
FT-IR product study on the photo-oxidation of dimethyl sulphide in the presence of NO x...
12
Atmospheric Environment 35 (2001) 3769–3780 FT-IR product study on the photo-oxidation of dimethyl sulphide in the presence of NO x }temperature dependence Cecilia Arsene a,b , Ian Barnes a, *, Karl H. Becker a , Raluca Mocanu b a Bergische Universit . at Gesamthochschule Wuppertal, Physikalische Chemie FB 9, Gauss Strasse 20, D-42097 Wuppertal, Germany b Faculty of Chemistry, Al.I. Cuza University Iasi, Carol I, 11, 6600 Iasi, Romania Received 28 November 2000; received in revised form 21 February 2001; accepted 7 March 2001 Abstract The products of the OH radical-initiated oxidation of dimethyl sulphide (DMS) have been investigated as a function of temperature (284, 295, and 306K) and different initial NO x (NO+NO 2 ) concentrations: initial NO was varied between 434 and 2821 ppb and NO 2 between 135 and 739ppb. The experiments were performed at 1000 mbar total pressure in synthetic air using the photolysis of H 2 O 2 as the OH-radical source and FT-IR spectroscopy to monitor reactants and products. The major sulphur-containing products identified were SO 2 , dimethyl sulphoxide (DMSO), dimethyl sulphone (DMSO 2 ), methane sulphonic acid (MSA), methane sulphonyl peroxynitrate (MSPN) and OCS. The variation of the product yields with temperature and NO x concentration are consistent with the occurrence of both addition and abstraction channels in OH radical-initiated oxidation of DMS. Distinct trends in the yields of the various products have been observed as a function of temperature, initial NO x conditions and also reaction time as NO is consumed in the system. Increasing the initial NO concentration was found to depress the DMSO, SO 2 and OCS formation yields and enhance those of DMSO 2 , MSA and MSPN. The yield–time behaviour of DMSO 2 is supportive of a formation mechanism involving addition of O 2 to a (CH 3 ) 2 S–OH adduct, formed via the addition channel, followed by sequential reactions with NO and O 2 . The mechanisms controlling the concentration–time profiles of the individual products under the present experimental conditions are discussed in detail and consideration is given to possible implications for the photo-oxidation of DMS under ambient conditions. # 2001 Elsevier Science Ltd. All rights reserved. Keywords: Organic sulphur; OH radical; Sulphur dioxide; Dimethyl sulphoxide; Troposphere 1. Introduction The main natural source of reactive sulphur in the atmosphere is dimethyl sulphide (DMS: CH 3 SCH 3 ) emitted from marine phytoplankton (Andreae, 1990; Spiro et al., 1992; Bates et al., 1992; Berresheim et al., 1995). In the absence of anthropogenic sources, oxida- tion of DMS is considered to be a dominant source of submicron particles in marine air (Andreae and Crutzen, 1997; Andreae et al., 1995; Bates et al., 1987; Nguyen et al., 1983). During the day DMS is oxidised mainly by OH radicals and during the night by NO 3 radicals. Based on field study measurements of reactive halogen species (X 2 , XO) and also the behaviour of NMHC it has been speculated that Cl atoms and BrO radicals, in particular, might also play a role in the chemistry of the marine boundary layer and consequently in the chem- istry of DMS oxidation (Andreae and Crutzen, 1997; Berresheim et al., 1995). The present status concerning the kinetics and chemistry of the various oxidant/DMS interactions can be found in several reviews (Barone et al., 1995; Berresheim et al., 1995; Campolongo et al., 1999; Ravishankara et al., 1997). Oxidation of DMS is a complex process, however, it is well established that the reaction of OH radicals *Corresponding author. Tel.: +49-202-439-2510; fax: +49- 202-439-2505. E-mail address: [email protected] (I. Barnes). 1352-2310/01/$ - see front matter # 2001 Elsevier Science Ltd. All rights reserved. PII:S1352-2310(01)00168-6
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of FT-IR product study on the photo-oxidation of dimethyl sulphide in the presence of NO x...

Atmospheric Environment 35 (2001) 3769–3780
FT-IR product study on the photo-oxidation of dimethylsulphide in the presence of NOx}temperature dependence
Cecilia Arsenea,b, Ian Barnesa,*, Karl H. Beckera, Raluca Mocanub
aBergische Universit .at Gesamthochschule Wuppertal, Physikalische Chemie FB 9, Gauss Strasse 20, D-42097 Wuppertal, GermanybFaculty of Chemistry, Al.I. Cuza University Iasi, Carol I, 11, 6600 Iasi, Romania
Received 28 November 2000; received in revised form 21 February 2001; accepted 7 March 2001
Abstract
The products of the OH radical-initiated oxidation of dimethyl sulphide (DMS) have been investigated as a function oftemperature (284, 295, and 306K) and different initial NOx (NO+NO2) concentrations: initial NO was varied between434 and 2821 ppb and NO2 between 135 and 739ppb. The experiments were performed at 1000mbar total pressure insynthetic air using the photolysis of H2O2 as the OH-radical source and FT-IR spectroscopy to monitor reactants and
products. The major sulphur-containing products identified were SO2, dimethyl sulphoxide (DMSO), dimethyl sulphone(DMSO2), methane sulphonic acid (MSA), methane sulphonyl peroxynitrate (MSPN) and OCS. The variation of theproduct yields with temperature and NOx concentration are consistent with the occurrence of both addition and
abstraction channels in OH radical-initiated oxidation of DMS. Distinct trends in the yields of the various products havebeen observed as a function of temperature, initial NOx conditions and also reaction time as NO is consumed in thesystem. Increasing the initial NO concentration was found to depress the DMSO, SO2 and OCS formation yields and
enhance those of DMSO2, MSA and MSPN. The yield–time behaviour of DMSO2 is supportive of a formationmechanism involving addition of O2 to a (CH3)2S–OH adduct, formed via the addition channel, followed by sequentialreactions with NO and O2. The mechanisms controlling the concentration–time profiles of the individual products underthe present experimental conditions are discussed in detail and consideration is given to possible implications for the
photo-oxidation of DMS under ambient conditions. # 2001 Elsevier Science Ltd. All rights reserved.
Keywords: Organic sulphur; OH radical; Sulphur dioxide; Dimethyl sulphoxide; Troposphere
1. Introduction
The main natural source of reactive sulphur in theatmosphere is dimethyl sulphide (DMS: CH3SCH3)
emitted from marine phytoplankton (Andreae, 1990;Spiro et al., 1992; Bates et al., 1992; Berresheim et al.,1995). In the absence of anthropogenic sources, oxida-
tion of DMS is considered to be a dominant source ofsubmicron particles in marine air (Andreae and Crutzen,1997; Andreae et al., 1995; Bates et al., 1987; Nguyen
et al., 1983). During the day DMS is oxidised mainly by
OH radicals and during the night by NO3 radicals.Based on field study measurements of reactive halogenspecies (X2, XO) and also the behaviour of NMHC it
has been speculated that Cl atoms and BrO radicals, inparticular, might also play a role in the chemistry of themarine boundary layer and consequently in the chem-
istry of DMS oxidation (Andreae and Crutzen, 1997;Berresheim et al., 1995). The present status concerningthe kinetics and chemistry of the various oxidant/DMS
interactions can be found in several reviews (Baroneet al., 1995; Berresheim et al., 1995; Campolongo et al.,1999; Ravishankara et al., 1997).Oxidation of DMS is a complex process, however,
it is well established that the reaction of OH radicals
*Corresponding author. Tel.: +49-202-439-2510; fax: +49-
202-439-2505.
E-mail address: [email protected]
(I. Barnes).
1352-2310/01/$ - see front matter # 2001 Elsevier Science Ltd. All rights reserved.
PII: S 1 3 5 2 - 2 3 1 0 ( 0 1 ) 0 0 1 6 8 - 6

with DMS proceeds through two channels: H-atomabstraction by OH from a methyl group and
OH-radical addition to the S atom. The relativeimportance of the addition versus abstraction channelsis temperature dependent (Hynes et al., 1986). Attack of
OH radicals at the CH3 produces water and themethylthiomethyl radical (CH3SCH2), while additionto the S atom produces the dimethylhydroxysulphuranylradical ((CH3)2S–OH), an adduct which can either
dissociate back to reactants or react further to formproducts.
OHþ CH3SCH3 ! CH3SCH2þH2O
ðabstraction pathwayÞ;
OHþ CH3SCH3 ! ðCH3Þ2SOH ðaddition pathwayÞ:
Major end-products of the OH-radical initiated DMSoxidation observed both in laboratory and the tropo-sphere include species such as sulphur dioxide (SO2),
dimethyl sulphoxide (DMSO: CH3S(O)CH3), dimethylsulphone (DMSO2: CH3S(O)2CH3), methane sulphonicacid (MSA: CH3S(O)2OH) and methane sulphonylperoxynitrate (MSPN: CH3S(O)2OONO2) (Arsene
et al., 1999; Barnes et al., 1996; Berresheim et al.,1995; Patroescu et al., 1999; Ravishankara et al., 1997;S�rensen et al., 1996).
It is reasonably well established that in the presence ofNO the CH3SCH2 radical reacts to produce CH3Sradicals:
CH3SCH2þO2þM ! CH3SCH2OOþM;
CH3SCH2OOþNO ! CH3SCH2OþNO2;
CH3SCH2OþM ! CH3SþHCHOþM:
Further reactions of CH3S produce SO2, MSA andMSPN. The fate of the (CH3)2S–OH adduct is not sowell established. Possible reactions of the (CH3)2S–OH
adduct with O2 include:
ðCH3Þ2SOHþO2 ! HO2þCH3SðOÞCH3;
! CH3OOþ CH3SOH;
! OHþ CH3SðOÞ2CH3;
! CH3þCH3SðOHÞOO;
! CH3SðOHÞðOOÞCH3:
The addition pathway is known to produce DMSO andthe results from a recent product study from thislaboratory (Arsene et al., 1999) support that the major
fate of the (CH3)2S–OH adduct under NOx-free condi-tions is reaction with O2 to form DMSO. This result wasin contradiction with a pulsed laser photolysis/pulsed
laser-induced fluorescence study on the reaction of OHwith DMS by Turnipseed et al. (1996) who reported abranching ratio of F=0.5� 0.15 for the production of
DMSO based on the conversion of the HO2 produced toOH by reaction with NO. As discussed in Arsene et al.
(1999) the reason for the discrepancy is not entirelyclear, however, the reason may well be due to reaction
conditions employed in the two studies, i.e. with andwithout NO in the reaction system.Although the ambient NOx levels probably do
not have a large effect on the product distributionof the atmospheric photo-oxidation of DMS (Baroneet al., 1995), it is well established from smogchamber experiments where much higher concen-
trations than those found in the atmosphere areemployed, that the NOx levels in these experimentsinfluence quite substantially the product distribution
(Patroescu et al., 1999). In order to extract meaningfulresults from smog chamber studies on DMS, which canbe applied to the photo-oxidation of DMS under
atmospheric conditions, it is necessary to have a fullknowledge of the influences of NOx on the experimentalsystem.
In an extension of the work within our laboratory ondifferent aspects of the photo-oxidation mechanism ofDMS, we report here a detailed product study of the OHradical initiated oxidation of DMS as a function of
temperature and different initial concentrations of NOx
(NO+NO2). The work is aimed at clarifying someaspects of the influence of NOx on the product
distribution from the photo-oxidation of DMS withthe final goal of constructing a chemical oxidationmechanism for DMS relevant for ambient atmospheric
conditions.
2. Experimental
The experiments were performed in a temperatureregulated 1080 l quartz reaction chamber at a total
pressure of 1000mbar using synthetic air as bath gas.The reaction chamber is equipped with 32 super actinicfluorescent lamps (Philips TL05/40W: 3205l5480 nm,
lmax ¼ 360 nm) and 32 low-pressure mercury lamps(Philips TUV 40W: lmax ¼ 254 nm). The experimentalset-up is described in detail elsewhere (Barnes et al.,1994a). Experiments have been performed at 284, 295
and 306� 2K on DMS/H2O2/air mixtures for differentinitial NOx conditions. The photolysis of H2O2 with thelow-pressure mercury lamps was used to generate the
OH radicals:
H2O2 þ hn ðl¼ 254 nmÞ ! OHþOH:
The concentration–time behaviour of the DMS, NOx
and products were monitored in situ using a multi-reflection White-type mirror system (5.6m base length,
498m total path length) mounted in the reactor chamberand coupled to a Fourier transform infrared (FT-IR)spectrometer (Bruker IFS 88) equipped with a KBr
beam splitter and HgCdTe (MCD) detector. Typicalinitial concentration of DMS and H2O2 were kept fairly
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803770

constant during the experiments at 6–7 ppm andapproximately 25–30 ppm, respectively, where
1 ppm=2.46� 1013molecules cm�3 at 298K. The initialNO concentrations were varied between 434 and2821 ppb. The NO was always associated with an initial
NO2 concentration due to oxidation of NO duringtransfer and injection into the reactor. The initial NO2
concentrations varied between 135 and 739 ppb.In typical experiments infrared spectra were recorded
at 1 cm�1 resolution and 10–15 spectra were collectedover irradiation periods of 20–30min. Concentrations ofall species, reactant and products were determined by
computer-aided spectral abstraction using calibratedreference spectra generated in this laboratory.
3. Results and discussion
The sulphur-containing products identified in theirradiation of DMS/H2O2/NOx reaction mixtures insynthetic air included DMSO, DMSO2, SO2, MSA andMSPN. Formation of OCS was also observed during the
experiments when the NOx concentration fell to lowlevels. Fig. 1(a–f ) shows plots of the measured yield–time profiles for all the sulphur-containing products
observed, as a function of the different temperatures andinitial NOx conditions investigated. The yields of theproducts shown in Fig. 1(a–f ) have not been corrected
for secondary loss processes such as further reactionwith other oxidants, e.g. OH, or wall loss. In order tofacilitate the discussion on trends in the product
behaviour, the yield–time profiles of DMSO, DMSO2,SO2, MSA, MSPN and OCS for 295K and 1000mbarsynthetic air as a function of NOx concentration areshown separately in Fig. 2(a–f ).
SO2 and DMSO undergo considerable further oxida-tion in the reaction system. In order to obtain the‘‘exact’’ formation yields for these compounds correc-
tions need to be performed for reaction with OHradicals and also loss to the reactor walls. Correctionshave been made using the mathematical procedure
described in Tuazon et al. (1986) and also outlined ina recent publication from this laboratory on theoxidation of DMS under NOx free conditions (Arsene
et al., 1999). The constants used for the corrections arethe same as those given in Arsene et al. (1999), thereforedetails are not given here.Figs. 3 and 4 show examples of plots of the DMSO
and SO2 concentrations corrected for wall loss andreaction with OH radicals against the consumption ofDMS, the slopes give the ‘‘true’’ formation yields of the
compounds. Except for the study at 284K, the plots forDMSO in Fig. 3 show quite distinct deviations fromlinearity. Two distinct linear periods are often evident,
an initial period followed by a transition to a secondperiod where the DMSO yield increases compared to the
first period. The onset of the increase occurs at the pointwhen the NO concentration approaches values below
the system detection limit (10 ppb). This behaviour isattributed to a change in the reaction mechanism, whichis discussed below. Similar behaviour was not observed
in the SO2 yield plots, however, since SO2 can be formedin both the abstraction and addition channels (Arseneet al., 1999) the effect here is probably not so great andnot observable within the error limits of the measure-
ments. The corrected formation yields of DMSO andSO2 are shown in Tables 1 and 2, respectively, for theinitial reaction period. The yields are compared to
fraction of the reaction estimated to proceed via theaddition channel.
3.1. General trends in products with temperature andinitial NOx conditions
3.1.1. DMSOThe yield–time behaviour of DMSO varied system-
atically with changes in temperature and initial NOx.The uncorrected yield–time profiles for DMSO (Figs. 1a
and 2a) show that at each temperature and initial NOx
concentration the yield initially increases, passesthrough a maximum and then decreases again. The
magnitude of the yield turning point increases withdecreasing temperature for similar initial NOx condi-tions. The DMSO yield is also strongly dependent on the
initial NO concentration, increasing the initial NOconcentration systemically depressed the DMSO yieldmaximum at each of the temperatures investigated. For
the measurements at 306K, the bell-shaped be-haviour observed at 284 and 295K is not so pro-nounced. After attainment of the yield maximum there isonly a further slow decay of DMSO. In the experiments
at 306K the oxidation of DMS also slows downsignificantly near the point of the DMSO maximum.This behaviour in DMS suggests termination of the
propagation of the OH radical production chain atthis point under the conditions of these experiments. Theprobable explanation for the termination is efficient
scavenging of the OH radicals via the reaction OH+NO2+M!HNO3+M at the very high levels of NO2
which are generated under the conditions of these
experiments. Therefore, the flatness of the DMSO curvesat 306K after the maximum is presently attributed totermination of its photo-oxidation under the conditionsof the experiment.
The increase in the DMSO yield with decreasingtemperature and the shape of the curves is analogous tothe behaviour reported recently in our paper on the
oxidation of DMS under NOx free conditions (Arseneet al., 1999). As stated in the Introduction the OHinitiated oxidation of DMS is known to proceed by both
addition and abstraction channels, the addition channelproducing the dimethylhydroxysulphuranyl radical
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–3780 3771

((CH3)2S–OH). This adduct can thermally de-compose either back to reactants or products or itcan react further with O2 to form products which
include DMSO.
ðCH3Þ2S2OHþD! CH3SCH3þOH;
ðCH3Þ2S2OHþO2! products including DMSO:
Based on the above mechanism the importance of the
addition channel and hence DMSO formation willincrease with decreasing temperature. This is in broad
agreement with the experimental observations reportedhere. The bell-shape of the yield–time curves is causedby a period of rapid production from OH+DMS
followed by a period of fast removal via OH+DMSOas this removal reaction gradually becomes morecompetitive with the production process.
However, the simple mechanism above cannot explainthe significant dependence of the DMSO yield on theinitial NO concentration. This is an effect that has notbeen previously reported. The simplest explanation for
this phenomenon is that the DMS–OH also undergoes a
Fig. 1. Plots of the yields of DMSO (a), DMSO2 (b), SO2 (c), MSA (d), MSPN (e), and OCS (f) as a function of temperature (284, 295
and 306K) and initial NOx concentration against reaction time.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803772

reversible addition of O2.
ðCH3Þ2S2OHþO2þM Ð CH3SðOHÞðO2ÞCH3þM:
Further reaction of this peroxy radical with NO formsan alkoxy radical which reacts further with O2 to formdimethyl sulphone (DMSO2).
CH3SðOHÞðO2ÞCH3þNO ! CH3SðOHÞðOÞCH3þNO2;
CH3SðOHÞðOÞCH3þO2! CH3SðOÞ2CH3þHO2:
Since formation of DMSO2 is also observed in NOx-freeproduct studies of DMS photo-oxidation (Arsene et al.,
1999), there are obviously other routes forming DMSO2.Heterogeneous reactions of DMS with H2O2 at thereactor walls can lead to formation of DMSO2, however,
in the present work this was insignificant at thetemperatures of the experiments. Self reaction of theCH3S(OH)(O2)CH3 radical or reaction with otherperoxy radicals could also lead to the formation of the
CH3S(OH)(O)CH3 alkoxy radical and subsequently
Fig. 2. Plots of the yields of DMSO (a), DMSO2 (b), SO2 (c), MSA (d), MSPN (e), and OCS (f) for 295K and 1000mbar synthetic air
as a function of different initial NOx concentrations against reaction time.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–3780 3773

Fig. 3. Plots of the DMSO concentration (corrected for wall
loss and reaction with OH radicals) against the consumption of
DMS at 284, 295 and 306K for different initial NOx
concentrations.
Fig. 4. Plots of the SO2 concentration (corrected for wall loss
and reaction with OH radicals) against the consumption of
DMS at 284, 295 and 306K for different initial NOx
concentrations.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803774

DMSO2, e.g.
CH3SðOHÞðO2ÞCH3þCH3SðOHÞðO2ÞCH3
! 2CH3SðOHÞðOÞCH3þO2;
CH3SðOHÞðO2ÞCH3þCH3O2
! CH3SðOHÞðOÞCH3þCH3OþO2:
It is quite probable that such routes are operative
under NOx-free conditions in smog chambers. Thereaction of OH with DMSO is also known to produceDMSO2 (Arsene et al., 1999; S�rensen et al., 1996)possibly via
OHþ CH3SðOÞCH3! CH3SðOÞðOHÞCH3;
CH3SðOÞðOHÞCH3þO2! CH3SðOÞ2CH3þHO2:
However, this formation route cannot explain theobserved behaviour of DMSO with NOx. The yieldmaximum of DMSO2 would be expected to be shifted tolonger reaction times compared to that of DMSO and
no effect on variation of NO would be expected.Reaction of NO2 with the OH–DMS adduct or evenan OH–DMSO adduct are also potential routes to
DMSO2 formation:
CH3SðOHÞCH3þNO2! CH3SðOHÞðOÞCH3þNO;
CH3SðOHÞðOÞCH3þO2ðNO2Þ
! CH3SðOÞ2CH3þHO2ðHONOÞ;
CH3SðOÞðOHÞCH3þNO2! CH3SðOÞ2CH3þHONO:
Table 1
Corrected yields for the formation of DMSO in the reaction of OH with DMS in 1000mbar of synthetic air as a function of
temperature for the initial reaction period (see text for details). The yields are compared to the fractions of the reaction occurring via
the addition pathway calculated using the results of Hynes et al. (1986) for the specific reaction conditions
Temperature (K) NO (ppb) NO2 (ppb) Corrected DMSO yields
(M% yield)
Contribution of addition
pathway (%)
284 434 135 34 52
937 356 26
1200 515 22
295 685 251 27 33
886 368 19
1070 505 17
306 1579 141 15 17
2123 689 11
2821 739 9
Table 2
Corrected yields for the formation of SO2 in the reaction of OH with DMS in 1000mbar of synthetic air as a function of temperature
for the initial reaction period (see text for details). The yields are compared to the fractions of the reaction occurring via the addition
pathway and abstraction pathways calculated using the results of Hynes et al. (1986) for the specific reaction conditions
Temperature (K) NO (ppb) NO2 (ppb) Corrected SO2
yields (M% yield)
Contribution of
addition pathway (%)
Contribution of
abstraction
pathway (%)
284 434 135 65 52 48
937 356 43
1200 515 35
295 685 251 41 33 67
886 368 36
1070 505 33
306 1579 141 41 17 83
2123 689 34
2821 739 33
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–3780 3775

However, if these were major routes to DMSO2
formation their importance would be expected to
increase with increasing reaction time since the NO2
concentration is increasing in the system; this behaviouris not observed.
The formation of DMSO2 via reaction of a DMS–OH–O2 peroxy radical with NO should depress the yieldof DMSO from the addition channel. As can be seen inTable 1, the DMSO formation yield in the initial stages
of the reaction is systemically depressed with increasinginitial NO concentration. Further, in the plots of thecorrected DMSO concentration against the consump-
tion of DMS (Fig. 3) the formation of DMSO increaseswhen the NO concentration falls to low levels. All ofthese facts support that a major mechanism for the
formation of DMSO2 in the reaction system is reactionof a DMS–OH–O2 peroxy radical with NO as describedabove. As discussed below the yield–time behaviour of
DMSO2 appears to be generally in line with thismechanistic conception.It is of interest at this stage to compare the corrected
formation yields of DMSO with the estimated fraction
of the reaction proceeding via the addition pathwaycalculated using the kinetic data of Hynes et al. (1986). Itshould be borne in mind, when comparing the corrected
DMSO yield with the estimation of the OH+DMSreaction proceeding via the addition pathway, that thereare quite substantial uncertainties involved in both the
correction of the DMSO yields, particularly at 306Kwhere the yields are low, and also in the estimates of thefraction of the reaction proceeding via addition asdiscussed in Arsene et al. (1999). Based on the estimates
in Table 1 it is evident that at 284 and 295K thebranching ratios for DMSO formation from CH3S(OH)-CH3+O2 are considerably less than unity under all the
conditions whereas at 306K the differences appear to benot quite so pronounced, particularly at the low NOlevels. The branching ratio decreases with increasing
initial NO and the effectiveness of the depression of theDMSO yield with NO would appear to drop off withincreasing temperature. This general type of behaviour
would be expected for a reaction sequence involvingformation of a thermally unstable DMS–OH–O2 peroxyradical adduct.This finding is in stark contrast to other results from
this laboratory (Arsene et al., 1999), where under NOx-free conditions the DMSO yield was near unity under allthe reaction conditions employed. The branching ratio
values for DMSO formation are, however, quite similarto the branching ratio of F=0.5� 0.15 at 234 and258K for HO2 production from CH3S(OH)-
CH3+O2!HO2+DMSO reported by Turnipseedet al. (1996) in a pulsed laser photolysis/pulsed laser-induced fluorescence study on the reaction of OH with
DMS. Turnipseed et al. measured HO2 in theirexperiments by converting it to OH with NO. In their
analysis of their system Turnipseed et al. may not onlyhave been converting HO2 to OH with NO but they may
also have been disturbing the equilibrium DMS–OH+O2ÐDMS–OH–O2 through the reaction DMS–OH–O2+NO!DMS–OH–O+NO2. Such a reaction if
occurring in their system would have resulted in areduction in the apparent branching ratio for DMSOformation. It is believed that the existence of theaddition of O2 to the DMS–OH adduct with subsequent
reaction with NO offers a plausible explanation for thedifferences in the DMSO yield found between systemsoperating with and without NO.
3.2. Formation of DMSO2
As for DMSO, with the exception of the measure-ments at 306K, the DMSO2 yield goes through adistinct maximum (Figs. 1b and 2b). The highest
DMSO2 yields are obtained at the lowest temperatureand the lowest yields at the highest temperature. In allcases, the DMSO2 yield increases with increasing initialNO concentration. DMSO2 reacts very slowly with OH
radicals (Falbe-Hansen et al., 2000), therefore, apartfrom slow loss to the reactors walls it is not expected toreact further in the system. The bell shape of the
DMSO2 yield curves for the 284 and 295K measure-ments (Figs. 1b and 2b) is what would be expected for aformation mechanism which is active in the initial stages
of the reaction and then ceases when one of the reactantsis exhausted, i.e. in this case NO. At 284 and 295K,therefore, the behaviour of DMSO2 is generally in
qualitative agreement with formation from reaction of aDMS–OH–O2 peroxy radical with NO being animportant formation pathway in the reaction system.For the DMSO2 profiles obtained at 306K the DMSO2
yield increases to a plateau rather than going through amaximum. The behaviour of the DMSO2 yield at 306Kis attributable to the considerable slowing in the DMS
oxidation just after the DMSO maximum as discussedabove. This halts production of DMSO2 and causes itsyield–time plot to approach a plateau. In general, the
overall behaviour of DMSO2 with temperature and NOconditions is not inconsistent with the occurrence of theDMS–OH–O2 peroxy radical mechanism presentedabove.
3.3. Formation of sulphur dioxide
The uncorrected yield–time curves of SO2 show asteep initial rise followed by a much slower increase to aplateau. Since it was difficult to set the initial NOx
conditions to the same level for each of the experimentsat the different temperatures, there is no general patternwith respect to the onset of the approach to the plateau,
which is governed mainly by the level of NO2 in thesystem (see later). However, at all 3 temperatures there is
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803776

a general decrease of the yield with increasing initialNOx concentration, the effect being most pronounced at
the lowest temperature. The corrected molar yields ofSO2 for the respective NOx conditions are listed in Table2. The yields of SO2 are much lower than those observed
in NOx-free systems (Arsene et al., 1999), but are similarto those which have been reported previously for DMSoxidation systems containing NOx (Patroescu et al.,1999; S�rensen et al., 1996).
In contrast to DMSO, deviations from linearity weregenerally not observed in the plots of the corrected SO2
concentration against consumption of DMS (Fig. 4).
This is not too surprising since SO2 is not a directproduct and the pathways leading to SO2 formation inthe presence of NOx are much more complex than those
of DMSO and involve the formation of thermallyunstable peroxynitrate intermediates. The complexityof the SO2 formation probably masks any deviations
from linearity. However, the increasing importance ofthe DMSO2 formation pathway with increasing initialNO should be reflected in decreases in the SO2 yield. Asseen in Table 2 the corrected SO2 yield decreases with
increasing initial NO concentration in line with mechan-istic expectations.
3.4. Formation of MSA and MSPN
The yield–time contours of MSA all pass through a
fairly sharp maximum followed by a slow decay causedby loss to the aerosol phase and uptake by the reactorwall. For all of the temperatures the same trend is
observed, i.e. the yield maximum increases with increas-ing initial NO concentration. At 284 and 295K themaximum yield varies between approximately 5 and
12M%. At 306K where the initial NO concentrationsare much higher the MSA yield maximums are muchhigher and vary between approximately 28 and 46M%.This behaviour is in line with NO and NO2 promoting
the formation of MSA as observed in previous studiesvia reactions such as
CH3SOOþNO ! CH3SOþNO2;
CH3SðOÞOOþNO ! CH3SðOÞOþNO2;
CH3SðOÞ2OOþNO ! CH3SðOÞ2OþNO2;
CH3SþNO2! CH3SOþNO;
CH3SOþNO2! CH3SO2þNO;
CH3SO2þNO2! CH3SðOÞ2OþNO
followed by
CH3SðOÞ2OþRH ! CH3SðOÞ2OHþR:
Like MSA, the time-yield contours of MSPN(CH3S(O)2OONO2) also go through a maximum
followed by a slow decay and at all three temperaturesthe yield maximum increases with increasing initial NOx
concentration. However, there are several distinctdifferences compared to the MSA profiles. In contrastto MSA, the molar yield of MSPN increases with
decreasing temperature, the opposite applies for MSA.The position of the MSPN yield maximum also shifts tolonger reaction times with decreasing temperature andbecomes broader. This behaviour is in agreement with
what would be expected for a thermally labile peroxynitrate compound.
CH3SðOÞ2OONO2 þM Ð CH3SðOÞ2OOþNO2þM:
In such an equilibrium, decreases in temperature andincreases in the NO2 concentration obviously favour
formation of the peroxy nitrate. The ratio of NO to NO2
is also important since the equilibrium is in competitionwith
CH3SðOÞ2OOþNO ! CH3SðOÞ2OþNO2:
which favours formation of MSA. The variation of theprofiles of MSA and MSPN in Figs. 1d and e are in
accord with the expected behaviour from the reactionsoutlined above.
3.5. Formation of OCS
The formation of OCS is only observed after an initialdelay in the system. As seen in Figs. 1f and 2f, the onset
of the formation is dependent on the initial NOx
concentrations, the time of the onset increasing withincreasing initial NOx concentration. At the high initial
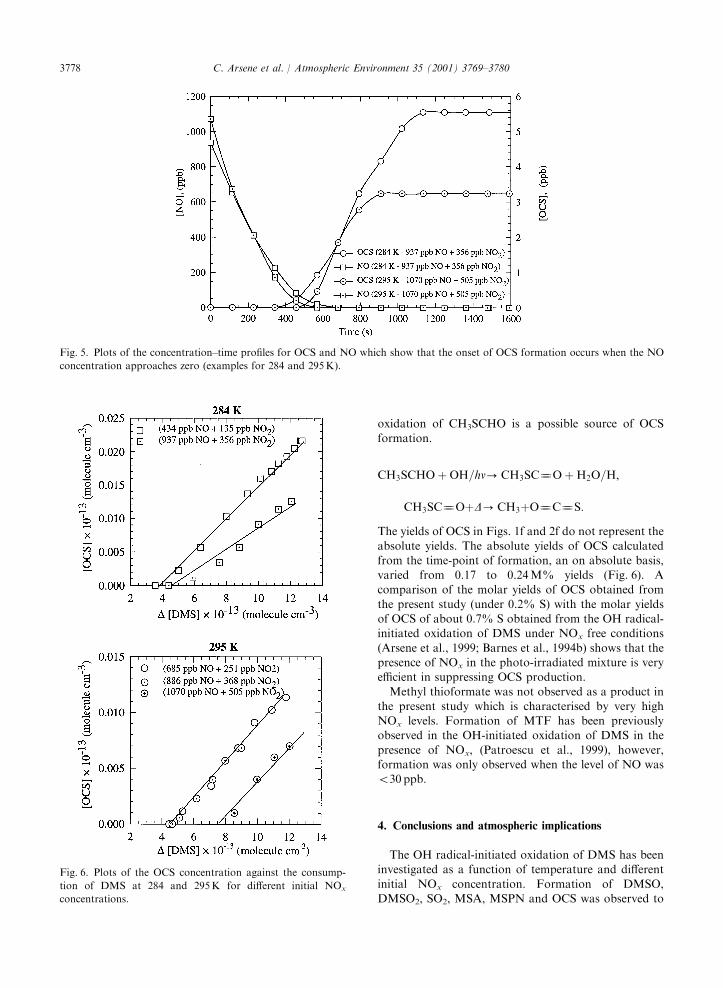
NOx concentrations of the experiments performed at306K formation of OCS is not observed. Analysis of theconcentration–time profiles shows that the onset of OCS
formation occurs as the NO concentration approacheszero (Fig. 5). This observation is line with other reportsof OCS formation from the oxidation of DMS
previously reported from this laboratory (Barnes et al.,1994b, 1996; Patroescu et al., 1999).Increasing the temperature should have the effect of
increasing the branching ratio for CH3SCH2 formation
in the OH radical-initiated oxidation of DMS, a radicalwhich is considered to be the precursor of the CH3Sradical and possibly methyl thiolformate (CH3SCHO).
Possible further reaction of CH3S radicals include
CH3Sþ CH3S ! CH3SHþH2C¼¼S;
CH3SþNO2! CH3SOþNO;
H2C¼¼SþOH=hn! HC¼¼SþH2O=H;
HC¼¼SþO2! O¼¼C¼¼SþOH:
The thermal decomposition of CH3SCO radicalsproduced in the photolysis or OH radical-initiated
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–3780 3777
oxidation of CH3SCHO is a possible source of OCS
formation.
CH3SCHOþOH=hn! CH3SC¼¼OþH2O=H;
CH3SC¼¼OþD! CH3þO¼¼C¼¼S:
The yields of OCS in Figs. 1f and 2f do not represent the
absolute yields. The absolute yields of OCS calculatedfrom the time-point of formation, an on absolute basis,varied from 0.17 to 0.24M% yields (Fig. 6). Acomparison of the molar yields of OCS obtained from
the present study (under 0.2% S) with the molar yieldsof OCS of about 0.7% S obtained from the OH radical-initiated oxidation of DMS under NOx free conditions
(Arsene et al., 1999; Barnes et al., 1994b) shows that thepresence of NOx in the photo-irradiated mixture is veryefficient in suppressing OCS production.
Methyl thioformate was not observed as a product inthe present study which is characterised by very highNOx levels. Formation of MTF has been previouslyobserved in the OH-initiated oxidation of DMS in the
presence of NOx, (Patroescu et al., 1999), however,formation was only observed when the level of NO was530 ppb.
4. Conclusions and atmospheric implications
The OH radical-initiated oxidation of DMS has beeninvestigated as a function of temperature and different
initial NOx concentration. Formation of DMSO,DMSO2, SO2, MSA, MSPN and OCS was observed to
Fig. 5. Plots of the concentration–time profiles for OCS and NO which show that the onset of OCS formation occurs when the NO
concentration approaches zero (examples for 284 and 295K).
Fig. 6. Plots of the OCS concentration against the consump-
tion of DMS at 284 and 295K for different initial NOx
concentrations.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803778

occur in the reaction systems. The level of NO in thereaction system was a critical factor in determining the
yields of DMSO and DMSO2 which were anti-corre-lated.The data support that the major channel for DMSO2
formation is probably reversible addition of O2 to theDMS–OH adduct formed in the addition channel of thereaction of OH with DMS followed by sequentialreactions with NO and O2.
DMS2OHþO2þM Ð OH2DMS2O2þM;
OH2DMS2O2þNO ! OH2DMS2OþNO2;
OH2DMS2OþO2! CH3SðOÞ2CH3þHO2:
This reaction sequence has been proposed previously ina slightly modified form by Yin et al. (1990). In themechanism of Yin et al. the reaction DMS–
OH+O2+M was not reversible and a thermal pathway
OH2DMS2O2! DMSOþHO2
was included. This paper does, however, represent thefirst study where experimental evidence for the occur-rence of such a reaction mechanism has been found.
It is difficult to extrapolate the present results totypical atmospheric concentrations of NO to obtainmeaningful information about the possible significance
of the above reaction sequence in the troposphere.Should the reaction sequence not be significant undertropospheric conditions this would imply a near unit
formation yield of DMSO from the addition channel ofthe reaction of OH with DMS. However, under the lowNOx conditions which often prevail in the remotemarine atmosphere there remains the possibility of
reaction of OH–DMS–O2 adduct with HO2 or otherperoxy radicals which may convert it to DMSO2,particularly at low temperature, for which no informa-
tion presently exists.The present study taken together with the results
published in Arsene et al. (1999) represents a very
detailed body of concentration–time product data on thereaction products of the OH-radical initiated oxidationof DMS as a function of temperature and NOx
conditions. A detailed modelling study of the results is
currently ongoing and should help to elucidate andbetter define the potential atmospheric importance ofmany of the speculative or uncertain aspects of the DMS
oxidation processes.
Acknowledgements
Financial support for this work by the BMBF within
the AFS project and the European Commission withinthe EL CID project is gratefully acknowledged.
References
Andreae, M.O., 1990. Ocean-atmosphere interactions in the
global biogeochemical sulfur cycle. Marine Chemistry 30,
1–29.
Andreae, M.O., Crutzen, P., 1997. Atmospheric aerosols:
biogeochemical sources and role in atmospheric chemistry.
Science 216, 1052–1058.
Andreae, M.O., Elbert, W., de Mora, S.J., 1995. Biogenic sulfur
emissions and aerosols over the tropical South Atlantic. 3.
Atmospheric dimethylsulfide, aerosols and cloud condensa-
tion nuclei. Journal of Geophysical Research 100, 11335–
11356.
Arsene, C., Barnes, I., Becker, K.H., 1999. FT-IR product
study of the photooxidation of dimethyl sulphide: Tempera-
ture and O2 partial pressure dependence. Physical Chemistry
Chemical Physics 1, 5463–5470.
Barnes, I., Becker, K.H., Mihalopoulos, N., 1994a. An FTIR
product study of the photooxidation of dimethyl disulfide.
Journal of Atmospheric Chemistry 18, 267–289.
Barnes, I., Becker, K.H., Patroescu, I.V., 1994b. The
tropospheric oxidation of dimethyl sulfide: a new source
of carbonyl sulfide. Geophysical Research Letter 21,
2389–2392.
Barnes, I., Becker, K.H., Patroescu, I.V., 1996. FTIR product
study of the OH initiated oxidation of dimethyl sulphide:
observation of carbonyl sulphide and dimethyl sulphoxide.
Atmospheric Environment 30, 1805–1814.
Barone, S.B., Turnipseed, A.A., Ravishankara, A.R., 1995.
Role of adducts in the atmospheric oxidation of dimethyl
sulfide. Faraday Discussions 100, 39–54.
Bates, T.S., Cline, J.D., Gammon, R.H., Kelly-Hansen, R.S.,
1987. Regional and seasonal variation of the flux of oceanic
dimethylsulfide to the atmosphere. Journal of Geophysical
Research 92, 2930–2938.
Bates, T.S., Lamb, B.K., Guenther, A., Dignon, J., Stoiber,
R.E., 1992. Sulfur emissions to the atmosphere from natural
sources. Journal of Atmospheric Chemistry 14, 315–337.
Berresheim, H., Wine, P.H., Davis, D.D., 1995. Sulfur in the
atmosphere. In: Singh, H.B. (Ed.), Composition, Chemistry,
and Climate of the Atmosphere. Van Nostrand Reinhold,
New York, pp. 251–307.
Campolongo, F., Saltelli, A., Jensen, N.R., Wilson, J., Hjorth,
J., 1999. The role of multiphase chemistry in the oxidation of
dimethylsulphide (DMS). A latitude dependent analysis.
Journal Atmospheric Chemistry 32, 327–356.
Falbe-Hansen, H., Sorensen, S., Jensen, N.R., Pedersen, T.,
Hjorth, J., 2000. Atmospheric gas phase reactions of
dimethylsulphoxide and dimethylsulphone with OH and
NO3 radicals, Cl atoms and ozone. Atmospheric Environ-
ment 34, 1543–1551.
Hynes, A.J., Wine, P.H., Semmes, D.H., 1986. Kinetics and
mechanism of OH reactions with organic sulfides. Journal of
Physical Chemistry 90, 4148–4156.
Nguyen, B.C., Bonsang, B., Gaudry, A., 1983. The role of the
ocean in the global atmospheric sulfur cycle. Journal of
Geophysical Research 88, 10903–10914.
Patroescu, I.V., Barnes, I., Becker, K.H., Mihalopoulos, N.,
1999. FT-IR product study of the OH-initiated oxidation of
DMS in the presence of NOx. Atmospheric Environment 33,
25–35.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–3780 3779

Ravishankara, A.R., Rudich, Y., Talukdar, R., Barone, S.B.,
1997. Oxidation of atmospheric reduced sulfur compounds:
perspective from laboratories. Philosophical Transactions of
the Royal Society of London 332, 171–182.
S�rensen, S., Falbe-Hansen, H., Mangoni, M., Hjorth, J.,
Jensen, N.R., 1996. Observation of DMSO and CH3S(O)OH
from the Gas Phase Reaction between DMS and OH.
Journal of Atmospheric Chemistry 24, 299–315.
Spiro, R.E., Jacob, D.J., Logan, J.A., 1992. Global inventory of
sulfur emissions with 18� 18 resolution. Journal of Geophy-sical Research 97, 6023–6036.
Tuazon, E.C., Mac Leod, H., Atkinson, R., Carter,
W.P.L., 1986. a-Dicarbonyl yields from the NOx}
air photooxidations of a series of aromatic hydro-
carbons in air. Environmental Science and Technology 20,
383–387.
Turnipseed, A.A., Barone, S.B., Ravishankara, A.R., 1996.
Reaction of OH with dimethyl sulfide. 2. Products and
mechanisms. Journal of Physical Chemistry 100, 14703–
14713.
C. Arsene et al. / Atmospheric Environment 35 (2001) 3769–37803780




















