
Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR®: Biodistribution, pharmacokinetic...
7
Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR ® : Biodistribution, pharmacokinetic features and in vivo antitumor activity Donatella Paolino a , Donato Cosco b , Leda Racanicchi c , Elena Trapasso d , Christian Celia b , Michelangelo Iannone e ,Efisio Puxeddu c , Giuseppe Costante a , Sebastiano Filetti f , Diego Russo b , Massimo Fresta b, ⁎ a Department of Experimental and Clinical Medicine, University “Magna Græcia” of Catanzaro, Campus Universitario “S. Venuta” - Building of BioSciences, Viale Europa, I-88100 Germaneto (CZ), Italy b Department of Pharmacobiological Sciences, University “Magna Græcia” of Catanzaro, Campus Universitario “S. Venuta” - Building of BioSciences, Viale Europa, I-88100 Germaneto (CZ), Italy c Department of Internal Medicine, University of Perugia, I-06126 Perugia, Italy d Pharmacochemistry Department, University of Messina, Viale Annunziata, I-98168 Messina, Italy e ARPA Calabria - Environmental Epidemiology Centre, Complesso “Ninì Barbieri”, I-88021 Roccelletta di Borgia (CZ), Italy f Department of Clinical Sciences, University of Roma ‘Sapienza’, I-00155 Roma, Italy abstract article info Article history: Received 18 October 2009 Accepted 16 February 2010 Available online 22 February 2010 Keywords: Unilamellar PEGylated liposomes ARO cell xenograft model Biodistribution Gemcitabine Pharmacokinetic The systemic efficacy of the chemotherapeutic agents presently used to treat solid tumors is limited by their low therapeutic index. Previously, our research group improved the in vitro antitumoral activity of gemcitabine, an anticancer agent rapidly deaminated to the inactive metabolite 2′,2′-difluorodeoxyuridine, entrapping it into unilamellar pegylated liposomes made up of 1,2-dipalmitoyl-snglycero-3-phosphocholine monohydrate/cholesterol/N-(carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3- phosphoethanolamine (6:3:1 molar ratio). In this work, we investigated the in vivo efficiency of the gemcitabine liposomal formulation (5 mg/kg) with respect to the antitumoral commercial product GEMZAR ® (50 mg/kg) on an anaplastic thyroid carcinoma xenograft model obtaining similar effects in terms of inhibition of tumor mass proliferation after 4 weeks of treatment. The investigation of the carrier biodistribution and the drug pharmacokinetic profile furnished the rationalization of the efficacy of the vesicular system containing the active compound 10-fold less concentrated; in fact, liposomes promoted the concentration of the drug inside the tumor and they increased its plasmatic half-life. In addition, no signs of blood toxicity were observed when vesicular devices of effective doses of the drug were used. © 2010 Elsevier B.V. All rights reserved. 1. Introduction Novel strategies are presently under investigation in order to reduce the toxic effects elicited by new and conventional active compounds effective against human cancer. Promising approaches are the use of lower doses of anticancer drugs, which may maintain the same efficacy thanks to their combinations which allow them to act against different molecular targets of tumor cells, and/or the use of drug delivery systems, which protects the drug against metabolic inactivation and improves its biopharmaceutical features, i.e. blood circulation longevity and uptake within tumor cells [1,2]. Gemcitabine (GEM) is a new fluorinated nucleoside analogue (2′,2′-difluorodeoxycytidine) which has emerged as a very potent anti-tumour drug against different solid malignancies [3]. Unfortu- nately, when effective anti-tumor doses of this compound were used, haematological toxicity and other side effects appeared (although they are lower than those of other antineoplastic drugs) [4,5]. In addition, GEM is rapidly converted into the inactive metabolite 2′-deoxy-2′,2′- difluorouridine by cytidine deaminase following systemic adminis- tration. This metabolite is rapidly excreted in the urine [6,7]. To reduce these therapeutic complications and to improve the drug biopharma- ceutical features, such as plasmatic half-life, several strategies have been proposed [8], i) the encapsulation of the drug in the aqueous liposomal compartment after a pH gradient generation [9,10] or ii) its entrapment in vesicle bilayers or polymeric nanoparticles as lipophilic derivates [11,12], iii) the inclusion in polyaspartylhydrazide copoly- mer-based supramolecular vesicular aggregates [13], iv) the conjuga- tion to squalenoil acid in order to obtain autoassembling nanodevices [14,15] or v) to PEG-folate prodrugs to improve its tumor localization [16]. In particular, liposomal device offers the possibility to easily modulate the biopharmaceutical properties of the active compound without use of drug chemical structural modification such as covalent bonds as a consequence of the opportunity to modify the technological characteristics of the carrier. Journal of Controlled Release 144 (2010) 144–150 ⁎ Corresponding author. Tel.: + 39 0961 369 4118; fax: + 39 0961 369 4237. E-mail address: [email protected] (M. Fresta). 0168-3659/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.jconrel.2010.02.021 Contents lists available at ScienceDirect Journal of Controlled Release journal homepage: www.elsevier.com/locate/jconrel
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR®: Biodistribution, pharmacokinetic...

Journal of Controlled Release 144 (2010) 144–150
Contents lists available at ScienceDirect
Journal of Controlled Release
j ourna l homepage: www.e lsev ie r.com/ locate / j conre l
Gemcitabine-loaded PEGylated unilamellar liposomes vs GEMZAR®: Biodistribution,pharmacokinetic features and in vivo antitumor activity
Donatella Paolino a, Donato Cosco b, Leda Racanicchi c, Elena Trapasso d, Christian Celia b,Michelangelo Iannone e, Efisio Puxeddu c, Giuseppe Costante a, Sebastiano Filetti f,Diego Russo b, Massimo Fresta b,⁎a Department of Experimental and Clinical Medicine, University “Magna Græcia” of Catanzaro, Campus Universitario “S. Venuta” - Building of BioSciences, Viale Europa,I-88100 Germaneto (CZ), Italyb Department of Pharmacobiological Sciences, University “Magna Græcia” of Catanzaro, Campus Universitario “S. Venuta” - Building of BioSciences, Viale Europa, I-88100 Germaneto (CZ), Italyc Department of Internal Medicine, University of Perugia, I-06126 Perugia, Italyd Pharmacochemistry Department, University of Messina, Viale Annunziata, I-98168 Messina, Italye ARPA Calabria - Environmental Epidemiology Centre, Complesso “Ninì Barbieri”, I-88021 Roccelletta di Borgia (CZ), Italyf Department of Clinical Sciences, University of Roma ‘Sapienza’, I-00155 Roma, Italy
⁎ Corresponding author. Tel.: +39 0961 369 4118; faE-mail address: [email protected] (M. Fresta).
0168-3659/$ – see front matter © 2010 Elsevier B.V. Adoi:10.1016/j.jconrel.2010.02.021
a b s t r a c t
a r t i c l e i n f oArticle history:Received 18 October 2009Accepted 16 February 2010Available online 22 February 2010
Keywords:Unilamellar PEGylated liposomesARO cell xenograft modelBiodistributionGemcitabinePharmacokinetic
The systemic efficacy of the chemotherapeutic agents presently used to treat solid tumors is limited by theirlow therapeutic index. Previously, our research group improved the in vitro antitumoral activity ofgemcitabine, an anticancer agent rapidly deaminated to the inactive metabolite 2′,2′-difluorodeoxyuridine,entrapping it into unilamellar pegylated liposomes made up of 1,2-dipalmitoyl-snglycero-3-phosphocholinemonohydrate/cholesterol/N-(carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (6:3:1 molar ratio). In this work, we investigated the in vivo efficiency of thegemcitabine liposomal formulation (5 mg/kg) with respect to the antitumoral commercial productGEMZAR® (50 mg/kg) on an anaplastic thyroid carcinoma xenograft model obtaining similar effects interms of inhibition of tumor mass proliferation after 4 weeks of treatment. The investigation of the carrierbiodistribution and the drug pharmacokinetic profile furnished the rationalization of the efficacy of thevesicular system containing the active compound 10-fold less concentrated; in fact, liposomes promoted theconcentration of the drug inside the tumor and they increased its plasmatic half-life. In addition, no signs ofblood toxicity were observed when vesicular devices of effective doses of the drug were used.
x: +39 0961 369 4237.
ll rights reserved.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Novel strategies are presently under investigation in order toreduce the toxic effects elicited by new and conventional activecompounds effective against human cancer. Promising approaches arethe use of lower doses of anticancer drugs, which may maintain thesame efficacy thanks to their combinations which allow them to actagainst different molecular targets of tumor cells, and/or the use ofdrug delivery systems, which protects the drug against metabolicinactivation and improves its biopharmaceutical features, i.e. bloodcirculation longevity and uptake within tumor cells [1,2].
Gemcitabine (GEM) is a new fluorinated nucleoside analogue(2′,2′-difluorodeoxycytidine) which has emerged as a very potentanti-tumour drug against different solid malignancies [3]. Unfortu-nately, when effective anti-tumor doses of this compound were used,
haematological toxicity and other side effects appeared (although theyare lower than those of other antineoplastic drugs) [4,5]. In addition,GEM is rapidly converted into the inactive metabolite 2′-deoxy-2′,2′-difluorouridine by cytidine deaminase following systemic adminis-tration. Thismetabolite is rapidly excreted in the urine [6,7]. To reducethese therapeutic complications and to improve the drug biopharma-ceutical features, such as plasmatic half-life, several strategies havebeen proposed [8], i) the encapsulation of the drug in the aqueousliposomal compartment after a pH gradient generation [9,10] or ii) itsentrapment in vesicle bilayers or polymeric nanoparticles as lipophilicderivates [11,12], iii) the inclusion in polyaspartylhydrazide copoly-mer-based supramolecular vesicular aggregates [13], iv) the conjuga-tion to squalenoil acid in order to obtain autoassembling nanodevices[14,15] or v) to PEG-folate prodrugs to improve its tumor localization[16]. In particular, liposomal device offers the possibility to easilymodulate the biopharmaceutical properties of the active compoundwithout use of drug chemical structural modification such as covalentbonds as a consequence of the opportunity tomodify the technologicalcharacteristics of the carrier.

145D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
In previous investigations, we efficiently encapsulated GEM inPEGylated liposomes (L-GEM) demonstrating the in vitro enhancedcytotoxic effect of this formulation with respect to the free drug. Inparticular, small unilamellar PEGylated liposomes showed a greaterantitumoral effect on different cancer cell lines due to a more effectivecellular uptake of gemcitabine that induced more evident apoptoticeffects [17,18]. The in vitro effective cytotoxic doses of L-GEM werewithin an interval ranging from 0.01 to 0.1 μM, which is compatiblewith a potential in vivo administration for the efficacious therapeutictreatment of cancer diseases.
In the present study we compare the in vivo antitumoral efficacyof our L-GEM formulation with respect to the commercial productGEMZAR®. In particular, the in vivo antitumor activity of GEMZAR®
administered at 50 mg/kg was tested in an immunodeficient NOD-SCID mouse xenograft model and compared to L-GEM administeredat a dose (5 mg/kg) 10-fold lower than GEMZAR®. Considering thatL-GEM formulation was designed to exhibit a long circulating be-haviour, the biodistribution of [3H]-hexadecyl-cholesterol-labelledL-GEM and the pharmacokinetic of L-GEMwith respect to GEMZAR®
were investigated. Finally, the histological sections of tumor massesobtained from mice following 1 month of treatment with thedifferent formulations were examined and the haematologicaleffects were also studied.
2. Materials and methods
2.1. Chemicals and biochemicals
The phospholipids, 1,2-dipalmitoyl-sn-glycero-3-phospocholinemonohydrate (DPPC) and N-(carbonyl-methoxypolyethylene glycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-mPEG 2000), were purchased from Genzyme (Suffolk, UK). Choles-terol (Chol), eosin B solution, hematoxylin solution and phosphatesaline tablets (for the preparation of phosphate buffer solution pH∼7.4 — PBS) were purchased from Sigma Chemicals Co. (St. Louis,MI, USA). [3H]cholesteryl hexadecyl ether ([3H]CHE, 40 Ci/mmol) and[5-3H]gemcitabine (5–20 Ci/mmol) were obtained from PerkinElmer-Italia (Monza, Italy) and Campro Scientific GmbH (Berlin,Germany), respectively. Gemcitabine hydrochloride (HPLCpurityN99%) was a gift of Eli-Lilly Italia S.p.a. (Sesto Fiorentino,Italy) while GEMZAR®was a gift of Prof. P. Tassone (Medical OncologyUnit, University of Catanzaro). Isotonic NaCl (0.9% w/v) solution(saline) was a Frekenius Kabi Potenza S.r.l. (Verona, Italy) product. Allother materials and solvents used in this investigation were ofanalytical grade (Carlo Erba, Milan, Italy).
2.2. Liposome preparation
Gemcitabine-loaded PEGylated unilamellar liposomes wereobtained as previously described [17]. Briefly, liposomes, made upof DPPC/Chol/DSPE-MPEG 2000 (6:3:1 molar ratio), a similar lipidcomposition of Doxil® [19] approved by the FDA, were prepared bydissolving 40 mg of this lipid mixture in a round-bottomed flask byusing a chloroform/methanol (3:1 v/v) solvent mixture. Organic sol-vents were removed by means of a R210 rotary evaporator (Büchi-Italia, Milan, Italy) under slow nitrogen flux, thus allowing theformation of a thin layer of film on the inner wall of a round-bottomedflask. Any trace of residual solvent was eliminated overnight at roomtemperature by using a Büchi T51 glass drying oven connected to avacuum pump. Lipid films were hydrated with a 250 mM ammoniumsulphate solution (2 ml) by stirring (700 rpm) at 45 °C for 45 min. Theobtained multilamellar liposomes were submitted to ten cycles offreezing (with liquid nitrogen) and thawing (with a water bath at45 °C), then were extruded through 400 nm and 200 nm pore sizepolycarbonate membrane filters (Costar, Corning Incorporated, NY,USA) by using a stainless steel extrusion device (Lipex Biomembranes,
Vancouver, B.C., Canada) and untrapped ammonium sulphate solutionwas removed by centrifugation (145,000×g) (Beckman Optima™
ultracentrifuge equipped with a TL S55 fixed angle rotor) at 4 °C for1 h. Our previous gel permeation chromatographic experiments [17]showed the absence of micelles due to the presence of DSPE-MPEG2000 at 10% mol. Finally, the obtained unilamellar liposomes weresuspended in an isotonic solution (2 ml) of gemcitabine hydrochlo-ride (1 mM) and kept at room temperature for 3 h. In this way a pHgradient was achieved with an acid environment in the intra-lipo-somal aqueous compartments and noteworthy drug entrapment wasachieved [20].
2.3. Physicochemical characterization of L-GEM
Mean size and size distribution (polydispersity index) of lipo-somes were evaluated by dynamic light-scattering experiments.Zetamaster (Malvern Instruments Ltd., Spring Lane South, Worcs,UK), a photo-correlation spectroscopy apparatus, was used for thedimensional analysis [21]. Vesicle morphology and lamellarity wasexamined by means of the freeze-fracture microscopy technique [21].
2.4. Antitumor activity in xenograft animals and histological analysis
Animal experiments were carried out in agreement with theprinciples and procedures outlined by the local Ethical Committee.ARO cells (1×106), an anaplastic thyroid carcinoma cell line, werecultured as previously described [17], were diluted in 100 μL of PBSand subcutaneously injected into the flank of immunodeficient NOD-SCID mice (Harlan Italy s.r.l., San Pietro al Natisone (UD), Italy). Whenthe tumor reached a 10-mm diameter, three groups of mice (n=5each) were treated i.v. (200 μl) with GEMZAR® (5 mg/kg or 50 mg/kg)or L-GEM (5 mg/kg) every three days. Control mice (n=5) received200 μl of saline, while blank mice (n=5) were treated with unloadedliposomes. The sizes of tumor masses were measured with a caliperand tumor volumes were calculated according to the formula:V=0.5×ab2, where a and b are the long and short diameter of thetumor, respectively. The body weight, feeding behaviour and motoractivity of mice were used as indicators of general health.
To evaluate the histological changes in neoplastic masses deter-mined by the treatments with the different formulations, after a fourweek treatment course, immunodeficient NOD-SCID mice weresacrificed by cervical dislocation (Approved by the JHU Animal Careand Use Committee on: April 17, 2003-revised 9/28/2006, 7/17/08AVMA Guidelines on Euthanasia June 2007. Available at www.jhu.edu/animalcare). Tumor masses were eradicated for the analysis,fixed with 4% (w/v) buffered formaldehyde (pH=7.4) at roomtemperature, dehydrated in alcohol and then embedded in paraffin.Sections with a thickness of 7–10 μm were sliced using a microtome(Leica RM2255) and were stained following the eosin B/hematoxylinmethod [22]. Stained samples were examined with a Leitz-orthoplanphotomicroscope equipped with a Leica DC200 photocamera. Imageswere acquired by Leica IM1000 software (Leica Microsystems,Heerbrugg, Switzerland).
2.5. L-GEM biodistribution
Biodistribution experiments were carried out in NOD-SCID mice(1 month old, 25–30 g), previously inoculated with ARO cells in orderto induce tumor formation. [3H]CHE (0.003% w/w corresponding to3 nmol of [3H]CHE) radiolabeled liposomes (200 μL, ∼4 mg of totallipid mixture) were injected into mice through the tail vein [23]. Atdifferent times (2, 4, 8, 12 and 24 h), the mice (three per point) weresacrificed by cervical dislocation and hearts, lungs, livers, spleens,kidneys, blood and tumor masses were collected. Complete organswere transferred in polypropylene liquid scintillation vials (20 mL-size) (Sigma-Aldrich Chemie, GmbH, Steinheim, Germany) andmixed

146 D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
with a quaternary ammonium hydroxide solution (2 ml) (BTS-450,Beckman Instruments, Inc., Fullerton, Netherlands) under continuousshaking for 4 h at 60 °C using an incubator shaker (Innova™ 4300,New Brunswick Scientific, Edison, NJ, USA) to allow completedissolution. At the end of the incubation time, various samples weredecolorized with 2 ml of 24% (v/v) H2O2 at room temperature for5 min, the liquid scintillation cocktail (7 ml) (Ready Organic™, Beck-man Coulter Inc., Fullerton, USA) was added, these mixtures werevigorously mixed and analyzed using a Wallac Win Spectral™ 1414liquid scintillation counter (PerkinElmer Life and Analytical Sciences,Inc. Waltham, MA, USA). A 1414 Win Spectral Wallac LCS Softwarewas used for data analysis. To eliminate the blood radioactivitycontained in the various organ samples, derived from the contributionof liposomes in the vascular space, as well as the tissue parenchyma(including the macrophages and the capillary endothelial cells)(Xtissue), a correction was made according to the following equation:
Xtissue = Xorgan− ðV0 × CðtÞÞ
where, Xorgan represented levels of radioactivity recovered from thevarious organ samples; V0 was the total volume of the vascular spaceand interstitial fluid, as determined by the radioactivity level in thewhole organ samples divided by the blood concentration 10 min afterthe i.v. injection of the [3H]CHE-L-GEM and C(t) was the bloodconcentration at the indicated time [23]. A further quenching cor-rection factor was obtained by measuring the radioactivity of blanktissues from non-injected mice spiked with known amounts of [3H]CHE (0.030 µCu/µmol lipids) [24]. Three different experiments werecarried out. In order to confirm the trend deriving from the bio-distribution of [3H]CHE labelled liposomes and to evaluate if the drugfollows the biological fate of the liposomal carrier, the sameexperiment was performed by using [5-3H]gemcitabine. In particular,6 nmol of the compound were encapsulated in the aqueous com-partment of the vesicular device and injected into mice throughthe tail vein. The biodistribution of free [5-3H]gemcitabine was alsoevaluated.
2.6. Evaluation of blood toxicity
The modification of blood biochemical indexes was evaluated tomeasure the blood toxicity index of free GEM and L-GEM. Four groups(five mice per group) were treated i.v. with 200 μl of GEM (5 mg/kgand 50 mg/kg), L-GEM (5 mg/kg) and unloaded PEGylated liposomesevery three days for 30 days. Then, samples (0.5 ml) were collectedvia the ocular vein plexus and immediately frozen at −80 °C. Suc-cessively, blood samples were defrosted and centrifuged (10 min atroom temperature) at 14,300 rpm by using an Eppendorf 5702 cen-trifuge (Eppendorf, Milan, Italy) for separating the serum. Differentblood parameters were then measured by using a biochemical auto-analyzer (Type 7170, Hitachi, Japan). The samples obtained by healthymice were used as control.
2.7. Pharmacokinetic experiments
Immunodeficient NOD-SCID mice were also used for pharmacoki-netic studies. GEMZAR® or L-GEM (1 mg drug/kg) were injected(100 μl) through the tail vein (groups of three animals per each timepoint). Blood samples (200 μl) were taken from the retro-orbitalplexus and frozen. Before analysis, samples were immediately de-frosted and centrifuged (14,300×g, 10 min) at room temperature andglacial acetic acid (50 μl) was added to plasma samples to decreasehydrogen bonding between nucleosides and proteins. Acetonitrile(1 ml) (HPLC grade) was added to plasma samples, which were thenvortex-mixed and centrifuged at 800×g for 15 min at 4 °C. Thesupernatant was removed and collected in a glass tube andacetonitrile (1 ml) was added to the pellet. Three cycles of vortex-
mixing and centrifugation procedure were carried out. Supernatantswere combined, evaporated to dryness under nitrogen flux at 42 °C(thermostated water bath) and stored at −20 °C. Before the HPLCanalysis, the residue was resuspended in water (1 ml) (HPLC grade),incubated for 5 min at 37 °C and then centrifuged at 12,000×g for10 min at 20 °C. The supernatant was removed, filtered through a0.22 μmpore size Anotop 10 syringe filter (Whatman, SpringfieldMill,UK) and placed in 4-ml HPLC glass vials for analytical determination.Analysis was carried out using a HPLC system (Varian Inc., Palo Alto,CA, USA) composed of a 200-2031Metachem online degasser, a M210binary pump, a ProStar 410 autosampler, a G1316A thermostatedcolumn compartment, and a 25 μl CSL20 Cheminert Sample Loopinjector. Data were acquired and processed with a Galaxie® chroma-tography manager software (Varian Inc.). Chromatographic separa-tion was carried out at room temperature using a GraceSmart RPC18 column (4.6×250 mm, 5 μm particle size, Alltech Grom GmbH,Rottenburg-Hailfingen, Germany). The mobile phase was water/acetonitrile (95:5 v/v). The flow rate was 1 ml/min and UV detectionwas performed at 269 nm.
No interference coming from plasma components was observedfor GEM and its metabolite (2I,2I-difluorodeoxyuridine) HPLC peaks.The chromatographic method provided a suitable separation of thepeaks of GEM and 2I,2I-difluorodeoxyuridine, which showed a re-tention time of 6.00 and 8.70 min, respectively. GEM quantificationwas carried out using an external standard curve in the linear con-centration range between 0.1 and 10 μg/ml. A standard solution ofGEM (1 mg/ml) was used for the construction of the standard curve.Plasmatic amounts of GEMwere determined using the standard curveaccording to the following equation:
AUC = 0:60112x + 0:02840
where x is the drug concentration (μg/ml) and AUC the area under thecurve (mAu×min). Pharmacokinetic calculations were performed byusing WinNonlin Professional, ver. 5.3 (Tharsight, Mountain View,CA). GEM plasmatic amounts were expressed as μg/ml. Experimentaldata are the mean of three different experiments.
2.8. Statistical analysis
All data were expressed as means ±SD. Differences between thevarious experimental points were evaluated with one-way analysis ofvariance (ANOVA) followed by the Turkey-Kramer multiple compar-ison test using GrafPAD Software for Science (San Diego, CA, USA).
3. Results and discussion
The generation of an acid pH-gradient [9,17,25] during the L-GEMpreparation procedure, between the inner aqueous compartments ofthe PEGylated unilamellar liposomes (acid pH environment) and thevesicular colloidal dispersion medium, provided a drug loadingcapacity of ∼90%, according to our previous findings [10,18]. Thesuitable encapsulation capacity of the L-GEM formulation as well asthe vesicle mean sizes (∼200 nm) (Fig. 1), the homogenous sizedistribution of the vesicle colloidal dispersion (polydispersity index of∼0.05), the z-potential value (−2 mV) and the presence of PEG on thesurface of liposomes make L-GEM a suitable candidate for the in vivodelivery of GEM to be used for the therapeutic treatment of cancerdiseases [26].
The technological parameters and the results previously obtainedin in vitro experiments [17] encouraged a further in vivo investigationto evaluate the real potentiality of using L-GEM as an effective for-mulation for the therapeutic treatment of solid cancer diseases. Inparticular, as shown in Fig. 2A, after 4 weeks of treatment, the volumeof the tumors in the mice treated with L-GEM (5 mg/kg) andGEMZAR® (50 mg/kg) was comparable (∼0.9 cm3), while that of
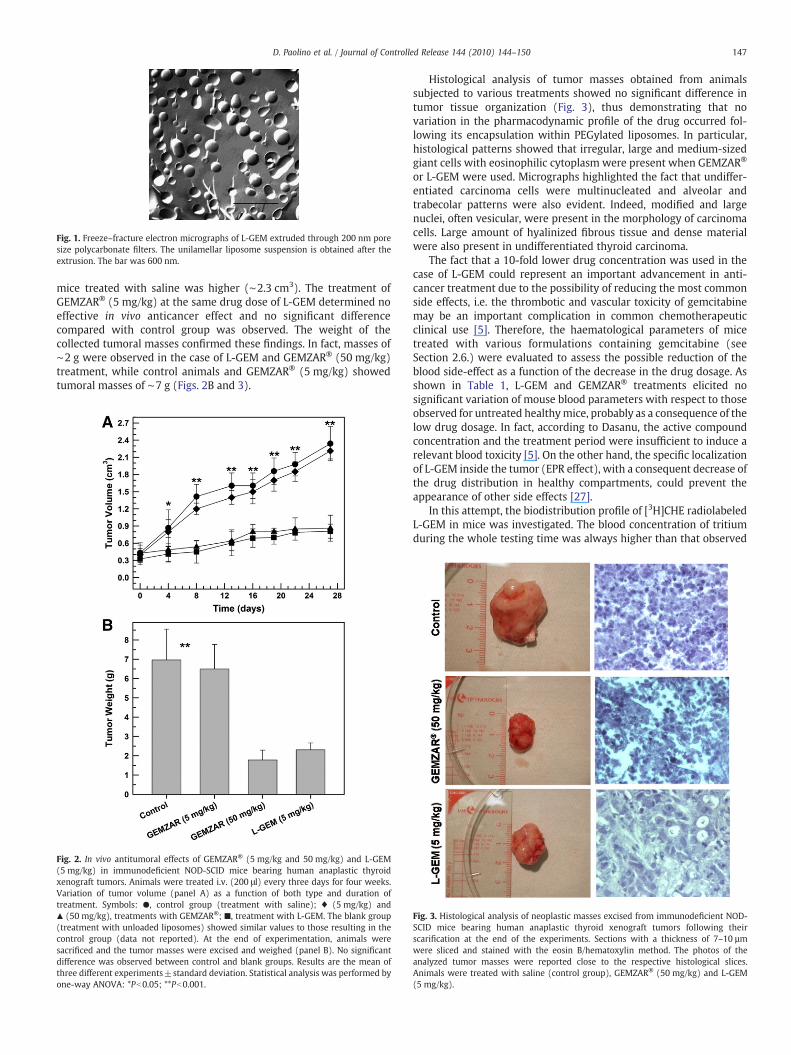
Fig. 1. Freeze–fracture electron micrographs of L-GEM extruded through 200 nm poresize polycarbonate filters. The unilamellar liposome suspension is obtained after theextrusion. The bar was 600 nm.
147D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
mice treated with saline was higher (∼2.3 cm3). The treatment ofGEMZAR® (5 mg/kg) at the same drug dose of L-GEM determined noeffective in vivo anticancer effect and no significant differencecompared with control group was observed. The weight of thecollected tumoral masses confirmed these findings. In fact, masses of∼2 g were observed in the case of L-GEM and GEMZAR® (50 mg/kg)treatment, while control animals and GEMZAR® (5 mg/kg) showedtumoral masses of ∼7 g (Figs. 2B and 3).
Fig. 2. In vivo antitumoral effects of GEMZAR® (5 mg/kg and 50 mg/kg) and L-GEM(5 mg/kg) in immunodeficient NOD-SCID mice bearing human anaplastic thyroidxenograft tumors. Animals were treated i.v. (200 μl) every three days for four weeks.Variation of tumor volume (panel A) as a function of both type and duration oftreatment. Symbols: ●, control group (treatment with saline); ♦ (5 mg/kg) and▲ (50 mg/kg), treatments with GEMZAR®; ■, treatment with L-GEM. The blank group(treatment with unloaded liposomes) showed similar values to those resulting in thecontrol group (data not reported). At the end of experimentation, animals weresacrificed and the tumor masses were excised and weighed (panel B). No significantdifference was observed between control and blank groups. Results are the mean ofthree different experiments±standard deviation. Statistical analysis was performed byone-way ANOVA: *Pb0.05; **Pb0.001.
Histological analysis of tumor masses obtained from animalssubjected to various treatments showed no significant difference intumor tissue organization (Fig. 3), thus demonstrating that novariation in the pharmacodynamic profile of the drug occurred fol-lowing its encapsulation within PEGylated liposomes. In particular,histological patterns showed that irregular, large and medium-sizedgiant cells with eosinophilic cytoplasm were present when GEMZAR®
or L-GEM were used. Micrographs highlighted the fact that undiffer-entiated carcinoma cells were multinucleated and alveolar andtrabecolar patterns were also evident. Indeed, modified and largenuclei, often vesicular, were present in the morphology of carcinomacells. Large amount of hyalinized fibrous tissue and dense materialwere also present in undifferentiated thyroid carcinoma.
The fact that a 10-fold lower drug concentration was used in thecase of L-GEM could represent an important advancement in anti-cancer treatment due to the possibility of reducing the most commonside effects, i.e. the thrombotic and vascular toxicity of gemcitabinemay be an important complication in common chemotherapeuticclinical use [5]. Therefore, the haematological parameters of micetreated with various formulations containing gemcitabine (seeSection 2.6.) were evaluated to assess the possible reduction of theblood side-effect as a function of the decrease in the drug dosage. Asshown in Table 1, L-GEM and GEMZAR® treatments elicited nosignificant variation of mouse blood parameters with respect to thoseobserved for untreated healthymice, probably as a consequence of thelow drug dosage. In fact, according to Dasanu, the active compoundconcentration and the treatment period were insufficient to induce arelevant blood toxicity [5]. On the other hand, the specific localizationof L-GEM inside the tumor (EPR effect), with a consequent decrease ofthe drug distribution in healthy compartments, could prevent theappearance of other side effects [27].
In this attempt, the biodistribution profile of [3H]CHE radiolabeledL-GEM in mice was investigated. The blood concentration of tritiumduring the whole testing time was always higher than that observed
Fig. 3. Histological analysis of neoplastic masses excised from immunodeficient NOD-SCID mice bearing human anaplastic thyroid xenograft tumors following theirscarification at the end of the experiments. Sections with a thickness of 7–10 µmwere sliced and stained with the eosin B/hematoxylin method. The photos of theanalyzed tumor masses were reported close to the respective histological slices.Animals were treated with saline (control group), GEMZAR® (50 mg/kg) and L-GEM(5 mg/kg).
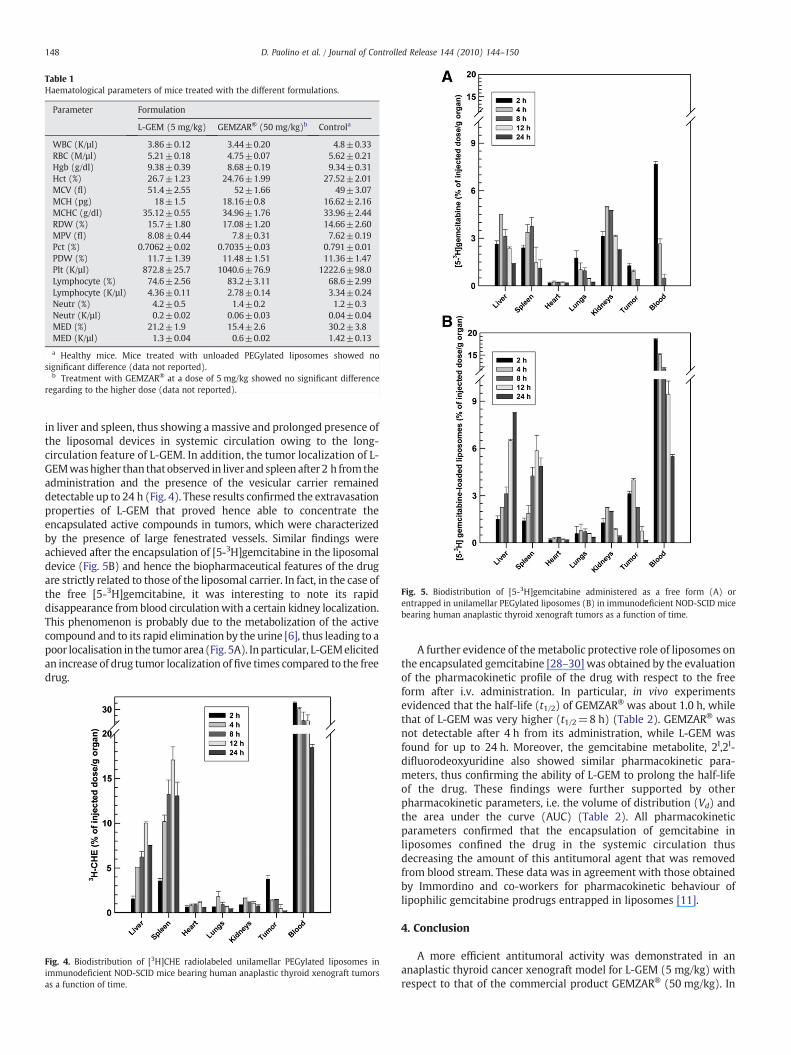
Table 1Haematological parameters of mice treated with the different formulations.
Parameter Formulation
L-GEM (5 mg/kg) GEMZAR® (50 mg/kg)b Controla
WBC (K/µl) 3.86±0.12 3.44±0.20 4.8±0.33RBC (M/µl) 5.21±0.18 4.75±0.07 5.62±0.21Hgb (g/dl) 9.38±0.39 8.68±0.19 9.34±0.31Hct (%) 26.7±1.23 24.76±1.99 27.52±2.01MCV (fl) 51.4±2.55 52±1.66 49±3.07MCH (pg) 18±1.5 18.16±0.8 16.62±2.16MCHC (g/dl) 35.12±0.55 34.96±1.76 33.96±2.44RDW (%) 15.7±1.80 17.08±1.20 14.66±2.60MPV (fl) 8.08±0.44 7.8±0.31 7.62±0.19Pct (%) 0.7062±0.02 0.7035±0.03 0.791±0.01PDW (%) 11.7±1.39 11.48±1.51 11.36±1.47Plt (K/µl) 872.8±25.7 1040.6±76.9 1222.6±98.0Lymphocyte (%) 74.6±2.56 83.2±3.11 68.6±2.99Lymphocyte (K/µl) 4.36±0.11 2.78±0.14 3.34±0.24Neutr (%) 4.2±0.5 1.4±0.2 1.2±0.3Neutr (K/µl) 0.2±0.02 0.06±0.03 0.04±0.04MED (%) 21.2±1.9 15.4±2.6 30.2±3.8MED (K/µl) 1.3±0.04 0.6±0.02 1.42±0.13
a Healthy mice. Mice treated with unloaded PEGylated liposomes showed nosignificant difference (data not reported).
b Treatment with GEMZAR® at a dose of 5 mg/kg showed no significant differenceregarding to the higher dose (data not reported).
Fig. 5. Biodistribution of [5-3H]gemcitabine administered as a free form (A) orentrapped in unilamellar PEGylated liposomes (B) in immunodeficient NOD-SCID micebearing human anaplastic thyroid xenograft tumors as a function of time.
148 D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
in liver and spleen, thus showing amassive and prolonged presence ofthe liposomal devices in systemic circulation owing to the long-circulation feature of L-GEM. In addition, the tumor localization of L-GEMwashigher than thatobserved in liver and spleen after 2 h fromtheadministration and the presence of the vesicular carrier remaineddetectable up to 24 h (Fig. 4). These results confirmed the extravasationproperties of L-GEM that proved hence able to concentrate theencapsulated active compounds in tumors, which were characterizedby the presence of large fenestrated vessels. Similar findings wereachieved after the encapsulation of [5-3H]gemcitabine in the liposomaldevice (Fig. 5B) and hence the biopharmaceutical features of the drugare strictly related to those of the liposomal carrier. In fact, in the case ofthe free [5-3H]gemcitabine, it was interesting to note its rapiddisappearance from blood circulationwith a certain kidney localization.This phenomenon is probably due to the metabolization of the activecompound and to its rapid elimination by the urine [6], thus leading to apoor localisation in the tumor area (Fig. 5A). Inparticular, L-GEMelicitedan increase of drug tumor localization offive times compared to the freedrug.
Fig. 4. Biodistribution of [3H]CHE radiolabeled unilamellar PEGylated liposomes inimmunodeficient NOD-SCID mice bearing human anaplastic thyroid xenograft tumorsas a function of time.
A further evidence of the metabolic protective role of liposomes onthe encapsulated gemcitabine [28–30] was obtained by the evaluationof the pharmacokinetic profile of the drug with respect to the freeform after i.v. administration. In particular, in vivo experimentsevidenced that the half-life (t1/2) of GEMZAR® was about 1.0 h, whilethat of L-GEM was very higher (t1/2=8 h) (Table 2). GEMZAR® wasnot detectable after 4 h from its administration, while L-GEM wasfound for up to 24 h. Moreover, the gemcitabine metabolite, 2I,2I-difluorodeoxyuridine also showed similar pharmacokinetic para-meters, thus confirming the ability of L-GEM to prolong the half-lifeof the drug. These findings were further supported by otherpharmacokinetic parameters, i.e. the volume of distribution (Vd) andthe area under the curve (AUC) (Table 2). All pharmacokineticparameters confirmed that the encapsulation of gemcitabine inliposomes confined the drug in the systemic circulation thusdecreasing the amount of this antitumoral agent that was removedfrom blood stream. These data was in agreement with those obtainedby Immordino and co-workers for pharmacokinetic behaviour oflipophilic gemcitabine prodrugs entrapped in liposomes [11].
4. Conclusion
A more efficient antitumoral activity was demonstrated in ananaplastic thyroid cancer xenograft model for L-GEM (5 mg/kg) withrespect to that of the commercial product GEMZAR® (50 mg/kg). In

Table 2Plasmatic pharmacokinetic parameters of GEM and its inactive metabolite, 2I,2I-difluorodeoxyuridine, after a single intravenous administration in immunodeficientNOD-SCID mice (with xenograft tumors).
Sample Pharmacokinetic parameters
T1/2(h)
Cmax
(μg/ml)Tmax
(h)Vd
(ml)bAUC(μg/ml×h)c
Free GEM 1.0±0.12 1.30±0.17 – 23.00±0.19 2.197±0.049L-GEMa 8.0±0.26 1.42±0.11 – 19.48±0.23 15.749±0.036Metabolitefrom GEM
4.0±0.24 2.51±0.15 0.5±0.2 11.62±0.14 21.992±0.098
Metabolitefrom L-GEMa
10.0±0.39 3.14±0.18 2.0±0.1 9.091±0.32 48.488±0.079
a All data of pharmacokinetic parameters have an ANOVA significance Pb0.001 withrespect to free GEM and metabolite from GEM.
b The apparent volume (Vd) was calculated according to the equation Vd=(ADD)t0/(PDC)t0, where ADD is the administered drug dose (1 mg/kg) at time t=0 and PDC isthe plasma drug concentration (µg ml−1) at time t=0, as extrapolated value on semi-log y-axis of the pharmacokinetic profile.
c The areas under the plasma concentration-time curve (AUC) (starting from the firstto the last sampling time) was calculated using the trapezoidal rule with extrapolationto infinity using the ratio Cn/Ke, where Cnwas the last measurable concentration and Keis the constant of drug elimination.
149D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
fact, the entrapment of gemcitabine within a PEGylated liposomalcarrier determined a noticeable improvement of the drug therapeuticindex by ameliorating the drug pharmacokinetic features, substan-tially increasing the drug's tumor localization. For these reasons, L-GEM has a potential therapeutic relevance for the treatment ofsolid tumors.
It should be also taken into consideration that innovative promisingapproaches to treating carcinomas, such as thyroid cancer, are based onsubstances designed to act against new molecular targets, i.e. thetyrosine-kinase inhibitor, which showed itself to be particularlyeffective in experimentalmodels, especiallywhen tested in combinationwith cytotoxicmolecules [31–33]. Considering our data, thepossible usein clinical trials of liposomes as a multidrug carrier [34–36] for theconcomitant delivery of various substances acting against differentmolecular targets is of particular interest. In this attempt, a suitableliposomal carrier containing gemcitabine and a tyrosine-kinase inhib-itor [37] is currently under investigation in our laboratory.
Acknowledgements
This workwas financially supported by grants from theMIUR Cofin2005 Program (Prof. Diego Russo), the MIUR PRIN 2006 Program(Prof. Massimo Fresta), the Fondazione Umberto Di Mario (Prof.Sebastiano Filetti) and the Italian Ministry of Health - RegioneCalabria (Dipartimento Tutela della Salute Politiche Sanitarie eSociali). The authors are very grateful to Dr. Lynn Whitted for herrevision of the language of this manuscript and toMs. Olga Amelio andMr. Angelo Rocca (ARPA Calabria - Environmental EpidemiologyCentre) for the administrative support.
References
[1] G. Cavallaro, L. Maniscalco, M. Licciardi, G. Giammona, Tamoxifen-loadedpolymeric micelles: preparation, physico-chemical characterization and in vitroevaluation studies, Macromol. Biosci. 4 (2004) 1028–1038.
[2] A.A. Gabizon, H. Shmeeda, S. Zalipsky, Pros and cons of the liposome platform incancer drug targeting, J. Liposome Res. 16 (2006) 175–183.
[3] L. Toschi, G. Finocchiaro, S. Bartolini, V. Gioia, F. Cappuzzo, Role of gemcitabine incancer therapy, Future Oncol. 1 (2005) 7–17.
[4] F.V. Fossella, S.M. Lippman, D.M. Shin, P. Tarassoff, M. Calayag-Jung, R. Perez-Soler,J.S. Lee, W.K. Murphy, B. Glisson, E. Rivera, W.K. Hong, Maximum-tolerated dosedefined for single-agent gemcitabine. A phase I dose–escalation study inchemotherapy-naïve patients with advanced non-small-cell lung cancer, J. Clin.Oncol. 15 (1997) 310–316.
[5] C.A. Dasanu, Gemcitabine: vascular toxicity and prothrombotic potential, ExpertOpin. Drug Saf. 7 (2008) 703–716.
[6] J.L. Abbruzzese, R. Grunewald, E.A. Weeks, D. Gravel, T. Adams, B. Nowak, S.Mineishi, P. Tarassoff, W. Satterlee, M.N. Raberet, A phase I clinical, plasma, andcellular pharmacology study of gemcitabine, J. Clin. Oncol. 9 (1991) 491–498.
[7] A.M. Storniolo, S.R.B. Allerheiligen, H.L. Pearce, Preclinical, pharmacologic, andphase I studies of gemcitabine, Semin. Oncol. 24 (2 suppl 7) (1997) S7-2-S7-7.
[8] L.H. Reddy, P. Couvreur, Novel approaches to deliver gemcitabine to cancers, Curr.Pharm. Des. 14 (2008) 1124–1137.
[9] M. Celano, M.G. Calvagno, S. Bulotta, D. Paolino, F. Arturi, D. Rotiroti, S. Filetti, M.Fresta, D. Russo, Cytotoxic effects of Gemcitabine-loaded liposomes in humananaplastic thyroid carcinoma cells, BMC Cancer 4 (2004) 63.
[10] M.G. Calvagno, C. Celia, D. Paolino, D. Cosco, M. Iannone, F. Castelli, P. Doldo, M.Fresta, Effects of lipid composition and preparation conditions on physical–chemical properties, technological parameters and in vitro biological activity ofgemcitabine-loaded liposomes, Curr. Drug Deliv. 4 (2007) 89–101.
[11] M.L. Immordino, P. Brusa, F. Rocco, S. Arpicco, M. Ceruti, L. Cattel, Preparation,characterization, cytotoxicity and pharmacokinetics of liposomes containinglipophilic gemcitabine prodrugs, J. Control. Release 100 (2004) 331–346.
[12] B. Stella, S. Arpicco, F. Rocco, V. Marsaud, J.M. Renoir, L. Cattel, P. Couvreur,Encapsulation of gemcitabine lipophilic derivatives into polycyanoacrylatenanospheres and nanocapsules, Int. J. Pharm. 344 (2007) 71–77.
[13] D. Paolino, D. Cosco, M. Licciardi, G. Giammona, M. Fresta, G. Cavallaro,polyaspartylhydrazide copolymer-based supramolecular vesicular aggregates asdelivery devices for anticancer drugs, Biomacromolecules 9 (2008) 1117–1130.
[14] P. Couvreur, B. Stella, L.H. Reddy, H. Hillaireau, C. Dubernet, D. Desmae1le, S.Lepetre-Mouelhi, F. Rocco, N. Dereuddre-Bosquet, P. Clayette, V. Rosilio, V.Marsaud, J.M. Renoir, L. Cattel, Squalenoyl nanomedicines as potential therapeu-tics, Nano Letters 6 (2006) 2544–2548.
[15] L.H. Reddy, C. Dubernet, S.L. Mouelhi, P.E. Marque, D. Desmaele, P. Couvreur, Anew nanomedicine of gemcitabine displays enhanced anticancer activity insensitive and resistant leukemia types, J. Control. Release 124 (2007) 20–27.
[16] G. Pasut, F. Canal, L. Dalla Via, S. Arpicco, F.M. Veronese, O. Schiavon, Antitumoralactivity of PEG–gemcitabine prodrugs targeted by folic acid, J. Control. Release 127(2008) 239–248.
[17] C. Celia, M.G. Calvagno, D. Paolino, S. Bulotta, C.A. Ventura, D. Russo, M. Fresta,Improved in vitro anti-tumoral activity, intracellular uptake and apoptoticinduction of gemcitabine-loaded pegylated unilamellar liposomes, J. Nanosci.Nanotechnol. 8 (2008) 2102–2113.
[18] C. Celia, N. Malara, R. Terracciano, D. Cosco, D. Paolino, M. Fresta, R. Savino,Liposomal delivery improves the growth-inhibitory and apoptotic activity of lowdoses of gemcitabine in multiple myeloma cancer cells, Nanomedicine: NBM 4(2008) 155–166.
[19] A. Gabizon, H. Shmeeda, Y. Barenholz, Pharmacokinetics of pegylated liposomalDoxorubicin: review of animal and human studies, Clin. Pharmacokinet. 42(2003) 419–436.
[20] A. Fritze, F. Hens, A. Kimpfler, R. Schubert, R. Peschka-Süss, Remote loading ofdoxorubicin into liposomes driven by a transmembrane phosphate gradient,Biochim. Biophys. Acta 1758 (2006) 1633–1640.
[21] D. Paolino, G. Lucania, D. Mardente, F. Alhaique, M. Fresta, Ethosomes for skindelivery of ammonium glycyrrhizinate: in vitro percutaneous permeationthrough human skin and in vivo anti-inflammatory activity on human volunteers,J Control. Release 106 (2005) 99–110.
[22] T.J. Shaw, M.K. Senterman, K. Dawson, C.A. Crane, B.C. Vanderhyden, Character-ization of intraperitoneal, orthotopic, and metastatic xenograft models of humanovarian cancer, Mol. Ther. 10 (2004) 1032–1042.
[23] D. Mudhakir, H. Akita, I.A. Khalil, S. Futaki, H. Harashima, Pharmacokineticanalysis of the tissue distribution of octaarginine modified liposomes in mice,Drug Metab. Pharmacokinet. 20 (2005) 275–281.
[24] A. Gabizon, D.C. Price, J. Huberty, R.S. Bresalier, D. Papahadjopoulos, Effect ofliposome composition and other factors on the targeting of liposomes toexperimental tumors: biodistribution and imaging studies, Cancer Res. 50(1990) 6371–6378.
[25] I.V. Zhigaltsev, N. Maurer, Q.F. Akhong, R. Leone, E. Leng, J. Wang, S.C. Semple, P.R.Cullis, Liposome-encapsulated vincristine, vinblastine and vinorelbine: a compar-ative study of drug loading and retention, J. Control. Release 104 (2005) 103–111.
[26] D.C. Drummond, O. Meyer, K. Hong, D.B. Kirpotin, D. Papahadjopoulos, Optimizingliposomes for delivery of chemotherapeutic agents to solid tumors, Pharmacol.Rev. 51 (1999) 691–743.
[27] A. Nagayasu, K. Uchiyama, H. Kiwada, The size of liposomes: a factor which affectstheir targeting efficiency to tumors and therapeutic activity of liposomalantitumor drugs, Adv. Drug Deliv. Rev. 40 (1999) 75–87.
[28] V. Heinemann, Y.Z. Xu, S. Chubb, A. Sen, L.W. Hertel, G.B. Grindey, W. Plunkett,Cellular elimination of 2′, 2′-difluorodeoxycytidine 5′-triphosphate: a mechanismof self-potentiation, Cancer Res. 52 (1992) 533–539.
[29] D.Y. Bouffard, J. Laliberté, R.L. Momparler, Kinetic studies on 2′, 2′-difluorodeox-ycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidinedeaminase, Biochem. Pharmacol. 45 (1993) 1857–1861.
[30] A. Matsuda, T. Sasaki, Antitumor activity of sugar-modified cytosine nucleosides,Cancer Sci. 95 (2004) 105–111.
[31] P. Kundra, K.D. Burman, Thyroid cancer signalling pathways and use of targetedtherapy, Endocrinol. Metab. Clin. North Am. 36 (2007) 839–853.
[32] G. Riesco-Eizaguirre, New insights in thyroid follicular cell biology and its impactin thyroid cancer therapy, Endocr. Rel. Cancer. 14 (2007) 957–977.
[33] R.C. Smallridge, L.A. Marlow, J.A. Copland, Anaplastic thyroid cancer: molecularpathogenesis and emerging therapies, Endocr. Rel. Cancer 16 (2009) 17–44.
[34] M.G. Calvagno, D. Cosco, C. Celia, D. Paolino, D. Russo, M. Fresta, Liposomalmultidrug carriers encapsulating gemcitabine and paclitaxel for the treatment of

150 D. Paolino et al. / Journal of Controlled Release 144 (2010) 144–150
multi-drug resistent tumors, “33rd CRS Annual Meeting & Exposition”, 22–26 July(2006) Austria Wien Center.
[35] D. Cosco, M.G. Calvagno, S. Schenone, M. Celano, C. Celia, M. Botta, D. Russo, M.Fresta, Cytotoxic effects of gemcitabine and tyrosine kinase inhibitor containingliposomes on human anaplastic thyroid carcinoma cells, “33rd CRS AnnualMeeting & Exposition”, 22–26 July (2006) Austria Wien Center.
[36] P.G. Tardi, R.C. Gallagher, S. Johnstone, N. Harasym, M. Webb, M.B. Bally, L.D.Mayer, Coencapsulation of irinotecan and floxuridine into low cholesterol-
containing liposomes that coordinate drug release in vivo, Biochim. Biophys. Acta1768 (2007) 678–687.
[37] M. Celano, S. Schenone, D. Cosco, M. Navarra, E. Puxeddu, L. Racanicchi, C. Brullo, E.Varano, S. Alcaro, E. Ferretti, G. Botta, S. Filetti, M. Fresta, M. Botta, D. Russo,Cytotoxic effects of a novel pyrazolopyrimidine derivative entrapped in liposomesin anaplastic thyroid cancer cells in vitro and in xenograft tumors in vivo, Endocr.Rel. Cancer 15 (2008) 499–510.




















