
Clinical and biologic implications of recurrent genomic aberrations in myeloma
Upload
clevelandclinicCategory
view
2download
0
Leukemia Research 26 (2002) 271–280
Growth inhibition and apoptosis of myeloma cells by the CDKinhibitor flavopiridol
Igor Semenov a, Canan Akyuz a,b, Vera Roginskaya a, Dharminder Chauhan c,Seth J. Corey a,d,*
a Department of Pediatrics (Hematology-Oncology), Children’s Hospital of Pittsburgh, 3705 Fifth A�enue, Pittsburgh, PA 15213, USAb Department of Pediatrics (Oncology), Faculty of Medicine, Hacettepe Uni�ersity, Ankara, Turkey
c Department of Adult Oncology, Dana-Farber Cancer Institute, Boston, MA, USAd Department of Pharmacology, Uni�ersity of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Received 27 December 2000; accepted 16 May 2001
Abstract
Although myeloma shows responsiveness in intensive chemotherapy, overall survival remains less than 40% at 2 years. Sincemyeloma appears to be dependent on cytokines, such as IL-6, we hypothesized that targeting signal transduction molecules couldeffectively treat myeloma. Two myeloma cell lines U266 and RPMI-8226 and CD38+ myeloma cells were studied by immunecomplex kinase assay or anti-phosphotyrosine blot for evidence of constitutive activation of tyrosine kinases. Growth arrest andapoptosis were evaluated in these two cell lines following their treatment with specific kinase inhibitors. We found that a varietyof Src and Janus kinases were present and constitutively active in U266 and RPMI-8226 cells. Inhibitors of both Src and Januskinases were inferior to the cyclin-dependent kinase inhibitor, flavopiridol, in inducing both growth arrest with GI50 of 100 nMand apoptosis in both cell lines and CD38+ myeloma cells. Although, flavopiridol did not affect cyclin D1 and cyclin A levels,it inhibited Mcl-1 and Bcl-2 protein levels and cyclin-dependent kinase 2 activity. Flavopiridol is a well-tolerated drug, currentlyin phase I–II trials for a variety of tumors. A clinical trial using flavopiridol should be performed in patients with myeloma. Itsmechanism of action may involve targets other than the cyclin-dependent kinases. © 2002 Elsevier Science Ltd. All rightsreserved.
Keywords: Myeloma; Flavopiridol; Kinase; Signal transduction therapeutics
www.elsevier.com/locate/leukres
1. Introduction
Multiple myeloma is the second most common hema-tologic malignancy and remains one of the mostdifficult to treat [1–4]. Conventional chemotherapy hasmade little impact, with �30% 2 year survival. Moreintensive regimens, including (tandem) autologous stemcell transplants, result in higher response and survivalrates [5,6], but the prognosis remains poor [7]. Asreported by the Intergroupe Francais du Myelome, the5 year event free survival was 28% in the high-dose
chemotherapy and transplantation treatment arm, com-pared with 10% in the conventional chemotherapy arm[5]. Transplantation may thus be considered an effec-tive, palliative strategy. A 1999 editorial in the NewEngland Journal of Medicine summed up current treat-ment by stating that new therapies are desperatelyneeded [8]. One new approach uses thalidomide as aputative anti-angiogenic agent. In patients with refrac-tory myeloma who were treated with thalidomide, therewas a �22% 12-month event free survival [9]. How-ever, questions now must be addressed to define theminimally effective dose of thalidomide and determineany additive benefit when combined with conventionalchemotherapy.
Due to myeloma’s resistance to standard cytotoxicchemotherapy and possible response to thalidomide, wehypothesized that newer therapies directed against sig-
Abbre�iations: CDK, cyclin-dependent kinase; IL-6, interleukin-6;PI, propidium iodide; PTK, protein tyrosine kinase.
* Corresponding author. Tel.: +1-412-692-5055; fax: +1-412-692-6473.
E-mail address: [email protected] (S.J. Corey).
0145-2126/02/$ - see front matter © 2002 Elsevier Science Ltd. All rights reserved.PII: S 0 1 4 5 -2126 (01 )00103 -5

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280272
nal transduction agents may be effective in myeloma.Myeloma is a plasma cell neoplasm, with signalingdysregulation becoming better identified [10,11]. In par-ticular, IL-6 is an anti-apoptotic growth factor formyeloma cells [10]. Intracellular signaling events havebeen shown to include Jak-Stat and Hck protein ty-rosine kinase (PTK) pathways. These pathways con-verge to promote cell cycle progression, which ismediated by the cyclin-dependent kinases (CDK’s) andtheir co-factors. Inhibition of PTK’s or CDK’s shouldlead to growth arrest. The dramatic responses seen inpatients with chronic myeloid leukemia who weretreated with the Bcr-Abl tyrosine kinase inhibitor,STI571, provides a remarkable proof-of-principle [12].
PTK inhibitors are numerous, with most of thempossessing a flavenoid structure. Sharing a similarstructure is flavopiridol, a novel potent inhibitor ofCDK’s [13,14]. Initial in vitro studies revealed thatflavopiridol leads to growth inhibition, G2/M phasearrest, and cytotoxicity in breast carcinoma cells byinhibiting Cdk1 (p34cdc2) [15–18]. Further studiesdemonstrated that flavopiridol also potently inhibitedCdk2 and Cdk4, which led to G1/S arrest. Earlierconsidered as a cytostatic agent, more recent studiesrevealed that it could induce apoptosis in normal andleukemic cell lines [18–20].
We analyzed an IL-6-independent myeloma cell line,U266, and an IL-6-responsive cell line, RPMI-8226[10]. We found that a variety of Src and Janus kinaseswere present and constitutively active, but when thesecell lines were treated with specific PTK inhibitors,growth arrest and apoptosis were negligible. The PI3-kinase inhibitor wortmannin was also found to beineffective. However, flavopiridol induced growth arrestand apoptosis in both cell lines at sub-micromolarconcentrations. These studies suggest that flavopiridolmay be a new effective agent in myeloma.
2. Materials and methods
2.1. Reagents
Polyclonal antibodies against Blk, Fyn, Hck, Lck,Lyn, cyclin A, and cyclin D1 were purchased fromSanta Cruz Biotechnology (Santa Cruz, CA). The anti-Src antibodies (Blk, Fyn, Hck, Lck, Lyn and Src) canbe used to immunoprecipitate tyrosine kinase activity[21]. Rabbit polyclonal anti-Jak1, Jak2, Jak3, and Mcl-1 and the murine monoclonal anti-phosphotyrosine4G10 were purchased from Upstate Biotechnology(Lake Placid, NY). Rabbit polyclonal antibody againstBcl-2 was purchased from Pharmingen (San Diego,CA). Flavopiridol was provided by Hoechst–MarionRoussel (Bridgewater, NJ), Wortmannin was purchasedfrom Sigma Chemical (St Louis, MO). AG490 was
provided by Dr Alexander Levitzki (Hebrew Univer-sity, Jerusalem, Israel). PD173 was provided by DrAlan Kraker (Parke-Davis, Ann Arbor, MI), 17-ally-lamino-17-demethoxygeldanamycin (17-AAG) by DrEdward Sausville (National Cancer Institute, Bethesda,MD), and SU5478 by Sugen (Redwood City, CA).Flavopiridol, wortmannin, AG490, PD 173, 17AAG,and SU5478 were dissolved in dimethyl sulfoxide(DMSO) and stored at −20 °C prior to use.
2.2. Myeloma cell lines
Two established myeloma cell lines (U266 andPRMI-8226) cultures were used in these experiments.The cell lines were purchased from the American TypeCulture Collection (ATCC, Manassas, VA) and grownin RPMI-1640 medium supplemented with 10% fetalcalf serum (FCS) (Hyclone, Orham, UT, USA), 2 mML-glutamine, 10 U/ml penicillin, and 10 U/ml strepto-mycin (Life Technologies, Grand Island, NY). Cellcounts were performed manually on a hemacytometer.
2.3. Purification of CD38+ myeloma cells
CD38+ cells were isolated from heparinized bonemarrow samples obtained from patients who gave in-formed consent, as reported [22]. Briefly, mononuclearcells were obtained by ficoll separation, followed byimmunodepletion with mAbs against CD3, CD11b,CD14, CD33, CD45/45RA, CD32 and glycophorin.The remaining cells were characterized as CD38+, 45−.
2.4. Immune complex kinase and western blot analysis
Myeloma cells (1×106) were harvested from mid-loggrowth cultures, washed with PBS, then lysed in 200 ullysis buffer (50 mM HEPES, 150 mM NaCl, 1% Tri-tonX-100, 10% glycerol, 5 mM/EGTA, 15 mM MgCl2,20 mM/NaF, 50 mM �-glycerophosphate, 2 mMPMSF, 1 mM Na3VO4, 10 �g/ml leupeptin, and 10�g/ml aprotinin). Cell lysates were centrifuged at 14,000rpm for 15 min. Lysates were incubated overnight withantibodies against Lyn, Blk, Fyn, Src, Hck, Jak1, Jak2,and Jak3, and immunoprecipitates were obtained withprotein A-sepharose. Immunoprecipitates were washedthree times with PBS/1%NP-40 and twice with 10 mMTris, pH 7.4, 100 mM NaCl and 1 mM EDTA (TNE).An in vitro kinase reaction was begun with the additionof ATP Buffer (TNE and 10 mM MgCl2, 10 mMMnCl2, and 10 uCi [32�}-ATP). After 15 min, thereaction was stopped with addition of 2×Laemmlibuffer, and the sample was boiled for 5 min. Equivalentamount of samples was loaded onto 10% polyacry-lamide gels (for Lyn, Blk, Hck, Fyn, Src) and 7.5% gels(for Jak1, Jak2, and Jak3). For Jak immunoprecipi-tates, no kinase reactions were performed. Instead, the

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280 273
immunoprecipitates were treated with 2×Laemmlibuffer, boiled for 5 min, and the samples were loadedonto 7.5% gel. The gel was transferred on to PVDFmembranes then blocked by incubation in 1% bovineserum albumin (BSA). Western blots were probed withanti-phosphotyrosine mAb 4G10. In other blots, whereanti-Bcl-2, anti-cyclin D1, and anti-cyclin A antibodieswere used, the protein gels were 12.5% acrylamide.Detection was performed by enhanced chemilumines-cence reagents. (Amershan, Arlington Heights, IL).
2.5. In �itro cyclin-dependent kinase 2 assay
Cells (2×106 cells for each samples) exposed toflavipiridol (100 nM, 1 and 10 uM) and DMSO werewashed with ice-cold PBS and lysed in NP-40 lysisbuffer (50mM Tris–HCl, pH 7.4, 150 mM NaCl, 20mM EDTA, 0.5% NP-40, 1 mM PMSF, 5 �g/ml apro-tinin, and 5 �g/ml leupeptin) on ice 10 min. Lysatescontaining 500 �g of total cellular protein and 1 �g ofpolyclonal antibody to Cdk2 (SC-163, Santa Cruz Bio-technology Inc., Santa Cruz, CA) were incubated forovernight and immunoprecipitates were obtained withprotein A-sepharose. Immune complexes were washedthree times with histone wash buffer (25 mM Tris pH7.5, 70 mM NaCl, 10 mM MgCl, 1 nM DTT) andthen histone assay solution (320 �l histone washbuffer, 18 �l of histone H1 solution, 7 ul of cold 500mM ATP, 7 ul of 10 uCi/�l 32P-�-ATP) was added.After 30 min, the reaction was stopped by the additionof 30 �l of 2×Laemmli buffer, the samples wereboiled for 5 min and loaded onto a 12% gel. The gelwas fixed, dried, and exposed for autoradiography. Inother cases, cells were lysed, CDK’s were immunopre-cipitated, and flavopiridol in varying doses was addedto the kinase reaction.
2.6. Cells �iability and proliferation
U266 and RPMI-8226 cells were harvested frommid-log growth and resuspended at 5×105 cells perwell in 96-well plates. Various concentrations offlavopiridol, wortmannin, AG490, PD173, 17-AAG,SU5478, doxorubicin, and etoposide were added. As acontrol, the diluent DMSO was added at 1:1000 (v/v).Total cell number and percent viability were deter-mined at 24 and 48 h by trypan blue staining andenumeration with hemacytometer.
2.7. Flow cytometric analysis of cell cycle andapoptosis
About 1×106 cells were washed with PBS and thenexposed to different dilutions of flavopiridol (100 nM,1 �M, 10 �M) or doxorubicin for 16 h. Cells were
collected and washed two times with cold PBS andresuspended in 1×binding buffer at a concentrationof 1×106 cells/ml. Fluorescein isothiocyanate (FITC)-conjugated annexin-V (5 �l) and 10 �l propidium io-dide (PI) were then added to 1×105 cells, and sampleswere incubated for 15 min at room temperature. Thesamples were then analyzed by flow cytometry (Bec-ton-Dickinson Excalibur, Mountain View, CA) withthe software program ModFitLT V 2.0 or CellQuest(Becton-Dickinson). For cell cycle analysis, cells werewashed in cold PBS and fixed in 70% ethanol. DNAwas stained by incubating the cells in PBS containing50 �g/ml PI and 1 mg/ml RNAse A for 30 min at37 °C. The DNA content of the cells was analyzed byflow cytometry.
3. Results
3.1. Analysis of kinase acti�ity in myeloma cell lines
Earlier studies have demonstrated that cytokinesmay promote myeloma growth. Classic hematopoietincytokines operate via receptors, which must recruitcytosolic protein tyrosine kinases. To determine poten-tial drug targets, we surveyed two myeloma cell lines,RPMI-8226 and U266, for tyrosine kinases. In vitrokinase assays were performed with specific anti-Srcfamily of PTK antibodies. The Src kinases assayedwere the ones found predominantly in lymphoid tis-sues: Blk, Lck, Lyn, Hck, and Fyn. Src was alsoassayed. For identification of Janus family kinases,anti-phosphotyrosine blots were performed on proteinsimmunoprecipitated with specific anti-Jak antibodies.Multiple members of the Src and Janus families ofPTK’s were found to be constitutively active in bothRPMI-8226 and U266 (Fig. 1a and b). Both cell linesshowed activity of Lyn and Fyn, albeit both wereweakly active in RPMI-8226 cells. The U266 cellsshowed weak Src activity. Doublets for Lyn and Fynrepresent alternatively-spliced forms. Jak family ki-nases were found to be constitutively tyrosine phos-phorylated in both cell lines, although tyrosinephosphorylated Jak3 was not present in RPMI-8226cells (Fig. 1b). CD38+ myeloma cells, isolated frompatients with active multiple myeloma, were assayedfor constitutive tyrosine phosphorylation of Jak andLyn. Of these PTKs, Lyn and only Jak1 were tyrosinephosphorylated (Fig. 1c). Both cell lines showed inhi-bition of Lyn kinase by the Src kinase inhibitorPD173 with IC50�100 nM (Fig. 2a). Jak2 activity wasinhibited at 10 �M AG490 in RPMI-8226, but it wasnot inhibited at �100 �M in U266 cells (Fig. 2b).These results suggested that a Src kinase inhibitormight be considered for treatment of myeloma,whereas a Jak inhibitor might be less potent.

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280274
3.2. Growth arrest in myeloma cell lines treated withkinase inhibitors
We next sought to determine whether signal trans-duction inhibitors, such as a Src kinase inhibitor, mightaffect growth inhibition in myeloma cells. Mid-loggrowth U266 and RPMI-8226 cells were incubated withindicated concentrations of the Src kinase inhibitorPD173 and the Jak2 inhibitor AG490, as well as the PI3-kinase inhibitor wortmannin, and the CDK inhibitorflavopiridol. In addition, cells were also incubated witheither doxorubicin or the geldanamycin analogue 17-al-
Fig. 2. Dose-response effects of Src and Janus PTK inhibitors inmyeloma cells. U266 and RPMI-8226 cells were incubated for 60 minat 37 °C with various concentrations of the Src kinase inhibitorPD173 (a) or the Jak2 inhibitor AG490 (b). Lysates were thenprepared and analyzed by either kinase assay for Lyn or anti-phos-photyrosine blot for Jak2. Percentage activity of DMSO-treated Lynor Jak2 kinase activity is provided.
Fig. 1. (a) Anti-phosphotyrosine blot and in vitro kinase assay forSrc-related PTK in myeloma cell lines. Lysates were prepared fromU266 and RPMI-8226 cells in mid-log phase growth. Specific anti-Srcfamily members antibodies were used to immunoprecipitate Src-re-lated PTK. Immunoprecipitates were run out on a 10% gel, trans-ferred onto PVDF, and then blotted with mAb anti-phosphotyrosine4G10. (b). Anti-phosphotyrosine blot of immunoprecipitated Jakproteins from myeloma cells. Similarly, lysates were prepared, andproteins immunoprecipitated with anti-Jak1, Jak2, or Jak3 antibod-ies. The blot was then probed with a monoclonal antibody againstphosphotyrosine residues (mAb 4G10). (c). Anti-phosphotyrosineblot of immunoprecipitated Jak and Lyn proteins from CD38+ cells.Lysates were prepared from CD38+ myeloma cells, and Jak or Lynproteins immunoprecipitated, electrophoresed, and blotted with mAb4G10 as described in (b).
lylamino-17-demethoxygeldanamycin (17-AAG). Cellviability was determined by hemacytometer counting ofcells stained with trypan blue. As shown in Fig. 3a,flavopiridol was the most potent agent in causinggrowth inhibition of both myeloma cell lines. The GI50
for the two myeloma cell lines was approximately 100nM. Subsequent studies revealed that at a concentra-tion of 10 �M, flavopiridol inhibited the myeloma celllines by approximately 80% relative to control cellsafter 48 h of drug exposure. No viable RPMI-8226 cellswere seen after 48 h treatment with 10 �M flavopiridol.CD38+ myeloma cells were also sensitive to flavopiri-dol-induced cell death (Fig. 3b). Although both celllines showed exquisite sensitivity of Lyn kinase to theSrc inhibitor PD173, there was no dose-dependent inhi-bition of growth. At 100 uM AG490, both cell linesshowed inhibition. To determine whether growth inhi-bition was due to cell cycle arrest, asynchronouslygrowing U266 cells were studied. A 3-fold increase inthe percentage of cells arrested at G2/M was observedin cells treated with flavopiridol for 16 h (Table 1).
3.3. Fla�opiridol-induced apoptosis
While kinase inhibitors are thought to be cytostatic,we and others have shown that PTK or CDK inhibitorsmay also induce apoptosis. To determine flavopiridol’seffects on apoptosis, treated cells were stained with PIand Annexin V-Fluorescein Isothiocyanate (AnnexinV-FITC) and analyzed for DNA content by flow cy-

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280 275
tometry. About 1×106 cells each well were treated withdifferent concentrations of flavopiridol (100 nM and 1�M) or doxorubicin. After treatment for 16 h, cellswere collected, washed, and stained with PI and an-nexin-V FITC. They were then analyzed for DNAcontent by flow cytometry. Early apoptosis was con-
firmed by staining cells with Annexin-V (lower rightquadrants in Fig. 4), which binds to phosphatidylserinein the outer leaflet of the lipid bilayer. Apoptosis couldbe seen as early as 8 h (data not shown), and by 24–48h, there was marked cell death (i.e. Annexin-V positive/PI positive). These changes were confirmed by light
Fig. 3. Growth Inhibition of myeloma cell lines by different kinase inhibitors (a). U266 and RPMI-8226 cells growing in mid-log phase wereresuspended with the indicated concentrations of agents or 0.1% DMSO as a negative control in 12-well tissue culture plates. Cell viability wasdetermined by trypan blue exclusion at 24 and 48 h. Multiple independent experiments were performed. Data shown are averages�S.D. for the48 h time point. (b). CD38+ cells were treated with 0.1% DMSO or varying concentrations of flavopiridol (1 nM–1 �M). Cell viability wasdetermined at 24 and 48 h by trypan blue exclusion. Two to three independent experiments were done. Data shown are averages�S.D. for the48 h time point.

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280276
Table 1Cell cycle analysis of flavopiridol-treated U266 cells
Cell cycle FlavopiridolDMSO
100 nM 1 �M
67G0–G1 59792215 24S10 16G2/M 5
Cells were taken out of mid-log growth and treated with indicatedconcentrations of DMSO (1/1000, v/v), flavopiridol. Cells were thenlabeled with PI and analyzed by flow cytometry. Data are expressedas % of total cells.
as Cyclin D1 or Bcl-2 [18,23,24]. We treated cells withflavopiridol and blotted for cyclin D1 or cyclin A. Atconcentrations associated with growth inhibition, nochange in either cyclin D1 or cyclin A levels were seenby immunoblotting (Fig. 6a). Similarly, we examinedlevels of Bcl-2 in cells treated with flavopiridol. Bcl-2levels were unchanged at 6 h (data not shown). How-ever, longer incubation (e.g. 24 h) resulted in decreasedBcl-2 levels in both U266 and RPMI-8226 cells (Fig.6b). We also observed flavopiridol-induced decrease inMcl-1, a Bcl-2 related anti-apoptotic protein found inhematopoietic cells (Fig. 6c). Mcl-1 is a particularlyattractive target in myeloma because expression ap-pears to be tightly regulated by IL-6 [24]. To determinewhether flavopiridol inhibited CDK, we immunoprecip-itated CDK2 from U266, RPMI-8226, and CD38+
myeloma cells and performed an in vitro histone phos-phorylation assay. When cells were treated with 100nM flavopiridol for 4 h, CDK activity diminished in allthree cells tested, with 67, 20 and 71% inhibition inU266, RPMI-8226, and CD38+ myeloma cells (Fig.6d). These studies suggest that the critical target(s) forflavopiridol-induced growth inhibition and apoptosismay be multiple and elusive.
microscopy. Morphological changes, such as nuclearcondensation and breakdown, were seen as early as 12h and at flavopiridol concentrations of �100 nM (Fig.5 panels B and C, E and F).
3.4. Biochemical effects of fla�opiridol on myelomacells
Earlier reports suggest that flavopiridol’s mechanismsof action may be due to its effects on other targets, such
Fig. 4. Flow cytometric analysis of apoptotic myeloma cells. U266 cells growing in mid-log phase were treated for indicated time periods withcontrol diluent DMSO, doxorubicin 1 �g/ml, flavorpiridol (100 nM and 1 �M) Cells were harvested, promptly stained with Annexin V (FITC)and analyzed on a Becton-Dickinson Excalibur. Scattergrams from a representative experiment is shown. Data shown are representative of 3independent experiments.
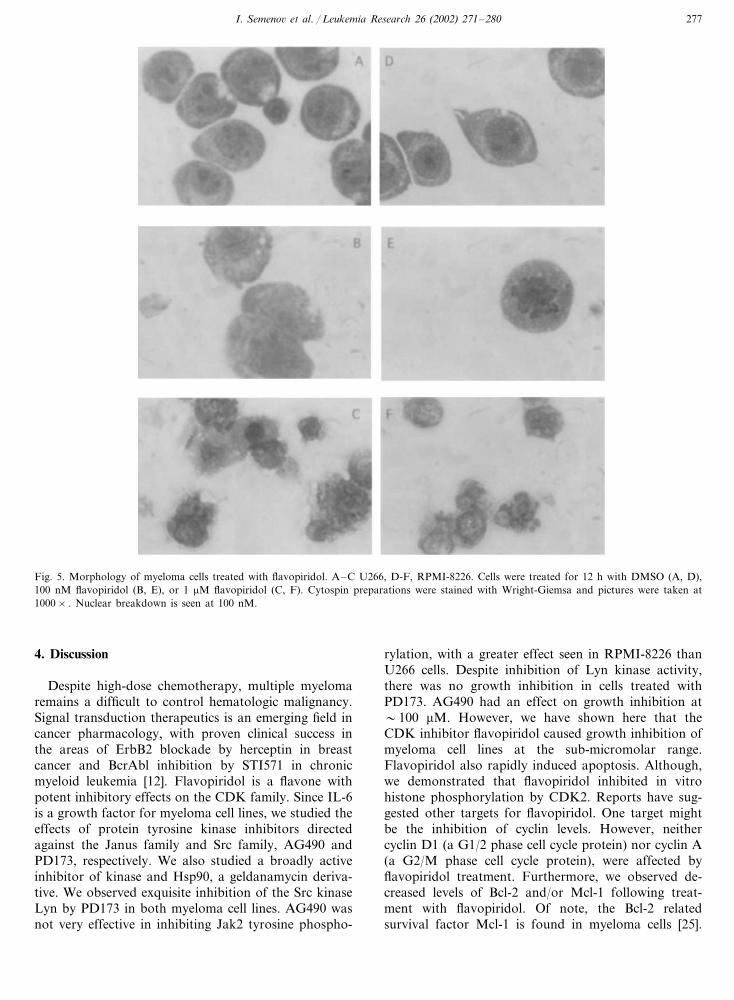
I. Semeno� et al. / Leukemia Research 26 (2002) 271–280 277
Fig. 5. Morphology of myeloma cells treated with flavopiridol. A–C U266, D-F, RPMI-8226. Cells were treated for 12 h with DMSO (A, D),100 nM flavopiridol (B, E), or 1 �M flavopiridol (C, F). Cytospin preparations were stained with Wright-Giemsa and pictures were taken at1000× . Nuclear breakdown is seen at 100 nM.
4. Discussion
Despite high-dose chemotherapy, multiple myelomaremains a difficult to control hematologic malignancy.Signal transduction therapeutics is an emerging field incancer pharmacology, with proven clinical success inthe areas of ErbB2 blockade by herceptin in breastcancer and BcrAbl inhibition by STI571 in chronicmyeloid leukemia [12]. Flavopiridol is a flavone withpotent inhibitory effects on the CDK family. Since IL-6is a growth factor for myeloma cell lines, we studied theeffects of protein tyrosine kinase inhibitors directedagainst the Janus family and Src family, AG490 andPD173, respectively. We also studied a broadly activeinhibitor of kinase and Hsp90, a geldanamycin deriva-tive. We observed exquisite inhibition of the Src kinaseLyn by PD173 in both myeloma cell lines. AG490 wasnot very effective in inhibiting Jak2 tyrosine phospho-
rylation, with a greater effect seen in RPMI-8226 thanU266 cells. Despite inhibition of Lyn kinase activity,there was no growth inhibition in cells treated withPD173. AG490 had an effect on growth inhibition at�100 �M. However, we have shown here that theCDK inhibitor flavopiridol caused growth inhibition ofmyeloma cell lines at the sub-micromolar range.Flavopiridol also rapidly induced apoptosis. Although,we demonstrated that flavopiridol inhibited in vitrohistone phosphorylation by CDK2. Reports have sug-gested other targets for flavopiridol. One target mightbe the inhibition of cyclin levels. However, neithercyclin D1 (a G1/2 phase cell cycle protein) nor cyclin A(a G2/M phase cell cycle protein), were affected byflavopiridol treatment. Furthermore, we observed de-creased levels of Bcl-2 and/or Mcl-1 following treat-ment with flavopiridol. Of note, the Bcl-2 relatedsurvival factor Mcl-1 is found in myeloma cells [25].

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280278
Flavopiridol may also be affecting other critical sur-vival/apoptotic molecules, such as caspases [23,26].These findings do not, however, detract from its po-tential use as an anti-myeloma agent, as the mecha-nism of action for thalidomide is also elusive. Serumlevels of flavopiridol are sufficient to inhibit cell
growth in vitro [26]. Dose-limiting toxicity has beendiarrhea [27,28]. Based on pre-clinical studies[20,23,29], flavopiridol has gone into clinical trial forsolid tumors and chronic lymphocytic leukemia. Datapresented here indicate that myeloma may be an ex-cellent target.
Fig. 6. Biochemical Effects of Flavopiridol. (a) Cyclin levels. U266 and RPMI-8226 cells were treated for 6 h with DMSO (control), 0.1, 1, and10 �M flavopiridol, and lysates were prepared in Laemmli buffer. After separating equivalent amount of proteins on a 12.5% gel, they weretransferred onto PVDF membrane and blotted with antibodies against cyclin D1 or cyclin A and developed by ECL. (b) Bcl-2 levels. U266 andRPMI-8226 cells were treated for 24 h with DMSO or 100, 500, and 1000 nM flavopiridol. Cells were counted and standardized, and whole celllysates were electrophoresed in a 12% SDS-polyacrylamide gel. Proteins were transferred onto PVDF membrane, which was blotted with antibodyagainst Bcl-2 and developed by ECL (c) Mcl-1 levels. U266 and RPMI-8226 cells were treated for 6 h with DMSO or 100 nM, 1 �M, and 10 �Mflavopiridol for electrophoresed in 10% SDS-polyacrylamide gel. Proteins were transferred onto PVDF membrane, which was blotted withantibody against Mcl-1 and developed by ECL (d) CDK2 Assay. U266, RPMI-8226, and CD38� myeloma cells were treated for 4 h with DMSO,100 nM, and 1 uM flavopiridol. CDK2 was then immunoprecipitated from lysates prepared from U266, RPMI-8226 cells, and CD38+ myelomacells. An in vitro phosphorylation assay was performed using Histone HI as an exogenous substrate and autoradiography performed.Densitometric analysis was done, and the % activity of DMSO treated cells is provided.

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280 279
Acknowledgements
This work has been supported by grants from theNational Institutes of Health (C.A.) and from theLeukemia and Lymphoma Society and the NationalInstitutes of Health (S.J. Corey). The authors thankMatt Sampson for assistance in manuscript prepara-tion. I. Semenov initiated the concept and design, col-lected and analyzed the data, provided technicalsupport, and helped with the revision. C. Akyuz con-tributed to the concept and design, drafted the originalmanuscript, and provided technical support. V. Rogin-skaya provided technical support and assisted with dataanalysis. D. Chauhan provided study materials andtechnical support and contributed to the revision. S.Corey contributed to the study design, provided techni-cal and logistical support, assisted with data interpreta-tion, and helped to draft and revise the paper.
References
[1] Barlogie B. Advances in therapy of multiple myeloma: lessonsfrom acute leukemia. Clin Cancer Res 1997;3:2605–13.
[2] Alexanian R, Dimopoulos M. The treatment of multiplemyeloma. New Engl J Med 1994;330:484–9.
[3] Japan Myeloma Study Group, Tsuchiya J, Murakami H, KanohT, Kosaka M, Sezaki T, Mikuni C, Kawato M, Takagi T,Togawa A, Isobe T, et al. Ten-year survival and prognosticfactors in multiple myeloma. Br J Haematol 1994;87:832–4.
[4] Durie BG, Salmon SE. A clinical staging system for multiplemyeloma. Correlation of measured myeloma cell mass withpresenting clinical features, response to treatment, and survival.Cancer 1975;36:842–54.
[5] Intergroupe Francais du Myelome, Attal M, Harousseau JL,Stoppa AM, Sotto JJ, Fuzibet JG, Rossi JF, Casassus P,Maisonneuve H, Facon T, Ifrah N, Payen C, Bataille R. Aprospective, randomized trial of autologous bone marrow trans-plantation and chemotherapy in multiple myeloma. New Engl JMed 1996;335:91–7.
[6] Barlogie B, Jagannath S, Vesole DH, Naucke S, Cheson B,Mattox S, Bracy D, Salmon S, Jacobson J, Crowley J, Tricot G.Superiority of tandem autologous transplantation over standardtherapy for previously untreated multiple myeloma. Blood1997;89:789–93.
[7] Tricot G, Jagannath S, Vesole DH, Crowley J, Barlogie B.Relapse of multiple myeloma after autologous transplantation:survival after salvage therapy. Bone Marrow Transplant1995;16:7–11.
[8] Raje N, Anderson K. Thalidomide–a revival story. New Engl JMed 1999;341:1606–9.
[9] Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddle-mon P, Munshi N, Anaissie E, Wilson C, Dhodapkar M, ZeddisJ, Barlogie B. Antitumor activity of thalidomide in refractorymultiple myeloma. New Engl J Med 1999;341:1565–71.
[10] Hallek M, Neumann C, Schaffer M, Danhauser-Riedl S, vonBubnoff N, de Vos G, Druker BJ, Yasukawa K, Griffin JD,Emmerich B. Signal transduction of interleukin-6 involves ty-rosine phosphorylation of multiple cytosolic proteins and activa-tion of Src-family kinases Fyn, Hck, and Lyn in multiplemyeloma cell lines. Exp Hematol 1997;25:1367–77.
[11] Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma:
increasing evidence for a multistep transformation process.Blood 1998;91:3–21.
[12] Druker BJ, Lydon NB. Lessons learned from the development ofan abl tyrosine kinase inhibitor for chronic myelogenousleukemia. J Clin Invest 2000;105:3–7.
[13] Senderowicz AM, Sausville EA. Preclinical and clinical develop-ment of cyclin-dependent kinase modulators. J Natl Cancer Inst2000;92:376–87 In process citation.
[14] De Azevedo WF Jr, Mueller-Dieckmann HJ, Schulze-GahmenU, Worland PJ, Sausville E, Kim SH. Structural basis forspecificity and potency of a flavonoid inhibitor of human CDK2,a cell cycle kinase. Proc Natl Acad Sci USA 1996;93:2735–40.
[15] Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone thatinduces cell death in noncycling A549 human lung carcinomacells. Cancer Res 1996;56:4856–61.
[16] Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ.Flavopiridol induces G1 arrest with inhibition of cyclin-depen-dent kinase (CDK) 2 and CDK4 in human breast carcinomacells. Cancer Res 1996;56:2973–8.
[17] Chien M, Astumian M, Liebowitz D, Rinker-Schaeffer C,Stadler WM. In vitro evaluation of flavopiridol, a novel cellcycle inhibitor, in bladder cancer. Cancer Chemother Pharmacol1999;44:81–7.
[18] Konig A, Schwartz GK, Mohammad RM, Al-Katib A,Gabrilove JL. The novel cyclin-dependent kinase inhibitorflavopiridol downregulates Bcl-2 and induces growth arrest andapoptosis in chronic B-cell leukemia lines. Blood 1997;90:4307–12.
[19] Parker BW, Kaur G, Nieves-Neira W, Taimi M, Kohlhagen G,Shimizu T, Losiewicz MD, Pommier Y, Sausville EA, Senderow-icz AM. Early induction of apoptosis in hematopoietic cell linesafter exposure to flavopiridol. Blood 1998;91:458–65.
[20] Arguello F, Alexander M, Sterry JA, Tudor G, Smith EM,Kalavar NT, Greene JF Jr, Koss W, Morgan CD, Stinson SF,Siford TJ, Alvord WG, Klabansky RL, Sausville EA. Flavopiri-dol induces apoptosis of normal lymphoid cells, causes immuno-suppression, and has potent antitumor activity in vivo againsthuman leukemia and lymphoma xenografts. Blood1998;91:2482–90.
[21] Corey SJ, Burkhardt AL, Bolen JB, Geahlen RL, Tkatch LS,Tweardy DJ. Granulocyte colony-stimulating factor receptorsignaling involves the formation of a three-component complexwith Lyn and Syk protein-tyrosine kinases. Proc Natl Acad SciUSA 1994;91:4683–7.
[22] Tai YT, Teoh G, Shima Y, Chauhan D, Treon SP, Raje N,Hideshima T, Davies FE, Anderson KC. Isolation and charac-terization of human multiple myeloma cell enriched populations.J Immunol Methods 2000;235:11–9.
[23] Byrd JC, Shinn C, Waselenko JK, Fuchs EJ, Lehman TA,Nguyen PL, Flinn IW, Diehl LF, Sausville E, Grever MR.Flavopiridol induces apoptosis in chronic lymphocytic leukemiacells via activation of caspase-3 without evidence of bcl-2 modu-lation or dependence on functional p53. Blood 1998;92:3804–16.
[24] Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ,Pestell R, Albanese C, Sausville EA, Senderowicz AM. Down-regulation of cyclin D1 by transcriptional repression in MCF-7human breast carcinoma cells induced by flavopiridol. CancerRes 1999;59:4634–41.
[25] Puthier D, Derenne S, Barille S, Moreau P, Harousseau JL,Bataille R, Amiot M. Mcl-1 and Bcl-xL are co-regulated by IL-6in human myeloma cells. Br J Haematol 1999;107:392–5.
[26] Motwani M, Delohery TM, Schwartz GK. Sequential dependentenhancement of caspase activation and apoptosis by flavopiridolon paclitaxel-treated human gastric and breast cancer cells. ClinCancer Res 1999;5:1876–83.
[27] Stadler WM, Vogelzang NJ, Amato R, Sosman J, Taber D,Liebowitz D, Vokes EE. Flavopiridol, a novel cyclin-dependent

I. Semeno� et al. / Leukemia Research 26 (2002) 271–280280
kinase inhibitor, in metastatic renal cancer: a University ofChicago Phase II Consortium study. J Clin Oncol 2000;18:371–5.
[28] Innocenti F, Stadler WM, Iyer L, Ramirez J, Vokes EE, RatainMJ. Flavopiridol metabolism in cancer patients is associated
with the occurrence of diarrhea. Clin Cancer Res 2000;6:3400–5.[29] Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase
inhibitors flavopiridol and 7-hydroxy-staurosporine down- regu-late antiapoptosis proteins in B-cell chronic lymphocyticleukemia. Blood 2000;96:393–7.
Copyright © 2022 FDOKUMEN




















