
Gene expression profile based classification models of psoriasis
8
Gene expression profile based classification models of psoriasis Pi Guo a,b,c,1 , Youxi Luo a,b,1 , Guoqin Mai a,b , Ming Zhang f,g , Guoqing Wang d,e, ⁎⁎, Miaomiao Zhao a,b , Liming Gao a,b , Fan Li d,e , Fengfeng Zhou a,b, ⁎ a Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, PR China b Key Lab for Health Informatics, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, PR China c Department of Public Health, Shantou University Medical College, No. 22 Xinling Road, Shantou, Guangdong 515041, PR China d Department of Pathogeny Biology, Norman Bethune Medical College, Jilin University, Changchun, Jilin 130021, PR China e Key Laboratory of Zoonosis Research, Ministry of Education, Jilin University, Changchun, Jilin 130021, PR China f Department of Epidemiology and Biostatistics, Faculty of Infectious Diseases, University of Georgia, Athens, GA 30605, USA g Institute of Bioinformatics, University of Georgia, Athens, GA 30605, USA abstract article info Article history: Received 25 March 2013 Accepted 1 November 2013 Available online 13 November 2013 Keywords: Gene expression profiles Classification Psoriasis Psoriasis is an autoimmune disease, which symptoms can significantly impair the patient's life quality. It is mainly diagnosed through the visual inspection of the lesion skin by experienced dermatologists. Currently no cure for psoriasis is available due to limited knowledge about its pathogenesis and development mechanisms. Previous studies have profiled hundreds of differentially expressed genes related to psoriasis, however with no robust psoriasis prediction model available. This study integrated the knowledge of three feature selection algorithms that revealed 21 features belonging to 18 genes as candidate markers. The final psoriasis classification model was established using the novel Incremental Feature Selection algorithm that utilizes only 3 features from 2 unique genes, IGFL1 and C10orf99. This model has demonstrated highly stable prediction accuracy (averaged at 99.81%) over three independent validation strategies. The two marker genes, IGFL1 and C10orf99, were revealed as the upstream components of growth signal transduction pathway of psoriatic pathogenesis. © 2013 Elsevier Inc. All rights reserved. 1. Introduction Psoriasis is a widely spread chronic autoimmune disease with major symptoms on skins and joints [1,2]. Psoriasis may increase the risks of stroke and myocardial infarction [3,4] and shares the same effective drug, p40-neutralizing antibodies, with the neurodegenerative Alzheimer's disease [5]. The prevalence of psoriasis varies considerably in different geographic regions and ethnic groups, with higher rate in the Caucasian population (e.g. 2.2% in the United States) and lower rate in the Asian (e.g. less than 1% in China) [6,7]. Along with the cutaneous manifestations, psoriasis is accompanied by inflammatory ar- thritis in up to 40% cases [8]. Its phenotypic symptoms may also include epidermal hyperplasia, keratinocyte differentiations, angiogenesis, infil- tration of T lymphocytes, dendritic cells, neutrophils, cytokines as well as chemokines [9–11]. The current diagnosis of psoriasis heavily relies on the visual observation of the skin lesion biopsy by an experienced dermatologist [12], which procedure is inefficient and labor intensive. Majority of the large-scale psoriasis studies have employed microar- ray or qPCR techniques to compare the gene expression pattern in psori- atic lesion samples with healthy controls. Gudjonsson et al. systematically screened ~54,000 probe sets in the Affymetrix HG-U133 Plus 2 platform, and detected 179 unique genes for 223 probe sets with differential expression in the uninvolved psoriatic skin samples [13]. The lipid metab- olism was demonstrated to be most associated with psoriasis, and three lipid metabolic related transcription factors, peroxisome proliferator- activator receptor alpha (PPARA), sterol regulatory element-binding protein (SREBF), and estrogen receptor 2 (ESR2), seem to regulate the dysregulated genes in the uninvolved psoriatic samples. Reischl et al. chose the Affymetrix HG-U133A platform to profile the expression pat- terns of ~22,000 probe sets in the human genome, and detected 179 genes for 203 probe sets with more than 2-fold expression changes in the psoriatic lesion skins [14]. Their data strongly suggested that the stem cell proliferation regulation Wnt pathway is associated with the pso- riasis development. But it still remains elusive whether the Wnt pathway plays a causative role or merely the human immune response to the pso- riatic lesion. The human immune response to the environmental stimulus, the type I interferon system, was screened in both the psoriatic lesion and uninvolved skin samples on the Affymetrix HG-U133 Plus 2 platform [15]. There were 1408 up-regulated and 1465 down-regulated probe sets detected in the psoriatic lesion skin samples, among which multiple T cell marker genes and two type I interferon inducible genes (STAT1 and ISG15) had ~3 fold or higher in expression changes. Swindell et al. Genomics 103 (2014) 48–55 ⁎ Correspondence to: Fengfeng Zhou, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, PR China. Fax:+86-755- 86392299. ⁎⁎ Correspondence to: Guoqing Wang, Department of Pathogeny Biology, Norman Bethune Medical College, Jilin University, Changchun, Jilin 130021, PR China. E-mail addresses: [email protected], [email protected] (F. Zhou). URL: http://www.healthinformaticslab.org/ffzhou/ (F. Zhou). 1 These authors contribute equally to this work. 0888-7543/$ – see front matter © 2013 Elsevier Inc. All rights reserved. http://dx.doi.org/10.1016/j.ygeno.2013.11.001 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Gene expression profile based classification models of psoriasis

Genomics 103 (2014) 48–55
Contents lists available at ScienceDirect
Genomics
j ourna l homepage: www.e lsev ie r .com/ locate /ygeno
Gene expression profile based classification models of psoriasis
Pi Guo a,b,c,1, Youxi Luo a,b,1, Guoqin Mai a,b, Ming Zhang f,g, Guoqing Wang d,e,⁎⁎, Miaomiao Zhao a,b,Liming Gao a,b, Fan Li d,e, Fengfeng Zhou a,b,⁎a Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, PR Chinab Key Lab for Health Informatics, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, PR Chinac Department of Public Health, Shantou University Medical College, No. 22 Xinling Road, Shantou, Guangdong 515041, PR Chinad Department of Pathogeny Biology, Norman Bethune Medical College, Jilin University, Changchun, Jilin 130021, PR Chinae Key Laboratory of Zoonosis Research, Ministry of Education, Jilin University, Changchun, Jilin 130021, PR Chinaf Department of Epidemiology and Biostatistics, Faculty of Infectious Diseases, University of Georgia, Athens, GA 30605, USAg Institute of Bioinformatics, University of Georgia, Athens, GA 30605, USA
⁎ Correspondence to: Fengfeng Zhou, Shenzhen InstiChinese Academy of Sciences, Shenzhen, Guangdong 5186392299.⁎⁎ Correspondence to: Guoqing Wang, Department oBethune Medical College, Jilin University, Changchun, Jilin
E-mail addresses: [email protected], FengfengZhou@gURL: http://www.healthinformaticslab.org/ffzhou/ (F.
1 These authors contribute equally to this work.
0888-7543/$ – see front matter © 2013 Elsevier Inc. All rihttp://dx.doi.org/10.1016/j.ygeno.2013.11.001
a b s t r a c t
a r t i c l e i n f oArticle history:Received 25 March 2013Accepted 1 November 2013Available online 13 November 2013
Keywords:Gene expression profilesClassificationPsoriasis
Psoriasis is an autoimmunedisease,which symptoms can significantly impair the patient's life quality. It ismainlydiagnosed through the visual inspection of the lesion skin by experienced dermatologists. Currently no cure forpsoriasis is available due to limited knowledge about its pathogenesis and development mechanisms. Previousstudies have profiled hundreds of differentially expressed genes related to psoriasis, however with no robustpsoriasis prediction model available. This study integrated the knowledge of three feature selection algorithmsthat revealed 21 features belonging to 18 genes as candidate markers. The final psoriasis classification modelwas established using the novel Incremental Feature Selection algorithm that utilizes only 3 features from 2unique genes, IGFL1 and C10orf99. This model has demonstrated highly stable prediction accuracy (averagedat 99.81%) over three independent validation strategies. The two marker genes, IGFL1 and C10orf99, wererevealed as the upstream components of growth signal transduction pathway of psoriatic pathogenesis.
© 2013 Elsevier Inc. All rights reserved.
1. Introduction
Psoriasis is a widely spread chronic autoimmune disease withmajorsymptoms on skins and joints [1,2]. Psoriasis may increase the risks ofstroke and myocardial infarction [3,4] and shares the same effectivedrug, p40-neutralizing antibodies, with the neurodegenerativeAlzheimer's disease [5]. The prevalence of psoriasis varies considerablyin different geographic regions and ethnic groups, with higher rate inthe Caucasian population (e.g. 2.2% in the United States) and lowerrate in the Asian (e.g. less than 1% in China) [6,7]. Along with thecutaneous manifestations, psoriasis is accompanied by inflammatory ar-thritis in up to 40% cases [8]. Its phenotypic symptoms may also includeepidermal hyperplasia, keratinocyte differentiations, angiogenesis, infil-tration of T lymphocytes, dendritic cells, neutrophils, cytokines as wellas chemokines [9–11]. The current diagnosis of psoriasis heavily relieson the visual observation of the skin lesion biopsy by an experienceddermatologist [12], which procedure is inefficient and labor intensive.
tutes of Advanced Technology,8055, PR China. Fax:+86-755-
f Pathogeny Biology, Norman130021, PR China.mail.com (F. Zhou).Zhou).
ghts reserved.
Majority of the large-scale psoriasis studies have employed microar-ray or qPCR techniques to compare the gene expression pattern in psori-atic lesion sampleswith healthy controls. Gudjonsson et al. systematicallyscreened ~54,000 probe sets in the Affymetrix HG-U133 Plus 2 platform,and detected 179 unique genes for 223 probe sets with differentialexpression in the uninvolved psoriatic skin samples [13]. The lipidmetab-olism was demonstrated to be most associated with psoriasis, and threelipid metabolic related transcription factors, peroxisome proliferator-activator receptor alpha (PPARA), sterol regulatory element-bindingprotein (SREBF), and estrogen receptor 2 (ESR2), seem to regulate thedysregulated genes in the uninvolved psoriatic samples. Reischl et al.chose the Affymetrix HG-U133A platform to profile the expression pat-terns of ~22,000 probe sets in the human genome, and detected 179genes for 203 probe sets with more than 2-fold expression changes inthe psoriatic lesion skins [14]. Their data strongly suggested that thestemcell proliferation regulationWntpathway is associatedwith thepso-riasis development. But it still remains elusive whether theWnt pathwayplays a causative role or merely the human immune response to the pso-riatic lesion. Thehuman immune response to the environmental stimulus,the type I interferon system,was screened in both the psoriatic lesion anduninvolved skin samples on the AffymetrixHG-U133 Plus 2 platform [15].There were 1408 up-regulated and 1465 down-regulated probe setsdetected in the psoriatic lesion skin samples, among which multiple Tcell marker genes and two type I interferon inducible genes (STAT1 andISG15) had ~3 fold or higher in expression changes. Swindell et al.

49P. Guo et al. / Genomics 103 (2014) 48–55
[16,17] compared the gene expression files of psoriasiform versus normalsamples betweenhuman andmouse. The calculated lists of top 5000 fold-change ranked up-regulated anddown-regulated probe sets showed con-sistent overlaps among different mouse psoriasiform phenotypes. Unfor-tunately, the lists of marker genes barely overlapped between differentstudies, leaving little information to construct a psoriatic classificationmodel.
We hypothesized that a robust disease classification model can beconstructed based on marker genes. This study employed the microarraybased gene expression profiles, in which three widely-used feature selec-tion algorithms were applied to screen the psoriasis associated features(probe sets). Majority of the 21 features were known to be associatedwith psoriasis. We further conducted a comprehensive performanceevaluation of the neural network based binary classifiers trained withthe Incremental Feature Selection strategy. The classifier based on thethree features of IGFL1 and C10orf99 outperformed the rest of models,with over 99.43% accuracy in all three validation tests.
2. Methods
2.1. Data collection and preprocessing
Twomicroarray data sets with accession numbers GSE14905 [15] andGSE13355 [16,17] were retrieved from the Gene Expression Omnibus(GEO) Database [18]. The two data sets provide gene expression profilesfor the same disease type (psoriasis) and the corresponding controls,but have no overlap of samples. This makes them an ideal paired set fordetecting the consistent psoriasis biomarkers. There are 176 participatingsubjects in total, including 91 psoriatic patients and 85 healthy controls,denoted by P = {P1, P2,…, P91} and N = {N1, N2,…, N85}. Samples fromboth data setswere processed by the standard protocol for the AffymetrixHuman Genome U133 Plus 2.0 platform. The detailed information of thesamples used in this study is listed in the Supplementary Table S1.
The raw fluorescence intensity data within CEL files were processedby background correction, log2 transformation, and quantile normaliza-tion using the Robust Multichip Analysis (RMA) algorithm [19], as im-plemented in the program Affymetrix Expression Console™, whichwas downloaded from http://www.affymetrix.com/estore/index.jsp.The systematic batch biases were corrected by the Distance WeightedDiscrimination (DWD) algorithm [20]. DWD eliminates source effectsacross different studies by finding a hyper-plane that separates thetwo systematic biases and adjusts the microarray data by projectingthem on the hyper-plane through subtracting out the DWD planemultiplied by the batch mean.
We removed the probe sets with less discriminating power bymeasuring the overall variance. This procedure was performed invarFilter of the R package Bioconductor genefilter. We chose the defaultsetting of the Inter-Quartile Range function IQR for the parametervar.func, and .5 as the var.cutoff. The function IQR was chosen for itsrobust outlier detection. This filtering step was applied on the DWD-processed data sets, and 27,336 probe sets of the 176 participatingsubjects were retained for further analysis.
This study investigated the binary classification problem betweenN1 = 91 psoriatic patients and N2 = 85 healthy controls. The totalnumber of samples was N = 91 + 85 = 176. The expression level ofeach probe set was denoted as Fi, where i ϵ {1, 2,…, 27,336}. The follow-ing sections aimed to detectm features Fj′, where j ϵ {1, 2,…,m} from theM = 27,336 features of the binary classificationmodel.We investigatedvarious feature selection algorithms to detect psoriasis related featuresfrom 27,336 probe sets, with classification accuracy as the mostimportant feature. The following three algorithms were chosen fortheir complementary performance features. ROC screens for the mostsignificant phenotype-associated individual features, by investigatingeach feature's parameter-independent association [21]. SVM-RFE fur-ther optimizes the inter-feature associations, which can top-rank theimportant features that have weak individual phenotype-association
[22]. Boruta removes the featureswith less contribution to classificationaccuracy by introducing random variables for the competition [23,24].We believe, the consensus features derived from the three algorithmsrepresent a reliable list of biomarker candidates. The R implementedversion of the three algorithms was used in this study.
2.2. SVM-RFE feature selection algorithm
SVM-RFE is a Recursive Feature Elimination (RFE) strategy utilizingthe weight vector produced by the classification model Support VectorMachine [25]. RFE is a widely used strategy that can recursively reducethe number of features by removing those features with the leastphenotype correlation rankings [22,26,27]. The Support VectorMachinealgorithmwas invented by Vladimir N Vapnik in 1979 for the classifica-tion problem [28], and it produces aweight vector for the input featuresafter being trained to optimize the classification accuracy between thepsoriatic and healthy samples. The generated weight vector will beused to rank the features, and the RFE strategywill recursively eliminatethe least ranked features. The R package mSVM-RFE was chosen as theimplementation of this algorithm [22].
2.3. ROC feature selection algorithm
The receiver operating characteristic (ROC) is a graphical plot in thesignal detection theory that illustrates the relationship between the truepositive rate (TPR) versus the false positive rate (FPR) of a binaryclassifier [29]. The ROC curve is one of the best measurements to rankthe features between multiple groups of tissues [30]. We ranked thefeatures based on the ROC-based binary classification performance ofthe psoriatic and healthy samples. In brief, the two expression distribu-tions of a given feature and/or genewere calculated for the psoriatic andhealthy samples, respectively. Next, the area under the ROC curve(AUC) was calculated to rank the features. The R package rocc wasused as the implementation of this algorithm [21].
2.4. Boruta feature selection algorithm
The Boruta feature selection algorithm is a random forest basedwrapper strategy that removes features proven to be less informativethan random probes [31]. The classification algorithm, random forest,was chosen because of its inexpensive calculation and parameter freefor manual tuning. A random forest [22] collects votes over multipledecision tree based weak classifiers, which are independently trainedover the binary classification data set between the psoriatic and healthysamples. The importance of a probe set is measured by how the classifi-cation performance is decreased due to the random permutation of theprobe set among samples. The trained random forest classifier providesan importance estimate for all features [22]. The R package implementa-tion of Boruta was used for this study [24].
2.5. Classification model
This study utilized a feed-forward neural network classifierwith onehidden layer of nodes [32] to distinguish the psoriatic samples from thehealthy controls. A neural network is amathematicalmodelwith a func-tion f : X → Y, where X is a collection of input neurons and Y is a collec-tion of output neurons. X = {Fj′}, where j ϵ {1, 2,…, m}, is the featuresselected by the aforementioned feature selection algorithms. There aretwo output nodes, Y = {0, 1}, representing the predicted results of ahealthy or psoriatic sample, respectively. The input and output neuronsare connected by the hidden layers of neurons. In order to avoid modelover-fitting, the number of hidden layers was set to one in this study.The rest of the parameters used the default values.
50 P. Guo et al. / Genomics 103 (2014) 48–55
2.6. Measurement of prediction performance
Aneural network based binary classifierwas evaluated using the fol-lowing three performance measurements [33,34]. Let P = {P1, P2,…,P91} and N = {N1, N2,…, N85} as the positive and negative data sets,respectively. TP is the number of psoriatic samples predicted to bepsoriatic, and FN is the number of psoriatic samples incorrectly predict-ed to be healthy. TN and FP are the numbers of healthy samples predict-ed to be healthy and psoriatic, respectively. The classification model'ssensitivity and specificity are defined to be Sn = TP/(TP + FN) andSp = TN/(TN + FP), respectively. The model's overall accuracy isAc = (TP + TN)/(TP + FN + TN + FP).
A neural network classification model may be over-fitted if the num-ber of features used in the model is similar to or larger than that of thetraining samples. This can be overcome by restricting the number of se-lected features and evaluating the classification model's cross-validationperformance. This study measures how a classification model performsby the commonly used 10-fold cross-validation strategy, as similar to[35–37]. Its principle is to randomly split the 91 psoriatic and 85 healthysamples into ten equal-sized subsets P = {P1, P2,…, P10} and N = {N1,N2,…, N10}, respectively. For the pair of psoriatic and healthy sample sub-sets Pk and Nk, where k ϵ {1, 2,…, 10}, a single-hidden-layer feed-forwardneural network classifier was trained over the data sets P\Pl and N\Nl,
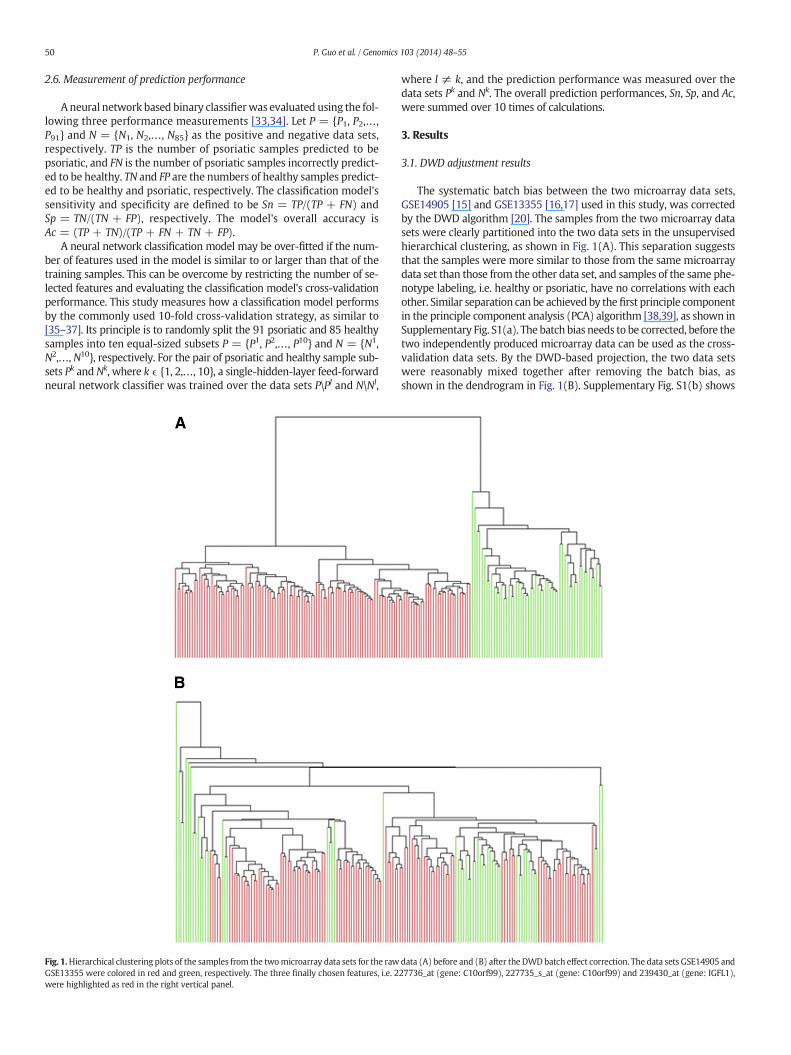
Fig. 1.Hierarchical clustering plots of the samples from the twomicroarray data sets for the rawGSE13355 were colored in red and green, respectively. The three finally chosen features, i.e. 2were highlighted as red in the right vertical panel.
where l ≠ k, and the prediction performance was measured over thedata sets Pk and Nk. The overall prediction performances, Sn, Sp, and Ac,were summed over 10 times of calculations.
3. Results
3.1. DWD adjustment results
The systematic batch bias between the two microarray data sets,GSE14905 [15] and GSE13355 [16,17] used in this study, was correctedby the DWD algorithm [20]. The samples from the two microarray datasets were clearly partitioned into the two data sets in the unsupervisedhierarchical clustering, as shown in Fig. 1(A). This separation suggeststhat the samples were more similar to those from the same microarraydata set than those from the other data set, and samples of the same phe-notype labeling, i.e. healthy or psoriatic, have no correlations with eachother. Similar separation can be achieved by thefirst principle componentin the principle component analysis (PCA) algorithm [38,39], as shown inSupplementary Fig. S1(a). The batch bias needs to be corrected, before thetwo independently produced microarray data can be used as the cross-validation data sets. By the DWD-based projection, the two data setswere reasonably mixed together after removing the batch bias, asshown in the dendrogram in Fig. 1(B). Supplementary Fig. S1(b) shows
data (A) before and (B) after the DWDbatch effect correction. The data sets GSE14905 and27736_at (gene: C10orf99), 227735_s_at (gene: C10orf99) and 239430_at (gene: IGFL1),

51P. Guo et al. / Genomics 103 (2014) 48–55
that at least the first four principle components and their pairs cannotseparate the samples from the two different microarray data sets, thussupporting the hypothesis that the batch bias was corrected.
3.2. Gene sets determined by feature selection algorithms
Different feature selection algorithms rank the features with differentphenotype-association measurements, and we hypothesized that fea-tures chosen by all the three feature selection algorithms, i.e. SVM-RFE,ROC and Boruta, have stable discrimination power. We chose 80 featurestop-ranked by each of the three algorithms SVM-RFE, ROC and Boruta.The intersection of the three top-ranked feature lists consisted of 21features, corresponding to 18 human genes, as shown in Fig. 2. Theprobe set IDs, official gene symbols, and functional annotations of theabove chosen features/probe-sets were detailed in SupplementaryTable S2.
We further investigated the discriminating power of the combinedlist of all the 21 features. The expression patterns of the 21 featureswere visualized as the unsupervised hierarchical clustering heatmapof all the 176 samples, as shown in Fig. 3. The 176 samples can be easilyseparated into their correct labels, with only two psoriatic samples in-correctly predicted as healthy. This visual separation gave a detectionaccuracy Ac = 174/176–98.86% for the 176 samples. The 10-foldcross-validation of the neural network binary classifier rendered thesame prediction result. Fig. 3 also demonstrated that all of the 21features (probe sets) were up-regulated in the psoriatic samples.
3.3. Robustness test of the binary classifier
The prediction robustness of the binary classifier was evaluatedusing the Incremental Feature Selection (IFS) strategy. The selected 21features were ranked by the averaged scores calculated from the threefeature selection algorithms as {f1, f2,…, f21}. We calculated the overallaccuracy Ac using the features {f1, f2,…, f1}, (i ≤ 21) over three indepen-dent validations. The number of features iwas chosen, if the average ac-curacy AvgAc over the three validations reaches the peak as shown inFig. 4. The three validation strategies are described below. For the twoindependently generated microarray data sets, GSE14905 andGSE13355, we firstly conducted a 10-fold cross-validation of the binaryclassifier over the combined data set, denoted here as 10FCV. In the sec-ond validation strategy, we used GSE14905 as the training data set andGSE13355 the test set to validate the classifier, denoted here as
Fig. 2. The overlapping features top-ranked by the three feature selection algorithms. Eachof the three algorithms, i.e. Boruta, SVM-RFE and ROC, chooses 80 features. There are 21features that are in the overlapping region of the three algorithms.
tGSE14905. The third validation strategy tGSE13355 was defined simi-larly as tGSE14905. The latter two validations evaluated the stability ofthe classification model on data sets with batch effects.
Thebinary classifier achieved thebest overall accuracies (99.81%) overthe three validation strategies, as shown in Fig. 4. The top one ranked fea-ture, 227736_at (gene C10orf99), achieved 100% in Ac for both 10FCV andtGSE13355 validations, but didn't work equally well on the tGSE14905validation. The second best feature, 227735_s_at, also resides withinC10orf99. But the classifier based on the two features, 227736_at and227735_s_at, performed worse than 227736_at itself over the 10FCV val-idation. The top three ranked features, 227736_at and 227735_s_at (geneC10orf99) and 239430_at (gene IGFL1), produced a binary classifierwhich performed well with 99.43%, 100% and 100% in overall accuracies(Ac) for the three validation strategies. A classifier based onmore featuresproduced fluctuated overall accuracies between 80% and 100% (Fig. 4).The three features were highlighted in bold and blue font in Supplemen-tary Table S2, whose functions are discussed in details below.
3.4. Enrichment analysis of differentially expressed genes
The Gene Ontology (GO) annotations were screened for the termsenriched in the 21 psoriasis associated features (18 human genes), asshown in Table 1. The enrichment analysis was conducted using thesystem DAVID with default parameters (Evalue cutoff = E−2) [40].The Pvalues of these multiple tests were statistically corrected byBonferroni and Benjamini algorithms [41]. Only the GO categorieswith Pvalues ≤ 0.05 after both multi-test corrections were kept for fur-ther analysis, as shown in Table 1. Genes involved in the developmentand differentiation processes of the skin cells, in particular thekeratinocyte and epidermal cells, were significantly enriched in the 21features that were chosen by three different feature selectionalgorithms. This result is consistent with the clinical observation that un-controlled differentiation of the skin cells is psoriasis's major symptom[42,43]. There is also a sub-cellular compartment, cornified envelope(GO:0001533 with Pvalue = 5.65E−03 after both correction algo-rithms), which was enriched with 3 psoriasis associated genes.
4. Discussion
4.1. A robust prediction signature for psoriasis
This study aimed to establish a robust psoriasis prediction model,based on the gene expression profiles of human samples. Multiple setsof genes were proposed to be associated with psoriasis, or differentiallyexpressed in psoriasis samples [13–15,44]. Such gene sets consist ofdozens or even hundreds of candidate markers, which may potentiallybe used for psoriasis diagnosis. However, psoriasis classification modelstudy is scarce. To our knowledge, this study is thefirst of this kind in con-structing a feed-forward neural network based psoriasis binary classifier.
We hypothesized that the association between the marker featuresand psoriasis is strong enough to pass the filtering criteria of different fea-ture selection algorithms. Thereforewe applied threewidely used featureselection algorithms, i.e. SVM-RFE, ROC and Boruta, to screen the 27,336features that passed the data quality filtering criteria and have statisticalsignificant variations between the disease and control samples. The top80 features ranked by each of the three feature selection algorithmswith default parameters, i.e. SVM-RFE, ROC and Boruta, were retainedfor further analysis. The intersection of these three lists resulted in 21 fea-tures (probe sets) that belong to 18 human genes. Majority of the 21 fea-tureswere known to be associatedwith the initiation anddevelopment ofpsoriasis, as shown in Table 1 and Supplementary Table S2.
A comprehensive classification validation was conducted to investi-gate how a subset of these 21 features may be combined to build abinary classifier, which can perform equally well on different datasets.We incrementally added one feature at a time to the feature subsetby the ranking order. We evaluated the feed-forward neural network
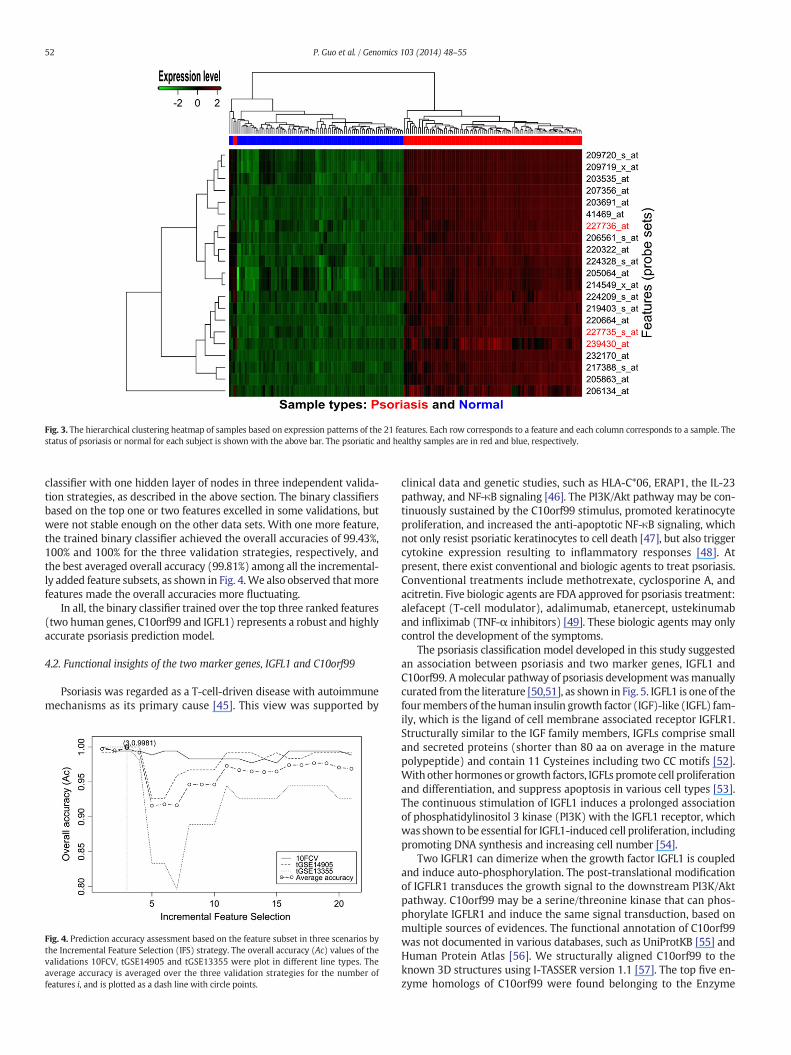
Fig. 3. The hierarchical clustering heatmap of samples based on expression patterns of the 21 features. Each row corresponds to a feature and each column corresponds to a sample. Thestatus of psoriasis or normal for each subject is shown with the above bar. The psoriatic and healthy samples are in red and blue, respectively.
52 P. Guo et al. / Genomics 103 (2014) 48–55
classifier with one hidden layer of nodes in three independent valida-tion strategies, as described in the above section. The binary classifiersbased on the top one or two features excelled in some validations, butwere not stable enough on the other data sets. With one more feature,the trained binary classifier achieved the overall accuracies of 99.43%,100% and 100% for the three validation strategies, respectively, andthe best averaged overall accuracy (99.81%) among all the incremental-ly added feature subsets, as shown in Fig. 4.We also observed thatmorefeatures made the overall accuracies more fluctuating.
In all, the binary classifier trained over the top three ranked features(two human genes, C10orf99 and IGFL1) represents a robust and highlyaccurate psoriasis prediction model.
4.2. Functional insights of the two marker genes, IGFL1 and C10orf99
Psoriasis was regarded as a T-cell-driven disease with autoimmunemechanisms as its primary cause [45]. This view was supported by
Fig. 4. Prediction accuracy assessment based on the feature subset in three scenarios bythe Incremental Feature Selection (IFS) strategy. The overall accuracy (Ac) values of thevalidations 10FCV, tGSE14905 and tGSE13355 were plot in different line types. Theaverage accuracy is averaged over the three validation strategies for the number offeatures i, and is plotted as a dash line with circle points.
clinical data and genetic studies, such as HLA-C*06, ERAP1, the IL-23pathway, and NF-κB signaling [46]. The PI3K/Akt pathway may be con-tinuously sustained by the C10orf99 stimulus, promoted keratinocyteproliferation, and increased the anti-apoptotic NF-κB signaling, whichnot only resist psoriatic keratinocytes to cell death [47], but also triggercytokine expression resulting to inflammatory responses [48]. Atpresent, there exist conventional and biologic agents to treat psoriasis.Conventional treatments include methotrexate, cyclosporine A, andacitretin. Five biologic agents are FDA approved for psoriasis treatment:alefacept (T-cell modulator), adalimumab, etanercept, ustekinumaband infliximab (TNF-α inhibitors) [49]. These biologic agents may onlycontrol the development of the symptoms.
The psoriasis classification model developed in this study suggestedan association between psoriasis and two marker genes, IGFL1 andC10orf99. Amolecular pathway of psoriasis development wasmanuallycurated from the literature [50,51], as shown in Fig. 5. IGFL1 is one of thefourmembers of the human insulin growth factor (IGF)-like (IGFL) fam-ily, which is the ligand of cell membrane associated receptor IGFLR1.Structurally similar to the IGF family members, IGFLs comprise smalland secreted proteins (shorter than 80 aa on average in the maturepolypeptide) and contain 11 Cysteines including two CC motifs [52].With other hormones or growth factors, IGFLs promote cell proliferationand differentiation, and suppress apoptosis in various cell types [53].The continuous stimulation of IGFL1 induces a prolonged associationof phosphatidylinositol 3 kinase (PI3K) with the IGFL1 receptor, whichwas shown to be essential for IGFL1-induced cell proliferation, includingpromoting DNA synthesis and increasing cell number [54].
Two IGFLR1 can dimerize when the growth factor IGFL1 is coupledand induce auto-phosphorylation. The post-translational modificationof IGFLR1 transduces the growth signal to the downstream PI3K/Aktpathway. C10orf99 may be a serine/threonine kinase that can phos-phorylate IGFLR1 and induce the same signal transduction, based onmultiple sources of evidences. The functional annotation of C10orf99was not documented in various databases, such as UniProtKB [55] andHuman Protein Atlas [56]. We structurally aligned C10orf99 to theknown 3D structures using I-TASSER version 1.1 [57]. The top five en-zyme homologs of C10orf99 were found belonging to the Enzyme
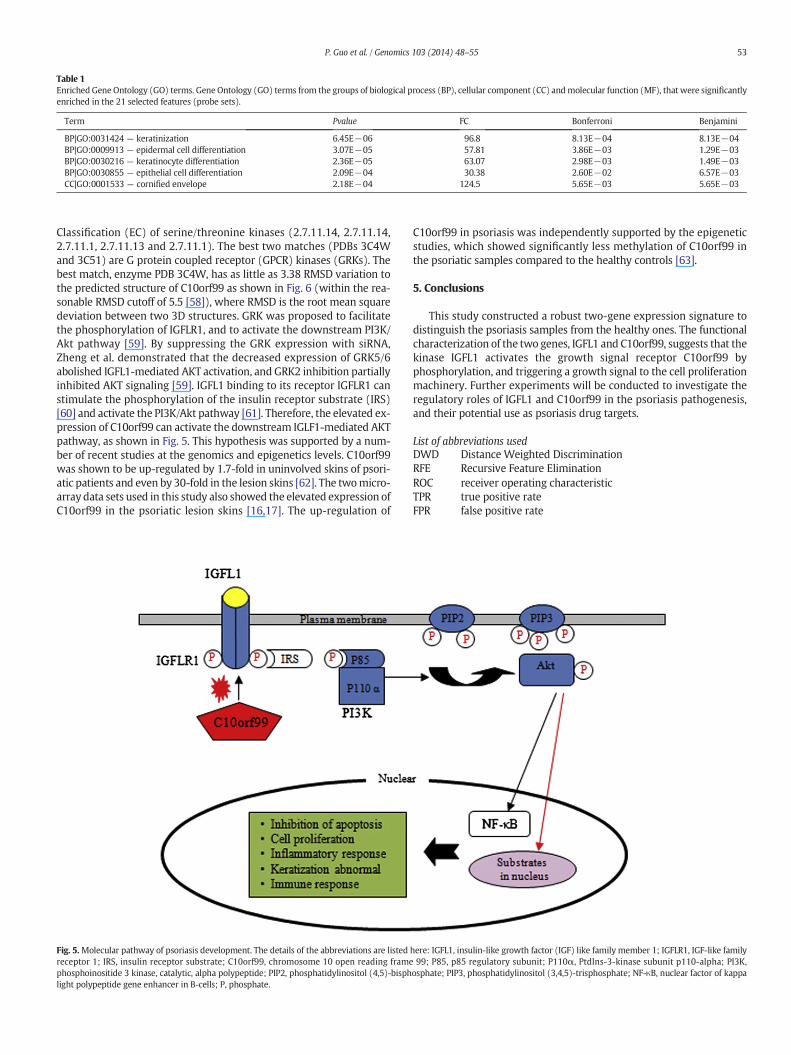
Table 1Enriched Gene Ontology (GO) terms. Gene Ontology (GO) terms from the groups of biological process (BP), cellular component (CC) andmolecular function (MF), that were significantlyenriched in the 21 selected features (probe sets).
Term Pvalue FC Bonferroni Benjamini
BP|GO:0031424 — keratinization 6.45E−06 96.8 8.13E−04 8.13E−04BP|GO:0009913 — epidermal cell differentiation 3.07E−05 57.81 3.86E−03 1.29E−03BP|GO:0030216 — keratinocyte differentiation 2.36E−05 63.07 2.98E−03 1.49E−03BP|GO:0030855 — epithelial cell differentiation 2.09E−04 30.38 2.60E−02 6.57E−03CC|GO:0001533 — cornified envelope 2.18E−04 124.5 5.65E−03 5.65E−03
53P. Guo et al. / Genomics 103 (2014) 48–55
Classification (EC) of serine/threonine kinases (2.7.11.14, 2.7.11.14,2.7.11.1, 2.7.11.13 and 2.7.11.1). The best two matches (PDBs 3C4Wand 3C51) are G protein coupled receptor (GPCR) kinases (GRKs). Thebest match, enzyme PDB 3C4W, has as little as 3.38 RMSD variation tothe predicted structure of C10orf99 as shown in Fig. 6 (within the rea-sonable RMSD cutoff of 5.5 [58]), where RMSD is the root mean squaredeviation between two 3D structures. GRK was proposed to facilitatethe phosphorylation of IGFLR1, and to activate the downstream PI3K/Akt pathway [59]. By suppressing the GRK expression with siRNA,Zheng et al. demonstrated that the decreased expression of GRK5/6abolished IGFL1-mediated AKT activation, and GRK2 inhibition partiallyinhibited AKT signaling [59]. IGFL1 binding to its receptor IGFLR1 canstimulate the phosphorylation of the insulin receptor substrate (IRS)[60] and activate the PI3K/Akt pathway [61]. Therefore, the elevated ex-pression of C10orf99 can activate the downstream IGLF1-mediated AKTpathway, as shown in Fig. 5. This hypothesis was supported by a num-ber of recent studies at the genomics and epigenetics levels. C10orf99was shown to be up-regulated by 1.7-fold in uninvolved skins of psori-atic patients and even by 30-fold in the lesion skins [62]. The twomicro-array data sets used in this study also showed the elevated expression ofC10orf99 in the psoriatic lesion skins [16,17]. The up-regulation of
Fig. 5.Molecular pathway of psoriasis development. The details of the abbreviations are listedreceptor 1; IRS, insulin receptor substrate; C10orf99, chromosome 10 open reading framephosphoinositide 3 kinase, catalytic, alpha polypeptide; PIP2, phosphatidylinositol (4,5)-bisphlight polypeptide gene enhancer in B-cells; P, phosphate.
C10orf99 in psoriasis was independently supported by the epigeneticstudies, which showed significantly less methylation of C10orf99 inthe psoriatic samples compared to the healthy controls [63].
5. Conclusions
This study constructed a robust two-gene expression signature todistinguish the psoriasis samples from the healthy ones. The functionalcharacterization of the two genes, IGFL1 and C10orf99, suggests that thekinase IGFL1 activates the growth signal receptor C10orf99 byphosphorylation, and triggering a growth signal to the cell proliferationmachinery. Further experiments will be conducted to investigate theregulatory roles of IGFL1 and C10orf99 in the psoriasis pathogenesis,and their potential use as psoriasis drug targets.
List of abbreviations usedDWD Distance Weighted DiscriminationRFE Recursive Feature Elimination
ROC receiver operating characteristicTPR true positive rateFPR false positive ratehere: IGFL1, insulin-like growth factor (IGF) like family member 1; IGFLR1, IGF-like family99; P85, p85 regulatory subunit; P110α, PtdIns-3-kinase subunit p110-alpha; PI3K,
osphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; NF-κB, nuclear factor of kappa

Fig. 6. The predicted structure of C10orf99. The alignment of C10orf99 to the bestmatchedenzyme, GRK, with the RMSD variance is only 3.38.
54 P. Guo et al. / Genomics 103 (2014) 48–55
AUC area under the ROC curvePCA principle component analysisIFS Incremental Feature SelectionGO Gene Ontology(IGF)-like (IGFL) insulin growth factor(GPCR) kinases (GRKs) G protein coupled receptor
Authors' contributions
FZ conceived and designed the study. PG, MZ, YL, and LG performedthe classification training and cross-validation. GM, GW and FLperformed the functional analysis and clinical characterization of theresults. FZ, PG and GM drafted the manuscript. All authors read andapproved the final manuscript.
Competing interests
Authors declare no potential competing interests.
Acknowledgments
This study was supported in part by the Shenzhen Research GrantZDSY20120617113021359, China 973 program (2011CB512003 and2010CB732606-6) and NSFC 31000447. Computing resources werepartly provided by the Dawning supercomputing clusters at SIAT CAS.We would like to thank the Health Informatics Laboratory (HILab)members for their helpful discussions. Constructive comments fromthe two anonymous reviewers are also appreciated.
Appendix A. Supplementary data
The data sets supporting the results of this study are cited in the textand additional files. The Supplementary Figs. S1–S2 and SupplementaryTables S1–S2 are available at http://www.healthinformaticslab.org/supp/. Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ygeno.2013.11.001.
References
[1] J. Villasenor-Park, D. Wheeler, L. Grandinetti, Psoriasis: evolving treatment for acomplex disease, Cleve. Clin. J. Med. 79 (2012) 413–423.
[2] T.W. Chu, T.F. Tsai, Psoriasis and cardiovascular comorbidities with emphasis in Asia,G. Ital. Dermatol. Venereol. 147 (2012) 189–202.
[3] L. Puig, Cardiovascular risk and psoriasis: the role of biologic therapy, ActasDermosifiliogr. 103 (2012) 853–862.
[4] J.M. Gelfand, E.D. Dommasch, D.B. Shin, R.S. Azfar, S.K. Kurd, X. Wang, A.B. Troxel, Therisk of stroke in patients with psoriasis, J. Invest. Dermatol. 129 (2009) 2411–2418.
[5] J. Vom Berg, S. Prokop, K.R. Miller, J. Obst, R.E. Kalin, I. Lopategui-Cabezas, A.Wegner,F. Mair, C.G. Schipke, O. Peters, Y. Winter, B. Becher, F.L. Heppner, Inhibition ofIL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitivedecline, Nat. Med. 18 (2012) 1812–1819.
[6] R.G. Langley, G.G. Krueger, C.E. Griffiths, Psoriasis: epidemiology, clinical features,and quality of life, Ann. Rheum. Dis. 64 (Suppl. 2) (2005) ii18–ii23(discussionii24-15).
[7] Psoriasis-Aid.com, Psoriasis Statistics, 2012.[8] D.D. Gladman, Natural history of psoriatic arthritis, Baillieres Clin. Rheumatol. 8
(1994) 379–394.[9] F. Chamian, M.A. Lowes, S.L. Lin, E. Lee, T. Kikuchi, P. Gilleaudeau, M.
Sullivan-Whalen, I. Cardinale, A. Khatcherian, I. Novitskaya, K.M. Wittkowski, J.G.Krueger, Alefacept reduces infiltrating T cells, activated dendritic cells, and inflam-matory genes in psoriasis vulgaris, Proc. Natl. Acad. Sci. U. S. A. 102 (2005)2075–2080.
[10] F. Chamian, J.G. Krueger, Psoriasis vulgaris: an interplay of T lymphocytes, dendriticcells, and inflammatory cytokines in pathogenesis, Curr. Opin. Rheumatol. 16 (2004)331–337.
[11] J.K. Kulski, W. Kenworthy, M. Bellgard, R. Taplin, K. Okamoto, A. Oka, T. Mabuchi, A.Ozawa, G. Tamiya, H. Inoko, Gene expression profiling of Japanese psoriatic skinreveals an increased activity in molecular stress and immune response signals,J. Mol. Med. 83 (2005) 964–975.
[12] Wikipedia, Psoriasis, 2012.[13] J.E. Gudjonsson, J. Ding, X. Li, R.P. Nair, T. Tejasvi, Z.S. Qin, D. Ghosh, A. Aphale, D.L.
Gumucio, J.J. Voorhees, G.R. Abecasis, J.T. Elder, Global gene expression analysisreveals evidence for decreased lipid biosynthesis and increased innate immunityin uninvolved psoriatic skin, J. Invest. Dermatol. 129 (2009) 2795–2804.
[14] J. Reischl, S. Schwenke, J.M. Beekman, U. Mrowietz, S. Sturzebecher, J.F. Heubach, In-creased expression of Wnt5a in psoriatic plaques, J. Invest. Dermatol. 127 (2007)163–169.
[15] Y. Yao, L. Richman, C.Morehouse,M. de los Reyes, B.W. Higgs, A. Boutrin, B.White, A.Coyle, J. Krueger, P.A. Kiener, B. Jallal, Type I interferon: potential therapeutic targetfor psoriasis? PLoS One 3 (2008) e2737.
[16] W.R. Swindell, A. Johnston, S. Carbajal, G. Han, C. Wohn, J. Lu, X. Xing, R.P. Nair, J.J.Voorhees, J.T. Elder, X.J. Wang, S. Sano, E.P. Prens, J. DiGiovanni, M.R. Pittelkow,N.L. Ward, J.E. Gudjonsson, Genome-wide expression profiling of fivemouse modelsidentifies similarities and differences with human psoriasis, PLoS One 6 (2011)e18266.
[17] R.P. Nair, K.C. Duffin, C. Helms, J. Ding, P.E. Stuart, D. Goldgar, J.E. Gudjonsson, Y. Li, T.Tejasvi, B.J. Feng, A. Ruether, S. Schreiber, M. Weichenthal, D. Gladman, P. Rahman, S.J.Schrodi, S. Prahalad, S.L. Guthery, J. Fischer, W. Liao, P.Y. Kwok, A. Menter, G.M.Lathrop, C.A. Wise, A.B. Begovich, J.J. Voorhees, J.T. Elder, G.G. Krueger, A.M. Bowcock,G.R. Abecasis, P. Collaborative Association Study of, Genome-wide scan reveals associa-tion of psoriasis with IL-23 and NF-kappaB pathways, Nat. Genet. 41 (2009) 199–204.
[18] T. Barrett, D.B. Troup, S.E. Wilhite, P. Ledoux, C. Evangelista, I.F. Kim, M.Tomashevsky, K.A. Marshall, K.H. Phillippy, P.M. Sherman, R.N. Muertter, M.Holko, O. Ayanbule, A. Yefanov, A. Soboleva, NCBI GEO: archive for functionalgenomics data sets—10 years on, Nucleic Acids Res. 39 (2011) D1005–D1010.
[19] R.A. Irizarry, B. Hobbs, F. Collin, Y.D. Beazer-Barclay, K.J. Antonellis, U. Scherf, T.P.Speed, Exploration, normalization, and summaries of high density oligonucleotidearray probe level data, Biostatistics 4 (2003) 249–264.
[20] M. Benito, J. Parker, Q. Du, J. Wu, D. Xiang, C.M. Perou, J.S. Marron, Adjustment ofsystematic microarray data biases, Bioinformatics 20 (2004) 105–114.
[21] M. Lauss, A. Frigyesi, T. Ryden, M. Hoglund, Robust assignment of cancer subtypesfrom expression data using a uni-variate gene expression average as classifier,BMC Cancer 10 (2010) 532.
[22] I. Guyon, J. Weston, S. Barnhill, V. Vapnik, Gene selection for cancer classificationusing support vector machines, Mach. Learn. 46 (2002) 389–422.
[23] M.B. Kursa, W.R. Rudnicki, Feature selection with the Boruta package, Journal, 2010.[24] A. Liaw, M.Wiener, Classification and regression by randomForest, R News 2 (2002)
18–22.[25] Y. Ding, D.Wilkins, Improving the performance of SVM-RFE to select genes inmicro-
array data, BMC Bioinforma. 7 (Suppl. 2) (2006) S12.[26] C. Furlanello, M. Serafini, S. Merler, G. Jurman, Entropy-based gene ranking without
selection bias for the predictive classification of microarray data, BMC Bioinforma. 4(2003) 54.
[27] C. Furlanello, M. Serafini, S. Merler, G. Jurman, An accelerated procedure for recur-sive feature ranking on microarray data, Neural Netw. 16 (2003) 641–648.
[28] V.N. Vapnik, The Nature of Statistical Learning Theory, 2 ed. Springer-Verlag, NewYork, 1999.
[29] J.A. Swets, Signal Detection Theory and ROC Analysis in Psychology and Diagnostics:Collected Papers, Lawrence Erlbaum Associates, Inc., Hillsdale, NJ, England, 1996.
[30] M.S. Pepe, G. Longton, G.L. Anderson, M. Schummer, Selecting differentiallyexpressed genes from microarray experiments, Biometrics 59 (2003) 133–142.
[31] M.B. Kursa, W.R. Rudnicki, Feature selection with the Boruta package, J. Stat. Softw.36 (2010) 1–13.

55P. Guo et al. / Genomics 103 (2014) 48–55
[32] D.A. Elizondo, R. Birkenhead, M. Gongora, E. Taillard, P. Luyima, Analysis and test ofefficient methods for building recursive deterministic perceptron neural networks,Neural Netw. 20 (2007) 1095–1108.
[33] K. Li, M. Yang, G. Sablok, J. Fan, F. Zhou, Screening features to improve the classprediction of acute myeloid leukemia and myelodysplastic syndrome, Gene 512(2013) 348–354.
[34] F. Zhou, Y. Xu, cBar: a computer program to distinguish plasmid-derived fromchromosome-derived sequence fragments in metagenomics data, Bioinformatics26 (2010) 2051–2052.
[35] X. Zhou, J. Mao, J. Ai, Y. Deng, M.R. Roth, C. Pound, J. Henegar, R. Welti, S.A. Bigler,Identification of plasma lipid biomarkers for prostate cancer by lipidomics andbioinformatics, PLoS One 7 (2012) e48889.
[36] A.P. Thrift, B.J. Kendall, N. Pandeya, D.C. Whiteman, A model to determineabsolute risk for esophageal adenocarcinoma, Clin. Gastroenterol. Hepatol. 11(2013)138–144.e2.
[37] X. Hu, H. Cammann, H.-A. Meyer, K. Miller, K. Jung, C. Stephan, Artificial neuralnetworks and prostate cancer—tools for diagnosis and management, Nat. Rev.Urol. 10 (2013) 174–182.
[38] S. Pegolo, G. Gallina, C. Montesissa, F. Capolongo, S. Ferraresso, C. Pellizzari, L. Poppi,M. Castagnaro, L. Bargelloni, Transcriptomic markers meet the real world: findingdiagnostic signatures of corticosteroid treatment in commercial beef samples,BMC Vet. Res. 8 (2012) 205.
[39] H. Akanuma, X.Y. Qin, R. Nagano, T.T. Win-Shwe, S. Imanishi, H. Zaha, J. Yoshinaga, T.Fukuda, S. Ohsako, H. Sone, Identification of stage-specific gene expression signa-tures in response to retinoic acid during the neural differentiation of mouse embry-onic stem cells, Front. Genet. 3 (2012) 141.
[40] G. Dennis Jr., B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane, R.A. Lempicki,DAVID: Database for Annotation, Visualization, and Integrated Discovery, GenomeBiol. 4 (2003) P3.
[41] Y. Benjamini, Y. Hochberg, Controlling the false discovery rate: a practical and powerfulapproach to multiple testing, J. R. Stat. Soc. Ser. B Methodol. 57 (1995) 289–300.
[42] D. Globe,M.S. Bayliss, D.J. Harrison, The impact of itch symptoms in psoriasis: resultsfrom physician interviews and patient focus groups, Health Qual. Life Outcomes 7(2009) 62.
[43] M.G. Boy, C. Wang, B.E. Wilkinson, V.F. Chow, A.T. Clucas, J.G. Krueger, A.S. Gaweco,S.H. Zwillich, P.S. Changelian, G. Chan, Double-blind, placebo-controlled,dose-escalation study to evaluate the pharmacologic effect of CP-690,550 in patientswith psoriasis, J. Invest. Dermatol. 129 (2009) 2299–2302.
[44] E. Guttman-Yassky, M. Suarez-Farinas, A. Chiricozzi, K.E. Nograles, A. Shemer, J.Fuentes-Duculan, I. Cardinale, P. Lin, R. Bergman, A.M. Bowcock, J.G. Krueger,Broad defects in epidermal cornification in atopic dermatitis identified throughgenomic analysis, J. Allergy Clin. Immunol. 124 (2009) 1235–1244(e1258).
[45] J.G. Bergboer, P.L. Zeeuwen, J. Schalkwijk, Genetics of psoriasis: evidence for epistaticinteraction between skin barrier abnormalities and immune deviation, J. Invest.Dermatol. 132 (2012) 2320–2321.
[46] J.G. Bergboer, P.L. Zeeuwen, J. Schalkwijk, Genetics of psoriasis: evidence for epistaticinteraction between skin barrier abnormalities and immune deviation, J. Invest.Dermatol. 132 (2013) 2320–2331.
[47] S. Madonna, C. Scarponi, S. Pallotta, A. Cavani, C. Albanesi, Anti-apoptotic effects ofsuppressor of cytokine signaling 3 and 1 in psoriasis, Cell Death Dis. 3 (2012) e334.
[48] M. Pasparakis, Role of NF-kappaB in epithelial biology, Immunol. Rev. 246 (2012)346–358.
[49] E. Krulig, K.B. Gordon, Ustekinumab: an evidence-based review of its effectivenessin the treatment of psoriasis, Core Evid. 5 (2010) 11–22.
[50] E.D. Roberson, Y. Liu, C. Ryan, C.E. Joyce, S. Duan, L. Cao, A.Martin,W. Liao, A.Menter,A.M. Bowcock, A subset of methylated CpG sites differentiate psoriatic from normalskin, J. Invest. Dermatol. 132 (2012) 583–592.
[51] A.A. Lobito, S.R. Ramani, I. Tom, J.F. Bazan, E. Luis, W.J. Fairbrother, W. Ouyang, L.C.Gonzalez, Murine insulin growth factor-like (IGFL) and human IGFL1 proteins areinduced in inflammatory skin conditions and bind to a novel tumor necrosis factorreceptor family member, IGFLR1, J. Biol. Chem. 286 (2011) 18969–18981.
[52] P. Emtage, P. Vatta, M. Arterburn, M.W. Muller, E. Park, B. Boyle, S. Hazell, R.Polizotto, W.D. Funk, Y.T. Tang, IGFL: a secreted family with conserved cysteineresidues and similarities to the IGF superfamily, Genomics 88 (2006) 513–520.
[53] S.M. Jones, A. Kazlauskas, Growth-factor-dependent mitogenesis requires twodistinct phases of signalling, Nat. Cell Biol. 3 (2001) 165–172.
[54] T. Fukushima, Y. Nakamura, D. Yamanaka, T. Shibano, K. Chida, S. Minami, T. Asano,F. Hakuno, S. Takahashi, Phosphatidylinositol 3-kinase (PI3K) activity bound toinsulin-like growth factor-I (IGF-I) receptor, which is continuously sustained byIGF-I stimulation, is required for IGF-I-induced cell proliferation, J. Biol. Chem. 287(2012) 29713–29721.
[55] M. Magrane, U. Consortium, UniProt Knowledgebase: a hub of integrated proteindata, Database (Oxford) 2011 (2011) bar009.
[56] F. Ponten, J.M. Schwenk, A. Asplund, P.H. Edqvist, The Human Protein Atlas as aproteomic resource for biomarker discovery, J. Intern. Med. 270 (2011) 428–446.
[57] A. Roy, A. Kucukural, Y. Zhang, I-TASSER: a unified platform for automated proteinstructure and function prediction, Nat. Protoc. 5 (2010) 725–738.
[58] Y. Zhang, J. Skolnick, Tertiary structure predictions on a comprehensive benchmarkof medium to large size proteins, Biophys. J. 87 (2004) 2647–2655.
[59] H. Zheng, C. Worrall, H. Shen, T. Issad, S. Seregard, A. Girnita, L. Girnita, Selective re-cruitment of G protein-coupled receptor kinases (GRKs) controls signaling of theinsulin-like growth factor 1 receptor, Proc. Natl. Acad. Sci. U. S. A. 109 (2012)7055–7060.
[60] T. Ogihara, T. Isobe, T. Ichimura, M. Taoka, M. Funaki, H. Sakoda, Y. Onishi, K. Inukai,M. Anai, Y. Fukushima, M. Kikuchi, Y. Yazaki, Y. Oka, T. Asano, 14-3-3 protein bindsto insulin receptor substrate-1, one of the binding sites of which is in thephosphotyrosine binding domain, J. Biol. Chem. 272 (1997) 25267–25274.
[61] Y. Benomar, A.F. Roy, A. Aubourg, J. Djiane, M. Taouis, Cross down-regulation ofleptin and insulin receptor expression and signalling in a human neuronal cellline, Biochem. J. 388 (2005) 929–939.
[62] Y. Liu, Identification of Genetic and Epigenetic Risk Factors for Psoriasis and PsoriaticArthritis in: Graduate School of Arts and Sciences, Washington University in St.Louis, St. Louis, MO, 2011. 193.
[63] S.H. Chu, D.F. Feng, Y.B. Ma, H. Zhang, Z.A. Zhu, Z.Q. Li, P.C. Jiang, Promoter methyl-ation and downregulation of SLC22A18 are associated with the development andprogression of human glioma, J. Transl. Med. 9 (2011) 156.




















