Androgen Receptor in the Progression of Human ProstateTumor Cells to Androgen Independence and Insensitivity John M. Kokontis, Stephen Hsu, Chih-pin Chuu, Mai Dang, Junichi Fukuchi, Richard A. Hiipakka, and Shutsung Liao* Ben May Institute for Cancer Research and the Department of Biochemistry and Molecular Biology, The University of Chicago,Chicago, Illinois BACKGROUND. Various studies have implicated the androgen receptor (AR) in the pro- gression of androgen-dependent human prostate cancer cells to androgen-independent and androgen-insensitive phenotypes, but the exact role of AR in progression is unclear. METHODS. To mimic the clinical situation and test the role of AR in progression, we cultured androgen-dependent LNCaP 104-S prostate tumor cells in the presence of the antiandrogen Casodex (bicalutamide) to derive resistant (CDXR) clones. In a second step, we cultured CDXR cells in the presence of the androgen R1881, which generated androgen- and Casodex- insensitive (IS) cells. These cells were then characterized with regard to AR function and the effect of ectopic AR expression or AR knockdown on androgen sensitivity. RESULTS. CDXR cells showed increased AR expression and transcriptional activity. CDXR cell proliferation was unaffected by Casodex but was repressed by androgen in vitro and in vivo. IS cells, on the other hand, had greatly reduced AR expression and activity compared to CDXR cells. Knockdown of AR expression in CDXR cells produced cells that were insensitive to androgen. Conversely, re-expression of AR in IS cells regenerated cells that were repressed by androgen. Knockdown of AR expression in 104-S cells produced cells that remained stimulated by androgen, while overexpression of AR in 104-S cells generated an androgen-repressed phenotype but did not confer androgen-independent growth. CONCLUSIONS. Increased AR expression determines whether prostate cancer cells are repressed by androgen, but is not required for androgen independence. These results may have implications for anti-AR therapy for prostate cancer. Prostate 65: 287 – 298, 2005. # 2005 Wiley-Liss, Inc. KEY WORDS: prostate cancer; progression; androgen; androgen receptor; LNCaP INTRODUCTION Progression of prostate tumors that are dependent upon androgen for survival and growth to tumors that are androgen-independent and -insensitive remains incompletely understood at the molecular level. Anti- androgen or androgen deprivation therapy of prostate cancer, pioneered by Charles Huggins [1], is initially effective in repressing prostate tumor growth. How- ever, endocrine therapy rarely succeeds in killing all tumor cells and progression of androgen-dependent tumor cells to androgen-independent cells occurs often. Several mechanisms have been identified that may participate in the transition of prostate tumor cells Abbreviations: CS-FBS, charcoal-stripped fetal bovine serum; AR, androgen receptor; 5a-DHT, 5a-dihydrotestosterone; R1881, 17b- hydroxy-17a-methylestra-4,9,11-trien-3-one; IPTG, isopropyl thioga- lactoside; PSA, prostate specific antigen; CDXR, Casodex-resistant; IS, androgen-insensitive. This study is dedicated to the memory of Dr. Charles B. Huggins. Grant sponsor: NIH (to SL); Grant numbers: CA58073, AT00850; Grant sponsor: Robert Earp Trust. *Correspondence to: Shutsung Liao, The Ben May Institute for Cancer Research, The University of Chicago, Box MC6027, 5841 S. Maryland Ave., Chicago, Illinois 60637. E-mail: [email protected] Received 8 November 2004; Accepted 21 April 2005 DOI 10.1002/pros.20285 Published online 13 July 2005 in Wiley InterScience (www.interscience.wiley.com). ȣ 2005 Wiley-Liss, Inc.
Transcript of He thong quan ly diem sinh vien

The Prostate 65:287^298 (2005)
Role ofAndrogenReceptor inthe ProgressionofHumanProstateTumorCells toAndrogen
Independence and Insensitivity
John M. Kokontis, Stephen Hsu, Chih-pin Chuu, Mai Dang, Junichi Fukuchi,Richard A. Hiipakka, and Shutsung Liao*
BenMay Institute for Cancer ResearchandtheDepartmentof BiochemistryandMolecular Biology,TheUniversityof Chicago,Chicago, Illinois
BACKGROUND. Various studies have implicated the androgen receptor (AR) in the pro-gression of androgen-dependent human prostate cancer cells to androgen-independent andandrogen-insensitive phenotypes, but the exact role of AR in progression is unclear.METHODS. Tomimic the clinical situation and test the role of AR in progression, we culturedandrogen-dependent LNCaP 104-S prostate tumor cells in the presence of the antiandrogenCasodex (bicalutamide) to derive resistant (CDXR) clones. In a second step, we cultured CDXRcells in the presence of the androgen R1881, which generated androgen- and Casodex-insensitive (IS) cells. These cells were then characterized with regard to AR function and theeffect of ectopic AR expression or AR knockdown on androgen sensitivity.RESULTS. CDXR cells showed increased AR expression and transcriptional activity. CDXRcell proliferationwasunaffectedbyCasodexbutwas repressedbyandrogen invitro and invivo.IS cells, on the other hand, had greatly reduced AR expression and activity compared to CDXRcells. Knockdown of AR expression in CDXR cells produced cells that were insensitive toandrogen. Conversely, re-expression of AR in IS cells regenerated cells that were repressed byandrogen. Knockdown of AR expression in 104-S cells produced cells that remained stimulatedby androgen, while overexpression of AR in 104-S cells generated an androgen-repressedphenotype but did not confer androgen-independent growth.CONCLUSIONS. Increased AR expression determines whether prostate cancer cells arerepressed by androgen, but is not required for androgen independence. These resultsmay haveimplications for anti-AR therapy for prostate cancer. Prostate 65: 287–298, 2005.# 2005 Wiley-Liss, Inc.
KEY WORDS: prostate cancer; progression; androgen; androgen receptor; LNCaP
INTRODUCTION
Progression of prostate tumors that are dependentupon androgen for survival and growth to tumors thatare androgen-independent and -insensitive remainsincompletely understood at the molecular level. Anti-androgen or androgen deprivation therapy of prostatecancer, pioneered by Charles Huggins [1], is initiallyeffective in repressing prostate tumor growth. How-ever, endocrine therapy rarely succeeds in killing alltumor cells and progression of androgen-dependenttumor cells to androgen-independent cells occursoften. Several mechanisms have been identified thatmay participate in the transition of prostate tumor cells
Abbreviations: CS-FBS, charcoal-stripped fetal bovine serum; AR,androgen receptor; 5a-DHT, 5a-dihydrotestosterone; R1881, 17b-hydroxy-17a-methylestra-4,9,11-trien-3-one; IPTG, isopropyl thioga-lactoside; PSA, prostate specific antigen; CDXR, Casodex-resistant;IS, androgen-insensitive.
This study is dedicated to the memory of Dr. Charles B. Huggins.
Grant sponsor: NIH (to SL); Grant numbers: CA58073, AT00850;Grant sponsor: Robert Earp Trust.
*Correspondence to: Shutsung Liao, The Ben May Institute forCancer Research, The University of Chicago, Box MC6027, 5841 S.Maryland Ave., Chicago, Illinois 60637.E-mail: [email protected] 8 November 2004; Accepted 21 April 2005DOI 10.1002/pros.20285Published online 13 July 2005 in Wiley InterScience(www.interscience.wiley.com).
� 2005 Wiley-Liss, Inc.

to clinical hormone-independence. These includeandrogen receptor (AR) gene amplification, AR muta-tion, and bypass of androgenic activation of AR or ofAR signaling itself for cell survival and proliferation(for reviews, see refs. [2–4]). We reported previouslythat clonally derived androgen-dependent LNCaP 104-S cells, after long-term androgen deprivation in vitro,can give rise to 104-R1 and 104-R2 cells, the growth ofwhich is repressed by androgen [5–8]. We demon-strated that these hormone independent cells exhibitedincreased AR expression and transcriptional activityand their proliferation was repressed by low concen-trations of androgen. The 5a-reductase inhibitor finas-teride and the antiandrogen Casodex (bicalutamide)blocked the repressive effects of testosterone. Andro-gen-repressed LNCaP 104-R1 cells can revert to anandrogen-stimulated phenotype when treated withandrogen in vitro [7] and a similar phenomenon isobserved in LNCaP 104-R1 cells grown as tumors incastrated athymic mice [8].
In this report, we sought to clarify the role ofAR in the progression of LNCaP 104 tumor cellsfrom hormone-dependence to hormone-independence.Recent reports have demonstrated sustained andheightened AR expression, function and sensitivity toandrogen in hormone-independent or recurrent pros-tate cancer cells [9–16]. Therefore, it was of interest toestablish whether continued and elevated AR ex-pression was necessary for the hormone-independentgrowth we observed in the LNCaP 104 progressionmodel. For this purpose we derived new androgen-independent and -repressed clonal sublines fromandrogen-dependent 104-S cells by selecting forgrowth in the presence of Casodex. We extended theprogression model by re-exposing Casodex-resistant(CDXR) cells to androgen to generate cells (IS) thatwerecompletely androgen and antiandrogen-insensitive.We then studied the effect of enforced reduction orre-expression ofAR in these cells and inprogenitor 104-S cells. Our results indicate that elevatedARexpressionmediates androgenic repression of cell growth but isnot responsible, at least by itself, for androgen-independent growth.
MATERIALSANDMETHODS
Materials
The LNCaP 104-S, 104-R1, and 104-R2 sublinesand the AN-21 anti-AR polyclonal antibody were des-cribed previously [5,7]. A polyclonal anti-prostatespecific antigen (PSA) antibody was from DAKO(Glostrup, Denmark), a monoclonal anti-p27Kip1 anti-body was from Transduction Laboratories/BD Bio-sciences (Lexington, KY), a monoclonal anti-actin
antibody was from Chemicon (Temecula, CA). R1881(17b-hydroxy-17a-methylestra-4,9,11-trien-3-one) wasfrom Perkin Elmer/NENLife Science Products (Boston,MA). Casodex1 (ICI 176,334; (2RS)-40-cyano-3-(4-fluorophenylsulfonyl)-2-hydroxy-2-methyl-30-(triflu-oromethyl)-propionanilide; bicalutamide) was fromAstraZeneca Pharmaceuticals (Wilmington, DE).
Cell Culture andGeneration of Casodex-ResistantandAndrogen-Insensitive Clones InVitro
LNCaP 104-S, 104-R1, and 104-R2 cells were pas-saged and maintained as described previously [7].For the selection of Casodex-resistant (CDXR) cells,2� 106 LNCaP 104-S cells were plated in DMEM sup-plemented with 10% charcoal-stripped fetal bovineserum (CS-FBS) and 5mMCasodex, andwere grown for3 weeks. Six colonies were amplified into clonal sub-lines called CDXR1 through CDXR6, which were usedin subsequent characterization of proliferative behav-ior andAR expression. For the generation of androgen-insensitive (IS) cells, the CDXR1, CDXR2, and CDXR3sublines were cultured in DMEM supplemented with10% CS-FBS and 10 nM R1881 for a period of 3 weeksbefore replating.
Cell Cycle DistributionAnalysis
Cell cycle distribution analysis onLNCaP104-S, 104-R1, CDXR, or IS cells was performed as described pre-viously [7]. One day after plating, cells were treatedwith R1881 or Casodex and grown for an additional4 days with a change of medium on day 2. After over-night fixation at�208C in 70%ethanol/30%phosphate-buffer saline (PBS), cells were treated with 0.1 mg/mlRNAseA inPBS for 30min at 378C.After pelleting, cellswere stained with 50 mg/ml propidium iodide in PBS.Cell cycle profiles and distributions were determin-ed by flow cytometric analysis of 2� 104 cells usingthe CellQuest program (v. 3.1f) on a BD Facscan flowcytometer (BD Biosciences, San Jose, CA). Cell cycledistribution analysis was performed using ModFit LTsoftware (Verity Software House, Topsham, ME).
Fluorometric Cell GrowthAssay
For measurement of cell growth, the DNA fluoro-metric method of Rago et al. [17] was used. Cells(5� 103/well) were plated in 96-well plates. The nextday cells were treated, 6 wells per treatment, withR1881 at 0.1 or 10 nM, Casodex at 5 mM, tetracyline at2 mg/ml, or isopropyl-b-D-thiogalactopyranosideside(IPTG) at 4 mg/ml and grown for 5 additional dayswith a medium change on day 4 after plating. After
288 Kokontis et al.

removal of growth medium, 100 ml of water wasadded and the plates were frozen at�908C until assay.Cells were thawed and 100 ml of a solution containing20 mg/ml Hoechst 33258, 2M NaCl, 10 mM Tris HClpH 7.4, and 1 mM EDTA was added. After thoroughmixing, plates were read in a Wallac 1420 fluorometricplate reader using a 355/460 nm excitation/emissionfilter.
Growthof CDXR3 and IS3 TumorsinAthymicMice
Six to eight-week old castratedmale athymic BALB/c mice (NCI-Frederick) were injected subcutaneouslywith 1� 106 CDXR3 or IS3 cells suspended in 0.5 mlof Matrigel (BD Bioscience). Four weeks after in-jection, half of tumor-bearing mice were implantedwith pellets containing 20 mg testosterone propionate.Pellets were replaced with fresh pellets after 30 days.Tumor growth was determined weekly by calipermeasurement. Tumor volume was calculated by theformula (length�width�depth)/2.
Real-Time PCR
Total RNA was isolated using Trizol reagent(Invitrogen Life Technologies, Carlsbad, CA). Contam-inating DNA was removed using DNase I (DNA-free,Ambion,Austin, TX). cDNAwas synthesized from2 mgtotal RNA using an Omniscript RT synthesis kit(Qiagen, Valencia, CA). For real-time PCR, a Quanti-Tect Probe PCR kit (Qiagen) and a Prism 7700 cycler(Applied Biosystems, Foster City, CA) were used in adual-labeled probe protocol. The following primerand probe sequences, selected using the Primer Ex-press program (AppliedBiosystems), were used forARreal-time PCR: forward primer, 50-CGCCCCTGATCT-GGTTTTC; reverse primer, 50-TTCGGACACACTG-GCTGTACA; FAM-labeled probe, 6FAM-50-TGAGTA-CCGCATGCACAAGCTCCG-30-TAMRA. The primerand FAM-labeled probe sequences used for PSA real-time PCR were described by Gelmini et al. [18]. ARibosomal RNA Control kit (Applied Biosystems) wasused to normalize transcript levels among samples.
RNAInterference forARKnockdown
Complementary 64-base oligonucleotides were syn-thesized (IntegratedDNATechnologies, Inc., Coralville,IA) containing a 19-base C-terminal AR sequence (50-GCACTGCTACTCTTCAGCA) in inverted repeat ori-entation [19]. After annealing, the 64-mer duplex wasinserted into BglII/HindIII-digested pH1RP RNAiexpression vector [20]. After transfection, G418-resis-tant 104-S and CDXR3 colonies were expanded and
screened for reduced AR expression by Western blotanalysis. For tetracycline-induced knockdown of AR,a 19 bp TETO2 operator sequence was introducedimmediately 30 of the TATA box of the H1 promoter byPCRamplification [21]. The 64-baseARRNAi constructwas then inserted into the BglII/HindIII-digestedpH1RP-TETO2 vector. Prior to transfection withthe pH1RP-TETO2-AR vector, 104-S and CDXR3 cellswere stably transfected with the pcDNA6/TR vector(Invitrogen) for TET repressor expression. BlasticidinS-resistant colonies were screened for TET repres-sor expression using an anti-TET repressor antibody(MoBiTec; Goettingen, Germany).
AROverexpression
Overexpression of AR was achieved by infecting IScells with the pLNCX2 retrovirus (BD-Clontech, PaloAlto, CA) carrying a 3 kb BglII human AR cDNA frag-ment derived from the pSG5-hAR vector [22]. Re-trovirus was generated using the Phoenix-amphopackaging cell line (G. Nolan, Stanford University).IPTG-induced AR expression in 104-S cells was carriedout using the 30SS (Stratagene) and pLNXRO2 [23]vectors using methods described previously [7].
RESULTS
Progression of LNCaP104-SCellstoCDXRCells
During a 3-week exposure of androgen-dependent104-S cells to 5 mM Casodex in medium supplementedwith CS-FBS, most cells stopped proliferating anddetached from the dishes. Parallel cultures of thesecells, fixed and stained with 4,6-diamidino-2-pheny-lindole (DAPI), did not show typical apoptotic nuclearcondensation and fragmentation. Discrete coloniessoon appeared at low frequency. Direct counting ofcolony number and Poisson estimation of colony fre-quency by tallying growth verses non-growth in platedaliquots of known cell number showed that CDXR cellswere present in the 104-S cell population at a frequencyof about 1 in 1.4� 105 cells at passage 8. Six colonieschosen at random were amplified into sublines, de-signated CDXR1 through CDXR6. A diagram showingthe lineages of cells we have derived from the LNCaP104-S clone is shown in Figure 1a. Proliferation ofCDXR cells, as measured by the percentage of cells in Sphase, was independent of and repressed by androgenaswell as insensitive to Casodex, similar to 104-R1 cells(Fig. 1b). CDXR cells treated with androgen accumu-lated in the G1 phase of the cell cycle (data not shown).The percentage of sub-G1 apoptotic cells under allconditions was negligible. Although Casodex did not
Role of Androgen Receptor in Progression 289

inhibit growth of CDXR cells, Casodex blocked growthrepression by androgen (Fig. 1b).
Acquisition of CompleteAndrogenInsensitivitybyCDXRClones
CDXR1, CDXR2, and CDXR3 clones were culturedin 10 nM R1881 for several weeks to select for cells thatcould proliferate in the presence of androgen. CDXRcells initially underwent G1 arrest after the addition of10 nMR1881. Starting at day 6, however, themajority ofcells underwent apoptosis as determined by DAPIstaining and detached from the plate over the next 6–8 days. A similar result was reported by Joly-Pharabozet al. [24] using androgen-independent LNCaP cellsthat they derived. By day 14, actively growing colonieshad formed that were then pooled separately for eachCDXR subline. We found that cell proliferation in thethree resultant sublines, called IS1, IS2, and IS3, wasunaffected by R1881, while the three CDXR progenitorsublines were repressed by androgen (Fig. 2a). Growthof IS cells, like that of CDXR cells, was also unaffected
byCasodex.When total cell accumulationwasmeasur-ed by fluorometric assay of DNA content after 5 daysof growth, similar results were obtained (Fig. 2b). IS3colonies arose fromCDXR3populations (passage 7) at afrequency of approximately 1 in 1.9� 103 cells plated,a frequency over 70-fold higher than the frequency ofCDXR cells in a 104-S population at a similar passagenumber.
ARExpression in CDXRand ISCells
AR protein expression in the six CDXR sublinesgrown in the presence and absence of R1881 wascompared to AR expression in 104-S, 104-R1, and 104-R2 cells byWestern blot analysis (Fig. 3a). All six CDXRsublines expressed AR protein at a level greater thanthat of 104-S cells, as also observed in 104-R1 and104-R2cells [5,7]. Western blot analysis of AR protein expres-sion in IS cells showed that AR levels were reducedconsiderably in IS1, IS2, and IS3 cells compared withCDXR1, CDXR2, and CDXR3 cells (Fig. 3b). ExpressionofARmRNAwasmeasured in 104-S, 104-R1, theCDXR
Fig. 1. a: Derivation of androgen-independent and -insensitivesublines from LNCaP 104-S cells after androgen (A) deprivation(top) and combined androgen deprivation/Casodex treatment fol-lowedbyandrogenre-exposure(bottom).b:PercentageofLNCaP104-S,104-R1or CDXR cells in S phase determined by flow cyto-metry of propidium iodide-stained cells. Cells were incubated inmedium containing ethanol vehicle (control), 0.1 nM R1881 and/or5 mMCasodex (CDX) for 96 hr prior to trypsinization and fixation.Values represent themean� standard error derived from three tofiveindependentexperiments.AsterisksindicateP value<0.01(*)or<0.05 (**) incomparisonwithuntreatedcells.
Fig. 2. a: Percentage ofCDXRand IS cells in Sphase determinedby flowcytometryofpropidiumiodide-stainedcells.Cellswereincu-bated inmedium containing ethanol vehicle (control), 0.1nMR1881,10 nMR1881or 5 mMCasodex for 96 hr prior to trypsinization andfixation.Values represent themean� standard error derived fromthree to five independent experiments. b: Fluorometric assay ofLNCaP104-S,104-R1,CDXR3,andIS3cellgrowth.Cells(5�103well)were plated in 96-well plates in150 ml DMEM/CS-FBS using 6 wellsper treatment.The next day150 ml ofmedium containing hormonewas added to give a final concentration of 0.1nMR1881,10 nMR1881,or 5 mMCasodex, and cellsweregrown for 5 daysbefore assay (see‘‘Materials and Methods’’). Values represent the mean� standarderror.AsterisksindicatePvalue<0.01(*)or<0.05(**)incomparisonwithuntreatedcells.
290 Kokontis et al.
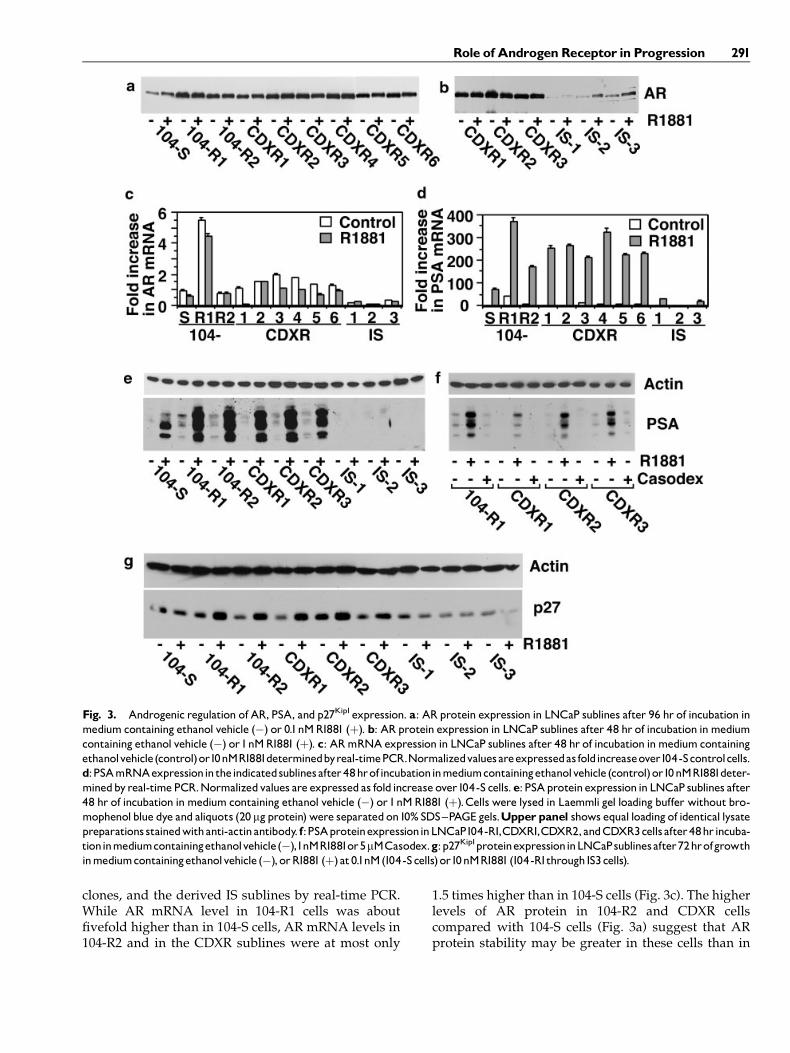
clones, and the derived IS sublines by real-time PCR.While AR mRNA level in 104-R1 cells was aboutfivefold higher than in 104-S cells, AR mRNA levels in104-R2 and in the CDXR sublines were at most only
1.5 times higher than in 104-S cells (Fig. 3c). The higherlevels of AR protein in 104-R2 and CDXR cellscompared with 104-S cells (Fig. 3a) suggest that ARprotein stability may be greater in these cells than in
Fig. 3. Androgenic regulation of AR, PSA, and p27Kip1expression. a: ARprotein expression in LNCaP sublines after 96 hr of incubation inmedium containing ethanol vehicle (�) or 0.1nMR1881 (þ). b: ARprotein expression in LNCaP sublines after 48 hr of incubation inmediumcontaining ethanol vehicle (�) or1nMR1881 (þ). c: ARmRNAexpression in LNCaP sublines after 48 hr of incubation inmedium containingethanolvehicle(control)or10nMR1881determinedbyreal-timePCR.Normalizedvaluesareexpressedas foldincreaseover104-Scontrolcells.d:PSAmRNAexpressionin theindicatedsublinesafter48hrofincubationinmediumcontainingethanolvehicle (control)or10nMR1881deter-minedbyreal-timePCR.Normalizedvalues are expressed as fold increase over104-S cells.e: PSAproteinexpression in LNCaP sublines after48 hr of incubation inmedium containing ethanol vehicle (�) or1nMR1881 (þ).Cells were lysed in Laemmli gel loading buffer without bro-mophenolblue dye and aliquots (20 mgprotein)were separatedon10% SDS^PAGE gels.Upperpanel shows equal loading of identical lysatepreparations stainedwithanti-actinantibody. f:PSAproteinexpressioninLNCaP104-R1,CDXR1,CDXR2,andCDXR3cellsafter48hrincuba-tioninmediumcontainingethanolvehicle(�),1nMR1881or5mMCasodex.g:p27Kip1proteinexpressioninLNCaPsublinesafter72hrofgrowthinmediumcontainingethanolvehicle (�), orR1881 (þ) at0.1nM(104-Scells)or10nMR1881(104-R1through IS3cells).
Role of Androgen Receptor in Progression 291

104-S cells. Expression of AR mRNA in the three ISsublines was greatly decreased compared to the CDXRcell progenitors.
PSAExpression in CDXRand ISCells
Transcriptional activity of AR in CDXR and IS cellswas assessed by examining androgen induction of PSAmRNA levels by real-time PCR. PSAmRNA levels after48 hr of treatmentwith androgenwere three to fourfoldhigher in the CDXR sublines than in 104-S cells, whilePSAmRNA levels were five and twofold higher in 104-R1 and 104-R2 cells, respectively, than in 104-S cells(Fig. 3d). PSA mRNA levels were low in the three ISsublines. The basal level of PSA mRNA in 104-R1 cellswas significantly higher than in 104-S cells and could bereduced by treatment with Casodex (not shown). Thismay indicate that AR in 104-R1 cells can scavenge lowlevels of endogenous androgen, but this ability is pro-bably not important in driving cell proliferation sinceCasodex had no effect on 104-R1 cell cycle progression(Figs. 1b,2b). Induction of PSA protein by androgen inthe IS sublines was undetectable (Fig. 3e). Casodex hadno effect by itself on PSA expression in CDXR cells,indicating that Casodexdoes not act as anARagonist inthese cells (Fig. 3f).
p27Kip1Expression inCDXRand ISCells
Previously we showed that androgenic control ofp27Kip1 expression at a post-transcriptional level waspartly responsible for androgen-induced G1 arrest in104-R1 and 104-R2 cells [7]. We examined p27Kip1 pro-tein expression in IS cells to determine whether in-duction of p27Kip1 is lost in these cells after androgentreatment. Expression of p27Kip1 in androgen-treatedand untreated IS cells was indeed lower than that in104-R1, 104-R2, and CDXR cells (Fig. 3g). This resultsuggests that loss of androgenic control of p27Kip1 ex-pression may enable IS cells to grow rapidly in thepresence of androgen.
Sensitivityof CDXR3 and IS3 TumorGrowthtoTestosterone
CDXR3 and IS3 tumors were grown in the flanksof male castrated athymic mice to test the effect ofandrogen on tumor growth in vivo. Four weeks afterinjection of cells, half of the tumor-bearing mice wereimplanted subcutaneously with pellets containing20 mg testosterone propionate. Tumor size wasmonitored over the next 30 days for all tumors and upto 150 days for the CDXR3 tumors in mice with testo-sterone implants. Testosterone repressed CDXR3tumor growth initially but had no effect on IS3 tumorgrowth (Fig. 4). After about 60 days post-implantation,
one group of CDXR3 tumors (N¼ 3) began to re-growand reached pre-implantation volume by 150 days.Another group (N¼ 5), composed of tumors of gen-erally smaller size at the time of pellet implantation,regressed completely. Removal of testosterone pelletsfrom the mice with regenerated tumors followed byinjection of Casodex did not affect tumor volume,indicating that the tumors were hormone-insensitive(not shown). These results corroborate observationswith these sublines grown in vitro (Figs. 1 and 2). Ex-pression of AR was significantly reduced in CDXR3tumors that had re-grown after implantation of testo-sterone pellets (Fig. 4, inset), consistent with the lower-ed expression of AR in IS cells.
KnockdownofARExpression inCDXR3CellsAbolishes Sensitivity toAndrogen
Using vector-derived interfering RNA [19], we de-termined whether CDXR3 cells would tolerate knock-down of AR expression. CDXR3 cells were transfectedwith a pH1RP vector [20] containing a 19-base C-terminal AR sequence or an empty pH1RP vector ascontrol. After colony selection and screening, threeclones (6,15,17) showing the least expression of ARwere used for further analysis (Fig. 5a). Induction ofPSA and p27Kip1 by androgen was also lost in theseclones. Flow cytometric analysis revealed that theclones had lost sensitivity to R1881 while CDXR3 cells
Fig. 4. CDXR3 and IS3 tumor growth in castratedmale athymicmice after subcutaneous implantation (þT) of a testosterone pro-pionate (20mg) pellet.Control animals receivednopellet.‘‘G’’ indi-catesre-growthgroup;‘‘R’’ indicatesregressedgroup.Thenumberoftumors per group ranged from 3 to 8.Values represent themean�standarderror.InsetisWesternblotshowingARproteinexpressionin104-S tumorcells (lane1),CDXR3 tumorcells (lane 2) andin twoCDXR3tumorsthatre-grewfollowingimplantationof testosteronepellets (lanes 3 and 4).Upper band represents a non-specifically-stainedprotein.
292 Kokontis et al.

transfectedwith empty vectorwere repressed byR1881(Fig. 5b). Similar results were obtained when total cellaccumulation was measured by fluorometric assayafter five days of growth in the absence or presence ofR1881 (Fig. 5c). Transfection and selection of colonieswas repeated in the presence of DHT to determinewhether an androgen-dependent phenotype couldbe generated in this context. However, clones withlowered AR expression exhibited only an androgen-insensitive phenotype (not shown). With constitutiveknockdown of AR, the possibility exists that colonies
underwent unknown changes that conferred growth inthe absence of AR expression. To assess the effect ofconditional knockdown of AR, which lessens this pos-sibility, the pH1RP-TETO2-AR vector was transfect-ed into CDXR3 cells expressing the TET repressor.Tetracycline-induced knockdown of AR expression inCDXR3 cells (Fig. 5d) resulted in no disruption of cellcycle and blocked androgen-induced arrest (Fig. 5e),similar to constitutive knockdown of AR. We con-clude that elevated AR expression is responsible forandrogenic repression of growth and that continuous
Fig. 5. KnockdownofARexpressioninCDXR3cellsbyinterferingRNA.a:WesternblotsshowingexpressionofAR,PSA,p27Kip1,andactininCDXR3cells transfectedwithemptypH1RP vector andin threeCDXR3cellclones transfectedwithpH1RP-ARknockdownvector.Cellsweregrownfor4days in thepresence of10nMR1881 (þ) orethanolvehicle (�).CellswerelysedinLaemmligel loadingbufferwithoutbromophenolbluedyeandaliquots(40mgprotein)wereseparatedon10%or12%(p27kip1)SDS^PAGEgels.b:PercentageofCDXR3cellsinSphasedeterminedby flow cytometry of propidium iodide-stained cells.CDXR3 cells transfectedwith empty pH1RP vector or the pH1RP-ARvector grown for4daysinthepresenceof10nMR1881orethanolvehicle(control)prior to trypsinizationandfixation.Valuesrepresentthemean� standarderrorof four independent experiments. Asterisk indicates P value<0.01 in comparison with untreated cells. c: Fluorometric cell growth assay ofCDXR3pH1RP-ARclones after 5 days ofgrowth in thepresence of10 nMR1881orethanolvehicle (control). Asterisk indicatesP value<0.01incomparison with untreated cells. d: Western blot showing tetracycline (Tet) induction of AR knockdown in CDXR3 pH1RP-TetO2-AR cells(clones1and2).Cellsweregrownfor4days in thepresence or absence of2mg/ml tetracycline (Tet).e:Percentage ofCDXR3pH1RP-TetO2-ARcells (clones1and 2) in Sphase determinedby flowcytometryofpropidiumiodide-stainedcells.Cellsweregrown for 4 days in thepresence of10nMR1881(R)and/or2mg/mltetracycline (Tet)orethanolvehicle (Con)prior to trypsinizationandfixation.
Role of Androgen Receptor in Progression 293

elevated AR expression in CDXR cells is not requiredfor androgen-independent cell growth.
Overexpression of ARin IS3 Cells RegeneratestheAndrogen-Repressed Phenotype
We infected IS3 cells with a pLNCX2 retroviralvector carrying the wild-type hAR cDNA to determinewhether an androgen-repressed phenotype could beregenerated. After selection and screening, four clonesthat exhibited the highest level of AR expression(clones 1, 2, 4, and 6; Fig. 6a) were used to assess theeffect of androgen on growth. Androgenic inductionof PSA and p27Kip1 was restored in these cells. Flowcytometric cell cycle analysis showed that proliferationof all four clones was inhibited by R1881, while controlIS3 cells remained insensitive to R1881 (Fig. 6b). Fur-thermore, the level of repression correlated with thelevel of AR expression among the clones. Fluorometricassay of cell growth showed similar results (Fig. 6c).Overexpression ofAR in IS cells, therefore, regeneratedan androgen-repressed phenotype.
KnockdownofARExpression and Inductionof AROverexpression in104-SCells
Previous reports have shown that reduction of ARsignaling, either through antisense oligonucleotides[25–27] or by decoy ARE-containing oligonucleotide[28], suppresses growth and induces apoptosis, respec-tively, in androgen-dependent LNCaP cells. To deter-mine the effect of reduced AR expression on 104-S cellgrowth, we used the pH1RP-TETO2-AR vector to con-ditionally knockdown AR expression. Interestingly,tetracycline-induced knockdown of AR (Fig. 7a) re-sulted in no change in responsiveness to androgenmeasured by either flow cytometric cell cycle analysis(Fig. 7b) or by fluorometric cell growth assay (Fig. 7c).However, it should be noted that AR was not com-pletely eliminated by tetracycline treatment. Completeblockade of AR expression would be expected to arrestcells similarly to Casodex.
We then attempted to generate an androgen-independent phenotype in 104-S cells by overex-pressing AR. However, infection of 104-S cells with
Fig. 6. EctopicoverexpressionofARinIS3cells.a:Westernblots showingexpressionofAR,PSA,p27Kip1,andactinincontrol IS3cells andIS3cellsinfectedwithpLNCX2-ARretrovirus.Cellsweregrownfor3daysinthepresenceof10nMR1881(þ)orethanolvehicle(�).b:PercentageofIS3 cells in Sphase determinedby flowcytometryofpropidiumiodide-stainedcells. IS3 cells and IS3 cell clones infectedwithpLNCX2-ARre-trovirus grown for 4 days in the presence of10 nMR1881or ethanol vehicle (Control) prior to trypsinization and fixation.Values representthe mean� standard error of four independent experiments. Asterisk indicates P value <0.01 in comparison with untreated (Control)cells.c:Fluorometric cellgrowthassayof IS3 clonesoverexpressingARafter5days ofgrowthin thepresence of10nMR1881orethanolvehicle(Control).AsteriskindicatesP values<0.01incomparisonwithuntreatedcells.
294 Kokontis et al.

retroviral vectors encodingAR followed by selection inthe presence or absence of androgen resulted in only afew antibiotic-resistant clones that exhibited no in-creased expression of AR. We therefore used an IPTG-inducible system to conditionally overexpress AR. One
clone that exhibited substantial IPTG-induced AR ex-pression, clone 22 (Fig. 7d), was grown in the presenceor absence of R1881 and IPTG. Induction of AR ex-pression by IPTGdid not confer hormone-independentgrowth measured by either flow cytometry (Fig. 7e) or
Fig. 7. ConditionalknockdownandoverexpressionofARin104-Scells.a: ARproteinexpression in two clones ofTETrepressor-expressing104-Scells transfectedwiththepH1RP-TETO2-ARknockdownvectorafter4daysofgrowthinpresenceorabsenceof2mg/mltetracyline(Tet).b:Percentageof104-ScellsinSphasedeterminedby flowcytometryofpropidiumiodide-stainedcells.104-ScellclonestransfectedwithpH1RP-TETO2-ARvectorgrownfor4daysinthepresenceof0.1nMR1881(R)and/or2mg/mltetracyline(Tet)orethanolvehicle(Con)prior to trypsini-zation and fixation.Values represent themean� standard error of three to five independentexperiments. Asterisk indicates P value<0.01incomparisonwith LNXRO2AR-22 cells treatedwith R1881alone. c: Fluorometric cell growth assay of104-S clones transfectedwith pH1RP-TETO2-ARvector.Cells (5�103 perwell)weregrown for 5 days in thepresence of 0.1nMR1881 (R) and/or 2mg/ml tetracyline (Tet) or ethanolvehicle(Con)prior toassay.d:ARproteinexpressionincontrol104-Scells transfectedwiththe30SSvector(expressing thelacrepressor)aloneor also infected with the lac repressor-controlled, AR expressing LNXRO2-AR retrovirus.Cells were grown for 4 days in the presence orabsenceof4mMIPTG.e:Percentageof104-ScellsinSphasedeterminedby flowcytometryofpropidiumiodide-stainedcells.104-Scells trans-fectedwith the 30SSvector alone andalso infectedwith theLNXRO2-ARretrovirusweregrown for 4days in thepresence of 0.1nMR1881 (R)and/or4mMIPTGorethanolvehicle(Con)prior totrypsinizationandfixation.Valuesrepresentthemean� standarderrorof threeindependentexperiments. Asterisk indicates P value<0.01in comparisonwith LNXRO2AR-22 cells treatedwithR1881alone. f:Fluorometric cellgrowthassayof104-Scells transfectedwiththe30SSvectoraloneandalsoinfectedwiththeLNXRO2-ARretrovirus.Cells(5�103perwell)weregrownfor5daysinthepresenceof0.1nMR1881(R)and/or4mMIPTGorethanolvehicle(Con)prior toassay.AsteriskindicatesP value<0.01incompar-isonwithcells treatedwithR1881alone.
Role of Androgen Receptor in Progression 295

fluorometric cell growth assay (Fig. 7f). Quite the oppo-site, overexpression of AR reduced R1881-induced cellproliferation in these cells and generated an androgen-repressed phenotype. IPTG repressed cell growthslightly in parental 104-S 30SS cells (Fig. 7f). This mayhave been due to a slight induction ofAR expression byIPTG in these cells (Fig. 7d). These observations explainwhy 104-S cells did not tolerate constitutive over-expression of AR: the cells cannot proliferate in eitherthe presence or absence of androgen. Based on theseresults, overexpression of AR by itself cannot conferandrogen-independent growth. Increased AR expres-sion may be locked in as a compensatory response toandrogendeprivation ormaybe linked indirectly to thechanges that are responsible for independent growth.Collectively, our results show that the level of ARexpression determines whether prostate cancer cellsare repressed by androgen, but other additional factorsor events are required for cells to become androgen-independent.
DISCUSSION
We have developed a progression model whereinandrogen-dependent prostate tumor cells progress toandrogen-independent and then to androgen-insensi-tive stages in two discrete steps. In the first step,androgen-independent but androgen-repressed CDXRcells arise at low frequencywhen androgen-dependentcells are cultured in the presence of antiandrogen.CDXR cells are typified by increased expression andactivity of AR. In the second step, progression to com-plete androgen insensitivity occurs at higher frequencywhen androgen is added to eliminate androgen-repressed cells. The genetic or epigenetic changes inCDXR cells responsible for androgen-independentgrowth have not been determined in the present study.It is clear from our results, however, that events otherthan increased AR expression must occur in LNCaP104-S cells during the transition to androgen indepen-dence. Conditional overexpression of AR in 104-S cellsgenerated an androgen-repressed phenotype but didnot confer androgen-independent growth. Mutation ofthe LNCaPAR gene to forms that utilize Casodex as anagonist was proposed as a mechanism responsible forCasodex withdrawal syndrome [29]. However, the ob-servation that androgen but not Casodex induces PSAexpression in the 104-R1 and CDXR sublines (Fig. 3f)indicates that AR mutation is not the mechanism res-ponsible for progression to Casodex resistance. We didnot detect the mutations in the ligand-binding domainof AR in 104-R1/R2 cells or CDXR cells that Hara et al.[29] detected in their Casodex-resistant LNCaP cells.Nor did we detect the mutations in the AR N-terminaldomain reported by Han et al. [30,31] that confer
ligand-independent transcriptional activity. Our re-sults also differ from those of Culig et al. [32] and Chenet al. [16] who reported agonistic effects of Casodex inandrogen-independent LNCaP cells. Chen et al. alsoreported that LNCaP cells readily overexpress AR con-stitutively after retroviral infection, which our clonalLNCaP104-S cells didnot tolerate. In the studybyChenet al. LNCaP cells overexpressing AR apparently pro-liferated by scavenging low levels of available andro-gen. CDXR as well as 104-R1 and 104-R2 cells appearnot to do this, however, because the growth of thesecells is insensitive to the antagonist Casodex. The ob-servation that CDXR cells can easily tolerate loss of ARindicates that continuous elevatedAR expression is notrequired in these cells.However,we cannot rule out thepossibility that these cells require ligand-independentsignaling from AR expressed at very low levels. Ourresults contrast with those of Zegarra-Moro et al. [33]who reported that antibody- or ribozyme-mediatedblockade of AR activity in hormone-refractive LNCaPcells repressed cell growth. The mechanism thatLNCaP 104-S cells utilize to progress to androgen-independency therefore appears to be different thanthe mechanisms reported by others. LNCaP and pros-tate tumor cells in general most likely have multiplemechanisms available to achieve growth in a low-androgen environment.
Knockdown of AR in CDXR3 cells resulted in cellsthat were insensitive to androgen and conversely, re-expression ofAR in IS3 cells regenerated cells thatwererepressed by androgen. These results indicate thatthe level of AR expression is the sole determinant ofwhether cells are repressed by androgen. Evidence forthe androgen-repressed phenotype outside of theLNCaP cell line is accumulating [34,35]. The ARCaPcell line is another prostate tumor cell line that isrepressed by androgen [36]. Hara et al. [14] recentlynoted suppression of MDA PCa 2b-hr prostate cancercell growth by high dose androgen after long termandrogen deprivation. Overexpression of AR in PC-3cells has been shown to confer growth repression byandrogen [37–39]. Bruchovsky et al. [40] noted pos-sible androgenic repression of tumor growth in aninterim analysis of the Canadian Prospective Trialof intermittent androgen deprivation therapy. Andro-gen has also been reported to repress cell growthduring androgen-induced differentiation of immorta-lizedhuman and rodent prostate epithelial cells in vitro[41,42]. Bruchovsky et al. [43] described the repressionby androgen of rat ventral prostate epithelial cellproliferation in the hormonal regulation of prostatehomeostasis. The intermediate androgen-independentbut androgen-repressed stage of LNCaP 104 tumor cellprogression may therefore have a real but under-recognized counterpart in clinical prostate tumor
296 Kokontis et al.

progression. Estrogenic repression of breast cancer inpostmenopausalwomen has been recognized formanyyears. Diethylstilbestrol treatment of breast cancerin postmenopausal women was standard endocrinetherapy before the introduction of tamoxifen [44] andwas shown to confer higher survival than tamoxifen[45]. Song et al. [46,47] reported that estrogen-inducedapoptosis inmammary carcinoma cells previously sub-jected to long-term estrogen deprivation was mediatedthrough Fas signaling.
Androgen-insensitive LNCaP IS cells were derivedfrom CDXR cells treated with R1881. IS cells are com-pletely insensitive to androgen stimulation or repres-sion. In this respect, LNCaP IS cells resemble cellsderived previously by Soto et al. [48] from androgen-repressed LNCaP cells after exposure to androgen. 104-R1Ad cells (Fig. 1a), which were derived from 104-R1cells treated with androgen, are androgen-responsive[7,8]. However, treatment of LNCaP 104-R2 cells withandrogen yielded cells that were insensitive to andro-gen, similar to IS cells (unpublished data). The 104-R1subline may therefore represent a discrete hormone-independent stage that is reversible, while the 104-R2 and CDXR sublines progress to an androgen-insensitive stage when challengedwith androgen. Thisbehavior was also observed in vivo. We showed thatCDXR3 tumors in athymic mice regressed followingtestosterone treatment, but could eventually give rise toandrogen-insensitive tumors (Fig. 4). In a clinical set-ting, these observations suggest that the use of a com-plete androgen blockade through antiandrogen andGnRH agonist treatment might result in the emergenceof androgen-independent and androgen-repressed andthen androgen-insensitive cells at an accelerated ratecompared to the use of androgen deprivation alone.Perhaps consistent with this, a recent prostate cancerprevention trial showed that finasteride increased therisk of high-grade prostate tumors while it reduced theoverall risk of prostate cancer [49]. In addition, ourresults have implications for reduction of AR expres-sion as a potential therapy for prostate cancer [50],which our results suggest recurrent cells may easilyevade. Attention may need to be directed toward ARreplacement in IS-like cells that have lostARexpressionto maintain sensitivity to androgen.
REFERENCES
1. Huggins C, Stevens RE, Hodges CV. Studies on prostatic cancer.II. The effects of castration on advanced carcinoma of theprostate gland. Arch of Surg 1941;43:209–223.
2. Kokontis JM, Liao S. Molecular action of androgen in thenormal and neoplastic prostate. In: Litwack G, editor. Vitaminsand hormones. Volume 55. New York: Academic Press; 1999.pp 219–308.
3. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001;1(1):34–45.
4. CuligZ,KlockerH,BartschG,HobischA.Androgen receptors inprostate cancer. Endocr Relat Cancer 2002;9(3):155–170.
5. Kokontis J, Takakura K, Hay N, Liao S. Increased androgenreceptor activity and altered c-myc expression in prostate cancercells after long-term androgen deprivation. Cancer Res 1994;54:1566–1573.
6. Umekita Y, Hiipakka RA, Kokontis JM, Liao S. Human prostatetumor growth in athymic mice: Inhibition by androgens andstimulation by a 5a-reductase inhibitor. Proc Natl Acad Sci USA1996;93:11802–11807.
7. Kokontis JM, Hay N, Liao S. Progression of LNCaP prostatetumor cells during androgen deprivation: Hormone-indepen-dentgrowth, repressionofproliferationbyandrogenand role forp27Kip1 in androgen-induced cell cycle arrest. Mol Endocrinol1998;12:941–953.
8. Chuu CP, Hiipakka RA, Fukuchi J, Kokontis JM, Liao S.Androgen causes growth suppression and reversion of andro-gen-independent prostate cancer xenografts to an androgen-stimulated phenotype in athymic mice. Cancer Res 2005;65:2082–2084.
9. Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM.Androgen receptor stabilization in recurrent prostate cancer isassociated with hypersensitivity to low androgen. Cancer Res2001;61(7):2892–2898.
10. Gregory CW,He B, Johnson RT, Ford OH,Mohler JL, French FS,Wilson EM. A mechanism for androgen receptor-mediatedprostate cancer recurrence after androgen deprivation therapy.Cancer Res 2001;61(11):4315–4319.
11. LinjaMJ, SavinainenKJ, SaramakiOR,TammelaTL,VessellaRL,Visakorpi T. Amplification and overexpression of androgenreceptor gene in hormone-refractory prostate cancer. Cancer Res2001;61(9):3550–3555.
12. Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgenreceptor gene amplification and protein expression in hormonerefractory prostate cancer. Br J Cancer 2003;89(3):552–556.
13. Zhang L, Johnson M, Le KH, Sato M, Ilagan R, Iyer M, GambhirSS, Wu L, Carey M. Interrogating androgen receptor functionin recurrent prostate cancer. Cancer Res 2003;63(15):4552–4560.
14. Hara T, Nakamura K, Araki H, Kusaka M, Yamaoka M.Enhanced androgen receptor signaling correlates with theandrogen-refractory growth in a newly established MDA PCa2b-hr human prostate cancer cell subline. Cancer Res 2003;63(17):5622–5628.
15. Bakin RE, Gioeli D, Sikes RA, Bissonette EA, Weber MJ.Constitutive activation of the Ras/mitogen-activated proteinkinase signaling pathway promotes androgen hypersensitivityin LNCaP prostate cancer cells. Cancer Res 2003;63(8):1981–1989.
16. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R,Rosenfeld MG, Sawyers CL. Molecular determinants of resis-tance to antiandrogen therapy. Nat Med 2004;10(1):33–39.
17. RagoR,Mitchen J,WildingG.DNAfluorometric assay in 96-welltissue culture plates using Hoechst 33258 after cell lysis byfreezing in distilled water. Anal Biochem 1990;191(1):31–34.
18. Gelmini S, Tricarico C, Petrone L, Forti G, Amorosi A, DedolaGL, Serio M, Pazzagli M, Orlando C. Real-time RT-PCR for themeasurement of prostate-specific antigen mRNA expression inbenign hyperplasia and adenocarcinoma of prostate. Clin ChemLab Med 2003;41(3):261–265.
Role of AndrogenReceptor in Progression 297

19. Brummelkamp TR, Bernards R, Agami R. A system for stableexpression of short interfering RNAs in mammalian cells.Science 2002;296(5567):550–553.
20. Fukuchi J, Hiipakka RA, Kokontis JM, Nishimura K, Igarashi K,Liao S. TATA-Binding protein-associated factor 7 regulatespolyamine transport activity and polyamine-induced apoptosis.J Biol Chem 2004;279:29921–29929.
21. van de Wetering M, Oving I, Muncan V, Pon Fong MT, BrantjesH, van Leenen D, Holstege FC, Brummelkamp TR, Agami R,Clevers H. Specific inhibition of gene expression using a stablyintegrated, inducible small-interfering-RNA vector. EMBO Rep2003;4(6):609–615.
22. Kokontis J, Ito K, Hiipakka RA, Liao S. Expression and functionof normal and LNCaP androgen receptors in androgen-insensitive human prostatic cancer cells: Altered hormone andantihormone specificity in gene transactivation. Receptor 1991;1:271–279.
23. ChangB-D, Roninson I. Inducible retroviral vectors regulatedbylac repressor in mammalian cells. Gene 1996;183:137–142.
24. Joly-Pharaboz M, Ruffion A, Roch A, Michel-Calemard L,Andre J, Chantepie J, Nicolas B, Panaye G. Inhibition ofgrowth and induction of apoptosis by androgens of a variantof LNCaP cell line. J Steroid Biochem Mol Biol 2000;73(5):237–249.
25. Eder IE, Culig Z, Ramoner R, Thurnher M, Putz T, Nessler-Menardi C, Tiefenthaler M, Bartsch G, Klocker H. Inhibition ofLNCaP prostate cancer cells by means of androgen receptorantisense oligonucleotides. Cancer Gene Ther 2000;7(7):997–1007.
26. Eder IE, Hoffmann J, Rogatsch H, Schafer G, Zopf D, Bartsch G,Klocker H. Inhibition of LNCaP prostate tumor growth in vivoby an antisense oligonucleotide directed against the humanandrogen receptor. Cancer Gene Ther 2002;9(2):117–125.
27. Eder IE, Haag P, BasikM,Mousses S, Bektic J, BartschG, KlockerH. Gene expression changes following androgen receptorelimination in LNCaP prostate cancer cells. Mol Carcinog 2003;37(4):181–191.
28. Kuratsukuri K, Sugimura K, Harimoto K, Kawashima H,Kishimoto T. ‘‘Decoy’’ of androgen-responsive element inducesapoptosis in LNCaP cells. Prostate 1999;41(2):121–126.
29. Hara T,Miyazaki J, ArakiH, YamaokaM,KanzakiN,KusakaM,MiyamotoM.Novelmutations of androgen receptor: A possiblemechanism of bicalutamide withdrawal syndrome. Cancer Res2003;63(1):149–153.
30. Han G, Foster BA, Mistry S, Buchanan G, Harris JM, Tilley WD,GreenbergNM.Hormone status selects for spontaneous somaticandrogen receptor variants that demonstrate specific ligand andcofactor dependent activities in autochthonous prostate cancer.J Biol Chem 2001;276(14):11204–11213.
31. Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ,Tilley W, Greenberg NM. Mutation of the androgen receptorcauses oncogenic transformation of the prostate. Proc Natl AcadSci USA 2005;102:1151–1156.
32. Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A,Bartsch G, Utermann G, Schneider MR, Parczyk K, Klocker H.Switch from antagonist to agonist of the androgen receptorbicalutamide is associatedwithprostate tumourprogression in anew model system. Br J Cancer 1999;81(2):242–251.
33. Zegarra-Moro OL, Schmidt LJ, HuangH, Tindall DJ. Disruptionof androgen receptor function inhibits proliferationof androgen-refractory prostate cancer cells. Cancer Res 2002;62(4):1008–1013.
34. Prehn RT. On the prevention and therapy of prostate cancer byandrogen administration. Cancer Res 1999;59(17):4161–4164.
35. Algarte-Genin M, Cussenot O, Costa P. Prevention of prostatecancer by androgens: Experimental paradox or clinical reality.Eur Urol 2004;46(3):285–295.
36. Zhau HY, Chang SM, Chen BQ, Wang Y, Zhang H, Kao C, SangQA, Pathak SJ, Chung LW. Androgen-repressed phenotype inhuman prostate cancer. Proc Natl Acad Sci USA 1996;93:15152–15157.
37. Yuan S, Trachtenberg J, Mills GB, Brown TJ, Xu F, Keating A.Androgen-induced inhibition of cell proliferation in an andro-gen-insensitive prostate cancer cell line (PC-3) transfectedwith ahuman androgen receptor complementary cDNA. Cancer Res1993;53:1304–1311.
38. Heisler LE, EvangelouA, LewAM, Trachtenberg J, Elsholtz HP,Brown TJ. Androgen-dependent cell cycle arrest and apoptoticdeath in PC-3 prostatic cell cultures expressing a full-lengthhuman androgen receptor. Mol Cell Endocrinol 1997;126:59–73.
39. Litvinov IV, Antony L, Isaacs JT. Molecular characterization ofan improved vector for evaluation of the tumor suppressorversus oncogene abilities of the androgen receptor. Prostate2004;61(4):299–304.
40. BruchovskyN,KlotzLH, SadarM,Crook JM,HoffartD,GodwinL, Warkentin M, Gleave ME, Goldenberg SL. Intermittentandrogen suppression forprostate cancer:Canadianprospectivetrial and related observations. Mol Urol 2000;4(3):191–200.
41. LingMT,ChanKW,ChooCK.Androgen inducesdifferentiationof a human papillomavirus 16 E6/E7 immortalized prostateepithelial cell line. J Endocrinol 2001;170(1):287–296.
42. WhitacreDC,ChauhanS,DavisT,GordonD,CressAE,MiesfeldRL. Androgen induction of in vitro prostate cell differentiation.Cell Growth Differ 2002;13(1):1–11.
43. Bruchovsky N, Lesser B, Van Doorn E, Craven S. Hormonaleffects on cell proliferation in rat prostate. In:MunsonPL,GloverJ, Diczfalusy E, Olson RE, editors. Vitamins and hormones.Volume 33. New York: Academic Press; 1975. pp 61–102.
44. Carter AC, Sedransk N, Kelley RM, Ansfield FJ, Ravdin RG,TalleyRW,PotterNR.Diethylstilbestrol:Recommendeddosagesfor different categories of breast cancer patients. Report of theCooperative Breast Cancer Group. JAMA 1977;237(19):2078–2079.
45. Peethambaram PP, Ingle JN, Suman VJ, Hartmann LC, LoprinziCL. Randomized trial of diethylstilbestrol vs. tamoxifen inpostmenopausal women with metastatic breast cancer. Anupdated analysis. Breast Cancer Res Treat 1999;54(2):117–122.
46. Song RX, Mor G, Naftolin F, McPherson RA, Song J, Zhang Z,Yue W, Wang J, Santen RJ. Effect of long-term estrogendeprivation on apoptotic responses of breast cancer cells to17beta-estradiol. J Natl Cancer Inst 2001;93(22):1714–1723.
47. Song RX, Santen RJ. Apoptotic action of estrogen. Apoptosis2003;8(1):55–60.
48. Soto AM, Lin T-M, Sakabe K, Olea N, Damassa DA, Son-nenschein C. Variants of the human prostate LNCaP carcinomacell line as tools to study discrete components of the androgen-mediated proliferative response. Oncol Res 1995;7:545–558.
49. Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ,Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM,Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA Jr.The influence of finasteride on the development of prostatecancer. N Engl J Med 2003;349:215–224.
50. Isaacs JT, IsaacsWB. Androgen receptor outwits prostate cancerdrugs. Nat Med 2004;10(1):26–27.
298 Kokontis et al.




















