
Gene sequence and crystal structure of the aldehyde oxidoreductase from Desulfovibrio desulfuricans...
12
Gene Sequence and Crystal Structure of the Aldehyde Oxidoreductase from Desulfovibrio desulfuricans ATCC 27774 Jorge Rebelo 1,2 , Sofia Macieira 1,3 , Joa ˜o M. Dias 1,2 , Robert Huber 3 Carla S. Ascenso 1,4 , Frank Rusnak 4 , Jose ´ J. G. Moura 1 , Isabel Moura 1 and Maria J. Roma ˜o 1,2 * 1 Departamento de Quı ´mica Centro de Quı ´mica Fina e Biotecnologia, Faculdade de Cie ˆncias e Tecnologia Universidade Nova de Lisboa 2825-114, Caparica, Portugal 2 ITQB, Apt. 127, 2780, Oeiras Portugal 3 Max-Planck-Institut fu ¨r Biochemie, am Klopferspitz 18a D-82152, Martinsried Germany 4 Department of. Biochemistry and Molecular Biology, Mayo Clinic and Foundation Rochester, MN, 55905, USA The aldehyde oxidoreductase (MOD) isolated from the sulfate reducer Desulfovibrio desulfuricans (ATCC 27774) is a member of the xanthine oxi- dase family of molybdenum-containing enzymes. It has substrate speci- ficity similar to that of the homologous enzyme from Desulfovibrio gigas (MOP) and the primary sequences from both enzymes show 68 % identity. The enzyme was crystallized in space group P6 1 22, with unit cell dimen- sions of a b 156.4 A ˚ and c 177.1 A ˚ , and diffraction data were obtained to beyond 2.8 A ˚ . The crystal structure was solved by Patterson search techniques using the coordinates of the D. gigas enzyme. The overall fold of the D. desulfuricans enzyme is very similar to MOP and the few differences are mapped to exposed regions of the molecule. This is reflected in the electrostatic potential surfaces of both homologous enzymes, one exception being the surface potential in a region identifiable as the putative docking site of the physiological electron acceptor. Other essential features of the MOP structure, such as residues of the active-site cavity, are basically conserved in MOD. Two mutations are located in the pocket bearing a chain of catalytically relevant water molecules. As deduced from this work, both these enzymes are very closely related in terms of their sequences as well as 3D structures. The comparison allowed confirmation and establishment of features that are essential for their function; namely, conserved residues in the active-site, catalytically relevant water molecules and recognition of the physiological electron acceptor docking site. # 2000 Academic Press Keywords: molybdopterin; aldehyde-oxidoreductase; crystallography; molybdo enzymes *Corresponding author Introduction Molybdenum enzymes are an important class of proteins that mediate the metabolism of nitrogen, carbon and sulfur and includes two enzyme groups, nitrogenases and oxotransferases (also designated as oxomolybdenum or mononuclear molybdenum enzymes) (Hille, 1996; Roma ˜o et al., 1997a). In crude cell extracts prepared from sulfate redu- cers, mononuclear molybdenum containing pro- teins with a wide range of functions are present (Moura & Barata, 1994). Examples are nitrate reductase, an enzyme catalyzing the first step in the conversion of nitrate (through nitrite) to ammo- nia (Bursakov et al., 1995; Dias et al., 1999), formate dehydrogenase, an enzyme able to convert formate to CO 2 (Costa et al., 1997) and aldehyde oxido- reductases that convert aldehydes to carboxylic acids (Barata et al., 1993). These enzymes function E-mail address of the corresponding author: [email protected] Abbreviations used: MCD, molybdopterin cytosine dinucleotide; MGD, molybdopterin guanine dinucleotide; MOP, aldehyde oxidoreductase from Desulfovibrio gigas; MOD, aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774; MPT, mononucleotide form of molybdopterin; FAD, flavin adenine dinucleotide; Moco, pyranopterin-1,2-ene- dithiolate cofactor or molybdopterin; CODH, carbon monoxide dehydhrogenase; XDH, xanthine dehydhrogenase. doi:10.1006/jmbi.2000.3552 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 297, 135–146 0022-2836/00/010135–12 $35.00/0 # 2000 Academic Press
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Gene sequence and crystal structure of the aldehyde oxidoreductase from Desulfovibrio desulfuricans...

doi:10.1006/jmbi.2000.3552 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 297, 135±146
Gene Sequence and Crystal Structure of the AldehydeOxidoreductase from Desulfovibrio desulfuricansATCC 27774
Jorge Rebelo1,2, Sofia Macieira1,3, JoaÄo M. Dias1,2, Robert Huber3
Carla S. Ascenso1,4, Frank Rusnak4, Jose J. G. Moura1, Isabel Moura1
and Maria J. RomaÄo1,2*
1Departamento de QuõÂmicaCentro de QuõÂmica Fina eBiotecnologia, Faculdade deCieÃncias e TecnologiaUniversidade Nova de Lisboa2825-114, Caparica, Portugal2ITQB, Apt. 127, 2780, OeirasPortugal3Max-Planck-Institut fuÈ rBiochemie, am Klopferspitz 18aD-82152, MartinsriedGermany4Department of. Biochemistryand Molecular Biology, MayoClinic and FoundationRochester, MN, 55905, USA
E-mail address of the [email protected]
Abbreviations used: MCD, moldinucleotide; MGD, molybdopterdinucleotide; MOP, aldehyde oxidDesulfovibrio gigas; MOD, aldehydDesulfovibrio desulfuricans ATCC 2mononucleotide form of molybdoadenine dinucleotide; Moco, pyradithiolate cofactor or molybdoptemonoxide dehydhrogenase; XDHdehydhrogenase.
0022-2836/00/010135±12 $35.00/0
The aldehyde oxidoreductase (MOD) isolated from the sulfate reducerDesulfovibrio desulfuricans (ATCC 27774) is a member of the xanthine oxi-dase family of molybdenum-containing enzymes. It has substrate speci-®city similar to that of the homologous enzyme from Desulfovibrio gigas(MOP) and the primary sequences from both enzymes show 68 % identity.The enzyme was crystallized in space group P6122, with unit cell dimen-sions of a � b � 156.4 AÊ and c � 177.1 AÊ , and diffraction data wereobtained to beyond 2.8 AÊ . The crystal structure was solved by Pattersonsearch techniques using the coordinates of the D. gigas enzyme. The overallfold of the D. desulfuricans enzyme is very similar to MOP and the fewdifferences are mapped to exposed regions of the molecule. This is re¯ectedin the electrostatic potential surfaces of both homologous enzymes, oneexception being the surface potential in a region identi®able as the putativedocking site of the physiological electron acceptor. Other essential featuresof the MOP structure, such as residues of the active-site cavity, are basicallyconserved in MOD. Two mutations are located in the pocket bearing achain of catalytically relevant water molecules.
As deduced from this work, both these enzymes are very closely relatedin terms of their sequences as well as 3D structures. The comparisonallowed con®rmation and establishment of features that are essential fortheir function; namely, conserved residues in the active-site, catalyticallyrelevant water molecules and recognition of the physiological electronacceptor docking site.
# 2000 Academic Press
Keywords: molybdopterin; aldehyde-oxidoreductase; crystallography;molybdo enzymes
*Corresponding authorIntroduction
Molybdenum enzymes are an important class ofproteins that mediate the metabolism of nitrogen,
ding author:
ybdopterin cytosinein guanineoreductase frome oxidoreductase from7774; MPT,pterin; FAD, ¯avinnopterin-1,2-ene-rin; CODH, carbon, xanthine
carbon and sulfur and includes two enzymegroups, nitrogenases and oxotransferases (alsodesignated as oxomolybdenum or mononuclearmolybdenum enzymes) (Hille, 1996; RomaÄo et al.,1997a).
In crude cell extracts prepared from sulfate redu-cers, mononuclear molybdenum containing pro-teins with a wide range of functions are present(Moura & Barata, 1994). Examples are nitratereductase, an enzyme catalyzing the ®rst step inthe conversion of nitrate (through nitrite) to ammo-nia (Bursakov et al., 1995; Dias et al., 1999), formatedehydrogenase, an enzyme able to convert formateto CO2 (Costa et al., 1997) and aldehyde oxido-reductases that convert aldehydes to carboxylicacids (Barata et al., 1993). These enzymes function
# 2000 Academic Press

Table 1. Data collection and processing statistics
All (last shell)
Resolution (AÊ ) 24.4-2.8 (2.9-2.8)Measured reflections 190,328Unique reflections 31,573Completeness (%) 98.2 (98.0)Rmerge
a (%) 16.9 (56.1)I/s(I) 6.7 (2.4)
a Rmerge (I) � �(jI(k) ÿ hIij)/� I(k), summation over all mea-surements of individual diffraction intensity values I(k). hIirepresents the corresponding mean values.
136 D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase
by transfer of an oxygen atom to/from a physio-logical acceptor/donor molecule and, for the vastmajority, a Mo � O unit is present in their active-sites (RomaÄo et al., 1997a). The molybdenum atomis bound to an organic moiety (pyranopterin-1,2-ene-dithiolate (molydopterin-Moco)) and it is gen-erally associated with iron-sulfur centers and/or a¯avin or heme centers.
The aldehyde oxidoreductase is a major enzymecomponent in sulfate reducers (RomaÄo et al., 1997a;Moura & Barata, 1994) and was ®rst puri®ed fromDesulfovibrio gigas based on its optical absorptionspectral characteristics and enzymatic activity. Thepresence of molybdenum was ®rst detected byEPR spectroscopy (Moura et al., 1978), suggestingits biological function (Turner et al., 1987). The ®rstMo-mononuclear enzyme structure to be solvedwas the D. gigas aldehyde oxidoreductase (MOP)(RomaÄo et al., 1995). MOP belongs to the xanthineoxidase family (Hille, 1996; RomaÄo & Huber, 1998),has an MCD (molybdopterin cytosine dinucleotide)MoVI � O, � O/S,-OH2 active-site and no proteinligand to the metal. As additional cofactors, itbinds two distinct [2Fe-2S] centers, which areinvolved in intramolecular electron transfer, and,contrary to xanthine oxidase, it lacks ¯avin adeninedinucleotide (FAD) and the corresponding domain.A mechanistic proposal for the xanthine oxidasefamily of enzymes was possible on the basis ofcrystallographic studies of several inhibitor,reduced, and resulfurated forms of the D. gigasenzyme, analyzed by crystallography (Huber et al.,1996).
A homologous enzyme was subsequently puri-®ed to homogeneity from extracts of D. desulfuri-cans (ATCC 27774), a sulfate reducer that can usesulfate or nitrate as alternative terminal respiratorysubstrates. The protein was described as a homodi-mer (2 � 98 kDa subunits) with one molybdenumatom associated with one MCD cofactor, lacking¯avin and containing two [2Fe-2S] centers (unpub-lished results). Visible and EPR spectroscopy indi-cate a close similarity with the D. gigas enzymeand activity studies performed for different alde-hydes indicate a broad speci®city. We use MOP torefer to the D. gigas enzyme and MOD for theD. desulfuricans enzyme.
We report the primary sequence and crystalstructure of the D. desulfuricans aldehyde oxido-reductase (MOD) solved and re®ned at 2.8 AÊ resol-ution, which enables a comparison with thehomologous D. gigas enzyme.
Results
Crystallization and crystal characterization
Crystals of MOD were obtained by vapor diffu-sion, using ammonium sulfate at pH 6.0 as precipi-tant and dioxane as additive. These crystals belongto the space group P6122, with unit cell dimensionsa � b � 156.4 AÊ and c � 177.1 AÊ . Diffraction wasobtained to beyond 2.5 AÊ resolution. A full data
set was measured using X-rays from a rotatinganode generator operating at 4.9 kW, yielding anoverall completeness of 98.2 % to a maximum res-olution of 2.8 AÊ . The solvent content is approxi-mately 60 % (v/v) assuming one monomer of98 kDa in the asymmetric unit (Mathews, 1968).Since the protein is known to be homodimeric insolution, the two subunits must be related by acrystallographic dyad. Other important data collec-tion statistics are summarized in Table 1.
Primary sequence determination
To obtain the amino acid sequence, severalattempts were made for cloning the gene using thePCR technique. Based on the MOD N-terminalsequence obtained by Edman sequencing (Edman& Begg, 1967), and on a highly conserved regionfound in molybdopterin enzymes, primers lucky1and lucky2 (see Materials and Methods) were syn-thesized and a DNA fragment of about 250 bp wasampli®ed. Nested primers based on this sequencewere used to generate two additional PCR frag-ments encompassing 578 bp of the 50-end of theMOD gene, including 67 bases of the 50-untrans-lated region. To extend the gene sequence towardsthe C terminus, information obtained from thecrystallographic data was used for oligonucleotideconstruction. After the initial MOD model wasre®ned with X-PLOR, a careful inspection of theelectron density map clearly revealed regionswhere the amino acid sequence was conserved inrelation to MOP. The peptide 859-DGPFGA-864, atthe C terminus, was selected to construct a degen-erate oligonucleotide, MOD864R, which togetherwith the homologous primer MOD1 was used toamplify a DNA fragment of 2319 bp. Informationon the regions ¯anking this sequence was obtainedby inverse PCR (Triglia et al., 1988; Ochman et al.,1988; Silver et al., 1989), using primers LASTFWDand MOD1Rev (see Materials and Methods). Theresulting 1300 bp product was used to determinethe remaining sequence of the MOD gene.
Assembly of the overlapping sequence fragmentsallowed the reconstitution of an open readingframe of 2721 bp, coding for a 907 amino acidpolypeptide chain with a deduced molecular massof 97,964.3 Da. This value is in good agreementwith electrophoresis data.

Table 3. Final model re®ned parameters
D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase 137
Structure solution and refinement
The MOD structure was solved using the closelyrelated MOP molecule as a search model. The highdegree of homology between the structures wasclear upon inspection of the ®rst electron densitymaps, when still no primary sequence was avail-able. Even with data to only 2.8 AÊ resolution itwas possible to carefully analyze the electron den-sity maps and locate the regions of the primarysequence conserved with respect to the MOP pro-tein. In the ®rst stages of the re®nement, an X-rayderived sequence was proposed for several seg-ments highly homologous to the MOP sequenceand dubious regions were left as polyalanine. There®nement and primary sequence determinationwere carried out in parallel and the re®nementadvanced as progress was made on the knowledgeof the primary sequence, which allowed insertionof new blocks of corrected amino acid sequence(see Table 2).
The side-chains of 11 exposed amino acid resi-dues, eight lysine residues (Lys132, 168, 232, 248,301, 478, 606 and 752), two glutamate residues(Glu572 and 612) and Arg475, which were notde®ned in the electron density, were omitted fromthe ®nal re®nement and phase calculation. Sevenother amino acid side-chains that were partiallyde®ned Glu163, Glu607, Lys246, Lys603, Lys801,Arg604 and Arg608 were set to an occupancy of0.5. The detailed progress of the re®nement as wellas statistical data of the re®ned MOD model aresummarized in Tables 2 and 3, respectively.
The ®nal overall geometric quality of the modelwas determined with PROCHECK (Laskowskiet al., 1993) with 88.4 % of all residues found inmost favored regions of the Ramachandran plot,11.1 % in the additional allowed regions and 0.3 %in the generously allowed regions. As found in theMOP protein, only two residues, Tyr142 (� � 69 � � ÿ 37 �) and Leu254 (� � 62 � � ÿ 68 �) are indisallowed regions but in both cases the electrondensity is very well de®ned. The Tyr142 (in MOPand in MOD) is placed on the N terminus of helix
Table 2. Stepwise R-factor and free R-factor improve-ment (%)
R Rfree
Rigid body refinement with 100N terminus residues known
26.8 32.6
Sequenced until residue 344and chain adjustments
25.8 32.5
Sequenced until residue 614insertions not included
25.0 31.3
Bulk solvent modellinginclusion of two insertions onprimary sequence
21.6 28.3
Sequenced until residue 713conformational correctionsand solvent inclusion
18.0 25.1
Sequenced until residue 864and solvent inclusion
16.4 23.6
Primary sequence completed 16.4 22.4
IV from domain Fe/S proximal and its hydroxylgroup establishes hydrogen bonding interactionswith the amide group from Cys137 and the carbo-nyl group of Pro839. The Tyr142 is totally con-served within the xanthine oxidase family ofenzymes (Figure 1). There are two X-Pro peptidebonds that adopt a cis conformation very clear inthe electron density maps, Phe408-Pro409, in a newinserted loop and Leu895-Pro896, which is alsopresent in the MOP structure. The MOD crystalstructure has an estimated average atomic pos-itional error of 0.2 AÊ (Luzzati et al., 1952).
Discussion
Description of the structure and comparisonwith MOP
The structure determination of the aldehydeoxidoreductase from Desulfovibrio desulfuricans(ATCC 27774) was carried out at a resolution of2.8 AÊ , which is lower than what we have reportedfor the homologous protein from D. gigas (1.8 AÊ
reported by Huber et al., 1996). When comparingboth structures, non-conserved residues are homo-geneously spread over the polypeptide chain.There are two single residue insertions and twosingle residue deletions on exposed loops of thestructure. The two insertions on the MOP sequenceoccur between residues 354 and 355 (355 in MOD)and between residues 406 and 407 (408 in MOD),and two deletions between residues 750 and 751(749 in MOP) and 757 and 758 (757 in MOP). Asexpected, the MOD enzyme has the same overallglobular structure as MOP (RomaÄo et al., 1995)with 27 % a-helix and 21 % b-sheet as de®ned byPROCHECK (Laskowski et al., 1993) (Figure 2) andthe same domain organization. Domain I (Fe/S distal, residues 1-76), has a fold typical of aplant-type ferredoxin (Rypniewski et al., 1991;
Number of protein atoms 7173Number of cofactor atoms 66Number of solvent atoms 262R-factor (%) 16.4Free R-factor (3 % of total data) 22.4Number of unique reflections used
( 24.4 to 2.8 AÊ ) 30614rmsd:Bond distances (AÊ ) 0.013Bond angles (deg) 1.81
A. Average temperature factors (AÊ 2)7173 (all) atoms 30.03628 main-chain atoms 28.43545 side-chain atoms 31.656 cofactor atoms 29.6262 solvent molecules 46.4
B. Structure qualityRamachandran plot outliers (%) 0.3Unusual peptide orientations (%) 3.3Non-rotamer side-chain conformations (%) 12.9Overall G-factor 0.2
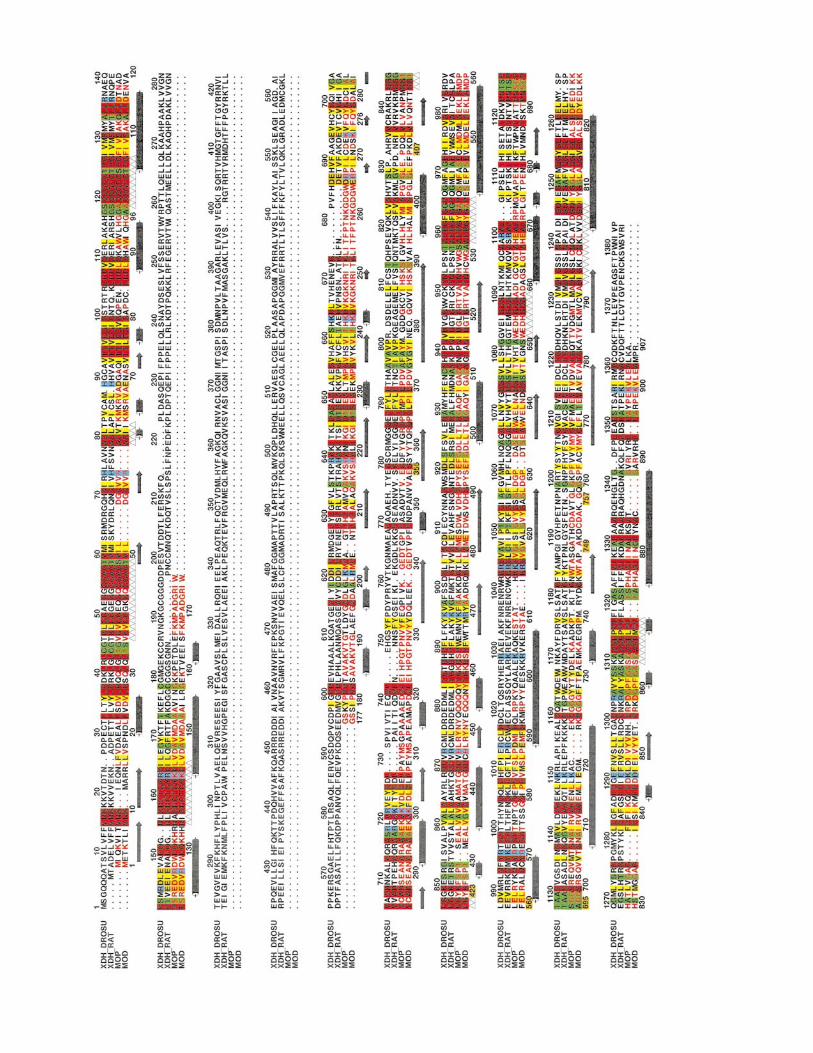

Figure 2. Stereo plot of the molecular structure of MOD with the four independent domains represented in, differ-ent colors and cofactors shown as colored spheres. I-Fe/S distal, residues 1-76, (green); II-Fe/S proximal, residues84-156, (red); connecting peptide, residues 158-195, (white); III- Mo1, residues 196-581, (yellow); IV- Mo2, residues582-907, (blue). This Figure was prepared with MOLSCRIPT (Kraulis, 1991) and Raster3D (Merrit & Murphy, 1994).
D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase 139
Fukuyama et al., 1995) with a ®ve-stranded b half-barrel plus an a-helix orthogonal to the strands,while domain II (Fe/S proximal, residues 84-156)is a four-helical bundle with the [2Fe-2S] centerbound at the N termini of the two central and lar-ger helices. These domains are involved in thebinding of the distal and proximal iron-sulfurredox centers, respectively (classi®ed with respectto their relative proximity to the molybdenumsite), and are followed by a 37 residue connectingpeptide (residues 158-195) leading to the contigu-ous third domain. Domain III (Mo1, residues 196-581) is rather elongated (ca 75 AÊ long and 28 AÊ
wide) and consists of two subdomains: a largerN-terminal part folded as a seven-stranded incom-plete b-barrel with one a-helix in its central cavityand two additional helices that ¯ank the barrel andare exposed to solvent. The smaller C-terminal sub-domain of Mo1 consists of a ®ve-stranded mixedparallel-antiparallel b-sheet ¯anked by two helicesapproximately parallel with the strand direction.Domain IV (Mo2, residues 582 to 907) is also orga-nized into two subdomains, each with a similarbasic fold consisting of a four-stranded b-sheetbent around a pair of helices. Both subdomainsresemble two large wings extending over 80 AÊ andwith the cofactor MCD lying at their intersection.The molybdenum catalytic site is located at the
Figure 1. Amino acid sequence alignment of MOD withdehydrogenases. XDH DROSU, xanthine dehydrogenaseP91711; XDH RAT, xanthine dehydrogenase from Rattus nhyde oxidoreductase from Desulfovibrio gigas, Swiss-Prot afrom Desulfovibrio desulfuricans ATCC 27774, EMBL sequewere performed with programs CLUSTALW and ALSCRIPserved hydrophobic residues, (yellow), conserved small ntively charged, brown, and positively charged, blue. Thehelices, (±&±) and the segments contacting the iron-sulfdues, (!), are shown.
extensive interface de®ned by these two domains,Mo1 and Mo2.
When we superimpose 879 common Ca atoms ofMOD and MOP, the average Ca distance is 0.59 AÊ
and the rmsd is 0.39 AÊ , which is not surprising inlight of the high degree of primary sequence iden-tity (68 %) between these enzymes (Figure 1). Thelargest differences, Ca deviations greater than 2 AÊ ,occur essentially at the C terminus and on a fewexposed loop regions (residues 66-67; 165-166, 170-172 (disordered loop), 319-320, 349-352 (one resi-due insertion in MOD), 642-644, 683-685, 722-730,750-751). The core and active-site of the moleculeare basically conserved and practically identical inboth structures. Also, the region surrounding theentrance to the funnel-like cavity that leads to theactive-site is remarkably conserved. Importantnon-polar residues at the half-length of the accesstunnel, Phe425, Phe494, Leu497 and Leu626, whichwe have described as swinging doors (RomaÄo et al.,1995), are also conserved in MOD (Phe427, Phe496,Leu499 and Leu628, respectively).
The MOD and MOP primary sequences arehomologous to eukaryotic xanthine dehydrogen-ases as well as to carbon monoxide dehydrogenase(CODH) from Oligotropha carboxidovorans (Dobecket al., 1999), which contain the ¯avin domainabsent from the aldehyde oxidoreductases. The cal-culation of the electrostatic potentials for MOD
MOP (numbering in the MOD row (orange)) and xanthinefrom Drosophila subobscura, Swiss-Prot accession number
orvegicus, Swiss-Prot accession number P22985; MOP, alde-ccession number Q46509; MOD: aldehyde oxidoreductase
nce database and accession number AJ251305. AlignmentsT, within the GCG package. Invariant residues, (red), con-
eutral residues, (green); conserved charged residues, nega-secondary structural elements of b-strands, (!) and a-
ur clusters, the molybdopterin, (~) and the cytosine resi-

140 D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase
and MOP (Figure 3) shows that there are differ-ences evenly spread on the molecular surfaces. Theupper right quadrant is dominated by a cluster ofpositively charged residues in both structures. Thissame region corresponds roughly to the dockingsite of the ¯avin domain of CODH and may markthe physiological electron acceptor docking site inMOP and MOD.
The MCD cofactor and the active-site
The MCD structure was parameterized using themodel from the very high-resolution structure ofMOP (RomaÄo et al., unpublished results). In there®ned structure, all cofactors had very clear den-sity and the individual atoms have comparabletemperature factors with the exception of the mol-ybdenum atom and its three oxygen ligands,which presented very high temperature factors forfull ocupancy. When the individual occupancieswere re®ned to lower values, the resulting modelof the Mo coordination sphere showed B-factors(35.3-42.2 AÊ 2) near the upper range observed forthe atoms of the pterinic moiety of the cofactor(16.2-35.8 AÊ 2). The ®nal electron density maps inthe active-site calculated with the re®ned occu-pancy were of much better quality. This obser-vation is in agreement with the known
Figure 3. Electrostatic surface potential for MOP and Mmolecular surfaces show the electrostatic potential colored fstant and T the absolute temperature.) The arrow points to
experimental evidence that enzymes from thexanthine oxidase group are often found with a cer-tain fraction in the demolybdo form (Moura &Barata, 1994). The thermal parameters found forMoco are not evenly distributed and the largestvalues are found for the atoms of the pterinic moi-ety (25-36 AÊ 2). This may re¯ect some disorder ofthis part of the cofactor present in its demolybdoform. In the re®ned structure, the molybdenumatom has been modelled in a square pyramidalgeometry with the two sulfur atoms of the dithio-lene unit, one water and one oxo ligand occupyingthe base and one additional oxo group on the apex(Figure 4).
On the MCD cofactor binding region, structuraldifferences are minimal when compared to theMOP case, and only two residue changes wereidenti®ed, Glu700 and Ser797, which have beenreplaced in MOD by Ser702 (and a water molecule)and Lys797, respectively. These mutations do notaffect the hydrogen-bonding pattern, which stabil-izes the MCD cofactor in the protein matrix(Figure 5).
Amino acid residues in the close vicinity of themolybdenum catalytic site are essentially con-served. Some of these residues were important forthe proposal of a reaction mechanism based on thepresence of a bound alcohol inhibitor (Huber et al.,
OD calculated with GRASP (Nicholls et al., 1991). Theserom 10kBT, (red) to 10 kBT, (blue). (kB is the Boltzman con-the distal, solvent-exposed, [2Fe-2S] center.

Figure 4. Representation of the active-site residues of MOD (in TURBO-FRODO color code) superimposed withthose of MOP (black). The numbering of MOP residues is shown in brackets. Only two water molecules are found onthe hydrophobic pocket of MOD instead of the three located in the MOP structure. The isopropanol inhibitor mol-ecule is not present in the MOD crystallizing solution and is therefore absent from the structure. This Figure was pre-pared with TURBO-FRODO (Roussel & Cambillau, 1992).
D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase 141
1996; RomaÄo et al., 1997b). Glu869, which is veryclose to the Mo site and is totally conserved in theXDH family, is involved in the catalytic cycle. Itactivates, as a general base, the coordinated watermolecule and probably binds transiently to themetal when the carboxylic acid product is released,before the vacant coordination position is reoccu-pied by one water molecule from the chain ofinterior water molecules. The isopropanol inhibitormolecule present in MOP is absent from the MODstructure, as expected, since it was not present inthe crystallization solution. However, a disorderedwater molecule most probably replaces it, which isseen in density but poorly de®ned and was there-fore not included in the ®nal model.
Another important aspect of the active-site cav-ity is the presence of the above-mentioned chain ofburied water molecules, proposed as catalyticallyrelevant. In the MOD structure, only two suchwater molecules were found instead of three as inMOP. The innermost water molecule, Wat3, pre-sent on MOP in a rather hydrophobic environment,is not seen in the MOD electron density maps. Thismay be due either to the low resolution of the dataor to the two mutations present in the pocket sur-rounding the water molecules. Phe505 and Ser624in MOP are replaced by Tyr507 and Ala626,respectively, in MOD (Figure 4). These mutationschange the hydrogen-bonding pattern of the loopTyr624-Gly627. While in MOP, water Wat3 is
stabilized by one hydrogen bond to Wat2 and tothe amide group from Gly623, in MOD, NH fromGly625 makes a favorable interaction to the OHfrom Tyr507 (NHÐOH 2.8 AÊ ). Additionally,Ser624, which in MOP stabilizes the loop confor-mation by interaction with the NH from Gly866(�3.0 AÊ ) is replaced by Ala626 in MOD.
On the basis of point mutations of the Aspergillusnidulans xanthine dehydrogenase (XDH), it waspossible to de®ne residues essential for substratepositioning (Glatigny et al., 1998). Some of thesemutations implied partial loss of function of XDHand corresponded to residues in the vicinity of theMOP/MOD substrate binding pocket. Particularlyinteresting is Arg503 (Arg501 in MOP), whosemutation in A. nidulans XDH (Arg911 to Glnor Gly) changes the hydroxylation position of2-hydroxypurine from C8 to C6. This Arg residueis totally conserved within the XDH family ofenzymes and was proposed to be essential to pos-ition purine substrates in the Mo center, in agree-ment with our earlier proposals that were based onthe crystallographic studies of MOP (Huber et al.,1996).
The two [2Fe-2S] redox centers
The two redox active iron-sulfur centers rep-resent distinct forms, which are structurally relatedbut very differently bound to the protein. For the

Figure 5. A representation of hydrogen-bonded contacts established between the MOD residues and the molybdop-terin cytosine dinucleotide and the two [2Fe-2S] centers. Residues are colored according to their location in one of thefour domains, Fe/S distal, (green); Fe/S proximal, (red); Mo1, (yellow) or Mo2, (blue). Contacts to a few buriedwater molecules are included. NHÐS hydrogen bonds surrounding both Fe/S clusters are marked. The residues thatare different in the MOP structure (E 41, Q 42, E 700 and S 797) are underlined.
142 D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase
distal cluster, a sequence Cys40-X1-X2-Gly-Gln-Cys45-Gly-Ala-Cys48- (as found in plant-type fer-redoxins) traps the center, which is closed byCys60, placed 11 residues ahead. For the proximalcluster, a totally different cysteine pattern isinvolved, as one of the iron atoms binds on aCys100-Gly-Phe-Cys103 motif. The cluster isenclosed by a similar pattern located 34 residuesahead, Cys137-Arg-Cys139. As a consequence ofthe different cysteine-chelating motif, these clustersexhibit different NHÐS bonding patterns(Figure 5). These NHÐS bonds are involved in theaccommodation of both Fe/S centers inside theprotein and modulate the redox potential as foundin other types of iron clusters (Backes et al, 1991;Sheridan et al., 1981).
The two Fe/S clusters are about 12.3 AÊ far apart(Fe2 atom of FeS distal to Fe1 atom of Fe/S proxi-mal). The Fe2 atom from the proximal iron-sulfurcenter is 14.7 AÊ away from the molybdenum atomand in contact with the pterin via residue Cys139.The side-chains of Val38 and Arg58 at the surfacepartially shield the distal cluster from the solvent.Not surprisingly, both clusters maintain their co-ordination as in MOP, in spite of a few mutationsin the close vicinity of the metals. Residues Ser104,Ala136 and Glu41, Gln42 and Ser49 have beenreplaced by Thr104, Ile136 and Gly41, Lys42 andThr49 for the proximal and the distal clusters,respectively. The most conspicuous mutation is on
residue 41 (Glu to Gly), which makes one NHÐScontact to a sulfur atom from the center. The side-chain of Glu41 is exposed. In residues 104 and 49,where a Thr residue in MOD replaces a Ser residuein MOP, the same hydrogen-bonding pattern ismaintained in both structures. The proximal clusterestablishes six NH±S hydrogen bonds. A directNH-S contact between the Sg atom of Cys139 andthe NH2 group at C2 of the pterin is the most prob-able electron transfer pathway (RomaÄo et al., 1995).The distal, iron-sulfur center has a different NH-Shydrogen bonding pattern, being stabilized by atotal of ten such interactions.
The dimer
As found in several other molybdenum hydroxy-lases (Hille, 1996), MOD is isolated as a homodi-mer although the subunits are functionallyindependent and, as in MOP, both subunits arerelated by a crystallographic dyad. In spite of thefact that MOP and MOD crystallize under totallydifferent conditions (30 % (v/v) isopropanol, 0.2 MMgCl2 (pH 7.2) (RomaÄo et al., 1993) versus 2.5 Mammonium sulfate (pH 6), respectively), both yieldhexagonal crystals (hexagonal bipyramides versushexagonal prisms, respectively) belonging to thesame space group P6122. The cell parameters aredifferent, however, and, due to different packingeffects, the location of the crystallographic a2 dimeris different in the hexagonal lattices.

D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase 143
In the dimer, the redox cofactors are relativelyfar apart in both subunits with the shortest dis-tance of 39.3 AÊ between the nearest iron atoms(from center Fe/S distal and its symmetry part-ner). The molecular dyad runs along the contact ofboth Fe/S distal domains through Gly32, Leu31,such that residues from Fe/S distal (I) and Mo1(III) domains contact symmetry partners fromdomains Fe/S distal* and Mo1*. These symmetri-cal contacts are mainly of a hydrophobic nature, asseen in Table 4, where contacts through hydrogenbonding interactions between both subunits aresummarized. An interesting feature observed onlyin the MOD dimer interface is the presence ofCys84 (Asn 84 in MOP) in close vicinity to its sym-metry mate (distance S-S* 3.1 AÊ ) but not forming adisul®de bridge.
In the related enzyme carbon monoxide dehy-drogenase from O. carboxidovorans (Dobeck et al.,1999), the dimer axis is different, but the redoxcofactors are also placed far apart in both subunits.This indicates that for all these enzymes of thexanthine oxidase family the two subunits are func-tionally independent.
Materials and Methods
Crystallization, data collection and processing
The protein was puri®ed by a procedure similar tothat used for the D. gigas enzyme (Barata et al., 1993)with some modi®cations and it was dialyzed against10 mM Tris-HCl (pH 7.6).
For protein crystallization, the vapor diffusion methodwas used at room temperature and the crystallizationsolution contained 1.8 M ammonium sulfate, 0.1 M Mes(pH 6.0) and 10 % (v/v) dioxane. This last additive wasessential for obtaining larger and better-diffracting crys-tals. Hanging drops were prepared with 4 ml of reservoirbuffer mixed with 4 ml of a 15 mg/ml protein solution in10 mM Tris-HCl (pH 7.6). Single crystals appear withinthree to four weeks and are red-brownish hexagonalprisms. X-ray-diffraction intensity data were collectedwith a conventional X-ray source, an Enraf-Nonius rotat-ing anode generator (Cu-Ka) operated at 4.5 kW and
Table 4. Contacting residues at the subunit interface ofthe MOD dimer
Subunit 1 Subunit 2a
Met1 Leu192Thr3 (Gln3) Thr377Met11 (Ile11) Leu192 (Thr192)
Ala187Arg14 (Asn14) Asn417Leu16 (Phe16) Met203
Tyr377Met203 Leu16Glu205 (Ala205) Ser289Tyr377 Thr3 (Gln3)Glu380 (Asp380) Pro19 (Ala19)
For comparison purposes, corresponding residues in theMOP dimer that are different from MOD are shown in parenth-eses.
a Crystallographically related.
equipped with a Mar Research Imaging Plate detector.Data were collected at room temperature from a singlecrystal mounted in a sealed capillary, leading to a maxi-mum of 2.5 AÊ resolution, from which only 2.8 AÊ resol-ution had acceptable quality.
The complete set of the detector images were indexed,and followed by parameter re®nement in DENZO. TheSCALEPACK package ended data processing by scalingand merging the data, yielding a ®nal ®le with acomplete set of data (Otwinowski & Minor, 1993). Datawere truncated and Friedel mates were merged (CCP4,1994) and a startup ®le with X-PLOR format was ®nallyavailable.
Structure solution
The MOD structure was solved by molecular replace-ment techniques using the program package AMoRe(Navaza, 1994). The model of the closely related alde-hyde oxidoreductase from D. gigas in the oxidized formwas used as a search model, after omitting cofactors andsolvent molecules. Using data in the resolution rangebetween 15.0 and 3.5 AÊ , a unique and clear solution wasobtained with an R-factor of 41.6 % and a correlationcoef®cient of 0.475. After rigid-body re®nement the R-factor dropped to 36.7 % with a correlation coef®cient of0.591. The ®nal values for the rotation and translationfunctions were a � 20.1 �, b � 19.4 �, g � 50.3 � (in Eulerangles) and tx � 0.1942, ty � 0.4149, tz � 0.4434 (fractionalco-ordinates), respectively.
Model building and refinement
The re®nement of the correctly positioned moleculewas initiated, in the ®rst cycles, still with the unmodi®edMOP sequence and was carried out with X-PLOR 3.81(BruÈ nger et al., 1992) using a least-squares re®nementalgorithm with FFT (Agarwal, 1978) and the parameterset reported by Engh & Huber (1991). The total recordedrange of data, 24.9 AÊ to 2.8 AÊ with a 2s cut-off level,were employed. In the beginning, only the ®rst 100amino acid residues from the N terminus were known,which led to a fall in the R-factor from 36.7 % to 26.8 %and in the free R-factor from 36.0 % to 32.6 %. Each cycleof re®nement involved map inspection and correctionfollowed by 100 steps of positional, and 50 steps of indi-vidual B-factor automated re®nement. The parameter ®lefor the molybdopterin cofactor was taken from the opti-mized geometry of the cofactors from the 1.28 AÊ resol-ution re®ned model of MOP (RomaÄo et al., unpublishedresults). Improved phases enabled the calculation ofclearer electron density maps, which contributed to thedesign of new PCR primers for completing the genesequence. When more than half of the sequence wasknown, bulk solvent correction was applied reducing theR-factors and free R-factors by about 3 %. Throughoutthe re®nement, Rfree (BruÈ nger, 1992) was calculated byusing a random independent set with 3 % of all collecteddata omitted from re®nement. The rebuilding andadjustments of the model, such as sequence replacement,to the displayed electron density maps of this structurewere performed using the graphical program TURBO(Roussel & Cambillau, 1992). Data on the progress of there®nement and statistical data on the re®ned MODmodel are summarized in Tables 2 and 3, respectively.

144 D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase
Sequencing of the gene encoding the MOD ofD. desulfuricans ATCC 27774
Oligonucleotide primers were synthesized based onthe amino acid sequence of the N terminus of MOD anda highly conserved region of molybdopterin enzymes,and used in a PCR reaction to amplify a DNA fragmentrepresenting about 250 bp near the 50-end of the MODgene. The amino acid sequence ALLVDV, obtainedby Edman degradative sequencing of the N term-inus, yielded one of the primers, lucky1:50-GCCCTGCTGGTNGACGT-30. A multiple sequencealignment of aldehyde oxidoreductases, aldehyde oxi-dases, and xanthine oxidases (Hille, 1996), identi®ed aregion approximately 100 amino acid residues from theN terminus that is absolutely conserved in this family ofenzymes. The conserved amino acid residues, QCGFC,were used to design a second primer, lucky2: 50-GCA-GAAGCCGCATTG-30 (complementary strand). PCRampli®cation followed by automated DNA sequencingof the PCR product yielded a DNA sequence that codedfor an open reading frame highly homologous to theD. gigas MOP gene (Thoenes et al., 1994).
This fragment served as a guide to obtain additionalDNA sequence of the MOD gene using two approaches.In one approach, nested primers based on the sequenceobtained above were synthesized and used according tomanufacturer's directions provided in the Universal Gen-ome Walker Kit (Clontech, Palo Alto, CA) to generatetwo additional PCR fragments encompassing 578 bp ofthe 50-end of the MOD gene including 67 bases of the 50-untranslated region. In a parallel approach, the DNAsequence obtained in the ®rst step was used to design ahomologous forward primer, MOD1:50-GCGTGGATC-CAGCACGGCGCGGCACAATGCGGCTTCTGC-30, andX-ray derived sequence DGPFGA, was used for design-ing a reverse primer, MOD864R:50-GCICCRAAIG-GICCRTC-30. A DNA fragment spanning to the 30-end ofthe MOD gene, representing 2319 bp, was ampli®ed byPCR using Taq DNA polymerase (MBI Fermentas).Direct sequencing of this product in both forward andreverse directions was done by primer walking using anautomated DNA sequencer (model 373, Applied Biosys-tems, Forster City, USA) and the PRISM ready reactiondye deoxy terminator cycle sequencing kit (AppliedBiosystems, Forster City). From the obtained DNAsequences, the ``inside-out'' primers LASTFWD: 50-CCCTCTATCAAGATGATTCC-30, and MOD1REV: 50-GCAGAAGCCGCATTGTGCCGCGCCGTGCTGGATCCACGC-30 were constructed for inverse PCR DNA ampli-®cation (Triglia et al., 1988; Ochman et al., 1988; Silveret al., 1989).
A restriction enzyme map of the known MOD genesequence was produced with the program MAP of theGCG package (Wilconsin Package V10.0; Genetic Com-puter Group, Madison, WI). The restriction enzyme NdeI(New England Biolabs) was chosen for digestion of analiquot of genomic DNA (5 mg) from D. desulfuricansATCC 27774 in a volume of 20 ml. After overnight incu-bation the enzyme was heat-inactivated for 20 minutesat 70 �C. Intramolecular ligation of the restriction frag-ments was performed using phage T4 DNA ligase (NewEngland Biolabs) for 16 hours at 16 �C in a total volumeof 100 ml. Using these DNA circles as templates, the pri-mers LASTFWD and MOD1REV, and Taq DNA poly-merase, a PCR product of about 1300 bp was obtained.The PCR product was directly sequenced, giving theremaining sequence of the MOD gene. Assembly of theoverlapping sequence fragments allowed us to reconsti-
tute an open reading frame of 2721 bp, coding for a 907amino acid residue polypeptide.
Software
Rough data processing was done using DENZO-SCA-LEPACK (Otwinowski & Minor, 1991), and the CCP4suite of programs (CCP4, 1994). Molecular replacementcalculations were done with AMoRe (Navaza, 1994),map calculations and re®nement were carried out withX-PLOR (BruÈ nger et al., 1987; BruÈ nger, 1990) and usingthe Engh & Huber (1991) force ®eld. Model building wasdone with TURBO-FRODO (Roussel & Cambilau, 1992).Sequence alignment and homology, and identity calcu-lations were made with CLUSTALW (Thompson et al.,1994) and Figure 1 was prepared with ALSCRIPT(Barton, 1993). The validation tool for geometric qualityassessment was PROCHECK (Laskowski et al., 1993).Hydrogen bond analysis of the cofactors in Figure 3 wasdone using HBLPLUS (McDonald et al., 1994). All modelpictures were drawn with either TURBO-FRODO (Rous-sel & Cambilau,1992) or MOLSCRIPT (Kraulis, 1991) andRaster3D (Merrit & Murphy, 1994).
Protein Data Bank accession code
The MOD DNA sequence has been deposited in theEMBL sequence database with the accession codeAJ251305. The atomic coordinates have been depositedin the RCSB Protein Data Bank with the accession code1DGJ.
Acknowledgments
This work was supported by PRAXIS/2/2.1/BIO/05/94, EC-TMR/FMRX-CT980204, PhD grant PRAXIS/BD/13530/97 (J.M.D.), PhD grant PRAXIS/BD/16009/98(S.M.) and MD grant PRAXIS/BM/12730/97 (J.R.).
References
Agarwal, R. C. (1978). A new least-squares re®nementtechnique based on the fast Fourier transform algor-ithm. Acta Crystallog. sect. A, 34, 791-809.
Backes, G., Mino, Y., LoÈhr, T. M., Meyer, T. E.,Cusanovich, M. A., Sweeney, W. V., Adam, E. T. &Sanders-LoÈhr, J. (1991). The environment of [4Fe-4S]clusters in ferredoxins and high-potential iron pro-teins. new information from X-ray crystallographyand raman spectrometry. J. Am. Chem. Soc. 113,2055-2064.
Barata, B. A. S., Legall, J. & Moura, J. J. G. (1993). Alde-hyde oxidoreductase activity in Desulfovibrio gigas:in vitro reconstitution of an electron-transfer chainfrom aldehydes to the production of molecularhydrogen. Biochemistry, 32, 11559-11568.
Barton, G. J. (1993). ALSCRIPT, a tool to format multiplesequence alignments. Protein Eng. 6, 37-40.
BruÈ nger, A. T. (1990). X-PLOR, (Version 3.851), Yale Uni-versity Press, New Haven CT.
BruÈ nger, A. T. (1992). Free R-value: a novel statisticalquantity for assessing the accuracy of crystal struc-tures. Nature, 355, 472-475.
Bursakov, S. A., Liu, M-Y., Payne, W. J., LeGall, J.,Moura, I. & Moura, J. J. G. (1995). Isolation andpreliminary characterization of a soluble nitrate

D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase 145
reductase from the sulfate reducing organism Desul-fovibrio desulfuricans ATCC 27774. Anaerobe, 1, 55-60.
Collaborative Computational Project Number 4 (1994).The CCP4 suite: programs for protein crystallogra-phy. Acta Crystallog. sect. D, 50, 760-763.
Costa, C., Teixeira, M., LeGall, J., Moura, J. J. G. &Moura, I. (1997). Formate dehydrogenase fromDesulfovibrio desulfuricans ATCC 27774: isolationand spectroscopic characterization of the active-sites(heme, iron-sulfur centers and molybdenum). J. Biol.Inorg. Chem. 2, 198-208.
Dias J., M., Than, M. E., Humm, A., Huber, R.,Bourenkov, G. P., Bartunik, H. D., Bursakov, S.,Calvete, J., Caldeira, J., Carneiro, C., Moura, J. J. G.,Moura, I. & RomaÄo, M. J. (1999). Crystal structureof the ®rst dissimilatory nitrate reductase at 1.9 AÊ
solved by MAD methods. Structure, 7, 65-79.Dobeck, H., Gremer, L., Meyer, O. & Huber, R. (1999).
Crystal structure and mechanism of CO dehydro-genase, a molybdo iron-sulfur ¯avoprotein contain-ing S-selanylcysteine. Proc. Natl Acad. Sci. USA, 96,8884-8889.
Edman, P. & Begg, G. (1967). A protein sequenator. Eur.J. Biochem. 1, 80-91.
Engh, R. & Huber, R. (1991). Accurate bond and angleparameters for X-ray protein structure re®nement.Acta Crystallog. sect. A, 47, 392-400.
Fukuyama, K., Ueki, N., Nakamura, H., Tsukihara, T. &Matsubara, H. (1995). Tertiary structure of [2Fe-2S]ferredoxin from Spirulina platensis re®ned at 2.5 AÊ
resolution: structural comparison of plant-type fer-redoxins and an electrostatic potential analysis.J. Biochem. 117, 1017-1123.
Glatigny, A., Hof, P., RomaÄo, M. J., Huber, R. &Scazzocchio, C. (1998). Altered speci®city mutationsde®ne residues essential for substrate positioning inxanthine dehydrogenase. J. Mol. Biol. 278, 431-438.
Hille, R. (1996). The mononuclear molybdenumenzymes. Chem. Rev. 96, 2757-2816.
Huber, R., Hof, P., Duarte, R. O., Moura, J. J. G., Moura,I., Liu, M.-Y., LeGall, J., Hille, R., Archer, M. &RomaÄo, M. J. (1996). A structure-based catalyticmechanism for the xanthine oxidase family of mol-ybdenum enzymes. Proc. Natl Acad. Sci. USA, 93,8846-8851.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots for protein struc-tures. J. Appl. Crystallog. 24, 946-950.
Laskowski, R. A., McArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck stereochemical quality of protein structures.J. Appl. Crystallog. 26, 283-291.
Luzzati, P. V. (1952). Traitement statõÂstique des erreursdans la deÂterminacion des structures cristalines.Acta Crystallog. sect. D, 5, 802-810.
Mathews, B. W. (1968). Solvent contents of protein crys-tals. J. Mol. Biol. 33, 491-497.
McDonald, I. K. & Thornton, J. M. (1994). Satisfyinghydrogen bonding potential in proteins. J. Mol. Biol.238, 777-793.
Merrit, E. A. & Murphy, M. E. P. (1994). Raster3D Ver-sion 2.0: a program for photorealistic moleculargraphics. Acta Crystallog. sect. D, 50, 869-873.
Moura, J. J. G. & Barata, B. A. (1994). Aldehyde oxido-reductases and other molybdenum-containingenzymes. Methods Enzymol. 243, 24-42.
Moura, J. J. G., Xavier, A. V., Carmmack, R., Hall, D. O.,Buschi, M. & LeGall, J. (1978). Oxidation-reduction
studies of the Mo(2Fe-2S) protein from Desulfovibriogigas. Biochem. J. 173, 419-425.
Navaza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157-163.
Nicholls, A., Sharp, K. & Honig, B. (1991). Protein fold-ing and association: insights from the interfacialand thermodynamic properties of hydrocarbons.Proteins: Struct. Funct. Genet. 11, 134-138.
Ochman, H., Gerber, A. S. & Hartl, D. L. (1988). Geneticapplications of an inverse polymerase chain reac-tion. Genetics, 120, 621-623.
Otwinowski, Z. & Minor, W. (1993). DENZO: A FilmProcessing Program for Macromolecular Crystallogra-phy, Yale University Press, New Haven CT.
RomaÄo, M. J. & Huber, R. (1998). Structure and functionof the xanthine-oxidase family of molybdenumenzymes. In Metal Sites in Proteins and Models: RedoxCentres, Structure and Bonding (Hill, H. A. O.,Sadler, A. J. & Thomson, A. J., eds), pp. 69-96,Springer-Verlag, Berlin.
RomaÄo, M. J., Barata, B. A. S., Archer, M., Lobeck, K.,Moura, I., Carrondo, M. A., LeGall, J., Lottspeich,F., Huber, R. & Moura, J. J. G. (1993). Subunit com-position, crystallization and preliminary crystallo-graphic studies of Desulfovibrio gigas aldehydeoxidoreductase containing molybdenum and [2Fe-2S] centers. Eur. J. Biochem. 215, 729-732.
RomaÄo, M. J., Archer, M., Moura, I., Moura, J. J. G.,LeGall, J., Engh, R., Schneider, M., Hof, P. & Huber,R. (1995). Crystal structure of the xanthine oxidase-related aldehyde oxido-reductase from D. gigas.Science, 270, 1170-1176.
RomaÄo, M. J., KnaÈblein, J., Huber, R. & Moura, J. J. G.(1997a). Structure and function of molybdopterincontaining enzymes. Prog. Biophys. Mol. Biol. 68,121-144.
RomaÄo, M. J., RoÈsch, N., KnaÈblein, J. & Huber,R. (1997b). The molybdenum site in the xanthineoxidase-related aldehyde oxidoreductase fromDesulfovibrio gigas and a catalytic mechanism forthis class of enzymes. J. Biol. Inorg. Chem. 2, 782-785.
Roussel, A. & Cambillau, C. (1992). TURBO-FRODO,molecular modelling package. In Silicon GraphhicsGeometry Partner Directory (Silicon, Graphics,ed.), pp. 77-78, Silicon Graphics, Mountain View,CA.
Rypniewski, W. R., Breiter, D. R., Benning, M. M.,Wesenberg, G., Oh, B. H., Markley, G. L., Rayment,I. & Holden, H. M. (1991). Crystallization and struc-ture determination to 2.5 AÊ resolution of the oxi-dized [2Fe-2S] ferredoxin isolated from Anabaena7120. Biochemistry, 30, 4126-4131.
Sheridan, R. P., Allen, L. C. & Carter, C. W., Jr (1981).Coupling between oxidation state and hydrogenbond conformation in high potential iron-sulphurprotein. J. Biol. Chem. 256, 5052-5057.
Silver, J. & Keerikatte, V. (1989). Novel use of thepolymerase chain reaction to amplify celular DNAadjacent to an integrated provirus. J. Virol. 63,1924-1928.
Thoenes, U., Flores, O. L., Neves, A., Devreese, B., VanBeeumen, J. J., Huber, R., RomaÄo, M. J., LeGall, J.,Moura, J. & Rodrigues-Pousada, C. (1994). Molecu-lar cloning and sequence analysis of the gene of themolybdenum-containing aldehyde oxido-reductaseof Desulfovibrio gigas. The deduced amino acid

146 D. desulfuricans ATCC 27774 Aldehyde Oxidoreductase
sequence shows similarity to xanthine dehydrogen-ase. Eur. J. Biochem. 220, 901-910.
Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994).CLUSTAL W: improving the sensitivity of progress-ive multiple sequence alignment through sequenceweighting, position-speci®c gap penalties andweight matrix choice. Nucl. Acids. Res. 22, 4673-4680.
Triglia, T., Peterson, M. G. & Kemp, D. J. (1988). A pro-cedure for in vitro ampli®cation of DNA segmentsthat lie outside the boundaries of known sequences.Nucl. Acids Res. 16, 8186.
Turner, N., Barata, B., Bray, R. C., Deistung, J., LeGall, J.& Moura, J. J. G. (1987). The molybdenum iron-sul-phur protein from Desulfovibrio gigas as a form ofaldehyde oxidase. Biochem. J. 243, 755-761.
Edited by D. C. Rees
(Received 3 September 1999; received in revised form 21 December 1999; accepted 22 January 2000)




















