
Improving Salmonella vector with rec mutation to stabilize the DNA cargoes
Upload
independentCategory
view
1download
0
Published Ahead of Print 19 March 2007. 10.1128/MCB.00153-07.
2007, 27(12):4328. DOI:Mol. Cell. Biol. Tannishtha Reya and Jeffrey C. RathmellWofford, Leah N. Dimascio, Olga Ilkayeva, Ameeta Kelekar,
A.E. Herman, Sarah R. Jacobs, Heather L. Wieman, Jessica Yuxing Zhao, Brian J. Altman, Jonathan L. Coloff, Catherine Signaling Pathway To Stabilize Mcl-1Mediate a Glucose-Sensitive Antiapoptotic
β and 3αGlycogen Synthase Kinase 3
http://mcb.asm.org/content/27/12/4328Updated information and services can be found at:
These include:
REFERENCEShttp://mcb.asm.org/content/27/12/4328#ref-list-1at:
This article cites 62 articles, 24 of which can be accessed free
CONTENT ALERTS more»articles cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
on M
arch 24, 2014 by guesthttp://m
cb.asm.org/
Dow
nloaded from

MOLECULAR AND CELLULAR BIOLOGY, June 2007, p. 4328–4339 Vol. 27, No. 120270-7306/07/$08.00�0 doi:10.1128/MCB.00153-07Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Glycogen Synthase Kinase 3� and 3� Mediate a Glucose-SensitiveAntiapoptotic Signaling Pathway To Stabilize Mcl-1�
Yuxing Zhao,1 Brian J. Altman,1 Jonathan L. Coloff,1 Catherine E. Herman,1,2 Sarah R. Jacobs,1Heather L. Wieman,1 Jessica A. Wofford,1 Leah N. Dimascio,1 Olga Ilkayeva,1,2
Ameeta Kelekar,3 Tannishtha Reya,1 and Jeffrey C. Rathmell1,2,4*Department of Pharmacology and Cancer Biology,1 Sarah W. Stedman Nutrition and Metabolism Center,2 and Department of
Immunology,4 Duke University Medical Center, Durham, North Carolina 27710, and Department ofLaboratory Medicine and Pathology, University of Minnesota, Minneapolis, Minnesota 554553
Received 26 January 2007/Returned for modification 2 March 2007/Accepted 9 March 2007
Glucose uptake and utilization are growth factor-stimulated processes that are frequently upregulated incancer cells and that correlate with enhanced cell survival. The mechanism of metabolic protection fromapoptosis, however, has been unclear. Here we identify a novel signaling pathway initiated by glucose catab-olism that inhibited apoptotic death of growth factor-deprived cells. We show that increased glucose metab-olism protected cells against the proapoptotic Bcl-2 family protein Bim and attenuated degradation of theantiapoptotic Bcl-2 family protein Mcl-1. Maintenance of Mcl-1 was critical for this protection, as glucosemetabolism failed to protect Mcl-1-deficient cells from apoptosis. Increased glucose metabolism stabilizedMcl-1 in both cell lines and primary lymphocytes via inhibitory phosphorylation of glycogen synthase kinase3� and 3� (GSK-3�/�), which otherwise promoted Mcl-1 degradation. While a number of kinases canphosphorylate and inhibit GSK-3�/�, we provide evidence that protein kinase C may be stimulated byglucose-induced alterations in diacylglycerol levels or distribution to phosphorylate GSK-3�/�, maintain Mcl-1levels, and inhibit cell death. These data provide a novel nutrient-sensitive mechanism linking glucose me-tabolism and Bcl-2 family proteins via GSK-3 that may promote survival of cells with high rates of glucoseutilization, such as growth factor-stimulated or cancerous cells.
Maintenance of tissue homeostasis requires balanced celldeath and proliferation. A key factor that determines this bal-ance is the availability of cell extrinsic growth signals (9, 42). Inthe absence of such signals, a cell death program that is regu-lated by proteins of the Bcl-2 family is initiated. In the hemato-poietic system, cytokines, such as interleukin-3 (IL-3), pro-vide signals to prevent the activation of programmed cell death(27). Prior to commitment to death, however, cytokine-de-prived cells undergo a program of cellular atrophy (45). Duringatrophy, surface glucose transporters, such as Glut1, becomeinternalized and degraded in lysosomes (14, 45), leading toreduced glycolytic flux and mitochondrial membrane potential(53). Ultimately, loss of glucose metabolism leads to cell death(7, 8, 20, 45). Conversely, maintenance or enhanced glucoseuptake has been shown to protect hematopoietic cells (44, 53),neurons (47), and cardiomyocytes (32). Thus, control of glu-cose metabolism may play a key role in regulation of cell fate.While amino acid sensing is well known to occur via mTOR/Raptor protein complexes (58), nutrient-sensing pathways thatmay respond to changes in glucose metabolism and affect cellsurvival are poorly understood.
In contrast to normal cells, in which glucose metabolism isregulated by growth factors (16, 53), cancer cells often bothmaintain enhanced glucose utilization and resist cell deatheven in the absence of growth factors (19, 21). An increased
rate of glycolysis is an attribute of cancer cells that has beenappreciated for many years (55), with tumor cells exhibitinghigh rates of glucose uptake and utilization but moderate ratesof mitochondrial oxidation (49). More recently, the high ratesof glucose uptake into cancer cells have been exploited inpositron emission tomography to image many types of tumorsin patients (18). Increased glycolysis can stem from tumorhypoxia (19), and a number of oncogenes (4, 10, 20, 40, 44, 52)have been shown to directly promote glucose metabolism. Inaddition, genes directly involved in glucose utilization, such asglucose transporter 1 (Glut1) and hexokinases 1 and 2 (HK1and HK2), which catalyze the uptake and phosphorylation ofglucose to glucose-6-phosphate, respectively, are often overex-pressed in cancer cells and have been correlated with poorprognosis (50, 59). Despite ample evidence for altered glucosemetabolism in cancer cells, the specific impact that this form ofmetabolism may have on cell fate has been obscured by themyriad of other signaling and transcriptional events that canoccur in cellular transformation.
The ability to maintain glucose metabolism may play a keyrole in cancer cell resistance to death upon growth factordeprivation. We have shown that overexpression of Glut1 andHK1 is sufficient to delay loss of plasma membrane integrity ofgrowth factor-withdrawn cells (44). These findings suggestedthe existence of a glucose-dependent signaling pathway thatcould delay or prevent apoptosis. While glucose-sensitive sig-naling to initiate insulin secretion in pancreatic beta cells isrelatively well understood (22), glucose-dependent signalingpathways that may regulate apoptosis are less certain. In sup-port of such a pathway, pretreatment of cells with high extra-
* Corresponding author. Mailing address: Department of Pharma-cology and Cancer Biology, DUMC Box 3813, Durham, NC 27710.Phone: (919) 681-1084. Fax: (919) 668-6044. E-mail: [email protected].
� Published ahead of print on 19 March 2007.
4328
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

cellular glucose concentrations or hyperglycemia has beenshown to enhance de novo synthesis of diacylglycerol and pro-mote protein kinase C (PKC) activation to protect cardiomyo-cytes and neurons from ischemic injury. The mechanism of thisprotection, however, is not clear (29, 38, 46, 57).
The Bcl-2 family of proteins includes both pro- and anti-apoptotic proteins (12) and are likely candidates to mediateregulation of cell death by glucose metabolism. Some evidencehas previously suggested that Bcl-2 family proteins may besensitive to cell metabolic status because the proapoptoticBcl-2 family member Bax can become activated upon glucosedeprivation (7, 53). The mechanism linking the Bcl-2 familyand glucose metabolism, however, is not certain yet likely uti-lizes critical regulatory Bcl-2 family proteins. In hematopoieticcells, induction and translocation to mitochondria of the pro-apoptotic BH3-only protein Bim have been shown to play animportant role in many cell death pathways (6). The antiapop-totic Bcl-2 family protein Mcl-1 also plays a vital role in cellsurvival and binds Bim on mitochondria to inhibit Bim-inducedactivation of the proapoptotic protein Bax. Conditional genetargeting has shown that Mcl-1 is required for the survival ofboth B and T cells (36). Mcl-1 levels are highly regulated byproteolytic degradation, and phosphorylation of Mcl-1 byGSK-3 in cytokine-deprived cells has recently been shown toplay an important role in targeting Mcl-1 for ubiquitinationand degradation (31, 62).
Cellular activation or transformation leads to signaling andtranscriptional as well as metabolic events that may affect Bcl-2family members and cell death. To address how the increasedglucose uptake and catabolism characteristic of activated lym-phocytes or cancer cells may influence the Bcl-2 family in theabsence of other signaling or transcriptional events, we haveanalyzed the death of cells in which only glucose uptake andmetabolism are promoted. Using this approach, we describe anovel glucose-responsive antiapoptotic signaling pathway. Inthis pathway, enhanced glucose uptake led to inhibitory phos-phorylation of GSK-3, most likely via PKC activity, to stabilizethe antiapoptotic Bcl-2 family protein Mcl-1. This novel nutri-ent-sensing pathway may provide a critical mechanism to con-nect glucose utilization to the Bcl-2 family of proteins viadiacylglycerol-induced activation of PKC isoforms to inhibitGSK-3 and set a threshold for apoptosis of cells with high ratesof glucose metabolism.
MATERIALS AND METHODS
Cells. FL5.12 cells, BAF3 cells, and 32D cells were cultured in RPMI mediumsupplemented with 0.5 ng/ml recombinant mouse IL-3 (BD Pharmingen andPepro Tech, Rocky Hill, NJ) as previously described (5). Glut1/HK1-expressingFL5.12 cells were generated after selection of clones stably expressing Glut1 andHK1 and have been previously described (44). Transient transfections wereperformed by nucleofection (Kit V; Amaxa Biosystems). Stable expression of theshort hairpin RNA interference vector (shRNAi) was achieved by retroviraltransfection with pKDGFP shRNAi. Stable clones were identified after cellsorting for green fluorescent protein (GFP)-positive cells and Western analysisfor gene expression. For growth factor withdrawal, cells were washed three timesin phosphate-buffered saline (PBS) prior to resuspension in RPMI mediumlacking IL-3. To inhibit protein synthesis, cells were cultured in medium con-taining 25 �g/ml of cycloheximide. Glycolysis was measured as described previ-ously (43).
Plasmid constructs. Bim isoforms were cloned from FL5.12 mRNA usingSuperScript One-Step reverse transcription-PCR with Platinum Pfx (Invitrogen)according to the manufacturer’s protocol and subcloned into pEF6 (Invitrogen).
Hemagglutinin (HA)-tagged Mcl-1 (generously provided by H. J. Brady, Uni-versity College, London) was cloned into pEF6. Bim and Mcl-1 shRNAi plasmidswere constructed using previously described approaches (15). shRNAi sequenceswere as follows: Bim, GATCTGCGCCCGGAGATACGGATTCCTCGAGCAATCCGTATCTCCGGGCGCAGATC; Mcl-1, AATGGTTCGATGAAGCTTTCCCTCGAGCGAAAGCTTCATCGAACCATT (34), and GFP, GATCCGTTCAACTAGCAGACCATTCCTCGAGCAATGGTCTGCTAGTTGAACGGATC. Sequences were cloned under control of the hU6 promoter in pCR2.1 andpKD vectors as described previously (15). The GSK-3� shRNAi construct waskindly provided by David Turner (University of Michigan) (60). EGFP-PKD-CRD and EGFP-PKD-CRD-P155/287G constructs were kindly provided byMartin Spitaler and Doreen Cantrel (University of Dundee, United Kingdom).pCMV-FLAG-ubiquitin was kindly provided by Michael Ehlers (Duke Univer-sity).
Mass spectral analysis of organic acids. Intracellular organic acids were an-alyzed as described previously (24). Briefly, cells were washed in PBS, lysed in 0.1M HCl, and cleared by centrifugation. A cocktail of stable-isotope internalstandards was added, and lysates were then extracted in ethyl acetate, dried, andconverted to methyl esters, with the addition of trimethyl silyl esters with ethoxy-amine hydrochloride. Organic acids were then analyzed by capillary gas chro-matography/mass spectrometry.
Cell fractionation. To isolate mitochondria, cells were washed once in PBS,resuspended in mitochondrial isolation buffer (20 mM HEPES [pH 7.5], 10 mMKCl, 5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, proteaseinhibitors), and forced by syringe to pass through a 5-�m polycarbonate filtermembrane. Unbroken cells and nuclei were pelleted by centrifugation at 800 �g for 10 min at 4°C. The supernatant was subjected to an additional centrifuga-tion at 16,000 � g for 10 min at 4°C. The pellet was collected as heavy membraneand mitochondrial fractions, and the supernatant was used as the cytosolicfraction.
Clonogenic assay. Cells were withdrawn from IL-3 for 0, 12, 18, 24, and 48 h;washed; and recultured in medium containing IL-3 in 96-well plates at concen-trations of 1/3, 1, or 3 cells per well. The frequency of colonies on each plate after7 days was determined as the mean and standard error of colony number relativeto cell input for each plate (n � 5).
Western blots. Cells were lysed in radioimmunoprecipitation assay buffer withprotease inhibitors (BD Pharmingen) and phosphatase inhibitors (Sigma) on icefor 10 min and precleared by centrifugation. Protein concentrations were deter-mined by bicinchoninic acid protein assay (Bio-Rad), and 25 �g of protein wassubjected to 4 to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis(Bio-Rad). Antibodies used were rabbit anti-Bax (Cell Signaling), rabbit anti-Bim (BD Pharmingen), rabbit anti-Bcl-2 (BD Pharmingen), mouse anti-cyto-chrome c (BD Pharmingen), rabbit anti-Glut1 (Abcam), rabbit anti-phospho-GSK-3(Ser9/21) (Cell Signaling), rabbit anti-GSK-3� (Cell signaling), mouseanti-phospho-Akt (Ser473), rabbit anti-Mcl-1 (Santa Cruz and Biolegend), rabbitanti-Cox IV, rabbit antiubiquitin (Santa Cruz), rat anti-HA (Roche), rabbitanti-FLAG (Sigma), and mouse antiactin (Sigma). Rabbit anti-phospho-Mcl-1(Ser159) was a kind gift of D. Green (St. Jude Children’s Research Hospital,Memphis, TN) and U. Maurer (Institut fur Molekulare Medizin und Zellfor-schung, Germany). Secondary antibodies were anti-rabbit and anti-mousehorseradish peroxidase-labeled antibodies (BD Pharmingen) and anti-rabbitinfrared-labeled antibody and were detected with ECL-Plus (Amersham, Bio-sciences) or the Odyssey infrared imaging system (Licor). All images were uni-formly contrasted, and some were digitally rearranged for ease of viewing (re-arranged lanes indicated by spaces).
Immunoprecipitation. Cells were harvested and lysed in CHAPS buffer {50mM Tris [pH 7.5], 10% glycerol, 1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS], 150 mM NaCl, 1 mM EDTA, proteasome inhib-itor}. Lysates were precleared, antibodies (rabbit anti-Bax [clone 6A7; BD Phar-mingen], rabbit anti-Bim [BD Pharmingen], and rat anti-HA [Roche]) wereadded, and the mixtures were rotated at 4°C for 1 h. Anti-rabbit immunoglobulinG beads (ebioscience) or protein A/G beads (Santa Cruz) were then added to thelysates and incubated overnight. Beads were washed with 1% CHAPS bufferthree times, and proteins were eluted by boiling the beads in sodium dodecylsulfate loading buffer.
Fluorescence microscopy. To image GSK-3 phosphorylation in primary bonemarrow cells, cells were cytospun, fixed on the slides in 4% paraformaldehyde,washed in PBS–0.1% Tween, and blocked with 20% normal goat serum. Slideswere stained with anti-nerve growth factor receptor (NGFR)–biotin (BD Phar-mingen) and anti-phospho-GSK-3 (Cell Signaling) or an isotype control, fol-lowed by anti-rabbit–Alexa 546 (Molecular Probes), antistreptavidin–FITC (BDPharmingen), and DAPI (4�,6�-diamidino-2-phenylindole). To image cells ex-pressing GFP fusion proteins, cells were fixed with 1% paraformaldehyde in PBS
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4329
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

and viewed with a Zeiss LSM410 confocal microscope (Zeiss, Thornwood, NY)and Metamorph software.
Glut1-transgenic mice and primary cell culture. Full-length rat Glut1 wascloned into the pLck.E2 vector, which we have used previously for T-lymphocyte-specific transgene expression (43). Transgenic animals were made in and were agenerous gift of the Abramson Family Cancer Research Institute transgenicfacility at the University of Pennsylvania. Mice were backcrossed five generationsto C57BL6/J mice and cared for at Duke University. T cells were purified bynegative selection, and glucose uptake and in vitro survival in neglect weremeasured as described previously (3, 43). Bone marrow cells were cultured in 5ng/ml recombinant IL-3-containing RPMI 1640 medium. Media with IL-3 werechanged every 2 days. On day 4, cells were infected in retrovirus supernatant(MSCV-Glut1-IRES-NGFR or vector control) at a concentration of 1 � 106
cells/ml for 24 h at 37°C, and they were analyzed by immunofluorescence 2 dayslater.
RESULTS
Glut1 and HK1 signal to delay Bcl-2 family-mediated com-mitment to cell death after growth factor withdrawal in amanner that requires glucose hydrolysis. To determine howchanges in glucose metabolism may signal to affect cell deathpathways, we analyzed the effects of Glut1 and HK1 expression(Glut1/HK1 cells) (44) on cell death by using the FL5.12
hematopoietic precursor cell line. FL5.12 cells are nontrans-formed and are strictly dependent on the cytokine IL-3 formaintenance of glucose uptake and survival. Expression vec-tors for Glut1 and HK1 (pSFFV-Glut1 and pcDNA3-HK1,respectively) were previously cotransfected into FL5.12 cells,and stable clones were identified by Geneticin selection andimmunofluorescence labeling (44). Analysis of Glut1/HK1cells, therefore, provided a means to directly identify glucose-stimulated antiapoptotic signaling pathways independent of othertransformation- or oncogene-induced antiapoptotic mechanisms.Expression of Glut1 and HK1 promoted and maintained en-hanced glycolysis during IL-3 withdrawal (Fig. 1A), and massspectrometry-based quantification of intracellular organic acidsshowed dramatically increased production of lactate that wasmaintained in the absence of IL-3 (Fig. 1B). In contrast, Krebscycle intermediates were only modestly increased or were re-duced in Glut1/HK1 cells. Although expression of Glut1 and HK1does not direct coordinate control over entire metabolic path-ways, Glut1/HK1 cells displayed a highly glycolytic phenotype inthe presence of IL-3 and shortly after IL-3 withdrawal, similar towhat has been observed in many cancer cells (49).
FIG. 1. Glut1/HK1 expression increases glucose metabolism and delays commitment to cell death. (A) Glycolytic flux was measured in control(Neo) and Glut1/HK1 cells in the presence or absence of IL-3. (B) Tandem mass spectrometry-based analysis of organic acids in control andGlut1/HK1 cells cultured in the presence or absence of IL-3 for 6 h. (C) Clonogenicity of multiple individual control, Glut1/HK1, and Bcl-xL cellscultured in the absence of IL-3 for the indicated times, followed by readdition of IL-3. (D) Control and Glut1/HK1 cells were IL-3 withdrawn andcultured in 10 mM glucose, 10 mM 2DOG plus 1 mM methylpyruvate, or 5 mM glucose plus 5 mM 2DOG, and viability was determined by PIexclusion. (E) Bax activation was analyzed by immunoprecipitation (I.P.) of active Bax from cells cultured in the presence or absence of IL-3 for10 h followed by Western blotting for total Bax. (F) Cytochrome c release in control (Neo), Glut1/HK1, and Bcl-xL cells after IL-3 withdrawal.Cells were cultured in the presence or absence of IL-3 for the indicated times, fractionated to isolate cytosolic and mitochondrial proteins, andanalyzed by Western blotting for cytochrome c. Values are means and standard deviation (n � 3) (A, B, and D) or means and standard errors ofthe means (n � 5) (C). N, Neo control; GH, Glut1/HK1; X, Bcl-xL.
4330 ZHAO ET AL. MOL. CELL. BIOL.
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

We have previously shown that Glut1/HK1-expressing cellsexhibit delayed cell permeability to the vital fluorescent dyepropidium iodide (PI) upon IL-3 withdrawal (44). Permeabilityto PI, however, may be delayed by maintenance of cellularenergetics and continued production of ATP (56) and thusmay not reflect a true change in the commitment point ofdeath. To more rigorously test the survival capacity of Glut1/HK1 cells upon IL-3 withdrawal, we performed limiting-dilu-tion clonogenicity analysis on individual control or Glut1/HK1clones following restoration of IL-3 to cells withdrawn fromIL-3 for various periods of time (Fig. 1C). Consistent withresults obtained by PI exclusion (44), control cells rapidly losttheir ability to recover and grow with readdition of IL-3,whereas Glut1/HK1 maintained significant clonogenic survival,albeit not to the same extent as cells expressing the antiapop-totic Bcl-2 family protein Bcl-xL. The survival advantage ofGlut1/HK1 cells over control cells required a hydrolyzableglucose source, as the nonhydrolyzable 2-deoxyglucose (2DOG)failed to enhance cell survival even in the presence of methylpyruvate (a cell-permeable form of pyruvate to provide a mito-chondrial substrate) (Fig. 1D). These findings suggest that glu-cose may initiate an antiapoptotic signaling pathway requiringglucose hydrolysis to delay commitment to cell death.
Commitment to cell death upon growth factor withdrawal inhematopoietic cells occurs at the point of mitochondrial dis-ruption by Bax and Bak and release of cytochrome c into thecytosol. To identify the stage at which glucose metabolism maydelay cell death commitment, Bax activation and cytochrome crelease were analyzed in Glut1/HK1 cells. Control and Glut1/HK1 cells were deprived of IL-3 for 10 h, and Bax was immu-noprecipitated from cell lysates by using an active Bax con-formation-specific antibody (Fig. 1E). No active Bax wasdetectable in cells cultured in the presence of IL-3. Upon IL-3withdrawal, activated Bax was readily observed in control cellsbut not in Glut1/HK1 cells. Cytosolic and mitochondrial cellfractions from control, Glut1/HK1, and Bcl-xL cells were alsoanalyzed by immunoblotting to observe mitochondrial disrup-tion and release of cytochrome c (Fig. 1F). Mitochondria incontrol cells began to lose integrity 10 h after IL-3 withdrawal,with decreased cytochrome c in the mitochondrial fractionsand the appearance of cytochrome c in cytoplasmic fractions.In contrast, cytochrome c release was delayed in Glut1/HK1cells, and cytosolic cytochrome c was readily detectable onlyafter 12 h of IL-3 withdrawal. By this time, control cells hadreleased the majority of the cytochrome c into the cytosol.Cells overexpressing Bcl-xL maintained mitochondrial integritythroughout the time analyzed. Caspase activation in Glut1/HK1 cells was also reduced upon IL-3 withdrawal, as precipi-tation of caspase 3 in Glut1/HK1 cells with biotinylated zVAD,a broad caspase inhibitor that binds to active caspases, yieldedonly 40% of that obtained in control cells upon IL-3 with-drawal (data not shown). These results indicate that increasedglucose metabolism delayed commitment to cell death aftergrowth factor withdrawal upstream of Bax activation and cy-tochrome c release.
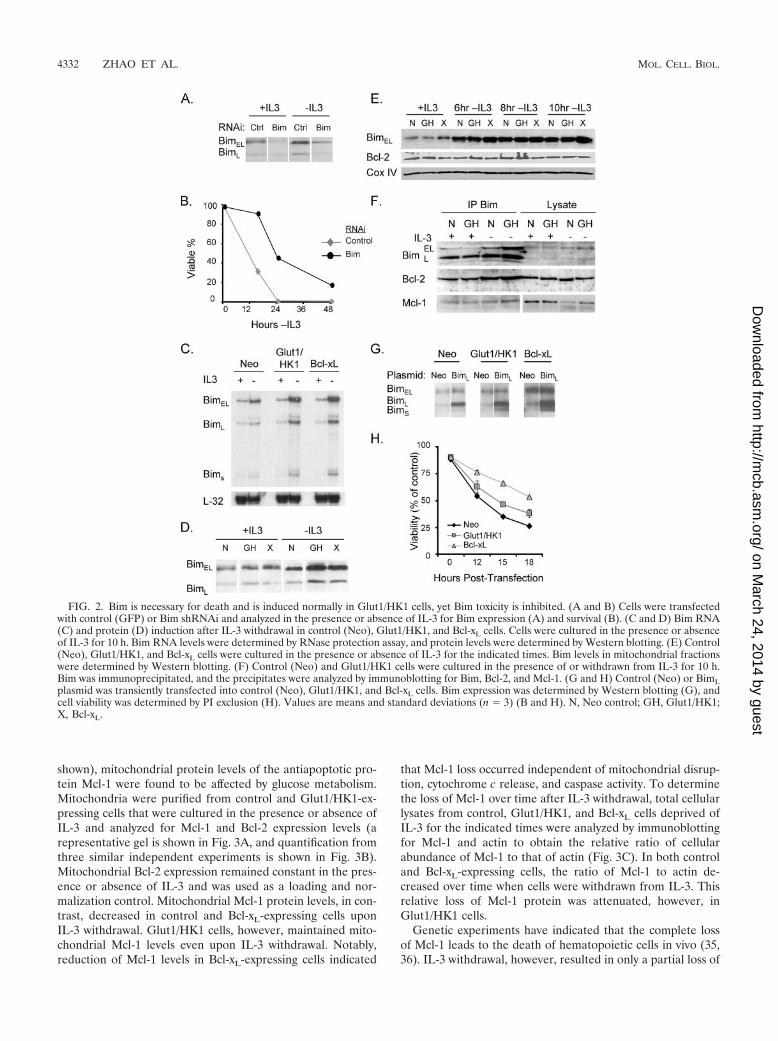
Bim is necessary and sufficient for cytokine withdrawal-induced apoptosis, yet glucose metabolism promotes resis-tance to Bim toxicity. Antiapoptotic glucose signaling in Glut1/HK1 cells delayed Bax activation and cytochrome c releaseupon IL-3 withdrawal, suggesting regulation of the Bcl-2 family
members by glucose hydrolysis. Gene-targeting studies haveshown that the proapoptotic BH3-only Bcl-2 family memberBim plays a pivotal role in apoptosis of hematopoietic cellsupon cytokine withdrawal (6). In FL5.12 cells, the three iso-forms of Bim (BimEL, BimL, and BimS) were induced and wereimportant for efficient apoptosis upon IL-3 withdrawal, as cellswith decreased Bim expression by shRNAi partially resistedIL-3 deprivation-induced apoptosis (Fig. 2A and B). To test ifglucose metabolism affected Bim induction, we cultured con-trol, Glut1/HK1, and Bcl-xL cells in IL-3 or withdrew cells fromIL-3 for 10 h. Transcriptional induction of Bim RNA wasanalyzed by RNase protection (Fig. 2C), and total Bim proteinlevels were determined by immunoblotting (Fig. 2D). All threeBim isoforms were induced at the RNA level, and BimEL andBimL were upregulated at the protein level in all three celllines upon IL-3 withdrawal. Endogenous BimS protein wasundetectable in all conditions (data not shown). For reasonsthat are not clear, Glut1/HK1 and Bcl-xL cells had somewhathigher levels of total cellular Bim RNA and protein inductionfollowing IL-3 deprivation. Nevertheless, apoptosis was de-layed in Glut1/HK1 cells (Fig. 1C). Mitochondrial levels ofBim are critical for Bim to promote apoptosis, and increasedglucose metabolism may have affected Bim translocation tomitochondria (41). To analyze Bim translocation, mitochon-dria were isolated from control, Glut1/HK1, and Bcl-xL cellsdeprived of IL-3 for the indicated times and were immuno-blotted for Bim protein levels. Despite somewhat higher totalBim levels, Glut1/HK1 and Bcl-xL cells showed similar Bimtranslocation to mitochondria compared to control cells, (Fig.2E). To determine if Bim protein associations were altered byGlut1/HK1 expression, Bim was immunoprecipitated fromcontrol and Glut1/HK1 cells and analyzed by immunoblottingfor associations with Bcl-2 and Mcl-1 (Fig. 2F). While IL-3withdrawal increased the amount of immunoprecipitated Bim,no differences between control and Glut1/HK1 cells were ob-served in Bcl-2 coimmunoprecipitation. Mcl-1 was also precip-itated with Bim in both cell lines, although total Mcl-1 levelswere reduced in IL-3-withdrawn control cells.
Bim induction was greater and Bim translocation to mito-chondria and association with other Bcl-2 family proteins ap-peared normal in Glut1/HK1 cells, yet Glut1/HK1 cells re-sisted cell death upon IL-3 withdrawal. To determine ifincreased glucose metabolism could prevent Bim cytotoxicitydownstream of induction and translocation to mitochondria,control or BimL-expressing plasmids were transfected into con-trol, Glut1/HK1, and Bcl-xL cells in the presence of IL-3 andcell viability was monitored. Expression of BimL was similar inall three cell lines, with moderately higher expression in Glut1/HK1 and Bcl-xL cells relative to control cells (Fig. 2G). BimL
led to significant death of control cells, while Bcl-xL-expressingcells were more resistant. Glut1/HK1 cells showed intermedi-ate resistance to BimL (Fig. 2H). Similar results were obtainedwith BimS (data not shown). These data suggested that in-creased glucose metabolism acted on the Bcl-2 family proteinsat the mitochondria to prevent Bim toxicity and promote cellsurvival.
Increased glucose metabolism stabilizes Mcl-1 to protectcells against IL-3 withdrawal-induced apoptosis. AlthoughRNase protection showed no differences in mRNA levels ofmultiple Bcl-2 family members upon IL-3 withdrawal (data not
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4331
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
shown), mitochondrial protein levels of the antiapoptotic pro-tein Mcl-1 were found to be affected by glucose metabolism.Mitochondria were purified from control and Glut1/HK1-ex-pressing cells that were cultured in the presence or absence ofIL-3 and analyzed for Mcl-1 and Bcl-2 expression levels (arepresentative gel is shown in Fig. 3A, and quantification fromthree similar independent experiments is shown in Fig. 3B).Mitochondrial Bcl-2 expression remained constant in the pres-ence or absence of IL-3 and was used as a loading and nor-malization control. Mitochondrial Mcl-1 protein levels, in con-trast, decreased in control and Bcl-xL-expressing cells uponIL-3 withdrawal. Glut1/HK1 cells, however, maintained mito-chondrial Mcl-1 levels even upon IL-3 withdrawal. Notably,reduction of Mcl-1 levels in Bcl-xL-expressing cells indicated
that Mcl-1 loss occurred independent of mitochondrial disrup-tion, cytochrome c release, and caspase activity. To determinethe loss of Mcl-1 over time after IL-3 withdrawal, total cellularlysates from control, Glut1/HK1, and Bcl-xL cells deprived ofIL-3 for the indicated times were analyzed by immunoblottingfor Mcl-1 and actin to obtain the relative ratio of cellularabundance of Mcl-1 to that of actin (Fig. 3C). In both controland Bcl-xL-expressing cells, the ratio of Mcl-1 to actin de-creased over time when cells were withdrawn from IL-3. Thisrelative loss of Mcl-1 protein was attenuated, however, inGlut1/HK1 cells.
Genetic experiments have indicated that the complete lossof Mcl-1 leads to the death of hematopoietic cells in vivo (35,36). IL-3 withdrawal, however, resulted in only a partial loss of
FIG. 2. Bim is necessary for death and is induced normally in Glut1/HK1 cells, yet Bim toxicity is inhibited. (A and B) Cells were transfectedwith control (GFP) or Bim shRNAi and analyzed in the presence or absence of IL-3 for Bim expression (A) and survival (B). (C and D) Bim RNA(C) and protein (D) induction after IL-3 withdrawal in control (Neo), Glut1/HK1, and Bcl-xL cells. Cells were cultured in the presence or absenceof IL-3 for 10 h. Bim RNA levels were determined by RNase protection assay, and protein levels were determined by Western blotting. (E) Control(Neo), Glut1/HK1, and Bcl-xL cells were cultured in the presence or absence of IL-3 for the indicated times. Bim levels in mitochondrial fractionswere determined by Western blotting. (F) Control (Neo) and Glut1/HK1 cells were cultured in the presence of or withdrawn from IL-3 for 10 h.Bim was immunoprecipitated, and the precipitates were analyzed by immunoblotting for Bim, Bcl-2, and Mcl-1. (G and H) Control (Neo) or BimLplasmid was transiently transfected into control (Neo), Glut1/HK1, and Bcl-xL cells. Bim expression was determined by Western blotting (G), andcell viability was determined by PI exclusion (H). Values are means and standard deviations (n � 3) (B and H). N, Neo control; GH, Glut1/HK1;X, Bcl-xL.
4332 ZHAO ET AL. MOL. CELL. BIOL.
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

Mcl-1. We sought, therefore, to determine how moderatechanges in Mcl-1 protein level that occur upon IL-3 withdrawalmay influence sensitivity to apoptosis. Mcl-1 protein levelswere decreased using shRNAi to approximately 50% in controland Glut1/HK1 cells to mimic the levels of Mcl-1 that occur ingrowth factor withdrawal (Fig. 3D). In the presence of cyto-kine, this reduced level of Mcl-1 in Mcl-1 shRNAi-treated cellswas not sufficient to promote spontaneous activation of Bax orcell death (Fig. 3E and data not shown). Upon withdrawalfrom cytokine, however, cells expressing shRNAi for Mcl-1displayed an increased rate of Bax activation and more rapidcell death. This rapid cell death required Bim induction, assimultaneous shRNAi of Bim led to a partial rescue of Mcl-1-deficient cells (data not shown). Importantly, Mcl-1-deficientGlut1/HK1 cells underwent programmed cell death at thesame high rate as Mcl-1-deficient control cells. Increased glu-cose catabolism, therefore, appeared to delay apoptosis upongrowth factor withdrawal via regulation of the antiapoptoticBcl-2 family protein Mcl-1 to prevent a sudden change in Mcl-1levels that may allow Bim-mediated Bax activation and apop-tosis.
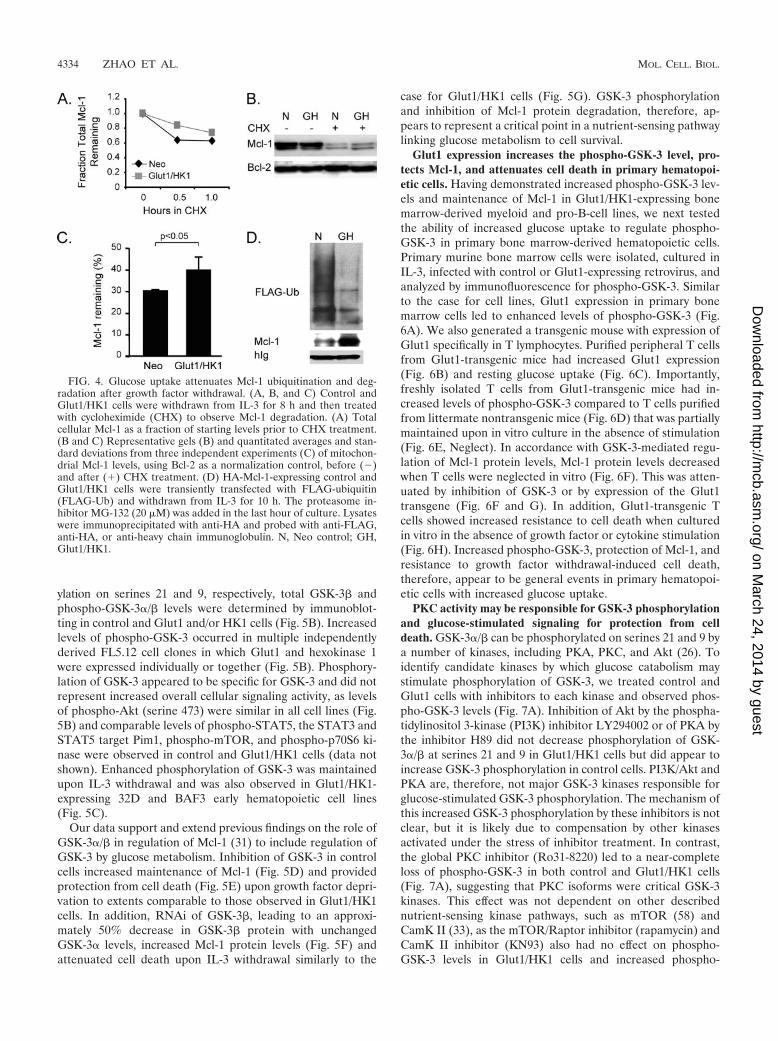
Expression of Glut1 and HK1 attenuates Mcl-1 protein deg-radation after growth factor withdrawal. Ubiquitination andproteasomal degradation play critical roles in control of Mcl-1levels (62). To determine the effect of Glut1/HK1 expressionon the degradation of Mcl-1, we cultured control and Glut1/
HK1 cells in the absence of IL-3 for 8 h, treated cells withcycloheximide to block new protein synthesis, and observedMcl-1 degradation. In whole-cell (Fig. 4A) or mitochondrial (arepresentative gel is shown in Fig. 4B, and quantification fromthree similar independent experiments is shown in Fig. 4C)extracts, the Mcl-1 was more rapidly degraded in control cellsthan in Glut1/HK1 cells. To further confirm delayed Mcl-1degradation, control and Glut1/HK1 cells expressing HA-tagged Mcl-1 were transfected with FLAG-tagged ubiquitinand withdrawn from IL-3. Immunoprecipitation of HA–Mcl-1followed by immunoblotting for FLAG-ubiquitin showed thatMcl-1 ubiquitination was reduced in Glut1/HK1 cells (Fig.4D). Antiapoptotic glucose signaling, therefore, appeared toinhibit growth factor withdrawal-induced apoptosis via a glu-cose-sensitive attenuation of Mcl-1 protein ubiquitination anddegradation.
Inhibition of GSK-3 kinase activity by glucose metabolismprotects Mcl-1 levels and attenuates cell death. Mcl-1 proteindegradation is regulated in part through Mcl-1 phosphoryla-tion by GSK-3 on serine 159 and subsequent Mcl-1 ubiquiti-nation by the BH3 domain containing ubiquitin ligase Mule(31, 62). We therefore analyzed phosphorylation of Mcl-1 S159and found it decreased relative to that in control cells in twoindependent clones expressing Glut1 alone or with HK1 (Fig.5A), suggesting reduced GSK-3 kinase activity in glycolyticcells. As GSK-3�/� activity is negatively regulated by phosphor-
FIG. 3. Maintenance of Mcl-1 protein level is critical for protection of Glut1/HK1 cells upon IL-3 withdrawal. (A and B) Control, Glut1/HK1,and Bcl-xL cells were cultured in the presence or absence of IL-3 for 10 h, and mitochondria were isolated. (A and B) Representative gel (A) andquantitated averages and standard deviations of mitochondrial Mcl-1 levels using Bcl-2 as a normalization control (B). (C) Control (Neo),Glut1/HK1, and Bcl-xL cells were withdrawn from IL-3, and the total cellular Mcl-1/actin ratio was determined over time. (D and E) Control andGlut1/HK1 cells were transiently transfected with control (GFP) or Mcl-1 shRNAi, and then Mcl-1 protein level was determined by immuno-blotting (D) and cell viability was observed by PI exclusion (E). Values are means for cell survival from triplicate samples; error bars indicatestandard deviations. N, Neo control; GH, Glut1/HK1; X, Bcl-xL.
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4333
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
ylation on serines 21 and 9, respectively, total GSK-3� andphospho-GSK-3�/� levels were determined by immunoblot-ting in control and Glut1 and/or HK1 cells (Fig. 5B). Increasedlevels of phospho-GSK-3 occurred in multiple independentlyderived FL5.12 cell clones in which Glut1 and hexokinase 1were expressed individually or together (Fig. 5B). Phosphory-lation of GSK-3 appeared to be specific for GSK-3 and did notrepresent increased overall cellular signaling activity, as levelsof phospho-Akt (serine 473) were similar in all cell lines (Fig.5B) and comparable levels of phospho-STAT5, the STAT3 andSTAT5 target Pim1, phospho-mTOR, and phospho-p70S6 ki-nase were observed in control and Glut1/HK1 cells (data notshown). Enhanced phosphorylation of GSK-3 was maintainedupon IL-3 withdrawal and was also observed in Glut1/HK1-expressing 32D and BAF3 early hematopoietic cell lines(Fig. 5C).
Our data support and extend previous findings on the role ofGSK-3�/� in regulation of Mcl-1 (31) to include regulation ofGSK-3 by glucose metabolism. Inhibition of GSK-3 in controlcells increased maintenance of Mcl-1 (Fig. 5D) and providedprotection from cell death (Fig. 5E) upon growth factor depri-vation to extents comparable to those observed in Glut1/HK1cells. In addition, RNAi of GSK-3�, leading to an approxi-mately 50% decrease in GSK-3� protein with unchangedGSK-3� levels, increased Mcl-1 protein levels (Fig. 5F) andattenuated cell death upon IL-3 withdrawal similarly to the
case for Glut1/HK1 cells (Fig. 5G). GSK-3 phosphorylationand inhibition of Mcl-1 protein degradation, therefore, ap-pears to represent a critical point in a nutrient-sensing pathwaylinking glucose metabolism to cell survival.
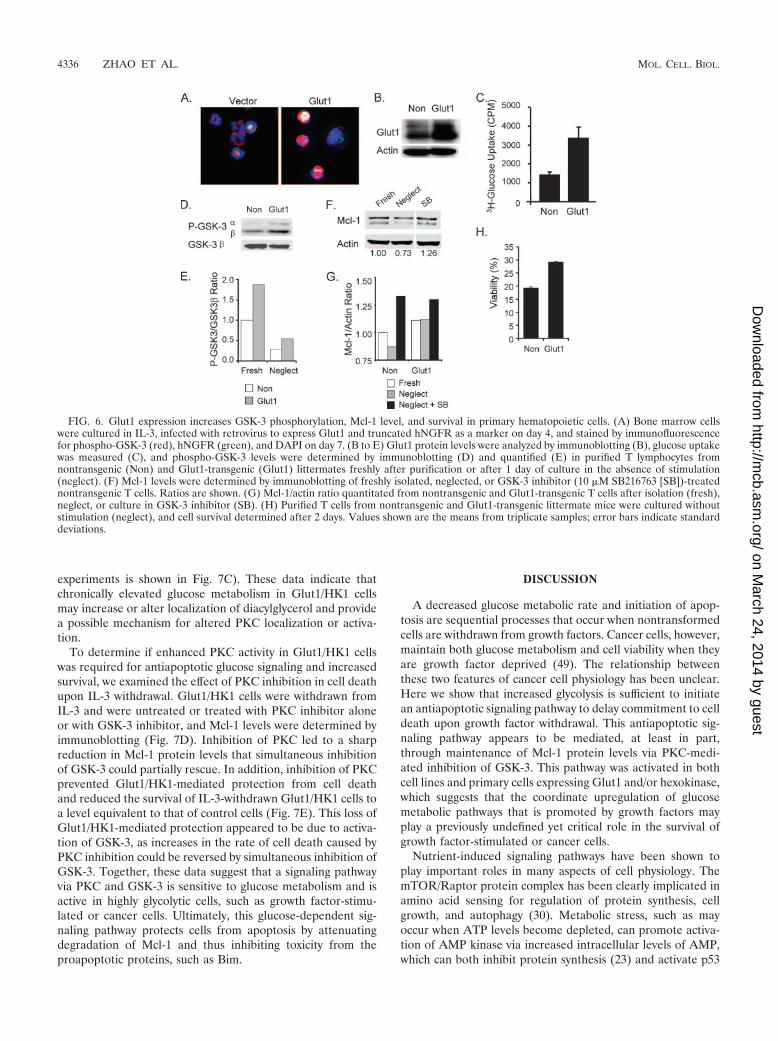
Glut1 expression increases the phospho-GSK-3 level, pro-tects Mcl-1, and attenuates cell death in primary hematopoi-etic cells. Having demonstrated increased phospho-GSK-3 lev-els and maintenance of Mcl-1 in Glut1/HK1-expressing bonemarrow-derived myeloid and pro-B-cell lines, we next testedthe ability of increased glucose uptake to regulate phospho-GSK-3 in primary bone marrow-derived hematopoietic cells.Primary murine bone marrow cells were isolated, cultured inIL-3, infected with control or Glut1-expressing retrovirus, andanalyzed by immunofluorescence for phospho-GSK-3. Similarto the case for cell lines, Glut1 expression in primary bonemarrow cells led to enhanced levels of phospho-GSK-3 (Fig.6A). We also generated a transgenic mouse with expression ofGlut1 specifically in T lymphocytes. Purified peripheral T cellsfrom Glut1-transgenic mice had increased Glut1 expression(Fig. 6B) and resting glucose uptake (Fig. 6C). Importantly,freshly isolated T cells from Glut1-transgenic mice had in-creased levels of phospho-GSK-3 compared to T cells purifiedfrom littermate nontransgenic mice (Fig. 6D) that was partiallymaintained upon in vitro culture in the absence of stimulation(Fig. 6E, Neglect). In accordance with GSK-3-mediated regu-lation of Mcl-1 protein levels, Mcl-1 protein levels decreasedwhen T cells were neglected in vitro (Fig. 6F). This was atten-uated by inhibition of GSK-3 or by expression of the Glut1transgene (Fig. 6F and G). In addition, Glut1-transgenic Tcells showed increased resistance to cell death when culturedin vitro in the absence of growth factor or cytokine stimulation(Fig. 6H). Increased phospho-GSK-3, protection of Mcl-1, andresistance to growth factor withdrawal-induced cell death,therefore, appear to be general events in primary hematopoi-etic cells with increased glucose uptake.
PKC activity may be responsible for GSK-3 phosphorylationand glucose-stimulated signaling for protection from celldeath. GSK-3�/� can be phosphorylated on serines 21 and 9 bya number of kinases, including PKA, PKC, and Akt (26). Toidentify candidate kinases by which glucose catabolism maystimulate phosphorylation of GSK-3, we treated control andGlut1 cells with inhibitors to each kinase and observed phos-pho-GSK-3 levels (Fig. 7A). Inhibition of Akt by the phospha-tidylinositol 3-kinase (PI3K) inhibitor LY294002 or of PKA bythe inhibitor H89 did not decrease phosphorylation of GSK-3�/� at serines 21 and 9 in Glut1/HK1 cells but did appear toincrease GSK-3 phosphorylation in control cells. PI3K/Akt andPKA are, therefore, not major GSK-3 kinases responsible forglucose-stimulated GSK-3 phosphorylation. The mechanism ofthis increased GSK-3 phosphorylation by these inhibitors is notclear, but it is likely due to compensation by other kinasesactivated under the stress of inhibitor treatment. In contrast,the global PKC inhibitor (Ro31-8220) led to a near-completeloss of phospho-GSK-3 in both control and Glut1/HK1 cells(Fig. 7A), suggesting that PKC isoforms were critical GSK-3kinases. This effect was not dependent on other describednutrient-sensing kinase pathways, such as mTOR (58) andCamK II (33), as the mTOR/Raptor inhibitor (rapamycin) andCamK II inhibitor (KN93) also had no effect on phospho-GSK-3 levels in Glut1/HK1 cells and increased phospho-
FIG. 4. Glucose uptake attenuates Mcl-1 ubiquitination and deg-radation after growth factor withdrawal. (A, B, and C) Control andGlut1/HK1 cells were withdrawn from IL-3 for 8 h and then treatedwith cycloheximide (CHX) to observe Mcl-1 degradation. (A) Totalcellular Mcl-1 as a fraction of starting levels prior to CHX treatment.(B and C) Representative gels (B) and quantitated averages and stan-dard deviations from three independent experiments (C) of mitochon-drial Mcl-1 levels, using Bcl-2 as a normalization control, before (�)and after (�) CHX treatment. (D) HA-Mcl-1-expressing control andGlut1/HK1 cells were transiently transfected with FLAG-ubiquitin(FLAG-Ub) and withdrawn from IL-3 for 10 h. The proteasome in-hibitor MG-132 (20 �M) was added in the last hour of culture. Lysateswere immunoprecipitated with anti-HA and probed with anti-FLAG,anti-HA, or anti-heavy chain immunoglobulin. N, Neo control; GH,Glut1/HK1.
4334 ZHAO ET AL. MOL. CELL. BIOL.
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

GSK-3 levels in control cells (Fig. 7A), possibly due to com-pensatory increases in PKC activity similar to those of otherkinase inhibitors. To further examine a role for PKC isoformsin phospho-GSK-3�/�, control and Glut1/HK1 cells were sub-jected to treatment with a variety of PKC inhibitors with dif-fering specificities (Fig. 7A). With the exception of Go6983and Rottlerin, which inhibit primarily conventional PKCs andPKC, respectively, each provided at least partial inhibition ofGSK-3 phosphorylation, suggesting that PKC isoforms mayplay prominent roles in phosphorylation of GSK-3 in responseto enhanced glucose metabolism.
Diacylglycerol acts as secondary messenger for conventionaland novel PKC activation, and its level and distribution can be
altered by glucose metabolism (28, 38, 39). To determine ifincreased glucose metabolism affected diacylglycerol to pro-mote PKC activity, cells were transfected with a diacylglycerolbinding reporter construct. Control and Glut1/HK1 cells weretransfected with a diacylglycerol binding domain of the PKC-related kinase PKD (PKC�)-CRD fused to GFP (PKD-CRD-GFP) or with a mutant in which the PKD-CRD was altered toattenuate its ability to bind diacylglycerol (PKD-CRD-GFPP155/287G). Cell membrane localization of PKD-CRD-GFPwas significantly higher in Glut1/HK1 cells than in controlcells, whereas no difference was observed in PKD-CRD-GFPP155/287G-transfected cells (representative confocal imagesare shown in Fig. 7B, and quantitation from three independent
FIG. 5. GSK-3 phosphorylation is increased in Glut1- and HK1-expressing cells and regulates both Mcl-1 degradation and cell death.(A) Mcl-1-expressing control, Glut1, or Glut1/HK1 cells were analyzed for phospho-S159 and total Mcl-1. Ratios are shown. (B) Multiple FL5.12clones expressing Glut1 and/or HK1 were analyzed for phospho-Akt, phospho-GSK-3, and total GSK-3� by immunoblotting. (C) Control andGlut1/HK1 FL5.12, 32D, and BAF3 cells were cultured in IL-3 or withdrawn from IL-3 for 6 hours, and phospho-GSK-3 and total GSK-3� weredetermined by immunoblotting. Ratios are shown. (D and E) Cells were treated with GSK-3 inhibitor (10 �M SB216763), followed byimmunoblotting for Mcl-1 and actin (ratios are shown) (D) and determination of cell viability upon IL-3 withdrawal (E). (F and G) FL5.12 cellswere transiently transfected with shRNAi for GFP or GSK-3�. Cells were analyzed by immunoblotting for GSK-3�, Mcl-1, and actin in thepresence or absence of IL-3 (ratios are shown) (F), and cell survival upon IL-3 withdrawal was determined by PI exclusion (G). Values are meansfrom triplicate samples; error bars indicate standard deviations. M, hMcl-1; N, Neo control; G, Glut1; H, HK1; GH, Glut1/HK1.
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4335
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
experiments is shown in Fig. 7C). These data indicate thatchronically elevated glucose metabolism in Glut1/HK1 cellsmay increase or alter localization of diacylglycerol and providea possible mechanism for altered PKC localization or activa-tion.
To determine if enhanced PKC activity in Glut1/HK1 cellswas required for antiapoptotic glucose signaling and increasedsurvival, we examined the effect of PKC inhibition in cell deathupon IL-3 withdrawal. Glut1/HK1 cells were withdrawn fromIL-3 and were untreated or treated with PKC inhibitor aloneor with GSK-3 inhibitor, and Mcl-1 levels were determined byimmunoblotting (Fig. 7D). Inhibition of PKC led to a sharpreduction in Mcl-1 protein levels that simultaneous inhibitionof GSK-3 could partially rescue. In addition, inhibition of PKCprevented Glut1/HK1-mediated protection from cell deathand reduced the survival of IL-3-withdrawn Glut1/HK1 cells toa level equivalent to that of control cells (Fig. 7E). This loss ofGlut1/HK1-mediated protection appeared to be due to activa-tion of GSK-3, as increases in the rate of cell death caused byPKC inhibition could be reversed by simultaneous inhibition ofGSK-3. Together, these data suggest that a signaling pathwayvia PKC and GSK-3 is sensitive to glucose metabolism and isactive in highly glycolytic cells, such as growth factor-stimu-lated or cancer cells. Ultimately, this glucose-dependent sig-naling pathway protects cells from apoptosis by attenuatingdegradation of Mcl-1 and thus inhibiting toxicity from theproapoptotic proteins, such as Bim.
DISCUSSION
A decreased glucose metabolic rate and initiation of apop-tosis are sequential processes that occur when nontransformedcells are withdrawn from growth factors. Cancer cells, however,maintain both glucose metabolism and cell viability when theyare growth factor deprived (49). The relationship betweenthese two features of cancer cell physiology has been unclear.Here we show that increased glycolysis is sufficient to initiatean antiapoptotic signaling pathway to delay commitment to celldeath upon growth factor withdrawal. This antiapoptotic sig-naling pathway appears to be mediated, at least in part,through maintenance of Mcl-1 protein levels via PKC-medi-ated inhibition of GSK-3. This pathway was activated in bothcell lines and primary cells expressing Glut1 and/or hexokinase,which suggests that the coordinate upregulation of glucosemetabolic pathways that is promoted by growth factors mayplay a previously undefined yet critical role in the survival ofgrowth factor-stimulated or cancer cells.
Nutrient-induced signaling pathways have been shown toplay important roles in many aspects of cell physiology. ThemTOR/Raptor protein complex has been clearly implicated inamino acid sensing for regulation of protein synthesis, cellgrowth, and autophagy (30). Metabolic stress, such as mayoccur when ATP levels become depleted, can promote activa-tion of AMP kinase via increased intracellular levels of AMP,which can both inhibit protein synthesis (23) and activate p53
FIG. 6. Glut1 expression increases GSK-3 phosphorylation, Mcl-1 level, and survival in primary hematopoietic cells. (A) Bone marrow cellswere cultured in IL-3, infected with retrovirus to express Glut1 and truncated hNGFR as a marker on day 4, and stained by immunofluorescencefor phospho-GSK-3 (red), hNGFR (green), and DAPI on day 7. (B to E) Glut1 protein levels were analyzed by immunoblotting (B), glucose uptakewas measured (C), and phospho-GSK-3 levels were determined by immunoblotting (D) and quantified (E) in purified T lymphocytes fromnontransgenic (Non) and Glut1-transgenic (Glut1) littermates freshly after purification or after 1 day of culture in the absence of stimulation(neglect). (F) Mcl-1 levels were determined by immunoblotting of freshly isolated, neglected, or GSK-3 inhibitor (10 �M SB216763 [SB])-treatednontransgenic T cells. Ratios are shown. (G) Mcl-1/actin ratio quantitated from nontransgenic and Glut1-transgenic T cells after isolation (fresh),neglect, or culture in GSK-3 inhibitor (SB). (H) Purified T cells from nontransgenic and Glut1-transgenic littermate mice were cultured withoutstimulation (neglect), and cell survival determined after 2 days. Values shown are the means from triplicate samples; error bars indicate standarddeviations.
4336 ZHAO ET AL. MOL. CELL. BIOL.
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

to inhibit cell cycle progression (25). Here we provide evidenceof a novel glucose-sensitive signaling pathway. This pathwaywas stimulated by expression of Glut1 and/or HK1 and ap-peared to require glucose catabolism, as the nonhydrolyzableglucose analog 2DOG was incapable of providing this glucose-induced survival signal. Elevated glucose metabolism led tophosphorylation of GSK-3�/� on serines 21 and 9 in cells tomaintain Mcl-1 protein levels and inhibit Bim-induced apop-tosis in hematopoietic cells. While PKC activation in responseto hyperglycemia has been thought to play a potential anti-apoptotic role (1, 28, 38, 39), to our knowledge this is the firstdescription of a similar pathway in cells with intrinsically highglucose uptake, such as growth factor-stimulated or cancercells, and the first to connect this pathway to GSK-3 and theBcl-2 family of proteins.
The approach utilized here to examine the effects of glucosemetabolism on cell death in the absence of other signaling oroncogenic events revealed a pathway that may be of broadimportance in both cell survival and growth. While the anti-apoptotic effects of Glut1 and HK1 expression were not longterm, this survival advantage nevertheless provided a signifi-cant clonogenic survival advantage over control cells (Fig. 1C)and likely underestimated the overall role that sustained glu-cose utilization may play in preventing cell death. Indeed,cytokine withdrawal ultimately leads to the loss of expression
of both glycolytic and pentose phosphate pathway enzymes,which is not prevented by expression of Glut1 and HK1 (datanot shown and reference 44). Thus, in contrast to the case forgrowth factor-stimulated or cancer cells, which coordinatelyupregulate and maintain entire metabolic pathways (49), ex-pression of Glut1 and HK1 alone allows only a temporaryincrease in glucose metabolism after IL-3 withdrawal. In addi-tion, while this analysis focused on the effects of this glucosesignaling pathway on hematopoietic cell death, PKC isoformsand GSK-3 have multiple functions, and glucose-stimulatedPKC activity and GSK-3 inhibition may affect other aspects ofcell survival and growth to promote cancer progression orproliferation of growth factor-stimulated cells of multiple tis-sues. Addressing the contribution of glucose-stimulated signal-ing will be an important consideration for future studies ofgrowth and survival of cancer cells as well as of stimulatedlymphocytes, which show a robust increase in glucose uptakeand utilization after antigenic stimulation (2, 13, 17).
There has been additional evidence to suggest that glucose-mediated pathways may regulate apoptosis. In hepatocytes andpancreatic �-cells, glucokinase may reside in a protein complexwith the BH3-only protein Bad, where Bad phosphorylation byPKA can regulate glucose-stimulated insulin secretion and celldeath induced by glucose deprivation (11). It has also recentlybeen shown that NADPH levels can regulate phosphorylation
FIG. 7. PKC activity is necessary for GSK-3 phosphorylation and may be stimulated by altered diacylglycerol distribution in Glut1/HK1 cells.(A) Control and Glut1/HK1 FL5.12 cells were cultured in IL-3 in the presence of various kinase inhibitors (PKC, 10 �M Ro31-8220 [Ro]; PI3K,10 �M LY294002 [LY]; PKA, 10 �M H89; CamK II, 10 �M KN93; mTOR, 25 nM rapamycin [Rap]; PKC, 10 �M GF 109203X; PKC, 2 �MGo6976; PKC, 10 �M Go 6983; PKC, 20 �M Rottlerin [Rott]) for 1 h, and phospho-GSK-3 levels were determined by immunoblotting. (B andC) Control and Glut1/HK1 cells transiently transfected to express PKD-CRD-GFP or PKD-CDR-GFP P155/287G were imaged by confocalmicroscopy (B), and cells with membrane-targeted GFP were blindly quantified (means and standard deviations from three independentexperiments are shown) (C). (D) Glut1/HK1 cells were withdrawn from IL-3 for 3 hours in vehicle alone (Ctrl), PKC inhibitor (2 �M Ro31-8220[Ro]), or PKC inhibitor plus GSK-3 inhibitor (10 �M SB216763 [SB]), and Mcl-1 and actin levels were detected by immunoblotting. Ratios areshown. (E) Control and Glut1/HK1 cells were withdrawn from IL-3 and cultured in the absence or presence of PKC (10 �M Ro31-8220 [Ro]) orGSK-3 (10 �M SB216763 [SB]) inhibitor, and cell survival was observed over time. Ratios are shown. N, Neo control; GH, Glut1/HK1.
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4337
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

of caspase 2 and mitochondrial release of cytochrome c viaCamK II in Xenopus egg extracts (33). Each of these models isnot mutually exclusive with the antiapoptotic glucose-signalingpathway proposed here. In particular, NADPH levels may playa key role in metabolic signaling to PKC if acyl coenzyme Asynthesis is required for de novo diacylglycerol generation. Aswe did not, however, observe any effect of PKA or CamK IIinhibition on phospho-GSK-3 levels in Glut1/HK1 cells andGlut1/HK1 expression increased clonogenicity upon IL-3 with-drawal, our results likely identify a novel PKC-mediated glu-cose signaling pathway. It is also possible that Glut1/HK1 in-hibited cell death in part by enhanced availability of metabolicsubstrates and generation of cytosolic NADH and ATP thatmay be cytoprotective as energy sources. The glucose-initiatedsignaling pathway described here may act in concert with andin the context of each of these other pathways and directmetabolic effects to prevent cell death of glycolytic cells.
Altered diacylglycerol levels or distribution and preventionof GSK-3 phosphorylation by PKC inhibitors suggest that thisnutrient-signaling pathway is initiated by glucose-mediated ef-fects on diacylglycerol and PKC localization or activity. Con-ventional and novel PKC isoforms are regulated by phosphor-ylation, calcium, and diacylglycerol (51). While they are notcalcium or diacylglycerol regulated, atypical PKCs (PKC andPKC�) are regulated by other kinases, including PKCs (61), orlipids (37, 48). In principle, increased glucose metabolism mayaffect intracellular calcium levels or kinase and phosphatasepathways that promote PKC activity. Using a biosensor fordiacylglycerol, we show here that increased glucose metabo-lism leads to altered diacylglycerol levels or localization. In-creased extracellular levels of glucose have been shown toenhance de novo synthesis of diacylglycerol in a variety of celltypes, and this can stimulate activity of multiple PKC isoforms(38, 54, 57) that can be protective in both cardiac and neuronalischemia (29, 46). Basal de novo generation of diacylglyceroland related lipids depends on both glucose and the availabilityor synthesis of fatty acids and thus incorporates several aspectsof cell metabolism to indicate nutrient status. This pathwaymay, therefore, be ideally situated to regulate nutrient-stimu-lated survival and may regulate basal PKC activity not just incells in the presence of high extracellular glucose but also incells with high intrinsic glucose uptake and utilization.
For decades it has been known that cancer cells have in-creased rates of glycolysis and that growth factors and lympho-cyte activation stimulate glucose metabolism. Due to mutationor activation of specific transcriptional programs that may af-fect apoptosis, however, it has been impossible to discern theeffects of glycolysis on apoptosis. In this study, we directlyanalyzed the effects of increased glucose utilization on apop-tosis by expressing only essential glycolytic genes in nontrans-formed cell lines and primary lymphocytes. Our results dem-onstrate the existence of a novel antiapoptotic signalingpathway stimulated by glucose metabolism that can set athreshold for apoptotic death. While regulation of Mcl-1 levelsappeared to be the critical regulator of cell death in the sys-tems used here, GSK-3 has also been shown to influence avariety of targets to regulate cell function and fate (26). Thus,glucose-regulated PKC activation and inhibition of GSK-3 mayaffect a variety of cell types through distinct mechanisms. Thisstudy indicates that the long-acknowledged glycolytic metabo-
lism of cancer cells and activated lymphocytes may directlyinfluence cell survival and reveals a novel pathway for possibletherapeutic intervention.
ACKNOWLEDGMENTS
We thank Sally Kornbluth (Duke University), Christopher Newgard(Duke University), Leta Nutt (Duke University), David R. Plas (Uni-versity of Cincinatti), W. Kimryn Rathmell (University of North Caro-lina at Chapel Hill), Dennis Thiele (Duke University), Craig B.Thompson (University of Pennsylvania), and Tso-Pang Yao (DukeUniversity) for helpful comments. We thank the Sarah W. StedmanCenter for Nutrition and Metabolism for technical assistance. Glut1-transgenic mice were a generous gift of Craig Thompson and theAbramson Family Cancer Research Institute (University of Pennsyl-vania). We are grateful for reagents provided by H. J. Brady (Univer-sity College, London), D. Turner (University of Michigan), M. Spitalerand D. Cantrell (University of Dundee), M. Ehlers (Duke University),U. Maurer (Institut fur Molekulare Medizin und Zellforschung), andD. Green (St. Jude Children’s Research Hospital).
This work was funded by a Howard Temin KO1 Career Develop-ment Award from the National Cancer Institute (to J.C.R.), a SidneyKimmel Foundation for Cancer Research Scholar Award (to J.C.R.),a V Foundation for Cancer Research Scholar Award (to J.C.R.), andR01 AI063345 (to J.C.R.).
REFERENCES
1. Babazono, T., J. Kapor-Drezgic, J. A. Dlugosz, and C. Whiteside. 1998.Altered expression and subcellular localization of diacylglycerol-sensitiveprotein kinase C isoforms in diabetic rat glomerular cells. Diabetes 47:668–676.
2. Bental, M., and C. Deutsch. 1993. Metabolic changes in activated T cells: anNMR study of human peripheral blood lymphocytes. Magn. Reson. Med.29:317–326.
3. Bentley, J., D. Itchayanan, K. Barnes, E. McIntosh, X. Tang, C. P. Downes,G. D. Holman, A. D. Whetton, P. J. Owen-Lynch, and S. A. Baldwin. 2003.Interleukin-3-mediated cell survival signals include phosphatidylinositol3-kinase-dependent translocation of the glucose transporter GLUT1 to thecell surface. J. Biol. Chem. 278:39337–39348.
4. Bentley, J., I. Walker, E. McIntosh, A. D. Whetton, P. J. Owen-Lynch, andS. A. Baldwin. 2001. Glucose transport regulation by p210 Bcr-Abl in achronic myeloid leukaemia model. Br. J. Haematol. 112:212–215.
5. Boise, L. H., M. Gonzalez-Garcia, C. E. Postema, L. Ding, T. Lindsten, L. A.Turka, X. Mao, G. Nunez, and C. B. Thompson. 1993. bcl-x, a bcl-2 relatedgene that functions as a dominant regulator of apoptotic cell death. Cell74:597–608.
6. Bouillet, P., D. Metcalf, D. C. Huang, D. M. Tarlinton, T. W. Kay, F.Kontgen, J. M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relativeBim required for certain apoptotic responses, leukocyte homeostasis, and topreclude autoimmunity. Science 286:1735–1738.
7. Chi, M. M., J. Pingsterhaus, M. Carayannopoulos, and K. H. Moley. 2000.Decreased glucose transporter expression triggers BAX-dependent apopto-sis in the murine blastocyst. J. Biol. Chem. 275:40252–40257.
8. Ciofani, M., and J. C. Zuniga-Pflucker. 2005. Notch promotes survival ofpre-T cells at the beta-selection checkpoint by regulating cellular metabo-lism. Nat. Immunol. 6:881–888.
9. Conlon, I., and M. Raff. 1999. Size control in animal development. Cell96:235–244.
10. Dang, C. V. 1999. c-Myc target genes involved in cell growth, apoptosis, andmetabolism. Mol. Cell. Biol. 19:1–11.
11. Danial, N. N., C. F. Gramm, L. Scorrano, C. Y. Zhang, S. Krauss, A. M.Ranger, S. R. Datta, M. E. Greenberg, L. J. Licklider, B. B. Lowell, S. P.Gygi, and S. J. Korsmeyer. 2003. BAD and glucokinase reside in a mito-chondrial complex that integrates glycolysis and apoptosis. Nature 424:952–956.
12. Danial, N. N., and S. J. Korsmeyer. 2004. Cell death: critical control points.Cell 116:205–219.
13. Doughty, C. A., B. F. Bleiman, D. J. Wagner, F. J. Dufort, J. M. Mataraza,M. F. Roberts, and T. C. Chiles. 2006. Antigen receptor-mediated changes inglucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinasesignaling in the glycolytic control of growth. Blood 107:4458–4465.
14. Edinger, A. L., and C. B. Thompson. 2002. Akt maintains cell size andsurvival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell13:2276–2288.
15. Fox, C. J., P. S. Hammerman, R. M. Cinalli, S. R. Master, L. A. Chodosh,and C. B. Thompson. 2003. The serine/threonine kinase Pim-2 is a transcrip-tionally regulated apoptotic inhibitor. Genes Dev. 17:1841–1854.
16. Frauwirth, K. A., J. L. Riley, M. H. Harris, R. V. Parry, J. C. Rathmell, D. R.
4338 ZHAO ET AL. MOL. CELL. BIOL.
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from

Plas, R. L. Elstrom, C. H. June, and C. B. Thompson. 2002. The CD28signaling pathway regulates glucose metabolism. Immunity 16:769–777.
17. Frauwirth, K. A., and C. B. Thompson. 2004. Regulation of T lymphocytemetabolism. J. Immunol. 172:4661–4665.
18. Gambhir, S. S. 2002. Molecular imaging of cancer with positron emissiontomography. Nat. Rev. Cancer 2:683–693.
19. Gatenby, R. A., and R. J. Gillies. 2004. Why do cancers have high aerobicglycolysis? Nat. Rev. Cancer 4:891–899.
20. Gottlob, K., N. Majewski, S. Kennedy, E. Kandel, R. B. Robey, and N. Hay.2001. Inhibition of early apoptotic events by Akt/PKB is dependent on thefirst committed step of glycolysis and mitochondrial hexokinase. Genes Dev.15:1406–1418.
21. Hanahan, D., and R. A. Weinberg. 2000. The hallmarks of cancer. Cell100:57–70.
22. Henquin, J. C. 2000. Triggering and amplifying pathways of regulation ofinsulin secretion by glucose. Diabetes 49:1751–1760.
23. Inoki, K., T. Zhu, and K. L. Guan. 2003. TSC2 mediates cellular energyresponse to control cell growth and survival. Cell 115:577–590.
24. Jensen, M. V., J. W. Joseph, O. Ilkayeva, S. Burgess, D. Lu, S. M. Ronne-baum, M. Odegaard, T. C. Becker, A. D. Sherry, and C. B. Newgard. 2006.Compensatory responses to pyruvate carboxylase suppression in islet beta-cells: preservation of glucose-stimulated insulin secretion. J. Biol. Chem.281:22342–22351.
25. Jones, R. G., D. R. Plas, S. Kubek, M. Buzzai, J. Mu, Y. Xu, M. J. Birnbaum,and C. B. Thompson. 2005. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18:283–293.
26. Jope, R. S., and G. V. Johnson. 2004. The glamour and gloom of glycogensynthase kinase-3. Trends Biochem. Sci. 29:95–102.
27. Khaled, A. R., and S. K. Durum. 2002. Lymphocide: cytokines and thecontrol of lymphoid homeostasis. Nat. Rev. Immunol. 2:817–830.
28. Koya, D., and G. L. King. 1998. Protein kinase C activation and the devel-opment of diabetic complications. Diabetes 47:859–866.
29. Malliopoulou, V., C. Xinaris, I. Mourouzis, A. D. Cokkinos, N. Katsilambros, C.Pantos, E. Kardami, and D. V. Cokkinos. 2006. High glucose protects embry-onic cardiac cells against simulated ischemia. Mol. Cell. Biochem. 284:87–93.
30. Martin, D. E., and M. N. Hall. 2005. The expanding TOR signaling network.Curr. Opin. Cell Biol. 17:158–166.
31. Maurer, U., C. Charvet, A. S. Wagman, E. Dejardin, and D. R. Green. 2006.Glycogen synthase kinase-3 regulates mitochondrial outer membrane per-meabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21:749–760.
32. Morissette, M. R., A. L. Howes, T. Zhang, and J. Heller Brown. 2003.Upregulation of GLUT1 expression is necessary for hypertrophy and survivalof neonatal rat cardiomyocytes. J. Mol. Cell Cardiol. 35:1217–1227.
33. Nutt, L. K., S. S. Margolis, M. Jensen, C. E. Herman, W. G. Dunphy, J. C.Rathmell, and S. Kornbluth. 2005. Metabolic regulation of oocyte cell deaththrough the CaMKII-mediated phosphorylation of caspase-2. Cell 123:89–103.
34. Oishi, K., S. Kamakura, Y. Isazawa, T. Yoshimatsu, K. Kuida, M. Nakafuku,N. Masuyama, and Y. Gotoh. 2004. Notch promotes survival of neural pre-cursor cells via mechanisms distinct from those regulating neurogenesis.Dev. Biol. 276:172–184.
35. Opferman, J. T., H. Iwasaki, C. C. Ong, H. Suh, S. Mizuno, K. Akashi, andS. J. Korsmeyer. 2005. Obligate role of anti-apoptotic MCL-1 in the survivalof hematopoietic stem cells. Science 307:1101–1104.
36. Opferman, J. T., A. Letai, C. Beard, M. D. Sorcinelli, C. C. Ong, and S. J.Korsmeyer. 2003. Development and maintenance of B and T lymphocytesrequires antiapoptotic MCL-1. Nature 426:671–676.
37. Pandian, S. S., A. A. Sneddon, C. S. Bestwick, S. McClinton, I. Grant, K. W.Wahle, and S. D. Heys. 2001. Fatty acid regulation of protein kinase Cisoforms in prostate cancer cells. Biochem. Biophys. Res. Commun. 283:806–812.
38. Peter-Riesch, B., M. Fathi, W. Schlegel, and C. B. Wollheim. 1988. Glucoseand carbachol generate 1,2-diacylglycerols by different mechanisms in pan-creatic islets. J. Clin. Investig. 81:1154–1161.
39. Pinton, P., T. Tsuboi, E. K. Ainscow, T. Pozzan, R. Rizzuto, and G. A. Rutter.2002. Dynamics of glucose-induced membrane recruitment of protein kinaseC beta II in living pancreatic islet beta-cells. J. Biol. Chem. 277:37702–37710.
40. Plas, D. R., J. C. Rathmell, and C. B. Thompson. 2002. Homeostatic controlof lymphocyte survival: potential origins and implications. Nat. Immunol.3:515–521.
41. Puthalakath, H., D. C. Huang, L. A. O’Reilly, S. M. King, and A. Strasser.1999. The proapoptotic activity of the Bcl-2 family member Bim is regulatedby interaction with the dynein motor complex. Mol. Cell 3:287–296.
42. Raff, M. C. 1992. Social controls on cell survival and cell death. Nature356:397–400.
43. Rathmell, J. C., R. L. Elstrom, R. M. Cinalli, and C. B. Thompson. 2003.Activated Akt promotes increased resting T cell size, CD28-independent Tcell growth, and development of autoimmunity and lymphoma. Eur. J. Im-munol. 33:2223–2232.
44. Rathmell, J. C., C. J. Fox, D. R. Plas, P. Hammerman, R. M. Cinalli, andC. B. Thompson. 2003. Akt-directed glucose metabolism can prevent Baxconformation change and promote growth factor-independent survival. Mol.Cell. Biol. 23:7315–7328.
45. Rathmell, J. C., M. G. Vander Heiden, M. H. Harris, K. A. Frauwirth, andC. B. Thompson. 2000. In the absence of extrinsic signals, nutrient utilizationby lymphocytes is insufficient to maintain either cell size or viability. Mol.Cell 6:683–692.
46. Raval, A. P., K. R. Dave, D. Mochly-Rosen, T. J. Sick, and M. A. Perez-Pinzon. 2003. Epsilon PKC is required for the induction of tolerance byischemic and NMDA-mediated preconditioning in the organotypic hip-pocampal slice. J. Neurosci. 23:384–391.
47. Ravikumar, B., A. Stewart, H. Kita, K. Kato, R. Duden, and D. C. Rubin-sztein. 2003. Raised intracellular glucose concentrations reduce aggregationand cell death caused by mutant huntingtin exon 1 by decreasing mTORphosphorylation and inducing autophagy. Hum. Mol. Genet. 12:985–994.
48. Selbie, L. A., C. Schmitz-Peiffer, Y. Sheng, and T. J. Biden. 1993. Molecularcloning and characterization of PKC iota, an atypical isoform of proteinkinase C derived from insulin-secreting cells. J. Biol. Chem. 268:24296–24302.
49. Semenza, G. L., D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E.Laughner, R. Ravi, J. Simons, P. Taghavi, and H. Zhong. 2001. The metab-olism of tumours: 70 years later. Novartis Found Symp. 240:251–260, 260–264.
50. Smith, T. A. 2000. Mammalian hexokinases and their abnormal expression incancer. Br. J. Biomed. Sci. 57:170–178.
51. Tan, S. L., and P. J. Parker. 2003. Emerging and diverse roles of proteinkinase C in immune cell signalling. Biochem. J. 376:545–552.
52. Valverde, A. M., P. Navarro, M. Benito, and M. Lorenzo. 1998. H-ras inducesglucose uptake in brown adipocytes in an insulin- and phosphatidylinositol3-kinase-independent manner. Exp. Cell Res. 243:274–281.
53. Vander Heiden, M. G., D. R. Plas, J. C. Rathmell, C. J. Fox, M. H. Harris,and C. B. Thompson. 2001. Growth factors can influence cell growth andsurvival through effects on glucose metabolism. Mol. Cell. Biol. 21:5899–5912.
54. Vary, T. C., and C. J. Lynch. 2006. Meal feeding stimulates phosphorylationof multiple effector proteins regulating protein synthetic processes in rathearts. J. Nutr. 136:2284–2290.
55. Warburg, O. 1956. On the origin of cancer cells. Science 123:309–314.56. Waterhouse, N. J., J. C. Goldstein, O. von Ahsen, M. Schuler, D. D. Newmeyer,
and D. R. Green. 2001. Cytochrome c maintains mitochondrial transmembranepotential and ATP generation after outer mitochondrial membrane permeabi-lization during the apoptotic process. J. Cell Biol. 153:319–328.
57. Whiteside, C. I., and J. A. Dlugosz. 2002. Mesangial cell protein kinase Cisozyme activation in the diabetic milieu. Am. J. Physiol. Renal Physiol.282:F975–F980.
58. Wullschleger, S., R. Loewith, and M. N. Hall. 2006. TOR signaling in growthand metabolism. Cell 124:471–484.
59. Younes, M., R. W. Brown, M. Stephenson, M. Gondo, and P. T. Cagle. 1997.Overexpression of Glut1 and Glut3 in stage I nonsmall cell lung carcinomais associated with poor survival. Cancer 80:1046–1051.
60. Yu, J. Y., J. Taylor, S. L. DeRuiter, A. B. Vojtek, and D. L. Turner. 2003.Simultaneous inhibition of GSK3alpha and GSK3beta using hairpin siRNAexpression vectors. Mol. Ther. 7:228–236.
61. Zhang, J., P. Z. Anastasiadis, Y. Liu, E. A. Thompson, and A. P. Fields. 2004.Protein kinase C (PKC) betaII induces cell invasion through a Ras/Mek-,PKC iota/Rac 1-dependent signaling pathway. J. Biol. Chem. 279:22118–22123.
62. Zhong, Q., W. Gao, F. Du, and X. Wang. 2005. Mule/ARF-BP1, a BH3-onlyE3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulatesapoptosis. Cell 121:1085–1095.
VOL. 27, 2007 GSK-3 MEDIATES ANTIAPOPTOTIC GLUCOSE SIGNALING 4339
on March 24, 2014 by guest
http://mcb.asm
.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















