
Fast Two-Dimensional NMR Spectroscopy of High Molecular Weight Protein Assemblies
9
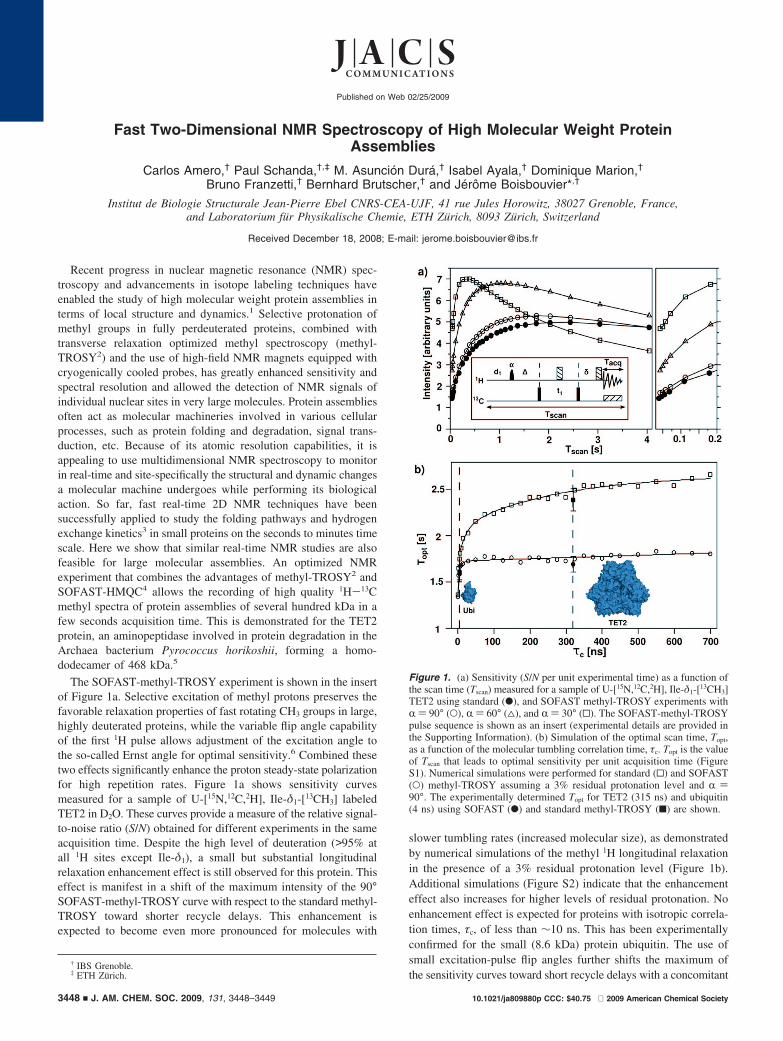
Fast Two-Dimensional NMR Spectroscopy of High Molecular Weight Protein Assemblies Carlos Amero, † Paul Schanda, †,‡ M. Asuncio ´n Dura ´, † Isabel Ayala, † Dominique Marion, † Bruno Franzetti, † Bernhard Brutscher, † and Je ´ro ˆ me Boisbouvier* ,† Institut de Biologie Structurale Jean-Pierre Ebel CNRS-CEA-UJF, 41 rue Jules Horowitz, 38027 Grenoble, France, and Laboratorium fu ¨r Physikalische Chemie, ETH Zu ¨rich, 8093 Zu ¨rich, Switzerland Received December 18, 2008; E-mail: [email protected] Recent progress in nuclear magnetic resonance (NMR) spec- troscopy and advancements in isotope labeling techniques have enabled the study of high molecular weight protein assemblies in terms of local structure and dynamics. 1 Selective protonation of methyl groups in fully perdeuterated proteins, combined with transverse relaxation optimized methyl spectroscopy (methyl- TROSY 2 ) and the use of high-field NMR magnets equipped with cryogenically cooled probes, has greatly enhanced sensitivity and spectral resolution and allowed the detection of NMR signals of individual nuclear sites in very large molecules. Protein assemblies often act as molecular machineries involved in various cellular processes, such as protein folding and degradation, signal trans- duction, etc. Because of its atomic resolution capabilities, it is appealing to use multidimensional NMR spectroscopy to monitor in real-time and site-specifically the structural and dynamic changes a molecular machine undergoes while performing its biological action. So far, fast real-time 2D NMR techniques have been successfully applied to study the folding pathways and hydrogen exchange kinetics 3 in small proteins on the seconds to minutes time scale. Here we show that similar real-time NMR studies are also feasible for large molecular assemblies. An optimized NMR experiment that combines the advantages of methyl-TROSY 2 and SOFAST-HMQC 4 allows the recording of high quality 1 H- 13 C methyl spectra of protein assemblies of several hundred kDa in a few seconds acquisition time. This is demonstrated for the TET2 protein, an aminopeptidase involved in protein degradation in the Archaea bacterium Pyrococcus horikoshii, forming a homo- dodecamer of 468 kDa. 5 The SOFAST-methyl-TROSY experiment is shown in the insert of Figure 1a. Selective excitation of methyl protons preserves the favorable relaxation properties of fast rotating CH 3 groups in large, highly deuterated proteins, while the variable flip angle capability of the first 1 H pulse allows adjustment of the excitation angle to the so-called Ernst angle for optimal sensitivity. 6 Combined these two effects significantly enhance the proton steady-state polarization for high repetition rates. Figure 1a shows sensitivity curves measured for a sample of U-[ 15 N, 12 C, 2 H], Ile-δ 1 -[ 13 CH 3 ] labeled TET2 in D 2 O. These curves provide a measure of the relative signal- to-noise ratio (S/N) obtained for different experiments in the same acquisition time. Despite the high level of deuteration (>95% at all 1 H sites except Ile-δ 1 ), a small but substantial longitudinal relaxation enhancement effect is still observed for this protein. This effect is manifest in a shift of the maximum intensity of the 90° SOFAST-methyl-TROSY curve with respect to the standard methyl- TROSY toward shorter recycle delays. This enhancement is expected to become even more pronounced for molecules with slower tumbling rates (increased molecular size), as demonstrated by numerical simulations of the methyl 1 H longitudinal relaxation in the presence of a 3% residual protonation level (Figure 1b). Additional simulations (Figure S2) indicate that the enhancement effect also increases for higher levels of residual protonation. No enhancement effect is expected for proteins with isotropic correla- tion times, τ c , of less than ∼10 ns. This has been experimentally confirmed for the small (8.6 kDa) protein ubiquitin. The use of small excitation-pulse flip angles further shifts the maximum of the sensitivity curves toward short recycle delays with a concomitant † IBS Grenoble. ‡ ETH Zu ¨rich. Figure 1. (a) Sensitivity (S/N per unit experimental time) as a function of the scan time (T scan ) measured for a sample of U-[ 15 N, 12 C, 2 H], Ile-δ 1 -[ 13 CH 3 ] TET2 using standard (b), and SOFAST methyl-TROSY experiments with R) 90° (O), R) 60° (4), and R) 30° (0). The SOFAST-methyl-TROSY pulse sequence is shown as an insert (experimental details are provided in the Supporting Information). (b) Simulation of the optimal scan time, T opt , as a function of the molecular tumbling correlation time, τ c . T opt is the value of T scan that leads to optimal sensitivity per unit acquisition time (Figure S1). Numerical simulations were performed for standard (0) and SOFAST (O) methyl-TROSY assuming a 3% residual protonation level and R) 90°. The experimentally determined T opt for TET2 (315 ns) and ubiquitin (4 ns) using SOFAST (b) and standard methyl-TROSY (9) are shown. Published on Web 02/25/2009 10.1021/ja809880p CCC: $40.75 2009 American Chemical Society 3448 9 J. AM. CHEM. SOC. 2009, 131, 3448–3449
-
Upload
uaemmorelos -
Category
Documents
-
view
2 -
download
0
Transcript of Fast Two-Dimensional NMR Spectroscopy of High Molecular Weight Protein Assemblies
Fast Two-Dimensional NMR Spectroscopy of High Molecular Weight ProteinAssemblies
Carlos Amero,† Paul Schanda,†,‡ M. Asuncion Dura,† Isabel Ayala,† Dominique Marion,†
Bruno Franzetti,† Bernhard Brutscher,† and Jerome Boisbouvier*,†
Institut de Biologie Structurale Jean-Pierre Ebel CNRS-CEA-UJF, 41 rue Jules Horowitz, 38027 Grenoble, France,and Laboratorium fur Physikalische Chemie, ETH Zurich, 8093 Zurich, Switzerland
Received December 18, 2008; E-mail: [email protected]
Recent progress in nuclear magnetic resonance (NMR) spec-troscopy and advancements in isotope labeling techniques haveenabled the study of high molecular weight protein assemblies interms of local structure and dynamics.1 Selective protonation ofmethyl groups in fully perdeuterated proteins, combined withtransverse relaxation optimized methyl spectroscopy (methyl-TROSY2) and the use of high-field NMR magnets equipped withcryogenically cooled probes, has greatly enhanced sensitivity andspectral resolution and allowed the detection of NMR signals ofindividual nuclear sites in very large molecules. Protein assembliesoften act as molecular machineries involved in various cellularprocesses, such as protein folding and degradation, signal trans-duction, etc. Because of its atomic resolution capabilities, it isappealing to use multidimensional NMR spectroscopy to monitorin real-time and site-specifically the structural and dynamic changesa molecular machine undergoes while performing its biologicalaction. So far, fast real-time 2D NMR techniques have beensuccessfully applied to study the folding pathways and hydrogenexchange kinetics3 in small proteins on the seconds to minutes timescale. Here we show that similar real-time NMR studies are alsofeasible for large molecular assemblies. An optimized NMRexperiment that combines the advantages of methyl-TROSY2 andSOFAST-HMQC4 allows the recording of high quality 1H-13Cmethyl spectra of protein assemblies of several hundred kDa in afew seconds acquisition time. This is demonstrated for the TET2protein, an aminopeptidase involved in protein degradation in theArchaea bacterium Pyrococcus horikoshii, forming a homo-dodecamer of 468 kDa.5
The SOFAST-methyl-TROSY experiment is shown in the insertof Figure 1a. Selective excitation of methyl protons preserves thefavorable relaxation properties of fast rotating CH3 groups in large,highly deuterated proteins, while the variable flip angle capabilityof the first 1H pulse allows adjustment of the excitation angle tothe so-called Ernst angle for optimal sensitivity.6 Combined thesetwo effects significantly enhance the proton steady-state polarizationfor high repetition rates. Figure 1a shows sensitivity curvesmeasured for a sample of U-[15N,12C,2H], Ile-δ1-[13CH3] labeledTET2 in D2O. These curves provide a measure of the relative signal-to-noise ratio (S/N) obtained for different experiments in the sameacquisition time. Despite the high level of deuteration (>95% atall 1H sites except Ile-δ1), a small but substantial longitudinalrelaxation enhancement effect is still observed for this protein. Thiseffect is manifest in a shift of the maximum intensity of the 90°SOFAST-methyl-TROSY curve with respect to the standard methyl-TROSY toward shorter recycle delays. This enhancement isexpected to become even more pronounced for molecules with
slower tumbling rates (increased molecular size), as demonstratedby numerical simulations of the methyl 1H longitudinal relaxationin the presence of a 3% residual protonation level (Figure 1b).Additional simulations (Figure S2) indicate that the enhancementeffect also increases for higher levels of residual protonation. Noenhancement effect is expected for proteins with isotropic correla-tion times, τc, of less than ∼10 ns. This has been experimentallyconfirmed for the small (8.6 kDa) protein ubiquitin. The use ofsmall excitation-pulse flip angles further shifts the maximum ofthe sensitivity curves toward short recycle delays with a concomitant
† IBS Grenoble.‡ ETH Zurich.
Figure 1. (a) Sensitivity (S/N per unit experimental time) as a function ofthe scan time (Tscan) measured for a sample of U-[15N,12C,2H], Ile-δ1-[13CH3]TET2 using standard (b), and SOFAST methyl-TROSY experiments withR ) 90° (O), R ) 60° (4), and R ) 30° (0). The SOFAST-methyl-TROSYpulse sequence is shown as an insert (experimental details are provided inthe Supporting Information). (b) Simulation of the optimal scan time, Topt,as a function of the molecular tumbling correlation time, τc. Topt is the valueof Tscan that leads to optimal sensitivity per unit acquisition time (FigureS1). Numerical simulations were performed for standard (0) and SOFAST(O) methyl-TROSY assuming a 3% residual protonation level and R )90°. The experimentally determined Topt for TET2 (315 ns) and ubiquitin(4 ns) using SOFAST (b) and standard methyl-TROSY (9) are shown.
Published on Web 02/25/2009
10.1021/ja809880p CCC: $40.75 2009 American Chemical Society3448 9 J. AM. CHEM. SOC. 2009, 131, 3448–3449

sensitivity increase of ∼40%. Similar results are obtained for asecond sample of U-[15N,12C,2H] Ala-�-[13CH3]-labeled TET2.
The SOFAST-methyl-TROSY experiment yields a significantsensitivity gain, especially in the context of very fast dataacquisition, such as is required for real-time NMR studies, whereshort recycle delays of a few milliseconds are used. In this fast-pulsing regime the S/N is increased by a factor of ∼3 compared toa conventional methyl-TROSY pulse scheme. Figure 2 shows
SOFAST-methyl-TROSY spectra of the TET2 protease recordedin a total acquisition time of ∼3 s on a 800 MHz spectrometerequipped with a cryogenic triple-resonance probe. 80 µM samplesof the 468 kDa oligomeric TET2 protease were used with selectivemethyl group isotope labeling of either 30 Ala7 or 34 Ile (δ1
position) residues.8 High quality methyl NMR spectra are obtainedfor both samples, indicating that this method provides usefulspectroscopic probes, both close to the polypeptide backbone (Alamethyls) and at the extremity of long and generally more flexibleside chains (Ile δ1 methyls).
The average S/N for individual peaks in these spectra is ∼10:1,as can be appreciated from the 1D traces shown in Figure 2. Underthese conditions, the SOFAST-methyl-TROSY experiments are stillin the sampling limited regime where the acquisition time is dictatedby the number of data points that need to be sampled along the t1
time evolution dimension. Therefore alternative sampling andprocessing techniques9 may be used in to reduce the number ofrepetitions (scans) and, thus, to achieve even shorter experimentaltimes for recording 2D methyl correlation spectra. Examples ofmethyl spectra of the TET2 protein recorded using limited time-
domain sampling in both the 1H and 13C dimensions and 2Dmaximum entropy10 spectral reconstruction (instead of Fouriertransformation) are shown in Figure S4 of the Supporting Informa-tion. Such studies allow the time resolution of real-time NMR tobe reduced to less than a second.3 These data illustrate thatSOFAST-methyl-TROSY enables monitoring fast molecular kinet-ics of reactions catalyzed by macromolecular machineries simul-taneously at multiple (methyl) sites within the molecule. Such real-time NMR studies may provide a comprehensive picture of themolecular mechanisms involved in enzymatic function.
In summary we have shown that selective excitation of CH3
groups accelerates the recovery of methyl proton polarization inhigh molecular weight protein assemblies in the presence of smallresidual protonation levels. Combined with Ernst angle excitation,this effect allows the recycle delay between successive scans to bereduced to a few milliseconds. SOFAST-methyl-TROSY NMRcombines the advantages of longitudinal and transverse spin-relaxation optimization to enable acquisition of high quality methyl1H-13C correlation spectra of protein assemblies of several hundredsof kDa in a few seconds acquisition time. This time requirementcan be further reduced to <1 s using sparse time-domain samplingschemes combined with advanced nonlinear data processingmethods. SOFAST-methyl-TROSY-based real-time NMR offersnew opportunities for the study of structural and dynamic changesoccurring in molecular nanomachines while they perform theirbiological function in Vitro. This technique may also prove usefulfor real-time investigation of macromolecular folding and self-assembly. Therefore we believe that the NMR method presentedhere provides an important new analytical tool for the investigationand characterization of complex biomolecular assemblies and theirimplication in various biochemical processes in the cell.
Acknowledgment. J.B. acknowledges funding from HFSP(CDA) and CNRS (PIR interface physics/biology/chemistry), andB.B. and B.F. acknowledge funding from ANR. The authors thankDrs. M. Plevin, P. Gans, and R. Sounier for stimulating discussions.
Supporting Information Available: Description of Methyl SO-FAST-TROSY pulse sequence, protocols for preparation of proteinsamples, simulations of methyl proton polarization recovery for differentprotonation levels, and NMR spectra. This material is available free ofcharge via the Internet at http://pubs.acs.org.
References
(1) Fiaux, J.; Bertelsen, E. B.; Horwich, A. L.; Wuthrich, K. Nature 2002,418, 207–211. Wider, G. Methods Enzymol. 2005, 38, 2–398. (b) Sprangers,R.; Kay, L. E. Nature 2007, 445, 618–622. (c) Foster, M. P.; McElroy,C. A.; Amero, C. D. Biochemistry 2007, 46, 331–340.
(2) Tugarinov, V.; Hwang, P. M.; Ollerenshaw, J. E.; Kay, L. E. J. Am. Chem.Soc. 2003, 125, 10420–10428.
(3) Schanda, P.; Forge, V.; Brutscher, B. Proc. Natl. Acad. Sci. U.S.A. 2007,104, 11257–11262. Schanda, P.; Brutscher, B.; Konrat, R.; Tollinger, M.J. Mol. Biol. 2008, 380, 726–741.
(4) Schanda, P.; Brutscher, B. J. Am. Chem. Soc. 2005, 127, 8014–8015.Schanda, P.; Kupce, E.; Brutscher, B. J. Biomol. NMR 2005, 33, 199–211.
(5) Dura, M. A.; Receveur-Brechot, V.; Andrieu, J.; Ebel, C.; Schoehn, G.;Roussel, A.; Franzetti, B. Biochemistry 2005, 44, 3477–3486.
(6) Ernst, R. R.; Bodenhausen, G.; Wokaun, A. Principles of NuclearMagnetic Resonance in One and Two Dimensions; Oxford University Press:1987.
(7) Isaacson, R. L.; Simpson, P. J.; Liu, M.; Cota, E.; Zhang, X.; Freemont,P.; Matthews, S. J. Am. Chem. Soc. 2007, 129, 15428–15429. (a) Ayala,I.; Sounier, R.; Use, N.; Gans, P.; Boisbouvier, J. J. Biomol. NMR 2009,43, 111–119.
(8) Gardner, K. H.; Kay, L. E. J. Am. Chem. Soc. 1997, 119, 7599–600. (a)Tugarinov, V.; Kanelis, V.; Kay, L. E. Nat. Protoc. 2006, 1, 749–754. (b)Sounier, R.; Blanchard, L.; Wu, Z.; Boisbouvier, J. J. Am. Chem. Soc. 2007,129, 472–473.
(9) Freeman, R.; Kupce, E. J. Biomol. NMR 2003, 27, 101–113.(10) Hoch, J. C.; Stern, A. S. Methods Enzymol. 2002, 338, 159–178.
JA809880P
Figure 2. SOFAST-methyl-TROSY spectra acquired on ∼80 µM samplesof the 468 kDa TET2 protease in D2O (∼1 mM of protomer). (a) Ala methylspectra7 recorded on the U-[15N,12C,2H], Ala-�-[13CH3]-TET2 sample. (b)Ile-δ1 methyl spectra8 recorded on the U-[15N,12C,2H], Ile-δ1-[13CH3]-TET2sample. Both spectra have been acquired in 3.4 s at 37 °C on a 800 MHzspectrometer, with a 1 ms d1 delay, 30 ms acquisition time (Tacq), and 16complex points in the 13C dimension (t1). The variable flip angle was set to25° (a) and 18° (b). Additional 1D traces are shown on the left.
J. AM. CHEM. SOC. 9 VOL. 131, NO. 10, 2009 3449
C O M M U N I C A T I O N S

S 1
Fast Two-Dimensional NMR Spectroscopy of High Molecular Weight Protein Assemblies
Carlos Amero, Paul Schanda#, M. Asunción Durá, Isabel Ayala, Dominique Marion, Bruno Franzetti, Bernhard Brutscher, Jérôme Boisbouvier*.
Institut de Biologie Structurale Jean-Pierre Ebel CNRS-CEA-UJF, 41 rue Jules Horowitz, 38027 Grenoble, France #Laboratorium für Physikalische Chemie, ETH Zürich, 8093 Zürich, Switzerland
E-mail: [email protected]
Supporting Information
Samples preparation: -Production of U-[2H, 15N, 12C], Ile-δ1-[13CH3] proteins E. coli BL21(DE3) carrying the plasmid of the overexpressed protein were grown in three stages during 24 h to adapt them progressively to M9/D20 media containing 1 g/L 15ND4Cl, and 2 g/L D-glucose-d7. In the final culture, the bacteria were grown at 37°C in M9 media prepared with 99.85% D2O. When the O.D. (600 nm) reached 0.8, 100 mg/L of 2-keto-4-13C-3,3-d2-butyrate was added.1 After 1 hour, protein expression was induced by the addition of IPTG to a final concentration of 0.5 mM, then cells were grown at 37°C during 4 hours before harvesting.
-Production of U-[2H, 15N, 12C], Ala-β-[13CH3] proteins. E. coli BL21(DE3) carrying the plasmid of the overexpressed protein were grown in three
stages during 24 h to adapt them progressively to M9/D20 media containing 1 g/L 15ND4Cl, 2.5 g/L glycerol-d5 (Cambridge Isotope Laboratories, Inc.) and 1 g/L D-glucose-d7. In the final culture, the bacteria were grown at 37°C in M9 media prepared with 99.85% D2O. When the O.D. (600 nm) reached 0.8, a D2O solution containing methyl protonated alanine (prepared with the protocol described elsewhere2) and different perdeuterated precursors was added. After addition the concentrations in culture medium were: 800 mg/L of 2-[2H], 3-[13C] L-alanine; 2.5 g/L of succinate-d4 (Cambridge Isotope Laboratories, Inc.); 200 mg/L for 2-ketoisovalerate-d7 (CDN Isotopes, Inc.); and 60 mg/L for isoleucine-d10 (Cambridge Isotope Laboratories, Inc.). After 1 hour, protein expression was induced by the addition of IPTG to a final concentration of 0.5 mM, then cells were grown at 37°C during 4 hours before harvesting.
-Purification of proteins:
Ubiquitin was purified by Ni-NTA chromatography a single step. Typical final yield was 60 mg/L of methyl specific protonated purified ubiquitin. The final NMR sample consisted of 1mM of ubiquitin, 10mM of MES (pH=7.4 uncorrected) dissolved in 300 µL D2O.
TET2 was purified using two anion exchange chromatography steps (DEAE Sepharose CL-6B, and Resource Q 6 mL, GE Healthcare) followed by gel filtration (Superdex 200pg, GE Healthcare). Typical final yield was 20 mg/L of methyl specific protonated purified TET2. Samples prepared in this manner are fully active measured by hydrolytic activity using Leu-4-nitroanitide. The final NMR sample consisted of ~80 µM TET2 dodecamer in 20 mM Tris (pH=7.4 uncorrected), 20mM NaCl dissolved in 300 µL D2O.

S 2
Theoretical section: In order to simulate the 1H longitudinal relaxation of methyls in specifically protonated proteins, the evolution of the proton magnetization was calculated using full relaxation matrix approach:
€
−ddt
I1Z (t)I2Z (t).
InZ (t)
=
ρ1 σ12 . σ1nσ 21 ρ2 . σ 2n. . . .
σ n1 σ n2 . ρn
I1Z (t) − I1Z (0)I2Z (t) − I2Z (0)
.InZ (t) − InZ (0)
(1) IiZ(t) is the longitudinal magnetization of proton i. For simulation of the evolution of proton magnetization between two successive scans in a methyl-TROSY experiment, IiZ(0) was set to 0 for all protons. For the SOFAST methyl-TROSY experiment, IiZ(0) was set to 0 for methyl sites selectively protonated (i.e. Ile-δ1 only) and to 1 for other residual protons. In this simplified approach dipolar cross-correlated relaxation effects were neglected, and only intra-methyl (carbon-proton and proton-proton) dipolar interactions and proton-proton dipolar interactions between each methyl proton and remote protons were considered. The auto relaxation and cross relaxation rates constants ρi and σij are given by:
€
ρi = RHiCimethyl + RHiH jj≠i
∑ (2)
€
RHiCimethyl =
ξHC4
J(ωH −ωC ) + 3J(ωH ) + 6J(ωH +ωC )( ) (3)
€
RHiH j=ξHH4
J(0) + 3J(ωH ) + 6J(2ωH )( ) (4)
€
σ ij =ξHH4
6J(2ωH ) − J(0)( ) (5)
€
ξab =µo4π
2 h2π
2 γa
2γb2
rab6
(6) where RHC
methyl and RHH are the relaxation rates due to proton-carbon (intra methyl only) and proton-
proton dipolar interactions (intra-methyl and remote interactions). The spectral densities J(ω) are given by :
€
J(ω) =251−αS f
2( ) τ
1+ ωτ( )2+αS f
2 τ c1+ ωτ c( )2
(7)

S 3
where α=(P2(cosθ))2 and 1/τ=1/τC+1/τe. The factor α takes into account the fast methyl rotation. θ is the angle between the dipole vector and the rotation axis, α=1/9 for intra-methyl proton-carbon dipolar interaction and 1/4 for intra-methyl proton-proton dipolar interaction. Sf
2 and τe are the order parameter and effective correlation time reflecting other forms of local dynamics. Sf
2 =0.6, τe =10 ps were used for intramethyl (proton-proton and proton-carbon) interactions and αSf
2 =1 was used for dipolar interactions between remote protons. For simulation we have used TET2 as a model structure (PDB code 1Y0R). Ile-δ1 methyls were considered protonated with an occupancy level of 100%. All exchangeable hydrogen sites were perdeuterated. Residual protonation was generated by random introduction of spins in 3% of non-exchangeable hydrogen sites. For each value of correlation time τC, the equation (1) was integrated numerically and the available proton longitudinal magnetization IiZ for each Ile-δ1 methyl site was plotted as a function of Tscan. T1 was calculated as the time where IiZ(t) reaches 63% (≈1-e-1) of its thermal equilibrium value. The T1 for each methyl was averaged for 10 sets of random distribution of residual protons. The average values of Topt (≈1.25 T1) for the 32 Ile-δ1 methyl sites of TET2 are reported in Figure 1b for different values of τC. The correlation time (τC) of TET2 was fixed to 315 ns, based on a comparison of relaxation rates measured on Malate synthase G (82 kDa, τC=55 ns) in similar condition of buffer and temperature. Comparison between experimental and simulated recovery curves for TET2 are presented in Figure 1 and S1.
Figure S1. Comparison between experimental data for TET2-Ile (open circles) and simulated data (dash line) for the SOFAST-methyl-TROSY experiment with α=90°. a) Recovery curve following 90°-SOFAST-methyl-TROSY sequence. b) Sensitivity curve (S/N per unit experimental time) as a function of the scan time. Simulations were performed using TET2 structure assuming 3% residual proton level.
Figure S2 presents simulation of the optimal scan delay Topt as a function of correlation time τC,
for different level of residual protonation ranging from 1% to 10% of non-exchangeable protons sites.
These simulations indicate that the recovery of methyl magnetization in SOFAST-methyl-TROSY
experiment is only weakly affected by the presence of extra residual protons. The recovery of fast

S 4
rotating methyl 1H magnetization in SOFAST-methyl-TROSY is dominated by intramethyl 1H-1H and 13C-1H dipolar interactions (Eq. 3-4, 7).
In the other hand, for large methyl specific protonated proteins, the longitudinal relaxation of
methyl in standard methyl-TROSY is significantly slower in presence of residual protons. This result is
due to magnetization transfer via dipolar interactions (NOEs) between slow relaxing residual protons
and fast relaxing methyl groups.
Higher levels of residual protonation are not considered in these simulations, in order to preserve
the favorable transversal relaxation properties of methyl proton in high molecular weight proteins.
These results demonstrate the applicability of the SOFAST-methyl-TROSY approach for different level
of residual protonation.
Figure S2. Simulation of the optimal scan time (Topt) as a function of the molecular tumbling correlation time τC. Numerical simulations were performed with different residual proton level (1%, 3%, 5%, 7% and 10%). Solid lines correspond to standard Methyl-TROSY and dashed lines to SOFAST-methyl-TROSY.

S 5
Figure S3. Pulse scheme of the SOFAST-methyl-TROSY experiment. Filled and open pulse symbols indicate 90° and 180° rf pulses, except for the 1H excitation pulse applied with flip-angle α. The variable flip-angle pulse has a PC9 shape,3 and a pulse length, δ = 8 ms. The high power band-selective 1H pulse has a BIP-720-50-20 shapes.4 The delay, d1, was set to 1 ms, the transfer delay, Δ, is set to 1/(2JHC) and the delay, Δ', is set to Δ' = Δ − δ/2. The acquisition time Tacq was 30 ms for spectra in Figure 2 and Figure S2c,d and 10ms for Figure S2e,f. The 1H and 13C frequencies are positioned at the center of the methyl region of interest. The pulsed field gradient pulse applied along the z-axis (PFGZ) has a duration of 200 µs followed by a recovery delay of 100 µs. The phase cycle is: ϕ = x, -x.

S6
Figures S4: Comparison of 2D 1H-13C methyl-TROSY spectra. The data were recorded at 37°C for ~ 80 µM samples of the 468 kDa homo dodecameric TET2 protease in D2O buffer (800 MHz). a) Standard methyl-TROSY recorded in 15 min on U-[15N,12C,2H], Ala-β-[13CH3] TET2 sample. With 16 complex points in the 13C dimension (t1max = 15 ms). b) Standard methyl-TROSY recorded in 15 min on U-[15N,12C,2H], Ile-δ1-[
13CH3] TET2 sample. With 16 complex points in the 13C dimension (t1max = 16 ms). c) SOFAST-methyl-TROSY experiment recorded in 3.4 sec on U-[15N,12C,2H], Ala-β-[13CH3] TET2 sample. The variable flip-angle pulse was set to 25°, the 1H acquisition time was 30 ms, the d1 delay was set to 1 ms, 16 complex points were collected in the 13C dimension (t1max = 15 ms). d) SOFAST-methyl-TROSY experiment recorded in 3.4 sec on U-[15N,12C,2H], Ile-δ1-[
13CH3] TET2 sample. The variable flip-angle pulse was set to 18°, the 1H acquisition time was 30 ms, the d1 delay was set to 1 ms, 16 complex points were collected in the 13C dimension (t1max = 16 ms). e) SOFAST-methyl-TROSY experiment recorded in 1.3 sec on U-[15N,12C,2H], Ala-β-[13CH3] TET2 sample. The variable flip-angle pulse was set to 15°, the 1H acquisition time was reduced to 10 ms, the d1 delay was set to 1 ms, 10 complex points were collected in the 13C dimension (t1max = 9 ms). Processing was done using 2D maximum entropy reconstruction with a line narrowing of 30 Hz in both dimensions as implemented in RNMRTK5 software. f) SOFAST-methyl-TROSY experiment recorded in 1.6 sec on U-[15N,12C,2H], Ile-δ1-[
13CH3] TET2 sample. The variable flip-angle pulse was set to 14°, the 1H acquisition time was reduced to 10 ms, the d1 delay was set to 1 ms, 12 complex points were collected in the 13C dimension (t1max = 12 ms). Processing was done using 2D maximum entropy reconstruction with a line narrowing of 10 Hz Hz in both dimensions as implemented in RNMRTK5 software.

S7
REFERENCES (1) Gardner, K.; Kay, L. J. Am. Chem. Soc 1997, 119, 7599-7600.; Tugarinov, V.; Kanelis, V.; Kay, L. E. Nat Protoc 2006, 1, 749-754.; Sounier, R.; Blanchard, L.; Wu, Z.; Boisbouvier, J. J. Am. Chem. Soc. 2007, 129, 472-473. (2) Ayala, I.; Sounier, R.; Usé, N.; Gans, P.; Boisbouvier, J. J. Biomol. NMR in press. (3) Smith, M. A.; Hu, H.; Shaka, A. J. Journal of Magnetic Resonance 2001, 151, 269-283. (4) Kupce, E.; Freeman, R. Journal of magnetic resonance 1994, 108, 268-273. (5) Mobli, M.; Stern, A. S.; Hoch, J. C. J. Magn. Reson. 2006, 182, 96-105.; Mobli, M.; Maciejewski, M. W.; Gryk, M. R.; Hoch, J. C. J. Biomol. NMR 2007, 39, 133-139.




















