
Please ensure that you have carefully encoded your subject ...

and Evolution. All rights reserved. For permissions, please e-mail: [email protected] The Author 2006. Published by Oxford University Press on behalf of the Society for Molecular Biology
Evolutionary relationships of apusomonads inferred from taxon-rich analyses of six
nuclear-encoded genes
Research Article
Eunsoo Kim1, Alastair G. B. Simpson2, Linda E. Graham1
1Department of Botany, University of Wisconsin-Madison, 132 Birge Hall, 430 Lincoln
Dr., Madison, WI 53706, USA
2Department of Biology, Dalhousie University, Halifax. N.S., B3H 4J1, Canada
Correspondence to: Eunsoo Kim, E-mail: [email protected], Phone: 608-262-0657
Fax: 608-262-7509
Contact information: Eunsoo Kim, Address: Department of Botany, University of
Wisconsin-Madison, 132 Birge Hall, 430 Lincoln Dr., Madison, WI 53706, USA
Running head: Phylogenetics of apusomonads
Key words: Apusomonas, Apusomonadidae, Opisthokonta, Amoebozoa, protists,
evolution
MBE Advance Access published September 18, 2006 by guest on M
arch 10, 2016http://m
be.oxfordjournals.org/D
ownloaded from

ABSTRACT
The phylogenetic relationships of the biflagellate protist group Apusomonadidae have
been unclear despite the availability of some molecular data. We analyzed sequences
from six nuclear encoded genes—SSU rRNA, LSU rRNA, α-tubulin, β-tubulin, actin,
and Hsp90—to infer the phylogenetic position of Apusomonas proboscidea Aléxéieff
1924. To increase the taxon richness of the study, we also obtained new sequences from
representatives of several other major eukaryotic groups: Chrysochromulina sp. NIES
1333 (Haptophyta), Cyanophora paradoxa (Glaucophyta), Goniomonas truncata
(Cryptophyceae), Leucocryptos marina (Kathablepharidae), Mesostigma viride
(Streptophyta, Viridiplantae), Peridinium limbatum (Alveolata), Pterosperma cristatum
(Prasinophytae, Viridiplantae), Synura sphagnicola (Stramenopiles), and
Thaumatomonas sp. (Rhizaria). In most individual gene phylogenies, Apusomonas
branched close to either of two related taxa - Opisthokonta (including animals, fungi, and
choanoflagellates) or Amoebozoa. Combined analyses of all four protein-coding genes,
or all six studied genes strongly supported the hypothesis that Apusomonadidae is closely
related to Opisthokonta (or to all other eukaryotic groups except Opisthokonta, depending
the position of the eukaryotic root). Alternative hypotheses were rejected in AU tests at
the 5% level. However, the strong phylogenetic signal supporting a specific affiliation
between Apusomonadidae and Opisthokonta largely originated from the α-tubulin data.
If α-tubulin is not considered, topologies in which Apusomonadidae are sister to
Opisthokonta or are sister to Amoebozoa were more or less equally supported. One
current model for deep eukaryotic evolution holds that eukaryotes are divided into
primary ‘unikont’ and ‘bikont’ clades and are descended from a ‘uniflagellate’ common
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

ancestor. Together with other information, our data suggest instead that ‘unikonts’
(=Opisthokonta and Amoebozoa) are not strictly monophyletic and are descended from
biflagellate ancestors.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

INTRODUCTION
Our understanding of eukaryotic phylogeny has improved in recent years as the
result of increasing sequence data from diverse taxonomic groups. For example, a
molecular gene analysis revealed that many morphologically diverse protists form a
superclade known as Rhizaria (Nikolaev et al. 2004). Most eukaryotes are now placed
into one of about 15 major eukaryotic lineages whose monophyly is generally
undisputed. Some authors reduce these lineages to six ‘supergroups’ (Simpson and Roger
2004; Adl et al. 2005; Keeling et al. 2005), although the monophyly of some of these
supergroups is currently under debate. More contentiously still, it has been proposed that
all major lineages fall into just two primary clades – ‘unikonts’ and ‘bikonts’ (Stechmann
and Cavalier-Smith 2003, Richards and Cavalier-Smith, 2005) However, a number of
protist taxa cannot be assigned unambiguously to any of these major eukaryotic lineages,
despite the presence of electron microscopical data and at least some sequence data
(Simpson and Roger 2004; Adl et al. 2005). These unassigned taxa are pivotal for
understanding eukaryotic diversification and for testing macroevolutionary hypotheses
such as the unikont/bikont bifurcation (Stechmann and Cavalier-Smith 2003b; Cavalier-
Smith, Chao, Oates 2004). Apusomonadidae, the focus of our study, is one such group.
The Apusomonadidae (‘apusomonads’) are a group of free-living heterotrophic
biflagellates consisting of two genera—Amastigomonas and Apusomonas. Apusomonads
glide along surfaces and feed on bacteria, which are usually engulfed using ventral
pseudopodia (Vickerman, Darbyshire, Ogden 1974). Their cells are covered with a
thickened submembranous ‘theca’ except in the ventral feeding region (Molina and
Nerad 1991). The cells possess two heterodynamic flagella: one anteriorly-directed and
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

one posteriorly-oriented (Vickerman, Darbyshire, Ogden 1974). The anterior flagellum is
encased by a membranous sleeve, a trait that is a synapomorphic feature for
Apusomonadidae (Patterson 2000a). The two basal bodies are inserted at right angles and
three rootlets are associated with these (Molina and Nerad 1991). One rootlet is a
multilayered structure (MLS), while two other rootlets, each consisting of 3–5 and 5–12
microtubules, underline each side of the ventral groove (Karpoff and Zhukov 1986;
Molina and Nerad 1991). Mitochondria have tubular cristae (Karpoff and Zhukov 1986;
Molina and Nerad 1991). A distinguishing feature of Apusomonas is the mastigophore, a
long extension of the two ventral grooves, from which the two flagella originate
(Patterson 2000b). SSU rRNA phylogenes clearly established the monophyly of
Apusomonadidae (Cavalier-Smith and Chao 2003b).
Thus far, two nuclear encoded genes—SSU rRNA and heat shock protein (Hsp)
90—have been used to infer the phylogenetic relationships of Apusomonadidae
(Cavalier-Smith and Chao 1995; Stechmann and Cavalier-Smith 2003). Based on SSU
rRNA gene phylogenies, Cavalier-Smith and Chao (1995) initially suggested that
Apusomonas is closely related to Opisthokonta, the clade that includes animals, fungi,
and choanoflagellates, with this hypothesis receiving moderate bootstrap support. The
same relationship was also recovered in later SSU rRNA studies, but with lower
bootstrap support (eg. Atkins, McArthur, Teske 2000; Fig 1 in Cavalier-Smith and Chao
2003b; Fig 4 in Cavalier-Smith, Chao, Oates 2004). However, other analyses have not
recovered a close association between Apusomonadidae and Opisthokonta (eg. Fig 2 in
Cavalier-Smith 2002; Simpson et al. 2002; Fig 2 in Cavalier-Smith, Chao, Oates, 2004;
Berney, Fahrni, Pawlowski 2004). Analyses based on Hsp90 gene sequences including
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

that of Amastigomonas marina did not strongly support any particular phylogenetic
position for apusomonads (Stechmann and Cavalier-Smith 2003a). Furthermore, the
dihydrofolate reductase-thymidylate synthase (DHFR-TS) gene fusion was identified in
Amastigomonas debruynei, suggesting a placement of apuosomonads in the large ‘bikont’
clade supposedly identified by this fusion character, and therefore remote from the
‘unikont’ clade (Opisthokonts and Amoebozoa), whose members lack this gene fusion
(Stechmann and Cavalier-Smith 2002; 2003b).
In this study, we sequenced five nuclear encoded genes from Apusomonas
proboscidea (LSU rRNA, α-tubulin, β-tubulin, actin, and Hsp90), and performed various
phylogenetic analyses with the goal of resolving the phylogenetic position of
Apusomonadidae. We also analyzed existing SSU rRNA gene data from apusomonads.
New sequences were also obtained from several distantly related protists in known
eukaryotic groups, in order to increase the taxon richness of our study. Organisms
included were Chrysochromulina sp. NIES 1333 (Haptophyta), Cyanophora paradoxa
(Glaucophyta), Goniomonas truncata (Cryptophyceae), Leucocryptos marina
(Kathablepharidae), Mesostigma viride (Streptophyta, Viridiplantae), Pterosperma
cristatum (Prasinophytae, Viridiplantae), Peridinium limbatum (Alveolata), Synura
sphagnicola (Stramenopiles), and Thaumatomonas sp. (Rhizaria). We carefully chose
taxa with sequences that are not particularly long-branched and/or that are early-
diverging members of some major eukaryotic lineages, and performed both Maximum
Likelihood (ML) and Bayesian analyses of individual and combined gene data sets.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

MATERIALS AND METHODS
Cultures
Apusomonas proboscidea Aléxéieff (CCAP 1905/1) was purchased from the
Culture Collection of Algae and Protozoa (CCAP), Argyll, Scotland. Chrysochromulina
sp. (NIES 1333), Leucocryptos marina (Braarud) Butcher (NIES 1335), Mesostigma
viride Lauterborn (NIES 296) and Pterosperma cristatum Schiller (NIES 936) were
obtained from the Microbial Culture Collection at the National Institute for
Environmental Studies (MCC NIES), Ibaraki, Japan. Cyanophora paradoxa Korshikov
and Glaucocystis nostochinearum Itzigsohn var. nostochinearum were obtained from
Carolina Biological Supply Company, Burlington, North Carolina. Goniomonas truncata
(Fresenius) Stein, Peridinium limbatum (Stokes) Lemmermann, Synura sphagnicola
(Korshikov) Korshikov, and Thaumatomonas sp., were cultured from various lakes in
Wisconsin, USA following single cell isolation (Stein 1975) and were maintained in
appropriate culture media—in most cases, a mixture of sterilized filtered lake water and
soil extract. Cultures were maintained at 15° C. Cultures were identified both by light
microscopic features and SSU rRNA gene sequences. The isolate identified as
Thaumatomonas sp. had SSU rRNA gene sequence 99.8% similar to database sequences
for Thaumatomonas sp. (SA), based on an NCBI BLAST sequence similarity search.
DNA/RNA Preparation
Cells were concentrated by centrifugation and genomic DNA was extracted using
the DNeasy Plant Mini Kit (Qiagen, Valencia, CA), according to the manufacturer’s
suggested protocols. Cells of Glaucocystis, Mesostigma, Peridinium, Pterosperma, and
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Synura were disrupted in liquid nitrogen with a plastic pestle. Vigorous vortexing in the
lysis solution was sufficient for colorless protist cells. In some cases, use of degenerate
PCR primers for amplifications of protein-coding genes from genomic DNA resulted in
multiple bands presumably representing non-specific amplification, and RT-PCR was
necessary. Total RNA was purified from Apusomonas, Chrysochromulina, Goniomonas,
Leucocryptos, and Thaumatomonas, using the RNeasy Mini Kit (Qiagen, Valencia, CA),
and cDNA was synthesized from oligo dT primers using the Access RT-PCR kit
(Promega, Madison, WI).
PCR Amplification, Cloning, and Sequencing
PCR primers for amplifying SSU rRNA, LSU rRNA, α-tubulin, β-tubulin, actin,
and Hsp90 gene sequences were designed based on sequence alignments as well as
previous studies (Table 1) (Simpson, Lukes, Roger 2002; Simpson, Inagaki, Roger 2006).
For some protein coding genes, a two-step nested PCR technique was applied. The
standard 50 µl reaction mixture consisted of 2.5 unit of Takara Ex Taq (Takara, Tokyo),
1X Ex Taq buffer, 0.2 mM of each dNTP, 0.6 µM of each primer, and 5% glycerol.
When PCR primers were degenerate at several positions, the standard PCR cyclic
reactions consisted of a denaturation step at 95°C for 3 min; 13 cycles of 1 min at 95°C, 1
min at 58°C (1°C decrease each cycle), and 1.5 min at 68°C; 20 cycles of 30 sec at 94°C,
1 min at 45°C, and 1.5 min at 68°C; and a final 10 min at 68°C. For SSU rRNA and LSU
rRNA gene amplifications, the standard PCR cyclic reactions consisted of a denaturation
step at 95°C for 3 min; 30 cycles of 1 min at 95°C, 1 min at 45°C, and 1~3 min at 72°C;
and a final 15 min at 72°C. For Hsp90 from Apusomonas, a 330bp fragment near the 5’
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

end of the coding region was amplified by nested PCR and used to design an exact-match
primer. A near-complete coding region was then amplified by a nested PCR with this
exact-match primer. The 20 µl reaction mixtures consisted of 1 unit of Taq polymerase
(Sigma-Aldrich, St. Louis, MO), 1X buffer (1.5mM MgCl2), 0.2 mM of each dNTP and
1 µM of each primer. The cycling for the final PCR consisted of a denaturation step at
94°C for 2 min; 35 cycles of 20s at 94°C, 1 min at 54°C, and 2.5 min at 72°C; and a final
5 min at 72°C. PCR-amplified fragments were either gel-purified, or were cleaned using
Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI), and then were
cloned into pCR 4-TOPO or pCR 2.1-TOPO vector (Invitrogen, Carlsbad, CA) or
pGEM®-T Easy vector (Promega, Madison, WI). Plasmids were isolated from multiple
positive bacterial clones using the QIAquick Miniprep Kit (Qiagen, Valencia, CA) or
Sigma miniprep kit (Sigma-Aldrich, St. Louis, MO). Multiple clones were partially
sequenced, and at least one clone from each reaction was selected for complete
sequencing. To eliminate the possibility of contamination, the identities of all gene
sequences were verified as described in online Supplementary Material (Method S1).
GenBank accession numbers of new sequences obtained in this study are listed in online
Supplementary Material (Table S1).
Sequence Alignments
Protein coding genes were translated to amino acids, which were manually
aligned using MacClade ver. 4.05 (Maddison and Maddison 2001). For the alignment of
SSU rRNA genes, CLUSTAL X (Thompson et al. 1997) was used to produce an
approximate alignment, after which the sequences were aligned more accurately based on
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

eukaryotic SSU rRNA secondary structure models (Wuys et al. 2002) from the European
database. Yves Van de Peer (Ghent University) kindly provided the LSU rRNA gene
sequence alignment used in a previous study (Ben Ali et al. 2001), which incorporated
both primary and secondary structure information. We added our new sequences and
other sequences available from GenBank. Ambiguously aligned positions were excluded
for all analyses. Initial individual gene alignments included more sequences than the final
versions. This allowed detection of possible paralogy and selection of short-branched
homologous copies. Through various preliminary phylogenetic analyses, mostly using
neighbor joining and maximum parsimony methods, long-branched or potentially non-
orthologous sequences were identified and excluded. For example, sequences of most
animals and embryophytes, and some fungi were removed from our α-tubulin, β-tubulin,
or actin gene alignments because multiple paralogs were present in these multicellular
organisms. Multiple gene copies, present in some of the included taxa, were closely
related to each other, to the exclusion of all other sequences analyzed. We carefully chose
taxa included in the final alignments to increase overall taxonomic representation of the
study, yet minimize potential problems associated with long-branch attraction artifacts
(Philippe 2000). In some cases, gene sequences from closely related taxa were
concatenated for the combined gene analyses. No alignment position or taxon was
included in the analysis if more than 20% of the total data were missing. In addition, in
the combined gene alignments, no taxon was included if more than 40% of the individual
gene data were missing. Sequence alignments are deposited at TreeBASE
(www.treebase.org).
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Because we restricted sequence length and percentage of included positions
across taxa, missing data did not exceed 3% in any one alignment. Only 1.6% of
sequence data were missing in the combined four protein-coding gene sequence analysis.
All included amino acid sequences passed a chi-square test of compositional
homogeneity at the 5% level, as implemented in TREE-PUZZLE. However, some LSU
rRNA gene sequences (Chrysochromulina, Cryptosporidium Cyanidioschyzon,
Mesostigma, Phaeocystis, Prymnesium) failed compositional homogeneity tests.
Phylogenetic Analysis
Protein-coding gene alignments were analyzed at the amino acid level. Maximum
likelihood (ML) analysis of amino acid sequences was performed using PROML in the
PHYLIP package ver. 3.7 (Felsenstein 2004). A JTT+Γ+I model of protein evolution was
applied with the user-defined Hidden Markov Model (HMM) option for modeling
among-site rate variation. The rates and probabilities for the HMM were estimated from
the neighbor-joining trees using TREE-PUZZLE ver. 5.2 (Schmidt et al. 2002). For each
ML tree search, the input order of sequences was randomized and the process was
repeated 100 times with ‘global rearrangements’. Bootstrap values were obtained from
100 re-samplings, each search with one round of random taxon addition followed by
global rearrangements.
PAUP* 4.0b (Swofford 2002) was utilized for the ML analysis of SSU and LSU
rRNA gene sequences. Modeltest ver. 3.7 (Posada and Crandall 1998) was used to find
the best fitting model of nucleotide evolution and to estimate substitution rates, base
frequencies, Γ distribution parameter (α), and proportion of invariable sites. For each ML
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

tree search, the input order of sequences was randomized and the process was repeated
100 times with the tree bisection and reconnection branch-swapping algorithm. Bootstrap
values were obtained from 100 re-samplings, each search with one round of random
taxon addition and the nearest-neighbor interchange branch-swapping algorithm.
Bayesian inference of phylogeny was performed using MrBayes ver. 3.1
(Huelsenbeck and Ronquist 2001). For DNA sequence analyses, the GTR+Γ+I model of
evolution was applied. For protein sequence analyses, the WAG+Γ+I model of evolution
was used. Preliminary Markov Chain Monte Carlo (MCMC) runs with about 10,000
generations of trees were used to find the optimal temperature values, which seemed to
be an important factor in chain mixing (data not shown). At least two independent
MCMC runs were then completed and were compared to assess the reliability of each
run. A total of 1,000,000-2,000,000 generations of trees were selected and evaluated, and
every hundredth tree was sampled for further analysis. The burn-in period was evaluated
using Gnuplot ver. 4.0 (Williams and Kelly 1998).
The approximately unbiased (AU) test was performed to compare three
competing hypotheses related to the phylogenetic position of A. proboscidea relative to
Opisthokonta and Amoebozoa. Tree topologies reflecting these hypotheses were
generated by rearrangement of the ML tree for the dataset (if required). Site likelihoods
for each topology were calculated using TREE-PUZZLE. The AU test was performed
using CONSEL ver. 0.1h (Shimodaira and Hasegawa 2001). The output file from TREE-
PUZZLE was converted to a CONSEL-compatible format using a Python script kindly
provided by Jessica Leigh (Department of Biochemistry and Molecular Biology at
Dalhousie University).
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

RESULTS
Evolutionary relationships of Apusomonas proboscidea
The phylogenetic position of Apusomonas was well resolved in the ML analysis
of the combined four protein-coding genes (Fig.1). The clade comprising Apusomonas
and Opisthokonta was recovered, and strongly supported, with an ML bootstrap value of
99%. Amoebozoa was sister to the Apusomonadidae+Opisthokonta clade with 99% ML
bootstrap support.
The same relationship between Apusomonas and Opisthokonta was found in the
ML trees based on individual gene sequence analyses of α-tubulin and β-tubulin with
90% and 41% bootstrap values respectively (see Supplementary Material online). The
specific and strongly supported relationship between Apusomonas and Opisthokonta is
still recovered if diplomonads, Carpediemonas, parabasalids and Andalucia incarcerata
are added to the analysis (These sequences are normally the closest relatives to
Opisthokonta in α-tubulin phylogenies—data not shown). Monophyly of Opisthokonta
was not recovered in the actin analysis, but Apusomonas weakly formed a clade with the
opisthokont Amoebidium (27% ML bootstrap value).
In the analysis of Hsp90 gene sequences, both Amastigomonas and Apusomonas
weakly allied with Stramenopiles (13% ML bootstrap value). In this case, neither an
Opisthokonta-Apusomonadidae-Amoebozoa clade, nor an Opisthokonta-Amoebozoa
clade was found (see Supplementary Material online). When slightly longer-branched
sequences were removed, Apusomonadidae instead formed a weak clade with
Amoebozoa and Opisthokonta (for more information about sequences removed, see
Supplementary Material online). Apusomonadidae were sister to Dictyostelium in the ML
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

tree, making Amoebozoa paraphyletic, but this relationship was not significantly
supported (17% ML bootstrap value).
In the combined SSU-LSU rRNA gene phylogeny, Opisthokonta,
Apusomonadidae, and Amoebozoa formed a clade in both the ML and Bayesian analyses
(Fig. 2). Apusomonas branched weakly with the concatenated ‘amoebozoan’ sequence
(44% ML bootstrap value). However, if additional (long-branched) Amoebozoan
sequences were included, Apusomonas formed a very weak clade with Opisthokonta
instead (data not shown). SSU rRNA gene analysis also suggested that Apusomonadidae
were related to the naked lobose amoebozoan Vexillifera, but without significant support.
In the LSU rRNA gene phylogenies, Apusomonas weakly branched within unresolved
clades of biflagellates, not closely related to the included amoebozoan (Mastigamoeba).
Bayesian analysis of the six combined gene sequences indicated that Apusomonas
is closely related to Opisthokonta, with Amoebozoa falling as the sister group to
Apusomonadidae+Opisthokonta (Fig 4). Most deep divergences including these clades
were resolved with Bayesian posterior probabilities of 1. The more conservative ML
bootstrapping approach could not be applied to this mixed amino acid/nucleotide data set.
Our data were used to compare the following three hypotheses. Hypothesis I is
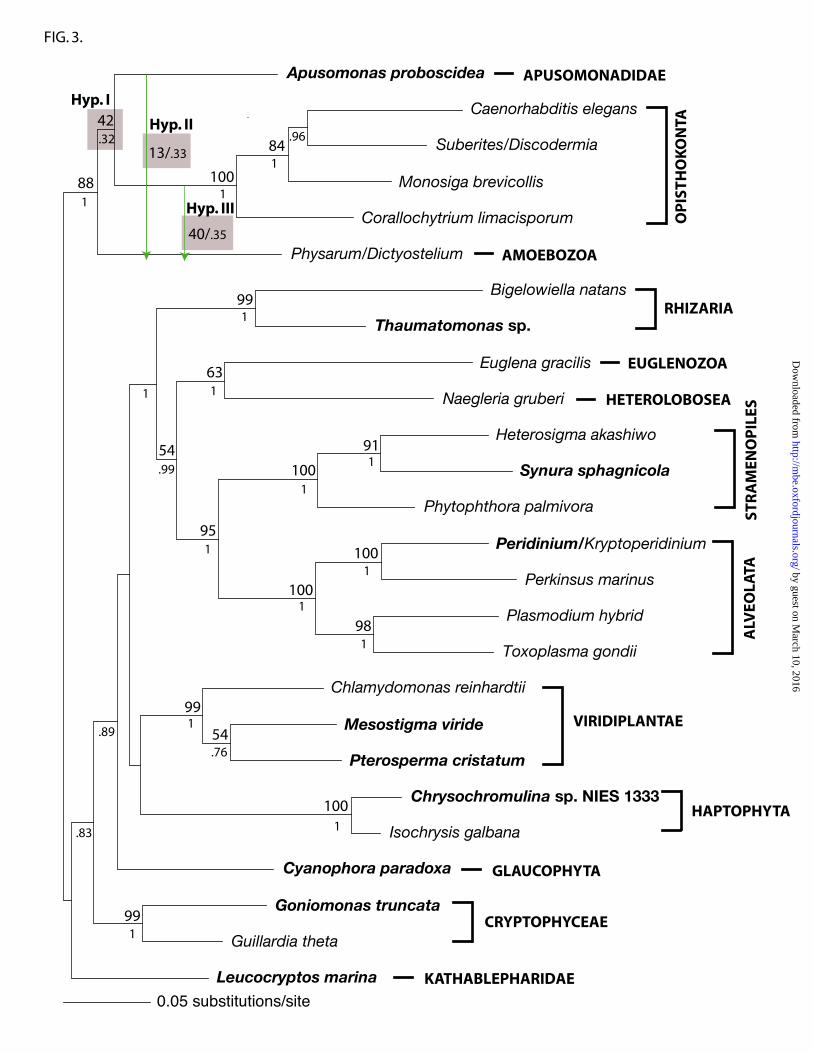
that Apusomonadidae is sister to Opisthokonta (the hypothesis most strongly supported
by the analyses described above). Hypothesis II is that Apusomonadidae and Amoebozoa
are sisters (supported by some of our phylogenies). Hypothesis III is that Opisthokonta
and Amoebozoa are sisters to the exclusion of Apusomonadidae (consistent with a
‘unikont’ clade). In the AU test based on the combined four proteins, hypotheses II and
III were rejected (p=2 X 10-4, 4 X 10-4 respectively) (see Supplementary Material online).
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

The AU test based on α-tubulin alone strongly rejected hypotheses II and III (both p=1 X
10-7). However, the analysis of the combined β-tubulin, actin, and Hsp90 gene sequences
did not reject hypotheses II and III (p=0.539, 0.472 respectively). These results suggest
that the strong rejection signals in the combined protein-coding gene analysis (Fig. 1)
mostly originated from the α-tubulin data set. AU tests based on the combined six gene
sequences also rejected hypotheses II and III (p=2 X 10-4, 4 X 10-5 respectively) (see
Supplementary Material online).
Evolutionary relationships of other protist groups
Our combined protein coding gene sequence analyses found moderate to strong
support (88-95%) for a sister relationship between Alveolata and Stramenopiles. Close
affinity of Kathablepharidae to Cryptophyceae was recovered in both SSU and LSU gene
phylogenies (see Supplementary Material). The combined SSU and LSU gene phylogeny
recovered a Kathablepharidae-Cryptophyceae clade with 100% bootstrap support (Fig 2).
Interestingly, none of the protein phylogenies suggested monophyly of
Kathablepharidae+Cryptophyceae, and the position Kathablepharidae was not resolved
with over 50% ML bootstrap support in any of the protein gene phylogenies, although the
six gene analysis placed it in a clade with Cryptophyceae (Fig 4). Two genes—SSU and
Hsp90—supported a sister relationship between Kathablepharidae+Cryptophyceae and
Glaucophyta, although ML bootstrap was moderate, or weak (see Supplementary
Material).
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

DISCUSSION
The evolutionary position of Apusomonadidae
With the goal of clarifying the evolutionary position of Apusomonadidae, we
determined five new Apusomonas gene sequences—those for LSU rRNA, α-tubulin, β-
tubulin, actin, and Hsp90, the first four of which are new to Apusomonadidae. Most
individual gene analyses suggested that Apusomonadidae are closely related to
Opisthokonta or Amoebozoa. α-tubulin, β-tubulin, and actin genes separately provided
evidence of Opisthokonta+Apusomonadidae clade, assuming that the eukaryotic root
does not lie between the two groups. Hsp90 genes, when analyzed without including
some excavate sequences, supported an Apusomonadidae plus Amoebozoa clade.
However, the position of Apusomonadidae based on Hsp90 gene sequences was sensitive
to taxon sampling, and Apusomonadidae sometimes appeared as sister to biflagellate
clades, as found by Stechmann and Cavalier-Smith (2003a). Our LSU rRNA gene
phylogenies likewise did not support the affinity of Apusomonadidae to any particular
group, and the position of Apusomonadidae was sensitive to taxon sampling.
The position of Apusomonadidae in SSU rRNA gene phylogenies has been
unstable. In most cases, inferred topologies were poorly supported except for the earlier
study by Cavalier-Smith and Chao (1995), in which a sister relationship between
Apusomonadidae and Opisthokonta was relatively well supported in maximum
parsimony analyses (84%) and least-squares analyses of Jukes-Cantor distances (96%).
However, in subsequent studies, support for the Apusomonadidae-Opisthokonta clade
was weaker (for example Cavalier-Smith and Chao 2003b), or alternative topologies were
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

inferred (for examples Berney, Fahrni, Pawlowski 2004; Fig 2 in Cavalier-Smith, Chao,
Oates 2004).
When α-tubulin gene sequences were included, our analyses strongly supported
an Apusomonadidae-Opisthokonta clade, and AU tests rejected the alternative hypotheses
of an Apusomonadidae-Amoebozoa clade or an Opisthokonta-Amoebozoa clade. When
α-tubulin sequences were excluded, an Apusomonadidae-Opisthokonta clade was
recovered in the ML tree (Fig 3), but two alternative hypotheses—an Apusomonadidae-
Amoebozoa clade and an Opisthokonta-Amoebozoa clade (with Apusomonadidae as
their sister group)—could not be rejected. None of the individual or combined gene data
sets analyzed in this study rejected the Apusomonadidae-Opisthokonta clade when AU
tests were applied. Overall, an Apusomonadidae-Opisthokonta clade is the best supported
hypothesis.
Apusomonadidae is not likely to branch within Opisthokonta because members of
Opisthokonta included in our study formed a strong clade (Fig 1–3). In addition,
Steenkamp, Wright, and Baldauf (2005) reported that Apusomonadidae lack an amino
acid insertion in elongation factor 1-α, a synapomorphic character for Opisthokonta.
While Hampl et al. (2005) and Simpson, Inagaki and Roger (2006) noted that
tubulin gene (α-tubulin in particular) phylogenies conflicted with other gene trees with
respect to the positions of some excavates, we did not find any significant conflict
between the α-tubulin tree and any of the other five gene trees with respect to the
position of Apusomonadidae. Determination of the utility of α-tubulin, or any other
genes as phylogenetic markers for analysis of deep eukaryote divergences would require
the comparative analysis of many gene sequences from diverse eukaryotic groups.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Some components of our study suggested a close relationship between
Amoebozoa and Apusomonadidae. Amoebozoa includes diverse unicellular and
multicellular eukaryotic organisms for which the only morphological commonality is
amoeboid movement (Walochnik, Michel, Aspöck 2004) (the non-amoeboid flagellates
Phalansterium and Multicilia being likely exceptions [Nikolaev et al. 2006]). Several
molecular analyses have suggested monophyly of Amoebozoa (see Fahrni et al. 2003 for
review), however, without robust statistical support, except when taxon sampling was
low (Bapteste et al. 2002). While there are several major subclades in Amoebozoa, so far
a significant amount of genomic data have only been obtained for a few amoebozoan
taxa, which like Entamoeba, are typically long-branched. This is the reason for the
limited taxon sampling for Amoebozoa in our study. Additional sequence data for other
amoebozoan taxa, such as Phalansterium, Tubulinea, and Fabellinea, are needed to
further explore the relatedness of Apusomonadidae to Amoebozoa.
Richards and Cavalier-Smith (2005) suggested that Opisthokonta and Amoebozoa
(‘unikonts’) form a monophyletic group based on five putative synapomorphies
concerning myosin gene types and sequence features. However, this study did not include
several important eukaryotic taxa, such as Rhizaria and Apusomonadidae. Since our
multigene analyses place Apusomonadidae cladistically within the ‘unikont’ clade,
testing for the presence of ‘unikont’-specific myosin genes and indels in
Apusomonadidae would be particularly valuable. Likewise, determination of whether
Apusomonadidae undergo flagellar transformation, and which flagellum is mature would
be useful because flagellar transformation (in which the posterior flagellum is the mature
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

one) is proposed to be a key character distinguishing ‘bikonts’ from ‘unikonts’ (Cavalier-
Smith 2003).
Cavalier-Smith and Chao (2003) united apusomonads with the obscure flagellate
Ancyromonas in a more inclusive taxon Apusozoa, based on the presence of a thecae-like
layer on the dorsal surface of Ancyromonas and a weak affinity between the groups in
some SSU rRNA phylogenies. This relationship requires further confirmation, once more
data are available from Ancyromonas.
Implications of an ‘Apusomonadidae-Opisthokonta’ clade
The two most likely relationships of Apusomonadidae suggested in our
study—the Apusomonadidae-Opisthokonta clade and the Apusomonadidae-Amoebozoa
clade—conflict with two existing hypotheses related to deep eukaryotic divergences. The
first of these hypotheses is that Opisthokonta and Amoebozoa shared a common unikont
ancestor (i.e. with a single flagellum and one basal body), while other eukaryotic groups
were ancestrally bikont (having two flagella and two basal bodies) (Cavalier-Smith
2002). The second widely-cited hypothesis is that bikonts—including
Apusomonadidae—are monophyletic, based primarily on the presence or absence of the
DHFR-TS gene fusion (Stechmann and Cavalier-Smith 2002). As explained below,
neither hypothesis is well supported by existing morphological/genomic data and both are
contra-indicated by our results.
Morphological and our molecular phylogenetic data conflict with a hypothesis of
unikont ancestry for Opisthokonta and/or Amoebozoa: Though flagellate cells of
Opisthokonta (those of choanoflagellates, chytridiomycetes, certain Ichthyosporea
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

[=Mesomycetozoea], and male gametes of animals) possess a single flagellum, such cells
typically have a non-flagellated second basal body (Barr 1981; Karpov and Leadbeater
1997), consistent with biflagellate ancestry. The ancestral flagellate condition of
Amoebozoa is more ambiguous because flagellate amoebozoans are relatively
uncommon, and evolutionary relationships among the major subclades of Amoebozoa are
poorly understood (Fahrni et al. 2003; Smirnov et al. 2005). On the one hand, flagellate
members of pelobionts, some Protostelia, Phalansterium, and Multicilia possess a single
flagellum and one basal body per kinetid (a unit consisting of one or more flagellar basal
bodies and any associated fibers, roots, and cytoskeleton). On the other hand, those of
Myxogastria and some Protostelia have two basal bodies and usually two flagella (Olive
1975). Spiegel (1981) suggested that the common ancestor of Protostelia likely possessed
two basal bodies, and that some members of Protostelia had lost their second basal body.
Hence, the ‘unikont’ hypothesis is not particularly well supported by the available
morphological data, even before considering the position of apusomonads.
Because the apusomonads have two basal bodies and two flagella, their possible
positioning within the Opisthokont-Amoebozoa clade makes a biflagellate common
ancestor for this clade more parsimonious. Our molecular study therefore further weakens
the hypothesis of unikont ancestry for Opisthokonta and Amoebozoa (Fig 5). In view of
available morphological and molecular evidence, categorizing the minimum
Opisthokonta+Amoebozoa clade as ‘unikonts’ seems unjustified on present data.
Conflict between our molecular phylogenetic results and interpretations of the
DHFR-TS gene fusion data: Philippe et al. (2000) and Stechmann and Cavalier-Smith
(2002) proposed that possession of two separate, monofunctional DHFR and TS (thyA)
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

genes was the archaic condition for eukaryotes, since bacteria, when they possess these
two genes, also produce two separate proteins. Based on this premise, Stechmann and
Cavalier-Smith (2002) used the presence or absence of gene fusion between DHFR and
TS (thyA) to help infer the position of the eukaryotic root. Stechmann and Cavalier-Smith
(2002, 2003b) did not find the DHFR-TS fusion gene in Opisthokonta and Amoebozoa,
but noted that studied representatives of Alveolata, Apusomonadidae, Euglenozoa,
Rhizaria, Stramenopiles, and Viridiplantae have bifunctional DHFR-TS fusion genes.
Therefore, these authors suggested that eukaryotes with this ‘derived’ gene fusion form a
monophyletic group (‘bikonts’), within which the eukaryote root cannot lie. However,
this concept is questionable for several reasons.
First, as Embley and Martin (2006) noted, DHFR-TS fusion data are currently
available for relatively few taxonomic groups, and cannot not be used to infer positions
of lineages such as diplomonads and parabasalids that are devoid of these genes. The
assumption that the DHFR-TS gene fusion represents the derived condition in eukaryotes
is another issue. Since multiple cases of replacement and separation of DHFR and TS
genes have been documented, particularly in bacteria (Philip, Creevey, and McInerney
2005), separate positioning of DHFR and TS (thyA) genes may not represent the archaic
condition in eukaryotes. Alternatively, the DHFR-TS fusion gene could be viewed as the
archaic condition in eukaryotes because the DHFR-TS fusion gene is transcribed into a
single mRNA molecule like bacterial DHFR and TS (thyA) genes, which usually occur in
a single operon (although the gene order is reversed). In contrast, DHFR and TS (thyA)
genes are separately transcribed in Opisthokonta and the amoebozoan Hartmannella
(Stechmann and Cavalier-Smith 2003b). These separate DHFR and TS (thyA) genes may
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

have been derived from re-fission of the fused gene. While Stechmann and Cavalier-
Smith (2002) suggested that the reversal of the DHFR-TS gene fusion is improbable,
several examples of fusion and re-fission of other genes over evolutionary time have now
been documented in eukaryotes (Arisue, Hasegawa, Hashimoto 2005; Krauss et al. 2006;
Waller, Slamovits, Keeling 2006). Phylogenetic analyses of the DHFR or TS (thyA) gene
sequences also did not positively support a hypothesis of recent common origin of the
fused genes (Lazar, Zhang, Goodman 1993; Schlichtherle, Roos, Van Houten 1996). In
addition, the DHFR and TS genes may have been subjected to multiple lateral gene
transfer (LGT) events. For example, the amoebozoans Dictyostelium (Leduc et al. 2004)
and Physarum have apparently replaced their TS (thyA) genes with non-homologous TS
(thyX) genes. Lastly, there is a lack of strong independent evidence for the ‘bikont’ clade
supposedly identified by DHFR-TS fusion. Stechmann and Cavalier-Smith (2002)
propose that the presence of flagellar transformation is a second synapomorphy for the
bikont clade (see also Cavalier-Smith 2002), however this idea is complicated by the
unambiguous presence of a form of flagellar transformation in the biflagellate ‘unikont’
Physarum (Wright, Moisand, and Mir 1980). Collectively, these considerations suggest
that the proposal that the DHFR-TS gene fusion represents a single derived evolutionary
event within the diversification of extant eukaryotes is questionable.
Our study, which supports an Apusomonadidae+Opisthokonta clade, places a
fusion-bearing taxon within the only non-fusion-bearing clade of eukaryotes. If our
phylogenetic placement of Apusomonadidae is correct, this implies one of two
possibilities: i) The DHFR-TS fusion was laterally transferred at least once (or that the
fusion event occurred more than once), and hence is an unreliable phylogenetic marker,
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

or, ii) The DHFR-TS fusion represents an unique evolutionary event, but this took place
before the divergence of extant eukaryotes, and hence is an ancestral character state
(plesiomorphy) for all living eukaryotes. Therefore our study adds substantial additional
doubt as to the validity of the DHFR-TS fusion as a marker for deep eukaryote
diversification, and the monophyly of the group identified by the fusion (‘bikonts’).
Evolutionary relationships of other groups
Our new sequences for representatives of Alveolata, Cryptophyceae,
Glaucophyta, Haptophyta, Stramenopiles, Kathablepharidae, Rhizaria, and Viridiplantae,
allowed us to evaluate additional relationships among major eukaryotic lineages. Our
LSU rRNA gene phylogeny confirmed the previous result of SSU rRNA phylogeny, that
Kathablepharidae and Cryptophyceae are sister taxa (Okamoto and Inouye 2005).
Alveolata and Stramenopiles were sisters in multiple protein gene phylogenies in our
analyses (Fig 1, 3), consistent with previous multi-protein analyses (Baldauf et al. 2000;
Harper, Waanders, Keeling 2005; Simpson, Inagaki, Roger 2006). In the combined SSU
and LSU rRNA phylogeny (Fig 2), however, Alveolata branched weakly with Rhizaria,
which was also observed in some previous SSU rRNA gene analyses (for example Fig 1
and 2 in Cavalier-Smith and Chao 2003a)
None of our analyses suggested the monophyly of “chromalveolates”, including
Alveolata, Cryptophyceae (+Kathablepharidae), Haptophyta, and Stramenopiles
(Cavalier-Smith 1999), particularly with respect to the Kathablepharidae-Cryptophyceae
clade. The “chromalveolate hypothesis”, advocating a single red algal plastid origin for
“chromalveolates” and the monophyly of these groups, is currently hotly debated
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

(Falkowski et al. 2004; Grzebyk et al. 2004; Keeling et al. 2004). Because our individual
gene analyses of SSU rRNA and Hsp90 as well as previous Hsp70 phylogeny (Rensing et
al. 1997) suggested, albeit without strong support, close affinity of the Kathablepharidae-
Cryptophyceae clade (or Cryptophyceae) to Glaucophyta, additional genomic data from
Kathablepharidae and Cryptophyceae would be useful to further evaluate their
phylogenetic relationship to other putative “chromalveolate” groups.
Supplementary Material
Supplementary Figures S1–S6 represent ML trees based on analyses of individual
gene sequences. Table S1 shows GenBank accession numbers of newly obtained
sequences in this study. Table S2 shows AU test results. Method S1 includes
supplementary method information.
Acknowledgements
This research was supported by grant MCB-9977903 from the National Science
Foundation, a Davis Summer Research Fellowship (Department of Botany at the
University of Wisconsin-Madison), an Anna Grant Birge Memorial Award (University of
Wisconsin-Madison), and NSERC grant 298366-04 to AGBS. Y. Van de Peer at Ghent
University kindly provided LSU rRNA sequence alignments. The authors also thank B.
Larget (University of Wisconsin-Madison) for access to a computation facility, J. Graham
(University of Wisconsin-Madison) for the Peridinium limbatum culture, L. Wilcox
(University of Wisconsin-Madison) for obtaining samples from aquatic habitats, and J.
Leigh (Dalhousie University) for a Python script.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

References
Adl SM, Simpson GB, Farmer MA, et al. (28 co-authors). 2005. The new higher level
classification of eukaryotes with emphasis on the taxonomy of protists. J. Eukaryot.
Microbiol. 52:399–451.
Arisue N, Hasegawa M, Hashimoto T. 2005. Root of the eukaryota tree as inferred from
combined likelihood analyses of multiple molecular sequence data. Mol. Biol. Evol.
22:409–420.
Atkins MS, McArthur AG, Teske AP. 2000. Ancyromonadida: a new phylogenetic
lineage among the protoza closely related to the common ancestor of metazoans, fungi,
and choanoflagellates (Opisthokonta). J. Mol. Evol. 51:278–285.
Baldauf SL, Roger AJ, Wenk-Sierfert, Doolittle WF. 2000. A kingdom-level phylogeny
of eukaryotes based on combined protein data. Science 290:972–977.
Bapteste E, Brinkmann H, Lee JA, et al. (11 co-authors). 2002. The analysis of 100 genes
supports the grouping of three highly divergent amoebae; Dictyostelium, Entamoeba, and
Mastigamoeba. P. Natl. Acad. Sci. USA. 99:1414–1419.
Barr DJS. 1981. The phylogenetic and taxonomic implications of flagellar roolet
morphology among zoosporic fungi. Biosystems 14:359–370.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Ben Ali A, De Baere R, Van der Auwera G, De Wachter R, Van de Peer Y. 2001.
Phylogenetic relationships among algae based on complete large-subunit rRNA
sequences. Int. J. Syst. Evol. Micr. 51:737–749.
Berney C, Fahrni J, Pawlowski J. 2004. How many novel eukaryotic ‘kingdoms’? Pitfalls
and limitations of environmental DNA surveys. BMC Biol. 2:13.
Cavalier-Smith T. 1999. Principles of protein and lipid targeting in secondary
symbiogeneis: Euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote
family tree. J. Eukaryot. Microbiol. 46:347–366.
Cavalier-Smith T. 2002. The phagotrophic origin of eukaryotes and phylogenetic
classification of protozoa. Int. J. Syst. Evol. Micr. 52:297–354.
Cavalier-Smith T. 2003. Protist phylogeny and the high-level classification of protozoa.
Eur. J. Protistol. 39:338–348.
Cavalier-Smith T, Chao EE. 1995. The opalozoan Apusomonas is related to the common
ancestor of animals, fungi, and choanoflagellates. P. Roy. Soc. Lond. B. Bio. 261: 1–6.
Cavalier-Smith T, Chao EE. 2003a. Molecular phylogeny of centrohelid Heliozoa, a
novel lineage of bikont eukaryotes that arose by ciliary loss. J. Mol. Evol. 56:387–396.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Cavalier-Smith T, Chao EE. 2003b. Phylogeny of Choanozoa, Apusozoa, and other
protozoa and early eukaryote megaevolution. J. Mol. Evol. 56:540–563.
Cavalier-Smith T, Chao EE, Oates B. 2004. Molecular phylogeny of Amoebozoa and the
evolutionary significance of the unikont Phalansterium. Eur. J. Protistol. 40:21–48.
Embley TM, Martin W. 2006. Eukaryotic evolution, changes and challenges. Nature
440:623–630.
Fahrni J, Bolivar I, Berney C, Nassonova E, Smirnov A, Pawlowski J. 2003. Phylogeny
of lobose amoebae based on actin and small-subunit ribosomal RNA genes. Mol. Biol.
Evol. 20:1881–1886.
Falkowski PG, Katz ME, Knoll AH, Quigg A, Raven JA, Schofield O, Taylor FJR. 2004.
The evolution of modern eukaryotic phytoplankton. Science 305:354–360.
Felsenstein, J. 2004. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by
the author. Department of Genome Sciences, University of Washington, Seattle.
Grzebyk D, Katz ME, Knoll AH, Quigg A, Raven JA, Schofield O, Taylor FJR,
Falkowski PG. 2004. Response to Comment on “The evolution of modern eukaryotic
phytoplankton”. Science 306:2191c.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Hampl V, Horner DS, Dyal P, Kulda J, Flegr J, Foster PG, Embley TM. 2005. Inference
of the phylogenetic position of oxymonads based on nine genes: support for Metamonada
and Excavata. Mol. Biol. Evol. 22:2508–2518.
Harper JT, Waanders E, Keeling PJ. 2005. On the monophyly of chromalveolates using a
six-protein phylogeny of eukaryotes. Int. J. Syst. Evol. Micr. 55:487–496.
Huelsenbeck, JP. Ronquist F. 2001. MrBayes: Bayesian inference of phylogeny.
Bioinformatics 17:754–755.
Karpov SA, Leadbeater BSC. 1997. Cell and nuclear division in a freshwater
choanoflagellate, Monosiga ovata Kent. Eur. J. Protistol. 33:323–334.
Karpoff SA, Zhukov BF. 1986. Ultrastructure and taxonomic position of Apusomonas
proboscidea Alexeieff. Arch. Protistenkd. 131:13–26.
Keeling PJ, Archibald JM, Fast NM, Palmer JD. 2004. Comment on “The evolution of
modern eukaryotic phytoplankton”. Science 306:2191b.
Keeling PJ, Burger G, Durnford DG, Lang BF, Lee RW, Pearlman RE, Roger AJ, Gray
MW. 2005. The tree of eukaryotes. Trends Ecol. Evol. 20:670–676.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Krauss V, Fassl A, Fiebig P, Patties I, Sass H. 2006. The evolution of the histone
methyltransferase gene Su(var)3-9 in metazoans includes a fusion with and a re-fission
from a functionally unrelated gene. BMC Evol. Biol. 6:18.
Lazar G, Zhang H, Goodman HM. 1993. The origin of the bifunctional dihydrofolate
reductase-thymidylate synthase isogenes of Arabidopsis thaliana. Plant J. 3:657–668.
Leduc D, Graziani S, Lipowski G, Marchand C, Maréchal PL, Liebl U, Myllykallio H.
2005. Functional evidence for active site location of tetrameric thymidylate synthase X at
the interphase of three monomers. P. Nat. Acad. Sci. USA. 101:7252–7257.
Maddison DR, Maddison WP. 2001. MacClade 4: Analysis of phylogeny and character
evolution. Version 4.03. Sunderland, MA: Sinauer Associates.
Molina FI, Nerad TA. 1991. Ultrastructure of Amastigomonas bermudensis ATCC 50234
sp. nov. Eur. J. Protistol. 27:386–396.
Nikolaev SI, Berney C, Fahrni JF, Bolivar I, Polet S, Mylnikov AP, Aleshin VV, Petrov
NB, Pawlowski J. 2004. The twilight of Heliozoa and rise of Rhizaria, an emerging
supergroup of amoeboid eukaryotes. P. Nat. Acad. Sci. USA. 101:8066–8071.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Nikolaev SI, Berney C, Petrov NB, Mylnikov AP, Fahrni JF, Pawlowski J. 2006.
Phylogenetic position of Multicilia marina and the evolution of Amoebozoa. Int. J. Syst.
Evol. Micr. 56:1449–1458.
Okamoto N, Inouye I. 2005. The Kathablepharidaes are a distant sister group of the
cryptophyta: a proposal for Kathablepharidaeophyta divisio nova/ Kathablepharida
phylum novum based on SSU DNA and beta-tubulin phylogeny. Protist 156:163–179.
Olive LS. 1975. The mycetozoans. NY: Academic Press.
Patterson DJ. 2000a. Apusomonadidae. Apusomonadidae Karpov & Mylnikov 1989.
Version 18 September 2000. http://tolweb.org/Apusomonadidae/2387/2000.09.18 in The
Tree of Life Web Project, http://tolweb.org
Patterson DJ. 2000b. Apusomonas Aléxéieff 1924. Rostromonas Karpov S. A. & Zhukov,
B. F. 1980. Version 18 September 2000.
http://tolweb.org/Apusomonas/20484/2000.09.18 in The Tree of Life Web Project,
http://tolweb.org
Philip GK, Creevey CJ, McInerney JO. 2005. The Opisthokonta and the Ecdysozoa may
not be clades: stronger support for the grouping of plant and animal than for animal and
fungi and stronger support for the Coelomata than Ecdysozoa. Mol. Biol. Evol.
22:1175–1184.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Philippe H. 2000. Opinion: Long branch attraction and protist phylogeny. Protist
151:307–316.
Philippe H, Lopez P, Brinkmann H, Budin K, Germot A, Laurernt J, Moreira D, Muller
M, Le Guyader H. 2000. Early-branching or fast-evolving eukaryotes? An answer based
on slowly evolving positions. P. Roy. Soc. Lond. B. Bio. 267:1213–1221.
Posada D, Crandall KA. 1998 Modeltest: testing the model of DNA substitution.
Bioinformatics 14:817–818.
Richards TA, Cavalier-Smith T. 2005. Myosin domain evolution and the primary
divergence of eukaryotes. Nature 436:1113–1118.
Waller RF, Slamovits CH, Keeling PJ. 2006. Lateral gene transfer of a multigene region
from cyanobacteria to dinoflagellates resulting in a novel plastid-targeted fusion protein.
Mol. Biol. Evol. 23:1437–1443.
Schlichtherle IM, Ross DS, Van Houten JL. 1996. Cloning and molecular analysis of the
bifunctional dihydrofolate reductase-thymidylate synthase gene in the ciliated protozoan
Paramecium tetraurelia. Mol. Gen. Genet. 250:665–673.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Schmidt HA, Strimmer K, Vingron M, Von Haeseler A. 2002. TREE-PUZZLE:
maximum likelihood phylogenetic analysis using quartets and parallel computing.
Bioinformatics 18:502–504.
Shimodaira H, Hasegawa M. 2001. CONSEL: for assessing the confidence of
phylogenetic tree selection. Bioinformatics 17:1246–1247.
Simpson AGB, Inagaki Y, Roger AJ. 2006. Comprehensive multigene phylogenies of
excavate protists reveal the evolutionary positions of “primitive” eukaryotes. Mol. Biol.
Evol. 23:615–625.
Simpson AGB, Lukes J, Roger AJ. 2002. The evolutionary history of kinetoplastids and
their kinetoplasts. Mol. Biol. Evol. 19:2071–2083.
Simpson AGB, Roger AJ. 2004. The real ‘kingdoms’ of eukaryotes. Curr. Biol.
14:R693–R696.
Simpson AGB, Roger AJ, Silberman JD, Leipe DD, Edgcomb VP, Jermin LS, Patterson
DJ, Sogin ML. 2002. Evolutionary history of “early-diverging” eukaryotes: The excavate
taxon Carpediemonas is a close relative of Giardia. Mol. Biol. Evol. 19:1782–1791.
Smirnov A, Nassonova E, Berney C, Fahrni J, Bolivar I, Pawlowski J. 2005. Molecular
phylogeny and classification of the lobose amoebae. Protist 156:129–142.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Spiegel FW. 1981. Phylogenetic significance of the flagellar apparatus in protostelids
(Eumycetozoa). Biosystems 14:491–499.
Stechmann A, Cavalier-Smith T. 2002. Rooting the eukaryote tree by using a derived
gene fusion. Science 297:89–91.
Stechmann A, Cavalier-Smith, T. 2003a. Phylogenetic analysis of eukaryotes using heat-
shock protein Hsp90. J. Mol. Evol. 57:408–419.
Stechmann A, Cavalier-Smith T. 2003b. The root of the eukaryote tree pinpointed. Curr.
Biol. 13:R665–R666.
Steenkamp ET, Wright J, Baldauf SL. 2006. The protistan origins of animals and fungi.
Mol. Biol. Evol. 23:93–106.
Stein JR. Ed. 1973. Handbook of phycological methods: Culture methods and growth
measurements. Cambridge University Press.
Swofford DL. 2002. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other
Methods). Version 4. Sunderland, MA: Sinauer Associates.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The ClustalX
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

windows interface: flexible strategies for multiple sequence alignment aided by quality
analysis tools. Nucleic Acids Res. 24:4876–4882.
Vickerman K, Darbyshire JF, Ogden CG. 1974. Apusomonas proboscidea Aléxéieff
1924, an unusual phagotrophic flagellate from soil. Arch. Protistenkd. 116:254–269.
Walochnik J, Michel R, Aspöck H. 2004. A molecular biological approach to the
phylogenetic position of the genus Hyperamoeba. J. Eukaryot. Microbiol. 51:433–440.
Williams T, Kelly C. 1998. Gnuplot: an interactive plotting program. Dartmouth College,
Hanover, NH.
Wright M, Moisand A, Mir, L. 1980. Centriole maturation in the amoebae of Physarum
polycephalum. Protoplasma 105:149-160.
Wuyts J, Van de Peer Y, Winkelmans T, De Wachter R. 2002. The European database on
small subunit ribosomal RNA. Nucleic Acids Res. 30:183–185.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

TABLE 1. Primers for PCR amplifying and sequencing nuclear-encoded SSU rRNA, LSU rRNA, α-tubulin, β-tubulin, actin, andHsp90 genes. Primer positions are relative to location within Arabidopsis thaliana (LSU rRNA) or Chlamydomonas reinhardtii (allothers).
Primer Other names 5′ end 3′ end Primer sequence Referencenu-SSU Primersnu-SSU-0024-5′ NSF4/21 0004 0024 CTG GTT GAT CCT GCC AGT AGT This studynu-SSU-0033-5′ NSF13/21 0013 0033 CCT GCC AGT AGT CAT AYG CTT This studynu-SSU-0977-5′ NSF963/18 0960 0977 TTR ATC AAG AAC GAA AGT This studynu-SSU-1173-3′ NSR1197/24 1196 1173 CCC GTG TTG AGT CAA ATT AAG CCG This studynu-SSU-1757-3′ NSR1784/21 1777 1757 CAG GTT CAC CTA CGG AAA CCT This studynu-SSU-1768-3′ NSR1795/21 1788 1768 TGA TCC TTC YGC AGG TTC ACC This studynu-LSU Primersnu-LSU-0046-5′ NLF184/23 0024 0046 ACC CGC TGA AYT TAA GCA TAT CA This studynu-LSU-0058-5′ NLF196/23 0036 0058 TAA GCA TAT CAM TAA GCG GAG GA This studynu-LSU-1152-5′ NLF1280/23 1130 1152 TTT GGT AAG CAG AAC TGG CGA TG This studynu-LSU-1262-3′ NLR1431/23 1284 1262 AGT TGT TAC ACA CTC CTT AGC GG This studynu-LSU-2199-5′ NLF2343/24 2176 2199 TGA TTT CTG CCC AGT GCT CTG AAT This studynu-LSU-2383-3′ NLR2571/22 2404 2383 CTC AAC AGG GTC TTC TTT CCC C This studynu-LSU-3100-3′ NLR3287/23 3122 3100 GGA TTC TGR CTT AGA GGC GTT CA This studyα-Tubulin Primersnu-αTUB-0044-5′ TUAF22/23 0022 0044 CAC ATC GGN CAR GCC GGN RTC CA This studynu-αTUB-0083-5′ TUAF58/25 0058 0083 TGC TGG GAG CTN TAC TGC CTN GAG CA This studynu-αTUB-1219-3′ TUAR1248/26 1244 1219 TCC TCC ATN CCY TCN CCN ACR TAC CA This studynu-αTUB-1237-3′ TUAR1268/26 1262 1237 GCY TCR GAR AAY TCN CCY TCC TCC AT This studyβ-Tubulin Primersnu-βTUB-0050-5′ TUBF28/23 0028 0050 GGN CAG TGY GGN AAC CAG ATY GG This studynu-βTUB-0065-5′ funiv 0040 0065 AAY CAR ATY GGY KC/ideoxyI/ AAR TTY TGG GA This studynu-βTUB-1207-3′ buniv 1232 1207 GCY TC/ideoxyI/ GWR AAY TCC AWY TCG TCC AT This studynu-βTUB-1261-3′ TUBR1294/23 1283 1261 GCN TCC TGG TAC TGY TGR TAC TC This studyActin Primersnu-ACTIN-0054-5′ ACTf-13(s) 0037 0054 GAC AAY GGN WCN GGM ATG TG This study
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

nu-ACTIN-0056-5′ ACTf-12 0034 0056 TGC GAC AAY GGN TCN GGM ATG GT This studynu-ACTIN-0060-5′ ACTf-13 0037 0060 GAC AAY GGN TCN GGM ATG GTS AAG This studynu-ACTIN-0071-5′ ACTf-17 0049 0071 GGM ATG TGY AAG GCN GGN TTY GC This studynu-ACTIN-0377-5′ ACT120f 0355 0377 GAR AAR ATG ACN CAR ATH ATG TT This studynu-ACTIN-0400-3′ ACT139b 0419 0400 GCY TGD ATN GCN ACR TAC AT This studynu-ACTIN-1108-3′ ACTb-376 1132 1108 AGA AGC AYT T/ideoxyI/C KGT GNA CRA TNG A This studynu-ACTIN-1111-3′ ACTb-377 1133 1111 TAG AAG CAY TTN CKG TGN ACR AT This studyHSP90 Primersnu-HSP90-0041-5′ HspFA 0022 0041 GAR ACN TTY GCN TTY CAR GC This studynu-HSP90-0083-5′ 100XF 0052 0083 CAG CTG ATG TCC CTG ATC ATY AAY ACN TTY TA Simpson, Lukes, Roger
(2002)nu-HSP90-0389-5′ HspFB 0367 0389 CAR TTY GGT GTB GGY TTY TAC TC This studynu-HSP90-0602-5′ HspFC 0581 0602 TSA AGG ACC TSR TCA AGA AGC A This studynu-HSP90-1390-3′ HspRD 1410 1390 CTC NCC RGT GAT GWA GTA GAT This studynu-HSP90-1732-3′ HspRB2 1754 1732 CGY TCC ATR TTN GCN GAC CAN CC This studynu-HSP90-1741-3′ HspRA 1761 1741 CAT GAT NCG YTC CAT RTT NGC This studynu-HSP90-1759-3′ 880XR 1781 1759 TCG CGC AGR GCY TGN GCR TTC AT Simpson, Inagaki,
Roger (2006)nu-HSP90-1809-3′ HspRC 1833 1809 GGG GTT GAT TTC CAT NGT YTT CTT G This studynu-HSP90-1813-3′ 910XR 1835 1813 TCG GGG TTG ATY TCC ATN GTY TT Simpson, Inagaki,
Roger (2006)nu-HSP90-1990-3′ 970XR 2015 1990 TCG AGG GAG AGR CCN ARC TTR ATC AT Simpson, Lukes, Roger
(2002)
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

FIGURE LEGENDS
Fig. 1. ML tree (JTT+Γ+I, 8 rate categories) inferred from four nuclear encoded protein
coding gene sequences. The data set included 26 taxa and 1594 amino acid positions. The
root was arbitrarily placed between the Apusomonadidae-Opisthokonta-Amoebozoa
clade and the other taxa. Bayesian analysis (WAG+Γ+I, 8 rate categories) also found a
similar topology. ML bootstrap values and Bayesian posterior probabilities are indicated
at the corresponding nodes. Bootstrap values ≥50% and posterior probabilities ≥0.5 are
shown. Dashes represent bootstrap values <50%. Taxa from which new sequences were
obtained in our study are labeled in boldface.
Fig. 2. ML tree (GTR+Γ+I, 8 rate categories) was inferred from the combined nuclear
encoded SSU rRNA and LSU rRNA gene data set. The data set included 48 taxa and
3287 nucleotide positions—1283 from SSU rRNA and 2004 from LSU rRNA. Statistical
support values are listed in the same way as Fig. 1. The only exception is for the node
leading to Apusomonas. The SSU rRNA gene sequence of Mastigamoeba balamuthi
was substituted with that of Vexillifera armata to reduce the overall branch length of
Amoebozoa. When the SSU rRNA sequence of M. balamuthi was used, Mastigamoeba
branched with the Cryptophyceae+Kathablepharidae clade, while Apusomonas remained
branching close to Opisthokonta.
Fig. 3. ML tree inferred from the same data set used in Fig. 1 without α-tubulin gene
sequences. The same position for Apusomonas (Hypothesis I) was recovered. Statistical
support values for hypothesis II and III are also indicated.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Fig. 4. Bayesian consensus tree based on six genes. A GTR+Γ+I model (8 rate
categories) of nucleotide evolution was applied to the concatenated SSU and LSU rRNA
gene sequences and a WAG+Γ+I model (8 rate categories) of protein evolution was
applied to the combined four protein-coding genes. Bayesian posterior probabilities are
shown at the corresponding nodes.
Fig. 5. A simplified unrooted tree showing the main conclusion of this study. Regardless
of the position of the eukaryote root, ‘unikonts’ do not form a monophyletic group.
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Apusomonas proboscidea
Caenorhabditis elegans
Suberites/Discodermia
Monosiga brevicollis
Corallochytrium limacisporum
Physarum/Dictyostelium
Bigelowiella natans
Thaumatomonas sp.
Euglena gracilis
Naegleria gruberi
Heterosigma akashiwo
Synura sphagnicola
Phytophthora palmivora
Peridinium/Kryptoperidinium
Perkinsus marinus
Plasmodium sp.
Toxoplasma gondii
Chlamydomonas reinhardtii
Mesostigma viride
Pterosperma cristatum
Cyanophora paradoxa
Goniomonas truncata
Guillardia theta
Chrysochromulina sp. NIES 1333
Isochrysis galbana
Leucocryptos marina
0.05 substitutions/site
77
89
99
99
99
100
100
100
97
77
98
100
100
99
100
88
1
1
1
1
1
1
1
1
1
1
1
1
1
–/.93
.94
.96
1
.71
.98
1
1
.91
–/.96
FIG . 1.
OP
ISTH
OK
ON
TA
APUSOMONADIDAE
RHIZARIA
AMOEBOZOA
EUGLENOZOA
HETEROLOBOSEA
STR
AM
ENO
PIL
ESA
LVEO
LATA
VIRIDIPLANTAE
GLAUCOPHYTA
CRYPTOPHYCEAE
KATHABLEPHARIDAE
HAPTOPHYTA
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Apusomonas proboscideaVexillifera/Mastigamoeba
Blastocladiella emersonii
Pneumocystis cariniiSaccharomyces cerevisiae
Tricholoma matsutakeUmbelopsis ramanniana
Chytriomyces hyalinusOedogoniomyces sp.Nuclearia simplex
Hydra circumcincta
Leucosolenia sp.Mnemiopsis leidyi
Suberites ficusIchthyophonus hoferi
Monosiga brevicollisSalpingoeca infusionum
Aureococcus anophagefferensNannochloropsis salina
Heterosigma akashiwoScytosiphon lomentaria
Tribonema aequaleSynura sphagnicola
Cylindrotheca closteriumHyphochytrium catenoides
Phytophthora sp.Bigelowiella natans
Thaumatomonas sp.Cryptosporidium parvum
Toxoplasma gondiiPerkinsus andrewsi
Prorocentrum micansPhaeocystis antarcticaPrymnesium patelliferum
Bangia atropurpureaCyanidioschyzon merolae
Chlamydomonas sp.Chlorella ellipsoidea
Pterosperma cristatumFunaria hygrometrica
Gnetum gnemonOryza sativa
Mesostigma virideCyanophora paradoxa
Glaucocystis nostochinearumGoniomonas truncata
Guillardia thetaLeucocryptos marina
0.01 substitutions/site
44/.9
73/1
65/1
100
80
100
100/1
92/1
100
57/.94
65/.97100/1
99
100
98
80/.99
98100
100
95/1
100
98/1
95/158/.93
1
73/.87
1 –/.86
87/175/1
53/1
–/1
66/.99
1
1
1
–/.98
1
1
.85
1
–/1
–/.94
/1
–/.99 100/197/1
1
100/182/1
74/.99–/.97
1
100/1
74/1
FIG. 2.
HAPTOPHYTA
ALVEOLATA
STRAMENOPILES
RHIZARIA
RHODOPHYTA
VIRIDIPLANTAE
CRYPTOPHYCEAE
KATHABLEPHARIDAE
GLAUCOPHYTA
OPISTHOKONTA
APUSOMONADIDAE
AMOEBOZOA
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from
Apusomonas proboscidea
Caenorhabditis elegans
Suberites/Discodermia
Monosiga brevicollis
Corallochytrium limacisporum
Physarum/Dictyostelium
Bigelowiella natans
Thaumatomonas sp.
Euglena gracilis
Naegleria gruberi
Heterosigma akashiwo
Synura sphagnicola
Phytophthora palmivora
Peridinium/Kryptoperidinium
Perkinsus marinus
Plasmodium hybrid
Toxoplasma gondii
Chlamydomonas reinhardtii
Mesostigma viride
Pterosperma cristatum
Chrysochromulina sp. NIES 1333
Isochrysis galbana
Cyanophora paradoxa
Goniomonas truncata
Guillardia theta
Leucocryptos marina
0.05 substitutions/site
84
100
42
88
99
100
99
91
100
63
54
99
100
98
100
95
54
1
1
1
1
1
1
1
1
1
.99
1
1
.76
.89
.83
1
1
1
1
.96.32
FIG. 3.
40/.35
13/.33
Hyp. I
Hyp. II
Hyp. III
APUSOMONADIDAE
OP
ISTH
OK
ON
TA
AMOEBOZOA
RHIZARIA
EUGLENOZOA
HETEROLOBOSEA
ALV
EOLA
TAST
RA
MEN
OP
ILES
VIRIDIPLANTAE
GLAUCOPHYTA
CRYPTOPHYCEAE
KATHABLEPHARIDAE
HAPTOPHYTA
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Apusomonas proboscidea
Coprinopsis/Cryptococcus/Tricholoma
Monosiga brevicollis
Discodermia/Suberites
Dictyostelium/Mastigamoeba/Physarum/Vexillifera
Bigelowiella natans
Thaumatomonas sp.
Heterosigma akashiwo
Synura sphagnicola
Cylindrotheca/Thalassiosira
Phytophthora sp.
Kryptoperidinium/Peridinium/Prorocentrum
Perkinsus sp.
Toxoplasma gondii
Chrysochromulina sp. NIES 1333
Chlamydomonas sp.
Pterosperma cristatum
Mesostigma viride
Cyanophora paradoxa
Goniomonas truncata
Guillardia theta
Leucocryptos marina
0.05 substitutions/site
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
.72
1
FIG . 4.
KATHABLEPHARIDAE
HAPTOPHYTA
CRYPTOPHYCEAE
GLAUCOPHYTA
VIRIDIPLANTAEA
LVEO
LATA
STR
AM
ENO
PIL
ES
RHIZARIA
OP
ISTH
OK
ON
TA
APUSOMONADIDAE
AMOEBOZOA
by guest on March 10, 2016
http://mbe.oxfordjournals.org/
Dow
nloaded from

Apusomonadidae ('bikonts')
Opisthokonta ('unikonts')
Amoebozoa('unikonts')
Other eukaryotes ('bikonts')
FIG 5. by guest on M
arch 10, 2016http://m
be.oxfordjournals.org/D
ownloaded from
Copyright © 2022 FDOKUMEN



















![Binder 200, Small families [Trematoda Taxon Notebooks]](https://static.fdokumen.com/doc/165x107/6324444cb104cba27a091035/binder-200-small-families-trematoda-taxon-notebooks.jpg)
