
Folic acid conjugated amino acid-based star polymers for active targeting of cancer cells
9
Published: August 22, 2011 r2011 American Chemical Society 3469 dx.doi.org/10.1021/bm200604h | Biomacromolecules 2011, 12, 3469–3477 ARTICLE pubs.acs.org/Biomac Folic Acid Conjugated Amino Acid-Based Star Polymers for Active Targeting of Cancer Cells Adrian Sulistio, † Justin Lowenthal, ‡ Anton Blencowe, † Marie N. Bongiovanni, †, § Lydia Ong, †, § Sally L. Gras, †, § Xiaoqing Zhang, ^ and Greg G. Qiao* ,† † Department of Chemical and Biomolecular Engineering, University of Melbourne, Parkville, Melbourne, VIC 3010, Australia ‡ Department of Biomedical Engineering, Yale University, New Haven, Connecticut 06511, United States § The Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Melbourne, VIC 3010, Australia ^ CSIRO Material Science and Engineering, Private Bag 33, Clayton South MDC 3169, VIC, Australia b S Supporting Information ’ INTRODUCTION The development of polymeric nanocarriers as targeted drug delivery vehicles for the treatment of cancer offers significant therapeutic benefits over other strategies, such as conventional chemotherapy or radiation theraphy. 1 For example, polymeric vesicles can improve drug solubility in an aqueous medium, provide increased payload capacity, prolong the circulation time of the drug in the bloodstream, reduce drug toxicity to healthy cells, 2 and passively target tumor tissue due to enhanced per- meability and retention (EPR) effects as well as actively targeting cancer cells. 1,3,4 Such targeted approaches allow for reduced drug dosage while improving the efficacy of the treatment and minimizing the side effects to the patient. Various nanocarriers have been developed, including polymeric micelles (self-assembled from amphiphilic polymers), 510 layer-by-layer (LbL) capsules, 11 dendrimers, 12 and star polymers. 13,14 It has been demonstrated that star polymers possessing hydrophobic cores and radiating hydrophilic arms can combine the advantages of polymeric micelles, dendrimers, and LbL capsules by providing enhanced encapsulation capabilities for hydrophobic drugs in the core, 15 while avoiding issues associated with the sudden dissociation (under shear or dilution below the critical micelle concentration (cmc)) of polymeric micelles and the time-consuming multistep synthesis of LbL capsules and dendrimers. 1416 Moreover, star polymers are known to possess unique properties such as low solution viscosities 17,18 and enhanced functionalities, 1923 which makes this class of macro- molecules an attractive candidate for drug delivery applications. However, only a limited number of these polymeric nanocarriers are reported to be biocompatible and biodegradable. 24 Recently, we reported the successful synthesis of core cross- linked star (CCS) polymers composed exclusively of amino acid building blocks. 25 These star polymer were composed of poly(L- lysine) arms radiating from a poly(L-cystine) core and could be site specifically functionalized to possess hierarchical functional- ities spanning from the core, along the arms, to the periphery. Furthermore, the core building block, L-cystine, has a centrally located disulfide bond that can be cleaved by reducing agents such as glutathione; hence, the resulting star polymers possess bioreducable cores. Subsequently, it was demonstrated that these star polymers are also capable of sequestering hydrophobic drugs, such as pirarubicin, within their interior. In this study, the viability of this CCS polymer as an active targeting drug delivery vehicle for the treatment of cancer was Received: May 3, 2011 Revised: August 18, 2011 ABSTRACT: Amino acid-based core cross-linked star (CCS) polymers (poly(L-lysine) arm poly(L-cystine) core ) with peripheral allyl functionalities were synthesized by sequential ring-opening polymerization (ROP) of amino acid N-carboxyanhydrides (NCAs) via the arm-first approach, using N-(trimethylsilyl) allylamine as the initiator. Subsequent functionalization with a poly(ethylene glycol) (PEG)folic acid conjugate via thiol ene click chemistry afforded poly(PEG-b-L-lysine) arm poly(L- cystine) core stars with outer PEG coronas decorated with folic acid targeting moieties. Similarly, a control was prepared with- out folic acid, using just PEG. A fluorophore was used to track both star polymers incubated with breast cancer cells (MDA-MB-231) in vitro. Confocal microscopy and flow cytometry revealed that the stars could be internalized into the cells, and higher cell internalization was observed when folic acid moieties were present. Cytotoxicity studies indicate that both stars are nontoxic to MDA-MB-231 cells at concentrations of up to 50 μg/mL. These results make this amino acid-based star polymer an attractive candidate in targeted drug delivery applications including chemotherapy.
Transcript of Folic acid conjugated amino acid-based star polymers for active targeting of cancer cells

Published: August 22, 2011
r 2011 American Chemical Society 3469 dx.doi.org/10.1021/bm200604h | Biomacromolecules 2011, 12, 3469–3477
ARTICLE
pubs.acs.org/Biomac
Folic Acid Conjugated Amino Acid-Based Star Polymers for ActiveTargeting of Cancer CellsAdrian Sulistio,† Justin Lowenthal,‡ Anton Blencowe,† Marie N. Bongiovanni,†,§ Lydia Ong,†,§ Sally L. Gras,†,§
Xiaoqing Zhang,^ and Greg G. Qiao*,†
†Department of Chemical and Biomolecular Engineering, University of Melbourne, Parkville, Melbourne, VIC 3010, Australia‡Department of Biomedical Engineering, Yale University, New Haven, Connecticut 06511, United States§The Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Melbourne, VIC 3010, Australia^CSIRO Material Science and Engineering, Private Bag 33, Clayton South MDC 3169, VIC, Australia
bS Supporting Information
’ INTRODUCTION
The development of polymeric nanocarriers as targeted drugdelivery vehicles for the treatment of cancer offers significanttherapeutic benefits over other strategies, such as conventionalchemotherapy or radiation theraphy.1 For example, polymericvesicles can improve drug solubility in an aqueous medium,provide increased payload capacity, prolong the circulation timeof the drug in the bloodstream, reduce drug toxicity to healthycells,2 and passively target tumor tissue due to enhanced per-meability and retention (EPR) effects as well as actively targetingcancer cells.1,3,4 Such targeted approaches allow for reduceddrug dosage while improving the efficacy of the treatment andminimizing the side effects to the patient. Various nanocarriershave been developed, including polymeric micelles (self-assembledfrom amphiphilic polymers),5�10 layer-by-layer (LbL) capsules,11
dendrimers,12 and star polymers.13,14
It has been demonstrated that star polymers possessinghydrophobic cores and radiating hydrophilic arms can combinethe advantages of polymeric micelles, dendrimers, and LbLcapsules by providing enhanced encapsulation capabilities forhydrophobic drugs in the core,15 while avoiding issues associatedwith the sudden dissociation (under shear or dilution below thecritical micelle concentration (cmc)) of polymeric micelles andthe time-consuming multistep synthesis of LbL capsules and
dendrimers.14�16 Moreover, star polymers are known to possessunique properties such as low solution viscosities17,18 andenhanced functionalities,19�23 which makes this class of macro-molecules an attractive candidate for drug delivery applications.However, only a limited number of these polymeric nanocarriersare reported to be biocompatible and biodegradable.24
Recently, we reported the successful synthesis of core cross-linked star (CCS) polymers composed exclusively of amino acidbuilding blocks.25 These star polymer were composed of poly(L-lysine) arms radiating from a poly(L-cystine) core and could besite specifically functionalized to possess hierarchical functional-ities spanning from the core, along the arms, to the periphery.Furthermore, the core building block, L-cystine, has a centrallylocated disulfide bond that can be cleaved by reducing agentssuch as glutathione; hence, the resulting star polymers possessbioreducable cores. Subsequently, it was demonstrated that thesestar polymers are also capable of sequestering hydrophobicdrugs, such as pirarubicin, within their interior.
In this study, the viability of this CCS polymer as an activetargeting drug delivery vehicle for the treatment of cancer was
Received: May 3, 2011Revised: August 18, 2011
ABSTRACT: Amino acid-based core cross-linked star (CCS)polymers (poly(L-lysine)armpoly(L-cystine)core) with peripheralallyl functionalities were synthesized by sequential ring-openingpolymerization (ROP) of amino acid N-carboxyanhydrides(NCAs) via the arm-first approach, using N-(trimethylsilyl)allylamine as the initiator. Subsequent functionalization with apoly(ethylene glycol) (PEG)�folic acid conjugate via thiol�ene click chemistry afforded poly(PEG-b-L-lysine)armpoly(L-cystine)core stars with outer PEG coronas decorated with folicacid targeting moieties. Similarly, a control was prepared with-out folic acid, using just PEG. A fluorophore was used to track both star polymers incubated with breast cancer cells (MDA-MB-231)in vitro. Confocal microscopy and flow cytometry revealed that the stars could be internalized into the cells, and higher cellinternalization was observed when folic acid moieties were present. Cytotoxicity studies indicate that both stars are nontoxic toMDA-MB-231 cells at concentrations of up to 50 μg/mL. These results make this amino acid-based star polymer an attractivecandidate in targeted drug delivery applications including chemotherapy.

3470 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
explored by functionalizing the star with a targeting moiety andinvestigating its in vitro cell distribution with a breast cancer cellline (MDA-MB-231). The star polymer was synthesized via aone-pot, arm-first approach using sequential ring-openingpolymerization (ROP) of amino acid N-carboxyanhydride (NCA)derivatives initiated with N-(trimethylsilyl)allylamine.26 Z-L-Ly-sine was chosen as the building block for the arms since thependent amine functionalities available after deprotection couldpotentially be used to conjugate chemotherapeutic drugs viaacid-labile linkers, such as aryl imines,27 or tracing agents forimaging (vide infra). The incorporation of allyl functionalities atthe star periphery allowed for postpolymerization functionaliza-tion with thiolated PEG derivatives via “thiol�ene” click chem-istry. Thus, the use of heterodifunctional α-thiol, ω-folic acidPEG provided a facile route to PEGylation and simultaneouslyintroduce targeting moieties. This functionalization served twopurposes: (i) PEGylation of the star would shield the peptidecomponents from rapid enzymatic degradation and suppresstheir interaction with plasma protein and cells, hence reducingtheir uptake by the liver and increasing their circulation time inthe bloodstream;12,16 and (ii) the folic acid (FA) moieties impartactive targeting capabilities due to their high binding affinity forfolate receptors (FRs) (Kd ∼ 10�10 mol L�1),28�30 which areoverexpressed on the surfaces of endothelial cancer cells.31
Targeting these receptors can facilitate cell internalization, pre-sumably via receptor-mediated endocytosis.32 Gel permeationchromatography (GPC), dynamic light scattering (DLS), and 1HNMR spectroscopic analysis were used to characterize thePEGylated CCS polymers and their precursors. The cellularuptake of the star polymers in MDA-MB-231 cells was assessedvia confocal microscopy and flow cytometry, and their cytotoxi-city at various concentrations was determined using a cellviability assay.
’EXPERIMENTAL SECTION
Materials. Z-L-Lys(Z)-OH (Bachem), L-cystine (Aldrich), N-(trime-thylsilyl)allylamine (96%, Aldrich), benzyl chloroformate (95%, Aldrich),sodium bicarbonate (NaHCO3) (99%, Ajax Fine Chemicals), magnesiumsulfate (MgSO4) (>98%, anhydrous, Merck), phosphorus pentachloride(PCl5) (>99%, Merck), HO-PEG-NHCO-C2H4-S-Trt (Mw = 3 kDa)(Rapp Polymere), MeO-PEG-OH (Mw = 2 kDa) (Aldrich), folic acid(FA) (g97%, Sigma),N-(3-(dimethylamino)propyl)-N0-ethylcarbodiimidehydrochloride (EDCI) (>98%, Fluka), 4-(dimethylamino)pyridine(DMAP) (99%, Aldrich), 2,2-dimethoxy-2-phenylacetophenone (DMPA)(99%, Aldrich), aminomethylpyrene hydrochloride (95%, Aldrich), hydro-bromic acid (33% in acetic acid, Aldrich), trifluoroacetic acid (TFA) (98%,Aldrich), hydrochloric acid (37%, Scharlau), lithium bromide (99.9%,Aldrich), tetrahydrofuran (THF) (99.9%, HPLC grade, RCI Labscan),3-mercaptopropanoic acid (g99%, Aldrich), sulfuric acid (H2SO4) (99.8%,BDH), sodium chloride (NaCl) (AR grade, Chem-Supply) and N,N-dimethylformamide (DMF) (99.8%, anhydrous, Aldrich) were used asreceived. n-Hexane (99.95%, Merck), ethyl acetate (99.5%, Chem-Supply),dimethyl sulfoxide (DMSO) (99.9%, Chem-Supply), chloroform (99.8%,Chem-Supply), andmethanol (99.7%,ChemSupply) were used as received.1,4-Dioxane (>98%, Fluka) and diethyl ether (99%, Chem-Supply) wereused tomake a binary solvent (2:3 v/v) and dried for 48 h prior to use over 3Å molecular sieves (Aldrich). Acetonitrile (99.99%, HPLC grade, B&J) wasstored over 3 Å molecular sieves. MALDI ToF MS matrices (dithranol(98.5%)) and cationization agents (NaTFA (98%), KI (99%)) werepurchased from Fluka and Aldrich, respectively, and were used as received.Dichloromethane (99.5%, Chem-Supply) was distilled from CaH2 underargon.Milli-Qwater (18.2MΩ 3 cm) was obtained from aMillipore Synergy
Water System. Acetone-d6 (99.9%), DMSO-d6 (99.9%), DMF-d7 (99.5%),and D2O (99.9%) were purchased from Cambridge Isotope Laboratoriesand used as received. Breast cancer cells (MDA-MB-231, American TypeCulture Collection (ATCC)), Dulbecco’s Modified Eagle’s Medium withL-glutamine and without folic acid (DMEM) (Sigma), Dulbecco’s phos-phate buffered saline (PBS) (Sigma), penicillin�streptomycin solution(Sigma), fetal bovine serum (Invitrogen), trypsin (Sigma), 6-well and 96-well cell culture plates, and T25 cell culture flasks (Corning) were used forcell culture. Alexa Fluor 647 carboxylic acid succinimidyl ester (Invitrogen)was used to stain the polymer. Alexa Fluor 488 wheat germ agglutinin(Invitrogen) andProLongGold antifade reagent dopedwith 40,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen) were used to stain thecells. Tyton X (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)) (Promega) was used to performcytotoxicity assays. Z-L-Lysine-NCA (lys NCA), di-Z-L-cystine ((Z-Cys-OH)2), L-cystine NCA (cys NCA), and aminomethylpyrene (AMP) weresynthesized according to previously published literature procedures.25
Instrumentation. GPC analysis was performed on a Shimadzuliquid chromatography system fitted with a Wyatt DAWNHELEOS LSdetector (λ = 658 nm), a Shimadzu RID-10 refractometer (λ = 633 nm),and a Shimadzu SPD-20A UV�vis detector, using three identical PLgelcolumns (5 μm, MIXED-C) in series and HPLC grade DMF with 0.05M LiBr as the mobile phase (1 mL/min). The oven temperature was setto 70 �C to maintain an acceptable pressure across the system, and thedetectors were temperature controlled to 25 �C. Astra software (WyattTechnology Corp.) was used to determine the molecular weightcharacteristics using known dn/dc values for poly(Z-L-lysine) (PZLL)in DMF (dn/dcPZLL = 0.101mL/g (25 �C)). 1HNMR spectroscopy wasperformed using a Varian Unity400 (400 MHz) spectrometer with thedeuterated solvent as reference and a sample concentration of ca. 20mg/mL. DLS measurements were performed on a Malvern high-perfor-mance particle sizer (HPPS) with a He�Ne laser (633 nm) at an angleof 173� and a temperature of 25( 0.1 �C—initial sample concentrationsof 10 mg/mL in DMF were used, and then serial dilutions wereperformed until stable spectra were obtained. All sample solutions werefiltered using 0.45 μm filters. MALDI ToF MS was performed on aBruker Autoflex III mass spectrometer operating in positive/linearmode. The analyte, matrix (dithranol), and cationization agent (KI orNaTFA) were dissolved in THF at concentrations of 10, 10, and 1 mg/mL, respectively, and then mixed in a ratio of 10:1:1. 0.3 μL of thissolution was then spotted onto a ground steel target plate, and thesolvent was allowed to evaporate prior to analysis. FlexAnalysis (Bruker)was used to analyses the data. UV�vis spectrophotometry was per-formed on a Shimadzu UV-2101PC spectrometer using quartz cuvetteswith a 1 cm path length. A Cyflow Space (Partec GmbH) flow cytometerusing an excitation wavelength of 633 nm was used to measure theintensity of the CCS polymers; at least 10 000 particles were analyzed ineach experiment. A FLUOstar OPTIMA (BMG Labtech, Germany)plate reader was used to measure the MTS absorbance at λ = 492 nm.Confocal laser scanning microscopy was performed on a Leica TCS SP2(Leica Microsystems, Heidelberg, Baden-W€urttemberg, Germany)powered by Ar/Kr and He/Ne lasers. The sample was viewed usingan oil immersion 63� lens (1.32 numerical aperture), and the pinholediameter was maintained at 1 airy unit. DAPI was excited at λ = 310 nm,Alexa Fluor 488 at λ = 488 nm, and Alexa Fluor 647 at λ = 633 nm. Theemission filters were set at λ = 410�450 nm for DAPI, λ = 500�580 nmfor Alexa Fluor 488, and λ = 650�710 nm for Alexa Fluor 647. Imageswere recorded at a depth of 10�20 μm from the surface of the glasscoverslip. Leica confocal software was used to acquire images of 512 �512 pixels that were the average of eight scans.Synthesis of Heterodifunctional α-Thiol,ω-Folic Acid PEG
(FA-PEG3000-SH). FA (0.071 g, 0.16 mmol) and DCC (0.034 g, 0.17mmol) were dissolved in DMSO (15 mL) under argon, followed byadditionofDMAP(0.019 g, 0.15mmol) andHO-PEG-NHCO-C2H4-S-Trt

3471 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
(0.050 g, 0.017 mmol). The reaction mixture was stirred in the dark atroom temperature for 24 h and then centrifuged to remove the insolublebyproduct, dicyclohexylurea. The supernatant was concentrated in vacuoto ca. 3.0 mL in volume and precipitated into DCM (50 mL). Theinsoluble, unreacted FA was removed via centrifugation and the super-natant concentrated in vacuo. The resulting residue was redissolved inDCM (2 mL) and precipitated into diethyl ether (20 mL), and theprecipitated product, FA-PEG-S-Trt, was collected via centrifugationand dried in vacuo (35 mg). UV�vis spectrophotometry and MALDI-ToF MS were used to determine the conjugation efficiency (ca. 30%),and the mixture was not separated. MALDI-ToFMS:Mw = 3803, PDI =1.00. 1H NMR (400 MHz, d6-DMSO) δH 2.10 (t, 2H, J = 7.0 HzCH2CO), 2.19 (t, 2H, J = 7.0 Hz, CH2S), 3.12 (m, 2H, CH2N), 3.47 (m,160H, 80CH2O) 3.65 (t, 2H, J = 6.0Hz, CH2C), 4.51 (t, 2H, J = 6.0 Hz,CH2O), 7.19�7.32 ppm (m, 15H, 15ArH). (It is expected that the FAresonances are indistinguishable from the baseline since the 1H NMRspectrum only revealed resonances corresponding to PEG-S-Trt.) TheTrt protecting group was removed by dissolving the FA-PEG-S-Trt(15 mg) in TFA (2.0 mL) to afford a clear yellow solution that turnedcolorless when the deprotection was complete by the dropwise additionof triethylsilane. The product FA-PEG3000-SH (containing 70% un-functionalized HO-PEG3000-SH) was isolated by precipitation intodiethyl ether (30 mL), followed by centrifugation and drying in vacuo(10 mg) for 1 h. This was used immediately for subsequent reactions toprevent oxidation of the thiols.Synthesis of Thiol PEG (MeO-PEG2000-SH). Thiolated PEG
without folic acid conjugated was synthesized by acid-catalyzed ester-ification. MeO-PEG2000-OH (3.50 g, 1.75 mmol) was heated to 90 �Cunder argon, and then 3-mercaptopropanoic acid (0.24 g, 2.28 mmol)and H2SO4 (0.034 g, 0.35 mmol) were added. The reaction mixture wasstirred for 16 h, cooled to room temperature, and diluted with DCM(80 mL). The solution was washed with 1:1 saturated NaHCO3:H2O(80 mL� 5) and saturated NaCl (80 mL), dried (MgSO4), filtered, andconcentrated in vacuo to afford MeO-PEG2000-SH, 3.0 g (80%). MAL-DI-ToF MS: Mw = 2022, PDI = 1.01. 1H NMR (400 MHz, CDCl3)δH 2.66 (t, 2H, J = 6.0 Hz, CH2CO), 2.75 (q, 2H, J = 6.0 Hz, CH2S),3.37 (s, 3H, CH3O), 3.49�3.77 (m, 90H, 45CH2O), 4.28 ppm (t, 2H,J = 4.0 Hz, CH2O).Synthesis of Poly(Z-L-lysine)armpoly(L-cystine)core CCS Poly-
mer 1. The method used was similar to that previously reported in theliterature with slight modification.25 Briefly, lys NCA (1.00 g, 3.26 mmol)was dissolved in anhydrous DMF (10 mL) in a flame-dried, argon-purgedtwo-necked flask.N-(Trimethylsilyl)allylamine (M/I = 40, 13.7 μL, 0.0817mmol) was added, and themixturewas stirred at room temperature for 12 hto afford poly(Z-L-lysine) (PZLL) (Mw = 9.67 kDa, PDI = 1.12). CysNCA
(MI/CL = 30, 0.716 g, 2.45 mmol) was added, and the mixture was stirredfor a further 6 h to afford the crude CCS polymer (Mw = 358 kDa, PDI =1.73, f = 22). This solution was added to AMP (0.283 g, 2.35 mmol)dissolved in DMF (20 mL), stirred for 3 h, and then precipitatied inmethanol (250mL). The precipitate was isolated via centrifugation, washedwith chloroform (100mL) andTHF (100mL), and dried in vacuo to affordCCS polymer 1, 1.05 g (ca. 70%). GPC-MALLS:Mw = 668.3 kDa, PDI =1.83, f = 36. 1H NMR (400 MHz, d6-DMSO) δH 1.10�2.06 (m, CH2),3.01 (br s, CH2N), 3.72�4.31 (m, CHN), 5.03 (br s, CH2O), 7.01�7.50ppm (m, ArH). Refer to Supporting Information for calculation of f.Synthesis of PEGylated Poly(PEG-b-Z-L-lysine)armpoly(L-
cystine)core CCS Polymers 2PEG and 2PEG-FA. In two 4 mL vialsfitted with stirrer bars, CCS polymer 1 (20 mg, 29.9 nmol) was dissolvedin DMF (3 mL). To one, MeO-PEG2000-SH (2.5 equiv per arm, 5.26mg, 2.52 μmol) was added, and to the other, FA-PEG3000-SH (2.5 equivper arm, 7.57 mg, 2.52 μmol) was added. Photoinitiator DMPA (0.32mg, 1.26 μmol) was added to each vial, the vials were sealed, and thereaction mixtures were stirred under UV irradiation (λ = 256 nm) for3 h. The resulting solutions were precipitated into 1:1 ethyl acetate:diethyl ether (50mL) and the precipitates isolated via centrifugation anddried in vacuo to afford PEGylated CCS polymers 2PEG (18 mg, 81%)and 2PEG-FA (16 mg, 68%), respectively. GPC MALLS: (2PEG) Mw =710.2 kDa, PDI = 1.85, f = 36; (2PEG-FA)Mw = 811.5 kDa, PDI = 1.75,f = 36. 1H NMR (400 MHz, d6-DMSO, identical spectra were obtainedfor both 2PEG and 2PEG-FA) δH 1.23�1.86 (m,CH2), 2.91 (br s, CH2N),3.46 (br s, CH2O), 3.72�4.26 (m, CHN), 4.96 (br s, CH2O),6.99�7.32 ppm (m, ArH).Synthesis of Poly(PEG-b-L-lysine)armpoly(L-cystine)core CCS
Polymers 3PEG and 3PEG-FA. Separately, CCS polymers 2PEG and2PEG-FA (10 mg) were dissolved in TFA (50 μL), and 33% HBr in aceticacid was then added (200 μL). After stirring for 1 h at room temperaturethe mixture was precipitated into diethyl ether (2.5 mL). The residuewas isolated via centrifugation, redissolved in water (2 mL), andprecipitated into THF (30 mL); this step was repeated three timesand the residue dried in vacuo (0.1 mbar) to afford CCS polymers 3PEG(5.2 mg) and 3PEG-FA (3.2 mg) as off-white solids. 1H NMR (400 MHz,D2O, identical spectra were obtained for both 3PEG and 3PEG-FA) δH1.20�1.64 (m,CH2), 2.86 (br s, CH2N), 3.65 (br s, CH2O), 4.18�4.20(m, CHN).Fluorescent Tagging of CCS Polymers 3PEG and 3PEG-FA
with Alexa Fluor 647. The CCS polymers 3PEG and 3PEG-FA weredissolved in water (0.5 mg/mL), and Alexa Fluor 647 carboxylic acidsuccinimidyl ester dissolved in DMSO (1 mg/mL) was added (2.5 μL/mgof polymer). The mixtures were stirred for 2 h at room temperatureand then precipitated into THF (10 times the reaction volume). The
Scheme 1. Synthesis of PEG�Folic Acid Conjugate via DCC-Mediated Coupling
3472 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
resulting solid was isolated via centrifugation and dried in vacuo to affordthe fluorescently tagged derivatives.Cell Culture. MDA-MB-231 human breast cancer cells were
cultured in Dulbecco’s modified Eagle’s medium supplemented withpenicillin (100 units/mL), streptomycin (100 mg/mL), fetal bovineserum (10% v/v), and 2 mM L-glutamine at 37 �C, 5% CO2, and 95%relative humidity. Confluent cultures were rinsed in Dulbecco’s phos-phate buffered saline (DPBS) and split (1:3) every 3 days using trypsin�ethylenediaminetetraacetic acid (EDTA) (1� liquid, 1 mL per T25 flask).Cellular Uptake of CCS Polymers 3PEG and 3PEG-FA. For
confocal microscopy experiments, sterilized glass coverslips were placedin a 6-well tissue culture plate and coated with fibronectin from bovineplasma (10 μg/mL in PBS) for 1 h. The excess fibronectin was removedby washing with PBS. MDA-MB-231 cells were detached from cultureusing trypsin�EDTA and seeded onto the coated coverslips at a densityof 10 000 cells/well and then allowed to attach at 37 �C for 1 h. The cellswere then treated with 50 μL of 3PEG and 3PEG-FA polymer solutions(1 mg/mL in PBS), and the polymers were incubated with the cells for3 h at 37 �Cand 5%CO2. The coverslips were gently washedwithwarmedDPBS then fixed in 4% w/v paraformaldehyde in PBS for 20min. The cellmembrane was stained with AlexaFluor 488 conjugated wheat germagglutinin overnight. Coverslips were washed with PBS and then mountedonto glass slides using a DAPI-doped glycerol antifade medium.
For flow cytometry experiments, the cells were seeded directly in a6-well culture plate and treated with the star polymers as described forthe confocal microscopy experiments. Following an incubation period of3 h, the cells were detached from the plate using trypsin�EDTA (100μL),pelleted at 500g for 5 min, and then resuspended in PBS (1 mL) and lefton ice. Samples were analyzed by flow cytometry performed on a CyflowSpace flow cytometer using an excitation wavelength of λ = 633 nm.10 000 counts were collected for each run. Forward and side scatter datawere used to fix a gate around live cell population. This gate was usedto identify the population which demonstrated a higher fluorescenceintensity compared to the cells alone.Measurement of Cell Cytotoxicity. Breast cancer cells (MDA-
MD-231) were detached from the culture vessel using trypsin�EDTA,and the cell density was adjusted to 60 000 cells/mL with DMEM. A 96-well plate was then seeded with the cells in quadruplicate (3000 cells/well) and left at 37 �C for 30 min before addition of the star polymers.For concentration-dependent toxicity testing different concentrations(0, 2.5, 5.0, 12.5, 5.0, and 100 μg/mL) of star polymers 3PEG and 3PEG-FAwere added to the cells and incubated for 72 h. For time-course studiescells were incubated with polymers at 5.0 μg/mL for 24, 48, and 72 h,where each time point was conducted on a separate plate. After theindicated time period MTS assays were conducted according to themanufacturers protocol. The cells were incubated with theMTS solutionfor 3 h followed by measurement of absorbance at λ = 492 nm in amicrotiter plate reader. A student t test was performed to determinestatistical significance of differences between treatments (p < 0.05).Each experiment was repeated on two separate occasions, and cellsthat were untreated or lysed with 2 μL of Triton X-100 prior to theaddition of MTS solution were used as controls for live and dead cells,respectively.
’RESULTS AND DISCUSSION
Synthesis of Folic Acid�PEGConjugate.Thiolated and folicacid (FA) conjugated PEG (FA-PEG3000-SH) was synthesizedvia reaction of HO-PEG3000-S-Trt with FA using DCC-mediatedcoupling, followed by deprotection of the trityl (Trt) group. Ithas previously been demonstrated that the coupling reactionoccurs predominately between the hydroxyl group of the PEGand the γ-carboxylic acid of FA,33,34 which allows the conjuga-ted FA to maintain a high affinity toward folate receptors on
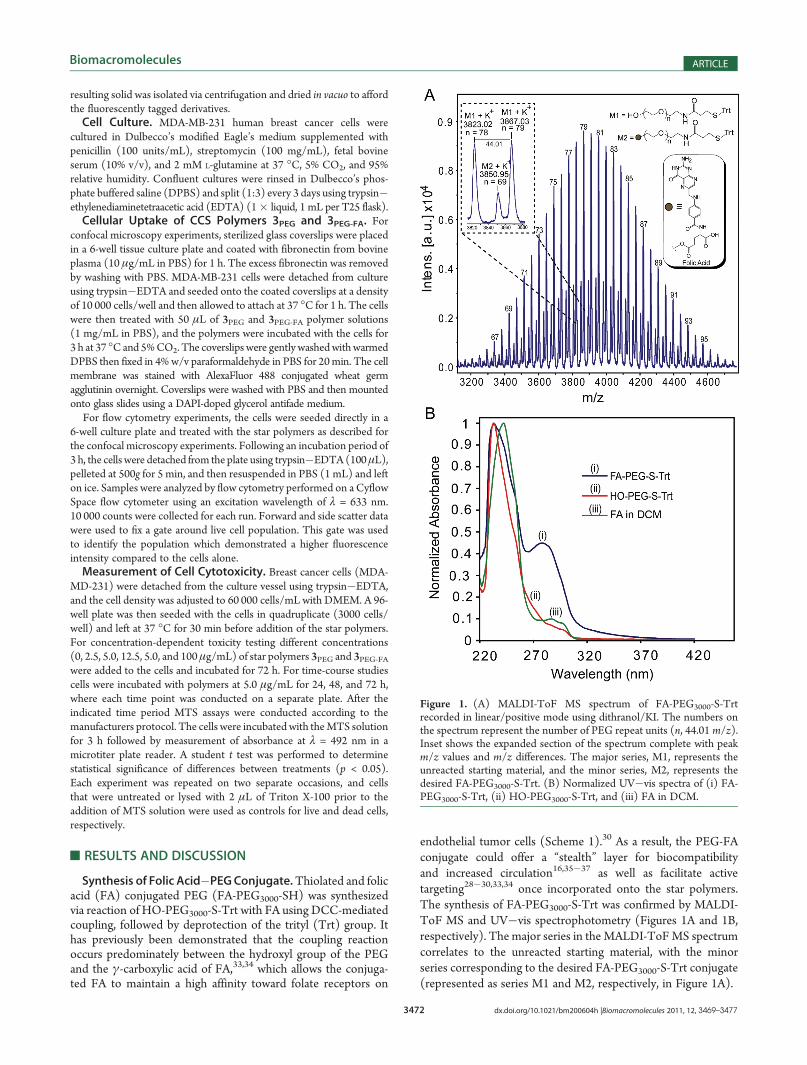
endothelial tumor cells (Scheme 1).30 As a result, the PEG-FAconjugate could offer a “stealth” layer for biocompatibilityand increased circulation16,35�37 as well as facilitate activetargeting28�30,33,34 once incorporated onto the star polymers.The synthesis of FA-PEG3000-S-Trt was confirmed by MALDI-ToF MS and UV�vis spectrophotometry (Figures 1A and 1B,respectively). The major series in the MALDI-ToFMS spectrumcorrelates to the unreacted starting material, with the minorseries corresponding to the desired FA-PEG3000-S-Trt conjugate(represented as series M1 and M2, respectively, in Figure 1A).
Figure 1. (A) MALDI-ToF MS spectrum of FA-PEG3000-S-Trtrecorded in linear/positive mode using dithranol/KI. The numbers onthe spectrum represent the number of PEG repeat units (n, 44.01 m/z).Inset shows the expanded section of the spectrum complete with peakm/z values and m/z differences. The major series, M1, represents theunreacted starting material, and the minor series, M2, represents thedesired FA-PEG3000-S-Trt. (B) Normalized UV�vis spectra of (i) FA-PEG3000-S-Trt, (ii) HO-PEG3000-S-Trt, and (iii) FA in DCM.

3473 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
The normalized UV�vis spectra of the starting materials(HO-PEG3000-S-Trt and FA) and resulting product (FA-PEG3000-S-Trt) in DCM (Figure 1B) revealed an absorbanceat λ = 280 nm and a broadening of the peak between λ =220�270 nm for the FA-PEG3000-S-Trt, which matches the FAabsorbance profile. Additionally, pure FA is only slightly solublein DCM and displays a low absorbance at λ = 280 nm corre-sponding to the aromatic chromophore. Therefore, the morepronounced peak at λ = 280 nm for the product confirms thesuccessful incorporation of FA and synthesis of FA-PEG3000-S-Trt.9 From both MALDI-ToF MS and UV�vis spectrophoto-metry, the extent of FA conjugation was calculated to be ca. 30%,which was deemed sufficient to provide active targeting oncecoupled to the star polymer.38,39 Thus, the mixture of FAfunctionalized and unfunctionalized PEG was not further pur-ified and was used directly for PEGylation of the CCS polymer(vide infra). 1H NMR spectroscopic analysis was also used tocharacterize the product; however, due to the low ratio of FAend-groups to the main chain repeat units, the FA protonresonances could not be distinguished from the baseline. TheTrt protecting group was subsequently removed by reaction ofFA-PEG3000-S-Trt with triethylsilane in TFA to afford thedesired thiolated derivative, FA-PEG3000-SH (also containing70% unfunctionalized HO-PEG3000-SH), which was used for thesubsequent reaction immediately after isolation to preventoxidation. In order to confirm the deprotection of the Trt group,1H NMR spectroscopy was performed on a separate sample ofHO-PEG3000-S-Trt, which revealed the successful removal of theTrt protecting groups when subjected to the same deprotectionconditions (Supporting Information, Figure S1).40
For control experiments a thiolated PEG derivative (MeO-PEG2000-SH) without conjugated FA was synthesized by reacting
MeO-PEG2000-OHwith 3-mercaptopropanoic acid. The absence ofthe FA targeting moiety on this PEG derivative allowed thedifference in the in vitro cellular interaction between star polymerswith and without FA to be studied. The synthesis ofMeO-PEG2000-SH was confirmed via MALDI-ToF MS and 1H NMR spectro-scopic analysis (Supporting Information Figures S2A and S2B,respectively), which revealed quantitative conversion.Synthesis of CCS Polymers 1.The synthesis of star polymer 1
was achieved via ROP of amino acid NCAs using an arm-first,one-pot strategy as outlined in Scheme 2. The ROP of lys NCAusing N-(trimethylsilyl)allylamine as the initiator yielded livingPZLL (Mw = 9.67 kDa, PDI = 1.12), which served as themacroinitiator (MI) for star formation (Figure 2A, PZLL).Subsequent addition of the cross-linker (CL) cys NCA([CL]/[MI] = 30) afford an intermediate star polymer, havingaMw of 358 kDa, PDI of 1.73, and f (average number of arms) of22. The GPC differential refractive index (DRI) chromatogramof the intermediate (Figure 2A, intermediate) revealed a slightshoulder to the right of the star peak, with a retention time similarto the precursor PZLL (ca. 22.5 min), indicating the presence ofsome unincorporated PZLL derivatives.From the authors’ previous studies,25 it was determined that
some pendant NCAs remained unreacted in the core after starformation, which allowed for postpolymerization functionaliza-tion with aminomethylpyrene (AMP) to afford CCS polymer 1.After isolation in MeOH and washing with chloroform and THFto remove the excess AMP, GPC analysis revealed an increase inMw to 668.3 kDa (PDI = 1.89, f = 36) (Figure 2A, 1).Determination of the pyrene loading via UV�vis spectrophoto-metry provided a value of 261 mol/mol star, which equates toan increase in molecular weight of ca. 60 kDa. Therefore, theincrease in the Mw observed by GPC (ca. 300 kDa) can be
Scheme 2. Synthesis of Amino Acid-Based CCS Polymers via a One-Pot, Arm-First Strategy

3474 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
attributed mainly to the occurrence of star�star coupling duringthe functionalization and isolation process.25 The disappearanceof the shoulder observed in the GPC DRI chromatogram of theintermediate (Figure 2A, intermediate) indicated that the freePZLL derivatives (∼22.5 min) had either been incorporated intothe star or removed during the washing procedure with THF, inwhich PZLL was slightly soluble.DLS analysis was performed at each reaction stage to follow
the change in the hydrodynamic radius (dH) of the polymers(Figure 2B). The macroinitiator PZLL was found to have a dH of8.4 nm, which increased to 86 nm upon formation of theintermediate star. DLS analysis of the intermediate star alsoconfirmed the presence of some unincorporated poly(Z-L-lysine-b-L-cystine) (P(ZLL-b-LC)) copolymer, with a dH of 15 nm. Afterpostpolymerization functionalization with AMP the dH of star 1was determined to be 110 nm, and unincorporated P(ZLL-b-LC)was no longer observed, which correlates well with the GPCresults.PEGylation of CCS Polymer 1. The peripheral allyl groups of
star 1 derived from the amine initiator used for the ROP of aminoacid NCAs and star formation25,26 allowed for facile functiona-lization of the stars via thiol�ene click chemistry (Scheme 2).Since star 1 is composed entirely of peptide chains, it would beprone to bloodstream enzymatic degradation, plasma opsoniza-tion, and monocyte phagocytic system (MPS) uptake, which isundesirable for targeted drug delivery vehicles. PEGylation is awidely used technique to shield therapeutic agents from thehost’s immune system and was employed in this study to protectthe stars against rapid degradation into their amino acid con-stituents and prolong their circulation time in the bloodstream.Two types of PEGylated stars, 2PEG and 2PEG-FA, were synthe-sized via thiol�ene click of star 1 with thiolated PEG derivatives
MeO-PEG-SH and FA-PEG3000-SH, respectively, in the pre-sence of a photoinitiator, where the former does not possesstargeting capabilities and the latter has 30% FA-conjugated fortargeted drug delivery.GPC DRI chromatograms of both stars 2PEG and 2PEG-FA
revealed retention times higher than those of the precursorstar 1 (Figure 2A, 2PEG and 2PEG-FA, respectively). The dn/dcvalues of densely branched CCS polymers and linear polymersof the same monomeric constitution have been reported to becomparable.41 Therefore, using the dn/dc value calculated forPEGylated linear PZLL (dn/dc = 0.065 mL/g), GPC analysisprovided increases in the molecular weight for both stars 2PEGand 2PEG-FA. For 2PEG theMw was determined to be 710.2 kDa(PDI = 1.85), which correlates to ca. 58% PEGylation of thearms, whereas for 2PEG-FA the Mw of 811.5 kDa (PDI = 1.75)correlated to >99% PEGylation. The slight peak shift to higherretention time might result from changes in the physicalproperties of the star polymers after PEGylation, which affectstheir interaction with the GPC columns. The appearance ofpeaks between 20 and 25 min was attributed to the unreactedexcess PEG, and their dimerized derivatives formed as a resultof disulfide bond formation. However, the extent of PEGyla-tion calculated from 1H NMR spectroscopic analysis providedvalues of ca. 30 and 50% for 2PEG and 2PEG-FA, respectively(Supporting Information, Figure S3). The discrepanciesbetween the GPC and 1H NMR spectroscopy calculations areexpected to arise from a combination of resonance broadening
Figure 2. (A) GPCDRI chromatograms and (B) DLS traces in DMF ofPZLL macroinitiator, intermediate CCS polymer prior to core functio-nalization with AMP, CCS polymer 1, and PEGylated CCS polymers2PEG and 2PEG-FA. Each DLS result is the average of at least threeindependent measurements.
Figure 3. MALDI-ToF MS spectrum of control degradation experi-ment using FA-PEG3000-S-Trt in HBr, recorded in linear/positivemode using dithranol/KI. The numbers on the spectrum representthe number of PEG repeat units (n, 44.01 m/z). Inset shows theexpanded section of MS spectrum complete with peak m/z values andm/z differences. The major series, M10 and M20, represent the startingmaterial and PEG-FA conjugate without the Trt protecting groups,respectively, and the minor series M1 represents the unreacted startingmaterial.
3475 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
and overlap in the NMRmeasurements and the dn/dc value usedin GPC calculations, which assumes that all of the star’s arms arePEGylated. DLS analysis also revealed increases in size afterPEGylation, with dH values of 238 and 262 nm for stars 2PEG and2PEG-FA, respectively (Figure 2B). This marked increase afterPEGylation may result from aggregation.42 However, this sizeregime ensures that the drug delivery vehicles will not be able toenter the healthy/normal vasculature but only to the “leaky”tumor vasculature due to enhanced permeability and retention(EPR) effects, which provides passive targeting abilities inaddition to the active targeting provided by the conjugated folicacid moieties.Deprotection of PEGylated CCS Polymers 2.The removal of
the pendent Cbz protecting groups along the arms of stars 2PEG and2PEG-FA was achieved using HBr to afford water-soluble CCSpolymers 3PEG and 3PEG-FA, respectively, with pendent aminefunctionalities available along the arms. 1H NMR spectroscopicanalysis revealed ca. 80% removal of the protecting groups for both3PEG and 3PEG-FA (Supporting Information, Figure S4). To ensurethat the conjugated FA present on 3PEG-FA was stable during theacid-mediated deprotection, a control experiment was conducted byexposing FA-PEG3000-S-Trt to HBr for 6 h, which was longer thanthe time required to deprotect star 2PEG-FA (1 h). MALDI-ToFMSof the control experiment after 6 h showed that the conjugated FAremained intact during the process despite the fact that 85% of theTrt protecting groups were removed (Figure 3).Cellular Uptakeof CCSPolymers 3PEG and3PEG-FA.Confocal
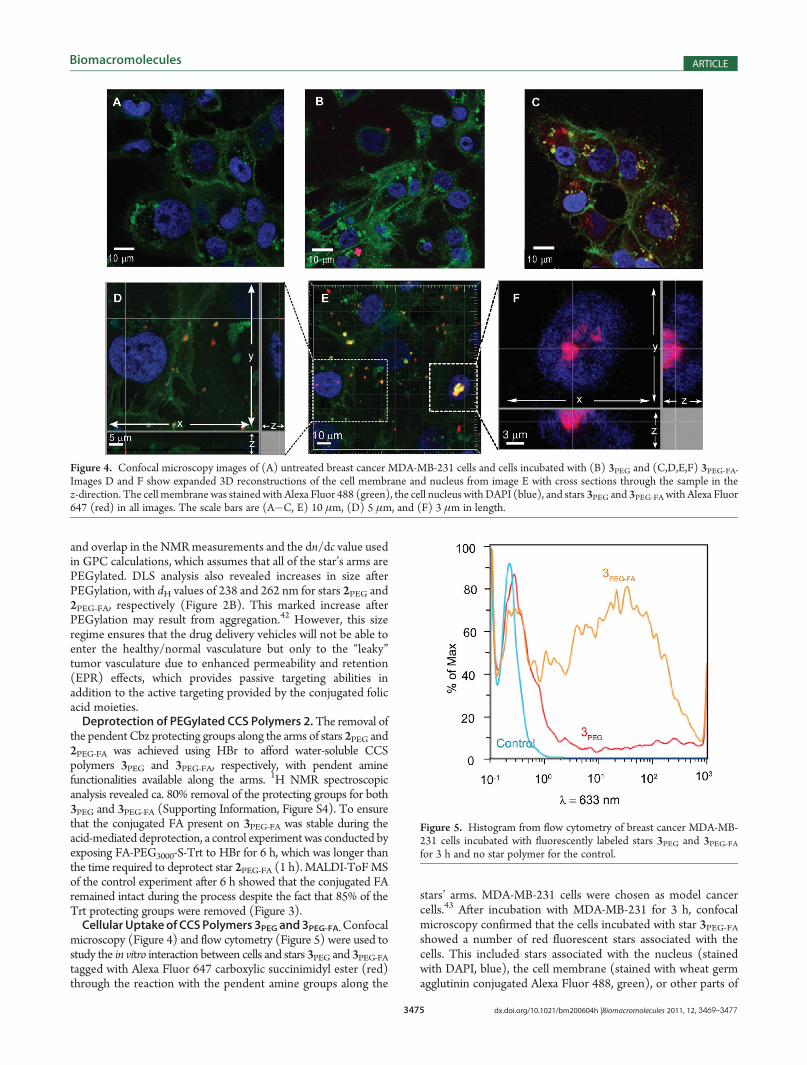
microscopy (Figure 4) and flow cytometry (Figure 5) were used tostudy the in vitro interaction between cells and stars 3PEG and 3PEG-FAtagged with Alexa Fluor 647 carboxylic succinimidyl ester (red)through the reaction with the pendent amine groups along the
stars’ arms. MDA-MB-231 cells were chosen as model cancercells.43 After incubation with MDA-MB-231 for 3 h, confocalmicroscopy confirmed that the cells incubated with star 3PEG-FAshowed a number of red fluorescent stars associated with thecells. This included stars associated with the nucleus (stainedwith DAPI, blue), the cell membrane (stained with wheat germagglutinin conjugated Alexa Fluor 488, green), or other parts of
Figure 4. Confocal microscopy images of (A) untreated breast cancer MDA-MB-231 cells and cells incubated with (B) 3PEG and (C,D,E,F) 3PEG-FA.Images D and F show expanded 3D reconstructions of the cell membrane and nucleus from image E with cross sections through the sample in thez-direction. The cell membrane was stained with Alexa Fluor 488 (green), the cell nucleus with DAPI (blue), and stars 3PEG and 3PEG-FA with Alexa Fluor647 (red) in all images. The scale bars are (A�C, E) 10 μm, (D) 5 μm, and (F) 3 μm in length.
Figure 5. Histogram from flow cytometry of breast cancer MDA-MB-231 cells incubated with fluorescently labeled stars 3PEG and 3PEG-FAfor 3 h and no star polymer for the control.
3476 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
the cell suggesting cellular association and uptake of the starpolymer 3PEG-FA (Figure 4C,E). The 3D reconstructions inFigure 4D,F show expanded 3D views of the nucleus and areaaround the nucleus for two cells highlighted in Figure 4E andclearly confirm that some of the 3PEG-FA appear within thecross section of the cell (Figure 4D and Supporting Informa-tion Figure S5) as well as the cell nucleus (Figure 4F). Incomparison, the cells incubated with 3PEG had considerablyfewer fluorescent stars (Figure 4B), and these appearrestricted to the membrane within 3D reconstructions of thecell (Supporting Information Figure S6). This result was ex-pected since star 3PEG did not possess a targeting ligand, andtherefore interactions with the cells were nonspecific comparedto the receptor mediated attachment and internalization of star3PEG-FA.Flow cytometry was used to confirm and quantify the
association MDA-MB-231 cells with Alexa Fluor 647-labeledstars. Figure 5 shows that after 3 h coincubation only 13% ofcells incubated with 3PEG were associated with these particlescompared to 55% for cells incubated with 3PEG-FA (Figure 5),consistent with observations by confocal microscopy. Thehistograms show a broad distribution of the Alexa Fluor 647-positive cells most likely as a result of uneven distribution of
the stars across all cells due to aggregation (also evident fromthe confocal images (Figure 4C�F)), which has been pre-viously observed even in PEGylated systems.42 Nevertheless,these results further confirm that star 3PEG-FA showed pre-ferential attachment to the cells due to the presence ofperiphery conjugated FA, and a greater number of these starpolymers appear internalized.Cell Cytotoxicity Study.The viability of both stars 3PEG and
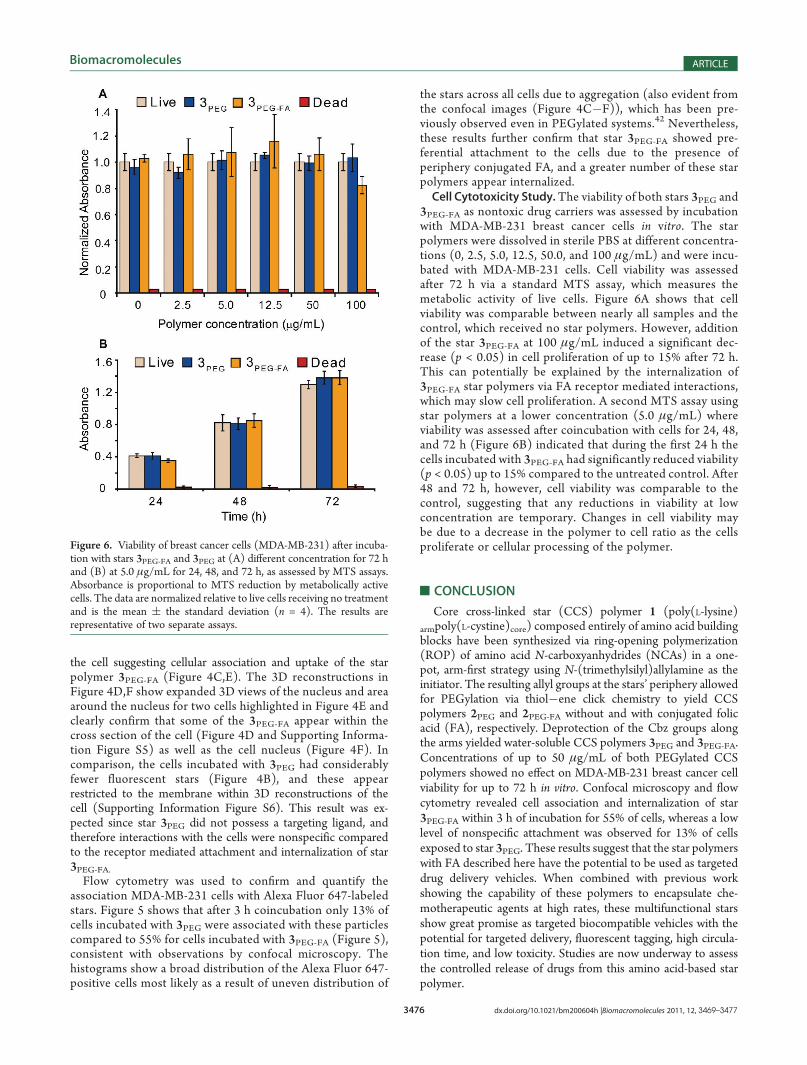
3PEG-FA as nontoxic drug carriers was assessed by incubationwith MDA-MB-231 breast cancer cells in vitro. The starpolymers were dissolved in sterile PBS at different concentra-tions (0, 2.5, 5.0, 12.5, 50.0, and 100 μg/mL) and were incu-bated with MDA-MB-231 cells. Cell viability was assessedafter 72 h via a standard MTS assay, which measures themetabolic activity of live cells. Figure 6A shows that cellviability was comparable between nearly all samples and thecontrol, which received no star polymers. However, additionof the star 3PEG-FA at 100 μg/mL induced a significant dec-rease (p < 0.05) in cell proliferation of up to 15% after 72 h.This can potentially be explained by the internalization of3PEG-FA star polymers via FA receptor mediated interactions,which may slow cell proliferation. A second MTS assay usingstar polymers at a lower concentration (5.0 μg/mL) whereviability was assessed after coincubation with cells for 24, 48,and 72 h (Figure 6B) indicated that during the first 24 h thecells incubated with 3PEG-FA had significantly reduced viability(p < 0.05) up to 15% compared to the untreated control. After48 and 72 h, however, cell viability was comparable to thecontrol, suggesting that any reductions in viability at lowconcentration are temporary. Changes in cell viability maybe due to a decrease in the polymer to cell ratio as the cellsproliferate or cellular processing of the polymer.
’CONCLUSION
Core cross-linked star (CCS) polymer 1 (poly(L-lysine)armpoly(L-cystine)core) composed entirely of amino acid buildingblocks have been synthesized via ring-opening polymerization(ROP) of amino acid N-carboxyanhydrides (NCAs) in a one-pot, arm-first strategy using N-(trimethylsilyl)allylamine as theinitiator. The resulting allyl groups at the stars’ periphery allowedfor PEGylation via thiol�ene click chemistry to yield CCSpolymers 2PEG and 2PEG-FA without and with conjugated folicacid (FA), respectively. Deprotection of the Cbz groups alongthe arms yielded water-soluble CCS polymers 3PEG and 3PEG-FA.Concentrations of up to 50 μg/mL of both PEGylated CCSpolymers showed no effect on MDA-MB-231 breast cancer cellviability for up to 72 h in vitro. Confocal microscopy and flowcytometry revealed cell association and internalization of star3PEG-FA within 3 h of incubation for 55% of cells, whereas a lowlevel of nonspecific attachment was observed for 13% of cellsexposed to star 3PEG. These results suggest that the star polymerswith FA described here have the potential to be used as targeteddrug delivery vehicles. When combined with previous workshowing the capability of these polymers to encapsulate che-motherapeutic agents at high rates, these multifunctional starsshow great promise as targeted biocompatible vehicles with thepotential for targeted delivery, fluorescent tagging, high circula-tion time, and low toxicity. Studies are now underway to assessthe controlled release of drugs from this amino acid-based starpolymer.
Figure 6. Viability of breast cancer cells (MDA-MB-231) after incuba-tion with stars 3PEG-FA and 3PEG at (A) different concentration for 72 hand (B) at 5.0 μg/mL for 24, 48, and 72 h, as assessed by MTS assays.Absorbance is proportional to MTS reduction by metabolically activecells. The data are normalized relative to live cells receiving no treatmentand is the mean ( the standard deviation (n = 4). The results arerepresentative of two separate assays.

3477 dx.doi.org/10.1021/bm200604h |Biomacromolecules 2011, 12, 3469–3477
Biomacromolecules ARTICLE
’ASSOCIATED CONTENT
bS Supporting Information. Figures S1�S6. This materialis available free of charge via the Internet at http://pubs.acs.org.
’AUTHOR INFORMATION
Corresponding Author*E-mail: [email protected]. Tel: +61 3 8344 8665. Fax:+61 3 8344 4153.
’ACKNOWLEDGMENT
The authors thank Dr. Angus Johnston and Ms. Sher Leen Ngfor their help with confocal microscopy and flow cytometry. A.S.is a recipient of CSIRO Food Future Flagship Scholarship. TheMALDI ToF MS used in these studies was supported under theAustralian Research Council’s Linkage Infrastructure, Equipmentand Facilities (LIEF) funding scheme (LE0882576).
’REFERENCES
(1) Murakami, M.; Cabral, H.; Matsumoto, Y.; Wu, S.; Kano, M. R.;Yamori, T.; Nishiyama, N.; Kataoka, K. Sci. Transl. Med. 2011, 64, 64ra2.(2) Zhou, H.; Yu, W.; Guo, X.; Liu, X.; Li, N.; Zhang, Y.; Ma, X.
Biomacromolecules 2010, 11, 3480–3486.(3) Vicent, M. J.; Duncan, R. Trends Biotechnol. 2006, 24, 39–47.(4) Maeda, H.; Greish, K.; Fang, J. Adv. Polym. Sci. 2006, 193,
103–121.(5) Kataoka, K.; Harada, A.; Nagasaki, Y. Adv. Drug Delivery Rev.
2001, 47, 113–131.(6) Sun, H.; Guo, B.; Cheng, R.; Meng, F.; Liu, H.; Zhong, Z.
Biomaterials 2009, 30, 6358–6366.(7) Ryu, J.; Roy, R.; Ventura, J.; Thayumanavan, S. Langmuir 2010,
26, 7086–7092.(8) Chan, Y.; Wong, T.; Byrne, F.; Kavallaris, M.; Bulmus, V.
Biomacromolecules 2008, 9, 1826–1836.(9) Pan, D.; Turner, J. L.; Wooley, K. L. Chem. Commun. 2003,
2400–2401.(10) Nystroem, A. M.; Xu, Z.; Xu, J.; Taylor, S.; Nittis, T.; Stewart,
S. A.; Leonard, J.; Wooley, K. L. Chem. Commun. 2008, 3579–3581.(11) Johnston, A. P. R.; Cortez, C.; Angelatos, A. S.; Caruso, F. Curr.
Opin. Colloid Interface Sci. 2006, 11, 203–206.(12) Liu, M.; Kono, K.; Fr�echet, J. M. J. J. Controlled Release 2000,
65, 121–131.(13) Cho, H. Y.; Gao, H.; Srinivasan, A.; Hong, J.; Bencherif, S. A.;
Siegwart, D. J.; Paik, H.; Hollinger, J. O.; Matyjaszewski, K. Biomacro-molecules 2010, 11, 2199–2203.(14) Wiltshire, J.; Qiao, G. Aust. J. Chem. 2007, 60, 699–705.(15) Schramm, O. G.; Meier, M. A. R.; Hoogenboom, R.; Erp, H. P.;
Gohy, J. F.; Schubert, U. S. Soft Matter 2009, 5, 1662–1667.(16) Kojima, C.; Kono, K.; Maruyama, K.; Takagishi, T. Bioconjugate
Chem. 2000, 11, 910–917.(17) Ho, A.; Gurr, P.; Mills, M.; Qiao, G. Polymer 2005, 46,
6727–6735.(18) Goh, T. K.; Coventry, K.; Blencowe, A.; Qiao, G. Polymer 2008,
49, 5095–5104.(19) Blencowe, A.; Goh, T. K.; Best, S.; Qiao, G. Polymer 2008,
49, 825–830.(20) Wiltshire, J.; Qiao, G. Macromolecules 2008, 41, 623–631.(21) Helms, B.; Guillaudeu, S. J.; Xie, Y.; McMurdo, M.; C. J.
Hawker, C. J.; Fr�echet, J. M. J. Angew. Chem., Int. Ed. 2005, 44,6384–6387.(22) Spiniello, M.; Blencowe, A.; Qiao, G. J. Polym. Sci., Polym. Chem.
2008, 46, 2422–2432.(23) Blencowe, A.; Tan, J. F.; Goh, T. K.; Qiao, G. Polymer 2009,
50, 5–32.
(24) Liu, Z.; Cheng, R.; Meng, F.; Cui, J.; Ji, S.; Zhong, Z. Angew.Chem., Int. Ed. 2009, 48, 9914–9918.
(25) Sulistio, A.; Widjaya, A.; Blencowe, A.; Zhang, X.; Qiao, G.Chem. Commun. 2011, 47, 1151–1153.
(26) Lu, H.; Cheng, J. J. Am. Chem. Soc. 2008, 130, 12562–12563.(27) Matesic, L.; Locke, J. M.; Vine, K. L.; Ranson, M.; Bremner,
J. B.; Skropeta, D. Bioorg. Med. Chem. 2011, 19, 1771–1778.(28) Lu, Y.; Low, P. S. Adv. Drug Delivery Rev. 2002, 54, 675–693.(29) Ross, J. F.; Chaudhuri, P. K.; Ratnam, M. Cancer 1994, 73,
2432–2443.(30) Sudimack, J.; Lee, R. J. Adv. Drug Delivery Rev. 2000, 41,
147–162.(31) Weitman, S. D.; Lark, R. H.; Coney, L. R.; Fort, D. W.; Frasca,
V.; Zurawski, V. R., Jr.; Kamen, B. A. Cancer Res. 1992, 52, 3396–3401.(32) Canal, F.; Vicent, M. J.; Pasut, G.; Schiavon, O. J. Controlled
Release 2010, 146, 388–399.(33) Chen, S.; Zhang, X. Z.; Cheng, S. X.; Zhuo, R. X.; Gu, Z. W.
Biomacromolecules 2008, 9, 2578–2585.(34) Saul, J. M.; Annapragada, A.; Natarajan, J. V.; Bellamkonda,
R. V. J. Controlled Release 2003, 92, 49–67.(35) Greenwald, R. B.; Conover, C. D.; Choe, Y. H. Crit. Rev. Ther.
Drug Carrier Syst. 2000, 17, 101–161.(36) Greenwald, R. B.; Choe, Y. H.;McGuire, J.; Conover, C. D.Adv.
Drug Delivery Rev. 2003, 55, 217–250.(37) Berna, M.; Dalzoppo, D.; Pasut, G.; Manunta, M.; Izzo, L.;
Jones, A. T.; Duncan, R.; Veronese, F. M. Biomacromolecules 2006, 7,146–153.
(38) Bae, Y.; Nishiyama, N.; Lataoka, K. Bioconjugate Chem. 2007,18, 1131–1139.
(39) De, P.; Gondi, S. R.; Sumerlin, B. S. Biomacromolecules 2008,9, 1064–1070.
(40) Badyal, J. P.; Cameron, A. M.; Cameron, N. R.; Coe, D. M.;Cox, R.; Davis, B. G.; Oates, L. J.; Oyea, G.; Steel, P. G. Tetrahedron Lett.2001, 42, 8531–8533.
(41) Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K. Macromolecules2005, 38, 9018–9027.
(42) Wenig, L.; Kilcher, G.; Tirelli, N. Macromol. Biosci. 2007, 7,987–998.
(43) Jhaveri, M. S.; Rait, A. S.; Chung, K. N.; Trepel, J. B.; Chang,E. H. Mol. Cancer Ther. 2004, 12, 1505–1512.




















