
Functional Variation of HIV-1 Rev Response Element In a Longitudinally Studied Cohort (P 367-373)
117
Journal of Medical Virology Volume 75, Issue 3, Pages 367-481 (March 2005) Functional variation of HIV-1 Rev response element in a longitudinally studied cohort (p 367-373) Angsana Phuphuakrat, Robert M. Paris, Sorachai Nittayaphan, Suda Louisirirotchanakul, Prasert Auewarakul Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20279 Heterogeneous nature of HIV-1 recombinants spreading in Spain (p 374-380) Africa Holguín, Amparo Álvarez, Vincent Soriano Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20280 Polymorphism and drug-selected mutations in the reverse transcriptase gene of HIV-2 from patients living in southeastern France (p 381-390) Philippe Colson, Mireille Henry, Natacha Tivoli, Hervé Gallais, Jean-Albert Gastaut, Jacques Moreau, Catherine Tamalet Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20296 Clinical and virological characteristics of lamivudine resistance in chronic hepatitis B patients: A single center experience (p 391-398) Jian Sun, Zhanhui Wang, Shiwu Ma, Guobing Zeng, Zhiyong Zhou, Kangxian Luo, Jinlin Hou Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20281 Anesthetist to patient transmission of hepatitis C virus associated with non exposure-prone procedures (p 399-401) J. Mawdsley, C.G. Teo, M. Kyi, M. Anderson Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20282 Hepatitis C virus core, NS3, NS5A, NS5B proteins induce apoptosis in mature dendritic cells (p 402-411) Samila Siavoshian, Jean Daniel Abraham, Christine Thumann, Marie Paule Kieny, Catherine Schuster Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20283 TRUGENE sequencing versus INNO-LiPA for sub-genotyping of HCV genotype-4 (p 412-420) Abdel Rahman N. Zekri, Hanaa M. Alam El-Din, Abeer A. Bahnassy, Amal M.R. El-Shehabi, Heba El-Leethy, Ashraf Omar, Hussein M. Khaled Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20293 Growth of herpes simplex virus in epidermal keratinocytes determines cutaneous pathogenicity in mice (p 421-426) Yoshihiro Yoshida, ZhiHong Li, Masahiko Kurokawa, Takashi Kawana, Masami Imakita, Kimiyasu Shiraki Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20284
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Functional Variation of HIV-1 Rev Response Element In a Longitudinally Studied Cohort (P 367-373)

Journal of Medical Virology Volume 75, Issue 3, Pages 367-481 (March 2005)
Functional variation of HIV-1 Rev response element in a longitudinally studied cohort (p 367-373) Angsana Phuphuakrat, Robert M. Paris, Sorachai Nittayaphan, Suda Louisirirotchanakul, Prasert Auewarakul Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20279
Heterogeneous nature of HIV-1 recombinants spreading in Spain (p 374-380) Africa Holguín, Amparo Álvarez, Vincent Soriano Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20280
Polymorphism and drug-selected mutations in the reverse transcriptase gene of HIV-2 from patients living in southeastern France (p 381-390) Philippe Colson, Mireille Henry, Natacha Tivoli, Hervé Gallais, Jean-Albert Gastaut, Jacques Moreau, Catherine Tamalet Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20296
Clinical and virological characteristics of lamivudine resistance in chronic hepatitis B patients: A single center experience (p 391-398) Jian Sun, Zhanhui Wang, Shiwu Ma, Guobing Zeng, Zhiyong Zhou, Kangxian Luo, Jinlin Hou Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20281
Anesthetist to patient transmission of hepatitis C virus associated with non exposure-prone procedures (p 399-401) J. Mawdsley, C.G. Teo, M. Kyi, M. Anderson Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20282
Hepatitis C virus core, NS3, NS5A, NS5B proteins induce apoptosis in mature dendritic cells (p 402-411) Samila Siavoshian, Jean Daniel Abraham, Christine Thumann, Marie Paule Kieny, Catherine Schuster Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20283
TRUGENE sequencing versus INNO-LiPA for sub-genotyping of HCV genotype-4 (p 412-420) Abdel Rahman N. Zekri, Hanaa M. Alam El-Din, Abeer A. Bahnassy, Amal M.R. El-Shehabi, Heba El-Leethy, Ashraf Omar, Hussein M. Khaled Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20293
Growth of herpes simplex virus in epidermal keratinocytes determines cutaneous pathogenicity in mice (p 421-426) Yoshihiro Yoshida, ZhiHong Li, Masahiko Kurokawa, Takashi Kawana, Masami Imakita, Kimiyasu Shiraki Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20284

Detection of herpesvirus-6A in a case of subacute cerebellitis and myoclonic dystonia (p 427-429) Elisa Borghi, Elisabetta Pagani, Roberta Mancuso, Serena Delbue, Marilena Valli, Romina Mazziotti, Lucio Giordano, Roberto Micheli, Pasquale Ferrante Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20285
Drug-induced hypersensitivity syndrome due to cyanamide associated with multiple reactivation of human herpesviruses (p 430-434) Naoko Mitani, Michiko Aihara, Yuko Yamakawa, Masako Yamada, Norihiko Itoh, Nobuhisa Mizuki, Zenro Ikezawa Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20295
Detection of HPV 16 and HPV 18 DNA in the blood of patients with cervical cancer (p 435-439) Patti Kay, Bruce Allan, Lynette Denny, Margaret Hoffman, Anna-Lise Williamson Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20294
Molecular epidemiology of adenovirus strains isolated from patients with ocular disease in the area of Thessaloniki, Greece (1998-2002) (p 440-446) Filanthi Frantzidou, Aikaterini Pavlitou, Asimina Mataftsi, Kamal Dumaidi, Nikolaos Georgiadis Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20286
Detection of BK virus and simian virus 40 in the urine of healthy children (p 447-454) John A. Vanchiere, Zoe S. White, Janet S. Butel Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20287
New human coronavirus, HCoV-NL63, associated with severe lower respiratory tract disease in Australia (p 455-462) Katherine E. Arden, Michael D. Nissen, Theo P. Sloots, Ian M. Mackay Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20288
Detection of human coronavirus NL63 in young children with bronchiolitis (p 463-465) Takashi Ebihara, Rika Endo, Xiaoming Ma, Nobuhisa Ishiguro, Hideaki Kikuta Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20289
Genetic characterization of the M RNA segment of a Balkan Crimean-Congo hemorrhagic fever virus strain (p 466-469) Anna Papa, E. Papadimitriou, B. Bo ovi , A. Antoniadis Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20290
Rapid and sensitive detection of mumps virus RNA directly from clinical samples by real-time PCR (p 470-474) Kazue Uchida, Michiyo Shinohara, Shin-ichi Shimada, Yukari Segawa, Rie Doi, Atushi Gotoh, Ryo Hondo Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20291
Outbreak of acute gastroenteritis associated with group A rotavirus and genogroup I sapovirus among adults in a mental health care facility in Japan (p 475-481) Hainian Yan, Toshiaki Abe, Tung Gia Phan, Tuan Anh Nguyen, Tatuya Iso, Yasunori Ikezawa, Kiyo Ishii, Shoko Okitsu, Hiroshi Ushijima Published Online: 12 Jan 2005 DOI: 10.1002/jmv.20292

Journal of Medical Virology 75:367–373 (2005)
Functional Variation of HIV-1 Rev ResponseElement in a Longitudinally Studied Cohort
Angsana Phuphuakrat,1 Robert M. Paris,2 Sorachai Nittayaphan,2 Suda Louisirirotchanakul,1
and Prasert Auewarakul1*1Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand2Armed Forces Research Institute of Medical Sciences, Bangkok, Thailand
We showed previously that HIV-1 Rev ResponseElement (RRE) contains a certain degree ofstructural variation, and in a set of limitedsamples, RRE from HIV-1 natural isolates werefound to have functional variability. The signifi-cance of the RRE heterogeneity is addressedfurther by analyzing the functional variation ofRREs in a longitudinal cohort. While the RREactivity at early time points was not a goodpredictor of disease outcome, the RRE activityat late time points was correlated with rates ofCD4þ count decline. These data suggest that RREheterogeneity may be important in viral patho-genesis and disease progression. J. Med. Virol.75:367–373, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: RRE; disease progression;CD4þ count
INTRODUCTION
RRE is a cis-acting element that binds to the viralprotein Rev, which mediates the transport of incomple-tely spliced RNAs across the nuclear membrane[Hammarskjold et al., 1989; Malim et al., 1989]. Becausethese RNA species serve as templates for structural andaccessory protein translation as well as genome forprogeny virus, Rev and RRE are necessary for viralreplication. RRE–Rev interaction also facilitates RNAstability and translation [Arrigo et al., 1989; Felberet al., 1989; Arrigo and Chen, 1991; Schwartz et al.,1992]. HIV-1 RRE is a 234-nt region, lying immediatelydownstream to the junction between gp120 and gp41,and upstream to the tat, rev, nef second splice acceptor.Because of this specific location, all the incompletelyspliced transcripts contain RRE. RRE also binds somecellular proteins [Xu et al., 1996; Powell et al., 1997; Liet al., 1999; Reddy et al., 1999]. All proteins identified todate have been shown to have some roles in RNAprocessing. Thus, structural variation in the RRE regionis expected to alter RRE binding to both viral andcellular proteins, and hence, may modulate viral geneexpression and replication.
Progression of human immunodeficiency virus type 1(HIV-1) disease is characterized by declining CD4þ cellcount and increasing quantity of the virus. Despite thedefined pattern of disease progression, time to developAIDS can be different in each infected individual.Various factors have been described to contribute tothe rate of disease progression. These include bothviral and host factors. For host factors, mutation ofchemokine receptors, variation in amount of chemokineproduction, and particular HLA alleles have beenassociated with susceptibility to HIV infection anddisease progression (reviewed in Theodorou et al.,2003]. For viral factors, although no common viralgenetic determinants could account for all differentrates of disease progression, infection with a nef-defective virus was found to be strongly associated withan attenuated disease phenotype [Deacon et al., 1995;Kirchhoff et al., 1995]. However, infected individualswho harbor susceptible genetic factors and were infectedwith normal circulating strains of virus still developAIDS with a wide range of progression rates.
HIV-1 is highly variable. Variations of HIV sequencesthat cause genetic defects and slow disease progressionhave been shown in many studies. These regions includegag, env, vif, vpr, vpu, tat, and rev [Iversen et al., 1995;Michael et al., 1995; Connor et al., 1996; Zhang et al.,1997b; Huang et al., 1998; Yamada and Iwamoto, 2000].LTR variation associated with attenuated virus hasbeen also described, albeit rare [Zhang et al., 1997a].Therefore it is unlikely that a single common viralgenetic determinant accounts for diverse rates ofdisease progression in HIV-1 infection.
Grant sponsor: Thailand Research Fund; Grant sponsor:Ellison Foundation.
*Correspondence to: Prasert Auewarakul, M.D., Dr. Med.,Department of Microbiology, Faculty of Medicine Siriraj Hospital,Mahidol University, Bangkok 10700, Thailand.E-mail: [email protected]
Accepted 27 October 2004
DOI 10.1002/jmv.20279
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

During the course of the HIV infection, viral loadincreases over time. Among infected individuals, achange in chemokine receptor usage from CCR5 toCXCR4 is correlated with disease progression. However,in some HIV-infected cases [de Roda Husman et al.,1999], and the majority of infections with HIV-1 subtypeC [Ping et al., 1999; Cecilia et al., 2000; Morris et al.,2001], changing chemokine usage during disease pro-gression does not occur. Theoretically, variation of RREduring disease progression may contribute to differentrates of virus replication and, hence, different ratesof disease progression. Recently, we showed that RREsequences are structurally heterogeneous [Phuphuak-rat and Auewarakul, 2003], and we observed that RREsfrom natural isolates were functionally heterogeneous.In this study we further address the significance of thisfunctional variability of RRE in an HIV-infected patientcohort.
MATERIALS AND METHODS
Subjects
Twenty-one HIV-1 infected subjects were selectedfrom individuals enrolled between 1993 and 1997 ina natural history cohort study conducted by theArmed Forces Research Institute of Medical Sciences(AFRIMS) in Bangkok, Thailand. Five subjects wereseroconverters, and the other 16 were seroprevalentcases who were asymptomatic at the time of enrollment.These subjects were followed every 6 months to 1 yearwith the duration of follow-up ranging from 3 to 7 years.At each visit, CD4þ count was quantified by flow cyto-metry, and peripheral blood mononuclear cell (PBMC)and plasma samples were frozen and archived. Sixvolunteers received antiretroviral drug monotherapy,three subjects received dual therapy, and none hadreceived HAART during the period from which sampleswere utilized for this study.
RRE Clones
DNA was isolated from PBMCs of HIV infectedpatients using the QIAmp DNA blood mini kit (Qiagen,Germantown, MD). The RRE regions were amplified bynested PCR using pfu polymerase (Promega, Madison,WI) with outer primers 50O-RRE (50-AACCACTAGGAA-TAGCACCC-30) and 30O-RRE (50-TAATTGCTAATTT-CTCTCTCCC-30), and inner primers RRESalI (50-TCTGACAGTCGACAAAAGAGCAGTGGGAATAGG-30)and RRENotI (50-TGACAGCGGCCGCAGCAGCCC-CAAAGT-30), yielding amplified fragments flankingwith 50SalI and 30NotI restriction sites. The fragmentswere cloned into pGEM1-T Easy (Promega). Due to thevariability of viral quasispecies, four clones from eachsubject were selected, and RRE segments were excisedwith SalI and NotI to clone into the widely used HIV-1molecular clone, pHXB2, replacing the SalI-NotI frag-ment that contained tat, rev, and env ORFs (SalI sitewas at nucleotide 5787 and NotI site was introduced atnucleotide 8796). Because the tat, rev, and env ORFs
were deleted from the tested constructs (replaced byRRE fragment), Tat and Rev were supplied in trans by apCMVtat/rev/env construct. This construct was gener-ated by replacing the beta-galactosidase gene of pCMVb(Clontech, Palo Alto, CA) by tat, rev, and env ORFs froma molecular clone of an HIV-1 primary isolate, whichefficiently expressed functional Tat and Rev (data notshown).
Transfection, p24 Assay,and GFP Expression Detection
RRE constructs in an amount of 0.5 mg were co-transfected with 0.5 mg of pCMVtat/rev/env into theCOS7 cell line by using a cationic lipid reagent-mediatedtransfection technique (DMRIE-C Reagent, Invitrogen,Carlsbad, CA). Mock transfection was performed bytransfecting an equal amount of pGEM1-T Easy. Tonormalize the transfection efficiencies, 0.1 mg of pEGFP-N1 (Clontech) was co-transfected as an internal control.The transfection was performed according to the manu-facturer’s protocol with some modifications. Briefly, in24-well tissue culture plate, 5�104 cells were plated theday before transfection. On the day of transfection, theplasmids were diluted into 250 ml of serum-free DMEM.In a separate tube, 4 mg of DMRIE-C reagent was dilutedin 250 ml of similar medium. The two solutions were thencombined, incubated at room temperature for 20 min,and replaced culture medium. The cells were thenincubated at 378C for 4 hr. Transfection solution wasthen replaced with growth medium. Transfected cellsand supernatants were collected at 3 days after trans-fection. The supernatants were assayed for p24 by anELISA kit (Organon Teknika, Durham, NC), and GFPexpression was detected by flow cytometry (FACScan,Becton Dickinson, San Jose, CA). The p24 levels werethen normalized by the transfection efficiencies asmeasured by percent GFP positive cells. Data withtransfection efficiency below the 5th centile or above95th centile were excluded from the analysis. Themedian of p24 expression from the four clones in apatient was used to represent RRE function.
Multiplex Polymerase ChainReaction-Restriction Fragment LengthPolymorphism (Multiplex PCR-RFLP)
of Host Genetic Polymorphisms
Since host genetic factors may confound the asso-ciation between viral genetic variability and HIVdisease progression, we screened for known host poly-morphisms associated with delayed HIV disease pro-gression, which included CCR5-m303, CCR2-64I, andSDF1-30A [Michael, 1999]. We did not detect CCR5D32because it is known to be absent in Asian population[Huang et al., 1996]. A Multiplex PCR-RFLP for CCR5-m303, CCR2-64I, and SDF1-30A was performed accord-ing to an established protocol described elsewhere(Louisirirotchanakul et al., 2004). Briefly, multiplexhot start PCR was carried out using primers CCR5 aþand CCR5 a�; CCR21 and CCR23; S2 and S4 to amplify
368 Phuphuakrat et al.

parts of CCR5, CCR2, and SDF genes, respectively. Thesizes of the amplified products were 1194, 645, and 207bp for CCR5, SDF, and CCR2, respectively. Theproducts were then genotyped by cutting separatelywith HinCII, BsaBI, and MspI overnight to detectpolymorphism of CCR5, CCR2, and SDF genes, respec-tively. For CCR5 m303, HinCII yielded two fragments(806 and 388 bp) for the wild type but no digestionoccurred of the mutant. For CCR2 64I, the BsaBIdigestion yielded 187 and 20 bp fragments forthe mutant but no digestion occurred with the wildtype. For SDF1 3’A, there were 406 and 239 bp productsafter MspI digestion for the wild type, and there was nodigestion of the mutant.
Statistical Analysis
CD4þ slopes from each patient were used to comparedisease progression with RRE activity and calculated bylinear regression for panel data from all CD4þ counts atevery visit. Continuous data such as age, CD4þ count,CD4þ slope were analyzed by the Mann–Whitney Utest. Categorical variables were analyzed using Fisher’sexact test. Statistical analyses were performed withSPSS version 11.5.
RESULTS
Cohort Characteristics
CD4þ count at each time point of 21 HIV-1 infectedindividuals in the cohort was used to generate the CD4þ
slope of each patient. The slope was then used to dividethe cohort into two groups as faster progressors andslower progressors. We arbitrarily used a CD4þ countslope of �75 cells/ml/year to separate faster progressorsand slower progressors, which is comparable to amedian CD4þ slope in a Thai HIV natural history cohort[Kilmarx et al., 2000]. The slopes of subjects in the twogroups are shown in Figure 1. The difference betweenslopes of the two groups is clear-cut with averages of�118 and 14 cells/ml/year for the faster progressor andslower progressor groups, respectively.
Analysis of other cohort characteristics includedinitial CD4þ counts between faster progressors andslower progressors, which were not significantly differ-ent, while the CD4þ counts of follow up samples isstatistically different (P¼0.005). The duration betweenbaseline and follow up CD4þ count determinations weresimilar between the two groups. The groups were alsocomparable for both age and gender (Table I).
Analysis of RRE Function
In this study, two possible outcomes were hypothe-sized: (1) there might be some patients who wereinfected with HIV-1 that contained more efficient RREand others who were infected with HIV-1 that containedless efficient RRE, and CD4þ declined faster in theformer group; (2) there might be functional variability ofRRE during the progression of disease, i.e., the moresevere type of RRE developed during progressive CD4þ
decline.In the co-transfection experiments, p24 expression
was used as a marker for RRE activity. Because p24 isa product of an intron-containing transcript, RRE isneeded for its expression. RRE clones showed somedegree of variation in the p24 expression levels withinthe same sample, but this variation was significan-tly less than the variation among different samples(P< 0.01, F-test). The variation of RRE activity wasobserved at the early time point when the subjects wereenrolled into the cohort. However, this variation did notshow a significant difference between faster progres-sor and slower progressor groups (P¼ 0.903, Mann–Whitney U test, 2-tailed, Fig. 2A). In the follow upsample, statistical significance was borderline(P¼ 0.051, Mann–Whitney U test, 2-tailed). However,it was found that variation in RRE activity was diversein the faster progressor group, while the activity in theslower progressor group was found to aggregate into twogroups of low activity and moderate activity (Fig. 2B).The lack of an obvious correlation between RREactivities and CD4þ slopes suggested that other host orviral genetic factors might contribute to variation indisease progression.
Fig. 1. Comparison of CD4þ slopes for each subcohort. The CD4þ slope from each patient in the studycohort is shown.A: Faster progressor group, (B) slower progressor group. The slope for each individual wasdefined by GEE analysis, and plotted based on dates of CD4þ determination.
HIV-1 RRE Variation in a Cohort 369

Attempts were made to eliminate confounding factorsby excluding host genetic polymorphisms that areknown to influence disease progression rates. Thesehost genetic variations included CCR5-m303, CCR2-64Iheterozygous or homozygous, and SDF1-30A homozy-gous. The variant CCR5-m303 was not found in thiscohort. However, five of the infected individuals werefound to be heterozygous or homozygous for either orboth the CCR2-64I, and/or homozygous of SDF1-30Apolymorphisms. The subjects with these chemokine/chemokine receptor variants were then excluded fromthe analysis. After excluding these samples, there wasstill no correlation between RRE activities in earlysamples and rates of CD4þdecline (Fig. 3A). However, atthe late time point, RRE activities were found to scatterin moderate to high activity in the faster progressorgroup, while those of slower progressors were found toamass in low and moderate activity (Fig. 3B). Statisticanalysis showed that RRE activities were significantlydifferent between the two groups (P¼0.004, Mann–Whitney U test, 2-tailed). This suggested that aside fromhost factors, RRE is one of the viral factors that mightplay a role in modulating HIV disease outcome.
The design of this analysis could not reject or acceptthe hypothesis that patients infected with HIV-1 thatcontained more efficient RRE would lead to faster CD4þ
declines. However, in the follow up samples, when hostgenetic factors associated with delayed disease progres-sion were excluded, RRE activities were associated withrates of CD4þ decline. This finding supports the alter-nate hypothesis that RRE variation occurs during thecourse of disease, but such variation may occur in anydirection. To show further correlation of RRE activitywith rate of CD4þ decline, RRE activities of the samplesafter excluding the host genetic variants describedabove were plotted on p24 expression versus CD4þ
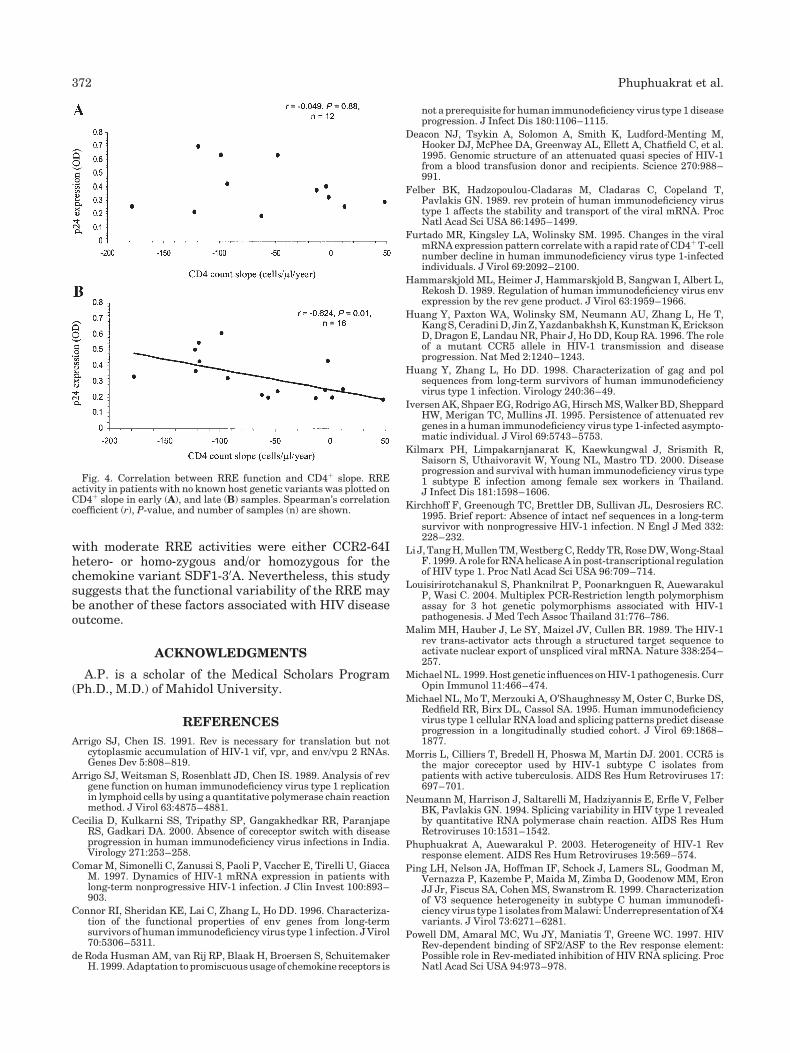
slopes. As expected, a linear correlation was not found inthe early time point (P¼ 0.88), whereas the activities infollow up samples were found to be moderately inverselycorrelated with CD4þ slopes (Fig. 4, Spearman’s corre-lation coefficient¼�0.624, P¼ 0.01).
DISCUSSION
HIV-1 disease progression involves complex interplaybetween various viral and host factors. Despite manystudies on variation of various viral genes and regions inHIV-1 on disease progression, functional variability ofthe HIV-1 RRE during disease progression has neverbeen investigated. The RRE functional variation wasstudied in a small cohort and showed that the HIV-1RRE activities from the follow up samples of infectedpatient were associated with the rates of CD4þ decline.
Previous data have shown that the splicing pattern ofHIV-1 is complex and there are differences in splicingpatterns among viral isolates [Neumann et al., 1994].The switching of HIV-1 mRNA splicing pattern from apredominately spliced to a predominately unsplicedpattern in freshly isolated peripheral blood mononuc-lear cells has been shown to be associated with disease
TA
BL
EI.
Coh
ort
Ch
ara
cter
isti
cs
Coh
ort
(n¼
21)
Ch
ara
cter
isti
c
Mea
na
CD
4þ
cou
nt
(cel
l/ml
)M
ean
bC
D4þ
slop
eM
ean
foll
owu
p(y
ears
)A
ge
(yea
rs)
No.
ofp
ati
ents
ofin
dic
ate
dse
x
Male
Fem
ale
Fast
erp
rogre
ssor
s(n
¼8)
615.4
6�
142.3
4(p
re),
128�
115.2
5(p
ost)
�118�
0.0
74.3
9�
1.1
725.6
3�
4.1
03
5
Slo
wer
pro
gre
ssor
s(n
¼13)
522.1
6�
95.0
7(p
re),
409.6
5�
216.6
8(p
ost)
14�
36
5.4
2�
1.5
528.2
3�
6.8
27
6
P-v
alu
ec0.1
98
(pre
),0.0
05
(pos
t)�
0.0
10.4
77
0.4
89
0.4
77
d
aS
how
nas
mea
n�
stan
dard
dev
iati
onfo
rall
mea
nvalu
esin
the
table
.P
re,base
lin
esa
mp
le;p
ost,
foll
ow-u
psa
mp
le.
b(C
ells
per
mic
roli
ter)
yea
r�1.
c By
Man
n–
Wh
itn
eyte
stu
nle
ssin
dic
ate
dot
her
wis
e.dB
yF
ish
er’s
exact
test
.
370 Phuphuakrat et al.

progression [Furtado et al., 1995; Michael et al., 1995;Comar et al., 1997]. These data suggested that modula-tion of splicing and Rev-dependent RNA transportmight be involved in viral replication kinetics, whichcould influence disease progression. Our data supportthis hypothesis. The lack of correlation between earlyRRE activities and disease progression suggests that thepattern of RRE evolution rather than the properties ofthe originally infecting viruses was the determiningfactor.
Still, an interesting question on the causal relation-ship of RRE activity and disease progression remains to
be answered. In this study, it cannot be determinedconclusively whether RRE variation or disease progres-sion is a cause or an outcome. RRE may either evolve to amore efficient type causing higher viral load and diseaseworsening. Alternatively, after deterioration of theimmune system, viruses with a more competent type ofRRE dominate.
As mentioned above, it is clear that RRE as well as anyother single factor cannot be the sole determinant ofprogression rate. Other host and viral factors mayexplain the deviation of some subjects from the correla-tion, for example, four of the slower progressor subjects
Fig. 2. Analysis of RRE function. A: RRE function in early samples as measured by p24 ELISA. B: RREfunction in late samples of the cohort. Each point represents median of the four RRE clones tested in eachpatient. Horizontal line represents the median of each group.
Fig. 3. Analysis of RRE function between groups not known to host genetic variants. As in Figure 2, butfive patients with known host factor variants were excluded from the analysis. RRE function in early(A), and late (B) samples. Horizontal line represents median of each group.
HIV-1 RRE Variation in a Cohort 371
with moderate RRE activities were either CCR2-64Ihetero- or homo-zygous and/or homozygous for thechemokine variant SDF1-30A. Nevertheless, this studysuggests that the functional variability of the RRE maybe another of these factors associated with HIV diseaseoutcome.
ACKNOWLEDGMENTS
A.P. is a scholar of the Medical Scholars Program(Ph.D., M.D.) of Mahidol University.
REFERENCES
Arrigo SJ, Chen IS. 1991. Rev is necessary for translation but notcytoplasmic accumulation of HIV-1 vif, vpr, and env/vpu 2 RNAs.Genes Dev 5:808–819.
Arrigo SJ, Weitsman S, Rosenblatt JD, Chen IS. 1989. Analysis of revgene function on human immunodeficiency virus type 1 replicationin lymphoid cells by using a quantitative polymerase chain reactionmethod. J Virol 63:4875–4881.
Cecilia D, Kulkarni SS, Tripathy SP, Gangakhedkar RR, ParanjapeRS, Gadkari DA. 2000. Absence of coreceptor switch with diseaseprogression in human immunodeficiency virus infections in India.Virology 271:253–258.
Comar M, Simonelli C, Zanussi S, Paoli P, Vaccher E, Tirelli U, GiaccaM. 1997. Dynamics of HIV-1 mRNA expression in patients withlong-term nonprogressive HIV-1 infection. J Clin Invest 100:893–903.
Connor RI, Sheridan KE, Lai C, Zhang L, Ho DD. 1996. Characteriza-tion of the functional properties of env genes from long-termsurvivors of human immunodeficiency virus type 1 infection. J Virol70:5306–5311.
de Roda Husman AM, van Rij RP, Blaak H, Broersen S, SchuitemakerH. 1999. Adaptation to promiscuous usage of chemokine receptors is
not a prerequisite for human immunodeficiency virus type 1 diseaseprogression. J Infect Dis 180:1106–1115.
Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M,Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, et al.1995. Genomic structure of an attenuated quasi species of HIV-1from a blood transfusion donor and recipients. Science 270:988–991.
Felber BK, Hadzopoulou-Cladaras M, Cladaras C, Copeland T,Pavlakis GN. 1989. rev protein of human immunodeficiency virustype 1 affects the stability and transport of the viral mRNA. ProcNatl Acad Sci USA 86:1495–1499.
Furtado MR, Kingsley LA, Wolinsky SM. 1995. Changes in the viralmRNA expression pattern correlate with a rapid rate of CD4þ T-cellnumber decline in human immunodeficiency virus type 1-infectedindividuals. J Virol 69:2092–2100.
Hammarskjold ML, Heimer J, Hammarskjold B, Sangwan I, Albert L,Rekosh D. 1989. Regulation of human immunodeficiency virus envexpression by the rev gene product. J Virol 63:1959–1966.
Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, He T,Kang S, Ceradini D, Jin Z, Yazdanbakhsh K, Kunstman K, EricksonD, Dragon E, Landau NR, Phair J, Ho DD, Koup RA. 1996. The roleof a mutant CCR5 allele in HIV-1 transmission and diseaseprogression. Nat Med 2:1240–1243.
Huang Y, Zhang L, Ho DD. 1998. Characterization of gag and polsequences from long-term survivors of human immunodeficiencyvirus type 1 infection. Virology 240:36–49.
Iversen AK, Shpaer EG, Rodrigo AG, Hirsch MS, Walker BD, SheppardHW, Merigan TC, Mullins JI. 1995. Persistence of attenuated revgenes in a human immunodeficiency virus type 1-infected asympto-matic individual. J Virol 69:5743–5753.
Kilmarx PH, Limpakarnjanarat K, Kaewkungwal J, Srismith R,Saisorn S, Uthaivoravit W, Young NL, Mastro TD. 2000. Diseaseprogression and survival with human immunodeficiency virus type1 subtype E infection among female sex workers in Thailand.J Infect Dis 181:1598–1606.
Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC.1995. Brief report: Absence of intact nef sequences in a long-termsurvivor with nonprogressive HIV-1 infection. N Engl J Med 332:228–232.
Li J, Tang H, Mullen TM, Westberg C, Reddy TR, Rose DW, Wong-StaalF. 1999. A role for RNA helicase A in post-transcriptional regulationof HIV type 1. Proc Natl Acad Sci USA 96:709–714.
Louisirirotchanakul S, Phanknilrat P, Poonarknguen R, AuewarakulP, Wasi C. 2004. Multiplex PCR-Restriction length polymorphismassay for 3 hot genetic polymorphisms associated with HIV-1pathogenesis. J Med Tech Assoc Thailand 31:776–786.
Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. 1989. The HIV-1rev trans-activator acts through a structured target sequence toactivate nuclear export of unspliced viral mRNA. Nature 338:254–257.
Michael NL. 1999. Host genetic influences on HIV-1 pathogenesis. CurrOpin Immunol 11:466–474.
Michael NL, Mo T, Merzouki A, O’Shaughnessy M, Oster C, Burke DS,Redfield RR, Birx DL, Cassol SA. 1995. Human immunodeficiencyvirus type 1 cellular RNA load and splicing patterns predict diseaseprogression in a longitudinally studied cohort. J Virol 69:1868–1877.
Morris L, Cilliers T, Bredell H, Phoswa M, Martin DJ. 2001. CCR5 isthe major coreceptor used by HIV-1 subtype C isolates frompatients with active tuberculosis. AIDS Res Hum Retroviruses 17:697–701.
Neumann M, Harrison J, Saltarelli M, Hadziyannis E, Erfle V, FelberBK, Pavlakis GN. 1994. Splicing variability in HIV type 1 revealedby quantitative RNA polymerase chain reaction. AIDS Res HumRetroviruses 10:1531–1542.
Phuphuakrat A, Auewarakul P. 2003. Heterogeneity of HIV-1 Revresponse element. AIDS Res Hum Retroviruses 19:569–574.
Ping LH, Nelson JA, Hoffman IF, Schock J, Lamers SL, Goodman M,Vernazza P, Kazembe P, Maida M, Zimba D, Goodenow MM, EronJJ Jr, Fiscus SA, Cohen MS, Swanstrom R. 1999. Characterizationof V3 sequence heterogeneity in subtype C human immunodefi-ciency virus type 1 isolates from Malawi: Underrepresentation of X4variants. J Virol 73:6271–6281.
Powell DM, Amaral MC, Wu JY, Maniatis T, Greene WC. 1997. HIVRev-dependent binding of SF2/ASF to the Rev response element:Possible role in Rev-mediated inhibition of HIV RNA splicing. ProcNatl Acad Sci USA 94:973–978.
Fig. 4. Correlation between RRE function and CD4þ slope. RREactivity in patients with no known host genetic variants was plotted onCD4þ slope in early (A), and late (B) samples. Spearman’s correlationcoefficient (r), P-value, and number of samples (n) are shown.
372 Phuphuakrat et al.

Reddy TR, Xu W, Mau JK, Goodwin CD, Suhasini M, Tang H, FrimpongK, Rose DW, Wong-Staal F. 1999. Inhibition of HIV replication bydominant negative mutants of Sam68, a functional homolog of HIV-1 Rev. Nat Med 5:635–642.
Schwartz S, Felber BK, Pavlakis GN. 1992. Distinct RNA sequences inthe gag region of human immunodeficiency virus type 1 decreaseRNA stability and inhibit expression in the absence of Rev protein.J Virol 66:150–159.
Theodorou I, Capoulade C, Combadiere C, Debre P. 2003. Geneticcontrol of HIV disease. Trends Microbiol 11:392–397.
Xu Y, Reddy TR, Fischer WH, Wong-Staal F. 1996. A Novel hnRNPSpecifically Interacts with HIV-1 RRE RNA. J Biomed Sci 3:82–91.
Yamada T, Iwamoto A. 2000. Comparison of proviral accessory genesbetween long-term nonprogressors and progressors of humanimmunodeficiency virus type 1 infection. Arch Virol 145:1021–1027.
Zhang L, Huang Y, Yuan H, Chen BK, Ip J, Ho DD. 1997a. Genotypicand phenotypic characterization of long terminal repeat sequencesfrom long-term survivors of human immunodeficiency virus type 1infection. J Virol 71:5608–5613.
Zhang L, Huang Y, Yuan H, Tuttleton S, Ho DD. 1997b. Geneticcharacterization of vif, vpr, and vpu sequences from long-termsurvivors of human immunodeficiency virus type 1 infection.Virology 228:340–349.
HIV-1 RRE Variation in a Cohort 373

Journal of Medical Virology 75:374–380 (2005)
Heterogeneous Nature of HIV-1 RecombinantsSpreading in Spain
Africa Holguın,* Amparo Alvarez, and Vincent Soriano*
Department of Infectious Diseases, Hospital Carlos III, Madrid, Spain
HIV-1 infections due to non-B subtypes are in-creasing rapidly in number and spreading acrossEurope.ThegeneticnatureofHIV-1non-Bvariantscontaining subtype G sequences at the protease(PR)-coding region are described from 48 unre-lated subjects living in Spain. Phylogeneticanalyses of the HIV-1 reverse transcriptase (RT)and envelope (env) genes (including the V3 loop)were performed. Up to 32 (66.6%) of samplescarried inter-subtype recombinant viruses.Although double recombinants were foundmostfrequently (G/A in 20; G/B in 8; G/K in 2), twoindividuals harbored triple recombinant viruses(GPR/BRT/Aenv and GPR/KRT/Aenv, respectively). Only33 (68.7%) and 9 (18.7%) sequences clusteredwith clade G when examining the RT and envgenes, respectively. Nearly 70% of samples withpol sequences (PR/RT) belonging to subtype Gharboredenv sequencesascribed toother clades:A (55.6%), B (11.1%), or K (3.7%). Of note, mostrecombinant viruses clustered with CRF02_AG,although CRF14_BG recombinants were alsofound. This studydemonstrates thatmost virusescirculating in Spain with clade G sequences atthe pol-coding region are in fact inter-subtyperecombinants, with CRF02_AG being the mostprevalent virus. J. Med. Virol. 75:374–380,2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: HIV-1 subtypes; recombinants;protease; reverse transcriptase;envelope; genetic variability
INTRODUCTION
Both the high error rate of the reverse transcriptase(RT) and the occurrence of recombination events aremostly responsible for the high genetic heterogeneity ofthe human immunodeficiency virus type 1 (HIV-1) invivo [Coffin, 1995]. Currently, HIV-1 can be divided intothree distinct and highly divergent groups: M (major),O (outlier), andN (non-M, non-O). Severalmajor geneticvariants can be recognized within HIV-1 group M,including nine subtypes (A, B, C, D, F, G, H, J, K) and atleast 15 major circulating recombinant forms (CRFs)
[Kuiken et al., 2000]. CRFs are defined as intersubtyperecombinants for which at least three epidemiologicallyunlinked variants are monophyletic, sharing an iden-tical genetic structure. Unique recombinant forms(URF) are also widely found worldwide. ClassificationofHIV-1 into subtypes is basedprimarily on the analysisof sequences coding for the envelope (env) gene. How-ever, the pol-coding region has also been validated forthis purpose [Hue et al., 2004] and is currently usedmuch more since drug resistance testing is undertakenroutinely in a large scale.
The occurrence of HIV-1 recombination in naturehas an important influence upon HIV-1 populationdynamics throughout the world. More than one-thirdof HIV-1 strains described so far in the HIV databasemight be recombinant forms, presumably generatedafter co-circulation of different subtypes in the samegeographical area. This is a frequent phenomenon inAfrica [Janssens et al., 1997; Peeters et al., 2003], whereat least 9 million people are infected with CRF02_AGviruses, a variant which contains subtype G and Asequences within the same genome [McCutchan, 2000].It accounts for 50%–80%of infections inWest andWest-CentralAfrica [Cornelissenetal., 2000;Montalvonetal.,2000; Peeters et al., 2000; Nyambi et al., 2002],suggesting a longstanding presence of these recombi-nants in the global epidemic.
In Spain, as in North America and other WesternEuropean countries, subtype B is the most prevalentHIV-1 variant.However, recent reports have indicatedarapid spread of non-B variants. This is mainly due topopulation movements, such as migration, travel, and
Grant sponsor: Asociacion Investigacion y Educacion en SIDA(AIES); Grant sponsor: Red de Investigacion en SIDA (RIS)project 173, Ministerio de Ciencia y Tecnologıa; Grant number:SAF2003-03551; Grant sponsor: Fondo de Investigaciones Sani-tarias; Grant number: FIS PI030004.
*Correspondence to: Vincent Soriano and Africa Holguın,Calle Nueva Zelanda 54, 4o B, 28035 Madrid, Spain.E-mail: [email protected]
Accepted 15 October 2004
DOI 10.1002/jmv.20280
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

risk behaviors of HIV-1-infected individuals originatingin countries where those variants are highly prevalent.Not surprisingly, the highest HIV-1 diversity in theEuropeanUnion is found in countrieswith the strongesthistorical, economical, and/or political links with Africa[review in Holguın et al., 2003]. In the last 10 years thenumber of immigrants has increased significantly inSpain, with the largest proportion of subjects comingfrom Africa and South America. Accordingly, nearlytwo-thirds of foreigners with HIV-1 infection in ourHIV/AIDS clinic carry non-B subtypes, subtype Gviruses being the most frequent among West-CentralAfrican immigrants, at least when the protease (PR)-coding region is examined [Holguın et al., 2002a].
National public health authorities have been alertedon the rapid spread of non-B viruses, particularly in thelight of confronting potential problems using currentdiagnostic tests [Arnold et al., 1995; Candotti et al.,2000; Baldrich-Rubio et al., 2001] and viral loadmeasurements [Holguın et al., 1999, 2001; Jenny-Avitaland Beatrice, 2001]. Moreover, given the high rateof polymorphisms at the PR in non-B subtypes atcodons associated with resistance to protease inhibitors[Holguın et al., 2002a,b], a possible reduced suscept-ibility to these compounds might exist [Descamps et al.,1998; Holguın et al., 2002b], complicating the therapeu-tic management of patients infected with these viruses.
PATIENTS AND METHODS
Plasma samples collected from a total of 48 subjectswere examined. They belonged to a repository of clinicalspecimens from individuals previously known to carryHIV-1 non-B variants with PR/RT sequence fragmentsbelonging to subtype G [Holguın et al., 2000a, 2001,2002a]. A more extensive genetic characterization ofthese viruses was undertaken by direct sequencing andphylogenetic analyses of nested PCR purified productsobtained from plasma RNA. The three non-overlappingsequences thatwere assessed included thePR (positionsfrom 2253 to 2549 in HXB2 isolate; 297-bp), reversetranscriptase (RT) (positions 2682 to 3121 in HXB2isolate; 440-bp), and env (positions from 7059 to 7374 inHXB2 isolate; 316-bp, covering the V3 region).
For the amplification of the env region, outer primersED3 and ED14 were used for the first PCR reaction,followed by a second round with inner primers ED5 andED12, as previously described [Shafer et al., 1997].Standard reaction conditions were: incubation at 948Cfor 5min, one cycle at 948C for 1min, 558C for 1min, and728C for 1 min, followed by 32 cycles at 948C for 15 sec,558C for 45 sec, and728C for 1min,withafinal extensionat 728C for 10 min. Some env sequences were amplifiedusing a different set of primers [Gehring et al., 1997].
Reference sequences belonging to all HIV-1 group Msubtypes and CRF02_AG and CRF14_BG, recombinantcarrying partial or total subtype G sequences at PR-coding region, were used as references in the phy-logenetic analysis and obtained from the GenBankdatabase. The tree topology was derived using the
Neighbor-Joining program. All trees were rooted withYBF30 (HIV-1 group N). Alignment of DNA sequenceswas performed using the Clustal X method. A pairwisedistance matrix was estimated using the Kimura two-parameter model within the DNADIST program, asimplemented in the PHYLIP software package. Boot-strap re-sampling (1000 data sets) of the multiplealignment was done to test the statistical robustness ofthe tree. A sample was ascribed to a specific subtypeafter the presence of a significant bootstrap, supportedby values equal or greater than 70% for the node joiningwith reference samples for that subtype. All sequenceswere deposited at the GenBank.
In addition, some specimens were analyzed using theSimPlot program (http://www.med.jhu.e-du/deptmed/sray/download/), to extend further the genetic charac-terization of potential recombinant sequences. More-over, all sequences were also examined using a rapidsubtyping method, provided by the Stanford University(http://hivdb.stanford.edu/hiv) [Shafer et al., 1998].
RESULTS
All specimens tested were collected from unrelatedindividuals living in Spain from 1997 to 2003. Forty-one (85.4%) were individuals of African origin, comingfrom 15 different countries, mostly from West Africa.Among them, 25% came from Equatorial Guinea, aformer Spanish colony located in West Africa andadjacent to Cameroon. In addition, three (6.2%) subjectswere Spaniards, two (4.2%) came from South America(Ecuador), and one (2.1%) from Portugal. The mean ageof the study populationwas 33 years, and 52.1% subjectswere male. Most had acquired HIV-1 infection throughheterosexual relationships in their country of origin.Table I summarizes the main characteristics of thestudy population and indicates the subtype assignmentconsidering the distinct HIV-1 genomic regions.
TheRT and env regions couldnot be amplified in 7 and13 out of 48 specimens, respectively. Overall, 32 (66.6%)specimens resulted to be inter-subtype recombinants.Although double recombinants were found most fre-quently (G/A in 20; G/B in 8; G/K in 2), two individualsharbored triple recombinant viruses (GPR/BRT/Aenv andGPR/KRT/Aenv, respectively).
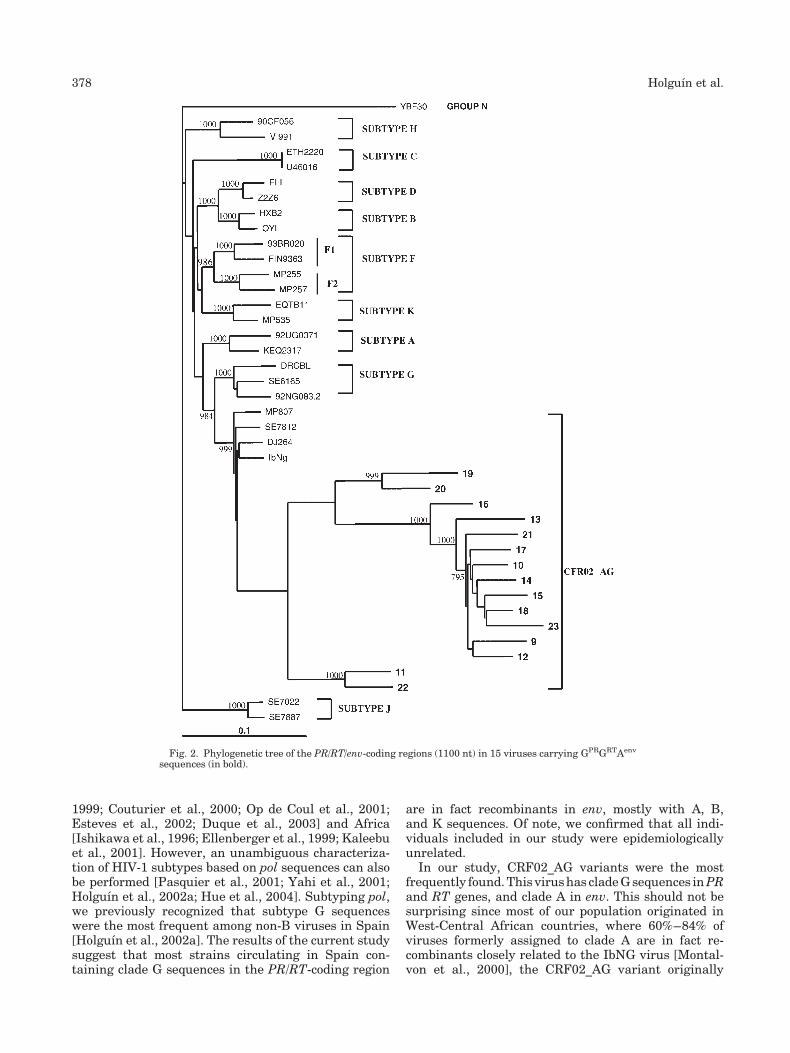
Only 33 (68.7%) and 9 (18.7%) sequences clusteredwith clade G when examining the RT and env genes,respectively. Figure 1 shows the phylogenetic tree of thePR/RT-coding region (840 nt) in those 33 viruses withcladeGsequences. Among them, env amplification couldbe obtained in 27 (Table I). The following subtypes wererecorded: G (29.6%), A (55.6%), B (11.1%), and K (3.7%).Therefore, nearly 70% of samples with PR/RT seq-uencesbelongingtosubtypeGwererecombinantsat env.Three sequences (nos. 26, 30, and 31) clustered withCRF14_BG, a recombinant virus recently found amongintravenous drug addicts in Northwestern Spain[Thomson et al., 2001] (Fig. 1).
The phylogenetic analysis including pol and envsequences (1100 nts) conducted in the 15 samples
HIV-1 Recombinants in Spain 375

carrying GPR/GRT/Aenv viruses showed that CRF02_AGvariants were the predominant virus in our studypopulation (Fig. 2). Interestingly, three samples (nos.3, 7, and 8) harboring clade G sequences at the threeregions clustered together and apart from pure subtypeG and GA recombinants. Further analysis in othergenomic regions are ongoing to determine the geneticnature of these viruses.Overall, 81.8% (27/33) of viruses with clade G
sequences at the PR/RT-coding regions were found insubjects coming from West African countries, mostly
from Liberia (n¼ 11) and Equatorial Guinea (n¼ 10).Among the rest, three were Spaniards, two came fromSouth America, and one from Portugal. The threeCRF14_BG specimens were found in one Spaniard(no. 31), a Portuguese (no. 26) and one individual fromCape Verd (no. 30). Two had been infected heterosexu-ally, whereas the other was a former intravenous druguser.
Clade K sequences at env (sample no. 35) and RT(samples nos. 37 and 38) were also confirmed using boththe SimPlot program and the Stanford software [Shafer
TABLE I. Main Demographics of the Study Population, and Subtype Assignment by Phylogenetic Analysis of the protease,RT,and env (V3)-Coding Regions, With the Corresponding GenBank Accession Numbers
No. SpecimenaGender/
ageCountry of
birthRoute ofinfection PR
Accessionnumber RT
Accessionnumber env
Accessionnumber
1 00SP.LP3 M/43 Liberia Htsex G AF455603 G AF455633 G AF4662052 99SP.1446016092 F/35 EG Htsex G AF354034 G AF455634 G AF4662063 00SP.19804 F/25 Africa (U) Htsex G AF354004 G AF455635 G AF4662074 98SP.7031-HC9 M/28 Liberia Htsex G AF125289 G AF188339 G AF1252995 98SP.10401-HC19 M/31 Nigeria Htsex G AF188351 G AF188346 G AF1883576 01SP.LP38 M/29 Togo Htsex G AF455630 G AF455660 G AF4662347 01SP.R3683 F/23 Ecuador Htsex G AF466244 G AF455661 G AF4662358 01SP.LP41 M/31 Spain IDUþHtsex G AF455632 G AF455653 G AF4662279 01SP.22158 M/41 EG Htsex G AF455665 G AF455636 A AF46620810 01SP.22470 F/34 EG Htsex G AF455666 G AF455637 A AF46620911 98SP.8416-HC12 F/24 EG Htsex G AF125292 G AF188342 A AF18835312 00SP.19770 F/40 EG Htsex G AF354007 G AF455638 A AF46621013 99SP.LP7 F/33 Liberia U G AF455606 G AF455639 A AF46621114 00SP.LP9 F/33 Liberia U G AF455607 G AF455640 A AF46621215 00SP.LP13 M/54 Liberia Htsex G AF455611 G AF455641 A AF46621316 00SP.LP18 M/45 Ghana Htsex G AF455615 G AF455642 A AF46621417 99SP.LP15 F/38 Sierra
LeoneHtsex G AF455612 G AF455643 A AF466215
18 00SP.LP21 M/36 Mauritania Htsex G AF455618 G AF455644 A AF46621619 98SP.7072-HC11 M/28 Ghana Htsex G AF125291 G AF188341 A AF12530120 98SP.7074-HC8 F/26 Mali Htsex G AF125288 G AF188338 A AF12529821 99SP.M1911 F/33 Zaire Htsex G AF247022 G AF455645 A AF46621722 97SP.4730-HC14 F/34 EG Htsex G AF125294 G AF455646 A AF46621823 00SP.19156 F/55 EG Htsex G AF354043 G AF455647 A AF46622124 98SP.6900-HC15 F/31 Liberia Htsex G AF125295 G AF188343 B AF12530225 00SP.21590 U GB IDU G AF455669 G AF455648 n.d. —26 97SP.4264.HC10 U Portugal ADVP G AF125290 G AF188340 B AF12530027 00SP.R3620 M/34 Ecuador Homo G AF354037 G AF455649 n.d. —28 00SP.LP6 M/38 Liberia Htsex G AF455605 G AF455650 n.d. —29 99SP.LP16 F/33 Sierra
LeoneU G AF455613 G AF455655 n.d. —
30 99SP.M2388 F/32 Cape Verd Htsex G AF247036 G AF455656 n.d. —31 01SP.22627 M/U Spain Htsex G AF455668 G AF455658 B AF46623232 99SP.15283 M/35 EG Htsex G AF354038 B AF455651 n.d. —33 00SP.SK2 M/17 Gambia Htsex G AF466718 B AF466719 n.d. —34 00SP.LP20 F/39 Ghana U G AF455617 B AF466246 n.d. —35 99SP.M2773 M/36 Cameroon Htsex G AF247037 G AF455659 K AF46623336 98SP.1067015541 F/25 Liberia Htsex G AF354030 B AF455652 A AF46622637 01SP.24316 M/27 Cameroon Htsex G AF455667 K AF455654 A AF46622838 99SP.M2444 F/35 EG Htsex G AF247023 K AF455664 n.d. —39 00SP.17749 F/U Spain U G AF354006 G AF455657 n.d. —40 01SP.22781 F/21 U IDUþHtsex G AF455670 B AF455662 n.d. —41 00SP.LP22 M/30 Liberia U G AF455619 A AF455663 n.d. —42 99SP.11269 M/32 Liberia Htsex G AF354020 n.d. — A AF46621943 97SP.5113-HC13 F/32 EG Htsex G AF125293 n.d. — A AF46622044 98SP.7019-7420 F/24 Ivory Coast Htsex G AF354027 n.d. — A AF46623845 96SP.1593 F/U Kenya Htsex G AF354019 n.d. — A AF46623946 96SP.2597 M/33 Liberia Htsex G AF466721 n.d. — G AF46624547 96SP.2703 F/37 Zaire Htsex G AF354021 n.d. — B AF46672248 01SP.R3594 M/24 Nigeria Htsex G AF354035 n.d. — n.d. —
n.d., subtype not determined due to repeated negative amplification results; U, unknown; EG, Equatorial Guinea; GB, Guinea Bissau; IDU,intravenous drug user; Htsex, heterosexual; and Homo, homosexual.aNomenclature: The first 2 numbers correspond to the year of isolation. SP, Spain. After dot, sample code.
376 Holguın et al.

et al., 1998] (data not shown). Patients nos. 35 and37 came fromCameroon, where a broad variety ofHIV-1subtypes has been described, whereas patient no. 38originated in Equatorial Guinea.
DISCUSSION
Tracking different HIV-1 subtypes and CRFs is ofcrucial importance for understanding trends in theAIDS pandemic. In Spain, even after the unprecedentedlarge number of immigrants arriving in Spain over thelast 10 years, many of whom came from HIV endemicregions in South America andWest Africa, HIV-1 non-Bclades still represent less than 3% of circulating viruses[Garcıa-Albert et al., 2001; Martın et al., 2002]. How-
ever, HIV-subtyping is still not implemented as aroutine in many hospitals. Therefore, infections withnon-B variants can be underestimated. In fact, non-Bcladeswere found innearly two-thirds ofHIV-1-infectedforeigners attended in our hospital until 2001, themajority being of African origin [Holguın et al., 2002a].Variants carrying subtype G sequences at the PR genewere predominant. Growing rates of infections causedby non-B viruses, including G strains, are currently ofgreat concern inmanyotherEuropean countries [reviewin Holguın et al., 2003].
When examining only env-coding regions, subtype Ahas been the subtype most frequently found in epide-miological studies conducted in Europe [Boni et al.,
Fig. 1. Phylogenetic tree of the PR/RT-coding region (840 nt) in 33 viruses with clade G sequences (inbold). CRF14_BG viruses with arrows.
HIV-1 Recombinants in Spain 377
1999; Couturier et al., 2000; Op de Coul et al., 2001;Esteves et al., 2002; Duque et al., 2003] and Africa[Ishikawa et al., 1996; Ellenberger et al., 1999; Kaleebuet al., 2001]. However, an unambiguous characteriza-tion of HIV-1 subtypes based on pol sequences can alsobe performed [Pasquier et al., 2001; Yahi et al., 2001;Holguın et al., 2002a; Hue et al., 2004]. Subtyping pol,we previously recognized that subtype G sequenceswere the most frequent among non-B viruses in Spain[Holguın et al., 2002a]. The results of the current studysuggest that most strains circulating in Spain con-taining clade G sequences in the PR/RT-coding region
are in fact recombinants in env, mostly with A, B,and K sequences. Of note, we confirmed that all indi-viduals included in our study were epidemiologicallyunrelated.
In our study, CRF02_AG variants were the mostfrequently found.This virushas cladeGsequences inPRand RT genes, and clade A in env. This should not besurprising since most of our population originated inWest-Central African countries, where 60%–84% ofviruses formerly assigned to clade A are in fact re-combinants closely related to the IbNG virus [Montal-von et al., 2000], the CRF02_AG variant originally
Fig. 2. Phylogenetic tree of the PR/RT/env-coding regions (1100 nt) in 15 viruses carrying GPRGRTAenv
sequences (in bold).
378 Holguın et al.

described in Nigeria [Howard and Rasheed, 1996]. InsomeWest African countries, such as Gabon, more than70% of circulating strains exhibit a discordant subtypein pol and env sequences, in accordance with this view[Pandrea et al., 2002].
In Spain, both G/A and G/B recombinants have beenreported previously among foreigners and nativeswho acquired HIV-1 infection through sexual contact[Holguın et al., 2000b, 2003] or among native intrave-nous drug users (IDUs) in Galicia [Perez-Alvarez et al.,2001; Thomson et al., 2001]. Our data represents thefirst indirect evidence for sexual transmission ofCRF14_BG variants. Two of the three individualscarrying CRF14 viruses denied any drug abuse, andwere most likely infected in Africa (one of them in CapeVerd Islands). We can not exclude the possibility thatthis CRF could have appeared originally in Africa,being transferred later to the west costs of the Ibericpeninsula, where those recombinants were firstlydescribed among intravenous drug users [Thomsonet al., 2001]. In Spain, CRF02_AG variants most likelyhave been circulating before CRF14_BG strains, whichmay have been introduced more recently and rapidlyspreading among intravenous drug addicts [Thomsonet al., 2001; Esteves et al., 2002; Duque et al., 2003].
In summary, this study shows that viruses circulatingin Spain containing clade G sequences at the PR-codingregion are mostly recombinants, CRF02_AG being themost frequent variant, but also appearing CRF14_BGand several URFs (unique recombinant forms). Anunknown proportion of viruses from Africans originallyassigned to cladeBorAbasedon env sequences,might infact be recombinants and carry clade G sequences at polgenes. Overall, our results point out that CRFs arehighly prevalent in Spain, being transmitted hetero-sexually in most instances. The spread of HIV-1 inter-subtype recombinants may have serious implicationson efforts to control the AIDS pandemic by futurevaccination trials, and might affect HIV-1 diagnosis,plasma viral load measurements, and even the activityof some antiretroviral drugs.
ACKNOWLEDGMENTS
We thank Dr. Marıa Jose Pena (Hospital Dr. Negrın,Las Palmas de Gran Canaria, Spain), Dr. Jorge delRomero (Centro Sandoval, Madrid, Spain), andDr. Belen Aracil (Hospital de Mostoles, Madrid, Spain)for providing some of the clinical specimens reportedin this article. Finally, the authors also thank JulieSheldon for her assistance in the final preparation of themanuscript in English.
REFERENCES
Arnold C, Barlow K, Kaye S, Loveday C, Balfe P, Clewley J. 1995. HIVtype 1 sequence subtype G transmission from mother to infant:Failure of variant sequence species to amplify in the RocheAmplicor test. AIDS Res Hum Retroviruses 11:999–1001.
Baldrich-Rubio E, Anagonou S, Stirrups K, Lafia E, Candotti D, Lee H,Allain J. 2001. A complex HIV type 1 A/G/J recombinant virusisolated from a seronegative patient with AIDS from Benin, WestAfrica. J Gen Virol 82:1095–1106.
Boni J, Pyra H, Gebhardt M, Perrin L, Burgisser P, Matter L, Fierz W,Erb P, Piffaretti J, Minder E, Grob P, Burckhardt J, Zwahlen M,Schupbach J. 1999. High frequency of non-B subtypes in newlydiagnosed HIV-1 infections in Switzerland. J Acquir Immun DeficSyndr 22:174–179.
Candotti D, Adu-Sarkodie Y, Davies F, Baldrich-Rubio E, Stirrups H,Allain J. 2000.AIDS inanHIV-seronegativeGhanaianwomanwithintersubtypeA/G recombinantHIV-1 infection. JMedVirol 62:1–8.
Coffin J. 1995. HIV population dynamics in vivo: Implicationsfor genetic variation, pathogenesis and therapy. Science 267:483–489.
Cornelissen M, Van Den Burg R, Zorgdrager F, Goudsmit J. 2000.Spread of distinct HIV type 1 AG recombinant lineages in Africa.J Gen Virol 81:515–523.
Couturier E, Damond F, Roques P, Fleury H, Barin F, Brunet J, Brun-Vezinet F, Simon F. 2000. HIV-1 diversity in France, 1996–1998.AIDS 14:289–296.
DescampsD, Apetrei C, Collin G, Damond F, Simon F, Brun-Vezinet F.1998. Naturally occurring decreased susceptibility of HIV-1 sub-type G to protease inhibitors. AIDS 12:1109–1111.
Duque V, Holguın A, Melico-Silvestre A, Gonzalez-Lahoz J, Soriano V.2003. HIV-1 recombinant B/G subtypes circulating in Coimbra,Portugal. Clin Microbiol Infect 9:422–425.
Ellenberger D, Pieniazek D, Nkengasong J, Luo C, Devare S, MauriceC, Janini M, Ramos A, Fridlund C, Hu D, Coulibaly I, Ekpini E,Wiktor S, Greenberg A, Schochetman G, Rayfield M. 1999. Geneticanalysis of HIV in Abidjan, Ivory Coast reveals predominance ofHIV-1 subtype A and introduction of subtype G. AIDS Res HumRetroviruses 15:3–9.
Esteves A, Parreira R, Venenno T, Franco M, Piedade J, Germano DeSousa J, Canas-FerreiraW. 2002.Molecular epidemiology of HIV-1infection in Portugal: High prevalence of non-B subtypes. AIDSResHum Retroviruses 18:313–325.
Garcıa-Albert L, Ortız M, Garcıa-Saiz A, Garcia-Saiz A. Group for theStudy of Subtype Prevalence in Spain. 2001. HIV type 1 non-Bsubtype prevalence in Spain, 1997–1998. AIDS Res Hum Retro-viruses 17:1317–1320.
Gehring S, Maayan S, Ruppach H, Balfe P, Juraszczyk J, Yust I,Vardinon N, Rimlawi A, Polak S, Bentwich Z, Rubsamen-Waigmann H, Dietrich U. 1997. Molecular epidemiology of HIV inIsrael. J AIDS Hum Retrovirol 15:296–303.
Holguın A, Soriano V. 2002b. Resistance to antiretroviral agents inindividualswithHIV-1 non-B subtypes.HIVClinTrials 3:403–411.
Holguın A, de Mendoza C, Soriano V. 1999. Comparison of three com-mercial methods for quantification of plasma viremia in clinicalspecimens belonging to non-B subtypes. Eur J ClinMicrobiol InfectDis 18:256–259.
Holguın A, Rodes B, Soriano V. 2000a. Protease gene analysis ofHIV-1 non-B subtypes in Spain. AIDS Res Hum Retroviruses 16:1395–1403.
Holguın A, Rodes B, Soriano V. 2000b. Recombinant HIV-1 circulatingin Spain. AIDS Res Hum Retroviruses 16:505–511.
HolguınA,AracilB, AlvarezA,BarrosC,SorianoV. 2001.Prevalence ofHIV-1 non-B subtypes in foreigners living in Madrid, Spain, andcomparison of the performance of the Amplicor HIV-1 Monitorversion 1.0 and the new automated version 1.5. J Clin Microb39:1850–1854.
Holguın A, Alvarez A, Soriano V. 2002a. High prevalence of subtype Gand spectrum of natural polymorphisms at the protease geneamong HIV-infected immigrants attended in Madrid. AIDS 16:1163–1170.
Holguın A, Alvarez A, Pena M, Artiles F, Molina L, Soriano V. 2003.High prevalence of non-B subtypes among HIV-positive immi-grants in the Canary Islands, Spain. Implications for public healthin Europe. HIV Clin Trials 4:184–192.
Howard T, Rasheed S. 1996. Sequence structure and nucleotidesequence analysis of a new HIV-1 subtype A strain from Nigeria.AIDS Res Hum Retroviruses 12:1413–1425.
Hue S, Clewley J, Cane P, Pillay D. 2004. HIV-1 pol gene variation issufficient for reconstruction of transmission in the era of anti-retroviral therapy. AIDS 18:719–728.
IshikawaK, JanssensW, Brandful J, Heyndrickx L, Takebe Y, AmpofoW, Sata T, Yamazaki S, Osei-Kwasi M, Yamamoto N, Koyanagi Y,Van der Groen G, Kurata T. 1996. Genetic and phylogeneticanalysis of HIV-1 env subtypes in Ghana, West Africa. AIDS ResHum Retroviruses 12:1575–1578.
HIV-1 Recombinants in Spain 379

Janssens W, Buve A, Nkengasong J. 1997. The puzzle of HIV-1subtypes in Africa. AIDS 11:705–712.
Jenny-Avital E, Beatrice S. 2001. Erroneously low or undetectableplasma HIV-1 RNA load, determined by PCR, in West African andAmerican patients with non-B subtype HIV-1 infection. Clin InfectDis 32:1227–1230.
Kaleebu P, Ross A, Morgan D, Yirrell D, Oram J, Rutebemberwa A,Lyagoba F, Hamilton L, Biryahwaho B, Whitworth J. 2001.Relationship between HIV-1 Env subtypes A and D and diseaseprogression in a rural Ugandan cohort. AIDS 15:293–299.
Kuiken C, Foley B, Hahn B, et al. 2000. HIV sequence compendium.Los Alamos, NM: Theoretical Biology and Biophysics Group, LosAlamos National Laboratory.
Martın JC, Holguın A, Soriano V. 2002. Prevalence of different HIV-1subtypes in an urban clinic in Madrid. Sex Transm Inf 78:e1.
McCutchan F. 2000. Understanding the genetic diversity of HIV-1.AIDS 14:S31–S44.
Montalvon C, Toure-Kane C, Liegeois F, Mpoudi E, Bourgeois A,Vergne L, Perret J, Boumah A, Saman E, Mboup S, Delaporte E,Peeters M. 2000. Most env and gag subtype A HIV-1 virusescirculating inWestandCentralAfrica are similar to those prototypeAGrecombinant virus IbNg. JAcquir ImmunDeficSyndr 23:363–374.
Nyambi P, Heyndrickx L, Vereecken K, Burda S, De Houwer K,Coppens S, UrbanskiM,Williams C, Ndumbe P, JanssensW. 2002.Predominance of infection with HIV-1 circulating recombinantform CRF02_AG in major Cameroonian cities and towns. AIDS16:295–305.
Op de Coul E, Coutinho R, van der Schoot A, van Doornum GJ,Lukashov VV, Goudsmit J, Cornelissen M. 2001. The impact ofimmigration on env HIV-1 subtype distribution among hetero-sexuals in the Netherlands: Influx of subtype B and non-B strains.AIDS 15:2277–2286.
Pandrea I, Robertson D, Onanga R, Gao F, Makuwa M, Ngari P,Bedjabaga I,RoquesP, SimonF,Apetrei C. 2002. Analysis of partialpol and env sequences indicates a high prevalence of HIV-1
recombinant strains circulating in Gabon. AIDS Res Hum Retro-viruses 18:1103–1116.
Pasquier C, Millot N, Njouom R, Sandres K, Cazabat M, Puel J, IzopetJ. 2001. HIV-1 subtyping using phylogenetic analysis of polgenesequences. J Virol Methods 94:45–54.
Peeters M, Esu-Williams E, Vergne L, Montavon C, Mulanga-KabeyaC, Harry T, Ibironke A, Lesage D, Patrel D, Delaporte E. 2000.Predominance of subtype A and G HIV type 1 in Nigeria, withgeographical differences in their distribution. AIDS Res HumRetroviruses 16:315–325.
Peeters M, Toure-Kane C, Nkengasong J. 2003. Genetic diversity ofHIV in Africa: Impact on diagnosis, treatment, vaccine develop-ment and trials. AIDS 17:2547–2560.
Perez-Alvarez L, Thomson M, Villahermosa L, de Parga EV,Rodriguez A, Cuevas T, Delgado E, Manjon N, Miralles C,Medrano L, Taboada J, Najera R. 2001. HIV-1 subtype G and BGrecombinant viruses in Spanish natives: Evidence of characteristicmutations in reverse transcriptase and protease. AIDS 15:1907–1910.
Shafer R, Eisen J,Merigan T, Katzenstein D. 1997. Sequence and drugsusceptibility of subtype C reverse transcriptase from HIV-1seroconverters in Zimbabwe. J Virol 71:5441–5448.
Shafer R, Jung D, Betts B, Xi Y, Gonzales M. 1998. HIV reversetranscriptase and protease sequence database. Nucleic Acid Res28:346–348.
ThomsonM,Delgado E,ManjonN, OcampoA, Villahermosa L,MarinoA, Herrero I, Cuevas T, Vazquez-de Parga E, Perez-Alvarez L,Medrano L, Taboada J, Najera R. 2001. HIV-1 genetic diversity inGalicia, Spain: BG intersubtype recombinant viruses circulatingamong injecting drug users. AIDS 15:509–516.
YahiN, Fantini J, Tourres C, Tivoli N, KochN, Tamalet C. 2001. Use ofdrug resistance sequence data for the systematic detection of non-BHIV-1 subtypes: How to create a sentinel site for monitoringthe genetic diversity of HIV-1 at a country scale. J Infect Dis 183:1311–1317.
380 Holguın et al.

Journal of Medical Virology 75:381–390 (2005)
Polymorphism and Drug-Selected Mutations in theReverse Transcriptase Gene of HIV-2 From PatientsLiving in Southeastern France
Philippe Colson,1 Mireille Henry,1 Natacha Tivoli,1 Herve Gallais,2 Jean-Albert Gastaut,3
Jacques Moreau,4 and Catherine Tamalet1*1Federation Hospitaliere de Microbiologie Clinique et d’Hygiene, Laboratoire de Virologie,Centre Hospitalo-Universitaire Timone, Faculte de Medecine, Universite de la Mediterranee, Marseille, France2Service de Maladies Infectieuses, Hopital Conception, Marseille, France3Service d’Hematologie, Hopital Sainte-Marguerite, Marseille, France4Service de Maladies Infectieuses, Hopital Nord, Marseille Cedex, France
Few data are available about the susceptibilityand the genotypic resistance pattern of humanimmunodeficiency virus type 2 (HIV-2) to nucleo-side reverse transcriptase inhibitors (NRTIs).The HIV-2 reverse transcriptase (RT) gene from25 HIV-2-infected patients followed-up in Mar-seilles and the surrounding area was analyzed.The aims of this study were to characterize thepolymorphismofHIV-2RT in theabsenceof drug,to determinewhether it naturally harbors codonsassociated with drug-resistance in HIV-1, andto identify mutations emerging under NRTI-selective pressure. Fourteen patients had neverundergone antiretroviral therapy and 11 receivedNRTI. Seventy sequences were analyzed. In un-treated patients, 12 spots of high natural poly-morphism (at positions 10, 11, 20, 43, 104, 121,135, 162, 176, 180, 200, and 227)were observed; 4of themwere specific of HIV-2 (10, 176, 180, 227).Moreover, results showed four positions thatcould be associated with natural resistance toNRTI (75I, 118I, 219E, and perhaps 215S), inaddition to those described previously for non-nucleoside reverse transcriptase inhibitors(NNRTIs) (181I, 188L, 190A). In HIV-2-infectedpatients receiving NRTI-containing therapies,specific genotypic patterns were observed witha high frequency of mutation Q151M (in 45% ofpatients) often associated with 70R, 115F, 214L,and/or 223R, which might compose an HIV-2multi-NRTI resistance complex. Four newly orrarely described NRTI-selected mutations wereobserved: I5V, K35R, F214L, andK223R. As inHIV-1, substitution M184V was found in 3TC-treatedpatients. In conclusion, these findings highlightthe need for specific guidelines for determininggenotypic resistance and treatment of HIV-2. J.Med. Virol. 75:381–390, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: HIV-2; reverse transcriptase;variability;drug-resistancemuta-tions; natural polymorphism
INTRODUCTION
Human immunodeficiency virus type 2 (HIV-2) isthe second causative agent of the acquired immunode-ficiency syndrome (AIDS). It is endemic in West Africa,where it was first identified in 1986 [Clavel et al., 1986],with a prevalence from 1% to 28% according to thecountries [Reeves and Doms, 2002]. Outside thisgeographical area, HIV-2 infections are mostly linkedepidemiologically to West Africa, as in Marseilles andthe surrounding area in southeastern France, whereHIV-2 is responsible for about 0.6% of cases of HIVinfections. HIV-2 and HIV-1 infections differ in theirvirological and clinical features [Bock and Markovitz,2001; Reeves and Doms, 2002], and the reverse trans-criptases of the two viruses have about 40% divergencein amino acid sequence [Guyader et al., 1987].
Regarding drug resistance, HIV-2 was found notsusceptible to non-nucleoside reverse transcriptaseinhibitors (NNRTIs) on the basis of in vitro data [Condraet al., 1992; Shaharabany and Hizi, 1992; Bacolla et al.,1993]. However, because of the limited geographicalspread of HIV-2, few data onHIV-2-infected individualsare available concerning the efficacy of antiretroviraltherapies including other drugs and the potentialemergence of resistance [Schutten et al., 2000; Sorianoet al., 2000; Smith et al., 2001; Houston et al., 2002].
*Correspondence to: Catherine Tamalet, Laboratoire de Vir-ologie, CHRU Timone, 264 rue Saint-Pierre, 13385 Marseillecedex 05, France. E-mail: [email protected]
Accepted 15 October 2004
DOI 10.1002/jmv.20296
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

Thus, to date, the therapeutic management of HIV-2-infected patients has been based mostly on HIV-1treatment guidelines. Moreover, recent clinical studiesshowed that patients under antiretroviral combinationregimen with or without protease inhibitors have asurprisingly high rate of virological failure [Adje-Toureet al., 2003; Brandin et al., 2003; Van der Ende et al.,2003]. Therefore, the present study was undertaken ona Marseilles cohort of 32 HIV-2-infected patients[Colson et al., 2004b] with the following aims: (i) tocharacterize the polymorphic regions in the HIV-2reverse transcriptase (RT) gene in theabsence of nucleo-side reverse transcriptase inhibitors (NRTIs), (ii) todetermine whether amino acids conferring resistance toNRTI in HIV-1 are present naturally in HIV-2, and (iii)to identify HIV-2-mutations selected under NRTI-selective pressure.
MATERIALS AND METHODS
Patients and Sequences
The Marseilles cohort included 32 HIV-2-infectedpatients being treated at different hospitals in Mar-seilles and the surrounding area. Twenty-four HIV-2reverse transcriptase sequences were available from14 naive patients, and 46 sequences from 11 patientsreceiving anNRTI-containing regimen. FourteenHIV-2sequences for 4 NRTI-naive patients and 43 sequencesfor 8NRTI-treated patientswere available from sequen-tial samples. For twoNRTI-treated patients, a sequencewas also available before initiation of antiretroviraltherapy. The 70 sequences studied included 17 DNAand 53 RNA sequences corresponding to different timepoints.
Nucleic Acid Extraction and Purification
Whole blood was collected in tubes containing EDTA.Plasma was aliquoted after a centrifugation step andstored at �808C. Viral RNA was concentrated from500 ml of plasma by ultracentrifugation for 1 hr at17,000� g and then extracted using the QIAamp ViralRNA Kit (Qiagen, Courtaboeuf, France). The viral RNAwas eluted in 50 ml of elution buffer. Peripheral bloodmononuclear cells (PBMC) were separated from bloodsamples collected in EDTA by Ficoll-Hypaque centrifu-gation (Eurobio, Les Ullis, France). Aliquots containing1� 106 to 5�106 PBMC measured by cell count werefrozen as dry pellets at �808C. The PBMC pellets werethawed, and totalDNAwas extractedusing theQIAampDNA minikit (Qiagen). Prepared RNA and DNA wereanalyzed directly or stored at �808C.
Nucleic Acid RT-PCR or PCR Amplificationand DNA and RNA pol Sequencing
Amplification and sequencing of proviral DNA andRNA, from PBMC and plasma respectively, wereperformed as described previously [Colson et al.,2004b]. In brief, a 10-ml sample of extracted viral RNAorproviralDNAwasused initially. Protease and reverse
transcriptase genomic regions of HIV-2 pol gene wereamplified in the samePCRproduct of 1753 bpwith outerprimers H2Mp1 (nucleotide (nt) 1859; the primerlocation was defined with reference to HIV-2 ROD[GenBank accession number M15390]) and H2Mp2(nt 3612) using SuperScriptTM One-Step RT-PCR withPlatinium Taq (Invitrogen Life technologies, Carlsbad,CA). Nested-PCR was performed using 1–10 ml of theprevious amplification product and inner primersH2Mp3 (nt 2020) and H2Mp4 (nt 3527) with Rochepolymerase (Roche, Mannheim, Germany) to obtain agenomic fragment of 1507 bp. The PCR products wereanalyzed in 1.5% agarose gel with ethidium bromide.The resulting PCR-amplified DNA fragments werepurified using Multiscreen PCR (Millipore, Molshein,France) as specified by the manufacturer. The PCRproduct was used as the DNA template for nucleotidesequencing analysis of the HIV-2 protease-RT codingregion with eight primers (H2Mp3; H2Mp6, nt 2482;H2Mp9, nt 2932; H2Mp5, nt 2441; H2Mp7, nt 2555;H2Mp8, nt 2857; H2Mp10, nt 3151; H2Mp4). Cyclesequencing of both strands was performed on theGeneAmp PCR system 9600 instrument (Applied-Biosystems, Branchburg,NJ)with theBigDyeTerminatorcycle-sequencing kit (Applied-Biosystems). Excess dye-labeled terminators were removed from the extensionproducts on SephadexG50 Superfine placed onNAHVN4550 plates (Millipore), and the purified products weresequenced on ABI Prism 3100 genetic analyzer(Applied-Biosystems). The nucleotide sequences of theprotease gene and the 240 first codons of the RT genewere aligned and translated with Auto Assembler andSequence Navigator software programs (Applied-Biosystems) by using as references the sequences ofHIV-2 ROD and HIV-1 HXB2 [GenBank accessionnumbers M15390 and K03455, respectively].
Variability Study
TheASVARAPprogram [http://ifr48.free.fr/recherche/jeu_cadre/jeu_rickettsie.html] was used; it analyzesautomatically amino acid variability at each positionin a set of sequences. Alignments of sets of sequenceswere created with ClustalX v.1.8 [Thompson et al.,1997], and then theprogramcalculated theproportion ofsequences harboring an amino acid different from theonemost frequently found at this position in the studiedset of sequences.
Statistical Analysis
Chi-square or Fisher tests were used to assess thesignificance of differences between sets of sequences.
RESULTS
Patients and HIV-2 ReverseTranscriptase Sequences
Themain epidemiological, immunological, and clinicalfeatures of the 32 patients are summarized in Table I.Twenty-six individuals had been infected several years
382 Colson et al.

TABLE
I.Epidem
iological,Clinical,andIm
munolog
icalCharacteristics
ofthe32HIV
-2In
fected
Patien
ts
Patien
tAge
(yr)
Gen
der
Geo
graphic
origin
(even
tual
epidem
iologicallink
withhighen
dem
icarea)
Transm
ission
group
Disea
sestagea
Duration
offollow
-up
(mon
ths)
First
CD4þ
cell
countavailable
during
follow
-up
(cells/m
m3)
Low
est
CD4þ
cell
countduring
follow
-up
(cells/m
m3)
HIV
-2su
btype
(pol
gen
e)Antiretrov
iral
therapy
Nucleo
side
reverse
transcriptase
inhibitorswhen
administeredb
Protease
inhibitors
when
administeredb
MRT-1
38
FAlgeria
Heterosex
ual
B71
210
188
AYes
AZT,3TC,
d4T,ABC
NFV
MRT-2
31
MIvoryCoa
stHeterosex
ual
A9
37
37
BNo
——
MRT-3
46
FGuinea
-Bissa
uHeterosex
ual
A65
616
429
N.A.
No
——
MRT-4
40
FFrance
Heterosex
ual
A34
445
326
ANo
——
MRT-5
35
FFrance
(Con
go)
Heterosex
ual
A22
676
442
ANo
——
MRT-6
35
FFrance
(sub-Saharan
Africa)
Heterosex
ual
A20
599
349
N.A.
No
——
MRT-7
47
MFrance
(sub-Saharan
Africa)
Heterosex
ual
A61
451
228
AYes
AZT,3TC,
ABC,ddI
RTV,NFV
MRT-8
52
FIvoryCoa
stTransfusion
A119
308
96
AYes
AZT,ddI,ddC,
3TC,d4T,ABC
SQV,RTV,
NFV,ID
VMRT-9
46
FSen
egal
Heterosex
ual
A21
404
504
ANo
——
MRT-10
51
MSen
egal
Heterosex
ual
A16
213
213
AYes
AZT,3TC,ABC
—MRT-11
40
MFrance
IVDU/
Heterosex
ual
A54
1883
1192
ANo
——
MRT-12
38
FBurk
inaFaso
Heterosex
ual
A75
200
200
AYes
AZT,3TC
—MRT-13
27
FSen
egal
Heterosex
ual
A9
580
Nodata
ANo
——
MRT-14
41
MGuinea
-Bissa
uUnknow
nA
6453
453
ANo
——
MRT-15
61
FNodata
Heterosex
ual
A60
498
476
ANo
——
MRT-16
61
MGuinea
-Bissa
u/Ivory
Coa
st/
Burk
inaFaso
Heterosex
ual
C12
62
61
BYes
AZT,3TC,ABC
NFV
MRT-17
37
MIvoryCoa
st/
Burk
inaFaso
Heterosex
ual
A133
464
243
BNo
——
MRT-18
50
MCen
tralAfrican
Rep
ublic
Heterosex
ual
A11
1019
984
ANo
——
MRT-19
40
MBurk
inaFaso
Heterosex
ual
A8
578
578
ANo
——
MRT-20
47
MGuinea
-Bissa
uHeterosex
ual
C105
805
137
AYes
AZT,ddI,
3TC,d4T
IDV,NFV
MRT-21
40
MSen
egal
Heterosex
ual
A57
1204
504
ANo
——
MRT-22
57
MSen
egal
Heterosex
ual
A55
126
76
AYes
AZT,ddC,3TC,
d4T,ddI
NFV,LPV/r
MRT-23
47
MGuinea
-Bissa
uHeterosex
ual
C75
375
175
AYes
AZT,ddI,
3TC,d4T
IDV,NFV
MRT-24
41
FGuadelou
pe
Heterosex
ual/
Transfusion
A38
156
144
AYes
AZT,3TC,d4T,
ddI,ABC
RTV,ID
V,
NFV
MRT-25
45
FFrance
(Cape
Verdeislands)
Heterosex
ual
B52
69
54
AYes
AZT,ddI,3TC,
d4T,ABC,ddC,
TDF
IDV,NFV,
LPV/r,APV,
RTV
MRT-26
56
FGuinea
-Bissa
uHeterosex
ual
A130
763
572
ANo
——
MRT-27
37
FGhaana
Heterosex
ual
C20
136
60
N.A.
Yes
AZT,3TC,d4T
IDV,LPV/r
MRT-28
72
FSen
egal
Heterosex
ual
A21
507
507
ANo
——
MRT-29
45
MSen
egal
Heterosex
ual
C12
10
4A
Yes
3TC,d4T
NFV
MRT-30
40
FBurk
inaFaso
Heterosex
ual
B139
600
189
AYes
3TC,AZT
—MRT-31
47
MBurk
inaFaso
Unknow
nC
(dea
d)
45
sNodata
BYes
d4T
NFV
MRT-32
79
MSen
egal
Heterosex
ual
A2
609
481
ANo
——
F,female;M,male;IV
DU,intraven
ousdru
guse;N.A.,not
available.
aAccordingto
Cen
ters
forDisea
seCon
trol
(CDC,Atlanta,GA)classification
(1993).
bAZT,zidov
udine;
3TC,lamivudine;
d4T,stavudine;
ddC,za
lcitabine;
ddI,didanosine;
ABC,abacavir;TDF,tenofov
irdisop
roxilfumarate;NFV,nelfinavir;RTV,ritonavir;SQV,sa
quinavir;ID
V,
indinavir;LPV/r,lopinavir
plusritonavir;andAPV,amprenavir.

beforehand and six were diagnosed during the previousyear. Twenty-nine patients had been followed-up untilstudy date; two were lost to follow-up, and one died.Median length of follow-up was 42 months (range, 2–139 months). Most patients were from or had epidemio-logical links with sub-Saharan Africa and were infectedby heterosexual contact. Twenty-three patients were atstage A, three at stage B, and six at stage C according tothe Centers for Disease Control (CDC) classification(1993). Median baseline and lowest CD4þ cell-countswere respectively 580 and 479 cells/mm3 for untreatedindividuals and 205 and 141 for NRTI-treated ones.Sequencingandphylogenetic analyses ofHIV-2RTwereperformed for 29 patients;HIV-2 strainswere of subtypeA in25 cases andof subtypeB in4 cases.Finally, 70HIV-2 RT sequences from 25 patients were studied. Foranalysis of natural polymorphism and natural re-sistance, 24 sequences from 14 of the 17 treatment-naive patients were used. For analysis of amino acidchanges selected under NRTI-selective pressure, 46sequences from 11 of the 15 patients treated with NRTIwere used. For these 11, median duration of treatmentwas 58 months (range 6–137 months). All receivedlamivudine (3TC), 10 zidovudine (AZT), 8 stavudine(d4T), 7 didanosine (ddI), 5 abacavir (ABC), and 1 teno-fovir (TDF). Ten of these 11 NRTI-treated patients alsoreceived protease inhibitors. Three patients received amonotherapy of AZT as first antiretroviral regimen,but other compounds where then added before thefirst time-point at which a sample was available forexamination. All patients received at least once bi- ortri-therapies (in two cases) of NRTI, which includedAZT, d4T, 3TC, ddC, ddI, or ABC.
Natural Polymorphism in HIV-2 RT
Sequences obtained from untreated patients wereused to examine natural polymorphism. An amino acidposition was considered polymorphic when at leasttwo different amino acids were found. Amino acidvariability at a given position was established bycalculating the percentage of sequences harboring anamino acid different from the onemost frequently foundat this position in the set of sequences. Twenty-foursequences were analyzed from 14 untreated patients(Fig. 1a). Concomitantly, amino acid variability in HIV-1 sequences from untreated patients was analyzed.Eighty-two (34%) of the 240 positions studied in HIV-2RT were polymorphic. The highest amino acid varia-bility (>30%) was at positions 10, 11, 20, 43, 104, 121,135, 162, 176, 180, 200, and 227 (Fig. 1a). In the Mar-seilles HIV sequence database [Tamalet et al., 2003],HIV-2 and HIV-1 consensus sequences from untreatedpatients differed at 81 amino acid positions. Eightpositions (10, 22, 28, 82, 86, 176, 180, 227) with aminoacid variability higher than 20% in HIV-2 (of whichfour had variability >30%) were only slightly variablein HIV-1 from untreated patients according to theMarseilles and StanfordHIVRTandProtease SequenceDatabases [Shafer et al., 1999; Rhee et al., 2003].
Conversely, three positions (83, 177, 207) with aminoacid variability >20% in HIV-1 sequences were totallyconserved in HIV-2 strains of the present study. Fouramino acid positions (122, 135, 162, 200) had similarvariability in HIV-1 and HIV-2 (>20%). Interestingly,in HIV-2 and HIV-1 RT, four regions were highlyconserved (variability<10%) with identical amino acidsat most positions: they concerned positions 91–100,107–120, 143–161, and 182–194 including the catalyticsite (except position 189 due to HIV-2 strains of subtypeB). In addition, in the present study, HIV-2 strainsof subtype A or B from untreated patients displayeddifferent amino acids at positions 8 (I or V), 21 (L/Q or I),27 (T or S), 32 (E orL), 66 (K orR), 90 (I orV), 107 (T or S),122 (K/E or P), 163 (T or S), 166 (Q/N or K), 169 (E or D),194 (TorS), 203 (LorS), 211 (G/SorN/D), 212 (LorM/V),218 (D or E), and 237 (T or K).
Analysis of Positions AssociatedWith Potential Natural Drug-Resistance
For 14 treatment-naive patients, HIV-2 sequences atthe 28 positions classically associated with HIV-1reverse transcriptase inhibitor (RTI) resistance werestudied according to the guidelines of the Internatio-nal AIDS Society [http://www.iasusa.org/resistance_mutations/resistance.pdf], and/or the French AIDSResearch Agency (ANRS) [http://www.hivfrenchresis-tance.org]. Amino acids at six positions corresponded inmost (>74%) or all sequences to those conferringresistance to NRTI in HIV-1: 75I, 118I, 219E, and toNNRTI: 181I, 188L, 190A (Table II). Of note, amino acid215S, which is a transitional mutant in HIV-1 RT, wasfound in all HIV-2 sequences from untreated patientsbut 215Y or 215F were never observed.
Identification of SuspectedNRTI-Resistance Mutations
Sequences from untreated and treated patients werecompared to identify mutations selected under NRTI-containing regimen, basedon thepresence of at least twoof the following criteria: (i) a significantly higher aminoacid variability at a given position inNRTI-treated thanin naive patients (Fig. 1); (ii) a significant differencebetween the frequency of amino acids at a given positionin naive and NRTI-treated patients (Table III); (iii)progressive selection of mutated codons observed inlongitudinal studies under selective pressure in at leasttwo NRTI-treated patients; and/or (iv) simultaneouspresence as a mixture of codons considered wild-typeand mutated in at least one sequence. Given thesecriteria, the following substitutions were suspected tooccur under the selective pressure of NRTI: I5V, K35R,Y115F, Q151M, M184V, F214L, and K223R (Fig. 1,Table III). Substitutions at positions 115, 151, and 184were identical to those observed in HIV-1. M184Valways emerged in presence of 3TC in nine of ninepatients examined. In the three cases for which a 184Mbaseline sequence was available at initiation of 3TC,184V occurred in 5 months or less. Interestingly, for
384 Colson et al.

one patient in whom 3TC was interrupted, 184M re-emerged. The emergence of Q151M mutant strainswas observed in patients who received various antire-troviral drugs such as AZT, d4T, ddI, ABC, ddC, or 3TC.For two patients, longitudinal analysis showed that115F or 223R HIV-2 RT mutants emerged simulta-neously with 151M. Amino acid 214L emerged simulta-neously with 151M in HIV-2 from two patients, andin one case it occurred when 151M replaced 151L. Inanother case, F214L occurred at least 2months after theQ151M mutation. At position 227 in the primer gripregion [Post et al., 2003], a tyrosine was observed in91% of sequences from HIV-2-treated patients and in50% of sequences from untreated patients (P< 0.05).Note, however, that amino acid substitution F227Y wasselected in only one patient during the longitudinalanalysis. No deletion or insertion was found at positions67–70 of RT. The following HIV-1 substitutions rarelyoccurred in HIV-2 RT: A62V and K65R from two NRTI-treated patients, and K70R from one NRTI-treatedpatient.
Associations or Exclusions of Mutations
In all available HIV-2 RT sequences from patientstreated or not with antiretroviral therapies, the poten-tial associations or exclusions of amino acids at eachposition inHIV-2RTwere studied. In all sequenceswithamino acids 115F and/or 223R, amino acid 184V waspresent. All sequences with amino acids 70R, 75M,115F, and/or 223R were 151M. Of note, amino acid 70Rwas found in only three sequences from treatedpatients;these sequences harbored amino acids 5V, 65R, 115F,151M, 184V, 214F, 223K, and 227Y. Variability differedsignificantly at various positions, depending onwhetheramino acid 151 was mutated or not: 70 (13% vs. 0%),75 (13% vs. 0%), 115 (30% vs. 0%), 214 (50% vs. 2%), 223(20% vs. 0%), and 227 (0% vs. 45%) (P< 0.05). A similarobservation was made for position 184 at positions 115(30% vs. 0%), 214 (40% vs. 6%), and 227 (10% vs. 38%)(P< 0.05). Note that globally the amino acid variabilityof theRT sequenceswas significantly lesserwhenaminoacid 151was amethionine thanwhen itwas a glutamine
Fig. 1. Amino acid variability at each site of HIV-2 reversetranscriptase for untreated individuals (a) and patients treatedwith nucleoside reverse transcriptase inhibitor-containing regimen(b). Gray rods indicate positions where variability exceeded 30%.aVariability was considered the percentage of sequences that harboredat agivenpositionanamino acid thatwasnot themost frequently found
in the studied set of sequences. Amino acid positions of interest (highlyvariable or potentially associated with resistance to nucleoside reversetranscriptase inhibitors) are in bold font. Hachured rods indicatepositions suspected of being associated with resistance to nucleosidereverse transcriptase inhibitors in HIV-2.
Polymorphism and Resistance in HIV-2 Reverse Transcriptase 385

TABLE
II.Frequen
cyof
AminoAcidsin
HIV
-2Rev
erse
Transcriptase
From
Untrea
tedandNRTI-TreatedPatien
tsatPositionsAssociatedWithNRTIandNNRTI-Resistance
inHIV
-1
Virus
HIV
-2reverse
transcriptase
aminoacidatpositiona
41
44
62
65
67
69
70
74
75
77
100
103
106
108
115
116
118
151
181
184
188
190
210
215
219
225
230
236
WtHIV
-1amino
acidsb
ME
AK
DT
KL
VF
LK
VV
YF
VQ
YM
YG
LT
KP
MP
Dru
g-resistant
HIV
-1amino
acidsc
LD
VR
ND/Ins
RV
IL
IN
AI
FY
IM
C/I
V/I
L/C/H
S/A
WY/F
Q/E
HL
L
HIV
-2su
btypeA
consensu
s
sequen
ced
m(t)
EA
KD
NK
LI
FL
KI
V(i)
YF
IQ
i(v)
ML
AN
SE
PM
P
HIV
-2su
btypeB
consensu
s
sequen
ced
ME
AK
DN
KL
IF
l(a)
k(x)
IV
YF
i(v)
QI
ML
AN
SE
PM
P
Aminoacids
observed
in
HIV
-2strains
from
untrea
ted
patien
ts(%
of
sequen
ces)
M(100)
E(100)
A(100)
K(100)
D(96)
D/G
(4)
N(100)
K(100)
L(100)
I(100)
F(100)
L(100)
K(88)
R(12)
I(100)
V(100)
Y(100)
F(100)
I(100)
Q(100)
I(83)
V(13)
I/V
(4)
M(100)
L(1
00)
A(1
00)
N(100)
S(100)
E(7
5)
D(25)
P(100)
M(100)
P(96)
Q(4)
Aminoacids
observed
in
HIV
-2strains
from
patien
ts
receiving
NRTI-
containing
antiretrov
iral
regim
en(%
of
sequen
ces)
M(100)
E(98)
N/Q
(2)
A(94)
A/V
(4)
V(2
)
K(91)
K/R
(7)
R(2
)
D(100)
N(100)
K(94)
K/R
(4)
R(2
)
L(100)
I(92)
M(8)
F(100)
L(100)
K(94)
R(6)
I(100)
V(100)
Y(83)
F(1
1)
F/Y
(4)
F/S(2)
F(100)
I(100)
M(5
9)
Q(39)
L(2)
I(94)
V(6)
V(6
1)
M(37)
M/I
/V(2
)
L(1
00)
A(1
00)
N(100)
S(85)
T(9)
F(4
)
P(2)
E(8
7)
D(13)
P(100)
M(100)
P(100)
Ins,insertionatRTcodon
67–70;NRTI,nucleosidereverse
transcriptase
inhibitors;NNRTI,non
-nucleosidereverse
transcriptase
inhibitors.
aBelow
each
aminoacidpositionisthelist
ofaminoacidsthatare
observed
andin
parentheses
isthepercentageofsequen
cesconcern
ed.T
hepredom
inantaminoacidforea
chpositionisgiven
first.Amino
acidsthatare
themostfreq
uen
tlyfoundor
alw
ayspresentin
HIV
-2strainsandassociatedwithdru
g-resistance
inHIV
-1are
inbold.
bWt,Wild-type;
sequen
cefrom
thereference
HIV
-1strain
HXB2(G
enBankaccession
number
K03455).
c Accordingto
theIn
tern
ation
alAID
SSociety
(IAS;http://w
ww.iasu
sa.org/resistance_m
utation
s/resistance.pdf).
dAvailable
atURL:http://hiv-w
eb.lanl.gov/con
tent/hiv-db/ALIG
N_C
URRENT/ALIG
N-INDEX.htm
l;non
predom
inantaminoacidsare
indicatedin
parentheses.

TABLE
III.
Frequen
cyof
AminoAcidsin
HIV
-2Rev
erse
Transcriptase
From
Untrea
tedandNRTI-Con
tainingReg
imen
TreatedPatien
tsatPositionsSusp
ectedof
Being
AssociatedWithResistance
toNucleo
sideRev
erse
Transcriptase
Inhibitors
Virus
HIV
-2reverse
transcriptase
aminoacidatpositiona
5**
b35*
115*
151**
184**
214*
223(*)
WtHIV
-1aminoacidsc
IV
YQ
ML
KDru
g-resistantHIV
-1aminoacids,
when
described
d—
—F
MV/I
——
HIV
-2su
btypeA
consensu
ssequen
cee
i(l/v)
k(r)
YQ
MF
K
HIV
-2su
btypeB
consensu
ssequen
cee
i(v)
KY
QM
FK
Aminoacidsob
served
inHIV
-2strainsfrom
patien
tswithou
tanyantiretrov
iraltherapy
(%of
sequen
ces)
I(88)
V(1
2)
K(100)
Y(100)
Q(100)
M(100)
F(100)
K(100)
Amino
acids
observed
inHIV
-2strainsfrom
patien
tsreceivingNRTI-containing
antiretrov
iralregim
en(%
ofsequen
ces)
V(4
8)I(33)
V/
I(17)L(2)
K(78)
R(1
5)
K/R
(7)
Y(83)
F(1
1)
F/
Y(4
)F
/S(2
)M
(59)Q(39)L(2)
V(6
1)M(37)
M/I
/V(2
)F(65)
L(2
4)
F/
L(9
)F/C(2)
K(87)
K/R
(13)
NRTI,nucleosidereverse
transcriptase
inhibitors.
aBelow
each
aminoacidpositionisthelist
ofaminoacidsthatare
observed
andin
parentheses
isthepercentageof
sequen
cesconcern
ed.T
hepredom
inantaminoacidforea
chpositionisgiven
first.
Aminoacidsfoundto
beselected
under
NRTI-containingantiretrov
iraltherapyare
inbold.
bStatisticaldifference
betweenpresence
ofaminoacidsin
HIV
-2RTfrom
untrea
tedandNRTI-trea
tedpatien
ts,*P<0.05;**P
<10�3;(*)
P<0.05when
consideringpatien
tsreceivingzidov
udine-
containingantiretrov
iralregim
en.
c wt,Wild-type;
sequen
cefrom
thereference
HIV
-1strain
HXB2(G
enBankaccession
number
K03455).
dAccordingto
theIn
tern
ation
alAID
SSociety
(IAS;http://w
ww.iasu
sa.org
/resistance_m
utation
s/resistance.pdf).
eAvailable
atURL:http://hiv-w
eb.lanl.gov
/con
tent/hiv-db/ALIG
N)_CURRENT/ALIG
N-INDEX.htm
l;non
predom
inantaminoacidsare
indicatedin
parentheses.
Polymorphism and Resistance in HIV-2 Reverse Transcriptase 387

(mean amino acid variability per site, 4.6 vs. 6.6;P<0.05). For example, positions 21, 86, 121, and 176weremore conservedwhenaminoacid151wasM(100%,97%, 100%, and 87% of sequences, respectively) thanwhen it was Q (68%, 55%, 43%, and 43% of sequences,respectively) (P<0.05 in all cases).
DISCUSSION
To gain insight onHIV-2 resistance to antiretrovirals,the genotypic patterns of 70 HIV-2 RT from 25 patientsfollowed-up in southeastern France were analyzed.New information about the natural polymorphism
of HIV-2 RT included 12 highly polymorphic spots(at positions 10, 11, 20, 43, 104, 121, 135, 162, 176,180, 200, and 227) detected in the HIV-2 RT fromuntreated patients. Some of these natural polymorph-isms were located at specific positions in HIV-2 RT. Incontrast, in other regions such as from amino acid 91 to100 and 143 to 161, both HIV-2 and HIV-1 RT wereconserved. This finding suggests a common structuralbackbone for enzymes of both viruses. Obviously, theseconserved areas should be taken into account in thedesign of new antiviral compounds.Natural resistance to NNRTI is a well-described
phenomenon in HIV-2, based on phenotypic, bio-chemical, and structural considerations [Condra et al.,1992; Shaharabany and Hizi, 1992; Bacolla et al., 1993;Hizi et al., 1993; Witvrouw et al., 1999; Smith et al.,2001; Ren et al., 2002]. In HIV-1, the amino acidsubstitutions Y181I, Y188L, and G190A confer resis-tance to nevirapine and efavirenz [http://www.iasusa.org/resistance_mutations/resistance.pdf] and they arethenaturalaminoacids inHIV-2RTsequences (Table II).Applying the same approach as for NNRTI, one mightconsider HIV-2 strains to be naturally resistant to someNRTI. Thus, HIV-2 sequences from untreated patientsexhibited amino acids 75I, 118I, and 219E, whichpotentially confer low-level resistance to some NRTI inHIV-1 (Table II). Amino acid 215S was naturally foundin all cases, but was only described as transitional toresistance inHIV-1 [deRondeet al., 2001;Garcia-Lermaet al., 2001]. Yet, in vitro studies revealed that HIV-2and HIV-1 isolates were similarly susceptible to NRTI,hence excluding the possibility of natural resistance toNRTI [Coates et al., 1992; Cox et al., 1994; Perach et al.,1995;Balzarini et al., 1996, 1997;Witvrouwet al., 2004].In contrast, recent results showed that HIV-2 was 200-fold less susceptible to AZT than HIV-1 [Reid et al.,2004], which suggests HIV-2 strains are naturallyresistant to this compound.TheHIV-2 RT drug-selected patterns observed in this
study differed from those described for HIV-1 strains.The HIV-2 sequences from treated patients showedseven amino acid substitutions, I5V, K35R, Y115F,Q151M, M184V, F214L, and K223R, emerging underNRTI-selective pressure (Table III). Of interest, onlythree of them, at positions 115, 151, and 184, also occurin HIV-1-infected patients. In line with recent reports[Rodes et al., 2000; Adje-Toure et al., 2003; Brandin
et al., 2003; Van der Ende et al., 2003],M184Vmutationwas found in HIV-2 strains from 9 of 11 (82%) 3TC-treated patients. Furthermore, the role of M184V inHIV-2-drug resistance was supported by a few pheno-typic studies [Van der Ende et al., 2000; Smith et al.,2001; Adje-Toure et al., 2003]. Interestingly, M184V islocated in theYMDDmotif of theRTcatalytic site,whichis conserved and identical in HIV-2, HIV-1, and in thehepatitis B virus (HBV) [Das et al., 2001]. Moreover, itoccurs under 3TC-selective pressure in the threeviruses. Given the high frequency of HIV-2-HBV co-infections in the Marseilles cohort (30%) [Colson et al.,2004a], this would contra-indicate the use of 3TCmonotherapy in HIV-2-HBV co-infected patients, asrecommended in the setting of HIV-1-HBV co-infections[de Franchis et al., 2003]. Q151M mutant strains,observed in less than 5% of HIV-1-infected treatedpatients [Van Vaerenbergh et al., 2000], were found in5 of 11 (45%) HIV-2-infected treated patients, but neverin naive patients. This observation agreeswith previousstudies, which found 151M sequences in 17%–40% ofHIV-2-infected patients under combination therapyincluding AZT, ddI, or d4T [Rodes et al., 2000; Adje-Toure et al., 2003; Brandin et al., 2003; Van der Endeet al., 2003]. Of note, a Q151Lmutation was observed inone patient having received dual therapies includingAZT, 3TC, ddI, or d4T. 151L preceded 151M and couldbe considered a transitional amino acid as in HIV-1[Kosalaraksa et al., 1999]. The Q151M mutation isresponsible for significantly lower susceptibility of HIV-2 or SIV to various NRTI, according to phenotypicstudies [Van Rompay et al., 1997; Van der Ende et al.,2000; Adje-Toure et al., 2003]. Furthermore, in thisstudy as in previous reports, 151M variants wereassociated with therapeutic failure, including drop inCD4-cell counts and/or persistently detectable HIV-2RNA in plasma [Rodes et al., 2000; Adje-Toure et al.,2003; Brandin et al., 2003; Van der Ende et al., 2003].In HIV-1, 151M is associated with 62V, 75I, 77L, and116Y in amulti-NRTI resistance (MNR) complex [http://www.iasusa.org/resistance_mutations/resistance.pdf].In contrast, in the present study, 151M was preferen-tially associated with 70R, 115F, 214L, and/or 223R,which occurred (in all but one case, concerning 214L) inHIV-2 151Mvariants andmay constitute a specificHIV-2 MNR complex. Together, these data raise theimportant question of whether Q151M induces resis-tance to NRTI by itself or whether it needs accompany-ingmutations such as those described above. Answeringthis question would require further investigations,including fitness analysis.
The Y115F mutation was observed in 18% of NRTI-treated patients. As this substitution usually emergesunder abacavir selective pressure in HIV-1 [Tisdaleet al., 1997], it should be noted that, in this study,mutation Y115F was observed in HIV-2 from a patientwho received abacavir. The classical HIV-1 thymidineanalogue mutations (TAM) [Yahi et al., 2000] were notfound in the present study, except the K70R sub-stitution, in one patient who previously received AZT-
388 Colson et al.

containing regimen. In the present study, 219D washarbored by 25% of sequences from untreated patientsand 13% of sequences from treated patients (Table II);the E219D substitution was previously observed inHIV-2 EHO [Witvrouw et al., 1999], and in two HIV-2-infected patients in another study, in which it wasconsideredaprimaryAZTresistancemutation [Brandinet al., 2003]. The absence of deletion or insertion at RTcodons 67–70 in the present study might be related tothe limited number of treated patients, given the lowfrequency (<5%) of these mutations in HIV-1 [Tamaletet al., 2000].
Until the study date, NRTI-selected mutation K223Rwas never observed, and mutations I5V, K35R, andF214L were reported only once [Brandin et al., 2003].Amino acid changes I10V, A11T, K20R, R43K, I75M,Y162H, and S176P were observed, as in other studies[Van der Ende et al., 1996, 2003; Rodes et al., 2000;Brandin et al., 2003], but according to the present data,they cannot be related to NRTI-selective pressure.Conversely, mutations K40R, N69S, K70N/S, R147V,T163A,K177M,F221Y, andR228H, previously reportedinHIV-2-infectedpatients,werenot found in thepresentstudy [VanderEnde et al., 1996;Rodes et al., 2000;Adje-Toure et al., 2003; Brandin et al., 2003].
In summary, the findings suggest possible naturalresistance to NRTI at codons 75, 118, and 219.MutationQ151M was found with high frequency in NRTI-treated patients, and it could constitute a major re-sistance pathway. Contrasting with the HIV-1 setting,the classicalmembers of theMNRQ151M complexwerenot found, nor were the classical TAM. An importantoutcome of the present study is the possible involvementof four amino acids (70R, 115F, 214L, 223R) in a specificHIV-2 MNR complex. Therefore, HIV-2 could elicitpathways different than those of HIV-1 for NRTIresistance, except M184V, which emerged in 3TC-treated patients. Finally, four drug-resistance muta-tions described in this study were rarely or neverreported to date: I5V, K35R, F214L, and K223R. Theseresults require confirmation in additional patients andin investigations including site-directed mutagenesisand fitness experiments. The findings warrant specificantiretrovirals against HIV-2 strains and specific inter-pretation rules ofHIV-2genotypic resistancemutations.
REFERENCES
Adje-Toure CA, Cheingsong R, Garcia-Lerma JG, Eholie S, BorgetMY,Bouchez JM, Otten RA,Maurice C, Sassan-MorokroM, Ekpini RE,Nolan M, Chorba T, Heneine W, Nkengasong JN. 2003. Antire-troviral therapy inHIV-2-infectedpatients: changes inplasmaviralload, CD4þ cell counts, and drug resistance profiles of patientstreated in Abidjan, Cote d’Ivoire. AIDS 17:S49–S54.
Bacolla A, Shih CK, Rose JM, Piras G, Warren TC, Grygon CA,IngrahamRH, Cousins RC, Greenwood DJ, Richman D, Cheng YC,Griffin JA. 1993. Amino acid substitutions in HIV-1 reversetranscriptase with corresponding residues from HIV-2. Effect onkinetic constants and inhibition by non-nucleoside analogs. J BiolChem 268:16571–16577.
Balzarini J, Wedgwood O, Kruining J, Pelemans H, Heijtink R,De Clercq E, McGuigan C. 1996. Anti-HIV and anti-HBV activityand resistance profile of 20,30-dideoxy- 30-thiacytidine (3TC) and its
arylphosphoramidate derivative CF 1109. Biochem Biophys ResCommun 225:363–369.
Balzarini J, Kruining J, Wedgwood O, Pannecouque C, Aquaro S,Perno CF, Naesens L, Witvrouw M, Heijtink R, De Clercq E,McGuigan C. 1997. Conversion of 20,30-dideoxyadenosine (ddA)and 20,30-didehydro-20,30-dideoxyadenosine (d4A) to their corre-sponding aryloxyphosphoramidate derivatives markedly potenti-ates their activity against human immunodeficiency virus andhepatitis B virus. FEBS Lett 410:324–328.
Bock PJ, Markovitz DM. 2001. Infection with HIV-2. AIDS 15:S35–S45.
Brandin E, Lindborg L, Gyllensten K, Brostrom C, Hagberg L, GisslenM, Tuvesson B, Blaxhult A, Albert J. 2003. pol gene sequencevariation in Swedish HIV-2 patients failing antiretroviral therapy.AIDS Res Hum Retroviruses 19:543–550.
Clavel F, Guetard D, Brun-Vezinet F, Chamaret S, Rey MA, Santos-FerreiraMO,LaurentAG,DauguetC,KatlamaC,RouziouxC, etal.1986. Isolation of a new human retrovirus from West Africanpatients with AIDS. Science 233:343–346.
Coates JA, Cammack N, Jenkinson HJ, Jowett AJ, JowettMI, PearsonBA, PennCR,Rouse PL, VinerKC, Cameron JM. 1992. (-)-20-deoxy-30-thiacytidine is a potent, highly selective inhibitor of humanimmunodeficiency virus type 1 and type 2 replication in vitro.Antimicrob Agents Chemother 36:733–739.
ColsonP,HenryM,SimonS,TivoliN,GallaisH,MoreauJ,Gastaut JA,Tamalet C. 2004a. High prevalence of HBV genotype E infectionin HIV-2-infected patients living in southeastern France. 11thConference on Retroviruses and Opportunistic Infections, SanFrancisco, Calif., USA: abstr. 833.
Colson P, Henry M, Tourres C, Lozachmeur D, Gallais H, Gastaut JA,Moreau J, Tamalet C. 2004b. Polymorphism and drug-selectedmutations in the protease gene of human immunodeficiency virustype 2 from patients living in Southern France. J Clin Microbiol42:570–577.
Condra JH, Emini EA, Gotlib L, Graham DJ, Schlabach AJ, WolfgangJA, Colonno RJ, Sardana VV. 1992. Identification of the humanimmunodeficiency virus reverse transcriptase residues that con-tribute to the activity of diverse nonnucleoside inhibitors. Anti-microb Agents Chemother 36:1441–1446.
Cox SW, Aperia K, Albert J, Wahren B. 1994. Comparison of thesensitivities of primary isolates of HIV type 2 and HIV type 1 toantiviral drugs and drug combinations. AIDS Res Hum Retro-viruses 10:1725–1729.
Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG,Arnold E. 2001. Molecular modeling and biochemical characteriza-tion reveal the mechanism of hepatitis B virus polymeraseresistance to lamivudine (3TC) and emtricitabine (FTC). J Virol75:4771–4779.
de Franchis R, Hadengue A, Lau G, Lavanchy D, Lok A, McIntyre N,Mele A, Paumgartner G, Pietrangelo A, Rodes J, Rosenberg W,Valla D, EASL Jury. 2003. EASL International ConsensusConference on Hepatitis B. 13–14 September, 2002 Geneva,Switzerland. Consensus statement (long version). J Hepatol 39:S3–S25.
de Ronde A, van Dooren M, van Der Hoek L, Bouwhuis D, de Rooij E,van Gemen B, de Boer R, Goudsmit J. 2001. Establishment of newtransmissible and drug-sensitive human immunodeficiency virustype 1 wild types due to transmission of nucleoside analogue-resistant virus. J Virol 75:595–602.
Garcia-Lerma JG, Nidtha S, Blumoff K, Weinstock H, Heneine W.2001. Increased ability for selection of zidovudine resistance in adistinct class ofwild-typeHIV-1 fromdrug-naive persons. ProcNatlAcad Sci USA 98:13907–13912.
Guyader M, EmermanM, Sonigo P, Clavel F, Montagnier L, Alizon M.1987. Genome organization and transactivation of the humanimmunodeficiency virus type 2. Nature 326:662–669.
Hizi A, Tal R, Shaharabany M, Currens MJ, Boyd MR, Hughes SH,McMahon JB. 1993. Specific inhibition of the reverse trans-criptase of human immunodeficiency virus type 1 and the chimericenzymes of human immunodeficiency virus type 1 and type 2 bynonnucleoside inhibitors. Antimicrob Agents Chemother 37:1037–1042.
Houston SC, Miedzinski LJ, Mashinter LD. 2002. Rapid progression ofCD4 cell decline and subsequent response to salvage therapy inHIV-2 infection. AIDS 16:1189–1191.
Kosalaraksa P, Kavlick MF, Maroun V, Le R, Mitsuya H. 1999.Comparative fitness of multi-dideoxynucleoside-resistant human
Polymorphism and Resistance in HIV-2 Reverse Transcriptase 389

immunodeficiency virus type 1 (HIV-1) in an in vitro competitiveHIV-1 replication assay. J Virol 73:5356–5363.
PerachM, Rubinek T, Hizi A. 1995. Resistance to nucleoside analogs ofselective mutants of human immunodeficiency virus type 2 reversetranscriptase. J Virol 69:509–512.
Post K, Guo J, Howard KJ, Powell MD, Miller JT, Hizi A, Le Grice SF,Levin JG. 2003. Human immunodeficiency virus type 2 reversetranscriptase activity in model systems that mimic steps in reversetranscription. J Virol 77:7623–7634.
Reeves JD, Doms RW. 2002. Human immunodeficiency virus type 2.J Gen Virol 83:1253–1265.
Reid P, MacInnes H, Cong M, Heneine W, Garcia-Lerma JG. 2004.Natural resistance of HIV-2 to zidovudine. 11th Conference onRetroviruses and Opportunistic Infections, San Francisco, Calif.,USA, abstr. 691.
Ren J, Bird LE, Chamberlain PP, Stewart-Jones GB, Stuart DI,Stammers DK. 2002. Structure of HIV-2 reverse transcriptaseat 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc Natl Acad Sci USA 99:14410–14415.
Rhee SY,GonzalesMJ,Kantor R, Betts BJ, Ravela J, Shafer RW. 2003.Human immunodeficiencyvirus reverse transcriptase andproteasesequence database. Nucleic Acids Res 31:298–303.
Rodes B, Holguin A, Soriano V, Dourana M, Mansinho K, Antunes F,Gonzalez-LahozJ. 2000.Emergence of drug resistancemutations inhuman immunodeficiency virus type 2-infected subjects under-going antiretroviral therapy. J Clin Microbiol 38:1370–1374.
Schutten M, van der Ende ME, Osterhaus AD. 2000. Antiretroviraltherapy in patients with dual infection with human immunodefi-ciency virus types 1 and 2. N Engl J Med 342:1758–1760.
Shafer RW, Stevenson D, Chan B. 1999. Human immunodeficiencyvirus reverse transcriptase and protease sequence database.Nucleic Acids Res 27:348–352.
Shaharabany M, Hizi A. 1992. The catalytic functions of chimericreverse transcriptases of human immunodeficiency viruses type 1and type 2. J Biol Chem 267:3674–3678.
Smith NA, Shaw T, Berry N, Vella C, Okorafor L, Taylor D, AinsworthJ, Choudhury A, Daniels RS, El-Gadi S, Fakoya A,Moyle G, OxfordJ, Tedder R, O’Shea S, de Ruiter A, Breuer J. 2001. Antiretroviraltherapy for HIV-2 infected patients. J Infect 42:126–133.
Soriano V, Gomes P, Heneine W, Holguin A, Doruana M, Antunes R,Mansinho K, Switzer WM, Araujo C, Shanmugam V, Lourenco H,Gonzalez-Lahoz J, Antunes F. 2000. Human immunodeficiencyvirus type 2 (HIV-2) in Portugal: Clinical spectrum, circulatingsubtypes, virus isolation, and plasma viral load. J Med Virol61:111–116.
Tamalet C, Yahi N, Tourres C, Colson P, Quinson AM, Poizot-Martin I,Dhiver C, Fantini J. 2000. Multidrug resistance genotypes (inser-tions in the beta3-beta4 finger subdomain and MDR mutations)of HIV-1 reverse transcriptase from extensively treated patients:Incidence and association with other resistance mutations. Viro-logy 270:310–316.
Tamalet C, Fantini J, Tourres C, Yahi N. 2003. Resistance of HIV-1to multiple antiretroviral drugs in France: A 6-year survey (1997–2002) based on an analysis of over 7000 genotypes. AIDS 17:2383–2388.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG.1997. The CLUSTAL_X windows interface: Flexible strategies formultiple sequence alignment aided by quality analysis tools.Nucleic Acids Res 25:4876–4882.
Tisdale M, Alnadaf T, Cousens D. 1997. Combination of mutations inhuman immunodeficiency virus type 1 reverse transcriptaserequired for resistance to the carbocyclic nucleoside 1592U89.Antimicrob Agents Chemother 41:1094–1098.
Van der Ende ME, Schutten M, Ly TD, Gruters RA, Osterhaus AD.1996. HIV-2 infection in 12 European residents: Virus character-istics and disease progression. AIDS 10:1649–1655.
Van der EndeME, Guillon C, Boers PH, Ly TD, Gruters RA, OsterhausAD, SchuttenM. 2000. Antiviral resistance of biologic HIV-2 clonesobtained from individuals on nucleoside reverse transcriptaseinhibitor therapy. J Acquir Immune Defic Syndr 25:11–18.
Van der Ende ME, Prins JM, Brinkman K, Keuter M, Veenstra J,Danner SA, Niesters HG, Osterhaus AD, Schutten M. 2003.Clinical, immunological and virological response to differentantiretroviral regimens in a cohort of HIV-2-infected patients.AIDS 17:S55–S61.
Van Rompay KK, Greenier JL, Marthas ML, Otsyula MG, Tarara RP,Miller CJ, Pedersen NC. 1997. A zidovudine-resistant simianimmunodeficiency virusmutant with a Q151Mmutation in reversetranscriptase causes AIDS in newborn macaques. AntimicrobAgents Chemother 41:278–283.
Van Vaerenbergh K, Van Laethem K, Albert J, Boucher CA, Clotet B,Floridia M, Gerstoft J, Hejdeman B, Nielsen C, Pannecouque C,PerrinL,PirilloMF,RuizL,Schmit JC,SchneiderF,SchoolmeesterA, Schuurman R, Stellbrink HJ, Stuyver L, Van Lunzen J,Van Remoortel B, VanWijngaerden E, Vella S, Witvrouw M, YerlyS, De Clercq E, Destmyer J, Vandamme AM. 2000. Prevalence andcharacteristics of multinucleoside-resistant human immunodefi-ciency virus type 1 among European patients receiving combina-tions of nucleoside analogues. Antimicrob Agents Chemother44:2109–2117.
Witvrouw M, Pannecouque C, Van Laethem K, Desmyter J, De ClercqE, Vandamme AM. 1999. Activity of non-nucleoside reversetranscriptase inhibitors against HIV-2 and SIV. AIDS 13:1477–1483.
Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E,HeneineW. 2004. Susceptibility of HIV-2, SIV and SHIV to variousanti-HIV-1 compounds: Implications for treatment and postexpo-sure prophylaxis. Antivir Ther 9:57–65.
Yahi N, Tamalet C, Tourres C, Tivoli N, Fantini J. 2000. MutationL210W of HIV-1 reverse transcriptase in patients receivingcombination therapy. Incidence, association with other mutations,and effects on the structure of mutated reverse transcriptase. JBiomed Sci 7:507–513.
390 Colson et al.

Journal of Medical Virology 75:391–398 (2005)
Clinical and Virological Characteristics ofLamivudine Resistance in Chronic Hepatitis BPatients: A Single Center Experience
Jian Sun, Zhanhui Wang, Shiwu Ma, Guobing Zeng, Zhiyong Zhou, Kangxian Luo, and Jinlin Hou*
Department of Infectious Diseases, Nanfang Hospital, Southern Medical University, Guangzhou, China
We have investigated the characteristics oflamivudine-resistant strains in patients withchronic hepatitis B in Guangdong, China, wherethe predominant genotypes are B and C. Twohundred forty-seven patients treated with lami-vudine in Nanfang Hospital were followed-up.Patients with hepatitis B e antigen (HBeAg)positive and hepatitis B virus (HBV)-DNA levelsover 7.5�106 copies/ml at baseline had a shortertime to the selection of YMDD mutant (P¼0.02and 0.00, respectively). The detection of YMDDmutant precedes HBV-DNA breakthrough andalanine transaminase (ALT) flare in about 2 and3 months, respectively. The ALT flare after theappearance of YMDD mutants was more evidentin HBeAg positive patients than HBeAg negativepatients (P¼ 0.02). After emergence of YMDDmutant, the HBV-DNA level was significantlyhigher in genotype C patients compared withgenotype B patients (P¼ 0.02). No significantdifference of YMDD mutant pattern was foundbetween patients with genotype B and C. Fourkinds of new mutants were found in over twopatients including rtL80I, rtG172E, rtG174C, andrtG172E/rtG174C. In vitro transfection and real-time analysis showed that rtG172E, rtG174C, andrtG172E/rtG174C mutants had a decreased repli-cation competence compared with wild type(33%, 27%, and 15% of the wild type HBV,respectively). Our result suggest that genotypicmonitoring of YMDD mutant is important for themanagementofpatients treatedwith lamivudine.J. Med. Virol. 75:391–398, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: hepatitis B virus; polymerasegene; lamivudine;mutant; drugresistance
INTRODUCTION
Chronic hepatitis B is a severe form of liver diseasethat affects at least 300 million people worldwide [Alterand Mast, 1994]. Some may develop cirrhosis and
hepatocellular carcinoma during the long history ofinfection [Schalm et al., 1990; Lee, 1997; Beasley, 1998].However, treatment of hepatitis B is still unsatisfactory.Interferon alpha (IFN-a) has been approved for thetreatment of chronic hepatitis B [Hoofnagle and DiBisceglie, 1997], but it is poorly tolerated and effectivein only 20%–40% patients. The approval of lamivudinehas revolutionized the treatment of chronic hepatitis B.Results of clinical trials showed that most patients,including liver transplant recipients, can benefit fromlamivudine administration with hepatitis B virus(HBV)-DNA suppression, normalization of alanine trans-aminase (ALT), and improvement in liver histology,compared with patients who received placebo [Dienstaget al., 1995; Lai et al., 1998; Dienstag et al., 1999a,b;Perrillo et al., 1999; Tassopoulos et al., 1999].
The development of lamivudine-resistant hepatitis Bvirus (HBV) mutants is becoming an increasinglyimportant problem. Genotypic resistant mutations havebeen detected in 14%–32% of patients at 1 year of treat-ment [Lai et al., 1998; Dienstag et al., 1999a; Schalmet al., 2000], increasing to 38%, 49%, and 66% at 2, 3,and 4 years of continued treatment, respectively [Leunget al., 1999; Chang et al., 2000; Liaw et al., 2000].The lamivudine-resistant viruses have a characteristicamino acid substitution in the tyrosine-methionine-aspartate-aspartate (YMDD)-motif of HBV polymerase.The methionine is either replaced by an isoleucine
Abbreviations: HBV, hepatitis B virus; HBsAg, hepatitis Bsurface antigen; anti-HBs, hepatitis B surface antibody; HBeAg,hepatitis B e antigen; PCR, polymerase chain reaction; HCV,hepatitis C virus; ALT, alanine transaminase; HIV, humanimmunodeficiency virus; RT, reverse transcriptase.
Grant sponsor: Major State Basic Research (973) Program ofthe People’s Republic of China (to JH); Grant number:G1999054106; Grant sponsor: National Natural Science Founda-tion of China (to JH); Grant numbers: 30225042, 30070695.
*Correspondence to: Jinlin Hou, MD, Department of InfectiousDisease, Nanfang Hospital, Southern Medical University,Guangzhou 510515, China. E-mail: [email protected]
Accepted 25 October 2004
DOI 10.1002/jmv.20281
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

(rtM204I) or a valine (rtM204V) [Ling et al., 1996;Naoumov et al., 1996; Tipples et al., 1996; Honkoop et al.,1997; Schalm, 1997]. In addition, the rtM204V mutationis often accompanied by a leucine to methionine(rtL180M) change [Fu and Cheng, 1998; Niesters et al.,1998]. In vitro studies showed that the combination ofrtL180M and rtM204V mutations are more resistantthan rtM204V alone and similar to that of rtM204I[Allen et al., 1998; Xiong et al., 1998; Ono-Nita et al.,1999a]. Accumulating data indicates that these kinds oflamivudine resistant mutants have a decreased replica-tion competence in tissue culture as well as in vivo [Fuand Cheng, 1998; Melegari et al., 1998; Ono-Nita et al.,1999b; Ling and Harrison, 1999]. At the same time,acute exacerbation of hepatitis may occur after theemergence of YMDD mutant [Liaw et al., 1999]. Fatalhepatic failure after emergence of YMDD mutant hasalso been reported [Wang et al., 2002].
However, there is little information on the relativeprevalence of viral resistance and the clinical conse-quence of viral resistance in patients with hepatitis B inmainland China, where the predominant genotypesare B and C. In some instances, mutations affectingother regions of the HBV polymerase gene and theimpact of polymerase gene mutations on the overlappingsurface gene have not been reported. These observa-tions prompted us to conduct a systematic study on thecharacteristics of lamivudine resistance in patients withhepatitis B in mainland China.
PATIENTS AND METHODS
Patients
Two hundred forty-seven consecutive chronic hepati-tis B patients were followed-up by two hepatologistsin our department. There were 41 women and 206 menand the mean age was 34.14 years (range 11–75 years).All patients were seropositive for hepatitis B surfaceantigen (HBsAg) and HBV-DNA before treatment butseronegative for hepatitis C virus (HCV), HDV, andhuman immunodeficiency virus (HIV) infection. Theywere treated with lamivudine at a dose of 100 mg daily.
Follow-Up
Patients were followed at 1–3 months intervals duringtreatment for serum ALT levels and HBV serologicaltests including HBV markers, HBV-DNA level, andpolymerase gene YMDD mutants (rtM552I/V). Serumsamples were prospectively stored at �408C.
Serological Assays
Serological markers for HBV (HBsAg, HBeAg, andanti-HBe), HCV, HDV, and HIV were tested by commer-cially available immunoassays (Abbott Laboratories,Chicago, IL). Serum HBV-DNA was quantitated byLightCycler-based real-time fluorescence quantitativepolymerase chain reaction (PCR) with a detection limitof 1�103 copies/ml. PCR amplification was performedwith a set of PCR primers and a probe, corresponding to
the surface gene of the HBV. The PCR primers were:HBV-S-F: 50-CCTCTT CATCCTGCTGCT-30, HBV-S-R:50-AACTGAAAGC CAAACAGTG-30. The probe was 50-TCCCATCCCATCATCCTGGGCTTT-30. The PCR wereperformed according the manufacturer’s instructions(PG Biotech, Shenzhen, China) using Lightcyclerdetection system [He et al., 2002].
Definitions
Clinical breakthrough was classified into two groups:viral breakthrough and biochemical breakthrough.Viral breakthrough was defined by a sustained increaseof at least two log copies/ml HBV-DNA level by twoconsecutive measurements at least 1 month apart. Bio-chemical breakthrough was defined by an ALT elevationto over 2.5 times the upper limit of normal [ULN] in twomeasurements at least 1 month apart.
Extraction of HBV-DNA
Two hundred microliters serum samples were incu-bated for 4 hr at 378C in a buffer (50 mmol/L Tris,150 mmol/L NaCl, 2.5 mmol/L EDTA, 0.5% SDS) con-taining 0.5 mg/ml proteinase K, followed by phenol–chloroform extraction and precipitated with ethanol.The pellet was resuspended in 20 ml H2O and used forPCR amplification.
Detection of YMDD Mutant
A sensitive PCR-RFLP method was used for thedetection of YMDD mutants as described previously[Allen et al., 1999]. Briefly, primer pairs P24 (50-TTC-CCCCACTGTCTGGCTTTCAGTCAT-30) and P29 (50-ATACCCAAAGACAAAAGAAAA-30) were used for PCRamplication. Primer P24 anneals just upstream from themethionine codon of the YMDD motif and changes thetyrosine codon from TAT to CAT. This change created anNdeI site (CA #TATG) when the HBV-DNA template wasof wild type sequence (ATG for methionine). If the HBV-DNA template had a variation at methionine, eitherrtM204I (ATTfor isoleucine)or rtM204V (GTGforvaline)change, or other changes such as ATC or ATA, then theNdeI restriction site was absent from the amplified DNA.HBV-DNA extracted from patient sera was subjected toPCR amplication with primer pair P24 and P29. A 5 mlaliquot of PCR product was digested with NdeI for 3 hr at378C. The resulting DNA fragments were analyzed byelectrophoresis. To confirm the result of RFLP and theexistenceof rtL180M change, all the sampleswith YMDDmutant detected by RFLP were sequenced with primerP29 following PCR amplication with primer S1 (50-ATGGAGAACATCACATCAGGA-30) and P29.
Genotyping of Samples With YMDD Mutant
Genotyping of samples with YMDD mutant was car-ried out by PCR-RFLP as previously described [Lindhet al., 1997]. Briefly, the extracted DNA was amplifiedfor the fragment of the HBV genome between nucleotidepositions 256 and 796. The polymerase chain reaction
392 Sun et al.

products were subsequently treated with restrictionenzymes. After incubation, the samples were run on a3% agarose gel and stained by ethidium bromide. Sixgenotypes (A–F) of HBV were identified by the restric-tion patterns of DNA fragments.
Sequence Changes of Polymerase GeneBefore Treatment and at the Time
of Emergence of YMDD Mutant
The sequence of the polymerase gene from 19 patientswas determined directly from PCR products. The HBVpolymerase gene sequence from nucleotide position300–910, which includes domains A–E of the viralpolymerase gene, was analyzed. The extracted DNA wasamplified with primers BS1 (50-CCTGCTGGTGGCTCC-AGTTCC-30, nucleotide position 56–76) and Pol2 (50-CGGGGCAACGGGGTAAAGGTT-30,nucleotideposition1,150–1,138). A nested PCR was performed on somesamples which did not give a successful amplification,using primers S1: 50-ATGGAGAACATCACATCAGGA-30 at nucleotide position 157–177 and SP1: 50-CTAAAACATTGCTTGAGTTT-30 at position 950–930.Before sequencing, the PCR products were purifiedusing the QIA quick PCR purification kit (Qiagen, Inc.,Chatsworth, CA) and semiquantified by agarose gelelectrophoresis. Samples were sequenced using anautomated fluorescent DNA sequencer with primerSP1. Some selected second round PCR products werecloned into pGEM-T (Promega, Madison, WI) afterbeing gel-purified. Five positive clones derived fromeach sample were sequenced as mentioned above.
Site-Directed Mutagenesis
Replication competent plasmid p3.8II containing a1.3-times genome-length wild type HBV (3,713 bp, nt1,404–1,901, adr subtype, donated by Prof. Wang Yuanfrom Shanghai Institute of Biochemistry) was used astemplate [Fu et al., 1996]. Site-directed mutagenesiswas performed using Quickchange Site-Directed Muta-genesis kit (Stratagene, La Jolla, CA). The nucleotidesequences of all primers used in the site-directedmutagenesis experiments are listed in Table I. Thenucleotide sequences of all plasmids were verified byDNA sequencing.
Cell Culture and Transfection
HepG2 cells were cultured in DMEM supplementedwith 10% fetal bovine serum. Twenty-four hours before
transfection, HepG2 Cells were seeded into dishes at adensity of 5�105 cells per 60 mm dish. Plasmid DNAused for transfection was purified with the QiagenMaxiprep kits. Twenty-five microgram plasmid wasused to transfect HepG2 cells in 60-mm dishes using thecalcium phosphate precipitation method. Ten-micro-gram reporter plasmid DNA expressing secreted alka-line phosphatase (SEAP) was cotransfected into eachculture as an internal control to normalize the transfec-tion efficiency among dishes. The transfected cells werecollected 72 hr after transfection. All transfection ex-periments were done three times on separate days.
Purification of Intracellular Core Particles
Purification of intracellular core particles was con-ducted as reported by Lin et al. [2001]. Briefly, cells werewashed twice with chilled phosphate-buffered salinebuffer and lysed with 600 ml of lysis buffer (10 mM Tris-HCl [pH 7.9], 1 mM EDTA, 1% NP-40, 8% sucrose) at378C for 10 min. After centrifugation at 12,000g for2 min, cleared supernatants were collected, and 6 ml of1 M magnesium acetate (MgOAc), 12 ml of 5 mg/mlDNase I, and 3 ml of 20 mg/ml RNase A were added,followed by incubation at 378C for 1 hr to digest re-maining DNA and RNA. After a quick spin at 12,000g for1 min, supernatants were collected, 16 ml of 0.5 M EDTAand 130 ml of 35% polyethylene glycol 8000 in 1.75 MNaCl were added. The mixture was kept on ice for 1 hr toprecipitate core particles. After spinning at 9,000g for5 min, pellets were resuspended in 1 ml ddH2O.
Real-Time Analysis of IntracellularCore Particles
HBV-DNA was quantitated by LightCycler-basedreal-time fluorescence quantitative PCR system [Heet al., 2002]. HBV-DNA was extracted from 200 mlprecipitated core particles with the high pure viralnucleic acid extraction kit. The amount of intracellularcore particles extracted from each plate of transfectedcells was normalized using the expression of SEAP insupernatant as an internal control.
Statistical Analysis
Quantitative values expressed as mean and rangewere compared with the student’s t-test or with theMann–Whitney test. The Kaplan–Meier estimate andLog-rank analysis was used to identify factors asso-ciated with the time to hepatitis B e antigen (HBeAg)
TABLE I. Primers Used for Site-Directed Mutagenesis
Primers Nucleotide sequence
M3(rtG172E/rtG174C) 50-GGAAAATTCCTATGGAAGTGTGCCTCAGCCCGTTTCTCCT-30
M3R 50-AGGAGAAACGGGCTGAGGCACACTTCCATAGGAATTTTCC-30
M4(rtG172E) 50-GGAAAATTCCTATGGAAGTGGGCCTCAGCCCGTT-30
M4R 50-AACGGGCTGAGGCCCACTTCCATAGGAATTTTCC-30
M5(rtG174C) 50-TTCCTATGGGAGTGTGCCTCAGCCCGTTTCTCCT-30
M5R 50-AGGAGAAACGGGCTGAGGCACACTCCCATAGGAA-30
The underlined nucleotides are mismatched sequence.
Lamivudine Resistance in Chinese Hepatitis B Patients 393

seroconversion and emergence of YMDD mutants. Cox’sproportional hazard model was used for multivariateanalysis. Qualitative value were correlated with Fisherexact test. Statistical analysis was done with thesoftware SPSS.
RESULTS
Baseline Characteristics of the Patients
At baseline, the mean ALT value was 170.35 IU/L(range 22–939 IU/L) and was not statistically differentbetween HBeAg positive (mean 162.60 IU/L, range 22–877 IU/L) and HBeAg negative patients (mean185.40 IU/L, range 34–939 IU/L) (Mann–Whitney test,P¼0.99). The mean HBV-DNA level was 6.42�107 copies/ml (range 3�103–1.2� 109 copies/ml) andwas significantly higher in HBeAg positive (mean8.76�107 copies/ml, range 3�103–1.2� 109 copies/ml)than HBeAg negative patients (mean 1.89� 107 copies/ml, range 3� 103–4.52� 108 copies/ml) (Mann–Whitney test, P¼ 0.00). No mutation in the YMDDmotif of the viral polymerase gene was detected atbaseline.
Antiviral Response
After 6 months of treatment, the mean level ofHBV-DNA decreased from 6.42� 107 copies/ml (range3� 103–1.2� 109 copies/ml) at baseline to 1.74�105 copies/ml (range 0–6�106 copies/ml) and the meanALT level decreased from 170.35 IU/L (range 22–939 IU/L) at baseline to 40.90 IU/L (range 9–207 IU/L).HBV-DNA became undetectable by real-time PCR(<1�103 copies/ml) in 42% of patients (102/247) andserum ALT normalized (�40 IU/L) in 76% patients(176/242) after 6 months of treatment. Of the 162HBeAg-positive patients before treatment, 44 serocon-verted to anti-HBe with a mean duration of 15 monthstreatment (range 3–48 months). The cumulative HBeAgseroconversion rate was 16.08% after 1 year treatment,33.75% and 47.00% after 2 and 3 years treatment,respectively. Log-rank analysis showed that patientswith high pretreatment ALT level (�2 ULN) was asso-ciated with a shorter time to HBeAg seroconversion(P¼ 0.04).
Time and Frequency of Appearanceof YMDD Mutant
YMDD mutants were found in 42 out of 247 patients.In these 42 patients, the detection of YMDD mutant(mean 533 days, range 274–1,189) precedes HBV-DNA breakthrough (mean 593 days, range 305–1,372)(Student’s t-test, P¼ 0.00). The cumulative incidence ofYMDD mutation was 8.33% after 1 year, 25.79% and41.30% at 2 and 3 years treatment, respectively.
Relationship Between Pretreatment Parametersand the Time to Appearance of YMDD Mutant
Pretreatment parameters including HBV-DNA level,ALT level and HBeAg status were analyzed in relation to
the time of appearance of YMDD mutant. Log-rankanalysis showed that a high pretreatment HBV-DNAlevel greater than 7.5�106 copies/ml and HBeAgpositive at baseline were associated with a more rapidselection of YMDD mutant (Log-rank test, P¼0.02 and0.00, respectively). A multivariate analysis (includingpretreatment HBeAg status, HBV-DNA, and ALT level)showed that only HBeAg positive at baseline wasassociated with a more rapid occurrence of YMDDmutants (OR 3.19, 95% CI 1.05–9.72, P¼0.04). Themedian time to lamivudine resistance was 15 months ingenotype B patients (range 9–39) and 18 months ingenotype C patients (range 9–36) (Mann–Whitney test,P¼0.35).
Clinical Characteristics of Viral Resistance
Biochemical and virological breakthrough occurred in66.67% (28/42) and 85.71% (36/42) of patients, respec-tively, after the emergence of YMDD mutants. The peakHBV-DNA (mean 593 days, range 305–1,372) occurredearlier than ALT flare (mean 634 days, range 305–1,464) (Student’s t-test, P¼0.01). The peak ALT andHBV-DNA levels observed after emergence of viralresistance were compared with pretreatment values.It showed that the peak HBV-DNA level (mean2.87�107 copies/ml, range 0–1.80� 108 copies/ml) afterthe appearance of YMDD mutants was lower than thatof baseline (mean 1.90�108 copies/ml, range 3�103–1.20�109 copies/ml) (Mann–Whitney t-test, P¼ 0.01).After emergence of YMDD mutant, the peak HBV-DNAlevel was significantly higher in genotype C patients(mean 3.25�107 copies/ml, range 0–1.80�108 copies/ml) compared with genotype B patients (mean 2.31�107 copies/ml, range 0–1.70�108 copies/ml) (Mann–Whitney test, P¼0.02). However, the baseline HBV-DNA level did not differ significantly between genotypeB and C patients (Mann–Whitney test, P¼0.35). Thepeak ALT level after the emergence of YMDD mutant(mean 192.38 IU/L, range 11–653 IU/L) was not statis-tically different from that of baseline (mean 186.19 IU/L,range 81–392 IU/L) (Mann–Whitney test, P¼ 0.64).However, the peak ALT after the appearance of YMDDmutants was higher in HBeAg positive (mean 209.38 IU/L, range 11–653 IU/L) than HBeAg negative patients(mean 66.60 IU/L, range 15–201 IU/L) (Mann–Whitneytest, P¼ 0.02). After emergence of YMDD mutant, thepeak ALT level did not differ significantly betweengenotype C patients and genotype B patients (Mann–Whitney test, P¼0.72).
Relationship Between Genotypeand Pattern of YMDD Mutation
Among 42 patients with YMDD mutant, 17 patientswere infected with genotype B and 25 patients wereinfected with genotype C. As five patients were sequenc-ed after cloning and showed the presence of mixture ofwild type, YIDD, and YVDD mutant. So there were notincluded in this statistics analysis. Mutants rt204I wereseen in seven patients in group B and in 12 patients in
394 Sun et al.

group C, while mutants rt204V were seen in eight pa-tients in group B and ten patients in group C (two-sidedFisher exact test, P¼0.74). L180M change occurredin six and seven patients in genotype B and genotypeC patients, respectively (two-sided Fisher exact test,P¼0.73).
Sequence Changes Before Treatment and AfterEmergence of YMDD Mutant
The polymerase gene of 19 patients were sequencedbefore lamivudine treatment and after emergence ofYMDD mutant following PCR amplification. Five pa-tients who were infected with a mixture of wild type andYMDD mutants were further sequenced after cloning.Five positive clones derived from each sample weresequenced and the results are presented in Figure 1. Tenof the 19 patients who had mutations in the YMDD motifwere observed to have additional changes in the P gene.
Four of the changes rtL80I, rtG172E, rtG174C, andrtG172E/rtG174C were detected in over two patientssimultaneously (Fig. 1). These changes are conserved in202 published whole gene sequences of HBV with dif-ferent genotypes (data not shown). Mutation rtL80Ioccurred in combination with the YMDD mutation.However, mutations rtG172E, rtG174C, and rtG172E/rtG174C were not accompanied by YMDD mutants inmost instances.
Replication Competence of New FoundPolymerase Gene Mutants
It was shown recently that rtG172 and rtG174 areconserved amino acids through all RNA-dependentDNA polymerases and are the sites for reverse tran-scriptase (RT) interactions with template and primer,which are similar to the YMDD motif [Stuyver et al.,2001]. Therefore, mutations located in these sites may
Fig. 1. Sequences of the A–E domains of the polymerase gene inpatients at baseline and after emergence of YMDD mutant. B–D arecharacteristic sequence of different genotypes from GenBank withtheir accession number. Arabic number represents different patient.The sequence of baseline sample is marked with A and compared withthat at the time of detection of YMDD mutant. Sequences of sevenpatients are shown here. Five samples with YMDD mutant were
sequenced after cloning. The number after the hyphen represents theclone number. In these seven patients, four changes including rtL80I,rtG172E, rtG174C, and rtG172E/rtG174C were detected in over twopatients simultaneously. These changes are conserved in 202 pub-lished whole gene sequences of HBV with different genotypes (data notshown).
Lamivudine Resistance in Chinese Hepatitis B Patients 395

influence the replication of HBV. To address this ques-tion, in vitro replication experiment was performed.Cells transfected with wild type plasmid and plasmidswith rtG172E, G174C, and rtG172E/rtG174C mutationwere harvested 72 hr after transfection and lysedwithout breaking HBV particles. The DNase digestionremoves any contaminated DNA before isolation of HBVcore particles. The supernatant of culture medium wasused for the quantitation of SEAP expression level ac-cording to the manufacturer’s instruction (Clontech,Palo Alto, CA). The relative result of intracellular HBV-DNA copy number to SEAP expression level representthe replication capacity of different plasmid. It showedthat rtG172E, rtG174C, and rtG172E/rtG174C repli-cated inefficiently compared with the wild type HBV(33%, 27%, and 15% of the wild type HBV, respectively)(Fig. 2).
DISCUSSION
Lamivudine administration can achieve potent sup-pression of HBV replication, normalization of ALT andimprovement in liver histology. However, the emer-gence of drug resistance is becoming an importantquestion. The recent development of a sensitive PCRbased assay for detection of lamivudine resistant HBVmutants provided us with a new tool to monitor drugresistance. Therefore, PCR-RFLP was employed for thedetection of emergence of drug resistance in patientstreated with lamivudine.
In this study, we studied 247 chronic hepaitits Bpatients treated with lamivudine in mainland China.Lamivudine showed potent antiviral effect in thesepatients. After 6 months of therapy, HBV-DNA became
undetectable by real time PCR in 42% of patients andALT normalized in 76% of them. Log-rank analysisshowed that patients with ALT level no less than twotimes ULN at baseline was associated with a shortertime to HBeAg seroconversion. As reported previously,patients high pretreatment ALT level (�2 ULN) have agreater chance of HBeAg seroconversion [Perrillo et al.,2002] and patients without HBeAg seroconversionduring treatment will relapse more often after lamivu-dine withdrawal than patients with HBeAg seroconver-sion [Dienstag et al., 2003]. This suggested that patientswith ALT lower than two times ULN at baseline shouldnot be considered for lamivudine treatment in mostinstances.
Previous clinical trials have shown that high HBV-DNA levels are predictive factors of viral resistanceto lamivudine compared with patients who did notdevelop viral resistance [Lai et al., 1997]. Here, we foundthat HBeAg positivity and high HBV-DNA level (over7.5�106 copies/ml) at baseline were associated with amore rapid occurrence of YMDD mutations. This wasnot surprising as HBeAg positivity and high HBV-DNAlevels, both markers of higher replication, are associat-ed with the rapid generation of viral genome mutations.We did not observe the difference of the median timeto lamivudine resistance between genotype B and Cpatients.
The detection of YMDD mutant precedes HBV-DNAbreakthrough and HBV-DNA breakthrough precedesALT flare in about 2 and 1 month(s), respectively. This isvery important clinically because after the emergence ofYMDD mutant, doctors can make full use of this intervalto take measures and try to prevent the occurrence ofsevere hepatitis flare, for example, close monitoring oruse of adefovir as hepatitis flare was occasionally severe,especially in patients with cirrhosis. We proposed thatpatients treated with lamivudine for over 6 monthsshould be monitored tri-monthly for YMDD mutant sothat we can detect YMDD mutant as early as possible.That will be helpful for the management of thesepatients.
Biomedical and virological breakthrough occurredin over half of the patients after emergence of YMDDmutant. The mean HBV-DNA after appearance ofYMDD mutant was still lower than that of baseline,which suggested that lamivudine still showed antiviraleffect for patients with YMDD mutant, or the mutantreplicated inefficiently. The ALT flare after selection ofYMDD mutant was more evident in HBeAg positivethan HBeAg negative patients which may be related tothe more active liver disease in HBeAg positive patients.However, the small sample size in the latter group (onlyfive patients developed ALT flare in HBeAg negativepatients) make us difficult to draw a conclusion from theresult. Recently, acute exacerbations after emergence ofYMDD mutants were described in patients with HBeAg-positive chronic hepatitis B, which were followed byHBeAg seroconversion [Liaw et al., 1999]. HBeAg sero-conversion after breakthrough was not observed in ourstudy. As no serious flare was recorded and adefovir is
Fig. 2. The replication competence of different plasmid. Wild typeplasmid and plasmids carrying rtG172E, rtG174C, and rtG172E/rtG174C mutations were transfected into HepG2 cells. Cells wereharvested 72 hr after transfection. HBV core particles were isolatedfrom transfected cells and quantitated by real-time PCR. The SEAPexpression in the supernatant of culture medium was quantitatedaccording to the manufacturer’s instructions (Clontech). The relativeresult of HBV-DNA copy number to SEAP expression level representthe replication capacity of plasmid. The value of wild type plasmid wasdefined as 1.0.
396 Sun et al.

not available in mainland China, all the patients withYMDD mutant were suggested by the treating physicianto continue lamivudine. Thus, most patients were stillon lamivudine treatment. But a few patients chose towithdraw lamivudine and others changed to interferontreatment. All these patients are still followed-up. Theimpact of interferon treatment, continued lamivudine,and lamivudine withdrawal on the disease progressionin these patients will be investigated further.
The mutational pattern of lamivudine resistant mu-tants differs between genotypes A and D. MutationrtM204I was more prevalent in genotype D, whilertM204V and rtL180M was more prevalent in genotypeA [Zollner et al., 2004]. There was no significant dif-ference of the pattern of YMDD mutant betweengenotype B and C patients in this study. At baseline,HBV-DNA level did not differ between genotype B and Cpatients. However, after emergence of YMDD mutant,the HBV-DNA level was significantly higher in genotypeC patients than that in genotype B patients. The under-lying mechanism has not been clarified. This mayberelated to the different replication competence of YMDDmutants with different genotypes. The in vitro transfec-tion and replication analysis of plasmids with differentgenotypes (A–F) carrying YMDD mutation is understudy to address this question.
HBV polymerase gene sequences at baseline werecompared with those after the emergence of YMDDmutant. The result showed that mutations outside ofthe YMDD motif can also be selected after lamivudinetreatment. Four types of mutations (rtL80I, rtG172E,rtG174C, and rtG172E/rtG174C) were found in over twopatients. These mutations are conserved through 202published HBV genome sequences. As already report-ed, rtG172 and rtG174 are located in the conserved Bdomain and are the sites for RT interaction with primerand template [Stuyver et al., 2001]. These mutants mayinfluence, therefore, the activity of RT. So they wereselected for in vitro transfection analysis. The rtG172E,rtG174C, and rtG172/G174C mutants were shown toreplicate inefficiently compared with wild type (lowerthan one third of wild type). As most patients (4/5)with these new mutations experienced HBV-DNAbreakthrough and ALT flare after emergence of YMDDmutant. Furthermore, these new mutants were notfound in pretreatment samples which was shown inFigure 1. So the clinical and virological characteristicsof new mutants need to be studied in more detail.The dynamics of new mutants in these patients, theirimpact on liver diseases will be further investigated inthe follow-up study.
In conclusion, the results of this study revealed impor-tant findings on the monitoring of lamivudine resistantmutant because it was found that: (1) HBeAg positiveand HBV-DNA over 7.5�106 copies/ml at baselinewere associated with a rapid occurrence of YMDDmutation during lamivudine treatment. (2) The emer-gence of YMDD mutant precedes the occurrence of viralbreakthrough and ALT flare. (3) ALT flare was moreevident after the emergence of YMDD mutant in HBeAg
positive patients, which may be related to the moreactive liver disease in these patients. (4) HBV-DNA levelwas significantly higher in genotype C patients com-pared with genotype B patients after emergence ofYMDD mutant. (5) The mutational pattern of YMDDmutant was not different between patients with geno-type B and C. (6) Apart from the YMDD mutations, othermutations in domains A–E of the P gene may be selectedafter lamivudine treatment. Some of the mutations caninfluence the replication capacity of hepatitis B virus.However, the drug susceptibility of new mutants andtheir impact on HBsAg and virion particles productionneed to be investigated further.
REFERENCES
Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA,Tyrrell DL, Brown N, Condreay LD. 1998. Identification andcharacterization of mutations in the hepatitis B virus resistant tolamivudine. Lamivudine clinical Investigation Group. Hepatology27:1670–1677.
Allen MI, Gauthier J, DesLauriers M, Bourne EJ, Carrick KM,Baldanti F, Ross LL, Lutz MW, Condreay LD. 1999. Two sensi-tive PCR-based methods for detection of hepatitis B virus variantsassociated with reduced susceptibility to lamivudine. J ClinMicrobiol 37:3338–3347.
Alter MJ, Mast EE. 1994. The epidemiology of viral hepatitis in theUnited States. Gastroenterol Clin North Am 23:437–455.
Beasley RP. 1998. The major etiology of hepatocellular carcinoma.Cancer 61:1942–1956.
Chang TT, Lai CL, Liaw YF, Guan R, Lim SG, Lee CM, Ng KY.2000. Incremental increases in HBeAg seroconversion and con-tinued ALT normalization in Asian chronic HBV (CHB) patientstreated with lamivudine for four years [abstract]. Antivir Ther5:44.
Dienstag J, Perillo R, Schiff E, Bartholomew M, Vicary C, Rubin M.1995. A preliminary trial of lamivudine for chronic hepatitis Binfection. N Engl J Med 333:1657–1661.
Dienstag JL, Schiff ER, Wright TL, Perrilo RP, Hann HW, Goodman Z,Crowther L, Condreay LD, Woessner M, Rubin M, Brown NA.1999a. Lamivudine as initial treatment for chronic hepatitis B inthe United States. N Engl J Med 341:1256–1263.
Dienstag JL, Schiff ER, Mitchell M, Casey DE, Jr., Gitlin N, Lissoos T,Gelb LD, Condreay L, Crowther L, Rubin M, Brown N. 1999b.Extended lamivudine retreatment for chronic hepatitis B: Main-tenance of viral suppression after discontinuation of therapy.Hepatology 30:1082–1087.
Dienstag JL, Cianciara J, Karayalcin S, Kowdley KV, Willems B, PlisekS, Woessner M, Gardner S, Schiff E. 2003. Durability of serologicresponse after lamivudine treatment of chronic hepatitis B.Hepatology 37:748–755.
Fu L, Cheng YC. 1998. Role of additional mutations outside the YMDDmotif of hepatitis B virus polymerase in L(�)SddC (3TC) resistance.Biochem Pharmacol 55:1567–1572.
Fu L, Wu X, Kong YY, Wang Y. 1996. Regulation of Hepatitis B Virusgene expression by its two enhancers. Sheng Wu Hua Xue Yu ShengWu Wu Li Xue Bao (Shanghai) 28:590–599.
He ML, Wu J, Chen Y, Lin MC, Lau GK, Kung HF. 2002. A new andsensitive method for the quantification of HBV cccDNA by real-timePCR. Biochem Biophy Res Commun 295:1102–1107.
Honkoop P, Niesters HG, de Man RA, Osterhaus AD, Schalm SW. 1997.Lamivudine resistance in immunocompetent chronic hepatitis B.J Hepatol 26:1393–1395.
Hoofnagle JH, Di Bisceglie AM. 1997. Treatment of chronic viralhepatitis. N Engl J Med 336:347–356.
Lai CL, Liaw YF, Leung NW, Deslauriers M, Barnard J, Sanathanan L,Gray DF. 1997. Genotypic resistance to lamivudine in a prospective,placebo-controlled multicentre study in Asia of lamivudine therapyfor chronic hepatitis B infection: Incidence, kinetics of emergence,and correlation with disease parameters [abstract]. Hepatology26:522A.
Lai CL, Chien RN, Leung NW, Chang TT, Guan R, Tai DI, Ng KY,Wu PC, Dent JC, Barber J, Stephenson SL, Gray DF. 1998. A one
Lamivudine Resistance in Chinese Hepatitis B Patients 397

year trial of lamivudine for chronic hepatitis B. N Engl J Med 339:61–68.
Lee WM. 1997. Hepatitis B virus infection. N Engl J Med 337:1733–1745.
Leung NW, Lai CL, Chang TT, Guan R, Lee CM, Ng KY, Wu PC.1999. Three year lamivudine therapy in chronic HBV [abstract].J Hepatol 30:59.
Liaw YF, Chien RN, Yeh CT, Tsai SL, Chu CM. 1999. Acuteexacerbation and hepatitis B virus clearance after emergence ofYMDD motif mutation during lamivudine therapy. Hepatology30:567–572.
Liaw YF, Leung NW, Chang TT, Guan R, Tai DI, Ng KY, Chien RN,Dent J, Roman L, Edmundson S, Lai CL. 2000. Effects of extendedlamivudine therapy in Asian patients with chronic hepatitis B.Gastroenterology 119:172–180.
Lin X, Yuan ZH, Wu L, Ding JP, Wen YM. 2001. A single amino acid inthe reverse transcriptase domain of hepatitis B virus affects virusreplication efficiency. J Virol 75:11827–11833.
Lindh M, Andersson AS, Gusdal A. 1997. Genotypes, nt 1858 variants,and geographic origin of hepatitis B virus—large-scaled analysisusing a new genotyping method. J Infect Dis 175:1285–1293.
Ling R, Harrison TJ. 1999. Functional analysis of mutations conferringlamivudine resistance on hepatitis B virus. J Gen Virol 80:601–606.
Ling R, Mutimer D, Ahmed M, Boxall EH, Elias E, Dusheiko GM,Harrison TJ. 1996. Selection of mutations in the hepatitis B viruspolymerase during therapy of transplant recipients with lamivu-dine. Hepatology 3:711–713.
Melegari M, Scaglioni PP, Wands JR. 1998. Hepatitis B virus mutantsassociated with 3TC and famciclovir administration are replicationdefective. Hepatology 27:628–633.
Naoumov NV, Smih C, Williams R. 1996. Emergence and characteriza-tion of lamivudine resistant hepatitis B virus variant [abstract].Hepatology 24:282A.
Niesters HG, Honkoop P, Haagsma EB, de Man RA, Schalm SW,Osterhaus AD. 1998. Identification of more than one mutation inthe hepatitis B virus polymerase gene arising during prolongedlamivudine treatment. J Infect Dis 177:1382–1385.
Ono-Nita SK, Kato N, Shiratori Y, Lan KH, Yoshida H, Carrilho FJ,Omata M. 1999a. Susceptibility of lamivudine-resistant hepatitis Bvirus to other reverse transcriptase inhibitors. J Clin Invest103:1635–1640.
Ono-Nita SK, Kato N, Shiratori Y, Masaki T, Lan KH, Carrilho FJ,Omata M. 1999b. YMDD motif in hepatitis B virus DNA polymerase
influences on replication and lamivudine resistance: A study byin vitro full-length viral DNA transfection. Hepatology 29:939–945.
Perrillo R, Rakela J, Dienstag J, Levy G, Martin P, Wright T, CaldwellS, Schiff E, Gish R, Villeneuve JP, Farr G, Anschuetz G, CrowtherL, Brown N. 1999. Multicenter study of lamivudine therapy forhepatitis B after liver transplantation. Lamivudine TransplantGroup. Hepatology 29:1581–1586.
Perrillo RP, Lai CL, Liaw YF, Dienstag JL, Schiff ER, Schalm SW,Heathcote EJ, Brown NA, Atkins M, Woessner M, Gardner SD,Perrillo RP. 2002. Predictors of HBeAg loss after lamivudinetreatment for chronic hepatitis B. Hepatology 36:186–194.
Schalm SW. 1997. Clinical implications of lamivudine resistance byHBV. Lancet 9044:3–4.
Schalm SW, Thomas HC, Hadziyannis SJ. 1990. Chronic hepatitis B.In: Popper H, Schaffner FM, editors. Progress in liver disease.New York: Saunders. pp 443–462.
Schalm SW, Heathcote J, Cianciara J, Farrell G, Sherman M, WillemsB, Dhillon A, Moorat A, Barber J, Gray DF. 2000. Lamivudine andalpha interferon combination treatment of patients with chronichepatitis B infection: A randomized trial. Gut 46:562–568.
Stuyver LJ, Locarnini SA, Lok A, Richman DD, Carman WF, DienstagJL, Schinazi RF. 2001. Nomenclature for antiviral-resistant humanhepatitis B virus mutations in the polymerase region. Hepatology33:751–757.
Tassopoulos NC, Volpes R, Pastore G, Heathcote J, Buti M, Goldin RD,Hawley S, Barber J, Condreay L, Gray DF. 1999. Efficacy oflamivudine in patients with hepatits B e antigen-negative/hepatitisB virus DNA-positive (precore mutant) chronic hepatitis B.Hepatology 29:889–896.
Tipples GA, Ma MM, Fischer KP, Bain VG, Kneteman NM, Tyrell DL.1996. Mutations in HBV RNA-dependent DNA polymerase confersresistance to lamivudine in vivo. Hepatology 3:714–717.
Wang JH, Lu SN, Lee CM, Lee JF, Chou YP. 2002. Fatal hepatic failureafter emergence of the hepatitis B virus mutant during lamivudinetherapy in a patient with liver cirrhosis. Scand J Gastroenterol37:366–369.
Xiong X, Flores C, Yang H, Toole JJ, Gibbs CS. 1998. Mutations inhepatitis B DNA polymerase associated with resistance to lamivu-dine do not confer resistance to adefovir in vitro. Hepatology 28:1669–1673.
Zollner B, Petersen J, Puchhammer-Stockl E, Kletzmayr J, SterneckM, Fischer L, Schroter M, Laufs R, Feucht HH. 2004. Viral featuresof lamivudine resistant hepatitis B genotypes A and D. Hepatology39:42–50.
398 Sun et al.

Journal of Medical Virology 75:399–401 (2005)
Case Report
Anesthetist to Patient Transmission ofHepatitis C Virus Associated WithNon Exposure-Prone Procedures
J. Mawdsley,1* C.G. Teo,2 M. Kyi,3 and M. Anderson4
1Barts and Royal London Hospital, Queen Mary College of Medicine and Dentistry, London, United Kingdom2Sexually Transmitted and Blood Borne Virus Laboratory, Health Protection Agency, London, United Kingdom3West Middlesex University Hospital, London, United Kingdom4Chelsea and Westminster Hospital, London, United Kingdom
A 44-year-old lady was diagnosed with acutehepatitis C virus (HCV) infection 8 weeks afterhysterectomy at which the attending anesthetistwas known to be hepatitis C seropositive. Com-parative nucleotide sequence analysis and phy-logenetic comparison proved that transmissionhad occurred from the anesthetist to the patient.The patient had received general anesthesia withendotracheal intubation and peripheral intrave-nous cannulation. No exposure-prone anestheticprocedures had been performed. This is the firstcase described in UK involving transmissionfromananesthetist to apatient during anesthesiawhere no exposure prone procedures werecarried out. It is the first example in which theanesthetist was known to be seropositive forhepatitis C prior to the operation. J. Med. Virol.75:399–401, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: hepatitis C virus; transmission;anesthetist
CASE DESCRIPTION
A 44-year-old lady presented to her general physicianwith a 3-day febrile illness associated with painlessjaundice. She was referred to the hepatology clinic at herlocal hospital. By the time she attended the clinic amonth later her symptoms had resolved.
Eight weeks prior to becoming jaundiced, a hyster-ectomy with bilateral salpingoophrectomy had beencarried out for menorrhagia attributed to adenomyosisand uterine leiomyomatosis. Hormone replacementtherapy was started after the operation, but was stoppeddue to the development of jaundice. It was restarted2 weeks prior to the attendance at the hepatology clinicwith no recurrence of the jaundice. She had taken noother medications or herbal remedies.
The patient drank approximately 10 U of alcohol perweek. She had never injected drugs and had not beensexually active for the last 5 years. There was norelevant travel history. She had never received a bloodtransfusion but had donated blood in 1995.
Examination in the clinic was normal with no evi-dence of jaundice or stigmata of chronic liver disease. Ablood sample, taken at the time of her acute illness,showed a marked hepatitic picture: bilirubin 138 mmol/L(normal range (NR): 0–17 mmol/L), aspartate transami-nase 724 IU/L (NR: 11–55 IU/L), gamma-glutamyltranspeptidase 372 IU/L (NR: 6–51 IU/L), and alkalinephosphatase 937 IU/L (NR: 110–300 IU/L). At this timeserum anti-hepatitis C virus (HCV) IgG, hepatitis Bsurface antigen and anti-hepatitis A IgM and IgG testswere negative.
A second sample taken 4 weeks later at the time of herattendance at the hepatology clinic showed that liverfunction tests had become normal, but serum anti-HCVIgG was now detectable, as determined by enzyme im-munoassay (EIA) (Ortho Diagnostic Systems, Raritan,NJ). On repeat serological testing 6 months later, anti-HCV IgG was now also detectable by a second EIA(Sanofi Diagnostics, Marnes-la-Coquette, France) and aradioimmunosorbant assay (Ortho Diagnostic systems).It was concluded that the patient’s illness was due toacute hepatitis C.
Abbreviations: HCV, hepatitis C virus; NR, normal range; UK,United Kingdom; 50NCR, 50non coding region; NS5, non structuralprotein 5; CDC, Center for Disease Control and Prevention, USA.
*Correspondence to: J. Mawdsley, Department of Adult andPaediatric Gastroenterology, Digestive Disease Research Centre,Royal London Hospital, Turner Street, London E1 2AD.E-mail: [email protected]
Accepted 14 October 2004
DOI 10.1002/jmv.20282
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

The anesthetist, attending the total abdominal hys-terectomy 3 months earlier, was known to be seroposi-tive for anti-HCV IgG and was HCV RNA positive. Hehad emigrated to the UK from Egypt 20 years earlier.His hepatitis C seropositivity had been diagnosedseveral years earlier, during investigations for abnor-mal liver function tests which were found ultimately tobe due to biliary colic. It was thought most likely that hehad acquired the infection in Egypt, as he had no otherrisk factors for infection. He had been allowed tocontinue to practice according to the UK Departmentof Health guidelines. Hepatitis C RNA levels are notavailable from the time of the transmission incident, butwere 11 million copies per ml when tested several yearslater.
The other members of the operating team were alsoscreened but were all anti-HCV seronegative. Theoperation itself had been uncomplicated with no high-risk anesthetic procedures undertaken and no bloodtransfusions given. The anesthetist had inserted a cuff-ed oral endotracheal tube, to give intermittent positivepressure ventilation, and had also sited a peripheralcannula. He had no open wounds prior to the operation,had not sustained any injuries during the procedureand, as was his normal practice, had washed his handsbefore the operation and had worn gloves throughout.
To investigate the possibility of transmission from theanesthetist to the patient, a comparative nucleotidesequence analysis was undertaken of cDNA amplified byreverse-transcriptase PCR from the HCV genomecarried in serum samples from both individuals. Twoblood samples were taken at separate times from thepatient and the anesthetist to minimize the chance ofconfounding due to PCR contamination. Sequences inthe 50 non-coding region (50NCR) and the region codingfor the highly variable non structural protein 5 (NS5)were examined. The sequence of the 50NCR fragment(139 base-pairs in length) was identical in both subjects,and permitted assignment of HCV to genotype 4. Type 4sequences are identified infrequently among Cauca-sians in the UK, but are prevalent in people originatingfrom the Middle East [Harris et al., 1999].
Pair-wise analysis of the more variable 185 base pairsegment in the NS5 region showed the two sequences tobe greater than 99.9% homologous. Phylogenetic com-parisons revealed the two sequences to be clusteredtogether, segregated from other genotype 4 sequencesarchived in the United Kingdom Health ProtectionAgency sequence database. The molecular findings indi-cate that the HCV carried by the anesthetist and by thepatient are very closely related, consistent with trans-mission from the anesthetist to the patient.
The most recent blood tests, taken 2 years after theacute infection, have shown the patient to be seroposi-tive for anti-HCV IgG but negative for HCV RNA. Herbiochemical tests of liver function have remained in thenormal range.
The anesthetist retired shortly afterwards on thegrounds of ill health. The Hospital involved consideredperforming a look back procedure and consulted the UK
Health Protection Agency. However, this was ultimatelyfelt to be impractical as it would have involved contact-ing all the patients he had attended during his 20 yearcareer.
DISCUSSION
Transmission of HCV is described from an anesthetistto a patient. The patient had no exposure risks forcontracting the virus other than her recent surgery. Thetiming of the acute illness, 2 months after the operation,is consistent with infection acquired during surgery.She was infected by HCV belonging to a genotype that isuncommon in the UK, and the very close evolutionarydistances of the 50NCR and NS5 sequences amplifiedfrom the HCV genome in the blood of the anesthetist andthe patient indicate transmission from one to the other.
Transmission of hepatitis C from infected healthcareworkers to patients during surgery has been describedpreviously [Esteban et al., 1996; Duckworth et al., 1999].HCV-infected surgeons are usually the source. Theprincipal route is thought to be via abrasions and cutssustained by the surgeon during surgery. Infection ofpatients in the course of anesthesia is more rarelyreported. Patient-to-patient infection due to repeateduse of contaminated anesthetic ampoules has been ob-served [Tallis et al., 2003]. The reusable part of thebreathing apparatus was implicated as the vehicle oftransmission in another series of patient-to-patienttransmissions of HCV [Chant et al., 1994]. It wassuggested that once the apparatus had been contami-nated with HCV, the virus was transmitted to otherpatients via abrasions sustained in the upper airwayduring laryngeal mask insertion.
Reports of transmission from infected anesthetists oranesthetic assistants to patients are even rarer thanreports of transmission involving infected surgeons.Stefan-Ross et al. [2000] described transmission from achronically infected patient to an anesthetic assistant,and from him to five other patients. In this series an openwound on the assistant’s hand was considered to be themost likely route of transmission, particularly as he hadnot worn gloves. Cody et al. [2002] described transmis-sion of HCV from an anesthetist to a patient during anoperation in which the only anesthetic proceduresperformed were endotracheal intubation and peripheralintravenous cannulation. The anesthetist was incubat-ing HCV after having been exposed to an HCV-infectedpatient 9 weeks earlier. In both of these incidents theanesthetist and assistant were not known to be infectedwith HCV. In contrast, in the case described above,the anesthetist was known to be hepatitis C positiveprior to the operation and hence adhered to all existinginfection protocols formulated to minimize the risk oftransmission.
No other cases of HCV transmission unassociatedwith performance of exposure-prone procedures haveyet been described. As HCV can be transmitted viawound secretions or through minute skin abrasions[Chouela et al., 1996], it is plausible that transmission
400 Mawdsley et al.

may have occurred by HCV shed from abrasions andthen inoculated to the patient during intravenouscannulation, or via microabrasions sustained duringendotracheal intubation. The US Centre for DiseaseControl and Prevention (CDC) has recommended that,in all cases involving transmission from medical person-nel with no obvious route, the possibility that themember of staff might be injecting residual opiates andthen reusing the vial should be considered. However, inthis case the anesthetist had no history of intravenousdrug use and strongly denied this possibility.
Our findings have implications for the control of HCVtransmission from the health professional to the patient.Current UK Department of Health guidelines relatingto known HCV-seropositive healthcare workers statethat they should not perform exposure-prone proce-dures [UK Health Departments, 2002] including proce-dures in which ‘‘the workers gloved hands may be incontact with sharp instruments, needle tips or sharptissues inside a patient’s open body cavity, wound orconfined anatomical space where the hands or fingertipsmay not be completely visible at all times’’ [UK HealthDepartments, 1994]. In 1996, the Association of Anaes-thetists of Great Britain and Ireland published thisadvice regarding transmission of blood borne viruses:‘‘anesthetists necessarily put their fingers into patients’mouths in order to perform a number of procedures.However, the group can find no evidence either in theliterature or from experience of its members and theircolleagues that anesthetists sustain soft tissue injuriesduring these procedures’’ [Association of Anaesthetistsof Great Britain and Ireland, 1996]. It was thereforerecommended that the insertion of the anesthetist’sfingers into patients’ mouths during intubation shouldnot be regarded as an exposure-prone procedure.Furthermore, with regard to intravenous cannulation,the UK Department of Health guidelines state that‘‘setting up and maintaining iv lines ’’ is not an exposure-prone procedure [UK Health Departments, 1994]. Thisled the Association of Anaesthetists of Great Britain andIreland to conclude that, provided exposure-proneprocedures were avoided, ‘‘a hepatitis infected anesthe-tist may continue in clinical practice’’ [Association ofAnaesthetists of Great Britain and Ireland, 1992].Similarly, the CDC state that ‘‘no recommendationsexist to restrict professional activities of health-careworkers with HCV infection. As recommended for allhealth-care workers, those who are HCV-positiveshould follow strict aseptic technique and standardprecautions, including appropriate use of hand washing,
protective barriers, and care in the use and disposal ofneedles and other sharp instruments’’ [Centers forDisease Control and Prevention, 1998].
The findings from our case, and those reported byCody et al. [2002] suggest that anesthetist-patienttransmission can occur through anesthetic techniques,even when there are no exposure-prone proceduresinvolved and when all infection protocols are observed.However, given the high number of operative proce-dures performed per year and the fact that this is thefirst case reported in the UK and the third worldwide,the risk of transmission should be considered to be verylow. Nonetheless, in the light of these findings, guide-lines for the operating room conduct of HCV-seroposi-tive anesthetic staff may need to be revised. Thedefinition of what constitutes an exposure prone pro-cedure may need to be reviewed, and procedures such asintravenous cannulation and endotracheal intubation,which were previously thought to carry no risk, mayneed to be re-evaluated.
REFERENCES
Association of Anaesthetists of Great Britain and Ireland. 1992. HIVand other blood borne viruses. Guidance for Anaesthetists.
Association of Anaesthetists of Great Britain and Ireland. 1996. HIVand other blood borne viruses. Guidance for Anaesthetists.
Centers for Disease Control and Prevention. 1998. Recommendationsfor prevention and control of hepatitis C virus (HCV) infection andHCV-related chronic disease. MMWR 47:1–39.
Chant K, Kociuba K, Munro R. 1994. Investigation of possible patient-to-patient transmission of hepatitis C in a hospital. NSW PublicHealth Bull 5:47–51.
Chouela E, Abeldano A, Panetta J. 1996. Hepatitis C virus antibody(anti-HCV): Prevalence in psoriasis. Int J Dermatol 35:797–799.
Cody SH, Nainan OV, Garfein RS. 2002. Hepatitis C virus transmissionfrom an anesthesiologist to a patient. Arch Intern Med 162:345–350.
Duckworth GJ, Heptonstall J, Aitken C. 1999. Transmission ofhepatitis C virus from a surgeon to a patient. Commun Dis PublicHealth 2:188–192.
Esteban JI, Gomez J, Martell M. 1996. Transmission of hepatitis Cvirus by a cardiac surgeon. N Engl J Med 334:555–560.
Harris KA, Gilham C, Mortimer PP, Teo CG. 1999. The most prevalenthepatitis C virus genotypes in England and Wales are 3a and 1a.J Med Virol 58:127–131.
Stefan-Ross R, Viazov S, Gross T. 2000. Transmission of hepatitis Cvirus from a patient to an anaesthiology assistant to five patients. NEngl J Med 343:1851–1854.
Tallis GF, Ryan GM, Lambert SB. 2003. Evidence of patient-to-patienttransmission of hepatitis C virus through contaminated intrave-nous anaesthetic ampoules. J Viral Hepat 10:234–239.
UK Health Departments. 1994. AIDS/HIV infected health careworkers: Guidance for the management of infected healthcareworkers.
UK Health Departments. 2002. Hepatitis C infected health careworkers.
Anesthetist to Patient Transmission of Hepatitis C Virus 401

Journal of Medical Virology 75:402–411 (2005)
Hepatitis C Virus Core, NS3, NS5A, NS5B ProteinsInduce Apoptosis in Mature Dendritic Cells
Samila Siavoshian,1 Jean Daniel Abraham,1 Christine Thumann,1 Marie Paule Kieny,2
and Catherine Schuster1*1INSERM U544, Strasbourg, France2WHO/IVR, Geneve, Switzerland
Although reasons for hepatitis C virus (HCV) per-sistence are still unknown, specific cellularimmune responses appear to influence thepathogenesis and outcome of the infection.Apoptosis of cells infected by virusesmayappearsuicidal to the viruses that induce programmedcell death of its host. However, apoptosis hasbeen suggested to be a response to virus in-fection as a mean of facilitating virus dissemina-tion. Annexin V-propidium iodide staining andDNA fragmentation, were used to show that ex-pression of the core, NS3, NS5A, or NS5B proteininduces apoptosis in mature dendritic cells. Inaddition, immunoblotting was used to demon-strate that expression level of p21waf1/cip1 proteindecreased in cells expressing one of these HCVproteins. No expression of p53 could be detectedand expression of Akt was independent of HCVproteins expression. These results suggest thatthe effect of these HCV proteins on HCV asso-ciated pathogenesis may be linked (at leastpartially) to its ability to modulate apoptosispathways in mature dendritic cells. J. Med.Virol. 75:402–411, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: hepatitis C virus; apoptosis;p53; p21
INTRODUCTION
Hepatitis C virus (HCV) is one of the most commonetiologic agents of chronic liver diseases [Choo et al.,1989; Kuo et al., 1989]. The HCV genome consists of apositive-stranded RNA molecule of approximately 9,500nucleotides with a single large open reading frame(ORF). Translation of this ORF results in the productionof a polyprotein precursor which is cleaved co- and post-translationally by viral and host cell proteases to yieldthe individual structural and non-structural proteins(NS). NS proteins are encoded by the 30 two thirds of theviral genome and are termed NS2, NS3, NS4A, NS4B,NS5A, and NS5B [Hijikata et al., 1993; Tomei et al.,1993; Tanji et al., 1995; Behrens et al., 1996]. The capsid
protein or core and the two envelope glycoproteins E1and E2, which constitute HCV structural proteins, areencoded by the 50 third of the genome [Hijikata et al.,1991; Mizushima et al., 1994].
Infection with HCV becomes persistent in most in-fected humans and may lead to the development ofchronic hepatitis, cirrhosis, and hepatocellular carci-noma [Watson, 1999].
Apoptosis of cells infected by viruses may appearsuicidal to the viruses that induce programmed celldeath of its host. However, apoptosis has been suggestedto be a response to virus infection as a mean of facili-tating virus dissemination [Everett and McFadden,1999].
The aim of the present study is to investigate thepotential apoptosis effects of the core, NS3, NS5A, andNS5B proteins in dendritic cells, another target for HCV[Navas et al., 2002]. Dendritic cells are professionalantigen presenting cells and play a key role for theinduction of the immune response [Banchereau andSteinman, 1998]. In most tissues, dendritic cells arepresent in an immature state; these immature dendriticcells are able to capture antigens and then, after matu-ration into mature dendritic cells and concomitantmigration to lymphoid areas, they are able to stimulatelymphocytes. During this process, the surface expres-sion of adhesion and co-stimulatory molecules (CD40,CD80, CD86), as well as the major histocompatibilitycomplex (MHC) molecules are up-regulated [Bancher-eau et al., 2000]. In addition, mature dendritic cellssecrete high levels of interleukin-12 (IL-12), a cytokinethat promotes the maturation of cytotoxic T cells.
Grant sponsor: ARC, France; Grant number: 76035703; Grantsponsor: Fondation BNP-Paribas; Grant number: EPB/GP/CL011011.
*Correspondence to: Catherine Schuster, INSERM U544, 3 RueKoeberle, 67000 Strasbourg, France.E-mail: [email protected]
Accepted 26 October 2004
DOI 10.1002/jmv.20283
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

Recent studies have focused on putative interactionsof HCV proteins with dendritic cells and subsequentmodifications of dendritic cells functions. Indeed, inHCV patients, a reduction in the expression of CD86, aclassical stimulation molecule expressed in dendriticcells and a diminished production of IL-12, correlatedwith impaired allostimulatory functions of dendriticcells were reported [Kanto et al., 1999; Kakumu et al.,2000]. Other researchers have observed that alteredstimulation capacity is correlated with chronic infectionand that patients who cleared HCV infection did notpresent any modification in these dendritic cells proper-ties [Auffermann-Gretzinger et al., 2001; Bain et al.,2001]. Sarobe et al. [2002] demonstrated that immaturedendritic cells expressing core and E1 proteins werepoor stimulators of allogenic reactions, when these cellswere used to stimulate T lymphocytes [Sarobe et al.,2002]. Moreover, HCV structural proteins impair dend-ritic cell maturation induced with TNF-a and inhibitin vivo induction of cellular immune responses [Sarobeet al., 2003].
The eukaryotic cell division cycle has been found to begoverned by two families of proteins, which are asso-ciated to form heterodimers: the cyclins and the cyclindependent kinases (cdk) [Sherr, 1993]. The activities ofthe cdks are regulated by a family of cdk-inhibitoryproteins [Serrano et al., 1993; Xiong et al., 1993].p21waf1/cip1 interacts with cdk–cyclin complexes [Harperet al., 1993; Xiong et al., 1993] and causes G1 or G2 arrest[Harper et al., 1995; Dulic et al., 1998].
Akt is a proto-oncogene encoding a serine-threoninekinase. Activation of Akt inhibits apoptosis induced bygrowth factor withdrawal or irradiation in neural cells,fibroblasts, and lymphocytes [Hemmings, 1997; Cofferet al., 1998]. It has been shown that antisense oligonu-cleotides against p21waf1/cip1 block cell cycle, inhibit Aktinduction, and enhance cell death in differentiatingmyocyte cultures [Fujio et al., 1999] suggesting thatthe expression of Akt protein is under the control ofp21waf1/cip1 protein.
The p53 tumor suppressor protein is a transcriptionfactor required for the transactivation of a number ofgenes involved in growth control [Wiman, 1997]. Indeed,p53 transcriptionally activates Bax and represses Bcl2expression, which results in the activation of the caspasecascade leading to apoptosis [Somasundaram, 2000].In addition, p53 transcriptionally activates p21waf1/cip1
and represses cyclin B expression, which results inthe inhibition of cell growth [Somasundaram, 2000].However, p21waf1/cip1 expression can be activated by ap53 independent pathway [Cox, 1997].
Beside its structural role, the HCV core protein hasbeen shown to possess a number of regulatory functions[McLauchlan, 2000]. Indeed, Honda et al. [2000] havefound that core protein sensitized CHO-K1 cells toapoptotic stimuli and significantly induced the expres-sion of the c-myc protein as well as other apoptosisinducing proteins downstream of c-myc, like p53,p21waf1/cip1 and Bax [Honda et al., 2000]. However, in-hibition of p21waf1/cip1 expression by HCV core protein
has been shown in other studies [Ray et al., 1997; Rayet al., 1998, Yoshida et al., 2001; Jung et al., 2001;Dubourdeau et al., 2002; Han et al., 2002; Lee et al.,2002].
The viral non-structural protein NS3 is a multi-functional protein, which is necessary for virus re-plication [Kwong et al., 2000]. Kwun et al. [2001]demonstrated that the NS3 protein can specificallyrepress the promoter activity of p21waf1/cip1 in a dose-dependent manner. Furthermore, these authors haveshown that the NS3-expressing cell line growth rate wasat least twice as fast as that of the parent NIH3T3 cells,indicating that repression of p21waf1/cip1 is actuallyreflected by a stimulation of cell growth [Kwun et al.,2001].
NS5A is a phosphoprotein, which plays probably a rolein the replication cycle [Reyes, 2002]. It was shown thatNS5A interacts directly with p53 and represses tran-scription of the cell-cycle regulatory gene p21waf1/cip1
[Ghosh et al., 1999; Majumder et al., 2001; Lan et al.,2002; Qadri et al., 2002].
NS5B has been demonstrated to constitute the viralRNA-dependent RNA polymerase [Behrens et al., 1996].It was shown that NS5B protein (as well as core, NS3,and NS5A proteins) inhibit cell proliferation indepen-dently from p53 expression in hepatocarcinoma celllines [Siavoshian et al., 2004].
In the present paper, the potential apoptosis effects ofHCV proteins in the mature dendritic cells was in-vestigated from the viewpoint of cell-cycle proteinswhich, are involved in apoptosis.
MATERIALS AND METHODS
Cell Culture
Human Huh7 hepatoma cells and 293 cells weremaintained in Dulbecco’s modified Eagle’s mediumsupplemented with 10% fetal calf serum, at 378C in a5% CO2 incubator. All tissue culture reagents were fromInvitrogen (Cergy Pontoise, France).
Generation of Human Dendritic Cells
Dendritic cells were differentiated from mono-cytes obtained by cytapheresis from healthy donors(Etablissement Francais du Sang, Strasbourg, France).This technique allows direct separation of peripheralblood mononuclear cells from total blood. Purifiedmonocytes were isolated by counter-current elutriationof peripheral blood mononuclear cells with more than95% purity, as characterized by Giemsa staining or flowcytometry using an anti-CD14 phycoerythrin-labeledantibody (Pharmingen, BD Biosciences, San Diego, CA).Dendritic cells were generated as described previously[Sallusto and Lanzavecchia, 1994] with some modifica-tions. Briefly, monocytes were maintained in cultureat 5.105 cells/ml for 6 days in RPMI 1640 medium(Invitrogen) containing L-glutamine (2 mM), penicillinG sodium (100 U/ml), and streptomycin sulfate (100 mg/ml) supplemented with 5% fetal calf serum (Invitrogen),
Mature Dendritic Cells Apoptosis by HCV Proteins 403

100 ng/ml (1,000 U/ml) of granulocyte-macrophagecolony-stimulating factor (Peprotech, Rocky Hill, NJ)and 20 ng/ml of interleukin 4 (IL-4, Peprotech). Cellswere fed again with one third fresh medium containingcytokines on day 1 and with fresh cytokines on day 4. Atday 6, non-adherent cells, corresponding to immaturedendritic cells were analyzed by cytometry.
Maturation of Immature Dendritic Cells
Immature dendritic cells were maintained in culturein the presence of medium containing RPMIþ 5% fetalcalf serumþ lipopolysaccharide (Sigma-Aldrich, St.Quentin Fallavier, France) at 1 mg/ml for 48 h and thephenotype of mature dendritic cells was appreciated bycytometry.
Flow Cytometry Analysis of ImmatureDendritic Cells and Mature Dendritic Cells
Analysis of the expression of dendritic cells surfacemolecules was done with a panel of monoclonal anti-bodies recognizing markers characteristic of the imma-ture dendritic cells and mature dendritic cells stages.Cells (0.2�106 cells/well) were incubated for 15–30 minat þ48C in 96-well round-bottom plates with thefollowing antibodies diluted in FACS Flow (BectonDickinson) at 20 ml/106 cells: anti-CD14, anti-CD1a,anti-HLA DR, anti-CD40, anti-CD80, anti-CD83, anti-CD86; all antibodies were phycoerythrin (PE)-labeledand provided by Pharmingen-Becton Dickinson. Speci-fic isotypes (anti-IgG1-PE and anti-IgG2-PE) were usedas controls. After incubation, cells were washed threetimes with FACS flow and fixed for at least 20 min atþ48C in 400 ml of paraformaldehyde (Merck, Eurolab,Darmstadt, Germany) at 1.5% in PBS (Invitrogen). Dataacquisition was done for 5,000 cells by using a FACScancytometer (Becton Dickinson) and surface expression ofthe different molecules was analyzed with the CELLQuest Software (Becton Dickinson).
Recombinant Adenovirus Construction,Production, and Titration
All recombinant adenoviral genomes were generatedas infectious plasmids by homologous recombination inE. coli as described previously [Chartier et al., 1996].The two plasmids (pTG 13387 and pTG 6624) containingthe adenoviral genome were kindly provided by TRANS-GENE S.A. (Strasbourg, France). Briefly, sequencesderived from an HCV 1b genotype (HCV-J) cDNAcorresponding to the core protein (aminoacids 1–191),to the NS3 protein (aminoacids 1,027–1,657), to theNS5A protein (aminoacids 1,973–2,420), to the NS5Bprotein (2,421–3,011), and the Green Fluorescent Pro-tein (GFP) from pEGFP-C1 vector (Clontech Labora-tories, Inc., Palo Alto, CA) were amplified by PCR andinserted in the adenoviral shuttle plasmid (pTG 13387)containing a CMV-driven expression cassette surround-ed by adenoviral sequences (nucleotides 1–458 andnucleotides 3,511–5,788) to allow homologous recombi-
nation with adenoviral sequences on the backbonevector (pTG 6624) [Chartier et al., 1996]. The resultingfull-length viral genomes contain a deletion in E3(nucleotides 28,592–30,470), whereas the adeno E1region (nucleotides 459–3,327) is replaced by the ex-pression cassette containing, from 50 to 30, the CMVimmediate-early enhancer/promoter, a chimeric humanb-globin/IgG intron, the HCV or the GFP heterologoussequences, and the bovine growth hormone polyadeny-lation signal. All the constructs were sequenced using aDNA Li-Cor 4200 sequencer (Science Tech).
The recombinant adenoviruses were generated bytransfection into the 293 complementation cell line ofthe corresponding viral genomes released from theirrespective plasmids by PacI digestion. Virus propaga-tion and amplification were undertaken by successivepassages on 293 cells. Recombinant adenovirus purifi-cation as well as titration of infectious units by indirectimmunofluorescence of the viral DNA binding proteinwere carried out as described previously [Lusky et al.,1998]. Briefly, after clearing of the cell lysate, viruseswere purified by a two-rounds CsCl density centrifuga-tion. Purified viruses were stored in 1 M sucrose, 10 mMTris-HCl, pH 8.5, 1 mM MgCl2, 150 mM NaCl, and theirtitration was performed by detection of the DBP at 16 hrafter infection of 293 cells, using purified a72K B6-8hybridoma supernatant provided by TRANSGENE S.A.[Reich et al., 1983].
Transduction of Immature Dendritic CellsWith Adenovirus Followed byLipopolysaccharide Addition
Immature dendritic cells harvested at day 6 of culturewith cytokines were transduced with the purified non-replicative recombinant adenoviruses encoding HCVcore, NS3, NS5A, or NS5B. Adenovirus expressing GFPor ‘‘empty’’ adenovirus were used as controls as well asnon-infected cells. The infection protocol was derivedfrom Dietz and Vuk-Pavlovic [1998]. Briefly, 1.25 mg ofLipofectamin (Invitrogen Life Technologies) was addedto 2.108 adenovirus infectious units in a total volume of100mlRPMI (Invitrogen LifeTechnologies). After 15minof incubation at room temperature, immature dendriticcells diluted at 107 cells/ml in RPMI without serum wereincubated with lipofectamin-pretreated adenovirus at amultiplicity of infection of 200 for 2 hr at 378C withfrequent gentle shaking. Non-adsorbed virus waseliminated by washing cells twice with RPMI; cells weremaintained in culture at 378C for 24, 48, or 72 hr at aconcentration of 0.5� 106 cells/ml in medium containingin a 1/1 ratio, RPMIþ 2.5% fetal calf serumþ1 mg/mllipopolysaccharide and monocyte conditioned mediumcorresponding to medium from cultured monocytes atday 6. It is noteworthy that more than 70% of cells werefound to express GFP following infection.
Immunoblotting
Cell lysates were prepared at 48 hr post-infection foranalysis of HCV proteins expression and 72 hr post-
404 Siavoshian et al.

infection for analysis of cell-cycle protein expression.After washing the cells once with PBS, cells wereincubated for 1 hr at 48C in 500 ml lysis buffer consistingin 10 mM Tris-HCl pH 7.4, 20 mM NaCl, 5 mM MgCl2,0.5% Nonidet P40, and 0.1 mM phenylmethylsulfonyl-fluoride. After centrifugation at 10,000g during 10 minat 208C, the protein content of the extracts wasdetermined spectrophotometrically using the Bradfordassay. Thirty micrograms of total proteins were re-solved on a 12% SDS–PAGE gel along with prestainedprotein molecular weight standards (Bio-Rad, Marnes laCoquette, France). Gels were blotted onto PVDFmembranes (Amersham, ORSAY, France). Upon com-pletion of the transfer, the blots were blocked with PBScontaining 5% non-fat milk overnight at 48C. Mem-branes were then incubated with the primary antibodiesa 1:100 dilution in 1% milk/PBS for 1 hr at roomtemperature. Rabbit polyclonal antibodies anti-Akt,p21waf1/cip1 and p53 were obtained from Santa CruzBiotechnology (Santa Cruz, CA). Anti-HCV NS3 andNS5B polyclonal antibodies were generously providedby R. Bartenschlager (Institute for Virology, Heidelberg,Germany). Anti-HCV core (27D5G5) and NS5A (4F3H2)monoclonal antibodies were generously provided by C.Jolivet (BioMerieux, Lyon, France). Blots were washedwith PBS three times followed by incubation with biotin-conjugated anti-rabbit (Sigma) or anti-mouse (Sigma)antibodies diluted 1:500 in 1% milk/PBS for 1 hrat room temperature. After additional washes, blotswere incubated with streptavidin–peroxydase complex(Amersham) diluted 1/500 in 1% milk/PBS for 1 hr atroom temperature, and proteins were detected using achemiluminescence assay system (Amersham).
Annexin V-FITC, Propidium Iodide Staining,and DNA Fragmentation Analysis
Apoptosis of dendritic cells was determined 72 hr post-infection by staining with annexin V-FITC and propi-dium iodide (Annexin Apoptosis Detection kit, Euro-medex) according to the manufacturer’s instructions.FACS analysis was carried out on a FACScan cytometer(Becton-Dickinson). DNA fragmentation of apoptoticHCV infected dendritic cells was then studied using aDNA ladder assay according to the manufacturer’sinstructions (Apoptotic DNA ladder Detection kit,CHEMICON, CA) at 72 hr post-infection.
RESULTS
Phenotype of Dendritic Cells at theTime of Transduction
Monocytes maintained for 6 days in the presence ofgranulocyte-macrophage colony-stimulating factor andIL-4 were differentiated into an homogeneous non-adherent cell population with typical dendritic cellsmorphology. These cells were characterized as imma-ture dendritic cells using flow cytometry with a panel ofantibodies. The classical immature dendritic cell phe-notype was obtained: cells lost the expression of CD14
which is highly expressed on monocytes at the beginningof the culture and expressed high levels of CD1a, aspecific epidermal dendritic cells (Langerhans cells)marker and low levels of the MHC class II molecule,HLA-DR. Co-stimulation molecules were expressed atlow levels as shown for CD40 and CD80 or moderatelevels for CD86. CD83, the typical marker of maturedendritic cells, was negative on immature dendritic cells(Fig. 1A).
Dendritic cells generated from immature dendriticcells in the presence of lipopolysaccharide (1 mg/ml)during 48 hr displayed a mature dendritic cell pheno-type: negative expression of CD14, down-regulated ex-pression of CD1a, high expression of HLA-DR, moderatelevels of CD40, high levels of CD80, CD86 and CD83expression (Fig. 1B).
In the presence of medium lacking lipopolysaccharideand complemented with either fetal calf serum or humanserum, complete maturation did not occur as revealed bycytometry analysis (data not shown). Labeling of cellswith anti-HLA-DR or anti-CD86 antibodies showed,however, an intermediate phenotype between immatureand mature dendritic cells. In the presence of humanserum, cells were adherent to the surface of the plates(data not shown). Since the dendritic cells could not bemaintained under a strict immature phenotype, we onlythe effect of core, NS3, NS5A, and NS5B proteins werestudied in lipopolysaccharide matured dendritic cells.
Expression of HCV Proteins
The expression of core, NS3, NS5A, and NS5B pro-teins in infected cells was examined at 48 hr post-transduction using immunoblotting (Fig. 2). As shownin Figure 2, expression of the core, NS3, NS5A, andNS5B proteins was detected at the correct molecularweight in mature dendritic cells.
Core, NS3, NS5A, or NS5B Induce Apoptosisin Mature Dendritic Cells
Apoptotic cells are characterized by the presence offragmented DNA in their nucleus. This fragmentedDNA is extracted and analyzed on a 1% agarose gel. Toexamine the apoptotic changes of mature dendritic cellsexpressing core, NS3, NS5A, or NS5B protein, DNAladdering of infected and control mature dendritic cellswas evaluated at 72 hr post-infection. As shown inFigure 3, the presence of fragmented DNA was detectedonly in mature dendritic cells expressing the core, NS3,NS5A, or NS5B proteins.
The apoptotic cell death of mature dendritic cellsexpressing core, NS3, NS5A, or NS5B protein was thenstudied by annexin V-FITC/propidium iodide staining at72 hr post-infection. Annexin V-FITC/propidium iodidedouble-positive cells represent mature dendritic cells in alate apoptotic state. Figure 4 shows an increased labelingin cells expressing the core, NS3, NS5A, or NS5B proteincompared to control cells. It is noteworthy that emptyadenovirus was used for this experiment as a control andnot GFP expressing adenovirus, as GFP would have
Mature Dendritic Cells Apoptosis by HCV Proteins 405
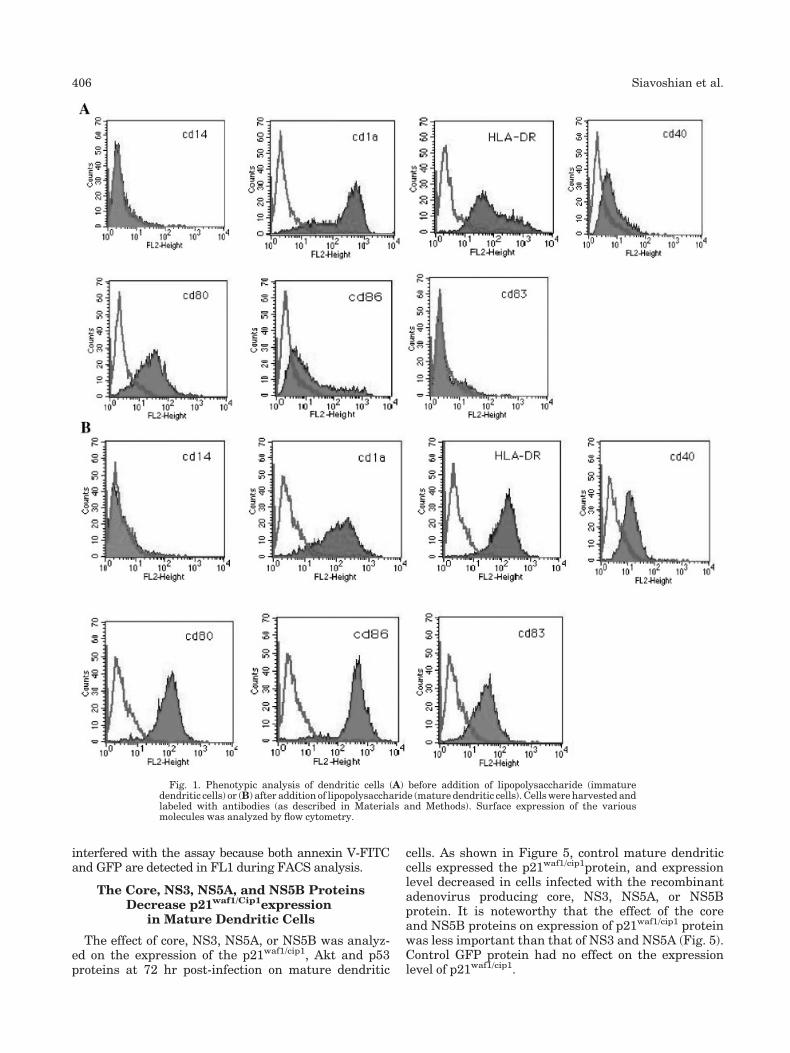
interfered with the assay because both annexin V-FITCand GFP are detected in FL1 during FACS analysis.
The Core, NS3, NS5A, and NS5B ProteinsDecrease p21waf1/Cip1expression
in Mature Dendritic Cells
The effect of core, NS3, NS5A, or NS5B was analyz-ed on the expression of the p21waf1/cip1, Akt and p53proteins at 72 hr post-infection on mature dendritic
cells. As shown in Figure 5, control mature dendriticcells expressed the p21waf1/cip1protein, and expressionlevel decreased in cells infected with the recombinantadenovirus producing core, NS3, NS5A, or NS5Bprotein. It is noteworthy that the effect of the coreand NS5B proteins on expression of p21waf1/cip1 proteinwas less important than that of NS3 and NS5A (Fig. 5).Control GFP protein had no effect on the expressionlevel of p21waf1/cip1.
Fig. 1. Phenotypic analysis of dendritic cells (A) before addition of lipopolysaccharide (immaturedendritic cells) or (B) after addition of lipopolysaccharide (mature dendritic cells). Cells were harvested andlabeled with antibodies (as described in Materials and Methods). Surface expression of the variousmolecules was analyzed by flow cytometry.
406 Siavoshian et al.

Control mature dendritic cells expressed the Aktprotein and the expression level of this protein remainedunchanged in mature dendritic cells expressing GFP asa control, core, NS3, NS5A, or NS5B protein (Fig. 5). Inaddition, the p53 protein was not detected in controlmature dendritic cells whereas it was detected in Huh7cells. Following expression of core, NS3, NS5A, or NS5B,p53 protein remained undetected (Fig. 5).
DISCUSSION
In this study, the potential apoptosis effects of core,NS3, NS5A, or NS5B protein was examined in maturedendritic cells. The results obtained after DNA frag-mentation and annexin V-propidium iodide stainingshow that core, NS3, NS5A, and NS5B proteins induceapoptosis in mature dendritic cells.
The apoptotic effect of HCV core protein has beendemonstrated in other cell lines. Indeed, it was shownpreviously that core protein expression induced apopto-sis in response to anti-fas monoclonal antibody [Ruggieriet al., 1997] and enhanced the susceptibility of hepato-cytes to TNF-a mediated apoptosis [Zhu et al., 1998].Moreover, Chinese Hamster Ovary cells stably expres-
sing core protein were shown to undergo apoptosis inresponse to serum starvation [Honda et al., 2000].Chung et al. [2003] demonstrated that HCV coreenhances sodium phenylbutyrate induced apoptosis inhepatocellular carcinoma cells, which is independent ofp53 expression [Chung et al., 2003].
Recently, an apototic effect of HCV NS3 protein wasdescribed. Indeed, it was shown that NS3 induces dys-function and apoptosis of three main subsets of cytotoxiclymphocytes, which are prevalently found in chronicallyHCV-infected liver tissue [Thoren et al., 2004].
Although anti-apoptotic effect of NS5A was describedin other publications [Ghosh et al., 2000; Lan et al., 2002;Majumder et al., 2002; Chung et al., 2003], the resultsobtained in this study show that NS5A induce apoptosisin mature dendritic cells. The reasons of this discre-pancy could be the type of experiment, cell lines, andHCV strains used. In fact, the anti-apoptotic effect ofNS5A protein was studied mainly in the hepatocarci-noma cell lines [Ghosh et al., 2000; Lan et al., 2002;Chung et al., 2003] in response to TNF-a [Ghosh et al.,2000; Majumder et al., 2002], or sodium phenylbutyrate[Chung et al., 2003].
The effect of HCV core, NS3, NS5A, and NS5B pro-teins on cell-cycle regulatory proteins was then studiedby Western blot analysis. The data show that uninfectedmature dendritic cells express p21waf1/cip1 protein and
Fig. 2. Detection of the core, NS3, NS5A, and NS5B proteins byimmunoblotting. Lysates of mature dendritic cells expressing core,NS3, NS5A, or NS5B protein were submitted to electrophoresis on 12%SDS–PAGE, followed by immunoblotting using antibodies directedagainst HCV proteins (core, NS3, NS5A, NS5B) and enhancedchemiluminescence detection as described in Materials and Methods.Protein bands are shown by arrows. Mock, non-transduced cells; GFP,cells transduced by adenovirus GFP; core, cells transduced byadenovirus core; NS3, cells transduced by adenovirus NS3; NS5A,cells transduced by adenovirus NS5A; and NS5B, cells transduced byadenovirus NS5B, respectively.
Fig. 3. Demonstration of DNA laddering in mature dendritic cellsexpressing core, NS3, NS5A, or NS5B protein. The DNA of maturedendritic cells transduced for 72 hr without (control) or with recombi-nant adenoviruses expressing GFP, core, NS3, NS5A, or NS5B proteinwere extracted (as described in Materials and Methods) and analyzedon an 1% agarose gel.
Mature Dendritic Cells Apoptosis by HCV Proteins 407

that HCV core, NS3, NS5A, and NS5B proteins down-regulate p21waf1/cip1 expression. These results are con-sistent with the inhibition of p21waf1/cip1 expression byHCV core observed by others [Ray et al., 1998; Junget al., 2001; Yoshida et al., 2001; Dubourdeau et al.,2002; Han et al., 2002; Lee et al., 2002]. Indeed, thesestudies showed that the HCV core protein represses thetranscriptional activity of the p21waf1/cip1 promoter.Kwun et al. [2001] have also reported that NS3 proteincould specifically repress the promoter activity ofp21waf1/cip1 in a dose-dependent manner [Kwun et al.,2001]. The same observation has been made for HCVNS5A protein. Indeed, the authors demonstrated thatan association of NS5A and p53 allows transcriptional
modulation of the p21waf1/cip1 gene [Ghosh et al., 1999;Majumder et al., 2001; Lan et al., 2002; Qadri et al.,2002].
Although the modulation of expression of Akt proteinby p21waf1/cip1 was described by Fujio et al. [1999],the result obtained in this study show that maturedendritic cells express the Akt protein and that the ex-pression of this protein remains unchanged before andafter expression of HCV proteins. These results suggestthat expression of the Akt protein is not regulated by thep21waf1/cip1 in mature dendritic cells.
The expression of p53 was not detected in maturedendritic cells. This could be explained by the short half-life of p53, which is often undetectable in normal cells
Fig. 4. Propidium iodide (PI) and annexin V-FITC (ann-FITC) uptake of mature dendritic cells ex-pressing the core, NS3, NS5A, or NS5B protein. At 72 hr post-transduction, mature dendritic cells werestained with PI and ann-FITC (as described in Materials and Methods) and analyzed using flow cytometry.Ann-FITC and PI were plotted along the X and Y axis, respectively.
408 Siavoshian et al.

[Somasundaram, 2000]. However, the absence of mod-ulation of p53 expression by HCV proteins suggeststhat, in mature dendritic cells, the apoptosis effect ofHCV proteins could be independent of p53 expression.Moreover, the expression of other proteins under thecontrol of p53 such as Bax and Bcl2 which can activatethe caspase cascade during apoptosis was analyzedusing Western blot. Bax and Bcl2 were absent in controlmature dendritic cells, following expression of HCVproteins, Bax and Bcl2 proteins remained undetected(data not shown).
Taken together, these results suggest that the modu-lation of p21waf1/cip1 expression by HCV core, NS3,NS5A, and NS5B proteins could induce apoptosis inmature dendritic cells. Indeed, in a number of studies, ithas been shown that p21waf1/cip1 induction can decreaseapoptosis in various cell system. In fact, in culturedmyoblasts induced to differentiate in low serum con-dition, overexpression of p21waf1/cip1 decreased thenumber of apoptotic deaths [Wang and Walsh, 1996].In addition, RKO human colorectal carcinoma cells wereprotected from prostaglandin A2 mediated apoptosis byoverexpressing p21waf1/cip1 [Gorospe and Holbrook,1996]. It was shown that p21waf1/cip1 antisense oligo-nucleotides partially block p21waf1/cip1 expression lead-ing to programmed cell death during differentiationof a neuronal line [Poluha et al., 1996]. In otherstudies, however, marked up-regulation of endogenousp21waf1/cip1 has been reported to be coupled with in-duction of p53-dependent or independent apoptosis inresponse to certain agents [El-Deiry et al., 1994; Shaoet al., 1995; Sheikh et al., 1996].
Although, a down-regulation of the p21waf1/cip1 expres-sion by HCV proteins was observed in the maturedendritic cells expressing HCV proteins, the modifica-tion of p21waf1/cip1 expression alone is not sufficient toexplain the induction of apoptosis in mature dendriticcells. In fact, the degree of p21waf1/cip1 down-regulationin core and NS5B expressing cells is minimal compared
to the NS3 and NS5A proteins while the induction ofapotosis by core and NS5B is comparable to NS3 andNS5A proteins. These results suggest that other apop-tosis pathways could be activated by HCV proteins.Further studies are needed to elucidate this hypothesis.
The results obtained in this study show that the HCVproteins (core, NS3, NS5A, and NS5B) induce apoptosisin mature dendritic cells and decrease the expression ofp21waf1/cip1 protein in these cells. These results suggestthat the effect of these HCV proteins in the HCV asso-ciated pathogenesis may be linked (at least partially) toits ability to modulate apoptosis pathways in the maturedendritic cells.
ACKNOWLEDGMENTS
We thank Dr. Kunitada Shimotohno for providing thepCMV/C980 and pCMV/N729-3010 plasmid constructs.Dr. Colette Jolivet (BioMerieux, Lyon, France) andDr. Ralph Bartenschlager (Heidelberg, Germany) areacknowledged for the generous gift of antibodies. SS andJDA were recipients of grants from BioMerieux (France)and the Association pour la Recherche contre le Cancer(ARC, France).
REFERENCES
Auffermann-Gretzinger S, Keeffe EB, Levy S. 2001. Impaired dendriticcell maturation in patients with chronic, but not resolved, hepatitisC virus infection. Blood 97:3171–3176.
Bain C, Fatmi A, Zoulim F, Zarski JP, Trepo C, Inchauspe G. 2001.Impaired allostimulatory function of dendritic cells in chronichepatitis C infection. Gastroenterology 120:512–524.
Banchereau J, Steinman RM. 1998. Dendritic cells and the control ofimmunity. Nature 392:245–252.
Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ,Pulendran B, Palucka K. 2000. Immunobiology of dendritic cells.Annu Rev Immunol 18:767–811.
Behrens SE, Tomei L, De Francesco R. 1996. Identification andproperties of the RNA-dependent RNA polymerase of hepatitis Cvirus. EMBO J 15:12–22.
Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M.1996. Efficient generation of recombinant adenovirus vectors byhomologous recombination in Escherichia coli. J Virol 70:4805–4810.
Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M.1989. Isolation of a cDNA clone derived from a blood-borne non-A,non-B viral hepatitis genome. Science 244:359–362.
Chung YL, Sheu ML, Yen SH. 2003. Hepatitis C virus NS5A as apotential viral Bcl-2 homologue interacts with Bax and inhibitsapoptosis in hepatocellular carcinoma. Int J Cancer 107:65–73.
Coffer PJ, Jin J, Woodgett JR. 1998. Protein kinase B (c-Akt): A multi-functional mediator of phosphatidylinositol 3-kinase activation.Biochem J 335:1–13.
Cox LS. 1997. Multiple pathways control cell growth and transforma-tion: Overlapping and independent activities of p53 and p21Cip1/WAF1/Sdi1. J Pathol 183:134–140.
Dietz AB, Vuk-Pavlovic S. 1998. High efficiency adenovirus-mediatedgene transfer to human dendritic cells. Blood 91:392–398.
Dubourdeau M, Miyamura T, Matsuura Y, Alric L, Pipy B, RousseauD. 2002. Infection of HepG2 cells with recombinant adeno-virus encoding the HCV core protein induces p21(WAF1) down-regulation—effect of transforming growth factor beta. J Hepatol37:486–492.
Dulic V, Stein GH, Far DF, Reed SI. 1998. Nuclear accumulation ofp21Cip1 at the onset of mitosis: A role at the G2/M-phase transition.Mol Cell Biol 18:546–557.
El-Deiry WS, Harper JW, O’Connor PM, Velculescu VE, Canman CE,Jackman J, Pietenpol JA, Burrell M, Hill DE, Wang Y. 1994. WAF1/
Fig. 5. Effect of core, NS3, NS5A, or NS5B protein on the expressionof the p21waf1/cip1, Akt, and p53 proteins in mature dendritic cells. Equalvolumes of whole cell extracts containing 30 mg of proteins wereseparated by electrophoresis and blotted. Proteins were detected withantibodies to p21waf1/cip1, Akt and p53 as indicated. Huh7 cells wereused as a positive control for the detection of p53 expression.
Mature Dendritic Cells Apoptosis by HCV Proteins 409

CIP1 is induced in p53-mediated G1 arrest and apoptosis. CancerRes 54:1169–1174.
Everett H, McFadden G. 1999. Apoptosis: An innate immune responseto virus infection. Trends Microbiol 7:160–165.
Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. 1999.Cell cycle withdrawal promotes myogenic induction of Akt: A posi-tive modulator of myocyte survival. Mol Cell Biol 19:5073–5082.
Ghosh AK, Steele R, Meyer K, Ray R, Ray RB. 1999. Hepatitis C virusNS5A protein modulates cell cycle regulatory genes and promotescell growth. J Gen Virol 80:1179–1183.
Ghosh AK, Majumder M, Steele R, Meyer K, Ray R, Ray RB. 2000.Hepatitis C virus NS5A protein protects against TNF-alphamediated apoptotic cell death. Virus Res 67:173–178.
Gorospe M, Holbrook NJ. 1996. Role of p21 in prostaglandin A2-mediated cellular arrest and death. Cancer Res 56:475–479.
Han HJ, Jung EY, Lee WJ, Jang KL. 2002. Cooperative repression ofcyclin-dependent kinase inhibitor p21 gene expression by hepatitisB virus X protein and hepatitis C virus core protein. FEBS Lett518:169–172.
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. 1993. The p21Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816.
Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P,Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, Fox MP,Wei N. 1995. Inhibition of cyclin-dependent kinases by p21. Mol BiolCell 6:387–400.
Hemmings BA. 1997. Akt signaling: Linking membrane events to lifeand death decisions. Science 275:628–630.
Hijikata M, Kato N, Ootsuyama Y, Nakagawa M, Shimotohno K. 1991.Gene mapping of the putative structural region of the hepatitis Cvirus genome by in vitro processing analysis. Proc Natl Acad SciUSA 88:5547–5551.
Hijikata M, Mizushima H, Akagi T, Mori S, Kakiuchi N, Kato N,Tanaka T, Kimura K, Shimotohno K. 1993. Two distinct proteinaseactivities required for the processing of a putative nonstructuralprecursor protein of hepatitis C virus. J Virol 67:4665–4675.
Honda M, Kaneko S, Shimazaki T, Matsushita E, Kobayashi K, Ping LH,Zhang HC, Lemon SM. 2000. Hepatitis C virus core protein inducesapoptosis and impairs cell-cycle regulation in stably transformedChinese hamster ovary cells. Hepatology 31:1351–1359.
Jung EY, Lee MN, Yang HY, Yu D, Jang KL. 2001. The repressiveactivity of hepatitis C virus core protein on the transcription ofp21(waf1) is regulated by protein kinase A-mediated phosphoryla-tion. Virus Res 79:109–115.
Kakumu S, Ito S, Ishikawa T, Mita Y, Tagaya T, Fukuzawa Y, YoshiokaK. 2000. Decreased function of peripheral blood dendritic cells inpatients with hepatocellular carcinoma with hepatitis B and C virusinfection. J Gastroenterol Hepatol 15:431–436.
Kanto T, Hayashi N, Takehara T, Tatsumi T, Kuzushita N, Ito A,Sasaki Y, Kasahara A, Hori M. 1999. Impaired allostimulatorycapacity of peripheral blood dendritic cells recovered from hepatitisC virus-infected individuals. J Immunol 162:5584–5591.
Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH,Miyamura T, Dienstag J, Alter MJ, Stevens CE, Tegtmeier GE,Bonino F, Colombo M, Lee WS, Kuo C, Berger K, Shuster JR,Overby LR, Bradley DW, Houghton H. 1989. An assay forcirculating antibodies to a major etiologic virus of human non-A,non-B hepatitis. Science 244:362–364.
Kwong AD, Kim JL, Lin C. Structure and function of hepatitis C virusNS3 helicase. 2000. Curr Top Microbiol Immunol 242:171–196.
Kwun HJ, Jung EY, Ahn JY, Lee MN, Jang KL. 2001. p53-dependenttranscriptional repression of p21(waf1) by hepatitis C virus NS3.J Gen Virol 82:2235–2241.
Lan KH, Sheu ML, Hwang SJ, Yen SH, Chen SY, Wu JC, Wang YJ,Kato N, Omata M, Chang FY, Lee SD. 2002. HCV NS5A interactswith p53 and inhibits p53-mediated apoptosis. Oncogene 21:4801–4811.
Lee MN, Jung EY, Kwun HJ, Jun HK, Yu DY, Choi YH, Jang KL. 2002.Hepatitis C virus core protein represses the p21 promoter throughinhibition of a TGF-beta pathway. J Gen Virol 83:2145–2151.
Lusky M, Christ M, Rittner K, Dieterle A, Dreyer D, Mourot B, SchultzH, Stoeckel F, Pavirani A, Mehtali M. 1998. In vitro and in vivobiology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J Virol 72:2022–2032.
Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. 2001. Hepatitis Cvirus NS5A physically associates with p53 and regulates p21/waf1
gene expression in a p53-dependent manner. J Virol 75:1401–1407.
Majumder M, Ghosh AK, Steele R, Zhou XY, Phillips NJ, Ray R, RayRB. 2002. Hepatitis C virus NS5A protein impairs TNF-mediatedhepatic apoptosis, but not by an anti-FAS antibody, in transgenicmice. Virology 294:94–105.
McLauchlan J. 2000. Properties of the hepatitis C virus core protein:A structural protein that modulates cellular processes. J ViralHepat 7:2–14.
Mizushima H, Hijikata M, Asabe S, Hirota M, Kimura K, ShimotohnoK. 1994. Two hepatitis C virus glycoprotein E2 products withdifferent C termini. J Virol 68:6215–6222.
Navas MC, Fuchs A, Schvoerer E, Bohbot A, Aubertin AM, Stoll-KellerF. 2002. Dendritic cell susceptibility to hepatitis C virus genotype 1infection. J Med Virol 67:152–161.
Poluha W, Poluha DK, Chang B, Crosbie NE, Schonhoff CM, KilpatrickDL, Ross AH. 1996. The cyclin-dependent kinase inhibitor p21(WAF1) is required for survival of differentiating neuroblastomacells. Mol Cell Biol 16:1335–1341.
Qadri I, Iwahashi M, Simon F. 2002. Hepatitis C virus NS5A proteinbinds TBP and p53, inhibiting their DNA binding and p53interactions with TBP and ERCC3. Biochim Biophys Acta 1592:193–204.
Ray RB, Steele R, Meyer K, Ray R. 1997. Transcriptional repression ofp53 promoter by hepatitis C virus core protein. J Biol Chem 272:10983–10986.
Ray RB, Steele R, Meyer K, Ray R. 1998. Hepatitis C virus core proteinrepresses p21WAF1/Cip1/Sid1 promoter activity. Gene 208:331–336.
Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonalantibodies which recognize native and denatured forms of theadenovirus DNA-binding protein. Virology 128:480–484.
Reyes GR. 2002. The nonstructural NS5A protein of hepatitis C virus:An expanding, multifunctional role in enhancing hepatitis C viruspathogenesis. J Biomed Sci 9:187–197.
Ruggieri A, Harada T, Matsuura Y, Miyamura T. 1997. Sensitization toFas-mediated apoptosis by hepatitis C virus core protein. Virology229:68–76.
Sallusto F, Lanzavecchia A. 1994. Efficient presentation of solubleantigen by cultured human dendritic cells is maintained bygranulocyte/macrophage colony-stimulating factor plus interleu-kin 4 and downregulated by tumor necrosis factor alpha. J Exp Med179:1109–1118.
Sarobe P, Lasarte JJ, Casares N, Lopez-Diaz de Cerio A, Baixeras E,Labarga P, Garcia N, Borras-Cuesta F, Prieto J. 2002. Abnormalpriming of CD4 (þ) T cells by dendritic cells expressing hepatitis Cvirus core and E1 proteins. J Virol 76:5062–5070.
Sarobe P, Lasarte JJ, Zabaleta A, Arribillaga L, Arina A, Melero I,Borras-Cuesta F, Prieto J. 2003. Hepatitis C virus structuralproteins impair dendritic cell maturation and inhibit in vivoinduction of cellular immune responses. J Virol 77:10862–10871.
Serrano M, Hannon GJ, Beach D. 1993. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature366:704–707.
Shao ZM, Dawson MI, Li XS, Rishi AK, Sheikh MS, Han QX, OrdonezJV, Shroot B, Fontana JA. 1995. p53 independent G0/G1 arrest andapoptosis induced by a novel retinoid in human breast cancer cells.Oncogene 11:493–504.
Sheikh MS, Garcia M, Zhan Q, Liu Y, Fornace AJ, Jr. 1996. Cell cycle-independent regulation of p21Waf1/Cip1 and retinoblastomaprotein during okadaic acid-induced apoptosis is coupled withinduction of Bax protein in human breast carcinoma cells. CellGrowth Differ 7:1599–1607.
Sherr CJ. 1993. Mammalian G1 cyclins. Cell 73:1059–1065.
Siavoshian S, Abraham JD, Kieny MP, Schuster C. 2004. HCV core,NS3, NS5A, and NS5B proteins modulate cell proliferationindependently from p53 expression in hepatocarcinoma cell lines.Arch Virol1 49:323–336.
Somasundaram K. 2000. Tumor suppressor p53: Regulation andfunction. Front Biosci 5:424–437.
Tanji Y, Hijikata M, Satoh S, Kaneko T, Shimotohno K. 1995. HepatitisC virus-encoded nonstructural protein NS4A has versatile func-tions in viral protein processing. J Virol 69:1575–1581.
Thoren F, Romero A, Lindh M, Dahlgren C, Hellstrand K. 2004. Ahepatitis C virus-encoded, nonstructural protein (NS3) triggersdysfunction and apoptosis in lymphocytes: Role of NADPH oxidase-derived oxygen radicals. J Leukoc Biol 76:1180–1186.
410 Siavoshian et al.

Tomei L, Failla C, Santolini E, De Francesco R, La Monica N. 1993. NS3is a serine protease required for processing of hepatitis C viruspolyprotein. J Virol 67:4017–4026.
Wang J, Walsh K. 1996. Resistance to apoptosis conferred by Cdkinhibitors during myocyte differentiation. Science 273:359–361.
Watson RW. 1999. The rising incidence of hepatocellular carcinoma. NEngl J Med 341:451–452.
Wiman KG. 1997. p53: Emergency brake and target for cancer therapy.Exp Cell Res 237:14–18.
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. 1993.p21 is a universal inhibitor of cyclin kinases. Nature 366:701–704.
Yoshida I, Oka K, Hidajat R, Nagano-Fujii M, Ishido S, Hotta H. 2001.Inhibition of p21/Waf1/Cip1/Sdi1 expression by hepatitis C viruscore protein. Microbiol Immunol 45:689–697.
Zhu N, Khoshnan A, Schneider R, Matsumoto M, Dennert G, Ware C,Lai MM. 1998. Hepatitis C virus core protein binds to thecytoplasmic domain of tumor necrosis factor (TNF) receptor 1 andenhances TNF-induced apoptosis. J Virol 72:3691–3697.
Mature Dendritic Cells Apoptosis by HCV Proteins 411

Journal of Medical Virology 75:412–420 (2005)
TRUGENE Sequencing Versus INNO-LiPA forSub-Genotyping of HCV Genotype-4
Abdel Rahman N. Zekri,1* Hanaa M. Alam El-Din,1 Abeer A. Bahnassy,2 Amal M.R. El-Shehabi,3
Heba El-Leethy,1 Ashraf Omar,4 and Hussein M. Khaled5
1Virology and Immunology Unit, Cancer Biology Department, National Cancer Institute, Cairo, Egypt2Tissue Culture Unit, Pathology Department, National Cancer Institute, Cairo, Egypt3Biochemictry Department, Kasr El-Aini School of Medicine, Cairo, Egypt4Tropical Medicine Department, Kasr El-Aini School of Medicine, Cairo, Egypt5Medical Oncology Department, National Cancer Institute, Cairo University, Cairo, Egypt
Hepatitis C virus genotypes and subtypes deter-mination is an important factor forunderstandingtheepidemiologyof thevirus, in thepre-treatmentevaluation of the patients and in defining bettertreatment strategies. In the present study, wecompared two commercially available assaysfor HCV genotyping: the reverse hybridizationbased Innogenetics INNO-LiPA HCV II and thedirect sequencing by TRUGENE assay. The studyincluded 31 HCV-RNA positive Egyptian patients;18 patients with chronic active hepatitis, 8 withHCC, and 5 with cirrhosis. Using the TRUGENEgenotyping test, all the samples had genotype4 (100%) and subtyped as 4a in 18/31(58%), 4c in10/31 (32%), 4e in 1/31 (3%), 4a/c in 1/31 (3%), and4g in 1/31 (3%). Using the INNO-LiPA assay,30 samples had genotype 4 (97%), and 1 samplehad genotype 1e (3%). One sample showedmixed infection with type 4f and type 1. Only sixsamples were subtypable by INNO-LiPA, threewere genotype 4c/d, and the other three were 4f,4e, and 1e. Seven samples gave reactivity inthe INNO-LiPA of lines 5, 6, 16, 17, 18, which areconsidered untypable by the interpretation chartbut considered to be a rare HCV genotype 4 bythe manufacturer. At the genotype level, therewas a 97% concordance between TRUGENEsequencing and INNO-LiPA, but at the subtypelevel the concordance rate was 3% only. Weconclude that the TRUGENE genotyping assay isa reliable test for HCV genotyping for the detec-tion ofmajor types and subtypes detection, whileINNO-LiPA is agood test at the genotype level butunreliable for subtyping especially in the Egyp-tian population. This is mainly due to the highdiversity of genotype 4, which is the most pre-valent genotype in Egypt. J. Med. Virol. 75:412–420, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: hepatitis C virus; sequence;Egypt; genotype 4
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped positivesingle-stranded RNA virus that causes chronic hepati-tis and end-stage liver disease worldwide. It is also amajor cause of hepatocellular carcinoma [Sherlock andDooley, 2002]. There is a high incidence of anti-HCVseropositivity in the Egyptian population, with anoverall age-adjusted prevalence of HCV antibodies of21.9% [Frank et al., 2000]. HCV demonstrates a highdegree of sequence variability resulting in the formationof quasispecies. However, the levels of heterogenecitydiffer considerably among the various regions of thevirus, ranging from19% in the 50 untranslated region (50
UTR) to 50% ormore in the E1 region [Chan et al., 1992;Honda et al., 1994; Bukh et al., 1995; Simmonds, 1995].Based on the analysis of variability in the viral genome,many genotypes and subtypes are defined. Themethodsdeveloped to investigate HCV genotypes are based onmolecular biology or serological techniques. The firstincludesdirectnucleicacid sequencing [Simmondsetal.,1993], reverse hybridization line probe assay (LiPA)[Stuyver et al., 1996], subtype-specific reverse tran-scription [Okamoto et al., 1992], DNA restrictionfragment length polymorphism [Nakao et al., 1991],whereas serotyping identifies type specific antibodies
*Correspondence to: Abdel Rahman N. Zekri, M.Sc., Ph.D.,Virology and Immunology Unit, Cancer Biology Department,National Cancer Institute, Fom El-Khalig, Cairo 11796, Egypt.E-mail: [email protected]
Accepted 12 November 2004
DOI 10.1002/jmv.20293
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

against HCV genotype dependant pitopes [Dixit et al.,1995].
The prevalent genotype in Egypt is type 4, with thepresence of other genotypes. Genotype 4 was the com-monest (73%) followed by genotype 1 (26%) whereasmixed infection was found in 15.7% of the studiedsamples [Zekri et al., 2000].
Many studies have used the sequence analysis derivedfrom the 50 UTRofHCV to determine the genotype of thevirus [Chen and Weck, 2002; Anderson et al., 2003;Haushofer et al., 2003]. The two most commonly usedassays are the TRUGENE HCV 50 non-coding (50 NC)genotyping sequencing kit, which is a standard directsequencing kit for genotyping determination, and thereverse hybridization Line Probe Assay (INNO-LiPAHCV II; INNOGENETICS, N.V; Zwijnaarde, Belgium).
To date, there are only few comparison studies ofthese two tests, and most of them are reported fromEurope [Ross et al., 2000; Ansaldi et al., 2001; Halfonet al., 2001;Nolte et al., 2001] andNorthAmerica [Zhenget al., 2003] but none from Egypt where there is a highprevalence of HCV-4.
Therefore, the present study was conducted tocompare HCV genotyping by TRUGENE HCV directsequencing analysis of the 50 NC region and the reversehybridization by INNO-LiPA analysis of the sameregion for samples obtained from different parts of theEgyptian community. The concordance, accuracies, andsensitivities of both assays were assessed in this clinicalevaluation.
PATIENTS AND METHODS
The study included 31 subjects with anti-HCVpositive and high levels of SGPT & SGOT for more than6 months. All cases were subjected to complete historytaking and thorough clinical examination. Liver func-tion tests and liver biopsy were performed. Sampleswere tested for anti-HCVusinga commercially availableEIA.
Detection of HCV
1—RNA extraction. HCVRNAwas extracted fromthe sera by the silica method as previously described[Boom et al., 1990].
2—RT-PCR of HCV. RT and PCR were performedwith a primer pair selected from the highly conserved5-UTR of HCV genome [Choo et al., 1989]. All stepswere done as previously described [Zekri et al., 1995].The following sequenceswere used as antisense primersfor c-DNA synthesis HCV-6 [5-ACC-TCC nucleotides(NT) 319–324]. The internal primers were RB6A andRB6B for amplification of 266 bp of the 5-UTR, RB6A [5-GTG AGG AAC TAC TGT CTT CAC G-3 (NT 47–68)],and RB6B [5-ACT CGC AAG CAC CCT ATC AGG (NT292–312)-3] [Zekri et al., 1995].
All samples were analyzed twice for HCV RNA by theRT-PCR on different days with identical results. Uponcompletion of the amplification reaction, 10 ml of eachPCR reaction product was analyzed by electrophoresis
through a 1.2%agarose gel in Tris-Acetate-EDTAbuffer(pH 8.0) and ethidium bromide staining. DNA wastransferred from the gel onto nitrocellulose filter usingalkaline buffer (4N NaOH). The transferred DNA wascross linkedby incubation for 2–3hr at 808Cand theblotwas then hybridized with an internal probe [Zekri et al.,1995].
HCV Genotyping
All clinical samples were genotyped with both theLiPA and TRUGENE50 NCkits from aliquots of a singleRT-PCR sequence analysis.
1—INNO-LiPA II, genotyping. The line probeassay was used to assess HCV genotypes using kitsprovided by INNOGENETICS, N.V. The 5-UTR regionwas amplified using nested PCR with biotinylatedprimers. The labeled amplicon was allowed to hybridizeand mounted on a strip. After stringent washing, strep-tavidin labeled with alkaline phosphatase was used totrace thehybridizedproducts, andnitroblue tetrazoliumand 5-bromo-4-chloro-3-indoyl-phosphate were usedas a substrate according to the manufacturer’s instruc-tions. The probe reactivity patterns were interpretedusing the chart provided by the manufacturers [Zekriet al., 2000].
2—HCV genotyping by sequencing. The TRU-GENEHCV 50 NC genotyping kit, Visible Genetics, Inc.(Toronto, Ontario, Canada) was used in conjunctionwith the OpenGene DNA sequencing system. All HCVPCR products were run on a 1% agarose gel electro-phoresis. A positive control of known standard sequenceand a negative HCV control provided with the kit wereutilized as controls in each run. The specific band wascut from the gel and purified by silica protocol [Boomet al., 1990], and sequenced by CLIP sequencing whichallows both directions of the target amplicon to besequenced simultaneously in the same tube using twodifferent dye-labeled primers (Cy5.0 andCy5.5) for eachreaction.
This method provides sequence information forboth positive and negative DNA strands from a singlereaction. The forward and reverse sequences are com-bined to form a query sequence. The query sequence isthen compared to previously characterized isolates inthe TRUGENE HCV 50 NC Module of the OpenGenesoftware system in order to determine the HCV geno-type of the sample. Gene Objects software analyzeschromatograms from each sample; the final 50 UTRsequence was obtained from the comparison of bothsequenced strands. This information is compared withdeposited HCV sequences by Gene LibrarianTM soft-ware with a minimal concordance of 98%. The genotypeassignments of these sampleswere confirmedbyBLASTsearches.
Nucleotide sequence accession numbers. Thenucleotide sequences reported here are deposited inThe National Center for Biotechnology Information/National Institute of Health GenBank nucleotide se-quence database (accession no. AY624965–AY624986).
TRUGENE Sequencing Versus INNO-LiPA 413
RESULTS
All of the 31 HCV-RNA positive samples tested bythe TrueGene sequencing were genotype 4 (100%).They were subtyped as 4a (65%), 4c (29%), 4a/c (3%),and 4g (3%) (Table I).
Figure 1 shows 50 NC region sequence analysis andFig. 2 showed the phylogenetic analysis of all studiedsamples in conjunction with three known GenBanknucleotide sequences with Blast database. Comparisonbetween the sequences obtained in the present studyand all previously reported 50 UTR sequences revealed anew 50 UTR mutation within HCV genotype-4 andconfirm the heterogeneity of this type in the Egyptianpopulation. On the other hand, most of these mutationswere randomly distributed, however; they still main-tained the postulated secondary structure of the 50 UTR
TABLE I. Comparison of HCV Genotyping ResultObtained With the LiPA and TRUGENE 50 NC Kit for
31 Clinical Specimens
LiPA type
TRUGENE sequencing
4a 4c 4e 4g 4a/c Total
4 12 4 1 1 184c/d 2 1 34e 1 11e 1 14fþ 1 1 1NTa 2 5 7Total 18 10 1 1 1 31
aNT, not typeable by LiPA.
Fig. 1. The alignment of all cases typed by both INNO-LiPA and TRGUENE sequence analysis withmultiple sequence alignment of HCV-genotype-4 from the gene data bank (X78866, X78865, AF142056)using CLUSTALW analysis.
414 Zekri et al.

genomic RNA and revealed the strong conservation ofthe genotypes.
By the INNO-LiPA, the samples were found to begenotype 4 in 23 (74%) samples and genotype 1 in 1 (3%)sample. Seven samples (23%) gave reactivity of lines 5,6, 16, 17, 18 which are considered untypable by theinterpretation chart. However, by contacting the man-ufacturer, they considered them to be rare genotype 4patterns, which will most probably be added to thefuture update of the interpretation chart. The geno-type assignments of these samples were confirmed bythe blast search. The concordance between TrueGenesequencing and INNO-LiPA was 74% (23/31 samples)at the main genotype level (Fig. 1A). By adding theseven untypable samples, which were considered by themanufacturer as type 4, the concordance rises to (97%).The cladograms of these sequences with known type 4sequences from the GenBank are shown in Figure 1B.
Subtypingwas possible in six cases only by the INNO-LiPA assay, of which three (9.7%) were 4c/d, one (3%) 4f,
one (3%) 4e, and one (3%) 1e (Table I). Figure 1C showsthe cladogram of five of these caseswith type 4. Only onecase showed mixed infection by INNO-LiPA, however itshowed genotype-4a by TrueGene.
One sample only (AY624963) showed discordancebetween the two tests at the genotype level. This samplewas classified as genotype 1e by INNO-LiPA and asgenotype 4a by TRUGENE. The cladogram of thissequencealongwith type1 sequences fromtheGenBankis shown in Figure 1D.
The demographic, clinical, and genotype features ofthe patients in relation to liver biopsy data are shownin Table II. The incidence of ascites, jaundice, andsplenomegaly was significantly higher in HCC thanin CAH patients (P¼ 0.000, 0.02, 0.03, respectively).HCC patients were mainly from Metropolitan CairoArea (P¼0.03), whereas those with CAH were mainlyfrom Delta area (P¼<0.001).
Table II also shows the comparison between genotype4a and 4c in relation to different study parameters.
Fig. 1. (Continued )
TRUGENE Sequencing Versus INNO-LiPA 415
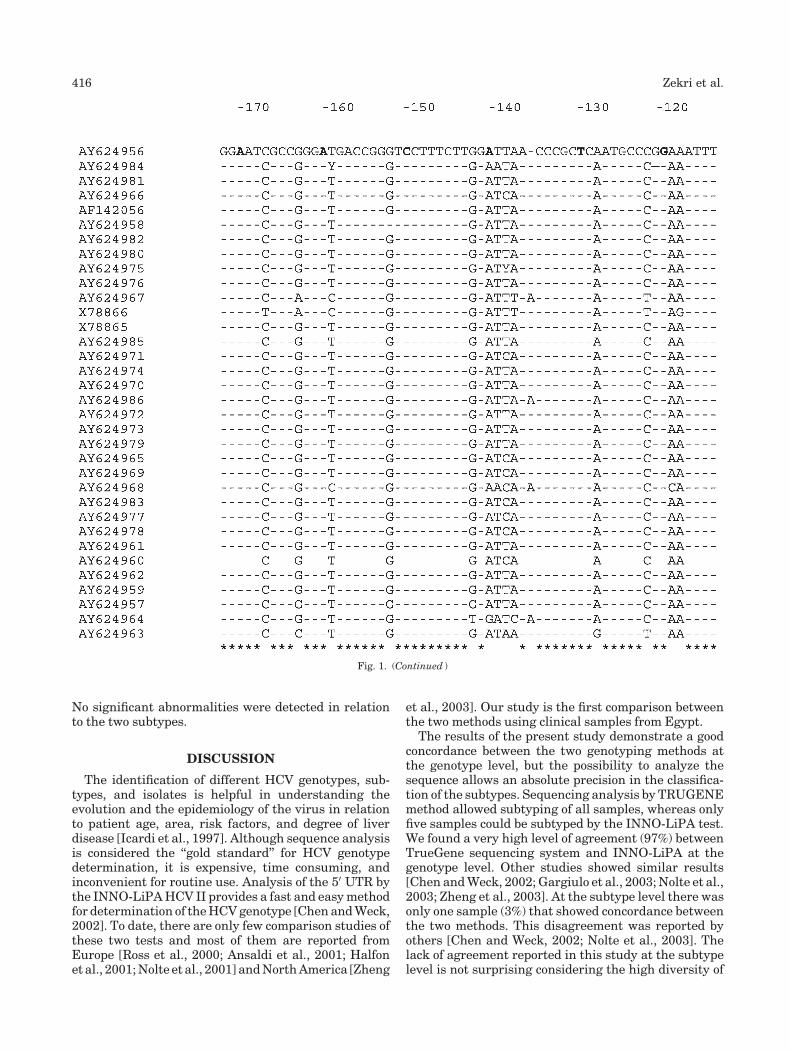
No significant abnormalities were detected in relationto the two subtypes.
DISCUSSION
The identification of different HCV genotypes, sub-types, and isolates is helpful in understanding theevolution and the epidemiology of the virus in relationto patient age, area, risk factors, and degree of liverdisease [Icardi et al., 1997]. Although sequence analysisis considered the ‘‘gold standard’’ for HCV genotypedetermination, it is expensive, time consuming, andinconvenient for routine use. Analysis of the 50 UTR bythe INNO-LiPAHCV II provides a fast and easymethodfor determination of theHCVgenotype [ChenandWeck,2002]. To date, there are only few comparison studies ofthese two tests and most of them are reported fromEurope [Ross et al., 2000; Ansaldi et al., 2001; Halfonet al., 2001;Nolte et al., 2001] andNorthAmerica [Zheng
et al., 2003]. Our study is the first comparison betweenthe two methods using clinical samples from Egypt.
The results of the present study demonstrate a goodconcordance between the two genotyping methods atthe genotype level, but the possibility to analyze thesequence allows an absolute precision in the classifica-tion of the subtypes. Sequencing analysis by TRUGENEmethod allowed subtyping of all samples, whereas onlyfive samples could be subtyped by the INNO-LiPA test.We found a very high level of agreement (97%) betweenTrueGene sequencing system and INNO-LiPA at thegenotype level. Other studies showed similar results[Chen andWeck, 2002;Gargiulo et al., 2003;Nolte et al.,2003; Zheng et al., 2003]. At the subtype level there wasonly one sample (3%) that showed concordance betweenthe two methods. This disagreement was reported byothers [Chen and Weck, 2002; Nolte et al., 2003]. Thelack of agreement reported in this study at the subtypelevel is not surprising considering the high diversity of
Fig. 1. (Continued )
416 Zekri et al.

genotype 4, especially in the Egyptian population aspreviously shown by others [Zekri et al., 2001] compar-ing sero- and geno-typing methods, The detection of alarge number of individual mutated strains by hybridi-zation assay such as INNO-LiPA in a single test isdifficult since it requires very high stringent conditions[Ansaldi et al., 2001]. Also the lack of agreementbetween the subtypes is not unusual considering thatbothmethods target the50NCregion.The50NCregion isamong the most conserved regions of the viral genome,and in several cases, only one or two nucleotide changesdistinguish unique subtypes [Smith et al., 1995]. It hasbeen suggested that clinical laboratories should not callHCV subtypes from analysis of the 50 NC regardless ofthe method employed because of the inherent inaccu-racy of the calls [Nolte et al., 2003]. Also they stated thatthere are no recognized subtype-specific differences indisease progression or response to therapy that wouldwarrant these designations.
In our study, subtypes 4a and 4c were the mostprevalent; this was similar to that previously reported
[Zekri et al., 2000]. We could not correlate between thedifferent subtypes and clinical picture, gender, resi-dence, or liver biopsy results due to the low number ofcases studied. In Egypt, the response of genotype 4to interferon treatment was reported in many studies[El-Zayadi et al., 1999; Esmat et al., 2002; Thakeb et al.,2003], however there are no available data of theresponse of subtypes 4a and 4c to interferon treatment.
Regarding the patient with cirrhosis that showednon-concordant results, it was classified as type 1e byINNO-LiPA and genotype 4a by the Visible Geneticssequencing system. A similar result was previouslyobtained [Ansaldi et al., 2001]. The mistyping could beascribed to the close phylogenetical similarity betweengenotype 1 and 4, since the difference in the regionanalyzed is only six nucleotides [Ansaldi et al., 2001].
From this study, we concluded that TRUGENEsequencing for HCV genotyping and INNO-LiPA pro-vide reliable results at the type level and have almostsimilar analytical sensitivities regarding genotype 4.TRUGENE method cannot, however, detect mixed
Fig. 1. (Continued )
TRUGENE Sequencing Versus INNO-LiPA 417

Fig. 2. The first cladogram shows all cases (A); the second shows the seven INNO-LiPA untypable (B);the third shows the five genotype 4 subtypable by INNO-LiPA (C); and the fourth (D) shows the case withgenotype 1 with four other HCV genotype 1 from the gene data bank (Z36518–Z36520).
418 Zekri et al.
infection as the INNO-LiPA method. Although, theTRUGENE 50 NC method is more technically complexto perform than LiPA, it is more detailed informativemethod in the level of the subtype especially inEgyptianpopulations with a very high heterogeneity of thesutype-4. It is also the best method for the epidemiolo-gical investigations. In addition, TRUGENE 50 NCsystem can be easily updated, as new HCV sequenceinformation becomes available of the HCV genome. Astreatment of HCV is dependent on main genotype andnot subtype, INNO-LiPA test can be used for geno-typing of HCV until more studies can detect significantdifference in response to interferon in different subtypesin genotype 4, the most prevalent in Egypt. Futureanalysis is needed in order to correlate subtyping withTRUGENE with response to interferon treatment inEgyptian patients with type 4, since all previouslypublished data in this field was done by INNO-LiPAsequencing method.
REFERENCES
Anderson J, Simonetti J, Fisher D, Williams J, Yamamura Y,Rodriguez N, Sullivan DG, Gretch DR, McMahon B, Williams KJ.2003. Comparison of different HCV viral load and genotypingassays. J Clin Virol 28:27–37.
Ansaldi F, Torre F, Bruzzone B, Piciotto A, Crovari P, Icardi G. 2001.Evaluation of new hepatitis C virus sequencing assay as a routinemethod for genotyping. J Med Virol 63:17–21.
Boom R, Sol CJA, Salimans MMM, Jausen CL, Wertheim-Van DillenPME, Van der Noordaa J. 1990. Rapid and simple method forpurification of nucleic acids. J Clin Microbiol 28:495–503.
Bukh J,Miller R, Purcell R. 1995. Genetic heterogenicity of hepatitis Cvirus: Quasispecies and genotypes. Semin Liver Dis 15:41–63.
Chan SW, McOmish F, Holmes EC, Dow B, Peutherer JF, Follett E,Yap PL, Simmonds P. 1992. Analysis of a newhepatitis C virus typeand its phylogenetic relationship to existing variants. J Gen Virol73:1131–1141.
Chen Z, Weck K. 2002. Hepatitis C virus genotyping: Interrogation ofthe 50 untranslated region cannot accurately distinguish genotypes1a and 1b. J Clin Microbiol 40:3127–3134.
Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M.1989. Isolation of cDNA clone derived from a blood-borne non-A,non-B viral hepatitis genome. Science 244:359–362.
Dixit V, Quan S, Martin P, Larson D, Brezina M, DiNello R, Sra K,Lau JY, Chien D, Kolberg J, et al. 1995. Evaluation of a novelserotyping system for hepatitis C virus: Strong correlation withstandard genotyping methodologies. J Clin Microbiol 33:2978–2983.
El-Zayadi A, Selim O, Haddad S, Simmonds P, Hamdy H, Badran HM,Shawky S. 1999. Combination treatment of interferon alpha-2band ribavirin in comparison to interferon monotherapy in treat-ment of chronic hepatitisCgenotype 4patients. Ital JGastroenterolHepatol 31:472–475.
Esmat G, Abuzied A, Abdel-Aziz F, et al. 2002. Treatment with peg-interferon alfa 2b (peg-IFN) plus ribavirin compared to interferonalfa 2b (IFN alfa-2b) plus ribavirin on subjects with chronichepatitis C infected with HCV genotype 4. Hepatology , AASLDAbstr 364A.
Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR,Magder LS, El Khoby T, Abdel-Wahab Y, Aly Ohn ES, Anwar W,Sallam I. 2000. The role of parenteral anti-schistosomaltherapy in the spread of hepatitic C virus in Egypt. Lancet 355:887–891.
Gargiulo F, De FrancescoM, Pinsi G, Pallara C, Terlenghi L, PerandinF, Manca N. 2003. Determination of HCV genotype by directsequence analysis of quantitative PCR products. J Med Virol69:202–206.
HalfonP, Trimoulet P,BourliereM,KhiriH, deLedinghenV,CouzigouP, Feryn JM, Alcaraz P, Renou C, Fleuy HJA, Ouzan D. 2001.Hepatitis C virus genotyping based on 50 noncoding sequenceanalysis (Trugene). J Clin Microbil 39:1771–1773.
Haushofer A, Berg J, Hauer R, Trubert-Exinger D, Stekel HG, KesslerHH. 2003. Genotyping of hepatitis C virus. Comparison of threeassays. J Clin Virol 27:276–285.
Honda M, Kaneko S, Sakai A, Unoura M, Murakami S, Kobayashi K.1994. Degree of diversity of hepatitis C virus quasispecies andprogression of liver disease. Hepatology 20:1144–1150.
Icardi G, Bonanni P, Wicks R, Faraldi L, Roccatagliata A, Santo M,Orione L, De Conca V, Crovari P. 1997. Characterization ofhepatitis C virus genotypes in Italian subjects at different riskinfection. In: Rizzetto M, Purcell H, Gerin JL, Verme G, editors.Viral hepatitis and liver disease. Torino:EdizioneMinerva. pp598–600.
Nakao T, Enomoto N, Takada N, Takada A, Date T. 1991. Typing ofhepatitis C virus genomes by restriction fragment length poly-morphism. J Gen Virol 72:2105–2112.
Nolte RS, Green A, Fiebelkon K, Caliendo A. 2001. Clinical evaluationof two methods for genotyping hepatitis C virus based on50noncoding (NC) region sequence analysis. J Mol Diag 3:204.
Nolte FS, Green AM, Fiebelkorn KR, Caliendo AM, Sturchio C,Grunwald A, Healy M. 2003. Clinical evaluation of two methodsfor genotyping hepatitis C virus based on analysis of the 50 non-coding region. J Clin Microbiol 41:1558–1564.
TABLE II. Demographic, Clinical And Genotype Features of the Study Groups
Parameter
Diagnosis
P-value
Genotype
P-valueCAH
(n¼ 18)Cirrhosis(n¼ 5) HCC (n¼ 8) 4a (No. %) 4c (No. %)
SexM/F 18/0 4/1 5/3 0.01 18/2 8/1 0.928Age (mean�SD) 47� 10 56� 10 53� 12 0.08 51.5� 11.7 46.3� 8.7 0.22
Liver stateHepatomegaly 14 (78%) 3 (60%) 8 (100%) 0.12 17 (71%) 7 (29%) 0.634Ascites 1 (5.5%) 2 (40%) 8 (100%) 0.000 8 (72.7%) 3 (27.3%) 0.732Jaundice 2 (11%) 1 (20%) 5 (62.5%) 0.02 5 (62.5%) 3 (37.5%) 0.642L.L. oedema 1 (5.5%) 2 (40%) 3 (37.5%) 0.12 3 (75%) 1 (25%) 0.779Splenomegaly 8 (44%) 5 (100%) 8 (100%) 0.03 15 (79%) 4 (21%) 0.109
ResidenceMCA 3 (16.7%) 1 (20%) 6 (75%) 6 (60%) 4 (40%)Delta 14 (78%) 2 (40%) 2 (25%) 12 (71%) 5 (29%)Others 1 (5.5%) 2 (40%) 0 (0%) 2 (100%) 0 (0%)P-value <0.001 0.95 0.03 0.523
CAH, chronic active hepatitis; HCC, hepatocellular carcinoma; L.L. oedema, lower limb oedema; MCA, Metropolitan Cairo Area.P-value <0.05 is significant.
TRUGENE Sequencing Versus INNO-LiPA 419

Okamoto H, Sugiyama Y, Okada S, Kurai K, Akahane Y, Sugai Y,Tanaka T, Sato K, Tsuda F, Miyakawa Y, et al. 1992. Typinghepatitis C virus by polymerase chain reaction with type-specificprimers: Application to clinical surveys and tracing infectioussources. J Gen Virol 73:673–679.
Ross RS, Viazov SO, Holtzer CD, Beyou A, Monnet A, Mazure C,Roggendorf M. 2000. Genotyping of hepatitis C virus isolates usingCLIP sequencing. J Clin Microbiol 38:3581–3814.
Sherlock S, Dooley J. 2002. Diseases of liver and biliary system, 11thedn. Oxford UK: Blackwell scientific publication. Chapter (18).pp 305–320.
SimmondsP. 1995.Variability of hepatitisCvirus.Hepatology21:570–583.
Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B,Beall E, Yap PL, Kolberg J, Urdea MS. 1993. Classification ofhepatitis C virus into six major genotypes and a series of subtypesby phylogenetic analysis of the NS-5 region. J Gen Virol 74:2391–2399.
Smith DB, Mellor J, Jarvis LM, Davidson F, Kolberg J, Urdea M,Yap PL, Simmonds P. 1995. Variation of the hepatitis C virus 50
noncoding region: Implications for secondary structure, virusdetection, and typing. J Gen Virol 76:1749–1761.
Stuyver L,Wyseur A, vanArnhemW,Hernandez F,Maertens G. 1996.Second-generation line probe assay for hepatitis C virus geno-typing. J Clin Microbiol 34:2259–2266.
Thakeb F, Omar M, El-Awady M, et al. 2003. Randomized controlledtrial of peg-interferon alfa-2a plus ribavirin for chronic hepatitisC virus genotype 4 among Egyptian patients. Hepatology38(Suppl 1):252 (Abstract).
Zekri A, Bahnassay A, Khaled H, Mnsour O, Attia MA. 1995.Comparative analysis of different PCR techniques for detectionof HCV in hepatocellular carcinoma patients. Cancer J 8:331–335.
Zekri AR, Bahnassy AA, Shaarawy SM, Mansour OA, Maduar MA,Khaled HM, El-Ahmadi O. 2000. Hepatitis C virus genotyping inrelation to neu-oncoprotein overexpression and the development ofHepatocellular carcinoma. J Med Microbiol 49:89–95.
Zekri A, Bahnassy A, Ramadan A, El-Bassuoni A, Badran A, MadwarMA. 2001. Hepatitis C virus genotyping versus serotyping inEgyptian patients. Infection 29:24–26.
Zheng X, Pang M, Chan A, Roberto A, Warner D, Yen-Liberman B.2003. Direct comparison of hepatitis C virus genotypes tested byINNO-LiPA HCV II and TRUGENE HCV genotyping methods.J Clin Virol 28:214–216.
420 Zekri et al.

Journal of Medical Virology 75:421–426 (2005)
Growth of Herpes Simplex Virus in EpidermalKeratinocytes Determines CutaneousPathogenicity in Mice
Yoshihiro Yoshida,1 ZhiHong Li,1 Masahiko Kurokawa,1 Takashi Kawana,2
Masami Imakita,3 and Kimiyasu Shiraki1*1Department of Virology, Toyama Medical and Pharmaceutical University, Toyama, Japan2Department of Gynecology, Teikyo University, Tokyo, Japan3Department of Pathology, Rinku General Medical Center, Izumisano Municipal Hospital, Osaka, Japan
Herpes simplex viruses (HSV)-1 and -2 isolatedfrom genital lesions were examined for cutane-ouspathogenicity and its correlationwith cellulartropism. HSV-1 caused vesiculation, erosion/ulcer, and zosteriform lesions successively, butskin lesions of HSV-2 developed without vesicu-lation in some mice, and with statistically signi-ficantly less frequent vesiculation than HSV-1.Thus, the virological type of HSV was correlatedwith its cutaneous pathogenicity. The growthcharacteristics of HSV-1 and -2 were comparedin cultured human embryonic lung (HEL) fibro-blasts, human lung cancer A549 cells, humanneonatal epidermal keratinocytes, human neo-natal dermal fibroblasts, HeLa cells, and Verocells. HSV-2 produced plaques that were 72%times the size of HSV-1 plaques in epidermalkeratinocytes but 230%–500% the size in theother cells. The difference between HSV-1 and-2 in the ratio of plaque size to virus yieldin epidermal keratinocytes was much larger(502 times) than the ratio of the other cells(5.57–28.8 times). Keratinocytes are the majorconstituent of the epidermal layer of the skinand the cells in which vesiculation and erosion/ulceration occur histologically. Therefore, thesmaller spread of HSV-2 in keratinocytes of theepidermal layer and the greater spread in othercells of the dermal layer might reflect its lesserinvasiveness in theepidermal layer despite largerinvasiveness in the dermal layer, which is re-flected in the low incidenceof erosion/ulcer of theskin compared toHSV-1. Thus, the growthofHSVin epidermal keratinocytes appeared to correlatewith the cutaneous pathogenicity causing vesi-culation in the skin.J.Med.Virol. 75:421–426,2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: herpes simplex virus; herpessimplex virus infection; kerati-
nocytes; skin lesions; genitalherpes; tropism
INTRODUCTION
Herpes simplex virus (HSV) has been divided intotypes 1 and 2 according to immunological, biological,and biophysical properties such as plaque formation,temperature sensitivity, cell tropism, reactivation, andclinical manifestations [Anderson, 1940; Parker andBanatvala, 1967; Nahmias and Dowdle, 1968; Plummeret al., 1968, 1974]. HSV-1 and -2 produce small and largepocks or plaques, respectively [Parker and Banatvala,1967; Plummer et al., 1974], and HSV-2 encephalitiscauses more and severer sequelae than HSV-1 encepha-litis [Whitley et al., 1991; Kimura et al., 2002], but thereason for the difference in pathogenicity is not clear.Little research on the differences in their biologicalcharacters with respect to pathogenicity has beenreported.
While screening the antiviral activity of anti-HSVagents in mice infected with HSV-1 or -2, a differencewas noted in the cutaneous pathogenicity of the viruses.
Grant sponsor: Ministry of Education, Culture, Sports, Scienceand Technology of Japan (Grants-in-Aid); Grant numbers:135508094, 14657074.
ZhiHong Li’s present address is Beijing JunKey MedPharmConsulting Co. Ltd., Beijing, China.
Masahiko Kurokawa’s present address is Department ofBiochemistry, School of Pharmaceutical Science, Kyushu Uni-versity of Health and Welfare, Nobeoka 882-8508, Japan.
*Correspondence to: Kimiyasu Shiraki, MD, Professor andChairman, Department of Virology, Toyama Medical and Phar-maceutical University, 2630 Sugitani, Toyama 930-0194, Japan.E-mail: [email protected]
Accepted 3 November 2004
DOI 10.1002/jmv.20284
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

Development of skin lesions without vesiculation in anHSV-1 cutaneous infection model was quite unusual inour experience [Kurokawa et al., 1992, 1998, 1999, 2001;Nagasaka et al., 1995; Nakano et al., 1998]. In order tocharacterize the different cutaneous pathogenicities,the biological differences in the viruses were examined.Fresh clinical isolates were used to characterize biolo-gical differences such as cytopathology, plaque size,histological findings on chorioallantoic membrane, andneurotropism [Nahmias and Dowdle, 1968] because thepassage history of a strain affects these characteristics.Twenty-one fresh clinical isolates from genital lesionswere used without information on their virologicaltyping and adjusted their passage history to one pas-sage in Vero cells for isolation and one passage inhuman embryonic lung (HEL) cells for virus prepara-tion. When these were categorized into two groups bythe difference in vesiculation in the development ofskin lesions in mice, this grouping correlated signi-ficantly with the virological typing of HSV-1 and -2.Based on the consistency between virological typing andcutaneous pathogenicity, the growth characteristics ofthese strains were compared in various cells, includinghuman dermal fibroblasts and keratinocytes, and foundthat HSV-2 showed less efficient spreading of in-fection in keratinocytes and more efficient spread intothe other cells than HSV-1. Histologically, a vesicularlesion is interstitial edema in the epidermal layer com-posed of degenerated keratinocytes. Erosion/ulcerationis the loss of the epidermal layer of the skin or mucosa.Therefore, emphasis was placed focused on the viralgrowth of HSV-1 and -2 in epidermal keratinocytes,which constitute the epidermal layer of the skin andmucosa. The difference in the growth of HSV-1 and -2 inepidermal keratinocytes correlated with the differencein their cutaneous pathogenicity in the epidermal anddermal layers of the skin.
MATERIALS AND METHODS
Viruses and Cells
HSV-1 7401H, as a standard HSV-1 strain, and 21clinical HSV isolates from genital lesions were propa-gated in HEL cells [Kawana et al., 1974; Hasegawa et al.,2001]. HEL cells were infected with HSV strains for 1 hrand incubated in Eagle’s minimum essential mediumsupplemented with 2% fetal bovine serum at 378C[Kurokawa et al., 1992; Nagasaka et al., 1995; Hase-gawa et al., 2001]. The infected cultures were frozen andthawed three times and centrifuged at 3,000 rpm for 15min. After determination of their virus titers in lungcancer A549 cells, each supernatant was stored as virusstock at �858C until use.
Cells used were HEL cells, A549 cells, primary humanneonatal epidermal keratinocytes, human neonataldermal fibroblasts, HeLa cells, and Vero cells. Primaryhuman epidermal keratinocytes and dermal fibroblastswere purchased from Bio Whittaker (Walkersville, MD)and propagated according to the manufacturer’s ins-tructions [Shiraki et al., 2003].
Typing of HSV Isolates
The typing of isolated viruses was carried out by aMicrotrack test kit and PCR of the DNA polymerasegene region [Kawana et al., 1974; Kimura et al., 2002].
Growth Character and Plaque Formationof HSV Strains
Various cells were infected with the HSV-1 7401Hstrain and 21 clinical isolates, and the virus yields andplaque formation were compared. HSV strains wereinoculated into the monolayer of various cells in plasticflasks (25 cm2) at a concentration of 200 plaque-formingunits (PFU) per flask, and then the cultures were in-cubated and harvested at the indicated times. Afterthree cycles of freezing and thawing, followed by centri-fugation at 3,000 rpm for 15 min, their supernatantswere subjected to the plaque assay for the determinationof virus yields.
Various cell cultures in 60 mm Petri dishes wereinoculated with 30 PFU of each HSV strain and overlaidwith nutrient methylcellulose medium for 72 hr. Thecells were fixed with formalin and stained with methy-lene blue. Then the plaque size was measured with theNIH Image program (version 1.62, National Institute ofHealth, Bethesda, MD).
Cutaneous Infection
Female BALB/c mice (6 weeks old, 17–19 g, JapanSLC Icn., Hamamatsu, Japan) were housed five per cageand acclimated for at least 3–4 days before starting anexperimental procedure. The animal experimentationguidelines of the Toyama Medical and PharmaceuticalUniversity were followed in the animal studies. The hairof the right midflank of each mouse was clipped withhair clippers (National Electronics, Osaka, Japan), andthe skin was depilated with a chemical depilatory (HairRemover, Shiseido Co., Ltd., Tokyo, Japan). The hairlessskin of each mouse was scratched with a bundle of 27-gauge needles and infected by spreading it with a 10 mldrop containing 2�105 PFU [Simmon and Nash, 1984;Kurokawa et al., 1992, 2001; Nagasaka et al., 1995]. Thedevelopment of the skin lesions was observed threetimes daily, and the severity of the lesions was scored asfollows: 0, no lesion; 2, vesicles in the local region; 4,erosion/ulceration in the local region; 6, mild zosteriformlesion; 8, moderate zosteriform lesion; 10, severe zosteri-form lesion; 12, death.
Statistical Analyses
Fischer’s exact probability test was used to examinethe correlation between skin lesion groups and typinggroups. Student’s t-test was used to evaluate the signi-ficant differences between groups in the mean valuesof virus yields, plaque sizes, and their ratio. The re-peated measure analysis of variance (repeated measureANOVA) with Dunn’s procedure as a multiple compar-ison procedure was used to analyze the interaction
422 Yoshida et al.

TA
BL
EI.
Com
pari
son
Bet
wee
nV
irol
ogic
al
an
dV
esic
ula
rT
yp
ing,
an
dP
laqu
eS
izes
For
med
by
Vari
ous
HS
VS
train
s72
hr
Aft
erIn
fect
ion
Str
ain
Vir
olog
ical
typ
eV
esic
ula
rle
sion
s(t
yp
e)H
um
an
embry
onic
lun
g(H
EL
)A
549
Ver
oD
erm
al
fibro
bla
sts
Ep
ider
mal
ker
ati
noc
yte
s
7401H
110/1
0(1
)3.4
�1.2
0.1
�0.0
0.0
9�
0.0
3.3
�0.7
6.6
�1.8
TL
I-56
110/1
0(1
)1.5
�0.2
0.1
�0.0
0.1
0�
0.1
0.9
�0.2
8.1
�2.3
TL
K-9
41
10/1
0(1
)2.5
�0.6
0.1
�0.1
0.0
8�
0.0
1.1
�0.2
4.5
�0.9
TH
K-1
67
11
10/1
0(1
)2.3
�0.5
0.4
�0.0
0.0
4�
0.0
2.3
�0.6
11.2�
2.4
TV
K-1
71
19/1
0(1
)1.7
�0.5
0.2
�0.0
0.0
5�
0.0
2.0
�0.4
4.9
�1.2
TK
M-2
22
19/1
0(1
)2.7
�1.1
0.1
�0.0
0.2
6�
0.1
1.9
�0.8
5.1
�1.7
WT
N-2
69
12/1
0(2
)2.3
�0.8
0.1
�0.0
0.0
3�
0.0
1.0
�0.5
6.8
�2.3
HM
A-2
74
14/1
0(2
)2.6
�0.9
0.1
�0.0
0.0
4�
0.0
1.5
�0.5
5.5
�1.4
KG
A-2
78
110/1
0(1
)3.5
�0.7
0.1
�0.0
0.0
7�
0.0
2.6
�1.0
7.1
�1.3
YM
T-2
84
110/1
0(1
)1.7
�0.6
0.1
�0.0
0.0
7�
0.0
1.6
�0.5
7.2
�2.1
DIK
-303
11/1
0(2
)1.9
�0.6
0.1
�0.0
0.1
0�
0.1
1.5
�0.4
6.7
�1.9
TK
I-1672
18/1
0(2
)1.5
�0.4
0.2
�0.1
0.0
9�
0.0
2.3
�0.8
4.0
�0.6
Su
mm
ary
ofH
SV
-1st
rain
sT
yp
eco
nsi
sten
cyM
ean
of12
stra
ins
8st
rain
s/12
stra
ins
2.3
�0.6
0.1
�0.0
0.1
�0.0
1.8
�0.5
6.5
�1.7
TV
N-3
32
6/1
0(2
)6.8
�2.1
2.1
�0.3
0.4
5�
0.1
5.1
�1.4
3.7
�0.9
TV
O-6
62
7/1
0(2
)8.0
�1.4
2.5
�0.5
0.5
2�
0.3
2.2
�0.7
4.5
�0.9
TV
M-1
27
27/1
0(2
)6.5
�1.7
1.7
�0.6
0.4
1�
0.1
3.7
�1.1
3.5
�1.8
TV
Y-1
49
23/1
0(2
)8.5
�1.5
3.2
�0.5
0.6
3�
0.2
3.3
�1.0
5.3
�1.2
TV
I-159
210/1
0(1
)7.8
�1.6
2.1
�0.5
0.1
1�
0.0
5.2
�1.1
5.4
�1.5
TV
K-1
69
29/1
0(1
)7.1
�1.6
1.7
�0.4
0.4
9�
0.2
4.7
�1.3
6.3
�1.5
NK
G-2
53
28/1
0(2
)8.0
�1.2
2.5
�0.4
0.6
6�
0.2
4.8
�1.3
6.0
�1.9
KT
G-9
77
25/1
0(2
)8.5
�2.0
2.6
�0.4
0.6
1�
0.2
8.2
�1.7
5.0
�1.2
TK
H-1
170
28/1
0(2
)5.8
�2.3
1.9
�0.7
0.5
5�
0.1
3.5
�1.0
3.2
�0.7
SIT
-1396
25/1
0(2
)5.9
�2.2
2.5
�0.8
0.6
8�
0.2
6.4
�2.8
3.7
�1.5
Su
mm
ary
ofH
SV
-2T
yp
eco
nsi
sten
cyM
ean
of10
stra
ins
8st
rain
s/10
stra
ins
7.3
�1.8
2.3
�0.5
0.5
�0.0
4.7
�1.3
4.7
�1.3
Pla
qu
esi
zera
tio
(HS
V-2
/HS
V-1
)3.1
2.3
5.0
2.6
0.7
2H
SV
-1ver
sus
-2P<
0.0
1*
P<
0.0
01**
P<
0.0
01**
P<
0.0
01**
P<
0.0
01**
P¼
0.0
5**
Pla
qu
esi
zes
are
exp
ress
edas
the
mea
n�
SE
(mm
2).
*Com
pari
son
ofsk
inle
sion
sbet
wee
nH
SV
-1an
d-2
by
Fis
her
’sex
act
pro
babil
ity
test
.**
Com
pari
son
ofth
ep
laqu
esi
zebet
wee
nH
SV
-1an
d-2
by
Stu
den
t’st-
test
.
Cutaneous Pathogenicity of HSV-2 423
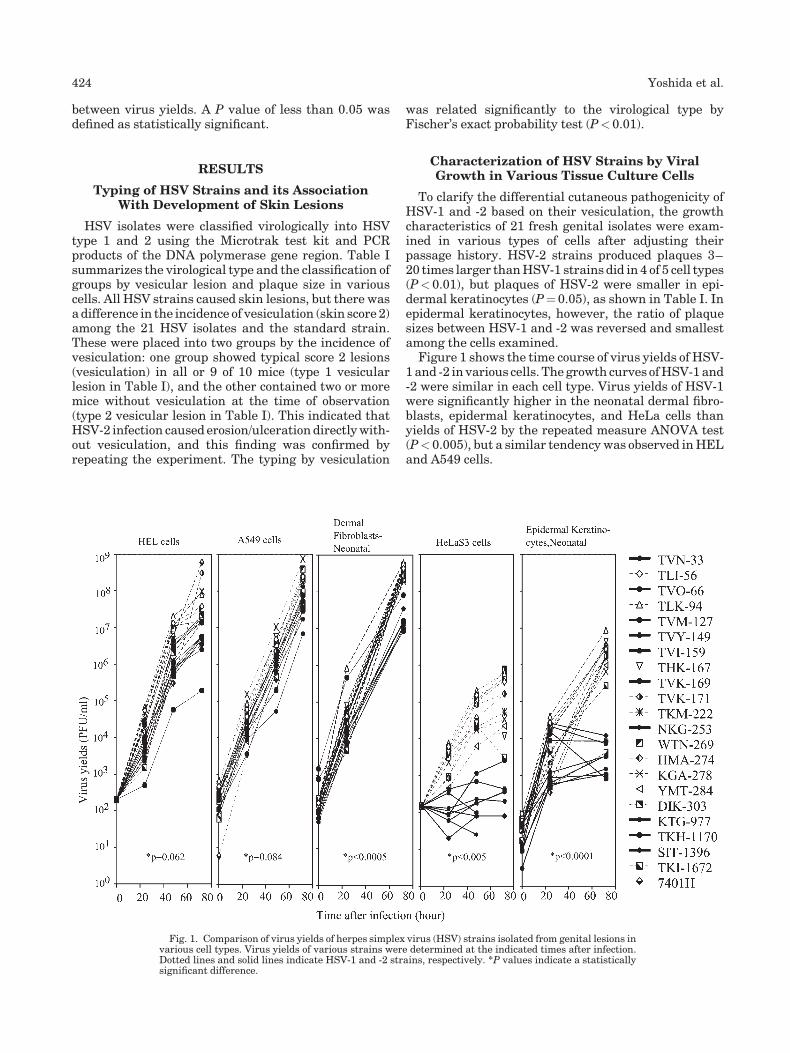
between virus yields. A P value of less than 0.05 wasdefined as statistically significant.
RESULTS
Typing of HSV Strains and its AssociationWith Development of Skin Lesions
HSV isolates were classified virologically into HSVtype 1 and 2 using the Microtrak test kit and PCRproducts of the DNA polymerase gene region. Table Isummarizes the virological type and the classification ofgroups by vesicular lesion and plaque size in variouscells. All HSV strains caused skin lesions, but there wasa difference in the incidence of vesiculation (skin score 2)among the 21 HSV isolates and the standard strain.These were placed into two groups by the incidence ofvesiculation: one group showed typical score 2 lesions(vesiculation) in all or 9 of 10 mice (type 1 vesicularlesion in Table I), and the other contained two or moremice without vesiculation at the time of observation(type 2 vesicular lesion in Table I). This indicated thatHSV-2 infection caused erosion/ulceration directly with-out vesiculation, and this finding was confirmed byrepeating the experiment. The typing by vesiculation
was related significantly to the virological type byFischer’s exact probability test (P<0.01).
Characterization of HSV Strains by ViralGrowth in Various Tissue Culture Cells
To clarify the differential cutaneous pathogenicity ofHSV-1 and -2 based on their vesiculation, the growthcharacteristics of 21 fresh genital isolates were exam-ined in various types of cells after adjusting theirpassage history. HSV-2 strains produced plaques 3–20 times larger than HSV-1 strains did in 4 of 5 cell types(P<0.01), but plaques of HSV-2 were smaller in epi-dermal keratinocytes (P¼ 0.05), as shown in Table I. Inepidermal keratinocytes, however, the ratio of plaquesizes between HSV-1 and -2 was reversed and smallestamong the cells examined.
Figure 1 shows the time course of virus yields of HSV-1 and -2 in various cells. The growth curves of HSV-1 and-2 were similar in each cell type. Virus yields of HSV-1were significantly higher in the neonatal dermal fibro-blasts, epidermal keratinocytes, and HeLa cells thanyields of HSV-2 by the repeated measure ANOVA test(P<0.005), but a similar tendency was observed in HELand A549 cells.
Fig. 1. Comparison of virus yields of herpes simplex virus (HSV) strains isolated from genital lesions invarious cell types. Virus yields of various strains were determined at the indicated times after infection.Dotted lines and solid lines indicate HSV-1 and -2 strains, respectively. *P values indicate a statisticallysignificant difference.
424 Yoshida et al.

Table II shows the ratio of plaque size to virus yield invarious cells due to HSV-1 and -2 at 72 hr after infection.The ratios of plaque size to virus yield of HSV-2 were 5–500 times larger than those of HSV-1 in all cell types(P<0.05). Growth in epidermal keratinocytes showed themost contrast (502 times) between HSV-1 and -2 amongthe cells (5.57–28.8 times) examined, indicating thatepidermal keratinocytes might play a crucial differentialrole in the cutaneous pathogenesis of HSV-1 and -2.
DISCUSSION
HSV-1 and -2 cause similar diseases and are difficultto differentiate from each other by their clinical mani-festations. Fresh clinical isolates from genital lesionswere used and adjusted the passage history in order toexamine the correlation between their cutaneous patho-genicity and biological properties. The growth charac-teristics of HSV-1 and -2 are summarized in Table II.Although HSV-2 produced lower virus yields than HSV-1 [Plummer et al., 1968], HSV-2 spread more rapidly incultured cells, as characterized by the larger ratio ofplaque formation to virus production, than HSV-1. Thisefficient spread of HSV-2 may be reflected in the severityand sequelae of encephalitis caused by HSV-2 [Plummerand Hackett, 1966; Whitley et al., 1991; Kimura et al.,2002]. The difference in the ratio of plaque size to virusyield may be due to the interaction of cell-specific meta-bolism and a functional difference in viral proteinsbetween HSV-1 and -2 rather than a difference in thecellular receptors for HSV infection [Spear, 2004]. Itis not clear what causes the difference in growthcharacteristics, especially in keratinocytes, but multiplefunctional gene products may be responsible for the co-ordinated differences between HSV-1 and -2 in varioussteps of replication, depending on the cell type.
The cutaneous pathogenicity was examined in micewithout the HSV type information, and subsequenttyping of tested HSV strains indicated that HSV-2 pro-duced vesicular lesions significantly less frequentlythan HSV-1. This less frequent vesiculation due toHSV-2 was further assessed by its cell tropism to variouscells, especially in epidermal keratinocytes.
Epidermal keratinocytes constitute the epidermallayer of the skin, and vesicular lesions and erosion/ulceration are formed in the epidermal layer. Vesicular
lesions in the skin of mice histologically show balloon-ing, degeneration and acantholysis of the keratinocytes,intercellular edema and vesiculation in the epidermalkeratinocyte layer, and erosion/ulceration of the epider-mal layer. The plaque size of HSV-2 was smaller thanthat of HSV-1 in primary epidermal keratinocytes andlarger in the other cells, including primary dermalfibroblasts. This property of plaque formation in var-ious cells indicates why HSV-2 infection spreads less(0.01–0.06 times from the ratio of plaque size to virusyield) in the epidermal keratinocyte layer, despite thelarger spread in the dermal layer, and explains thedelay in vesicle formation and the rapid destruction ofthe epidermal layer, followed by the greater destructionof the epidermal and dermal layers to cause erosion/ulceration. We observed larger and deeper histologicalchanges in HSV-2 skin lesions than in HSV-1 skinlesions (data not shown). This process suggested thatHSV infection of the mucosa, which is composed of athin epidermal keratinocyte layer without thick corni-fication, might be eroded by the destruction of theoverlying keratinocyte layer, explaining the small dif-ference between HSV-1 and -2 in the development oferosion/ulceration in the mucosa [Nakano et al., 1998].In contrast, the keratinocyte layer of the skin mayundergo vesiculation before erosion/ulceration due tothe destruction of the cornified epidermal layer. Thismay cause the difference in the nature of skin lesionsin the mucosa and the thick cornified skin. Thus thetropism of HSV-1 and -2 to epidermal keratinocytes mayplay an important role in determining the developmentof skin lesions in both mouse and human. The differencein the development of skin lesions in vivo is not as clearas that in tissue culture, indicating that host factorssuch as the immune system may affect the formation ofskin lesions, as reported by Spruance and Chow [1980].In this study, the differences between HSV-1 and -2 incell tropism toward epidermal keratinocytes comparedto other cells and the spread per virus yield in these cellswas reasonably well correlated with the development ofvesicular lesions using fresh clinical isolates with anadjusted passage history.
ACKNOWLEDGMENTS
We thank Katherine Ono for editing the manuscript.
TABLE II. Comparison of Ratio of Plaque Size (mm2) to Virus Yield (Plaque-Forming Units(PFU)) at 72 hr Between HSV-1 and -2
Cell HSV-1 HSV-2 P values
A549 2.78� 1.10 (1) 56� 140 (20.1) <0.05HEL 92.0� 27.8 (1) 512� 314 (5.57) <0.0001Keratinocytes (�104) 0.47� 0.12 (1) 236� 66 (502) <0.001Dermal fibroblasts 5.88� 0.90 (1) 41� 55.0 (6.97) <0.0001Vero 0.651� 0.17 (1) 18.8� 6.2 (28.8) <0.01
The ratios of plaque size (mm2) to virus yield (PFU) are expressed as the mean�SE, and the figures inparentheses indicate the relative ratio of HSV-2 to 1. P values indicate the comparison of HSV-1 and -2 byStudent’s t-test.
Cutaneous Pathogenicity of HSV-2 425

REFERENCES
Anderson K. 1940. Pathogenesis of herpes simplex virus infection inchicken embryos. Am J Pathol 16:137–156.
Hasegawa T, Kawana T, Okuda T, Tsukada T, Horii M, Shiraki K. 2001.Analysis of herpes simplex virus isolated from the genital lesionssince 1977 in Japan. J Med Virol 63:57–63.
Kawana T, Shinkai K, Yoshino K. 1974. Typing of herpes simplexvirus strains of genital and nongenital origins. Jpn J Microbiol 18:235–241.
Kimura H, Ito Y, Futamura M, Ando Y, Yabuta Y, Hoshino Y,Nishiyama Y, Morishima T. 2002. Quantitation of viral load inneonatal herpes simplex virus infection and comparison betweentype 1 and type 2. J Med Virol 67:349–353.
Kurokawa M, Ochiai H, Nagasaka K, Neki M, Xu H, Kadota S, SutardjoS, Matsumoto T, Namba T, Shiraki K. 1992. Antiviral traditionalmedicines against herpes simplex virus (HSV-1), poliovirus, andmeasles virus in vitro and their therapeutic efficacies for HSV-1infection in mice. Antiviral Res 22:175–188.
Kurokawa M, Hozumi T, Basnet P, Nakano M, Kadota S, Namba T,Kawana T, Shiraki K. 1998. Purification and characterization ofeugeniin as an anti-herpes virus compound from Geum japonicumand Syzygium aromaticum. J Pharmacol Exp Therapeut 284:728–735.
Kurokawa M, Basnet P, Ohsugi M, Hozumi T, Kadota S, Namba T,Kawana T, Shiraki K. 1999. Anti-herpes simplex virus activity ofmoronic acid purified from Rhus javanica in vitro and in vivo.J Pharmacol Exp Therapeut 289:72–78.
Kurokawa M, Hozumi T, Tsurita M, Kadota S, Namba T, Shiraki K.2001. Biological characterization of eugeniin as an anti-herpessimplex virus type 1 compound in vitro and in vivo. J Pharmacol ExpTherapeut 297:372–379.
Nagasaka K, Kurokawa M, Imakita M, Terasawa K, Shiraki K. 1995.Efficacy of Kakkon-to, a traditional herb medicine, in herpessimplex virus type 1 infection in mice. J Med Virol 46:28–34.
Nahmias AJ, Dowdle WR. 1968. Antigenic and biologic differences inherpesvirus hominis. Prog Med Virol 10:110–159.
Nakano M, Kurokawa M, Hozumi T, Saito M, Ida M, Morohashi T,Namba T, Kawana T, Shiraki K. 1998. Suppression of recurrentgenital herpes simplex virus type 2 infection by Rhus javanica inguinea pigs. Antiviral Res 39:25–33.
Parker J, Banatvala J. 1967. Herpes genitalis—Clinical and virologicalstudies. Br J Vener Dis 43:212–216.
Plummer G, Hackett S. 1966. Herpes simplex virus and paralysis ofanimals. Br J Exp Pathol 47:82–85.
Plummer G, Warner JL, Bowling CP. 1968. Comparative studies ofherpes simplex viruses type 1 and type 2 ‘‘herpes simplex’’ viruses.Br J Exp Pathol 49:202–208.
Plummer G, Goodheart CR, Miyagi M,SkinnerGRB, Thouless ME, WildyP. 1974. Herpes simplex viruses: Discrimination of types andcorrelation between different characteristics. Virology 60:206–206.
Shiraki K, Yoshida Y, Asano Y, Yamanishi K, Takahashi M. 2003.Pathogenetic tropism of varicella-zoster virus to primary humanhepatocytes and attenuating tropism of Oka varicella vaccinestrain to neonatal dermal fibroblasts. J Infect Dis 188:1875–1877.
Simmon A, Nash AA. 1984. Zosteriform spread of herpes simplex virusas a model of recrudescence and its use to investigate the role ofimmune cells in prevention of recurrent disease. J Virol 52:816–821.
Spear PG. 2004. Herpes simplex virus: Receptors and ligands for cellentry. Cell Microbiol 6:401–410.
Spruance SL, Chow FS. 1980. Pathogenesis of herpes simplex labialis.I. Replication of herpes simplex virus in cultures of epidermal cellsfrom subjects with frequent recurrence. J Infect Dis 142:671–675.
Whitley R, Arvin A, Prober C, Corey L, Burchett S, Plotkin S, Starr S,Jacobs R, Powell D, Nahmias A, Sumaya C, Edwards K, Alford C,Caddell G, Soong S-J, and the National Institute of Allergy andInfectious Diseases Collaborative Antiviral Study Group. 1991. Pre-dictors of morbidity and mortality in neonates with herpes simplexvirus infections. New Eng J Med 324:450–454.
426 Yoshida et al.

Journal of Medical Virology 75:427–429 (2005)
Case Report
Detection of Herpesvirus-6A in a Case ofSubacute Cerebellitis and Myoclonic Dystonia
Elisa Borghi,2 Elisabetta Pagani,1 Roberta Mancuso,1 Serena Delbue,1 Marilena Valli,1
Romina Mazziotti,1 Lucio Giordano,3 Roberto Micheli,3 and Pasquale Ferrante1,2*1Laboratory of Biology, Don C. Gnocchi Foundation ONLUS, IRCCS, Milan, Italy2Chair of Virology, Department of Biomedical Science and Technology, University of Milan, Milan, Italy3Department of Child Neurology, Spedali Civili, Brescia, Italy
This is a case study of a child who developedroseola infantum first, then varicella, and waslater affected by acute cerebellar syndrome,severe truncal ataxia, and myoclonic dystonia.Human herpesvirus 6 (HHV-6) A and B weredetected in the cerebrospinal fluid (CSF) andperipheral blood, respectively, uponataxiaonset.The intricacy of this case suggests multifacetedconclusions ranging from the need for a multi-directional approach to neurological diseases,to confirmation of a more pronounced neuro-tropism of HHV-6A and a possible role of virusesin myoclonic dystonia syndrome, although thislast hypothesis should be confirmed by largerstudies. J. Med. Virol. 75:427–429, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: human herpesvirus 6; truncalataxia; myoclonic dystonia;HHV-6A and B variants
INTRODUCTION
Humanherpesvirus 6 (HHV-6) is a b-herpesvirus thatwas isolated initially from the peripheral blood ofpatients with acquired immunodeficiency syndrome orwith unrelated lymphoproliferative disorders. HHV-6primary infection occurs during childhood and isassociated with exanthema subitum and febrile illness,more rarely with pneumonitis and other conditions,such as febrile seizures and encephalitis or encephalo-pathy [Campadelli-Fiume et al., 1999; Caserta et al.,2001]. HHV-6 infection is common, as shown by aseroprevalence that exceeds 90% in the healthy adultpopulation worldwide [Campadelli-Fiume et al., 1999;Caserta et al., 2001]. The presence of viral DNA inperipheral blood mononuclear cells of healthy peoplesupports the view that HHV-6 is capable of establishinglatent infection in these cells or replication at a low levelthat is kept under control by the immune system.
Neurological diseases such as demyelinating diseases,and epileptic seizures have rarely been observed duringprimary HHV-6 infection, but frequently in immuno-compromised subjects [Caserta et al., 1994; Campadelli-Fiume et al., 1999; Caserta et al., 2001].
Several studies have also implicated HHV-6 as apossible etiological factor in other neurological diseases,such as multiple sclerosis [Berti et al., 2000]. Twovariant groupsofHHV-6, termedHHV-6AandHHV-6B,have been identified. They appear to differ in theirepidemiology, in vivo and in vitro tropism and theirpathogenic potential [Di Luca et al., 1996; Campadelli-Fiume et al., 1999]. Both HHV-6A and HHV-6B havebeen isolated from the PBMCs of healthy adults,suggesting that both variants could establish persistentinfection in PBMCs. The possibility of dual infectionwith both variants has been also reported [Di Lucaet al., 1996]. The prevalent strain of HHV-6 isolatedfrom children with exantema subitum and other febrileillnesses has been variant B, whereas the neuroviru-lence of the A variant, in children and adults, is thoughtto be more pronounced [Di Luca et al., 1996; Hall et al.,1998].
PATIENTS AND METHODS
The patient is a girl who was 3 years old in March2001.One year after roseola infantum and 1month afterchickenpox, shedevelopedanacute cerebellar syndromeandmildmyoclonic-dystonia affecting the right leg. Thechild underwent treatment with corticosteroids andacyclovir, with almost complete recovery regarding the
Grant sponsor: Italian Ministry of Health.
*Correspondence to: Pasquale Ferrante, MD, Laboratory ofBiology, Don C. Gnocchi Foundation, IRCCS, via Capecelatro, 66,I-20148, Milan, Italy. E-mail: [email protected]
Accepted 2 November 2004
DOI 10.1002/jmv.20285
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

ataxic symptoms. Two months later, the child wasreadmitted to the hospital, presenting severe trun-cal ataxia, with difficulty in maintaining a standingposition.The neurological examination, performed in March
2001, showed a severe truncal ataxia and a paroxysmalkynesigenic myoclonic dystonia, most of the timesinduced by attempt to walk.Brain computed tomographic (CT) scan, as well as
cerebral and spinal magnetic resonance imaging (MRI)with and without gadolinium DTPA, were both normal.A CSF examination showed a slightly high link
(IgG) index (0.81; normal value <0.7), whereas theTourtelotte index was normal. Both indexes estimatethe amount of intra-blood brain barrier IgG syntesizedper day; therefore, since the high level of albumin in theCSF is a quantitativemarker of a damaged blood–brainbarrier, our patient revealed only a mild inflamma-tory process of the central nervous system. No virologi-cal analyses were performed on this first set of CSFsamples.Upon readmission to the hospital (June, 2001),
cerebrospinal fluid (CSF) and peripheral blood sampleswere collected and five times thereafter during theeleven months of follow-up.The first serum was tested to verify the presence
of HHV-6 and VZV antibodies using commerciallyavailable ELISA tests (HHV-6 IgG antibody ELISA,Advanced Biotechnologies, Inc., MD; Varicella Zostervirus ELISA, Genzyme Virotech Gmbh, Russelsheim,Germany).The samples were analyzed for the presence of
genomic DNA of herpes simplex virus types 1 and 2(HSV 1/2), varicella zoster virus (VZV), Epstein–Barrvirus (EBV), human cytomegalovirus (HCMV), humanherpesvirus-6 (HHV-6), human polyomavirus JC (JCV),and BK (BKV), using specific nested polymerase chainreaction (n-PCR) methods. The primers and protocolshave been previously described [Secchiero et al., 1995;Ferrante et al., 1997]. PCR sensitivity was 1.5 Geq forVZV and <10 copies for HHV-6.To differentiate the HHV-6A and HHV-6B variants,
the positive PCR products were subjected to restrictionenzyme analysis with Xho I (Boehring MannheimGmbH, Mannheim, Germany). This enzyme is able torecognize the CTCGAG sequence present only in HHV-6A (nt position 102,911–102,916), cutting the amplifiedproduct into two fragments of 139 and 120 bp, whereasthe HHV-6B variant is visualized as one fragmentof 259 bp. We also performed a nucleotide sequenceanalysis of the HHV-6 DNA amplified in the first CSFand peripheral blood samples, using the ThermoSequenaseTM CyTM5 Dye Terminator Kit (AmershamBiosciences AB, Uppsala, Sweden), and the ALFexpressTM automatedDNA sequencer (AmershamBios-ciences AB), according to the manufacturer’s protocol.Sequence homology comparison was performed usingBLAST at the National Center for Biotechnology In-formation and alignment was performed with ClustalWat the European Bioinformatic Institute. T
ABLE
I.Rep
resentation
ofClinicalCou
rse,Neu
roradiologicalFindingsWithMagneticReson
ance
Imaging(M
RI)andCranialTom
ography(C
T),andVirolog
icalResults
inRelation
totheAntiviralTherapy
March2001:truncalataxia
withsu
spectedpost-varicella
cerebellitis
June
2001
July
2001
Sep
tember
2001
Nov
ember
2001
Decem
ber
2001
May
2002
MRI
Neg
ative
ND
ND
ND
ND
ND
CT
Neg
ative
ND
ND
ND
ND
ND
Ataxia
þþ
þ�
��
�Myoclonus
þþ
þþþ
þ/�
þþ
Dyston
iaþ
þþ
þþ
þ/�
þþ
HHV6
CSF
HHV-6A
HHV-6A
Neg
ative
HHV-6A
Neg
ative
Neg
ative
PB
HHV-6B
HHV-6B
Neg
ative
HHV-6B
HHV-6B
ND
""
"Foscarn
et1st
cycle
Foscarn
et2ndcycle
Foscarn
et3rd
cycle
ND,not
don
e.
428 Borghi et al.

RESULTS
The virological investigations performed upon read-mission detected the presence of HHV-6 DNA, but notVZV DNA, while HHV-6 and VZV IgG antibodies weredetected in the serum. Using restriction enzyme anal-ysis and sequence analysis, HHV-6 variant B was foundin the peripheral blood andHHV-6 variantA in theCSF.The child was then treated with Foscarnet, with re-solution of the ataxia and persistence of the myoclonicdystonia. During the antiviral therapy, HHV-6 becameundetectable in both the CSF and peripheral blood.During the 4 months after therapy, a recrudescence ofmyoclonus and dystonia was observed concomitantlywith the detection of HHV-6 variants in subsequentperipheral blood (variant B) and CSF (variant A)samples (Table I). To avoid additional stress and pos-sible psychological repercussions, and also consideringthat thefirst examswerenegative, theMRIandCTwerenot included in the follow-up protocol.
In May 2002, a clinical examination was undertaken,during which slight myoclonic dystonia was still ob-servable (more evident during running), but the CSFsample collected on this occasion was negative for thepresence of HHV-6.
DISCUSSION
In this case, the presence of HHV-6A was detected inthe CSF, but not VZV or other viruses. HHV-6 primaryinfection occurred in this child in association withexanthema subitum (roseola infantum), 1 year beforeshe developed chickenpox (varicella). Thus VZV in-fection could have triggered HHV-6 reactivation, or areinfection with a different HHV-6 variant could beresponsible for the neurological symptoms, as the pre-sence of a dual infection with both HHV-6 variants wasdetected. The exclusive presence of HHV-6A in the CSFand HHV-6B in the peripheral blood lends furthersupport to the suspicion that HHV-6A is highly neuro-tropic and that this variant can persist in CSF, althoughHHV-6B proves to be more frequent among healthychildren and adults [Hall et al., 1998].
The data obtained from this case study of HHV-6involving subacute neurological disorders lead us tosome interesting conclusions. First of all, laboratoryinvestigations are clearly needed even when clinicaldata seem to provide clear etiological indications. In thiscase, there was a clinical diagnosis of post-varicellacerebellitis, but the laboratory results determined thepresence of HHV-6. Hence the importance of a correct,laboratory-supported diagnosis, and particularly whena specific drug is available to help a patient recover fromthe disease.
From the virological point of view, although it cannotbe excluded that VZV may have been, directly or
indirectly, the etiological agent in this case, it is in-teresting to note that the presence ofmyoclonic dystoniawas associated with a dual infection and particularlywith the presence of HHV-6A in the CSF. Myoclonicdystonia [Pueschel et al., 1992; Quinn, 1996] is a move-ment disorder characterized by the association of slow,sustained involuntary twisting movements of dystoniaand the shock-like involuntary muscle jerks of myoclo-nus. The etiology of the sporadic form of myoclonicdystonia remains unknown, although brain and, inparticular, cerebellar damage, drugs and infections ofthe central nervous system have been pointed out aspossible causative elements.
In conclusion, the observations noted in this casereport seem to indicate a need for a reevaluation ofHHV-6A as a possible etiological factor in otherneurological diseases, including movement disorderssuch as myoclonic dystonia. However, the associationof HHV-6A with this neurological disorder must beelucidated by prospective studies of suitable numbers ofchildren.
ACKNOWLEDGMENTS
This work was partially supported by IRCCS grants‘‘Ricerca Corrente 2004’’ and ‘‘ Ricerca Finalizzata 2003’’from Italian Ministry of Health.
REFERENCES
Berti R, Soldan SS, Akhyani N, McFarland HF, Jacobson S. 2000.Extended observations on the association of HHV-6 and multiplesclerosis. J Neurovirol 6:S85–S87.
Campadelli-Fiume G, Mirandola P, Menotti L. 1999. Human herpes-virus 6: An emerging pathogen. Emerg Infect Dis 5:353–366.
Caserta MT, Hall CB, Shnabel K, McIntyre K, Long C, Costanzo M,Dewhurst S, Insel R, Epstein LG. 1994. Neuroinvasion andpersistence of human herpesvirus 6 in children. J Infect Dis 170:1586–1589.
Caserta MT, Mock DJ, Dewhurst S. 2001. Human Herpesvirus 6. ClinInfect Dis 38:829–833.
Di Luca D, Mirandola P, Ravaioli T, Bigoni B, Cassai E. 1996. Dis-tribution of HHV-6 variants in human tissues. Infect Agents Dis5:S203–S214.
Ferrante P, Omodeo-Zorini E, Zuffolato MR, Mancuso R, Caldarelli-Stefano R, Puricelli S, Mediati M, Losciale L, Caputo D. 1997.HumanT-cell lymphotropic virus tax andEpstein–Barr virus DNAin peripheral blood of multiple sclerosis patients during acuteattack. Acta Neurol Scand S169:79–85.
Hall CB, Caserta MT, Schnabel KC, Long C, Epstein LG, Insel RA,Dewhurst S. 1998. Persistence of human herpesvirus 6 according tosite and variant: Possible greater neurotropism of variant A. ClinInfect Dis 26:132–137.
Pueschel SM,FriedmanJH,ShettyT. 1992.Myoclonic dystonia. ChildsNerv Syst 8:61–66.
Quinn NP. 1996. Essential myoclonus and myoclonic dystonia. MovDisord 11:119–124.
Secchiero P, Carrigan DR, Asano Y, Benedetti L, Crowley RW,Komaroff AL, Gallo RC, Lusso P. 1995. Detection of humanherpesvirus 6 in plasma of children with primary infection andimmunosuppressed patients by polymerase chain reaction. J InfectDis 171:273–280.
HHV-6A Cerebellitis and Myoclonic Dystonia 429

Journal of Medical Virology 75:430–434 (2005)
Case Report
Drug-Induced Hypersensitivity Syndrome Due toCyanamide Associated With Multiple Reactivationof Human Herpesviruses
Naoko Mitani,1 Michiko Aihara,1 Yuko Yamakawa,1 Masako Yamada,1 Norihiko Itoh,2*Nobuhisa Mizuki,2 and Zenro Ikezawa3
1Department of Dermatology, Yokohama City University Medical Center, Yokohama, Japan2Department of Ophthalmology, Yokohama City University School of Medicine, Yokohama, Japan3Department of Dermatology, Yokohama City University School of Medicine, Yokohama, Japan
Drug-induced hypersensitivity syndrome (DIHS),characterized by serious adverse systemic reac-tions in addition to skin rash, has unknownpathogenesis. Its association with human her-pesvirus (HHV),mainly HHV-6, has been reportedrecently. A 46-year-old Japanese man is des-cribed inwhomageneralizederuptiondevelopedabout 1month after taking cyanamide, a drug foralcoholism. This was associated with the follow-ing manifestations: high fever, lymphadenopa-thy, facial edema, marked leukocytosis witheosinophilia and atypical lymphocytes, lympho-cytopenia, liver and renal dysfunction, and lowIgG level. He was treated with 8 mg betametha-sone daily and his condition improved, but heneeded low-dose corticosteroid for almost 1 yearbecause of several episodes of recurrence. HHV-6, HHV-7, herpes simplex virus (HSV), and cyto-megalovirus (CMV) specific IgG titers showedmore than a four-fold rise sequentially. Signifi-cant numbers of copies of HHV-6 andHHV-7 DNAwere detected in the peripheral white blood cellsby real-time polymerase chain reaction (PCR).HHV-6 and CMVDNAwere detected in the serumby nested PCR. A patch test for cyanamide waspositive. Thediagnosis of DIHSdue to cyanamide,which has never been reported as a causal drug ofDIHS, accompanied by reactivation of not onlyHHV-6, but also HHV-7, CMV, andHSV,wasmade.Disturbanceof the immunesystemwassuggestedby the persistent low level of IgG, and consecutiveviral reactivation may have participated in theprolonged course in this case. J. Med. Virol.75:430–434, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: adverse drug reactions; hypo-gammaglobulinaemia; poly-
merase chain reaction; patchtest; lymphocyte stimulationtest
INTRODUCTION
Cyanamide is used in the treatment of alcoholism inJapan, Europe, and North America with comparativelyfew side-effects [Niederhofer et al., 2003]. Only a fewcases of adverse skin reactions, including allergiccontact dermatitis, lichen planus-like eruptions, exfo-liative dermatitis [Kawana, 1997], fixed drug eruption,and Stevens–Johnson syndrome, have been reported.
Recently, it has been recognized that drug-inducedhypersensitivity syndrome (DIHS), characterized byserious adverse systemic reactions in addition to a skinrash, is associated frequently with reactivation ofhuman herpesvirus 6 (HHV-6) [Descamps et al., 1997;Suzuki et al., 1998; Tohyama et al., 1998]. Reactivationof cytomegalovirus (CMV) [Aihara et al., 2001] orEpstein–Barr virus (EBV) [Descamps et al., 2003] hasalso been reported. This syndrome develops within 2–6weeks after taking the causal drug, and tends to beprolonged for 2 weeks to several months after itsdiscontinuation. The major causal drugs reported areanticonvulsants [Callot et al., 1996; Carroll et al., 2001;
*Correspondence to: Norihiko Itoh, D.V.M., Ph.D., Departmentof Ophthalmology, Yokohama City University School of Medicine,3–9, Fukuura, Kanazawa-ku, Yokohama 236-0004, Japan.E-mail: [email protected]
Accepted 4 November 2004
DOI 10.1002/jmv.20295
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.
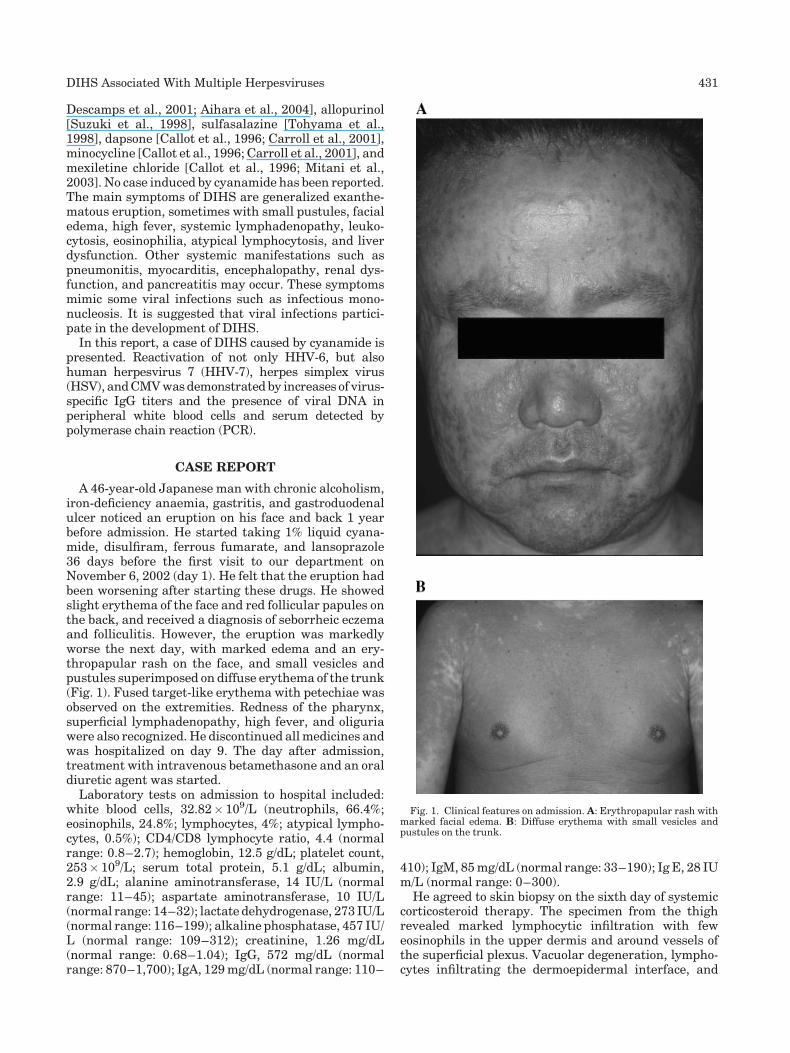
Descamps et al., 2001; Aihara et al., 2004], allopurinol[Suzuki et al., 1998], sulfasalazine [Tohyama et al.,1998], dapsone [Callot et al., 1996; Carroll et al., 2001],minocycline [Callot et al., 1996; Carroll et al., 2001], andmexiletine chloride [Callot et al., 1996; Mitani et al.,2003]. No case induced by cyanamide has been reported.The main symptoms of DIHS are generalized exanthe-matous eruption, sometimes with small pustules, facialedema, high fever, systemic lymphadenopathy, leuko-cytosis, eosinophilia, atypical lymphocytosis, and liverdysfunction. Other systemic manifestations such aspneumonitis, myocarditis, encephalopathy, renal dys-function, and pancreatitis may occur. These symptomsmimic some viral infections such as infectious mono-nucleosis. It is suggested that viral infections partici-pate in the development of DIHS.
In this report, a case of DIHS caused by cyanamide ispresented. Reactivation of not only HHV-6, but alsohuman herpesvirus 7 (HHV-7), herpes simplex virus(HSV), andCMVwasdemonstrated by increases of virus-specific IgG titers and the presence of viral DNA inperipheral white blood cells and serum detected bypolymerase chain reaction (PCR).
CASE REPORT
A 46-year-old Japanese man with chronic alcoholism,iron-deficiency anaemia, gastritis, and gastroduodenalulcer noticed an eruption on his face and back 1 yearbefore admission. He started taking 1% liquid cyana-mide, disulfiram, ferrous fumarate, and lansoprazole36 days before the first visit to our department onNovember 6, 2002 (day 1). He felt that the eruption hadbeen worsening after starting these drugs. He showedslight erythema of the face and red follicular papules onthe back, and received a diagnosis of seborrheic eczemaand folliculitis. However, the eruption was markedlyworse the next day, with marked edema and an ery-thropapular rash on the face, and small vesicles andpustules superimposed on diffuse erythema of the trunk(Fig. 1). Fused target-like erythema with petechiae wasobserved on the extremities. Redness of the pharynx,superficial lymphadenopathy, high fever, and oliguriawere also recognized. He discontinued allmedicines andwas hospitalized on day 9. The day after admission,treatment with intravenous betamethasone and an oraldiuretic agent was started.
Laboratory tests on admission to hospital included:white blood cells, 32.82� 109/L (neutrophils, 66.4%;eosinophils, 24.8%; lymphocytes, 4%; atypical lympho-cytes, 0.5%); CD4/CD8 lymphocyte ratio, 4.4 (normalrange: 0.8–2.7); hemoglobin, 12.5 g/dL; platelet count,253� 109/L; serum total protein, 5.1 g/dL; albumin,2.9 g/dL; alanine aminotransferase, 14 IU/L (normalrange: 11–45); aspartate aminotransferase, 10 IU/L(normal range: 14–32); lactate dehydrogenase, 273 IU/L(normal range: 116–199); alkaline phosphatase, 457 IU/L (normal range: 109–312); creatinine, 1.26 mg/dL(normal range: 0.68–1.04); IgG, 572 mg/dL (normalrange: 870–1,700); IgA, 129mg/dL (normal range: 110–
410); IgM, 85mg/dL (normal range: 33–190); Ig E, 28 IUm/L (normal range: 0–300).
He agreed to skin biopsy on the sixth day of systemiccorticosteroid therapy. The specimen from the thighrevealed marked lymphocytic infiltration with feweosinophils in the upper dermis and around vessels ofthe superficial plexus. Vacuolar degeneration, lympho-cytes infiltrating the dermoepidermal interface, and
Fig. 1. Clinical features on admission. A: Erythropapular rash withmarked facial edema. B: Diffuse erythema with small vesicles andpustules on the trunk.
DIHS Associated With Multiple Herpesviruses 431
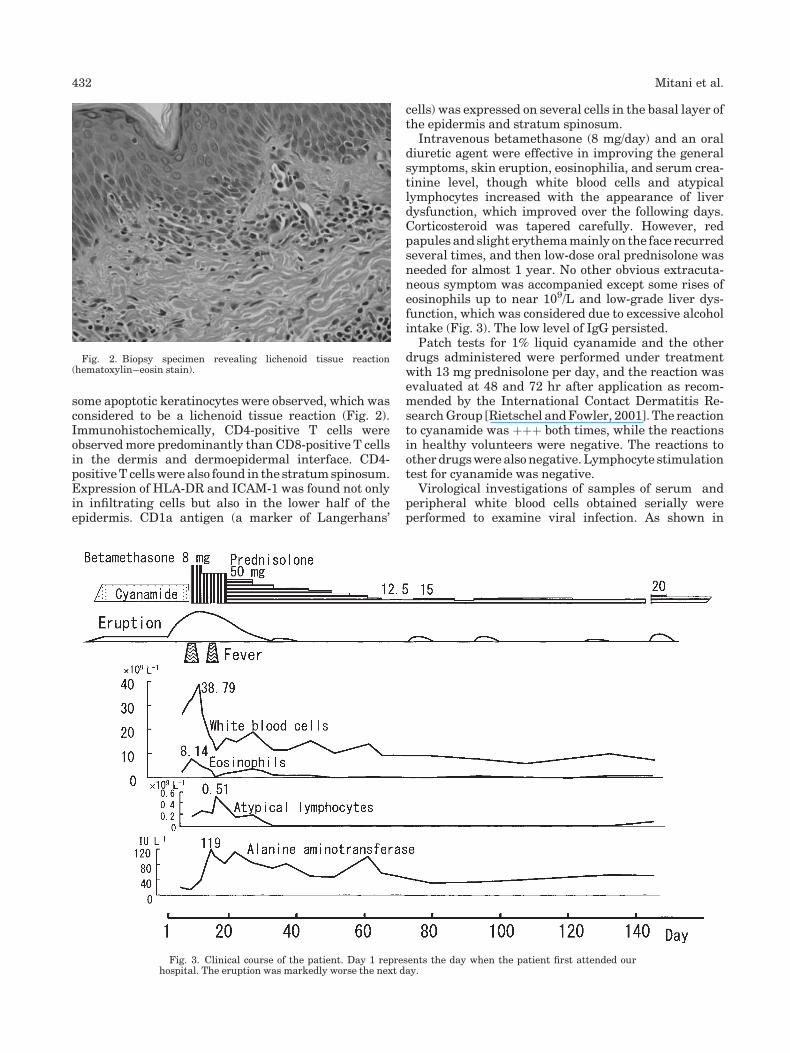
some apoptotic keratinocytes were observed, which wasconsidered to be a lichenoid tissue reaction (Fig. 2).Immunohistochemically, CD4-positive T cells wereobservedmore predominantly than CD8-positive T cellsin the dermis and dermoepidermal interface. CD4-positiveT cellswere also found in the stratumspinosum.Expression of HLA-DR and ICAM-1 was found not onlyin infiltrating cells but also in the lower half of theepidermis. CD1a antigen (a marker of Langerhans’
cells) was expressed on several cells in the basal layer ofthe epidermis and stratum spinosum.
Intravenous betamethasone (8 mg/day) and an oraldiuretic agent were effective in improving the generalsymptoms, skin eruption, eosinophilia, and serum crea-tinine level, though white blood cells and atypicallymphocytes increased with the appearance of liverdysfunction, which improved over the following days.Corticosteroid was tapered carefully. However, redpapules and slight erythemamainly on the face recurredseveral times, and then low-dose oral prednisolone wasneeded for almost 1 year. No other obvious extracuta-neous symptom was accompanied except some rises ofeosinophils up to near 109/L and low-grade liver dys-function, which was considered due to excessive alcoholintake (Fig. 3). The low level of IgG persisted.
Patch tests for 1% liquid cyanamide and the otherdrugs administered were performed under treatmentwith 13 mg prednisolone per day, and the reaction wasevaluated at 48 and 72 hr after application as recom-mended by the International Contact Dermatitis Re-searchGroup [Rietschel andFowler, 2001]. The reactionto cyanamide was þþþ both times, while the reactionsin healthy volunteers were negative. The reactions tootherdrugswerealsonegative.Lymphocyte stimulationtest for cyanamide was negative.
Virological investigations of samples of serum andperipheral white blood cells obtained serially wereperformed to examine viral infection. As shown in
Fig. 2. Biopsy specimen revealing lichenoid tissue reaction(hematoxylin–eosin stain).
Fig. 3. Clinical course of the patient. Day 1 represents the day when the patient first attended ourhospital. The eruption was markedly worse the next day.
432 Mitani et al.
Table I, specific IgG titers forHHV-6,HHV-7, CMV, andHSV increased consecutively. Serological tests forhepatitis B virus and hepatitis C virus were negative.As shown in Table II, real-time PCR assays for HHV-6[Tanaka et al., 2000] andHHV-7DNA [Hara et al., 2002]in peripheral white blood cells showed significantnumber of copies on day 17 but not on other days.Nested PCR for HHV-6, HHV-7, and CMVDNA [Nagaeet al., 1998] was also performed on serum samples, fromwhich DNA was extracted after ultracentrifugation(105g, 4 hr, 48C) to maximize sensitivity. HHV-6 DNAwas detected on day 13, but not on other days. HHV-7DNA was not detected. CMV DNA was detected on day17, 38, and 45.
DISCUSSION
Cyanamide (H2N-C:N), an inhibitor of aldehydedehydrogenase, is used as a pharmacological adjunctin the aversive treatment of chronic alcoholism. Besidesother previous reports of cyanamide-induced eruption,Kawana [1997] described six cases of exfoliative derma-titis and noted that cyanamide-induced eruptionsmightnot be rare. Few of those patients had fever, lymphade-nopathy, leukocytosis, or eosinophilia, but no liverdysfunction except one, who had had liver cirrhosisbefore the administration of cyanamide. A relationshiphas not been described between viral infection andadverse reaction due to cyanamide.
The present case of DIHS caused by cyanamide wasassociated with reactivation of HHV-7, CMV, and HSVin addition to HHV-6. In this case, a skin eruptiondeveloped after the administration of cyanamide for37 days, accompanied by severe systemic symptoms and
low IgG level. Viral reactivation was confirmed byincreases of virus-specific IgG titers and the presenceof viral DNA in serum or peripheral white blood cells byPCR. The relation between viral reactivation and theclinical manifestations or treatment with systemiccorticosteroid was unclear. However, the relapse offever and increases of alanine aminotransferase andatypical lymphocytes were recognized in the sameperiod when HHV-6, HHV-7, and CMV DNA weredetected in white blood cells and serum. Lymphocyto-penia may have been related to HHV-6, as in a patientdescribed previously with a fatal CMV infection due tosevere and prolonged lymphocyte depletion associatedwith HHV-6 reactivation [Yoshikawa et al., 2002].
HHV-6,HHV-7, andCMV,which belong to thehumanbeta-herpesviruses, infectmanyhealthyadults latently,like other human herpesviruses, and are reactivatedespecially in immunocompromised patients such asorgan transplant recipients. On the other hand, theimmunomodulatory effects of beta-herpesviruses havebeen shown in vivo and in vitro: HHV-6 depletes CD4 Tlymphocytes, downregulates CD3 and CD46 transcrip-tion, alters expression of cytokines and receptors,decreases lymphocyte proliferation, and upregulatescytotoxicity of natural killer cells, while CMV impairsthe ability of lymphocytes andmonocytes to produce andrespond to cytokines, and impairs the function ofantigen-specific cytotoxic T cells and natural killer cells[Boeckh and Nichols, 2003; Dockrell, 2003].
The mechanism of viral reactivation in DIHS has notbeen elucidated. In this patient, the persistent low levelof IgGmighthave contributed tomultiple reactivation ofherpesviruses, which in turn might have contributed tothe severity of thedisease, prolongation, or recurrence of
TABLE I. Virus Specific IgG Titers
Method Day 7 Day 10 Day 28 Day 38 Day 66 Day 133
HSV EIA ND 29.6 21.7 19.9 >128 NDVZV EIA ND 8.3 10.2 6.1 13.9 NDHHV-6 FA 1:10 ND 1:2,560 1:2,560 ND 1:1,280HHV-7 FA 1:20 ND 1:80 1:160 ND 1:80CMV EIA ND 7.4 10.4 21.1 67.7 NDEBV VCA ELISA ND 1.3 0.8 0.4 1.1 ND
Day 1 represents the day when the patient first attended our hospital.The eruption was markedly worse the next day.HSV, herpes simplex virus; VZV, varicella-zoster virus; HHV-6, human herpesvirus 6; HHV-7, human herpesvirus 7; CMV, cytomegalovirus;EBV, Epstein–Barr virus; VCA, virus capsid antigen; EIA, enzyme immunoassay. Values correspond to units/ml; FA, fluorescent antibodymethod; ELISA, enzyme-linked immunosorbent assay. Values correspond to units/ml.ND, not done.
TABLE II. PCR Analysis for HHV-6, HHV-7, and CMV
Samples Method Day 10 Day 13 Day 17 Day 20 Day 31 Day 38 Day 45 Day 53
HHV-6 White blood cells Real-time PCRa ND ND 900 20 <20 <20 <20 NDSerum Nested PCR � þ � ND ND � � �
HHV-7 White blood cells Real-time PCRa ND ND 1,600 <20 <20 <20 ND NDSerum Nested PCR � � � ND ND � � �
CMV Serum Nested PCR � � þ ND ND þ þ �
PCR, polymerase chain reaction.�, not detected; þ, detected.aCounts of DNA copies per 106 cells.
DIHS Associated With Multiple Herpesviruses 433

symptoms in this patient. Hypogammaglobulinaemiaassociated with DIHS has been reported [Callot et al.,1996; Descamps et al., 2001; Aihara et al., 2003],although it is known that anticonvulsants, the majorcausal drugs of DIHS, sometimes induce hypogamma-globulinaemia even without symptoms of DIHS. In thispatient, it seems that drugs did not induce the low levelof IgG because it continued over half a year afterwithdrawal of the causal drugs.Excessive alcohol intakeand low nourishment might have caused hypogamma-globulinaemia. Other than the low level of IgG, thedecrease of peripheral lymphocytes and CD8 cells,cytotoxic T cells, or other possible immunosuppressiveconditions might have contributed to viral reactivation.In addition tomalnourishment, the immunomodulatoryeffects of herpesvirus as mentioned above might havecaused or exacerbated an immunosuppressed state.It has been reported that reactivation of HHV-6 and
HHV-7 increases the risk of subsequent CMV infectionafter organ transplantation [Boeckh and Nichols, 2003;Dockrell, 2003]. In addition, bothHHV-6andHHV-7areknown to infect and reside in circulating CD4þlymphocytes [Lusso et al., 1994], and HHV-7 infectionhas been reported to trigger reactivation of HHV-6[Tanaka-Taya et al., 2000]. Taken together, in thispatient, herpesvirus might have been reactivated oneafter another under an immunosuppressed condition,which was induced by many factors.The marked reaction to the patch test for cyanamide
under administration of 13 mg prednisolone per day,which was performed about 6 months after withdrawalof cyanamide, suggests that a strong allergic reaction tothe drug persists even after a long time.In immunocompromised patients, it might be possible
that somedrugs that have never been reported as causaldrugs of DIHS, induce DIHS.Further investigations are needed to clarify the
mechanism by which herpesviruses are reactivatedin patients with DIHS in relation to disturbance ofimmunocompetence.
REFERENCES
Aihara M, Sugita Y, Takahashi S, Nagatani T, Arata S, Takeuchi K,Ikezawa Z. 2001. Anticonvulsant hypersensitivity syndrome asso-ciated with reactivation of cytomegalovirus. Br J Dermatol144:1231–1234.
AiharaY, Ito SI, Kobayashi Y, YamakawaY, AiharaM,Yokota S. 2003.Carbamazepine-induced hypersensitivity syndrome associatedwith transient hypogammaglobulinaemia and reactivation ofhuman herpesvirus 6 infection demonstrated by real-time quanti-tative polymerase chain reaction. Br J Dermatol 149:165–169.
Aihara M, Mitani N, Kakemizu N, Yamakawa Y, Inomata N, Ito N,Komatsu H, Aihara Y, Ikezawa Z. 2004. Human herpesvirusinfection in drug-induced hypersensitivity syndrome, toxic epider-mal necrolysis and Stevens–Johnson syndrome. Allergol Int 53:23–29.
Boeckh M, Nichols WG. 2003. Immunosuppressive effects of beta-herpesviruses. Herpes 10:12–16.
Callot V, Roujeau JC, BagotM,Wechsler J, ChosidowO, Souteyrand P,Morel P, Dubertret L, Avril MF, Revuz J. 1996. Drug-inducespseudolymphoma and hypersensitivity syndrome. Arch Dermatol132:1315–1321.
Carroll MC, Yueng-Yue KA, Esterly NB, Drolet BA. 2001. Drug-inducedhypersensitivity syndrome inpediatric patients. Pediatrics108:485–492.
Descamps V, Bouscarat F, Laglenne S, Aslangul E, Veber B, DescampsD, Saraux JL, GrangeMJ,GrossinM,Navratil E, Crickx B, BelaichS. 1997. Human herpesvirus 6 infection associated with antic-onvulsant hypersensitivity syndrome and reactive haemophagocy-tic syndrome. Br J Dermatol 137:605–608.
Descamps V, Valance A, Edlinger C, Fillet AM, Grossin M, Lebrun-Vignes B, Belaich S, Crickx B. 2001. Association of humanherpesvirus 6 infection with drug reaction with eosinophilia andsystemic symptoms. Arch Dermatol 137:301–304.
Descamps V,MaheE,HouhouN, Abramowitz L, Rozenberg F, Ranger-Rogez S, Crickx B. 2003. Drug-induced hypersensitivity syndromeassociated with Epstein–Barr virus infection. Br J Dermatol 148:1032–1034.
Dockrell DH. 2003. Human herpesvirus 6: Molecular biology andclinical features. J Med Microbiol 52:5–18.
Hara S, Kimura H, Hoshino Y, Tanaka N, Nishikawa K, Ihira M,Yoshikawa T, Morishima T. 2002. Detection of herpesvirus DNA inthe serum of immunocompetent children. Microbiol Immunol 46:177–180.
Kawana S. 1997. Drug eruption induced by cyanamide (carbimide): Aclinical and histopathologic study of 7 patients. Dermatology195:30–34.
Lusso P, Secchiero P, Crowley RW, Garzino-Demo A, Berneman ZN,Gallo RC. 1994. CD4 is a critical component of the receptor forhuman herpesvirus 7: Interference with human immunodeficiencyvirus. Proc Natl Acad Sci USA 91:3872–3876.
Mitani N, Sugiyama A, Aihara M, Ikezawa Z. 2003. A case of drug-induced hypersensitivity syndrome due to mexiletine chlorideimproved by low dose prednisolone: A case report and the literaturereview of drug-induced hypersensitivity syndrome due to mexile-tine chloride. Jpn J Dermatoallergol 11:71–77 (in Japanese withEnglish summary).
Nagae Y, Nakagawa Y, Tano Y, Mori Y, Aono T. 1998. The diagnosticsignificance of polymerase chain reaction for ocular samples in viralretinitis. J Jpn Ophthalmol Soc 102:509–514 (in Japanese withEnglish summary).
NiederhoferH, StaffenW,Mair A. 2003. Comparison of cyanamide andplacebo in the treatment of alcohol dependence of adolescents.Alcohol Alcohol 38:50–53.
Rietschel RL, Fowler JF, editors. 2001. Fisher’s contact dermatitis. 5thedition. Philadelphia: Lippincott Williams & Wilkins. pp 23–24.
Suzuki Y, Inagi R, Aono T, Yamanishi K, Shiohara T. 1998. Humanherpesvirus 6 infection as a risk factor for the development of severedrug-induced hypersensitivity syndrome. Arch Dermatol 134:1108–1112.
Tanaka N, Kimura H, Hoshino Y, Kato K, Yoshikawa T, Asano Y,Horibe K, Kojima S, Morishima T. 2000. Monitoring four herpes-viruses in unrelated cord blood transplantation. Bone MarrowTransplant 26:1193–1197.
Tanaka-TayaK,KondoT,NakagawaN, InagiR,MiyoshiH, SunagawaT,OkadaS,YamanishiK. 2000.Reactivation of humanherpesvirus6 by infection of human herpesvirus 7. J Med Virol 60:284–289.
Tohyama M, Yahata Y, Yasukawa M, Inagi R, Urano Y, Yamanishi K,Hashimoto K. 1998. Severe hypersensitivity syndrome due tosulfasalazine associated with reactivation of human herpesvirus 6.Arch Dermatol 134:1113–1117.
Yoshikawa T, Ihira M, Asano Y, Tomitaka A, Suzuki K, Matsunaga K,Kato Y, Hiramitsu S, Nagai T, Tanaka N, Kimura H, Nishiyama Y.2002. Fatal adult case of severe lymphocytopenia associated withreactivation of human herpesvirus 6. J Med Virol 66:82–85.
434 Mitani et al.

Journal of Medical Virology 75:435–439 (2005)
Detection of HPV 16 and HPV 18 DNA in the Bloodof Patients With Cervical Cancer
Patti Kay,1 Bruce Allan,1 Lynette Denny,4 Margaret Hoffman,3 and Anna-Lise Williamson1,2*1Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Observatory,Cape Town, South Africa2National Health Laboratory Service, Groote Schuur Hospital, Cape Town, South Africa3School of Public Health and Family Medicine, Faculty of Health Sciences, University of Cape Town, Observatory,Cape Town, South Africa4Department of Obstetrics and Gynaecology, University of Cape Town, Observatory, Cape Town, South Africa
Persistent infection of the uterine cervix withhigh-risk human papillomaviruses (HPV) is cau-sally associated with cancer of the cervix. A fewstudies have reported the presence of HPV DNAin the blood of women with cervical neoplasia.The aim of this study was to determine if HPVDNA could be detected in whole blood of womenwith a range of cervical pathologies and withHPV 16 or 18 cervical infections and if there isa correlation between cervical lesion grade andthe appearance of HPV DNA in the circulatorysystem. Forty-five women with histologicallygraded cervical cancer were confirmed to havecervical HPV16 or 18 infections. Eleven (24.4%) ofthesewomen had detectable HPV 16 or 18 in theirblood. The HPV types detected in the bloodmatched those detected at the cervix. No HPV16 or 18 DNA was detected in the blood of32 women with pre-cursor cervical lesions ornormal cervical pathology but who had cervicalHPV 16 or 18 infections. One of 77 women withnormal cervical pathology and no cervical HPVinfection was positive for HPV 16 DNA in herblood. The results indicate that HPV DNA canbe detected in the blood of women with moreadvanced cervical carcinomas but not in theblood of womenwith pre-cursor cervical lesions.The results of our study indicate that the role ofHPVDNA in the circulatory systemappearsnotbeof diagnostic significance and HPV DNA is onlydetectable in women with more advanced cervi-cal cancers. J. Med. Virol. 75:435–439,2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: human papillomavirus; blood;cervical neoplasias; prognosis
INTRODUCTION
Cancer is driven by an accumulation of geneticchanges that occur over a period of time leading to the
development of malignant clonal lesions in the affectedpatient [Sidransky, 1997]. Blood is the only fluid that isin direct contact with all the body’s organs and thereforeoffers an attractive and non-invasive means of cancerdetection. An early study by Leon et al. [1977] revealedthe presence of circulating tumor DNA in the serum ofpatients with various types of cancer and that patientswith metastatic lesions had even higher levels of circu-lating DNA. Quantitative studies [Shapiro et al., 1983]indicated that patients with malignant disease of thegastrointestinal tract had higher levels of circulatingDNA when compared to those patients with benign dis-orders of the gastrointestinal tract.
Infection with high-risk human papillomavirus (HPV)types has been shown to be causally associated withcervical cancer with 99.7% of cervical cancers harboringHPV DNAWalboomers et al. [1999]. Globally, cancer ofthe cervix is the second most common cancer in women.Stage specific survival of cervical cancerpatientshasnotimproved since the 1960s despite advances in patienttreatment [Schwartz et al., 2001]. Seventy to eighty-fivepercent of cervical cancer patients with stage I and IIalesions are cured [NIH Consensus Statement Online,1996] whereas the 5-year survival rate of women withadvanced non-metastatic cervical carcinomas is only40%–60%.
The presence of viral DNA offers a specific tumormarker for viral associated cancers. Evidence suggeststhat Epstein–Barr virus (EBV) DNA in the plasma ofpatientswithnasopharyngeal cancer is derived from thetumor [Mutirangura et al., 1998] and shows promise of
Grant sponsor: Cancer Association of South Africa.
*Correspondence to: Prof. Anna-Lise Williamson, Institute ofInfectious Disease and Molecular Medicine, Faculty of HealthSciences, University of Cape Town, Observatory 7925, CapeTown, South Africa. E-mail: [email protected]
Accepted 2 November 2004
DOI 10.1002/jmv.20294
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

being a non-invasive tumor marker [Lo et al., 1999; Lo,2001]. In contrast, the evidence of circulatingHPVDNAas a tumor marker is more controversial as a literaturesearch reveals that few papers have been publishedon the prevalence of HPV DNA in the blood of womenwith cervical cancer and there are differences in resultsbetween studies.There is only one published study investigating the
detection of HPV DNA in the blood of 11 women with arange of cervical pathologies [Kedzia et al., 1992]. Asecond study conducted by Pao et al. [1991] investigated25 women who did not have cervical cancer but wereinfected with HPV. There are three other publishedstudies that have investigated the detection of circulat-ing HPV DNA [Liu et al., 2001; Pornthanakasem et al.,2001; Dong et al., 2002]. All three studies only in-vestigated patients with cervical cancers and did notinclude a wide range of cervical precursor lesions.In this study, we aimed to determine if HPV DNA
could be detected in the blood ofwomenwhowere knownto have cervical HPV infection and whose cervicalpathology ranged from grade IVB lesions to no pathol-ogy, and to determine if there was a correlation with thecervical lesion grade and the detection of HPV DNA inblood of these women.
MATERIALS AND METHODS
Study Population
The participants in this study were selected retro-spectively from a larger cervical cancer case-controlstudy [Hoffman et al., 2003] conducted in the urbanand peri-urban areas of the Cape Town region in theWestern Cape province of South Africa during betweenJanuary 1998 and December 2001. Only invasivecervical cancers were included in the cases thus ex-cluding four cases of cancer in situand1A.Controlswerewomenwithout cancer and were matched with cases forage, race and place of residence. A total of 227 womenwere included in the present study and divided intothree groups based on the following criteria:
. Group A consisting of women with cervical cancerstaged in accordance with the International Federa-tion of Gynecology and Obstetrics (FIGO) classifica-tion system.This group included 118women forwhichparaffin embedded cervical tissue were available.Characteristics of these women are shown in Table I.
. Group B consisting of 32 women with HPV 16 or HPV18 DNA at the cervix as determined by PGMY PCRand typing by line blot assay kindly provided byRocheMolecular Systems, Inc., Pleasanton, CA [Gravittet al., 1998, 2000]. Cervical cells were assessed byPapanicolou smear (PAP) in accordance with theBethesda system. Of the 32 group B women, 22 werepositive for HPV 16 DNA at the cervix, nine werepositive for HPV 18 DNA, and one was infected withbothHPV 16 and 18DNATwenty womenwere shownto have no cervical pathology, four presented withatypical squamous cells of unknown significance(ASCUS), three with low grade squamous intrae-pithelial lesions (LGSIL), and five with high gradesquamous intraepithelial lesions (HGSIL) (Table II).
. Group C consisting of 77 women shown to have noabnormal cervical pathology bymeans of a PAP smearand who were HPV DNA negative at the cervix byDigene HPV hybrid capture [Shapiro et al., 2003] aswell as by PGMY PCR and typing by line blot assay(kindly provided by Roche Molecular Systems, Inc.).
A detailed questionnaire was completed for eachparticipant by trained nurse interviewers. Data in-cluded age, race, number of sexual partners, parity,urban/rural living, number of PAP smears, and age offirst intercourse. Cervical cells for HPV DNA detectionwere collected using a hybrid capture cervical sampler,placed in a tube with 1 ml specimen transport medium(Digene Corporation, Gaithersburg, MD) and stored at�208C until required. Punch biopsies were obtainedfrom cervical cancer cases, fixed in formalin, andparaffin wax embedded for histological examination.Whole blood was drawn from all participants in a 5-mlcollection tube containing EDTA as an anti coagulant.Blood samples were stored at �808C until required.
TABLE I. Characteristics of Group A Women Included in theStudy With Cervical Cancer
No. of women(n¼ 118)
FIGOgrading
Age range(24–59)
Medianage (43.3)
16 IB 30–57 40.619 IBI 28–56 41.38 IBII 34–47 4020 IIB 24–59 44.423 IIIB 29–59 47.117 IVA 31–58 46.215 IVB 27–57 43.7
TABLE II. Characteristics and Clinical Staging of Group B Women
No. of women(n¼ 32)
PAPsmear
HPV 16 pos(n¼ 23a)
HPV 18 pos(n¼ 10a)
Age range(22–59)
Medianage (41.8)
20 Negative 14 7 26–57 40.54 ASCUS 3 1 42–59 48.53 LSIL 1 2 22–41 31.75 HSIL 5 0 44–58 51.2
aincludes one dual infection of HPV 16 and 18.
436 Kay et al.

DNA Extraction
Bloods. DNAwas extracted fromwhole blood usinga Qiagen DNA Blood Mini kit (Qiagen, Valencia, CA).Manufacturer’s instructions were followed with theexceptions that double the amount of blood (400 ml)was used as starting material and DNA was eluted inhalf the volume of provided elution buffer (100 ml).Extracted DNA was stored at �808C until required.
Paraffin embedded biopsies. Paraffin wax wasremoved from the fixed tissues for group A cases byadding 1,200 ml xylene. After vigorous vortexing, bio-psies were centrifuged at 20,000g. The supernatent wasremoved and 1,200 ml 96% ethanol added to the pelletto remove residual xylene. The biopsies were gentlyvortexed and centrifuged at 20,000g at room tempera-ture for 5min. The ethanolwas removedand the ethanolwash step repeated. Ethanol was evaporated off byincubating the open microfuge tubes at 378C for 15 minor until all ethanol had evaporated. Lysis buffer (180 ml;as supplied by manufacturer) was added to each biopsyand the DNA extracted as per manufacturer’s instruc-tions using a Qiagen DNA mini kit (Qiagen).
Polymerase Chain Reaction Amplification
HPV 16 and HPV 18 E6 nested type specificPCR. HPV 16 and HPV 18 type specific PCR wasperformed on DNA extracted from the bloods of GroupsA, B, and C and paraffin embedded tissue from Group Awomen using E6 nested PCR primers [Lai et al., 1996].
The outer reactionswere performed in a100ml volumeand the inner nested reactions in a 50 ml volume asfollows: PCR buffer as supplied by JMRHoldings, I unitSupertherm Taq polymerase (JMR Holdings), 1.5 mMmagnesium chloride, 200 mM each deoxyribonucleo-side triphosphate (AbGene) and 50 pmol each primer.Twenty microliters extracted DNA was added to theouter PCR reaction and 5 ml of the outer PCR productwas used as template for the inner nested reaction.Thermocycling conditions were the same for both outerand inner reactions of both sets of type specific primers.Thermocycling was performed in a Perkin Elmer 9700as follows: 958C for 3 min; 30 cycles of 958C for 30 sec,558C for 30 sec, 728C for 1min, and afinal extension stepof 728C for 5min. PCRproductswere electrophoresed ona 1.8% agarose gel containing 0.5 mg ethidium bromideand the appropriate size PCR product was visualized byexposure to ultraviolet light.
As nested PCR is highly sensitive, precautions weretaken to avoid cross contamination of samples. PCRreaction mixes, addition of template to the outer reac-tion and the nested reactions were performed in threeseparate areas, each with its own dedicated equipmentand multiple negative controls were included with eachbatch of PCR.
To ensure that adequate DNA had been extractedfrom the blood samples and paraffin embedded cervicaltissue, PCR using primers for the human CCR5 gene[Michael et al., 1999] was performed as described pre-viously [Kay et al., 2003].
RESULTS
HPV DNA was detected in 45/118 (38%) paraffinembedded cervical tissue samples using type specificPCR primers for HPV 16 and 18. Forty (89%) of the45 women with cervical HPV DNA from group A werepositive for HPV 16 and 5 (11%) were positive for HPV18 DNA at the cervix.
The CCR5 gene was detected by PCR in the bloods ofall three groups indicating that all blood samples hadadequate DNA for testing.
Eleven of the 45 (24.4%) group A women positive forHPV 16 or 18 DNA at the cervix, had detectable HPVDNA in their blood. The mean age of these women was45.8 years (range 33–55). HPV 16 DNA was detected in10 and HPV 18 in 1 of the bloods (Table III). Four of the15 women with stage IIB, 6/21 with stage IIIB and 1/2with stage IVB cancer presentedwithHPVDNA in theirblood (Fig. 1). In all cases, the HPV type detected in theblood correlated with the HPV type infecting the cervix.
None of the group Bwomen had detectable HPVDNAin their blood despite the fact these women had beenselected on the basis that theywereHPV 16 and/or HPV18 positive at the cervix.
One of the Group C women had HPV DNA 16 in herblood. This was an unexpected finding as the Group Cwomen had been selected for this study on the basis thattheywereHPVDNAnegative at the cervix by bothHPVhybrid capture and PGMY PCR/reverse line blot assayand had no cervical pathology by means of PAP smear.
There was a statistically significant difference in thedetection of HPV DNA in the blood of group A womenwhen compared to theGroupC (P value¼0.0019) and togroup B women (P value¼0.0004).
DISCUSSION
In this present study, it was found that 24.4% ofwomen with advanced cervical cancer had detectableHPV 16 or 18 DNA in their blood. Other studies usingPCR to detect circulating HPV DNA have showndetection levels ranging from 6.9% [Dong et al.,2002] to 12% [Pornthanakasem et al., 2001] and 20%[Liu et al., 2001]. The results of our study, therefore,showed a higher prevalence rate to the Dong and
TABLE III. Characteristics of Women With Detectable HPVDNA in Their Blood
Figo stage Age Race HPV type
IIB 33 MR 1653 B 1649 MR 1855 B 16
IIIB 37 B 1650 MR 1637 B 1644 B 1650 B 1646 B 16
IVB 50 B 16
MR, mixed race; B, Black-African.
Detection of HPV DNA in Blood 437

Pornthanakasem studies, but were in keeping with theLiu study and confirms that HPV DNA is detectable inthe blood of women with advanced cervical cancer. Thedifferences in study results could be due to differences inPCR sensitivity as we chose PCR primers that target aportion of the HPV E6 transforming gene which is in-variably expressed in cervical cancer cells and the PCRprimerswere used in anested reaction to ensure optimalsensitivity.We were unable to detect HPV DNA in the blood of
women with cervical HPV infection and pre-cursorlesions (group B) in contrast to a study by Pao et al.[1991] in which it was found that 52% (13/25) of womenhad detectable HPV 11, 16, or 18 DNA in their peri-pheral mononuclear cells. However, a small study byKedzia et al. [1992] on 11womenwith a range of cervicalpathologies indicated that 4/5 women with clinicallydiagnosed cervical carcinoma had detectable HPV DNAin their blood whereas all six women with lower gradelesions or inflammation were negative for HPV in theirblood. These findings are similar to those of our study.The mechanisms of release of tumor DNA into thebloodstream are not yet fully understood and it is notknown why cancer patients have larger quantities ofcirculating DNA. But the results of our study and theKedzia study suggest that entry of HPV DNA into theblood takes place at a later stage of cervical disease.The detection ofHPV16DNA in the blood of one of the
women with no cervical HPV infection or cervicalpathology was an unexpected finding, although thishas been previously reported [Dong et al., 2002]. Theresult could be due to PCR contamination, but this isunlikely as indicated by the PCR controls. There is aremote possibility of anHPV associatedmalignancy at anon-cervical site.The significance of detecting HPV DNA in the circu-
latory system is unclear as the main route of spread ofcervical cancer is directly into the adjacent tissue or viathe draining lymphnodes [DiSaia andCreasman, 1997].Lymphatic spread initially involves the parametrialnodes and finally metastases into the periaortic nodes.In a high percentage of patients, death from cervicalcancer is due to local extension. In an autopsy study byHenriksen [1949], it was found that only 27% of womenhadmetastasesabove theaortic chain.Metastases to the
lungs occurs in less than 15% of women with cervicalcancer [DiSaia and Creasman, 1997] and it could behypothesized that women with circulating HPV DNAcould be at higher risk of developing metastases to thelung or other distant organs. Results of a study by Tsenget al. [1999] suggests that the detection of HPVE6mRNA in the blood of womenwith advanced cervicalcancer could act as a early marker identifying patientsat risk for metastases.
In conclusion, the results of this study confirmed thatHPV 16 and 18 DNA is detectable in the blood of womenwith cervical cancer and indicated that the release ofthisHPVDNAoccurs at a later stage of disease. Itwouldappear from the results of our study that the detection ofcirculating HPV DNA has no diagnostic value as only asmall number of women were shown to have circulatingHPV DNA and the appearance of this DNA in the bloodwas only detected in late stage cancers. However, thedetection of circulating HPV DNA may have prognosticvalue in that it could serve as a marker for women whoare at risk for distant blood borne metastases, butfurther long term follow up studies are required toverify these findings as the dynamics of metastasis arecomplex.
ACKNOWLEDGMENTS
WeacknowledgeDr. S. Shapirowhowas the PrincipalInvestigator of the larger case control study from whichwe drew our samples. We are indebted to the gynecol-ogists of Groote Schuur hospital for providing thebiopsies and the staff of the School of Public Healthand Primary Health Care for collection of blood samplesand cervical cells. We are also indebted to the patholo-gists from the Department of Anatomical Pathology atthe University of Cape Town for grading of the biopsiesand the cytologists for grading of the PAP smears. Wealso thank Roche Molecular Systems for providing thePGMY primers and line blot assay to type HPV incervical samples.
REFERENCES
DiSaia PJ, Creasman WT. 1997. Clinical gynecologic oncology. 5thedition. Missouri: Mosby-Year Book, Inc.
Dong SM, Pai SI, Rha S-H, Hildesheim A, Kurman RJ, Schwartz PE,Mortel R, McGown L, Greenberg MD, Barnes WA, Sidransky D.2002. Detection and quantitation of human papillomavirus DNA inthe plasma of patients with cervical carcinoma. Cancer EpidemiolBiomark Prev 11:3–6.
Gravitt PE, Peyton CL, Apple J, Wheeler CM. 1998. Genotyping of27 human papillomavirus types by using L1 consensus PCRproducts by a single-hybridisation, reverse line blot detectionmethod. J Clin Microbiol 36:3020–3027.
Gravitt PE, Peyton CL, Alessi TQ, Wheeler CM, Coutlee F,Hildesheim A, SchiffmanMH, Scott DR, Apple RJ. 2000. Improvedamplification of genital human papillomaviruses. J Clin Microbiol38:357–361.
Henriksen E. 1949. The lymphatic spread of carcinoma of the cervixand of the body of the uterus: A study of 420necropsies. AmJObstetGynecol 58:924.
Hoffman M, Cooper D, Carrara HRO, Rosenberg L, Kelly J, Stander I,Williamson A-L, du Toit G, Shapiro S. 2003. Limited Pap screeningassociatedwith reduced risk of cervical cancer in SouthAfrica. Int JEpidemiol 32:573–577.
Kay P, Soeters R, Nevin J, Denny L, DeHaeck CMC, Williamson A-L.2003. High prevalence of HPV 16 in South African women with
Fig. 1. Detection of human papillomaviruses (HPV) 16/18 positivebloods in patients with HPV 16/18 associated cervical cancer.
438 Kay et al.

cancer of the cervix and cervical intraepithelial neoplasia. J MedVirol 71:265–273.
Kedzia H, Gozdzicka-Jozefiak A, Wolna M, Tomczak E. 1992.Distribution of human papillomavirus 16 in the blood of womenwith uterine cervical cancer. Eur J Gynecol Oncol 13:522–526.
Lai YM, Yang F-P, Pao CC. 1996. Human papillomavirus deoxyribo-nucleic acid and ribonucleic acid in plasma and sperm cells. FertilSteril 65:1026–1030.
Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. 1977. Free DNA in theserum of cancer patients and the effect of therapy. Cancer Res37:646–650.
Liu VWS, Tsang P, Yip A, Ng T-Y, Wong L-C, Ngan HYS. 2001. Lowincidence of HPV DNA in sera of pretreatment cervical cancerpatients. Gynecol Oncol 82:269–272.
Lo YM. 2001. Quantitative analysis of Epstein–Barr virus DNA inplasmaand serum:Applications to tumor detection andmonitoring.Ann NY Acad Sci 945:68–72.
Lo YM, Chan LY, Chan AT, Leung SF, Lo KW, Zhang J, Lee JC, HjelmNM, Johnson PJ, Huang DP. 1999. Quantitative and temporalcorrelation between circulating cell-free Epstein–Barr virus DNAand tumor recurrence in nasopharyngeal carcinoma. Cancer Res59:1188–1191.
Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, Birx DL,Sheppard HW. 1999. The role of viral phenotype and CCR-5 genedefects in HIV-1 transmission and disease progression. Nat Med3:338–340.
Mutirangura A, Pornthanakasem W, Theamboonlers A, SriuranpongV, Lertsanguansinchi P, Yenrudi S, Voravud N, Supiyaphun P,Poovorawan Y. 1998. Epstein–Barr viral DNA in the serum ofpatients with nasopharyngeal carcinoma. Clin Cancer Res 4:665–669.
NIH Consensus Statement Online. 1996. April 1–3; 43:1–3. http://consensus.nih.gov/cons/102/102_intro.htm
Pao CC, Lin S-S, Lin C-Y, Maa J-S, Lai C-H, Hsieh T-T. 1991.Identification of human papillomavirus DNA sequences in periph-eral blood mononuclear cells. Am J Clin Pathol 540–546.
Pornthanakasem W, Shotelersuk K, Termrungruanglert W, VoravudN, Niruthisard K, Mutirangura A. 2001. Human papilloma-virus DNA in plasma of patients with cervical cancer. BMC Cancer1:2.
Schwartz SM, Daling JR, Shera KA, Madelaine MM, McKnight B,Galloway DA, Porter PL, McDougall JK. 2001. Human Papilloma-virus and prognosis of invasive cervical cancer: A population-basedstudy. J Clin Oncol 19:1906–1915.
Shapiro B, Chakrabarty M, Cohen EM, Leon SA. 1983. Determinationof circulating DNA levels in patients with benign or malignantgastrointestinal disease. Cancer 51:2116–2120.
Shapiro S, Carrara H, Allan BR, Hoffman M, Rosenberg L, Kelly JP,Cooper DD, Williamson AL. 2003. Hypothesis: The act of taking aPapanicolaou smear reduces the prevalence of human papilloma-virus infection: A potential impact on the risk of cervical cancer.Cancer Causes Control 14:953–957.
Sidransky D. 1997. Nuclei acid-based methods for the detection ofcancer. Science 278:1054–1058.
Tseng C-J, Pao CC, Lin J-D, Soong Y-K, Hong J-H, Hsueh S. 1999.Detection of human papillomavirus types 16 and 18 mRNA inperipheral blood of advanced cervical cancer patients and itsassociation with prognosis. J Clin Oncol 17:1391–1396.
Walboomers JMM, Jacobs MV, Manos MM, Bosch FX, Kummer JA,Shah KV, Snijders PJF, Peto J, Meijer JLM, Munoz N. 1999.Human papillomavirus is a necessary cause of invasive cervicalcancer worldwide. J Pathol 189:12–19.
Detection of HPV DNA in Blood 439

Journal of Medical Virology 75:440–446 (2005)
Molecular Epidemiology of Adenovirus StrainsIsolated From Patients With Ocular Disease inthe Area of Thessaloniki, Greece (1998–2002)
Filanthi Frantzidou,1* Aikaterini Pavlitou,1 Asimina Mataftsi,2 Kamal Dumaidi,1
and Nikolaos Georgiadis2
1A’ Department of Microbiology, School of Medicine, Aristotle University of Thessaloniki, Greece2Eye Clinic, AHEPA Hospital, School of Medicine, Aristotle University of Thessaloniki, Greece
Thirty strainsof adenovirus (Ads) associatedwithocular disease have been isolated over a periodof 4 years in Thessaloniki, Northern Greece.Eleven strains were isolated from sporadicpatients with conjunctivitis or keratoconjunctivi-tis in Thessaloniki city between 1998 and 2000.Nineteen strains were isolated from patientswith keratoconjunctivitis during an outbreak ofAds in the area of Thessaloniki (Thessaloniki andSerres cities) in 2002. PCR-sequence methodusing primers targeted against the hypervariableregions (HVRs) of hexon gene, as well as theneutralization test were used for typing the Adisolates and assessing a possible relation amongthese strains, and their genetic variability. Ad4with very close homology to variant Z-G 95-873was the most frequent genotype causing spora-dic conjunctivitis over a period of 4 years. Twoother strains, one Ad2, and one Ad3 were similarto the prototype ones, and a third one showsclose homology to the variant of prototype Ad15,theMorrisonstrain. Thegenome typingof twentytwo Ad8 isolates showed very close homologyin their amino acid and nucleotide sequences tothe variant of Ad8, strain 1127 (accession no.X74663). Four were isolated from patients withkeratoconjunctivitis in 1998, 1999, 2000 and 18during the outbreak in 2002. As far as strain 1127is concerned, all the Ad8 isolates showed thesame changes in the HVR 1 and HVR 2 except oneisolate in 1998, which showed some changesoutside the HVRs. During the outbreak of Ad8keratoconjunctivitis, it was not possible to iden-tify the exact source of infection (nosocomial or/and outpatients). Finally, Ad4 variant Z-G 95-873and Ad8 which is closely related to the strain1127, were found to be the predominant adeno-viruses circulating in Northern Greece during1998–2002. J. Med. Virol. 75:440–446, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: Greece; adenovirus; (kerato)-conjunctivitis; outbreak; poly-merase chain reaction withsequencing analysis; genometyping
INTRODUCTION
To date, 51 different serotypes of adenoviruses (Ads)have been identified [de Jong et al., 1999]. They areclassified into six subgenera,A–F, on thebasis of nucleicacid differences, fiber protein characteristics, and bio-logical properties [Wadell et al., 1986; Murphy et al.,1995]. The principal adenovirus types 2, 3, 4, 6, 7, 8, 19,and37havebeen found to be responsible for themajorityof ocular infections. Acute conjunctivitis may occur aspart of a pharyngo-conjunctivitis syndrome or as sepa-rate entity. The most frequently encountered types areadenovirus 3 (subgenus B), and adenovirus 4 (subgenusE). Other types such as adenovirus 1, 2, and 6 (subgenusC) and 9, 10, 15, 17, 20, 22, 29 (subgroup D) and ad-enovirus 7,16 of subgroup B have been reported to causeconjunctivitis. Adenovirus type 4 is a significant cause ofoutbreak of respiratory disease amongmilitary recruits,but it has also been associated with follicular conjuncti-vitis [Aoki et al., 1982; Cooper et al., 1993; Schepetiuket al., 1993]. InWestern countries adenoviral conjuncti-vitis is frequently reported as swimming pool conjuncti-vitis or nosocomial conjunctivitis. Certain members ofsubgenus D, namely the Ad8, Ad19a, and Ad37, causeoutbreaks of keratoconjunctivitis and rapid identifi-cation of these serotypes can help in prevention and
*Correspondence to: Filanthi Frantzidou, A’ Department ofMicrobiology, School of Medicine, Aristotle University of Thessa-loniki, Thessaloniki 54124. E-mail: [email protected]
Accepted 25 November 2004
DOI 10.1002/jmv.20286
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

control [Jawetz et al., 1955; Darougar et al., 1977; Kempet al., 1983].
Diagnosis of adenoviral infections is based on virusisolation in tissue culture, antibodies studies or antigendetection by immunofluoerescence [Wadell et al., 1999].However, virus isolation in tissue culture is laborious,time consuming, especially as Ad 8 [Hanna and Jawetz,1962;Wigandetal., 1983],Ad40andAd41 [deJongetal.,1992] are fastidious with slow and inefficient growth incell culture. The need for rapid and sensitive detectionhas led to polymerase chain reaction (PCR) being thebest established among all other methods. Adenovirusserotyping can be performed by neutralization orhemagglutination inhibition test or by sequencing and/or restriction fragment length polymorphism analysis(RFLP). The neutralization test is a type specificmethod, since it based on the use of antibodies directedagainst epitope(s) on the hexon [Norby, 1969]. RFLP isalso a powerful and practical tool to differentiate theadenoviral serotypes. Recently, PCR assay and sequen-cing analysis have been used for the diagnosis andserotyping ofAds, especially the fastidious serotypes, forwhich DNA restriction analysis is not possible [Morriset al., 1995; Cooper et al., 1999; Takeuchi et al., 1999].The combination of PCR and sequencing analysis ismore sensitive and rapid than virus isolation in tissueculture [Pring-Akerblom and Adrian, 1994]. RFLP mayfail in serotyping a virus in the presence of a pointmutation on the restriction site, whereas sequencinganalysis overcomes this disadvantage. Studies on sequ-ences coding for thehexonproteins in several adenoviralstrains showed that while most of the pedestal regionsare conserved, variable regions exist in L1 and L2[Kinlochet al., 1984;Pring-AkerblomandAdrian, 1994].Seven discrete hypervariable regions (HVRs) on thehexon that make up serotype-specific loops on thesurface of the protein have been identified [Adrianet al., 1989; Toogood et al., 1989; Crawford-Miksza andSchnurr, 1996a]. Six variable regions exist inL1and onein L2. Mutations in the HVRs can be extensive andmaylead to antigenic shift, drift and the evolution of a newserotype [Wadell et al., 1980; Crawford-Miksza andSchnurr, 1996b].
The numbers of studies that have looked into theepidemiology of adenoviral eye infections in Greeceare scarce. In an outbreak of keratoconjunctivititisin Northern Greece (1992–1993), Ad8 was the onlyserotype (typed by neutralization test) to be isolated[Frantzidou et al., 1994]. During the last few years, anincrease in the isolation of Ads at the ThessalonikiUniversity Virus Laboratory was observed, and mostisolates originated from local eye hospitals. This studyreports the detection and genetic characterization ofadenovirus strains isolated from patients with ocularinfection in Thessaloniki city inNorthernGreece, duringthe period 1998–2000, as well as findings from an out-break of Ad8 that occurred in the Thessaloniki area(Thessaloniki andSerres) fromMarch toDecember 2002.
The neutralization test and PCR-gene sequencemethod using primers targeting HVRs of hexon were
carried out in order to type the adenovirus isolates,assess their genetic variability and possible relationamong the strains.
MATERIALS AND METHODS
Patients and Virus Strains
During the years 1998–2002, 70 clinical ocularsamples were studied for Ads. The samples originatedfrom patients with acute conjunctivitis or keratoconjuc-tivitis and sent to the laboratory fromAHEPAHospital,Saint Dimitrios Public Hospital in Thessaloniki, and aprivate ophthalmology clinic in Serres, a city 80 kmaway from Thesssaloniki. Twenty-five samples camefrom sporadic cases in the city of Thessaloniki duringthe period from March 1998 to December 2000, and45 samples froman outbreak that occurred in the area ofThessaloniki from March to December 2002.
The outbreak lasted 10 months, but was recognizedat the end of April. During the outbreak 91 affectedsubjects were identified, of which: (1) 57 were out-patients seen at the AHEPA hospital, 3 being membersof the same family. One patient was subjected to eyeexamination in the AHEPA hospital 10 days before hisinfection. (2) Six were doctors working at the eye clinic,(3) three were inpatients in the neurosurgical clinic ofthe AHEPA hospital, probably representing nosocomialinfections, and (4) 25 were outpatients seen in a privateeye clinic inSerres.Clinical and epidemiological data forpatients were derived from the hospital files and fromthe private ophthalmologist. Out of 91 reported patientspresenting with keratoconjunctivitis during the out-break samples were obtained and investigated in only45 cases.
Thessaloniki and Serres are two cities located innorthern part of Greece. Thessaloniki is a large city andconsidered as the capital of North with a population of1.5 million, and Serres is a city located to the north ofThessaloniki with 50,000 inhabitants. The people in thetwo cities are in a regular and continuous movement.
Thirty strains of adenovirus (11 from sporadic casesand 19 from the outbreak) were isolated and investi-gated further. Each isolate was obtained from differentpatient. Patients who proved negative for an adenoviralinfection were investigated for other pathogens (Herpessimplex, Chlamydia trachomatis, and other commonbacteria).
Virus Culture—Neutralization Test
Cotton-tipped conjunctival swabs from patients sus-pected to have an adenoviral infection were collected invirus transport medium and inoculated onto Hep-2 andA-549 cells. Infected cells were identified by immuno-fluorescent-antibody technique with murine monoclo-nal antibody, specific to a common epitope on theadenovirus hexon protein, conjugated to FITC (DacoLtd., Ely, Cambridge, UK). One hundred TCID50 of eachisolate was used for virus neutralization tests, whichwere performed on A549 cells. Anti-rabbit sera used in
Molecular Epidemiology of Adenovirus Strains 441

this study were obtained from Denka Seiken Co. Ltd.(Tokyo, Japan).
PCR- Sequence MethodDNA Extraction
Samples preparation and DNA extraction frominfected cells were done using the QIAamp blood Kitprotocol (QIAGEN Cmbh, Hilden, Germany) accordingto the instructions of the manufacturer.
Primers and PCR Procedure
Adenovirus is a double-stranded DNA virus, withthe hexon accounting for about 2.8 kbp [Kinloch et al.,1984]. The set of primers included HX5-1 (forwardprimer), 50-AAGATGGCCACCCCCTCGATGATGCCG-CAGT-30, and HX3-1 (reverse primer), 50-CACTTATG-TGGTGGCGTTGCCGGCCGAGAACGG-30 (InvitrogenLtd., Paisley, Scotland). These primers amplify theregion that corresponds to 1–2,829 bp in the hexon basesequence of adenovirus type 3 [Takeuchi et al., 1999].The PCR protocol of Takeuchi was modified to accountfor the Herculase enzyme used. Briefly, 2 ml DNAextracted from infected cells was used in a total reactionvolume of 30 ml of PCR. The reaction solution contained1xHerculase buffer (Stratagene, Jolla, CA), 400 mMcon-centrations of each deoxynucleoside triphosphate(dNTPs), (Gibco BRL, Inchiman, UK), 50 pmoles of eachprimer, and 2.5 U of Herculase DNA polymerase.Thermal cycling was performed in Peltier ThermalCycler PTC-200 (MJ Research, Watertown, MA) for atotal of 40 cycles of denaturing at 948C for 30 sec,annealing at 688C for 5 sec, and extension at 728C for10 min. Five to 10 ml of PCR product was analyzed in a1.5% TBE agarose gel, stained with ethidium bromide.
Sequence Analysis
PCR products of samples were purified by usingstandard protocol (Concert Rapid Purification System,GibcoBRL) and analyzed bymeans of an auto sequencer(MWG Biotech, Ebersberg, Germany) and the follow-ing primers were used for sequencing analysis: senseprimersS-29: 50-GCCAGCACRTWCTTTGACAT-30, and
S-51: 50CCCAACAGACCCAAYTACA T-30, and anti-sense primer S-54: 50-CCAGCATTGCGGTGGTGRTT-30 [Takeuchi et al., 1999]. These primers were success-fully used to sequence a nucleotide portion of 1,630 bp ofthe hexon gene that contains all seven HVRs. Thenucleotide sequence of amplified hexon gene for eachisolate of Ads was analyzed using BLAST program ofNBCI (National center for Biotechnology Information).
The genotype determined by the PCR-sequencemethod was based on homology of our sequence datawith published sequences in GenBank.
RESULTS
Overall, 30 strains of Ads were isolated. All isolateswere obtained from distinct patients. Patients fromwhom adenovirus was not isolated were also foundnegative for infection byHerpes simplex virus, Chlamy-dia trachomatis, and other common bacteria. From1998 to 2000, eleven strains of adenovirus were isolatedfrom 25 sporadic cases of conjunctivitis or keratocon-junctivitis in Thessaloniki city. The year of isolation,hospital, symptoms, results of typing by neutralizationtest and sequencing analysis are given in Table I.
The portion of the hexon gene used for nucleotideanalysis was 1,630 bp and the estimation of geneticvariation was restricted to the hexon gene regionsequenced and analyzed. The eleven strains from 1998to 2000 show the following homology in their amino acidand nucleotide sequences with adenovirus isolatespublished in GenBank: adenovirus type 2 (Ad2) shows94% and 99% homology with Ad2 possessing the ac-cession no. JO1917 in GenBank, respectively. Adeno-virus type 3 (Ad3) shows 91.8% and 98.6% homologywith Ad3 possessing accession no. X76549, respectively.The isolate that was not typed by neutralization test,due to unavailability of antiserumshows84%and 95.5%homology with adenovirus type 15H9 (Morrison) pos-sessing accession no. X76707, respectively. The fourstrains of adenovirus type 4 (Ad4) that were isolatedfrom patients with conjunctivitis or follicular conjuncti-vitis show 99.6% and 99.7 % homology respectively withthe isolate Z-G95-873 (accession no. F065064). In thesestrains of Ad4 in relation to isolate Z-G95-873, a
TABLE I. Sporadic Cases of Ocular Disease Investigated for Adenoviruses in Thessaloniki City in 1998–2000
Patientnumber
Year ofisolation Hospital Symptoms
Neutralizationtest (NT)
Nucleotide homology withstrains published in GenBank
1 1998 AHEPA Follicular conjunctivitis Ad4 99.7% with AF0650642 1998 AHEPA Keratoconjunctivitis Ad8 98.7% with X746633 1999 S. Dimitrios Conjunctivitis Ad4 99.7% with AF0650644 1999 S. Dimitrios Follicular conjunctivitis Ad4 99.7% with AF0650645 1999 S. Dimitrios Conjunctivitis Ad4 99.7% with AF0650646 1999 S. Dimitrios Conjunctivitis NDa 95.5 % with X767077 1999 S. Dimitrios Keratoconjunctivitis Ad8 98.7% with X746638 2000 S. Dimitrios Conjunctivitis Ad3 98.6% with X765499 2000 S. Dimitrios Conjunctivitis Ad2 99% with J0191710 2000 AHEPA Keratoconjunctivitis Ad8 98.8% with X7466311 2000 AHEPA Keratoconjunctivitis Ad8 98.8% with X74663
aAntiserum not available.
442 Frantzidou et al.

replacement of 428 amino acid in HVR1 (P 428 T) andanother replacement of 457 amino acid outside theHVRs were observed. Among the four strains of ad-enovirus type 8 that had been isolated during the above-mentioned period, one isolate in 1998 shows 89.7% and98.7% homology in its amino acid and nucleotidesequence respectively with the isolate 1127 (accessionno. X74663), while the isolates of Ad8 in 1999 and 2000show 91% and 98.8% homology with the isolate 1127,respectively.
During the outbreak of keratoconjunctivitis in 2002in the area of Thessaloniki, 19 strains of Ads wereisolated from 45 ocular samples. Except from one isolateof adenovirus type 4, which is identical to Ad4 strainsisolated in the previous years, the remaining 18 isolateswere typed by neutralization test as adenovirus type 8.Themonth of isolation, distribution, patient’s class, andhomology in nucleotide sequence with the isolate 1127(accession no. X74633) are given in Table II. Among 18isolates 13 came from the AHEPA hospital in Thessa-loniki and 5 from a private clinic in Serres. The isolatesfrom Thessaloniki and Serres cities share 91% and98.7%–98.8% homology in their amino acid and nucleo-tide sequences respectively with the isolate 1127, andwere identical to Ad8 isolates in 1999 and 2000.
Figure 1 shows the comparison of the deduced aminoacid sequences of seven HVRs among four representa-tive strains of adenovirus type 8 isolated inThessaloniki(1998, 2000, and 2002) and Serres (2002).
The nucleotide sequences of the four isolates ofadenovirus type 8 showed 98.7%–98.8% homology withthe isolate 1127 (X74663). Furthermore, the deducedamino acid sequences starting from position 22 of theisolate 1127 (X74663) showed 89.7%–91% homology.All four analyzed strains showed the same amino acid
changes in the HVR1 and HVR2. In addition to thesechanges, the Ad8 (1998) showed some changes outsidethe HVRs in hexon gene.
DISCUSSION
In this study, 30 isolates of adenovirus from differentpatients with ocular disease over a period of 4 yearsin the Thessaloniki area were typed by the neutraliza-tion test and PCR-sequence method using primerstargeted to the HVRs of hexon gene. PCR is moresensitive and rapid than virus isolation in tissue culturefor diagnosis [Pring-Akerblom and Adrian, 1994].Although, RFLP analysis could better reflect genomicvariations among isolates belonging to a same serotype,the sequencemethodwasapplied in this study because avariety of adenovirus isolates belonging to differentserotypes had to be analyzed. Sequencing analysis hasan advantage on RFLP since a point mutation in therestriction site may lead to failure of serotyping therespective virus by RFLP. From 1998 to 2000, elevenstrains of adenovirus belonging to types 2, 3, 4, 8, and 15were isolated from sporadic cases of conjunctivitis.During the epidemic of keratoconjunctivitis in 2002, 18strains Ad8 and 1 strain Ad4 were isolated.
Adenovirus type 4 (Ad4) is a known cause of con-junctivitis.OutbreaksofAd4havebeenreported inAsia,Europe, and Australia [Aoki et al., 1982; Cooper et al.,1993; Schepetiuk et al., 1993]. Two strikingly dissimilargenome types of Ad4 prototype and Ad4a have beendemonstrated by DNA restriction enzyme analysis(RFLP) to be implicated [Li and Wadell, 1988a; Itakuraet al., 1991]. TheAd4agenome type is isolated commonlyin Japan as cause of conjunctivitis [Aoki and Tagara,2002]. In this study,Ad4was isolated fromonepatient in1998, three patients in 1999, and one patient in 2002.
TABLE II. Cases of Ocular Disease Investigated for Adenoviruses During an Outbreak of Keratoconjunctivitis in ThessalonikiArea From March to December 2002
Patientnumber
Month ofisolation Location Patient’s class
Neutralizationtest
Nucleotide homology withstrains published in GenBank
12 May Serresa Outpatient Ad4 99.7% with AF06506413 April Thessalonikib Outpatient Ad8 98.8% with X7466314 April Thessalonikib Doctor in eye clinic Ad8 98.8% with X7466315 April Thessalonikib Doctor in eye clinic Ad8 98.8% with X7466316 April Thessalonikib Inpatient Ad8 98.8% with X7466317 April Thessalonikib Inpatient Ad8 98.8% with X7466318 April Serresa Outpatient Ad8 98.7% with X7466319 April Serresa Outpatient Ad8 98.7% with X7466320 May Serresa Outpatient Ad8 98.7% with X7466321 May Serresa Outpatient Ad8 98.7% with X7466322 June Thessalonikib Outpatient Ad8 98.8% with X7466323 September Thessalonikib Outpatient Ad8 98.8% with X7466324 September Thessalonikib Outpatient Ad8 98.8% with X7466325 September Thessalonikib Outpatient Ad8 98.8% with X7466326 September Thessalonikib Outpatient Ad8 98.8% with X7466327 September Thessalonikib Outpatient Ad8 98.8% with X7466328 October Thessalonikib Outpatient Ad8 98.8% with X7466329 November Thessalonikib Outpatient Ad8 98.8% with X7466330 December Thessalonikib Outpatient Ad8 98.8% with X74663
aPrivate clinic.bAHEPA hospital.
Molecular Epidemiology of Adenovirus Strains 443

Ocular symptoms due to Ad4 showed a moderateseverity and ranged from mild to severe. All isolates ofAd4 were identical to each other and showed 99.6% and99.7% homology in their amino acid and nucleotidesequences with the variant Z-G 95-873 (accession no.AF065064), respectively. In a study of strain variation ofAd4 serotype it was found that the evolution of Ad4 wascomplex, with continuous genetic drift punctuated byreplacement with a new strain [Crawford-Miksza et al.,1999]. The strain Z-G 95-873 appeared in the UnitedStates in 1995 and all isolates ofAd4 recovered since thattime were this new strain. The variant Ad4 AF065064was found to reside innorthernGreece during theperiod1998–2002. However, because of absence of data con-cerning adenovirus type 4 before 1998, it is not possibleto know when this variant first appeared in the area.Adenovirus type 3 (Ad3) is a common cause of con-
junctivitis worldwide, and an alternation of different
genotypes of Ad3 has occurred in different countries[Guo et al., 1988; NIH, 1995; Itoh et al., 1999; Saitoh-Inagava et al., 1999]. More than twenty genotypes ofAd3 have been identified by RFLP analysis [Li andWadell, 1988b; Itoh et al., 1999]. The Ad3 genotypeisolated in this study was related closely to Ad3 isolatewith accession no. Ad3X76549. O’ Donnell et al. [1993]reported that Ad3 prototype was isolated more fre-quently from patients with conjunctivitis than otherAd3 genotypes during the period from 1981 to 1988 inGlasgow, except in 1986 when Ad3a was the most fre-quent Ad3 genotype that had been isolated. In addition,the Ad3 prototype has been reported to be predominantin Europe, Brazil, Africa, and Australia [Wadell, 1990].
The adenovirus type 2 (Ad2) isolate in our series wasclosely related to Ad2 with accession no. JO1917. Thisstrain of Ad2 was isolated from a child with pharyngo-conjunctivitis. The association of Ads with this clinical
Fig. 1. Comparison of deduced amino acid sequences of seven hypervariable regions (HVRs) among thegenome types of adenovirus type 8 isolated in Thessaloniki (The 1998, The 2000, The 2002) and Serres (Ser2002). The sequences of loop 1 and loop 2 have been aligned to obtain maximal homology. Deduced aminoacids of Ad8 isolate 1127 accession numberX74663was obtained fromGenBank. The shaded regions showthe changes of amino acid.
444 Frantzidou et al.

presentation has been reported in the 50s [Bell et al.,1955]. Finally, the Ad15 isolatewas a variant of theAd15prototype and showed 84% and 95% homology withAd15H9withaccessionno.X76707 (Morrison).ThisAd15was isolated from a sporadic case of mild conjunctivitis.
Ad8 is a classic and very contagious agent causingepidemic keratoconjunctivitis, which may be trans-mitted by ocular examination instruments [Richmondet al., 1984]. Ad8 has amuch higher tropism for conjunc-tival cells than Ad19 and Ad37 and produces severeclinical manifestations and pathological alterations.This type persists in the population and causes sporadicepidemics in many countries [Kemp and Heirholtzer,1986; Chastel et al., 1988; de Jong et al., 1992; Corsaroet al., 1999; Tanaka et al., 2000; Adhikary et al., 2003].Many investigators have used genomic characterizationby different restriction endonucleases to establishgenomic variants of Ad8, and around 20 genotypes havebeen identified [Fujii et al., 1983, 1984; Kemp et al.,1983; Wadell, 1984]. Genome typing can be also donewith subgenus- or type-specific PCR primers [Adrianet al., 1994; Tanaka et al., 2000; Adhikary et al., 2003].Recently, some methods have suggested that the com-bination of PCR and RFLP analysis facilitate the typingprocedure [Kidd et al., 1996; Saitoh-Inagava et al.,1996].However, thesemethodsare incomplete or limited,with results difficult to interpret. Although the RFLPanalysis couldbetter reflect thevariationamong isolatesbelonging to the same serotype, in this study, the PCR-sequence method was used for various reasons; not allneutralizing antisera for Ads were available; no refer-ence strain for all the Ads was available; a varietyof adenovirus isolates belonging to different serotypeshad to be analyzed. Finally, data concerning theAds thatcirculate in this geographical region was unavailable.
The DNA sequence of hexon HVRs of all our Ad8isolates (18 strains in the 2002 outbreak and 4 strainsin 1999 and 2000) were identical and showed 98.7%–98.8% homology to Ad8 strain 1127 (accession no.X74633). The isolate 1127, which is closely related toAd8A, Ad8B, and Ad8E [Adhikary et al., 2003], wasfirst isolated from patients with keratoconjunctivitisin Germany by Wigand et al. [1983] and was laterdeposited in GenBank under the accession no. X74663[Pring-Akerblom and Adrian, 1994].
During the outbreak, itwasnotpossible to identify theexact source of infection (nosocomial or outpatients),because Ad8 was isolated from doctors working in theeye clinic, inpatients in another clinic at the AHEPAhospital, as well as from outpatients with keratocon-junctivitis presented at the AHEPA hospital and at aprivate clinic in Serres, all at the same time.However, itis worthwhile mentioning that not all patients whopresented with keratoconjunctivitis to the eye clinicswere investigated virologically. The transmission withinthe eye clinic in AHEPA hospital was monitored oncecontrolmeasureswere applied. Thessaloniki and Serrescities are located in the northern part of Greece. Thepeople in the two cities are in a regular and continuousmovement. Thus, it is possible that the outbreak started
in one city and transferred to another by a patient orhealth workers, e.g. doctors, nurses.
An adenovirus strain identical to prototype Ad8 (Trimstrain) was isolated from three patients with kerato-conjunctivitis residing inAthens,Greece, in 1980–1982,by using a neutralization test and restriction enzymelength polymorphism analysis [Kemp and Heirholtzer,1986]. In 1992 to 1993 an outbreak of keratoconjuncti-vitis due to Ad8 occurred in the area of Thessaloniki[Frantzidou et al., 1994]. In this outbreak, Ad8 was iso-lated in tissue culture from eight patients and identifiedonly by serum neutralization test, therefore, we cannotknow if this strain was the prototype of Ad8. The isolateAd8 in this study, which is very closely related to strain1127 of Ad8, was firstly identified to be endemic in thearea of Thessaloniki, in 1998 by our group. The isolatesof Ad8 in 1999, 2000, and 2002 during the outbreakwereidentical. The isolate in 1998 showed some changesoutside the HVRs. As it is well known that the genomicvariants of Ad8 may allow a virus to persist in thepopulation over a long period [Kemp and Heirholtzer,1986; Ishiik et al., 1987; Adrian et al., 1994], there isa possibility that the strains of the outbreak of Ad8keratoconjunctivitis in 1992–1993 were identical orsimilar to strain 1127 (accession no. X74633).
In conclusion, this study showed that the predomi-nant typesofAds inpatientswithocular infectionsovera4-year period were: (1) a variant of Ad4, strain Z-G 95-853 (accession no. AF064065) in sporadic cases ofconjunctivitis, and (2) a variant of Ad8, which showedvery close homology to the strain 1127 (accession no.X74663) was the causative agent in sporadic cases ofkeratoconjunctivitis in 1998, 1999, 2000 and in anoutbreak of keratoconjunctivitis in 2002. The PCR-sequence method is an efficient, accurate, and rapidmeans of diagnosis and typing of the adenovirus and hassignificant clinical and epidemiological implications.Finally, this study provides important information onthe relationships between the existing adenoviralserotypes, their HVRs sequences, and presence ofsporadic cases and of a keratoconjunctivitis outbreakthat occurred in this region of the world.
REFERENCES
AdhikaryAK,NumagaJ,KaburakiT,KawashimaH,AraieM, IkedaY,Ogino T, Suzuki E, UshijimaH,Mukoyama A, Matsuno S, Inada T,Okabe N. 2003. Genetic characterization of adenovirus type 8isolated in Hiroshima city over a 15 years period. J Clin Pathol56:120–125.
Adrian T, Best B, Hierholzer JC,Wigand R. 1989. Molecular epidemio-logy and restriction site mapping of adenovirus type 3 genometypes. J Clin Microbiol 27:1329–1334.
Adrian T,Wolf U, LauerHG,WigandR. 1994. Restriction sitemappingof adenovirus type 8 genome types. Res Virol 141:611–624.
Aoki K, Tagara T. 2002. A twenty-one years surveillance of adenoviralconjunctivitis Sapporo, Japan. Int Ophthalmol Clin 42:49–54.
Aoki K, Kato M, Ohtsuka H, Ishii K, Nakazono N, Sawada H. 1982.Clinical and etiological study of adenoviral conjunctivitis, withspecial reference to adenovirus types 4 and 19 infections. Br JOphthalmol 66:776–780.
Bell JA, Rowe WB, Engler JI, Parrott RH, Huebner RJ. 1955.Pharyngoconjunctivitis fever: Epidemiological studies of a recentlyrecognized disease entity. JAMA 175:1083–1092.
Molecular Epidemiology of Adenovirus Strains 445

Chastel C, Adrian T, Demazure M, Legrand-Quillien MC, Colim J,Wigand R. 1988. Molecular epidemiology of two consecutiveoutbreak of adenovirus 8 keratoconjunctivitis. J Med Virol 24:199–204.
Cooper RJ, Bailey AS, Kikkough R, Richmond SJ. 1993. Genomeanalysis of adenovirus 4 isolated over a six-year period. JMed Virol39:62–66.
Cooper RJ, Yeo CY, Bailey AS, Tullo AB. 1999. Adenovirus polymerasechain reaction assay for rapid diagnosis of conjunctivitis. InvestOphathalmol Vis Sci 40:90–95.
Corsaro D, Gut JP, Le Fau a. 1999. Molecular epidemiology of ocularisolates of adenovirus 8 obtained over nine years. J Clin Pathol52:860–861.
Crawford-Miksza LK, Schnurr DP. 1996a. Analysis of 15 adenovirushexon proteins reveals the location and structure of sevenhypervariable regions containing serotype specific residues. J ClinMicrobiol 70:1836–1844.
Crawford-Miksza LK, Schnurr DP. 1996b. Adenovirus serotype evolu-tion is driven by illegitimate recombination in the hypervariableregions of the hexon protein. Virology 224:357–367.
Crawford-Miksza LK, Nang RN, Schnurr DP. 1999. Strain variation inadenovirus serotypes 4 and 7a causing acute respiratory disease.J Clin Microbiol 37:1107–1112.
Darougar S, QuinlanMP, Gibson JA, Jones BR, McSwiggan DA. 1977.Epidemic keratoconjunctivitis and chronic papillary conjunctivitisin London due to adenovirus type 19. Br J Opthalmoll 61:76–85.
de Jong JC, Demazure M, Legrant-Quillien MC, Le Lay G, Golin J,Wermenbol AG, Verweij-Uyterwaal MW, van der Avoort HG,Chastel C. 1992. New developments in molecular epidemiology ofadenovirus 8 keratoconjunctivitis. J Med Virol 38:102–107.
de Jong JC,Wermenbol AG, Verweij-UyterwaalMW,KeesW, SlaterusKW, Van Doornum GJJ, Wertheim-van Dillen P, Van DoornumGJJ, Khoo SH, Hierholzer JC. 1999. Adenoviruses from humanimmunodeficiency virus-infected individuals, including two strainsthat represent new candidate serotypes Ad50 and Ad51 of speciesB1 and D, respectively. J Clin Microbiol 37:3940–3945.
Frantzidou F, Kyriazopoulou B, Souliou E, Diza E. 1994. An outbreakof Adenovirus type 8 keratoconjunctivitis in the region of Thessa-loniki. Acta Microbiol Hellenica 39:644–650.
Fujii S, Nakazono N, Sawada H, Ishii K, Kato M, Aoki K, Ohtsuka H,Fujinapa K. 1983. Restriction endonuclease cleavage analysis ofadenovirus 8: Two new subtypes from patients with epidemickeratoconjunctivitis in Sapporo, Japan. Jpn JMed Sci Biol 36:307–313.
Fujii S, NakazonoN, Ishii K, Lin C, SheuM, ChenC, FujinagaK. 1984.Molecular epidemiology of adenovirus 8 (Ad8) in Taiwan: Foursubtypes recovered during the period 1980–1981 from patientswith epidemic keratoconjunctivitis. Jap JMed Sci Biol 37:161–169.
Guo DF, Shibata R, Shinagawa M, Sato G, Aoki K, Sawada H. 1988.Genomic comparison of adenovirus type 3 isolates fro patients withacute conjunctivitis in Japan, Australia, and the Philippines.Microbiol Immunol 32:833–842.
HannaL,JawetzE. 1962.Attempt to enchance infectivity of adenovirustype 8 for cell culture. Proc Soc Exp Biol 110:707–709.
IshiikK,NakazonoN, FujinagaK,Fugii S,KatoM,OhtsukaH,AokiK,ChenCW, LinKH,OumBS, Lee SH, ChunCH, Yoshii T, YamazakiS. 1987. Comparative studies on aetiology of viral of three countriesof East Asia-Japan, Taiwan, South Korea. Int J Epidemiol 16:98–103.
ItakuraS,AokiK,SawadaH, IshiguroN,ShinagawaM.1991.Changesin subgenome types of adenovirus type 4 isolated from patientswith ocular disease between 1985 and 1989 in Sapporo, Japan.J Clin Microbiol 29:1740–1743.
ItohN,KeicoT,AokiK,HinokumaR,NakagawaH,Tackeuchi S,UchioE, Shiao S, Ohno S. 1999. Four new genotypes of adenovirus type 3isolated from patients with conjunctivitis in Japan. J Med Virol59:73–77.
Jawetz E, Kimura SJ, Hanna L, Coleman VR, Thygeson P, Nicholas A.1955. Studies in the etiology of epidemic keratoconjunctivitis. Am JOphtalmol 40:200–211.
KempMC,Heirholtzer JC. 1986.Threeadenovirus type8genome typesdefined by restriction enzyme analysis: Prototype stability ingeographically separated populations. J Clin Microbiol 23:469–474.
Kemp MC, Heirholtzer JC, Cabradilla CP, Obijeski JF. 1983. Thechanging etiology of epidemic keratoconjunctivitis: Antigenic andrestriction enzyme analysis of adenovirus types19 and 37 isolatedover a ten-year period. J Infect Dis 148:24–33.
Kidd AH, Jonsson M, Garwicz D, Kajon A, Wermenbol AG, VerweijMW, de Jong JC. 1996. Rapid subgenus identification of humanadenovirus isolates by a general PCR. J Clin Microbiol 34:622–627.
Kinloch R, Mackay N, Mautner V. 1984. Adenovirus hexon sequencecomparison of subgroupCserotypes 2 and5. JBiolChem259:6431–6436.
Li Q, Wadell G. 1988a. The degree of genetic variability amongadenovirus type 4 strains isolated fromman and chimpanzee. ArchVirol 101:65–77.
Li QG,Wadell G. 1988b. Comparison of 17 genome types of adenovirustype3 identified among strains recovered fromsix continents. JClinMicrobiol 26:1009–1015.
Morris DJ, Bailey AS, Cooper RJ, Turner PC, Jackson R, Corbitt G,Tullo AB. 1995. Polymerase chain reaction for rapid detection ofocular adenovirus infection. J Med Virol 46:126–132.
Murphy FA, Fauquet CM, Bishop DHL, Ghabrial SA, Jarvis AW,Martelli GP, Mayo MA, Summers MD, editors. 1995. Virustaxonomy:Classification andnomenculture of viruses. Sixth report.Vienna, Austria: Springer-Verlag. pp 128–133.
NIH. 1995. Viruses isolated from the eye, Japan 1990–1994. Infectagents surveillance Rep 16:97–98.
Norby E. 1969. The structural and functional diversity of adenoviruscapsid components. J Gen Virol 5:221–236.
O’ Donnell B, McCruden EA, Desselberger U. 1993. Molecularepidemiology of adenovirus conjunctivitis in Glasgow 1881–1991.Eye 7:8–14.
Pring-AkerblomP, Adrian T, 1994. Type and group specific polymerasechain reaction for adenovirus detection. Res Virol 145:25–35.
Richmond S, Burman R, Crosdale E, Cropper L, Longson D, Enoch BE,Dodd CL. 1984. A large outbreak of keratoconjunctivitis due toadenovirus type 8. J Hyg 93:285–291.
Saitoh-InagavaW,OshimaA,AokiK, ItohN, IsobeK,UchioE,OhnoS,NakajimaH, Hata K, Ishiko H. 1996. Rapid diagnosis of adenoviralconjunctivitis by PCR and restriction fragment length polymor-phism analysis. J Clin Microbiol 34:2113–2116.
Saitoh-Inagava W, Aoki K, Uchio E, Itoh N, Ohno S. 1999. Ten years’surveillance of viral conjunctivitis in Sapporo, Japan. Graefes ArchClin Exp Ophthalmol 237:35–38.
Schepetiuk SK, Norton R, Kok T, Irving LG. 1993. Outbreak ofadenovirus type 4 conjunctivitis in South Australia. J Med Virol41:316–318.
Takeuchi S, Itoh N, Uchio E, Aoki K, Ohno S, 1999. Serotyping ofAdenoviruses on conjunctival scrapings by PCR. J Clin Microbiol3:1839–1845.
Tanaka K, Itoh N, Saitoh-Inagawa W, Uchio E, Takeuchi S, Aoki K,SorianoE,NishiM,DurigonEL,StewienKE,OhnoS. 2000.Geneticcharacterization of adenovirus strains from patients with acuteconjunctivitis in the city of Sao Paulo, Brazil. J Med Virol 61:143–149.
Toogood CIA. Murali R, Burentt RM, Hay Rt. 1989. The adenovirustype 40 hexon: Sequence, predicted structure and relationship toother adenovirus hexon. J Gen Virol 70:3203–3214.
Wadell G. 1984. Molecular epidemiology of human adenoviruses.Curr Top Microbiol Immunol 110:191–220.
Wadell G, 1990. Adenoviruses. In: Zuckerman AJ, Banatvala JE,Pattison JR, editors. Principles and practice of clinical virology.Chichester: John Wiley and Sons Ltd. pp 286–287.
Wadell G, Hammarsjold ML, Winberg G, Varsanyi T, Sundell G. 1980.Genetic variability of adenoviruses. Ann NY Acad Sci 354:16–42.
Wadell G, Allard A, Evander M, Li Q. 1986. Genetic variability andevolution of adenoviruses. Chem Scripta 26B:325–335.
Wadell G, Allard A, Hierholzer JC. 1999. Adenvoviruses, p970-982. In:Murray PR, Baron EJ, Pflaller MA, Tenover F, Yolken N, editors.Manual of clinical microbiology, 17th edn. Washinton, DC: Amer-ican Society for Microbiology.
WigandR,GeiderblomH,OzelM,DistlerH,Adrian T. 1983. Characterstics of Mastadenovirus h8, the causative agent of epidemickeratoconjunctivitis. Arch Virol 76:307–319.
446 Frantzidou et al.

Journal of Medical Virology 75:447–454 (2005)
Detection of BK Virus and Simian Virus 40in the Urine of Healthy Children
John A. Vanchiere,1* Zoe S. White,2 and Janet S. Butel2
1Department of Pediatrics, Section of Infectious Diseases, Houston, Texas2Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas
Seroprevalence studies indicate that most pri-mary infections with BK virus (BKV) and JC virus(JCV) occur in the first and second decades oflife, respectively. Relatively little is known aboutthe transmission of these agents, including theprimary source of human exposure, the portal ofentry, and the pathophysiology of life-long viralpersistence. We sought to determine if simianvirus 40 (SV40) excretion could be detected in theurine of healthy children and to define the age-related prevalence of polyomavirus shedding inthis population. A point prevalence study of poly-omavirus shedding was conducted in healthychildren using rigorous enrollment criteria. Urinesamples were collected from healthy children,age from 3 to 18 years, during routine evaluationat two urban pediatric clinics. Qualitative PCRanalysiswasperformedusingprimers that detecta conserved region of the T-antigen gene of BKV,JCV, and SV40. The identity of polyomavirusesdetected was determined by DNA sequenceanalysis and/or PCR amplification of otherregions of the viral genomes. Seven of 72 (9.7%)urine samples were positive for polyomaviruses:three with BKV (ages 4, 6, 13), two with SV40(ages 6, 16), two with BKV and SV40 co-excretion(ages 6, 15), and none with JCV. DNA sequenceanalysis confirmed the identity of viruses de-tected. These results suggest that the timing ofSV40 infections in humansmay be similar to thatof BKV and that urine fromhealthy children couldcontribute to theubiquityofBKV infectionearly inlife. J. Med. Virol. 75:447–454, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: polyomavirus; viruria; pedia-tric; persistence
INTRODUCTION
BK virus (BKV) and JC virus (JCV) are members ofthe family Polyomaviridae that infect humans earlyin life and persist asymptomatically, typically causingdisease only in association with immune compromise
[Shah, 1996]. Seroprevalence data indicate that BKVand JCV have worldwide distributions, with BKVprimary infections occurring in early childhood andthose with JCV occurring later in school-aged children[Padgett and Walker, 1973; Taguchi et al., 1982;Knowles, 2001]. The precise mechanism(s) of transmis-sion remains unclear. Although human polyomavirusinfections usually are asymptomatic, BKV has beenfound in association with upper respiratory symptomsandurinary tract disease, albeit infrequently [Goudsmitet al., 1981, 1982; Sundsfjord et al., 1994]. There is littleevidence to suggest thatBKVplays a causal role in thesecommon syndromes [Padgett andWalker, 1973; Padgettet al., 1983; Sundsfjord et al., 1994]. The human kidneyis the primary reservoir of BKV, and urinary excretionoccurs intermittently in less than 5% of children andadults [Knowles, 2001]. BKV excretion is a sensitiveindicator of immune status, with viruria being morecommon during times of immune suppression, such aspregnancy, systemic lupus erythematosis, or chemo-therapy [Jin et al., 1993; Chang et al., 1996; Sundsfjordet al., 1999]. JCV can also be found in the urine ofhealthy subjects [Chesters et al., 1983; Kitamura et al.,1990; Agostini et al., 1996] and, in healthy adults,viruria with JCV is considerably more common thanwith BKV [Tsai et al., 1997; Chang et al., 2002; Linget al., 2003]. Excretion of JCV is strikingly age-dependent, with the prevalence of viruria after thesecond decade of life roughly equal to the age of thepatient group [Kitamura et al., 1990;Changet al., 2002].The basis of thismarked difference in the control of BKVand JCV excretion remains unknown.
Grant sponsor: National Institutes of Health (to JAV); Grantnumber: DK62090; Grant sponsor: National Institutes of Health(to JSB); Grant number: CA104818.
*Correspondence to: John A. Vanchiere, MD, PhD, Section ofPediatric Infectious Diseases, Baylor College of Medicine, OneBaylor Plaza, #302A, Houston, TX 77030.E-mail: [email protected]
Accepted 22 October 2004
DOI 10.1002/jmv.20287
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

Simian virus 40 (SV40) is a polyomavirus of rhesusmacaque origin and a potent tumor-inducing virus thatwas introduced into the human population as an un-known contaminant of some poliovirus and adenovirusvaccines used between 1955 and 1963 [Shah andNathanson, 1976; Butel and Lednicky, 1999]. SV40continues to circulate in the human population, as evi-denced by the detection of SV40 DNA in human tumorand kidney samples [Bergsagel et al., 1992; Carboneet al., 1997, 2000; Li et al., 2002a,b; Garcea andImperiale, 2003; Vilchez et al., 2003], including in somefrom individuals born after the use of contaminatedvaccines. The prevalence of SV40 infections in differentpopulations and geographic areas is unknown.We previously detected SV40 neutralizing antibodies
in hospitalized children and found an associationbetween the presence of SV40-specific antibodies andrenal transplantation [Butel et al., 1999b]. Severalantibody-positive children were identified who had noobvious immune deficiency, suggesting that immuno-competent children also may be infected with SV40.Molecular studies subsequently confirmed the presenceof SV40 DNA in renal biopsy material from severalchildren who had received renal allografts [Butel et al.,1999a]. Recent studies by Li et al. [2002a,b] havedocumented the possible association of SV40 with renaldisease in adults, and several studies have detectedSV40 viremia and viruria in healthy adults [Li et al.,2002a; Barbanti-Brodano et al., 2004].We hypothesized that children, like healthy adults,
will excrete polyomaviruses sporadically in their urine.Further, if SV40 does indeed circulate in the humanpopulation, we speculated that SV40 DNA may be de-tectable in the urine of healthy children, subject to theconstraints of an unknown prevalence of infection anda probable low frequency of urinary excretion. Wesought to determine the prevalence of polyomaviruriain healthy children using a PCR technique designed toamplify a conserved region of the JCV, BKV, and SV40genomes, followed by identification of the specific virusdetected. This method allows for detection of co-infec-tions with all three polyomaviruses that infect humans.Finally, we examined the genetic variation of the poly-omavirus strains detected in these healthy children.
MATERIALS AND METHODS
Study Subjects
Healthy children, ages 3–18 years, were recruitedfromprivatepediatric grouppractices inHouston,Texasat the time of routine well-child examinations. Childrenwhomet all of the following criteriawere asked to enroll:(1) ‘‘well’’ on the day of enrollment; (2) no fever antibioticuse during the past 7 days; (3) no systemic steroid useduring the past 30 days; (4) no hospitalizations withinthe past 2 years; (5) no history of blood or platelet trans-fusions; and (6) no chronic medical illnesses or chronicsystemic medications. Children with mild asthma orattention deficit disorder were allowed to participate inthe study if criteria 1 through 5 were met. Parents were
asked to complete a brief questionnaire regarding eachsubject’s medical history, focused primarily on renal orurinary tract disease. Voided urine samples were col-lected and urinalysis was performed using a Multistix10 SG test strip (Bayer, Elkhart, IN). Additionally, thefirst 50 samples were tested for the presence ofmicroalbumin using the ChemstripMicral (Roche Diag-nostics, Indianapolis, IN). Samples were stored at 48Cuntil being transported to the laboratory for processing(<4 hr). Fourteen-milliliter aliquots of urine werecentrifuged at 2,000 rpm for 10 min and supernatantswere discarded. Unwashed urine pellets were resus-pended in 200 ml of Tris-saline and stored at �208C.Subjects received $5 (USD) for participation in thestudy.
DNA Extraction and PCR
Strict measures for avoidance of cross-contaminationwere maintained throughout the study, including theuse of separate, plasmid-free rooms, and centrifugesfor processing of clinical samples, isolation of DNA, cellculture, PCR set-up, and PCR analysis. Separate cellculture laminar flowhoods and incubatorswere used formaintenance of infected and uninfected cells. DNA wasisolated from urine pellets by proteinase K digestion(20 mg/mlworking concentration) for 2–3 hr, followed byphenol–chloroformextraction andethanol precipitation[Ling et al., 2003]. Approximately 10% of each DNAsample (representing 1.4 ml of urine) was used for eachPCR reaction performed. DNA samples were screenedusing the AG1/AG2 primers (human A-gamma globingene) to assess the suitability of DNA for PCR testingand the PYV.for/PYV.rev primers, which amplify a con-served region of the T-antigen gene fromJCV, BKV, andSV40 (Table I). PCR reaction components weremixed at48C to final concentrations of 0.5–1 mM for primers,0.05–0.1 U/ml for heat-stable DNA polymerase (Ampli-Taq DNA Polymerase, Applied Biosystems, Inc., FosterCity, CA), 1� for PCR reaction buffer containingMgCl2,final concentration 1.5 mM (Applied Biosystems, Inc.),and 20 mM for dNTPs (Promega, Madison, WI)assembled in a final volume of 50 ml. Conditions forPCR amplification using the AG1/AG2 and PYV.for/PYV.rev primerswere: 45 cycles of denaturation at 968Cfor 30 sec, annealing at 608C for 30 sec, and extension at728C for 45 sec [Bergsagel et al., 1992; Lednicky et al.,1997]. Plasmid clones of BKV-Dunlop strain, JCV-Mad-1 strain, and SV40-21N (laboratory-modified SV40 776with a unique sequence insertion within the regulatoryregion) were used as positive controls for PCR reactions.Samples that produced appropriate sized DNA ampli-mers with PYV.for and PYV.rev primers, as determinedby direct visualization on ethidium bromide-stainedagarosegels,were further testedusingregulatoryregionprimers specific for JCV, BKV, and SV40 (Table I).Conditions for PCR amplification using the regulatoryregion primers were: 45 cycles of denaturation at 968Cfor 30 sec, annealing at 638C for 30 sec, and extension at728C for 45 sec [Lednicky et al., 1997]. PCR primers
448 Vanchiere et al.

BKTC1 and BKTC2 (based on the sequence of BKV-Dunlop strain, Genbank # V01108 ) were designed toamplify a portion of the carboxy-terminus of the T-antigen of BKV, analogous to the amplicon produced bySV40-specific primers TA1 and TA2 [Lednicky et al.,1997, 57/id]. PCR products of the regulatory regions andT-antigen genes were visualized on ethidium bromide-stained agarose gels after electrophoresis.
DNA Sequence Determination and Analysis
PCR products were treated with shrimp alkalinephosphatase (Amersham, Piscataway, NJ) and exonu-clease I (New England Biolabs, Inc., Beverly, MA) for45min at 378C, followed by enzyme inactivation at 858Cfor 10 min. DNA sequencing was carried out by LoneStar Labs, Inc. (Houston, TX) utilizing the ABI Prism1
Automated DNA sequencer 377XL and Big Dye Termi-nator Ready Reaction Cycle Sequencing Kit (AppliedBiosystems, Inc.) according to the manufacturer’sdirections. Consensus viral sequences were alignedand compared with known polyomavirus sequencesusing Gene Runner, version 3.05 (Hastings Software,Inc., Hastings, NY).
Statistical Analysis
Statistical analysis of potentially confounding demo-graphic and medical data was performed using a t-testfor comparison of means (Microsoft Excel 2000, Micro-soft Corporation) and the Fisher Exact test for compar-ison of frequencies (StatCalc, EpiInfo Version 6.0,Centers for Disease Control and Prevention, Atlanta).
RESULTS
Study Enrollment and Sample Collection
One hundred and two children were enrolled over a2-week period during July 2003 from two urban pedia-
tric offices in Houston, TX. The mean age of enrolleeswas 9.46 years (range 3.1–16.9 years). Thirty-nine(38.2%) of the enrollees were female and 78 (76.5%)were Caucasian. Of the samples collected, 25 were in-adequate for polyomavirus testing based on the inabilityto amplify a cellular gene used as the positive control forDNA integrity. The majority of these 25 samples con-tained significant precipitates and/or mucus that madeDNA extraction difficult, even though several differentmethods were attempted. Samples from five subjectswere excluded from the analysis because they werefound not to meet the screening criteria when examinedby their pediatrician. These five children included threewith otitis externa, one with a viral upper respiratorysyndrome, and one with resolving otitis media on thefifth day of oral antibiotic therapy. None of the fivepatients excluded after enrollment were found to havepolyomaviruria.
Detection of Polyomaviruses in Urine
Seven of 72 children (9.7%) were found to have poly-omaviruria, based on thePCRproduction of anampliconusing the PYV.for/PYV.rev primer set (Fig. 1). Therewas no difference in the mean age of polyomavirus-excreting (10.0 years; range 4.9–16.8 years) comparedto that of non-excreting subjects (9.4 years; range 3.1–16.9 years, P¼0.77). Among polyomavirus-excretingsubjects, 3 of 7 (42.9%) were female, whereas 38 of 65(58.5%) of non-excreting subjectswere female (P¼0.45).The distribution of race/ethnicity among subjects wasno different in comparison of polyomavirus-excreting(85.7% Caucasian) and non-excreting subjects (76.9%Caucasian). Only one study subject had a history ofgenitourinary or renal disease, and that child waspolyomavirus-positive. Six of 65 (9.2%) non-excretingand 0 of 7 (0%) polyomavirus-excreting subjects had a
TABLE I. PCR Primers Used for Analysis of Urine-Derived DNAs From Children
Primer Position Amplicon (bp) Annealing (8C)
Cellular DNA [Lednicky et al., 1997]AG1 50-CTC AGA CGT TCC AGA AGC GAG TGT-30 Human genome 379 60AG2 50-AAA CGG CTG ACA AAA GAA GTC CT-30
Polyomavirus conserved sequence (T-antigens) [Bergsagel et al., 1992]PYV.for 50-TAG TGC CAA CCT ATG GAA CAG A-30 Virus-specific 173–182 60PYV.rev 50-GAA AGT CTT TAG GGT CTT CTA CC-30
BK virus (BKV) regulatory region [Markowitz et al., 1991]BK1 50-GGCCTCAGAAAAAGCTTCCACACCCTTACTACTTGA-30 nt 50–85 355 60BK2 50-CTT GTC GTG ACA GCT GGC GCA GAA C-30 nt 415–391
Simian virus 40 (SV40) regulatory region [Lednicky et al., 1997]RA1 50-AAT GTG TGT CAG TTA GGG TGT G-30 nt 266–245 317 63RA2 50-TCC AAA AAA GCC TCC TCA CTA CTT-30 nt 5195–5218
JC virus (JCV) regulatory region [Lednicky et al., 2003]JC1 50-CCT CCA CGC CCT TAC TAC TTC TGA G-30 nt 5086–5110 308 60JC2 50-AGC TGG TGA CAA GCC AAA ACA GCT CT-30 nt 272–247
BKV T-antigen carboxy-terminus (this report)BKTC1 50-GGT AGA AGA GGT TAG GGT GTT TGA TGG-30 nt 2547–2573 452 60BKTC2 50-GGC TGG ATT CTG AGA TAA GTA TGT ATA CTT TTC-30 nt 2999–2968
SV40 T-antigen carboxy-terminus [Lednicky et al., 1997]TA1 50-GAC CTG TGG CTG AGT TTG CTC A-30 nt 3070–3048 441 60TA2 50-GCT TTA TTT GTA ACC ATT ATA AG-30 nt 2630–2652
BKV and SV40 in Urine of Children 449

family history of renal disease (P¼1.00). Analysis of theage distribution of polyomaviruria revealed statisticallyinsignificant differences in the prevalence of polyoma-virus excretion within the subgroups: 4 of 23 (17.4%)in the 3- to 6-year-old group, 0 of 25 (0.0%) in the 7- to11-year-old group, and 3 of 24 (12.5%) in the 12- to17-year-old group.When sub-grouped by gender, all polyomavirus-
excreting females (n¼4) were found to be under theage of 7. Statistically, females younger than the meanage of all females (9.63 years) were more likely thanthose older than the mean age of the females to excretepolyomaviruses (4of 14 foryounger femalesvs. 0 of 17 forolder females, P¼ 0.032 by Fisher Exact Test). Con-versely, polyomavirus-excreting males were found to beage 13 or older. Althoughmales older than themean ageof all males (>9.33 years) were more likely to excretepolyomaviruses than younger males, this differencefailed to reach statistical significance (0 of 26 for youngermales vs. 3 of 15 for older males, P¼0.062 by FisherExact Test).We hypothesized that polyomavirus excretion may
be associated with proteinuria or microalbuminuria inhealthy subjects. To test this hypothesis and to excludesubclinical renal disease in the study participants, dip-
stick urinalysis was performed within 10 min of samplecollection. Proteinuria and hemoglobinuria (>trace)were detected in four and two subjects, respectively;none of these abnormalities were detected in childrenwith polyomaviruria. Among the 42 samples testedwiththemore sensitive microalbuminuria test strip, 23 werepositive (>20 mcg/dl): 3 from 3 subjects with polyoma-viruria and 20 from 39 subjects without polyomaviruria(P¼0.24).
Viral Identification and StrainCharacterization by Sequence Analysis
Specific polyomaviruses were identified by sequenceanalysis of the PYV amplicons (produced by PCR ampli-fication with primers PYV.for and PYV.rev). Based onthe PYV region, four childrenwere found to be excretingBKV and three were excreting SV40; JCV was notdetected (Table II). Additional PCR amplifications ofother viral genomic regions next were attempted usingDNA from the samples that had been confirmed positivefor a polyomavirus. Primers BK1/BK2, RA1/RA2, andJC1/JC2 were used to attempt amplification of the re-gulatory regions for polyomaviruses detected. Regula-tory region sequences were amplified from five of theseven polyomavirus-positive samples (subjects 3–7).Four of the regulatory regions were concordant withthe earlier identification of the specific virus present(SV40 or BKV) based on the PYV region. The discordantsample (from subject 4) had a BKV sequence from thePYV amplicon and an SV40 sequence from the regula-tory region amplicons. From this sample, the carboxy-terminus of the T-antigen (C-Tag) genes for both BKVand SV40 were amplified with virus-specific primers(BKTC1/BKTC2 and TA1/TA2), confirming the simul-taneous excretion of both viruses by this patient. TheC-Tag region of BKV also was amplified from the urinesamples of subjects 3, 5, and 6, while that of SV40 wasamplified only from the urine of subject 4. Of note is thedetection of the PYV region and regulatory region ofSV40 and the C-Tag region of BKV in subject 6, sug-gesting that this patient was also excreting both poly-omaviruses at the time of sample collection.
Fig. 1. Representative ethidium bromide-stained agarose gelsshowing (A) PYV.for/PYV.rev amplicons (180 bp) and (B) BKTC1/BKTC2amplicons (450 bp).M,marker;þ, PCRpositive control;�, PCRnegative control. Numbers refer to study subjects described in Table II.
TABLE II. Specific Viral DNA Sequences Identified in Subjects With Polyomaviruria*
Subject Age (years) GenderVirus
detectedPolyomavirus genome regionsconfirmed by sequence analysis
1 4.9 F BKV PYV2 6.0 F SV40 PYV3 6.0 F BKV PYV, RR, T-ag-C4 6.8 F BKV PYV, T-ag-C
SV40 RR, T-ag-C5 13.8 M BKV PYV, RR, T-ag-C6 15.8 M SV40 PYV, RR
BKV T-ag-C7 16.8 M SV40 PYV, RR
*F, female; M, male; PYV, amplicon from conserved region near the amino terminus of the T-ag gene,obtained using the PYV.for/PYV.rev primer set (BK virus (BKV), JCV, and SV40 can be distinguished bysequence analysis of this amplicon); RR, regulatory region (designated asNCCR for BKV); T-ag-C, carboxyterminus of the T-antigen gene.
450 Vanchiere et al.

The DNA sequence amplified by the PYV.for/PYV.revuniversal primer set is useful for discriminating amongJCV, BKV, and SV40, but it is uninformative withrespect to assessment of strain variation for a particularvirus. Amplicons from more informative viral genomicregions (regulatory region and/or C-Tag) were analyzedfor strain characterization. The regulatory region se-quences [designated non-coding control region (NCCR)for BKV] from BKV-positive samples were comparedto that of the strain BKV-WWT (GenBank # M34048).Both BKV-NCCR sequences detected in this study werearchetypal in structure, with no regulatory region rear-rangements [Moens and Rekvig, 2001]. Both patientsamples (subjects 3 and 5) had nucleotide changes atpositions 65, 253, and 330, which are known polymor-phic sites in the BKV-NCCR. The BKV-NCCR amplifiedfrom subject 3 had seven additional nucleotide changesrelative to BKV-WWT, whereas the NCCR detectedfrom subject 5 had two additional nucleotide changes.The SV40 regulatory region sequences detected fromthree positive samples had duplicated enhancer regions[Lednicky and Butel, 2001]. In two cases (subjects 6 and7), the viral regulatory region contained aC-to-A changeat nucleotide position 5237 (relative to strain 776),
making those viruses identical to the HuKi-3 variant ofSV40 previously detected in the kidney allograft of achild [Butel et al., 1999a]. The third SV40-positivesample (subject 4) had a regulatory region identical insequence to that of strain 776 of SV40 (GenBank #J02400).
The DNA sequence of the C-Tag regions from BKV-positive samples were determined by direct sequencingof the PCR products (Fig. 2). The natural isolates had2, 3, or4nucleotide changes in this region, relative to twoBKV laboratory strains, Dunlop and MM. The SV40 C-Tag sequence from subject 4 was like that of SV40-776.
DISCUSSION
Using a qualitative PCR assay that detects all threehuman polyomaviruses, we observed polyomavirusexcretion in 9.7% of healthy children and did not findsignificant differences in the demographic characteris-tics or medical history of those with or without viruria.These data extend the age-associated prevalence ofpolyomavirus urinary excretion into the period of viralacquisition and provide evidence that SV40 is causinginfections in the human population. The use of very
Fig. 2. Alignment of BK virus (BKV) C-terminal T-antigen sequences detected in the urine of healthychildren compared to BKV strains Dunlop and MM. The nucleotide positions (based on BKV-Dunlop) areindicated above the first and last nucleotides of the alignment. ‘‘.’’ indicates identity with BKV-Dunlop.Samples HC-A50, HC-A29, HC-A59, and HC-A46 represent subjects 6, 3, 4, and 5, respectively, listed inTable II.
BKV and SV40 in Urine of Children 451

strict criteria for defining ‘‘healthy’’ in this study isimportant because it eliminates the potentially con-founding factors of immune activation (due to inflam-matory disease), immune suppression (due to systemicsteroid use), and transfusion-acquired infection. Whilethe immune status of the host is known to influencethe excretion of polyomaviruses, especially for BKV[Sugimoto et al., 1989; Chang et al., 1996; Tsai et al.,1997], it is unclear whether the mode of transmission orthe state of systemic immune activation can influencethe excretion of polyomaviruses.Our detection of BKV in the urine of 5.5% of healthy
children is consistent with other studies of BKV excre-tion [reviewed inKnowles, 2001], although prior studieshave included predominantly hospitalized children.Using a PCR technique that would detect only BKV, apoint prevalence study of healthy children, ages 3–7 years, found that 8 of 211 (3.8%) were excreting BKV[Di Taranto et al., 1997]. Information on the healthstatus of those subjects was not reported. Whethertransient alterations in the health status of children(e.g., minor bacterial infections, viral upper respiratoryor gastrointestinal disease, or short-term steroid ther-apy) influence the duration of BKV shedding afterprimary infection or the incidence of viral reactivationis unknown. In order to eliminate these potential con-founding factors, we elected to use more stringentcriteria for the inclusion of healthy children.The BKV DNA sequences detected here are similar,
but not identical, to the few reported wild-type BKVsequenceswith respect to the structure of the regulatoryregion and the carboxy-terminus of the T-antigen gene.The detection of SV40 DNA in the urine of 5.5% ofhealthy children is consistentwith thefinding ofSV40 inthe urine of healthy adults [Li et al., 2002a] and adds tothe growing body of data supporting the circulation ofSV40 in the human population. Importantly, variationin the DNA sequences of the BKV and SV40 strainsdetected in this study as compared to known laboratorystrains argues strongly against laboratory contamina-tion as an explanation for our findings. The inability toamplify the regulatory region and/or C-Tag region fromsome samples that yielded sequence-proven, virus-specific PYV amplicons is consistent with our experi-ence, and may be related to variations in viral load,the relative sensitivities of the different primer pairs,genetic differences among viral strains, and/or sizes ofthe amplicons produced. Whether cross-virus interfer-encemay occur in the PCR amplification of dual infectedsamples is unclear. JCV was not detected among thesamples tested, and this is consistent with previousreports of low JCV excretion rates (<5%) among in-dividuals less than 20 years old [Chang et al., 2002; Linget al., 2003; Jeong et al., 2004]. This is probably due, inlarge part, to the timing of JCV infection, which tendsto bemore common during the second and third decadesof life.Co-infections with BKV and SV40 were detected, as
observedpreviously [Butel et al., 1999a; Li et al., 2002b],but this time in two healthy children. The prior cases of
BKV and SV40 co-infection were in patients followingrenal transplantation, one involving an adult and onea child. Whether this combination of viruses puts ahealthy child at risk for future renal or malignant dis-ease remains unknown.
There are several limitations of this study. We do notknowwhat proportion of children in the study had silentinfections with different polyomaviruses, cannot ascer-tain whether the observed excretions were associatedwith primary infection or viral reactivation, cannotevaluate the duration of polyomavirus shedding byindividual subjects, and donot knowwhat the frequencyof episodes of excretion would be if samples were col-lected at several times. For example, in a longitudinalstudy of 30 healthy adults extending over 14 months(with most volunteers providing 7 collections), no BKVviruria was detected [Ling et al., 2003]. JCV viruriaoccurred at least once in 46.7% of the volunteers.Volunteers ranged in age from 22 to 68 years, andJCV excretion occurred predominantly in those over theage of 40 years. However, throughout the longitudinalstudy, if the results of single-collection time pointswere considered independently, JCV shedding fre-quencies for the study group varied from 20% to 39%.Some individuals had multiple positive samples overthe course of the study, others had only a single posi-tive urine specimen, and others were always negative.Whether similar patterns of polyomavirus excretionoccur in childhood remains unknown.
Based on the current knowledge of human polyoma-virus infection, we believe that the prevalence of poly-omavirus excretion will likely be influenced by (1) theincidence of infection, which increases with age; (2) thelikelihood of excretion, which may be greatest during aprimary infection, prior to control of virus replication bythe immune response; (3) the likelihood of reactivation,which increases with age for JCV, but not for BKV(based on studies of older adults); and (4) the immunestatus and/or immunogenetics of the subjects. Withspecific reference to understanding the common occur-rence of BKV infection during infancy and early child-hood, if urine is the primary source of infectious virusexposure, the sum of these influences suggests that(1) adults are unlikely to be the common source of BKVexposure and (2) young children are a highly likelysource, given the likelihood of excretion after primaryinfection. The data presented here are consistent withthe hypothesis that exposure to human urine, especiallyfromyoung children,maybean importantmechanismofBKV infection.Our observations suggest that the timingof SV40 infection in humans may more closely mimicthat of BKV than JCV, although larger studies will berequired to validate this conclusion. Similarly, largerstudies will be required to define the potential role ofhost genetic and/or medical factors in polyomavirusinfection, reactivation, and excretion.
Polyomaviruses are notoriously difficult to cultiv-ate in vitro and molecular biologic techniques havebecome important tools for the study of polyomaviruspersistence due to their high sensitivity and specificity.
452 Vanchiere et al.

The PCR methodology, coupled with sequence anal-ysis, employed here allows the detection of multiplepolyomaviruses in clinical samples. There is consider-able variability among reported studies with respectto techniques utilized for acquisition, extraction, andamplification of polyomavirus DNA. In a comparison ofknown BKV-positive clinical samples, we have foundthat urine pellet-derived DNA is a more sensitivesubstrate for qualitative PCR detection than clarifiedor unclarified urine (data not shown). Additionally, theutilization of unwashed urine pellets as the startingmaterial for DNA extraction necessarily includes asmall volume of urine, which may contribute to the poolof detectable virus.
The study of polyomaviruses during the period ofacquisition is important because thatmay be the time ofmost vigorous viral replication in immune competenthosts, the time when different tissues get seeded withvirus, and a time of possible selection of variants withgreater replication orpathogenic potential. Such studiesare especially important for SV40, given its relativelyrecent and multi-focal introduction into the humanpopulation. Prospective long-term studies of SV40-infected subjects and their close contacts will be re-quired to determine the significance of SV40 infection inotherwise healthy children.
ACKNOWLEDGMENTS
We thank the physicians and nurses of HoustonPediatric Associates (Houston, TX) and the PediatricMedical Group (Houston, TX) for assistance in recruit-ment of patients.
REFERENCES
Agostini HT, Ryschkewitsch CF, Stoner GL. 1996. Genotype profile ofhuman polyomavirus JC excreted in urine of immunocompetentindividuals. J Clin Microbiol 34:159–164.
Barbanti-Brodano G, Sabbioni S, Martini F, Negrini M, Corallini A,Tognon M. 2004. Simian virus 40 infection in humans andassociation with human diseases: Results and hypotheses. Virology318:1–9.
Bergsagel DJ, Finegold MJ, Butel JS, Kupsky WJ, Garcea RL. 1992.DNA sequences similar to those of simian virus 40 in ependymomasand choroid plexus tumors of childhood. N Engl J Med 326:988–993.
Butel JS, Lednicky JA. 1999. Cell andmolecular biology of simian virus40: Implications for human infections and disease. J Natl CancerInst 91:119–134.
Butel JS, Arrington AS, Wong C, Lednicky JA, Finegold MJ. 1999a.Molecular evidence of simianvirus 40 infections in children. J InfectDis 180:884–887.
Butel JS, Jafar S, Wong C, Arrington AS, Opekun AR, Finegold MJ,Adam E. 1999b. Evidence of SV40 infections in hospitalizedchildren. Hum Pathol 30:1496–1502.
CarboneM, Rizzo P, PassHI. 1997. Simian virus 40, poliovaccines, andhuman tumors: A review of recent developments. Oncogene 15:1877–1888.
Carbone M, Rizzo P, Pass H. 2000. Simian virus 40: The link withhuman malignant mesothelioma is well established. AnticancerRes 20:875–877.
Chang D,WangM, OuWC, Lee MS, Ho HN, Tsai RT. 1996. Genotypesof human polyomaviruses in urine samples of pregnant women inTaiwan. J Med Virol 48:95–101.
Chang H,WangML, Tsai RT, Lin HS, Huan JS,WangWC, Chang DC.2002. High incidence of JC viruria in JC-seropositive olderindividuals. J Neurovirol 8:447–451.
Chesters PM, Heritage J, McCance DJ. 1983. Persistence of DNAsequences of BK virus and JC virus in normal human tissues and indiseased tissues. J Infect Dis 147:676–684.
Di Taranto C, Pietropaolo V, Orsi GB, Jin L, Sinibaldi L, Degener AM.1997. Detection of BK polyomavirus genotypes in healthy andHIV-positive children. Eur J Epidemiol 13:653–657.
Garcea RL, Imperiale MJ. 2003. Simian virus 40 infection of humans.J Virol 77:5039–5045.
Goudsmit J, BaakML, Sleterus KW, van der Noordaa J. 1981. Humanpapovavirus isolated from urine of a child with acute tonsillitis. BrMed J (Clin Res Ed) 283:1363–1364.
Goudsmit J,Wertheim-vanDP, van Strien A, van der Noordaa J. 1982.The role of BK virus in acute respiratory tract disease and thepresence of BKV DNA in tonsils. J Med Virol 10:91–99.
Jeong BH, Lee KH, Choi EK, Kim K, Kim YS. 2004. Genotyping of theJC virus in urine samples of healthy Korean individuals. J MedVirol 72:281–289.
Jin L, Gibson PE, Booth JC, Clewley JP. 1993. Genomic typing of BKvirus in clinical specimensbydirect sequencing of polymerase chainreaction products. J Med Virol 41:11–17.
Kitamura T, Aso Y, KuniyoshiN,HaraK, Yogo Y. 1990.High incidenceof urinary JC virus excretion in nonimmunosuppressed olderpatients. J Infect Dis 161:1128–1133.
KnowlesWA. 2001. The epidemiology of BKvirus and the occurrence ofantigenic and genomic subtypes. In: Khalili K, Stoner GL, editors.Human polyomaviruses: Molecular and clinical perspectives.New York: Wiley-Liss, Inc., pp 527–559.
Lednicky JA, Butel JS. 2001. Simian virus 40 regulatory regionstructural diversity and the association of viral archetypalregulatory regions with human brain tumors. Semin Cancer Biol11:39–47.
Lednicky JA, Stewart AR, Jenkins JJ III, FinegoldMJ, Butel JS. 1997.SV40 DNA in human osteosarcomas shows sequence variationamong T-antigen genes. Int J Cancer 72:791–800.
Lednicky JA, Vilchez RA, Keitel WA, Visnegarwala F, White ZS,Kozinetz CA, Lewis DE, Butel JS. 2003. Polyomavirus JCVexcretion and genotype analysis in HIV-infected patients receivinghighly active antiretroviral therapy. AIDS 17:801–807.
Li RM, Branton MH, Tanawattanacharoen S, Falk RA, Jennette JC,Kopp JB. 2002a. Molecular identification of SV40 infection inhuman subjects and possible association with kidney disease. J AmSoc Nephrol 13:2320–2330.
LiRM,MannonRB,KleinerD,TsokosM,BynumM,KirkAD,KoppJB.2002b. BK virus and SV40 co-infection in polyomavirus nephro-pathy. Transplantation 74:1497–1504.
LingPD,LednickyJA,KeitelWA,PostonDG,WhiteZS,PengRS,LiuZ,Mehta SK, Pierson DL, Rooney CM, Vilchez RA, Smith EO, ButelJS. 2003. The dynamics of herpesvirus and polyomavirus reactiva-tionandshedding inhealthyadults:A14-month longitudinal study.J Infect Dis 187:1571–1580.
Markowitz RB, Eaton BA, Kubik MF, Latorra D, McGregor JA,Dynan WS. 1991. BK virus and JC virus shed during pregnancyhave predominantly archetypal regulatory regions. J Virol 65:4515–4519.
Moens U, Rekvig OP. 2001. Molecular biology of BK virus and clinicaland basic aspects of BK virus renal infection. In: Khalili K,Stoner GL, editors. Human polyomaviruses: Molecular and clinicalperspectives. New York: Wiley-Liss, Inc., pp 359–408.
Padgett BL, Walker DL. 1973. Prevalence of antibodies in human seraagainst JC virus, an isolate from a case of progressive multifocalleukoencephalopathy. J Infect Dis 127:467–470.
PadgettBL,WalkerDL,DesquitadoMM,KimDU.1983.BKvirusandnon-haemorrhagic cystitis in a child [letter]. Lancet 1:770.
Shah KV. 1996. Polyomaviruses. In: Fields BN, Knipe DM, HowleyPM, Chanock RM, Melnick JL, Monath TP, Roizman B, StrausSE, editors. Fields virology. Philadelphia: Lippincott-Raven,pp 2027–2043.
Shah K, Nathanson N. 1976. Human exposure to SV40: Review andcomment. Am J Epidemiol 103:1–12.
Sugimoto C, Hara K, Taguchi F, Yogo Y. 1989. Growth efficiency ofnaturally occurring BK virus variants in vivo and in vitro. J Virol63:3195–3199.
BKV and SV40 in Urine of Children 453

SundsfjordA,SpeinAR,LuchtE,FlaegstadT,SeternesOM,TraavikT.1994. Detection of BK virus DNA in nasopharyngeal aspirates fromchildrenwith respiratory infections but not in saliva from immuno-deficient and immunocompetent adult patients. J Clin Microbiol32:1390–1394.
SundsfjordA,OseiA,RosenqvistH,VanGhelueM,SilsandY,HagaHJ,Relvig OP, Moens U. 1999. BK and JC viruses in patients withsystemic lupus erythematosus: Prevalent and persistent BKviruria, sequence stability of the viral regulatory regions, andnondetectable viremia. J Infect Dis 180:1–9.
Taguchi F, Kajioka J, Miyamura T. 1982. Prevalence rate and age ofacquisition of antibodies against JC virus and BK virus in humansera. Microbiol Immunol 26:1057–1064.
Tsai RT, Wang M, Ou WC, Lee YL, Li SY, Fung CY, Huang YL,Tzeng TY, Chen Y, ChangD. 1997. Incidence of JC viruria is higherthan that of BK viruria in Taiwan. J Med Virol 52:253–257.
Vilchez RA, Kozinetz CA, Arrington AS, Madden CR, Butel JS.2003. Polyomavirus SV40 in human cancers. Am J Med 114:675–684.
454 Vanchiere et al.

Journal of Medical Virology 75:455–462 (2005)
New Human Coronavirus, HCoV-NL63,Associated With Severe Lower RespiratoryTract Disease in Australia
Katherine E. Arden,1,2 Michael D. Nissen,1,2,3,4 Theo P. Sloots,1,2,3,4 and Ian M. Mackay1,2*1Clinical Virology and Molecular Microbiology Research Units, Sir Albert Sakzewski Virus Research Centre,Royal Children’s Hospital, Queensland, Australia2Clinical and Medical Virology Centre, University of Queensland, Queensland, Australia3Division of Microbiology, Queensland Health Pathology Service, Royal Brisbane Hospitals Campus, Queensland,Australia4Department of Paediatrics and Child Health, Royal Children’s Hospitals, Queensland, Australia
A new human coronavirus, HCoV-NL63, was as-sociated recently with bronchiolitis. The currentstudy aimed to examine retrospectively storedspecimens for the presence of HCoV-NL63 usingnested RT-PCR assays targeting the 1a and 1bgenes. The study population was composed ofpatients with acute respiratory diseasewarrantingpresentation to Queensland hospitals. HCoV-NL63 was detected in the nasopharyngeal aspi-rates (NPA) of 16 of 840 specimens representing766 patients (2%). HCoV-NL63 positive individualswere diagnosed most commonly with lowerrespiratory tract (LRT) disease (81%). The clinicaldiagnosis was commonly supported by an abnor-mal chest X-ray (56%) together with respiratorydistress (50%), wheeze (44%), and rales (25%) onfirst presentation with HCoV-NL63 infection. Allpatients positive for HCoV-NL63 required admis-sion to hospital. Among 38% of HCoV-NL63 posi-tive specimens a second pathogen was detected.Sequencing of amplicon from gene 1b revealedmore than 99% nucleotide homology with theviral type strains while sequencing ampliconfrom gene 1a permitted the grouping of viralstrains. It was shown that HCoV-NL63 is asso-ciated with severe LRT disease in an Australianhospital setting during the cooler months of theyear. We propose that HCoV-NL63 is a global andseasonal pathogen of both children and adultsassociated with severe LRT illness. J. Med.Virol. 75:455–462, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: human coronavirus; emerging;respiratory; pediatric; disease
INTRODUCTION
Among humans, RNA viruses are the most frequentagent to cause the common cold; usually a self-limiting
upper respiratory tract (URT) illness [Heikkinen andJarvinen, 2003]. Viruses are also a common cause oflower respiratory tract infection (LRTI) and includethe diseases bronchitis, bronchiolitis, laryngotracheo-bronchitis, and pneumonia, which may necessitatemechanical ventilation. Human rhinoviruses (HRV),respiratory syncytial virus (hRSV), and parainfluenzaviruses (PIV) are the viruses isolated most commonly[Selwyn, 1990; Hall, 2001; Hayden, 2004]. Featuresof mild URT disease include fever, cough, and coryza,while LRT infections may also exhibit the moresevere signs of dyspnoea, rales, respiratory distress,and wheeze. Most recently, the discoveries of humanmetapneumovirus (hMPV) and SARS coronavirus(SARS-CoV) have exemplified the benefits of a molecu-lar approach to rapidly identify and characterizepreviously unknown microbial causes of serious re-spiratory illness and to determine their epidemiology.Nonetheless, up to 60% of respiratory tract infectionscontinue to go undiagnosed among hospitals and clinicsworldwide [Nicholson et al., 1997].
The human coronaviruses (HCoV) are envelopedviruses, which carry a plus-sense RNA genome appro-ximately 30 kb in length. The prototype HCoV, 229E,and OC43, were identified in the 1960s [Hamre andProcknow, 1966] but until the discovery of the SARS-CoV, received relatively little attention as humanpathogens beyond their causal role in the common cold
Grant sponsor: Woolworth’s Fresh Futures Appeal through theRoyal Children’s Hospital Foundation; Grant number: 912-011.
*Correspondence to: Ian M. Mackay, Sir Albert SakzewskiVirus Research Centre and Clinical Medical Virology Centre,Royal Children’s Hospital and University of Queensland, HerstonRoad, Herston, Queensland 4029, Australia.E-mail: [email protected]
Accepted 14 October 2004
DOI 10.1002/jmv.20288
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

and associated viral wheeze [Hendley et al., 1972; Vileet al., 1995]. It is entirely possible that the prevalenceof these viruses has been underestimated because ofthe widespread use of serological assays to diagnoseinfection and the fastidious nature of viral growthin vitro [Simons et al., 1996; van Elden LJR et al.,2004]. However, recent reports have associated HCoVwith gastrointestinal problems [Vabret et al., 2003],acute disseminated encephalomyelitis [Yeh et al., 2004],and more severe respiratory tract infection, particularlyin the elderly and the immunocompromised [Falseyet al., 2002; Pene et al., 2003], pointing to a greaterrole for HCoVs in disease than previously thought.Apart from the laboratory adapted type strains, relatedvariants are known to exist yet there are few reportsdetailing the extent of genetic variation within the threeserogroups within the genus Coronavirus.
A fourth human coronavirus entitled NL63 (HCoV-NL63; [van der Hoek et al., 2004]), together with theSARS-CoV, also suggest the genus Coronavirus con-tains members capable of much more significant disease[Rota et al., 2003]. A second group from the Netherlandsrecently described the detection of HCoV-NL63 fromfour infants and a child suffering from respiratory tractdisease [Fouchier et al., 2004]. Detections of HCoV-NL63 by van der Hoek and colleagues occurred in fourchildren and three adults, with a peak detection rate of7% among ill visitors to a Medical Center in Amsterdamduring mid-winter. No virus was detected during thewarmer months. Interestingly, the data indicatedthe possible existence of several viral lineages withinthe HCoV-NL63-like genomes. Because very little isknown about this pathogen, an Australian patientpopulation was investigated for the presence of HCoV-NL63. Once found, this study determined the clinicalfeatures of disease associated with infection and ex-amined local strains for genetic variability from the typestrain.
METHODS
The study population comprised 840 specimens from766 ill patients who had presented to Queenslandhospitals or general practitioners during November,2001 to February, 2004 with acute respiratory tractdisease suspected of having an infectious etiology.Specimens were predominantly nasopharyngeal aspi-rates (NPA; 93%), but also included bronchoalveolarlavages (3.4%), endotracheal aspirates (1.4%), andbronchial washings (2.0%) collected either at the timeof visit or following hospital admission. The majority ofpatients visited the Royal Children’s Hospital, RoyalBrisbane, and Women’s Hospitals in Brisbane, Queens-land while others visited the Ipswich, Logan, Redlands,Redcliffe, and Cairns Hospitals in Queensland. Non-consecutive specimens were selected by season, withoutprior knowledge of patient details or microbiologicalstatus.The subjects ranged inage from 3 days to80 years(median¼ 1.3 years), with children 5 years of age oryounger comprising 77.6% of the study population.Specimens from winter (n¼492), spring (n¼183),
summer (n¼86), and autumn (n¼79) were tested.Prior to this retrospective study, all specimens had beentested for common microbial respiratory pathogensand stored at �708C. The laboratory assays includedculture-amplified direct fluorescent assay and subse-quent RT-PCR [Syrmis et al., 2004] of negative speci-mens for hRSV, human adenoviruses (hAdV), PIV typesI, II, and III, and influenza viruses A (FluA) and B[Syrmis et al., 2004]. An in-house RT-PCR was used tosimultaneously detect all four subtypes of hMPV. PCRassays for the HCoV OC43, and 229E (Table II) andBordetella pertussis [Kosters et al., 2002] were alsoperformed. Selective culture media were used to growbacterial pathogens including Pseudomonas aerugi-nosa, Streptococcus species, Hemophilus influenzae,Neisseria species, Staphylococcus species, Legionellapneumophila, and Candida albicans. No microbes weredetected in 557 specimens taken from 506 patients (66%)while a suspected pathogen was detected in the remain-ing 283 specimens representing 260 patients (34%).
To detect HCoV-NL63, two previously describednested assays ([van der Hoek et al., 2004]; Table II)were used to examine 1 ml of purified RNA. Testing ofRNA consisted of a single-tube RT-PCR amplificationfollowed by a nested PCR using 1 ml of the first roundproduct (HotStarTaq, QIAGEN, Australia). Positiveresults from assay 1 were confirmed using assay 2(Fig. 1). The two assays were in agreement.
Fig. 1. Agarose gel electrophoresis of the amplicons from the twonested RT-PCR reactions. Lane L, 100 bp molecular weight DNAladder. Lane 1, assay 1, round 1. Lane 2, assay 1, round 2. Lane 3,assay 2, round 1. Lane 4, assay 2, round 2.
456 Arden et al.

All RT-PCR assays were performed using the OneStepRT-PCR kit (QIAGEN) incorporating 0.6 mM of eachprimer and subjecting the reaction mixes to a 20 minincubation at 508C followed by 15 min at 958C. PCR wasperformed for 45 cycles at 948C for 30 sec; 558C for 30 sec;and 728C for 30 sec.
Infectious virus was isolated by inoculating 200 ml ofNPA from positive patients onto monkey kidney cells(passage 5) in the presence of 5 mg/ml of trypsin (Gibco,USA). Cultures were maintained for 14 days andexamined each day for cytopathic effects and for viralRNA by RT-PCR.
RESULTS
HCoV-NL63 was detected in the NPA specimensfrom 16 patients (Table I), comprising 13 males and3 females. All patients positive for HCoV-NL63 requiredhospital admission and one ultimately died frompossible complications. Ten positive patients (63%) hadno other microbe identified nor any underlying medicalcondition, strongly suggesting that HCoV-NL63 was thesole cause of disease. Virus was isolated from Case 14 asdetermined by the production of pleomorphic cells inculture, cell detachment and a positive RT-PCR signalfrom the culture supernatant using assay 2.
Six patients infected with HCoV-NL63 (38%) werealso infected with one other respiratory pathogen; eitherPIV III (Cases 1 and 2), hMPV (Case 16), hRSV (Case 11),Bordetella pertussis (Case 5), or Legionella pneumo-phila (Case 12). The age of infected patients rangedfrom 1 month to 62 years. The full medical records of all16 subjects were examined. They all exhibited featuresof an acute respiratory tract infection at the time ofdetection including cough (81%), fever (69%), respira-tory distress (50%), and coryza/vomiting/wheeze (44%).Minor clinical features of infection included; pharyngitis(38%), lethargy (31%), anorexia/rales (25%), irritability(19%), cervical lymphadenopathy/diarrhoea/dyspnoea/otitis media/rash/rhinorrhoea/rigors (13%), and con-junctivitis/cyanosis/headache/hematochezia/hepatitis/jaundice/poor sleep/stridor (6%). The duration of clinicalsymptoms ranged from 1 to 10 days prior to the collectionof an NPA. The most common clinical diagnosis wasLRTI (81%), in particular, bronchiolitis (38%). The samestudy population was also screened for the presence ofother seasonal respiratory viruses including hRSV,hAdV, PIV I to III, and FluA. Rates of detection were11% (85 patients), 4% (32 patients), 7% (51 patients), and4% (33 patients), respectively. When available, a clinicaldiagnosis upon presentation indicated LRT disease in46%, 25%, 35%, and 30% of patients, respectively.
Chest radiographs were obtained from 11 individuals(9 children and 2 adults) of which 9 (56%) were abnormal,demonstrating increased parahilar peribronchial mark-ings consistent with a non-specific viral LRTI (Fig. 2).
Two individuals had pre-existing lung disease due toeither chronic neonatal lung disease (Case 7) or chronicobstructive pulmonary disease (Case 12). One child hadDown’s syndrome (Case 6). One patient (Case 13) wasseverely immunosuppressed due to a bone marrow
transplant and was acutely neutropaenic secondary tochemotherapy for relapsed chronic lymphocytic leuke-mia. He had been a long-term inpatient for 24 daysprior to the detection of HCoV-NL63 and therefore was aprobable nosocomial case. On two occasions inducedsputum was negative for Pneumocystis jiroveci by directfluorescent antigen and polymerase chain reaction.He died 25 days later from a secondary fungal septicillness due to Scediosporum apiosperum. While HCoV-NL63 was not the direct cause of death, a gradual declinein his respiratory status could be traced back to the timeof first detection of the virus.
Three children (Cases 5, 7, and 16) had other familymembers who were unwell with upper respiratory tractinfections (URTI) at the time of HCoV-NL63 detection.Another two children attended day care centers (Cases 2and 8), identifying possible sites of exposure to infection.
The new coronavirus was detected in 11 of 493 winterspecimens (2.2%), which represents 5.6% of the total 171microbial diagnoses made during this period. Addition-ally, HCoV-NL63 was detected in two specimens fromautumn and three from spring. Other microbes detectedin the specimens taken during winter included hRSV(25.4% of microbial detections), hAdV (14.7%), FluA(13.2%), hMPV (12.7%), PIV III (9.1%), and Streptococ-cus and Staphylococcus species (5.1%). The study foundthat the peak month for incidence of HCoV-NL63 wasJuly (mid-winter), at 3.1% of specimens compared to7.0% of specimens in the Netherlands. Overall, the pre-valence of HCoV-NL63 in the study population was 1.9%(2.1% of total patients), compared to 1.3% and 2.9% inthe Netherlands [Fouchier et al., 2004; van der Hoeket al., 2004]. In keeping with the expected incidence ofHCoV, there were no examples of infection during thesummer months despite testing a greater number ofspecimens from summer than autumn [Hendley et al.,1972].
The 237 bp amplicon resulting from RT-PCR of theHCoV-NL63 gene 1b of five positive specimens wassequenced (GenBank accession numbers AY600442–AY600446). The Queensland strains shared 99.5%–100% nucleotide identity with the same region of theHCoV-NL63 type strain. All strains shared 100%predicted amino acid homology with the strains fromboth previous reports. Three Australian strains con-tained the same single synonymous nucleotide changecompared to HCoV-NL63 and one other Australianstrain contained a different synonymous change in anearby location. We next sequenced the 525 bp am-plicon resulting from amplification of the 1a gene.Comparison of 425 bp of these sequences (GenBankaccession numbers AY632576–AY632651) indicatedthat strains shared 98%–100% nucleotide identity and96%–100% predicted amino acid homology (Fig. 3).
DISCUSSION
This study demonstrates for the first time thatchildren and adults outside the Netherlands are sub-ject to infection by HCoV-NL63. Additionally, given
Australian Incidence and Features of HCoV-NL63 457

TA
BL
EI.
Pati
ents
Pos
itiv
efo
rH
CoV
NL
63
Case
Age
Sex
Date
ofco
llec
tion
aC
lin
ical
featu
res
Du
rati
onof
sym
pto
ms
(day(s
))b
Oth
erin
form
ati
onC
lin
ical
dia
gn
osis
c
11
yea
r4
mon
ths
FS
epte
mber
2001
An
orex
ia,
cerv
ical
lym
ph
ad
enop
ath
y,
cory
za,
cou
gh
,fe
ver
,ot
itis
med
ia,
ph
ary
ngit
is,
rale
s
3C
o-in
fect
ion
-PIV
III,
CX
R-b
ilate
ral
para
hil
ar
per
ibro
nch
ial
infi
ltra
tes
an
dh
yp
erin
flate
d
LR
TI/
pn
eum
onia
210
mon
ths
MS
epte
mber
2001
Cou
gh
,cy
an
osis
,le
tharg
y,
resp
irato
ryd
istr
ess,
wh
eeze
1C
o-in
fect
ion
-PIV
III,
CX
R-b
ilate
ral
para
hil
ar
per
ibro
nch
ial
infi
ltra
tes.
Att
end
sd
ay
care
LR
TI/
lary
ngot
rach
eob
ron
chit
is
37
mon
ths
MM
ay
2003
Cou
gh
,fe
ver
,p
hary
ngit
is,
rale
s,re
spir
ato
ryd
istr
ess,
stri
dor
,vom
itin
g3
CX
R-i
ncr
ease
dri
gh
tu
pp
erlo
be
mark
ings
LR
TI/
lary
ngot
rach
eob
ron
chit
is
41
mon
thM
May
2003
Cou
gh
,fe
ver
,re
spir
ato
ryd
istr
ess,
vom
itin
g10
CX
R-r
igh
tlu
ng
opaci
tyB
ron
chio
liti
s5
1m
onth
MJu
ne
2003
Cou
gh
,p
hary
ngit
is,
rhin
orrh
oea,
vom
itin
g7
Co-
infe
ctio
n-
B.
per
tuss
is;
CX
R-
bil
ate
ral
para
hil
ar
per
ibro
nch
ial
infi
ltra
tes,
mot
her
un
wel
lw
ith
UR
TI
Per
tuss
is
62
yea
rs4
mon
ths
MJu
ne
2003
Cer
vic
al
lym
ph
ad
enop
ath
y,
cou
gh
,fe
ver
,le
tharg
y,
ph
ary
ngit
is,
rhin
orrh
oea,
wh
eeze
7C
XR
-bil
ate
ral
para
hil
ar
per
ibro
nch
ial
infi
ltra
tes
LR
TI
72
yea
rs3
mon
ths
FJu
ly2003
An
orex
ia,
con
jun
ctiv
itis
,co
ryza
,fe
ver
,ir
rita
bil
ity,
leth
arg
y,
ph
ary
ngit
is3
Sib
lin
gu
nw
ell
wit
hU
RT
IU
RT
I/p
erio
rbit
al
cell
uli
tis
82
yea
rsF
Ju
ly2003
Cor
yza
,co
ugh
,vom
itin
g2
Att
end
sd
ay
care
LR
TI/
ast
hm
a/‘‘
per
tuss
is’’
94
mon
ths
MJu
ly2003
Cor
yza
,co
ugh
,re
spir
ato
ryd
istr
ess,
vom
itin
g,
wh
eeze
1D
own
’ssy
nd
rom
eL
RT
I/bro
nch
ioli
tis
10
9m
onth
sM
Ju
ly2003
Cou
gh
,fe
ver
,ir
rita
bil
ity,
poo
rsl
eep
,re
spir
ato
ryd
istr
ess,
vom
itin
g,
wh
eeze
4C
hro
nic
neo
nata
llu
ng
dis
ease
onsu
pp
lem
enta
lox
ygen
LR
TI/
bro
nch
ioli
tis
11
9m
onth
sM
Ju
ly2003
An
orex
ia,co
ryza
,co
ugh
,fe
ver
,ot
itis
med
ia,
rale
s,re
spir
ato
ryd
istr
ess,
wh
eeze
4C
o-in
fect
ion
-hR
SV
,C
XR
-bil
ate
ral
para
hil
ar
per
ibro
nch
ial
infi
ltra
tes
LR
TI/
bro
nch
ioli
tis
12
55
yea
rsM
Ju
ly2003
An
orex
ia,
cou
gh
,d
ysp
noe
a,
fever
,le
tharg
y,
rigor
s,w
hee
ze6
CO
PD
,co
-in
fect
ion
-leg
ion
ella
pn
eum
oph
ila,
CX
R-R
lobar
pn
eum
onia
LR
TI/
legio
nel
lap
neu
mon
ia
13
62
yea
rsM
Ju
ly2003
Ch
oles
tati
ch
epati
tis
&ja
un
dic
e,d
iarr
hoe
a,
dysp
noe
a,
fever
,h
aem
ato
chez
ia,
leth
arg
y,
pet
ech
ial
rash
,ri
gor
s
6C
XR
-bil
ate
ral
pn
eum
onic
infi
ltra
tes,
bon
em
arr
owtr
an
spla
nt,
CL
Lre
lap
se,
neu
trop
aen
ic,
dec
ease
d
LR
TI
14
1yea
r5
mon
ths
MA
ugu
st2003
Cor
yza
,fe
ver
,h
ead
ach
e,ir
rita
bil
ity,
ph
ary
ngit
is1
CS
Fgra
m(þ
),cu
ltu
res
(-)
UR
TI/
men
ingit
is
15
5m
onth
sM
Au
gu
st2003
An
orex
ia,
cory
za,
cou
gh
,re
spir
ato
ryd
istr
ess,
wh
eeze
7C
XR
nor
mal
LR
TI/
bro
nch
ioli
tis
16
6m
onth
sM
Sep
tem
ber
2003
Cou
gh
,d
iarr
hoe
a,
fever
,ra
les,
rash
,re
spir
ato
ryd
istr
ess,
vom
itin
g,
wh
eeze
7C
o-in
fect
ion
-hM
PV
,C
XR
nor
mal,
fam
ily
mem
ber
su
nw
ell
wit
hU
RT
IsL
RT
I/bro
nch
ioli
tis
aD
ate
ofco
llec
tion
ofn
aso
ph
ary
ngea
lasp
irate
.bD
ura
tion
ofsy
mp
tom
sp
rior
tom
edic
al
pre
sen
tati
on.
cL
RT
I,lo
wer
resp
irato
rytr
act
infe
ctio
n;
UR
TI,
up
per
resp
irato
rytr
act
infe
ctio
n;
CX
R,
ches
tX
-ray;
CO
PD
,ch
ron
icob
stru
ctiv
ep
ulm
onary
dis
ease
;C
LL
,ch
ron
icly
mp
hoc
yti
cle
uk
aem
ic;
CS
F,
cere
bro
spin
al
flu
id.
458 Arden et al.
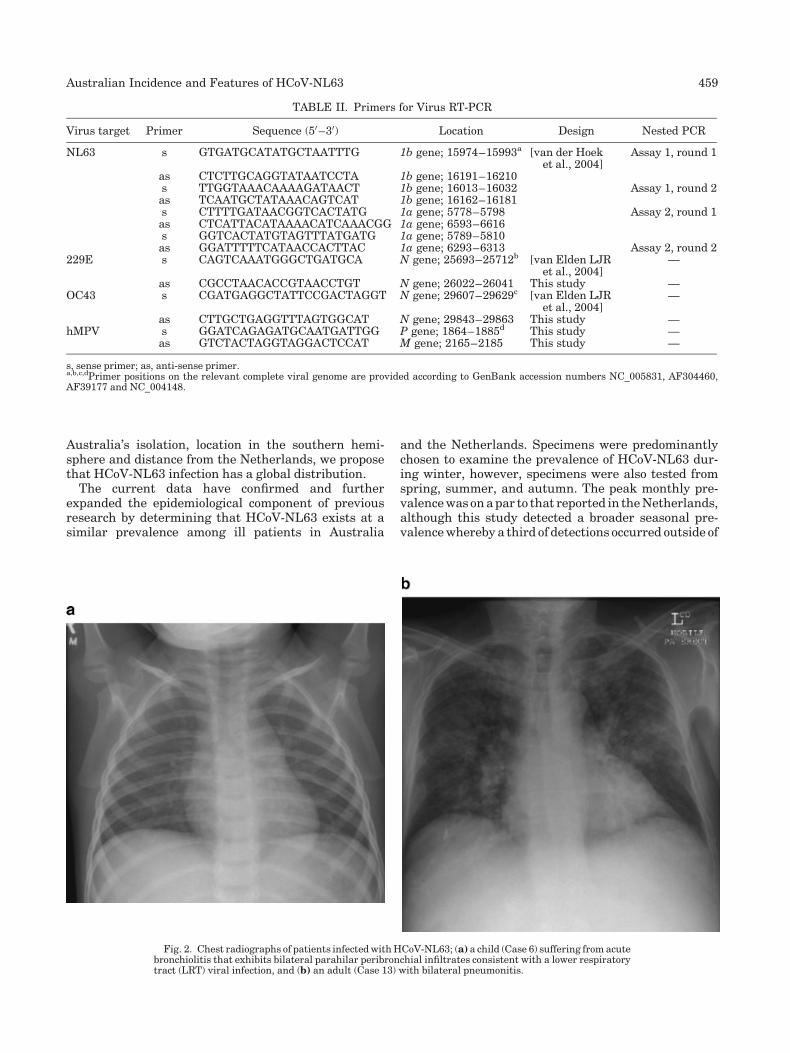
Australia’s isolation, location in the southern hemi-sphere and distance from the Netherlands, we proposethat HCoV-NL63 infection has a global distribution.
The current data have confirmed and furtherexpanded the epidemiological component of previousresearch by determining that HCoV-NL63 exists at asimilar prevalence among ill patients in Australia
and the Netherlands. Specimens were predominantlychosen to examine the prevalence of HCoV-NL63 dur-ing winter, however, specimens were also tested fromspring, summer, and autumn. The peak monthly pre-valence was on a par to that reported in the Netherlands,although this study detected a broader seasonal pre-valence whereby a third of detections occurred outside of
TABLE II. Primers for Virus RT-PCR
Virus target Primer Sequence (50–30) Location Design Nested PCR
NL63 s GTGATGCATATGCTAATTTG 1b gene; 15974–15993a [van der Hoeket al., 2004]
Assay 1, round 1
as CTCTTGCAGGTATAATCCTA 1b gene; 16191–16210s TTGGTAAACAAAAGATAACT 1b gene; 16013–16032 Assay 1, round 2as TCAATGCTATAAACAGTCAT 1b gene; 16162–16181s CTTTTGATAACGGTCACTATG 1a gene; 5778–5798 Assay 2, round 1as CTCATTACATAAAACATCAAACGG 1a gene; 6593–6616s GGTCACTATGTAGTTTATGATG 1a gene; 5789–5810as GGATTTTTCATAACCACTTAC 1a gene; 6293–6313 Assay 2, round 2
229E s CAGTCAAATGGGCTGATGCA N gene; 25693–25712b [van Elden LJRet al., 2004]
—
as CGCCTAACACCGTAACCTGT N gene; 26022–26041 This study —OC43 s CGATGAGGCTATTCCGACTAGGT N gene; 29607–29629c [van Elden LJR
et al., 2004]—
as CTTGCTGAGGTTTAGTGGCAT N gene; 29843–29863 This study —hMPV s GGATCAGAGATGCAATGATTGG P gene; 1864–1885d This study —
as GTCTACTAGGTAGGACTCCAT M gene; 2165–2185 This study —
s, sense primer; as, anti-sense primer.a,b,c,dPrimer positions on the relevant complete viral genome are provided according to GenBank accession numbers NC_005831, AF304460,AF39177 and NC_004148.
Fig. 2. Chest radiographs of patients infected with HCoV-NL63; (a) a child (Case 6) suffering from acutebronchiolitis that exhibits bilateral parahilar peribronchial infiltrates consistent with a lower respiratorytract (LRT) viral infection, and (b) an adult (Case 13) with bilateral pneumonitis.
Australian Incidence and Features of HCoV-NL63 459

winter; during late autumn or early spring. This mayreflect the environmental impact of Queensland’stropical and sub-tropical climate.
Strains detected in Australia show a high degreeof genetic homology to the known strains and toCanadian sequences submitted to GenBank during thepreparation of this manuscript. The lack of variability inthe predicted amino acid sequence in amplicon derivedfrom the 1b gene is in keeping with the essential natureof the coronavirus non-structural proteins including thereplicase and proteases. However, the 1a gene exhibitedgreater variability suggesting the existence of two mainviral lineages. Further studies will be required tocharacterize the degree of genetic diversity amongless conserved genes from viral strains worldwide.Until this can be defined and the applicability of currentdiagnostic molecular assays truly evaluated, the use ofmore than one PCR screening assay, as presented byboth earlier reports is a sensible approach for studyingthe epidemiology of HCoV-NL63.
Within the study population, composed of patientswith a range of respiratory illnesses, HCoV-NL63 wasfound to be more frequently associated with severe lowerrespiratory tract (LRT; 81%) disease than either of thetwo earlier studies (38% and 20%; [Fouchier et al., 2004;
van der Hoek et al., 2004]. This assessment of diseaseseverity is based on the fact that all patients positivefor HCoV-NL63 required admission to hospital, themajority had an abnormal chest X-ray, many hadclinical features of LRTI (dyspnoea, rales, respiratorydistress, and wheeze) and one immunocompromisedpatient died. While the nature of the study populationprevented a complete description of the annual in-cidence of HCoV-NL63 infection, the clinical featuresand diagnoses of patients infected with HCoV-NL63were shown to be considerably more severe than thosecommonly attributed to infection of subjects by HCoV-229E and HCoV-OC43.
HCoV-NL63 was detected in the NPA of 10 patients(63% of detections) with no other known pathogenpresent, strongly supporting the role of this new virusin clinically diagnosed LRT disease. Nonetheless, thevirus has been detected from tissue obtained directlyfrom LRT in only one instance [van der Hoek et al.,2004]. Therefore severe URT infection with HCoV-NL63, which indirectly causes LRT disease cannot beexcluded. Many diagnoses of viral LRT disease are basedon the combined clinical data and laboratory detection ofvirus from URT tissue. A study using specimens col-lected from the LRT without possible contamination bytissue from the URT is essential to more accuratelydetermine the etiology of HCoV-NL63 disease. Smallanimal and/or primate experiments are also necessaryto determine the pathogenesis and incubation period forthis new respiratory pathogen.
The new coronavirus was also co-detected withanother pathogen in six subjects infected with eitherPIV III, hRSV, hMPV, Bordetella pertussis, or Legio-nella pneumophila [0.7% of all specimens, (0.8% of allpatients); 38% of HCoV-NL63 infections]. Interestingly,no co-infections were seen with hAdV despite its highseasonal co-incidence. The previous studies detectedHCoV-NL63 as a co-infection in 25% and 20% of thepatients; comparable to this study’s 38% co-infectionrate [Fouchier et al., 2004; van der Hoek et al., 2004].These data may indicate a significant characteristic ofHCoV-NL63 epidemiology since such a high rate of co-infection is uncommon. Furthermore, fewer of theHCoV-NL63 infected patients in the study had under-lying medical conditions than reported by Fouchier et al.[2004]. All studies to date have found HCoV-NL63infections in patients with underlying disease, whetherthey are immunocompromised due to chemotherapy orHIV infection or have another medical condition such aschronic obstructive pulmonary disease. Further studywill be required to determine whether patients with co-detected microbes have more severe disease or a worselong-term prognosis than patients with a single detectedmicrobe using clinically validated severity scores. It isnoteworthy, however, that HCoV-NL63 infection wasmore commonly associated with LRT disease thanhRSV, HAdV, PIV, or FluA infection among ourpopulation. One of the dually infected patients was alsoa probable case of nosocomial infection (Case 13),highlighting the need for rapid detection of the virus,
Fig. 3. Phylogenetic analysis of Queensland (Q), Dutch (Patient ‘‘x’’),and Canadian (CAN) 1a gene sequences from HCoV-NL63 strainspresented on a topology tree prepared in MEGA 2.1. Branch lengthsare irrelevant. The nucleotide distance matrix was generated usingthe p-distance estimation. Nodal confidence values indicate the resultsof boot strap resampling (n¼ 500). Two main sequence clusters areapparent (Type A and B) with subgroups indicated (A1, A2, B1, and B2).
460 Arden et al.

as well as isolation or cohorting of HCoV-NL63 cases inhealth care institutions.
Two HCoV-NL63 positive patients (Cases 5 and 8)were clinically labeled as ‘‘pertussis’’ because of a paro-xysmal cough, although the laboratory diagnosis wasconfirmed in only one of these (Case 5). Also, theassociation of HCoV-NL63 with acute laryngotracheo-bronchitis or ‘‘croup’’ (Cases 2 and 3) and PIV IIIinfection (Cases 1 and 2) is of clinical interest. HCoV-NL63 should therefore be considered as a possibleetiological agent when features of acute paroxysmalcough or croup are present.
Interestingly, two HCoV-NL63 positive patients(Cases 13 and 16) experienced gastrointestinal tract(GIT) symptoms, which are commonly attributed toanimal coronaviruses and were reported recently in57% of human cases during an HCoV-OC43 outbreak[Vabret et al., 2003]. Case 13 was intensively investi-gated for GIT pathogens including rotavirus, withoutdetection. However, norovirus co-infection could notbe excluded. Neither norovirus nor rotavirus couldbe excluded from Case 16 as these assays were notperformed.
The new coronavirus accounted for 5% of all themicrobes detected in the study population, however,66% of the specimens remain undiagnosed. HRV werenot tested here, however, there is an increasing bodyof literature linking HRV infection to severe lowerrespiratory disease suggesting that they may constitutea proportion of the pathogens present in the remainingundiagnosed group [Hayden, 2004].
While it remains to be proven, we believe that thisvirus is another example of a ‘‘new’’ microbe that hasbeen infecting and causing disease among humansfor many years and that the intensive application ofmolecular biology to specimens from patients withrespiratory illness has succeeded in identifying aviral cause, where traditional methods of diagnosticculture and serology have failed. Taking into accountthe seasonal spread of infections over the study period,the recent detection of HCoV-NL63 in specimens datingback to 1988 by Fouchier et al. [2004] and the extentof genetic homology to HCoV-229E, it does not appearthat HCoV-NL63 arose from a recent animal virusmutation as may have occurred for the SARS-CoV[Fouchier et al., 2004; Marra et al., 2004]. Therefore,the result of infection by HCoV-NL63 may moreaccurately be labeled a redefined infectious disease(RID) rather than an emerging infectious disease, whichis defined as disease that has newly emerged in apopulation or significantly increased in incidence orgeographic range [Morse, 1995]. While HIV, Escherichiacoli O157:H7, and the SARS-CoV are examples thatcause the former, hMPV exemplifies a cause of RID [vanden Hoogen et al., 2001].
It is apparent from our study population that HCoV-NL63 may be a cause of severe LRT disease. It shouldbe considered in the differential diagnosis of hospita-lized children, the elderly and those with pre-existingimmunocompromise or chronic respiratory disease
during the cooler months of the year. Ongoing studieswill address the transmission, incidence, and molecularepidemiology of HCoV-NL63 in the non-hospitalizedcommunity to determine whether the pathogen circu-lates among the community in a similar seasonalpattern causing less severe disease. The identificationof this respiratory pathogen will help clinicians improvemethods of patient management, reduce antibiotic use,and implement better infection control procedureswithin the hospital environment. The development ofa single well-characterized and evaluated molecularassay to detect all HCoV-NL63-like viruses is essentialbefore the full impact of this pathogen on communityacquired respiratory disease and the significance ofco-infection with characterized microbes on clinicaloutcome among infected patients can be elucidated.
ACKNOWLEDGMENTS
This study was funded by a grant no. 912-011 from theWoolworth’s Fresh Futures Appeal through the RoyalChildren’s Hospital Foundation. We thank FrankFiumara, John Gavranich, Ann Gillett, Ross Messer,Gail Phythian, and Eric van der List for supplying theclinical information on their patients. We also thankDavid Seibert for provision of clinical specimens.
REFERENCES
Falsey AR, Walsh EE, Hayden FG. 2002. Rhinovirus and coronavirusinfection-associated hospitalizations among older adults. J InfectDis 185:1338–1341.
Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC,Simon JH, Osterhaus AD. 2004. A previously undescribed corona-virus associated with respiratory disease in humans. Proc NatlAcad Sci USA 101:6212–6216.
Hall CB. 2001. Respiratory syncytial virus and parainfluenza virus. NEngl J Med 344:1917–1928.
Hamre D, Procknow JJ. 1966. A new virus isolated from the humanrespiratory tract. Proc Soc Exp Biol Med 121:190–193.
Hayden FG. 2004. Rhinovirus and the lower respiratory tract. Rev MedVirol 14:17–31.
Heikkinen T, Jarvinen A. 2003. The common cold. Lancet 361:51–59.
Hendley JO, Fishburne HB, Gwaltney JM, Jr. 1972. Coronavirusinfections in working adults. Am Rev Resp Dis 105:805–811.
Kosters K, Reischl U, Schmetz J, Riffelmann M, Wirsing von Konig CH.2002. Real-time LightCycler for detection and discrimination ofBordetella pertussis and Bordetella parapertussis. J Clin Microbiol40:1719–1722.
Marra MA, Jones SJM, Astell CR, Holt RA, Brooks-Wilson A,Butterfield YSN, Khattra J, Asano JK, Barber SA, Chan SY,Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL,Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK,Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus F,Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N,Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L,Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldman H,Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA,Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M,Petric M, Skowronski DM, Upton C, Roper RL. 2004. The genomesequence of the SARS-associated coronavirus. Science 300:1399–1404.
Morse SS. 1995. Factors in the emergence of infectious diseases. EmergInfect Dis 1:7–15.
Nicholson KG, Kent J, Hammersley V, Cancio E. 1997. Acute viralinfections of upper respiratory tract in elderly people living in thecommunity: Comparative, prospective, population based study ofdisease burden. Br Med J 315:1060–1064.
Australian Incidence and Features of HCoV-NL63 461

Pene F, Merlat A, Vabret A, Rozenberg F, Buzyn A, Dreyfus F, CariouA, Freymuth F, Lebon P. 2003. Coronavirus 229E-related pneumo-nia in immunocompromised patients. Clin Infect Dis 37: 929–932.
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP,Penaranda S, Bankamp B, Maher K, Chen M-H, Tong S, Tamin A,Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, PeretTCT, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S,Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M,Fouchier R, Gunther S, Osterhaus ADME, Drosten C, PallanschMA, Anderson LJ, Bellini WJ. 2003. Characterization of a novelcoronavirus associated with severe acute respiratory syndrome.Science 300:1394–1398.
Selwyn BJ. 1990. The epidemiology of acute respiratory tract infectionin young children: Comparison of findings from several developingcountries. Rev Infect Dis 12:S870–S888.
Simons JN, Desai SM, Schultz DE, Lemon SM, Mushahwar IK. 1996.Translation initiation in GB viruses A and C: Evidence for internalribosome entry and implications for genome organization. J Virol70:6126–6135.
Syrmis MW, Whiley DM, Thomas M, Mackay IM, Williamson J,Siebert DJ, Nissen MD, Sloots TP. 2004. A sensitive, specific andcost-effective multiplex reverse-transcriptase-PCR assay for thedetection of seven common respiratory viruses in respiratorysamples. J Mol Diagn 6:125–131.
Vabret A, Mourez T, Gouarin S, Petitjean J, Freymuth F. 2003. Anoutbreak of coronavirus OC43 respiratory infection in Normandy,France. Clin Infect Dis 36:985–989.
van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R,Fouchier RAM, Osterhaus ADME. 2001. A newly discovered humanpneumovirus isolated from young children with respiratory tractdisease. Nat Med 7:719–724.
van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, BerkhoutRJM, Wolthers KC, Wertheim-van DIllen PME, Kaandorp J,Spaargaren J, Berkhout B. 2004. Identification of a new humancoronavirus. Nat Med 368–373.
van Elden LJR, van Loon AM, van Alphen F, HendriksenKAW, Hoepelman AIM, van Kraaij MGJ, Oosterheert J-J,Schipper P, Schuurman R, Nijhuis M. 2004. Frequent detectionof human coronaviruses in clinical specimens from patients withrespiratory tract infection by use of a novel real-time reverse-transcription polymerase chain reaction. J Infect Dis 189:652–657.
Vile RG, Diaz RM, Mille r N, Mitchell S, Tuszyanski A, Russell SJ. 1995.A model of viral wheeze in nonasthmatic adults: Symptoms andphysiology. Virology 214:307–313.
Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H. 2004. Detectionof coronavirus in the central nervous system of a child with acutedisseminated encephalomyelitis. Pediatrics 113:E73–E76.
462 Arden et al.

Journal of Medical Virology 75:463–465 (2005)
Detection of Human Coronavirus NL63 inYoung Children With Bronchiolitis
Takashi Ebihara, Rika Endo, Xiaoming Ma, Nobuhisa Ishiguro, and Hideaki Kikuta*
Department of Pediatrics, Hokkaido University Graduate School of Medicine, Sapporo, Japan
HCoV-NL63, the fourth human coronavirus, hasbeen isolated recently from childrenwith respira-tory tract infections, including upper respiratoryinfection, bronchiolitis, and pneumonia. Thevirus has been also detected in immunocompro-mised adults with respiratory tract infections. Atotal of 118 nasopharyngeal swab samples from118 hospitalized young children aged less than2 years with bronchiolitis who were not infectedwith human respiratory syncytial virus, influenzaAorB, or humanmetaneumoviruswere selected.Three (2.5%) of the 118 samples were positive forHCoV-NL63 by reverse transcription-polymerasechain reaction tests. HCoV-NL63 may be one ofthe causative agents of bronchiolitis in youngchildren. J. Med. Virol. 75:463–465, 2005.� 2005 Wiley-Liss, Inc.
KEY WORDS: HCoV-NL63; acute bronchioli-tis; acute laryngotracheobron-chitis
INTRODUCTION
Coronaviruses, a genus of the Coronaviridae family,have been identified in humans and animals and aredivided genetically and serologically into four groups[Holmes and Lai, 1996]. Three strains of coronaviruses,HCoV-229E, HCoV-OC43, and severe acute respiratorysyndrome-associated coronavirus (SARS-CoV), havebeen associated with various respiratory illnesses inhumans ranging from common cold to severe pneumo-nia. HCoV-229E and HCoV-OC43, which were discov-ered as causes of the common cold in the 1960s [Holmesand Lai, 1996], belong to group 1 and group 2, res-pectively. Both strains have been identified occasionallyas agents of lower respiratory tract infections in infants,older adults, and immunocompromised hosts [McIntoshet al., 1974; Nicholson et al., 1997; Glezen et al., 2000].SARS-CoV, which was identified as a pathogen causingsevere respiratory illnesses in 2003, belongs to group 4[Ksiazek et al., 2003].
A new human coronavirus, NL63 (HCoV-NL63), wasdiscovered first from a 7-month-old Dutch child withbronchiolitis and conjunctivitis by van der Hoek et al.
[2004]. Fouchier et al. [2004] also identified the newhuman coronavirus (HCoV-NL) from an 8-month-oldboy with pneumonia. Since no official name has beengiven to the new human coronavirus by the Interna-tional Committee on Taxonomy of Viruses yet, the name‘‘HCoV-NL63’’ was used tentatively to designate thevirus in this study. Since their sequence studies showedthat HCoV-NL63 was related most closely to HCoV-229E, itwas classified into group1 [Fouchier et al., 2004;van der Hoek et al., 2004]. These two studies showedthat the virus seemed to be associated with non-fatalupper and lower respiratory tract infection in youngchildren and immunocompromised adults.
In this study, nasopharyngeal swab samples obtainedfrom hospitalized young children with bronchiolitiswere investigated for the presence of HCoV-NL63.
MATERIALS AND METHODS
Patients
From October 2002 to September 2003, a total of118 nasopharyngeal swab samples were collected from118 children aged less than 2 years who were diagnosedas having bronchiolitis and admitted to hospitals inSapporo, Japan. All of the samples were collected afterexcluding the possibility of infection with hRSV orinfluenza A or B by rapid antigen detection tests andexcluding the possibility of infection with humanmetaneumovirus (hMPV) by a reverse transcription-polymerase chain reaction (RT-PCR) test [Ebihara et al.,2004]. The median age of the children was 11.5 months.The male-to-female ratio was 1.7 to 1. All samples were
Grant sponsor: Ministry of Education, Culture, Sports, Scienceand Technology, Japan; Grant number: 14657179; Grant sponsor:21st Century Center of Excellence for Zoonosis Control from theMinistry of Education, Culture, Sports, Science and Technology,Japan.
*Correspondence to: Dr. Hideaki Kikuta, Department ofPediatrics, Hokkaido University Graduate School of Medicine,N-15, W-7, Kita-ku, Sapporo 060-8638, Japan.E-mail: [email protected]
Accepted 15 October 2004
DOI 10.1002/jmv.20289
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

collected after obtaining informed consent from thechildren’s parents.
RT-PCR Test and Sequencing
Total RNA was extracted from each sample by usingthe RNAzolTMB (TEL-TEST, Inc., Friendswood, TX)method according to themanufacturer’s protocol. cDNAwas synthesized from total RNA by using a First-StrandcDNA Synthesis Kit (Amersham Pharmacia Biotech,Piscataway, NJ). The PCR primers used for detection ofHCoV-NL63 have been published previously [van derHoek et al., 2004]. Two primer pairs were used for anested PCR to detect a 169-nucleotide replicase 1b genefragment of HCoV-NL63. A forward primer with asequence of 50-GTGATGCATATGCTAATTTG-30 and areverse primer with a sequence of 50-CTCTTGCAGG-TATAATCCTA-30 were used for the first PCR. Thesecond PCR was performed by using a forward primerwith a sequence of 50-TTGGTAAACAAAAGATAACT-30
and a reverse primer with a sequence of 50-TCAATGC-TATAAACAGTCAT-30. Sequencing of the PCRproductswas performed by using a BigDye terminator cyclesequencing ready reaction kit (Perkin-Elmer AppliedBiosystems, Foster, CA) with an ABI Prism 310 geneticanalyzer (Perkin-Elmer Applied Biosystems).
RESULTS
RNA sequences of HCoV-NL63 were detected insamples from 3 (2.5%) of the 118 children with bron-chiolitis. Three (13.6%) of 22 samples collected in
February and March were positive for HCoV-NL63(Table I). Direct sequencing of the PCR products of thethree samples showed that the sequences were identicalto the sequence of HCoV-NL63 (GenBank accessionnumber AY567487). Clinical and laboratory features ofthe three patients positive for HCoV-NL63 are shown inTable II. The three patients had no underlying disease.All three patients suffered from expiratory wheezing,fever, cough, and nasal discharge. Two patients pre-sented with tachypnea. One girl (case 2) developedhoarseness and barking cough after admission, and shewas diagnosed as also having laryngotracheobronchitis.Maximum temperature ranged from 39.0 to 40.38C. Theduration of fever and expiratory wheezing ranged from1 to 4 days and from 3 to 7 days, respectively. Radio-graphs of the chest showed emphysematous changes intwo patients.
DISCUSSION
Bronchiolitis is an acute, inflammatory respiratoryillness in children occurringmainly in the first 2 years oflife. hRSV is the virus detected most frequentlydetected virus in patients with bronchiolitis, reporteddetection rates being 50%–60% [Andreoletti et al., 2000;Papadopoulos et al., 2002; Jartti et al., 2004]. Respira-torypicornavirus is the second-most frequent agent, andhas been detected in 20%–40% of bronchiolitis cases[Andreoletti et al., 2000; Papadopoulos et al., 2002;Jartti et al., 2004]. hMPV, which has been recognizedrecently as a causative agent as bronchiolitis, was pre-sent in 11%–16% of bronchiolitis cases [Jartti et al.,2004; Xepapadaki et al., 2004]. The detection rate ofHCoV-229E and HCoV-OC43 in bronchiolitis cases hasbeen reported to be only 2% [Papadopoulos et al., 2002;Jartti et al., 2004]. However, there are other causativeagents of bronchiolotis that remain to be discovered. vander Hoek et al. [2004] reported an infant with bronch-iolitis caused byHCoV-NL63. In this study,HCoV-NL63was detected in 2.5% of the bronchiolitis cases, indicat-ing that HCoV-NL63may be one of the causative agentsof bronchiolitis.
TABLE I. Detection of HcoV-NL63 in 2002–2003
Periods Positive no./no. tested
October 2002–November 2002 0/20December 2002–January 2003 0/26February 2003–March 2003 3/22April 2003–May 2003 0/38June 2003–July 2003 0/8August 2003–September 2003 0/4
TABLE II. Clinical and Laboratory Features of Three Patients Positive for HcoV-NL63
Case 1 Case 2 Case 3
Sampling date 14 February, 2003 19 March, 2003 28 March, 2003Age 1 year 11 months 1 year 6 months 1 year 3 monthsSex F F FPresenting symptoms Rhinorhoea, cough,
wheezing, fever(max: 39.68C)
Rhinorhoea, barking cough,wheezing, tachypnea,hoarseness, fever(max: 40.38C)
Rhinorhoea, cough,wheezing, tachypnea,fever (max: 39.08C)
Duration of fever (days) 1 4 4Duration of wheezing (days) 3 3 7Clinical signs Expiratory wheezing Inspiratory and expiratory
wheezings, subcostalretraction
Expiratory wheezing
Chest X ray No abnormality Emphysematous change Emphysematous changeWBC (leukocytes/ml) 7,900 16,200 12,140CRP (mg/dl) 0.26 0.3 1.04
F, female; WBC, white blood cells; CRP, C reactive protein.
464 Ebihara et al.

Fouchier et al. [2004] reported that HCoV-NL63 wasdetected in 4 (2.9%) of 139 patients with respiratorytract infections of unknown etiology. Since the incidenceof HCoV-NL63 in patients with respiratory tract infec-tions varies depending on several factors such as age,season, year, and clinical diagnosis, it is difficult todetermine the true incidence of the virus. Previousstudies showed that outbreaks of HCoV-229E andHCoV-OC43 occur every 2–3 years [McIntosh et al.,1970; Hambre and Beem, 1972]. If outbreaks of HCoV-NL63 occur every 2–3 years and this 1-year study wasperformed in non-outbreak year, there is the possibilityof underestimation in this data. In studies by the twoDutchgroups, all of the samplespositive forHCoV-NL63were collected in winter. In this study, all the threepositive samples were collected in February andMarch,which is a winter season in Sapporo. This suggests thatHCoV-NL63 may have a seasonal peak in winter inJapan as well as in The Netherlands. Although van derHoeket al. [2004] suggested the cocirculation ofmultiplesubgroups ofHCoV-NL63, the sequences of threeHCoV-NL63 detected in this studywere completely identical tothat of HCoV-NL63 (GenBank accession numberAY567487).
Three nasopharyngeal samples from the threepatients were inoculated on LLC-MK2 cells. However,no cytopathic effect was observed during a 3-weekculture period (data not shown). The two Dutch groupsinitially carried out virus isolation of HCoV-NL63 ontertiary monkey kidney cells, and LLC-MK2 cells orVero cells were used for subsequent virus propagation[Fouchier et al., 2004; van der Hoek et al., 2004].Tertiary monkey kidney cells may be more sensitivethan LLC-MK2 cells for initial isolation of HcoV-NL63.
To our knowledge, this is the first report on HCoV-NL63 infections and the incidence of HCoV-NL63infection in children with bronchiolitis in Asia. Thisstudy suggests that HCoV-NL63 may be widespreadthroughout the world and is one of the causative agentsof acute respiratory wheezing in young children. Toclarify the clinical impact of HCoV-NL63, furthersurveillance in various age groups and various clinicalgroups is needed.
ACKNOWLEDGMENTS
Nasopharyngeal swab samples were kindly pro-vided by Dr. Naofumi Kajii of Ebetsu City Hospital,Dr. Hiroyuki Sawada of Hokkaido Social Insurance
Hospital, and Dr. Mutsuko Konno of Sapporo KoseiGeneral Hospital. We also thank Dr. KunihikoKobayashi, eremitus professor of Hokkaido University,for giving us suggestions and Stewart Chisholm forproofreading the article.
REFERENCES
Andreoletti L, LesayM,DeschildreA, Lambert V,DewildeA,Wattre P.2000. Differential detection of rhinoviruses and enteroviruses RNAsequences associated with classical immunofluorescence assaydetection of respiratory virus antigens in nasopharyngeal swabsfrom infants with bronchiolitis. J Med Virol 61:341–346.
EbiharaT,EndoR,KikutaH, IshiguroN, IshikoH,HaraM,TakahashiY, Kobayashi K. 2004. Human metapneumovirus infection inJapanese children. J Clin Microbiol 42:126–132.
Fouchier RA, Hartwig NG, Bestebroer TM, Niemeyer B, de Jong JC,Simon JH, Osterhaus AD. 2004. A previously undescribed corona-virus associated with respiratory disease in humans. Proc NatlAcad Sci USA 101:6212–6216.
Glezen WP, Greenberg SB, Atmar RL, Piedra PA, Couch RB. 2000.Impact of respiratory virus infections on persons with chronicunderlying conditions. JAMA 283:499–505.
HambreD,BeemM.1972.Virologic studies of acute respiratorydiseasein young adults. V. Coronavirus 229E infections during six years ofsurveillance. Am J Epidemiol 96:94–106.
Holmes KV, Lai MC. 1996. Coronaviridae. In: Fields BN, Knipe DM,Howley PM, editors. Fields virology, 3rd edn. Vol 1. Philadelphia:Lippincott-Raven Press. pp 1075–1093.
Jartti T, Lehtinen P, Vuorinen T, Osterback R, van den Hoogen B,Osterhaus AD, Ruuskanen O. 2004. Respiratory picornavirusesand respiratory syncytial virus as causative agents of acuteexpiratory wheezing in children. Emerg Infect Dis 10:1095–1101.
KsiazekTG,ErdmanD,GoldsmithCS,ZakiSR,PeretT,EmeryS,TongS, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE,HumphreyCD,ShiehWJ,Guarner J,PaddockCD,RotaP,FieldsB,DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ,Anderson LJ. 2003. A novel coronavirus associated with severeacute respiratory syndrome. N Engl J Med 348:1953–1966.
McIntosh K, Kapikian AZ, Turner HC, Hartley JW, Parrott RH,Chanock RM. 1970. Seroepidemiologic studies of coronavirusinfection in adults and children. Am J Epidemiol 91:585–592.
McIntosh K, Chao RK, Krause HE, Wasil R, Mocega HE, Mufson MA.1974. Coronavirus infection in acute lower respiratory tract diseaseof infants. J Infect Dis 130:502–507.
Nicholson KG, Kent J, Hammersley V, Cancio E. 1997. Acute viralinfections of upper respiratory tract in elderly people living in thecommunity: Comparative, prospective, population based study ofdisease burden. BMJ 315:1060–1064.
Papadopoulos NG, Moustaki M, Tsolia M, Bossios A, Astra E,Prezerakou A, Gourgiotis D, Kafetzis D. 2002. Association ofrhinovirus infection with increased disease severity in acutebronchiolitis. Am J Respir Crit Care Med 165:1285–1289.
van derHoekL, PyrcK, JebbinkMF,Vermeulen-OostW,BerkhoutRJ,WolthersKC,Wertheim-vanDillenPM,Kaandorp J, Spaargaren J,Berkhout B. 2004. Identification of a new human coronavirus. NatMed 10:368–373.
Xepapadaki P, Psarras S, Bossios A, Tsolia M, Gourgiotis D, Liapi-Adamidou G, Constantopoulos AG, Kafetzis D, Papadopoulos NG.2004. Human metapneumovirus as a causative agent of acutebronchiolitis in infants. J Clin Virol 30:267–270.
HCoV-NL63 in Children With Bronchiolitis 465

Journal of Medical Virology 75:466–469 (2005)
Genetic Characterization of the M RNA Segmentof a Balkan Crimean-Congo HemorrhagicFever Virus Strain
Anna Papa,1* E. Papadimitriou,1 B. Bozovic,2 and A. Antoniadis1
1A’ Department of Microbiology, School of Medicine, Aristotle University of Thessaloniki, Greece(WHO Collaborating Center for Reference and Research on Arboviruses and Haemorrhagic Fever Viruses)2Torlak Institute of Immunology and Virology, Belgrade, Yugoslavia
Crimean-Congo hemorrhagic fever (CCHF) viruscauses one of the most severe diseases inhumans, with a mortality rate of up to 30%. It istransmitted to humans by the bite of hard ticks orby contact with blood or tissues from humanpatients or infected livestock. Balkan Peninsula isan endemic region of the disease, and sporadiccases or even outbreaks are observed every year.The M RNA segment encodes for the glycopro-tein precursor of two surface glycoproteins Gnand Gc. Up to now complete M RNA CCHF virussequences have been published from strainsisolated in Nigeria, China, Pakistan, Tajikistan,and Russia. In the present study, the geneticcharacterization of the complete nucleotide se-quence of the M RNA segment of a Balkan CCHFvirus strain, Kosovo/9553/2001, isolated in sum-mer of 2001 from a human fatal case in Kosovo isreported. This is the first published complete Mnucleotide sequence of a CCHF virus strainisolated in Balkans. It was found that the Balkanstrain is similar to the Russian strain, both strainsdiffering from all other completely sequencedCCHF virus strains by approximately 22% at thenucleotide level forming an independent clade inthe phylogenetic tree. J. Med. Virol. 75:466–469, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: Crimean-Congo hemorrhagicfever; M segment; geneticcharacterization; glycoprotein;Kosovo
INTRTODUCTION
Crimean-Congo hemorrhagic fever (CCHF) virus(genus Nairovirus, family Bunyaviridae) causes one ofthe most severe diseases in humans, with a mortalityrate of up to 30%.CCHFwasfirst described in the 1940s,when more than 200 human cases occurred in theCrimean peninsula of Russia [Chumakov, 1947], and is
now endemic in different regions of Africa, Asia, andEurope. In Europe, human CCHF cases have beenreported in Albania, former Yugoslavia, Bulgaria, andEuropean Russia [Aristova et al., 2001; Papa et al.,2002a; Papa et al., 2002b; Yashina et al., 2003; Papaet al., 2004], where a number of cases occur every year,either sporadically or in an outbreak form. During thespring and summer of 2001 a large CCHF outbreakoccurred in Kosovo, and, in a smaller scale, in Albaniaand in Bulgaria [www.who.int/disease-outbreak-news/n2001/june/29june2001.html; Papa et al., 2002a, 2004].
CCHFV, like allmembers of the genusNairovirus, is anegative stranded RNA virus with a tripartite genomeconsisting of a small (S), a medium (M), and a large (L)segment, encoding the nucleocapsid (N) protein, theglycoprotein precursor, resulting in the two envelopeglycoproteins Gn and Gc (previously referred to as G2
and G1), and the putative RNA-dependent polymerase,respectively [Nichol, 2001]. The virus is transmitted tohumans by the bite of hard ticks (family Ixodidae),usually those belonging in the genera Hyalomma,Amblyomma, Dermacentor, and Rhipicephalus, or bycontact with blood or tissues from human patients orinfected livestock. There is a high risk for spread fromperson to person by exposure to infected blood, respira-tory secretions and excreta, resulting occasionally innosocomial outbreaks. After an incubation period of 3–7 days, clinical picture is sudden onset of fever, chills,myalgia, headache, and rapidly evolving severe illness,followed by a hemorrhagic state with bleeding from the
This work was performed at the A’ Department of Microbiology,School of Medicine, Aristotle University of Thessaloniki, Greece.
Grant sponsor: Hellenic Center for Infectious Diseases Control(HCIDC), Greek Ministry of Health and Welfare.
*Correspondence to: Prof. Anna Papa, MD, A’ Department ofMicrobiology, School of Medicine, Aristotle University of Thessa-loniki, 54124, Thessaloniki, Greece. E-mail: [email protected]
Accepted 2 November 2004
DOI 10.1002/jmv.20290
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

mucous membranes and petechiae, associated withthrombocytopenia and leukopenia [Swanepoel et al.,1987].
In the summer of 2001, two viral strains have beenisolated from a fatal CCHF case in Kosovo and theattendant doctor in Belgrade (strains Kosovo/9553/2001and Kosovo/9717/2001, respectively) [Papa et al., 2002].Phylogenetic analysis of partial S genome RNAsequences of the strains (identical each other) revealedthat they were clustering together with the Drosdovstrain, differing from it by 4% at the nucleotide level[Papa et al., 2002]. The Drosdov strain was isolated in1967 from a patient in the Astrakhan region in Russia,and was characterized genetically in 1997 (GenBankaccession number U88412) [Aristova et al., 2001].
As theglycoproteinsprobably influence thevertebrateand tick host usage, the cell tropism of the virus and thehighpathogenicity of thevirusduringhuman infections,knowledge of M segment genetic variability is of greatvalue. To date, several complete, and evenmore partial,S segment sequences have been identified. However,only a very limited number of complete M segmentsequences are available; these sequences are fromstrains isolated in Nigeria, China, Pakistan, Russia,andTajikistan [Morikava et al., 2002; Papa et al., 2002b;Sanchez et al., 2002; Seregin et al., 2004]. In the presentstudy, the genetic characterization of the complete MRNAsegment ofCCHFvirus strainKosovo/9553/2001 isdescribed.
MATERIALS AND METHODS
CCHF Virus Strain
TheCCHFvirus strain used in this studywasKosovo/9553/2001, which was isolated in 2001 fromwhole bloodsample of a fatal case inKosovo. The sixth passage of thevirus in Vero E6 cells was used in this study.
RNA Extraction, Reverse Transcription,Amplification, and Sequencing
Viral RNA was extracted from culture supernatantusing the Viral RNA extraction kit (Qiagen GmbH,Hilden, Germany). A number of PCRs were performedusing primers based on the sequence of IbAr10200CCHFvirus strain, previously designed for the sequencedetermination of the M RNA segment of the ChineseCCHF virus strains. Additional primers were alsodesigned on the basis of the newly obtained sequences.PCR products were purified using the QIAquick PCRPurification kit (Qiagen GmbH). Sequence reactionswere generated from the amplified cDNA by CLIPTM
sequencing. CLIP allows both directions of the cDNA tobe sequenced simultaneously in the same tube, usingtwo different dye labeled primers. Sequences wereobtained using the OpenGene automated DNA sequen-cing system (Visible Genetics, Inc., Toronto, Canada).
Alignment, Phylogenetic, and Protein Analysis
Clustal W [Thompson et al., 1994] was used to alignthe obtained nucleotide sequences with complete CCHF
M RNA sequences taken from the GenBank DataBase.Sequences used for the genetic comparisonwere those oftwo Chinese strains BA66019 (AF350448) and BA8402(AF350449) [Papa et al., 2002b], the Matin strain(AF467769) [Sanchez et al., 2002], the IbAr10200 strain(U39455) [Sanchez et al., 2002], and strains VLG/TI29414 (AY179961) and TADJ/HU8966 (AY179962)from Russia and Tajikistan, respectively [Seregin et al.,2004]. Both Chinese strains were isolated in XinjiangProvince, an autonomous region in north-westernChina. Strain BA66019 was isolated from a CCHFpatient in 1965, while strain BA8402 was isolated fromHyalomma asiaticum ticks in 1984. Matin strain wasisolated in from a patient in Pakistan in 1976; theIbAr10200 strain was originally isolated from Hya-lomma excavatum ticks from Sokoto, Nigeria, in 1976;VLG/TI29414 was isolated from Hyalomma margin-atum ticks in the Volgograd area in 2000; and TADJ/HU8966was isolated fromaCCHFpatient in Tajikistanin 1990.
A number of partial sequences from CCHF virusstrains from Russia and Central Asia were also used forcomparison with the respective genome region of theKosovo/9557 strain [Yashina et al., 2003; Kuhn et al.,2004].
Aligned sequences were investigated by phylogeneticinference analysis using the PHYLIP package version3.57c [Felsenstein, 1993]. DNADIST, PROTDIST, andFITCH programs were used to measure genetic dis-tances and construct trees; SEQBOOT was used togenerate bootstrap data files (100 replicates), andCONSENSE to compute the consensus tree.
Prediction of transmembrane helices (TMHs) inproteins was conducted with TMHMM version 2.0[Krogh et al., 2001]. SignalP (version 2.0) was used forthe prediction of signal sequence cleavage sites [Nielsenet al., 1997], while predictions of mucin-type GalNAc O-glycosylation sites were done with the NetOGlyc 2.0prediction server [Hansen et al., 1998].
RESULTS
The complete nucleotide sequence of theM segment ofCCHF virus Kosovo/9553 strain was 5364 nts in length(taking into account also the primers), encoding apolyprotein of 1688 amino acids (aa) with a predictedmolecular weight of 187127.7. The sequence has beensubmitted to GenBank and assigned the accessionnumber AY675511. A deletion of one aa is observedcomparing with Matin strain, as well as with strainsfromChina, Nigeria, and Tajikistan. This is the 82nd aanumbering ofMatin strain. The samedeletion is presentin all Russian CCHF virus strains, for which the re-spective genome fragment has been identified.
A phylogenetic tree was constructed based on thecomplete ORF nucleotide sequence, using Dugbe virus,another member of Nairovirus genus, as outgroup(Fig. 1). The genetic distances among the Kosovo/9553strain and other CCHF virus strains in the M segmentORF nucleotide sequence and the encoded amino acid
M RNA Segment of a Balkan CCHF Virus Strain 467

sequenceare shown inTable I.Kosovo/9553 strain formstogether with strain VLG/TI29414 an independent‘‘European clade.’’An exception in the clustering of the European
strains comprises the Greek CCHF virus strain, AP92[Papadopoulos and Koptopoulos, 1980], not yet asso-ciated with disease in humans. Preliminary study onpartial regions of theM segment of strain AP92, showedthat it differs from all other strains forming an inde-pendent clade (data not shown, studies in progress).This happens also in the phylogenetic tree based on SRNA segment sequences, where AP92 differs from theKosovo/9553 strain by 24.3% at the nucleotide level[Papa et al., 2002b].As was the case in the Chinese, Matin and IbAr10200
strains, the M segment encoded polyprotein consists oftwo distinct regions: the amino terminus, consisting of243–248 aa of the polyprotein, which is highly variable,
and a relatively conserved region, which encompassesthe remainder of the polyprotein [Papa et al., 2002c;Sanchez et al., 2002]. Strains from Kosovo and Russiaare similar also in the variable region (genetic difference7.8%–9%), differing from strains isolated in China,Tajikistan, Pakistan, and Nigeria by 46%–55%. In thisregionagreat number ofO-linked glycosylation sites arepresent, giving to the domain a mucin-like feature,which is found only in nairoviruses, and in no othergenera of the Bunyaviridae family. Concerning the con-served region of the genome, a lower divergence isobserved (genetic distance among Kosovo/9553 andRussian strains 5.9%–6.6%, comparing with 19%–22%with the rest strains).
The M segment encodes a polyprotein from whichPreGn (140 kDa) and PreGc (85 kDa) proteins areprocessed to constitute the precursors of mature Gn(37 kDa) and Gc (75 kDa), respectively [Sanchez et al.,2002]. In Kosovo/9553 strain, cleavage of the signalpeptide is predicted to occur betweenaa27and28 (AHG-QS) based on neural networks (NN) trained on eucar-yotic data. The same cleavage site is observed in theChinese strain BA8402 (SHG/LS), while the predictedcleavage site for BA66019 aa is between aa 21 and22 (LWS-LE), for IbAr10200 strainbetweenaa22and23(THG-SH), and for Matin strain between aa 24 and25 (SEG-IH).
There are five predicted TMHs within the glycopro-tein ORF. A hydropathy plot of the M segment proteinsequence shows at least five highly hydrophobic,potentially membrane-spanning regions. The theoreti-cal pI is 7.9. The polypeptide sequence is predicted tocontain 14 potential asparagine-linked glycosylationsites (Asp-X-Ser/Thr), 12 of them in the lumenal side. Aheavy concentration of O-glycosylation is seen withinthe variable domain, and a smaller one in the beginningof the conserved region.
The N termini of the Gn and Gc proteins of Matinstrain were found to begin at aa 525 and 1046 of theencoded polyprotein precursor, respectively, and tetra-peptides RRLL and RKPL preceded the mature Gn andGc, suggesting the likely use of SKI-1-like proteases forthe major glycoprotein proteolytic processing events,similar to Lassa fever and Guanarito arenaviruses[Sanchez et al., 2002]. Further studies indicate thatcorrect processing of the Gn of CCHF virus require thehydrophobic region aa 967–995, as well as the intactRRLL tetrapeptide at the N-terminus of Gn, and ismediated by SKI-1 protease, while Gc is preceded byRKPLand is independent of SKI-1 [Vincent et al., 2003].By alignment, tetrapeptides RRLL and RKPL are
Fig. 1. Phylogenetic tree based on 4773-nt region of themedium (M)RNA segment of Crimean-Congo hemorrhagic fever (CCHF) virusstrains tested in this study. Dugbe virus (NC_004158) was used asoutgroup. Horizontal distances are proportional to nucleotide differ-ence; vertical distances are for graphic display only. Bootstrap support(in %) is indicated at the respective branch.
TABLE I. Genetic Distances of Complete M Segment ORF Sequences Between Kosovo/9553 Strain and Other Crimean-CongoHemorrhagic Fever (CCHF) Virus Strains
VLG/TI2941Russia
TADJ/HU896Tajikistan
MatinPakistan
IbAr10200Nigeria
BA8402China
BA66019China
Kosovo/9553 (nt) 6.67 23.16 20.82 20.29 22.48 21.72Kosovo/9553 (aa) 6.11 18.16 17.23 17.10 16.96 17.30
nt, nucleotide; aa, amino acid.
468 Papa et al.

preceded mature Gn and Gc, respectively, in all CCHFvirus sequences tested in this study, exceptKosovo/9553andRussian strains,whichhaveRKLLinsteadofRRLL.However, RKLL is also a major cleavage recognitionsite for SKI-1 protease, which is a novel mammaliansubtilase that cleaves precursors within the cis/medial-Golgi at the motif (R/K)X(hydrophobic)Z#, where Z isany amino acid, preferentially Leu or Thr, but excludingVal, Pro, Glu, Asp, or Cys [Elagoz et al., 2002]. Dugbevirus has also tetrapeptide RKLL preceding Gn.
DISCUSSION
Although there are several complete, and even morepartial, S segment sequences identified, there is onlylimited information concerning the M RNA segmentsof CCHF virus strains. The M segment encodes twostructural glycoproteins, Gn and Gc, located in the Nand C termini of the polyprotein, respectively. In thisstudy the complete nucleotide sequence of the M RNAsegment of Kosovo/9553/2001 strain of CCHF virus,which was isolated from blood sample of a fatal case inKosovo in 2001, is described. These are the first datafrom a Balkan CCHF virus strain increasing ourknowledge in the molecular epidemiology of CCHF, notonly geographically, but also chronologically. A greatdegree of genetic diversity is observed among strainsisolated from different geographic areas, like Nigeria,Pakistan, Tajikistan, Russia, and China. Kosovo strainis clustering with the Russian strains differing from allother strains tested, comprising an independent clade inthe completeM segment phylogenetic tree.Whether thegenetic divergence is associatedwith different tick hostsremains to be elucidated.
As partialMRNA sequences fromRussian strains areavailable in the GenBank, a comparison of the Kosovostrain with them showed that they are quite similar,even in the variable N-terminal region, suggesting thatthere is a common European clade and a commonevolution route. Similar clustering is observed in the Ssegment CCHF sequences; however, a lower diversity isobserved in the S RNA segment than in the M segment.
Despite the great genetic divergence among CCHFstrains, some interesting features are common amongthem, such as proteolytic cleavage and the presence oftwo distinct regions of the encoded polyprotein: thevariable N-terminal and the conserved middle and C-terminal of the protein. Further studies are needed toelucidate the exact features of the encoded proteins andtheir role in the pathogenicity of the virus.
REFERENCES
Aristova VA, Kolobukhina LV, Shchelkanov MY, Lvov DK. 2001.Ecology and clinical features of Crimean-Congo hemorrhagicfever in Russia and neighboring countries. Vopr Virusol 45:7–15(in Russian).
Chumakov MP. 1947. A new virus disease—Crimean hemorrhagicfever. Nov Med 4:9–11.
ElagozA,Benjannet S,Mammarbassi A,WickhamL, SeidahNG. 2002.Biosynthesis and cellular trafficking of the convertase SKI-1/S1P.J Biol Chem 277:11265–11275.
Felsenstein J. 1993. PHYLIP (Phylogeny Inference Package) version3.5c. Distributed by the author. Seattle: Department of Genetics,University of Washington.
Hansen JE, Lund O, Tolstrup N, Gooley AA, Williams KL, Brunak S.1998. NetOGlyc: Prediction of mucin type O-glycosylation sitesbased on sequence context and surface accessibility. Glycoconj J15:115–130.
KroghA, LarssonG, vonHeijneG, SonnbammerELL. 2001. Predictingtransmembrane protein topology with a hidden Markov model:Application to complete genomes. J Mol Biol 305:567–580.
KuhnJH,SereginSV,MorzunovSP,Petrova ID,VyshemirskiiOI,LvovDK, Tyunnikov GI, Gutorov VV, Netesov SV, Petrov VS. 2004.Genetic analysis of the M RNA segment of Crimean-Congo hemorrhagic fever virus strains involved in the recentoutbreaks in Russia. Arch Virol online DOI 10.1007/s00705-004-0354-3.
Morikava S, Qing T, Xinqin Z, Saijo M, Kurane I. 2002. Geneticdiversity of the M RNA segment among Crimean-Congo hemor-rhagic fever virus isolates in China. Virology 296:159–164.
Nichol ST. 2001. Bunyaviruses. In: Knipe DM, Howley P, editors.Fields virology, vol. 2, Philadelphia, Pennsylvania: Lippincott,Williams and Wilkins. pp 1603–1633.
NielsenH, Engelbrecht J, Brunak S, vonHeijne G. 1997. Identificationof prokaryotic andeukaryotic signal peptides andprediction of theircleavage sites. Protein Eng 10:1–6.
Papa A, Bino S, Llagami A, Brahimaj B, Papadimitriou E, Pavlidou V,Velo E, Cahani G, Hajdini M, Pilaca A, Harxhi A, Antoniadis A.2002a. Crimean-Congo hemorrhagic fever in Albania, 2001. Eur JClin Microbiol Infect Dis 21:603–606.
Papa A, Bozovic B, Pavlidou V, Papadimitriou E, Pelemis M,Antoniadis A. 2002b. Genetic detection and isolation of Crimean-Congo hemorrhagic fever virus, Kosovo, Yugoslavia. Emerg InfectDis 8:852–854.
Papa A, Ma B, Kouidou S, Tang Q, Hang C, Antoniadis A. 2002c.Genetic characterization of the M RNA segment of CrimeanCongo hemorrhagic fever virus strains, China. Emerg Infect Dis8:50–53.
Papa A, Christova I, Papadimitriou E, Antoniadis A. 2004. Crimean-Congo hemorrhagic fever in Bulgaria. Emerg Infect Dis 10:1465–1467.
Papadopoulos O, Koptopoulos G. 1980. Crimean-Congo hemorrhagicfever (CCHF) in Greece: Isolation of the virus from Rhipicephalusbursa ticks and a preliminary serological survey. Zentbl BakteriolHyg Abt 1(Suppl 9):189–193.
Sanchez AJ, Vincent MJ, Nichol ST. 2002. Characterization of theglycoproteins of Crimean-Congo hemorrhagic fever virus. J Virol76:7263–7275.
Seregin SV, Samokhvalov EI, Petrova ID, Vyshemirskii OI,Samokhvalova EG, Lvov DK, Gutorov VV, Tyunnikov GI,Shchelkunov SN, Netesov SV, Petrov VS. 2004. Genetic character-ization of theMRNAsegment of Crimean-Congo hemorrhagic fevervirus strains isolated in Russia and Tajikistan. Virus Genes 28:187–193.
Swanepoel R, ShepherdAJ, LemanPA, Shepherd SP,McGillivrayGM,Erasmus MJ, Searle LA, Gill DE. 1987. Epidemiologic and clinicalfeatures of Crimean-Congo hemorrhagic fever in southern Africa.Am J Trop Med Hyg 36:120–132.
Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTALW: Improvingthe sensitivity of progressivemultiple sequence alignment throughsequence weighting, position-specific gap penalties, and weightmatrix choice. Nucleic Acids Res 22:4673–4680.
Vincent MJ, Sanchez AJ, Erickson BR, Basak A, Chretien M, SeidahNG, Nichol ST. 2003. Crimean-Congo hemorrhagic fever virusglycoprotein proteolytic processing by subtilase SKI-1. J Virol 77:8640–8649.
YashinaL, Vyshemirskii O, Seregin S, Petrova I, Samokhvalov E, LvovD, Gutorov V, Kuzina I, Tyunnikov G, Tang YW, Netesov S, PetrovV. 2003. Genetic analysis of Crimean-Congo hemorrhagic fevervirus in Russia. J Clin Microbiol 41:860–862.
M RNA Segment of a Balkan CCHF Virus Strain 469

Journal of Medical Virology 75:470–474 (2005)
Rapid and Sensitive Detection of Mumps Virus RNADirectly From Clinical Samples by Real-Time PCR
Kazue Uchida,1,2* Michiyo Shinohara,1 Shin-ichi Shimada,1 Yukari Segawa,1 Rie Doi,1 Atushi Gotoh,1
and Ryo Hondo2
1Division of Virology, Saitama Institute of Public Health, Saitama, Japan2Division of Veterinary Public Health, Nippon Veterinary and Animal Science University, Tokyo, Japan
A rapid, sensitive, and specific assay to detectmumps virus RNA directly from clinical speci-mens using a real-time PCR assay was devel-oped. The assay was capable of detecting fivecopies of standard plasmid containing cDNAfrom themumps virus F gene. No cross-reactionswere observed with other members of Paramy-xoviridae, or with viruses or bacteria known to bemeningitis pathogens. Seventy-three clinicalsamples consisting of throat swabs collectedfrom patients with parotitis, and cerebrospinalfluid (CSF) collected from patients with asepticmeningitis, were examined with a real-time PCRassay developed by the authors, reverse-transcription nested-PCR (RT-n-PCR), and virusisolation using cell culture. Like the RT-n-PCRassay, the real-time PCR assay could detectmumps virus RNA in approximately 70% of boththroat swabs and CSF samples, while, by tissueculture, mumps virus was isolated from onlyapproximately 20% of CSF and 50% of throatswab samples. In addition, the real-time PCRassay could be developed easily into a quantita-tive assay for clinical specimens containingmorethan 1,800 copies of mumps virus RNA/ml byusing serial dilutions of the standard plasmid.The results suggest that the real-time PCR assayis useful for identification of mumps virus infec-tions, not only in typical cases, but also insuspected cases, which show only symptoms ofmeningitis or encephalitis. J. Med. Virol. 75:470–474, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: mumps meningitis; TaqManPCR; quantitative assay
INTRODUCTION
Mumps virus is a member of the family Paramyxovir-idae, genus Rubulavirus [for review, see Rima et al.,1995; Elango et al., 1988], and its genome is a singlestrand of RNA with negative polarity, approximately15 kb in length, encoding seven proteins in the following
order: 30-nucleocapsid (N), phosphoprotein (P), matrix(M), fusion (F), small hydrophobic (SH), hemagglutinin-neuraminidase (HN), and large protein (L)-50. Mumpsvirus is a common human pathogen that causes child-hood parotitis and also, frequently, complications suchas meningitis, pancreatitis, and orchitis. The majorityof mumps infections are benign; however, the diseasecauses more serious and rare complications such asencephalitis, cerebellar ataxia [Cohen et al., 1992], andhearing loss [Okamoto et al., 1994]. Only a fewmethodshave hitherto been available for laboratory diagnosis ofmumps virus infections, one based on the detection ofmumps virus-specific IgM antibodies in serum, and theother, on virus isolation using tissue culture. Only a fewcommercial kits for serological tests such as enzymeimmunoassay are available, other than methods suchas hemagglutination inhibition, immunofluorescence,and complement-fixation assay, which are laborious.Moreover, mumps virus infections may occur withoutthe induction of the specific IgMantibodies, particularlyin patients vaccinated previously or in reinfection cases[Gut et al., 1995], and false positive results due to cross-reactivity with other paramyxoviruses complicate ser-ological diagnosis [Julkunen, 1984]. On the other hand,the procedure for mumps virus isolation from clinicalsamples lacks sensitivity and is time-consuming. A fewinvestigators have reported that the rates of isolationfrom CSF and nasopharyngeal swab (NPS) samplesobtained from patients with confirmed mumps virusinfections were about 30–40% and 50%, respectively[Kashiwagi et al., 1997; Poggio et al., 2000]. They alsodescribed the development of the reverse transcription(RT)-nested PCR (n-PCR) procedures for rapid andsensitive detection of mumps virus RNA directly fromclinical samples, and found that the rate of detection of
*Correspondence to: Kazue Uchida, Division of Virology,Saitama Institute of Public Health, 639–1, Kamiokubo, Sakura-ku, Saitama City, Saitama Prefecture, 338-0824, Japan.E-mail: [email protected]
Accepted 14 October 2004
DOI 10.1002/jmv.20291
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.

the virus could bemarkedly increased [Kashiwagi et al.,1997; Jin et al., 1999; Poggio et al., 2000].
The purpose of this study is to describe a rapidand sensitive method of detecting mumps virus RNAdirectly from clinical samples using a real-time PCRassay. The assay does not require nested PCR proce-dures to obtain sensitivity, restriction fragment lengthpolymorphism (RFLP), or nucleotide sequencing foridentification of the amplified products. The real-timePCR assay described below was evaluated by testingclinical samples consisting of throat swab samples andcerebrospinal fluid (CSF) samples. Also, the resultsare compared with those from virus isolation and theRT-n-PCR assay described previously.
MATERIALS AND METHODS
Clinical Samples and Mumps Virus Stock
The clinical samples used consisted of 46 throat swabsamples obtained from 46 patients with parotitis and27 CSF samples obtained from 27 patients with asepticmeningitis. Of the meningitis patients, 24 had salivarygland swelling, two patients had had mumps vaccina-tion 2 weeks before onset, and one patient did not showsalivary gland swelling. Throat swab samples, whichwere shown by isolation or by PCR to be positive forother viral or bacterial agents, were used to confirmthe specificity of the real-time PCR. They includeenteroviruses (poliovirus 1–3, coxsackievirus A and B,enterovirus 71, and echoviruses), influenza A virus(AH1N1, AH3N2), influenza B virus, measles virus,human parainfluenza virus 1–3, human respiratory-syncytial virus, Listeria monocytogenes, Neisseriameningitidis, Haemophilus influenzae, Streptococcusagalactiae.Between103and104 copies ofDNAofhumanherpesviruses 1–6 (Advanced Biotechnologies, Inc.,Columbia, MD) and Escherichia coli, Streptococcuspneumoniae, Staphylococcus aureus (American typeculture collection) were also tested. All of these clinicalsamples were collected from clinics and hospitals in theSaitama Prefecture, Japan, during the years 1997–2003. Throat swab samples were obtained by wiping thethroatwith cotton swabs andwere soaked in 2ml of 2.5%veal infusion broth (Becton Dickinson Company, Ltd.,Franklin Lakes, NJ) solution with antibiotics andbovine serum albumin. These samples were kept at�808C until used in virus isolation. They were thawedonce for virus isolation, then frozen again and kept at�808C until used in RNA extraction. For the mumpsvirus stock, SA963/Ja00 strain [Uchida et al., 2003] waspropagated in Vero cells, and the virus stock was storedin 0.5-ml aliquots at�808C. Plaque titration of the virusstockwasdoneusing confluentVero cells in 6-well tissueculture plates.
Virus Isolation
Virus isolation was performed using Vero and BS-C-1cell cultures. Cellmonolayers on a 24-well tissue cultureplate with 1 ml minimal essential medium containing0.001%acetylated trypsinwere inoculatedwith0.1ml of
the clinical samples. The cell monolayers were incu-bated at 378C for 7 days andwere examined daily for theappearance of cytopathic effects (CPE). The sampleswere passaged twice if no CPE were observed. Thepresence of mumps virus in cultures showing typicalCPE was confirmed by a neutralization test usingmumps virus antibody (Denka Seiken Co., Ltd., Tokyo,Japan). Samples without CPE after two blind passageswere considered negative for virus isolation.
RNA Extraction and cDNA Synthesis
Viral RNA was extracted from 140 ml of a clinicalsample using a QIAamp Viral RNA mini kit (QIAGENCorp., Inc., Hilden, Germany) according to the manu-facturer’s instructions. RNA was eluted with 60 ml ofdiethyl pyrocarbonate-treated water. The cDNA wassynthesized in 20 ml of a reaction mixture containing12 ml of eluted RNA, 200U of SuperScript II RNaseH(�)reverse transcriptase (Invitrogen Corp., Carlsbad, CA)and first strand buffer solution supplied with theenzyme, 0.5mMof each dNTPmix, 5mMdithiothreitol,50 pmol of random hexamers (Invitrogen), and 20 Uof RNasin ribonuclease inhibitor (Promega Corp.,Madison, WI). The mixture was incubated at 428C for2 hr, and the enzymewas inactivated at 958C for 10min.
Real-Time PCR and RT-n-PCR
For the real-time PCR assay, primers F1073 (50-TCTCACCCATAGCAGGGAGTTATAT-30) and R1151(50-GTTAGACTTCGACAGTTTGCAACAA-30) and aTaqMan probe (50-AGGCGATTTGTA GCACTGGATG-GAACA-30) were designed from the published sequenceof the mumps virus F gene from the SA956/Ja00 strain(DDBJ accession number AB085219) [Uchida et al.,2003] using Primer Express software version 1.5(Applied Biosystems, Foster City, CA). The probe wassynthesized with a reporter dye, 6-carboxyfluorescein(FAM), and a quencher dye, 6-carboxytetramethylrho-damine (TAMRA), covalently linked to the50 and30 ends,respectively (Applied Biosystems). The real-time quan-titative PCR was carried out in a final volume of 35 mlreaction mixture containing 4 ml of cDNA, 17.5 ml ofTaqMan Universal PCR Master Mix (Applied Biosys-tems), each primer at a concentration of 300 nM, and theprobe at 100 nM. PCR amplification was performedwith an ABI PRISM 7700 Sequence Detector (AppliedBiosystems) under the following conditions: initialincubation at 508C for 2 min and 958C for 10 min, andthen 45 cycles of amplification with denaturation at958C for 15 sec and annealing and extension at 598Cfor 1 min. Amplification data were collected andanalyzed with Sequence Detector software version 1.7(Applied Biosystems). In each operation, a recombinantplasmid containing one copy of the mumps virus F genewas used as a standard plasmid. To construct thestandard plasmids, RNA from the mumps virus strainSA956/Ja00 was transcribed to cDNA, then the cDNAwas amplified with primers F8 and F9, describedpreviously [Uchida et al., 2003]. The amplified product
Mumps Virus Detection by Real-Time PCR 471
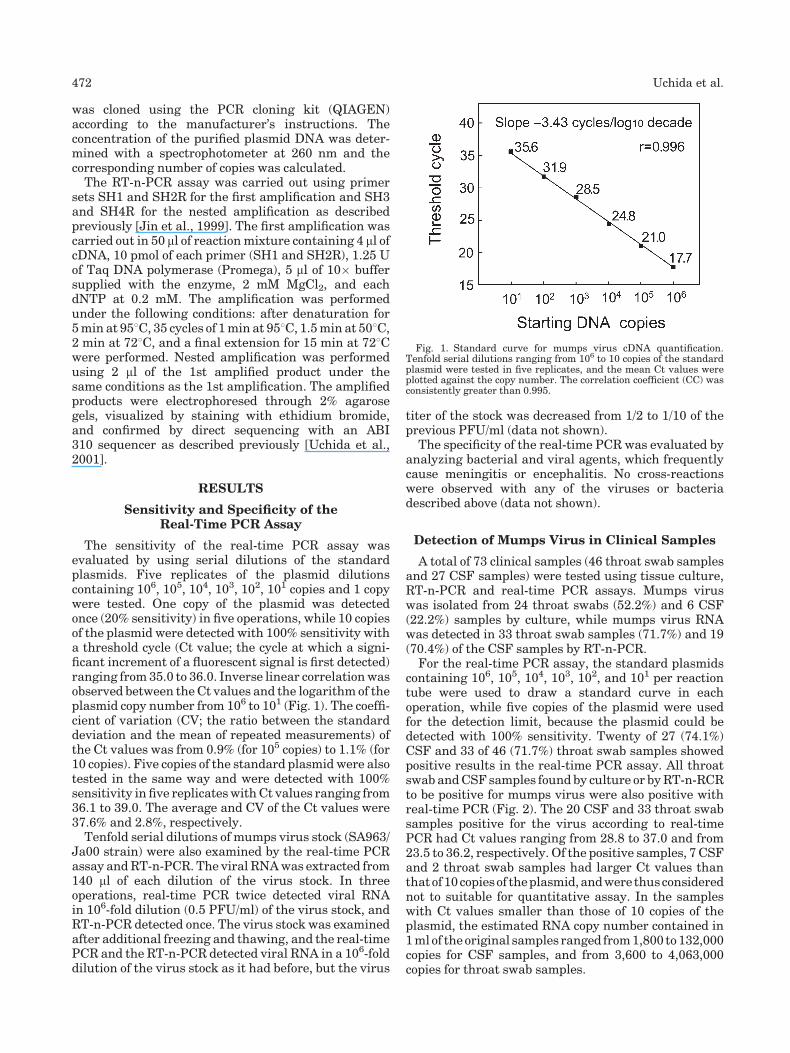
was cloned using the PCR cloning kit (QIAGEN)according to the manufacturer’s instructions. Theconcentration of the purified plasmid DNA was deter-mined with a spectrophotometer at 260 nm and thecorresponding number of copies was calculated.The RT-n-PCR assay was carried out using primer
sets SH1 and SH2R for the first amplification and SH3and SH4R for the nested amplification as describedpreviously [Jin et al., 1999]. The first amplification wascarried out in 50 ml of reactionmixture containing 4 ml ofcDNA, 10 pmol of each primer (SH1 and SH2R), 1.25 Uof Taq DNA polymerase (Promega), 5 ml of 10� buffersupplied with the enzyme, 2 mM MgCl2, and eachdNTP at 0.2 mM. The amplification was performedunder the following conditions: after denaturation for5min at 958C, 35 cycles of 1min at 958C, 1.5min at 508C,2 min at 728C, and a final extension for 15 min at 728Cwere performed. Nested amplification was performedusing 2 ml of the 1st amplified product under thesame conditions as the 1st amplification. The amplifiedproducts were electrophoresed through 2% agarosegels, visualized by staining with ethidium bromide,and confirmed by direct sequencing with an ABI310 sequencer as described previously [Uchida et al.,2001].
RESULTS
Sensitivity and Specificity of theReal-Time PCR Assay
The sensitivity of the real-time PCR assay wasevaluated by using serial dilutions of the standardplasmids. Five replicates of the plasmid dilutionscontaining 106, 105, 104, 103, 102, 101 copies and 1 copywere tested. One copy of the plasmid was detectedonce (20% sensitivity) in five operations, while 10 copiesof the plasmid were detected with 100% sensitivity witha threshold cycle (Ct value; the cycle at which a signi-ficant increment of a fluorescent signal is first detected)ranging from35.0 to 36.0. Inverse linear correlationwasobserved between theCt values and the logarithm of theplasmid copy number from 106 to 101 (Fig. 1). The coeffi-cient of variation (CV; the ratio between the standarddeviation and the mean of repeated measurements) ofthe Ct values was from 0.9% (for 105 copies) to 1.1% (for10 copies). Five copies of the standard plasmid were alsotested in the same way and were detected with 100%sensitivity in five replicateswithCt values ranging from36.1 to 39.0. The average and CV of the Ct values were37.6% and 2.8%, respectively.Tenfold serial dilutions of mumps virus stock (SA963/
Ja00 strain) were also examined by the real-time PCRassay andRT-n-PCR. The viral RNAwas extracted from140 ml of each dilution of the virus stock. In threeoperations, real-time PCR twice detected viral RNAin 106-fold dilution (0.5 PFU/ml) of the virus stock, andRT-n-PCR detected once. The virus stock was examinedafter additional freezing and thawing, and the real-timePCR and the RT-n-PCR detected viral RNA in a 106-folddilution of the virus stock as it had before, but the virus
titer of the stock was decreased from 1/2 to 1/10 of theprevious PFU/ml (data not shown).
The specificity of the real-time PCR was evaluated byanalyzing bacterial and viral agents, which frequentlycause meningitis or encephalitis. No cross-reactionswere observed with any of the viruses or bacteriadescribed above (data not shown).
Detection of Mumps Virus in Clinical Samples
A total of 73 clinical samples (46 throat swab samplesand 27 CSF samples) were tested using tissue culture,RT-n-PCR and real-time PCR assays. Mumps viruswas isolated from 24 throat swabs (52.2%) and 6 CSF(22.2%) samples by culture, while mumps virus RNAwas detected in 33 throat swab samples (71.7%) and 19(70.4%) of the CSF samples by RT-n-PCR.
For the real-time PCR assay, the standard plasmidscontaining 106, 105, 104, 103, 102, and 101 per reactiontube were used to draw a standard curve in eachoperation, while five copies of the plasmid were usedfor the detection limit, because the plasmid could bedetected with 100% sensitivity. Twenty of 27 (74.1%)CSF and 33 of 46 (71.7%) throat swab samples showedpositive results in the real-time PCR assay. All throatswabandCSF samples found by culture or byRT-n-RCRto be positive for mumps virus were also positive withreal-time PCR (Fig. 2). The 20 CSF and 33 throat swabsamples positive for the virus according to real-timePCR had Ct values ranging from 28.8 to 37.0 and from23.5 to 36.2, respectively. Of the positive samples, 7 CSFand 2 throat swab samples had larger Ct values thanthatof10copiesof theplasmid,andwere thusconsiderednot to suitable for quantitative assay. In the sampleswith Ct values smaller than those of 10 copies of theplasmid, the estimated RNA copy number contained in1mlof theoriginal samples ranged from1,800 to132,000copies for CSF samples, and from 3,600 to 4,063,000copies for throat swab samples.
Fig. 1. Standard curve for mumps virus cDNA quantification.Tenfold serial dilutions ranging from 106 to 10 copies of the standardplasmid were tested in five replicates, and the mean Ct values wereplotted against the copy number. The correlation coefficient (CC) wasconsistently greater than 0.995.
472 Uchida et al.
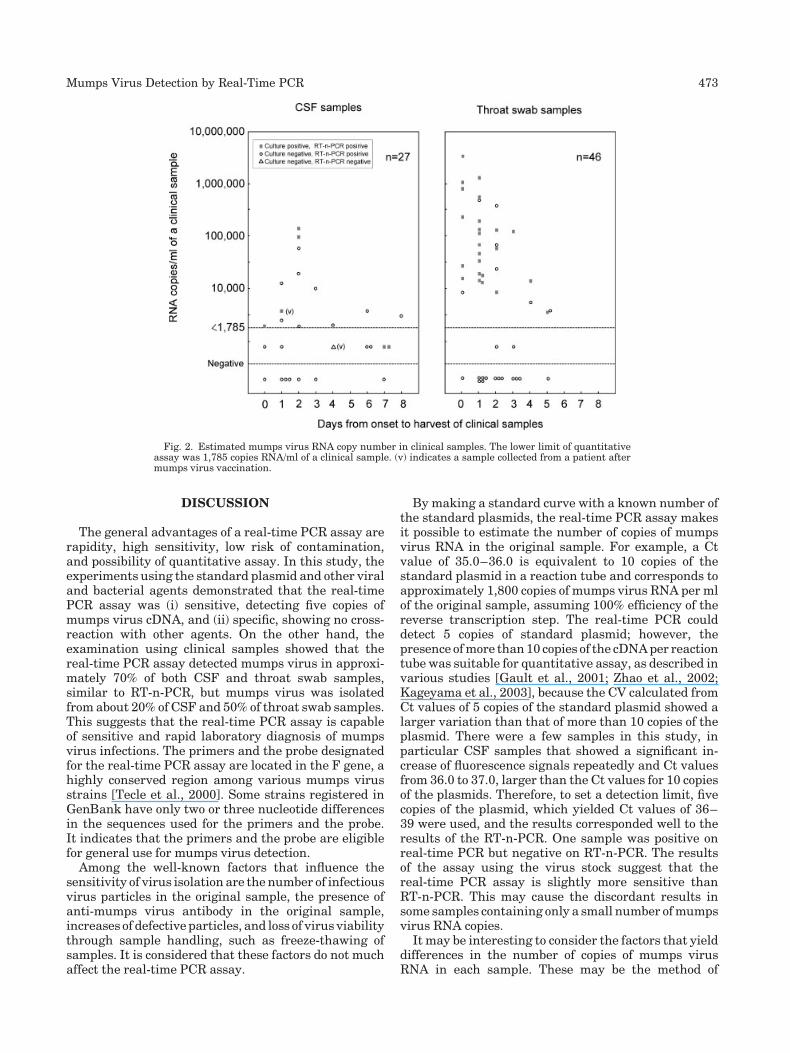
DISCUSSION
The general advantages of a real-time PCR assay arerapidity, high sensitivity, low risk of contamination,and possibility of quantitative assay. In this study, theexperiments using the standard plasmid and other viraland bacterial agents demonstrated that the real-timePCR assay was (i) sensitive, detecting five copies ofmumps virus cDNA, and (ii) specific, showing no cross-reaction with other agents. On the other hand, theexamination using clinical samples showed that thereal-time PCR assay detected mumps virus in approxi-mately 70% of both CSF and throat swab samples,similar to RT-n-PCR, but mumps virus was isolatedfrom about 20% of CSF and 50% of throat swab samples.This suggests that the real-time PCR assay is capableof sensitive and rapid laboratory diagnosis of mumpsvirus infections. The primers and the probe designatedfor the real-time PCR assay are located in the F gene, ahighly conserved region among various mumps virusstrains [Tecle et al., 2000]. Some strains registered inGenBank have only two or three nucleotide differencesin the sequences used for the primers and the probe.It indicates that the primers and the probe are eligiblefor general use for mumps virus detection.
Among the well-known factors that influence thesensitivity of virus isolation are the number of infectiousvirus particles in the original sample, the presence ofanti-mumps virus antibody in the original sample,increases of defective particles, and loss of virus viabilitythrough sample handling, such as freeze-thawing ofsamples. It is considered that these factors do not muchaffect the real-time PCR assay.
By making a standard curve with a known number ofthe standard plasmids, the real-time PCR assay makesit possible to estimate the number of copies of mumpsvirus RNA in the original sample. For example, a Ctvalue of 35.0–36.0 is equivalent to 10 copies of thestandard plasmid in a reaction tube and corresponds toapproximately 1,800 copies of mumps virus RNA per mlof the original sample, assuming 100% efficiency of thereverse transcription step. The real-time PCR coulddetect 5 copies of standard plasmid; however, thepresence ofmore than10 copies of the cDNAper reactiontube was suitable for quantitative assay, as described invarious studies [Gault et al., 2001; Zhao et al., 2002;Kageyama et al., 2003], because the CV calculated fromCt values of 5 copies of the standard plasmid showed alarger variation than that of more than 10 copies of theplasmid. There were a few samples in this study, inparticular CSF samples that showed a significant in-crease of fluorescence signals repeatedly and Ct valuesfrom 36.0 to 37.0, larger than the Ct values for 10 copiesof the plasmids. Therefore, to set a detection limit, fivecopies of the plasmid, which yielded Ct values of 36–39 were used, and the results corresponded well to theresults of the RT-n-PCR. One sample was positive onreal-time PCR but negative on RT-n-PCR. The resultsof the assay using the virus stock suggest that thereal-time PCR assay is slightly more sensitive thanRT-n-PCR. This may cause the discordant results insome samples containing only a small number ofmumpsvirus RNA copies.
It may be interesting to consider the factors that yielddifferences in the number of copies of mumps virusRNA in each sample. These may be the method of
Fig. 2. Estimated mumps virus RNA copy number in clinical samples. The lower limit of quantitativeassay was 1,785 copies RNA/ml of a clinical sample. (v) indicates a sample collected from a patient aftermumps virus vaccination.
Mumps Virus Detection by Real-Time PCR 473

obtaining throat swab samples, the period from onset toharvesting of samples, and differences of the severity inclinical symptoms in each patient. In this study, theCSF and throat swab samples were harvested from 0 to8 days and from 0 to 5 days after onset, respectively. In alimited number of the samples examined, the viral RNAconcentration in the CSF samples seemed to be higherwhen they were harvested 2 days after onset. On theother hand, the viral RNA concentration in throat swabsamples had a wide range from 3.6�103/ml to 4.0�106/ml, and the difference of the viral RNA concen-trations of samples due to the period from onset toharvest was not clear. Though these samples wereobtained from different patients, little information onthe severity of their clinical symptoms was obtained.More clinical samples will be required to reveal thecorrelation between the concentration of mumps virusRNA in the samples and features of the samples.Some important aspects of mumps virus infections
have been described, but information is still limited.These include the importance of mumps virus as acausative agent of central nervous system infectionssuch asmeningitis and encephalitis [Poggio et al., 2000]and other complications. The real-time PCR assaydescribed here is rapid and sensitive with a low risk ofcontamination, and is capable of development into aquantitative assay. It will be useful for identification ofnot only suspected cases of mumps, but also of atypicaland more significant cases including encephalitis andcerebellar ataxia, and in future investigations of themumps virus infections.
REFERENCES
CohenHA, Ashkenazi A, NussinovitchM, Amir J, Hart J, FrydmanM.1992. Mumps-associated acute cerebellar ataxia. Am J Dis Child146:930–931.
Elango N, Varsanyi TM, Kovamees J, Norrby E. 1988. Molecularcloning and characterization of six genes, determination of geneorder and intergenic sequences and leader sequence of mumpsvirus. J GenVirol 69:2893–2900.
Gault E,Michel Y, Dehee A, Belabani C, Nicolas J, Garbarg-Chenon A.2001. Quantification of human cytomegalovirus DNA by real-timePCR. J Clin Microbiol 39:772–775.
Gut JP, Lablache C, Behr S, Kirn A. 1995. Symptomatic mumps virusreinfections. J Med Virol 45:17–23.
Jin L, Beard S, Brown DWG. 1999. Genetic heterogeneity of mumpsvirus in the United Kingdom: Identification of two new genotypes.J Infect Dis 180:829–833.
Julkunen I. 1984. Serological diagnosis of parainfluenza virus infectionby enzyme immunoassay with special emphasis on purity of viralantigens. J Med Viol 14:177–187.
Kageyama T, Kogima S, Shinohara M, Uchida K, Fukushi S,Hoshino FB, Takeda N, Katayama K. 2003. Broadly reactive andhighly sensitive assay for Norwalk-like viruses based on real-timequantitative reverse transcription-PCR. J Clin Microbiol 41:1548–1557.
Kashiwagi Y, Kawashima H, Takekuma K, Hoshika A, Mori T,Nakayama T. 1997. Detection of mumps virus genome directlyfrom clinical samples and a simple method for genetic differentia-tion of theHoshino vaccine strain fromwild strains ofmumps virus.J Med Virol 52:195–199.
Okamoto M, Shitara T, Nakayama M, Takamiya H, Nishiyama K,Ono Y, Sano H. 1994. Acta sudden deafness accompanied byasymptomatic mumps. Otolaryngol Suppl 514:45–48.
Poggio GP, Rodriguez C, Cisterna D, Freire MC, Cello J. 2000.Nested PCR for rapid detection of mumps virus in cerebrospinalfluid from patients with neurological disease. J Clin Microbiol38:274–278.
Rima BK, Alexander DJ, Billeter MA, Collins PL, Kingsbury DW,Lipkind MA, Nagai Y, Orvell C, Pringle CR, ter Meulen V. 1995.The Paramyxoviridae. In: Murphy FA, Fauquet CM, Bishop DHL,Ghabrial SA, Jarvis AW, Martelli GP, Mayo MA, Summers MD,editors. Virus Taxonomy. Sixth Report of the Internationalcommittee on Taxonomy of Viruses. Vienna and New York:Springer-Verlag. pp 268–274.
Tecle T, Johansson B, Yun Z, Orvell C. 2000. Antigenic and geneticcharacterization of the fusion (F) protein of mumps virus strains.Arch Virol 145:1199–1210.
Uchida K, Shinohara M, Shimada S, Segawa Y, Hoshino Y. 2001.Characterization of mumps virus isolated in Saitama prefecture,Japan, by sequence analysis of the SH gene. Microbiol Immunol45:851–855.
Uchida K, Shinohara M, Shimada S, Segawa Y, Kimura K, Hoshino Y.2003. Characterization of the F gene of contemporary mumps virusstrains isolated in Japan. Microbiol Immunol 47:167–172.
Zhao Y, Yu M, Miller JW, Chen M, Bremer EG, Kabat W, Yogev R.2002. Quantification of human immunodeficiency virus type 1proviral DNA by using TaqMan Technology. J Clin Microbiol40:675–678.
474 Uchida et al.

Journal of Medical Virology 75:475–481 (2005)
Outbreak of Acute Gastroenteritis AssociatedWith Group A Rotavirus and Genogroup ISapovirus Among Adults in a Mental Health CareFacility in Japan
Hainian Yan,1 Toshiaki Abe,2 Tung Gia Phan,1 Tuan Anh Nguyen,1 Tatuya Iso,2 Yasunori Ikezawa,2
Kiyo Ishii,2 Shoko Okitsu,1 and Hiroshi Ushijima1*1Department of Developmental Medical Sciences, Institute of International Health, Graduate School of Medicine,The University of Tokyo, Bunkyo-ku, Tokyo, Japan2National Center for Persons with Severe Intellectual Disabilities, Takasaki, Japan
Anoutbreak of acute gastroenteritis consisting of57 cases occurred in a mental health care facilityin Takasaki city, Japan during 6th February and27th March 2002. A total of 18 fecal specimenscollected from 17 residents and one member ofthemedical staff during this outbreakwere testedfor the presence of viral enteropathogens byRT-PCR and latex agglutination. Group A rota-virus and sapovirus were detected in 5 out of 18fecal specimens (55.6%). To our knowledge, thisis thefirst findingofanoutbreakofgastroenteritisassociated with co-circulation of different kindsof viruses such as group A rotavirus and sapo-virus. All of group A rotaviruses were typedfurther as P[4]G2 strains. Both rotavirus andsapovirus were subjected to molecular analysisby sequencing. It was noteworthy that all rota-viruses and sapoviruses had high homologies,respectively, to each other and sapoviruses pre-sented a potential novel sapovirus genogroup I(GI) genotype, which was obviously differentfrom any GI genotypes (GI-a, b, c, and d). Theoutbreak associated with these viruses spreadgradually from dormitory to dormitory, suggest-ing a spread by person-to-person contact,although investigation on the route of transmis-sion of the outbreak is lacking. The findingsconfirm the presence of group A rotavirus andsapovirus are important in acute gastroenteritisamong adults in Japan. J. Med. Virol. 75:475–481, 2005. � 2005 Wiley-Liss, Inc.
KEY WORDS: multiplex PCR; genogroup;enteropathogen
INTRODUCTION
Acute gastroenteritis is a major cause of morbidityandmortality among infants and young children in both
developedanddeveloping countries.Amongother enter-opathogenic viruses, rotaviruses are recognized as themajor etiologic agents of gastroenteritis in children andyoung animals. Rotaviruses are classified into sevengroups (A–G) on the basis of distinct antigenic andgenetic properties [Bridger, 1994; Saif and Jiang, 1994].Human infectionhas been reportedwith groupA,B, andC rotaviruses. Of these, group A rotaviruses is the mostimportant, being amajor cause of severe gastroenteritisin infants and young children worldwide [Kapikianet al., 2001]. The two outer capsid proteins (the glyco-protein VP7; the protease-sensitive protein VP4) ofrotavirus particle identify G and P serotypes, respec-tively [Estes, 2001].Todate, at least15G-typesand21P-types have been found in humans and animals [Bridger,1994; Pang et al., 2000]. Apart from rotavirus as themajor etiologic agent of gastroenteritis in children andyoung animals, sapovirus is considered to be a signifi-cant global enteropathogen. Etiologic studies of acutegastroenteritis in infants conducted in Japan withsensitive reverse transcription-PCR methods showedthat sapovirus was one of the most common causes ofoutbreaks of viral gastroenteritis among infants inSapporo [Chiba et al., 1979, 2000]. This virus is alsoassociated with sporadic outbreaks of gastroenteritisworldwide and is recognized just as important as
Grant sponsor: Ministry of Education, Culture, Sports, Scienceand Technology, Japan; Grant sponsor: Ministry of Health, Laborand Welfare, Japan; Grant sponsor: Sumitomo Foundation,Japan; Grant sponsor: Japanese Food Hygiene Association.
*Correspondence to: Hiroshi Ushijima, Department of Devel-opmental Medical Sciences, Institute of International Health,Graduate School of Medicine, The University of Tokyo, 7-3-1Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.E-mail: [email protected]
Accepted 23 November 2004
DOI 10.1002/jmv.20292
Published online in Wiley InterScience(www.interscience.wiley.com)
� 2005 WILEY-LISS, INC.
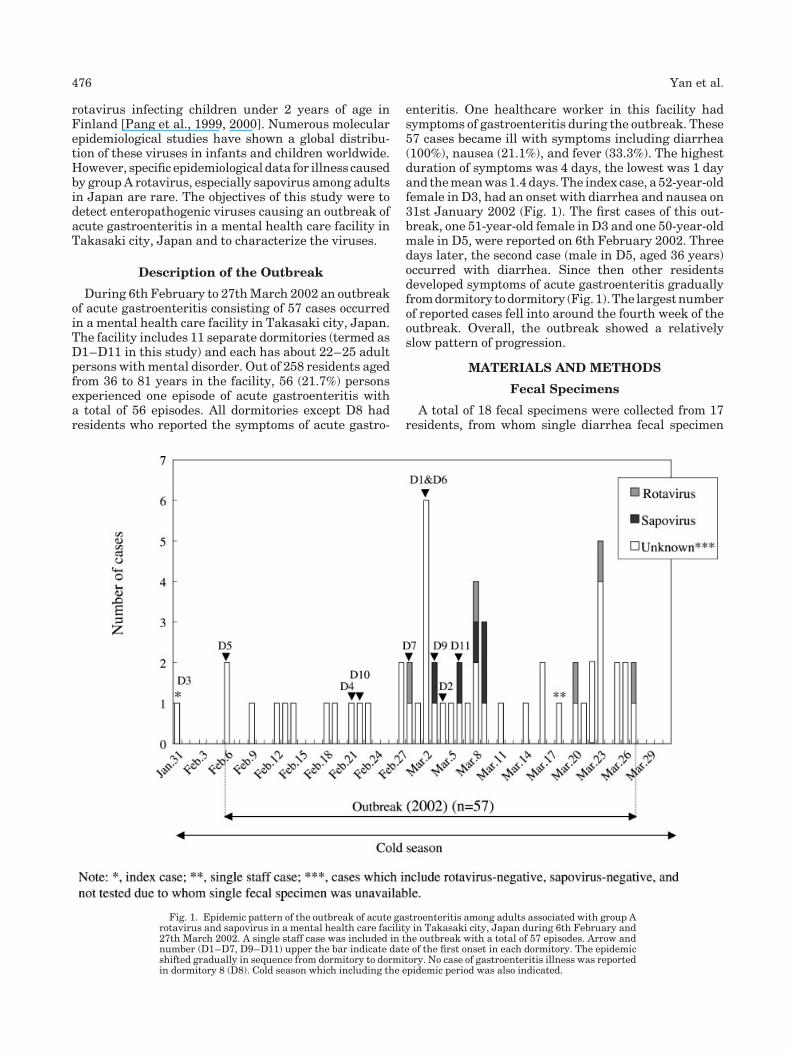
rotavirus infecting children under 2 years of age inFinland [Pang et al., 1999, 2000]. Numerous molecularepidemiological studies have shown a global distribu-tion of these viruses in infants and children worldwide.However, specific epidemiological data for illness causedby groupA rotavirus, especially sapovirus among adultsin Japan are rare. The objectives of this study were todetect enteropathogenic viruses causing an outbreak ofacute gastroenteritis in a mental health care facility inTakasaki city, Japan and to characterize the viruses.
Description of the Outbreak
During 6th February to 27thMarch 2002 an outbreakof acute gastroenteritis consisting of 57 cases occurredin a mental health care facility in Takasaki city, Japan.The facility includes 11 separate dormitories (termed asD1–D11 in this study) and each has about 22–25 adultpersons withmental disorder. Out of 258 residents agedfrom 36 to 81 years in the facility, 56 (21.7%) personsexperienced one episode of acute gastroenteritis witha total of 56 episodes. All dormitories except D8 hadresidents who reported the symptoms of acute gastro-
enteritis. One healthcare worker in this facility hadsymptoms of gastroenteritis during the outbreak. These57 cases became ill with symptoms including diarrhea(100%), nausea (21.1%), and fever (33.3%). The highestduration of symptoms was 4 days, the lowest was 1 dayand themeanwas1.4 days. The index case, a 52-year-oldfemale in D3, had an onset with diarrhea and nausea on31st January 2002 (Fig. 1). The first cases of this out-break, one 51-year-old female in D3 and one 50-year-oldmale in D5, were reported on 6th February 2002. Threedays later, the second case (male in D5, aged 36 years)occurred with diarrhea. Since then other residentsdeveloped symptoms of acute gastroenteritis graduallyfromdormitory todormitory (Fig. 1).The largestnumberof reported cases fell into around the fourth week of theoutbreak. Overall, the outbreak showed a relativelyslow pattern of progression.
MATERIALS AND METHODS
Fecal Specimens
A total of 18 fecal specimens were collected from 17residents, from whom single diarrhea fecal specimen
Fig. 1. Epidemic pattern of the outbreak of acute gastroenteritis among adults associated with group Arotavirus and sapovirus in a mental health care facility in Takasaki city, Japan during 6th February and27th March 2002. A single staff case was included in the outbreak with a total of 57 episodes. Arrow andnumber (D1–D7, D9–D11) upper the bar indicate date of the first onset in each dormitory. The epidemicshifted gradually in sequence from dormitory to dormitory. No case of gastroenteritis illness was reportedin dormitory 8 (D8). Cold season which including the epidemic period was also indicated.
476 Yan et al.

was available, and one member of the staff. Thesespecimens were tested for the presence of rotaviruses,noroviruses, sapoviruses, astroviruses, and adeno-viruses. The fecal specimens were diluted with distilledwater to 10% suspensions, and clarified by centrifuga-tion at 10,000g for 10 min. The supernatants werecollected and stored at�308C until use for the detectionof diarrheal viruses.
Extraction of Viral Genomes
The viral genomes were extracted from 10% fecalsuspensions using a spin column technique accord-ing to the manufacturer’s instructions (QIAGEN1,Hilden, Germany).
Multiplex RT-PCR
Multiplex RT-PCR was conducted for detecting twogroups of diarrhea viruses, in which, the first group ofviruses including human astrovirus, norovirus (GI,GII), and sapovirus and the second group includinggroup A, B, and C rotavirus and adenovirus. Theidentification of the first group of viruses was performedby using multiplex RT-PCR with specific primers asreported previously [Yan et al., 2003]. Briefly, G1-SKF/G1-SKR, andCOG2F/G2-SKRfornorovirus genogroup Iand II, respectively, SLV5317/SLV5749 for sapovirusand PreCAP1/82b for astrovirus. All of the four primerpairs amplify the capsid region of target viral genome,produce four size-specific amplicons of 330, 387, 434,719 bp for norovirus genogroup I and II, sapovirus andastrovirus, respectively. In order to detect the secondgroup of viruses, after viral extract was heated at 948Cfor 3 min followed by the RT step described elsewhere[Yan et al., 2003], four pairs of published primers (Beg9and VP7-1
0, B5-2 and B3-3, G8NS1 and G8NA2 for
amplifying VP7 gene of human group A, B, and Crotaviruses, respectively; Ad1 andAd2 for hexon gene ofall species fromA to F adenoviruses) were used [Gouveaet al., 1990; Xu et al., 2000; Kobayashi et al., 2001;Kuzuya et al., 2003]. These primers were specificallygenerated four different sizes of amplicons of 395, 814,352, and 482 bp for group A, B, and C rotaviruses andadenovirus, respectively. The PCR was performed at948C for 3 min followed by 35 cycles of 948C 30 sec, 558C30 sec, 728C 60 sec, and a final extension at 728C for7 min, and then held at 48C.
G and P Serotyping RT-PCR
Group A rotaviruses were characterized further for Gand P serotypes by using the method described pre-viously byGouvea et al. [1990] andGentsch et al. [1992],respectively. Briefly, for the protocol of RT-PCR for Gserotyping, the RNA of rotavirus was reverse tran-scribed and then further amplifiedwith Beg9 and End9,generating a full-length of VP7 gene in 1,062 bp. Thenested PCR was carried out using the first PCR productas the template with G-serotype specific mixed primers
(BT1, CT2, ET3, DT4, and FT9) for upstream primingandEnd9 for downstreampriming in an amplification ofVP7 genes of G1 to G4, and G9, respectively [Gouveaet al., 1990]; moreover, for the P serotyping RT-PCR,the RNA of rotavirus was reverse transcribed and thenamplified further with Con2 and Con3 primers for theamplification of the VP4 gene. In the second amplifica-tion, a mixture of primers, 1T-1, 2T-1, 3T-1, 4T-1, 5T-1,ND2, and Con3 primers were used for identificationof P[8], P[4], P[6], P[9], P[10], and P[11], respectively[Gentsch et al., 1992].
Latex Agglutination Test
A latex agglutination test was used as a confirmationtest for the detection of group A rotavirus by a commer-cial Rotalex kit (DaiichKagakuCo., Ltd., Tokyo, Japan).Fecal samples were processed according to the manu-facturer’s instructions. A drop of the fecal supernatantwas mixed with a drop of test latex on a slide, andreaction was observed after 2 min. Development ofdistinctagglutination intheRotalexreagentwastreatedas positive. If agglutination was seen in the negativecontrol latex, the test was considered uninterpretable.
Monoplex PCR for Amplificationof Sapovirus Polymerase Region
Sapovirus positives by multiplex PCR in fecal speci-mens were examined further by monoplex PCR withspecific primers SR80 and JV33 [Vinje et al., 2000] forgenerating a 320 bp amplicom in the polymerase region,with the same thermal cycler program as for the multi-plex PCR.
Nucleotide Sequencingand Phylogenetic Analysis
The nucleotide sequences of PCRproducts positive forrotavirus (VP7 gene) and sapovirus (polymerase geneand capsid gene) were determined with the Big-Dyeterminator cycle sequencing kit and an ABI Prism310 Genetic Analyzer (Applied Biosystems, Inc., Tokyo,Japan). Sequence analysis was performed using E-CLUSTAL W (Version 1.6). Reference strains andaccession numbers used in this study were as follows:Sapovirus GI strains: Sapporo/82/JP (U65427), Lyon/30388/98/F (AJ251991), Plymouth (X86559), Houston/86/US (U95643), Manchester (X86560), Potsdam/2000/DEU (AF294739), Parkville (U73124), Houston/27/90/US (U95644), Stockholm/318/97/SE (AF194182),Chiba/010658F/2001 (AJ412827), Chiba/000782F/2000(AJ412813), Chiba/010469F/2001 (AJ412820), Chiba/000527H/2000 (AJ412801); Sapovirus GII strains: Lon-don/29845/92/UK (U95645), Lyon/598/97/F (AJ271056),Chiba/010557S/2001 (AJ412821), Chiba/010604F/2001(AJ412826), Chiba/010592F/2001 (AJ412824), Chiba/010469F/2001 (AJ412820), Chiba/990727S/1999 (AJ-41795), Chiba/010004H/2001 (AJ412816), Chiba/010675F/2001 (AJ412828); and Sapovirus GIV strains:Chiba/000671T/1999 (AJ412805).
Gastroenteritis Outbreak Among Japanese Adults 477

RESULTS
Detection of Viruses Causing Diarrhea
The results shown in Table I revealed that virusescausing diarrhea were detected in 10 out of 18 (55.6%)fecal specimens tested. GroupA rotavirus and sapoviruswere detected in 5 (3 in D4 and 2 in D7), 5 (1 in D9 and 4in D11) out of 18 fecal specimens, respectively and thispresented27.8%and27.8%, respectively.GroupBandCrotaviruses, adenovirus, norovirus, and astrovirus werenot found in these patients. No virus was identified inthe fecal specimen from a member of the staff (Patientno. 9) bymultiplex PCR. All of group A rotaviruses wereidentified as P[4]G2 strains by the serotyping RT-PCR,and confirmed by latex agglutination test (Rotalex kit).
Nucleotide Sequencing and PhylogeneticAnalysis of Group A Rotavirus isolates
The rotavirus sequences clustered together in a G2serotype phylogenetically. The homology of thesenucleotide sequences with rotavirus G2 isolates in VP7gene were over 96%, among those, homologous to G2isolates Mvd9707, 9708, 9713, and 9716 (AF480270,AF480273, AF480268, and AF480275, respectively)showed 99%. All of the five isolates of rotavirus demon-strated a high identity (100%) to each other suggestedthat they probably came from the same source ofinfection.
Nucleotide Sequencing and PhylogeneticAnalysis of Sapovirus Isolates
The sapovirus sequences clustered into only distinctgenogroup I (knownas theManchester virus) both in thecapsid region (Fig. 2) and the polymerase region (datanot shown). However, these sapoviruses did not belongto any of the published clusters and represented apotential novel sapovirus GI genotype in the capsidregion according to the classification scheme of Okadaet al. [2002]. They had a rather low identity on thenucleotide aswell as the amino acidwith other referencestrains in the same genogroup previously registered intheDDBJDNAdatabase, ranged from83% to 89% in thecapsid region. Also, the homologies of these sapoviruseswith the prototype strain Sapporavirus/82 in the poly-merase region was 73% and 77% on the nucleotide andamino acid sequence, respectively. All sapoviruses hada high identity (100%) at the nucleotide as well as theamino acid to each other also suggesting that theyprobably came from the same source of infection. Thenucleotide sequence data for the capsid region fromstrains Takasaki 5, 7, 8, 11, and 13 has been submittedto the DDBJ DNA database and has been assignedaccession number AB180405, AB180406, AB180482,AB180483, AB180484, respectively.
DISCUSSION
The present study describes an outbreak of acutegastroenteritis in a mental health care facility in T
ABLE
I.Characteristics
of10ViralPositives
intheOutbreakof
Acu
teGastroen
teritisin
aMen
talHea
lthCare
Facility
inTakasa
kiCity,JapanDuring6th
Feb
ruary
and27th
March2002
Patien
tno.
Dormitory
code
Sex
Age(yea
r)Date
ofon
set
Duration
(day)
Symptoms
Laboratory
findings
Diarrhea
Fev
erNausea
RT-PCR
Rotalex
RV
G-typing
RV
P-typing
3D7
F50
28Feb
ruary
1þ
þþ
RA
þG2
P[4]
4D7
F61
8March
1þ
þþ
RA
þG2
P[4]
5D9
M53
3March
3þ
��
SV
�nd
nd
7D11
F44
6March
1þ
��
SV
�nd
nd
8D11
F54
8March
1þ
þ�
SV
�nd
nd
11
D11
F61
9March
1þ
��
SV
�nd
nd
13
D11
F67
9March
1þ
þþ
SV
�nd
nd
14
D4
M59
20March
3þ
þþ
RA
þG2
P[4]
15
D4
M47
23March
2þ
��
RA
þG2
P[4]
17
D4
M40
27March
1þ
��
RA
þG2
P[4]
No.,number;M,male;F,female;RA,groupArotavirus;SV,sa
pov
irus;nd,not
don
e;þ,positive;
�,neg
ative.
478 Yan et al.

Takasaki city, Japanduring6thFebruary to27thMarch2002 was caused by group A rotavirus and sapovirus.Out of the 18 patients from whom single fecal specimenwasavailable, about 55.6%might be due to thediarrhealviruses and 44.4% caused by other etiologic agents. Thefindings indicate that the rotavirus infections observedin this studywereunlikely tobedue to infectionbygroupA rotavirus. Numerous reports of group A rotavirusinfection in infants and children with acute gastro-enteritis worldwide have been published, however, ithas beenalso established that groupArotaviruses causeinfection in adults and that outbreaks occur commonlyparticularly in closed institutions [Cubitt and Holzel,1980; Holzel et al., 1980]. Among gastroenteritis out-
breaks in adults in the USA between November 1998and December 2000, 3 out of 263 outbreaks were linkedto rotavirus serotypeG2 [Griffin et al., 2002]. In a recentAustralian report, 3 out of 7 rotavirus-associated out-breaks mainly in mid-winter to early-spring wereidentified with G2, among 53 gastroenteritis outbreaksoccurred in aged-care facilities [Marshall et al., 2003].The results of this study were in agreement withprevious findings that group A rotavirus serotype G2was detected mainly during the cold season andaccounted for 27.8% (5/18) of acute gastroenteritis inadults patients (Patient nos. 3, 4, 14, 15, and 17). Ourfindings provide further evidence to support the hypoth-esis that natural immunity toG2 is inadequate in adults
Fig. 2. Phylogenetic tree of nucleotide sequences of five isolates of sapovirus. The tree was constructedfrompartial amino acid sequences of 5 isolates of sapovirus detected in the outbreak of acute gastroenteritisamong adults in mental health care facility, Takasaki city, Japan during 6th February and 27th March2002. Reference strains of sapovirus selected from DDBJ/GenBank under the accession number indicatedin the text. Sapoviruses found in the outbreak were indicated in the oval circle and presented a novelsapovirus GI genotype. The numbers in the branches indicate the bootstrap value.
Gastroenteritis Outbreak Among Japanese Adults 479

[Gentsch et al., 1996]. In Japan, the incidence of P[4]G2was detected to be mostly among school-aged children[Inoue et al., 2003]. The present study is a first report onan acute gastroenteritis outbreak associatedwith groupA rotavirus P[4]G2 among Japanese adults.To date, there have been several reports of outbreaks
of sapovirus that involved adults, e.g., HuCV/Sapporo/Japan, the prototype sapovirus was detected from anoutbreak inJapan that affected adults and children. TheParkville strainwas isolated fromanoutbreak involvingadults in England [Noel et al., 1997]. Interestingly, ourstudy has demonstrated that infections with sapovirusin a relatively high percentage of 27.8% (5/18) in thisoutbreak (Patient nos. 5, 7, 8, 11, and 13). Similarity inseasonal pattern of infections among infants andchildren with acute gastroenteritis, mostly sapoviruswas detected during the cold season [Phan et al., 2004].In our study, sapovirus was subjected to molecularanalysis by sequencing inbothpartial capisid regionandpolymerase region. A total of five sapovirus amino acidsequences were examined by phylogenetics and group-ed using the recent sapovirus capsid region classifica-tion scheme of Okada et al. [2002]. It was noteworthy topoint out that all sapoviruses demonstrated a highidentity (100%) at the nucleotide as well as the aminoacid to each other suggesting that they probably camefrom the same source of infection. Interestingly, as de-scribed above, these sapovirus sequences clustered intogenogroup I, but did not belong to any of the publishedclusters and represented a potential novel sapovirus GIgenotype. This finding also indicated that the sapovirusinfections observed in our study were unlikely to be dueto contamination by sapovirus.Viruses causing diarrhea can be transmitted by a
variety of routes, including fecal–oral, foodborne, andwaterborne [Brugha et al., 1999; Mead et al., 1999]. Inaddition, airborne droplet has also been suggested as aroute of transmission that might cause gastroenteritisoutbreak [Sawyer et al., 1988; Marks et al., 2003]. Someinvestigators indicated that in long-term care facilitiessuch as long-term hospital ward, elderly nursing home,retirement facilities, viruses causing diarrhea spreadeasily among residents and staffs through the careprocess and highly infectious agent contamination inthe excretions that remain on environmental surfaces[Caceres et al., 1998; Green et al., 1998; Kuusi et al.,2002]. In the present study, the route of transmissionremains unknown, however, the outbreak associatedwith viruses such as group A rotavirus and sapovirusspread gradually from dormitory to dormitory with adistinctive epidemic curve based on analysis of theprevalence pattern, suggesting a high possibility ofspread by person-to-person contact.The present study demonstrated an outbreak
caused by two distinct viruses circulating simulta-neously. Although similar findings have been reported[Gray et al., 1987; Lewis et al., 1989], this is thefirst finding of an outbreak of gastroenteritis associ-ated with co-circulation of group A rotavirus andsapovirus.
REFERENCES
Bridger JC. 1994.Non-groupArotavirus. In:KapikianAZ, editor. Viralinfection of the gastrointestinal tract, 2nd edn. New York: MarcelDekker. pp 369–408.
BrughaR,Vipond IB,EvansMR, SandiferQD,Roberts RJ, SalmonRL,Caul EO, Mukerjee AK. 1999. A community outbreak of food-bornesmall round-structured virus gastroenteritis caused by a contami-nated water supply. Epidemiol Infect 122:145–154.
Caceres VM, Kim DK, Bresee JS, Horan J, Noel JS, Ando T, Steed CJ,Weems JJ, Monroe SS, Gibson JJ. 1998. A viral gastroenteritisoutbreak associated with person-to-person spread among hospitalstaff. Infect Control Hosp Epidemiol 19:162–167.
Chiba S, Sakuma Y, Kogasaka R, Akihara M, Horino K, Nakao T,Fukui S. 1979. An outbreak of gastroenteritis associated withcalicivirus in an infant home. J Med Virol 4:249–254.
Chiba S, Nakata S, Numata-Kinoshita K, Honma S. 2000. Sapporovirus: History and recent findings. J Infect Dis 181:303–308.
CubittWD,HolzelH. 1980. An outbreak of rotavirus infection in a long-stay ward of a geriatric hospital. J Clin Pathol 33:306–308.
Estes MK. 2001. Rotavirus and their replication. In: Knipe DM,Howley PM, editors. Fields virology, 4th edn. Lippicott Williamsand Wilkins Press. pp 1747–1785.
Gentsch JR, Glass RI, Woods P, Gouvea V, Gorziglia M, Flores J,Das BK, BhanMK. 1992. Identification of group A rotavirus gene 4types by polymerase chain reaction. JClinMicrobiol 30:1365–1373.
Gentsch JR,Woods PA, RamachandranM, Das BK, Leite JP, Alfieri A,Kumar R, Bhan MK, Glass RI. 1996. Review of G and P typingresults from a global collection of rotavirus strains: implications forvaccine development. J Infect Dis 174:30–36.
Gouvea V, Glass RI, Woods P, Taniguchi K, Clark HF, Forrester B,Fang ZY. 1990. Polymerase chain reaction amplification and typingof rotavirus nucleic acid from stool specimens. J Clin Microbiol 28:276–282.
Gray JJ, Wreghitt TG, Cubitt WD, Elliot PR. 1987. An outbreak ofgastroenteritis in a home for the elderly associated with astrovirustype 1 and human calicivirus. J Med Virol 23:377–381.
Green J, Wright PA, Gallimore CI, Mitchell O, Morgan-Capner P,Brown DW. 1998. The role of environmental contamination withsmall round structured viruses in a hospital outbreak investigatedby reverse-transcriptase polymerase chain reaction assay. J HospInfect 39:39–45.
GriffinDD, FletcherM, LevyME, Ching-LeeM,NogamiR, Edwards L,Peters H, Montague L, Gentsch JR, Glass RI. 2002. Outbreaksof adult gastroenteritis traced to a single genotype of rotavirus.J Infect Dis 185:1502–1505.
Holzel H, Cubitt DW, McSwiggan DA, Sanderson PJ, Church J. 1980.An outbreak of rotavirus infection among adults in a cardiologyward. J Infect 2:33–37.
Inoue Y, Kitahori Y, Adachi O, Imai S. 2003. P genotypic identificationof human group A rotaviruses. Jpn J Infect Dis 56:179–180.
Kapikian AZ, Hoshino Y, Chanock RM. 2001. Rotaviruses. In: KnipeDM, Howley PM, editors. Fields virology, 4th edn. Philadelphia:Lippincott-Raven Press. pp 1787–1833.
Kobayashi N,Naik TN,Kusuhara Y, Krishnan T, Sen A, BhattacharyaSK, Taniguchi N, Alam MM, Urasawa T, Urasawa S. 2001.Sequence analysis of genes encoding structural and nonstructuralproteins of a human group B rotavirus detected in Calcutta, India.J Med Virol 64:583–588.
Kuusi M, Nuorti JP, Maunula L, Minh NN, Ratia M, Karlsson J,von Bonsdorff CH. 2002. A prolonged outbreak of Norwalk-likecalicivirus (NLV) gastroenteritis in a rehabilitation centre due toenvironmental contamination. Epidemiol Infect 129:133–138.
Kuzuya M, Fujii R, Hamano M, Ogura H. 2003. Outbreak of acutegastroenteritis caused by human group C rotavirus in a youtheducational center in Okayama Prefecture. Kansenshogaku Zasshi77:53–59.
Lewis DC, Lightfoot NF, Cubitt WD, Wilson SA. 1989. Outbreaks ofastrovirus type 1 and rotavirus gastroenteritis in a geriatric in-patient population. J Hosp Infect 14:9–14.
MarksPJ, Vipond IB,ReganFM,WedgwoodK,FeyRE,Caul EO. 2003.A school outbreak of Norwalk-like virus: Evidence for airbornetransmission. Epidemiol Infect 131:727–736.
Marshall J, Botes J, Gorrie G, BoardmanC, Gregory J, Griffith J, HoggG, Dimitriadis A, Catton M, Bishop R. 2003. Rotavirus detectionand characterisation in outbreaks of gastroenteritis in aged-carefacilities. J Clin Virol 28:331–340.
480 Yan et al.

Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C,Griffin PM, Tauxe RV. 1999. Food-related illness and death in theUnited States. Emerg Infect Dis 5:607–625.
Noel JS, Liu BL, Humphrey CD, Rodriguez EM, Lambden PR,Clarke IN, Dwyer DM, Ando T, Glass RI, Monroe SS. 1997.Parkville virus: A novel genetic variant of human calicivirus inthe Sapporo virus clade, associated with an outbreak of gastro-enteritis in adults. J Med Virol 52:173–178.
Okada M, Shinozaki K, Ogawa T, Kaiho I. 2002. Molecular epidemiol-ogy and phylogenetic analysis of Sapporo-like viruses. Arch Virol147:1445–1451.
Pang XL, Joensuu J, Vesikari T. 1999. Human calicivirus associatedsporadic gastroenteritis in Finnish children less than two years ofage followed prospectively during a rotavirus vaccine trial. PediatrInfect Dis J 18:420–426.
PangXL,HonmaS,Nakata S,Vesikari T. 2000.Human caliciviruses inacute gastroenteritis of young children in the community. J InfectDis 181:288–294.
Phan TG, Okame M, Tuan AN, Maneekarn N, Nishio O, Okitsu S,Ushijima S. 2004. Human astrovirus, norovirus (GI, GII), and
sapovirus infections in Pakistani children with diarrhea. J MedVirol 72:256–261.
Saif LJ, Jiang B. 1994. Non group A rotaviruses of humans andanimals. Curr Top Microbiol 85:339–371.
Sawyer LA,Murphy JJ, Kaplan JE, Pinsky PF, ChaconD,Walmsley S,Schonberger LB, Phillips A, Forward K, Goldman C. 1988. 25- to30-nm virus particle associated with a hospital outbreak of acutegastroenteritis with evidence for airborne transmission. Am JEpidemiol 127:1261–1271.
Vinje J, Deijl H, van der Heide R, Lewis D, Hedlund KO. Svensson L,Koopmans MP. 2000. Molecular detection and epidemiology ofSapporo-like viruses. J Clin Microbiol 38:530–536.
Xu W, McDonough MC, Erdman DD. 2000. Species-specific identifica-tion of human adenoviruses by a multiplex PCR assay. J ClinMicrobiol 38:4114–4120.
Yan H, Yagyu F, Okitsu S, Nishio O, Ushijima H. 2003. Detection ofnorovirus (GI, GII), sapovirus and astrovirus in fecal samples usingreverse transcription single-roundmultiplex PCR. J Virol Methods114:37–44.
Gastroenteritis Outbreak Among Japanese Adults 481














![S2-22-367-2001] BETWEEN REPCO (MALAYSIA) SDN BHD](https://static.fdokumen.com/doc/165x107/63157415c72bc2f2dd04b62d/s2-22-367-2001-between-repco-malaysia-sdn-bhd.jpg)





