
Parent versus child reporting of tobacco smoke exposure at home and in the c
Upload
independentCategory
view
3download
0
1282
Exposure to Environmental Tobacco SmokeIncreases Myocardial Infarct Size in Rats
Bo-qing Zhu, MD; Yi-ping Sun, MD; Richard E. Sievers, BS; Stanton A. Glantz, PhD;William W. Parmley, MD; Christopher L. Wolfe, MD
Background Exposure to environmental tobacco smoke(ETS) has been epidemiologically linked to death from isch-emic heart disease in nonsmokers. In this study, we evaluatedthe influence of 3 days, 3 weeks, and 6 weeks of ETS exposureon myocardial infarct size in a rat ischemia/reperfusion model.Methods and Results Sprague-Dawley rats exposed to ETS
(four Marlboro cigarettes per 15 minutes, 6 hours per day, 5days per week) for 3 days (n=24), 3 weeks (n=21), or 6 weeks(n= 12) and control rats (n=24, n=21, and n=12, respectively)were subjected to 35 minutes of left coronary artery occlusionand 2 hours of reperfusion. Infarct size and risk area weredetermined by triphenyltetrazolium chloride and phthalocya-nine blue staining, respectively. Air nicotine, carbon monox-ide, and total particulates were measured during ETS expo-sure. Serum lipids, plasma carbon monoxide hemoglobin(COHb), nicotine, and cotinine concentrations were measuredin additional groups (6 to 13 rats each) exposed to 3 days, 3weeks, or 6 weeks of ETS and controls. Average air nicotine,
E nvironmental tobacco smoke (ETS), the tobaccocombustion products inhaled by nonsmokers inthe proximity of burning tobacco, is hazardous
because it contains high concentrations of ammonia,benzene, nicotine, carbon monoxide, and many othercarcinogens and irritants.1,2 The effects of passive smok-ing on health have been reported to include acuteeffects, such as exacerbation of asthma and angina, aswell as chronic effects, such as increased risk of lungcancer, respiratory tract infection, and atherosclero-sis.3-6 ETS also has been linked to death from ischemicheart disease in nonsmokers. Epidemiological studiesconducted by a number of investigators demonstrateapproximately a 30% increase in risk of death fromischemic heart disease or myocardial infarction in non-smokers living with smokers.3,4,7,8Although no one has previously reported the effect of
ETS exposure on myocardial infarct size, a number ofstudies suggest that ETS exposure adversely affects themyocardial oxygen supply-and-demand relation thatwould predispose the heart to develop ischemia orexacerbate preexisting ischemia. Direct or indirect ex-posure to tobacco smoke has been shown to increase thehemodynamic determinants of myocardial oxygen de-
Received September 8, 1993; revision accepted December 4,1993.From the Cardiovascular Division, Department of Medicine,
and the Cardiovascular Research Institute, University of Califor-nia, San Francisco.Correspondence to Christopher L. Wolfe, MD, 1186-Moffitt
Hospital, University of California, San Francisco, CA 94143-0124.
carbon monoxide, and total particulate concentrations were1103 ,ug/m3, 92 ppm, and 60 mg/m3 for the ETS-exposed rats.Infarct size (infarct mass/risk areax100%) increased signifi-cantly in the ETS groups compared with the control groups ina dose-dependent manner (P=.023), with longer exposureassociated with larger infarct size. Infarct size nearly doubledwith 6 weeks of ETS exposure (61+5% versus 34±3% forcontrol, mean+SEM). Plasma COHb, nicotine, and cotininelevels increased significantly in the ETS groups in a dose-dependent manner (all P<.001).
Conclusions Exposure to passive smoking increases myo-cardial infarct size in a rat model of ischemia and reperfu-sion. This increase of infarct size exhibited a dose-responserelation. These results are consistent with epidemiologicalstudies demonstrating that ETS increases the risk of heartdeath. (Circulation. 1994;89:1282-1290.)Key Words * smoking * myocardial infarction * nicotine
. carbon monoxide
mand910 at the same time that it potentially reducesboth myocardial blood supply and oxygen delivery byenhancing the development of coronary atherosclero-sis,1011 causing coronary vasoconstriction,11'2 and re-ducing the oxygen-carrying capacity of blood throughincreased serum carboxyhemoglobin (COHb) levels.9Furthermore, through an increase in platelet aggrega-tion, the likelihood of coronary thrombosis is enhancedafter ETS exposure.13-18 Finally, ETS exposure appearsto reduce oxidative phosphorylation in cardiac cells,rendering them less capable of producing ATP.19,20These changes suggest that ETS exposure not onlyincreases the likelihood of developing an acute myocar-dial infarction but also may increase the amount ofmyocardial damage once an acute myocardial infarctionhas occurred.
In this study, the effects of 3 days, 3 weeks, and 6weeks of ETS exposure on myocardial infarct size wereexamined in an in vivo rat model of coronary arteryocclusion and reperfusion. In addition, we examined theeffects of ETS exposure on serum lipids, bleeding time,plasma COHb, nicotine, and cotinine.
MethodsExperimental GroupsSprague-Dawley rats (225 to 250 g) were randomly divided
into the experimental and control groups described below. Therats were housed in separate cages in 1.92x 1.92x0.97 m (3.58mi3), well-mixed ETS exposure chambers (model H 5500,BioClean, Duo Flo, Lab Products Inc) as previously de-scribed.6 The interior volume of the exposure chamber approx-imates that of a Mazda 626 sedan (3.7 m3).21 The rats were
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from

Zhu et al Smoking Increases Infarct Size 1283
housed in a room maintained at a constant temperature andkept on a 12-hour light-dark cycle. The 3-day study includedtwo groups: group ETS-3 days (n=24) was exposed to side-stream smoke from Marlboro filter cigarettes (four cigarettesper 15 minutes, 6 hours per day, for 3 days) with a smokingmachine (RM 1/G, Heinr Borgwald GmbH, Hamburg, Ger-many). At the end of each day, the smoking machine wasturned off and the ventilation system of the BioClean chamberturned on to rapidly reduce the ETS level in the exposurechamber. The control animals (n=24) were in a separate roombreathing clear air for 3 days. The 3-week and 6-week studieseach included two groups: group ETS-3 weeks (n=21) andgroup ETS-6 weeks (n= 12) were exposed to the same dose ofETS (6 hours per day, 5 days per week) for 3 or 6 weeks; thecontrol animals (n=21 and n= 12, respectively) were in aseparate room breathing clear air for 3 or 6 weeks. All ratswere fed a regular diet.
Rat Model of Acute Myocardial Ischemiaand ReperfusionA rat model of left coronary artery occlusion and reper-
fusion was used as previously described.22-24 After inductionof anesthesia (pentobarbital, 40 mg/kg IP), a tracheostomywas performed, and the animal was ventilated on a HarvardRodent Respirator (model 683). A reversible coronary ar-tery snare occluder was placed around the proximal leftanterior descending coronary artery via a midline sternot-omy. After the snare occluder was visually tested during abrief period of occlusion and reperfusion, the thoracotomywas closed. The animals were then subjected to the experi-mental protocol.
Experimental ProtocolAll six groups (12 to 24 rats each) were subjected to 35
minutes of left coronary artery occlusion followed by 120minutes of reperfusion. The occlusion/reperfusion protocolwas performed 10 to 20 minutes after the 6-hour ETS expo-sure period. The bleeding time was measured before the ratswere killed as described below. At the conclusion of theprotocol, infarct size was measured as described below.
Infarct SizingInfarct sizing was performed as described previously.25
The left coronary artery was reoccluded, and phthalocyanineblue dye was injected into the left ventricular cavity, allowingnormally perfused myocardium to stain blue. The heart wasthen excised, rinsed of excess dye, and sliced transverselyfrom apex to base into sections 2 mm thick. The tissuesamples were incubated in a 1% solution of triphenyltetra-zolium chloride for 10 to 15 minutes until viable myocardiumwas stained brick red. Infarcted myocardium fails to stainwith triphenyltetrazolium chloride. Tissue samples were thenfixed in a 10% formalin solution and weighed. Color photo-graphs of both sides of each transverse slice were obtainedwith an Olympus OM-2 camera with a 90-mm Macro lensand a 2x teleconverter. The regions showing blue-stained(nonischemic), red-stained (ischemic but noninfarcted), andunstained (infarcted) tissue were outlined on each colorphotograph and measured by planimetry in a blinded fash-ion. On each side, the fraction of left ventricular (LV) arearepresenting infarcted tissue (average of two photographs)was multiplied by the weight of that section to determine theabsolute weight of infarcted tissue. The infarct size for eachheart was expressed as
Infarct size/LV mass (%)=
5 infarct weight in each slicetotal LV weight
The risk area unstained by blue dye was expressed as
Risk area/LV mass (%)=
total weight of unstained sectiontotal LV weight
x 100%
Infarct size as a percentage of risk area was then calculated as
Infarct size/risk area (%)=
X infarct weight in each sliceX risk area weight of each slice
Four rats in group ETS-3 weeks were excluded because oftechnical problems with staining.Histopathological Evaluation
Histological sections through the left coronary artery weremade in the three control rats and three 6-week ETS-exposedrats. All sections were stained by hematoxylin and eosin (HE)and elastin van Giesen eosin (EVG) separately. Histopatho-logical findings under the light microscope were determined bytwo investigators blindly.
Hematological and Biochemical AnalysisDetermination of bleeding times was performed as de-
scribed previously by warming the rat tail for three minutes ina normal saline bath maintained at 37°C. The tail was thenremoved from the bath, and a standard 1/2-cm longitudinalprick was made into the distal tail, avoiding any macroscopi-cally obvious vessels, with a carbon steel blade. The incised tailwas then immediately placed into the saline bath, which wasgently agitated with a magnetic stirrer. The bleeding time wastaken as the time required for blood flow in the incision tocease.
Total serum cholesterol, triglycerides, high-density lipopro-tein (HDL) cholesterol, plasma COHb, nicotine, and cotininewere measured in additional rats after 3 days, 3 weeks, and 6weeks of ETS exposure and their controls (6 to 13 rats pergroup). The plasma concentrations of nicotine and cotininewere determined by gas chromatography with nitrogen-phos-phorus detection.26 This method has been modified for simul-taneous extraction and determination of cotinine by use ofcapillary gas chromatography.27 Total serum cholesterol andtriglyceride levels were determined by automated enzymaticmethods (Coulter DART cholesterol reagent and the DACOSand DACOS XL analyzers), and HDL cholesterol concentra-tions were measured after precipitation of other lipoproteinclasses with dextran and magnesium ions (HDL cholesterolprecipitant [catalog No. 236141], Ciba Corning DiagnosticCorp, Oberlin, Ohio).
Monitoring Smoke Exposure Inside the ChambersAir concentrations of carbon monoxide, total particulates,
respirable suspended particulates, and nicotine inside theexposure chambers were measured weekly. Average carbonmonoxide concentrations during the 6-hour exposure periodwere determined by a model L15 CO Personal ExposureSystem (Langan Products, Inc, San Francisco, Calif). Theaverage daily value was taken from 2160 samples during the6-hour exposure period. The total particulate concentrationwas measured using a Miniram PDM-3 optical scatteringparticle monitor (MIE, Inc, Bedford, Mass), which monitoredparticulate concentration every 10 seconds and computed theaverage total particulate concentration during the 6-hourexposure period. Respirable suspended particulates were mea-sured with a piezobalance respirable aerosol mass monitor(model 3500, Thermo-System, Inc, St Paul, Minn). In addition,air nicotine levels were measured with a passive diffusionmonitor located in the middle of the exposure chamber duringthe 6-hour exposure period.
_
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from
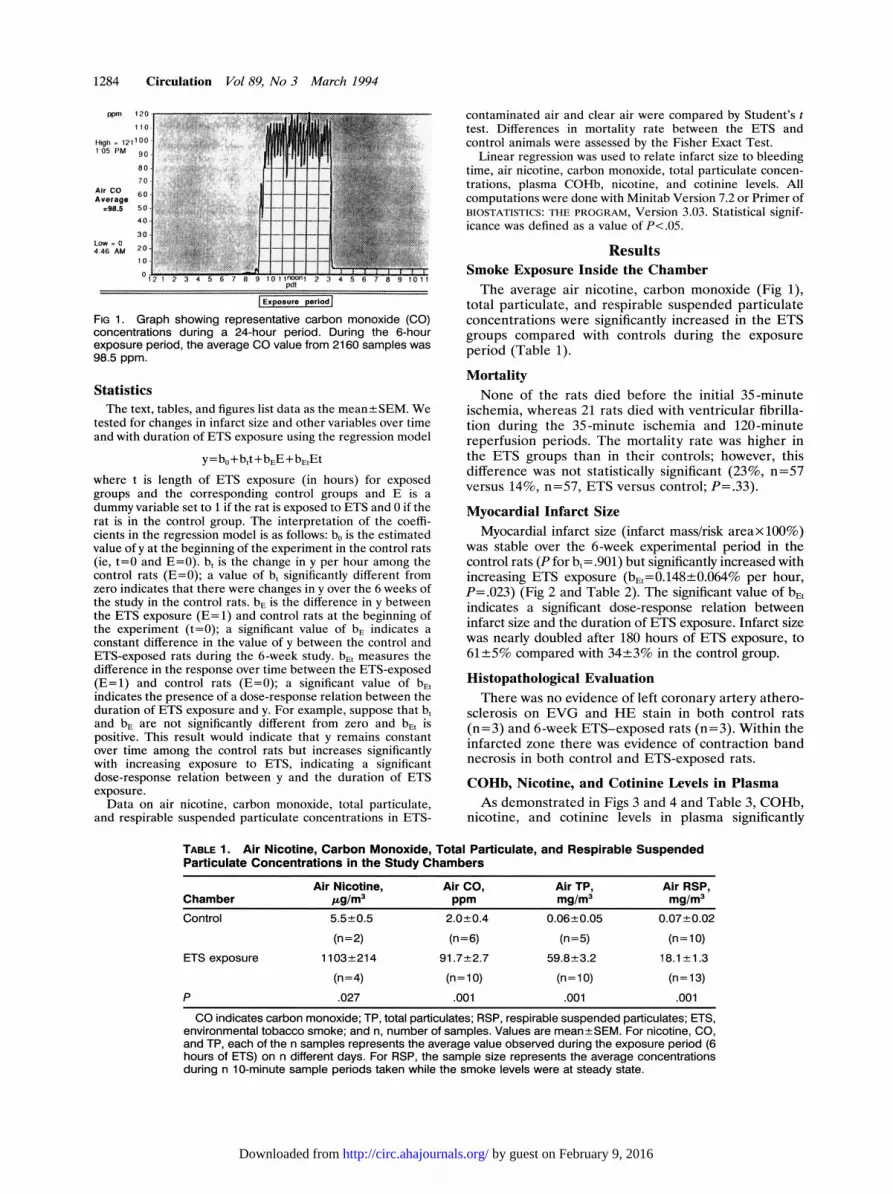
1284 Circulation Vol 89, No 3 March 1994
FIG 1. Graph showing representative carbon monoxide (CO)concentrations during a 24-hour period. During the 6-hourexposure period, the average CO value from 2160 samples was98.5 ppm.
StatisticsThe text, tables, and figures list data as the mean-+-SEM. We
tested for changes in infarct size and other variables over timeand with duration of ETS exposure using the regression model
y-b(+bt+bEE+bE,Etwhere t is length of ETS exposure (in hours) for exposedgroups and the corresponding control groups and E is a
dummy variable set to 1 if the rat is exposed to ETS and 0 if therat is in the control group. The interpretation of the coeffi-cients in the regression model is as follows: b0) is the estimatedvalue of y at the beginning of the experiment in the control rats(ie, t=0 and E=O). b, is the change in y per hour among thecontrol rats (E=O); a value of b, significantly different fromzero indicates that there were changes in y over the 6 weeks ofthe study in the control rats. bE is the difference in y betweenthe ETS exposure (E=1) and control rats at the beginning ofthe experiment (t=O); a significant value of bE indicates a
constant difference in the value of y between the control andETS-exposed rats during the 6-week study. bE, measures thedifference in the response over time between the ETS-exposed(E 1) and control rats (E=O); a significant value of bE,indicates the presence of a dose-response relation between theduration of ETS exposure and y. For example, suppose that b,and bE are not significantly different from zero and bE, ispositive. This result would indicate that y remains constantover time among the control rats but increases significantlywith increasing exposure to ETS, indicating a significantdose-response relation between y and the duration of ETSexposure.Data on air nicotine, carbon monoxide, total particulate,
and respirable suspended particulate concentrations in ETS-
contaminated air and clear air were compared by Student's ttest. Differences in mortality rate between the ETS andcontrol animals were assessed by the Fisher Exact Test.
Linear regression was used to relate infarct size to bleedingtime, air nicotine, carbon monoxide, total particulate concen-
trations, plasma COHb, nicotine, and cotinine levels. Allcomputations were done with Minitab Version 7.2 or Primer ofBIOSTATISTICS: TH E PROGRAM, Version 3.03. Statistical signif-icance was defined as a value of P<.05.
ResultsSmoke Exposure Inside the ChamberThe average air nicotine, carbon monoxide (Fig 1),
total particulate, and respirable suspended particulateconcentrations were significantly increased in the ETSgroups compared with controls during the exposureperiod (Table 1).
MortalityNone of the rats died before the initial 35-minute
ischemia, whereas 21 rats died with ventricular fibrilla-tion during the 35-minute ischemia and 120-minutereperfusion periods. The mortality rate was higher in
the ETS groups than in their controls; however, thisdifference was not statistically significant (23%, n-57versus 14%, n=57, ETS versus control; P=.33).
Myocardial Infarct SizeMyocardial infarct size (infarct mass/risk areax 100%)
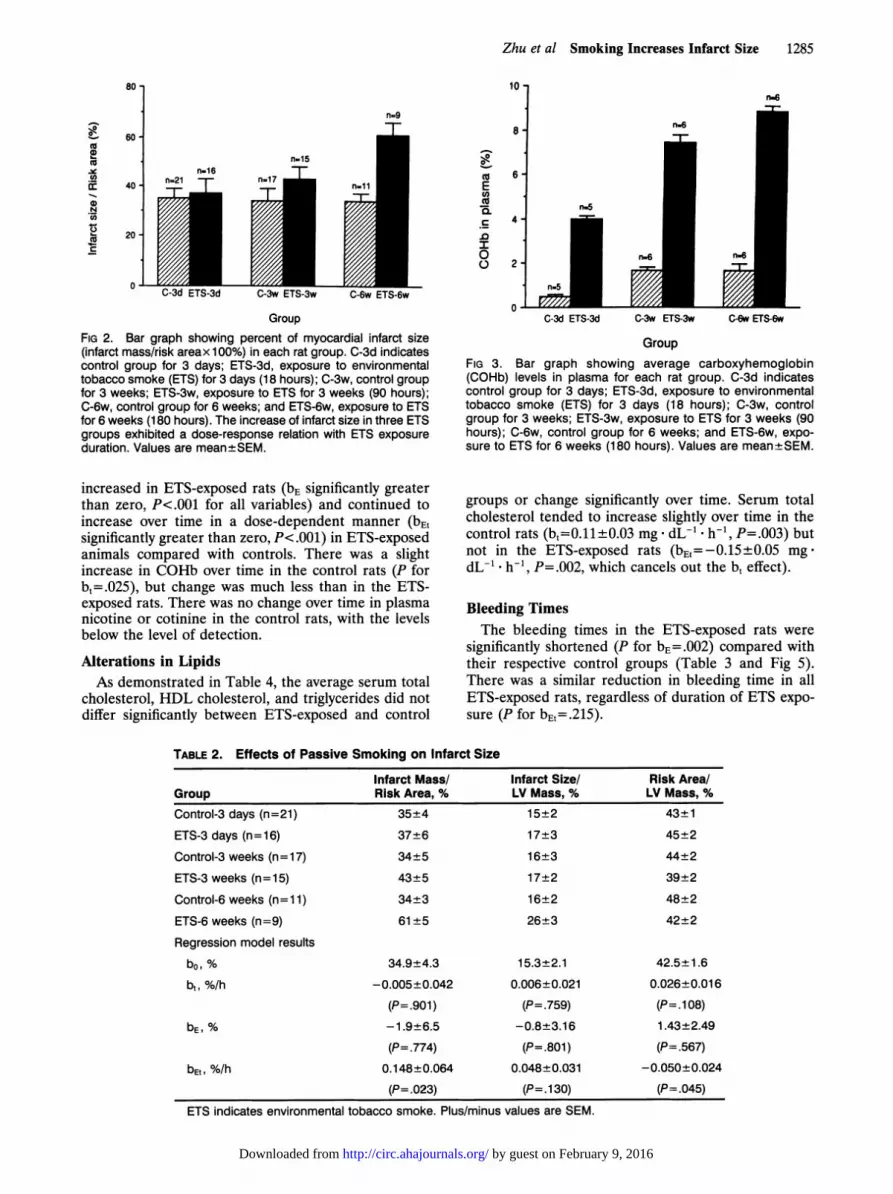
was stable over the 6-week experimental period in thecontrol rats (P for b =.901) but significantly increased withincreasing ETS exposure (bEL=0.148±0.064% per hour,P-.023) (Fig 2 and Table 2). The significant value of bE,indicates a significant dose-response relation betweeninfarct size and the duration of ETS exposure. Infarct sizewas nearly doubled after 180 hours of ETS exposure, to61+5% compared with 34±3% in the control group.
Histopathological EvaluationThere was no evidence of left coronary artery athero-
sclerosis on EVG and HE stain in both control rats(n-3) and 6-week ETS-exposed rats (n-3). Within theinfarcted zone there was evidence of contraction bandnecrosis in both control and ETS-exposed rats.
COHb, Nicotine, and Cotinine Levels in PlasmaAs demonstrated in Figs 3 and 4 and Table 3, COHb,
nicotine, and cotinine levels in plasma significantly
TABLE 1. Air Nicotine, Carbon Monoxide, Total Particulate, and Respirable SuspendedParticulate Concentrations in the Study Chambers
Air Nicotine, Air CO, Air TP, Air RSP,Chamber pg/mr3 ppm mg/m3 mg/m3Control 5.5+±-0.5 2.0+0.4 0.06+0.05 0.07+0.02
(n=2) (n=6) (n=5) (n=10)ETS exposure 1103+214 91.7-2.7 59.8+3.2 18.t+ 1.3
(n=4) (n=10) (n=10) (n=13)
P .027 .001 .001 .001
CO indicates carbon monoxide; TP, total particulates; RSP, respirable suspended particulates; ETS,environmental tobacco smoke; and n, number of samples. Values are mean-+-SEM. For nicotine, CO,and TP, each of the n samples represents the average value observed during the exposure period (6hours of ETS) on n different days. For RSP, the sample size represents the average concentrationsduring n 1 0-minute sample periods taken while the smoke levels were at steady state.
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from
Zhu et al Smoking Increases Infarct Size 1285
80 -
:-.c0-
CO
0)
I-
(D
N0)
_
ccc
60
40-
20 -
0
10-
n=9
8-
n-16n=21TT I
n.150
E(A0
n
.0
0
D)
GroupFIG 2. Bar graph showing percent of myocardial infarct size(infarct mass/risk areax 100%) in each rat group. C-3d indicatescontrol group for 3 days; ETS-3d, exposure to environmentaltobacco smoke (ETS) for 3 days (18 hours); C-3w, control groupfor 3 weeks; ETS-3w, exposure to ETS for 3 weeks (90 hours);C-6w, control group for 6 weeks; and ETS-6w, exposure to ETSfor 6 weeks (180 hours). The increase of infarct size in three ETSgroups exhibited a dose-response relation with ETS exposureduration. Values are mean±SEM.
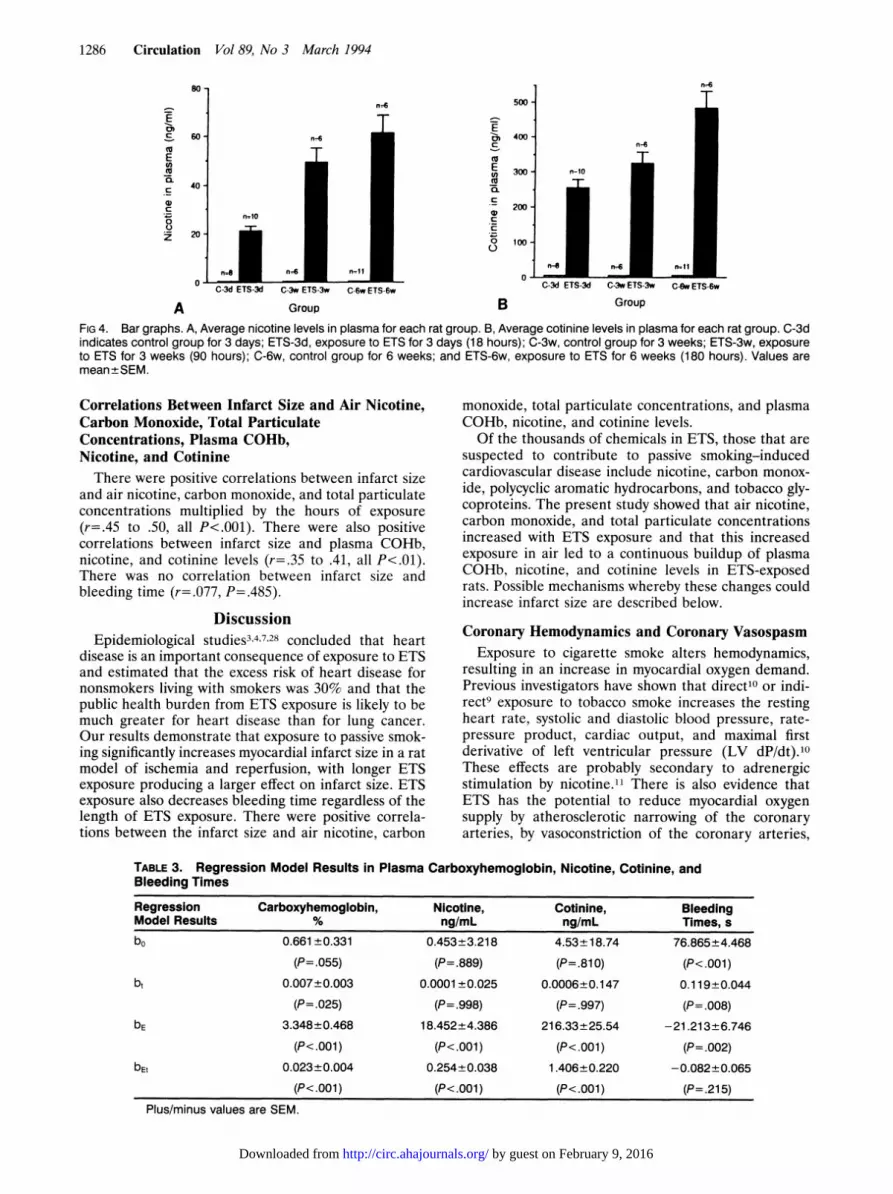
increased in ETS-exposed rats (bE significantly greaterthan zero, P<.001 for all variables) and continued toincrease over time in a dose-dependent manner (bE,significantly greater than zero, P<.001) in ETS-exposedanimals compared with controls. There was a slightincrease in COHb over time in the control rats (P forb,=.025), but change was much less than in the ETS-exposed rats. There was no change over time in plasmanicotine or cotinine in the control rats, with the levelsbelow the level of detection.
Alterations in LipidsAs demonstrated in Table 4, the average serum total
cholesterol, HDL cholesterol, and triglycerides did notdiffer significantly between ETS-exposed and control
6-
n.6
n-6
n-5
C-3d ET-3d C-3w tET-3W C-6w ETS-6w
Group
FIG 3. Bar graph showing average carboxyhemoglobin(COHb) levels in plasma for each rat group. C-3d indicatescontrol group for 3 days; ETS-3d, exposure to environmentaltobacco smoke (ETS) for 3 days (18 hours); C-3w, controlgroup for 3 weeks; ETS-3w, exposure to ETS for 3 weeks (90hours); C-6w, control group for 6 weeks; and ETS-6w, expo-sure to ETS for 6 weeks (180 hours). Values are mean±SEM.
groups or change significantly over time. Serum totalcholesterol tended to increase slightly over time in thecontrol rats (b,=0.11±0.03 mg * dL`-* h', P=.003) butnot in the ETS-exposed rats (bE,=-0.15±0.05 mg
dL-'. h`, P=.002, which cancels out the b, effect).
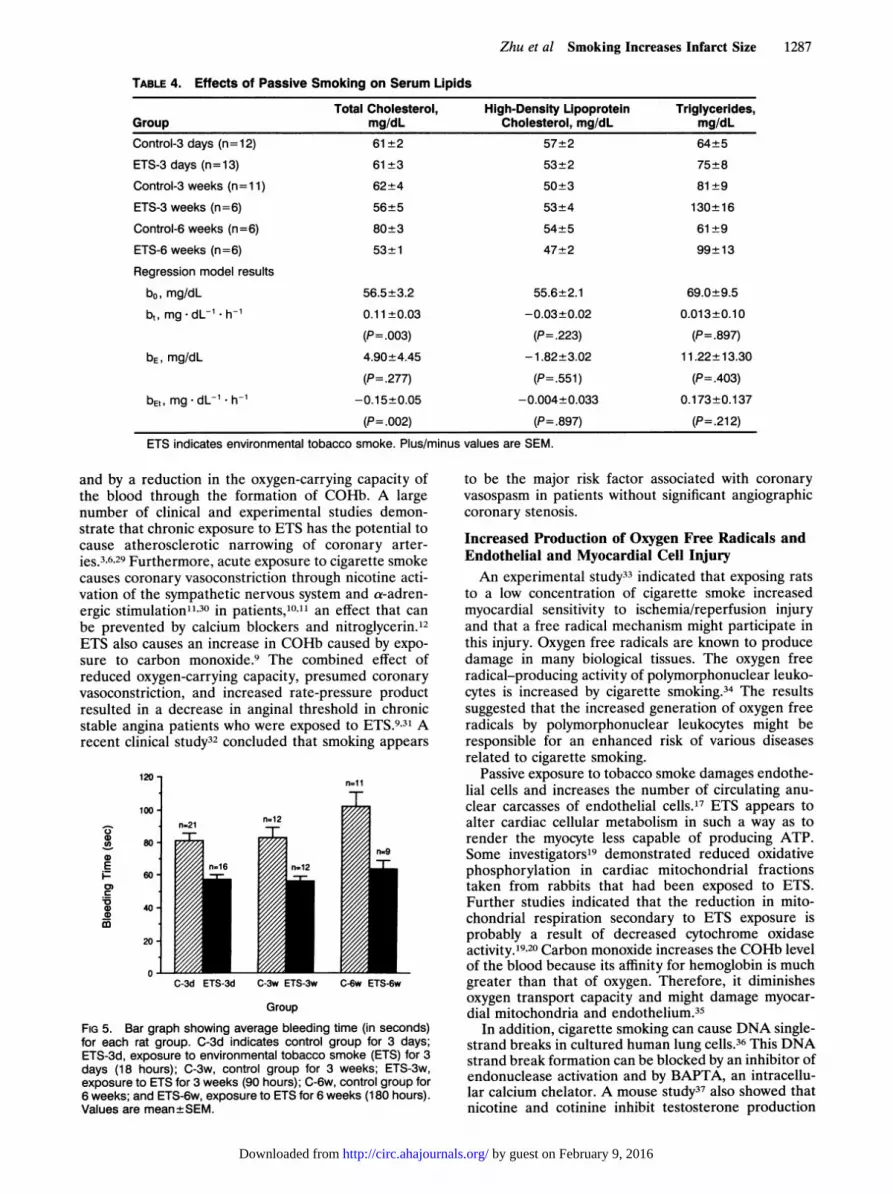
Bleeding TimesThe bleeding times in the ETS-exposed rats were
significantly shortened (P for bE=.002) compared withtheir respective control groups (Table 3 and Fig 5).There was a similar reduction in bleeding time in allETS-exposed rats, regardless of duration of ETS expo-sure (P for bEt=.215).
TABLE 2. Effects of Passive Smoking on Infarct Size
Infarct Mass/ Infarct Size/ Risk Area/Group Risk Area, % LV Mass, % LV Mass, %Control-3 days (n=21) 35±4 15±2 43±1ETS-3 days (n=16) 37±6 17±3 45±2
Control-3 weeks (n= 17) 34±5 16±3 44±2
ETS-3 weeks (n= 15) 43±5 17±2 39±2
Control-6 weeks (n=11) 34±3 16±2 48±2
ETS-6 weeks (n=9) 61±5 26±3 42±2
Regression model results
bo, % 34.9±4.3 15.3±2.1 42.5±1.6
bt, %/h -0.005±0.042 0.006±0.021 0.026±0.016
(P=.901) (P=.759) (P=.1 08)bE, % -1.9±6.5 -0.8±3.16 1.43±2.49
(P=.774) (P=.801) (P=.567)
bEt, %/h 0.148±0.064 0.048±0.031 -0.050±0.024
(P=.023) (P=. 1 30) (P=.045)ETS indicates environmental tobacco smoke. Plus/minus values are SEM.
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from
1286 Circulation Vol 89, No 3 March 1994
E
E0
C
C
z
A
C-3d ETS-3d C-3w ETS-3w C-6w ETS-6w
Group
E
cn
tn
cCL
.C
0
B Group
FIG 4. Bar graphs. A, Average nicotine levels in plasma for each rat group. B, Average cotinine levels in plasma for each rat group. C-3dindicates control group for 3 days; ETS-3d, exposure to ETS for 3 days (18 hours); C-3w, control group for 3 weeks; ETS-3w, exposureto ETS for 3 weeks (90 hours); C-6w, control group for 6 weeks; and ETS-6w, exposure to ETS for 6 weeks (180 hours). Values aremean+SEM.
Correlations Between Infarct Size and Air Nicotine,Carbon Monoxide, Total ParticulateConcentrations, Plasma COHb,Nicotine, and CotinineThere were positive correlations between infarct size
and air nicotine, carbon monoxide, and total particulateconcentrations multiplied by the hours of exposure(r=.45 to .50, all P<.001). There were also positivecorrelations between infarct size and plasma COHb,nicotine, and cotinine levels (r=.35 to .41, all P<.01).There was no correlation between infarct size andbleeding time (r=.077, P=.485).
DiscussionEpidemiological studies3,4'7-28 concluded that heart
disease is an important consequence of exposure to ETSand estimated that the excess risk of heart disease fornonsmokers living with smokers was 30% and that thepublic health burden from ETS exposure is likely to bemuch greater for heart disease than for lung cancer.Our results demonstrate that exposure to passive smok-ing significantly increases myocardial infarct size in a ratmodel of ischemia and reperfusion, with longer ETSexposure producing a larger effect on infarct size. ETSexposure also decreases bleeding time regardless of thelength of ETS exposure. There were positive correla-tions between the infarct size and air nicotine, carbon
monoxide, total particulate concentrations, and plasmaCOHb, nicotine, and cotinine levels.Of the thousands of chemicals in ETS, those that are
suspected to contribute to passive smoking-inducedcardiovascular disease include nicotine, carbon monox-ide, polycyclic aromatic hydrocarbons, and tobacco gly-coproteins. The present study showed that air nicotine,carbon monoxide, and total particulate concentrationsincreased with ETS exposure and that this increasedexposure in air led to a continuous buildup of plasmaCOHb, nicotine, and cotinine levels in ETS-exposedrats. Possible mechanisms whereby these changes couldincrease infarct size are described below.
Coronary Hemodynamics and Coronary VasospasmExposure to cigarette smoke alters hemodynamics,
resulting in an increase in myocardial oxygen demand.Previous investigators have shown that direct'O or indi-rect9 exposure to tobacco smoke increases the restingheart rate, systolic and diastolic blood pressure, rate-pressure product, cardiac output, and maximal firstderivative of left ventricular pressure (LV dP/dt).10These effects are probably secondary to adrenergicstimulation by nicotine.1" There is also evidence thatETS has the potential to reduce myocardial oxygensupply by atherosclerotic narrowing of the coronaryarteries, by vasoconstriction of the coronary arteries,
TABLE 3. Regression Model Results in Plasma Carboxyhemoglobin, Nicotine, Cotinine, andBleeding Times
Regression Carboxyhemoglobin, Nicotine, Cotinine, BleedingModel Results % ng/mL ng/mL Times, sbo 0.661±0.331 0.453±3.218 4.53±18.74 76.865±4.468
(P=.055) (P=.889) (P=.810) (P<.001)bt 0.007±0.003 0.0001 ±0.025 0.0006±0.147 0.119±0.044
(P=.025) (P= .998) (P=.997) (P=.008)bE 3.348±0.468 18.452±4.386 216.33±25.54 -21.213±6.746
(P<.001) (P<.001) (P<.001) (P=.002)bEt 0.023±0.004 0.254±0.038 1.406±0.220 -0.082±0.065
(P<.0011) (P<.001) (P<.001) (P=.215)
Plus/minus values are SEM.
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from
Zhu et al Smoking Increases Infarct Size 1287
TABLE 4. Effects of Passive Smoking on Serum Lipids
Total Cholesterol, High-Density Lipoprotein Triglycerides,Group mg/dL Cholesterol, mg/dL mg/dL
Control-3 days (n=12) 61±+2 57+2 64±5
ETS-3 days (n=13) 61±+3 53+2 75+8
Control-3 weeks (n=1 1) 62±4 50+3 81+9
ETS-3 weeks (n=6) 56±5 53±4 130±+16
Control-6 weeks (n=6) 80+3 54±5 61±9
ETS-6 weeks (n=6) 53±1 47+2 99±13
Regression model results
bo, mg/dL 56.5±3.2 55.6±2.1 69.0±9.5
bt, mg dL-1 . h-' 0.11±0.03 -0.03±0.02 0.013±0.10
(P=.003) (P=.223) (P=.897)
bE, mg/dL 4.90±4.45 -1.82±3.02 11.22±13.30
(P=.277) (P=.551) (P=.403)
bEt, mg* dL-1 * h-1 -0.15±0.05 -0.004±0.033 0.173±0.137
(P=.002) (P=.897) (P=.212)
ETS indicates environmental tobacco smoke. Plus/minus values are SEM.
and by a reduction in the oxygen-carrying capacity ofthe blood through the formation of COHb. A largenumber of clinical and experimental studies demon-strate that chronic exposure to ETS has the potential tocause atherosclerotic narrowing of coronary arter-ies.3,6,29 Furthermore, acute exposure to cigarette smokecauses coronary vasoconstriction through nicotine acti-vation of the sympathetic nervous system and a-adren-ergic stimulation1"30 in patients,10"' an effect that canbe prevented by calcium blockers and nitroglycerin.12ETS also causes an increase in COHb caused by expo-sure to carbon monoxide.9 The combined effect ofreduced oxygen-carrying capacity, presumed coronaryvasoconstriction, and increased rate-pressure productresulted in a decrease in anginal threshold in chronicstable angina patients who were exposed to ETS.931 Arecent clinical study32 concluded that smoking appears
120
100*
0Co0
E
0)
0
0D
80
60-
40
20-
0
n=1 1
T
n=21 n=12-1-
C-3d ETS-3d C-3w ETS-3w C-6w ETS-6w
Group
FIG 5. Bar graph showing average bleeding time (in seconds)for each rat group. C-3d indicates control group for 3 days;ETS-3d, exposure to environmental tobacco smoke (ETS) for 3days (18 hours); C-3w, control group for 3 weeks; ETS-3w,exposure to ETS for 3 weeks (90 hours); C-6w, control group for6 weeks; and ETS-6w, exposure to ETS for 6 weeks (180 hours).Values are mean±SEM.
to be the major risk factor associated with coronaryvasospasm in patients without significant angiographiccoronary stenosis.
Increased Production of Oxygen Free Radicals andEndothelial and Myocardial Cell InjuryAn experimental study33 indicated that exposing rats
to a low concentration of cigarette smoke increasedmyocardial sensitivity to ischemia/reperfusion injuryand that a free radical mechanism might participate inthis injury. Oxygen free radicals are known to producedamage in many biological tissues. The oxygen freeradical-producing activity of polymorphonuclear leuko-cytes is increased by cigarette smoking.34 The resultssuggested that the increased generation of oxygen freeradicals by polymorphonuclear leukocytes might beresponsible for an enhanced risk of various diseasesrelated to cigarette smoking.
Passive exposure to tobacco smoke damages endothe-lial cells and increases the number of circulating anu-clear carcasses of endothelial cells.17 ETS appears toalter cardiac cellular metabolism in such a way as torender the myocyte less capable of producing ATP.Some investigators19 demonstrated reduced oxidativephosphorylation in cardiac mitochondrial fractionstaken from rabbits that had been exposed to ETS.Further studies indicated that the reduction in mito-chondrial respiration secondary to ETS exposure isprobably a result of decreased cytochrome oxidaseactivity.19'20 Carbon monoxide increases the COHb levelof the blood because its affinity for hemoglobin is muchgreater than that of oxygen. Therefore, it diminishesoxygen transport capacity and might damage myocar-dial mitochondria and endothelium.35
In addition, cigarette smoking can cause DNA single-strand breaks in cultured human lung cells.36 This DNAstrand break formation can be blocked by an inhibitor ofendonuclease activation and by BAPTA, an intracellu-lar calcium chelator. A mouse study37 also showed thatnicotine and cotinine inhibit testosterone production
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from

1288 Circulation Vol 89, No 3 March 1994
and verapamil significantly reversed this inhibition.These results suggest that nicotine and cotinine eitheraffect intracellular calcium content or block the effectsof calcium on steroidogenesis in the mouse.
ThrombosisETS also has been shown to increase platelet aggrega-
tion, which might increase the likelihood of coronarythrombosis or vasoconstriction after acute exposure orthe development of coronary atherosclerosis afterchronic ETS exposure.3,13,14 Passive smoking also in-creases platelet-activating factor, platelet factor 4,,B-thromboglobulin, and fibrinogen concentration, whichprovides a marker of its effect on coronary heart dis-ease.38-40 Davis and coworkers15-18 demonstrated that aslittle as 20 minutes of ETS exposure resulted in signifi-cantly increased platelet aggregability in patients. Wefound similar increases in platelet activity, reflected as ashortened bleeding time. Consistent with our earlierwork,6 we found a large effect associated with even thelowest level of ETS exposure (in this case 18 hours over3 days).
Nicotine has been shown to inhibit the in vitro releaseof prostacyclin from the rings of rabbit or rat aorta andhuman veins. The suggested mechanism is the inhibitionof prostacyclin synthesis through the inhibition of cyclo-oxygenase. Nicotine could also affect platelets by releas-ing catecholamines, which lead to increased thrombox-ane A2.30 Passive smoking also increases blood viscosityand hematocrit because of relative hypoxia induced bychronic carbon monoxide exposure.30 Clinical stud-ies41,42 also showed that after abstention from chroniccigarette smoking, the blood thrombogenic factors andhemorheological parameters will be normalized.
Catecholamine Release and ArrhythmogenesisA clinical study43 demonstrated that smoking-associ-
ated increments in mean (±SEM) plasma norepineph-rine (227±23 to 324±39 pg/mL, P<.01) and epinephrine(44±4 to 113 ± 27 pg/mL, P<.05) and smoking-associatedincrements in pulse rate, blood pressure, blood glycerol,and blood lactate/pyruvate ratio were prevented by ad-renergic blockade. Therefore, it can be concluded thatsmoking-associated increments in these hemodynamicand metabolic variables are mediated through adrenergicmechanisms. Cigarette smoking can lead to catechol-amine release by nicotine. Increased circulating concen-trations of catecholamines enhance platelet adhesivenessand decrease the ventricular fibrillation threshold, whichis also affected by carbon monoxide.3044 We found ahigher mortality from ventricular fibrillation in ETS-exposed compared with control rats (23% versus 14%).This was not statistically significant, however (P=.33).The lack of significance may be a reflection of the smallnumbers of rats we studied. The power of these experi-ments to detect a doubling of the mortality rate is only36%.
Thus, passive smoking, including nicotine and carbonmonoxide, may contribute to acute myocardial ischemiaor sudden death through reduction of coronary bloodflow, increasing myocardial oxygen demands, increasingoxygen free radical generation, enhancement of throm-bosis, releasing catecholamines, and arrhythmogenesis.ETS exposure may not only increase the likelihood of
the amount of myocardial damage once an acute myo-cardial infarction does occur.
Dose and DurationThis study is the first to show that passive smoking
significantly increases myocardial infarct size in a ratmodel of ischemia and reperfusion, with greater myo-cardial damage directly related to greater exposure toETS. There were positive relations between myocardialinfarct size and the average concentrations of nicotine,carbon monoxide, and particulates. Compared withaverage air concentrations of nicotine and carbon mon-oxide in human heavy smoking environments (50 to 500,g/m3 and 5 to 50 ppm, respectively),6'45 the averageconcentrations of air nicotine and carbon monoxide inour present study were only about twofold higher (1103gg/m3 and 92 ppm). The duration of exposure, however,was short compared with even a rat's lifetime. Myocar-dial infarct size nearly doubled with just 180 hours ofETS exposure spread over 6 weeks.
Recently, Penn and Snyder46 reported that passivesmoking significantly enhanced arteriosclerotic plaquein cockerels who were exposed to sidestream smoke for6 hours a day, 5 days a week, for 16 weeks. This isequivalent to 0.4% of their projected lifespan. Assuminga human lifespan of 74 or 75 years, an equivalent periodof exposure to sidestream smoke would be 3 hours a dayfor 2.4 years.The exposure chamber we used had an interior
volume (3.58 m3) similar to that of a Mazda 626automobile (3.7 m3). Recently, Ott et a121 reported thatthe air concentration of carbon monoxide in a movingautomobile is 63 ppm when a smoker smokes eachcigarette for 7 minutes, four cigarettes per hour, and thewindows are closed. This real microenvironment wasthe same size as our exposure chamber and would besimilar to our rat study if four passengers each smokedfour cigarettes per hour.
Clinical RelevancePrevious clinical studies have demonstrated increased
cardiovascular morbidity associated with cigarettesmoking. In 978 patients with a first myocardial infarc-tion, Robinson et a147 reported that the smokers had alarger infarct size as estimated by serum peak creatinekinase concentrations (1382 versus 1146 LU/L, P=.010).This study did not report on the severity of coronarydisease. Multivariate analysis of possible risk factors forreinfarction identified at the time of initial infarction48showed current cigarette smoking to be the only predic-tive factor (reinfarction occurred in 12.5% of smokersversus 6.3% of nonsmokers, P=.04). One other study49that followed patients after a first acute myocardialinfarction reported that the risk of nonfatal reinfarctioncorrelated with a variety of clinical parameters, includ-ing the presence or absence of cigarette smoking. Smok-ing was found to increase the risk of first acute myocar-dial infarction in a dose-dependent manner, the riskincreasing 2% to 3% for each gram of tobacco smokeddaily.50 Patients with a previous myocardial infarction44had a significantly higher plasma carbon monoxide levelafter exercise in a smoking environment. The cardiacresponses, including peak exercise power, time to recov-ery of preexercise heart rate, and expired concentration
having an acute myocardial infarction but also increase of carbon monoxide, were significantly worsened by
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from

Zhu et al Smoking Increases Infarct Size 1289
passive smoking, especially in those subjects with previ-ous myocardial infarction. There are no previous dataregarding the direct effect of passive smoking on myo-cardial infarct size. The present study is the first study todemonstrate that passive smoking increases experimen-tal myocardial infarct size in rats.
ConclusionsThese data indicate that exposure to passive smoking
significantly increases myocardial infarct size in a ratmodel of ischemia and reperfusion with a dose-responserelation. These results are consistent with epidemiolog-ical studies demonstrating that ETS increases the risk ofheart disease death.
AcknowledgmentsThis study was supported in part by Grants-in-Aid from the
California Affiliate of the American Heart Association (grant88-N135), the National American Heart Association (grant92007600), and the Tobacco Related Disease Research Pro-gram (grant 1RT 145) and by the George D. Smith Fund.Grateful appreciation is extended to John Hudson and Bar-bara Maher of the Electronic Facilities, Cardiovascular Re-search Institute, University of California, San Francisco, andLee Langan Products, Inc, for their excellent technical assis-tance. We also thank Lisa Yu, Lucy Wu, Peyton Jacob, andMinjiang Duan in Dr Neal L. Benowitz's laboratory formeasuring plasma nicotine and cotinine levels and KatharineHammond of the University of Massachusetts Medical School,Worcester, for help in analysis of the air nicotine levels.
References1. US Public Health Service. The Health Consequences of Involuntary
Smoking: A Report of the Surgeon GeneraL Washington, DC:DHS(CDC)87-8398, 1986.
2. National Research Council. Environmental Tobacco Smoke: Mea-suring Exposure and Assessing Health Effects. Washington, DC:National Academy Press; 1986.
3. Glantz SA, Parmley WW. Passive smoking and heart disease:epidemiology, physiology, and biochemistry. Circulation. 1991;83:1-12.
4. Steenland K. Passive smoking and the risk of heart disease. JAMA.1992;267:94-99.
5. US Environmental Protection Agency. Respiratory Health Effects ofPassive Smoking: Lung Cancer and Other Disorders. EPA/600/6-90/006F, December 1992.
6. Zhu B-Q, Sun Y-P, Sievers RE, Isenberg WM, Glantz SA, ParmleyWW. Passive smoking increases experimental atherosclerosis incholesterol-fed rabbits. JAm Coll Cardiol. 1993;21:225-232.
7. Taylor AE, Johnson DC, Kazemi H. Environmental tobacco smokeand cardiovascular disease: a position paper from the Council onCardiopulmonary and Critical Care, American Heart Association.Circulation. 1992;86:1-4.
8. Wells A..An estimate of adult mortality in the United States frompassive smoking. Environ Int. 1988;14:249-265.
9. Aronow W. Effect of passive smoking on angina pectoris. N Engl JMed. 1978;299:21-24.
10. Nicod P, Rehr R, Winniford MD, Campbell WB, Firth BG, HillisLD. Acute systemic and coronary hemodynamic and serologicresponses to cigarette smoking in long-term smokers with athero-sclerotic coronary artery disease. J Am Coll Cardiol. 1984;4:964-971.
11. Winniford MD, Wheelan KR, Kremers MS, Ugolini V, van denBerg E Jr, Niggemann EH, Jansen DE, Hillis LD. Smoking-induced coronary vasoconstriction in patients with atheroscleroticcoronary artery disease: evidence for adrenergically mediatedalteration in coronary artery tone. Circulation. 1986;73:662-667.
12. Winniford MD, Jansen DE, Reynolds GA, Appril P, Black WH,Hillis LD. Cigarette smoking-induced coronary vasoconstriction inatherosclerotic coronary artery disease and prevention by calciumantagonists and nitroglycerin. Am J Cardiol. 1987;59:203-207.
13. Fuster V, Chesebro J. Antithrombotic therapy: role of platelet-inhibitor drugs, I: current concepts of thrombogenesis: role ofplatelets. Mayo Clin Proc. 1981;56:102-112.
14. Ross R. The pathology of atherosclerosis: an update. NEnglJMed.1986;314:488-500.
15. Davis J, Shelton L, Eigenberg D, Hignite C, Watanabe I. Effects oftobacco and non-tobacco cigarette smoking on endothelium andplatelets. Clin Pharmacol Ther. 1985;37:529-533.
16. Davis J, Hartman C, Lewis H Jr, Shelton L, Eigenberg D, Has-sanein K, Hignite C, Ruttinger H. Cigarette smoking-inducedenhancement of platelet function: lack of prevention by aspirin inmen with coronary artery disease. J Lab Clin Med. 1985;105:479-483.
17. Davis JW, Shelton L, Watanabe IS, Arnold J. Passive smokingaffects endothelium and platelets. Arch Intern Med. 1989;149:386-389.
18. Davis J, Shelton L, Zucker M. A comparison of some acute effectsof smoking and smokeless tobacco on platelet and endothelium.J Vasc Med Biol. 1990;2:289-293.
19. Gvozdjakova A, Bada V, Sany L, Kucharska J, Kruty F, Bozek,Trstanksy L, Gvozdjak J. Smoke cardiomyopathy: disturbance ofoxidative process in myocardial mitochondria. Cardiovasc Res.1984;18:229-232.
20. Gvozdjakova A, Kucharska J, Sany L, Bada V, Bozek, Gvozdjak J.Effect of smoking on the cytochrome and oxidase system of themyocardium. Bratisl Lek Listy. 1985;83:10-15.
21. Ott W, Langan L, Switzer P. A time series model for cigarettesmoking activity patterns: model validation for carbon monoxideand respirable particles in a chamber and an automobile.J Exposure Anal Environ Epidemiol. 1992;2(suppl 2):175-200.
22. Sievers RE, Schmiedl U, Wolfe CL, Brasch RC, Moseley ME,Parmley WW, Lipton MJ. A model of acute regional myocardialischemia and reperfusion in the rat. Magn Reson Med. 1989;10:172-181.
23. Wolfe CL, Moseley ME, Wikstrom MG, Sievers RE, WendlandMF, Dupon JW, Finkbeiner WE, Lipton MJ, Parmley WW, BraschRC. Assessment of myocardial salvage after ischemia and reper-fusion using magnetic resonance imaging and spectroscopy. Circu-lation. 1989;80:969-982.
24. Wolfe CL, Donnelly TJ, Sievers RE, Parmley WW. Myocardialprotection with verapamil during ischemia and reperfusion: disso-ciation between myocardial salvage and degree of ATP depletionduring ischemia. Cardiovasc Res. 1991;25:101-109.
25. Donnelly TJ, Sievers RE, Vissern FLJ, Welch WJ, Wolfe CL. Heatshock protein induction in rat heart: a role for improved myo-cardial salvage after ischemia and reperfusion? Circulation. 1992;85:769-778.
26. Jacob P, Wilson M, Benowitz NL. Improved gas chromatographicmethod for the determination of nicotine and cotinine in biologicfluids. J Chromatogr. 1981;222:61-70.
27. Jacob P, Yu L, Wilson M, Benowitz NL. Selected ion monitoringmethod for determination of nicotine, and deuterium-labelledanalogs: absence of an isotope effect in the clearance of (5)-nicotine-3',3'-d2 in humans. Biol Mass Spectrom. 1991;20:247-252.
28. Steenland K. Passive smoking and the risk of heart disease. JAMA.1992;267:94-99.
29. Gillis C, Hole D, Hawthorne V, Boyle P. The effect of environ-mental tobacco smoke in two urban communities in the west ofScotland. Eur J Respir Dis. 1984;65(suppl 133):121-126.
30. Benowitz NL. Nicotine and coronary heart disease. Trends Car-diovasc Med. 1991;1:315-321.
31. Khalfen E, Klochkov V. Effect of passive smoking on physicaltolerance of ischemic heart disease patients. Ter Arkh. 1987;59:112-115.
32. Sugiishi M, Takatsu F. Cigarette smoking is a major risk factor forcoronary spasm. Circulation. 1993;87:76-79.
33. van Jaarsveld H, Kuyl JM, Alberts DW. Exposure of rats to lowconcentration of cigarette smoke increases myocardial sensitivityto ischemia/reperfusion. Basic Res Cardiol. 1992;87:393-399.
34. Kalra J, Chaudhary AK, Prasad K. Increased production of oxygenfree radicals in cigarette smokers. Int J Exp Pathol. 1991;72:1-7.
35. Astrup P. The arterial wall in atherogenesis. In: Cavallero, ed.Atherogenesis. Padua, Italy: Piccin Medical Book; 1975:77-92.
36. Leanderson P, Tagesson C. Cigarette smoke-induced DNA damagein cultured human lung cells: role of hydroxyl radicals and endonu-clease activation. Chem-Biol Interact. 1992;81:197-208.
37. Patterson TR, Stringham JD, Meikle AW. Nicotine and cotinineinhibit steroidogenesis in mouse Leydig cells. Life Sci. 1990;46:265-272.
38. Miyaura S, Eguchi H, Johnston JM. Effect of a cigarette smokeextract on the metabolism of the proinflammatory autacoid,platelet-activating factor. Circ Res. 1992;70:341-347.
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from

1290 Circulation Vol 89, No 3 March 1994
39. Imaizumi T-A, Satoh K, Yoshida H, Kawamura Y, Hiramoto M,Takamatsu S. Effect of cigarette smoking on the levels of platelet-activating factor-like lipid(s) in plasma lipoproteins. Athero-sclerosis. 1991;87:47-55.
40. Dobson AJ, Alexander HM, Heller RF, Lloyd DM. Passivesmoking and the risk of heart attack or coronary death. Med JAust.1991;154:793-797.
41. Ernst E, Matrai A. Abstention from chronic cigarette smokingnormalizes blood rheology. Atherosclerosis. 1987;64:75 -77.
42. Feher MD, Rampling MW, Brown J, Robinson R, Richmond W,Cholerton S, Brain BJ, Sever PS. Acute changes in atherogenic andthrombogenic factors with cessation of smoking. J R Soc Med.1990;83:146-148.
43. Cryer PE, Haymond MW, Santiago JV, Shah SD. Norepinephrineand epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. N Engl J Med.1976;295:573-577.
44. Leone A, Mori L, Bertanelli F, Fabiano P, Filippelli M. Indoorpassive smoking: its effect on cardiac performance. Int J Cardiol.1991;33:247-252.
45. Repace JL. Indoor concentrations of environmental tobaccosmoke: field survey. In: O'Neill IK, Brunnemann KD, Dodet B,Hoffmann D, eds. Environmental Carcinogens: Methods ofAnalysisand Exposure Measurement (9, Passive Smoking). Lyon, France:IARC Scientific Publications; 1987:141-159.
46. Penn A, Snyder CA. Inhalation of sidestream cigarette smokeaccelerates development of arteriosclerotic plaques. Circulation.1993;88(pt 1):1820-1825.
47. Robinson K, Conroy RM, Mulcahy R. Smoking and acutecoronary heart disease: a comparative study. Br Heart J. 1988;60:465 -469.
48. Rivers JT, White HD, Cross DB, Williams BF, Norris RM. Rein-farction after thrombolytic therapy for acute myocardial infarctionfollowed by conservative management: incidence and effect ofsmoking. J Am Coll Cardiol. 1990;16:340-348.
49. Wiklund I, Oden A, Sanne H, Ulvenstam G, Wilhelmsson C,Wilhelmsen L. Prognostic importance of somatic and psychosocialvariables after a first myocardial infarction. Am J Epidemiol. 1988;128:786-795.
50. Nyboe J, Jensen G, Appleyard M, Schnohr P. Smoking and the riskof first acute myocardial infarction. Am Heart J. 1991;122:438-447.
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from

B Q Zhu, Y P Sun, R E Sievers, S A Glantz, W W Parmley and C L WolfeExposure to environmental tobacco smoke increases myocardial infarct size in rats.
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 1994 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/01.CIR.89.3.1282
1994;89:1282-1290Circulation.
http://circ.ahajournals.org/content/89/3/1282World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 9, 2016http://circ.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















