
Characterization of the cydAB-Encoded Cytochrome bd Oxidase from Mycobacterium smegmatis

doi:10.1016/j.jmb.2010.09.029 J. Mol. Biol. (2010) 404, 158–171
Contents lists available at www.sciencedirect.com
Journal of Molecular Biologyj ourna l homepage: ht tp : / /ees .e lsev ie r.com. jmb
Coevolution Predicts Direct Interactions betweenmtDNA-Encoded and nDNA-Encoded Subunits ofOxidative Phosphorylation Complex I
Moran Gershoni1†, Angelika Fuchs2†, Naama Shani1, Yearit Fridman1,Marisol Corral-Debrinski3, Amir Aharoni1, Dmitrij Frishman2⁎and Dan Mishmar1⁎1Department of Life Sciences and the Nation Institute of Biotechnology in the Negev, Ben-Gurion University of the Negev,Beer Sheva 84105, Israel2Technische Universität München, Wissenschaftszentrum Weihenstephan, Am Forum 1, 85354 Freising, Germany3Institut de la Vision, Université Pierre et Marie Curie Paris 6, Unité Mixte de Recherche, S 592, 17 rue Moreau,Paris F-75012, France
Received 29 October 2009;received in revised form5 September 2010;accepted 13 September 2010Available online22 September 2010
Edited by M. Sternberg
Keywords:coevolution;complex I;correlated mutation analysis;mitochondrial DNA;mitochondria
*Corresponding authors. E-mail ad† M.G. and A.F. contributed equaAbbreviations used: OXPHOS, ox
mitochondrial DNA; nDNA, nuclearexpected squared; ELSC, explicit likethe curve.
0022-2836/$ - see front matter © 2010 E
Despite years of research, the structure of the largest mammalianoxidative phosphorylation (OXPHOS) complex, NADH–ubiquinone oxi-doreductase (complex I), and the interactions among its 45 subunits arenot fully understood. Since complex I harbors subunits encoded bymitochondrial DNA (mtDNA) and nuclear DNA (nDNA) genomes, withthe former evolving ∼10 times faster than the latter, tight cytonuclearcoevolution is expected and observed. Recently, we identified threenDNA-encoded complex I subunits that underwent accelerated aminoacid replacement, suggesting their adjustment to the elevated mtDNA rateof change. Hence, they constitute excellent candidates for bindingmtDNA-encoded subunits.Here, we further disentangle the network of physical cytonuclear
interactions within complex I by analyzing subunits coevolution. Firstly,relying on the bioinformatic analysis of 10 protein complexes possessingsolved structures, we show that signals of coevolution identified physicallyinteracting subunits with nearly 90% accuracy, thus lending support to ourapproach. When applying this approach to cytonuclear interaction withincomplex I, we predict that the ‘rate-accelerated’ nDNA-encoded subunits ofcomplex I,NDUFC2 andNDUFA1, likely interact with the mtDNA-encodedsubunits ND5/ND4 and ND5/ND4/ND1, respectively. Furthermore, wepredicted interactions among mtDNA-encoded complex I subunits. Usingthe yeast two-hybrid system, we experimentally confirmed the predictedinteractions of human NDUFC2 with ND4, the interactions of humanNDUFA1 with ND1 and ND4, and the lack of interaction of NDUFC2 withND3 and NDUFA1, thus providing a proof of concept for our approach.
dresses: [email protected]; [email protected] to this study.idative phosphorylation; complex I, NADH–ubiquinone oxidoreductase; mtDNA,DNA; McBASC, McLachlan-based substitution correlation; OMES, observed minuslihood of subset covariation; ROC, receiver operator characteristic; AUC, area under
lsevier Ltd. All rights reserved.

159Coevolution in Oxidative Phosphorylation Complex I
Our study shows, for the first time, evidence for direct interactionsbetween nDNA-encoded and mtDNA-encoded subunits of humanOXPHOS complex I and paves the path towards deciphering subunitinteractions within complexes lacking three-dimensional structures. Oursubunit-interactions-predicting method, ComplexCorr, is available athttp://webclu.bio.wzw.tum.de/complexcorr.
© 2010 Elsevier Ltd. All rights reserved.
Introduction
Subunit interactions within large protein com-plexes, such as the nuclear pore and the proteasome,are readily apparent from their crystal structures.However, the three-dimensional structures of manybiologically important protein complexes have yetto be resolved, thus limiting our understanding oftheir subunit interactions and functionality. Onesuch protein complex is the membrane-boundoxidative phosphorylation (OXPHOS) complexNADH–ubiquinone oxidoreductase (complex I).1
Complex I, the first and largest of the OXPHOScomplexes (45 subunits in mammals), is the mostcommon mutational target for mitochondrialdysfunction.2 This protein complex increased almostthreefold in size from the so-called 14 ‘core’ subunitsin Escherichia coli to 45 subunits in Homo sapiens bygradual recruitment of subunits throughoutevolution.3 Unlike other OXPHOS complexes, cluesfor subunit interactions within the L-shaped mam-malian complex I exist for the mitochondrial matrixarm, but not for the membrane arm of the complex.4
Hence, much of the knowledge on subunit interac-tions and composition within the membrane arm ofthe complex originates from the investigation of itsbacterial ortholog, NDH1.5,6 Furthermore, studies ofcomplex I assembly have revealed subunit compo-sition within subcomplexes. Still, such findings onlyoffer general clues for specific interactions (Vogelet al.7 and references within). Since understandingsubunit interactions within complex I is importantfor deciphering its function and since disruptionof complex I assembly causes diseases,7 alterna-tive approaches are needed.Four of the five OXPHOS complexes (i.e., com-
plexes I, III, IV, and V) are composed of subunitsencoded by nuclear and mitochondrial genomes[nuclear DNA (nDNA) and mitochondrial DNA(mtDNA), respectively] that differ 10 times in termsof mutational rates.8 Accordingly, elevated aminoacid replacement rates indicating positive selectionhave been identified in nDNA-encoded subunitsthat closely interact with fast-evolving mtDNA-encoded subunits within OXPHOS complexes withexperimentally determined three-dimensional struc-ture, namely complex III, complex IV, and part ofcomplex V.9–12 In a recent rigorous sequenceanalysis, we identified three complex I nDNA-
encoded subunits that underwent acceleratedamino acid replacement during the course ofprimate evolution and are thus likely candidates tointeract with the fast-evolving mtDNA-encodedsubunits.13 Since cytonuclear subunit interactionsplay important roles in disease and evolution,14,15
we sought to decipher such direct interactionswithin complex I. Here, we applied combinedevolutionary and experimental approaches to ana-lyze the interaction of the fast-evolving nDNA-encoded subunits NDUFC2 and NDUFA1 with themtDNA-encoded subunits of complex I.While coevolving amino acid residueswere initially
used to predict intramolecular contacts,16 severalgroups have recently attempted to employ thisapproach to predict residue contacts at the interfaceof interacting proteins. It has been suggested that thespatial distances between coevolving residue pairsharbored by interacting proteins are significantlysmaller than the distances between random residuepairs.17,18 Analysis of correlatedmutations assisted inthe successful identification of interdomain or inter-protein docking configurations.19,20 Moreover, coe-volving amino acids were found to be prevalentamong interacting residue pairs within and betweenproteins (‘in silico two-hybrid system’).21 Takentogether, these findings indicate that although thecoevolution signal may not be sufficient to discernactual contacting residue pairs, it may be informativefor identifying physically interacting protein pairs.Hence, residue coevolution constitutes a promisingapproach to assessing protein–protein interactionswithin large complexes such as OXPHOS complex I.Here, using a data set of 10 multisubunit protein
complexes with resolved crystal structures, we havedetected a clear correlation between the presence ofhighly coevolving residues and physical interactionsbetween subunits. This enabled us to extract optimalparameters and to define the criteria for predictingcandidate interactions among mtDNA-encoded andnDNA-encoded subunits of complex I. We appliedthese criteria to all of the mtDNA-encoded subunitsof complex I and two candidate nDNA-encodedsubunits (NDUFC2 and NDUFA1) thought tointeract with them.13 This approach predictedinteractions between NDUFC2 and two OXPHOScomplex I mtDNA-encoded subunits (ND4 andND5), interactions between NDUFA1 and threemtDNA-encoded subunits (ND1, ND4, and ND5),

Table 1. Complexes with known crystal structures used for an analysis of coevolving residues
Protein DataBank ID Description Chains
Possiblepairsa Alignmentsb Interactionsc
1GW5 AP2 clathrin adaptor core A|B|M|S 6 6 61W63 AP1 clathrin adaptor core A|B|M|S 6 6 62BCJ Gq-GRK2-G complex A|B|G|Q 6 6 32J0S Exon junction complex A|C|D|T 6 3 31LDK Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase
complexA|B|C|D|E 10 10 5
2CV5 Nucleosome core particle A|B|G|H 6 6 52CK3 F1-ATPase A|D|G|H|I 10 6 41SFC Synaptic fusion complex A|B|C|D 6 6 61BGY Cytochrome bc1 complex A|B|C|D|E|F|G|H|I|J 45 34 181V54 Cytochrome c oxidase A|B|C|D|E|F|G|H|I|J|
K|L66 15 10
a Number of possible protein pairs consisting of two different subunits for the given complex.b Subset of protein pairs in a concatenated alignment with at least 10 sequences. Only these protein pairs could be used for correlated
mutation analysis.c Number of protein pairs in a concatenated alignment with at least 10 sequences and at least one residue contact. These protein pairs
formed the subset of interacting complex subunits used for evaluating the prediction method.
160 Coevolution in Oxidative Phosphorylation Complex I
and other interactions involving mtDNA-encodedsubunits. Utilizing the yeast two-hybrid system, weexperimentally confirmed the interactions of humanNDUFC2 with ND4, the interactions of NDUFA1with ND1 and ND4, and the lack of interaction ofNDUFC2 with ND3 and NDUFA1, thus providing aproof of concept for our approach. Furthermore, weidentified specific amino acid residues in NDUFC2and NDUFA1 that have undergone positive selec-tion. Our analysis thus sheds light on cytonuclearsubunit interactions within the enigmatic mamma-lian complex I. More generally, we present anexperimentally supported predictive method fordefining protein–protein interactions within largeprotein complexes with as yet unsolved structures.
Results and Discussion
Coevolving residues in subunit pairs predictphysical interactions in complexes withresolved structures
To assess the usefulness of the coevolution signalas a tool for predicting subunit interactions, wegenerated a nonredundant set of multisubunitprotein complexes with solved structures (Fig.S1). To this end, we selected protein structuresfrom the Protein Data Bank database22 harboringfour or more polypeptides (with each polypeptidebeing longer than 15 amino acids), which corre-spond to different Swiss-Prot‡ accession numbers.Protein complexes sharing at least one pair ofsubunits with other complexes in the data set wereexcluded from consideration. The remaining data
‡www.expasy.ch/sprot
set contained 10 complexes (Table 1) harboring atotal of 56 different protein subunits involved in 95pairwise interactions.To formulate the criteria for the use of coevolving
amino acid residues to assess protein–protein inter-actions, we employed three different predictionalgorithms: McLachlan-based substitution correla-tion (McBASC),23 observed minus expected squared(OMES),24 and explicit likelihood of subset covari-ation (ELSC).25 Briefly, the McBASC predictionalgorithm assigns higher importance to residueswith low rates of change, whereas both the OMESalgorithm and the ELSC algorithm underline posi-tions with high rates of change. In general, use of theMcBASC algorithm led to either slightly increasedcorrelation scores for interacting subunits (using theMcLachlan matrix) or even higher correlation scoresin noninteracting subunits (using the Miyatamatrix). In contrast, use of both the OMESalgorithm and the ELSC algorithm resulted in theassignment of clearly higher correlation scores forinteracting versus noninteracting proteins (Table 2).Specifically, the average maximal score for inter-acting proteins obtained by both OMES and ELSCalgorithms was nearly 1.5-fold higher than thevalues calculated for noninteracting proteins. Sincethe differences in the maximal coevolution scoreswere significant with both the OMES algorithm andthe ELSC algorithm (Kolmogorov–Smirnov test:pb0.01 and pb0.05, respectively; Mann–Whitneytest: pb0.001 and pb0.01, respectively), we con-cluded that both prediction methods consistentlydetected highly coevolving residue pairs withinphysically interacting subunits of large proteincomplexes. It is worth noting, consistent withprevious reports, that strongly coevolving residuepairs within predicted coevolving subunits are, inmost cases (99.8%), not in direct contact.24,26
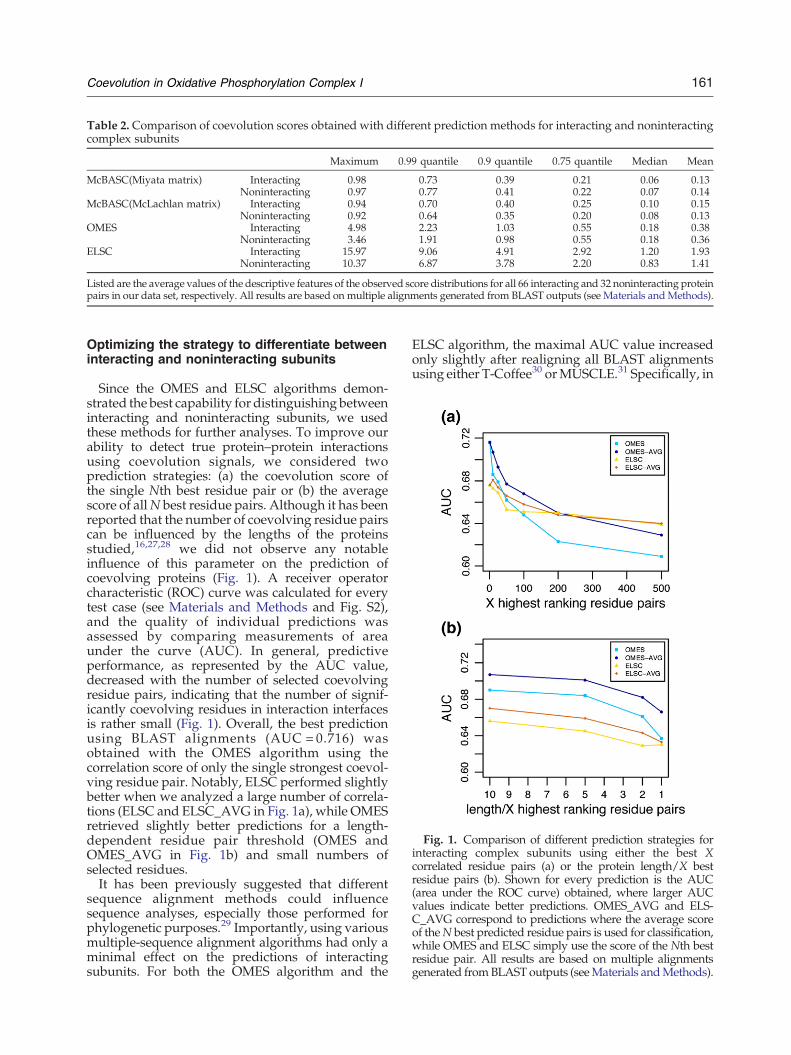
Fig. 1. Comparison of different prediction strategies forinteracting complex subunits using either the best Xcorrelated residue pairs (a) or the protein length/X bestresidue pairs (b). Shown for every prediction is the AUC(area under the ROC curve) obtained, where larger AUCvalues indicate better predictions. OMES_AVG and ELS-C_AVG correspond to predictions where the average scoreof theN best predicted residue pairs is used for classification,while OMES and ELSC simply use the score of the Nth bestresidue pair. All results are based on multiple alignmentsgenerated fromBLAST outputs (seeMaterials andMethods).
Table 2. Comparison of coevolution scores obtained with different prediction methods for interacting and noninteractingcomplex subunits
Maximum 0.99 quantile 0.9 quantile 0.75 quantile Median Mean
McBASC(Miyata matrix) Interacting 0.98 0.73 0.39 0.21 0.06 0.13Noninteracting 0.97 0.77 0.41 0.22 0.07 0.14
McBASC(McLachlan matrix) Interacting 0.94 0.70 0.40 0.25 0.10 0.15Noninteracting 0.92 0.64 0.35 0.20 0.08 0.13
OMES Interacting 4.98 2.23 1.03 0.55 0.18 0.38Noninteracting 3.46 1.91 0.98 0.55 0.18 0.36
ELSC Interacting 15.97 9.06 4.91 2.92 1.20 1.93Noninteracting 10.37 6.87 3.78 2.20 0.83 1.41
Listed are the average values of the descriptive features of the observed score distributions for all 66 interacting and 32 noninteracting proteinpairs in our data set, respectively. All results are based on multiple alignments generated from BLAST outputs (see Materials andMethods).
161Coevolution in Oxidative Phosphorylation Complex I
Optimizing the strategy to differentiate betweeninteracting and noninteracting subunits
Since the OMES and ELSC algorithms demon-strated the best capability for distinguishing betweeninteracting and noninteracting subunits, we usedthese methods for further analyses. To improve ourability to detect true protein–protein interactionsusing coevolution signals, we considered twoprediction strategies: (a) the coevolution score ofthe single Nth best residue pair or (b) the averagescore of allN best residue pairs. Although it has beenreported that the number of coevolving residue pairscan be influenced by the lengths of the proteinsstudied,16,27,28 we did not observe any notableinfluence of this parameter on the prediction ofcoevolving proteins (Fig. 1). A receiver operatorcharacteristic (ROC) curve was calculated for everytest case (see Materials and Methods and Fig. S2),and the quality of individual predictions wasassessed by comparing measurements of areaunder the curve (AUC). In general, predictiveperformance, as represented by the AUC value,decreased with the number of selected coevolvingresidue pairs, indicating that the number of signif-icantly coevolving residues in interaction interfacesis rather small (Fig. 1). Overall, the best predictionusing BLAST alignments (AUC = 0.716) wasobtained with the OMES algorithm using thecorrelation score of only the single strongest coevol-ving residue pair. Notably, ELSC performed slightlybetter when we analyzed a large number of correla-tions (ELSC and ELSC_AVG in Fig. 1a), while OMESretrieved slightly better predictions for a length-dependent residue pair threshold (OMES andOMES_AVG in Fig. 1b) and small numbers ofselected residues.It has been previously suggested that different
sequence alignment methods could influencesequence analyses, especially those performed forphylogenetic purposes.29 Importantly, using variousmultiple-sequence alignment algorithms had only aminimal effect on the predictions of interactingsubunits. For both the OMES algorithm and the
ELSC algorithm, the maximal AUC value increasedonly slightly after realigning all BLAST alignmentsusing either T-Coffee30 orMUSCLE.31 Specifically, in

162 Coevolution in Oxidative Phosphorylation Complex I
the case of OMES, the AUC of 0.716 (BLASTalignments) was increased to 0.725 (MUSCLE) and0.723 (T-Coffee). For ELSC, the maximal AUC scoresof 0.681 and 0.675 (MUSCLE and T-Coffee, respec-tively) closely matched the maximal AUC of 0.676found for alignments generated from BLAST out-puts. This observation could be attributed to the factthat the presence of a single strongly coevolvingresidue pair is only rarely affected by the method ofalignment generation even though the exact correla-tion score may change (Table S1). Our predictionmethod is thus highly robust against the chosenalignment tool; hence, the results presented beloware based on the original BLAST and ClustalWalignments.
Accuracy assessment for the prediction ofinteracting subunits using selectedscore thresholds
Based on the above described analyses, we furtherinvestigated the classifications obtained using onlythe score of the highest-ranking residue pair of eachinteracting protein pair (Table S1). We selectedseveral distinct score thresholds and classified all95 subunit pairs in our data set into interacting andnoninteracting subunits. For every score thresholdchosen, we calculated specificity (i.e., the fraction ofknown noninteracting subunits that were predictedas noninteracting), sensitivity (i.e., the fraction ofknown interacting subunits that were predicted asinteracting), and accuracy (i.e., the fraction of trulyinteracting subunits out of the sum of all subunitsthat were predicted to interact). Obviously, thehigher is the score threshold, the more specific andless sensitive a prediction becomes. Both OMES andELSC resulted in a prediction with 90.6% specificityand 33.3% sensitivity (OMES score threshold, 5.0;ELSC score threshold, 20) (Table 3). The best tradeoffbetween sensitivity and specificity was obtainedwith an OMES threshold of 3.5, resulting in
Table 3. Assessing the specificity, sensitivity, andaccuracy obtained while predicting interacting subunitswithin complexes
AlgorithmScore
thresholdSpecificity
(%)Sensitivity
(%)Accuracy
(%)
OMES 5.0 90.6 33.3 88.0OMES 4.0 84.4 39.4 83.9OMES 3.5 81.3 56.1 86.0OMES 3.2 62.5 72.7 80.0ELSC 20 90.6 33.3 88.0ELSC 15 84.4 37.9 83.3ELSC 12 78.1 44.0 80.6
The analysis was performed using different score thresholds forthe highest-scoring residue pair. All results are based onmultiple alignments generated from BLAST outputs (seeMaterials and Methods).
predictions with 56.1% sensitivity and 81.3% spec-ificity (for detailed results, see Table S2). Altogether,screening for coevolving residue pairs amongproteins allowed the detection of truly physicallyinteracting subunits with low false-positive signals.Nevertheless, our approach has somewhat limitedsensitivity, as some truly interacting proteins weremissed.
Predicting subunit interactions within themembrane arm of OXPHOS complex I
As demonstrated above, we have demonstratedthat identifying coevolving residues within largeprotein complexes can accurately predict at least asubset of physically interacting subunits. We thussought to predict subunit interactions within aprotein complex with a poorly resolved crystalstructure. We chose, as a test case, to focus on theinteractions between mtDNA-encoded and nDNA-encoded subunits of the first and largest proteincomplex of the mitochondrial energy-producingmachinery, namely complex I. The crystal structureof complex I has only been solved with lowresolution, and the network of subunit interactions,including the interactions between mtDNA-encoded and nDNA-encoded subunits (cytonuclearinteraction), is poorly understood.32 The choice tofocus on cytonuclear interactions in complex I stemsfrom their important role in diseases,33 but also inevolutionary processes such as adaptation10 and theemergence of new species.34
We recently identified three nDNA-encoded sub-units of complex I that underwent positive selectionand, as such, were selected as the best candidates tointeract with the fast-evolving mtDNA-encodedsubunits.13 To identify candidate subunits involvedin cytonuclear physical interaction within complex I,we predicted coevolving residues and analyzedthem using the OMES and ELSC algorithms. As afirst step, we applied our correlated mutationsapproach to identify the best candidates within theseven complex I mtDNA-encoded subunits (ND1–ND6 and ND4L) as interacting with one of thepositively selected subunits (NDUFC2) chosen as amodel for the current analysis. The approach wasalso applied to identify all possible interactionsamong mtDNA-encoded subunits. For every possi-ble pair of subunits, considering either NDUFC2 orany of the mtDNA-encoded subunits, the highest-scoring residue pairs were utilized for the identifi-cation of candidate interacting and noninteractingsubunits. Favoring specificity over sensitivity andusing OMES and ELSC, we identified the ND4 andND5 subunits as exhibiting the highest scores, thuscorresponding to the most likely mtDNA candidatesto interact with NDUFC2 (Table 4). Figure 2exemplifies a representative coevolving pair ofamino acids in NDUFC2 and ND5. Further
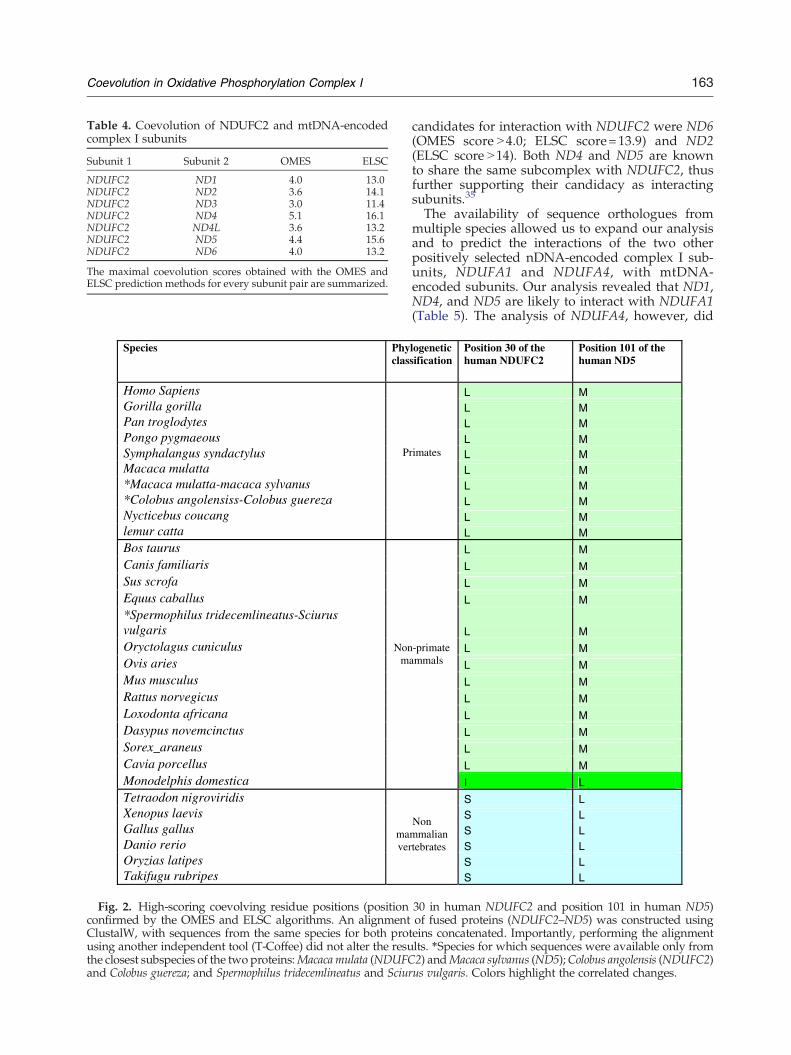
Table 4. Coevolution of NDUFC2 and mtDNA-encodedcomplex I subunits
Subunit 1 Subunit 2 OMES ELSC
NDUFC2 ND1 4.0 13.0NDUFC2 ND2 3.6 14.1NDUFC2 ND3 3.0 11.4NDUFC2 ND4 5.1 16.1NDUFC2 ND4L 3.6 13.2NDUFC2 ND5 4.4 15.6NDUFC2 ND6 4.0 13.2
The maximal coevolution scores obtained with the OMES andELSC prediction methods for every subunit pair are summarized.
Species Phyclas
Homo SapiensGorilla gorillaPan troglodytesPongo pygmaeousSymphalangus syndactylusMacaca mulatta*Macaca mulatta-macaca sylvanus*Colobus angolensiss-Colobus guerezaNycticebus coucanglemur catta
P
Bos taurusCanis familiarisSus scrofaEquus caballus*Spermophilus tridecemlineatus-SciurusvulgarisOryctolagus cuniculusOvis ariesMus musculusRattus norvegicusLoxodonta africanaDasypus novemcinctusSorex_araneusCavia porcellusMonodelphis domestica
Nonm
Tetraodon nigroviridisXenopus laevisGallus gallusDanio rerioOryzias latipesTakifugu rubripes
maver
Fig. 2. High-scoring coevolving residue positions (positionconfirmed by the OMES and ELSC algorithms. An alignmentClustalW, with sequences from the same species for both prousing another independent tool (T-Coffee) did not alter the resuthe closest subspecies of the two proteins:Macaca mulata (NDUFCand Colobus guereza; and Spermophilus tridecemlineatus and Sciur
163Coevolution in Oxidative Phosphorylation Complex I
candidates for interaction with NDUFC2 were ND6(OMES score N4.0; ELSC score=13.9) and ND2(ELSC score N14). Both ND4 and ND5 are knownto share the same subcomplex with NDUFC2, thusfurther supporting their candidacy as interactingsubunits.35
The availability of sequence orthologues frommultiple species allowed us to expand our analysisand to predict the interactions of the two otherpositively selected nDNA-encoded complex I sub-units, NDUFA1 and NDUFA4, with mtDNA-encoded subunits. Our analysis revealed that ND1,ND4, and ND5 are likely to interact with NDUFA1(Table 5). The analysis of NDUFA4, however, did
logenetic sification
Position 30 of thehuman NDUFC2
Position 101 of thehuman ND5
L ML ML ML ML ML ML ML ML M
rimates
L M
L M
L M
L M
L M
L M
L M
L M
L M
L M
L M
L M
L M
L M
-primateammals
I L
S LS LS LS LS L
Nonmmaliantebrates
S L
30 in human NDUFC2 and position 101 in human ND5)of fused proteins (NDUFC2–ND5) was constructed usingteins concatenated. Importantly, performing the alignmentlts. ⁎Species for which sequences were available only from2) andMacaca sylvanus (ND5);Colobus angolensis (NDUFC2)us vulgaris. Colors highlight the correlated changes.
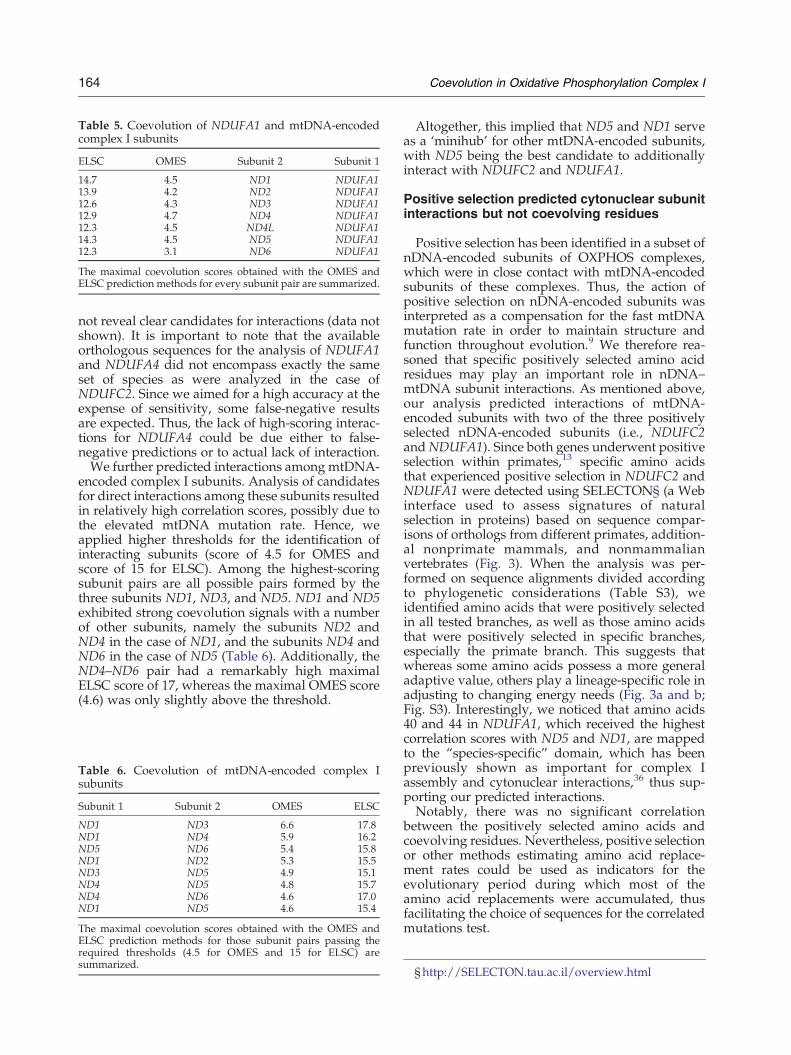
Table 5. Coevolution of NDUFA1 and mtDNA-encodedcomplex I subunits
ELSC OMES Subunit 2 Subunit 1
14.7 4.5 ND1 NDUFA113.9 4.2 ND2 NDUFA112.6 4.3 ND3 NDUFA112.9 4.7 ND4 NDUFA112.3 4.5 ND4L NDUFA114.3 4.5 ND5 NDUFA112.3 3.1 ND6 NDUFA1
The maximal coevolution scores obtained with the OMES andELSC prediction methods for every subunit pair are summarized.
164 Coevolution in Oxidative Phosphorylation Complex I
not reveal clear candidates for interactions (data notshown). It is important to note that the availableorthologous sequences for the analysis of NDUFA1and NDUFA4 did not encompass exactly the sameset of species as were analyzed in the case ofNDUFC2. Since we aimed for a high accuracy at theexpense of sensitivity, some false-negative resultsare expected. Thus, the lack of high-scoring interac-tions for NDUFA4 could be due either to false-negative predictions or to actual lack of interaction.We further predicted interactions among mtDNA-
encoded complex I subunits. Analysis of candidatesfor direct interactions among these subunits resultedin relatively high correlation scores, possibly due tothe elevated mtDNA mutation rate. Hence, weapplied higher thresholds for the identification ofinteracting subunits (score of 4.5 for OMES andscore of 15 for ELSC). Among the highest-scoringsubunit pairs are all possible pairs formed by thethree subunits ND1, ND3, and ND5. ND1 and ND5exhibited strong coevolution signals with a numberof other subunits, namely the subunits ND2 andND4 in the case of ND1, and the subunits ND4 andND6 in the case of ND5 (Table 6). Additionally, theND4–ND6 pair had a remarkably high maximalELSC score of 17, whereas the maximal OMES score(4.6) was only slightly above the threshold.
Table 6. Coevolution of mtDNA-encoded complex Isubunits
Subunit 1 Subunit 2 OMES ELSC
ND1 ND3 6.6 17.8ND1 ND4 5.9 16.2ND5 ND6 5.4 15.8ND1 ND2 5.3 15.5ND3 ND5 4.9 15.1ND4 ND5 4.8 15.7ND4 ND6 4.6 17.0ND1 ND5 4.6 15.4
The maximal coevolution scores obtained with the OMES andELSC prediction methods for those subunit pairs passing therequired thresholds (4.5 for OMES and 15 for ELSC) aresummarized.
Altogether, this implied that ND5 and ND1 serveas a ‘minihub’ for other mtDNA-encoded subunits,with ND5 being the best candidate to additionallyinteract with NDUFC2 and NDUFA1.
Positive selection predicted cytonuclear subunitinteractions but not coevolving residues
Positive selection has been identified in a subset ofnDNA-encoded subunits of OXPHOS complexes,which were in close contact with mtDNA-encodedsubunits of these complexes. Thus, the action ofpositive selection on nDNA-encoded subunits wasinterpreted as a compensation for the fast mtDNAmutation rate in order to maintain structure andfunction throughout evolution.9 We therefore rea-soned that specific positively selected amino acidresidues may play an important role in nDNA–mtDNA subunit interactions. As mentioned above,our analysis predicted interactions of mtDNA-encoded subunits with two of the three positivelyselected nDNA-encoded subunits (i.e., NDUFC2andNDUFA1). Since both genes underwent positiveselection within primates,13 specific amino acidsthat experienced positive selection in NDUFC2 andNDUFA1 were detected using SELECTON§ (a Webinterface used to assess signatures of naturalselection in proteins) based on sequence compar-isons of orthologs from different primates, addition-al nonprimate mammals, and nonmammalianvertebrates (Fig. 3). When the analysis was per-formed on sequence alignments divided accordingto phylogenetic considerations (Table S3), weidentified amino acids that were positively selectedin all tested branches, as well as those amino acidsthat were positively selected in specific branches,especially the primate branch. This suggests thatwhereas some amino acids possess a more generaladaptive value, others play a lineage-specific role inadjusting to changing energy needs (Fig. 3a and b;Fig. S3). Interestingly, we noticed that amino acids40 and 44 in NDUFA1, which received the highestcorrelation scores with ND5 and ND1, are mappedto the “species-specific” domain, which has beenpreviously shown as important for complex Iassembly and cytonuclear interactions,36 thus sup-porting our predicted interactions.Notably, there was no significant correlation
between the positively selected amino acids andcoevolving residues. Nevertheless, positive selectionor other methods estimating amino acid replace-ment rates could be used as indicators for theevolutionary period during which most of theamino acid replacements were accumulated, thusfacilitating the choice of sequences for the correlatedmutations test.
§http://SELECTON.tau.ac.il/overview.html

(a)
1 11 21 31 41 51 61 71 81 91 101 111
(b)
1 11 21 31 41 51 61
(c)
1 11 21 31 41 51 61 71 81 91 101 111
121 131 141
The selection scale:
Positive selection Purifying selection
Fig. 3. Patterns of positively selected amino acid positions in NDUFC2 and NDUFA1 versus THRSP. Analysis wasperformed and statistical significance was estimated using the default settings of SELECTON (http://SELECTON.tau.ac.il/). Color codes represent the relative degrees of either positive selection (yellow-orange spectrum) or negative selection(pink-purple spectrum). (a) Positive selection in NDUFC2 in primates. (b) Positive selection in NDUFA1 in primates. (c)THRSP is mostly negatively selected.
165Coevolution in Oxidative Phosphorylation Complex I
To control for the possibility that the positiveselection was merely a side effect of an acceleratedmutation rate in chromosomal location, we alsoperformed positive selection analysis for THRSP, agene whose sequence is located tail to tail withNDUFC2 (Fig. 3c). Our results indicate that THRSPis subjected to negative—rather than positive—selection. As negative selection or neutrality appliesto most protein-coding genes in humans, THRSPreflects the rule rather than the exception.37,38
Therefore, the positive selection effect noted is tobe specific for NDUFC2 and NDUFA1.
Yeast two-hybrid experiments confirm the directinteractions of NDUFC2 and NDUFA1 withmtDNA-encoded subunits
To verify the predicted protein–protein interac-tions, we used a yeast two-hybrid assay. Since thetraditional yeast two-hybrid assay is a qualitative(nonquantitative) assay and since we were interest-ed in cytonuclear interactions, we focused only onour strongest predictions for cytonuclear interac-tions. Accordingly, we amplified by reverse tran-scription PCR the cDNAs of human NDUFC2 orNDUFA1 from commercially available human mus-cle or colon RNA samples, respectively, and clonedthe transcripts into the pAD plasmid (see Materialsand Methods). Based on our bioinformatic predic-tions, we aimed at assessing the interactions ofmtDNA-encoded proteins with NDUFC2, with themtDNA-encoded proteins ND4 and ND5 represent-
ing the best candidates for interaction and with ND3predicted to have low odds of interaction withNDUFC2. Similarly, we tested the best candidates tointeract withNDUFA1, namelyND1,ND5, andND4(see the text below). Since all mtDNA-encodedproteins rely on the mtDNA genetic code, werecoded ND1, ND3, ND4, and ND5 to includegeneral cytoplasmic codons (see Materials andMethods). Each of these genes was cloned into thepBD plasmid. Since the construct harboring ND5proved toxic to the yeast in our hands, we continuedour experiments solely with plasmids encodingND3, ND4, ND1, NDUFC2, and NDUFA1. Firstly,we tested for the best predicted mtDNA-encodedprotein interactions with NDUFC2. The results ofthese experiments (Fig. 4) show yeast growth onrestrictive media (–L-W-H) in the combined pres-ence of NDUFC2 and ND4, consistent with a directinteraction of these subunits (Fig. 4b). In contrast, weobserved a lack of yeast growth when the combina-tion of NDUFC2 and ND3 was introduced, consis-tent with a lack of interaction between thesesubunits, exactly as predicted (Fig. 4). Secondly, inorder to assess whether the observed lack ofinteraction is true or due to lack of proteinexpression, we performed Western blot analysis.This analysis confirmed that all of the tested proteinswere expressed while growing the yeast in –L-Wrestrictive media (Fig. 4c); therefore, the observedlack of interaction is likely to be true. Finally, weapplied the yeast two-hybrid assay as describedabove to test for the highest-ranking interactions of

Fig. 4. NDUFC2 physically interacts with ND4 but notwith ND3. Yeast cells, cotransformed with plasmids pAD-NDUFC2 and pBD-ND3 (1), plasmid pBD-ND4 (2), anempty pAD vector with pBD-ND4 (3), and pAD-NDUFC2with an empty pBD plasmid, were serially diluted fivetimes and spotted onto selective media lacking leucineand tryptophan (a; –L-W) to ensure plasmid integrity.Physical interaction was detected by growth on plateslacking histidine (b; –L-W-H). (c) Western blot analysis ofpBD expression. Gel loaded with protein extract fromyeast transfected with (from left to right): (1)NDUFC2 andempty pBD; (2) empty pAD with ND4-BD; (3) NDUFC2with pBD-ND4; (4) NDUFC2 with pBD-ND3; (5) untrans-fected yeast.
Fig. 5. NDUFA1 physically interacts with ND4 or ND1but notwithNDUFC2. Similarly to the results demonstratedin Fig. 3a and b, yeast cells were cotransformed withplasmids pAD-NDUFA1 and pBD-NDUFC2 (1), plasmidspAD-NDUFA1 and pBD-ND4 (2), plasmids pAD-NDUFA1and pBD-ND1 (3), plasmid pAD-NDUFA1 and empty pBD(4), and plasmids pAD-NDUFC2 and pBD-ND4 (5). Thetransfected yeast cells underwent serial dilutions five timesand were spotted onto selective media lacking leucine andtryptophan (a; –L-W) to ensure plasmid integrity. Physicalinteraction was detected by growth on plates lackinghistidine (b; –L-W-H).
ND3
NDUFC2
?
ND1 ND5
ND4
ND6
ND2
ND4L
NDUFA1
Fig. 6. A proposed schematic model for the interactionnetwork of NDUFC2, NDUFA1, and mtDNA-encodedcomplex I subunits. The scheme is based on the compiledpredictions described in Tables 4, 5, and 6. This figuredemonstrates the networks of predicted interactions amongthe tested subunits. The illustration does not depict theactual size of the subunits. The thickness of the arrowscorresponds to the strength of the predictions, with thethickest arrows representing the best predicted interactionspredictions (based on the top-scoring correlated pairs ofresidues).Questionmark indicates a lackof clear predictionsfor the interaction of ND4L with any of the tested subunits.
166 Coevolution in Oxidative Phosphorylation Complex I
NDUFA1 with mtDNA-encoded subunits (ND4 andND1), as well as the predicted lack of interactionbetween NDUFA1 and NDUFC2, with the latterbeing cloned in pBD for this particular assay (Fig. 5).The results of these experiments show yeast growthon restrictive media (–L-W-H; Fig. 5b) in thecombined presence of NDUFA1 and ND4 or ND1,consistent with a direct interaction of these subunits.Taken together, these findings offer experimentalsupport for our bioinformatic predictions.
Proposed interaction network of NDUFC2,NDUFA1, and mtDNA-encoded complex Isubunits
Based on the bioinformatic predictions and yeasttwo-hybrid experiments, we propose a model for anetwork of interactions among the tested subunits(Fig. 6). This model is largely consistent withanalyses of complex I assembly intermediates 7.Firstly, bothND3 andND2were predicted to interactwith ND1, consistent with the transient associationof these very subunits in the presence of the complexI assembly factor NDUFAF1 (Vogel et al.7 and
references within). Secondly, we predicted thatNDUFA1, ND4, and ND5 would interact withseveral subunits, consistent with their known essen-tiality to the assembly of the membrane arm of

167Coevolution in Oxidative Phosphorylation Complex I
complex I (Vogel et al.7 and references within).Finally, ND5 was predicted to interact with severalmtDNA-encoded complex I subunits, consistentwith a recent structural analysis of its Thermusthermophilus ortholog Nqo12.5 However, in additionto these consistencies with the literature, our workput forward new insights into complex I cytonuclearsubunit interactions: both the predictions and theexperimental data presented in the current worksupport the interactions of NDUFA1 and NDUFC2with ND4 and the interactions of the former withND1, but also the lack of interactions of NDUFA1and NDUFC2. In addition, both NDUFA1 andNDUFC2 were predicted to interact with ND5.Since ND4, ND5, and NDUFA1, as mentionedabove, are essential for the assembly of the mem-brane arm of complex I, it would be tempting to testthe essentiality of NDUFC2 to the assembly process.Indeed, we noticed that transient knockdown ofNDUFC2 in cell culture resulted in cell growthretardation, providing clues consistent with thishypothesis (M.G. et al., unpublished results). Insummary, our findings not only support ourpredictive method for subunit interactions but alsoconstitute the first experimental evidence for directcytonuclear subunit interactions within the humanOXPHOS complex I. These findings pave the pathtowards understanding the molecular basis under-lying the known involvement of complex I cyto-nuclear interactions in diseases,33 reduced fitness,reproductive barriers, and speciation events.34
Conclusions
In this study, we have demonstrated the usefulnessof correlated mutations for identifying candidatephysically interacting subunits within large multi-subunit protein complexes. Our observations indicatethat interacting subunits within protein complexesoften possess a small number of highly coevolvingresidues that distinguish them from noninteractingsubunits. Hence, the presence of significantly coevol-ving residues could be used as a tool to identify atleast a subset of physically interacting subunits with arelatively high accuracy (up to nearly 90%). Specif-ically, of the four tested prediction algorithms, two(OMES and ELSC) retrieved predictions with highspecificity and accuracy. It is worth noting thatcertain complexes may differ remarkably in theirgeneral correlation level (Table S1). Alignments witha large number of sequences tend to retrieve highercorrelation scores than alignments of fewersequences.39 Additionally, the inclusion of ratherdiverse sequences resulted in increased correlationscores. Hence, there is a need to adjust the thresholdlevels individually for the identification of physicallyinteracting subunits in each newly investigatedcomplex. Ideally, once significantly more structures
of protein complexes are resolved, a future version ofour predictionmethodwill use complex-specific scorethresholds, supported by an empirically derived scalefor the number of expected interactions given acertain number of complex subunits.We applied our prediction approach to cytonuclear
subunit interactions in human mitochondrialOXPHOS complex I, a protein complex whosestructure is poorly understood. This allowed us toidentify candidate interactions among mtDNA-encoded subunits, as well as with the positivelyselected subunits NDUFC2 and NDUFA1, and topropose a model for these predictions (Fig. 6). Forthese two subunits, positive selection mainly occurredin primates, which led us to enrich for primatesequences and hence better detect signals of coevolu-tion. Since human complex I is far more complex thanits bacterial and yeast40 counterpart and since subunitinteractions in the membrane arm of the complex areyet to be resolved, our bioinformatic approachconstitutes the first step towards disentangling cyto-nuclear subunit interactions within this large multi-subunit complex. Using the yeast two-hybrid system,we confirmed the predicted interactions of NDUFC2withND4, the predicted interactions ofNDUFA1withND1 andND4, and the predicted lack of interaction ofNDUFC2 with ND3 and NDUFA1. To the best of ourknowledge, we provide the first experimental supportfor predictions of the direct interaction of mtDNA-encoded and nDNA-encoded proteins within humancomplex I. More generally, analysis of coevolvingresidues, underlining combined bioinformatic andexperimental approaches as described above, could beutilized to resolve the network of subunit interactionsin other large complexes. Understanding the networksof subunit interactions within complexes, in general,and in OXPHOS complex I, in particular, may shedlight on their functional roles in normal and diseaseconditions.
Materials and Methods
Blood samples
For RNA extraction, primate blood samples wereobtained from The Biblical Zoo (Jerusalem) and TheMonkey Park (Ben Shemen). The samples were taken onlyduring preplanned medical procedures. The blood sam-ples were collected in standard tubes containing anti-coagulation factors and kept in ice (no more than a fewhours) for RNA purification.
RNA purification
RNA purification was performed using the Versagenetotal RNA purification kit (Gentra), in accordance with themanufacturer's protocol. Briefly, red cells underwent lysisby RBC buffer [50 mM TRIS (pH 7.2), 0.5 mM EDTA] to

bOur final method, called ComplexCorr, is available athttp://webclu.bio.wzw.tum.de/complexcorrawww.ensembl.org
168 Coevolution in Oxidative Phosphorylation Complex I
facilitate the isolation of white blood cells by centrifuga-tion. Pellets of white blood cells were incubated in thepresence of a detergent/salt solution to eliminate endog-enous RNase activity. The RNA-containing supernatantwas loaded onto a purification column supplied by the kitfor RNA binding. The bound RNAwas washed in order todispose of proteins andDNA.Residual DNAwas removedby on-column DNase treatment. Finally, the purified RNAwas eluted with diethylpyrocarbonate-treated water.
cDNA preparation
Total cDNA was produced from blood RNA extractsusing the iScript cDNA synthesis kit (Bio-Rad), inaccordance with the manufacturer's protocol. The cDNAwas in turn used for PCR amplification and sequencing ofNDUFC2 and THRSP transcripts using specific primers(for primer list and PCR conditions, see Table S4).
Test data set and multiple-sequence alignments
To assess the coevolution signal as a predictive tool forsubunit interactions, we obtained a nonredundant set ofmultisubunit protein complexes with solved structure (Fig.S1). The final data set contained 10 complexes (Table 1)harboring a total of 56 different protein subunits. Thesesubunits were involved in 95 pairwise interactions, while thetotal number of possible interactions (given the way thesesubunits are distributed among the complexes) was 167.When a protein complex harbored multiple copies of thesame subunit, a representative subunit was selected for theanalysis corresponding to the protein chainwith themaximalnumber of observed interactions with other subunits.Homologous sequences for the analyzed polypeptides
were obtained with PSI-BLAST searches against theunfiltered NCBI‖ database, with three iterations andwith the E-value threshold for inclusion of relateddatabase sequences set to 1×10− 4. Following our previouswork dealing with the prediction of intramolecularcontacts in membrane-bound proteins,41 multiple align-ments for every subunit were directly constructed fromPSI-BLAST results by compiling all hits found using theiralignment to the reference sequence. Additionally, inaccordance with the previously published standards forthe prediction of intermolecular correlations,21 if morethan one sequence was obtained from a given species, thesequence most similar to the reference sequence was keptin the alignment. In general, species not contributingorthologous sequences to all analyzed subunits of a givencomplex were removed. Finally, to test the influence ofalignment quality on the resulting prediction, we rea-ligned all used sequences utilizing the multiple-alignmentprograms T-Coffee30 and MUSCLE.31
In order to predict correlated mutations between twoproteins, we first performed multiple orthodox alignmentsfor each protein separately and then constructed a jointalignment by concatenating sequences of the same organ-ism for every possible protein pair within each complex.From these alignments, sequences with a high level ofsimilarity to another sequence in the concatenated align-
‖www.ncbi.nlm.nih.gov
ment (N95%) and sequences with more than 25% gapswereremoved. Finally, concatenated alignments with fewer than10 sequences were removed from the analysis, resulting inconcatenated alignments for 95 protein pairs used forfurther analyses. For all 95 protein pairs, concatenatedsequence alignments were generated based on the originalBLAST, T-Coffee, and MUSCLE alignments.
Prediction of coevolving residues
As mentioned in Results and Discussion, we used threeprediction methods: McBASC, OMES, and ELSC. Thesemethods were selected because they performed best in acomparative study on fusion proteins.26 The McBASCmethod was applied using either the Miyata substitutionmatrix42 or the McLachlan substitution matrix.43 Witheach method, we extracted the maximum coevolutionscore, the quantiles, and the mean scores for each possiblepair of subunits. These values were grouped into two sets(corresponding to interacting and noninteracting sub-units) and compared. Several properties of the obtaineddistribution of correlation scores gained for each align-ment and prediction method were assessed for theirability to distinguish physically interacting complexsubunits from those that do not interact (see Results andDiscussion). To this end, the number of residue pairs witha Cβ–Cβ distance (Cα in the case of glycine) of less than 8 Åwas calculated from the corresponding Protein Data Bankstructure, and all protein pairs having at least one suchresidue pair were counted as interacting. Accordingly, 66protein pairs out of 95 in total were classified asinteracting, while 32 protein pairs were categorized asnoninteracting. For each test case, we calculated an ROCcurve (which plots the achieved true-positive rate againstthe false-positive rate), with any point above the diagonalcorresponding to better predictions than random (Fig. S2).The quality of different predictions was compared usingthe AUC measure, with AUC values above 0.5 indicatingpredictions better than randomb.
Sequence data of OXPHOS complex I mtDNA-encodedsubunits and nDNA-encoded subunits
Orthologous sequences of the complex I subunitNDUFC2, NDUFA1, and the mtDNA-encoded subunitsND1–ND6 and ND4L were obtained from the NCBI andENSEMBLa databases. To gain additional sequencevariability, we sequenced further orthologs from primatescurrently not present in public databases (Table S3).Primers for PCR amplification and sequencing arespecified in Table S4. In total, sequences from more than30 species were available (Table S3) and used to generatemultiple-sequence alignments by ClustalWb for each ofthe proteins separately; these alignments were subse-quently concatenatedc.
bwww.clustal.orgcAlignments are available at http://webclu.bio.wzw.
tum.de/oxphos

169Coevolution in Oxidative Phosphorylation Complex I
Prediction of interacting subunits within themitochondrial OXPHOS complex I
Subunit interactions were evaluated for all possiblecombinations of NDUFC2 or NDUFA1 with mtDNA-encoded subunits and for interactions among mtDNA-encoded subunits. As described above, a concatenatedalignment was constructed for every analyzed subunitpair by connecting sequences of the same species.Redundant sequences with a N95% identity to anothersequence in the alignment were removed. Coevolvingresidues were predicted using the OMES and ELSCmethods, which retrieved the best distinction betweeninteracting and noninteracting subunits in the previousanalysis. Finally, interacting subunits were predictedusing thresholds optimized on complexes with solvedstructures (see Results and Discussion).
Detecting positively selected amino acids within thecomplex I subunits NDUFC2 and NDUFA1
We generated multiple cDNA sequence alignments toidentify positively selected residue positions by ClustalWand SELECTON. The list of sequences used is provided inTable S3. Briefly, the hypothesis tested is whether positiveselection operates on NDUFC2 or NDUFA1, as contrastedwith a null hypothesis that assumes that there is nopositive selection. The SELECTON server allows fordetecting the selective forces even at single amino acidsites. The ratio of nonsynonymous (i.e., amino acidaltering) substitutions to synonymous (i.e., silent) sub-stitutions, known as the Ka/Ks ratio, is used to estimateboth positive selection and purifying selection at eachamino acid site. As a control for the analysis of positivelyselected amino acids, a corresponding alignment wasgenerated for THRSP, the 3′ end of which lies in a tail-to-tail orientation with the 3′ end of NDUFC2 both in birdsand in mammals.44
Yeast two-hybrid analysis
To assess predicted protein–protein interactions, wechose to use the yeast two-hybrid approach. To this end,we have amplified the cDNA of human NDUFC2 byreverse transcription PCR using commercially availablemuscle RNA (Ambion) as template, followed by incuba-tion with high-fidelity polymerase (plaque-forming units)and cloning into the pGEM T-vector (Promega). Since allmtDNA-encoded proteins used in this assay are coded bythe mtDNA genetic code, we recoded the humanND3 andND5 genes to comply with the general cytoplasmic codons(GeneScript) and used the recoded ND4 and ND1 genesthat were already available to us.45 The sequences of therecoded genes are available in Supplementary Materials.Analysis of protein–protein interaction was performedusing the commercial GAL4 Two Hybrid Phagemid vectorkit (Stratagene), in accordance with the manufacturer'sinstructions. Plasmids were constructed using conven-tional PCR recombination methods. In brief, the tran-scripts of NDUFC2 or NDUFA1 were fused to a plasmidcontaining the GAL4 activating domain (pAD), whileND3, ND4, NDUFC2, and ND1 were fused to a plasmidcontaining the GAL4 binding domain (pBD), generating
the plasmids pAD-NDUFC2, pAD-NDUFA1, pBD-ND3,and pBD-ND4, respectively, using a homologous recom-bination-based cloning procedure with six primersdescribed in Table S5. The yeast strain PJ694A (MATatrp1-901 leu2-3 112 ura3-52 his3-200 Δgal4 Δgal80 GAL2-ADE2 met2∷GAL7-lacZ LYS2∷GAL1-HIS3) was cotrans-formed with plasmid pAD-NDUFC2 and either plasmidpBD-ND3 or plasmid pBD-ND4. The same procedure wasapplied for cotransfection with plasmid pAD-NDUFA1and either plasmid pBD-ND1, plasmid pBD-ND4, orplasmid pBD-NDUFC2. Yeast cells harboring the plasmidswere cultured in synthetic medium lacking tryptophanand leucine. Cells with an appropriate optical density(3e−6cells/ml) were serially diluted, and positive interac-tions were detected by yeast growth on a medium lackinghistidine.
Protein extraction
Overnight yeast cultures were centrifuged at 3220 rcf,and the pellet was resuspended in 200 μl of 20%trichloroacetic acid. Cell lysate was obtained by vigorouspellet vortexing for 5 min with 425-μm to 600-μm acid-washed glass beads (Sigma). The cell lysate was centri-fuged in 16,000 rcf, and the pellet containing aggregatedproteins was resuspended in loading buffer following5 min of incubation in 95 °C for future electrophoresis.
Western blot analysis
In order to assess the protein expression of the fusedpBD constructs, we first extracted proteins from yeast, asdescribed above. Following resuspension of the precipi-tated proteins, we loaded the samples onto a 10%polyacrylamide gel and ran them for 1 h at 120 V. Thetransfer of proteins onto a nitrocellulose membrane wasperformed at 400 mA for 1 h using a Bio-Rad device. Weblocked the membrane with 5% milk in TBS-T [0.02 MTris-buffered saline (pH 7.6) and 0.1% Tween] for 1 h,followed by three 10-min washes in TBS-T. In order toperform the detection, we incubated the membrane withrabbit polyclonal IgG antibody raised against Gal4Binding Domain (a generous gift from Prof. MichalShapira of Ben-Gurion University of the Negev; SantaCruz Biotechnology). The membrane was incubatedovernight at 4 °C in the presence of a 1:500 dilution ofthe primary antibody and 1% milk in TBS-T. After three10-min washes with TBS-T, we incubated the membranewith the secondary antibody, peroxidase-labeled goatanti-rabbit IgG (KPL), which was diluted 1:50,000 in 1%milk–TBS-T solution. After three 10-min washes withTBS-T, we used the EZ-ECL chemiluminescence detectionkit (Biological Industries) for reaction with the peroxidaseconjugate. After 5 min of incubation with the substrate,signals were visualized using the LAS-300 Intelligent DarkBox (Rhenium; Fujifilm) after a 3-min exposure.
Supplementary information
Additional information as well as supplementary tablesand figures can be accessed at: http://www.webclu.bio.wzw.tum.de/oxphos/

170 Coevolution in Oxidative Phosphorylation Complex I
Links
SELECTON–web interface to assess signatures ofnatural selection in proteins (http://SELECTON.tau.ac.il/overview.html). Our final method, called Com-plexCorr, is available at http://webclu.bio.wzw.tum.de/complexcorr. Additional information as well assupplementary tables and figures could be accessedat: http://webclu.bio.wzw.tum.de/oxphos NCBI(www.ncbi.nlm.nih.gov) Swissport (www.expasy.ch/sprot) ENSEMBL (www.ensembl.org) ClustalW(www.clustal.org).
Acknowledgements
This work was funded by grants from the IsraelScience Foundation and the Bikurah FIRST Founda-tion (D.M.), by the Deutsche Forschungsge-meinschaft (A.F.), and by the EU BiosapiensNetwork of Excellence (D.F.). The authors also wishto thank the Negev Foundation for the scholarship ofM.G. The generation and evaluation of the ND4vector were financed by the French Agence Nationalpour la Recherche (ANR)/Maladies Rares (M.C.-D.).The authors express their special thanks to Drs. NiliAvni (The Biblical Zoo) and Tzachi Eizenberg (TheMonkey Park) for providing blood and tissuesamples of primates to the study, and to Prof. MichalShapira (Ben-Gurion University of the Negev) forproviding the antibodies for Western blot analysis.
References
1. Zickermann, V., Drose, S., Tocilescu, M. A., Zwicker,K., Kerscher, S. & Brandt, U. (2008). Challenges inelucidating structure and mechanism of protonpumping NADH:ubiquinone oxidoreductase (com-plex I). J. Bioenerg. Biomembr. 40, 475–483.
2. Schapira, A. H. (2006). Mitochondrial disease. Lancet,368, 70–82.
3. Gabaldon, T., Rainey, D. & Huynen, M. A. (2005).Tracing the evolution of a large protein complex in theeukaryotes, NADH:ubiquinone oxidoreductase (com-plex I). J. Mol. Biol. 348, 857–870.
4. Belogrudov, G. I. & Hatefi, Y. (1996). Intersubunitinteractions in the bovine mitochondrial complex I asrevealed by ligand blotting. Biochem. Biophys. Res.Commun. 227, 135–139.
5. Efremov, R. G., Baradaran, R. & Sazanov, L. A. (2010).The architecture of respiratory complex I. Nature, 465,441–445.
6. Torres-Bacete, J., Sinha, P. K., Castro-Guerrero, N.,Matsuno-Yagi, A. & Yagi, T. (2009). Features ofsubunit NuoM (ND4) in Escherichia coli NDH-1:topology and implication of conserved Glu144 forcoupling site 1. J. Biol. Chem. 284, 33062–33069.
7. Vogel, R. O., Smeitink, J. A. & Nijtmans, L. G. (2007).Human mitochondrial complex I assembly: a dynamic
and versatile process. Biochim. Biophys. Acta, 1767,1215–1227.
8. Lynch, M., Koskella, B. & Schaack, S. (2006). Mutationpressure and the evolution of organelle genomicarchitecture. Science, 311, 1727–1730.
9. Grossman, L. I., Wildman, D. E., Schmidt, T. R. &Goodman, M. (2004). Accelerated evolution of theelectron transport chain in anthropoid primates.Trends Genet. 20, 578–585.
10. Rand, D. M., Haney, R. A. & Fry, A. J. (2004).Cytonuclear coevolution: the genomics of coopera-tion. Trends Ecol. Evol. 19, 645–653.
11. Sackton, T. B., Haney, R. A. & Rand, D. M. (2003).Cytonuclear coadaptation in Drosophila: disruption ofcytochrome c oxidase activity in backcross genotypes.Evol. Int. J. Org. Evol. 57, 2315–2325.
12. Willett, C. S. & Burton, R. S. (2004). Evolution ofinteracting proteins in the mitochondrial electrontransport system in a marine copepod. Mol. Biol.Evol. 21, 443–453.
13. Mishmar, D., Ruiz-Pesini, E., Mondragon-Palomino,M., Procaccio, V., Gaut, B. & Wallace, D. C. (2006).Adaptive selection of mitochondrial complex I sub-units during primate radiation. Gene, 378, 11–18.
14. Mishmar, D. & Zhidkov, I. (2010). Evolution anddisease converge in the mitochondrion. Biochim.Biophys. Acta, 1797, 1099–1104.
15. Zhidkov, I., Livneh, E. A., Rubin, E. & Mishmar, D.(2009). mtDNA mutation pattern in tumors andhuman evolution are shaped by similar selectiveconstraints. Genome Res. 19, 576–580.
16. Gobel, U., Sander, C., Schneider, R. & Valencia, A.(1994). Correlated mutations and residue contacts inproteins. Proteins, 18, 309–317.
17. Pazos, F., Helmer-Citterich, M., Ausiello, G. &Valencia, A. (1997). Correlated mutations containinformation about protein–protein interaction. J. Mol.Biol. 271, 511–523.
18. Yeang, C. H. & Haussler, D. (2007). Detectingcoevolution in and among protein domains. PLoSComput. Biol. 3, e211.
19. Madaoui, H. & Guerois, R. (2008). Coevolution atprotein complex interfaces can be detected by thecomplementarity trace with important impact forpredictive docking. Proc. Natl Acad. Sci. USA, 105,7708–7713.
20. Filizola, M., Olmea, O. & Weinstein, H. (2002).Prediction of heterodimerization interfaces of G-protein coupled receptors with a new subtractivecorrelated mutation method. Protein Eng. 15, 881–885.
21. Pazos, F. & Valencia, A. (2002). In silico two-hybridsystem for the selection of physically interactingprotein pairs. Proteins, 47, 219–227.
22. Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G.,Bhat, T. N., Weissig, H. et al. (2000). The Protein DataBank. Nucleic Acids Res. 28, 235–242.
23. Olmea, O. & Valencia, A. (1997). Improving contactpredictions by the combination of correlated muta-tions and other sources of sequence information. Fold.Des. 2, S25–S32.
24. Fodor, A. A. & Aldrich, R. W. (2004). Influence ofconservation on calculations of amino acid covari-ance in multiple sequence alignments. Proteins, 56,211–221.

171Coevolution in Oxidative Phosphorylation Complex I
25. Dekker, J. P., Fodor, A., Aldrich, R. W. & Yellen, G.(2004). A perturbation-based method for calculatingexplicit likelihood of evolutionary co-variance inmultiple sequence alignments. Bioinformatics, 20,1565–1572.
26. Halperin, I., Wolfson, H. & Nussinov, R. (2006).Correlated mutations: advances and limitations. Astudy on fusion proteins and on the Cohesin–Dockerinfamilies. Proteins, 63, 832–845.
27. Grana, O., Baker, D., MacCallum, R. M., Meiler, J.,Punta, M., Rost, B. et al. (2005). CASP6 assessment ofcontact prediction. Proteins, 61, 214–224.
28. Izarzugaza, J. M., Grana, O., Tress, M. L., Valencia, A.& Clarke, N. D. (2007). Assessment of intramolecularcontact predictions for CASP7. Proteins, 69, 152–158.
29. Wong, K. M., Suchard, M. A. & Huelsenbeck, J. P.(2008). Alignment uncertainty and genomic analysis.Science, 319, 473–476.
30. Notredame, C., Higgins, D. G. & Heringa, J. (2000).T-Coffee: a novel method for fast and accuratemultiple sequence alignment. J. Mol. Biol. 302,205–217.
31. Edgar, R. C. (2004). MUSCLE: multiple sequencealignment with high accuracy and high throughput.Nucleic Acids Res. 32, 1792–1797.
32. Sazanov, L. A. & Walker, J. E. (2000). Cryo-electroncrystallography of two sub-complexes of bovine com-plex I reveals the relationship between the membraneand peripheral arms. J. Mol. Biol. 302, 455–464.
33. Potluri, P., Davila, A., Ruiz-Pesini, E., Mishmar, D.,O'Hearn, S., Hancock, S. et al. (2009). A novelNDUFA1 mutation leads to a progressive mitochon-drial complex I-specific neurodegenerative disease.Mol. Genet. Metab. 96, 189–195.
34. Gershoni, M., Templeton, A. R. & Mishmar, D. (2009).Mitochondrial bioenergetics as a major motive force ofspeciation. BioEssays, 31, 642–650.
35. Hirst, J., Carroll, J., Fearnley, I. M., Shannon, R. J. &Walker, J. E. (2003). The nuclear encoded subunits ofcomplex I from bovine heart mitochondria. Biochim.Biophys. Acta, 1604, 135–150.
36. Yadava, N., Potluri, P., Smith, E. N., Bisevac, A. &Scheffler, I. E. (2002). Species-specific and mutantMWFE proteins. Their effect on the assembly of afunctional mammalian mitochondrial complex I. J. Biol.Chem. 277, 21221–21230.
37. Bustamante, C. D., Fledel-Alon, A., Williamson, S.,Nielsen, R., Hubisz, M. T., Glanowski, S. et al. (2005).Natural selection on protein-coding genes in thehuman genome. Nature, 437, 1153–1157.
38. Nielsen, R., Hubisz, M. J., Hellmann, I., Torgerson, D.,Andrés, A. M., Albrechtsen, A. et al. (2009). Darwinianand demographic forces affecting human proteincoding genes. Genome Res. 19, 838–849.
39. Weigt, M., White, R. A., Szurmant, H., Hoch, J. A. &Hwa, T. (2009). Identification of direct residuecontacts in protein–protein interaction by messagepassing. Proc. Natl Acad. Sci. USA, 106, 67–72.
40. Hunte, C., Zickermann, V. & Brandt, U. (2010).Functional modules and structural basis of conforma-tional coupling in mitochondrial complex I. Science,329, 448–451.
41. Fuchs, A., Kirschner, A. & Frishman, D. (2009).Prediction of helix–helix contacts and interactinghelices in polytopic membrane proteins using neuralnetworks. Proteins, 74, 857–871.
42. Miyata, T., Miyazawa, S. & Yasunaga, T. (1979). Twotypes of amino acid substitutions in protein evolution.J. Mol. Evol. 12, 219–236.
43. McLachlan, A. D. (1971). Tests for comparing relatedamino-acid sequences. Cytochrome c and cytochrome c551. J. Mol. Biol. 61, 409–424.
44. Wang, X., Carre, W., Zhou, H., Lamont, S. J. &Cogburn, L. A. (2004). Duplicated Spot 14 genes in thechicken: characterization and identification of poly-morphisms associated with abdominal fat traits. Gene,332, 79–88.
45. Ellouze, S., Augustin, S., Bouaita, A., Bonnet, C.,Simonutti, M., Forster, V. et al. (2008). Optimizedallotopic expression of the human mitochondrial ND4prevents blindness in a rat model of mitochondrialdysfunction. Am. J. Hum. Genet. 83, 373–387.
Copyright © 2022 FDOKUMEN




















