
Fibroblasts in cancer
10
*Center for Matrix Biology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts 02215, USA. ‡ Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02215, USA. § Harvard-MIT Division of Health Sciences and Technology, Boston, Massachusetts 02215, USA. Correspondence to R.K. e-mail: rkalluri@BIDMC. Harvard.edu doi:10.1038/nrc1877 Published online 30 March 2006 Stroma Extracellular matrix, mesenchymal cells and a scaffold consisting of blood and lymphoid vessels, nerves and inflammatory cells. Cancer-associated fibroblast Activated fibroblasts within desmoplastic lesions that are associated with malignant tumours and often express α- smooth-muscle actin. Fibroblasts in cancer Raghu Kalluri* ‡§ and Michael Zeisberg* Abstract | Tumours are known as wounds that do not heal — this implies that cells that are involved in angiogenesis and the response to injury, such as endothelial cells and fibroblasts, have a prominent role in the progression, growth and spread of cancers. Fibroblasts are associated with cancer cells at all stages of cancer progression, and their structural and functional contributions to this process are beginning to emerge. Their production of growth factors, chemokines and extracellular matrix facilitates the angiogenic recruitment of endothelial cells and pericytes. Fibroblasts are therefore a key determinant in the malignant progression of cancer and represent an important target for cancer therapies. Genetic and cell-biology studies indicate that tumour growth is not just determined by malignant cancer cells themselves, but also by the tumour stroma 1 . Tumour progression is clearly dependent on angiogenesis 2–5 , and inflammatory cells also contribute to tumour growth 6,7 . It is becoming increasingly clear that fibroblasts 8,9 are also prominent modifiers of cancer progression. Our knowledge of the role of resting and activated fibro- blasts in cancer is still evolving, and evidence is increas- ing that a subpopulation of fibroblasts — the so-called cancer-associated fibroblasts (CAFs) — are important promoters of tumour growth and progression 10 . Fibroblasts were first described in the late 19th century, based on their location and their microscopic appear- ance 11,12 . They are the non-vascular, non-epithelial and non-inflammatory cells of the connective tissue 13 , and are its principal cellular component (BOX 1). They are embed- ded within the fibrillar matrix of the connective tissue and are, to a large extent, responsible for its synthesis. The important functions of fibroblasts include the deposition of extracellular matrix (ECM), regulation of epithelial differentiation, regulation of inflammation, and involvement in wound healing 14,15 . Fibroblasts synthesize many of the constituents of the fibrillar ECM such as type I, type III and type V collagen, and fibro- nectin 14,16 . They also contribute to the formation of basement membranes by secreting type IV collagen and laminin 17 . Fibroblasts are also an important source of ECM-degrading proteases such as matrix metalloproteinases (MMPs), which highlights their crucial role in maintain- ing an ECM homeostasis by regulating ECM turnover 17,18 . In addition, fibroblasts are important in maintaining the homeostasis of adjacent epithelia through the secretion of growth factors and direct mesenchymal–epithelial cell interactions 19 (BOX 2). As well as their function in healthy organs, fibroblasts have a prominent role in wound repair. They invade lesions, generate ECM to serve as a scaffold for other cells, and possess cytoskeletal elements that facilitate contractions of healing wounds 14 . As the principal source of ECM constituents, fibro- blasts are considered the main mediators of scar forma- tion and tissue fibrosis. Fibroblasts that are isolated from the site of a healing wound or from fibrotic tissue secrete higher levels of normal ECM constituents and prolifer- ate more than their normal counterparts isolated from healthy organs 20,21 . Such increased activity is referred to as ‘activation’ 20 (BOX 3; FIG. 1). Once the wound is repaired, the number of activated fibroblasts decreases significantly and the resting phenotype is thought to be restored 14 . It remains unknown whether the activated fibroblasts revert to a resting phenotype, or whether they undergo apoptosis followed by the repopulation of that particular region of the tissue by resting fibroblasts from the adjacent tissue 14 . Unlike wound healing, but similar to organ fibrosis, the fibroblasts at the site of a tumour remain perpetually activated. Are fibroblasts inducers of cancer progression and invasion? Or do they protect neoplastic lesions from becoming invasive carcinomas? What is the contribution of fibroblasts to metastasis? These questions remain largely unanswered, but data from co-culture and reconstitution experiments indicate that fibroblasts have a prominent role in defining the rate and extent of cancer progression. The tumour stroma In the early growth of tumours, cancer cells form a neoplastic lesion that is embedded in the micro- environment of a given tissue (usually epithelium) 22 but separated from the surrounding tissue and con- tained within the boundary of a basement membrane 22 . This is called the carcinoma in situ (CIS). The base- ment membrane, immune cells, capillaries, fibroblasts TUMOUR MICROENVIRONMENT REVIEWS 392 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer © 2006 Nature Publishing Group
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Fibroblasts in cancer

*Center for Matrix Biology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts 02215, USA.‡Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02215, USA.§Harvard-MIT Division of Health Sciences and Technology, Boston, Massachusetts 02215, USA.Correspondence to R.K.e-mail: [email protected]:10.1038/nrc1877Published online 30 March 2006
StromaExtracellular matrix, mesenchymal cells and a scaffold consisting of blood and lymphoid vessels, nerves and inflammatory cells.
Cancer-associated fibroblastActivated fibroblasts within desmoplastic lesions that are associated with malignant tumours and often express α-smooth-muscle actin.
Fibroblasts in cancerRaghu Kalluri*ठand Michael Zeisberg*
Abstract | Tumours are known as wounds that do not heal — this implies that cells that are involved in angiogenesis and the response to injury, such as endothelial cells and fibroblasts, have a prominent role in the progression, growth and spread of cancers. Fibroblasts are associated with cancer cells at all stages of cancer progression, and their structural and functional contributions to this process are beginning to emerge. Their production of growth factors, chemokines and extracellular matrix facilitates the angiogenic recruitment of endothelial cells and pericytes. Fibroblasts are therefore a key determinant in the malignant progression of cancer and represent an important target for cancer therapies.
Genetic and cell-biology studies indicate that tumour growth is not just determined by malignant cancer cells themselves, but also by the tumour stroma1. Tumour progression is clearly dependent on angiogenesis2–5, and inflammatory cells also contribute to tumour growth6,7. It is becoming increasingly clear that fibroblasts8,9 are also prominent modifiers of cancer progression. Our knowledge of the role of resting and activated fibro-blasts in cancer is still evolving, and evidence is increas-ing that a subpopulation of fibroblasts — the so-called cancer-associated fibroblasts (CAFs) — are important promoters of tumour growth and progression10.
Fibroblasts were first described in the late 19th century, based on their location and their microscopic appear-ance11,12. They are the non-vascular, non-epithelial and non-inflammatory cells of the connective tissue13, and are its principal cellular component (BOX 1). They are embed-ded within the fibrillar matrix of the connective tissue and are, to a large extent, responsible for its synthesis.
The important functions of fibroblasts include the deposition of extracellular matrix (ECM), regulation of epithelial differentiation, regulation of inflammation, and involvement in wound healing14,15. Fibroblasts synthesize many of the constituents of the fibrillar ECM such as type I, type III and type V collagen, and fibro-nectin14,16. They also contribute to the formation of basement membranes by secreting type IV collagen and laminin17. Fibroblasts are also an important source of ECM-degrading proteases such as matrix metalloproteinases (MMPs), which highlights their crucial role in maintain-ing an ECM homeostasis by regulating ECM turnover17,18. In addition, fibroblasts are important in maintaining the homeostasis of adjacent epithelia through the secretion of growth factors and direct mesenchymal–epithelial cell interactions19 (BOX 2). As well as their function in healthy organs, fibroblasts have a prominent role in wound repair.
They invade lesions, generate ECM to serve as a scaffold for other cells, and possess cytoskeletal elements that facilitate contractions of healing wounds14.
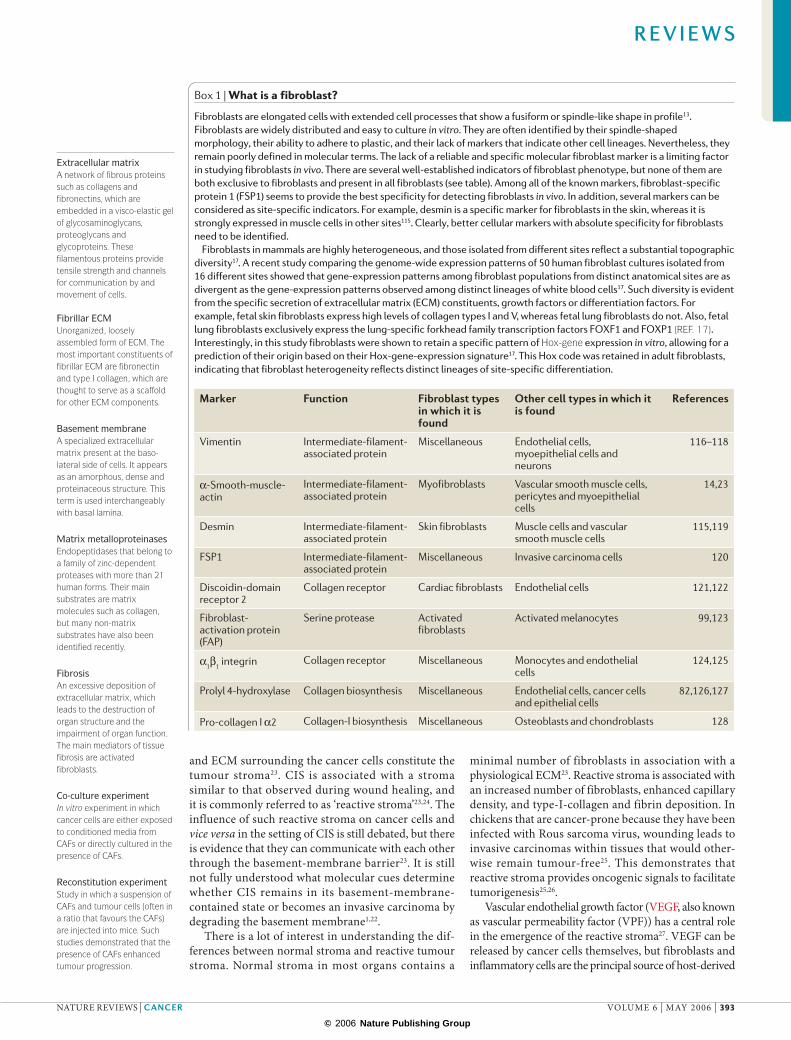
As the principal source of ECM constituents, fibro-blasts are considered the main mediators of scar forma-tion and tissue fibrosis. Fibroblasts that are isolated from the site of a healing wound or from fibrotic tissue secrete higher levels of normal ECM constituents and prolifer-ate more than their normal counterparts isolated from healthy organs20,21. Such increased activity is referred to as ‘activation’20 (BOX 3; FIG. 1). Once the wound is repaired, the number of activated fibroblasts decreases significantly and the resting phenotype is thought to be restored14. It remains unknown whether the activated fibroblasts revert to a resting phenotype, or whether they undergo apoptosis followed by the repopulation of that particular region of the tissue by resting fibroblasts from the adjacent tissue14.
Unlike wound healing, but similar to organ fibrosis, the fibroblasts at the site of a tumour remain perpetually activated. Are fibroblasts inducers of cancer progression and invasion? Or do they protect neoplastic lesions from becoming invasive carcinomas? What is the contribution of fibroblasts to metastasis? These questions remain largely unanswered, but data from co-culture and reconstitution experiments indicate that fibroblasts have a prominent role in defining the rate and extent of cancer progression.
The tumour stromaIn the early growth of tumours, cancer cells form a neoplastic lesion that is embedded in the micro-environment of a given tissue (usually epithelium)22 but separated from the surrounding tissue and con-tained within the boundary of a basement membrane22. This is called the carcinoma in situ (CIS). The base-ment membrane, immune cells, capillaries, fibroblasts
T U M O U R M I C R O E N V I R O N M E N T
R E V I E W S
392 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer
© 2006 Nature Publishing Group
Extracellular matrixA network of fibrous proteins such as collagens and fibronectins, which are embedded in a visco-elastic gel of glycosaminoglycans, proteoglycans and glycoproteins. These filamentous proteins provide tensile strength and channels for communication by and movement of cells.
Fibrillar ECM Unorganized, loosely assembled form of ECM. The most important constituents of fibrillar ECM are fibronectin and type I collagen, which are thought to serve as a scaffold for other ECM components.
Basement membraneA specialized extracellular matrix present at the baso-lateral side of cells. It appears as an amorphous, dense and proteinaceous structure. This term is used interchangeably with basal lamina.
Matrix metalloproteinasesEndopeptidases that belong to a family of zinc-dependent proteases with more than 21 human forms. Their main substrates are matrix molecules such as collagen, but many non-matrix substrates have also been identified recently.
FibrosisAn excessive deposition of extracellular matrix, which leads to the destruction of organ structure and the impairment of organ function. The main mediators of tissue fibrosis are activated fibroblasts.
Co-culture experimentIn vitro experiment in which cancer cells are either exposed to conditioned media from CAFs or directly cultured in the presence of CAFs.
Reconstitution experimentStudy in which a suspension of CAFs and tumour cells (often in a ratio that favours the CAFs) are injected into mice. Such studies demonstrated that the presence of CAFs enhanced tumour progression.
and ECM surrounding the cancer cells constitute the tumour stroma23. CIS is associated with a stroma similar to that observed during wound healing, and it is commonly referred to as ‘reactive stroma’23,24. The influence of such reactive stroma on cancer cells and vice versa in the setting of CIS is still debated, but there is evidence that they can communicate with each other through the basement-membrane barrier23. It is still not fully understood what molecular cues determine whether CIS remains in its basement-membrane-contained state or becomes an invasive carcinoma by degrading the basement membrane1,22.
There is a lot of interest in understanding the dif-ferences between normal stroma and reactive tumour stroma. Normal stroma in most organs contains a
minimal number of fibroblasts in association with a physiological ECM23. Reactive stroma is associated with an increased number of fibroblasts, enhanced capillary density, and type-I-collagen and fibrin deposition. In chickens that are cancer-prone because they have been infected with Rous sarcoma virus, wounding leads to invasive carcinomas within tissues that would other-wise remain tumour-free25. This demonstrates that reactive stroma provides oncogenic signals to facilitate tumorigenesis25,26.
Vascular endothelial growth factor (VEGF, also known as vascular permeability factor (VPF)) has a central role in the emergence of the reactive stroma27. VEGF can be released by cancer cells themselves, but fibroblasts and inflammatory cells are the principal source of host-derived
Box 1 | What is a fibroblast?
Fibroblasts are elongated cells with extended cell processes that show a fusiform or spindle-like shape in profile13. Fibroblasts are widely distributed and easy to culture in vitro. They are often identified by their spindle-shaped morphology, their ability to adhere to plastic, and their lack of markers that indicate other cell lineages. Nevertheless, they remain poorly defined in molecular terms. The lack of a reliable and specific molecular fibroblast marker is a limiting factor in studying fibroblasts in vivo. There are several well-established indicators of fibroblast phenotype, but none of them are both exclusive to fibroblasts and present in all fibroblasts (see table). Among all of the known markers, fibroblast-specific protein 1 (FSP1) seems to provide the best specificity for detecting fibroblasts in vivo. In addition, several markers can be considered as site-specific indicators. For example, desmin is a specific marker for fibroblasts in the skin, whereas it is strongly expressed in muscle cells in other sites115. Clearly, better cellular markers with absolute specificity for fibroblasts need to be identified.
Fibroblasts in mammals are highly heterogeneous, and those isolated from different sites reflect a substantial topographic diversity17. A recent study comparing the genome-wide expression patterns of 50 human fibroblast cultures isolated from 16 different sites showed that gene-expression patterns among fibroblast populations from distinct anatomical sites are as divergent as the gene-expression patterns observed among distinct lineages of white blood cells17. Such diversity is evident from the specific secretion of extracellular matrix (ECM) constituents, growth factors or differentiation factors. For example, fetal skin fibroblasts express high levels of collagen types I and V, whereas fetal lung fibroblasts do not. Also, fetal lung fibroblasts exclusively express the lung-specific forkhead family transcription factors FOXF1 and FOXP1 (REF. 17). Interestingly, in this study fibroblasts were shown to retain a specific pattern of Hox-gene expression in vitro, allowing for a prediction of their origin based on their Hox-gene-expression signature17. This Hox code was retained in adult fibroblasts, indicating that fibroblast heterogeneity reflects distinct lineages of site-specific differentiation.
Marker Function Fibroblast types in which it is found
Other cell types in which it is found
References
Vimentin Intermediate-filament-associated protein
Miscellaneous Endothelial cells, myoepithelial cells and neurons
116–118
α-Smooth-muscle-actin
Intermediate-filament-associated protein
Myofibroblasts Vascular smooth muscle cells, pericytes and myoepithelial cells
14,23
Desmin Intermediate-filament-associated protein
Skin fibroblasts Muscle cells and vascular smooth muscle cells
115,119
FSP1 Intermediate-filament-associated protein
Miscellaneous Invasive carcinoma cells 120
Discoidin-domain receptor 2
Collagen receptor Cardiac fibroblasts Endothelial cells 121,122
Fibroblast-activation protein (FAP)
Serine protease Activated fibroblasts
Activated melanocytes 99,123
α1β1 integrin Collagen receptor Miscellaneous Monocytes and endothelial cells
124,125
Prolyl 4-hydroxylase Collagen biosynthesis Miscellaneous Endothelial cells, cancer cells and epithelial cells
82,126,127
Pro-collagen I α2 Collagen-I biosynthesis Miscellaneous Osteoblasts and chondroblasts 128
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 6 | MAY 2006 | 393
© 2006 Nature Publishing Group

Hox genesThe Hox genes encode a family of homeodomain transcription factors that determine positional identity along the anterior–posterior and secondary axis in animals.
MyofibroblastsThe myofibroblast was initially identified by means of electron microscopy as a fibroblast with features that are more typical of smooth muscle cells, such as possessing bundles of microfilaments and maintaining gap junctions. Myofibroblasts are commonly detected by their expression of α-smooth-muscle actin.
NOD/SCID miceMice with multiple defects in innate immunity, including a largely reduced natural-killer-cell activity and a twofold-reduced bone-marrow cellularity.
VEGF28. VEGF induces microvascular permeability, which leads to the extravasation of plasma proteins such as fibrin, which in turn attract an influx of fibroblasts, inflammatory cells and endothelial cells27,29,30. These cells produce ECM that is rich in fibronectin and type I col-lagen, both of which are conducive to initiating tumour angiogenesis27,31,32 (BOX 1).
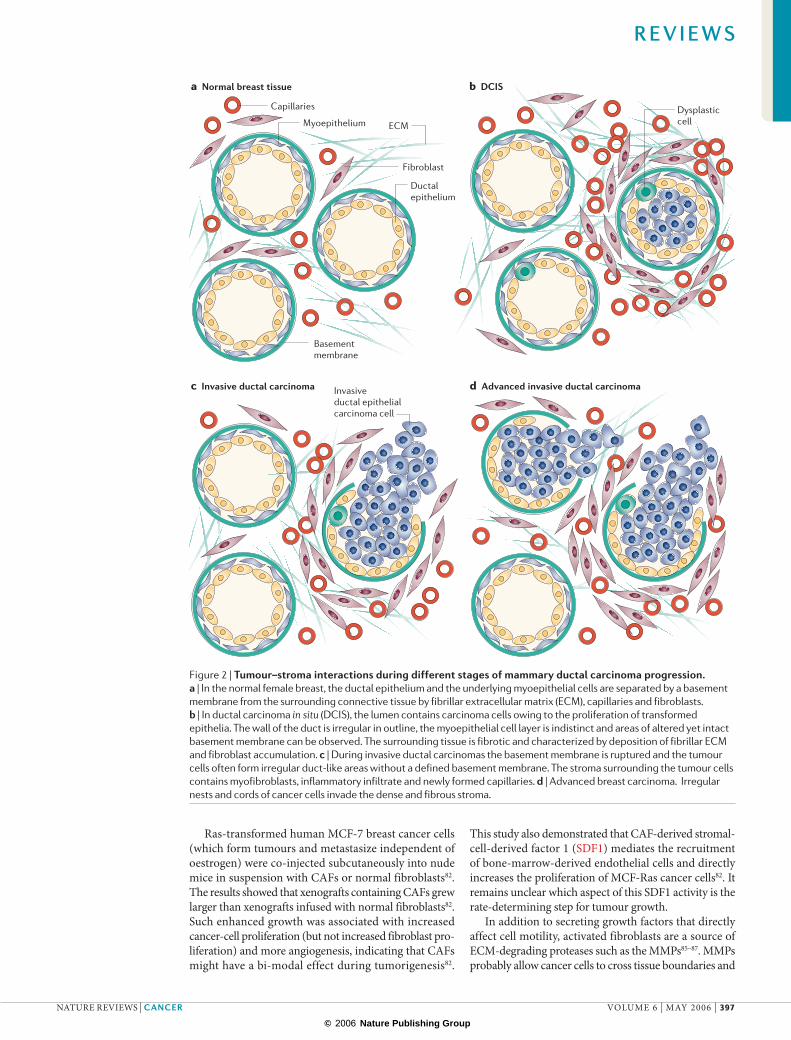
During cancer progression from CIS to invasive car-cinoma the tumour cells invade the reactive stroma23,33. FIGURE 2 exemplifies tumour development in the setting of mammary ductal carcinomas. Although the ductal epithelium and the underlying myoepithelial cells are separated from the surrounding connective tissue by a basement membrane in both normal female breast and in ductal carcinoma in situ (DCIS), the base-ment membrane in DCIS, although intact, is altered23. During invasive ductal carcinomas the basement mem-brane is degraded and the activated stroma (previously outside the basement-membrane zone), which contains myofibroblasts, inflammatory infiltrate and newly formed capillaries, comes into direct contact with the tumour cells23,33–35.
Invasive carcinoma is often associated with expan-sion of the tumour stroma and increased deposition of ECM35. Such increased deposition of ECM in tumours is known as desmoplasia and is similar to changes that
are observed during organ fibrosis23,34,36. However, whereas organ fibrosis is associated with a rarification of the microvasculature37, solid tumours are typically more vascularized than normal tissues38. Desmoplastic stroma contains increased amounts of fibrillar collagens, fibronectins, proteoglycans and tenascin C23. Tenascin C is absent in the normal adult mammary gland, but expression is regularly seen in mammary tumours39,40. Increased tenascin-C expression correlates with poor prognosis for both breast cancer and bladder cancer41,42, and it is associated with increased invasiveness40,43. Similarly, increased hyaluron in breast cancer tissue probably facilitates invasion44.
However, other constituents of the desmoplastic ECM have also been shown to inhibit tumour progres-sion. Injection of L-3,4 dehydroproline (which inhibits the formation of collagen fibrils) increases tumour cell invasion in a mouse model of B16F10 melanoma subcutaneous tumours45. Furthermore, ECM accumu-lation in tumours contributes to increased interstitial fluid pressure and hinders macromolecule and oxygen diffusion46,47, leading to tumour-cell necrosis.
In addition to influencing cancer-cell migration, the ECM also affects various regulatory pathways by modu-lating the release and availability of growth factors23 and by direct interactions with cells through integrins48. These studies indicate that fibroblasts can affect cancer progres-sion by secreting and organizing altered ECM within the tumour stroma. However, the overall effect of altered ECM in tumours and the effect of CAFs on angiogenesis during tumour progression are still poorly understood.
Carcinoma-associated fibroblastsThe notion that fibroblasts within the tumour stroma acquire a modified phenotype, similar to fibroblasts associated with wound healing, has been studied since the 1970s (REFS 49–53). Such ‘activated’ fibroblasts within the tumour stroma have been termed peri-tumoral fibroblasts, reactive stromal fibroblasts, CAFs or tumour-associated fibroblasts10,54. In breast carci-nomas, about 80% of stromal fibroblasts are thought to acquire this activated phenotype55. In the absence of electron microscopy validation, CAFs are commonly identified by their expression of α-smooth-muscle actin23,51,56 (BOX 1).
The signals that mediate the transition of a nor-mal fibroblast into a CAF are not fully understood. In culture, phenotypic features of CAFs can be induced by transforming growth factor-β (TGFβ), which medi-ates fibroblast activation during wound healing and organ fibrosis. Therefore, similar pathways might also be responsible for the emergence of CAFs in tumours57. Additional studies are needed to determine whether CAFs represent a unique fibroblast phenotype or whether they represent a common phenotype of fibroblast activation during injury.
Growth factors and fibroblastsThe tumour stroma comprises most of the tumour mass in many carcinomas23. Its volume and composition are partly governed by the response of fibroblasts to the
At a glance
• Fibroblasts are a key cellular component of tumours.
• Activated fibroblasts (which are sometimes referred to as myofibroblasts) that are found in association with cancer cells are known as carcinoma-associated fibroblasts (CAFs).
• The role of fibroblasts in the origin and initiation of cancer invasion is poorly understood, but recent evidence indicates that they can provide oncogenic signals to the transformed epithelia in a paracrine fashion.
• Some studies indicate that myofibroblasts might facilitate angiogenesis and cancer progression.
• Epithelial-to-mesenchymal transition is a source for converting epithelial cells into fibroblast-like cells in various tissues.
• Fibroblasts might also have a role in metastasis.
• Therapies against CAFs are being considered as a way to control cancer.
Box 2 | The role of fibroblasts in mammary development
In addition to their function in contributing to the extracellular-matrix microenvironment in connective tissues, fibroblasts have a role in the regulation of differentiation and homeostasis of adjacent epithelia. Such mesenchymal–epithelial interactions regulate the morphogenesis of epithelia, which is important in the organization and development of mammary glands18,19,129. This was demonstrated in a recent study that recapitulated the formation of mammary gland using human mammary epithelial cells in a mouse model80. Cleared breast pads of NOD/SCID mice were injected with immortalized human breast fibroblasts, which proliferated and invaded the fat pad, resulting in a ‘humanized’ stroma consisting of about 15% human fibroblasts after 8 weeks (REF. 80). When human mammary epithelial cell organoids (consisting of 100–1,000 mammary epithelial cells each) were co-injected with the human fibroblasts into the cleared fat pads (this method is known as a cleared-fat-pad transplantation system), they formed ductal, lobular and acinar structures80. This did not occur in the absence of co-injected human fibroblasts, despite the presence of mouse fibroblasts, indicating a species-specific role for adult fibroblasts in the differentiation of mammary glands80.
R E V I E W S
394 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer
© 2006 Nature Publishing Group

Cleared-fat-pad transplantation systemA cleared fat pad, devoid of any epithelium, into which mouse mammary epithelial cells are engrafted before puberty, causing an anatomically normal mammary gland to develop. However, human mammary epithelial cells injected into the cleared mouse fat pad do not form a mammary gland.
SenescenceA programme that is activated by normal cells in various situations of stress to prevent further proliferation. Senescence occurs in response to telomere capping, DNA damage, oxidative stress or activation of oncogenes.
Epithelial-to-mesenchymal transitionA process that is necessary for embryonic development, tumour progression and organ fibrosis. During EMT epithelial characteristics are lost and the mesenchymal phenotype is acquired.
pro-fibrotic growth factors that are released by the can-cer cells33 — for example, TGFβ, platelet-derived growth factor (PDGF) and fibroblast growth factor 2 (FGF2), all of which are key mediators of fibroblast activation and tissue fibrosis9. Increased expression of TGFβ in breast cancer correlates with the accumulation of fibrotic desmoplastic tissue58,59. It also correlates with the rate of disease progression60. It is not yet clear whether the desmoplastic response induces local CAF invasion, stimulated by TGFβ that is released by cancer cells, or whether CAF invasion reflects a wound-healing defence mechanism to ‘repair’ and ‘contain’ a cancerous lesion (such as DCIS) from becoming an invasive carcinoma9.
PDGF is another growth factor that is secreted by can-cer cells and correlates with cancer progression61. Most cancer cells do not express PDGF receptors, indicating that PDGF elicits its action primarily through para-crine mechanisms that involve other cell types, such as fibroblasts and endothelial cells62. As opposed to TGFβ, PDGF induces the proliferation of fibroblasts, but does not induce the acquisition of an activated phenotype that is associated with excessive ECM deposition63.
FGF2 was originally identified as a ‘basic fibroblast growth factor’, which stimulates the proliferation of 3T3 fibroblasts but not the expression of α-smooth-muscle actin23,64. Although FGF2 in the setting of cancer is predominantly recognized for its potential to induce angiogenesis65, several studies demonstrated a role for FGF2 in tissue fibrosis66, which indicates that it could be similarly involved in the formation of desmoplasia in tumours.
EMT as a source of CAFsIn epithelial-to-mesenchymal transition (EMT), epithelial cells lose cell–cell contacts and acquire mesenchymal properties67. Cancer cells undergoing EMT develop invasive and migratory abilities68. EMT of cancer cells is increasingly being recognized as an important deter-minant of tumour progression (discussed extensively in REFS 68,69). However, in addition to its role in tumour-cell invasiveness, EMT might modulate cancer progres-sion by serving as an additional source of fibroblast-like cells with an altered genome.
Several recent studies have analysed genetic alterations in both microdissected cancer cells and surrounding stromal cells (including fibroblasts) and have found a similar frequency of genetic alterations in both com-partments70–73. However, although genetic alterations are present in both cancer cells and stromal cells, the mutations are rarely identical73. In addition, there is a non-random X-chromosome-inactivation pattern in fibroblasts and cancer cells74. This indicates that although epithelial cancer cells and a subset of stromal fibroblasts might share a common origin, other cancer cells might undergo further genetic rearrangements under a distinct selection pressure that facilitates transition into mesenchymal-like cells74. These findings indicate that EMT of cancer cells does occur, but that it probably accounts for a fraction of the CAFs that are present in tumours.
EMT involving normal epithelial cells that are adjacent to malignant cancer cells might also contrib-ute to the emergence of CAFs69. In tissue fibrosis it is well-established that epithelial cells contribute to
Box 3 | Fibroblast activation
Normal fibroblasts are embedded within the fibrillar extracellular matrix (ECM) of the connective tissue and constitutively express vimentin and fibroblast-specific protein 1 (FSP1) (BOX 1). A normal fibroblast can acquire an ‘activated’ phenotype. To synthesize large amounts of ECM constituents, activated fibroblasts typically contain a large oval euchromatic nucleus with one or two nucleoli, rough endoplasmatic reticulum, and a prominent Golgi apparatus13. In inactive adult fibroblasts, the endoplasmic reticulum is smaller and the nucleus is flattened and heterochromatic13.
Fibroblast activation is induced by various stimuli that arise when tissue injury occurs103, including growth factors such as transforming growth factor-β (TGFβ), epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and fibroblast growth factor 2 (FGF2), which are released from injured epithelial cells and infiltrating mononuclear cells such as monocytes and macrophages103. In addition, fibroblasts are activated by direct cell–cell communication and contacts with leukocytes through adhesion molecules such as intercellular-adhesion molecule 1 (ICAM1) or vascular-cell adhesion molecule 1 (VCAM1)130. Fibroblast activation can also be achieved through reactive oxygen species, complement factor C1 or altered ECM composition103 (FIG. 1).
Activated fibroblasts express α-smooth-muscle actin, leading to the term ‘myofibroblasts’. In addition, activated fibroblasts also secrete increased levels of ECM-degrading proteases such as matrix metalloproteinase 2 (MMP2), MMP3 and MMP9, facilitating increased ECM turnover and altered ECM composition16. Activated fibroblasts often secrete increased amounts of growth factors such as hepatocyte growth factor (HGF), insulin-like growth factor (IGF), nerve growth factor (NGF), WNT1, EGF and FGF2, which can induce proliferative signals within adjacent epithelial cells93. Activated fibroblasts also have an important role as modulators of the immune response following tissue injury, through the secretion of cytokines such as interleukin-1 and chemokines such as monocyte chemotactic protein 1 (MCP1)131,132.
Activated fibroblasts are found in healing wounds and sclerosing tissues, and are also associated with cancers. Importantly, in the context of wound healing, fibroblast activation is reversed once the activating stimulus is attenuated. However, in tissue fibrosis, fibroblasts sustain their activated state, often until organ death. The molecular explanation for fibroblasts remaining activated even when the initial insult has regressed is not yet clear. Interestingly, fibroblasts that are isolated from fibrotic tissue maintain their activated phenotype when cultured in vitro21,82. Fibroblasts in such a sustained state of activation typically continue to secrete ECM constituents, growth factors and cytokines. This results in a self-perpetuating autocrine loop, which stimulates other fibroblasts and prevents resolution of the initial injury. Both cancer-associated fibroblasts and activated fibroblasts that are isolated from wounds or fibrotic areas maintain their phenotype over several passages in vitro, until they enter senescence. It has been suggested that epigenetic changes in response to stress cause the fibroblasts to maintain a state that is autonomous to microenvironmental stimuli82,103.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 6 | MAY 2006 | 395
© 2006 Nature Publishing Group
Collagen INucleus
VimentinActin fibres
FSP1
α1β1 integrin
Fibrillar ECM
a Fibroblast
Tenascin C
α-smooth-muscle-actin stress fibres
Growth factorsECM proteasesChemokinesFibronectin
b Activated fibroblast
SPARC
EDA-fibronectin
the accumulation of fibroblasts by undergoing EMT in response to stimuli from the microenvironment75,76. Proximal tubular epithelial cells of kidneys that are irreversibly marked by the expression of β-galactosidase showed that up to 30% of activated fibroblasts in kid-ney fibrosis originate from EMT of tubular epithelial cells77. Therefore, such EMT involving resident epi-thelia could similarly contribute to the pool of CAFs in cancer.
The role of CAFs in the initiation of cancerSkin fibroblasts of patients with a strong predisposition to breast cancer show an abnormal phenotype, includ-ing a reduced requirement of serum for proliferation78. Similarly, skin fibroblasts isolated from patients with breast cancer, malignant melanoma, familial poly-posis coli, retinoblastoma and Wilms tumours show an increased proliferation rate in vitro, indicating that altered fibroblast behaviour might increase the rate of cancer initiation78,79.
Several studies that used mouse models of geneti-cally altered fibroblasts demonstrated a direct involve-ment of resident fibroblasts in the initiation of cancer. A recent study used the capacity of human mammary epithelial cells to form ducts in a mouse breast stroma80, and demonstrates that overexpression of TGFβ and/or hepatocyte growth factor (HGF) in mouse fibrob-lasts induces the initiation of breast cancer within the normal human epithelium. A similar association of fibroblast-derived HGF and TGFβ expression with cancer initiation is demonstrated in FSP1-Cre TGFβRIIRflox/flox mice, which conditionally lack the
TGFβ type II receptor on fibroblast-specific protein 1 (FSP1, which is also known as S100A4 and MTS1)-positive fibroblasts (these fibroblasts are therefore unresponsive to TGFβ signalling)81. These mice have increased intra-epithelial neoplasia in the prostate and invasive squamous-cell carcinoma of the forestom-ach, associated with increased HGF expression in the surrounding fibroblasts81.
A further link between growth factors and CAFs in tumour initiation was indicated by a series of stud-ies comparing the effect of normal fibroblasts and of CAFs isolated from the primary tumour site82,83. When simian virus 40 (SV40)-transformed ‘normal’ prostate epithelial cells were grafted onto mice in combination with either normal fibroblasts or CAFs, only the CAFs led to an emergence of lesions resembling prostatic intraepithelial neoplasia83. When immortalized pros-tate epithelial cells were used, the presence of CAFs induced the growth of massive tumours in these mice, whereas tumours did not form in the presence of nor-mal fibroblasts83. These findings indicate that normal fibroblasts are required to maintain epithelial homeo-stasis, whereas CAFs probably initiate and promote tumorigenic alterations in epithelial cells.
The role of CAFs in the progression of cancerCAFs probably also promote tumour progression through specific communications with cancer cells. For example, CAFs facilitate the invasiveness of otherwise non-invasive cancer cells when co-injected into mice84. However, the details of how CAFs might promote cancer progression are only just emerging.
Figure 1 | Activated fibroblasts. a | Normal fibroblasts are embedded within the fibrillar extracellular matrix (ECM) of connective tissue, which consists largely of type I collagen and fibronectin. Fibroblasts interact with their surrounding microenvironment through integrins such as the α1β1 integrin. Typically, fibroblasts appear as fusiform cells with a prominent actin cytoskeleton and vimentin intermediate filaments. Although the detection of fibroblasts can be challenging because most known markers are not specific to fibroblasts, fibroblast-specific protein 1 (FSP1), which is a member of the family of S100 Ca 2+-binding proteins, is specific for fibroblasts in normal tissues. b | Fibroblasts can acquire an activated phenotype, which is associated with an increased proliferative activity and enhanced secretion of ECM proteins such as type I collagen and tenascin C, and also fibronectin that contains the extra domain a (EDA-fibronectin) and SPARC (secreted protein acidic and rich in cysteine). Phenotypically, activated fibroblasts are often characterized as expressing α-smooth-muscle actin. Numerous growth factors such as transforming growth factor-β (TGFβ), chemokines such as monocyte chemotactic protein 1 (MCP1), and ECM-degrading proteases have been shown to mediate the activation of fibroblasts.
R E V I E W S
396 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer
© 2006 Nature Publishing Group
a Normal breast tissue
c Invasive ductal carcinoma d Advanced invasive ductal carcinoma
Capillaries
Myoepithelium
Ductal epithelium
ECM
Fibroblast
b DCIS
Dysplasticcell
Basement membrane
Invasive ductal epithelial carcinoma cell
Ras-transformed human MCF-7 breast cancer cells (which form tumours and metastasize independent of oestrogen) were co-injected subcutaneously into nude mice in suspension with CAFs or normal fibroblasts82. The results showed that xenografts containing CAFs grew larger than xenografts infused with normal fibroblasts82. Such enhanced growth was associated with increased cancer-cell proliferation (but not increased fibroblast pro-liferation) and more angiogenesis, indicating that CAFs might have a bi-modal effect during tumorigenesis82.
This study also demonstrated that CAF-derived stromal-cell-derived factor 1 (SDF1) mediates the recruitment of bone-marrow-derived endothelial cells and directly increases the proliferation of MCF-Ras cancer cells82. It remains unclear which aspect of this SDF1 activity is the rate-determining step for tumour growth.
In addition to secreting growth factors that directly affect cell motility, activated fibroblasts are a source of ECM-degrading proteases such as the MMPs85–87. MMPs probably allow cancer cells to cross tissue boundaries and
Figure 2 | Tumour–stroma interactions during different stages of mammary ductal carcinoma progression. a | In the normal female breast, the ductal epithelium and the underlying myoepithelial cells are separated by a basement membrane from the surrounding connective tissue by fibrillar extracellular matrix (ECM), capillaries and fibroblasts. b | In ductal carcinoma in situ (DCIS), the lumen contains carcinoma cells owing to the proliferation of transformed epithelia. The wall of the duct is irregular in outline, the myoepithelial cell layer is indistinct and areas of altered yet intact basement membrane can be observed. The surrounding tissue is fibrotic and characterized by deposition of fibrillar ECM and fibroblast accumulation. c | During invasive ductal carcinomas the basement membrane is ruptured and the tumour cells often form irregular duct-like areas without a defined basement membrane. The stroma surrounding the tumour cells contains myofibroblasts, inflammatory infiltrate and newly formed capillaries. d | Advanced breast carcinoma. Irregular nests and cords of cancer cells invade the dense and fibrous stroma.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 6 | MAY 2006 | 397
© 2006 Nature Publishing Group

Epithelial cells
Endothelial cells
Pericytes
Cancer-associatedfibroblast
Invasive carcinoma
TGFβ
TGFβ
HGFTenascin C
Collagen I, IIIMCP1
VEGF
MMPsIL-1
Inflammatory cells
PericyteMesenchymal cells that wrap around endothelial cells in microvessels. They share the common basement membrane with the vascular endothelial cells.
Stellate cellsFibroblast-like cells in the liver that express glial fibrillary acidic protein and nestin, and are thought to be neural-crest-derived.
escape the primary tumour site. MMPs and other pro-teases also directly affect the motility and invasiveness of cancer cells. Such a direct effect has been elucidated for MMP3 (also known as stromelysin 1), which is highly expressed in fibroblasts. MMP3 directly cleaves the extra-cellular domain of E-cadherin, prompting normal mam-mary epithelial cells to disaggregate and undergo EMT, promoting cancer-cell invasiveness88. MMP1 also shows such a tumour-promoting effect. The protease-activated receptor PAR1, which is a tethered-ligand receptor, is acti-vated by proteolytic cleavage of the extracellular domain by MMP1, promoting cancer cell migration and invasion through PAR1-dependent Ca2+ signals85.
The role of CAFs in metastasisSome studies have indicated that activated fibroblasts at the metastatic site promote the proliferation of cancer cells there and, similar to the CAFs in the primary tumour, are conducive to tumour growth89. Such metastasis-associated fibroblasts could represent a variant of CAFs. Hepatic metastasis following the injection of B16M melanoma cells in mice89 showed that the formation of liver metas-tases is associated with the activation of stellate cells even before the tumour reached a diameter of 150 µm (REF. 89). This supports the notion that activated stellate cells might
be important in creating a niche for the cancer cells and supporting the initiation of angiogenesis90. A different study systematically evaluated the effect of fibroblasts on the proliferation of melanoma cells and found that only the melanoma cells that metastasized were influ-enced by fibroblasts91. This indicates that specific stimuli (which are not yet known) provided by fibroblasts create a niche that promotes the growth of subsets of cancer cells at distant sites. Tumour growth and metastasis in FSP1-deficient mice (FSP1-deficient fibroblasts show decreased motility) is significantly reduced92. Injection of wild-type fibroblasts into these mice partially reversed the observed phenotype, providing further evidence for the involvement of metastasis-associated fibroblasts in the emergence of metastasis92.
Fibroblasts and TGFβAs discussed above, EMT involving cancer cells is con-sidered an important determinant of cancer progres-sion, as it facilitates tumour invasion and metastasis68. Various exogenous stimuli control EMT of cancer cells68. These include MMPs, integrins and members of the TGFβ superfamily of proteins, which are growth factors that bind to tyrosine-kinase receptors, such as epider-mal growth factor (EGF) and FGF2. Downstream of tyrosine-kinase receptors, complex signalling networks mediate the induction of EMT. Most of the exogenous EMT stimuli are potentially contributed by CAFs10,93 (FIG. 3). Therefore, it is conceivable that CAFs have an important role in mediating EMT of carcinoma cells, thereby contributing to the progression of cancer.
As mentioned above, TGFβ has a role in the recruit-ment of fibroblasts. However, TGFβ is also the most prominent of the factors within the tumour micro-environment that are known to induce EMT of cancer cells94. Before the initiation of cancer, and during the early stages of carcinogenesis, TGFβ probably functions as a tumour suppressor. By contrast, during advanced stages of cancer, TGFβ signalling promotes cancer progression and metastasis60. Although the underlying molecular mechanisms are not fully understood, it is becoming increasingly clear that the dual effect of TGFβ on dif-ferent signalling pathways, and by extension the effect of CAFs, needs consideration33,95.
In normal tissues, the anti-proliferative and pro-apoptotic response of epithelial cells to TGFβ might limit the growth of normal epithelium and the emergence of malignant carcinomas22,94. With regard to fibroblasts, TGFβ facilitates fibroblast–epithelial-cell interactions, which further suppress cancer initiation93. In advanced cancer, on the other hand, the anti-proliferative effect of TGFβ is progressively lost — instead, it facilitates EMT of cancer cells, potentially promoting invasiveness and metastasis94. Signalling pathways that are implicated in TGFβ-mediated EMT involve Smad signalling, the phosphatidylinositol-3-kinase–Akt pathway and signal-ling through RHOA and p38 MAPK69,96 (FIG. 4). However, several studies indicate that TGFβ signalling involving Smads, and SMAD3 in particular, is the principal deter-minant of EMT, whereas the other pathways function synergistically but are dispensable94.
Figure 3 | Functions of activated fibroblasts in the tumour stroma. Fibroblasts communicate with cancer cells, resident epithelial cells, endothelial cells, pericytes and inflammatory cells through the secretion of growth factors and chemokines. Through the increased deposition of collagen types I and III and de novo expression of tenascin C they induce an altered extracellular-matrix microenvironment, which potentially provides additional oncogenic signals, probably leading to accelerated cancer progression. Fibroblasts mediate the inflammatory response by secreting chemokines such as monocyte chemotactic protein 1 (MCP1) and interleukins such as IL-1. Fibroblasts interact with the microvasculature by secreting matrix metalloproteinases (MMPs) and vascular endothelial growth factor (VEGF). Fibroblasts also provide potentially oncogenic signals such as transforming growth factor-β (TGFβ) and hepatocyte growth factor (HGF) to resident epithelia, and directly stimulate cancer-cell proliferation and invasion by secreting growth factors such as TGFβ and stromal-cell-derived factor 1 (SDF1).
R E V I E W S
398 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer
© 2006 Nature Publishing Group

Type I collagen EGF
SFC Ras
FAK
FGF HGF
Integrins
Raf
PI3K
Akt
MAPK
Smad RHOA
TGFβ
TGFβR
MMP3
APC–β-catenin–Axin
Free β-catenin
TKR
Triggers EMT
E-cadherin
SclerodermaFibrosis involving the skin.
Fibroblasts in the host defence against cancer?Numerous studies provide evidence for a cancer-promoting role of activated CAFs. Other studies that compare the effects of CAFs with that of ‘normal’ fibroblasts indicate that the latter inhibit cancer progression. Fibroblasts that overexpress TGFβ and HGF promote tumorigenic outgrowth of normal breast epithelium, but such outgrowth is inhibited by ‘normal’ stromal fibroblasts80. The loss of TGFβ signalling in fibroblasts results in prostate intraepi-thelial neoplasia (PIN), indicating that normal fibrob-lasts suppress such carcinogenic events81. However, it remains unclear how normal fibroblasts prevent tumorigenesis80,81,97. One possible mechanism for such a function of fibroblasts in host defence is modulation of the immune response. Fibroblasts are a signficant source of immune-modulatory cytokines such as inter-feron-γ, interleukin-6 and tumour-necrosis factor-α98, which can influence the mobilization of cytotoxic T lymphocytes, natural killer cells and macrophages. In this regard, it has also been suggested that normal fibroblasts help to prevent apoptosis of T cells98.
A significant hurdle in evaluating the physiological role of normal fibroblasts is the use of tumour xenografts, which relies on a protocol of mixing cancer cells and fibroblasts. The resulting ratio of cancer cells and fibro-blasts does not mirror the distribution of host-derived CAFs within the tumour site23, and distinct functions of those fibroblasts in the centre of the tumour rather than those at the border of the tumour-adjacent normal tissue might be overlooked. However, grafting fibroblasts on their own is difficult, as they often do not survive in the transplanted host133. Additionally, fibroblasts often become activated and further modified when cultured in vitro before injection134. Therefore, the role of nor-mal resident fibroblasts in cancer progression remains unclear. Studies using mouse models that allow specific ablation of normal host-fibroblasts are required to address this question.
Therapies to target activated fibroblastsAlthough we do not fully understand the mechanisms that regulate fibroblast activation and their accumulation in cancer, the evidence presented in this Review shows that it is conceivable that fibroblasts might serve as novel therapeutic targets in cancer. Such therapies can be given alone or in combination with chemotherapy, radiation or surgery. Fibroblast-directed therapy can be envisioned as either ‘to ablate’ or ‘to normalize’ CAFs.
The cell-surface serine protease known as fibroblast-activation protein (FAP) emerges as a promising candidate for specifically targetting CAFs99. FAP is not expressed by mature somatic tissues except for activated fibroblasts during wound healing and within tumour stroma100. A phase I dose-escalation study with sibrotuzumab (which is an antibody to human FAP) in patients with advanced colorectal carcinoma or non-small-cell lung carcinoma showed that sibrotuzumab bound specifically to the tumour sites without apparent side effects101.
Several growth factors and hormones have been used to decrease the number of normalizing activated fibro-blasts in tissue fibrosis102–104. However, it is unclear whether these can also be used in the setting of cancer because several of these molecules with anti-fibrotic properties are also promoters of cancer progression. For example, HGF inhibits the activation and progression of tissue fibrosis in several animal models of chronic disease104–106, and HGF overexpression prevents fibrosis in the liver and kid-neys107,108. However, numerous studies have implied that HGF promotes the malignant transformation of epithelial cells and also promotes cancer progression. The peptide hormone relaxin reduces the numbers of activated fibro-blasts and decreases fibrosis109,110. On this basis, clinical trials to test the efficacy of relaxin were initiated in patients with scleroderma109,111,112. However, increased relaxin serum concentrations correlate with a poor prognosis in patients with breast cancer, and relaxin directly increases the invasiveness of breast cancer cells113,114.
In summary, targeting CAFs as a therapeutic strat-egy against cancer is an intriguing concept that needs further study. The ideal therapeutic molecules will be ones that have been engineered to remove cancer-promoting properties but retain anti-fibrotic ones.
Figure 4 | Regulation of epithelial-to-mesenchymal transition by the microenvironment. Various stimuli within the tumour microenvironment promote epithelial-to-mesenchymal transition (EMT) of carcinoma cells. These include growth factors that bind to tyrosine-kinase receptors (TKR), such as fibroblast growth factor (FGF), epidermal growth factor (EGF) and hepatocyte growth factor (HGF), members of the transforming growth factor-β (TGFβ) superfamily that bind to TGFβ receptors (TGFβR), extracellular-matrix constituents that facilitate the integrin-mediated cell response, and factors that directly cause disruption of the E-cadherin–β-catenin complex such as matrix metalloproteinase 3 (MMP3), which cleaves E-cadherin. All of these exogenous stimuli can be contributed by cancer-associated fibroblasts (CAFs), indicating that they have an important role as promoters of cancer-cell EMT. Integrins, such as α2β1 integrin, induce EMT through focal-adhesion kinase (FAK)-mediated signalling. Activation of tyrosine-kinase receptors induces EMT predominantly through mitogen-activated protein kinase (MAPK). Activation of Ras induces Akt, potentially accounting for the increased resistance to apoptotic stimuli that is associated with EMT. TGFβ action is mediated by Smad signalling. Small GTPases such as RHOA have also been suggested to induce EMT of cancer cells in response to TGFβ. MMP3, but also MMP2 and MMP9, have been shown to cleave E-cadherin, which leads to the internalization of E-cadherin and the relocalization of transcriptionally active β-catenin to the nucleus, inducing EMT. PI3K, phosphatidylinositol 3-kinase.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 6 | MAY 2006 | 399
© 2006 Nature Publishing Group

1. Kalluri, R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nature Rev. Cancer 3, 422–433 (2003).A comprehensive review on ECM and tumour angiogenesis.
2. Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971).The first publication to indicate that angiogenesis is a target for controlling cancer.
3. O’Reilly, M. S. et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285 (1997).
4. Ishii, G. et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 309, 232–240 (2003).
5. Kim, K. J. et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362, 841–844 (1993).
6. Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
7. de Visser, K. E., Korets, L. V. & Coussens, L. M. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 7, 411–423 (2005).
8. Tlsty, T. D. & Hein, P. W. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr. Opin. Genet. Dev. 11, 54–59 (2001).An interesting review of tumour stromal cells and their role in oncogenic manipulation.
9. Elenbaas, B. & Weinberg, R. A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 264, 169–184 (2001).
10. Mueller, M. M. & Fusenig, N. E. Friends or foes — bipolar effects of the tumour stroma in cancer. Nature Rev. Cancer 4, 839–849 (2004).An in-depth review about tumour stroma.
11. Virchow, R. Die Cellularpathologie in Ihrer Begruendung auf Physiologische und Pathologische Gewebelehre (Hirschwald, A., Berlin, Germany, 1858).
12. Duvall, M. Atlas d’Embryologie (Masson, Paris, 1879).13. Tarin, D. & Croft, C. B. Ultrastructural features of
wound healing in mouse skin. J. Anat. 105, 189–190 (1969).
14. Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C. & Brown, R. A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nature Rev. Mol. Cell Biol. 3, 349–363 (2002).
15. Parsonage, G. et al. A stromal address code defined by fibroblasts. Trends Immunol. 26, 150–156 (2005).
16. Rodemann, H. P. & Muller, G. A. Characterization of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am. J. Kidney Dis. 17, 684–686 (1991).
17. Chang, H. Y. et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl Acad. Sci. USA 99, 12877–12882 (2002).This study compares primary fibroblasts that were isolated from various sites and sheds new light on the vast diversity of fibroblasts.
18. Simian, M. et al. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development 128, 3117–3131 (2001).
19. Wiseman, B. S. & Werb, Z. Stromal effects on mammary gland development and breast cancer. Science 296, 1046–1049 (2002).
20. Castor, C. W., Wilson, S. M., Heiss, P. R. & Seidman, J. C. Activation of lung connective tissue cells in vitro. Am. Rev. Respir. Dis. 120, 101–106 (1979).
21. Muller, G. A. & Rodemann, H. P. Characterization of human renal fibroblasts in health and disease: I. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am. J. Kidney Dis. 17, 680–683. (1991).
22. Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).A comprehensive review on cancer.
23. Ronnov-Jessen, L., Petersen, O. W. & Bissell, M. J. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol. Rev. 76, 69–125 (1996).
24. Dvorak, H. F., Form, D. M., Manseau, E. J. & Smith, B. D. Pathogenesis of desmoplasia. I. Immunofluorescence identification and localization of
some structural proteins of line 1 and line 10 guinea pig tumors and of healing wounds. J. Natl Cancer Inst. 73, 1195–1205 (1984).
25. Dolberg, D. S., Hollingsworth, R., Hertle, M. & Bissell, M. J. Wounding and its role in RSV-mediated tumor formation. Science 230, 676–678 (1985).
26. Sieweke, M. H., Thompson, N. L., Sporn, M. B. & Bissell, M. J. Mediation of wound-related Rous sarcoma virus tumorigenesis by TGF-β. Science 248, 1656–1660 (1990).
27. Brown, L. F. et al. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin. Cancer Res. 5, 1041–1056 (1999).
28. Fukumura, D. et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 94, 715–725 (1998).
29. Senger, D. R. et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219, 983–985 (1983).
30. Dvorak, H. F. et al. Distribution of vascular permeability factor (vascular endothelial growth factor) in tumors: concentration in tumor blood vessels. J. Exp. Med. 174, 1275–1278 (1991).
31. Feng, D. et al. Ultrastructural localization of the vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) receptor-2 (FLK-1, KDR) in normal mouse kidney and in the hyperpermeable vessels induced by VPF/VEGF-expressing tumors and adenoviral vectors. J. Histochem. Cytochem. 48, 545–556 (2000).
32. Leung, D. W., Cachianes, G., Kuang, W. J., Goeddel, D. V. & Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246, 1306–1309 (1989).
33. Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
34. Schedin, P. & Elias, A. Multistep tumorigenesis and the microenvironment. Breast Cancer Res. 6, 93–101 (2004).
35. Shekhar, M. P., Pauley, R. & Heppner, G. Host microenvironment in breast cancer development: extracellular matrix–stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res. 5, 130–135 (2003).
36. van Kempen, L. C., Ruiter, D. J., van Muijen, G. N. & Coussens, L. M. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur. J. Cell Biol. 82, 539–548 (2003).
37. Brown, L. F., Dvorak, A. M. & Dvorak, H. F. Leaky vessels, fibrin deposition, and fibrosis: a sequence of events common to solid tumors and to many other types of disease. Am. Rev. Respir. Dis. 140, 1104–1107 (1989).
38. Folkman, J., Watson, K., Ingber, D. & Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 339, 58–61 (1989).
39. Chiquet-Ehrismann, R., Mackie, E. J., Pearson, C. A. & Sakakura, T. Tenascin: an extracellular matrix protein involved in tissue interactions during fetal development and oncogenesis. Cell 47, 131–139 (1986).
40. Mackie, E. J. et al. Tenascin is a stromal marker for epithelial malignancy in the mammary gland. Proc. Natl Acad. Sci. USA 84, 4621–4625 (1987).
41. Ishihara, A., Yoshida, T., Tamaki, H. & Sakakura, T. Tenascin expression in cancer cells and stroma of human breast cancer and its prognostic significance. Clin. Cancer Res. 1, 1035–1041 (1995).
42. Brunner, A. et al. Prognostic significance of tenascin-C-expression in superficial and invasive bladder cancer. J. Clin. Pathol. 57, 927–931 (2004).
43. De Wever, O. & Mareel, M. Role of myofibroblasts at the invasion front. Biol. Chem. 383, 55–67 (2002).
44. Bertrand, P. et al. Hyaluronan (hyaluronic acid) and hyaluronectin in the extracellular matrix of human breast carcinomas: comparison between invasive and non-invasive areas. Int. J. Cancer 52, 1–6 (1992).
45. Barsky, S. H. & Gopalakrishna, R. Increased invasion and spontaneous metastasis of BL6 melanoma with inhibition of the desmoplastic response in C57 BL/6 mice. Cancer Res. 47, 1663–1667 (1987).
46. Netti, P. A., Berk, D. A., Swartz, M. A., Grodzinsky, A. J. & Jain, R. K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 60, 2497–2503 (2000).
47. Brown, E. B., Boucher, Y., Nasser, S. & Jain, R. K. Measurement of macromolecular diffusion coefficients in human tumors. Microvasc. Res. 67, 231–236 (2004).
48. Hynes, R. O. Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69, 11–25 (1992).
49. Ryan, G. B. et al. Myofibroblasts in an avascular fibrous tissue. Lab. Invest. 29, 197–206 (1973).
50. Ryan, G. B. et al. Myofibroblasts in human granulation tissue. Hum. Pathol. 5, 55–67 (1974).
51. Tsukada, T., McNutt, M. A., Ross, R. & Gown, A. M. HHF35, a muscle actin-specific monoclonal antibody. II. Reactivity in normal, reactive, and neoplastic human tissues. Am. J. Pathol. 127, 389–402 (1987).
52. Schor, S. L., Schor, A. M., Grey, A. M. & Rushton, G. Foetal and cancer patient fibroblasts produce an autocrine migration-stimulating factor not made by normal adult cells. J. Cell Sci. 90, 391–399 (1988).
53. Durning, P., Schor, S. L. & Sellwood, R. A. Fibroblasts from patients with breast cancer show abnormal migratory behaviour in vitro. Lancet 2, 890–892 (1984).
54. Barsky, S. H., Green, W. R., Grotendorst, G. R. & Liotta, L. A. Desmoplastic breast carcinoma as a source of human myofibroblasts. Am. J. Pathol. 115, 329–333 (1984).
55. Sappino, A. P., Skalli, O., Jackson, B., Schurch, W. & Gabbiani, G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 41, 707–712 (1988).
56. Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 (2003).
57. Ronnov-Jessen, L. & Petersen, O. W. Induction of α-smooth muscle actin by transforming growth factor-β1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 68, 696–707 (1993).
58. Lohr, M. et al. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 61, 550–555 (2001).
59. Aoyagi, Y. et al. Overexpression of TGF-β by infiltrated granulocytes correlates with the expression of collagen mRNA in pancreatic cancer. Br. J. Cancer 91, 1316–1326 (2004).
60. Akhurst, R. J. TGF-β antagonists: why suppress a tumor suppressor? J. Clin. Invest. 109, 1533–1536 (2002).
61. Bronzert, D. A. et al. Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc. Natl Acad. Sci. USA 84, 5763–5767 (1987).
62. Forsberg, K., Valyi-Nagy, I., Heldin, C. H., Herlyn, M. & Westermark, B. Platelet-derived growth factor (PDGF) in oncogenesis: development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl Acad. Sci. USA 90, 393–397 (1993).
63. Shao, Z. M., Nguyen, M. & Barsky, S. H. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene 19, 4337–4345 (2000).
64. Armelin, H. A. Pituitary extracts and steroid hormones in the control of 3T3 cell growth. Proc. Natl Acad. Sci. USA 70, 2702–2706 (1973).
65. Folkman, J. et al. A heparin-binding angiogenic protein — basic fibroblast growth factor — is stored within basement membrane. Am. J. Pathol. 130, 393–400 (1988).
66. Strutz, F. et al. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 57, 1521–1538. (2000).
67. Hay, E. D. An overview of epithelio–mesenchymal transformation. Acta Anat. 154, 8–20 (1995).
68. Thiery, J. P. Epithelial–mesenchymal transitions in tumor progression. Nature Rev. Cancer 2, 442–454 (2002).A good review on the role of EMT in cancer.
69. Kalluri, R. & Neilson, E. G. Epithelial–mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 112, 1776–1784 (2003).
70. Fukino, K. et al. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 64, 7231–7236 (2004).
71. Tuhkanen, H. et al. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int. J. Cancer 109, 247–252 (2004).
72. Ellsworth, D. L. et al. Genomic patterns of allelic imbalance in disease free tissue adjacent to primary breast carcinomas. Breast Cancer Res. Treat. 88, 131–139 (2004).
R E V I E W S
400 | MAY 2006 | VOLUME 6 www.nature.com/reviews/cancer
© 2006 Nature Publishing Group

73. Kurose, K. et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nature Genet. 32, 355–357 (2002).
74. Petersen, O. W. et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 162, 391–402 (2003).An interesting study indicating that cancer cells can contribute to the accumulation of stromal fibroblasts by undergoing a complete EMT.
75. Zeisberg, M. & Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 82, 175–181 (2004).
76. Zeisberg, M. et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature Med. 9, 964–968 (2003).The first study to indicate that targeting EMT can reduce the numbers of tissue fibroblasts and reverse fibrosis.
77. Iwano, M. et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 110, 341–350. (2002).Provides evidence that FSP1 protein is a fibroblast-specific protein, and provides further evidence relating to the role of EMT in organ fibrosis.
78. Kopelovich, L. Genetic predisposition to cancer in man: in vitro studies. Int. Rev. Cytol. 77, 63–88 (1982).
79. Schor, S. L. et al. Occurrence of a fetal fibroblast phenotype in familial breast cancer. Int. J. Cancer 37, 831–836 (1986).
80. Kuperwasser, C. et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl Acad. Sci. USA 101, 4966–4971 (2004).Demonstrates that fibroblasts aid mammary epithelia in forming functional breast tissue, and carcinogenesis.
81. Bhowmick, N. A. et al. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303, 848–851 (2004).Indicates that deletion of TGFβ type II receptor in FSP1-positive cells leads to the emergence of epithelial tumours.
82. Orimo, A. et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 (2005).Indicates a prominent role for fibroblasts in tumour angiogenesis facilitated by SDF1.
83. Olumi, A. F. et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 59, 5002–5011 (1999).A study that used co-culture methods to elucidate the role of CAFs in cancer progression.
84. Dimanche-Boitrel, M. T. et al. In vivo and in vitro invasiveness of a rat colon-cancer cell line maintaining E-cadherin expression: an enhancing role of tumor-associated myofibroblasts. Int. J. Cancer 56, 512–521 (1994).
85. Boire, A. et al. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313 (2005).
86. Stetler-Stevenson, W. G., Aznavoorian, S. & Liotta, L. A. Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 9, 541–573 (1993).
87. Sternlicht, M. D. et al. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 98, 137–146 (1999).
88. Lochter, A. et al. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 139, 1861–1872 (1997).
89. Olaso, E. et al. Tumor-dependent activation of rodent hepatic stellate cells during experimental melanoma metastasis. Hepatology 26, 634–642 (1997).
90. Olaso, E. et al. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology 37, 674–685 (2003).
91. Cornil, I. et al. Fibroblast cell interactions with human melanoma cells affect tumor cell growth as a function of tumor progression. Proc. Natl Acad. Sci. USA 88, 6028–6032 (1991).
92. Grum-Schwensen, B. et al. Suppression of tumor development and metastasis formation in mice lacking the S100A4mts1 gene. Cancer Res. 65, 3772–3780 (2005).
93. Bhowmick, N. A., Neilson, E. G. & Moses, H. L. Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337 (2004).
94. Siegel, P. M. & Massague, J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nature Rev. Cancer 3, 807–821 (2003).
95. Bierie, B. & Moses, H. L. TGF-β and cancer. Cytokine Growth Factor Rev. 17, 29–40 (2006).
96. Derynck, R., Akhurst, R. J. & Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nature Genet. 29, 117–129 (2001).
97. Cheng, N. et al. Loss of TGF-β type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-α-, MSP- and HGF-mediated signaling networks. Oncogene 24, 5053–5068 (2005).
98. Silzle, T., Randolph, G. J., Kreutz, M. & Kunz-Schughart, L. A. The fibroblast: sentinel cell and local immune modulator in tumor tissue. Int. J. Cancer 108, 173–180 (2004).
99. Rettig, W. J. et al. Regulation and heteromeric structure of the fibroblast activation protein in normal and transformed cells of mesenchymal and neuroectodermal origin. Cancer Res. 53, 3327–3335 (1993).
100. Wang, X. M., Yu, D. M., McCaughan, G. W. & Gorrell, M. D. Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology 42, 935–945 (2005).
101. Scott, A. M. et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 9, 1639–1647 (2003).
102. Zeisberg, M. et al. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Renal Physiol. 285, F1060–F1067 (2003).
103. Zeisberg, M., Strutz, F. & Muller, G. A. Role of fibroblast activation in inducing interstitial fibrosis. J. Nephrol. 13 (Suppl.), S111–S120. (2000).
104. Nakamura, T. et al. Hepatocyte growth factor prevents tissue fibrosis, remodeling, and dysfunction in cardiomyopathic hamster hearts. Am. J. Physiol. Heart Circ. Physiol. 288, H2131–H2139 (2005).
105. Kim, W. H., Matsumoto, K., Bessho, K. & Nakamura, T. Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am. J. Pathol. 166, 1017–1028 (2005).
106. Dai, C. et al. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J. Am. Soc. Nephrol. 15, 2637–2647 (2004).
107. Isaka, Y. et al. Electroporation-mediated HGF gene transfection protected the kidney against graft injury. Gene Ther. 12, 815–820 (2005).
108. Yang, J., Dai, C. & Liu, Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J. Am. Soc. Nephrol. 13, 2464–2477 (2002).
109. McDonald, G. A. et al. Relaxin increases ubiquitin-dependent degradation of fibronectin in vitro and ameliorates renal fibrosis in vivo. Am. J. Physiol. Renal Physiol. 285, F59–F67 (2003).
110. Heeg, M. H. et al. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 68, 96–109 (2005).
111. Samuel, C. S. et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 145, 4125–4133 (2004).
112. Seibold, J. R. et al. Recombinant human relaxin in the treatment of scleroderma. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 132, 871–879 (2000).
113. Binder, C. et al. Elevated concentrations of serum relaxin are associated with metastatic disease in breast cancer patients. Breast Cancer Res. Treat. 87, 157–166 (2004).
114. Binder, C., Hagemann, T., Husen, B., Schulz, M. & Einspanier, A. Relaxin enhances in-vitro invasiveness of breast cancer cell lines by up-regulation of matrix metalloproteases. Mol. Hum. Reprod. 8, 789–796 (2002).
115. Lazarides, E. & Balzer, D. R. Jr. Specificity of desmin to avian and mammalian muscle cells. Cell 14, 429–438 (1978).
116. Franke, W. W., Schmid, E., Osborn, M. & Weber, K. Different intermediate-sized filaments distinguished by immunofluorescence microscopy. Proc. Natl Acad. Sci. USA 75, 5034–5038 (1978).
117. Franke, W. W., Schmid, E., Osborn, M. & Weber, K. Intermediate-sized filaments of human endothelial cells. J. Cell Biol. 81, 570–580 (1979).
118. Mork, C., van Deurs, B. & Petersen, O. W. Regulation of vimentin expression in cultured human mammary epithelial cells. Differentiation 43, 146–156 (1990).
119. Schmid, E. et al. Distribution of vimentin and desmin filaments in smooth muscle tissue of mammalian and avian aorta. Exp. Cell Res. 137, 329–340 (1982).
120. Strutz, F. et al. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 130, 393–405 (1995).
121. Vogel, W., Gish, G. D., Alves, F. & Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 (1997).
122. Goldsmith, E. C. et al. Organization of fibroblasts in the heart. Dev. Dyn. 230, 787–794 (2004).
123. Ramirez-Montagut, T. et al. FAPα, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene 23, 5435–5446 (2004).
124. Gardner, H., Kreidberg, J., Koteliansky, V. & Jaenisch, R. Deletion of integrin α1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev. Biol. 175, 301–313 (1996).
125. Sudhakar, A. et al. Human α1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by α1β1 integrin. J. Clin. Invest. 115, 2801–2810 (2005).
126. Mussini, E., Hutton, J. J. Jr & Udenfriend, S. Collagen proline hydroxylase in wound healing, granuloma formation, scurvy, and growth. Science 157, 927–929 (1967).
127. Langness, U. & Udenfriend, S. Collagen biosynthesis in nonfibroblastic cell lines. Proc. Natl Acad. Sci. USA 71, 50–51 (1974).
128. Florin, L. et al. Cre recombinase-mediated gene targeting of mesenchymal cells. Genesis 38, 139–144 (2004).
129. Hogan, B. L. & Yingling, J. M. Epithelial–mesenchymal interactions and branching morphogenesis of the lung. Curr. Opin. Genet. Dev. 8, 481–486 (1998).
130. Clayton, A. et al. Cellular activation through the ligation of intercellular adhesion molecule-1. J. Cell Sci. 111, 443–453 (1998).
131. Strieter, R. M. et al. Monocyte chemotactic protein gene expression by cytokine-treated human fibroblasts and endothelial cells. Biochem. Biophys. Res. Commun. 162, 694–700 (1989).
132. Rollins, B. J., Stier, P., Ernst, T. & Wong, G. G. The human homolog of the JE gene encodes a monocyte secretory protein. Mol. Cell. Biol. 9, 4687–4695 (1989).
133. Duda, D. G. et al. Differential transplantability of tumor-associated stromal cells. Cancer Res. 64, 5920–5924 (2004).
134. Grupp, C. et al. A novel model to study renal myofibroblast formation in vitro. Kidney Int. 59, 543–553 (2001).
AcknowledgementsThe work from the author’s laboratories was supported by the National Institutes of Health (NIH) and the Center for Matrix Biology (R.K.). M.Z. is funded by a NIH training grant to BIDMC. We would like to extend our special thanks to H. Dvorak and M. A. Grant for critical reading of this manuscript.
Competing interests statementThe authors declare no competing financial interests.
DATABASESThe following terms in this article are linked online to:Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=geneEGF | FAP | FGF2 | FSP1 | HGF | MMP1 | MMP3 | SDF1 | Tenascin C | TGFβ | VEGFNational Cancer Institute: http://www.cancer.govbladder cancer | breast cancer | melanoma
FURTHER INFORMATIONRaghu Kalluri’s web page: http://www.hms.harvard.edu/dms/bbs/fac/kallurira.htmlAccess to this links box is available online.
R E V I E W S
NATURE REVIEWS | CANCER VOLUME 6 | MAY 2006 | 401
© 2006 Nature Publishing Group




















