
evans_thesis.pdf - University of Canterbury
201
X-RAY INVESTIGATION OF STRUCTURAL RELATIONSHIPS AND PHYSICAL PROPERTIES IN CHARGE-TRANSFER COMPLEXES. A thesis presented for the degree of Doctor of Philosophy in Chemistry in the University of Canterbury, Christchurch, New Zealand. by D.L. EVANS 1980
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of evans_thesis.pdf - University of Canterbury

X-RAY INVESTIGATION OF STRUCTURAL RELATIONSHIPS AND
PHYSICAL PROPERTIES IN CHARGE-TRANSFER COMPLEXES.
A thesis presented for the degree of
Doctor of Philosophy in Chemistry
in the University of Canterbury,
Christchurch, New Zealand.
by
D.L. EVANS 1980

PHYSICAL SCIENCES llBRA.B:'(
ABSTRACT
The crystal structures of four charge-transfer complexes and two organo-metallic compounds have been determined. The various geometries encountered are related to structural
isomerism and to physical properties. The charge-transfer
complexes phenanthrene pyromellitic acid dianhydride (PMDA),
fluorene PMDA, thianthrene PMDA and biphenyl trinitrofluorenone
have planar molecules except for thianthrene which is bent. The molecule N-(2-hydroxyphenyl)salicylaldimine dimethyl tin(IV)
is bipyramidal whilst the hexa-v-dithiocacodylato tetrazinc
sulphide isomers have a tetrahedral character.
The concept of the crystalline state as a hierarchical system is developed. The physical basis, and electrical properties of
charge-transfer complexes are reviewed, and the implications of structural features, and possible uses of these compounds in
modern signal processing technology is discussed.

TABLE OF CONTENTS
TITLE PAGE
ABSTRACT
CONTENTS
ACKNOWLEDGEMENTS
LIST OF TABLES
LIST OF FIGURES
ABBREVIATIONS
MOLECULAR STRUCTURES CITED
UNITS USED
CHAPTER 1
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
CHAPTER 2
2.1
2.1.1
2 .1. 2
2 .1. 3
2 .1. 4
2 .1. 5
2.1. 6
2.1. 7
2.1. 8
2.1. 9
2.2
2.2.1
2.2.2
INTRODUCTION
CRYSTALLOGRAPHIC STUDIES
BASE DATA
THE CRYSTALLINE STATE
THE PROPERTIES OF CRYSTALLINE MATERIALS
CHARGE-TRANSFER COMPLEXES
PYROMELLITIC ACID DIANHYDRIDE
ORGANO-METALLIC COMPOUNDS
COMPUTING DEVELOPMENT
FEATURES OF ELECTRON BEHAVIOUR IN CRYSTALS
INTRODUCTION
ELECTRON BEHAVIOUR
CRYSTAL AND MOLECULAR ORBITALS
CHARGE-TRANSFER COMPLEXES
INTERMOLECULAR CHARGE TRANSFER
RANDOM WALK THEORY FOR ELECTRONS IN CRYSTALS
BARRIERS TO ELECTRON MOTION
ELECTRON INTERACTIONS
HOPPING MODELS
POLARONS
EXCITONS
INTRODUCTION
EXCITON GENERATION
i
i
vi vii
ix
xi xiii
xvi
1
1
1
1
2
3
3
4
4
5
5
5
6
8
9
11
13
14
14
15
15
15
16

2.2.3 EXCITON DIFFUSION 17
2.2.4 EXCITON MOBILITY 17
2.2.5 EXCITON INTERACTIONS 18
2.2.6 EXCITON TRAPS 18
2.3 PHOTOCONDUCTIVITY 19
2.3.1 INTRODUCTION 19
2.3.2 CHARGE CARRIER MOBILITIES 22
2.3.3 MOBILITIES IN CHARGE-TRANSFER COMPLEXES 23
2.3.4 ELECTRON MOBILITY IN PHENANTHRENE PMDA 24
2.4 ESR SPECTROSCOPY OF CHARGE-TRANSFER COMPLEXES 25
CHAPTER 3 EXPERIMENTAL PROCEDURES 28
3.1 PRELIMINARY CRYSTALLOGRAPHIC STUDIES 28
3.2 DIFFRACTOMETER DATA COLLECTION 28
3.3 DATA PROCESSING 29
CHAPTER 4 SOLUTION AND REFINEMENT PROCEDURES 31
4.1 INTRODUCTION 31
4.2 ELECTRON DENSITY AND STRUCTURE FACTORS 31
4.3 PHASE DETERMINATION 32
4.3.1 PATTERSON METHODS 32
4.3.2 BASIS OF STATISTICAL METHODS 34
4.4 STATISTICAL METHODS IN PRACTICE 34
4.4.1 INPUT DATA 34
4.4.2 USE OF MULTAN 35
4.5 MODEL BUILDING AND REFINEMENT 36
4.5.1 STRUCTURE FACTOR CALCULATION 36
4.5.2 LEAST-SQUARES REFINEMENT 37
4.5.3 REFINEMENT OF HYDROGEN ATOM PARAMETERS 37
4.5.4 DIFFERENCE FOURIER SYNTHESIS 38
CHAPTER 5 THE CHARGE-TRANSFER COMPLEX PHENANTHRENE PMDA 39
5.1 INTRODUCTION 39
5.2 EXPERIMENTAL SECTION 39
5.3 STRUCTURE DETERMINATION AND REFINEMENT 40
ii

5.4
CHAPTER 6
6.1
6.2
6.3 6.4
CHAPTER 7
7.1
7.2
7.3
7.4
CHAPTER 8
DESCRIPTION AND DISCUSSION OF THE STRUCTURE 40
THE CHARGE-TRANSFER COMPLEX FLUORENE PMDA 48
INTRODUCTION 48
EXPERIMENTAL SECTION 48 STRUCTURE DETERMINATION AND REFINEMENT 49
DESCRIPTION AND DISCUSSION OF THE STRUCTURE 50
THE CHARGE-TRANSFER COMPLEX THIANTHRENE PMDA 59
INTRODUCTION 59
EXPERIMENTAL SECTION 59
STRUCTURE DETERMINATION AND REFINEMENT 60 DESCRIPTION AND DISCUSSION OF THE STRUCTURE 61
THE CHARGE-TRANSFER COMPLEX BIPHENYL
TRINITROFLUORENONE 69
8.1 INTRODUCTION 69
8.2 EXPERIMENTAL SECTION 69
8.3 STRUCTURE DETERMINATION AND REFINEMENT 70
8.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE 70
CHAPTER 9 REVIEW OF THE FOUR CHARGE-TRANSFER STRUCTURES 81
9.1 STRUCTURE GEOMETRY AND SYMMETRY 81
9.2 INTERMOLECULAR CHARGE TRANSFER 82 9.3 DIMENSIONALITY OF CHARGE CARRIER MOTION 82
9.4 SOLID STATE SIGNAL PROCESSING TECHNOLOGIES 84 9.5 DYNAMIC ELECTRONICS 85
9.6 FUTURE SIGNAL PROCESSING TECHNOLOGY 86
CHAPTER 10 THE STRUCTURE OF THE TRIDENTATE, FIVE COORDINATE
N-(2-HYDROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV) 87
10.1 INTRODUCTION 87
10.2 EXPERIMENTAL SECTION 87
10.3 STRUCTURE DETERMINATION AND REFINEMENT 88
iii

10.4
CHAPTER 11
11.1
11.2
11.3
11.4
CHAPTER 12
12.1
12 .1.1
12 .1. 2
12 .1. 3
DESCRIPTION AND DISCUSSION OF THE STRUCTURE
THE ISOMERIC STRUCTURES OF THE HEXA-P-DITHIO
CACO.DYLATO TETRAZINCSULPHIDE MOLECULES
INTRODUCTION
EXPERIMENTAL SECTION
STRUCTURE DETERMINATION AND REFINEMENT
DESCRIPTION AND DISCUSSION OF THE STRUCTURES
COMPUTER PROGRAMS
INVESTIGATION OF PEAKS IN PATTERSON MAPS (PATINV)
INTRODUCTION
INPUT
OBTAINING ATOMIC POSITIONS USING HARKER PLANES
AND LINES
91
95
95
97
97
98
116
116
116
116
116
12.1.4 VERIFICATION OF A MODEL'S CONSISTENCY WITH
THE PATTERSON MAP 119
12.1.5 MODEL EXTENSION BY ATOMIC SUPERPOSITION 121
12.2 LINE-PRINTER SHADED ELECTRON DENSITY MAPS (ARTSTC) 121
12.2.1 INTRODUCTION 121
12.2.2 PROGRAM FEATURES 122
12.2.3 PROGRAM CONTROL 123
12.2.4 PROGRAM ALGORITHM 123
12.3 SORTLIST 124
12.3.1 INTRODUCTION 124
12.3.2 STATISTICS SECTION 124
12.3.3 PUBLICATION LISTINGS OF STRUCTURE FACTORS 125
12.4 BONDSTAT 126
12.4.1 INTRODUCTION 126
12.4.2 PROGRAM DESCRIPTION 127
APPENDIX A STRUCTURE FACTOR TABLES 128
APPENDIX B SCALING OF INTENSITY DATA 136
REFERENCES 137
iv

COMPUTER PROGRAM LISTINGS
PAT INV
ARTSTC
SORTLIST
BOND STAT
SCALE
v
142
142
153
165
179
182

ACKNOWLEDGEMENTS
I am indebted to Dr. Ward T. Robinson for his able
supervision of my research, to Professor B.R. Penfold for his
introduction to X-Ray Crystallographic techniques. Thanks is due
to Dr. R. MacFarlane for his liaison with and introduction to
Dr. D. Haarer (IBM San Jose USA) who supplied the charge-transfer crystals. Appreciation is also due to Dr. J. R. Sams, University
of British Columbia who suggested the tin structure and supplied
the crystals for it.
The support of the research committee of the New Zealand University Grants Committee in the form of equipment grants is
hereby acknowledged.
Finally thanks are due to Professor J. Vaughan for an
appointment as a Teaching Fellow in the Chemistry Department.
vi

2.1.2
5.4a
5.4b
6.4a
6.4b
7.4a
7.4b
8.4a
8.4b
8.4c
8.4d
8.4e
10.3a
10.3b
LIST OF TABLES
CONFORMATIONAL ENERGIES OF SELECTED COMPOUNDS
ATOMIC PARAMETERS OF PHENANTHRENE PMDA
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION OF
PHENANTHRENE PMDA
ATOMIC PARAMETERS OF FLUORENE PMDA
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION OF FLUORENE
PMDA
8
46
47
57
58
ATOMIC PARAMETERS OF THIANTHRENE PMDA 67
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION OF THIANTHRENE
PMDA 68
INTERPLANAR ANGLES WITHIN BIPHENYL MOLECULES 76
INTERPLANAR ANGLES FOR TRINITROFLUORENONE MOLECULES 77
INTERPLANAR ANGLES WITHIN BIPHENYL AND
TRINITROFLUORENONE PAIRS 77
ATOMIC PARAMETERS OF BIPHENYL TRINITROFLUORENONE 79
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION
OF BIPHENYL TRINITROFLUORENONE 80
ATOMIC PARAMETERS OF (N-(2-HYDROXYPHENYL)
SALICYLALDIMINE DIMETHYL TIN(IV}
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION FOR
89
N-(2-HYDROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV) 90
10.4 BOND LENGTHS AND ANGLES FOR N-(2-HYDROXYPHENYL)
SALICYLALDIMINE DIMETHYL TIN(IV) 93
96
96
99
11.1a
11.lb
11.3a
11.3b
11.3c
11.3d
CONFORMATIONS OF CHAIRS AND BOATS
POSSIBLE CONFIGURATIONS IN A 2 RING CLASSIFICATION
ATOMIC PARAMETERS FOR [SZn {AsS (CH ) } ] (1) 4 2 3 2 6
ATOMIC PARAMETERS FOR [SZn {AsS (CH ) } ] (2) 4 2 3 2 6
ATOMIC PARAMETERS FOR [SZn {AsS (CH } } ] (3) 4 2 3 2 6
ATOMIC PARAMETERS FOR [SZn {AsS (CH } } ] (4) 4 2 3 2 6
12.1.3a VECTOR COUNT MATRIX
12.1.3b VECTOR COUNT SUBMATRIX
12.1.4a POSITIONAL CONSISTENCY MATRIX
12.1.4b MATRIX OF ESTABLISHED AND NEW POSITIONS
vii
100
101
102
119
119
120
120

12.3.2a FUNCTIONS CALCULATED IN THE WEIGHTING ANALYSIS
12.3.2b PROGRAM LIMITS FOR SORTLIST
12.3.3 CHARACTERISTICS OF SORTING INDICES
viii
124
125
126

LIST OF FIGURES
5.4.1 BOND DISTANCES AND ANGLES IN PHENANTHRENE
5.4.2 BOND DISTANCES AND ANGLES IN PMDA
5.4.3 CRYSTAL PACKING OF PHENANTHRENE AND PMDA MOLECULES VIEWED
DOWN THE STACK AXIS AND VIEWED EDGE-ON TO ONE OF THE PMDA
43
44
MOLECULES 45
6.3a
6.3b
6.4a
6.4b
SCHEMATIC DIAGRAM OF SUPERIMPOSED FLUORENE MOLECULES
LINE PRINTER SHADED ELECTRON DENSITY MAP OF PMDA
BOND DISTANCES AND ANGLES IN FLUORENE
BOND DISTANCES AND ANGLES IN PMDA
6.4c VIEWS EDGE ON TO A FLUORENE MOLECULE AND PARALLEL TO THE
y AXIS
7.4a
7.4b
7.4c
7.4d
8.4a
8.4b
8.4c
8.4d
8.4e
BOND DISTANCES AND ANGLES IN THIANTHRENE
BOND DISTANCES AND ANGLES IN PMDA (1)
BOND DISTANCES AND ANGLES IN PMDA (2)
VIEWS OF THE CRYSTAL PACKING OF THIANTHRENE AND PMDA
MOLECULES
DISTANCES AND ANGLES IN BIPHENYL (1)
DISTANCES AND ANGLES IN BIPHENYL (2)
DISTANCES AND ANGLES IN TRINITROFLUORENONE (1)
DISTANCES AND ANGLES IN TRINITROFLUORENONE (2)
VIEWS OF THE CRYSTAL PACKING OF BIPHENYL AND
51
52
53
54
55
62
63
64
65
71
72
73
74
TRINITROFLUORENONE 75
9.3 HOPPING PATHWAYS WITHIN THIANTHRENE PMDA 83
10.4 GENERAL VIEW OF THE COMPLEX ILLUSTRATING THE DISTORTED
TRIGONAL BIPYRAMIDAL ENVIRONMENT OF THE TIN ATOM 92
11.4a STRUCTURE OF [SZn {AsS (CH ) } ] JOHNSTONE ET AL 103 4 2 3 2 6
11.4b STRUCTURE OF [SZn {AsS (CH ) } ] BATES 104 4 2 3 2 6
11.4c STRUCTURE OF [SZn {AsS (CH ) } J MOLECULE 1 106 4 2 3 2 6
11.4d STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE 2 107 4 2 3 2 6
11.4e STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE 3 108 4 2 3 2 6
11.4f STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE 4 109 4 2 3 2 6
11.4f' STRUCTURE OF [SZn {AsS (CH ) } } ALT. VIEW MOLECULE 4 110 4 2 3 2 6
11.4g DISTANCES AND ANGLES FOR MOLECULE (1) 112
ix

11.4h DISTANCES AND ANGLES FOR MOLECULE {2)
11.4i DISTANCES AND ANGLES FOR MOLECULE (3}
11.4j DISTANCES AND ANGLES FOR MOLECULE (4}
12.1.3 VECTORS IN PATTERSON SPACE
X
113
114
115
118

LIST OF ABBREVIATIONS
A number of abbreviations are used throughout the text of
this thesis. Their full meaning is generally indicated in the text, and most of them are also listed here.
A acceptor
As arsenic
C carbon
Cl chlorine
CNDO complete neglect of differential overlap
CT charge-transfer Cu copper
D donor
DBC diffracted beam collimator DNA deoxyribose nucleic acid esd estimated standard deviation ESR electron spin resonance
F structure factor H Hamiltonian
H hydrogen HCNB hexacyanobenzene
Hfs hyper-fine splitting I intensity m mass of electron
e Mo molybdenum
N nitrogen
NIPC Naphthalene-Isopropylcarbazole picryl chloride 0 oxygen
p electron density
p linear momentum PCNT pentacyanotoluene
PMDA pyromellitic acid dianhydride
RNA ribose nucleic acid s angular momentum S sulphur
xi

S overlap integral
SCF self consistent field Sn tin
TCNB tetracyanobenzene
TCNQ tetracyanoquinodimethane
TNB trinitrobenzene TNF trinitrofluorenone
ZFS zero field splitting
Zn zinc
Zr zirconium
xii

MOLECULAR STRUCTURES CITED
A number of molecules are referred to in the body of this thesis. Most of their structures are detailed here, in an
alphabetical sequence, for the convenience of the reader.
anthracene
azulene
benzanthracene
biphenyl
chloranil
dimethyl
fluorene
hexamethylbenzene
CCX)
o:>
o-o
xiii

isopropylcarbazole
naphthalene
perylene
phenanthrene
picryl chloride
pyrene
Q;p CH-(:--CH
~ co
Cl N0
2
pyromellitic acid dianhydride
0 0 0 0
O Cl o0 Cl00
OsS:o CI?(OO 0
tetrachlorophthalia anhydride
thianthrene
thianthrene dioxide CXsS:o • 0
xiv
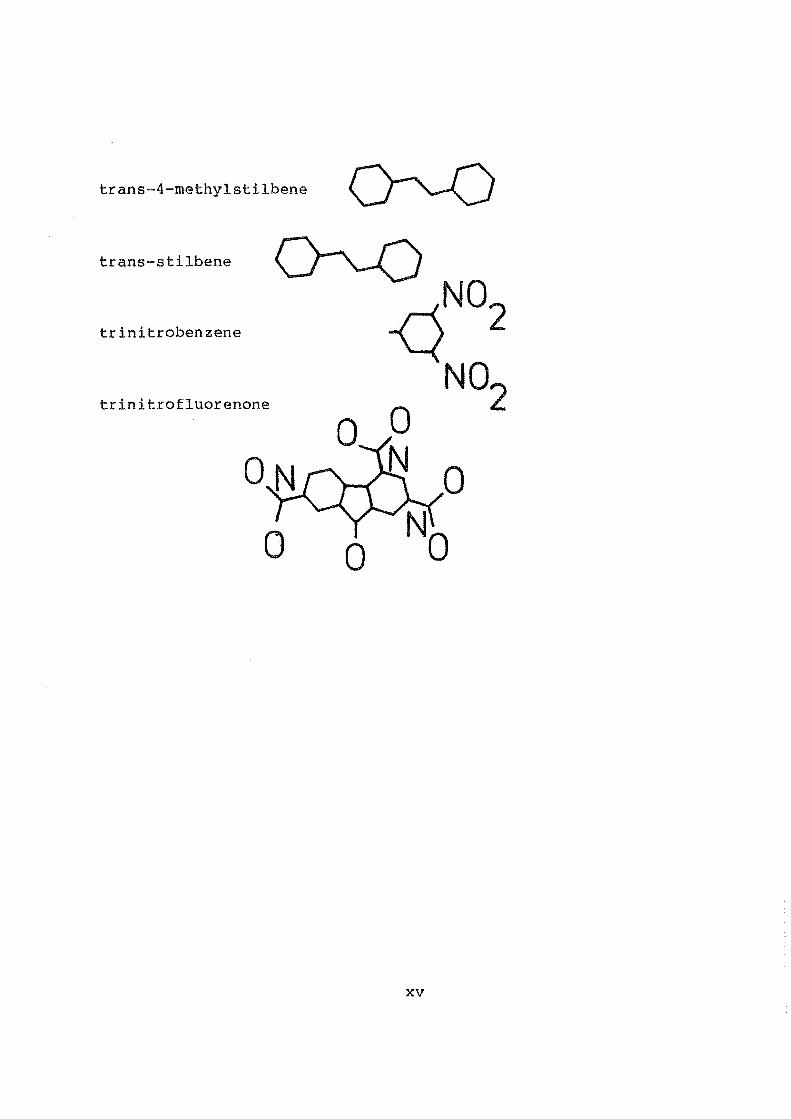
trans-4-methylstilbene
trans-stilbene
trinitrobenzene
trinitrofluorenone
0
XV

UNITS USED
A number of the units used or quoted in this thesis are
not according to the current standard SI convention but they do
remain in common usage. Accordingly they are listed here with
their standard equivalents.
0 -1 A nm xlO
-2 em m xlO
-1 2 em m xlO
-3 mm m xlO
-2 -4 Gauss wm xlO
xvi

CHAPTER 1
INTRODUCTION
1.1 CRYSTALLOGRAPHIC STUDIES
This thesis covers a range of crystallographic studies using
a variety of techniques. The structures of four ~-n chargetransfer (CT) complexes and two organo-metallic compounds are
determined. They are phenanthrene pyromellitic acid dianhydride (PMDA), fluorene PMDA, thianthrene PMDA, biphenyl trinitrofluorenone, and N-(2-hydroxyphenyl) salicylaldimine dimethyl tin(IV) and hexa-~-dithiocacodylato tetrazincsulphide.
Interesting disorder, isomerism and bonding features are
encountered. CT compounds have interesting electrical properties
discussed, but not investigated in this thesis. Models of, and features of the observed electrical behaviour in this class of
compound are discussed in Chapter 2.
1.2 BASE DATA
The crystallographic studies provide very important base
data, as knowledge of the arrangement of the molecules within the crystallographic unit cell is essential to a good understanding of physical properties. Knowing space group symmetry enables the
correct use of selection rules for calculating energy changes in
transitions between various states. Furthermore, the establishment of atomic parameters makes possible the calculation
of molecular orbitals in, what must be, the minimised potential energy state. Such calculations are not part of this thesis, but
the types of calculation which are made, are discussed.
1.3 THE CRYSTALLINE STATE
The concept of a crystal as a hierarchical type of structure,
having various types of states, disorder, and transitions is developed. It facilitates discussion of a crystal's physical
properties in terms of the molecular structures of which it may be comprised.
1

1.4 THE PROPERTIES OF CRYSTALLINE MATERIALS
A crystal consists of an array of identical chemical units.
The internal character of an ideal crystal can be described in
terms of a lattice, the smallest repeating unit of such a lattice
being termed a unit cell. The unit cell described by parameters a, b, c, a, S,y contains a number of identical chemical units
related to each other by symmetry operations and to those in
other cells by translations. For purposes of notation let such a cell be R, and a distinct site within a chemical unit be a, then
R denotes the site a in the cell R. The chemical units may be a
single atoms, parts of molecules, molecules or sets of chemically
identical (yet independent) or in some cases different molecules. The coordinates of atoms x,y,z within the unit cell are expressed
in terms of fractions of the unit cell dimensions. Bounding faces
and other planes in the crystal are described in terms of Miller indices hkl and are represented by points in reciprocal space. A real crystal approximates the ideal crystal described, deviating
from it either by cell or lattice imperfection.
The main bulk physical properties measurable are dimensions, density, bulk modulus, shear strength, thermal conductivity,
velocity of sound and refractive index. These can generally be
measured without significant interaction with the interior (contents} of the crystalline material.
The electrical properties involve charge carriers within the
crystal, namely electrons and holes, whose behaviour is determined
by the internal character of the crystal and interactions of external origin. The degree of interaction may be considerable in the measurement of electrical and spectral properties. External
influences affecting charge carrier motion are temperature, com
pression, electric, magnetic and strain fields and electromagnetic
radiation. A hole is defined as follows. In an array of neutral
atoms the ionisation of an atom by excitation of an electron leaves it positively charged. The vacant orbital into
which another electron could move is called a hole and may,
2

as an electron shifts from an adjacent atom or crystal orbital into it, behave as a positive charge carrier. If an electron and a hole move as a pair from atom to atom or molecule to molecule they are described as an exciton.
1.5 CHARGE-TRANSFER COMPLEXES
The stochiometrically 1-1, ~-~charge-transfer compounds are
a class of compounds currently subject to intensive study. Such physical properties as exciton motion (Mohwald & Sackmann 1974),
photoconductivity (Haarer & Mohwald 1975), electron mobility (Bergman & Jortner 19741 Mohwald, Haarer & Castro 1975) and fluorescence (Kepler, Caris, Avakien & Abramson 1963) are being investigated. Techniques in use include electron spin resonance
(ESR) (Dalal, Haarer, Bargon & Mohwald 1975), emission and absorption spectroscopy (Haarer & Karl 1973) and conductivity
measurements (Batt, Braun & Hornig 1968). A review of crystall
ographic work has been made by Herbstein (1971). The commonly
occurring features he discusses are (1) the donor-acceptor 0
distance of - 3.5 A, (2) stacking of the donor and acceptor
molecules in semi-infinite columns, (3) disorder, (4) centrosymmetric molecules, (5) the dihedral angle between the adjacent molecules and (6) the non-perpendicularity of the planes of the component molecules with respect to the stack axis.
1.6 PYROMELLITIC ACID DIANHYDRIDE
Interest in PMDA as an electron acceptor, has led to the
study of n-~ organic charge transfer compounds involving it and
various donor molecules. These include anthracene and perylene (Boeyens & Herbstein 1965), napthalene (Herbstein 1971), pyrene
(Herbstein & Snyman 1969), benzanthracene (Foster, Scrimgeour, Iball & Williams 1975), trans-stilbene (Kodama & Kumakura 1974a), trans-4-methylstilbene (Kodama & Kumakura 1974b), phenanthrene, fluorene and thianthrene (Evans & Robinson 1977, 1980a and 1980b).
3

1.7 ORGANO-METALLIC COMPOUNDS
N-(2-hydroxyphenyl) salicylaldimine dimethyl tin(IV) and
hexa-p-dithiocacodylato tetrazincsulphide were studied, to further
work by Professor B.R. Penfold and Dr. Ward T. Robinson,
respectively. The tin compound determination provided an
introduction to X-ray crystallography, and the arsenic compound
was the final structure determined and provided a suitably challenging finale.
1.8 COMPUTING DEVELOPMENT
The principal program developed was a shaded density line
printer plotting routine {ARTSTC), for illustrating electron density maps. A second major effort was a Patterson map invest
igating program {PATINV) combining a number of features which others have undoubtedly developed. A third, medium sized program,
(SORTLIST) to sort structure factors, present them in publication format, and to give statistical analyses of them is presented.
Two small programs SCALE and BONDSTAT written to meet certain contingencies are also given.
4

CHAPTER 2
FEATURES OF ELECTRON BEHAVIOUR IN CRYSTALS
2.1 INTRODUCTION
2.1.1 ELECTRON BEHAVIOUR
This section constitutes an overview of the behaviour of
electrons. A free electron mass m , charge e has angular momentums and linear momentum E <T£1>0) and its state may be
described by a wavefunction from which its energy may be deduced
using a Hamiltonian operator. In a crystal an electron may be regarded as 'free', or, as
in a crystal orbital, a molecular orbital or an atomic orbital. The electrons in a 'free' state are subject to the crystal field and, along with all other components of the crystal, to any
external fields. Electrons may also be coupled to the crystal lattice singly, as in excitons, polarons, and magnons, or in
pairs, as in superconductivity. If the set R{i) i=l,n
of phenomena in which electrons are 'free', bound (in orbital
states), or coupled to the lattice is considered then there are an associated set of energy states E(i) i=l,n. Energy transfer is
possible between these states, subject to various symmetry, parity, conservation and quantum mechanical rules. The set of possible types of transition are those associated with the set of Cartesian products P(ij) = R(i) X R(j) i,j=l,n, and, the set of
mechanisms,. (in which a number of members of P may participate) is given by the set of linear combinations L =Ec(k) .P(ij) {i,j,k=l,n)
c(k)=l indicating the presence of the kth member of set P(ij) in
the mechanism and c(k)=O its absence. That is, a distinct
mechanism may involve an electron in a finite sequence of energy transfers the initial and final states of the mechanism being observable.
In a crystal the number of mechanisms which are 'viable' is a function of the molecular/ionic or complex structure of the crystal. In addition to the host material forming the bulk
5

of the crystal, dislocations, guest-impurity species and other
point defects form an important constituent of the crystalline environment. In a sense, from the point of view of an electron, the host material may be regarded as an hierarchical environment, in which the order of the hierarchy is atomic, molecular, inter
molecular or 'free' or crystalline. Changes in energy in a
hierarchical system, within and between levels leads to a spectrum of discrete lines and bands with various levels of fine
and hyperfine structure. Quantum mechanical rules allow discrete
energy changes whilst continuous variation in the environment allows a certain spread in values about these otherwise
discrete values.
In addition to the internal environment external influences may be brought to bear, namely, electric, magnetic and strain fields, temperature and electromagnetic radiation.
These influences may either directly affect the single electron behaviour or do so indirectly via the crystal lattice or in
coupled phenomena jointly. The external influences may be
applied both singly or in combination.
The principal means of exciting electrons is with photons. It is specific in the sense that the photon-electron inter-
action is a many-one (although predominantly one-one) phenomenon. Sound and ultrasonic vibration, piezo-electric and pyro-electric
effects are utilized to a limited extent in spectroscopic studies. On the other hand temperature variation is very useful, magnetic
fields split degenerate spin states and electric fields greatly
affect conduction. Strain fields can break symmetry and allow
otherwise forbidden transitions.
2.1.2 CRYSTAL AND MOLECULAR ORBITALS
Crystal orbitals are described in terms of molecular
orbitals and these in turn in terms of atomic orbitals. Various models with differing approximations are in common use. The
calculation of molecular orbitals usually follows a Hartree
Fock or self consistent field (SCF) approach using linear
6

combinations of atomic orbitals (LCAO) with simplifying
assumptions such as complete neglect of differential overlap (CNDO).
The Huckel Theory uses an LCAO approach
1/J. = ~c .. ¢. 1 J 1) J
1/J being the ith molecular orbital i
¢. being the jth atomic orbital J
The constraint of total energy minimisation yields the set of Huckel equations
n ,L:. [H.. - ES. ,Jc .. = 0 , j=l,2, ••• n 1=J 1] 1] 1]
where H is the Hamiltonian of the orbitals i,j and s .. is the ij 1J
overlap 1ntegral of the two orbitals i and j. Hoffmann (1963)
extended the Huckel model by approximating the off-diagonal
terms Hij as H = 0.5k( H + H ) S ij ii jj ij
where a factor k has been introducea into Mulliken's (1949)
approximation for the product of two charge distributionsx and x. i J
X , X. = 0 • 5 S • • ( X , X, + X, X • ) 1 J 1] 1 1 J J
Hoffmann (1963) carried out calculat1ons of the energy for
various conformations of a number of molecules using Slater atomic orbitals and found that the conformation of that with minimum
energy tends to be close to that obtaining. Energies for a selection of compounds studied here are given in Table 2.1.2.
Molecular complexes have been studied in a number of ways. Chesnut and Moseley (1968) used a Hoffmann extended Huckel model simplified to the extent that they used only the 2p~ orbitals of
the molecular orbitals. Their aim was to study the relative
geometry of the pair of molecules in the complex. The intramolecular geometries were held fixed at the experimentally determined
values whilst the molecular relative geometry was varied in such a way as to keep the molecular planes parallel. The ~ only
approximation is satisfactory for molecular separations
7

0 > 2.5 A. The calculations indicate local minima close to the
experimentally observed geometry in most of the complexes studied.
Table 2.1.2
CONFORMATIONAL ENERGIES OF SELECTED COMPOUNDS
Formula Molecule Energy (eV)
C H benzene 25.068 6 6
c H azulene 019.095 10 8
c H biphenyl -841.681 12 10
c H anthracene -1158.974 14 10
c H phenanthrene -1158.974 14 10
2.1.3 CHARGE-TRANSFER COMPLEXES
The 1-1 charge-transfer complexes consist of stacks of
normally neutral alternating donor (D) and acceptor (A)
molecules. The crystal chemical unit contains a number of such
pairs. The planes of the D and A molecules are usually 0
approximately parallel to each other and about 3.5 A apart.
There is a limited amount of charge transfer between one molecule and the other and the characteristic TI-TI absorption in the
visible part of the spectrum evidences this. Mulliken (1952) introduced the simple concept of a wave
function, describing partial charge transfer between a donoracceptor pair, as
of the members of 1[;
being a linear
the pair.
= a1[; + b1[;
combination of the wavefunction
where a and b c
denote 2
D+ Athe extent
2 of mixing of the orbitals,
and S =I1jJ 1[; dv 0 1
complex A.B is
a + 2abS + b = 1
given that the ground state N of a molecular 1/J = a1[; + b1jJ + ••• N 0 1
This is a description for an isolated pair of atoms. For a pair
of molecules in a CT or EDA (electron-donor-acceptor) complex
ground states and excited states may be written (Mayoh & Prout 1972)
8

2 2 1/JG = aljJDA + biljJiD+A- for which a >> b
i 2 2
1jJ - a 'w + b'w a' << b' CT' DA i iD+A- i
respectively. Mulliken's (1956) "Overlap and Orientation
Principle" states that the dependence of the CT interaction on the relative orientation of adjacent plane-to-plane D and A molecules may affect their orientation. The stabilisation energy E of the ground state due to CT interaction is calculated
G using 2nd order perturbation theory. {Mayoh and Prout 1972).
E G
<lj! !H!lj! > = DA D+A-th atl (E - E ) ro+A- D+A- DA
2
The mixing coefficient b of each CT state into the ground i
state is given by
<w IHiw > b = DA D+A-
i (E - E )
D+A- DA
The matrix elements reduce to an integral in terms of molecular
orbitals and these to integrals involving atomic orbitals. CT
interactions only affect orientation when London dispersive,
charge dipole, dipole-dipole, dipole-induced dipole interactions
are insensitive to orientation as they are generally higher in energy than CT stabilisation energies. Mayoh and Prout (1972)
have shown that the CT interaction of the ground state, cannot be used as a guide to either the energy, or orientation sensitivity of the total CT stabilisation of a complex, although the majority of the complexes studied, have a donor-acceptor orientation, in which the CT stabilisation energy, is nearly maximised.
2.1.4 INTERMOLECULAR CHARGE TRANSFER
Middlemiss & Santry (1974) have looked at the intermolecular
charge transfer in more detail using a self consistent field
9

(SCF) model. The approximate SCF matrix equation for a molecular
crystal is FC = CE
where F is the Fock matrix for the entire crystal, C is the coefficient matrix for the expansion of the crystal orbitals
in terms of the atomic orbital basis set and E is the orbital energy matrix. The crystal density matrix is built up with linear
combinations of atomic orbitals, for individual molecules, grouped together, so that when the matrix is partitioned according to
molecules, submatrices on the diagonal are termed intramolecular RuRu
(denoted P).
'The first order submatrix for the Ruth molecule is
RuRu (1) p
.llJ.l =
occ vac u u RuRu u (O)u (0)
4 E E A C C i k ki llk JJi
h u (0) . . . • h h w ere C 1s the molecular orb1tal coeff1c1ent for t e ut Jlk
atomic orbital in the kth molecular orbital localized on a
molecule occupying the uth site of the Rth unit cell. ' RuRu
RuRu
A ki
RuRu ( 1) u ( 0 ) Fk. I ( E:.
1 1
A = 0 ii
u ( 0) E: )
k i 'I k
u ( 0) e is the ith zero order orbital energy for the uth molecule. i
RuRu ( 1) u u u ( 0) RuRu ( 1) ( 0) F =L:L: C F C
RuRu (1) where F
ld
ki .u v ,uk JJV vi is the first order change in the Ruth intra-
molecular Fock subatrix. The total first order chan1e in the electron population of the uth molecule is zero. uQ l) is
RuRu (1) given by the trace of P
occ vac u (1) u u u RuRu u (0) u {0)
Q = 4 E E L: A C C }J i k k i .uk )l i
10

occ vac u (0) (0} u u RuRu u
:::: 4 I: E A L: c c = 0 i k ki )J. ).lk }.l i
The second order change is u ( 2)
Q
occ vac occ vac u ( 2) y I: u{RuTyA }2_2 I: u I: y{TyRu 2
Q :::: 2 I: I: I: 1: I: Aki} T y i k ki T y i k
This is zero for Ru = Ty, that is for intramolecular terms, but
non-zero for intermolecular terms. Thus intermolecular interactions provide the leading contributions to intermolecular
charge transfer. So the circumstances in which charge transfer may occur can be summarised as follows.
(1) Between independent molecules of the same type within a
chemical unit.
(2} Between molecules of different types as in a CT complex. (3) When a structural distortion, such as is produced by an
asymmetric vibration of the unit cell molecules, breaks the equivalence of these molecules. Such a charge
transfer could make a significant contribution to the change of dipole moment associated with the vibrations.
Given that, subject to these conditions, inter and intra molecular charge transfer is possible, it is of interest to
examine, how charge carriers will flow in crystalline
materials. This can be measured macroscopically and described
in terms of a diffusion model. As our understanding improves
microscopic models are developed and these must give the same
or better results than the macroscopic models.
2.1.5 RANDOM WALK THEORY FOR ELECTRONS IN CRYSTALS
The possible movement of charge carriers around a
crystal can be studied by consideration of various aspects of random walks on periodic lattices. Montroll & Weiss (1965}
have considered a number of the aspects in the general case
where steps s of length Is! are made on a toroidal k-dimensional -k -
lattice with N lattice points s = (s ,s , ..• s ). The 1 2 k
11

principal assumptions and results of this model are given
here. The probability that a random walker is at step ~ after n steps is given by P (s) and in view of the periodic
n-boundary conditions
P ( s + j N, s + j N, ••. , s + j N) = P ( s) n 1 1 2 2 k k n -
where the j's are integers. If p(~) represents the probability that any step results in a vector displacement,
s, by a walker then the {P (s)} satisfy the recursion - n -relation
P (s) = L: p(s-s')P (s') n+l - s' - - n -
The Fourier expansion of p(s), termed the structure function
of the walker, is 2nir.s/N
~(2nr/N) = E p(s)e --s -
When walkers are conserved during the walk Ep(s) = 1 and s -
X(O) = 1. The random walk generating function <» n
P(s,z) = Ez P (s) o n -
is used and so by substitution
-k -2nir.s/N P (~, z) = N E e - -
r 1 - z)_(2nr/N) Considerable development of this model leads to a
number of results amongst which the following is of interest here.
The average number of distinct lattice points visited
after n steps S is given for 1, 2 and 3 dimensional lattices by
1/2 lD s - ( 8n/n)
n 2D s - nn/logn
n 3D s - n/P(O,l)
n where the value p ( 0, 1) depends
1.4 for cubic lattices. These
on the lattice type and is -
results are of interest in that
crystalline materials often have anisotropic properties and
where the degree of anisotropy is large the crystals may be best
regarded as one or two dimensional materials. The relative
12

values of S given here can therefore be used as n
discriminators of dimensionality when the physical properties involve processes which can be described by a random walk.
2.1.6 BARRIERS TO ELECTRON MOTION
An underlying assumption made in random walk theory
is that all sites are equally probable. However, in any
molecular crystal with its hierarchical environment and
lattice imperfections there are time and positional
influences prevailing on an electron which may not allow equivalent lattice points to be equally attractive to it.
In some instances an electron has its walk terminated - it
is trapped. In other circumstances there may be barriers
to certain lattice points. To cross these barriers thermal
excitation or tunnelling may be necessary. Such behaviour
has been examined by Chojnacki, Lorenz & Pigon (1973) in which
they account for changes in resistivity, activation and current
voltage characteristics in a doped crystal. The barrier height
is the ionisation energy of the donor (anion), and the barrier
has a width spanning an unspecified number of unit cells. The conductance is the product of the probability of
crossing the barrier and the barrier width 1. The
probability of crossing the barrier is the sum of the
probabilities of jumping and tunnelling the barrier which gives
the conductivity + -
a~ [exp(-E/kT)+(l-exp(-~kT)) (D- D )]1 +
where sis the effective height of the barrier, D and D are
the transmission coefficients in the direction of the
electric field and opposite to it. Values of 1 estimated
using rectangular barriers in the model are of the order of 0
35 A for various levels of dopant. This model assumes that
the effective mass of the electron is its free rest mass.
Barrier widths comparable to the intermolecular spacing are equivalent to a much greater effective mass (Fleming
13

197 2) •
2.1.7 ELECTRON INTERACTIONS
A further consideration is necessary when charge
carrier motion is depicted as being a random walk process.
There may be a large number of walkers who may interact, such interactions being called space charge effects. That is, the walkers may impose an ordering on each other, thereby reducing the random element of their individual
behaviour. Space charge effects, limited diffusion and trapping
can be either utilized, or avoided if possible, in experiments designed to examine charge carrier motion.
2.1.8 HOPPING MODELS
A number of organic charge-transfer crystals have been
studied in which mechanisms involving 'hopping' have been
discussed. The theory for models describing this (Mott, 1969, Brenig, Dahler & Wolfle 1971) is based on the
assumption of hopping processes between localised states distributed randomly in "energy-coordinate-space". This, (Brenig, Dobler & Heyszenau 1972) yields the following
temperature dependence for three dimensional hopping 1/4
lncr (T) = lna - (T /T) where T = ~3/N • 0 0 0 f
N is the density of states at the Fermi level and o.s~ is f
an average range parameter of the wave function of the
localised states. In the case of TCNQ complexes a twodimensional hopping model gives reasonable agreement with
the data. The behaviour is anisotropic and T0
= ~11 ~,~../Nf
where l}i .• and u.l are range parameters parallel and perpendicular to the chains of the complex. It is
interesting that subsequently Ehrenfreund et al (1972)
re-examined this data and showed that a one-dimensional
hopping model fits it even better than the two
dimensional one. This highlights the attention being paid
14

to this class of material. It is clearly of interest to
correlate the molecular structure of crystalline materials with their physical properties, particularly where the
dimensionality of the physical processes differs significantly from three.
2.1.9 POLARONS
A moving charge carrier may interact with the crystal
lattice and its vibrations. This can be described in terms
of a polaron. In polar crystals, the interaction of
an electron, with the polarization field generated by the lattice vibrations, increases the effective mass to m , the
1 polaron effective mass (Sewell 1958).
A tight binding model describes the situation in which a high effective mass pertains. As phonons exhibit a band
structure, their interactions with other charge carriers
leads to a band structure for polarons. The effective mass
is temperature dependent 0
m = m exp(T/T ) for kT >> hv, he/a 1 1 1
where h is Planck's constant, c the speed of light, k
Boltzmann's constant and a, a lattice parameter. This
equation describes the fact that the random thermal
motion of the ions opposes the transfer of their mean
positions. As a consequence of this the energy to induce current flow of polarons will exhibit this same temperature
dependence.
2.2 EXCITONS
2.2.1 INTRODUCTION
In superconductivity pairs of electrons are attracted to one another via the crystal lattice and move in concert.
An exciton on the other hand is a similar type of
phenomenon but with one of the electrons replaced by a hole. A hole has some of the same properties as a positron - positive charge in particular - but the
15

recombination of an electron and a hole is a much lower
energy process than an electron-positron interaction.
A number of scientists are searching for a material which makes a transition to a superconducting state at ambient temperatures. The exact nature of such a
transition is unknown but it would appear plausible that it could be from exciton to bound electron pair, or perhaps an
ordering, of pairs of excitons, giving paired electrons and
paired holes. The contribution of holes to superconductivity has not been widely reported nor perhaps investigated. If the transition is an ordering of excitons, resulting in
paired electrons, then a new approach to this interesting search could be made.
Currently the electrical properties of organic
crystals and charge-transfer complexes are under intensive
investigation, in order to widen our knowledge of their
electrical behaviour. It is becoming more widely
recognised that charge carrier motion is the behaviour
of electrons and holes both singly and jointly as excitons.
Excitons can be classified as singlet or triplet, depending
on their spin state. Their investigation and use as probes
(Mohwald, Erdle & Thaer 1977) is accordingly carried out using the techniques of electron spin resonance (ESR) •
2.2.2 EXCITON GENERATION
Excitons may be inherently present in a given
crystalline material, but they may be generated by
various techniques. In anthracene, which has been the most
widely investigated organic compound in this field, triplet
excitons (Avakien & Merrifield 1968) have been generated directly by absorption of 'red' light (ruby laser), by
recombination of injected holes and electrons in the crystal,
as well as by bombardment by X-rays, and high energy
particles. Singlet excitons can be generated optically and a proportion of these decay into triplet
16

excitons by intersystem crossing. Such singlet excitons
are generated by a double absorption of laser light (Singh et al. 1965), or by an intense xenon flash lamp. The
delayed fluorescence observed where double-photon absorption
dominates, is proportional to the fourth power of the incident laser intensity. The double-photon generation of excited singlets is a square dependent process, as is the
bimolecular triplet-triplet annihilation resulting in the delayed fluorescence.
2.2.3 EXCITON DIFFUSION
In order to examine exciton diffusion, triplets are
generated by irradiating a small area of the crystal, in such
a way that during their lifetime a substantial fraction of
them will have time to move outside the illuminated region, and, as a consequence to a region of lower excitation concentration. Such a decrease in concentration leads to a decrease in the subsequent luminescence intensity.
Ern et al. {1966) carried out dynamic experiments in which
the excitation was both temporally and spatially
inhomogeneous providing a full test for the time-dependent -5 2
diffusion equation. This gave a value of 2xl0 ern /sec for
the triplet exciton diffusion constant pertinent to
the ab plane of anthracene crystals at room temperature.
Determination of scattering time and r.m.s. exciton 0
velocity yields a scattering length of about 0.1 A, from
which it may be concluded that exciton motion in anthracene, is
best viewed as a hopping process making very strong interactions
interactions with the lattice.
2.2.4 EXCITON MOBILITY
Excitons may be mobile or immobile and it has been
shown {Haarer & Wolf 1970) that the exciton hopping
frequency u can be estimated from the line narrowing
17

(observed in ESR) accotdin~ to
u - ~ ( llH J.m ) j T AH
whi~e T is the life-time of the triplet state and where llH and llH are the line widths of the immobile and the mobile
triplet states, respectively. Such techniques as this enable
comparison of exciton diffusion mechanisms in different
crystals. Mohwald and Sackmann (1973 & 1974) have published
results for biphenyl-TCNB and anthracene-TCNB, finding
fundamental differences in the triplet exciton diffusion
processes therein. Anthracene-TCNB triplet excitons perform
a two-dimensional diffusion in the planes of the crystal
containing the donor molecules. The exciton diffusion
coefficient D is estimated according to 2
D = 1 u./2
where 1 is the averageJdistance between two donor molecules
in the donor plane.
2.2.5 EXCITON INTERACTIONS
Triplet excitons may interact with each other
giving an annihilation. They may also interact with charge
carriers releasing trapped carriers and enhancing
photoconductivity. Interactions with photons leading to
photoconductivity have been measured by Holzman et al. (1967).
The effects of magnetic fields on the rate of triplet-
triplet annihilation have been studied (Merrifield 1968). The
anisotropic behaviour is explained in terms of the nine
possible spin states of a pair of triplet excitons, the
rate of annihilation being proportional to the fractional
singlet character of each state.
2.2.6 EXCITON TRAPS
Triplet excitons may be trapped by impurities (Lupien
and Williams 1968) and by dislocations (Arnold, Whitten & Damask 1970). The existence of traps is demonstrated by
18

delayed fluorescence and triplet lifetime measurements.
Dislocations are easily induced by bending the crystal. For an unbent sample at room temperature the fluorescence in
anthracene is found to be proportional to the incident
excitation intensity, which is in accordance with
fluorescence arising from triplet-triplet annihilation.
The dislocation density N is proportional to the crystal
curvature r, and is given by N = 1/rb where b is the Burgers vector, and it is found that the increase in the
fluorescence decay constant is proportional to N. In anthracene four types of trap are postulated to exist and
to fit the fluorescence data they have depths between
0.02 and 0.3 eV.
2.3 PHOTOCONDUCTIVITY
2.3.1 INTRODUCTION
Photoconductivity is an interesting phenomenon in that it has
a number of very practical applications, principally in light metering devices. Its simplest description is the generation of
an electric current in a solid upon incidence of a photon.
Consider an organic crystal such as anthracene. Electrons need to
be promoted to the highest level in the hierarchical environment,
that is to crystal orbitals, or exciton states. So it is not
surprising that to a first approximation there is a threshhold energy for photoconductance, of wavelength typically about
0 4000 A, and that in the presence of different gases (Kepler
1960) - the molecules of which can form or cause to be formed
exciton affecting defects - the photoconductivity is different. The absorption of the incident photons is not necessarily
directly by the defects, nor the excitons, but in
anthracene (Bree, Carswell & Lyons 1955) is by the
anthracene molecules themselves, as the spectral dependence of
the photo-current generated follows pretty much the anthracene
absorption spectrum. Photoconductivity arises therefore as a two step process (Melz 1972), an electron excitation
19

to an excited state and thence to a continuum state. It is
generally agreed (Hughes 1971, Melz 1972, Chance & Braun
1973) that the free carrier yield in anthracene is
controlled by geminate recombination. In 1938 Onsager
modelled geminate recombination. The predicted electric
field dependence of the free carrier yield, for a
spherically symmetric distribution of geminate pair
configurations is linear and has a slope to intercept
ratio 3 2 2
S/I = e /8nEE k T 0
where e is the high-frequency dielectric constant and £
the permittivity of free space. 2
0 Batt, Braun & Hornig (1968)
observed the 1/T dependence and the linear field
dependence of the carrier quantum yield 3 2 2
~ = A(T) [1 + (e /2Dk T )E]
Geminate recombination describes the immediate
recombination of a proportion of the electrons and holes
following excitation, the photocurrent being due to
those which do not recombine. The electrons and holes with
a separation of r diffuse with the distribution g(r,Q), the
angle e being the angle made by~ with the electric field E.
The photo-generation efficiency for a particular electric
field E is 3
~(E) = ~ Jf(r,9,E)g(r,9)d r. 0
Melz (1972) studied photogeneration in the polymer
trinitrofluorenone-Poly(N-Vinylcarbazole). A film of this
substance was deposited on an anodized aluminium substrate,
and exposed to light pulses of 4 ~sec duration. In this
experiment
~(E) = C8v/eF where C is the capacitance/unit area of film, 8v the
potential drop due to the light impulses and F the 2
photons/em due to one light pulse.
Chance & Braun (1973) have also observed low-field
20

yield reductions which can be completely understood in terms
of the recombination of free carriers with the opposite sign, being trapped charges left behind in the excitation
region. As electrons migrate across the crystal they take up
a Boltzmann distribution n(x) near the crystal surface (x=O)
n(x) = (eE/kT)Ne -xeE/kT
where N is the number of free holes/em of irradiated
crystal surface. At time t = 0 the free electron distribution
can be assumed to be
-k X 0
C(x,O) = k e 0
where k is the absorption coefficient of the excitation 0
light. At time t the differential equation governing the
electron distribution is
dC(x,t)/dt = -Yn(x,t)C(x,t)
Y is the second order rate constant for the free electrontrapped hole interaction and n(x,t) the distribution of electrons at time t is given by
n(x,t) = NeE/kTe[-eE/kT(x+uEt) This is arived at by, imagining, moving the distribution of holes through a stationary distribution of electrons at u, the
speed of the electron mobility. Solution of these equations
leads to a prediction in agreement with the observed
quantum yields in the experimental range of the electric fields used.
Bergman and Jortner (1974) have made a review of photoconductivity in anthracene and studied its dependence
on incident intensity over a range of wavelengths. The
description of organic crystals must be hierarchical rather
than in terms of 'narrow' conduction and valence bands. Frenkel exciton states and the band states only
constitute the zero-order levels of the system. 'Coulomb
interactions couple the exciton and band states, non
adiabatic intramolecular terms couple vibronic components
21

of exciton states which correspond to different electronic
configurations, while nuclear coupling terms involving both
intermolecular and intramolecular nuclear displacements
will couple different vibronic components of a single
electronic configuration of a neutral or a band state.' 0
In the wavelength region 4400 - 4600 A one photon
excitation yields stable singlet excitons from which charge
carrier generation is either by exciton photo-ionization or
exciton collisions. The charge carrier yield m vs I 0
dependence varies as follows. In the region 4600 - 6120 A two
photon excitation takes place and the m vs I dependence is 0
mechanism dependent. In the region 4600 - 6120 A the dependence
is quadratic for low light intensities and quartic for
high intensities. These dependences can be assigned to the
production mechanisms - autoionization of metastable
excitons and collision ionization of excitons respectively. . 0 .
From 6180 - 6943 A a cubic m vs I dependence is observed,
attributable to photoionization of two-photon-excited
singlet states. As the m vs I dependences vary it is not
particularly meaningful to make m vs u measurements across
the experimental range, but rather it is better to examine
the cross-sections for the various photogeneration
mechanisms.
2.3.2 CHARGE CARRIER MOBILITIES
The mobilities of both holes and electrons in 2
anthracene are of the order of lcm /V-sec at room
temperature (LeBlanc 1961), and vary with the temperature -n
as T , 1 < n < 2. To model carrier mobility a tight binding
approximation is made using Bloch momentum eigenfunctions of
the perfect crystal, and the bands for electrons and holes
are anisotropic and of width - 0.56kT at room temperature.
Models of isotropic carrier scattering may assume either (i)
r(~) = -r constant free time, or (ii) T(~)xl~<~) I = )._ constant 0
free path for which the components of the mobility tensor
22

P,. are eT <v,v.>/kT and eA<v,v./jv(k) 1>/kT where the v, are l.J 0 l J 1 J - - 1
the components of ! and the brackets represent a statistical
average over the band. The velocity v is defined by
h!(~) = ~E(~)ja~ where k is the wave vector
These models lead to -12
l.SxlO sec and for A of
of the wavefunction ¢ • k
values for• of the order of 0 0
30 - 80 A. The drift mobility ~ of
electrons and holes may be measured by injecting charge
carriers into a crystal surface and measuring the time
to traverse the crystal t. 2
~ = d /Vt where d is the distance traversed and V the applied voltage.
When continuous carrier motion occurs space-charge-limited
currents may be observed, depending on the current density. If space-charge effects are to be studied then a continuous
source of carrier generation is used, whereas if they are to
be avoided then a pulsed source is used giving transient
currents to study. Such excitation is commonly made by pulsed
laser with pulses of duration < 5 ~sec.
The measured drift velocities in anthracene (Kepler &
Hoesterey 1974) do not show any deviation from a linear4
dependence on the electric field in the range 0 - 16xl0 V/cm.
The hole mobilities are 2.4 times greater then the electron mobilities.
2.3.3 MOBILITIES IN CHARGE-TRANSFER COMPLEXES
In the case of N-Isopropylcarbazole-Picryl
chloride (NIPC) (Sharp 1967) the Picryl chloride is a good
electron acceptor and acts as a trap. This is illustrated
by comparison of mobilities in a 1% and 50% picryl chloride
mixture with NI. The ratio of the linear densities of traps is 3.0 whilst the inverse ratio of the measured mobilities
is 3.6, suggesting a simple hopping model with the time
spent in traps being porportional to the trap strength. The
temperature dependence is exponential,
23

E/kT 1l = 1l e
0 the value of E determined yielding a trap depth of 0.1 eV.
The space-charge-limited current for one carrier in a
trap-free insulator is given by the expression (Rose 1955} -1~ 2 3
I = 4.4xl0 k V /d amp/em
where k is the dielectric constant of the insulator. The 2 3
V and 1/d dependences are obtained in NIPC at low voltages
and high light intensities. At lower light intensities the space-charge-limited condition is alleviated and an ohmic
behaviour is realized.
2.3.4 ELECTRON MOBILITY IN PHENANTHRENE PMDA
This has been studied in detail by Haarer, Mohwald & Castro -2
(1975). They find a mobility of 10 em /Vsec at room temperature -1
and an activation energy of 880 em , with an exponential
temperature dependence -fiE/kT
1l = 1l e 0
0 in the experimental range 250 < T < 450 K. The magnitude of 1l is
0 anisotropic, its value when being measured parallel to the stack
axis being approximately three times that measured perpendicular
to the stack axis. The anisotropic features of this material can
be attributed to the intermolecular interaction along the stack
axis being considerably larger than the interaction perpendicular
to the stack axis. Such features are triplet exciton energy transfer and charge-carrier transport. Haarer & Mohwald (1975)
report a field-induced charge-carrier trapping process, and explain the magnitude of the electric field at which this occurs in terms of the quasi-one-dimensional nature of phenanthrene PMDA.
The application of a hopping model as used by Kepler &
Hoesterey (1974} for anthracene seems justified. In a system with deep traps the mobility decays exponentially with time and the
characteristic decay time is called the CC-trapping time. In
phenanthrene PMDA the observed trapping processes follow a single
exponential decay with a constant decay time perpendicular to the stack axis, and with a decay time which gets shorter at higher
24

fields along the stack axis. This field dependent reduction of
the CC-trapping time is referred to as field induced trapping.
2.4 ESR SPECTROSCOPY OF CHARGE-TRANSFER COMPLEXES
The so-called weak CT-complexes, which are non-ionic in their
ground states, contain considerable ionic contributions to their lowest excited singlet and triplet states. The phosphorescent
spectra of these states are generally very broad and difficult to interpret, so, for observations of triplet behaviour it is more
appropriate to use ESR techniques. They enable measurement of the dipolar interaction of the triplet electrons as characterised by the ZFS-parameters (zero field splitting) D and E. The
dipolar interaction strongly depends on the distance between the
two unpaired spins. Such measurements provide a technique to establish the charge-transfer character of triplet states. When the triplet molecules are delocalized, over more than one molecule, the D-value is significantly reduced. Mohwald and Bohm (1976) report measurements on naphthalene tetracyanobenzene N-TCNB crystals doped with different guest acceptors. In the case of the
guest molecule hexacyanobenzene (HCNB} the hyper-fine splitting (Hfs} observed, is shown to be due to the ~ and a-protons of only
one naphthalene molecule, with coupling constants of 1.9 and 2.2
Gauss respectively corresponding to a CT-character of 33%. This
indicates a breakdown of the inversion symmetry in the triplet state, of the crystalline complex, the most probable explanation for this being, that the exciton-phonon interaction tends to stabilize in an asymmetric triplet state.
In the case of N-TCNB doped with pentacyanotoluene (PCNT) the Hfs are also well resolved and the CT-character is 26%. The
complex Napthalene-s-trinitrobenzene (TNB) forms a trap in the N-TNB doped crystals described by the ZFS parameters
-1 - -1 * -1 D = + 0.0670cm 1E = + O.Ol34cm 1D = 0.0709cm
which are rather high for a CT-complex, indicating a triplet state of low polarity. The Hfs shows 25 lines due to the coupling of three equal nitrogen atoms with three equivalent protons.
25

Napthalene-Chloranil shows an absorption and emission line, but
without any resolvable Hfs, indicative of a very short triplet lifetime (< lms) so that the Boltzmann equilibrium is not
established among the triplet sublevels. Using the D-value of chloranil the CT-character obtained is 76%, that is,
extremely polar. The Biomolecules riboflavin and flavin-adenosine
dinucleotide, which were also studied as guests, show small Hfs,
indicating an only slight interaction with the host donor
napthalene, suggesting thereby that CT-crystals are suitable matrices, in which to study ESR-spectra of large oriented molecules.
Dalal, Haarer, Bargon and Mohwald (1976) have examined
anthracene and phenanthrene as guest donor molecules in naphthalene TCNB. In the case of anthracene the degree of charge
2 transfer (b ) , in the triplet state, is almost identical with that
2 in the isolated molecule (b = 0.05), whereas phenanthrene shows
2 a significant degree of charge transfer (b = 0.47).
Triplet excitons, have been examined in 1,4 dibromona~halene crystals. Such crystals are stacked in linear arrays with the molecular planes being parallel, and so have similar
properties to charge-transfer complexes. The molecules are translationally inequivalent, and so the ESR spectra of two pairs
of lines, corresponding to this, yield only information on the
exciton diffusion perpendicular to the stack axis. Mohwald and
Sackmann (1973) have studied biphenyl-TCNB, in which the molecules
are translationally equivalent (triclinic space group). This
being so they cannot distinguish between behaviour parallel and
perpendicular to the stack axis. They are able to conclude, that
the lowest excited triplet state is a CT state, as the average ZFS
parameter of the crystal triplet state is considerably smaller
than the D-values of the donor and the acceptor, respectively. There is a strong narrowing of the ESR lines with increasing
0 temperature above 77 K, at which temperature the lines appear
26

to have been inhomogeneously broadened by the hyperfine
interaction of the triplet state electrons with the nuclei of the 0
donor and the acceptor. Above 77 K, at which temperature the
triplet excitons are nearly immobilized, the exciton motion
causes a random modulation of hyperfine interaction, leading to a motional narrowing of the ESR lines. Haarer (1969) has related the limiting line width ~H at negligable exciton motion to the
m 1/2 hopping frequency of the excitons by ~HN = ~H
m where N is the number of hyperfine states seen by a diffusing
exciton during its lifetime. Anthracene-PMDA (Haarer and Karl) has D and E values which clearly indicate that the ESR signal is
due to the locally excited donor molecule (anthracene), rather
than a CT triplet state. The ESR spectrum has both emissive and absorptive lines indicating a high degree of spin polarization.
ESR has been used by Erdle and Mohwald {1977) to investigate
an orientational phase transition of anthracene in the CT-complex 0 0
anthracene-TCNB. Above 196 K there is a libration of + 9 by the
anthracene, which freezes in during the transition, the crystal splitting into two sublattices. The triplet exciton being solely
located on the anthracene molecule is a good indicator. At high
temperatures a single narrow line is observed, which, at lower 0
temperatures (<196 K) has split into broad lines corresponding to
the two differently oriented sublattices. The phase change has
both first and second order components and is described by an even
function of the sixth order.
27

CHAPTER 3
EXPERIMENTAL PROCEDURES
3.1 PRELIMINARY CRYSTALLOGRAPHIC STUDIES
For each compound in a preliminary study, approximate
cell constants were obtained using a precession camera, the
crystal being mounted on a fine glass fibre using vaseline or
araldite. This enabled examination of zero level and upper
level reciprocal lattice nets to establish the space group
symmetry. Nicely shaped single crystals were selected in
each case particular care being taken to avoid twinned
crystals. The crystal dimensions of the crystal chosen for
data collection were measured, and its faces indexed, for
purposes of absorption correction calculations, should they
prove necessary.
3.2 DIFFRACTOMETER DATA COLLECTION
A suitable crystal was selected and two reflections,
identified on a precession photograph, were located with the
diffractometer thereby roughly establishing the crystal orientation. Subsequently, at least 12 higher angle reflections
of reasonable intensity were located by rapidly accumulating
intensity data from a thin shell of the reciprocal lattice.
These were then accurately centred using a 5.0 mm
diffracted beam collimator (DBC) under the control of the
computer and least-squares refinements of the cell constants
and the orientation matrix carried out. 0
The tube take-off angle was always maintained at 3.0
and tube alignment procedures (Furnas 1957) were such as to
obtain an X-ray beam of uniform intensity in the various
equivalent positions in which reflections can be measured.
The crystal mosaicity was measured by making open-counter
w scans using strong, low angle reflections. If the peak was
split or too broad the crystal was rejected.
The 9-29 scan technique under computer control was
28

used to collect the intensity data. The incident beam was filtered using the appropriate ~ filter. Symmetric step scans, centred on the reflection's position calculated
using the wavelength of the X-radiation, were carried out along with stationary counts of background radiation at both
ends of the step scan. The total time for each background count was generally set to be half that of the integrated step scan.
The crystal to DBC distance was 23 em and all intensity data
were collected using a 5.0 mm DBC.
When coincidence losses were significant the reflection intensity was re-measured with the incident beam being attenuated by suitable metal foils. These were calibrated at the end of the data collection by making long
measurements of selected high intensity reflections. The calibration obtained varied slightly between crystals of
different compounds.
Specific details and any changes or modifications are given in the discussion of the individual structures in later chapters.
3.3 DATA PROCESSING
The method of data processing was similar to that of
Corfield, Doedens and Ibers (1967). Throughout the scan range,
of any given reflection, background scattering is considered
to be a linear function of 9. The measured intensity and
estimated standard deviation are then formulated as
where
I = C-l/2(t /t (B +B )) c b 1 2
2 2 1/2 = [ ( c + 1/4 ( t It ) ( B + B ) + (pI) ] c b 1 2
cr(I)
I = intensity
C = scan count B = first background count
1
29

B = second background count 2
t = scan counting time c
t = background counting time b
cr(I) =estimated standard deviation in intensity
p (Grant, Killean & Lawrence 1969) is a factor
introduced to avoid the overweighting of strong reflections and is of the order of 0.05.
The value of p was varied, so that the error in an observation of unit weight, showed minimal dependence on the
magnitude of structure factors and Bragg angle, when refinement of the crystal structure was completed (Cruikshank 1965).
The weighting function used in the analyses was 2 2 2
w = 4F /[cr(F )] 0 0
2 where F is the observed structure factor and F = ki where
0 0 k is a constant for all reflections.
When equivalent reflections were observed, or the same one repeatedly observed for some reason, the estimated standard
2 deviation esd cr(F ) was taken either as the average of the
individual esd's, or, as a value estimated from the range
of intensities of equivalent reflections, whichever was larger. Finally all esd's were multiplied by (N/OBS);/
2
where N was the standard number of equivalent reflections measured, and OBS was the number actually recorded (Robinson
& Ibers 1967).
Absorption corrections, where they proved necessary,
were applied by using an analytical integration program ABSORB Templeton and Templeton (1973). Input to the program utilises
perpendicular distances of crystal faces to an arbitrary origin in the crystal and the Miller indices of those bounding faces.
30

CHAPTER 4
SOLUTION AND REFINEMENT PROCEDURES
4.1 INTRODUCTION
Structures described in this thesis were solved by
direct methods and Patterson techniques, and were refined by
least-squares. Details of the methods used are outlined in
this chapter whilst any details peculiar to individual structures are discussed later.
A crystal structure consists of an ordered set of atoms of various masses arranged in space, with certain periodicities defined by unit cell parameters. Generally the symmetry operations of the space group relate the asymmetric units
within the unit cell to each other. Crystallographic description, is usually confined to the unit cell, and primarily to the
asymmetric unit.
4.2 ELECTRON DENSITY AND STRUCTURE FACTORS
The electron density is given by:
1 I I -2wi(hx+ky+lz) ia p(x,y,z) = - E E E F{hkl) e e
v h k 1 2 2
2 2 -Bsin 9/~ where IF(hkl) I = ~ f . e , F(hkl) is defined as
1 01 the structure factor for the reflection from plane hkl, and f
oi is the scattering factor of atom i at rest.
2 B = 8wu is the isotropic temperature factor.
u is the root-mean-square amplitude of vibration.
e is the angle of incidence of the x-rays.
a is the phase angle {value 0 or w in centro-symmetric structures).
The electron density of each atom is represented as being spherically symmetric about its nucleus. Vibrational motion may
be represented by a single parameter B as isotropic motion, or by six parameters describing an ellipsoidal anisotropic motion.
31

2 The measured intensities are proportional to IF(hkl} I whilst
the phase changes on reflection are not measurable. The phases
are functions of the atomic positions and their elucidation is a
primary problem in crystallography.
4.3 PHASE DETERMINATION
4.3.1 PATTERSON METHODS
A formulation useful in determining atomic positions
is the Patterson Function (Patterson 1935).
1 1 1 P(u,v,w) = f F f p(x,y,z) p(x+u,y+v,z+w)Vdxdydz
0 0 0
where u,v,w is a point in Patterson-space and x,y,z a point in
crystal-space. (V is the crystal volume). This formulation can
be written in terms of a Fourier series. 1 2 -2~i(hu+kv+lw}
P(u,v,w) = - ED: IF(hkl) I e v hkl
This expression can be evaluated at any point u,v,w since it
is solely dependent on I(hkl) and independent of any phase angle.
Its topological features are interpreted in the following manner:
1. The magnitudes of local maxima are proportional to the
product of the numbers of electrons in the pairs of atoms
between which the vectors are formed. At the origin there are the contributions of N vectors (1
from each of the N atoms to itself). The remaining maxima consist of two sets of N{N-1)/2 peaks related to each
other by a centre of symmetry. As the density of peaks in a Patterson map is N times that of the molecular structure it represents, peaks often
overlap and are not easily resolved. Their resolution can
be improved by "sharpening". In a sharpened model the atoms are regarded as charges of equal value, located at
particular points.
2. Local maxima represent interatomic vectors.
The interatomic vectors fall into two classes, intramolecular and intermolecular. The intermolecular vectors
32

can be further classified as to whether or not they are
characterized by a difference between the symmetry operations of the real space group. Those that are,
are termed Harker vectors. Harker vectors can be used to determine atomic coordinates, and, as a consequence, phases. In practice the Patterson peaks of greatest magnitude, representing vectors between the heavier atoms of a
structure, are regarded as the most reliable. In a centrosymmetric structure the structure factor
2 2 -B.sin 9/}..
F(hkl) = Ef e J cos2~(hx,+ky,+lz,) If atom m i~ 1 heavy atom f isJmuc~ la~ger than f (j~m) and then
2 2 -B sin 9/ ~
m
m j
F(hkl) - f e cos2~(hx +ky +lz ) m m m m
That is, the sum is dominated by a single large term. The
sign, and hence phase of F(hkl) is determined by the sign of
cos2~(hx +ky +lz ) • This approximation defines, what is m m m
known as, the heavy atom method.
3. The Patterson map is comprised of N images of the molecular
structure, each displaced different distances from the true
molecular site. Once one atom position is obtained analytically, using
Harker peaks for instance, then the translation of the Patterson map, by a vector consisting of that atom's
coordinates, will bring one of the N images to the true
molecular position. This is known as a method of
superposition.
In summary, the measured intensities determine the Patterson peak
distribution and the intermolecular vectors thereby obtained, can enable the establishment of atomic positions and structure factor phases.
33

4.3.2 BASIS OF STATISTICAL METHODS
In the Heavy Atom Method use is made of an approximation
in which one large term dominates a summation. This concept of
a limited number of terms dominating a summation is also used in
other phase determining techniques.
Bochner and Chandrasekharan (1949) have shown that for structure
factors: 2 1
F(x) = V r r r F(x)h-h' k-k',l-l'F(x) h'k'l' '
Sayre (1952) realised that the electron density function d(X) 2
and its square d (x) are very nearly alike in shape and that it
is possible to write 2
F(p )hkl= ShklF(p)hkl
where S is a function to account for the change in shape. hkl
2 1 Whence F(p ) = -s E r E F(p) .F(p)
hkl v hkl h'k'l' h-h',k-k',l-1' h'k'l' In particular, if, for h' = h", the values of F(p)
h-h" k-k" 1-1" and F(h"k"l") are large, then their product will tend fo do~inate the summation and so the phase of F(hkl) is in the main determined
by them. The phase determining technique based on this equation
is an iterative process, successive steps determining phases which
are used to help determine further phases. Reliable methods have been developed, employing various approximations, and
incorporating statistical techniques which are used to indicate that certain phases are true.
4.4 STATISTICAL METHODS IN PRACTICE
4.4.1 INPUT DATA
Normalised structure factor magnitudes lEi known as E values 2
are used, and are calculated in such a way that <IE I> = 1.
They are defined by the expression:
34

I I [exp(A +A sin
29)
11211F I
E = o 1 o 2
1:Z::f, sin9/A 1
Where IF I is the measured structure factor, and f, the atomic 0 l
scattering factor, A and A being defined by the Wilson Plot 0 1
Method (Wilson 1942) using the expression:
2 2 loge<~m N i sin9/A>q = A
0 + A
1<sin 9>q
2 <Em IF I >q
R o
Here R is the number of reflections in range q, m is the multiplicity and 1: is a factor to allow for systematic absences.
Overall temperature (B) and scale (k') factors are
obtained as
2 B = A A /2
1
k' = exp(A /2) 0
4.4.2 USE OF MULTAN
Program MULTAN (Main, Woolfson & Germain 1971 & 1974) was used.
The crystal structures described herein all have centrosymmetric
space groups. MULTAN produces multiple solutions based on
different sets of phases for a set of starting reflections.
Frequently the solutions are the same, only apparently differing,
due to different centres in the unit cell being taken as the origin.
Up to 500 reflections with E > 1.4 were used. In each case
2000 triplets ¢ , ¢ , ¢ were sought and obtained. The set of h h' h-h'
phases having the best values for the figures of merit was
always the correct set. MULTAN has three stages, SIGMA2,
CONVERGE and FASTAN.
(i) SIGMA2 sets up all z:: phase relationships. 2
J{Jh ~ J{Jh' + ¢h-h' and these are given weight
35

-3/2 k = 2 a a IE E E I hh' 3 2 h h' h-h'
N n where a = L: z
n j=l j
(ii) CONVERGE determines the origin defining reflections and a
small group of other reflections which appear to provide a good starting point for phase determination.
(iii)FASTAN iteratively determines the final values for each set of phases using a refinement technique utilising
4.5
4.5.1
a tangent formula:
tan~ = w' cos{~ + h w E E
h' h h-h' h h-h' h'
L: w w IE E lsin(~ + ~ } h' h' h-h' h' h-h' ~h' ~h-h'
rzjh-h')
where w = tan(l/2 h) h
2 2 1/2 a, = IE I (T + B )
h h h h
MODEL BUILDING AND REFINEMENT
STRUCTURE FACTOR CALCULATION
B h
For N atoms located at positions (x,,y,,z,) the structure factor for the reflection from the planeJhkf i~
2 2 -B sin 9/A -2Tii(hx + ky, + lz )
N j j J j F(hkl) = L: f e e
j=l oj
in which atom j has vibrational motion B,, and scattering J factor value f . appropriate to the angle of the reflection
OJ -1 9 = sin . ~/2d(hkl)
The tabulated vales for the scattering factors used were those of Cromer and Waber (1965} and for hydrogen those of
Stewart, Davidson and Simpson (1965). For heavy elements in which the X-radiation falls near a natural absorption frequency, an anomalous phase change occurs and the
scattering factor becomes complex. The total scattering
36

is then expressed as f = f + Af' + iAf"
0
4.5.2 LEAST-SQUARES REFINEMENT
It is desirable to obtain atomic positions which minimise the
differences between the structure factors calculated from them
and the observed structure factors. Although the structure factors are not linear functions of the atomic parameters in the least-squares refinement the approximation is made that the equations are linear in the differences between the true
values of the parameters, and the a~proximate values used. The
quantity minimised is w(IF I-IF I> where the weighting function 0 c
2 2 2 w = 4F I cr (F ) •
0 0
The conventional R factor R , and the weighted R factor R 1 2
are defined as
Rl = L I I F I - I F I I I L I F I 0 c 0
2 2 112 R2 = ( E w ( I F I - I F I ) IE w I F I )
0 c 0
4.5.3 REFINEMENT OF HYDROGEN ATOM PARAMETERS
Churchill (1973) surveyed recent crystallographic papers and
concluded that •refinement of positional (and thermal) parameters of hydrogen atoms can be a meaningful procedure and leads to
self-consistent results even in the presence of heavy atoms•. Stewart, Davidson, and Simpson (1965) have shown that on theoretical grounds the use of free-atom spherically symmetric scattering factors for hydrogen, results in a reduction
0 . of 0.09 A for C-H, N-H and 0-H bonds determ1ned by x-ray
diffraction.
In this work hydrogen atom parameters have been
refined in those structures where such refinement did not
make the exercise too costly, otherwise they have been
included in calculated positions.
37

4.5.4 DIFFERENCE FOURIER SYNTHESIS
The electron density is the Fourier Transform of the
structure factors. Differences in the electron density distribution between the crystallographic model and reality
can therefore be expressed throughout the unit cell in terms of the observed and calculated structure factors F and F as
0 c 1 ia 2~i(hx+ky+lz)
p. (x,y,z) =-Z:H(IF I-IF l>e e dlff v hkl 0 c
This function reveals electron density as positive peaks. Its
main use is to locate atoms that have not already been found
and to assist in the improvement of the positioning of others.
It requires a set of phases which are predominantly correct.
The elucidation of further atomic sites leads to improvement of the set of phases. It yields positions which are good first
estimates for refinement of atomic parameters by the method of least-squares.
38

CHAPTER 5
THE CHARGE-TRANSFER COMPLEX PHENANTHRENE PYROMELLITIC
ACID DIANHYDRIDE
5.1 INTRODUCTION
In the complex phenanthrene-PMDA, phenanthrene acts as an
electron donor and PMDA as an electron acceptor. The physical
properties of this complex have been measured by Haarer & Mohwald
(1975). In their studies they have encountered a new phenomenon
which they have called field-induced charge-carrier trapping.
Their discussion of the photoconductivity in terms of a hopping
model enables them to argue that this new phenomenon is a
consequence of the quasi one-dimensional nature of the phenan
threne-PMDA structure.
5.2 EXPERIMENTAL SECTION
Crystals were supplied by D. Haarer. They are yellow
in colour and had been grown by the Bridgman method from
chromatographed and zone-refined phenanthrene and PMDA (Mohwald
& Castro, 1975) • Preliminary precession photographs of these
crystals exhibited monoclinic symmetry and the systematic
absences OkO k odd, hOl 1 odd uniquely indicate the space group
is P2 /c. The density of the crystal was measured by flotation, 1 -3
D = 1. 44g em Least-squares refinement of the cell constants m 0 0 as~= 7.046(3), b = 13.494(2), c = 19.396(7) A, B = 97.4(3)
- - -3 yields a calculated density of 1.44g em with four molecular
pairs in the unit cell. 0
Cu K~ radiation (~ = 1.5418 A) was used, reflections being
observed from a crystal of dimensions 0.5 x 0.6 x 0.8 mm having
the planes (012), (012), (100), (012), (211), {012) as its bounding faces.
Data were collected for one quadrant of reciprocal space with 0 e < 57 . The intensities of the diffracted X-rays were measured
0 with a 72 step single second 0.01 scan with 18 s background counts
at each end of the scan. The normal attenuators used, at times
39

proved inadequate, as the crystal was a very powerful diffractor.
Consequently additional foil was inserted into the diffracted beam and calibrated. The normal attenuation factors were 3.5, 9.5 and 33.3 and the foil gave an additional factor of 248. The
intensities of three standard reflections were monitored every
100 reflections and the data scaled accordingly. Lorentz and polarization corrections were applied and of the unique reflect
ions measured 1951 had intensities >Sa and 2105 had intensities >3a. No absorption corrections were made as test calculations
showed them to be unnecessary, the range of possible corrections
varying by no more than 5%. The linear absorption coefficient -1
is 8.8 em
5.3 STRUCTURE DETERMINATION AND REFINEMENT
As this complex is an all light atom structure direct methods
techniques were used. 25 of the 30 non-hydrogen atoms were
located by use of the program MULTAN (Main, Woolfson & Germain,
1971) using the 500 reflections with highest E values. The
positions of the remaining five atoms were calculated and three
cycles of refinement of positional and thermal parameters by full-matrix leas squares gave R = 0.203. A difference
1 Fourier calculation revealed additional electron density, near
established atomic positions, indicative of anisotropic
vibrational motion. Further refinement, with anisotropic thermal
parameters for all non-hydrogen atoms, reduced R to 0.092 and 1
refinement of the H atom positions with isotropic thermal
parameters finally gave R = 0.066. Shifts in all parameters 1
were less than 0.5 .af their standard deviations in the final
cycle of refinement.
A final difference Fourier synthesis revealed no anomalously large peaks.
5.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE
The structure of the complex consists of columns of
alternately stacked molecules of phenanthrene and PMDA. Figures
40

5.4.1 and 5.4.2 define the atom labelling scheme used throughout
this chapter. Figure 5.4.3 shows the spatial relations between the molecular pairs related by the symmetry elements of the space
group. The x axis of the crystal is described as the stack axis and the phenanthrene-PMDA repeat distance along the stack axis is
0 3.5 A whereas the perpendicular distance between these molecules
0 is 3.36 A. The angle between the normals to the molecular planes
0 of a phenanthrene-PMDA pair, shown in Figure 5.4.3 is 4.11 and
0 0 these same normals make angles of 15.52 and 14.35 , respectively,
with the stack axis. The non-parallelism of the phenanthrene and
PMDA molecules and the manner in which they overlap indicate
localized interactions (Goldberg, 1975). The extent of such
interactions is apparent in Figure 5.4.3. It is interesting to
note that when a molecular pair is viewed in projection the 0
atoms in PMDA do not lie above regions of electron density (be
they bonds or atoms) in the adjacent phenanthrene molecule. In
fact, in this view five of the six oxygen atoms lie 'outside'
the phenanthrene molecule. The sixth 0 atom lies centrally above
one of the end phenyl rings in phenanthrene. This feature is
common to complexes of PMDA with anthracene, naphthalene,
perylene, pyrene (Herbstein, 1971) and fluorene (Evans &
Robinson 1980a) • It appears then that the C atoms and their
concomitant H atoms are the atoms involved in the intermolecular
binding and charge interactions.
The bond distances and angles calculated from the atomic
coordinates (Table 5.4a) are given in Figures 5.4.1 and 5.4.2.
These figures illustrate the anisotropic thermal vibrations of the
atoms. Root-mean-square amplitudes of vibration are given in
Table 5.4b, and structure factors in Appendix Al. There is
no evidence of disorder in the structure. 0
The bond C(36)-C(46) is 1.442(3) A in length - longer
than any other c-c bond in phenanthrene. MO calculations
(Banerjee & Basak, 1975) indicate that TI-electron
delocalization in phenanthroids is the main cause of this
41

lengthening rather than steric overcrowding of H atoms.
42

Figure 5.4.1
BOND DISTANCES AND ANGLES IN PHENANTHRENE
Ct21J Ct22)
C(43) C!33}
Ct44) C(45) C!35) Cl34)
43

Figure 5.4.2
BOND DISTANCES AND ANGLES IN PMDA
0(12) 0(2)
0(11)
44

Figure 5.4.3
CRYSTAL PACKING OF PHENANTHRENE AND PMDA MOLECULES
VIEWED DOWN THE STACK AXIS AND VIEWED EDGE-ON TO
ONE OF THE PMDA MOLECULES
45

Atom
C(l)
C(2)
C(3)
C(4)
C(S)
C(ll) C(12)
C{13)
C(H)
C(15)
0(1)
0(2)
0(3)
0(11)
0(12)
0(13)
C(21)
C(22)
C(31)
C(32)
C(33)
C(34)
C(35)
C(36)
C(41)
C(42)
C(43)
C(44)
C(45)
c (46)
H (1)
H(ll) H(21)
H (22)
1!(32)
H(33)
H (34)
H(35)
B(42)
H(43)
ll ( 44)
H(45)
Table 5.4a
ATO.'IlC PARAMETERS FOR PI!E:NI\NTI!RENE PHDA
X
0.0231(3)
-0.0374 (3)
-0.0352 (3)
-0.1093(4)
-0.1099 (4)
0.0244 (3)
0.0659 (3)
0.0847 (3)
0.1613(4)
0.1592 (4)
-0.1365 (4)
-0.1505 (3)
-0.1315 (3)
0.1884 (3)
0.2011())
0.1827 (3)
0.4553 (4)
0.5257 (4)
0.5876 (3)
0.6642 (4)
0.7157(5)
0.6979 (4)
0.6262(4)
0.5695 (3)
0.4368(3)
0.3617 (4)
0.3435(4)
y
0.3929(2)
0,3199(2)
0,2207(2)
0.3282 (3)
0.1655(2)
0.1857 (2)
0.2577(2)
o. 3574 (2)
0,2487(2)
o. 4121 (2)
0. 3993 (2)
0.2342(2)
0. 0780 (2)
0,1773(2)
0.3433 (2)
0.4988(2)
0. 0894 (2)
0.0471(2)
0.1049 (2)
0.0615 (3)
0.1159 (3)
0.2193 (3)
0.6295(1)
0.5819(1)
o. 5984 (l)
0.5072(1)
0.5344(2)
0.6642 (1)
o. 7127 (1)
0.6959(1)
0,7863(1)
0.7594(1)
0.4702(1)
0.4820(1)
0. 5261 (1}
0.8225(1)
0. 8122 (1)
o. 7690 (1)
0,0734 (2)
0.1328 (2)
0.1938(1)
0.2578(2)
0. 314.1 (2)
0.3112 (2)
0.2658(2) 0.2501(1)
0.2098(2) 0.1893(1)
0,1941(2) 0.0657(1)
0.2406(3) 0.0019(1)
0,3417(3) -0.0030(2)
0.0232(6) 0.0051(1)
0.0204 (6)
0.0201(6)
0.0287 (8)
0.0220(7)
0.0246(6)
0.0205(6)
0.0206 (6)
0.0284 (7)
0.0220 (6)
0.0502(8)
0.0316(6)
0.0367 (6)
0,0446(7)
0.0316(5)
0.0342(6)
0.0266(7)
o. 0290 (7)
0.0209(6)
0.0281 (8)
0.0299 (8)
0.0305(8)
0.0261(7)
0,0181 (6)
0.0104(6)
0,0250(7)
0.0289 (8)
0,0069(2)
o. 0061 (2)
0.0122(3)
0.0113(3)
0.0046(1)
0.0056(2)
0. 0051 (1)
0.0088(2)
0.0081(2)
0.0162(3)
0.0155(2)
0. 0113 (2)
0.0125(2)
0.0116 (2)
0.0075 (l)
0. 0089 (2)
0.0056 (2)
0.0061(2)
0.0099 (2)
0.0154 (4)
0.0139 (3) 0.0083(2)
0. 0056 (1)
0.0083(2)
o. 0157 (3)
0. 0122 (3)
B 33
0.0035(1)
B 12
0.0005(2)
0,0027(1) -0.0002(2)
0.0026(1) -0.0015(2)
0.0031(1) 0.0032(3)
0.0037(1) -0.0015(3)
0.0037(1) -0.0001(2)
0.0025(1) 0,0001(2)
0.0033(1) -0.0002(2)
B 13
0,0026(2) 0.0005(1)
0.0024 (2) -0.0001(1)
0.0025 (2) -0.0009 (1)
0.0020(2) 0.0010(1)
0,0027 (2) -0.0021 (1)
0.0032(2) -0.0001(1) 0.0020(1) -0.0004(1)
0.0030 (2) -0.0009 (1)
0.0029 (1) 0.0034 (3) 0.0014 (2) 0.0006 (1)
0.0040(1) 0.0012(3) 0.0023(2) -0.0017(1)
0.0038(1) 0.0047(3)
0.0028(1) -0.0005(3)
0.0014(2) 0.0028(1)
0.0007(1) -0.0019(1)
0.0063(1) -0.0054(3) 0.0029(2) -0.0051(1)
0.0033(1) 0.0033(3) -0.0006(2) 0.0016(1)
0.0030(1) 0,0012(2) 0.0002(1) -0.0020(1)
0.0069(1) -0.0013(2) 0.0030(2) -0.0042(1)
0.0042(1) -0.0013(3) 0.0035(2) -0.0027(1)
0.0055(1) 0.0003(3) 0.0043(2) -0.0008(1)
0.0038(1) 0.0014(2)
0.0055(1) 0.0025(3)
0.0036(1) 0.0012(4)
0.0023(1) -0.0019(4)
0.0025 (1) -0.0006 (3)
0,0025(1) -0.0006(2)
0.0027(1) 0.0003(2)
0.0022(1) -0.0003(4)
0.0036(1) 0.0021(3)
0.0031(2)
0.0046(3)
0.0014 (2)
0.0008(2)
0.0010(1)
0.0035(1)
0.0039(2)
0.0007(1)
0.4012(4) 0.3995(3) 0.0536(2) 0.0289(8) 0,0087(2) 0.0039(1) 0.0014(3)
0.4760(4) 0.3585(2) 0.1151(2) 0.0266(7) 0.0062(2) 0.0031(1) -0,0000(2)
0.4943(3) 0,2545(2) 0.1239(1) 0.0179(5) 0,0063(2) 0.0023(1) -0.0004(2)
0.022(3) 0.454(2) 0.612(1) 3.0(5)
0.0020 (2) -0.0001 (1)
0.0023(2) -0.0001(1)
0.0027(2) -0.0008(1)
0.0014(2) -0.0009(1)
0.0022(2) 0.0025(1)
0.0028(2) 0.0021(1)
0.0030(2) 0.0006(1)
0,0023(1) -0.0001(1)
0.018(4) 0,129(2) 0.674(2) 4.0(6)
0.403(5)
0.525(4)
0.653(4)
o. 752(4)
0.716(4)
0.598(4)
0.319(4)
0,291(4)
0.393(4)
0.510(4}
0.037(3) 0.036(2)
-0.038(2) 0,131(2)
-0.012(3) 0.251(2)
0.087(2) 0.350(2)
0.253(2) 0,350(2)
0,333(2) 0.245(2)
0.203(2) -0.038(2)
0.369 (2) -0.046 (2)
0,456(2) 0,045(2)
0.385(2) 0.159(2)
6. 9 (9)
5.0 (7)
4. 9 (7)
4. 7 (7)
4. 7 (?)
4.2(7)
3.8(6)
4. 3 ( 6)
4.9(8)
3.8(6)
46

Table 5. 4b
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION 0
FOR PHENANTHRENE PMDA (A)
c (1) 0.213(3) 0.225(3) 0.271(3)
C(2) 0.199(3) 0.243(3} 0.253(3)
C(3} 0.188(3) 0.213(3) 0.266(3)
C(4) 0.231(4) 0.257(4) 0.346{4)
c ( 5) 0.213(3) 0.243{4) 0.346(4)
0{1) 0.237(3) 0.345(3) 0.413(3)
0 ( 2) 0.215(3) 0.281{3) 0.386(3)
0(3) 0.204(3) 0.298(3) 0.424(3)
C(11) 0.205(3) 0.222(3) 0.279(3)
C(l2) 0.195(3) 0.228(3) 0.239(3)
C(l3) 0.188(3) 0.219(3) 0.271(3)
C(l4) 0.228(4) 0.245(3) 0.306(3)
c (15) 0.205(3) 0.251(3) 0.313(4)
0(11) 0.226(3) 0.335(3) 0.359(3)
0(12) 0.210(3) 0.278(2) 0.349(3)
0 (13) 0.191(3) 0.288(2) 0.402(3)
C(21) 0.201(3) 0.253(3) 0.344(4)
C(22) 0.218(3) 0.253(3) 0.332(4}
c (31) 0.209(3) 0.222(3) 0.290{3)
C(32) 0.217(4) 0.254(4) 0.385(4)
c (33) 0.203(4) 0.272(4) 0.411(5)
C{34) 0.205(4) 0.272(4) 0.362(4)
C(35) 0.207(3) 0.255(3) 0.278(3)
C(36) 0.186(3) 0.225(3) 0.237(3)
C(41) 0.187(3) 0.244(3) 0.283(3)
c ( 42) 0.196(4) 0.249(4) 0.382(4)
c ( 43) 0.221(4) 0.263(4) 0.362(4)
C(44) 0.219(4) 0.261(3) 0.326(4)
c ( 45) 0.211(3) 0.244(3) 0.274(3)
C(46) 0.179(3) 0.227(3) 0.242(3)
47

CHAPTER 6
THE CHARGE-TRANSFER COMPLEX FLUORENE PYROMELLITIC
ACID DIANHYDRIDE
6.1 INTRODUCTION
It seems that fluorene PMDA is the first fluorene containing
charge-transfer complex to be studied. The structure of fluorene itself, a subject of controversy for twenty years, was finally
resolved by Burns and Iball (1955). Fluorene is related to the
carcinogenic dibenzfluorenes and benzanthracene, so it is interesting that structures of fluorene and benzanthracene with
PMDA have been elucidated at points close in time. There is interest in relating carcinogenic properties to charge transfer
properties as it is thought that complexing with DNA, RNA or a
protein could play an important role in the mechanism of
carcinogenesis (Foster, Scrimgeour, Iball & Williams 1975).
The physical properties of fluorene PMDA are as yet
unpublished. The unusual packing found for the molecular pairs and the statistical disordering of the fluorene molecules may
lead to electrical and spectral properties atypical of this class of compound.
6.2 EXPERIMENTAL SECTION
Crystals were supplied by D. Haarer. They are orange in
colour and were grown by the Bridgman Method from chromatograph
ad and zone-refined fluorene and PMDA (Haarer 1975}. Preliminary
precession photographs of these crystals exhibited monoclinic
symmetry and the systematic absences OkO k odd, hOl 1 odd,
uniquely indicate that the space group is P2 /c. The density of 1 -3
the crystal was measured by flotation D m
= 1.40g em Least-squares refinement of the cell constants (using 12 centred
. 0 reflect1ons} as a =10.146(2), b = 7.101(2), c = 13.004(2) A,
0 - - - -3 B = 108.29(1) yields a calculated density of 1.44g em with
2 molecular pairs in.the unit cell. The implication of this is
that each molecule has a centre of symmetry, which is possible
48

for PMDA, but not for fluorene, which therefore must be
statistically disordered. The only other possibility is that one cell constant should be doubled. This possibility was ruled
out by oscillation and Weissenberg photography using very long
exposure times. 0
Cu Ka radiation (~ = 1.5418A) was used. Data were collected 0
for one quadrant of reciprocal space with 9 < 76 using an Hilger
and Watts automatic diffractometer. The intensities of the
diffracted X-rays were measured using an 80-step, single second, 0
0.01 scan with 20 second background counts at each end of the
scan. Attenuators were used when the monitored pulses exceeded
8000/second. The attenuation factors were 3.44, 9.18 and 31.6.
The intensities of 3 standard reflections were monitored every 50
reflections and the data scaled accordingly. Lorentz and polarisation corrections were applied and, of the unique
reflections measured, 1177 had intensities >Sa and 1369 >3a. No
absorption corrections were made as test calculations showed them
to be unnecessary, the range of possible corrections varying by no
more than 5%. The linear absorption coefficient for Cu radiation -1
is 8.96cm for this compound.
6.3 STRUCTURE DETERMINATION AND REFINEMENT
Most of the structure was revealed by use of the direct
methods program MULTAN (Main, woolfson & Germain 1971) utilising
those reflections having E > 1.06. PMDA is located with the unit
cell origin at its centre of symmetry and its 8 unique atoms
(1/2 molecule) were revealed immediately. On the other hand
fluorene, which has no centre of symmetry, but is centred at
{1/2, 0, 0) appeared as the superposition of two fluorene
molecules - inverted with respect to each other. The orientation of the PMDA molecule was used to define a plane, with which it was
expected the plane of the fluorene molecule would be parallel. A
difference Fourier calculation, using phases determined by the
PMDA molecule, in the plane in which the fluorene molecule was
expected to lie, revealed two superimposed, inverted fluorene
49

molecules. Finding their locations was greatly facilitated by use
of a line-printer electron density plot (see section 12.2). Figure 6.3a shows the superimposed molecules schematically.
Figure 6.3b shows a line printer density map of the PMDA molecule
in this structure. The refinements of the two 1/2 weighted fluorene molecules have resulted in some unusual c-c bond lengths in the 6-membered rings, because the atom C(32) in one of the
superimposed molecules, and C(44) in the other are unresolvable. The parameters describing thermal motions of atoms in this area
are of uncertain value. Considerable use was made of difference Fourier calculations between least-squares refinements,
particularly for positioning the fluorene molecules. They also
revealed that the vibrational motion of the atoms was anisotropic,
particularly that of the oxygen atoms. Eventually least-squares
refinement with anisotropic thermal parameters applied to all
non-hydrogen atoms and isotropic parameters to hydrogen atoms converged with R = 0.055. Shifts in all parameters were less
1 than 0.2 of their standard deviation in the final cycle of
refinement. Secondary extinction was observed to be present
and the refined value of the Zachar sen extinction coefficient (Zachariasen 1963) is 0.11916 x 10 A final difference
Fourier synthesis revealed no anomalously large peaks.
6.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE
Figures 6.4a and 6.4b are projected views of the two
molecules PMDA and fluorene and serve to define the atom labelling scheme used in this chapter. Figure 6.4c shows spatial relation
ships between the molecules in the structure, emphasising both the stacking and interplanar angles between the molecules. Two
features of this structure are worth special note, the molecular packing and the statistically disordered orientation of the
fluorene molecules. The structure of fluorene PMDA can be viewed in two ways.
50

Figure 6.3a
SCHEMATIC DIAGRAM OF SUPERIMPOSED FLUORENE MOLECULES
\ \ \ \
\..
I
---\
-
51

Figure 6.3b
LINE-PRINTER SHADED ELECTRON DENSITY MAP OF PMDA
•• ,.,. •• ,11 ........ .
:., .. :,:e.~iHuui . .... '([U PA.JU.w[T(ftS A • 2Cl.U0 v • 20t00 t • 20•00 A\.f'MJ ·• 10,00 eth • tOtO-D Cillil'41. • tOd>O
l . •o,aoo • •(ifJe~ . "'(ltlfU
l . •01),.,_
• "'0t)l0 . •Od1)
• '"0•109
a •Ot~h
1 • •o.~rt ! • .. , .. >•• I • •0•2•v
I • ••• J,. .. "0•21•
t • •o.1o• I . •t1tUY
I • •-a t ,,.
I • •Qfl!.l!o
l . •o • t«J
l • "'OtUIS
l . "0• lll
I • •o•Ote
I • •o. oaJ I . "'OdiUI
I • •o' on t • •o•OH
I • •o.ou l • •CI• O(!ft
1 • OtOOO
~~ ~•OU
l • OtOU
l • 0.0'\J
I • !hOU
I • -DtCU
I • 0. 09tl
l • ihlll
I • c,. tze I • 0.10
l • "•ISti l • fhlh
I • o.u, I • Oc 20a
I • Ot21'1
I • th2J•
I • Ot2lt
l • C). 2611;
I •· Ot27v
I • 0'•2h
I • Ot )0'9
I • . fltlU
I • CdtO
~ . ()'tl'S!»
1 • O•lfo
I o OdiU
I . u.•ou
CO\.t!lot 5tA\.t • Oelf t1.Jii'HPIQfi$1tiC
0'•1": /(HUUC HM
' . ' · RO• $Cll.[ • Odt JHGStftOW:l/(M .
O•lOtLUOUIU,(T(Jt
! . • . •
SLcnow ttHilt • to.o
I . • • •
... .......... t ...... ...
......... ... • • 4. 4 ... ...
.......... 4.''. II~ ..... . .... :: i;;;u:: ::-::: r :::: .. ... ft •• f.' •• t t ttl t t t .......
......................................... .. -.......................... ... •• .... " .... t • • • • • It • • ••" • .. "'•"' .. • ~ • • t t t t t t • • •• "'"••·•• Ill tit t f C • •••
......... ~ f • t t-.' f ~ 4 ...... ..
.. :::::;:: :::: ::~: :::::
........... l'ttftttt•--...
........ ,.,.tIt t tt t t • • •• _..,,. .. • •• t iII • ...... .. .... .. .. . . . . ..... .. ..
........... ' •• f ..... .. .... ~ ............. ··-· ......... ~ t ........ ' .... .. .. ........................ .. .. ...................... .
................... ~--· ....... ... ...... .: ......... t ttl f' 'i ............... .. ............. .. ..... ,. ••• t t * ............ " t 1ft ............ .. ............................ ,.,.,11 t• ••t ·~~:!'~1tl• •t f I·~,.._,. .... ., .... ,.
"'ll'··········••ttl·-.. • I •• "'" • • •w• t., w 1; •• .. .. ,,., ..... rt-.••rta ••• ••qttt•••tt'lll't~l:!·••t••
•· :u ~ i :!} ;:; : r:t !:· .. .... 't ....... t ... . ..... '.f ... .. ..... !!!!* .. ........ .. ........ , .. .
••t1 •"tfr•ut••• .. .. ::; :: 1;: ~: r!: 1 ~:
~= ::i!!! !:: H: H H! !: .. ..... t t ••••• t ....... .. ··•t1••······ ":!!!!!!!:··
.. :::: i;; ;: ~;;::: i: ~:!! ~ ~ ;sl:::: :: ;; ;!! ::::::::::: ............. "'"'"'• f tJ•f •- •t.ft"'"•• t t •*"f "'~~! t t I Itt ':'l'"• ,,..,,. .......... .. .................. ,' "'t•• •t I' ljel•"'• • •·•t•!!~'' ••' att>~t·• t ~ ......... .. ·:::::::;: !.: ; 1 t ~::,:. : ~; ~~? ~:: ~~::~;~;:a;!;:!!:::::·
.. ........... ··-
..,., • • J t I t I f f ~ t • • .... • • tIt f f t t 1 t t t • .._ .. . .... ~- I t t ••• t I ••• t t ... ..
................ ~ t ,,.,,'In u .. , .. ~ e•n•!t •••!!:•t•••1i '""'"'"'· .... •• iit i i:; t ;; :::;~t.: I I I!!:-::?:~: :t : :::;::; !!:: ... ...... 't 1 t • .- .. , r' r r r ·• t t ... ..
:::!: U :;~;·~ ::::H;::: :: ..... ,, ........ t' t ....... t. •'!!"!~·· ( ...... t•!:••,tt'!t•t ..... .. .... , ,, t•• ""·'~'"!tit f t:!!l~~ ....... t , ..... , t tt •••tf•t 1'••1 t .... .. .. ...... ,,,.,,. •••~u~r•t• .. '""'"'•lt••e• 111 ,,,.,, ••
.... •ltt.tew ••• l!!ttl•• ••tt•r•cttaer•tt••
... tt "'" ~fvl.lr,tr.er.!'!!'l"l~tf •• tt t t t t •I i Itt! 1 t I t• .. ,._ .. ••t•t~~tthj•t•tt t It li"f"•tlti••~••IIHftll••- .. •• ••tte, lijt •t•it t tl .,, .. ,_.,, ........... lftltfttt·-.. •• •• t ,.,_ J t U lllllt~tft t ttl h~f "'tf t t ~ •••I Ht,.t t il"'"t , ........ ....... t••"'······-.. .... !!~!!!tt•·-
...... ,1,, ••• , .. ttt••••· ............................ , •• : ••• ~-~t•tl••·- .... .. ::::;! j~ ~!:! =~ ;;; : •!nr• :;!;:!! !! ; :! ! ! ; ; '~·: Hjl;; ;!: ; : ;:: .. . ,...,.,.._.tttlltHitlflttt'tH•I'.••fttt"•t•.!!'IF tit lfii'!H'"'"'''ittl••"'""'"
................................... • t f t;f.,t!l "llJ~!'!'~• till 4 •~•.t.tt t t .... ,t 1 • , ...... .................... -.... ·::u:;:~ 1:: ~~::!:~:!:!~z::!:;s;l:i:~!~~::::: .. .
.... _., ttttl•··· ..... t .......... tt••·
.... ,t,.J!tt•"'•l'lttt• ••t•tetltttt•~c•tt•· .. ............... ,, .. ,~ ..... ..
• • t • tetr' • u • tt • , .. , i ........ ..
::: t: !~;~:!! ~!i !: ; :::::: ...... tt ... t (J' .. t t•'- t 'c ..... .
.. ...... t t," ...... , 't .... . ........ t',' t t 11 ....... .. .................. ......... !:!::· ....
................ ..... t t t' t t ..... ••tt••••tt• .. -...... ,,t•"'•••t•• .. -
•• • • fIt t • •• • • • t t •·"' ••titlltllls•t•tttt•••
••ttJWt'ltff••~'lll•"'"'"" •• t • 'Itt< !!'ft'J • t I • ~ .... .. • t •I, !'I':'•• t • t 4 ..... ..... ,.,.,t t f ... , f ...
"'•-t t • •• t t I• .. ..... !!!:·· ............
• • • II C I • • .. ..... t ........ t * .... .. .... , .. , ......... ~ .... ..
••tt•xtr:•"'lt:i'-'ttlt••• "'* J t W'. 1:1"\. "'If f"'Joill t t f .. ,.. • ". t. t,,.' t , ........ "•t t .. .. . .. , ........................ .. •"'•I It • • • • • • • .... •• ~~~:; J t, ..
"'"" ••• tIt t tIt f.,,' •• .._ t I••• •• ••t t St t t t 1 t t 1 t til• ••• .... •• Itt t t• It Itt • • • •'""'
"'• • Itt ":~• t t t t tIt • • t t t t t t t f' • * t' t 11ft f f l •! t •1' • ... • • .... .
:::! J:! ~:~ :: :~:;:!: :!!;::!:! ::: ~! 1:: !! H: ~:: :::::· .,,.., • • Itt •• •• •tt f "" • ................ ., • t t • • t' !" .- •, f • f I • " ...... ..
....... t , ...... ,, ...... ,, ...................... ~ ... , ..... ,, ...... , , ....... .. .......... ,,._,, ...... t ... ,tt ............ .,_ ..... tttil. 1 "'••t' f• ..... .. -·····.,, ... I'll!"' .................... ,.! .. •·•·· ........... ,., tt~t 1 tt•"1tt••~••••l•"•"'Jll'l't-t•1t••r
·:::::::::!~: 1 •• ::~~!!!H~!H~=:;,t •• ,;,n~::i:: _ ........ t•·-~•~t•tn''''!-jlJjl*" r••~•j1tl•1•t• •t t............................. .. ·: •••••• J tfl••fffrt•~'' f•~l!'t!• tt•tfw:=t• f 1•• ................................. .
.. ..... !!!!!:::;;n:t : •• t .:::: !!~!::!;! ~== ;:~!!!!:::::: !!!! ~; ;::: .. · .......... ,, .. r:ct 1 ttt ''' r•tt t' t ••" • •·t·n· ··~"' 1 tt '''"''='~' t•1 t •·•
·::::!:;:::;t~;;:;:: ~ !:: :::: :;;;:t!! ~ r:~ :~: ::t:: :; ::i ::· *•••••• Jl i, J f I• u • .., .. It t• •• tt tit t f Iff t I J t tltl t ~~ ft••
""'""""••I t l tt If t n *H· f 'HI "(!'t fit t tttWII ~Yt till i~f t f •• • ...... it 1t tt' 1 t H t t f t tt ••t --~•Y r • ••ee:!~t ••••w!'•wt , .... '"'"*f t•'"t ftf f-.- •1 tf f ..... , f'\J~J:!\!!1" ~llff f tf f t(j!'JW 'tfff t , ..
..... ,, .. ,"'.' fltl'l• , ...... tt(• .... >o!'!'"•+•"'••t t ,_. ......... , ...... ..
.... ::;:n:~::u u z n:~:;nru 1 ur ;;:n !! ! ! ! : !! u • ... .. .. ...... :::;; !! ~: ;;~;~~:: ::: ~ ~~;g:: 1::' l: ll t::: ~; :::::::-..
... :::::::: !; ;;n: 1: t r:;; ~!!~! ;!!~;:: 11: ~:~; :;1 !::·:::::: .. .::::::::: f ::~ ;: t t I r I' l :~:!::; ;:; ; ;;; ~;;;; ~; :~! !:".. • .......... .. •• .. ""' .. ,.. .. • • l 1 t .. , t e t 1 t. J t t: 01 f • t t t It t t I J • , It • • ~ • • ..... .
.................. , 11• "tre~~t•r••" t t I • ., .. ,.. .... __ .......... .. .............. i' t t• ~~, .......... ,, t........... ... ........ ..
... ................ ,,, •• tit••• .... .. .. ........ _ .................... . ................. -........... . .................. .................... . .......... .- ....... .
a..::~::nn:tt::!::: ···••tt···-······· ........ .. ::::: n :: ::~ ~ '! :::::·
... ................... .. .. .................... . .. ......... ,.* ....... .. ........................... ........................ • •••• ,.* ......... ..
.. ...... , .............. .. •• • • itt tIt • • • • •••••• . ...... , ................. .. .. ..... *••····, ............. .. .......... ~ .......................... . . ...
•lettt••etttltltltltllttlll I I t • • I I I t I •
52

Figure 6.4a
BOND DISTANCES AND ANGLES IN FLUORENE
C!21l
Cl33l Cl43l
53

Figure 6.4b
BOND DISTANCES AND ANGLES IN PMDA
C(4)
0(2'1 0(2)
C(1')
54

Figure 6.4c
VIEWS EDGE ON TO A FLUORENE MOLECULE AND PARALLEL TO THE y AXIS
1.0J
Q0,1
55

As a charge-transfer complex of fluorene and PMDA molecules,
stacked in the a direction, the donor and acceptor molecules are - 0
unusually far apart (5.07 A). An alternative description is one
in which there are separate fluorene and PMDA stacks along the b direction, the intermolecular stacking being the more usual
0 0 distance of 3.522 A (c.f. 3.5 A Herbstein 1971). The average
0 fluorene-PMDA interplanar distance is 4.46 A. Atomic parameters
are given in Table 6.4a, root-mean-square amplitudes of vibration
in Table 6.4b and structure factors in Appendix A2.
The type of disorder obtaining in fluorene PMDA has been
observed in at least four other compounds - azulene {Robertson,
Shearer, Sim & Watson (1964), di-indenyliron {Trotter 1958}, acepleiadylene {Hanson 1960) and 2 amino-4-methyl-6-chloro
pyrimidin (Clews & Cochran 1948). In all these compounds, except the last, there are five and six membered rings fused as
in azulene, and in all except di-indenyliron the disorder
relates to a centre of inversion. In hindsight it is interesting
to note that Burns & !ball {1955) discounted such disorder when
they examined the crystal structure of fluorene itself, whereas
here, in the pure fluorene stacks, there is statistical inversion.
This is, of course, also observed in the fluorene PMDA stacks and
Figure 6.4c illustrates the case where reversal of the fluorene
orientation has taken place in successive cells.
56
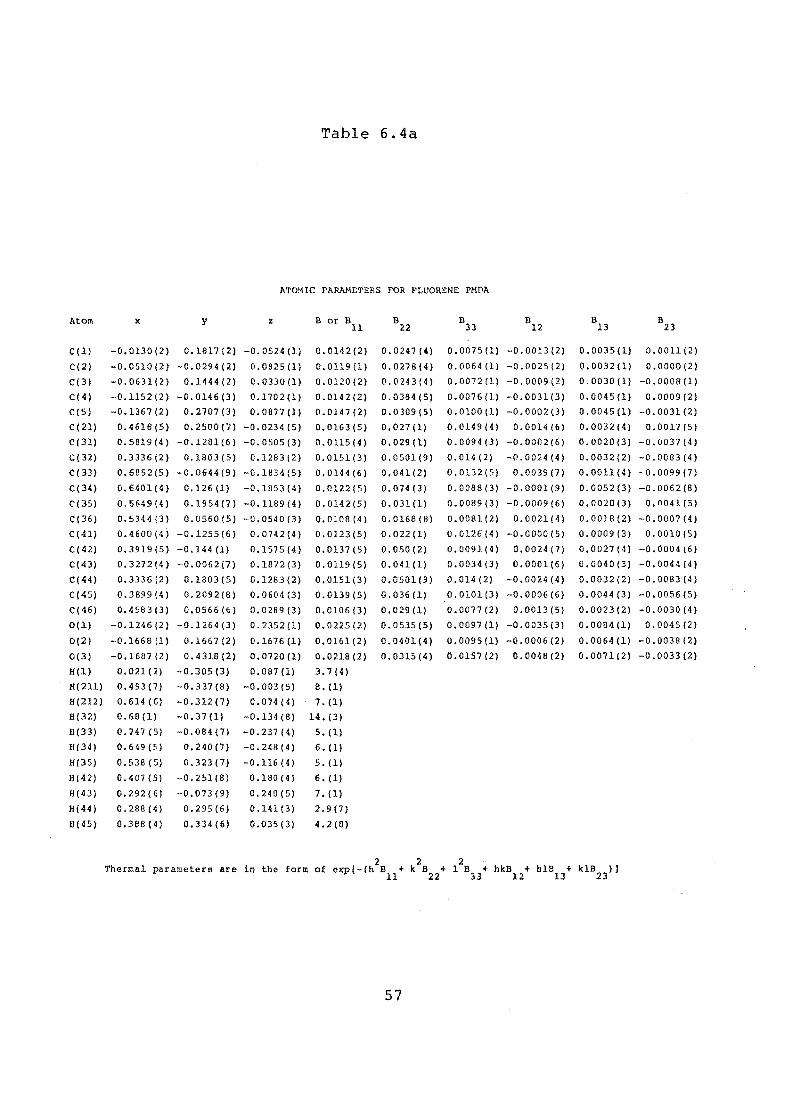
Table 6.4a
ATOMIC PARAMETERS FOR FLUORENE PMDA
Atom X y z B or s11
B B B B B 22 33 12 13 23
C(l) -0.0130 (2) 0.1817(2) -0.0524(1) 0.0142(2) 0.0247 (4) 0.0075(1) -0.0013(2) 0.0035{1) o. 0011 (2)
C(2) -0.0510(2) -0.0294(2) 0.0825(1) 0.0119 (1) 0,0278(4) 0.0064 (1) -0.0025(2) 0.0032(1) 0.0000(2)
C(3) -0,0631(2) 0.1444 (2) 0,0330(1) 0.0120(2) 0.0243(4) 0.0072(1) -0.0009(2) 0.0030(1) -0.0008(1)
C(4) -0.1152 (2) -0.0146 (3) 0.1702{1) 0.0142(2) 0.0384(5) 0.0076 (1) -0.0031(3) 0.0045(1) 0.0009(2)
C(S) -0.1367(2) 0.2707(3) 0.0877 {1) 0.0147 (2) 0.0309(5) 0.0100 (1) -0.0002(3) 0.0045(1) -0.0031(2)
C(21) 0.4616(5) 0.2500(7) -0.0234 (5) 0.0163(5) 0,027(1) 0.0149 (4) 0.0014 (6) 0.0032(4) 0.0017 (5)
C(31) 0.5819(4) -0.1281(6) -0.0505(3) 0.0115(4) 0.029(1) 0.0094(3) -0.0002(6) 0,0020(3) -0,0037(4)
C(32) 0.3336(2) 0.1803{5) 0.1283(2) 0.0151(3) 0.0501(9) 0.014 (2) -0.0024{4) 0.0032(2) -0.0083(4)
C(33) 0.6852(5) -0.0644(9) -0.1834(5) 0.0144{6) 0,041(2) 0.0132(5) 0.0039 (7) 0.0011(4) -0.0099{7)
C(34) 0.6401(4) 0.126(1) -0.1853(4) 0.0122(5) 0.074 (3) 0,0088(3) -0.0001 (9) 0.0052(3) -0.0062(8)
C(35) 0.5649{4) 0.1954(7) -0,1189(4) 0.0142{5) 0,031(1) 0.0089(3) -0.0009(6) 0.0020(3) 0.0041(5)
C(36) 0.5344(3) 0.0560(5) -0,0540(3) o. 0108 (4) 0.0168(8) 0.0081(2) 0.0021(4) 0.0018(2) -0.0007(4)
C{41) 0.4600(4) -0.1255(6) 0.0742(4) 0.0123(5) 0.022(1) 0.0126(4) -0.0000(5) 0.0009(3) 0.0010(5)
C(42) 0.3919(5) -0.144 (1) 0.1575 (4) 0.0137 (5) 0.050(2) 0.0091(4) 0.0024(7) 0,0027(4) -0.0004 (6)
C(43) 0.3272(4) -0.0062(7) 0.1872(3) 0.0119(5) 0.041(1) 0.0034(3) 0.0001(6) 0.0040(3) -0.0044(4)
C(44) 0.3336(2) 0.1803(5) 0.1283(2) 0.0151(3) 0,0501(9) 0.014 (2) -0.0024(4) 0.0032{2) -0.0083(4)
C(45) 0.3899(4) 0.2092(8) 0. 0604 (3) 0.0139 {5) 0.036(1) 0.0101(3) -0.0006(6) 0.0044(3) -0.0056(5)
C(46) 0.4583(3) 0,0566(6) 0.0289 (3) 0.0106(3) 0.029(1) 0. 0077 (2) 0,0013(5) 0.0023(2) -0.0030(4)
0(1) -0.1246(2) -0.1264(3) 0.2352(1) 0.0225{2) 0.0535(5) 0,0097(1) -0.0035(3) 0.0084(1) 0.0045(2)
0{2) -0.1666 (1) 0.1667 (2) 0,1676(1) 0.0161(2) 0.0401(4) 0.0095{1) -0.0006(2) 0.0064(1) -0.0038(2)
0(3) -0.1687 (2) 0.4316(2) o.ono (1J 0.0218(2) 0.0315(4) 0,0157(2) 0.0048(2) 0.0071(2) -0.0033{2)
H(1) 0.021 (2) -0.305 (3) 0.087 (1) 3.7(4) H(211) 0.493(7) -0.337 (8) -0.003(5) 6. (1)
H(212) 0.614(6) -o. 312 (7 > 0.074 (4) 7. (1) H(32) 0.68(1) -0.37(1) -0.134 (8) 14. (3)
ll{33) o. 747 {5) -0.084 {7) -0.237(4) 5, (l)
H(34) 0,649(5) 0,240(7) -0.248(4) 6. (1)
H(35) 0.538 (5) 0.323(7) -0.116(4) 5. (1)
H( 42) 0.407(5) -0. 251(8) 0.180 (4) 6. (1)
H(43) 0.292(6) -0.073(9) 0.240{5) 7. (1)
H(44) 0.288(4) 0.295(6) 0.141(3) 2.9{7)
ll(45) 0.386 (4) 0.334{6) 0.035(3) 4.2(8)
2 2 2 Thermal parameters are in the form of exp[-(h a
11 + k B + 1 B + hkB + blB + klB
23lJ
22 33 12 13
57

Table 6.4b
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION 0
FOR FLUORENE PMDA (A)
Min Int Max
C(l) 0.229(2) 0.252(2) 0.266(2)
c (2) 0.216(2) 0.231(2) 0.274(2)
C(3) 0.233(2) 0.235(2) 0.254(2)
c ( 4) 0.225(2) 0.257(2) 0.319(2)
C(5) 0.245(2) 0.265(2) 0.303(2)
c ( 21) 0.256(5) 0.281(5) 0.353(5)
C{31) 0.227(4) 0.246(6) 0.305(4)
c (32} 0.259(3) 0.292(3) 0.396(3)
C(33) 0.243(5) 0.252(5) 0.408(7)
C(34) 0.204(6) 0.264(5) 0.443(8)
C(35) 0.234(5) 0.259(5) 0.316(5)
C(36) 0.196(5) 0.227(4) 0.267(4)
C(41) 0.234(5) 0.239(6) 0.338(6)
C(42) 0.249(5) 0.274(6) 0.359(6)
C(43) 0.204(5) 0.243(5) 0.334(5)
C(44) 0.259(3) 0.292(3) 0.396(3)
C(45) 0.234(5) 0.257(4) 0.333(5)
C(46) 0.220(5) 0.227(4) 0.292(5)
0(1) 0.211(2) 0.334(2) 0.381(2)
0(2) 0.223(2) 0.285(2) 0.333(2)
0 (3) 0.248(2) 0.334(2) 0.355(2)
58

CHAPTER 7
THE CHARGE-TRANSFER COMPLEX THIANTHRENE PYROMELLITIC
ACID DIANHYDRIDE
7.1 INTRODUCTION
This chapter describes the structure of a complex which
appears to be the first involving thianthrene. PMDA plays its
usual role of electron acceptor (Evans & Robinson l980a). The
structure of the folded thianthrene molecule was first determined
by Lynton & Cox (1956) after Sutton (1955) had aroused their
interest in the c-s bond geometry by pointing out that the sulphur
atoms could use dw as well as pw orbitals in w-w bonding.
This type of complex, in which dw orbitals may be involved in
charge transfer processes, is apparently new. The non-planar
conformation of thianthrene causes difficulties in describing the
arrangement of molecules in this charge transfer-complex in terms
of a specific stack axis.
Physical properties of this complex have yet to be published.
7.2 EXPERIMENTAL SECTION
Crystals were supplied by D. Haarer. They are reddish
orange in colour and had been grown by the Bridgman method from
chromatographed and zone-refined thianthrene and PMDA (Haarer
1975). The lack of systematic absences and symmetry in
preliminary precession photographs, as well as Wilson statistics,
suggested the triclinic centrosymmetric space group Pl. The -3
density of the crystal was measured by flotation D = l.SOg em m
Least-squares refinement of the cell constants (using 12 centred 0
reflections) as a= 11.128(1), b = 12.309(1), c = 7.334(1) A, - - o-
a = 99.716(5), B= 82.617(5), Y = 107.450(5) yields a calculated -3
density of 1.54g em with two molecular pairs in the unit cell.
All data given in this chapter relate to this unconventional
cell. The conventional reduced cell constants a = 11.128, b = 0 - 0 -
13.899,£ = 7.334 A, a= 92.67, S=97.38, y= 122.35 can be
derived from those used by application of the transformation
59

-matrix 100/110/001.
0 Cu Ka radiation (~ = 1.5418A) was used. Data were collected
0 for one hemisphere of reciprocal space with 9<57 .The intensities
of the diffracted x-rays were measured using a 72-step, single 0
second, 0.01 scan with 18 second background counts at
each end of the scan. Attenuators were not required. The intensities of three standard reflections were monitored every 100
reflections and the data scaled accordingly. Lorentz and polarisation corrections were applied and, of the 2530 unique
reflections measured, 1507 had intensities >Sa and 1802
intensities >3a. Test calculations showed that absorption
corrections would be unnecessary as the range of possible
correction factors required was no more than 7%.
7.3 STRUCTURE DETERMINATION AND REFINEMENT
The direct methods program MULTAN (Main, Woolfson & Germain 1971) was used, utilizing the 376 reflections having E > 1.4. Of
the eight possible sets of phases the first two had the highest figures of merit. One of these had all phases the same whilst
the other proved to be the correct set. Twenty-eight of the thirty non-hydrogen atoms were located using a Fourier synthesis
based on the phased E values. The two remaining atoms were
located using a difference Fourier calculation based on structure
factors obtained following least-squares refinement of the
established structural parameters. Refinement, with isotropic
thermal parameters for the atoms, resulted in an agreement factor R of 0.146. A difference Fourier calculation showed some
1 evidence for anisotropic motion, particularly for the sulphur
atoms. Using anisotropic thermal parameters the agreement factor
R reduced to 0.086. Refinement with hydrogen atoms having 1
isotropic thermal parameters converged with R = 0.073. Owing to 1
the high number of variable parameters towards the end of the
refinement procedure the least-squares calculations had to be
done in blocks, and in order to help decide the order of refine
ment calculations of the standard deviations of the various
60

bond types within each molecule were carried out. The program BONDSTAT {see section 12.4) was used. Secondary extinction was detected and, in a final cycle of refinement, the value of
the Zachariasen extinction coefficient (Zachariasen 1963) -6
obtained was 0.16425 x 10 and R reduced to 0.064. 1
A final difference Fourier synthesis revealed no
anomalously large peaks.
7.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE
The structure of the complex consists of columns of
alternately positioned thianthrene and PMDA molecules. Two
PMDA molecules are present in the cell in crystallographically independent positions, about each of the centres of symmetry
(0, 0, 0) and (1/2, 1/2, 1/2). The two thianthrene molecules are related by the space group centre of inversion. Figures
7.4a,b and c define the atom labelling scheme used throughout
this chapter. Figure 7.4d shows the spatial relationships
amongst the various molecules in the unit cell. The dihedral 0
angle between the wings of the thianthrene molecule is 130.08 • 0
This can be compared with 128 for thianthrene (Lynton & Cox 0 0
1956), 131.1 for 2,7-dimethylthianthrene (Wei 1971) and 138 38
for thianthrene dioxide (Hosoya 1966) • The average e-s-c angle 0 0
is 100.5 and the average c-s distance is 1.767 A in excellent 0 0
agreement with the values of 100.1 and 1.758 A given by Lynton
& Cox (1956) and the bond length is shorter than the sum of the 0
covalent radii (1.812 A; radii given by Pauling 1960).
The orientation of the PMDA molecules is influenced by the
bent nature of thianthrene. Successive PMDA molecules are
aligned with 'opposite ends' of the particular thianthrene
molecule between them. The average distance between the parallel 0
(aligned) portions is 3.51 A and the angle between normals to 0
their planes is 4.72 . Figure 7.4c suggests that the [111]
61
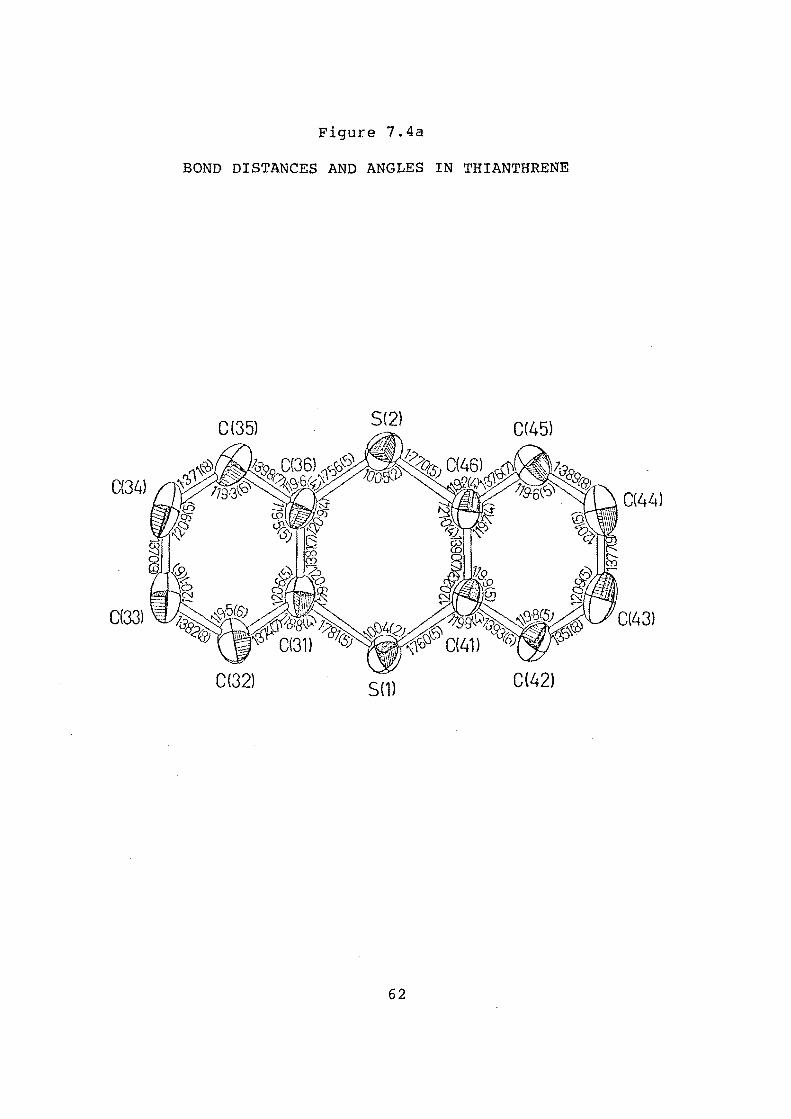
Figure 7.4a
BOND DISTANCES AND ANGLES IN THIANTHRENE
C(35l 5(2)
C{34} C(44l
C(33) C(43)
C{32) S(1)
62

Figure 7.4b
BOND DISTANCES AND ANGLES IN PMDA (1)
0(12')
C(11'1
63

Figure 7.4c
BOND DISTANCES AND ANGLES IN PMDA {2)
0{23')
C{24)
0(22') 0(22)
0(21') C(21')
64

Figure 7.4d
VIEWS OF THE CRYSTAL PACKING OF THIANTHRENE AND PMDA MOLECULES
001
1H28A
101
65

diagonal may be regarded as the stack axis of the charge-transfer
complex. However, as in fluorene PMDA (Evans & Robinson 1980a)
there is an alternative view in which the structure is regarded
as consisting of thianthrene and PMDA molecules stacked separately
parallel to the [001] direction. Atomic parameters are listed
in Table 7.4a, root-mean-square amplitudes of vibration in
Table 7.4b and structure factors in Appendix A3.
In contrast with other charge-transfer complexes it
is interesting to note that with the PMDA molecules lying parallel
to the 'wings' of the thianthrene molecule there is no straight
path which passes through parallel consecutive molecules in close
contact along a stack axis. These features are relevant to any
discussion of electron 'hopping' mechanism for electron and
exciton motion and photoconductive properties of this type of
complex. (Viz. Chapters 2 & 9)
66

Atom X y
Table 7.4a
ATOMIC PARI\HSTERS FOR THll\NTHRENE PHDA
!I 22
S(1) 0,6382(1) 0.2935(1) 0.0652(2) 0.0136(2) 0.0073(1) 0.0267(4) 0.0047(1) -0.0049(2) 0.0010(2)
S(2)
C(U)
C(42)
C(43)
C(44)
C(46)
C(31)
C(32)
C(33)
c P/4! C(3SJ
0.6022(1) 0.1910(1) 0.4665(2) 0.0106(2)
0.0091(5}
o. 0107 (5)
0.0084{5)
0,0085(5)
0.0085(5)
0.0088 (5)
0.0137(7)
0.0127 (7)
0.0107{6)
0.0095(5}
o. 7250 (4)
0.8086(5)
0.8706 (5)
0. 8546 (S)
0.7092(4)
0.6542(4)
0.6798(5)
0,3723(4)
0.4805 (4)
0.5432(4}
0.5004(5)
0.3275(4)
0.1543 (4)
0.2530(7)
0.2357(8)
0.383(1}
0.5487 (9)
0.4190(7}
0.0701(8}
0.0901 (5) -0.0946 (S)
0.6844(6) -0.0209 (5) -0.094(1)
0.6683(5) -0.0646(4) 0.070(1)
0.6467(5} -0.0004(4) 0.2354(9)
C(36) 0.6381(4)
C(11) 1.0242(5)
C(l3) 0.9777{4)
C(14) 0.9930(5)
C(15)
C(21)
C(22)
C(23)
C(24)
C(25)
0(11)
0(12)
0(13)
0(21}
0(22)
0.9556(5)
0.4806 (5)
0.4149 (4}
0.4329(4)
0.3126(5)
0.3439(5)
1.0051(5)
0.9668(4)
0.9326(5}
0.2670(4)
0.2753{4)
0.1107(4) 0.2360(8) 0.0074(5)
0.0559 (4) -0.1660 (7) 0.0097 (5}
0.0582(4) 0.1659(7) 0.0085(5)
0.2300 (5) 0.0622(9) 0.0100(6)
0.1425(5)
0.4473(4)
0.3978(4)
o. 4478'(41
0.2872(4)
0.3698(5)
0.3257 (9)
0.6692(7)
0. 5185 (7)
0.3538 (7)
0.4903(8)
0.2239(8)
0.3048(3) -0.0249(8)
0.2440(3) 0.2549(6)
0.1351(5)
0.2154(3)
0.2746(3)
0.4857 (6)
0.5914 (7)
0.3116(5}
0.0108{6)
0.0102(5)
0.0076(4)
0.0078(4)
0.0101(6)
0.0100(6)
0.0198{6)
0.0130 (5)
0.0241(7}
0.0142(5)
0.0108{4)
0.0084 (1)
0.0062(4}
0.0064 {4)
0.0069 (4)
0.0101(5)
0.0070(4)
0.0068(4)
0.0089 (5)
0.0079 (5)
0.0057(4)
0.0071(5)
0.0283(4)
0.025(1)
0.030(1)
0,045(2)
0.034(2)
0.026(1)
0.030(1)
0.029(1)
0.041(2)
0,055(2)
0.039{2)
0.0060(4) 0.032(1)
0.0097(5) 0.024(1)
0.0072(4) 0.022{1)
0.0072(5} 0.038(2)
0.0124(6)
0.0071(4)
0.0058(4)
0.0064(4)
0.0071(5)
0,0095(5)
0.0081(4}
0.0086(3)
0.0207(6)
0.0077(3)
0. 0072 (3}
0.029(2)
0.022(1)
0.026 (1}
0.024(1)
0.031(2)
0.027(1)
0.060(2)
0.037 (1)
0.021(1)
0.048(1)
0.033(1)
0(23) 0.3270(4) 0.3771(4) 0.0700(6) 0.0146(5) 0.0156(5) 0.025(1)
0.0017(1) 0.0027(2) 0.0039(2)
0.0038(4) -0.003(6) 0.0008(5)
0.0031(4) 0.0011(7) 0.0026(6)
0.0030(4) -0.0006(8) -0.0004(8)
0.0043(4} -0.0036(7) -0.0047(8}
0.0034(~) 0.0005(6) -0.0003(6)
0.0029(4) -0.0051(7) -0.0009(6)
0.0042(5) -0.0066(8) -0.0026(7)
0.0040(5) -0.0083(9) -0.0042(8)
0.0028(4) -0.008(1) -0.0016(9)
0.0013(4} -0.0030(8) 0.0047(7)
0.0013(3) -0.0032(6} 0.0018(6)
0.0029(4) 0.0003(6) 0.0031(6)
0.0027(4) -0.0022(6) -0.0002(6)
0.0022(4) 0,0008(8) 0.0017(8)
0.0047(5) -0.0048(8) -0.0016(8)
0.0035(4} 0.0002(6) 0.0006(6)
0.0028(3) 0.0006(6) 0.0011(6)
0.0029(3) 0.0001(6) -0.0004(5)
0.0035(4) 0.007(8} 0.0003(7)
0.0047(4) -0.0024(7) -0.0032(7)
0.0045(4) 0.0031(8) 0.0089(7)
0.0038{3) -0.0043(6) -0.0033(5)
0.0119(5) -0.0033(7) -0.0021(6)
0.0015(3) 0.0021{7) 0.0062(6}
0.0022(3) -0.0016(5) -0.0024(5)
0.0048(4) -0.0051(6) -0.0017(6)
H(ll1) 1.0427
H(ll2) 1.0022
H(21l) 0.4112
H(212) 0. 4700
H(42) 0.8185
H(43)
H(44)
H(45)
H(32)
H(33)
H(34)
H(35)
0.9203
0.8978
0. 76 20
0.6893
0.7001
0.6795
o. 6416
0.0816
-0.0207
0.4331
0.4210
0.5065
0.6235
0.5457
0.3550
0.1234
-0.0636 -0.1395
-0.0277
-0.2711
-0.2616
0.4117
0. 7614
0.0959
0,3784
0.6539
0. 67 56
-0.2047
-0.2137 0.0702
0.3520
6.0
6.0
6.0
6.0
6.0
6.0
6.0
6.0
6.0
6.0 6.0 6.0
2 2 2 Thermal parameters are in the form of exp(-(h B + k B + 1 B + hkB + h1B + k1B
23JJ
11 22 33 12 13
67

Table 7.4b 0
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION FOR THIANTHRENE PMDA (A)
Min Int Max
s ( 1) 0.200(2) 0.246(2) 0.296(2)
S(2) 0.213(2) 0.255(2) 0.295(2)
c ( 41) 0.177(7) 0.237(6) 0.261(7)
C(42) 0.201(7) 0.239(6) 0.289(7)
C(43) 0.200(7) 0.226(7) 0.352(8)
C(44) 0.200(7) 0.228(7) 0.341(8)
C(45) 0.211(7) 0.251(7) 0.290(7)
C(46) 0.186(6) 0.232(6) 0.274(7)
C(31) 0.204(7) 0.210(7) 0.295(7)
C(32) 0.222(7) 0.249(8) 0.319(7)
C(33) 0.212(7) 0.242(8) 0.360(8)
C(34) 0.192(8) 0.230(8) 0.390(8)
C(35) 0.211(7) 0.238(7) 0.323(7)
C(36) 0.196(7) 0.212(7) 0.290(6)
c (11) 0.228(6) 0.250(7) 0.263(7)
C(l2) 0.209(6) 0.224(7) 0.260(6)
c ( 13) 0.211(6) 0.215(7) 0.252(7)
C(l4) 0.221(7) 0.237(6) 0.323(8)
C(l5) 0.227(8) 0.255(7) 0.321(8)
C(21) 0.202(6) 0.242(6) 0.256(7)
c ( 22) 0.181(6) 0.214(6) 0.266(7)
C(23) 0.183(6) 0.220(6) 0.262(7)
C(24) 0.202(7) 0.243(7) 0.298(8)
C(25) 0.200(7) 0.238(7) 0.308(7)
0(11) 0.211(6) 0.326(5) 0.412(6)
0(12) 0.222(5) 0.265(5) 0.338(5)
0(13) 0.229(6) 0.309(6) 0.413(6)
0(21) 0.214(5) 0.293(5) 0.364(5)
0 ( 22) 0.208(5) 0.249(5) 0.316(5)
0(23) 0.238(5) 0.288(5) 0.344(5)
0 ( 23) 0.238(5) 0.288(5) 0.344(5)
68

CHAPTER 8
THE CHARGE-TRANSFER COMPLEX BIPHENYL TRINITROFLUORENONE
8.1 INTRODUCTION
Biphenyl trinitrofluorenone (B-TNF) is a charge-transfer
complex in which biphenyl as in the complex biphenyl tetra
cyanobenzene (Mohwald & Sackmann 1973), acts as the donor and
trinitrofluorenone as the acceptor. This accords with the behaviour of trinitrofluorenone in the complex trinitrofluorenone
hexamethylbenzene, as described by Brown, Cheung, Trefonas and Majeste (1974).
8.2 EXPERIMENTAL SECTION
Crystals were kindly supplied by D. Haarer. They are yellow
in colour and had been grown by the Bridgman method from zone
refined biphenyl and trinitrofluorenone (Haarer 1974). Systematic absences hOl 1 odd, OkO k odd uniquely indicate the space group
P2 /c. The density of the crystal was measured by flotation D = 1 -3 m
1.42g em Least-squares refinement of the cell constants (using
12 centred reflections) as a= 7.195(1), ~ = 28.108(9), £ = 0 0
21.065(7) A; ~ = 94.26(2) yields a calculated density of 1.47 -3
g em with eight molecular pairs in the unit cell. 0
Cu K X-radiation (~ = 1.5418 A) was used. Data were a o
collected for one quadrant of reciprocal space with G < 57 , with
an Hilger and Watts automatic diffractometer. The intensities of 0
the diffracted X-rays were measured with a 72 step 0.01 scan with
18 second background counts at each end of the scan. Attenuators
were used when the monitored pulses exceeded 8000/second. The
intensities of 3 standard reflections were monitored every 50
reflections, and the data scaled accordingly. Lorentz and polarisation corrections were applied and, of the unique reflections measured 3247 had intensities > 3o. Absorption
corrections were made and the maximum and minimum values were 1.55
and 1.171 respectively. The linear absorption coefficient is
69

-1 9.35cm Following this, further scaling, based on the
-equivalence of hkO and hkO reflections were carried out. A linear
variation in scale factor, was assumed for the hkl reflection
intensities, between successive hkO and h k+l 0 reflections. (See
Appendix B.) In this case such additional scale factors were
applied to partially offset the effects of detectable crystal movements.
8.3 STRUCTURE DETERMINATION AND REFINEMENT
Fifty-eight of the seventy non-hydrogen atoms were located by
use of the direct methods program MULTAN (Main, Woolfson & Germain
1971) utilizing the 249 reflections having E > 2.0. The twelve
remaining atoms were located using conventional difference Fourier
techniques. Refinement, in blocks with isotropic thermal
parameters for the atoms resulted in a value for R of 0.148. 1
Refinement of the 630 parameters required by a seventy atom
model (anisotropic thermal parameters) is very expensive. It
necessitates varying blocks of the parameters rather than the
model as a whole. The blocks used were those for whole molecules
or pairs of molecules. Program BONDSTAT was used to obtain
comparative estimates, of standard deviations, of equivalent sets
of bonds between molecules, to help determine the order of
refinement of blocks. The entire model was not refined to
convergence because of the cost involved, but to a point where
R = 0.108. When comparison is made with the similar structure 1
analyses reported in this thesis, the expected minimum value
of R1
is approximately 0.075. It is felt that with R1
at 0.108
this is a satisfactory state in which to leave this structure.
8.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE
The structure of the complex consists of alternately
positioned biphenyl and trinitrofluorenone molecules. Figures
8.4a, 8,4b, 8.4c, and 8.4d define the atom labelling scheme used.
Figure 8.4e shows the spatial relationships within the unit cell.
70

Figure 8.4a
DISTANCES AND ANGLES IN BIPHENYL (1)
C(132) C(131) C(141) C(142)
C(133) C(143)
C{134) C(135) C(145) C(144)
71

Figure 8.4b
DISTANCES AND ANGLES IN BIPHENYL (2)
C!232l C!231l C!24H C!242l
C!233l C!243l
C!235l Cf245l C!244l
72

0{111)
Figure 8.4c
DISTANCES AND ANGLES IN TNF (1)
0(131)
0(1}
73
0{122)

Figure 8.4d
DISTANCES AND ANGLES IN TNF (2)
0(231)
C(214)
0{2)
74
0(221}

Figure 8.4e
VIEWS OF THE CRYSTAL PACKING OF BIPHENYL AND TRINITROFLUORENONE
75

It is unusual to have two molecular pairs in the same
asymmetric unit. It does, however, give an opportunity to
obtain a measure of variation of the geometry of molecules in the
crystalline state. Consider the biphenyl molecules. Let their
molecular planes be denoted by B and their phenyl rings by a and
b. Table 8.4a contains interplanar angles for the biphenyl
molecules. Reference to Figures 8.4a and 8.4b indicates that the
deviation from planarity is principally a twist about the axial
direction defined by the bond C{36} - C{46}. The a and b planes
are folded towards each other slightly and this accounts for the
unequal sizes of the angles al - Bl, bl - Bl and a2 - B2, b2 - B2.
The axial twist is in contrast to that observed for biphenyl
in the crystalline state {Trotter 1961}, whereas it is in accord
with the situation prevailing in the vapour phase {Karle &
Brockway 1944}.
Table 8.4a
INTERPLANAR ANGLES WITHIN BIPHENYL MOLECULES
Biphenyl 1 Biphenyl 2 0
Interplanar Angles { }
Planes
a - b 6.99 8.33
a - B 3.44 4.15
b - B 3.55 4.18
For the trinitrofluorenone molecules denote the molecular
planes as TNF, the molecular plane devoid of the -NO groups TF 2
and the planes of the NO groups as eN, dN, and eN. Table 8.4b 2
contains the interplanar angles for the trinitrofluorenone
molecules.
76

Table 8.4b
INTERPLANAR ANGLES FOR TRINITROFLUORENONE MOLECULES
TNF 1 TNF 2 0
Planes Interplanar angles ( )
TF - TNF 1.66 2.63
TF - eN 17.77 12.69
TF - dN 127.62 156.13
TF eN 4.33 174.11 = -5.89
TNF eN 18.50 10.06
TNF - dN 128.16 156.30
TNF - eN 4.52 171.90 = -8.10
The orientations of the -NO groups show considerable 2
variation as can be seen in figure 8.4e. Table 8.4c contains
angles between biphenyl and trinitrofluorenone planes within each
molecular pair.
Table 8.4c
INTERPLANAR ANGLES WITHIN B AND TNF PAIRS
B-TNF 1 B-TNF 2 0
Planes Interplanar Angles ( )
a - TNF 4.71 3.72
a TF 3.31 6.34
b - TNF 4.41 4.72
b - TF 5.56 2.62
B - TNF 2.82 0.83
B - TF 2.82 2.49
The angle between normals to the two biphenyl molecules is 0 0
21.28 , between TFl and TF2 20.94 and between the trinitro-o
fluorenone molecules 23.45 • Table 8.4b shows that the greatest
differences occurring in the molecular geometry are the
differences in orientation of the three -NO groups, these being 2
77

of the order of five, thirty and ten degrees for the c, d and e
planes respectively. These variations are probably due to
crystal packing effects,rather than determining them, as losses
in 'conjugation energy' from nitro-group rotations are small (0.6 kcal/mole - Dashevskii, Struchkov & Akopyan 1966). The average
perpendicular distance between biphenyl and trinitrofluorenone 0
molecules is 3.36 A whereas the distance between them along the 0
stack axis is 3.598 A.
In certain analyses involving trinitrofluorenone a shortening
of the C(l)-C(lll), C(2)-C(211) bond lengths relative to the
equivalent bond lengths C(l)-C(l21), C(2)-C(221) has been observed
(Brown et al. 1974). In this analysis small shortenings of 0.04 0
and 0.02 A respectively in the two molecules are observed. The 0
long bonds C(ll6)-C(l26), C(216)-C(226) are 1.51 A in accord with
the corresponding bond length in the fluorene molecule of fluorene
PMDA.
Atomic parameters are listed in Table 8.4d and structure
factors in Appendix A4. Root-mean-square amplitudes of vibration
for trinitrofluorenone (2) are given in Table 8.4e. Values were
not calculated for the other molecules, but are expected to be of
similar magnitude.
78

Table 8.4d
1 or a11
C(llU 6.13)(2) O~l!lltSI O.l2l9{1} 0.02ltll 0,0019(;1!} O.MH!ll ¥0,002lt£1 -O.OOOlUl I),OOOH2.}
C(lll. 0.088(1} 0.21UU) Q,U7tU) O.nH(41 G,OOHUl O.OOHttl ·O,toll(7t -O,OOOlt1l O.OOO.)(~i
C{lll) 0,066(2) f>.l0l4(£} O.U11(1) O,Oltill O.GOllU) .O.OOH(tl .. Q.OOl0(1) Q,O.OOlfSi) ~O.OCHHtH
C(ll4) O,.OU(l) O,HHt5) O.l71l[7) O.Ul{l! O.OOllOI O,OOH!H ~0.0007Ui -0.01>04{!1) .. 0.0006(21
C(lH) O.llO(l) 0.1H1{S) 0.1091(1) O,OUtlJ O.OOl'H2i O.OOBfll -O.OOOf>UI ·0,0004!1! ~0.001)'5[21
C{lU, O,lHU) 0.290$-UI 0,21ll{U G.Olltll O.OOl)(l) O.OOl2fl} ... o.eoo~t'51 -0.0007!6) O.GOOl(H
c(l.U) 0,1t8!l1 0.2UJ(Sl O,UU(l) O,OH!l1 0.0011(21 o.oonn; O.MGltS} O.tHHJ-7!11 .. Q.M00\2)
C(lU O,:Bl (2) O.,lH (S) Q,HH fH O,Olt tl} O.OOH tll CLMH tll .Q, OOOH£l 0 ,DOlO tal ~o, 0006 PI
C{lO} 0,2fi0{2) 0.2'1Jlf5) O~Ots2t?l (J.O:Uilt Q,00l'if1) O.OO:U:tll -0.0002UI t.i.O<H21il 0,0000{2)
C(lH) 0.16112} tl,Jl'J9{S} 0.112~01 O.OHUl O.<Hlll(2l 0,0024{)1 O.OI:lOH'H 0,00\SPJ 0.000$(2)
<:(USJ 0.210(2) O.l2Hf5l 0.11'18('1 tl,OUill Q,OOiitH 0,002~!ll 0.0005-Ul O.OOll!ll O.Ot06(li
C(H') tL1U(l) O.l'IU((} O.llUUl O.i!UUJ O.OOllfll O.OOHUJ -0,0000(4} wO,OOI>H61 0.0001(2)
C(2JU 0.301(2) O.Oll1(S} 0.2ltl('1} O.OU.!li 0,0009(1} 0,001112:1 o.otiO)(.tl 0.0006t'l -O.MOl\2)
CUl2) O.lOH2.) O,OH6U) O.l128P) O.C2ljl) 0.0014(21 b.002liU O.OOll(~) 0~0001r7J .. (1.0001141
CUJ)) O.JSl(l) 0.0£12!5) 0,1Hif!'l 0,01'101 O.OO:U(l) C.Mlltli 0,0007!,) (1,0007!1} O.OO<HUl
C(2H} O.UlfS) 0.10Ui5) 0.1Utl61 0~019(3) O,OOlSU) O,OOB.tJl O,OQ06(S) !>.M06(6l 0.0006{lJ
<:C.2lS) O.tlt(2) O.lll?[5) 0.23(1?(£) (),02~{2} O.OOU(l) (),(lOll(); 0,000'$(~) O,OOl)C!l 0.0006!l1
C(2l4) O.lHU) 0.0754(.) O,)Ut(51 CLOHUI 0,00070) O.CQ17(H 0,0004(4) 0,0006(Si O,OOCOtl)
COU) O.l55(2l O.OUG{S) O.JHi(fi) 0,022{)} O,OOH(l) G.OOUU} 0.0000(!) 0,0006(6) -G.OM1C:~l
C(U2} O.lU{1l O.OUi(5) O.tH70l O.OlOfli O,CClttl) O~Ot-12{11 O,C.OtlUl 0~0020(1) 0,0007i1l
C(lHJ O.H2!l} 0.0869(6) O.<I7J2(8) D.Oll!l) O,OOBt:U O,OOHH} O,OOlJ(IJ .0,0012{9) -0.000112}
C(lof..C) 0.435(2) O.tlU(Ii.) O.H7S(I} O,Ol'1U) 0.0023(2) 0,0021(4} 0~0001(1) O,OOHUl -O.OtHl61))
CfHSl O,U7{2) -D.UJ1{5) O,JU£(?j 0.026{3} O.OOll(l) O,OIHSOl -O.OtHl(SJ 0.0016("1; -0.000({2)
C(iU) O,l77(2) Q,07U(4) O.lH7U} D.Ol1(2t 0.0009(1) O,OOH(':U O,OCO~H} 0.0008fEil 0.0001{1}
C(lll) O.U9{2) O.l01H4l O.lHl(~l O.tHS{ll O.OOO'i(l) G,DOllUl •O,MOtf51 O,OOOlf~i 0.0:001{2}
C012) 0.366(2) 0.30!15{4) O.l871(6} O.OHU} 0.0015-(:H 0.001lfll -0.0004!') ~0.000~(7) 0.0000f2l
C(lll} CL~HU"l O~lHJ(t} O.UOl!5i O.OU{l) 0".0015(2) O.{lOOi(l) .. c,OtHl~C5l 0.0001(1) -0.000){1}
CClH) CLS7&[2) 0.392S(t} 0.37Sl(U O.iH41ll 0,001)(2) 0,00170} O,OOOlt'l ·0,0004!8) ·0.0001121
CtllS) (1,£30(1) 0.1881(4) O.ll21(S) O.OH{)l 0.00090) to,OOHCll 0.0002(!) 0.0003(1) -0,000){2}
C(lHl O.U){l) O.H2H4J 0.2817(~} 0.016()1 0,0009(2) O.OOOS{l} C.OOOli!i) O.OOOH6l 0.0002(2)
C{lll) 0~£92(1) 0.1750(U O,H.C.O(~) 0.012(2} 0.0005(1) O,OCU(2l -o.OOOt{!!.) -0,"0002!U •0.0000!2}
c(ln) 0.717t2) O.HUt.tl O,l708($i O.!tl6ili o-.001H2) O.MUiH 0.0001(6} O.OOOH7i O,OoOa[2)
<:{123} O.B${2J O.l7a(4) 0.1H1{S) (1.016!31 0.0009(2) O,OOl'Hl} O.OOOH~l .. 0,(H):06('J) .. O.OV0612l
C(l2C) O.H5{2) 0.1220(4) 0.1125{5} 0.017(3) O.OO!)ll!2) 0.001~!31 O.:l-OO.tfS} 0.000((7) -O.OO(Hil)
C(USJ 0.1U(l) O.HU-!4) 0.1Ult5) 0.01712:) 0.0004{1) ().0011lH wO.OOOi{S) O,OOlOCti 0.0000{2):
C(llfi) 0.700(2) O.JH2{4) 0.21tS{SJ 0,016(3} 0.0007(:21 O,OOlllll .. Q.0005d~) -O.OCOl{~} ... Q,00\10{2)
CUJ 0.6U(2) 0.2$H(4) 0.2UJ£6i 0.0200) 0.0010(2) O,O{!B!l) O,!HiC3{6J 0.000-4{7) O-.Oo.OS{2)
O(l) 0.6H{l) O.ll16(l) 0,3041{4:} O.Olln!} 0,0008111 O.OOHill -0,00Hl{4} O.OOU:tSi O,ot\Ufll
K(ll} O,U4(2) .0,351,(4} 0.474G{S) 0.021fll 0,0019!21 0.001&!)) -C,0002{'1 O,OOUfU -Ct.OQ02Ul
0(111) O~.tH(l) O.l21i(l) O.~CH(t) C.Oli(l) 0,0023\2) O,OOlS{)J -0,0009(~1 ~.0022171 O.O!>Cllll
0(112) (l.tU!l) (),)982!4) 0.4936(5-l O.OHtll Q,0024t2) 0,0010(.{)" -0.0011(6} O,OOH!!I -0,0011(2)
t~(H) 0.1Uf1) o.2C3,(C) o.o~6;tsi o.n24(li o.ootlt2) o.oo2001 ... o.oOll!£1 o.ooo7f8l -o.oo0-6!2}
0{121) 0.&12(1) 0.2026(1) 0,0626!4) 0,031(2) O.OOUU) c.OOH(lj 0.0009t5} O,OOOJO. -0.001012)
O(lll) 0.770(1) (1.2U:7(l) 0.0070!"5) o.o-54(3} O.OOHC2l O.OOH{J) -0.001H5l 0.0019{71 -t>,0005i2)
lHlll 0.751{1) O,l9U(l) O,lHH31 O.OllOJ 0.0001(2) 0.0015t)} 0.001!{5) O.OOlOC7) O,OO!if{2}
O(lll) 0.6'72(1) O.H!C(l) 0.1188fS) 0,0.{1()) 0.0010(1) 0~0016{3) 0,001){51 0,0002{7) 0,0010{21
O(ll2l O.t58(l) O.Hefi(l) 0.205:1{4) O~t>l2{2) 0,0010!1) 0.0027(3} -O.Ot~c1)(4) O.OM'l(6) .. o.COO\f2l
C(lll) 0.1£0(1) O.UU(:'J) O.Ull!4l 0~01H2l 0,0008(1) 0.0016(2) -0.0001{4) G.(IQO~t~l .. o.OOOOtH
C{112} O.t70[1) O.H?Sil) Q,Ufi.l{t) 0.017(2} O.OOUfl} 0.001501 .. ().O{UlH5) ~0.0004{6) -0,0001!2)
C(2U) >Q.831{1) O,t58ti4) 0.5792((} 0.021()) O.OOl£tll 0.0010{1) -0.0011(5} tt,0006t61 0.0000!2)
C(lH) 0,196{1) 0,~02S()) 0.60Hi$) 0.025(3} 0.0012(2) O.Mll(ll -0,0008(~) 0.0012{7) O.OO<i2i2i
C{21S) 0.?90{1) 0.5081\l) 0,U90!41 G.OB{)J
C(2U) O.U2!1) O.HS1{Jl 0.10117!41 O.tH':iUl
C{l21) (1.879 [l) O.HlHl) 0, '192Jf4l O.OH(l)
C:{l22) 0.906(1) O.lHl(l) O.BJOHl 0.016(2)
o.oo09!1) o.ool1tl! o.ooo.H~l o.eoot{6J o.ooozc2J 0,0009{1) l.LM150) -0.0006(.) -O.OOOlHil -0.00010)
0.0008\li !1.0017(2} 0.00(19{4) O.OOi'li6J O:.C"00211)
O.COitOl (I,OOU(l! -O,OOOHU 0.0002(6) O.DOOS{2)
C:(22ll O.SlHI€1) 0.42$6{)) 0.'!0)2(.1 0.018.(2) 0.0016!2) 0,00lU2J -0.0002(5) -0.0001(6) 0.0002(1}
C{22t) O.U2(1) G.t726(l) 0.8952i4l O.OH(2} 'D.MlJ(l) 0 .. 00l6(2} -O.OOtHHS; -0.0001\61 C.OOOltll
C!2"2Sl o.utn1 o~U<JS(lJ o.tl2914i o.ono; o.ooo'liu o.oonc:n o.ooosc4J o.ooo9rsr o.ooo1ol Cf22'J 0~8ll0} 0.4Ull.l) 0.7H7{4l !1.01U2l tl.CtH')'il{l) 0.001)(2} -0.0000(4) -O.OOOH:il 0.0001(1!
Ct2~ 0,!9Sfl} 0~~880tJJ 0.7309!41 O.OH(ll 0,0010(11 0.0020{3) -O.OOO)(~l O.OOCli6l 0.0001(2~
0(2) O.ill{l) O.H6H2) 0.12511)) O.OJS{21 0.00011{1) 0.002~i;(l 0,0014((} ll.0007(S) 0.0001!li
K{21) O~USfH O • .C5H)(4) 0.5100{4) O.Cllf)) 0.0011!21 0.0017()1 -0.0021($} 0.0012i6) -0.000212)
Of2ll) O.il,ttl) O.HHIJ) 0.469ill(l) O.OH£3) 0.00Hi2l 0.002:?{~) -0.0002fS} O.t-028(6) -tLOOOJ/21
Oti12J 0.781{1) O.H'56(1) 0.476li!l "(},OS4t)l
Kil21 G.9Uill tl.-407014l 0."161Hi!4) 0,023(21
0(221} 0.\H rlJ c.J651 Pl 0.'115101 o.tH9 fJJ
0(221) 0.9H{l) O • .tlst(l) l.Ol20!ll O.OHl)l
Q,l}020Cl} O.OI.'Il7f2l .-0.002H51 -0,000Si6) O.GO!HllJ
O.fJD2lt2l 0.0020!)) 0:,0004{5} ·0.0009{6) 0.0006{2)
O.D-01911} (1,002lf2J O.C002iS1 -0,001216) O.f.I0\0{1)
iJ,OO;U:if2l 0~001S{2} 0.0007{5) O.O:HHl!6) 0.0001ill
Nt2l) O.H7(ll 0.5389(3) U~828:2fSJ O~Dillll O.OOl0'12i O.OOO<Jtll .O~OtH2!Sl -0.0006{8) {).0002{2}
O(llll 0,7'Hifll 0.564:2(2) 0.87'50!)) O • .OGOf)l 0.0012!11 O.COH!l) 0.0021{4! 0.{100)(6) -0.0004(1)
0(232} O,fi604.(9l D.5S26C2) 0.71!!4(3) \l.040t21 O.OH2fll 0.0020fl} 0.0024(ll -O.D027f4:l -0.0001!1)
79

Table 8.4e
ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION 0
OF TRINITROFLUORENONE ( 2} (A}
c ( 211} 0.18(1} 0.19(1} 0.20(1}
C(212} 0.17(1} 0.21(1} 0.24(1}
C(213} 0.15(1} 0.21(1} 0.27(1}
C(214} 0.18 (1} 0.23(1} 0.26(1}
C(215} 0.17(1} 0.21(1} 0.25(1}
C(216} 0.17(2} 0.19(1} 0.21(1}
C(221} 0.16(1} 0.17(1} 0.23(1}
C(222} 0.17(2} 0.20(1} 0.24(1}
C(223} 0.15(2} 0.22(1} 0.26(1}
C(224} 0.17(1} 0.20(1} 0.23(1}
C(225} 0.16(1} 0.17(1} 0.21(1}
C(226} 0.15(2} 0.18(1} 0.20(1}
c ( 2} 0.19(1} 0.20(1} 0.22(1}
0(2} 0.18(1} 0.238(9} 0.308(9}
N ( 21} 0.19(1} 0.23(1} 0.31(1}
0(211} 0.20(1} 0.31(1} 0.34(1}
0(212} 0.19(1} 0.27(1} 0.39(1}
N(22} 0.19(1} 0.26(1} 0.30(1}
0(221} 0.18(1} 0.31(1} 0.36(1}
0(222} 0.18(1} 0.32(1} 0.35(1}
N(23} 0.13(1} 0.20(1} 0.26(1}
0(231} 0.19(1} 0.25(1} 0.40(1}
0(232} 0.180(8} 0.210(8} 0.354(8}
80

CHAPTER 9
REVIEW OF THE CHARGE-TRANSFER STRUCTURES DETERMINED
9.1 STRUCTURE GEOMETRY AND SYMMETRY
The structures studied do show commonly occurring features
for this class of compound. For simplicity the order of
comparison used shall be that of the thesis chapters. All except
thianthrene PMDA have well defined stack axes. The donor-acceptor 0
perpendicular distances are 3.36, 4.46, 3.51 and 3.36 A. The
distance for fluorene PMDA is unusually long, however its donoro
donor and acceptor-acceptor distances are 3.52 A. The angles
between normals to the molecular planes in 0 0 0 0
9.84, 4.72, and 2.82 and 2.49. Both PMDA
0 the D-A pairs are 4.11,
and biphenyl molecules
can be centrosymmetric. PMDA molecules are found about centres of
inversion; (0,0,0) in the fluorene compound, and on two such
centres (crystallographically independent), (0,0,0) and
(1/2,1/2,1/2) in the thianthrene compound. None of phenanthrene,
fluorene, thianthrene and trinitrofluorenone molecules have a centre of inversion, although fluorene is statistically
disordered, in the unit cell of the compound. It is interesting
that for thianthrene PMDA the situation is mixed. There are two
crystallographically dependent molecules about centres of
inversion along with two thianthrene molecules (which could have
had some mirror plane symmetry) related to each other by the space
group centre of inversion. In contrast, in biphenyl trinitrofluorenone, there are two complete pairs of crystallographically
independent molecules in the asymmetric unit. Thianthrene PMDA
could be regarded in this way also, if only half molecules were considered. That is two half (connected) PMDA molecules and two
half (unconnected) thianthrene molecules. In biphenyl trinitrofluorenone the two systems run along
the same stack axis making similar angles of tilt to it, but tilted with respect to each other, normals to molecular planes
. 0 mak1ng an angle of N 21 to each other.
81

9.2 INTERMOLECULAR CHARGE TRANSFER
This may take place between independent molecules of the
same type within the same chemical unit or between molecules of
different types. So intermolecular charge transfer is possible
in each of these structures betweeen the two types of molecule
present in each case. In addition for thianthrene it is also
allowed between the independent PMDA molecules, and for biphenyl
TNF it is allowed between like members of the crystallographically
independent molecular pairs. Fluorene PMDA is likely to have
reduced D-A charge transfer due to the large D-A distance giving
reduced ~-~ orbital overlap. However fluorene-fluorene charge
transfer should be possible due to the random disordering of
the fluorene molecules breaking the cell to cell space group
equivalence.
9.3 DIMENSIONALITY OF CHARGE CARRIER MOTION
In a structure such as phenanthrene PMDA maximum charge
transfer could be expected where the D-A orbital overlap is
greatest, that is, along the stack axis. It could also be
expected, to a reduced extent, between D and A molecules on
nearest neighbour stacks and on more distant stacks. Hence a
large anisotropy could be expected between measurements parallel
and perpendicular to the stack axis, whilst between those
perpendicular to the stack axis there would be a small anisotropy
governed by variations in the donor-acceptor inter-stack
distances. That this is the case has been shown by Mohwald &
Haarer (1976), the anisotropy of the photocurrents parallel and
perpendicular to the stack axis being in the ratio of - 20:1. As
already discussed, a hopping model (pl4) on a quasi 1-D lattice
(pl2) is an appropriate description. The electron mobility in
fluorene PMDA, with its distinctive structure, could be expected
to be significaltly different in magnitude and degree of
anisotropy, but still predominantly pseudo one-dimensional.
Biphenyl is a fairly typical CT complex and so, as far as
mechanisms for charge transfer are concerned, could be expected
82

to behave similarly to phenanthrene PMDA. Thianthrene PMDA has the 'IT- 'IT overlap for charge transfer but the bent nature of
thianthrene may impede easy flow conduction along a specific direction. Several simple-minded models, in which electrons move from molecule to molecule, can be envisaged. They involve
hopping from D-A followed by translation within the second
molecule. This is illustrated by the pathways shown in Figure
9.3. Figure 9.3
HOPPING PATHWAYS WITHIN THIANTHRENE PMDA
The pathway of type A utilises hops between central, sulphur
containing thianthrene rings, through a central PMDA phenyl ring,
a translation between a thianthrene central and end ring, and an
end to end PMDA translation. Pathway type B involves end to end PMDA and thianthrene translations, and end ring - thianthrene PMDA
hops. If both of these pathways are feasible then others between
these two extremes may be possible. Also other routes are possible involving longer hops. The effect of imposing an
electric field is generally to assist electron motion in the
direction of the electric field. The different effects of electric fields on intermolecular and intramolecular charge
transfer, will alter the anisotropy of the measured mobility,
in ways which depend on the alignment of the electric field. If the effective resistance and dimensionality of a material can be
83

altered markedly by a transverse electric field, then it may have
useful properties for utilisation in components of electronic
devices.
The different hopping distances could give rise to steps in
the field dependence of the electron mobility in the directions in
which they are found. This would be equivalent to describing the
behaviour by a model containing a number of traps. That is to
say, some of the traps postulated in the literature may really be
manifestations of internal periodicities of the molecular
structures comprising the crystalline material.
9.4 SOLID STATE SIGNAL PROCESSING TECHNOLOGIES
The major such technology is that of electronics. Initial
developments were based on macroscopic properties of conduction,
resistance, capacitance and inductance using centimetre sized
components. The advent of the transistor, utilising microscopic
crystal defects in 'solid state' components, revolutionised
electronics. The subsequent miniaturisation of integrated cir
cuits has achieved a high level of refinement. Micro-processors
containing thousands of components, to perform numerous logical
functions, are produced on very small silicon chips. In this
process of refinement the basic functions of the various
components has remained unaltered. The study of new materials
and their properties has in some senses advanced ahead of this
technology. New materials are being used but a number of their
properties are only being superficially utilised. Most solid
state technology is based on electronics, and as its name
implies, electron motion and its management - storage, resistance,
oscillation and direction. Optical and acoustical signals as
input or output are transformed to electrical motion very early
or late in the sequence of processing. However photons and
phonons propagate well in the solid state, and can also
be managed, stored and transformed. Optical technologies
are being developed, utilising thin film light conduction,
diffraction, and excitation of electrons giving photons of
84

different frequency. The laser is the most popular example of
this technology. The optical 'pumping' used therein is in effect a frequency transformation with a simultaneous phase tune. Thin film optical guided waves have considerable potential for
use in optical integrated circuitry. Components for such circuitry may be either passive - lenses, prisms, or active -piezoelectric, electro-optic (Garwin 1975).
Where there is an interface between optical and electronic circuitry there is a need for a photoconducting material to
transform the optical signal to an electronic one. This makes the study of the structural and physical properties of such materials
an important exercise. An increase in understanding of their
properties and hierarchical structures will enable development
of devices using the microscopic features to much greater
advantage than at present. That is to say, on the one hand there
are many known discrete energy levels, and on the other hand there are many devices requiring discrete signals of specific magnitude
but at the moment only very simple discrete values are used. Such an example is that of computer storage, where a binary system is used corresponding to the two states of a magnetic dipole.
9.5 DYNAMIC ELECTRONICS
Up until recently the great majority of components used in
electronic circuitry have individually had a fixed function such
as resistance, inductance, capacitance, conduction/non-conduction.
Circuitry using such components could be termed Static Electronics. A new field which could be called Dynamic
Electronics, in which the function of individual components may vary through a number of modes in sequence, is beginning to
open up. A component would be able to have a variable function, that is, to act like a resistor, or capacitor, transistor or
other static type component on different occasions. The mode
of operation would become programmable. Transistors act like
valves, differentiating between current flows in opposite directions. A transistor whose direction of influence
85

could be altered would be useful. This is normally arranged for
by doping (introducing defects) one end of the transistor material
in order to make it an electron acceptor. CT complexes can
have defects introduced by bending/doping, and excitons produced
in them by optical irradiation. Mechanical strains and
vibrations can modify the effect of the dopant or dislocation\type
defect. A pair of such crystal complexes in concert could exhibit
differential electrical properties similar to a conventional
transistor, but be able to have their characteristics dynamically
modified. Two techniques for modification are application of
electric fields, and imposition of strains using piezo-electric
crystals, causing changes in the crystal's polarisation and
dislocation density respectively. Phosphorescence is modified by
the dislocation density, so vibrations in that density would yield
a pulsing phosphorescence. Phosphorescence is a time delayed
photonic emission. Variation of the delay would give a form of
time modulation of the signal input.
9.6 FUTURE SIGNAL PROCESSING TECHNOLOGY
The interface between input acoustical or optical signal and
electronics may disappear. In the case of light metering devices
the present photon - current circuitry - meter display could be
replaced by photon - integrated optics circuitry - visual display.
In more complex systems the processor may have input to waveform
circuitry followed by dynamic/static electronics. The possible
combinations are numerous, and techniques of electron and photon
management seem set to become much more sophisticated.
The study of new materials and their characterising
properties will, along with their integrated use, yield a new
amalgam of interesting phototronics, dynamic and static
electronics. Miniaturisation of such systems incorporating
organic compounds such as CT complexes for information data
processing, will give processors, closer in type to those
obviously present, yet only partially understood in the
biological world.
86

CHAPTER 10
THE STRUCTURE OF THE TRIDENTATE, FIVE COORDINATE
N-(2-HYDROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV)
10.1 INTRODUCTION
Among the organo-tin compounds pentacoordination is not
common. Examples include a number of bridged and polymeric
structures where the ligands are functioning as bidentates (Ho &
Zuckerman, 1973). However in all mononuclear structures so far
reported the ligands are monodentate. In N-(2-hydroxyphenyl) salicylaldimine dimethyl tin(IV) the ligand is potentially
tridentate and the structure therefore of considerable interest.
The compound was prepared by Ruddick (1973) in the course of
investigations of various Schiff base complexes.
10.2 EXPERIMENTAL SECTION
Well formed crystals of (CH ) Sn(C H O)CHN(OC H ) obtained by 3 2 6 4 6 4
recrystallisation from methanol were kindly supplied by Dr. J.R.
Sams, University of British Columbia.
Diffraction symmetry mmm and systematic absences of X-ray
reflections hkO for h+k odd, Okl for k odd, and hOl for 1
odd, established the space group uniquely as Pbcn. The
crystal density was measured by a flotation method using a mixture
of bromobenzene and 1,2,-dibromomethane. Least-squares refinement
of the cell contents (using 12 centred reflections) as a = 0 -
19.304(5), b = 10.534(3), c = 13.398(4) A yields a calculated - -3 -
density of 1.82g em with 8 molecules in the unit cell. 0
Zr filtered Mo radiation (~ = 0.7107 A) was used. Data were collected from the equivalent sets of reflections hkl and hkl in
0 that portion of reciprocal space for which 9<20 using an Hilger
and Watts automatic diffractometer. The intensities of the
diffracted X-rays were measured using an 80 step single second, 0
0.01 scan with 20 sec. background counts at each end of the scan.
Attenuators were used when the monitored pulses exceeded 8000/s, the attenuating factors being 3.16 and 8.28. The intensities of 3
87

standard reflections were monitored every 100 reflections and the
data scaled accordingly. Lorentz and polarisation corrections
were applied and, of the unique reflections measured 1024 had
intensities >a. Absorption corrections (by Gaussian integration} were applied, and the two equivalent data sets averaged.
10.3 STRUCTURE SOLUTION AND REFINEMENT
All non-hydrogen atoms were located by the application of
conventional heavy-atom procedures using the Sn atom, located from
the Patterson function, for initial phasing. In the final leastsquares refinement, all non-hydrogen atoms except the carbon atoms
comprising the two phenyl rings were refined anisotropically using
the conventional 6-parameter thermal ellipsoid model. The phenyl
carbon atoms were assigned single isotropic thermal parameters. Most of the hydrogen atoms were clearly revealed in difference
electron density maps. All phenyl hydrogens were included in
structure factor calculations in idealised positions assuming 0
C-H 1.08 A. In each methyl group one hydrogen position was well
defined and the remainder were included in idealised positions 0
assuming C-H 1.10 A and tetrahedral H-C-H angles. Refinement
converged with R = 0.044 and R = 0.043. Analysis of F and F 1 2 0 c
statistics indicated that for the most intense reflections, F 0
values were systematically too low and that therefore secondary
extinction was appreciable. This effect was corrected for, using
the angularly dependent function derived by Zachariasen (1967)
and incorporating it into the least-squares equations in the
manner suggested by Larson (1970). In the final difference map -3
there were no peaks of height greater than 0.51 eA or one
sixth of the average height of carbon atom peaks in earlier
difference syntheses.
The function minimized in the least-squares refinement showed
no systematic dependence on F or sin9, and a calculation of the 0
structure factors for the weak reflections not included in the
refinement revealed no anomalies. The final parameters for all
88

Table 10.3a
ATOMIC PARAMETERS FOR N- ( 2-fli'DROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV)
Atom X y z B or B B B B Bl3 B 11 22 33 12 23
Sn 0.0554(1) 0.1587(1) 0.0503(1) 0.0021(1) 0. 0080 (1) 0.0061(1) 0,0004(1) 0.0002(1) 0.0008(1)
0 (1) -0.0465(3) 0.0900(6) 0.0334(5) 0.0025(2) 0.0101(7) 0.0084(5) 0.0005(3) 0.0009(3) 0.0021(6)
0 (2) 0.1370(3) 0.7837(7) 0.0928(6) 0. 0022 (7 J 0.0089(8) 0.0148(3) 0.0004(4) 0.0001(3) 0.0043(7)
C(1) 0.0849(5) 0.040(1) 0.1686 (8) 0.0033(3) 0.010(1) 0.0069(8) 0.0008 (6) 0,0001(4) 0.0003(9)
c (2) 0.0791(5) 0.189 (1) -0.1076(8) 0.0029(4) 0.013(2) 0.0062(7) 0.0012(5) 0.0002(4) 0.0020(9)
N(1) -0.0120(4) 0.3181(8) 0.0997(6) 0.0032(3) 0.009(1) 0.0051(6) 0.0007(5) o. 0004 (3) 0.0009(7)
C(3) 0.0078(6) 0.430(1) 0.1214(8) 0.0040(4) 0.010 (1) 0.0046(8) 0.0008(7) 0.0005(4) 0.0001(9)
C(ll) -0.0987(5) 0.184.(1) 0.0667(6) 3.7(2)
c (12) -0.1658(5) 0.1186(9) 0.0644(7) 3. 8 (2)
C(13} -0.2196(6) 0.199(1) 0.0974(8) 5.0(2)
C(l4} -0.2052(5) 0.317(1) 0.1328(8) 4.9(2)
c (15) -0.1381(5) 0.362(1) 0.1355(7) 4.4(2)
C(l6) -0.0847(5) 0.284(1) 0.1018(7) 3. 5 (2)
c (21) 0.1385(5) o. 404 (1) 0.113 (8) 4.4(2)
c (22) 0.0776(4) 0.480(1) 0.1256(7) 3.9(2)
C{23} 0.0834(5) 0.608(1} 0.1514 (8) 5.0(2)
c (24) 0.1473(5) o. 665 (1) 0.1599(8) 5.4(3)
2 2 2 Thermal parameters are in the form of exp[-(h a
11+ k B + 1 B + hkB + hlB + k1B
23)}
22 33 12 13
89

Table 10.3b
0 ROOT-MEAN-SQUARE AMPLITUDES OF VIBRATION (A)
Sn 0.187(1) 0.214(1) 0.242(1)
0 (1) 0.19(1) 0.240(9) 0.29(1)
0 (2) 0.19(1) 0.21(1) 0.38(1)
c ( 1) 0.22(2) 0.25(2) 0.26(1)
c ( 2) 0.21(2) 0.23(1) 0.29(2)
c ( 3) 0.20(1) 0.21(1) 0.26(1)
N 0.20(2) 0.24(2) 0.28(2)
90

atoms are given in Table 10.3a while the root-mean-square
amplitudes of vibration of those atoms refined anisotropically are given in Table 10.3b. Structure factors are given in Appendix AS.
Anomalous dispersion corrections from Cromer (1965) were applied to the scattering factors for tin only.
10.4 RESULTS AND DISCUSSION
The crystal structure consists of monomeric units of N-(2-
hydroxyphenyl)-salicylaldimine dimethyl tin(IV) packed 8 per unit
cell. Figure 10.4 shows the overall molecular geometry and
indicates that the Schiff base is indeed functioning as a tridentate ligand. Principal intramolecular distances and angles
and their standard deviations are given in Table 10.4. The tri
dentate ligand is approximately planar but there are individual 0
atom deviations of up to 0.2 A from the mean plane through the
ligand and the tin atom. The departure from exact planarity can be described in terms of the orientations of the phenyl rings
0 whose planes are inclined at 10 to each other. They are twisted
so that C(l4) and C(l5) of the first ring are inclined towards C(2), and C(23) and C(24) of the second ring are inclined towards
c ( 1) •
As has been generally observed in pentacoordinate tin complexes (Ho & Zuckerman, 1973), the geometry around the tin
atom in c H NO Sn is best described as distorted trigonal bi-15 15 2
pyramidal. The oxygen atoms are at the apices and the methyl
carbon atoms and the nitrogen atom form the equatorial plane from 0
which the tin atom is displaced by 0.08 A. The geometry of the
Schiff base ligand does however lead to a considerable departure
from the ideal trigonal bipyramidal environment. Thus the 0 0
angle 0(1)-Sn-0(2) is reduced from the ideal 180 to 158.5 , as
a consequence of both oxygen atoms being bent towards the
nitrogen atom of the complex ligand and away from the two methyl groups. Also, in the equatorial plane, the angle C(l)-Sn-C{2)
0 has opened out to 140 • A similar effect has been observed in
91

Figure 10.4
GENERAL VIEW OF ONE MOLECULE OF THE COMPLEX ILLUSTRATING
THE DISTORTED TRIGONAL BIPYRAMIDAL ENVIRONMENT OF THE
TIN ATOM
C(25J
C(2)
92

Table 10.4
BOND LENGTHS AND ANGLES
N-(2-HYDROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV)
Bond Length Bond angle 0
Sn-0(1) 2.108(6)A C(l)-Sn-C(2) 139.6(4)
Sn-0(2) 2.130(7) C(l)-Sn-N 112.5(3)
Sn-C ( 1) 2.10(1) C(2)-Sn-N 107.3(4)
Sn-C(2) 2.12(1) C(l)-Sn-0(2) 158.2(2)
Sn-N 2.225(8) 0(1)-Sn-C(l) 97.0(3)
0(1)-C(ll) 1.35(1) O(l)-Sn-C(2) 98.6(3)
0(2)-C(21) 1.30(1) 0(1)-Sn-N 75.3(3)
N-C(3) 1.27(1) 0(2)-Sn-C(l) 88.0(4)
N-C(l6) 1.45(1) 0(2)-Sn-C(2) 90.2(3)
C(3)-C(22) 1.45(1) 0(2)-Sn-N 83.4 (3)
c-c ( pheny 1) Sn-0(1)-C(ll) 117.6(6)
mean of 12 bonds 1.39(1) Sn-0(2)-C(21) 132.5(6)
range 1.366 to 1.434 Sn-N-C(3) 126.2(8)
Sn-N-C(l6) 112.6(6)
N-C(3)-C(22) 128.8(8)
C(3)-N-C(l6) 121.2(9)
c-c-c (phenyl)
0
mean of 12 angles 120.0(9)
range 117.3 to 121.2
93

the [(CH) SnCl ]-anion (Einstein & Penfold, 1968) where, as in 3 2 3
the present compound, there are only two methyl groups and one
more highly electronegative ligand (a chlorine atom) in the equatorial positions.
Hitherto, nitrogen coordinated to tin in bipyramidal
pentacoordinate compounds, has invariably occupied axial positions. It is now well established that, for a given type
of bond in this type of coordination, axial bonds tend to be
longer than equatorial bonds (e.g. Ho & Zuckerman, 1973). The
Sn-N axial bonds listed by Chow (1971) are within the range 0
2.34-2.49 A with the exception of the bond in (CH } SnNCS 0 3 3
(Forder & Sheldrick, 1970) which is 2.15(6) A. While the large
error in this measurement makes any conclusion suspect it should
also be noted that the second axial bond in the compound, Sn-S 0
is very long (3.13 A) and must be very weak indeed. The 0
equatorial Sn-N length of 2.225(8) A observed in the present
compound may therefore be said to conform to the general
pattern in being significantly shorter than those axial Sn-N
bonds with which direct comparison is possible.
94

CHAPTER 11
THE ISOMERIC CONFORMATIONS OF THE
HEXA-V-DITHIOCACODYLATO TETRAZINCSULPHIDE
MOLECULES
11.1 INTRODUCTION
The hexa-~-dithiocacodylato tetrazincsulphide molecules are a
group of conformational isomers. The zinc atoms are tetrahedrally
arranged about a central sulphur atom. The central sulphur atom,
taken in turn with each of the six possible pairs of zinc atoms,
is at the centre of six fused cyclohexane type rings. If these
have conventional boat and chair conformations the conceivable
distributions range from six chairs or boats to three of each.
There are ten such possibilities detailed in Table ll.lb. Each
zinc atom is shared between three rings and this association
may prevent the occurrence of some of these possibilities.
The full symmetry of the tetrahedron imposes an overall
regime of symmetry on the molecule. That is to say, any
symmetry the molecule may possess must be a subset of that of the
tetrahedron. The presence of a three-fold axis of symmetry
would imply a six-boat, six-chair or three-boat,three-chair
configuration. Mirror plane symmetry would require at least one
one ring to be planar.
The four-fold inversion axis of symmetry of an exact
tetrahedron could only be found in a four-chair, two-boat (or vice
versa combination) • Thus symmetry considerations help identify
some possible conformational details in different isomers.
Furthermore the presence of symmetry is indicative of
portions of a molecule making equal contributions to the
potential energy of the whole molecule. Symmetrical molecules
have a lower entropy than asymmetrical ones, so are less likely to
occur. That they do occur, indicates that the minimisation of
potential energy dominates the tendency to maximisation of entropy
in such cases, and that the tendency to equipartition of potential
energy is also important. In a system such as this, where six
95

bridging ligands, may individually, alternately, take on chair and
boat configurations in the free state, a number of local potential
energy minima, corresponding to different isomers, may exist. This
is illustrated by the fact that different isomers have been cry
stallised for this complex. The probability that any particular
isomer will form depends on the environment in which crystallisa
tion takes place, and the number of ways of arranging ligands, in
chair and boat forms, which result in each possible isomer. The
second of these factors can be calculated in a strict combinat
orial sense and values are given in Table ll.la. Some of the
possible arrangements may be impossible to obtain, with sensible
chemical geometry and low potential energy.
Table ll.la
CONFORMATIONS OF CHAIRS AND BOATS (c=chair, b=boat)
Configuration 3c 3b
Number of ways 20
4c 2b
15
2c 4b
15
Sc 1b
6
lc Sb 6c
6 1
6b
1
The possible isomeric forms may be represented schematically.
The six edges of a regular tetrahedron are related, in pairs,
by its 3 s4
symmetry axes. When such a pairing is carried out for bridging ligands then the structural possibilities including
those discussed above may be represented as shown in Table 11.lb.
Table 11.lb
POSSIBLE CONFIGURATIONS IN A 2 RING TYPE CLASSIFICATION
Molecular Forms Asymmetric
With Possible Molecular
Point Symmetry Forms
bbb CCC CCC cbc bcb cbc CCC bee ebb bee bbb CCC bbb cbc bcb bbc ebb bbb bbb CCC
96

11.2 EXPERIMENTAL SECTION
The crystals are long, thin, needle-like in appearance and
transparent in colour and were grown by recrystallisation of the
hexagonal crystals obtained when the compound (CH3
)2As(S)SAs(CH
3)
2 (Bunsen, 1843; Cameron & Trotter 1964) is reacted with
Zn(Cl04
)2
.6H2o in ethanol or acetone. (Johnstone, Fergusson &
Robinson 1972). Preliminary precession photographs of these
crystals exhibited monoclinic symmetry and the systematic
absences hOl h+l odd, OkO k odd uniquely indicate that
the space group is P2 /n. 1
measured by flotation, D m
The density of the crystal was -3
= 2.00g em • Least-squares refinement
of the cell constants as a= 32.508(12), b = 18.489(7), c = o -o
30.589(12) A, ~ = 95.70(2) -3
yields a calculated density of 1.90g
em with four molecules in each asymmetric unit of the unit cell. 0
Zr filtered Mo radiation (A= .7107 A) was used. Data was
collected from the positive k and 1 region of reciprocal space 0
with 9 < 15 using an Hilger and Watts automatic diffractometer 48 0
step, single second, 0.01
at each end of the scan.
scan with 12 second background counts
The intensities of 3 standard
reflections were monitored every 100 reflections and the data
scaled accordingly. Lorentz and polarisation corrections were
applied. Of the 5176 reflections measured 735 had intensities
>3o, 1355 > 2o and 2742 > o •
11.3 STRUCTURE DETERMINATION AND REFINEMENT
A structure containing forty arsenic and zinc atoms, fifty
two sulphur atoms and forty-eight carbon atoms, that is, a total
of one hundred and forty non-hydrogen atoms having 560 variable
parameters is reasonably large. Furthermore, the quantity of high
quality data was not proportionately large. A variety of
techniques were applied in the solution of this structure.
Patterson and direct methods were applied in obtaining phases
giving acceptable structural parameters. The principal problem
arising was that of imperfect phasing giving rise to spurious
peaks yielding apparently polymeric structures. The computer
97

program PATINV (described in Chapter 12) was developed to help
search for the best solution strategy. Its first function was to
test the model for consistency with the Patterson Map, with which it agreed very well. The vectors between spurious 'atoms•, and between them and the molecules, were the same as those obtaining
within the molecules, so this test did not provide the desired discrimination. Using the atomic superposition option a model
identical to some of those already obtained by direct methods
resulted.
The structure was eventually elucidated using the 1974
edition of MULTAN {Main, Woolfson & Germain). 58 atoms were
located based on the phasing of the 500 reflections having highest
E values. Apart from carbon and hydrogen atoms the positions of
all but 4 of the remaining sulphur atoms were obtained by difference Fourier calculations following least-squares
refinements of the model the positions of the remaining atoms were calculated. The refinements had to be done in blocks.
Between cycles the variance of the bond lengths of the bond types present were calculated using the program BONDSTAT (see section
12.4). The strategy used, particularly in the late and expensive refinements, was to vary those blocks where the variance was
greatest. The blocks of parameters varied were those pertaining
to individual molecules. When temperature factors as well as
positional parameters were refined some of the temperature factors became negative, and the lowest value of R obtained was 0.20.
1 On the other hand, when temperature factors were held at the
constant values indicated in Tables 11.3a,b,c and d the lowest value of R obtained was 0.24. Structure factors for this latter
1 case are given in Appendix A6. The quality of the data did not
merit further expenditure.
11.4 DESCRIPTION AND DISCUSSION OF THE STRUCTURE
In 1972 Johnstone, Fergusson and Robinson reported the
formation of a three chair, three boat hexa-~-dithiocacodylato tetrazincsulphide complex and the preparation by recrystallisation
98

Table 11.3a
ATOMIC PARAHETERS FOR [SZn4 {AsS2 (c~3 J 2 } 6 J (1)
Atom X y z B
s (1) 0.265(3) 0.226(6) 0.677(4) 3.50
Zn(ll) 0.289(1) 0.339(2) 0.643(2) 2.00
Zn ( 21) 0.306(1) 0.139(2) 0.654(2} 2.00
Zn ( 31} 0.207(1) 0.215(2) 0.652(1) 2.00
Zn ( 41} 0.276(2) 0.241(2) 0.758(2) 2.00
As (11) 0.332(1) 0.409(2) 0.747(1) 2.00
As (21) 0.321(1) 0.073(2) 0.766(1) 2.00
As (31) 0.161(1) 0.261(2) 0.757(1) 2.00
As ( 41) 0.360(1) 0.251(2} 0.587(1) 2.00
As(51) 0.216(1) 0.046(2) 0.604(1) 2.00
As (61) 0.205(1) 0.374(2) 0.582(1) 2.00
s (111) 0.359(3) 0.384(6) 0.682(3) 3.50
s (141) 0.346(3) 0.318(6) 0.793(3} 3.50
S(221) 0.313(3) 0.032(6) 0,694(3) 3.50
s ( 241) 0.289(3) 0.131(6) 0.786(4) 3.50
s ( 331) 0.139(3) 0.281(6) 0.690(3) 3.50
s (341) 0.207(3) 0.319(6) 0.776(3) 3.50
S(521) 0.272(3) 0.099(6) 0.590(4) 3.50
S(531) 0.180(4) 0.105(6) 0.651(4) 3.50
S(611) 0.238(4) 0.410(6) 0.638(4) 3.50
S(631) 0.195(3) 0.260(6) 0.579(3) 3.50
c (111) o. 272 0.418 0.735 4.00
C(ll2) 0.355 0.502 0.768 4.00
c ( 211) 0.376 0.119 o. 773 4.00
C(212) 0.322 -O.Oll 0.806 4.00
c (311) 0.117 0.276 0.795 4.00
C(312) 0.178 0.158 0.760 4.00
c (411) 0.412 0.307 0.591 4.00
C(412) 0.360 0.188 0.535 4.00
C(Sll) 0.234 -0.046 0.631 4.00
C(512) 0.180 0.026 0.550 4.00
c ( 611) 0.236 0.399 0.534 4.00
C(612) 0.152 0.422 0.576 4.00
99 THE tH~RAttY '
UNIVERSITY C.\N7l:RBURY CHRISTCHURCH, N.Z.

Table 11.3b
ATOMIC PARAMETERS FOR [SZn {AsS (CH ) } ] 4 2 3 2 6
(2)
Atom X y z B
s ( 2) 0.418(4) 0.229(6) 0.001(4) 3.50
Zn(12) 0.422(1) 0.230(2) 0.077 (1) 2.00
Zn (22) 0.482(2) 0.241{2) -0.028(2) 2.00
Zn ( 3 2) 0.375(1) 0.342(2) -0.018(1) 2.00
Zn ( 4 2) 0.383(1) 0.121(3) -0.028(1) 2.00
As (12) 0.390(1) 0.051(2) 0.082(1) 2.00
As(22) 0.470(1) 0.095(2) -0.095(1) 2.00
As (32) 0.293(1) 0.229(2) -0.054(1) 2.00
As ( 42) 0.548(1) 0.234(2) 0.086(1) 2.00
As(52) 0.455(1) 0.412(2) -0.077(1) 2.00
As(62) 0.372(1) 0.394(2) 0.089(1) 2.00
S(ll2) 0.436(3) 0.113 (6) 0.118(3) 3.50
S(142) 0.406(4) 0.028(6) 0.028(4) 3.50
S(222) 0.508(3) 0.202(6) -0.105(3) 3.50
s ( 24 2) 0.402(4) 0.119(6) -0.095(4) 3.50
S(332) 0.301(3) 0.330(6) -0.020(4) 3.50
s (342) 0.315(3) 0.129(6)' -0.031(3) 3.50
S(412) 0.488(4) 0.303(6) 0.101(4) 3.50
S(422) 0.533(3) 0.168(6) 0.035(3) 3.50
S(522) 0.509(3) 0.360(6) -0.025(3) 3.50
S(532) 0.391(3) 0.387(6) -0.086(3) 3.50
S(612) 0.373(3) 0.293(6) 0.109(3) 3.50
S(632) 0.406(3) 0.437(6) 0.035(4) 3.50
C(l21) 0.379 -0.037 0.113 4.00
C(122) 0.340 0.109 0.073 4.00
C(221) 0.492 0.049 -0.039 4.00
C(222) 0.476 0.027 -0.143 4.00
C(321) 0.233 0.211 -0.064 4.00
C(322) 0.315 0.237 -0.111 4.00
C( 421) 0.567 0.178 0.139 4.00
C(422) 0.592 0.299 o. 072 4.00
c ( 521) 0.472 0.397 -0.135 4.00
C(522) 0.455 0.524 -0.062 4.00
C(621) 0.393 0.450 0.143 4.00
C(622) 0.313 0.419 0.076 4.00
100

Table 11.3c
ATOMIC PARAHETERS FOR [SZn4
{AsS2
(cH3
)2
}6
J (3)
Atom X y z B
S(3) 0.725(3) 0.272(6) 0.816{4) 3.50
Zn(13) o. 689 (1) 0.169(2) 0.844(1) 2.00
Zn (23) 0.801(1) 0.253(2) 0.849(1) 2.00
Zn(33) 0.708(1) 0,363(2) 0.857(2) 2.00
Zn(43) 0.726(2) 0.278(2) 0.742{2) 2.00
As(13) 0.661(1) 0.111(2) 0.732(1) 2.00
As (23) 0.833(1) 0.293(2) o. 741 (1) 2.00
As (33} 0.667(1) 0.438(2) 0.753(1) 2.00
As (43) 0.778(1) 0.073(2) 0.899(1) 2.00
As(53) 0.810(1) 0.397(2) 0.917(1) 2.00
As ( 63) 0.628(1) 0.277(2) 0.909(1) 2.00
s ( 113) 0.672(3) 0.071(6) 0.796(3) 3.50
S(143) 0.724(3) 0.160(6) 0.702(4) 3.50
s (223) 0.842(3) o. 311 (6) 0.820(4) 3.50
S(243) 0. 774 (3) 0.330(6) 0.709(3) 3.50
s (333) 0.654(3) o. 414. (6} 0.821(3) 3.50
S(343) 0.675(3) 0.344(6) 0.717(4) 3.50
S(413) o. 721(3} 0.120(6) 0.916(4) 3.50
S(423) 0.816(4) 0.117(6) 0.850(4) 3.50
S(523) 0.819(3) 0.296(6) 0.920(4) 3.50
S(533) 0.760(4) 0.442(6) 0.868(4) 3.50
S(613) 0.616(3) 0.208(6) 0.854(3) 3.50
S{633) 0.690(4) 0.328(6} 0.928(4) 3.50
c (131) 0.619 0.187 0.729 4.00
c (132) o. 641 0.030 0.695 4.00
C(231} 0.835 0.188 0.732 4.00
C(232) 0.878 0.339 o. 715' 4.00
C(331) 0.622 0,498 0. 725 4.00
C(332) o. 719 0.492 0.752 4.00
c ( 431) 0.812 0.078 0.956 4. 00
C(432) 0.769 -0.030 0.884 4.00
c (531) 0.863 0.439 0.903 4.00
C(532) 0.800 0.430 0.975 4.00
C(631) 0.618 0.224 0.960 4.00
C(632) 0.591 0.361 0.900 4.00
101

Table 11. 3d
ATOMIC PARAMETERS FOR [Szn4
{AsS2
(CH3
)2
}6
J (4)
Atom X y z B
S(4) 0.580(4) 0.261(6) 0.500(4) 3.50
Zn(14) 0.563(1) 0.268(2} 0.418(1) 2.00
Zn(24) 0.617(1) 0.367(3) 0.518H) 2.00
Zn(34) 0.608(1) 0.158(2) 0.521(1) 2.00
Zn(44) 0.520(2) 0.266(2) 0.524 (2) 2.00
As(14) 0.471(1) 0.292(2) 0.421(1) 2.00
As(24) 0.545(1) 0.417(2) 0.597{1) 2.00
As(34) 0.538(1) 0.092(2) 0. 587 (1) 2.00
As(44) 0.625 (1) 0.428(2) 0.405(1) 2.00
As(54) 0.698(1) 0.257(2) 0.565(1) 2.00
As(64) 0.597{1) 0.079(2) 0. 415 ( 1) 2.00
s (114) 0.516(4} 0.332(6) 0.395(4) 3.50
8(144) 0.465 (3) 0.322(6) o. 496 {3) 3.50
S(224) 0.610(3) 0.424(6) 0.589(4) 3.50
S(244) 0.514(3) 0.322(6) 0.600(3) 3.50
S(334) 0.589(4) 0.125(6) 0.594(4) 3.50
S(344) 0.485(3) 0.148{6) 0.546(3) 3.50
8 ( 414) 0.635(3) 0.348{6) 0.389(4) 3.50
S(424) 0.607(3) 0.464(6) 0.463(4) 3.50
S(524) 0.684(3) 0.354(6) 0.519(4) 3.50
S(534) 0.663(3) 0.188(6) 0.508(3) 3.50
8(614) 0.556(3) 0.164(6) 0.390(3) 3.50
8(634) 0.605(3) 0.044(6) 0.491(4) 3.50
c (141) 0.420 0.327 0.386 4,00
C{l42) 0.474 0.188 0.417 4.00
C(241) 0.535 0.466 0.652 4.00
C(242) 0.516 0.471 0.548 4.00
C(341) 0.545 -0.002 0.559 4.00
C(342) 0.519 0.078 0.645 4.00
C(441) 0.582 0.464 0.363 4.00
c (442) 0.676 0.478 0.400 4.00
c (541) 0.756 0.232 0.573 4.00
C(542) 0.673 0.259 0.619 4.00
C(641) 0.586 -0.009 0.380 4.00
C(642} 0.653 0.114 0.407 4.00
102

Figure 11. 4a
STRUCTURE OF [SZn {AsS (CH ) } ] JOHNSTONE ET AL. 4 2 3 2 6
(Three top rinqs are chairs, three bottom rings boats)
5(24)
$(61)
5(63)
103

Figure 11. 4b
STRUCTURE OF [SZn {AsS (CH ) } J BATES 4 2 3 2 6
(All rings are boats)
5(41')
5(4'1')
104

using other solvents, of three other crystalline forms. The
molecule, shown in Figure 11.4a had approximately C point group 3
symmetry. The structure, shown in Figure 11.4b, of a second
isomer consisting of six boats, has since been determined by Bates
(1978) and also has C (crystallographic) symmetry. If the 3
sulphur atoms are designated either central or ring then the 'bow-
stern' interatomic vectors of the boats are central sulphur
arsenic in three cases and ring sulphur-zinc in the three other
cases. Boats and chairs have been distinguished by mean-plane
calculations. The 1972 structure has twist-chairs in place of the
central sulphur-arsenic boats, with 'head-toe' interatomic
vectors in the same sense. The structure with space group P2 /n 1
and z = 16 discussed by Johnstone, Fergusson and Robinson is the
subject of study here. In each asymmetric unit it has three
molecules, shown in Figures 11.4c,d,e, with the three chair, three
boat C conformation and a fourth molecule, (see Figures 11.4f,f') 3
itself the third conformational isomer discovered, with S 4
symmetry. That is, this fourth molecule is the fourth type
indicated in Table ll.lb. Models can be made of the three
isomers whose structures have been determined as well as the
first of the asymmetric molecular forms in Table ll.lb. None
of these show undue mechanical strain, nor unacceptable contact
distances between the methyl groups. Of the crystalline forms
originally obtained by Johnstone et al. only the one with
hexagonal space group P6 has not been the subject of succesful 3
structure determination.
It may be further pointed out that as the space group P21/n
has a centre of inversion, molecules in different asymmetric units
must have enantiomorphic isomers in others, so there are twice as
many isomers as may at first appear to be the case. That two
different isomers should occur in the same asymmetric unit in the
proportion 3:1 is extremely unusual. On the other hand, as has
been discussed in 11.1, the compound is unusual in that it can
potentially adopt a number of forms five of which are potentially
105

Figure 11. 4c
STRUCTURE OF (SZn {AsS (CH ) } ] MOLECULE (1) 4 2 3 2 6
(Three top rings are chairs, three bottom rings boats)
S(241) S(141)
S(221)
106

5(222)
Figure 11.4d
STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE (2) 4 2 3 2 6
(Three top rings are chairs, three bottom rings boats)
5(242)
5(422)
As(52)
107
5( 1 42) 5(342)
5(632)

Figure 11.4e
STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE (3) 4 2 3 2 6
(Three top rings are chairs, three bottom rings boats)
S(243) S{143) S(343)
As(23)
S(223)
S(613)
As{53)
108

5(224)
Figure 11.4f
STRUCTURE OF [SZn {AsS (CH } } ] MOLECULE (4) 4 2 3 2 6
(Four chairs and two boats)
5(244) .S(144)
5(414) S(63L.)
S(53L.)
109

Figure 11. 4f'
STRUCTURE OF [SZn {AsS (CH ) } ] MOLECULE (4) ALT. VIEW 4 2 3 2 6
(Four chairs and two boats)
5(334)
5(424)
110
Zn(44)
5(144)
5(114) 5(414)

symmetric whilst others are definitely asymmetric. It is unlikely
that the gradients of potential energy towards formation of the
different isomeric forms will be of equal height or slope. If
P,. is defined as the probability of crystallisation of isomer
i1fn solvent (environment) j. P .. can then be represented as the
1] product of two factors P. and f .. where P. is the number of ways
in which an isomer may f~rm (as 1~iven in ~able ll.la) and f,. is a 1]
formation factor for isomer i in environment j. In some
environments it appears that for one particular value of i there
is a value of P close to 1.0, whilst in the case under study ij
here there are two isomeric components. For this environment
(recrystallisation of the hexagonal crystals from dichloro-
methane - di), j = di, for the three chair, three boat structure
P = 20f , and for the four chair, two boat structure ldi ldi
P = 15f The observed ratio for P : P is 3:1 which 2di 2di ldi 2di
taken with the above relationships implies that f /f = 9/4. ldi 2di
This indicates that the paths taken by the molecules to
potential energy minima on crystallisation as different isomers
are unequal, and gives a guide to the effective overall
differences.
Bond lengths and angles for the molecules are presented 'ring
by ring' in Figures 11.4g,h,i and j. The range of values of
bond lengths and angles reflects the poor quality of the data.
111

Figure 11.4g
DISTANCES AND ANGLES IN MOLECULE (1)
S(l) S(1)
9:411)
S!221) S(L.21)
As(11) As(t.1)
S(l) S(1)
As(21) As(511
S(1) S(1)
As(31) As(61)
S!2t.1) S(3t.1)
112

Figure 11. 4h
DISTANCES AND ANGLES IN MOLECULE (2}
$(112)
S(2) S(2)
S(t.12)
5[142) 5[222) 5[422)
5(2) ~ As(L.2) As(12)
5[2) S(L.22) 5522)
5[222) 5(332)
As(22)
5(2)
As(62)
113

Figure 11.4i
DISTANCES AND ANGLES IN MOLECULE (3)
Sl113)
S(3) S(3)
S(L.13) 2.
S(113) S(1L.3) S(223l S(t.231
As{13) As{l.3)
S{3) S(3)
51333) S{533l
As{23) As{ 53)
S{3) S(3)
5(21.3) 5(31.3)
114

Figure 11.4j
DISTANCES AND ANGLES IN MOLECULE {4)
5{t.) S(~l
S{lll.) 5(22~) S(t.2L.)
Asfit.l As(4L.)
S{t.) S(t.2L.I S{l.)
SI22L.) stw.l 5(33L.l S!53t.l
As(2t.) As(54l
5(4) S(l.)
S(53t.l
St33t.)
As(34) Asi3L.)
5(344)
115

CHAPTER 12
COMPUTER PROGRAMS
12.1 INVESTIGATION OF PEAKS IN PATTERSON MAPS (PATINV)
12.1.1 INTRODUCTION
This program forms the following tasks:
(1) Obtains possible atomic positions using Harker planes and
lines.
(2) Verifies the consistency of atomic positions of a proposed
model with the Patterson peaks.
(3) Extends models by atomic superposition (using only peak positions).
12 .1. 2 INPUT
(1) Title
(2) Cell constants and control parameters.
(3) Patterson peak positions.
(4) Proposed atomic positions.
12.1.3 OBTAINING ATOMIC POSITIONS USING HARKER PLANES AND LINES
The N real space symmetry operations are used to obtain the M
Patterson space symmetry operations by removing the translational
components (and adding a centre of symmetry if necessary).
The N space group symmetry operations are used to form the
N(N-1)/2 possible differences between equivalent positions which
may be on Harker lines or sections. This list is then reduced, to
the unique set in which none are related by Patterson space group
symmetry.
This set of Harker vectors is of the form h = a + x (i=l,3) i i i
and with each can be associated a vector z with components
z = o .. (i=l,37 j=l,3) where o .. is the Kronecker delta and .i ,lJ 1] 1 = J 1ff x = 0. (a = 0 or 0.5)
i i For any pair of vectors z , z where !z + z I > 12 atomic
-k -m -k ----m
116

positions can be found provided that vectors of classes k and m
exist in the Patterson map.
Consider the following Harker vector classes, and their
associated z vectors
h = 1/2 + 2x, 1/2, 1/2 - 2z then z = (1,0,1) k -k
and h = 0' 2y, 1/2 z = (0,1,0) m
I 1 (l,o,l) (o,l,o) 1
-m then lz + z = + = 3, and so Harker vectors
-k In of these 2 types will be able to yield possible atomic
coordinates.
For example, consider the two vectors in the Patterson map
v = 0.7 0.5 0.3 -k
and v = 0.0 0.6 0.5 -m
The solutions of 1/2 + 2x = 0.7
2y = 0.6
1/2 - 2z = 0.3
are calculated using modulo 1 arithmetic, as an ideal crystal
structure is invariant with respect to unit cell translations.
This leads to eight possible solutions each of which may be
generated from the vector £ = (0.1 0.3 0.1) by addition of 0.5
to any of the three coordinates.
The program lists the set of vectors ~ and the combinations
of z and z found to be acceptable when all possible pairs of -k -m
vectors in the map have been considered. Then the Patterson peak
list is searched and the peaks classified according to their z
character. The elements of classes of vectors taken two at a time
are then combined to yield possible atomic positions p, only one
of the eight possible positions being calculated at this point.
If 1~1 + ~n I > 12, as in the cases~·= (1,0,1) and~"= (0,1,1)
then it is necessary that p ' = p ". 3 3
A lot of the atomic positions 'found' will not be genuine,
so the number is reduced by accepting only those identical
positions,£ and£', which arise from two distinct pairs of z
classes. (Identity is established by equality of 2£ and 2£', to
overcome the ambiguity of 0.5 in each coordinate). Note that the
117

pair ~, Q is acceptable provided as always that !~! > 12 Having thus obtained a set {A} of n(A) possible atomic
positions, each of which has eight possible representations,
it is necessary to find the set (or sets) of such positions, which show the greatest consistency with the Patterson map vectors.
The set {A} is expanded to contain the 8n(A) possible vectors and contracted partially, where possible, by the elimination of
those which are related by the space group symmetry to obtain the
set A'. Differences are then taken between members of the set and
individually compared with the set of vectors which comprise the
Patterson map.
Consider an atom at A and another at B, then vectors A-> S(B)
are formed, where S represents the operation on B of the space
group symmetry operations. (S is the identity operation so 1
S (B) =B). For atoms A and B, a maximum N vectors may be 1
A
\
\
I
' '
Figure 12.1.3
VECTORS IN PATTERSON SPACE S (B)
].. ~·
\ \ ~.
S (B) 3
S (B) 2 - ~.
" ", S (B) ' 4 '::!.
found. The vectors are counted as they are located and a matrix
of these counts is printed out. It is symmetric about the
diagonal. Examination of the matrix will indicate a set of
atomic positions consistent among themselves with the Patterson peaks.
118

The matrix of Table 12.1.3a shows such a set. There are
three distinct sets of positions each with two unique
possibilities.
The
and
set
the
Table 12 .1. 3a
VECTOR COUNT MATRIX
x' x" x' x" x' x" 1 1 2 2 3 3
x' 4 4 2 2 4 3 1
x" 4 4 2 4 3 0 1 -
x' 2 2 2 3 4 4 2
x" 2 4 3 4 4 4 2
x' 4 3 4 4 4 4 3 - -
x" 3 0 4 4 4 2 3
of positions showing greatest consistency
submatrix obtained is presented in Table
Table 12.1.3b
VECTOR COUNT SUBMATRIX
4 4 3
4 4 4
3 4 4
are underlined
12.1.3b.
The positions are x", x", and x'. It is possible for more 1 2 3
than one such set to be found and the two are frequently related
by a translation. This may or may not be between equivalent
origins. If so any one will do, if not each model position can
be tested.
12.1.4 VERIFICATION OF MODEL'S CONSISTENCY WITH THE PATTERSON MAP
This falls into two modes. The first, tests consistency of
new atom positions, with those already established. A rectangular
matrix is produced, with all atom positions listed downwards and
the known atom positions listed across. Table 12.1.4a shows the
119

Table 12.1.4a
POSITIONAL CONSISTENCY MATRIX
X X X 1 2 3
X 4 4 4 1
X 4 4 3 2
X 4 3 2 3
a 4 4 1 1
a 4 2 4 2
a 0 2 1 3
a 4 4 2 4
case for three established positions and four proposed positions
of which a is clearly incorrect whilst a 1'
a 2'
and a are likely 3 4
to be correct.
The second mode is that in which the established and new
positions are all checked against each other. An example is given as Table 12.1.4b.
Table 12 .1. 4b
MATRIX OF ESTABLISHED AND NEW POSITIONS
X X X b b b b 1 2 3 1 2 3 4
X 4 4 3 3 2 1 0 1
X 4 4 2 3 4 0 1 2
X 3 2 4 4 3 1 2 3
b 3 3 4 2 4 0 1 1
b 2 4 3 4 3 2 2 2
b 1 0 1 0 2 4 4 3
b 0 1 2 1 2 4 4 4
The positions x , x , x , b , b form plenty of Patterson 1 2 3 1 2
vectors as do b and b • It is possible that by an allowed 3 4
translation b and b could be brought into better positions with 3 4
respect to the first five positions.
120

12.1.5 MODEL EXTENSION BY ATOMIC SUPERPOSITION
As the Patterson map contains N copies of the chemical
structure, each of which has an atom at the origin of the map,
the addition of an atomic position vector to the peak positions
will move one such structure to the correct chemical site. Those
Patterson peaks which show good agreement with the Patterson map when vectors are formed between them and several atomic positions,
already found, may yield further atomic positions. Each of the
atomic sites known at any point in time may be used for the
Patterson map translation, as some may enable better resolution of
'molecular fragments' than others. The usual geometrical tests
should be made for chemical sense. This process is one of
atomic superposition, and, as it only uses the peak positions,
it could be expected to be lighter in its use of computing
resources, than those using all calculated points in vector space.
As more atomic positions are found they are added in, and assist
in the location of further atomic sites.
The program, when running under this option, uses the
processes described in Task 2, and some empirically determined
criteria, to indicate likely atomic positions, amongst those
which are possibilities.
12.2 LINEPRINTER SHADED ELECTRON DENSITY MAPS (ARTSTC)
12.2.1 INTRODUCTION
A major task in crystallography is the computer simulation of
the contents of the unit cell of the crystal concerned, and visual
representations of the electron density can be a useful aid to
structure solution. The unit cell is, in general, a parallelepiped
defined by the vectors ~~~~£· The electron density is usually
calculated at discrete points in planar sections parallel to a
face of the unit cell. The shape of the sections is that of a
parallelogram.
This program represents the electron density by a shaded map,
in which various characters are produced by overprinting with a
121

lineprinter. For viewing electron density cross-sections such a representation is superior to alphanumeric (single character) or numeric plots. In the program the electron density is
interpolated between the calculated points in order to magnify
the printed map. Regions of negative 'electron density' are
ignored and the positive values are scaled for purposes of assigning shades to them.
12.2.2 PROGRAM FEATURES
1. Density cross-sections printed for each section.
2. Interpolation for increased clarity, and to minimise
topological distortion due to fixed print positions,
character height and width, and fixed line spacing.
4. Prints additional copies of all sections on request, and
then automatically supplies an overlay grid useful as an
aid to reading coordinates.
5. On request prints an overlay grid useful as an aid to
reading coordinates. 0
5. Prints cell dimensions and em A scale factors.
6. Fractional coordinates are listed for the calculated points.
0 0 7. Reproduces cell angles in the range 45 - 135 to within
0 0.1 •
8. Prints maps of any length, width only being limited by the width of the paper being printed on by the computer.
9. Contours can easily be made by tracing around areas
having the same shade.
10. Maximum and minimum density levels can be varied for specific purposes, and this varies the number of shades
spread across an interval of the map. 11. The darkest shade in each column of calculated values
is printed out at the bottom of each map as an aid to the
locating of points of high electron density.
12. The graduated scale of shades is indicated on each map.
122

12.2.3 PROGRAM CONTROL
User options By default User
Range of (electron density)
Number of copies
0 - 1000 1
-lOOO<p <p <1000 min max
1 - 8
Grid 0, 1 9
12.2.4 PROGRAM ALGORITHM
1. Assigns shades to the numbers 1 - 1000.
2. Reads file description.
3. Obtains tangent of section angle as a rational fraction.
4. Calculates normalising factor for electron density.
5. Decides on expansion of map and orientation on page.
6. Begins processing each section by reading electron
density values from file.
7. Calculates values for scales.
8. As the rows are read for interpolation a note is kept
of regions where p <p . so that they are not m1n
unnecessarily interpolated.
9. Carries out interpolation and places density values in
an array so that when the section is printed out row by
row the section angle will be reproduced accurately.
10. The values are transferred to a disk file.
11. Cell parameters and map printed out.
12. As each line is assembled the maximum number of overprints
for its characters is assessed and the values in
each of the calculated columns is also examined, in order
in order to find the maximum value.
13. For multiple copies of the maps the printing is repeated.
14. For a grid the program produces a map with dots at the
positions of the calculated values.
123

12.3 SORTLIST
12.3.1 INTRODUCTION
Program SORTLIST was written to replace two programs: RANGER and FILIST. It has two main functions (a) to carry
out a weighting analysis of the reflection data and (b) to produce
a table of structure factors in a form suitable for publication or for storage on microfiche.
12.3.2 STATISTICS SECTION
The quantities obtained in the weighting analysis are given
in Table 12.3.2a.
Table 12.3.2a
FUNCTIONS CALCULATED IN THE WEIGHTING ANALYSIS
Quantity
WSELSQ
R factor
Weighted R factor
Average value of F 0
Error in
observation of
unit weight.
Where
Functional form
2 2 (E(I!F I-IF 1>1 /cr(F)I )/n
0 c
R = ( E ( I I F I - I F I I ) /E F 1 0 c 0
2 2 2 2 1/2 R = [r{jjF I-IF II /cr(F) )/2::(F /cr(F) )]
2 0 c 0
MNFOBS = (EF )/n 0
ERROR= [E(jjF I-IF ll/cr(F))2
.N/n(N-m)]112
0 c
N = Total number of reflections.
m = Number of variables. n = Number in class.
2 cr(F) = cr(F )/2F
0 2 1/2
cr(Intensity) = [c + t /t (B +B ) + (pi) ] c b 1 2
c = counts/second
124

X
t = count time c
t b
B 1
B 2
=
=
=
background
Background
Background
I = Intensity.
count
count
count
p = Weighting factor.
time.
1 . 2
Program limits are given in Table 12.3.2b.
Table 12.3.2b.
PROGRAM LIMITS FOR SORTLIST
Range(X) Number of classes Default Value
sinG/}_ 0 - (sinG ) /).. 30 10
F 0
a( F)
H
K
L
max 0 - F 30 10
0 max
0 - (J 5 5 max
H - H H - H + 1 Range of min max max min
varied index
K - K K - K + 1 Range of min max max min
other two L - L L - L + 1 indices =
min max max min
Other information elucidated by the program is:
(1) The range of the reflection indices.
(2) The slowest varying index.
(3) The index sorted on.
=35
45
(4) Those reflections for which (F - F )/cr(F ) .ERROR > 3. 0 c 0
That is, those reflections for which agreement is poor.
(5) Duplicate reflections.
(6) A factor to scale p by in order to improve the weighting
scheme.
12.3.3 PUBLICATION LISTINGS OF STRUCTURE FACTORS
The table of observed and calculated structure factors is
printed out with six columns across a page. A number of columns
greater than 6 can be specified and a second portion of that table
125

is printed. If the table is large it can be split by defining a
maximum length for the columns. Normally data are sorted on the slowest varying index, which is the one with smallest integer
range. This can be over-ridden and either H, K or L specified. Table 12.3.3 indicates the program characteristics described.
Table 12.3.3
CHARACTERISTICS OF SORTING INDICES
Characteristic
Index sorted on
Number of columns Number of rows
12.4 BONDSTAT
12.4.1 INTRODUCTION
Default
Slowest varying
11 236
Otherwise
H, K or L
1 - 12 < 400
This program's function is to obtain interatomic bond lengths
within a connected chemical unit and to calculate average bond
lengths and their standard deviations for specified classes of
bonds. The bond lengths listed by the program are all those
occurring between specified upper and lower bounds. Within this
set those for which the statistics are calculated are indicated
by giving a list of characters to search for in specified positions of the atom name. Such a system demands a hierarchical
naming system, where the levels of the hierarchy could include atom type, molecule number and bond type. The program is so
designed that the ordering of the hierarchy is unimportant, in the
naming, as the order desired must be supplied as input. Such a
program as this is useful for making comparisons in those cases
where several molecules of the same type occur in an asymmetric unit.
126

12.4.2 PROGRAM DESCRIPTION
The input data, consist of a title, the number of bond
types for statistical calculations, the number of identifying
characters for the pair of atoms for each bond, bond length
bounds, the character position lists, cell constants and the
bond character lists. The descriptive part of this information is
printed out. Atomic parameter cards are read and atomic
coordinates orthogonalised.
Calculations of interatomic distances are made and those lying
between the bounds given are printed out. Those atoms for which
the characters in the nominated positions in their names coincide
with the lists given have bond lengths between themselves and
other such atoms included in the statistics calculations.
Lastly, for the nominated bond types, the mean bond lengths and
standard deviations are printed out.
Details of the necessary input for this program are given
in the listing on page 179.
127

APPENDIX A
STRUCTURE FACTOR TABLES
Observed and Calculated Structure Amplitudes in Electrons for A 1 PHENANTHRENE PMDA
A 2 FLUORENE PMDA
A 3 THIANTHRENE PMDA
A 4 BIPHENYL TNF
A 5 N-{2-HYDROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN{IV)
A 6 HEXA-~-DITHIOCACADYLATO TETRAZINCSULPHIDE
128

1 )
1 1
' '
I 1
' ' 1 I
- ' ' ' '
- l lj I. l
l I
I I I I 1 1 1 1
1 1
-,! ! - I I
1 '
' ' I I
1 1
....... :1 , ...
l l
Ll.i I I
l I ; l
PHENANTHRENE PMDA
I 1
I 1
I l
~ l l I l l l l ' ! i 1 l I 1
I I
' 1 1 1
: l l 1 1
I 1 1 1
129

!l H •: I n I-
I u r 1! H 1: l it !, i-• ~ ! != I jl II I I
!! !f !-! ' tl 0 L I! I! I= I H \< t-L j L 9-! U H s-L Z t-!
!! !t : ctZlOO
•••• 8 .. )I ••••
!! !l !: I ll !I li= I il !1 !: ! H I ! I ~1 t1 t.: ~ zl H c l unf-' hHjJ 8 t.Z t 'ioL ll. -,\ oz -t t'> u; -!! U f: I I! !i : H U s: : tz ~i ;: I) H i ! !, I, j: I l, !I !: ! I! H !i-1 I != i ,, I' !! 8 " !! •-' " ' I 8 L 1..-E tt t
11!1~! i!!'l~ 61 Ol ~ z 61' l ~ I 0
If !i l-! !! !! : 8 H I! f: ! H !I t: 8 II II != ! ~ 21 t-i II II l l 6l tt ( t J..t 6Z l 1
H H 1-I (t H z-t 11 Lt (-t
H !! t: l 81 81 &-T
!! h ( ! H tt ! 0 . ' - H II : i u tt a-o •••• L,. )I ••••
n ot c-6
i' I' j: t 9Z I..Z t-8 sz sz s-e 1! I' 1: I ti ~~ 1: ~ 1! tj i= ! i' I !'-1 U I! !-! .. 8 (-9
i! H 1: i 6t tz Oj-9 6t oz t-9
l' " !-! 0 t. ·-.. Lt H s-s H !I f: l l' I' r-1
H !! l ! It !t !-I u tl z-t 91 oz (-t
l! !! 1: I IZ tl 6-t 6 8 Zt-t
il !I ! I !I I I H I ! l 1! !1 !- a Z9 -
•••• 9 •ll: ••••
II !! !: I! !j t! t: 81 6 81 9-OT L Ill L-01
n I' r 2' ll !1 !-! n n F 1 II !! != I Ot H t-8 !it St 'ii-8
II ll •: I " " 1-I 'I oz ol-! H ! -! 9t H Z L
1' 31 ~-! H H -1.. Lt tt -L
I! !! ~~ I I! It !l: ! 6 6 • '
H l1 I i H H o ;
1: il !: i (6 " ,_ '
lt It j: ! I! !l p-! u !I , ~ i! ! l !i H I ! tt n f ~ l! !! !-! n n f: ~
il!il=! I\ i\ : ! '! ! -l 1, 1~ != ~ 9£ £ L t
il il ! i 11t !I != I l !! 1: I 81 !l I= I !! !i-1
!!lit! !I Jl I l 1! n ! 'I &i != , {: !I 1: ! " ll ·-( !I H !: !
1111 r1 il II !: II
h II !~ I! H i= II It !i-! f, !. p-! H !! ! j oz zz 'i
i! l! I I 'iL 9L 1 'i'i 9'i t-
!1 1l != I ~~ ~~ != I
H !! l: I i: i: i!: l ~~ U n g !I u !: 8 !! h !: 8 •••• c; ... ''''
II I! !~ II !! !! j: 81 H H ~: &I
I! !I I I U H != ! Lt 'it (-6
!, !! t: I !! " 1-I I 2' z: I !! I' !j= I I' H 1-I
II II 1
i u u l:i l!!lj=f u II li: I
I ! i t I! I! l ! 1' ~~ L
II b -! t 11 != f H 1! g t !I II II= I 1! fj p-I tl I I l 11 · 1! 1 I '! !. 1-; I 11 f: l ~~~ ~, t: l !! ! i= ! !l I! !l= l
!: 1: IF! !' !I I ! II il i !
I! 11 I ! I II !-! il !! f: ! ! !I != ! i!t 1!, 1: ! H !! li-!
If !f f I i ,i , I
!I n ! ! OL OL t-t
II H f: I ! ' !!' 1: I !!' !! != ! H H n: 1 " ~~ t1 I i! !i !' n n ~ !! H I I l!t l!t ! !!, !!, != I II U H ! io II !!-f a ,... n-t n n z1-E
!! I! 2 ! .! L I ' !: !' ! I L1 tf ' !II ip !: I !! I! I= ! 21 !l != ! ii !I 1: ! u tt ot-t 1t n n-1' I' !I u ~E a II !I ! !! I! I
II· J !:
0£1
!I" if 1: ' !! h ir I 11 11 ;-= lj nl nl 1: 1 1 1 ! l I== t !! I ! f! ' : I R
1 ll
1 ~ ~
tl ! !I= li, !j, 1-! II ! ~ II !r r :: r!:l ' ,. 1= al nl 1 8 !21 f2' : ! t 1!: I 11 f~ j-F I! !! ~~-~ I' lj t=-j! I I h, \ t ! -f :: .. !
1• }:~.~ !! t 'I' : h I ,, = 'l !
!! !! E II !l I I ~: ~~~~ Ji ~ II I I= IH l'l - II I ~ l 't ~ u !: l u ~-I II I! ~~ ll ii : I !: 1: ~ II ( l II' E ~ i 1 It, t !z !' -8 I -I'-1 ~ t ! -¥ !! ' -8 '
1 I (-I : t t L L --I
l!' fi 1: 18 !! tl w If If ff f: I ~~l ilt ' ! II ,~, w !1, fi.· I I &! != l !j ! I' II '!-hl ~l ' I I II tl, l H lj != I I I' ' : n• ' : !l !I, !11
11 11 11: 1 !1 !11 t I 11 !1 I 111 111 ~ 11 11 t 11: m ~ . n f! ! 1 11 11 E= ! i! 11 1 t u n ~-1-1 I! I! f= t tt. n, r= !
II !, ! I !' !i t: ! n a : ! H !! ! : ! 11 !, 1: ! U' U' f: ! !I I' : I i It != ! !It !II i= f I' !' II= ! H !I != ! 51 H '!: ' I : !t !!, 1: ! U' 1: tf ! I If ff 1: ! H I ! : 11 11 I= 1 ~ !I 'l= 1 !! n II= 1 t~ ~~ h ~r !!' 'l= ~ ,, ~~ ~1- ~~ !'I ,i-,. II ll ! : I !'' II != L ii ~~ I ; H I : f tt ! ! Ll ' ! !I I ! I! -I 8 ! I !I H I I' 8 ! !! u i I I !! I I It It !!' lr I h, h, I 1: ll: t i' il o-t !' !t' l ! lP !' U !, '!I II I 8, ~' ' d &! !: f 1t lit 1 1 u h I I 1sl ul Y: I ht Itt I 1 i' !F! I ~~ 1! l= t t .r ,_ !i " !-, n, !t, !: I n n i 1 t H Y-
V f 11~ I 11~ II~ ~~ Ill !II P: 1 If· II· ~-II· !'· ~ I ;n 1
1P~
!l 1! t 1 111 (1~ 11 11 rid li 11 11~ 11 Ill!-~ 1 II 1i fl= h t· !_ ' !1 1 -n 'l u-t If! m b 1 d 11 ~ tl1 ~ " !t '= I '! ~. i I Ll i ' ~ 'll " -2 ~· •t • 5 "' H' -. I I 1-I! I I, l I ! i i 1 r. h I ! fi, n, -1! ! ~ I !" 11' 1-s 11 n 1 ! m !t l f: I U u 1 ! :: n H I d 11 1= I r'f n i= s 1 l !i' ~ ! 1~ It t-_ ' fil 1 I tl' w --~ i ! -t " f: S ' ' " " II-! !Z I 1 t -
!! ~ 'I I ! ... ~ .. I!:.! I' II' ' j'" : .. ;... II' II' ~ :~ .. ~·. :!:.;
P ~-' n r n ,q ~ H ~· n~' 1 11
1
!,r " ~ fi_f J ~ L-u " I u u I' Ol ~ .. ,.. I L • ~ Ol
I( l.C Ot-Ot
i -I=
1 tit
F
1 'w •: t
id : ,,-: I= I 1 h -
li I I' I 1: 1: d p II iq j!! ill t f 1r ~
1~ ~=-f
i! I 11= I '!: lt I I ~ l1 i= I li u t=-,
~1 • 1 ,_
f : , s , 1 jt t~~ ~= I t .. l-
u n r= ~~ ~~ :j-f t! n t I h' !!' I I II! m ~ 1 U u s-- n n fj= ~~·~ r.HI ' s : f-8 H1 ei= 8 •••• 0 ••••••
~V:> SilO '1 H J'lV;) SIJO '1 II .:TJy;) S1IO '1 8 :rJY:> SUO 'I 8 :rJ1I:) !niO '1 I ::Tn:) 5'80 "t 8 :tn0 S'80 'I B :n"r.) hiO 'I a ;rn:) 6:&0 "I I ;nY.J 980 '] B :nY.) n10 'I D
va~ ::I'J3MOnl~

..... l(llf
• k r~s C&U'
! l
t i
rll : •t : I • l 1 ... . ., l' :: ·• ... ; =j I:: l l l i : ·H ... . .. . .. i :1 ~ i ~ :u i :; • ;j
THIANTHRENE
..
.. .. "l
:! ! l
•• 1
.. !I .. .. •t •I
;; :t :: ! :! :~ •1 II :t·:: g ;~ :{ :i =i j :, I
;j i N k O~.S Cll: II k ('0-J t:ll.!
11~ •\ 10 1 H 'il ~t ' ',', h
::'.·:~:' l:::.~~~:;:~o 11 •• IJ H ~J ::_! •• !:l
·.··~!.~::=· ~~ .. :~. ~l: .~~~ :.~.~ .... ;::_: Il.!.
!l :;\ ~!.! ] ll :_;l, " .!,' ,, ·g l ;; ~i
in , n ~ u I · ':.~ !_-~·-· , H ~~ : ~ ~. -.·~.·:· .;:1•• ·~.·:.::·~·::·.
1:.'-.;, '~i :.=:========_·.;:'::::::::: ~ n =~ :1
:11:1 ·j=IJ[if[ !·.!.! !·i: .. ::.
:!i Jr ~=_:.!: 1 1;=·l.·. r~.·~~· ~.l. =._i!. u g ! ,,, 201 :1 =! w :H 11 1 rn t•l-C
• 'ouc•u:
131
PMDA
ll ...... I
=;! ;!l ;!l :: ~~~ I:&
.. · ._i_ m .~.:~1:.·.;_-.: i ~i :,::,:, ... f ,B
: .• f ~.;··~~~~. !!, ·=_.;:. ·n •• ·.·• : •• ~:i:·.· '~l ! :j :!! t ! :H ~.l.i : .,a ~Q
••••L••••••
:: :1 1,!,~ i.· •• : :_l, =A ' ~·~.· n
l! ~~ '!l ·il
l :l I..! t•IJ
! :i ... . . , ... : :J : ., ! I : l :-·! l :. ' .. . .. '·• ~ :f ' . l l l I ' . t :u • •n
i ~i . ·• :"I : t ..
.. ·• .. ·> ., .,
J :I
H ' ' ' ' 1•1,
! ~~ .. , : ~A . ' !·:! '·• : =j i :,
I'.! ' ..

BIPt-ENYL TRINITROFll.K)RENOt£
I
I I I l I I I l I I I
I I I IIH. ••• UH
I I I I
I I I
. ! : II I I I
I ..... ~ .. : .. l I: 1:1 I I
! I i I I I I ! II I
II i '1'1 l I I j, I I I
: I 'i 'I I :
I I I I ,'I' I II : II • I I : I
I I I I II
I J' 1 '1• 1 d · jl .... j.~j,.iij
I ! I H ! l l II L I I H Ht
I I I 1 I
II 1 I I I !
: ' j I II !
I I I
I I I I I
I· ! I I
ll I I
I I
1. . I I
l I I I I I I I I l ...... ~ ....
I 1 i u I ~~ l l I f~ ·~l I
I I
132
••ul .. ) ••••
I I
I I I
I I I
I I

:p
c 0 u ...J >z ~ a.. ~
•··~·~<j •oz:~1!" c-··<
:::•-.o:r-~---c..cr-er ~"<~'..,.,_c-. .-... -
.,.,c;,....~-,~x,· ..::-:::o:r:
...-c:-c. ... t -.r
-.(
;(T
"t ':!"C
-... -~,..,_..._~-11..:;-......,~-,.._'%:_,,_
•...-t;: .... <"'c-N
o'>
l:-..,..(;-~.·.-•J•<-'•
N··----
---~1"<-N......-.--------------·---
.... ,..,.,,....c~ ~.ra;~•-..c..,..,._..,c,· c
.r--r-•.....,"'a
::--.r-r-.:--'r-c-o
.;:."•"
--" .. -
,z; -.a-
c.......rNc•c-:-.:---r-.r..(;-:::-e ,•-c
-.... .,....,..... .... _c,__,.._..,.,....,...."'"'•' c
c:"-.-
r.--
~·---...-..--•--••-.------
-····-,..·--·-----------------...:
,.....,..ro-.,.,;:.-,c
-"•-t-----"
·C-c
-c • .-.c
-"•<r<
:::"<"''=
.:-r-c-.r-..e
•-·c..,.,..
--
--:.:
.,..,...,..-.;-.r.r~·-~;...::..::"-"''"',_,....,.._,..a::a:;c:r r::cc----"'..-..-..,...,..,..,...,.....-...::..r-r-t-a;a;e -~-------------------
..t:c,...·---:r~r-~ --·
.... ---:-c,..-::':1!"-<=
"...,..-,. ----·.
----~
--...::a;..t:-,._..,.,_-cr:-........---c
:""c-
c:c
t-cccc---
-----------
c-ra;.....cr:::cac-,_,...,,_occ---... --c:r--e.r-C'cr-r-,..,.c-<
::-..C..er--cN
,..,...,..,
~ ::"::"~~=:~~::::;=::~::~~::-;:==(!;;;:;~::::::::::~~==~~~
.-• .-cr-~~--~~; .... e
r"'c~---a:c..,.~~;...,-
•r-...::e~:r.ro:r-c,.....,,_"',.._..:-a:-cr---r-r~-
-or... ..... -
-...... -
-.. ·-~.r
----~·.....-..
u: .ra:-r.r~.ra:r---r-c..,.,._a:.,.....,..,c.r~-c"'r--..,er-..c.rC'r--o&-o::a;N...-.r~a:
=:: .r..-r:-Ot~Naa;.-·tr---e,....,.,ta;.-.. .... .-.. ... ,
...~a-ocN.,.e.,.o.-..e.-er
c --
.................................... ------"~-----------"'~ -----
mco~-,...........-r-c-.ra:c-Nc-~-
..... -~~c~c--•~c""-N.r-
-----
----
w-,...,...,_,...,_..,...,......,..,...,...,.r.r.ror>
ro.&;o.c-tr-r--r--r-<
ema:<
:r<:rooo---"'"'-
-----------------------------N"'"""'~"'"'"'
..c -<
t"T
... ->&--e-a~·.,.co.r...::e-e.....-.-.r>&-
.r.rr-•"•-.cr-a-.,..c--.t--co-~-c~
"""'('" -~; ............ .,.,..,_
,..._"'.r
--------
N.....c~~;"'"'.r"'o::c.-..-•.r<:r oo.-....~o-C'<"'
--
--
-"'o~oor..-~mo-
.... o.-N-.,.ooro-~-N.,.o.-N---.r~.rm<:r~-N-mt:ro~..,.o-......,......::~-Nr--t:roo~
---
----
-----
.... ---
....
o~m<:r~--or-~.,.~~.,.ON
,.._,.._M,...\4:l......cl:l.,..l7'~-.rt-N""''t-.,.
~~-
..... ----~N.--......NN--
.r mNO.r-mm-.,.r-oNm~N~~
.... ,.._ .... _...,.ot:l'"'~~-"'-=-=~
. ......-..................... -~ ..... ---
..... ---... ~
o..,.r~~-~r--~ON~~
--
-U
o-t:l'~m-~-.ra-.r~<:r-~~""om•~~ON•~~-o~m<:r~ON-t:r•mm-~om.-~~...a .... ~~~~
g ~~~~=~=~~!~~~~""~ .... ~==~~""~~~~~ .... =~~~~'4:>~~~~~~=~~!=~~~
8 :::~~=~=:~~=E:~:~~;;~~~~!~=~~~~~§:~~~~~~~~~~:~~~
w: .... -
--
..................................... .,...,...~...,.,..~"'"'~"'"'~~"~~~\D'.r;JI,p~>CV>~'-">~r-r-r-r-r-r-r-r-r-r-r--r--=cz:>(Z)<Da:)/3<CI'O't:l'.,.._
w: .................. .., .... .
,....~.,.,1/'V'II'~CI:)a)
NNNNNNNNNNN~NNMN <
m"'~'"'o~-.r~.,..~~o-~,_.,..'-"''"'~"-'"'"""'.-
... .,.Nmm~-o~~N.-o
~.,._~'"''"'Nr-,_.,..~~-~~..,._..,.1'--'"',...,.CI).-O..,...,. _
_ O~~~.,._,.._~NN~
"""'~--N.-.,.-~=-----NN-:,.._-~ .... ~-N-~N N=~-N
.-....
"'NOO_,.._Nii"<l'<l'-'_f'..,.\POCI)N'"'.,._,..,...,...,..OO,...Nr-W~N<"'\C'oi'"'Of'+-4,..__11"10
. ;~~~~~:~~:~~~!~~=~~~~~13<~~~~~~~~~
N•~N ... ~
o-~..,.~~13<-~t---0-N'"'"".n~t-W""'"'..,.r-Q.-N~...,~~
.-............ ---
I --
....
~ ~~~~~~~~~~~~~~~~~~~?];:~~~~;~~~~;:~~
~ 5=sE=~~!~~~E~~~~~~~E:::;~~;~~~~~~E
13
3

N-(2- HYOROXYPHENYL) SALICYLALDIMINE DIMETHYL TIN(IV)
II « 08~ co~c .. ""' "- .. !..(,
-·~ (A&.( ~ • o•• (111\., ~ (Jij~ CALC • 0&1 tALC !I UtH Cl~t.l.
' I '" •• •• I .. •• l 1 1o ,. • l J u ., f2 •• • s JJ Jl I • • f H .. ~ ' ~n H • • 't: ~i i H n •••• L . ~ .... ~ lo ., " J 0 I<~ • • • ~ .. .. I> 0 •• ., J 1 1110 lOt • I IOU l•l . •I ~·
• 9 I. I. •• B "!2 ~· ~f • • I> ~! .,
! 16 •• i J lo .~~ y i 1o ·~
, H H 0 l .. • • .. I> J. •• • II J • J .. •0 t •• •1
I .. m <It 10 •I ., ;1 I) .. ,, J • liT I •• y . 10 l6 tl 'J " • lu :1 l lol 10 ' ~· I) ~ 1: 1: J • sH l ~~ • ) llJ 1 Jl 0 1: I } ~~ • f•• ilt':tO lv • I< t •• 0 . "I y ' ..
·~ I I. ,.
~ 1111 II) . ~· 'f .. l •I •l .
J H ~· IO •I 1< • 2 I• 12 I J ll t·J ~H ·~· 10 ~ )o 5. .. "' :r • <J u Ill ! lf~ IH . ) J) •• l ~ ll lo
'" 10 , .. . 12 . .'>1 4!:>1 IU . s •• u I' 0 lh 1•1 10 •• jY u . .. J7 . • lo I> 10 !~ ll • • u 10
•I ., .. • IUY Ill II ~v >I I r 0 I> • . ; •• >• lu >S • 1 .. 16 ~ f .. >I
J ~ •1; l~j II l\10 :o~ II I >O •• . I~; Ill> II IOJ lui • • IJ 1J " <o l . •I 11 I• II ~ 11 n . 8 lJ II lJ lS , I •• ·~ " • P• IJ> 11 . •• ·~ ,, • 9 6) •• ll . 'i ~~
, 2 ~n ll• J ~ 3o •' I • ••• 1•1 II , ., ·~ .. YV ·~ ~ 0 Pv ••• II ~ ) l • Jo Jl • H
,, II • l< ; ~ •• Jv !I ~~ • ~ • ,. IO l I l• ~~ l ' .. 10 •• ! ., OJ •• .. ,. • ~ H 16 J . >l l I 11> loJ ~~ ... j)J ....... . l > • Ho I•• ll co •• ~ , Jj .. J • hi " 1 ~
IH lot 12 ' .. II ~ 5 ll u li •• •1 s :t •I . I II J , .. I• ,, lL . lY ,. u l ! 12 IIY
' au IOl ., •< 6 n • I. l! lo J 5o ,. ll , IIV llo • Iff 117 > I• lu lJ It 19 • I 1 I • J « 9 ., •l ll I .. .. u IU , • "' ll I J j. <O • i •• ~~ • ~ ll .. • ~ h ~t ll I I> , • I•• II< • 0 <• lY I J . l• 1 • • ~J . • lo .. • ~~ I J " 1•1 1•1 I J•• J)9 • I II• II• I J , I• ' • • •• •• ~ l Ill IIJ l • JU I J J lv I ' I< .. • c cu •• IJ • l> -~ • ~ I• a , . .. •' • ' lv ,. ll '"" IO< I •• ., • J '"' IVY •• I <I a~~ • 2Z ,
' J; J> • ' ... ~· if jv n I ~ 10 IO . • •• .. I• J •• I~ I ~ II• II& • I ••• 1 i j ~ 1 ·~y lvl • .. I l>l 1>0 • 5 .. .. I• . l• 1 •• ., • ' <> .. l ~· •• I I•• I o7 l r Ol •• • I ., ov I!> I 2 J JZ 1 )
~= •• '
) '• ,, ~ )i) u I• ~ IU II• I • i.l , . • 9 •• •• ~~ . ~~ 11 1 . u l '• u ~. ., ~~ "I JV JU I ' •• I ~9 I 0 IJ• 1 Jo 10 l ' 5 lo I ., •> • '
~Ol ICY I~ .. ., .. t I 1•1 I I J I ·1t ,. • lo ) 1 • •• ~~ 1 J l• ly • •• .. I!> r • '" l 2 li• I<Y 1 2 ,. 11 I 7• I~ ' I JO •I 1 • 5o ,, • • H " lo •I ~· t l 15 H I • •• 05 • •I u u 1 5 lv .. • 6 ~" •• 16 ... <I < • lo 1 5 .. 0 • 2 IO• luo 7 • ld l>
' • .!~ It !1 ~ I' 11 t s 1J2 Uo 1
' ~~~ IO!> • •• • L .. 0 • 11 •• ~ •j 2> <J I l~o I i.O ,. t • , . 55 I ,. • 5 l> ~; • l
i~ JO I
J ;t_tJ'I 2ol IO < J) JY i ' J• J l • 0 ., YJ 0 • llo IIO • • ll ll " . I<
' •• •> I • .. ol 0 I l.V JO 0 2 Ill Ill • 7 •• 2~ • 5 .. •• ol ~ •I .. •• • 2 •J u u . ,, i'> y I 14 J IH 9 I '" .,
' 9 •• of •••• L • t • 2 ,. •• • l lv lo u . 5o .. • J i5 y 5 2• " ~ 2bU 2'>• J J .. .. . • •• ou I 11 lo H • • II ll IO l 2• t• • l ,. •L 0 " . .. JV1 J . IVJ Jy~ • 5 '>I >O I I l• • 5 •• u Ill ~ '" " • • llo lib u < lli I•• J \ "1 ~ • ,,
" I l u •5 IO •I <o 18 10 '• « • • 1' • I• J 0 . lc~ .. , J t ~1 ,. 0 1 .. •• I j •• OJ
·~ 2 99 lu 1 II l 3o J; • 6 I• II. u • i<4:;, 1•~ J •• 0 e lo lu I . JJ , . 10 • •• .. ' '>> , ' •J ~Co ~"· v ~ ,. ,,
~ • ,. •• • ~ 5J >• I ) I• I) IU , lO u H ., • f 9 I l ,, VI 0 iu ,. .. l •• •• • IJ <I I ~ •• lu 1Q • Jo •• • • 9 ~ lo . I J I 0 ~ \,l.J IOJ • z >0 ·~ .. J •• >I l ?: ~? II ; n •5 ll I> ·I·
' ~ .. .. I I IH l•l J ~1 II .. ~ '" ·~ I d •• 12 lo IJ lO 0 l ~y IYO I < J. ~J y ~\,lj . Ji • I Jj ,, l " •• 10 II l 2) " I J •• 11 10 2 ~y I J ,., <'I f II> II) IU "I •• <o 2 I .. •• II • •• •g I& • 1511 l•O 1 ( .. IV/ . •• •l IV 0 IIi II< ' i •I ., II ~ t~ • !OY lv• I • I> t; . • •• JU lw J OJ •• 2 ) •• •• I< I f)
• • 111 • L •ll• If t H ll ~ •I •' 5v > j •f: li2 10 • ~~ ~t ~ . JV •• ~~ s j~ •l •I ' ,,, 2~';' • •• lv ~ > J1 .. I• 0 u 12· IJI II J IZu ld l t >Jv .. ) . I» I JO IV • Jl J< ' • •• •• li. • l• 19 0 2 l• lv 11 f 16 h 2 J .. » , ~ tl ll lw 1 11 IJ l 1 5u •• ll 5 )Y 5• • . YT l :•t 11 ,, >1 l . IJJ I JO , • ,, ,. il 0 •• IOU II I>• lo• I J i u J2 ~ • '• '• ll .. Ol ~- ' ~ JJ J> > • .. • • .. I Jl .. l h 19 I J OJ ,. I I To '• ll 0 ~. Yo l • lv• .... > • " •• .. ~
,, >V .: n •J I J • .. Jl I ? IV 16 u 2 lo ... i r >• ;v • I 1<0 Iii .t ,. ,. J I J .. I •• •• I J II J l;i. n • •• ., ' . .. >v . l 'tS .,. .. • n •• ~ .. •• •• 2 lS 2S I 5 Zo Jl I .. •• J •I >I ,, • J I) II • I J II 0 111 I• l lo 16 l • 1• ) J) I JY Jy J " <~v l"'.!l' • . 1• fY II 7 I v 1u • lo 15 ·~ l ~· >o u .. •J l! II ll J " •• 10 • ~ ., •• I< 0 •< •• •I lUo hi'> 2 I IJ :•
~ Ill IIJ J J .. .,. • • IO lo ll I 01 II J .. , I •• J 2 ll " ,. Jl ,. J . .. .. • , .. ·~ ll j lli leu , .,, )5 . "L • 0 J h J) ,. • Zl <' ' > IO l< . • ·~ >I 1/. • lb lo . • 2J <l l . 5· >I n } ~! lo J . .. ·~ I l •• co l< 1 13 $~ • { lUJ ~j~ 0 j n H 2 • ~· ,. •• J , Jl J• 1 2 " IJ " ~ " 2vl " J I • J ~~ ~ '1 1Y J . JJ J• r . •a )b I J 0 II• lou ) ' HI leO 0 • :' " J J It i J u 0 ~ ~) ,. r .. •1 .. 2 '" I l > J J> .. v 6 u J ; h • 2 ll JJ I Itt. li• l 1} ~~ I J • .. "' ) . ~· •• l 0 •• u J ll I•
H • >• .. J I•• 1•1 I • .. ,., lJ • Ju) '1~ > < 12• 11• l I IH IJY • u I> . i H .. . " Jv I • •I •• " IJ I> , l 1J " I 2 lg ~·
. ! ll '1 ~n II• , .. .. ·l ~~ II I• I 11 ,, • ·l .. •• I ) l • • l J II B ~~ . • 1:' <• I Ju I• J 110 II) • u •• l2 I • ·~ 19 • • 1 J . u •o . { .. >v 2 2uJ 21) I• ~ ,, il • t 5< >0 I 5 16 l!2 5 ' l< :o . • . , .. . iv£ lo2 I'> "I l1 •• • l IH lJ5 I • Jl •• , • I> ., s " lv• IIJ ) •I =~ I> • l<o lo!<t • , .. •J I 1 61
l t~ • 0 •• ,. •••• L. • l ) l .. .,
' . ., I) 2 >C >I • t Jo J6 2 0 10• • I ~~ " ~ J ~· )) >l ,. I> ) it Ll t eo •• • I J> ·~ '
. .. 0 l .. ~1 ) ; llJ llu • •• ol I'> • ,. , . •l •• ol ~ J ., •J • lJ J G • 2• > ,, 11 I Jvl ItO lO 0 J> JJ 7 " •• luu ~· )S 1 I Jr N • 1 J 11 5 • 4t..v luJ 2 1 r lJ lo I 2) •• ~ l H •• ~ • • • •2 1 J .. <l • •• ·~ ' 1 i;) <> l •• >1 10 ~ ~~ l1 J 15 l ~ 11 li 1 If • 10 ll <I > • h <I 5 ll> Ill 10 )) 1 . S• ,. l t 1 <I
-~ l&u •a • v •• ••• • I• . 11 0 11 1:> I > •• 11 i 2• 2J 0 ll• II• •l • I . u •• . I Jl ·~ j/ 2 IJ . 1 • 9v y) 0 !Ill lvl ~ )( ·~ ) ~-,, • ; .. t. • 6 tJ IO ' .. •• •• J I 14 19 • l< . • ·~ ., • . lY< l•• IV •I 10 <I • u 2J .. • ~ .. • • • ~ H ..
~ lo •• • . >• >I lu l l•o I> .I ..... • • > • l '" J J •• •• y . , Jo •• • > 'i! 11 HI . •• o2 • l H lO l . Jl J2 • I 01 >' Sv •-i • < IV ~ <I d ,; 2 ?Yo JOl • . Jl J ~ i/.V ~~ 9 J ., •' • H >• • I ,. 5• •• • IY .~ II . ~~· l),J • > l• <I J
' II ro IO " •• .. • IO •• • 0 l> lo IU • 00 lo " • •.I >41 • . .. •I J ll 20 IU 2 'l ..
!f tf~ .. , . •v •• II J Ill I <0 u • m 1<• • 1 2/ ~J . i l1 H II I J l ., lv> v •• .. .. ,. ,. 1 I . .. . I Jt . ., •• f •J •• 1 I lvv l JO II J ll <0 • i !1 •• ' l JJ JO • • 16 I J 2o •• 7 I. .. •I II • ,. ,. I ,. • J >I >l • ~ .. lY •••• L. •It • p u 1 J 11~ Ill 11 ~ •• 19 I • IO ll • • IO I. • •I •• I $ ,. 1 .. .. II 1 •• <5 • 5 2vo 2uo • r ~;
,. 5 ~ 12• 12• 0 ! 7• •l • 2~ <o 1 .. •• " I ou ol I • I> .. IO •I Jj , H •2 0 2: I• 1 :f •• 1 .. .. I< s ., .. • 1 n •> lu u ;I •• ~ l H H l ,{ Jv •< • •• r .. .. lt I) • 1 • I 10 J .. •> • I . lf JJ
l • li' Jl 1 . ; .. ~. I< . .,
·~ I 9 Jl • • lu . ?J •• > • •• •l I ~ JJ H •I 15 I• • .. .. I< 5 10 f) l 1 IY lY lv ) i< 1.5 > 7 17 /I 2 I ., ·1
2 2•• 2J1 • • l¥> -lVb l< 1 •> •l < J IY7 I•• 10 7 IY Jl • 0 70 •• • l l• •• J ~~~ •I . 1 .. •• IJ 2 I J ,. t " lo II " •• ., • I 10 id • . lv 0 • •H 2v• • .. •• IJ • . , •I l . IUO •• ll I •• 10 • 2 lJ l2 l 5 .. ,. ~ 3• • II J II• lJ 5 .. ll l ~ l! lv II l )I JO • l •• • • J i .. •I
I , n " . l ,, 1V1 .. l .. •• <
' •it ·y II J u •• • . •• •1 . ., •o
1• • JV •I I• ' " <I < ll . .. •l • ~ •I •I • I ., .. t • l}t llo • "' '" •• j " II I. • 1$ 7o II > jy .. • '
H n ~ -· I< '· "J la • l.' 1 J J • .. ~ J I ~. ., II • 5• >O • •J JV > I l) •• • 2 • U• • ·~· I Jl ,. ~ ., OJ • 2 i1 u II I •• ol I 0 Jo J1 ) . •• ,. • H • • . ', tlo I• • IJ . J ) '" lo u J Ill II) r l u •• • I lo I< • l tc • I> I< ., ., )~ ,. J • Jl •• ~ ,,
sa I 2Y ~y • j 21 .. • • •• "" • •" g ., < ~Y . I. J ~ •• .. IJ • II• 1 •• ' 2o , . • , 1IY u1 • ,, I> . >I J ... ., I J. < ~· ,. I • ~~ JO I ,, .. • • Ia I> ., ; . . .. J 9 <• ,. I J J I' • r 5 o) 6 •l 2v d • • u I• ~~ ; ltJ lVI 10 r il :J ~ •I •• I J 6v 11 1 • •l .. • ' 2< « • 9 >o 10 ... . , I• • .; ll .. . I• ,, >I • •j I• 10 • l •• ll
l j 2!; •• 1.; J l J J• I• . •J .. . . 15 •• I• '" 18 • 0 lll II• 21. lv ; lv.r. IV< " I •• •• .. s lJ '" t• IOo lol • 2 •J •I 1 • 2~: ,., '" I> I~ " > ,, 10 t IJ l J I• I• • • l 11 H •••• L •ll
' .. IC • 11 I• y 15 '·' .. • • ~ 6 I Oil lo> lo , h 1 r . • il l> I> 10 11 • • ;s , . y u .. .. • I ·n IJJ II •• .. •••• L ~ "2, u .. t• ~· " • 0 ., Oj u .. ;v
' 2 Yl II <J •• ~ 1 >I • I ~l H l } H .. • J 2o ,, II )J >t u Jv• .!~!. • J I• II • l •• H • li • U• lo> :1 I<< !"-'«' . •l .; , • ll I!> • " •• l . { y • 21 I J •• ,,
• •• 101 ll ., .. v • .. ,. ) ~ H •l •• •I 11 • l u >• , . , • .. '" II f~ .. " • I< ) Jl 0 ~ lbl 2•• lu 0 II J llo l I 2u :1 l )J ,, I! lo I " I• IO ) • •• <l 0 •• ., )U 2 2o •• I ~ II , 1 12 J I<> II .. :7 I I l•l lvl . I •I •• u 0 .. •8 '" J IY 15 • J <• <>
f •• 0 12 . .. I 2 ,. ,. • 2 11 ,, I I •• •l 10 . ~~ ;g J " ;; , . • lOr; ~:;,1 lie I J/ . ., I J Ill 110 . J •• .. I " it J2 II 0 J I -'> ~ 3J .. li ' ,. II I . 10 tl • • 10• lvo I J JJ II ~ l• 29 l ~ 2• L<
j • 2o <> li. J .. or I > ll • • .. ., l li l ll ·~ .. ' J J) ,, 1 ~. • u II <• .. ll •I ., I • •l .. ' 'i .~~ lY I llJ 112 !} • H " • l Jl t• • .. ., l< ,,
J> I • II . r ... I ,, >l ? J I 7 • l• " 1.1 )I ... I ' II 10 7 1 ,. ,. I " •• 11 ~- JU • J •v .. • •) J> ~ ·l .. I .. I J 1U I Jo " • o2 •• r ' 121 I< I • dl o• 12 2 l1 I) • ! •• ... IJ .. .. I 1 ll l >• 1>1 11 l H ~= ; • " " • II J 11> ' 10 <l t A 1 ) ' c> ,. l J >< >I • >. Jl <Y 1 28 1 J II ll 0 • • lo ,. IJ ;o lo,; • 1 "' 1>1 . •J II • l Yi •• I J s I. • " u C& • ~ •• •< lJ '" >v < ,. ,, • I J> " (. ., 10 I• •• II • I In <I • , 2o •• d •< ~· < i Jl .. • 2 I•> lo6 l " 15 •• I •• .. • I> .<1 u I• y • • " <9 • • ji( "' t •1 I• 2 II 12

HEXA·-u-DITHIOCACADYLATO TETRAZINCSULPHIDE
135

APPENDIX B.
SCALING OF INTENSITY DATA.
It is normal practice to scale data, thus accounting for
variations in reflection intensity during data collection. A few
primary standard reflections, collected at regular intervals
during data collection are used for this purpose. If absorption
corrections are required, they can be applied using either
numerical or analytical computing techniques. The use of only a
few standards can be quite a crude method of correction of other
intensities which are well removed from them in reciprocal
space. Variations can also be detected and corrected using, as
secondary standards, those classes of reflections, in which
corresponding members must, by symmetry be of equal intensity.
When for instance it is legitimate to average the reflections hkO
and hkO, then it is common practice to do so.
Scaling of the general hkl reflections should be carried
out in a similar way in order to be consistent.
The quantity
I (hkO) 0 1
I(hkO) = I0
(hk0) (1 +I (hk0))2 = I0
(hk0)shk 0
is the average of I(hkO) and I(hkO) and defines a scale factor
s The scale factor s is defined in the same way. What hk hk+l
of
the intervening reflections ? If s were to equal s it would hk hk+l
seem reasonable to scale them by s also. For s and s hk hk hk+l
differing it is not an unreasonable first approximation to assume
that any variation between them is linear. Such linear variation
in scaling factors is introduced in the expression
I(hkl) =I (hkl) (s f.+ 0 hk 1
where f is the fraction 1/n(l), 1
reflections observed from planes
136
s (1-f)) hk+l 1
n(l) being the number of
between (hkO) and (h k+l 0) •

REFERENCES
ARNOLD, S., WHITTEN, W.B. & DAMASK, A.C. (1970). J. Chern. Phys.
53, 7, 2878-2884.
AVAKIAN, P. & MERRIFIELD, R.E. (1968). Melee. Cryst. ~~ 37-77.
BANERJEE, A.J. & BASAK, B.S. (1975). Acta Cryst. A31, S 121.
BATES, P.A. (1978). Private communication.
BATT, R.H., BRAUN, C.L. & HORNIG, J.F. (1968). J. Chern. Phys. ~~
1967-1968.
BERGMAN, A. & JORTNER, J. (1974). Phys. Rev. B 2_, 10, 4560-4574.
BOCHNER, S. & CHANDRASEKHARAN, K. Fourier Transforms. Princeton:
Princeton. University Press.
BOEYENS, J.C.A. & HERBSTEIN, J.H. (1965) J. Phys. Chern. 69,7,
2160 176.
BREE, A., CARSWELL, D.J. & LYONS, L.E. (1955). J. Chern. Soc.
(London} ~, 1728-1733.
BRENIG, W., DOBLER, G. & HEYSZENAU H. (1972). Phys. Lett.
BRENIG, W., DOHLER, G. & WOLFLE, P. (1971). Z. Phys. 246,
BROWN, J.N., CHEUNG, L.D., TREFONAS, L.M. & MAJESTE, R.J.
J. Cryst. Mol. Struct. !, 361-371.
39, 175.
l.
(1974) 0
BUNSEN, R.W. (1843). Ann. Chern • ..!§_, 2.
BURNS, D.M. & IBALL, J. (1955). Proc. Roy. Soc. (London) A227,
200-214.
CAMERON, N. & TROTTER, J. (1964). J. Chern. Soc. 497.
CHANCE, R.R. & BRAUN, L.C. (1958). Phil. Mag. l, 1361-1380.
CHANCE, R.R. & BRAUN, L.C. (1973). J. Chern. Phys. 59, 2269-2272.
CHESNUT, D.B. & MOSELEY, R.W. (1969). Theoret. Chirn. Acta. (Berl.)
13, 230-248.
CHOJNACKI, H., LORENZ, K. & PIGON K. (1973). Phys. Stat. Sol. 70,
211.
CHOW, Y.M. (1971) Inorg. Chern. 10, 1938.
CHURCHILL, M. R. ( 197 3) • Inorg. Chern. 12, 5, 1213-1214.
CLEWS, C.J .B. & COCHRAN, W. (1948). Acta Cryst • .!_, 4-11.
137

CORFIELD, P.W.R., DOEDENS, R.J. & IBERS, J.A. (1967). Inorg.
Chern. ~, 197.
CROMER, D.T. (1965) Acta Cryst. 18, 17.
CROMER, D.T. & WABER, J.T. (1965). Acta Cryst. 18, 104-109.
CRUIKSHANK, D.W.J. (1965). In 'Computing Methods in
Crystallography', Ed., J.S. Rollet, Pergamon, Oxford, pll4.
DALAL, N.S., HAARER, D., BARGON, J. & MOHWALD, H. (1976). Private
communication. DASHEVSKII, V. G. , STRUCHKOV, Y. T. & AKOPYAN, Z. A. (1966) •
J. Struct. Chern. (USSR) z, 555.
EHRENFREUND, E., ETEMAD, S., COLEMAN, L.B., RYBACZEWSKI, E.F.,
GARITO, A.F. & HEEGER, A.J. (1972). Phys. Rev. Lett. 29, 5,
269-272.
EINSTEIN, F.W.B. & PENFOLD, B.R. (1968). J. Chern. Soc. A 3019.
ERDLE, E. & MOHWALD, H. (1977). Private communication.
ERN, V., AVAKIAN, P. & MERRIFIELD, R.E. (1966). Phys. Rev. 148,
862.
EVANS, D.L. & PENFOLD, B.R. (1975). J. Cryst. Mol. Struct. ~,
93-100.
EVANS, D.L. & ROBINSON, Ward T. (1977). Acta Cryst. B2l, 2891-
2893.
EVANS, D.L. & ROBINSON Ward T. (1980a) Acta Cryst. In Press.
EVANS. D.L. & ROBINSON Ward T. (1980b) Acta Cryst. In Press.
FLEMING, R.J. (1972). J. Chern. Phys. 56, 4911.
FORDER, R.A. & SHELDRICK, G.M. (1970). J. Organometal. Chern. 21,
FROHLICH, H. & SEWELL, G.L. (1959). Proc. Phys. Soc. (London) Z!r 643.
FURNAS, T.C. (1957). Single Crystal Orienter Instruction Manual,
General Electric Co., Milwaukee, U.S.A.
GARWIN R.L. (1975). Selected Aspects of Modern Technology for
Computers, IBM Thomas J. Watson Research Centre New York
u.s.A. GOLDBERG, I. (1975). Theoret. Chim. Acta. (Berl.) 40, 271-281.
138

GRANT, D.F., KILLEAN, R.C.G. & LAWRENCE, J.L. (1969). Acta Cryst.
B25, 374.
HAARER, D. (1969). Thesis, University of Stuttgart.
HAARER, D. (1975). Private communication.
HAARER, D. & CASTRO G. (1975). Private communication prior to
publication.
HAARER, D. & KARL, N. (1973). Chern. Phys. Lett. 21, 1, 49-53.
HAARER, D. & MOHWALD, H. (1975). Phys. Rev. Lett. l!r 1447-1450.
HAARER, D. & WOLF, H.C. (1970). Mol. Cryst. Liqu. Cryst. 10, 359.
HANSON, A. W. ( 1960) • Acta Cryst. 13, 215-220.
HARKER, D. (1936). J. Chern. Phys. _!, 381.
HERBSTEIN, F.H. (1971). Perspectives in Structural Chemistry,
Vol. 4, 166-395. New York:John Wiley.
HERBSTEIN, F.H. & SNYMAN, J.A. (1969). Phil. Trans. Roy Soc., A,
264, 635-662.
HO, Y.K. & ZUCKERMANN, J.J. (1973). J. Organometal. Chern. ~, 1.
HOFFMANN, R. (1963). J. Chern. Phys. ~, 1397-1412.
HOLZMAN, P., MORRIS, R., JARNAGIN, R.C. & SILVER, M. (1967). Phys.
Rev. Lett. 19, 506.
HOSOYA, S. ( 1966) . Acta Cryst. 21, 21-26.
HUGHES, R.C. (1971). J. Chern. Phys. 55, 15, 5442-5447.
International Tables For X-Ray Crystallography, Vol. III.
(Kynoch Press, Birmingham 1962).
JOHNSTONE, D., FERGUSSON. J.E. & ROBINSON Ward T. (1972) Bull.
Chern. Soc. Jap. 3721.
KARLE, I.L. & BROCKWAY, L.O. (1944). J. Arner. Chern. Soc. ~~
1974-1979.
KEPLER, R. G. ( 196 0) Phys. Rev. 119, 4, 1226-1229.
KEPLER, R.G., CARIS, J.C., AVAKIAN, P. & ABRAMSON, E. (1963).
Phys. Rev. Lett. 10, 9, 400-402.
KEPLER, R.G. & HOESTEREY, D.C. (1974). Phys. Rev. B 2_, 6, 2743-
2745.
KODAMA, T. & KUMAKURA, S. (1974a). Bull. Chern. Soc. Jap. 47(5),
1081-1084.
139

KODAMA, T. & KUMAKURA, S. (1974b). Bull. Chern. Soc. Jap. 47(9),
2146-2151.
LARSON, A.C. in Crystallographic Computing, ed. F.R. Ahmed,
(Munksgaard, Copenhagen, 1970, p.291).
Phys. Rev. Lett. 10, 9, 400-402.
LEBLANC Jr., O.H. (1961). J. Chern. Phys. 35, 4, 1275-1280.
LUPIEN, Y. & WILLIAMS, D.F. (1968). Mol. Cryst. ~, 1.
LYNTON, H. & COX, E.G. (1956). J. Chern. Soc. 4886-4895.
MAIN, P., WOOLFSON, M.M. & GERMAIN, G. (1971). MULTAN.
Univ. of York, England and Univ. of Louvain, Belgium.
MAIN, P., WOOLFSON, M.M. & GERMAIN, G. (19~). MULTAN.
Univ. of York, England and Univ. of Louvain, Belgium.
MAYOH, B. & PROUT, C.K. (1972). J .C.S. Faraday Trans. II ~,
1072 082.
MELZ, P.J. (1972). J. Chern. Phys. ~' 4, 1694-1699.
MERRIFIELD, R.E. (1968}. J. Chern. Phys. ~, 4318.
MIDDLEMISS, K.M. & SANTRY, D.P. (1974). J. Chern. Phys. 12,
5400-5403.
MOHWALD, H. & BOHM, A. (1976). Z. Naturforsch 3la, 1324-1332.
MOHWALD, H. ERDLE, E. & THAER, A. (1977) ~(1), 79-87.
MOHWALD, H. & HAARER, D. (1976). Mol. Cryst. Liq. Cryst. ~,
215-218.
MOHWALD, H., HAARER, D. & CASTRO, G. (1975}. Private
communication.
MOHWALD, H. & SACKMANN, E. (1973). Chern. Phys. Lett. 21, 43 -48.
MOHWALD, H. & SACKMANN, E. (1974). Sol. Stat. Comm. 15, 445-448.
MONTROLL, E.W. & WEISS, G.H. (1965}. J. Math. Phys. &1 167-181.
MOTT, N.E. (1969). Phil. Mag. 19, 835.
MULLIKEN, R.S. {1949). J. Chim. Phys. 46, 675.
MULLIKEN, R.S. (1952). J. Amer. Chern. Soc • .zi, 1, 811-824.
MULLIKEN, R.S. (1956). Rec. Trav. Chim. Pays-Bas 75, 845.
ONSAGER, L. (1938}. Phys. Rev. 54, 554-557.
PATTERSON, A.L. (1935). Z. Krist. A90, 517.
140

PAULING, L. (1960). The Nature of the Chemical Bond. 3rd. ed.,
224, Ithaca: Cornell Univ. Press.
ROSE, A. (1955). Phys. Rev. 'fl._, 1538.
ROBERTSON, J. Monteath, SHEARER, H.M.M., SIM, G.A. & WATSON, D.G.,
(1962). Acta Cryst. ~, 1-8.
ROBINSON, Ward T. & IBERS, J.A. (1967). Inorg. Chern • .§_, 1208.
RUDDICK, J. (1973). Private communication.
SAYRE, D. (1952). Acta. Cryst. ~' 60-65.
SEWELL, G.L. (1958). Phil. Mag. ]_, 1361 - 1380.
SHARP, J.H. (1967). J. Phys. Chern. 71, 2587-2596.
SINGH, S., JONES, W.J., SIEBRAND, B.P. & SCHNEIDER, W.G. (1965).
J. Chern. Phys. ~, 330. STEWART, R.F., DAVIDSON, E.R. & SIMPSON, W.T. (1965). J. Chern.
Phys. 42, 3175-3187.
SUTTON, L.E. (1955) in 'Determination of Organic Structures by
Physical Methods'. ed. Brande & Nachad, Academic Press, Inc,
New York.
TEMPLETON, L.K. & TEMPLETON, D. (1973). Amer. Cryst. Assoc.
Summer Meeting, Connecticut, USA. Paper ElO.
TROTTER, J. (1958). Acta Cryst. 11, 355-360.
TROTTER, J. (1961). Acta Cryst. 14, 1135.
TROTTER, J. (1963). Acta Cryst. 16, 605-607.
WEI, C. H. (1971}. Acta Cryst. B.£7_, 1523-1527.
WILSON, A.J.C. (1942). Nature 150, 151.
ZACHARIASEN, W.H. (1963). Acta Cryst. 16, 1139-1144.
ZACHARIASEN, W.H. (1967). Acta Cryst. 23, 558.
141

c c c c c c c c c c c
***************************************************************** PROGRAM *PATINV* IS USED TO INVESTIGATE THE PEAKS OF A PATTERSON MAP.
****************************************************************** FUNCTIONS
WITH THE VECTORS IN THE PATTERSON MAP CHECKING A PROPOSED STRUCTURE TO SEE WHICH ATOMS ARE CONSISTENT EXTENSION OF THE LIST OF PATTERSON MAP PEAKS SOLUTION OF PATTERSON MAP GIVEN 3 KNOWN ATOMIC POSITIONS (E.G. SITES DETERMINED FROM HARKER PEAKS)
C ******STRATEGY****** C THE LARGER THE LIST OF PATTERSON PEAKS IS THE MORE FRUITFUL IS C THE USE OF THIS PROGRAM C (1) FIND ATOM POSITIONS FROM HARKER PEAKS. TEST THESE FOR INTERNAL C CONSISTENCY USE SELFCN=1 :c 12) TEST PROPOSED STRUCTURE USING KNOWN PEAKS TO FORM VECTORS C 3) TEST PROPOSED STRUCTURE AGAINST ITSELF AS IN (1) I.E. SELFCN=1 C ( ) TRY SOLVE C (5) USE OF KNPKXN,PATPXN MAY HELP c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c .c
OPERATING NSYM
INSTRUCTIONS NO. OF SYMETTRY CARDS FOR SPACE GROUP - ALL REQUIRED NO. OF PATTERSON PEAKS NOPTPK
NKNAT NOPK
TOLNC
PATEXN
ALLPAT
SELFCN
SOLVE
KNONUM
NO. OF KNOWN POSITIONS NUMBER OF POSITIONS IN PROPOSED STRUCTURE 3 TOLERANCE VALUES REQUIRED
SUGGEST 0.4fA6 0.4,B, O.~fC 0 NO EXTENSI N OF PATTERSON PEAK LIST 1 EXTENSIN OF PATTERSON PEAK LIST 1 EXTEND LIST OF 'KNOwN' PEAKS 0 NO ACTION 1 TREATS PATTERSON PEAKS AS PROPOSED STRUCTURE
NKNAT,NOPK MUST BE SET EQUAL TO NOPTPK 0 TESTS NOPK PEAKS AGAINST NKNAT KNOWN ATOMS FOR
CONSISTENCY WITH PATTERSON 1 TESTS A PROPOSED STRUCTURE FOR SELF CONSISTENCY
SET NOPK = NKNAT 0 NO ACTION 1 SUBTRACTS A SUCCESSION OF PEAKS FROM ALL PATTERSON
PEAKS AND MAKES VECTORS FROM THE NEW PEAKS AND THE (SUGGEST 3) KNOWN POSITIONS AND TESTS WITH THE PATTERSON
PEAKS FOR CONSISTENCY 2 AS ALLPAT=1 PLUS AS FOR SOLVE =1 AND GIVES SUGGESTED
ATOM POSITIONS IF SOLVE IS SET TO 2 SET NKNAT,NOPK=NOPTPK
0 NO ACTION N NUMBER OF KNOWN PEAKS - ONLY SET WHEN SOLVE=2
**FORMAT** 4I5,3F5.4,6I5 C2 THE NSYM SYMMETRY CARDS OF THE SPACE GROUP
**FORMAT** AS FOR CUCLS C3 THE NOPTPK PATTERSON PEAKS **FORMAT** 3F10.8
C4 THE NKNAT KNOWN ATOM POSITIONS **FORMAT**3F10.8
C5 **FORMAT**
DIMENSION DIMENSION DIMENSION DIMENSION
IF ALLPAT =1 OMIT POSITIONS OF ATOMS IN PROPOSED STRUCTURE 3F10.8 IF ALLPAT=1 OMIT IF SELFCN=1 OMIT IF KNPKXN=1 OMIT IF SOLVE=1 OMIT IF SOLVE=2 OMIT
HARKER(30,6)~HARKTP(30,3) ,ACCPRS(50,2) ,WPEAK(1500,4) IND ( 6 ) MUMP (..:: 5) NUMP(3f 1 TYPE(25,5) ,KNUM(100) ,TYPOLP(24) X(200,3J
142

DIMENSION CHECK1J1001 1 CHECK2(100),CHECK3{100),APEAK(100,3) DIMENSION RATING 100 DIMENSION PEAK{6 00, ),DIFPK(2000,3) ,FREPTP(1000) ,NUMPK(1000) DIMENSION TOLNC(3) DIMENSION NUMSPK(80 6 8o~ DIMENSION SPEAK1600 ,3 DIMENSION PPEAK 1000,3 ,FPEAK(300 3) ,JHALF(7 3) DIMENSION SYMRL 8,6) ,SYMPAT(8~.6)!RATPK(1000,J) ,KNATOM(300,3)
DIMENSION JMA( 1,MAP(51,51,24) ,AMULT(3) DIMENSION SYM(lO DATA AMULT/44.0, 2.0,46.0/ INTEGER PK,SELFCN,ALLPAT,PATEXN INTEGER SOLVE INTEGER TYPE,HARKTP INTEGER D INTEGER PAT REAL KNATOM DATA IND/2L2,4,4,6,6/ INTEGER MAl-'S DATA JHALF{1i0,0,1,1,0,1, :g'5'~'~:~'1'1i
50 F6RMATI4IS JF5.4 9!5) 60 FORMAT 1X fNSYM=' I2 'NOPTPK=' I3 'NKNAT=',I3 'NOPK=' I3,'TOLNCS',
*3F5.4 PATEXN=' I2 'KNPKXN=',r2 'ALLPAT=' I2,,SELFCN='I2 'SOLVE=', *I2,'KNONUM= • I2 'HARK= ',I2,'PATSYM= ',I2) '
100 FORMAT 30X,3FiO.G1 303 FORMAT 30X,3F10.6 304 FORMAT F15.8LI3,7 ,F14.8,3X,I3,3X,F15.8,6X,I3) 306 FORMAT 30X 3t·10.8) 200 FORMAT 1X,JF10.4,5X,I3) 203 FORMAT 1X,3F10.4,SX,I3,2X *A1) 205 FORMAT 1X,'PATTERSON PEAKS'L14X,'FREQUENCY') 206 FORMAT 1X, I PEAKS I, 24X' 'NUMB.i!iR I) 400 FORMAT 1X,F5.2,2X,I2, X',2X,F5.2,2X,I2,'Y',2X,F5.2,2X,I2,'Z 1
1 * 2X,F5.2,2X!I2,'X',2X,F5.2,2X,I2,'Y',2X,F5.2,2X,I2, 1 Z'J 803 FORMAT(1X, 1 NEW PEAKS FOUND, FROM PAT. PEAK NO., NO KNOWN ATOMS,
*AGREEMENT LEVEL') 801 FORMAT 1X,3F10.6,3(5X,I5)} 903 FORMAT 1X,'HARKER VECTORS')
8008 FORMAT 1X,'ACCEPTED PEAKS',17X,'RATING') 8007 FORMAT 1X,3F10.8,I6) 1000 FORMAT 1X,' ')
40003 FORMAT 1X,2I5) . PSYMR=
ESTAT=10 DIMENSION TITLE(14) READ(5,98642)TITLE
,98642 ~~~~tt!§~~4~f~ITLE .98643 FORMAT(~1Xf13A6,A2)
READ(5,782 )SYM 7821 FORMAT(9A1l
SYM ( 10) =.I* READ(5,50) NSYM,NOPTPK
6NKNAT,NOPK,(TOLNC(JV) ,JV=1,3) ,PATEXN,
*KNPKXN,ALLPAT,SELFCN,S LVELKNONUM,HARK,PATSYM,NREMOV READ(5~.50003) MAPS,KUMP,IC.i!iNT
50003 FORMAT{3I5) IF (SOLVE.EQ.2i ALLPAT=1 IF1ALLPAT.EQ.1 SELFCN=1 IF ALLPAT.EQ.1 PSYMR=O WR TE(6!60) NS M1 NOPTPK1 NKNAT
6NOPKL(TOLNC(JV) ,JV=1,3) ,PATEXN,
*KNPKXN,ALLPAT,SELFCN,SOLVE,KN NUM,HARK,PATSYM NNKNAT=NKNAT
c 'c***** READ IN SYM CARDS c
WRITE(6,399)
143

399 FORMAT(1X,'SPACE GROUP SYMMETRY',20X,'PATTERSON SYMMETRY') DO 103 JS = 1 NSYM READ ( 5' 304) ( SYMRL (JS ,N) ,N=1' 6)
c C***** CREATE PATTERSON SYM c
DO 104 JSP=2,6,2 SYMPAT(JS,JSP)=SYMRL(JS,JSP)
104 CONTINUE 103 CONTINUE
IF(ICENT.GT.O) PAT=1 DO 1103 JS=1,NSYM*SYM(1+PAT) IF(PATSYM.EQ.O) GO TO 11113 READ(5,304) (SYMPAT(JS,N) ,N=1,6)
11113 CONT!NUE WRITE(6L400) (SYMRL(JS,N) ,N=1,6), (SYMPAT(JS,JSP) ,JSP=1,6)
. 110 3 CONTINU.!:!i c .C***** READ IN PATTERSON PEAKS c
102 1102
9002 c
IF(ALLPAT.EQ.1) GO TO 1102 DO 102 JJ = 1,~0PTPK READ(5,303) (PATPK(JJ,M) ,M=1,3) CONTINUE CONTINUE
IF(HARK.EQ.1) GO TO 9002 GO TO 9004 CONTINUE
C***** CREATE HARKER GEOMETRIES FROM SYMMETRY ELEMENTS OF SPACE GROUP c
~~I§~~ 6 J~~l)NSYM-1 DO 901 JPP=JP+1,NSYM JHR=JHR+1 DO 902 JRR=1,6 HARKER(JHR,JRR)=SYMRL(JP,JRR)-SYMRL(JPP,JRR)
902 CONTINUE IF(JHR.EQ.1l GO TO 899 DO 9902 JW= ,JHR-1 DO 9903 JV=1,NSYM*SYM(1+PAT) IPN =0 DO 9904 JTV=1,6 IF{AMOD(ABSlHARKER(JHRfJTV)-HARKER{JWlJTV)*SYMPAT(JV,IND(JTV))
*)+0.001,10**(AMOD(JTV+ ,2))) .LT.0.002J IPN=IPN+1 9904 CONTINUE
IF(IPN.NE.6) GO TO 9903 JHR=JHR-1 GO TO 901
9903 CONTINUE 9902 CONTINUE
899 CONTINUE WRITE ( 6, 400) (HARKER ( JHR,JRS) , JRS=1, 6)
901 CONTINUE 900 CONTINUE
7002 c
NOHKTP=JHR
~~~~J~i*~9~lRKER TYPES')
C***** CHECK HARKER GEOMETRIES FOR CONSTANT COORDINATES c
907
7001
DO 906 JHT=1,NOHKTP DO 907 JSS=2,6,2 IF(ABS(HARKER(JHT,JSS)).LT.0.001) GO TO HARKTP(JHT,JSS/2)=1 CONTINUE WRITE(6f7001) (HARKTP(JHT,JSL) ,JSL=1,3) FORMAT( X,3I5)
144
907

906 CONTINUE c C***** DETERMINES WHICH CLASSES OF PATTERSON C***** OBTAIN POSITIONAL VECTORS c
c
7003 ~~~~~f~ik~9~tCEPTABLE TYPES') JCO=NOHKTP NMP=1 DO 984 IRT=1iJC0-1 DO 9997 JRZ= ,3 MUMP(JRZ)=O
9997 CONTINUE NUM=O DO 981 JRT=1,3 IF(HARKTP(IRT,JRT).NE.1) GO TO 981 MUMP(JRT)=1 NUM=NUM+l IF(NUM.NE.3) GO TO 981 TYPE~NMP,1~ = IRT
~~l¥~~~pi9§6)=T~PE(NMP,1) 986 FORMAT( X,I5)
NMP=NMP+1 GO TO 984
981 CONTINUE N=2 DO 983 KRT=IRT+1,JCO MUM=NUM TYPE~NMP,1)=IRT TYPE NMP,N)=KRT DO 9 2 JJRT=1,3 NUMP(JJRT)=MUMP(JJRT) IF(HARKTP.(KRT,JJRT) .NE.1) GO TO 982 IF(NUMP(JJRT).NE.1) GO TO 988 TYPOLP(NMP)=JJRT GO TO 982
988 CONTINUE NUMP(JJRT)=1 MUM=MUM+1 IF(MUM.NE.3) GO TO 982 WRITE(6i985) N~(TYPE(NMP,JRL) ,JRL=1,N)
985 FORMAT( X,*I5,~I5) TYPE(NMP, 5) =N
982 CONTINUE NMP=NMP+1
983 CONTINUE 984 CONTINUE
NOATYP=NMP-1
VECTORS CAN BE USED TO
,TYPOLP(NMP),NMP
C***** FROM PATTERSON PEAKS OBTAIN VALUES FOR VARIABLE COORDINATES OF C***** THOSE VECTORS WHICH HAVE AT LEAST ONE COORDINATE CONSTANT c
909
DO 908 JIP=1LNOHKTP KNUM(JIP) = ~X+1 JN=O DO 909 JSS=1 3 IF(HARKTP(JIP,JSS) .EQ.O) JN=JN+1 CONTINUE DO 920 JIZ=1,NOPTPK-NREMOV L=O PX=PX+1 DO 945 JSR=163 X(PX.cJSR)=O. IF(HARKTP(JIP,JSR) .EQ.1) GO TO 946 IF(AMOD(ABS(HARKER(JIP,2*JSR-1)-PATPK(JIZ,JSR))+0.001,1) .GT.0.002)
*GO TO 948 L=L+1
145

GO TO 945 946 CONTINUE
IF(HARKER(JIP,2*JSR).EQ.O) GO TO 935 X(PX,JSR)=(PATPK(JIZ,JSR)-HARKER(JIP,2*JSR-1))/HARKER(JIP,2*JSR)
935 CONTINUE 945 CONTINUE
IF(L.EQ.JN) WRITEJ6z911) (X(PX,JSR) ,JSR=11 3) ,(HARKTP(JIP,JIM) ,JIM=1 *,3) ,L,PX,JIPiJIZ, PATPK(JIZ~JLA) 6JLA=1,3J 911 FORMAT(1X,3F 0.7, OX,3I5,4I~,3Fl .6)
GO TO 920 948 PX=PX-1 920 CONTINUE 908 CONTINUE
KNUM(NOHKTP+1~=PX+1 WRITE(6i20001 NOHKTP,(KNUM(JAB) ,JAB=1,NOHKTP)
:20001 FORMAT( X,*I5 . MOP=O
DO 990 JLP=1iNOATYP DO 9993 I= ,3 IF(HARKTP(TYPE(JLP,1),I) .EQ.O) IT=I
9993 CONTINUE DO 994 MO=KNUM(TYPE(JLP,1})~KNUM(TYPE(JLP,1)+1)-1 IF{TYPE(JLP,5).NE.1) GO TO ~9000 MOP=MOP+1 DO 99002 J=1 3 WPEAK(MOP,J)bX(MO,J)
99002 CONTINUE
:~~~~~~Oi§9664r~~PEAK(MOP,JQ),JQ=1,4) 99004 FORMAT( X,4F10.4) 99000 CONTINUE
IF(TYPE(JLP,5) .NE.2) GO TO 9995 DO 995 JI=KNUM(TYPE-(JLP62)) ,KNUM(TYPE(JLP,2)+1)-1 IF(TYPOLP(JLP) .EQ.O) G TO 99600 DO 996 JZ=1,NSYM IF(AMOD(ABS{X(MO,TYPOLP~JLP)}-X(JI6TYPOLP(JLP))*SYMRL(JZ,2*TYPOLP( *JLP))+SYMRL(J~,2~TYPOLP JLP)-1))+T LNC(TYPOLP(JLP)) ,1).GT.2*TOLNC
*(TYPOLP(JLP))) GO TO 99 MOP=MOP+1 DO 993 I=1 3 IF(HARKTP(TYPE(JLPf1) ,I) .EQ.O) GO TO 993 WPEAK(MOP,I}=X(MO, ) WPEAK(MOP,4)=JLP
993 CONTINUE
:~¥~~~~oi~6aa6)x~~~Ef~~;g~~~t!t~i~ir;k6~;~~~J~;fi;BEPt~LP) ,IT 30000 FORMAT( X,3F10.4,5I5)
GO TO 995 996 CONTINUE
GO TO 995 99600 CONTINUE
MOP = MOP+1 DO 99601 JQR=1 3 WPEAK(MOP,JQR)~X(MO,JQR)+X(JI,JQR)
99601 CONTINUE WPEAKJMOP 4)=JLP WRITE 6i99602) (WPEAK(MOP,JRS) ,JRS=1,4)
99602 FORMA ( X,4F10.4) 995 CONTINUE
9995 CONTINUE IF(TYPE(JLP, 5).NE.3) GO TO 994 DO 1001 NMO=KNUM(TYPE(JLP,2)),KNUM(TYPE(JLP,2)+1)-1 MOP=MOP +1 DO 1002 NRM=1,3
1002 IF(HARKTP(TYPE(JLP,2),NRM) .EQ.1) WPEAK(MOP,NRM) =X(NMO,NRM) CONTINUE DO 1003 NMP=KNUM(TYPE(JLP,3)) ,KNUM(TYPE(JLP,3)+1)-1 MOP=MOP+1
146

c
DO 1004 NMM==1,3 IF(HARKTP(TYPE(JLP,3),NNM).EQ.1) WPEAK(MOP,NMM)==X(NMP,NMM)
1004 CONTINUE WPEAKJMOP 4)=JLP WRITE 6f1605) (WPEAK(MOP,NJ),NJ=1,3)
1005 FORMA ( X,3Fl0.4) 1003 CONTINUE 1001 CONTINUE
994 CONTINUE 990 CONTINUE
DO 1010 NA=l MOP-1 DO 1011 NB=NA+1,MOP DO 1012 NC=1,NSYM
NOND=O DO 1013 ND=1 3 IF(AMOD(2*ABS(WPEAK(NA,ND)-(WPEAK(NB1 ND)*SYMRL(NC
62*ND)+SYMRL
*(NC02*ND-1)))+2*TOLNC(ND) ,1).GT.4*TOLNC(ND)) GOT 1012 NON =NOND+l
1013 CONTINUE IF(WPEAK(NB 1 4)-WPEAK(NA~4) .GT.0.001) NOND=NOND+l IF(NOND.NE.4) GO TO 101~ NMOP=NMOP+1 DO 1014 NDD=1 3 IF{NMOP.GT.6060) GO TO 1010
PEAK(NMOP,NDD)=WPEAK(NA,NDD) 1014 CONTINUE 1012 CONTINUE 1011 CONTINUE 1010 CONTINUE
IF(MOP.EQ.O)NMOP=5 IF(MOP.EQ.O)MOP=5 IJZ=O
C***** OBTAINS LIST OF UNIQUE VECTORS c 40001 ~~~A~~~f~~98&iouE POSITIONS FROM PATTERSON')
DO 950 LL=1,NMOP DO 951 LM=1 IKZ
c
IF(IKZ.GT.124) GO TO 950 DO 952 PRM=1,NSYM*SYM(1+PAT) IJ=O DO 953 PRL=1,3 IFCAMOD ABS(PEAK(LLLPRL)-PPEAK(LM*8-7,PRL)*SYMPAT(PRM,2*PRL))+TOLN
*C(PRL ).LT.2*TOLNc(PRL)) IJ=IJ+1 953 CONTI
IF(IJ.EQ.3) GO TO 950 952 CONTINUE 951 CONTINUE
IJZ=IJZ+1 DO 954 IJR=1 3 PPEAK(IJZ*8-7,IJR)=PEAK(LL,IJR)
954 CONTINUE WRITE(6f960) (PPEAK(IJZ*8-7,IRT) ,IRT=1,3)
960 FORMAT( X,3F10.6) IKZ=IJZ
950 CONTINUE
C***** EXPANDS LIST 8FOLD TO ALLOW FOR AMBIGUITY OF 0.5 IN EACH C COORDINATE. c
DO 961 I0=1fiJZ DO 962 IOT= ,7 IOOT=I0*8+IOT-7 DO 963 IB=1,3 PPEAK(IOOT,IB)=PPEAK(I0*8-7,IB)+0.5*JHALF(IOT,IB)
963 CONTINUE 962 CONTINUE
147

961 CONTINUE c C***** OBTAIN LIST OF UNIQUE VECTORS .c 40002 WFORRMITEA( 6(fx40,9u0N2)QUE VECTORS r I FOLLOWING 8FOLD EXPANSION')
INZ=O DO 970 KI=1i8*IJZ DO 971 KII= ,IMZ DO 972 KYM=l,NSYM IJN=O DO 973 IC=1,3 IF(AMOD(ABS(PPEAK(KifiC)-FPEAK(KII~IC)*SYMRL(KYM,2*IC)+SYMRL(KYM,
*2*IC-1)Y+TOLNC(IC) ,1, .LT.2*TOLNC(Ic)} IJN=IJN+1 973 CONTINUE:
IF(IJN.EQ.3) GO TO 970 972 CONTINUE 971 CONTINUE
INZ=INZ+1 IF{INZ.GT.200} GO TO 9000 DO 974 IJN=1 3 FPEAK(INZ,IJN)=PPEAK(KI,IJN)
974 CONTINUE WRITE(6i978) (FPEAK(INZ,JOP) ,JOP=1,3)
978 FORMAr( X,3F10.6) IMZ=INZ
970 CONTINUE 9000 CONTINUE
DO 980 J=1,INZ DO 9981 JJ=l,3 KNATOM(J,JJ)=FPEAK(JLJJ) PEAK(J,JJ)=FPEAK(J,JJ)
9981 CONTINUE 980 CONTINUE
NOPK=INZ NKNAT=INZ IF(SELFCN.EQ.1) GO TO 3001 GO TO 10000
9004 c
CONTINUE
C***** READ IN KNOWN ATOMS c
c
6000 6001
2000 3000
101
GO TO 6001 NKNAT=KNONUM CONTINUE DO 101 J = l,NKNAT READ ( 5,100) (KNATOM (J, L)fL=1,3) IF(SELFCN.NE.1) GO TO 10 IF(PID.EQ.1) GO TO 101 DO 3000 :LL=l,3 PEAK(J,LL)=KNATOM(J,LL) IFCALLPAT.NE.1) GO TO 3000 PATPK(J,LL)=KNATOM(JLLL) WRITE(6f2000) PATPK(J,Lt) FORMAT( X,Fl0.6) CONTINUE CONTINUE IF(SELFCN.EQ.l.OR.ALLPAT.EQ.1)
C***** READ IN SUGGESTED PEAKS c
GO TO 3001
IF(SOLVE.EQ.1} GO TO 3001 DO 105 JPK=1 NOPK READ(5L306) (PEAK(JPK,LP) ,LP=1,3)
105 CONT!NuE 3001 CONTINUE
IF(KNPKXN.EO.O) GO TO 4001 NOPK=NOPTPKINSYM
148

c
NUMN01=1 NUMN02 =NOPTPK
DO 604 JP=1,NKNAT DO 602 JR=NUMN01 NUMN02 DO 601 JSYM=1,NSYM*SYM(1+PAT) DO 600 IZ=1 3
PEAK{(JR-1{*NSYM+JSYM,IZ) = PATPK(JR,IZ)*SYMPAT(JSYM,2*IZ) -*KNATOM(JP,IZ)
600 CONTINUE 601 CONTINUE 602 CONTINUE
4001 CONTINUE MKNAT=NKNAT NPNAT=NKNAT
IF(SOLVE.EQ.2.AND.PID.EQ.l) GO TO 5002 IF(SOLVE.NE.1) GO TO 5000
5002 CONTINUE DO 5604 JP=l
6NKNAT
DO 33 JU=1 8 DO 34 JJU=i,80 NUMSPK(JJU,JU)=O
34 CONTINUE 33 CONTINUE
DO 35 KU=1,100 CHECK1~KU) =0 CHECK2 KU)=O DO 36 KU=1 3 APEAK(KU,KKU) = 0
36 CONTINUE 35 CONTINUE
DO 37 KLM=1,1000 FREPTP(KLM) =0
37 CONTINUE DO 5603 JT=1,NOPTPK DO 5061 IT=1,3 PEAK(JTLIT)=PATPK(JT,IT)-KNATOM(JP,IT)
5061 CONTINU.!!i 5603 CONTINUE 5000 CONTINUE
C***** EXPAND PEAK LIST BY GENERATION OF EQUIVALENT PEAKS USING SYM. OPS c
c
DO 109 PK=1,NOPK DO 110 ISYM=1,NSYM DO 111 IS=1l3 SPEAK(NSYM*(PK-1)+ISYM,IS)=AMOD(PEAK(PK,IS)*SYMRL(ISYM,2*IS)+PSYMR
**SYMRL(ISYM,2*IS-1) ,1) 111 CONTINUE 110 CONTINUE 109 CONTINUE 500 CONTINUE
C*****CALCULATE DIFFERENCE BETWEEN KNOWN ATOM FOR ALL PEAKS IN .C***** EXPANDED PEAKS LIST 1 COMPONENT AT A TIME c
DO 106 NJ=1,NPNAT NUM=O NZUM=O DO 108 JZ=1,NOPK*NSYM IF(AMOD(JZ,NSYM).EQ.1) NUM=O DO 107 1:=1 3 DIFPK(JZ,If=AMOD(KNATOM(NJ I)-SPEAK(JZ,I) 1) JMA(AMOD{I+KUMP-1,3)+1)=AM6D(DIFPK(JZ,I)+1.6,1)*AMULT(AMOD(I+KUMP-
107*~6~f±&b~l.5 . IF(MAPS.EQ.Ol GO TO 11102 MAP(JMA(1) ,JMA(2) ,JMA(3))=MAP(JMA(1) ,JMA(2) ,JMA(3))+1
11102 CONTINUE
149

c C***** C***** C***** c
GO THROUGH THE PATTERSON PEAKS AND ALL THEIR SYM. EQUIVALENTS AND TEST FOR EQUALITY WITH THE DIFFERENCE VECTORS ABOVE COMPONENT BY COMPONENT.
c
DO 113 JY=1 NOPTPK DO 112 JSY=i~NSYM*SYM(1+PAT) DO 115 IL=1,.j IF(AMODfABS{DIFPK(JZ6IL}-PATPK(JY,IL)*SYMPAT(JSY,2*IL))+TOLNC(IL),
*1) .GT.2*TOLNC(IL)} G TO 112
C***** IF ALL CPTS. OF PATTERSON PEAK OR ITS C***** PEAK ADD 1 TO FREQUENCY OF OCCURRENCE c
EQUIVALENT EQUAL TO DIFF. OF PATTERSON PEAK
c IF(IL.EQ.3) FREPTP(JY)=FREPTP(JY}+1 IF (IL.EQ.3) NUM=NUM+l
C***** IF ALL 4 SYM. DIFF. ARE LOCATED THE PEAK IS LIKELY TO BE TRUE AND C***** ADD 1 TO ITS COUNT c
IF (NUM.EQ.NSYM} NUMPK((JZ+(NSYM-1))/NSYM)=NUMPK((JZ+(NSYM-1))/NSY *M}+l
IF(IL.EQ.3) GO TO 1108 115 CONTINUE 112 CONTINUE -
IF{SOLVE.EQ.2.AND.PID.EQ.0) CHECK3(JY)=NUMPK(JY) 113 CONTINUE
IF(PATEXN.EQ.O) GO TO 1108 DO 4561 JRZ=1,NZUM DO 4562 JRW=1,NSYM*SYM(1+PAT) DO 4563 JRY=1,3 IFCAMOD{ABS(DIFPK(JZ,JRY)-PATPK{NOPTPK+JRZ,JRY)*SYMPAT(JRW,2*JRY))
*+TOLNC(JRY) 1).GT.2*TOLNC(JRY)) GO TO 4562 IF(JRY.EQ.3f FREPTPCNOPTPK+JRZ)=FREPTP{JRZ+NOPTPK)+1 IF(JRY.EQ.3) GO TO 1108
4563 CONTINUE 4562 CONTINUE 4561 CONTINUE
NZUM=NZUM+1 DO 4564 ILL=1,3 PATPK(NOPTPK+NZUM,ILL)=DIFPK(JZ,ILL)
4564 CONTINUE FREPTP(NZUM+NOPTPK)=1
1108 CONTINUE IF{AMOD(JZ,NSYM).EQ.O) NUMSPK((JZ+NSYM-1)/NSYM,NJ)=NUM
108 CONTINUE: 106 CONTINUE
IF(MAPS.EQ.O) GO TO 39000 DO 50001 RMA=1,AMULT(3) WRITE(6 50000) KMA
50000 FORMATCiX,'SECTION NUMBER',I3) DO 50002 JMAA=1,50
60000 50002 50001 39000 c
WRITEC6f60000l(MAP(IMA,JMAA,KMA) ,IMA=1,50) FORMAT( X,50I ) CONTINUE CONTINUE CONTINUE
C***** WRITES OUT PATTERSIN PEAKS AND FREQUENCY OF ENCOUNTER. c
'C
~~I~~t 6J~~i)NOPTPK+NZUM WRITE ( 6, 206) (PATPK (JB, JBB) , JBB=1, 3) , FREPTP (JB)
201 CONTINUE IF(KNPKXN.EQ.1) GO TO 4002
C***** WRITES OUT PEAK AND NUMBER OF TIMES IT IS PRESENT. c
150

WRITE{6,206) WRITE 6,10001 WRITE 6f7882 NKNAT+2,(SYM(10) ,J=1,NKNAT+2)
7882 FORMAT( X,*A ) DO 202 JC=1 NOPK WRITE(6,203}{PEAK(JCfJCC)fJCC=1,3)LNUMPK(JC),NKNAT+2,SYM{10),
*(SYM(NUMSPK(JC,NS)+1J ,NS= ,NKNAT) ,oYM(10) IF (SOLVE.EQ.Ob GO TO 4999 A=NOMSPK(JC,AM D(JP,3)+1) B=NUMSPK(JC,AMOD(JP+1,3)+1) C=A+B IFjA.GT.1.AND.B.GT.1.AND.C.GT.NSYM/2) CHECK1(JC)=C IF A.GE.NSYM/2+1.0R.B.GT.NSYM/2+1) CHECK2(JC)=C IF CHECK2(JC).EQ.CHECK1(JC))CHECK2(JC)=O
4999 CO TINUE 202 CONTINUE
WRITE{6L7882)NKNAT+2,(SYM(lO),J=l,NKNAT+2) 4002 CONTINU.~!t
IF(SOLVE.EQ.l) GO TO 8000 IF (PID.EQ.1) GO TO 8000 IF(SOLVE.NE.l) GO TO 5065
5604 CONTINUE 5065 CONTINUE
IF(PID.EQ.1) GO TO 10000 PSYMR=1 PID=1 IF(SOLVE.EQ.2) GO TO 6000 IF(KNPKXN.EQ.O) GO TO 10000 NO=O M=O GO TO 605
804 CONTINUE NO=N0+1 IF(NO.EQ.NKNAT) GO TO 807
605 CONTINUE WRITE{6,803) N=O ¥~<~g~P~~J~}!~M~~~~i~~~~~1b;N~BM6o6 N=N+1 WRITE{6f801) (PEAK(JW,IB),IB=1,3) ,JW,NKNAT,NKNAT-NO DO 607 A=1,3 KNATOM(N+NUMN02,IA) =PEAK(JW,IA)
607 CONTINUE 606 CONTINUE
NNKNAT=NNKNAT+N GO TO 804
807 CONTINUE 604 CONTINUE
GO TO 8001 8000 CONTINUE
DO 8009 JH=1 NOPTPK IF(CHECK2(JHJ.GT.NSYM/2.AND.CHECK3(JH) .GE.NSYM/2-1) CHECK1(JH)=1+
*CHECK3(JH) 8009 CONTINUE
NUMA=O NUMACC=O ~~I§~d~'gg~i)NOPTPK IF(CHECK1~JTf.EQ.O) CHECK1(JT)=CHECK2(JT) IF(CHECK1 JT) .EQ.O.AND.CHECK2(JT).EQ.0) GO TO 8002 DO 8005 J =1,NUMACC DO 8004 IS=1,NSYM NUMZ=O DO 8003 IM=1 3 IF(AMOD{ABSJPEAK(JTliM)-{APEAK(JE 1 IM)*SYMRL(IS,2*IM)+SYMRL(IS,2*IM
*-1)))+T0LNC IM) 6l).LT.2*TOLNC(1M)J NUMZ=NUM~+1 IF(NUMZ.EQ. ) G TO 8002
151

8003 CONTINUE 8004 CONTINUE 8005 CONTINUE
NUMA= NUMA +1 NUMACC=NUMA DO 8006 IZ=1,3 APEAK(NUMA ,IZ}=PEAK(JT,IZ)
8006 CONTINUE
~~j~~?~~~~~7)~A~~~tJ8M~cK;f~f~f~I~~y~~f~tNG(NUMA 8002 CONTINU.t!i
KOUNT=O DO 6502 JT=1,NUMACC DO 6505 JE=1,MKNAT DO 6504 IS=1,NSYM NUMZ=O
¥~c~86(l~s7K~ATOM(JEliM)-(APEAK(JT,IM)*SYMRL(ISi2*IM)+SYMRL(IS, *2*!M-1)))+T0LNC(IM) ,1 .LT.2*TOLNC(IM))NUMZ=NUMZ+
IF(NUMZ.EQ.3) GO TO 6 02 6503 CONTINUE 6504 CONTINUE 6505 CONTINUE
KOUNT=KOUNT+1 DO 6506 IZ=1,3 KNATOM(MKNAT+KOUNT,IZ)=APEAK(JT,IZ)
6506 CONTINUE 6502 CONTINUE
MKNAT=MKNAT+KOUNT WRITE(6 6510)
6510 FORMAT(fX,'LIST OF KNOWN ATOM POSITIONS AT THIS POINT') DO 6511 JT=1 MKNAT WRITE(6f6512f (KNATOM(JT,IM) ,IM=1,3)
6512 FORMAT( OX,3Fl0.6) 6511 CONTINUE
NKNAT=MKNAT IF(MKNAT.GT.NPNAT)NPNAT=NPNAT+1
8001 CONTINUE GO TO 5604
10000 CONTINUE STOP END
152

c c c c c c c c c c c c c c c c c c c c c c c c c c c c c .c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c
PROGRAM ARTSTC **************
PROGRAM TO PRODUCE A SHADED ELECTRON DENSITY MAP FROM DATA PRODUCED IN A FOURIER PROGRAM. ANY NUMBER OF COPIES OF SECTIONS MAY BE PRODUCED. A GRID FOR DETERMINATION OF COORDINATES IS ALSO PRODUCED. MARK 2 VERSION SEPTEMBER 1975 WRITTEN BY D.L.EVANS UNIVERSITY OF CANTERBURY MARCH 1974 REVISED SEPTEMBER 1975 COPYRIGHT HELD BY THE AUTHOR.
FEATURES ******** 1. DENSITY PROFILE - PRINTED FOR EACH SECTION 2. INTERPOLATION - (1) FOR INCREASED SMOOTHING AND (2) TO MINIMISE
TOPOLOGICAL DISTORTION DUE TO FIXED PRINT POSITICN,CHARACTER WIDTH, HEIGHTf AND LINE SPACING CHARACTERISTICS.
3. ON REQUEST PR NTS A GRID USEFUL AS AN AID TO READING COORDINATES. .
4. PRINTS ADDITIONAL COPIES ON REQUEST (AND ALSO SUPPLIES A GRID). 5. PRINTS CELL DIMENSIONS AND CM. TO ANG. CONVERSION FACTORS, 6. FRACTIONAL COORDINATES ARE LISTED FOR THE CALCULATED POINTS. 7. REPRODUCES CELL ANGLES IN THE RANGE 45DEG. TO 135DEG. TO WITHIN 0.1DEGREE. 8. CAN PRINT MAPS OF ANY LENGTH,WIDTH LIMITED BY PAPER WIDTH. 9. CONTOURS CAN BE MADE BY TRACING AROUND REGIONS
CONTAINING THE SAME SHADING CHARACTER. 10. MAXIMUM AND MINIMUM DENSITY LEVELS CAN BE VARIED FOR SPECIFIC
PURPOSES6WHICH VARIES THE NUMBER OF SHADES SPREAD ACROSS REGIONS F THE MAP.
11. THE DARKEST SHADE IN EACH COLUMN OF CALCULATED VALUES IS PRINTED OUT AT THE BOTTOM OF EACH MAP AS AN AID TO LOCATION OF POINTS OF HIGH DENSITY.
12. THE COMPLETE LIST OF SHADES IS GIVEN IN THE HEADING OF EACH MAP.
PROGRAM CONTROL. ****************
VALUES OF CONTROL INTEGERS
USER OPTIONS BY DEFAULT RANGE OF DENSITY 0 TO 1000 NUMBER OF COPIES 1
USER -1000<RHOMIN<RHOMAX<1000
1 - 8 9 GRID 0,2 TO 8
** NOTE THAT THE NUMBER OF COPIES USING THE SAME CONTROL INTEGER
AND THE GRID ARE SPECIFIED
INPUT FORMAT 3I5 ***** ********** NUMPIC(NO. OF COPIES (OR 9 FOR A GRID WITH ONE COPY)) RHO MAX RHOMIN
DETAILS OF THE FILE STRUCTURE ***************************** THE FIRST RECORD CONTAINS INFORMATION ON LIMITS, INCREMENTS AND ORIENTATION AS DETAILED BELOW.
NX THE NUMBER OF POINTS ACROSS A LINE OF A SECTION. NY THE NUMBER OF ROWSLSECTION. NZ THE NUMBER OF SECTIONS.
NORIEN 100 SECTION COMPUTED ALONG THE X AXIS 10 SECTION COMPUTED ALONG THE Y AXIS
1 SECTION COMPUTED ALONG THE Z AXIS. ADDITIONAL NOTE CONCERNING NORIEN
NORIEN FASTEST LINE SECTION
153

c c c c c c c c c c ~c .c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c c
100 10
1
XMIN XMAX
YMIN YMAX
ZMIN ZMAX
VARYING (ACROSS PAGE) z
X y
INDEX (DOWN PAGE)
y z X
LIMITS ALONG THE ROW AXIS, IN FRACTIONAL CO-ORDINATES
LIMITS ALONG THE COLUMN AXIS, IN FRACTIONAL CO-ORDINATES
LIMITS ALONG THE STACK AXIS, IN FRACTIONAL CO-ORDINATES.
INDEX
X y z
THE SECOND AND SUBSEQUENT RECORDS OF THE FILE EACH HOLD ONE ROW OF VALUES FOR MAKING UP THE MAPS. THE PROGRAM IS DIMENSIONED FOR MAPS OF A MAXIMUM SIZE OF 105 X 105 VALUES PER SECTION.
THE LAST RECORD ON THE FILE CARRIES THE CELL DIMENSIONS (REALL COSINES) AND THE VALUE OF RMAX - THE VALUE OF THE HIGHEST PEAK IN ANY OF THE MAPS.
PRINCIPAL ARRAYS. ***************** SECT VALUES FOR A SECTION A1,A2 REMEMBER REGIONS OF ROWS TO BE INTERPOLATED MEM REMEMBERS REGIONS TO BE INTERPOLATED BETWEEN ADJACENT ROWS MEM2 REMEMBERS ROWS REQUIRING PRINTING(I.E. THOSE NOT COMPLETELY
BLANK. ) CELL CELL CONSTANTS. NPNP2 VALUES OF NP2 GIVEN NP WHERE NP IS EXPANSION FACTOR ACROSS
PAGE AND NP2 DOWN PAGE NNUP CONTAINS THE NUMBER OF ROWS EACH COLUMN IS TO BE 'MOVED UP'
TO OBTAIN THE CELL ANGLE APPROPRIATE TO THE MAP PRNTAA THE ARRAY WHICH HOLDS THE VALUES OF THE EXPANDED MAP
PRIOR TO PRINTING IT OUT. NOVER THE NUMBER OF TIMES A LINE IS OVERPRINTED ARTSTC DATA ARRAY HOLDING SHADING CHARACTERS VAL PAIRS OF ADJACENT ROWS OF SECTION FOR INTERPOLATION RINTER ROW INTERPOLATION OF VAL CINTER COLUMN INTERPOLATION OF VAL TINTER INTERPOLATION OF RINTER. INTEGER RINTER,VAL,PTNO,PLACE INTEGER SHADER INTEGER DPTNO REAL INDEX INTEGER CJ RJ INTEGER TITLE,PRNTAA,ROW INTEGER ROWN INTEGER UPNUM INTEGER SLSTART INTEGER G!{ID INTEGER BOTNUM INTEGER OUTROW
INTEGER TINTER,CINTER REAL JITTLE INTEGER BB,AA~cc6DD,EE DIMENSION MEM~(2 5) DIMENSION A1(64~2) ,A2(64,2) ,MEM(126} DIMENSION CELL(o) DIMENSION INDXA(3} DIMENSION NPNP2{5) DIMENSION XYZ(4) DIMENSION NNUP(l33) DIMENSION PLM(l25) DIMENSION ACR0SS(l25},AA(125) ,BB(125) ,CC(125),DD(125) ,EE(125)
154

c c c c
c c c c
NUMPIC=ISST(4) INITIALISES PROGRAM PARAMETERS
IF(NUMPIC .GE. 2) GRIDNM = 1 IF (NUMPIC .EQ.9) NUMPIC = 1 B = 1 c = 2
ASSIGNS 20 SHADESTO RANGES OF VALUES 0-1000 AND ASSESSES NUMBER OF OVERPRINTINGS FOR EACH SHADE
DO 100 NVALUE = 1,1000
155

c
299 298
297 200 100
1700
DO 200 SIZE = 1,20 IF (NVALUE .GT. D(SIZE) .AND. GO TO 297 CONTINUE MTONEFCNVALUE) = SIZE+1 NOVER(NVALUE)= G(SIZE+1) CONTINUE CONTINUE CONTINUE DO 1700 NVALUE = 1087,1103 SIZE = NVALUE-1066 MTONEF(NVALUE) = SIZE+1 NOVER(NVALUE) = G(SIZE+1) CONTINUE IVXX = 1
NVALUE .LE. D(SIZE+1)) GO TO 298
C READS FIRST RECORD OF FILE c
READ(IVXX,END=99) NX,NY,NZ,NORIEN,XMIN,XMAX,YMIN,YMAX,ZMIN,ZMAX 99 CONTINUE
c c c
1111 2000
c c c
1112 c c c c c c
1113 c c c
NXX = NX NYY = NY YYMAX = YMAX YYMIN = YMIN XXMAX = XMAX XXMIN = XMIN
READS FILE TO END TO ENABLE THE READING OF THE LAST RECORD
DO 2000 JKL = 1 NY*NZ READ(IVXX,END=1i11) DUMMY CONTINUE CONTINUE
READS LAST RECORD
READ(IVXX,END=1112) RMAX,(CELL(I) ,I=1,6) CONTINUE
REWINDS FILE
REWIND IVXX
READS FIRST RECORD TO BE READY FOR MAP RECORDS
READ(IVXX,END=1113) DUMMY CONTINUE
OBTAINS CELL ANGLES FROM THEIR COSINES
DO 771 I = 4 6 CELL(I) = ARCOS(CELL(I))*180.0/3.14159
771 CONTINUE c c c
c
456
457
INTERPRETS NORIEN
DO 456 NNM = 1 3 DIV = NORIEN/CiO**(NNM-1)) IF (DIV .EQ. 1.0) NNMM = ~NM CONTINUE DO 457 JKL = NNMM,NNMM+2 NA = JKL-NNMM+1 INDX(NA) = INDIC(MOD{JKL,3)+1) INDXA(NA) = MOD(JKL,3) + 1 CONTINUE NOPOIN = NX
C OBTAINS ANGLE FOR THE SECTIONS TO BE PRINTED
156

c
c ANGIN = CELL(7-NNMM) DIFANG = 50.0
C SETS PROGRAM PARAMETERS ACCORDING TO WHETHER ANGLE IS OBTUSE OR C ACUTE c
c c c
c c c c c c c
c c c
1802
1801
303
306 302 301
11002 11001 11000 11003
827
898
119
ACTANG = ANGIN-90.0 J1 = 0 J2 = 1 IF (ACTANG .LT. 0) GO TO 1802 GO TO 1801 CONTINUE J1 = 1 J2 = -1 CONTINUE ACTANG = ABS(ACTANG)
OBTAINS TANGENT OF ANGLE AS A RATIONAL FRACTION
DO 301 N = 1~.10 DO 302 M=1 1.j5 ANGLE(N,M) '= 180.0*(ATAN(1.2S*FLOAT(N)/M))/3.14159 IF (ABSCANGLE(N,M)-ACTANG) .LT. DIFANG) GO TO 303 GO '!'0 306 CONTINUE WTDANG = ANGLE(N,M) NANG = N MANG = M DIFANG = ABS(ANGLE(N,M) -ACTANG CONTINUE CONTINUE CONTINUE
DETERMINES SCALING FACTORS
MAKES DECISIONS AS HOW BEST TO EXPAND THE MAP,BY WHAT AMOUNT AND WHICH WAY TO ORIENT IT
SCALE1= 999.0LRMAX SCALE2 = 999.0/(RHOMAX-RHOMIN)
READS IN A SECTION OF THE MAP FROM THE FILE IF(NEND.EQ.O) GO TO 11003 DO 11000 K=1,NEND DO 11001 J=1,NY READCIVXX,END=11002) DUMMY CONTINUE CONTINUE CONTINUE CONTINUE IF(NOSECT.EQ.O) NOSECT=NZ DO 97 JSECT=NEND+1,NOSECT ROWN = 0 DO 827 JP = 1,5 JPJ = 6-JP JDIVX = 125/NXX JDIVY = 125LNYY IF (JDIVX .GE. JPJ) GO TO 829 IF (JDIVY .GE. JPJ) GO TO 898 CONTINUE GO TO 849 CONTINUE DO 227 KK = 1,NYY READ(IVXX,END=119) (SECT(K,KK),K=1,NXX) CONTINUE
157

c c c c
c c c c
c c c c
227 CONTINUE
829
115 116
849 839
311
313 416
312
400 402
NX = NYY NY = NXX NOPOIN = NX JSCACR = 1 JSCDOW = 3 YMAX = XXMAX XMAX = YYMAX YMIN = XXMIN XMIN = YYMIN LENDOW = CELL(MOD(5-NNMM,3)+1) LENACR = CELL(MOD(4-NNMM,3)+1)
NP IS THE EXPANSION FACTOR ACROSS THE PAGE NP2 IS THE EXPANSION FACTOR DOWN THE PAGE
NP = 5 IF (JDIVY .LT. 5) NP=JDIVY NP2 = NPNP2(NP} DIVl = FLOAT{NP)/NP2 ANG = 1.25*COS{(3.14159*ACTANG)/180.0) IF(ABS(DIVl-ANG)/DIVl .GT.ABS(l.O-ANG))NP2=NP GO TO 839 CONTINUE JSCDOW = 1 JSCACR = 3
OBTAINS CELL LENGTHS DOWN AND ACROSS THE PAGE
LENACR = CELL(MOD(5-NNMM,3)+1) LENDOW = CELL(MOD(4-NNMM,3)+1) DO 116 KK = 1 NY READ(IVXX,END~115) (SECT(KK,K) ,K=l,NX) CONTlNUE CONTINUE NP = 5 IF(JDIVX .LT.5) NP = JDIVX NP2 = NPNP2(NP) DIVl = FLOAT{NP)/NP2 ANG = 1.25*COS((3.14159*ACTANG)/180.0) IF(ABS(DIVl-ANG)/DIVl .GT.ABS(l.O-ANG))NP2=NP CONTINUE CONTINUE
CALCULATES THE NUMBER OF 'ROWS' TO MOVE EACH 'UPWARDS' TO OBTAIN THE ANGLE IN THE MAP
DO 1777 NPLACE = 1 1 (NX-l)*NP+l NM = FLOAT{MANG)/NANG + 0.5 IF(NANG .GE. 0.5*FLOAT(MANG)) GO TO 311 GO TO 312 CONTINUE NOOF2S = MANG-NANG NOOFlS = NANG-NOOF2S NL = FLOAT(MANG)/NOOF2S+0.5 DO 313 AL = 1,NL*NOOF2S,NL IF{MOD(NPLACE,MANG) .EQ. AL-l) GO TO 416 CONTINUE NUP = NUP+l CONTINUE GO TO 402 CONTINUE ¥~ {ggD~PLAeE~~~~~~G:~~. MM-1) NUP = NUP+l CONTINUE CONTINUE MPLACE = J1*((NX-l)*NP+2)+J2*(NPLACE)
158
'COLUMN' OF AN ARRAY

c c c
c c c c c c c
c c c c
1777 NNUP(MPLACE) = NUP CONTINUE SCALEY = (XMAX-XMIN)*10.0*LENACR/(2.54*((NX-l}*NP+1}) SCALEZ = (YMAX-YMIN)*8.0*LENDOW/(2.54*((NY-1)*NP2+1))
OBTAINS CM/ANG. CONVERSION SCALES
CHARCY = JYMAX-YMIN)*LENDOW/JJNY-1~*NP2) CHARCZ = XMAX-XMIN)*LENACR/ NX-1 *NP ) UPNUM = N *NP*FLOAT1NANG}/MA + 1 IF (NUMPIC .LT. 2) NUMPIC = 1
BEGINNING OF SECTION TO MAKE NUMPIC COPIES OF EXPANDED MAPS
DO 773 JJM = 1,NUMPIC
WRITES OUT HEADER INFORMATION FOR EACH SECTION INCLUDING CELL CONSTANTS,RMAX AND A LEGEND FOR THE SHADING
START1 = 56 START2 = START1 WRITE{6i1833)
1833 ~~~~rti~~i; '> 601 FORMAT( X I
1)
WRITE(6,562) fS~ART16 {ARTSTC{JJ, 502 FORMAT{(1H+,*X,20A1) IF (NZ .LT. 2) GO T 832
KKK) ,KKK=2,21),JJ=l,8)
SECTIN = (ZMAX-ZMIN)*(JSECT-1)/(NZ-1) + ZMIN GO TO 833
832 CONTINUE SECTIN = ZMIN
833 CONTINUE IF (GRID .EQ. 1) WRITE(6,1503)
1503 FORMAT(1X,4IX,'THIS IS AN OVERLAY GRID FOR DETERMINATION OF COORD! *NATES'}
IF (GR!D .NE. 1). WRITE(6~503} SCALEl,INDXf2)~SECTIN,RMAX so3 FORMAT(lx,•c = ',E10.3,4~x· ~ 'A1,' = i1F~.2,' * ',3sx, 'RMAX
*='E15.3)
600 ~~~f47i~09) ') WRITE(6,564) lSTART1r (ARTSTC{JJ,
504 FORMAT((1H+r*X,20A1J) WRITE(6,599J
KKK ) ,KKK=2,21) ,JJ=l,8)
599 ~~~~1t1~'~f' (~~LL(I) I=1,6) 772 FORMAT{iH,'CELL PARAMETERS A= 'F5.2,' B = 'F5.2,' c = 'F5.2,'
* ALPHA = 'F6.2 I BETA = 'F6.2 I GAMMA = 'F6.2) IF(ANGIN .LT. 89.0 .AND. JSECT .EQ. 1 .AND. (NUMPIC .EQ. 9 .OR.
*NUMPIC .EQ.1)) WTDANG = -WTDANG IF CABS(ANGIN-90.0~ .LT. 1.0) WTDANG = 0.0
775 ~~~f4~;;i~~~~t~~EPA~~~~R~TDA~gtb3N°Sg~~E = 'LF4.26
' ANGSTROMS/C *M HORIZ. ROW SCALE= ',F4.2,' ANGSTROMS/CM ~ECTI NANGLE= 'FS *.1f' COPY NUMBER',I3)
3 ~~~f4~i~7 ~~~,'. ',32X '· ') 776 WRITEl6 3~77) CHARCY CHARCZ
3777 FORMAT(iX,34X,1F5.3,fA/CHARACTER',17X,1F5.3,'A/CHARACTER')
PLACES THE ACROSS THE PAGE SCALE IN AN ARRAY TO ENABLE WRITTEN OUT 'VERTICALLY'
DO 700 PLACE = 1,NOPOIN ACROSS(PLACE) = (PLACE-1)*(XMAX-XMIN)/(NX-1) + XMIN IFCACROSS(PLACE) .GE. 0) GO TO 157 PLM(PLACE) = 1102 GO TO 159
159
IT TO BE

c c
!C
c c c c
c
157
159
700
4477 962
560 561
565 563
CONTINUE P~M(PLACE) = 1103 CONTINUE BBlPLACEl = ABS~ACROSS(PLACE~)*1000 ~ ~t~8~ ~ ~~~ ~~lE~i~~l~~£Atg5f;1o ~~ ~t~§~ ~ ~gg ~tAE~,c~)nE~~LACE))/10 CO TINUE DO 4477 JKLM = 1,150 DJ(JKLM)=1 CONTINUE CONTINUE
SETS PRINTOUT ARRAY TO VALUE CORRESPONDING TO BLANK
DO 561 JJJB=1,205 DO 560 JJJA = 1,126 PRNTAA(JJJA,JJJB) = 1100 CONTINUE CONTINUE DO 563 JACROS = 1 NOPOIN TITLE JACROS,1 = INDXA(JSCACR) + 1096 TITLE JACROS,2 = PLM(JACROS) TITLE JACROS,3 = 1101 TITLE JACROS,4 = EE{JACROS} +1087 TITLE JACROS,5 = DD JACROS +1087 TITLE JACROS,6 = AA JACROS +1087
PLACES ACROSS THE PAGE SCALE IN PRINTOUT ARRAY ON THE SAME SLANT AS THE MAP TO FOLLOW
~8w5~ 5u~~g~+RR5w~12 NPLACE = (JACROS-1)*NP + 1 ROWE = ROW-NNUP(NPLACE) PRNTAA(NPLACE,ROWE)=TITLE(JACROS,RROW) MEM2(R0WE)=1 CONTlNUE CONTINUE
C SETS PROGRAM PARAMETERS TO ENABLE THE EXPANDED MAP TO BE C CREATED IN SUCCESSIVE SECTIONS c
c c c c
OOTROW=O ROW = UPNUM + 1
NUMN = (200.0-UPNUM)/NP2 NUM1 = 1 NUM2=NUMN+1 IF (GRID .EQ. 1) NUM2 = NUMN BOTNUM = 200-MOD{200-UPNUM,NP2) NOROW=UPNUM+(NY-l)*NP2+1 IF(NY+1 .LT. NUM2) NUM2 = NY+1 GO TO 90
91 CONTINUE ROW =UPNUM-14 NOROW = ROW+NOROW-BOTNUM BOTNUM=(200.0/NP2)*NP2 NUMN=200.0/NP'2 NUM1 = 1+NUM2 NUM2 = NUM1 + NUMN IF (NY+1 .LT. NUM2) NUM2 = NY+1
90 CONTINUE
BEGINNING OF INTERPOLATION PROCEDURE
DO 98 KK = NUM1,NUM2
160

c c
c c c
c c c
4999 4998
5100 5101
5008
125
110 112
124
5000
122
MANIPULATES INFORMATION RETAINED FROM THE PREVIOUS ROW DEALT WITH
IF ~GRID .EQ. 1) GO TO 958
¥~{M:~f9 .~)1G~F~SGa998 A1 M,B =0.0 A2 M,B =0.0 CONTINUE CONTINUE DO 5100 M=1,KFLAG2 IF JM.EQ.O) GO TO 5101
~~lM:~l~~~~~~g~ A1 M,C =0.0 A2 M,C =0.0 CO TINUE CONTINUE KFLAG1=KFLAG2 JMM=O JMM=JMM+1 DO 110 K=A1(JMM61)fA2(JMM,1) IF(K.EQ.O) GO T 1 0 PLACE = K DO 125 PTNO = 1 NP-1 RINTER(PTNO,B,Kf = RINTER(PTNO,C,K) RINTER(PTN0,2,K)=1 NPLACE = (PLACE-1)*NP + 1 + PTNO ROWE=ROW-NNUP(NPLACE) PRNTAA(NPLACE,ROWE )=RINTER(PTN0,1,PLACE) MEM2 (ROWE) =1 CONTINUE NPLACE = (PLACE-1)*NP+1 VAL~B,K) = VAL(C,K) VAL 2,K)=1 ROW =ROW-NNUP(NPLACE) PRNTAA{NPLACE,ROWE )=VAL(1,K) MEM2(R0WE)=1 IF(JMM.EQ.KFLAG1) GO TO 110 IF(K.EQ.A2(JMM,1)) GO TO 5008 CONTINUE CONTINUE IF(KK.EQ.NY+1) GO TO 98
SCALES INPUT VALUES
DO 124 K = 1 NX VAL(C,K) = (SECT(KK,K)*SCALE1-RHOMIN)*SCALE2 CONTINUE
ASSESSES ROW FOR REGIONS WHICH ARE TO BE INTERPOLATED
K=O FLAG=O DO 122 PLACE = 1,NX JLN=PLACE IF lVALfC,PLACE} .GT. 1000) VAL(C,PLACE) = IF VAL C,PLACE .LT. 1) VAL(c6PLACE) = 1 IF VAL C,PLACE .GT.1) GO TO 5 00 FLA =0 GO TO 122 CONTINUE
A1 K,C)=(JLN-1l*(1-FLAG)+A1(K,C) IF!FLAG.EQ.O) K=K+1
f~ *~~)~~Al~KA6~fK,?~cl-FLAG)+A2(K,C)+1*FLAG IF JLN.EQ.NOP IN) A2(K,C)=NOPOI~ FLAG=1 CONTINUE
161
999

c c c
c c c c
c c c c
KFLAG2=K JMM=O
5002 JMM=JMM+l
INTERPOLATES ACROSS A ROW OF GRIDPOINTS
DO 102 PLACE=A1{JMM,C)fA2(JMM,C) IF (PLACE.EQ.O) GO TO 02 IF( VAL{C,PLACE).GT.DJ{PLACE))DJ(PLACE)=VAL(
*~6pt~~E~TNO = 1 NP-1 RINTER(PTNO,C,PLACE) =((NP-PTNO)*VAL(C,PLACE) + PTNO*VAL(C,PLACE+1
*~~/NJ~INTER{PTNO,C,PLACE) .LT. 1.0) RINTER{PTNO C PLACE) = 1 IF RINTER(PTNO,C,PLACE) .GT. 1000) RINTER(PTN6,C,PLACE) = 999
103 CON INUE IF~JMM.EQ.KFLAG2) GO TO 102 IF PLACE.EQ.A2(JMM,C)) GO TO 5002
102 CO TINUE
5004 4020 4021
5006 5005 5900
5009 5007 5901
DO 5004 JKL=1,NOPOIN MEM(JKL)=O CONTINUE IF(KK .LT. 2 ) GO TO 98 CONTINUE
REMEMBERS AREAS BETWEEN ROWS BY A LINEAR INTERPOLATION
IF(KFLAG1.EQ.0) GO TO 5900 DO 5005 N=1,KFLAG1 DO 5006 LN=A1(N,B) ,A2(N,B) MEM{LN)=1 CONTINUE CONTINUE CONTINUE IF(KFLAG2.EQ.O) GO TO 5901 DO 5007 N=1 KFLAG2 DO 5009 LL=A1(N,C) ,A2(N,C) MEM(LL)=1 CONTINUE CONTINUE CONTINUE
FOR WHICH VALUES ARE TO BE CALCULATED
CARRIES OUT THE INTERPOLATION CALCULATIONS FOR THE IN-BETWEEN ROWS AND PLACES THE RESULTS IN THE PRINTOUT ARRAY
CJ = 1 MPLACE = 1 IF(KFLAG1.EQ.O.AND.KFLAG2.EQ.O) GO TO 998 DO 104 PLACE = 1,NOPOIN IF(MEM(PLACE).EQ.O) GO TO 6104 DO 106 DPTNO = I,NP2 -1 CINTER(DPTNO,B,PLACE) =((NP2-DPTNO)*VAL(B,PLACE)+DPTNO*VAL(C,PLACE
*))/NP2 IF(CINTER~DPTN0,1,PLACEi .LT.1.0) CINTER(DPTNO,l,PLACE) = 1 IF(CINTER DPTN0,1LPLACE .GT.1000) CINTER(DPTN0,1,PLACE)= 999 ROWA=ROW+ PTNO-NNuP(MPL CE) PRNTAA(MPLACEfROWA )=CINTER{DPTN0,1,PLACE) CINTER{DPTNO, ,PLACE)=O MEM2{ROWA)=1 DO 117 PTNO = 1,NP-1 TINTER(DPTNO,PTNOLPLACE)=((NP2-DPTNO)*RINTER(PTNO,B,PLACE)+DPTNO*R *INTER(PTNO,C,PLAC~))/NP2
IF (TINTER(DPTNO,PTNO,PLACE) .LT. 1.0)TINTER(DPTNO,PTNO,PLACE)=l IF(TINTER(DPTNO,PTNO ,PLACE) .GT.1000) TINTER(DPTNO,PTNO,PLACE)= * 999 NPLACE = (PLACE-1)*NP + 1 + PTNO ROWA = ROW+DPTNO-NNUP(NPLACE)
162

117 106
1117 6104
PRNTAA(NPLACE,ROWA CONTINUE CONTINUE DO 1117 PTN0=1 NP-1 RINTER(PTNO,B,PLACE)=1.0 CONTINUE CONTINUE
)=TINTER(DPTNO,PTNO,PLACE)
104
958 •c 'c
MPLACE = MPLACE+NP VAL (1, PLACE} =1 CONTINUE GO TO 998 CONTINUE
c
c
TO PRODUCE A GRID FOR LOCATING PEAKS EASILY
DO 961 JJJAA = 1,(NX-1)*NP+1,NP ROWE= ROW-NNUP(JJJAA) PRNTAA(JJJAA,R0WE}=1101 MEM2(R0WE)=1
961 CONTINUE 998 CONTINUE
ROW = ROW+NP 2 98 CONTINUE
IF!NOROW.LE.200liEND=NOROW+4 IF NOROW.LE.200 SHADER=IEND IF NOROW.GT.200 IEND=BOTNUM IF NOROW.GT.200 SHADER=205 IF NOROW.GT.200 GO TO 778 NK=O DO 777 NPLACE =1,NOPOIN*NP,NP NK=NK+1 ROWE = SHADER-NNUP(NPLACE) PRNTAACNPLACE,ROWE)=DJ(NK) MEM2 (ROWE) =1
777 CONTINUE 778 CONTINUE
IF~NOROW.LE.200)ENDNUM=SHADER IF NOROW.GT.200)ENDNUM=BOTNUM-(UPNUM-15) CU NUM = UPNUM DO 197 ROW = 16ENDNUM OUTROW = ROW+ OTROW
192 CONTINUE CJ=1
C DETERMINING MAXIMUM DENSITY WITHIN 'ORIGINAL' COLUMNS c
IF(MEM2(ROW).EQ.0) GO TO 5780 DO 780 NPLACE=!,(NOPOIN-l)*NP+1 IF(PRNTAA(NPLACE, ROW).GT.CJ.AND.PRNTAA(NPLACE, ROW) .LT.1050)
*CJ=PRNTAA(NPLACE, ROW) 780 CONTINUE
5780 CONTINUE IF (ANGIN .LT. 90.0) UPNUM = 15 IF(NP2.Eg.1.AND.OUTROW.GE.UPNUM+1)GO TO 47 IF (MOD{ UTROW-UPNUM,NP2) .EQ. 1 .AND. OUTROW.GE.UPNUM+1) GO TO 47 GO TO 48
47 CONTINUE S1 = 120-({NX-1)*NP+1) INDEX= ((0UTROW-UPNUM)/NP2)*(YMAX-YMIN)/(NY-1)+YMIN IF {OUTROW .GE. NY*NP2+UPNUM) GO TO 1800
43 WRITE(6f43L S1iiNDX(JSCDOW) 1 INDEX FORMAT( X~ X,A ,' = ',F6.3 J GO TO 179':J
1800 WRITE{6..!.42) 1799 CONTINU~
48 CONTINUE IF (MOD(OUTROW-UPNUM,NP2) .NE. 1 .AND. OUTROW.GE.UPNUM+1) WRITE(6,
*42)
163

42 FORMAT(1X,' ') IFCOUTROW.LE.UPNUM)W~ITE{6,776)
776 FORMAT(1X, I ')
S = 132-{(NX-1)*NP+1) IF fMEM2£ROW).EQ.O) GO TO 779
*~~6I(~*~i)~&~+1f((ARTSTC(JJJ,MTONEF(PRNTAA(PLACE, *(N POIN-l)*NP+ )),JJJ=1,NOVER(CJ)) 107 FORMAT((1H+, *X,*A1)) 779 CONTINUE
DO 1979 NPLACE = 1,(NX-1)*NP+1 PRNTAA(NPLACE,ROW} = 1100
1979 CONTINUE 197 CONTINUE
OOTROW=OUTROW UPNUM = CUPNUM IF (UPNUM .EQ. 15) GO TO 196 DO 198 ROW = BOTNUM+16-UPNUMlBOTNUM NROW = ROW- (BOTNUM+15-UPNUM DO 195 NPLACE = 1,(NX-1l*NP+ PRNTAACNPLACE,NROW) = P~NTAA(NPLACE,ROW}
195 CONTINUE MEM2(NROW}=MEM2(ROW)
198 CONTINUE 196 CONTINUE
IF (NUM2 .LT. NY+1) GO TO 91 NUP = 0 IF (ABS(ANGIN-90.0) .LT. 1.0) GO TO 769
769 CONTINUE IF (GRID .EQ. 1) GO TO 96
773 CONTINUE 97 CONTINUE
IF ~GRIDNM .EQ. 1) GRID = 1 IF rGRIDNM .NE. 1) GO TO 96
859 ~~~~J'i~i;~ 1}
GO TO 962 96 CONTINUE
RETURN END
164
ROW))) ,PLACE=1,

PROGRAM SORTLIST
ALGOL SUBROUTINE $ SET LIBRARY $ LEVEL 3
~It~EFfE~~1~¥~~~P~f~~~~) ; REAL TEMP• ARRAY MLLf0:40,0:40];
] REAL PROCEDURE LABELL(L,M,LL) ;VALUE L,M,LL;REAL L,M,LL; BEGIN DEFINE BASEF=[47:BLENGTH ] #
BLENGTH=12 #; %%%%%%%%% THIS MUST BE 47-MAX1 %%%%%%% LABELL:=IF BOOLEAN((TEMP:=MLL[LL-1,M-1]). [L-1:1])THEN TEMP.BASEF+ONES
(TEMP. [L-1:L]) ELSE 0; END• PROCEDURE SETUP(N,MIN,MAX,DATA,IHKL) ;REAL N;ARRAY MIN,MAX,DATA,IHKL[*]; BEGIN ARRAY BUF[0:10]; DEFINE BASEF=r47:47-MAX1] #, FIRSTS0RTED=FILE2 # INDEXF=[MAX1:MAX1+1f #, MAX1=35 !' %%%%%%% IF YOU CHANGE THIS ALSO CHANGE BLENGTH IN LABELL MAX2=40 ,% IF YOU CHANGE THIS ALSO CHANGE GLOBAL DIMENSIONS OF MLL MAX3=40 ,% IF YOU CHANGE THIS ALSO CHANGE GLOBAL DIMENSIONS OF MLL ZEND=#• INTEGER ARRAY DIF,BMIN,BMAX[0:2];
REA~Et~D~;~~fiE~~?2~~K~~;~£6KEY,COUNT; BOOLEAN PROCEDURE SORTIN(RECD) ;ARRAY RECD[O];
BEGIN IF READ(FIRSTSORTED,11,RECD)OR RECD[6] GEQ 99 THEN
BEGIN SORTIN:=TRUE; END ELSE BEGIN RECD[FIRST] :=*+RECD[THIRD]*10000+RECD[SECOND]*100; END;
END; BOOLEAN PROCEDURE SORTCOMPARE(REC1~REC2)lARRAY REC1,REC2[0];
SORTCOMPARE: = REC1 [F!,RST] LEQ REc2 [FIR;:;T] ; PROCEDURE SORTOUT(BEOF,RECD) ;VALUE BEOF;
BOOLEAN BEOF1ARRAY RECD[O]; IF NOT BEOF THEN
BEGIN RECDfFIRST] :=(NEWKEY:=RECD[FIRST])-RECD[SECOND]*100-RECD[THIRD]
~10000;
IF<~ft~t28~J~E~~~6R8L~~~yf~8~x~~w~~ji§~~~E~~flf!~~&~Y7J ,RECD[8]) ELSE BEGIN
REPLACE DATA[6*COUNT]BY POINTER(RECD[O]) FOR 6 WORDS; REPLACE IHKL[5*COUNT]BY POINTER(RECD[6]) FOR 5 WORDS; COUNT:=*+1;
MLLfRECD[THIRD]-BMIN[THIRD-6lfRECD[SECOND]-BMIN[SECOND-6]] :=* & [RECD[FIRST]-BMIN[FIRST-6 :1];
EN~N~f..SE BEGIN NEWKEY:=COUNT· REPLACE DATA[G*COUNT]BY POINTER(BUF[O]) FOR 6 WORDS; REPLACE IHKL[5*COUNT]BY POINTER{BUF[6]) FOR 5 WORDS; COUNT:=O; FOR INDX:=O STEP 1 UNTIL BMAX[THIRD-6]-BMIN[THIRD-6]00 FOR INDEX2:=0 STEP 1 UNTIL BMAX[SECOND-6]-BMIN[SECOND-6]DO
BEGIN MLL[INDX,INDEX2] :=*&COUNT BASEF; COUNT:=*+ONES(MLL[INDX,INDEX2] .INDEXF);
165

END• IF COUNT NEQ NEWKEY THEN WRITE(FILE6,<"MISSCOUNT IN SETUP",2Il0>,
COUNT,NEWKEY) ~ END·
IF NOT FIRSTSORTED.OPEN THEN FIRSTSORTED.FILETYPE:=7; ~~~Df~5~~;3~r;~Do61s~¥~tfNDXl:=BMAX[INDX] :=BUF[INDX+6]; WHILE NOT R~AD(FIRSTSORTED,l ,BUF)AND BUF[6]LSS 99 DO
FOR INDX:=6,7,8 DO IF BMAX[INDX-6]LSS BUF[INDX1THEN BMAX[INDX-6] :=BUF[INDX] ELSE IF BMIN[INDX-6]GTR BUF INDX]THEN BMIN[INDX-6]:=BUF1INDX];
FIRST:=IF FIRST:=BMAX[O]-BMIN 0] LEO SECOND:= BMAX[l]-BMINfllTHEN IF FIRST LE8 BMAX[2]-BMIN[2J THEN 6 ELSE 8 ELSE IF SECOND LE BMAX[2]-BMIN[2] THEN 7 ELSE 8; SECOND:=CASE FIRST-6 OF (7,8~6)~ THIRD:=CASE FIRST-6 OF (8 6,t);
IF INDX:=FIRSTSORTED.RECORD GTR I~DEX2:=REAL(NOT FALSE}.BASEF THEN WRITE(FILE6l<Il0," RECORDS READ BUT DIMENSIONED FOR ONLY",IlO> INDX 1NDEX2J ELSE
IF DIFlOl :=BMAX~FIRST-61-BMINfFIRST-6]GTR MAX1 OR DIF 1 :=BMAX SECOND-6]-BMIN[SECOND-6] GTR MAX2 OR DIF 2 :=BMAX THIRD-6]-BMIN[THIRD-6]GTR MAX3 THEN WRITE(FILE6J<"RANG)"
,3 S,"EXCEEDED ALLOWABLE DIMENSIONS",3I5>,DIF[O] ,DIF[l] ,DIFL2], MAXlfMAX2 ,MAX3)
ELSE BEG N REWIND(FIRSTSORTED); OLDKEY:=O.S; SORT(SORTOUT,SORTIN,O,SORTCOMPARE,11,6000,100000); N:=COUNT; FOR INDX:=O,l,2 DO BEGIN MIN[INDX] :=BMIN[INDX] ;MAX[INDX] :=BMAX[INDX];
END~ END• END:
!SET SEPARATE AUTOBIND $ BIND = FROM SORTER
USE LABEL FOR LABELL
FORTRAN MAINPROGRAM
FILE c c c c c c c
2=XRC/IED,UNIT=DISK,BLOCKING=90,RECORD=11,AREA=900*5
UNIVERSITY OF CANTERBURY DEC. 1973. WRITTEN BY D. L.EVANS. FORTRAN PROGRAM WRITTEN FOR BURROUGHS B6700 PRODUCES STATISTICS BASED ON SINTHETA/LAMDA, FOBS,SIGMA1 MILLER INDICES AND PRINTS OUT A LISTING OF THE SORTED DATA
REAL MTLDAT REAL MNFOBS REAL LAMDA REAL JSUM REAL LIMIT REAL OR,ORW, ORSUMl, ORSUM2,0RWSM1,0RWSM2 INTEGER COUNT INTEGER VALM INTEGER ~~ ZAL ZB, ZC INTEGER HAWDA'l' INTEGER VAL INTEGER OCC INTEGER SUMA1 SUM INTEGER DATAA INTEGER C INTEGER DNL.~OTNO, SNP, DNN, PNPP, B A, AJ,COLMAX INTEGER PA.J:<AiVl DIMENSION LIMIT(3,30) DIMENSION MMM(3) DIMENSION SCALE(6)
166

c c c
c c c c c c c
900
901
9000
751 752
DIMENSION TITLE(14) DIMENSION DIFF(3i DIMENSION SUM(30 DIMENSION LAMDA( l DIMENSION JSUM(30 DIMENSION FSUM(30 DIMENSION FSQSUM~ 0) DIMENSION FCSQSM 30) DIMENSION NSUM(3 ) DIMENSION NUMPtN~3,30~ DIMENSION CLSVAL 3,30 DIMENSION NONCLS 3,30 DIMENSION MIN(3)
6 MAX(3)
DIMENSION OCC(20 0 3) DIMENSION DATA(6r.4600)fiHKL(5,4000) ,DATAA(4000)
gj~~~~jg~ ~~'~t~(62t~{ , 3gg~3) C4(3) C5(3) C6(3) DIMENSION HKLS0M(45) ,HKFSUM(~~) ,HKSQSM(45) ,HKQSUM(45) ,COUNT(45) DIMENSION ERROR(45) INDEX1 = 21 INDEX2 = 45 INDEX3 = 45
READS TITLE AND DATA CARDS
READ(5,900) TITLE FORMAT(13A6 A2) WRITE(6i901~ TITLE
~~~t~~9H65)A~6~f~SALAF,LAMID,NOCOL,COLMAX,SVP,NOVAR FORMAT(7!5) IF (NOCL .EQ. 0) NOCL = 10 LAMID = LAMID + 1 LAMDA~1~ = 0.7107 LAMDA 2 = 1.5418 LAMDA 3 = 1. 79 IF (NOCOL .EQ. 0) NOCOL = 11 IF (COLMAX .EQ. 0) COLMAX = 236 INDMIN = 100 DO 752 PARAM = 1 3 DO 751 VAL=1,2006 OCC(VAL~PARAM) = 0 CON't'INU.I:!i CONTINUE CALL SETUP(N,MIN,MAX,DATA,IHKL) NJ=N TOTNO = NJ N=N+1
READS NJ RECORDS FROM DISK
AN ILS FILE HAS 99 ON THE NJ+1TH RECORD AN !ED FILE HAS 999 ON THE NJ+1TH RECORD
80 IF JIHKLJ1,N) .EQ. 999) GO TO 7100 IF IHKL 1,N) .EQ. 99) GO TO 7222 GO 0 72 4
7222 !ED = 1
7224
7100 c c c
GO TO 7001 CONTINUE GO TO 7101 CONTINUE
READS NJ+1TH RECORD FOR SCALE FACTORS AND OTHER PARAMETERS
IF (NOVAR .EQ. 0 ) NOVAR = IHKL(4,N) NUMSCA = IHKL(5,N)
167

c c c
c c c
861
862
863 7101 7001
960
961
980
981
334
IF JNUMSCA .GT. 6b GO TO 862 IF NUMSCA .EQ. 0 GO TO 862 DO 61 NMNN = 1,N MSCA SCALE(NMNN) = DATA(NMNN,N) CONTINUE GO TO 7001 CONTINUE
IF NO SCALE FACTORS SETS ALL SCALE FACTORS TO ONE
DO 863 NNNM = 1,TOTNO SCALE(IHKL(4,NNNM)) = 1 CONTINUE CONTINUE CONTINUE IF (IED .EQ. 1) WRITE(6,960) FORMATl/,1H,' THE DATA COMES FROM AN ILS FILE') IF (!EO .EQ. 0) WRITE(6,961) FORMAT(/,1H,' THE DATA COMES FROM AN IED FILE') IF (SALAF .EQ. 1) WRITE(6,980) FORMAT{1H,' AND HAS BEEN SORTED, LISTED AND REFILED.') IF (SALAF .EQ. 0) WRITE(6,981) FORMAT(1H~' AND HAS BEEN SORTED AND LISTED') WRITE(6 3.:s4) NJ FORMAT(iH,' THE NUMBER OF REFLECTIONS SORTED AND LISTED
THE RANGE OF INDICES IS PRINTED OUT
333 ~~~~fl/J~f~,' INDEX RANGE')
~~I~~J6:11R~ ~!~~~~ :~~Q~~~ WRITE 6,278~ MIN~3~ 1 MAX~3~
78 FORMA 11,1X, I H ,2I5) 178 FORMAT lX,' K ',2I5) 278 FORMAT 1X,' L ',2I5)
A = SV DO 7010 I = 1~.3 DIFF(I) = MAX(I) -MIN(!} IF JDIFF(I) .LT. INDMIN INDMIN = DIFF(I) IF DIFF(I) .GE. INDMAX INDMAX = DIFF(I)
7010 CON INUE
IS',I5)
¥~ 6{B~~FI{I=} :Ei3 • INDMIN) JAB= I IF DIFF I .E • INDMAX) JAD = I IF DIFF I .N • INDMIN .AND. DIFF(I) .NE. INDMAX) JAC = I
6105 CONTINUE IF (JAB .EQ. 1) WRITE(6,9521)
9521 FORMAT(1HL ' THE SLOWEST VARYING INDEX IS H') IF (JAB .~Q. 2) WRITE(6,9522)
9522 FORMAT~1H, ' THE SLOWEST VARYING INDEX IS K')
9523 ~b~~~(1H~Qi ~hEW~l6~~gT9~i~~ING INDEX IS L') IF (A .EQ. 0) A = JAB SNP = A IF~A .EQ. 1} GO TO 6211 IF A .E8. 2 GO TO 6212 IF A .E • 3 GO TO 6213
6211 B = 2 c = 3
WRITE16J.9533) 9533 FORMAT 1n, 1 THE DATA HAS BEEN SORTED ON INDEX H')
GO TO 6 04 6212 B = 3
c = 1 WRITE16 1.9532)
9532 FORMAT 1H, ' THE DATA HAS BEEN SORTED ON INDEX K') GO TO 6 04
168

6213
9531 6204
2200
7008
20004
c c c c c c c c c c c c
20005
389 c c c
B = 1 c = 2
WRITE(6,9531) FORMAT (lH, 1 THE DATA HAS BEEN SORTED ON INDEX L') CONTINUE VALM = 0 NUMVAL = -1 CONTINUE NUMVAL = NUMVAL +1 DO 7008 N = l,NJ VAL= DATA(l,N)*lO.O**NUMVAL + 0.5 IF (VAL.GT. VALM) VALM = VAL CONTINUE
~~~~1~i~~9g6hv~EM~~LVA~~~~~¥~) IF ~VALM .LT. 100) GO TO 2~00 IF IED .EQ. 1) GO TO 527 DO 004 N = 1,1\JJ DO 389 PARAM = 1,2
FOR SORTING ON SIN-THETA/LAMDA THE VALUES OF IT ARE MULTIPLIED BY 1000 AND THE OCCURRENCE OF EACH SUCH VALUE IS ASCERTAINED. THE TOTAL NUMBER OF REFLECTIONS I DIVIDED BY THE NUMBER OF CLASSES-1 AND THE INTEGERS FOR SIN-THETA OVER LAMDA ARE RUN THROUGH UNTIL THE SUM OF THE OCCURRENCES EXCEEDS THE NUMBER OF REFLECTIONS IN A CLASS.
IF (PARAM .EQ.l) VAL= DATA(4,N)*1000.0 + 0.5
THE INTEGER VAL CAN BE SCALED UP BY A FACTOR OF 10 TO THE POWER OF NUMVAL IF FOBS IS LESS THAN 100.
IF (PARAM .EQ.2) VAL= DATA(l Nl*lO.O**NUMVAL + 0.5 IF(VAL.GT.1999) WRITE(6,20005fVAL,PARAM,NUMVAL FORMAT(lX 'VAL,PARAM NUMVAL',JI6) OCC(VAL,PARAM) = OCC(VAL,PARAM) + 1 CONTINUE
L IS THE SLOWEST VARYING INDEX AND ALL INDICES ARE MADE GREATER THAN ZERO BY ADDING (1-MIN(INDEX)).
7004 CONTINUE WRITE(6f20006) NJ,TOTNO
20006 FORMAT( X,'NJ TOTN0',2I6) c c c
c c c
383
20010
353
20003
NUM = NO. IN A CLASS
NUM = TOTNO/(NOCL-1) DO 383 MM = 1,20 NSUM(MM) = 0 CONTINUE DO 357 PARAM = 1,2 MM=l
DATA IS DIVIDED UP INTO CLASSES
NSUM(2)=NSUM(2)+0CC(2000,1) LSUM=NSUM(2) NUM=(TOTNO-tSUM)/NOCL DO 354 VAL=1,2000 NSUM(PARAM) = NSUM(PARAM) + OCC(VAL PARAM) IF(PARAM.E9.2) WRITE(6f20010) NSUM(~) ,(OCC(VAL,IM) ,IM=1,3) FORMAT(lX, NSUM OCC' 4 6) IF(NSUMlPARAM) .GE.MM*NUM+LSUM) GO TO 353 IF(NSUM PARAM) .EQ. TOTNO) GO TO 353 GO TO 3 4 LIMIT(PARAM MM) = VAL WRITE(6f20063) NSUM(PARAM) ,LIMIT(PARAM,MM) ,MM,VAL FORMAT( X,'NSUM LIMIT MM VAL',4I5)
169

c c c
c c c
777 778
354 4357
357
20000
5357 c c c 20001
c c c c
3333 c c c
NUMPLN IS THE NUMBER OF PLANES COUNTED
NUMPLN{PARAMfMM) = NSUM(PARAM) IF {MM .GT. ) GO TO 777
NONCLS IS NO. OF REFLECTIONS IN CLASS MM.
NONCLS(PARAM,1} = NUMPLN{PARAM,MM) GO TO 778 NONCLS(PARAM,MM) = NUMPLN{PARAM,MM) -CONTINUE IF(NSUM(PARAM) .EQ. TOTNO) GO TO 4357 MM=MM+1 CONTINUE MMM (PARAM) = MM CONTINUE
~~~~±~i~~9gg{,~) ~!~~ PARAM = 1 3 DO 5357 MM = 1,36 IF (PARAM .EQ. 3) NONCLS(PARAM,MM) = 0 JSUMiMMl= 0 FSUM MM = 0 FCSQ MJ ) = 0 FSQSUM MM) = 0 CONTIN E
BEGINNING OF STATISTICS CALCULATION.
WRITE(6f20001) MMM{PARAM)
NUMPLN(PARAM,MM-1)
E8~~~(Nx~·~~~T&3> IF f PARAM .EQ. 1) DATAA(N) = DATA(4,N)*1000.0 +0.5 IF PARAM .EO. 2} DATAA(N) = DATA(lfN)*10**NUMVAL + 0.5 IF PARAM .EQ. 3) DATAA(N) =DATA( ,N)**2 +0.5 JMM = MMM(PARAM) DO 359 MM = 1,JMM IF (PARAM .EQ.3) LIMIT(3,MM) = (MM)*DATAl1,N)*2.0*(DATA(2,N})+0.5 IF(PARAM .EQ. 3 .AND. MM .EQ.6} GO TO 3333
GOES THROUGH ALL REFLECTIONS TESTING TO WEE IF THEY LIE BETWEE N CLASS LIMITS.
IF ~DATAA(N) .GT. LIMIT(PARAM,MM)) GO TO 359 IF MM .EQ. 1} GO TO 352 IF DATAA(N) .LE. LIMIT(PARAM,MM-1)} GO TO 359
FOR THE SIGMA STSTISTICS THE NO, IN EACH CLASS IS COUNTED HERE.
c c c
352 IF (PARAM .EQ. 3) NONCLS(3,MM) = NONCLS{3,MM) + 1
359 350
766
20002
SUMS FOR STATS. ARE ACCUMULATED.
FSUM MM = FSUM MM) + ABS(DATA 1 N - DATA 3 N JSUMiMMl= JSUMiMM) + DATA(1 N} FCSQ M( ) = FC QSM(MMf+((ABS(DATA(l,N)-DAJAt3~~i))/DATA(2,N))**2 FSQSUM{MM} = FSQSUM(MM} + {DATA(1,N)/DATA(2,N)J**2 CONTINUE CONTINUE IF {PARAM .EQ. 2) GO TO 766 GO TO 767 CONTINUE DO 4129 MM = 1 JMM LIMITJ2,MM) = LIMIT(2~MM){(10**NUMVAL} WRITE 6f20002) LIMIT(~fMM ,MM FORMA { X,'LIMIT MM',2 5)
170

c 4129 CONTINUE
767 CONTINUE
C TABLE TITLES ARE WRITTEN OUT. c
c
IF (PARAM .EQ. 2) WRITE(6,610) 610 FORMAT(///,1XL'WEIGHTING ANALYSIS IN RANGES BASED ON MAGNITUDES OF
* STRUCTURE FAcTORS')
611 ~bR~K~~~1X~9wEfbH~¥~aEA~A~¥§is WITH RESPECT TO SINE THETA OVER LA *MBDA FOR ALL FOBS')
IF (PARAM .EQ. 3) WRITE(6,612) 612 FORMAT(///~1X,'WEIGHTING ANALYSIS IN RANGES BASED ON SIGMAS')
WRITE(6,37tj) 378 FORMAT(/
61X,' RANGE WDELSQ RW R MNFOBS
* ERR R NO. OF REFLECTIONS')
C OVERALL SUMS INITIALISED TO ZERO. c
c c c
c c c
c c c
ORSUM1 = 0 ORSUM2 = 0 ORWSM1 = 0 ORWSM2 = 0
STATS. CALC'D FROM SUMS A CLASS AT A TIME
IF(LSUM.GT.O) JMM=JMM+1 DO 355 MM = l,JMM IF (NONCLS(PARAM,MM) .E8. 0) GO TO 355 WDELSQ = FCSQSM(MM)/NON LS(PARAM,MM) RW = SQRT(FCSQSM(MM)/FSQSUM(MM)) R = FSUM(MM),JSUM(MM)
~~~8R~MM)J~MoA~~bt~8~~t~J~~~N~'~(TOTNO-NOVAR)*NONCLS(PARAM,MM))) IF(PARAM .EQ.3) GO TO 335 IF (MM .EQ. 1) WRITE£6,367) LIMIT(PARAM,l) ,WDELSQ,RW,R,MNFOBS,ERR
367*~~~~~~,?~~L~(P~RAM! ) I I3,5F10.3,7X,I3) IF (MM.GT. f) WRITE(6,36S) LIMIT(PARAM,MM-1) + 1,LIMIT(PARAM,MM
*) 6WDELSO,RW~R~MNFOBS~ERROR(MM) ,NONCLS(PARAM,MM) 368 F RMAT(IX,I~,~X,'-',~X,I3,5F10.3,7X,I3)
GO TO 337 335 CONTINUE
IF (MM .LT. 61WRITE(6,372) MM-1,MM,WDELSQ,RW,R,MNFOBS,ERROR(MM) ,NO *NCLS(PARAM,MM
372 FORMAT(/,1X,I ,'SIGMA-' I1 'SIGMA 'F6.3,4F10.3,6X I4) IF (MM .EQ. 61 WRITE(6,373f MM-1, WDELSQ,RW,R,MNF6BS,ERROR(MM) ,NO
*NCLS (PARAM,MM 373 FORMAT(/,1X,I ,'SIGMA- ',7X, F6.3,4F10.3,6X,I4) 337 CONTINUE
OVERALL SUMS ACCUMULATED.
ORSUM1 = ORSUM1 + FSUM(MM) ORSUM2 = ORSUM2 + JSUM(MM) ORWSM1 = ORWSM1 + FCSQSM(MM) ORWSM2 = ORWSM2 + FSQSUM(MM)
355 CONTINUE
OVERALL STATS. CALC'D.
OR = ORSUM1/0RSUM2 ORW = SQRT(ORWSM1/0RWSM2) OWDELS = ORWSM1/TOTNO OMNFOB = ORSUM2/TOTNO OERROR = SQRT(ORWSM1/(TOTNO-NOVAR)) JCAT=1 IF(LIMIT(2,1) .EQ.O)JCAT=2
171

c c c
379 WRITE(6,379} OWDELS,ORW,OR~OMNFOB~OERROR 1 TOTNO FORMAT(/,1X,' OVERALL l5F10.j 1 6X,I4J IF (PARAM .EQ. 2) GO TO 797~ GO TO 9797
7979 FACTOR=ERROR{JMM-1)/ERROR(JCAT) WRITE(6,458) FACTOn
458 FORMAT(/,1X,'THE FACTOR TO MULTIPLY P BY TO OBTAIN A NEW VALUE *IT IS '~F5.3,' AND IT IS THE QUOTIENT OF THE LAST')
WRITE(6 ~:~:59) 459 FORMAT(iX,'FULL ENTRY AND THE FIRST ENTRY IN THE ERROR TABLE')
9797 CONTINUE 358 CONTINUE
CNFSUM = 0 CNJSUM = 0 WCNSUM = 0 WCNQSM = 0
CONVENTIONAL R FACTORS USING SCALED FOBS CALC'D
NUMB = 0 DO 3157 N = 1,TOTNO IF (IHKL(4,N) .LE. 0 .OR. IHKL(4,N) .GT. 6) GO TO 288 GO TO 287
288 CONTINUE &~~~{~,~bMs ~ 1 IF fNUMB .GE. 2) GO TO 287
FOR
285 ~~~ifjri:~~<'*'>> 286 ~~~ifciR6~)THE SCALE FACTOR IDENTIFIER LIES OUTSIDE THE RANGE'/
*1X,'1-6 FRAT LEAST ONE OF THE REFLECTIONS AND HAS BEEN SET' / *1Xf'TO 1 IN SUCH CASES')
284 ~~~i4~ri~~~<'*'>> 287 CONTINUE
§~~~g~: §~~~g~ t ~~~~~BA~l(t~~r~~l¥!~1;N~~~*SCALE(IHKL(4,N)) WCNQSM = WCNQSM + ((ABS(DATA(1,N)-DATA(3,N~))*SCALE(IHKL(4,N))**2
*/DATA(2,N))**2 WCNSUM = WCNSUM + (DATA(1,N)*SCALE(IHKL(4,N))**2/DATA(2,N))**2
3157 CONTINUE CONVNR = CNJSUMjCNFSUM WRITE(6 3158) CONVNR
3158 FORMAT(f/L 1X~.'CONVE~TIONAL R FACTOR= ',F6.3) WGTDCR = SORT(WCNQSM/WCNSUM)
3159 c
WRITE(6,31S9) WGTDCR FORMAT(/,1X,'WEIGHTED R FACTOR= ',F6.3)
c c c c
FOR ANALYSIS ON MILLER INDICES THE MAX. VALUE OF INDICES IS ALTERED AS IS THE ORDER IN WHICH THEY ARE RUN THROUGH IN THE NESTED LOOPS.
WRITE(6,614) 614 FORMAT(/,1X,'WEIGHTING ANALYSIS FOR GIVEN VALUES OF MILLER INDICES
*') GO TO 10000 DO 526 Z = 1,3 DO 615 L = 1,INDEX3
~~E~fi~L~t~ = = 0 o HKFSUM L = 0
HKSQSM L = 0 HKQSUM L = 0
615 CONTINUE IF ~Z .EQ. 1} GO TO 605 IF Z .EQ. 2 GO TO 606 IF Z .EQ. 3 GO TO 607
172

c c c
c c c
605 CONTINUE ZA = A ZB = B zc = c GO TO 608
606 CONTINUE ZA = B ZB = C ZC = A GO TO 608
607 CONTINUE ZA = C ZB = A ZC = B
608 CONTINUE IF~ZA .EQ. 1~ WRITE{6,781} IF ZA .E8. 2 WRITE 6,782 IF ZA .E • 3 WRITE 6,783
781 FORMAT(/,1X,'H= WDELSQ * NO. OF REFLECTIONS')
782 FORMAT(/,1X~'K= WDELSQ * NO. OF RE~·LECTIONS')
783 FORMAT(/,1X,'L= WDELSQ * NO. OF REFLECTIONS')
WRITE(6f784) 784 FORMA'!' { X, I I)
DO 521 L = 1,MAXlZAl-MIN~ZA~ DO 522 M = 1,MAX ZB -MIN ZB DO 523 LL= 1lMAX ZC -MIN ZC IF ~ZA .EQ. A~ G T 601 IF ZA .EQ. B GO TO 602 IF ZA .EQ. C GO TO 603
601 CONTINUE LZ = L MZ = M LLZ = LL GO TO 604
602 CONTINUE LZ = LL MZ = L LLZ = M GO TO 604
603 CONTINUE LZ = M MZ = LL LLZ = L
604 CONTINUE
+ 1 + 1 + 1
RW
RW
RW
UNRECORDED LABEL VALUES ARE SKIPPED BY.
IF {LABEL{LZ,MZ,LLZ) .EQ. 0) GO TO 523 COUNT{L) = COUNT{L) + 1
SUMS ARE ACCUMULARED
R
R
R
MNFOBS
MNFOBS
MNFOBS
ERROR
ERROR
ERROR
HKLSUM1Li = HKLSUM{L) + DATA{1 LABEL{LZ MZ,LLZ)) HKFSUM L = HKFSUM{L) + {ABS{ DATA{1,LABEL{LZ,MZ,LLZ))- DATA{3,L
*ABEL{L , Z,LLZ)))) HKSQSM{L) = HKSQSM{L) + ({ABS( DATA{1,LABEL{LZ~MZ,LLZ))- DATA{3
*,LABEL LZ,MZ,LLZ)))) DATA 2,LABEL{LZ,MZ,LLZ)))**~ HKOSUM L) = HKOSUM{t) + ~ DATA{1,LABEL(LZ,MZ,LLZ))/ DATA{2,LABEL
*{LZ,MZ,LtZ)))**2 523 CONTINUE 522 CONTINUE 521 CONTINUE
ERRSUM = 0 DO 524 L = 1,{MAX{ZA)-MIN(ZA) +1) IF(HKLSUM{L) .EQ. 0) GO TO 524
173

c c c
c c c
c
525 524 526
IF (HKQSUM(L) .EQ. 0) GO TO 524
STATS. ARE CALC'D AND WRITTEN OUT A CLASS AT A TIME.
WDELSQ = HKSQSM(L)/COUNT(L) ERRSUg = ERRSUM + HKSQSM(L) RW = SQRT(HKS?SM~L)/HKQSUM(L))
~NFOB~K~Sll~t~O~H
1Lt28~J~4(L)
ERROR(L) = SQRT HKSQSM(L}*~OTNO/((TOTNO-NOVAR)*COUNT(L))) ACTL = t + (MIN ZA)-1) WRITE(6i525) AC L,WDELSQ,.RW,R,MNFOBS,ERROR(L) ,COUNT(L) FORMAT( H,I3,5F10.3,6X,I4) CONTINUE CONTINUE OERROR = SQRT(ERRSUM/(TOTNO-NOVAR))
'BAD' REFLECTIONS TESTED FOR AND WRITTEN OUT.
WRITE(6,532) 532 FORMAT(/L/L1XL'LIST OF REFLECTIONS WHERE FOBS-FCALC DIVIDED BY ERR
*OR IN OBSEKVA~ION OF UNIT WEIGHT IS GREATER THAN 3, FOR REFINEMEN *T ON FOBS'} WRITE(6,533)
533 FORMAT{/,1XL' H K L FOBS FCALC SINTHL TWO THE *TA NR~F NSIG ON F')
527 CONTINUE DO 6109 L = 1,(MAXJA1- MIN{A) +1) DO 6110 M = 1f(MAX B -MIN(B) + 1) DO 6111 LL = ,(MA ( ) -MINtC) + 1) IF (LABEL(L,MfLL) .EQ. 0) GO TO 6111 IF (IED .EQ. ) GO TO 531 IF(ABS((DATA(1,LABEL(L,M6LL))-DATA(3,LABEL(L,M,LL)))/DATA(2,LABEL(
*~6MT~L~~i·GE.3*0ERRO~) G TO 530 530 CONTINUE
WRITE(6,636) IHKL(l,LABEL(L,M,LL)) ,IHKL(2,LABEL(L,M,LL)) ,IHKL(3,LA *BEL(L,M LL)),DATA(1,LABEL *(L6M6LL{), DATA(3,.LABEL(L,.M,LL)) DATA(4~.LABEL(L,M,LL))! *36 . *ARSIN( DATA(4,LABELlLr.M,tL{)*LAMDA(LAMID))/3.1415~ ,IHKL *(5,.LABEL(L6M,LL)), ABS(DATA(1tLABEL(L,M,LL))-DATA(3,LABEL(L,M,LL)) *rf(OERROR* ATA(2fLABEL(L,M,LLJ))
636 rORMAT(1X,3I5,6F 0.2) 531 CONTINUE
IF (SALAF .EQ. 1) GO TO 4951 GO TO 4952
4951 CONTINUE WRITE(STDT,END=90) (DATA(JKJ,LABEL(L,M,LL)) ,JKJ = 1,6) ,(IHKL(IKI,L
*ABEL(LLM,LL)) ,IKI = 1,5) 90 CONTINuE
4952 CONTINUE 6111 CONTINUE 6110 CONTINUE 6109 CONTINUE
C NONCOL IS THE NUMBER IN A COLUMN. c
c c c
5000
4783
NONCOL = ((TOTN0+4.0*{DIFF(SNP)+1))/NOCOL + 0.9999} GO TO 45
RETURNS TO HERE IF AN INDEX TITLE COMES AT THE BOTTOM OF A COLUMN
CONTINUE DO 4781 JY=1,1300 DO 4782 JW = 1,12 DO 4783 JX = 1,4 PUBDAT(JX,JW,JY} = 0.0 CONTINUE
174

c
4782 4781
CONTINUE CONTINUE IF (NONCOL .LT. COLMAX) GO TO 44 COLMAX = COLMAX + 2 G TO 45
44 N OL = NONCOL + 2 45 CONTINUE
400
ANONCO = NONCOL NNPAGE = (ANONCO/COLMAX + 0.9999) LO = 1 MO = 1 LLO = 1 DO 3002 JD = 1,NNPAGE NOCOLL = NOCOL PUBDAT 2,1,1 = -1000.0 PUBDAT 2,1,2 = -1000.0 PUBDAT 2,1,3 = -1000.0 PUBDAT 2,1,4 = -1000.0 PUBDAT 1,1,3 = MIN(A) J = 5 IF(NONCOL .LE. COLMAX) GO TO 400 JCA = COLMAX*(JD-1~ + 1 IF fJD .LT. NNPAGE JDA = COLMAX*JD IF JD .EQ. NNPAGE JDA = NONCOL GO 0 500 JCA = 1
C DETERMINATION OF COLUMNS. c
c c c
JDA = NONCOL 500 CONTINUE
4904
4911
4912 4906 4950 4903 4902
PLACES DATA IN PUBDAT THE ARRAY PRINTED OUT WITH NOCOL COLUMNS.
I = 1 IF (JD .GT. 1) J = JCA DO 4901 L = LO,DIFF(A) + 1 DO 4902 M = MO,DIFF(B) + 1 MO = 1 DO 4903 LL = LLO,DIFF(C) + 1 LLO = 1 IF (LABEL(LLM,LL) .EQ. 0) GO TO 4950 PUBDAT!1,I,ul = M + MIN(Sl -1 PUBDAT 2,I,J = LL+ MIN(C -1 PUBDAT 3,I,J = DATA(1,LA EL{L,M,LL)) PUBDAT 41I,J = DATA(3,LABEL(L,M,LL)) J = J + JS = JDA -J IF (JS .LE. -1) GO TO 4904 GO TO 4906 CONTINUE J = JCA I = I + 1 JI = NOCOL-I ~b ~gi 4gf~· -1) GO TO 4911 CONTINUE LO = L MO = M LLO = LL+1 GO TO 4900
CONTINUE CONTINUE CONTINUE CONTINUE CONTINUE IF (L+MIN(A) .GT. MAX(A)) GO TO 4914 PUBDAT(2,1,J+1) = -1000.0
175

c c c
c c c
c
4914
4901 4900
4998 3002
9502
9512
9501
9511
600
700
1200
1201
1202
4001
4002
4003
5060 5001
5011
5021 6000
888
PUBDAT!2,I,J+2l = -1000.0 PUBDAT 2,I,J+3 = -1000.0 PUBDAT 2,I,J = -1000.0 PUBDAT 1,I,J+2 = L+MIN(A) CONTINUE J = J + 4 CONTINUE CONTINUE DO 4998 I = 1 NOCOL IF (PUBDAT(2,f,JDA-1) .EQ. -1000) GO TO 5000 IF (PUBDAT(2,I,JDA) .EQ. -1000) GO TO 5000 CONTINUE CONTINUE NPAGE = NNPAGE
WRITES ROW-COLUMN INFORMATION.
WRITE(6,9502) FORMAT~/f§i1H, 1 THE NUMBER OF ROWS IS') ~S~~J(~;.6~~x~o~~?L WRITE(6 95 1) FORMAT~iHs' THE NUMBER OF COLUMNS IS') ~S~~1(~~.;~~x~~~?L DO 3001 JA = 1,NPAGE IF (NONCOL .LE. COLMAX) GO TO 600 JC = COLMAX*(JA-1~ + 1 IF (JA .EQ. NPAGE JD = NONCOL IF (JA .LT. NPAGE JD = COLMAX*JA GO TO 700 JC = 1 JD = NONCOL CONTINUE DO 3003 U = 1,NOCOL,6 JT1 = 0 JT2 = 0 JT3 = 0 JT4 = 0 JT5 = 0 JT6 = 0
WRITES DATA PAGE TITLE AND ALIGNMENT SYMBOLS.
WRITE(6,1200) FORMAT(/{ffilX,6('.ALIGNMENT. ',9X) ,'.ALIGNMENT.') WRITE(6 20 ) FORMAT(f1'////,1X,6('.ALIGNMENT. ',9X) ,'.ALIGNMENT.') WRITE ( 6, 1202). FORMAT(//I//{//,1X,' '} IF (IED .EQ. ) GO TO 5060 IF(SNP .EQ. 1) WRITE (6,4001) J~~~~(:fu6:1~f3~k¥J~ ~6,4Bo2?BS CALC
i~~~~(:~6:1~f3~R¥J~ ~6,4Bo3?Bs CALC FORMAT(//L,1X,3X,6(' H K OBS CALC GO TO 6000 IF~SNP .EQ. 11 WRITEJ6,5001) J~ (~~~~:fu6:x~1xw~t~E(6,5B1f?Bs IRAW
J~~~~~~:,6:x~>xw~t~E~6,5B2f?Bs IRAW FORMAT(///,1X,3X,6(' H K IOBS !RAW CONTINUE WRITE(6i888) FORMAT ( X, I I )
176
I ) )
I ) )
I ) )
')) I ) )
I ) )

C FOR EACH OF THE SIX COLUMNS PER PAGE WRITES OUT PUBDAT. c
c c c c c c
1000
1100
1151
1110 2008
2009
2010 2000
1001
1101
1152
1111 2011
2012
2013 2001
1002
1102
1153
1112 2014
2015
2016 2002
1003
V = U+1 W = U+2 X = U+3 Y = U+4 Z = U+5
ADDS FURTHER SETS OF COLUMNS UNTIL NOCOL IS REACHED. IF NONCOL GREATER THAN 240 BEGINS A NEW PAGE. THE JTN COUNTERS ARE USED TO OBTAIN BLANKS AND TITLE WHEN THE
SORTING INDEX CHANGES.
DO 3000 J = JC 1 JD
~~rJ~uB~gt~fa68{1~us~RT(P~gg~u?~)~~N~~ ~ 1~?4) FORMAT I3
61X 1 IJ 1 1X,I4 1 lX 1 I4J
GO TO 00 JT1 = JT1 + 1 IF (JT1.EQ. 3) GO TO 1110 WRITE (C11.I151) FORMA'l' ( 1x ' ' ) IF (JTI .EQ. 4) JT1 = 0 GO TO 2000 IF (SNP .EO. 1) WRITE(C1 1 2008~ PUBDAT(1,U 1 J) FORMAT (I **** H =I I I2 I I **** ) IF (SNP .EO. 2) WRITE(C1,2009~ PUBDAT(1,U 1 J) FORMAT( I **** K =' II21 I **** ) IF (SNP .EO. 3) WRITE(C1,2010). PUBDAT(1,U,J) FORMAT (I **** L =I I I2 I I ****I) CONTINUE
~~I~~uBJg~~ta6Y{1~us~~T(P~gg~v~~)~~N~~ ~1~;4) FORMAT I3f1X 1 IJ 1 1X 1 I4,1X 1 I4J GO TO 00 JT2 = JT2 + 1 IF (JT2 .EQ. 3) GO TO 1111 WRITE(C2 1 1152) FORMAT ( 1X I I )
IF (JT2 .EQ. 4) JT2 = 0 GO TO 2001 IF (SNP .EO. 1) WRITE(C2,2011). PUBDAT(1 1 V1 J) FORMAT ( I :r; * * * H =I I I 2, I * * * * I ) IF (SNP .EO. 2) WRITE(C2 1 2012). PUBDAT(1 1 V 1 J) FORMAT(' **** K ='II2 I ****') IF (SNP .EO. 3) WRITE(C2,2013). PUBDAT(1 1 V1 J) FORMAT ( I :r; * * * L =I I I 2 I I * * * * I ) CONTINUE
~~IJ~UB~g~~fa6~{1~us~RT(P~gg~w?~)~~N~~ ~1~;4) FORMAT I3~1X 1 IJ 1 1X 1 I4 1 lX 1 I4J GO TO 00..:: JT3 = JT3 + 1 IF (JT3 .EQ. 3) GO TO 1112 WRITE(C3 1 1153) FORMA'l' ( 1X I I )
IF (JT3 .EQ. 4) JT3 = 0 GO '1'0 2002 IF (SNP .EO. 1) WRITE(C3 1 2014~ PUBDAT(1 1 W1 J) FORMAT( I **** H =' II2 I **** ) IF (SNP .EO. 2) WRITE(C3 1 2015). PUBDAT(1 1 W,J) FORMAT(' **** K =' 1 I2 1 ' ****') IF (SNP .EO. 3) WRITE(C3 1 2016). PUBDAT(1 1 W1 J) FORMAT (I **** L =I I I2 I I ****I) CONTINUE
~~I~~u'g~~tB6~{1~us~~T(P~gg~x~~)~~N~~ ~ 1~~4) FORMAT(I3 1 1X 1 IJ 1 1X 1 I4,1X 1 I4J
177

1103
1154
1113 2017
2018
2019 2003
1004
1104
1155
1114 2020
2021
2022 2004
1005
1105
1156
1115 2023
2024
2025 2005
999 3000 3003 3001
1203 10000
GO TO 2003 JT4 = JT4 + 1 IF (JT4 .EQ. 3) GO TO 1113 WRITE(C4,1I54) FORMAT(1X I I) IF (JT4 .EQ. 4) JT4 = 0 GO TO 2003 IF (SNP .EO. 1) WRITE(C4,2017) PUBDAT(1,X,J) FORMAT(' **** H =',I2 I ****') IF (SNP .EQ. 2) WRITE(C4,2018} PUBDAT(1,X,J) FORMAT(' **** K =' I2 I ****') IF (SNP .EO. 3) WRITE(C4,2019~ PUBDAT(1,X,J) FORMAT(' **** L =',I2,' ****) CONTINUE
J~I~~uBJg~~£g6Ir1~uB~RT(P~gg~Y~1>~~N~~ ; 1~;4> FORMAT I3,1X,IJ,1X,I4,IX,I4J GO TO OOq JT5 = JT5 + 1 IF (JT5 .EQ. 3) GO TO 1114 WRITE (C5 J.1I55) FORMAT ( 1x ' ' ) IF (JT5 .EQ. 4) JT5 = 0 GO TO 2004 IF (SNP .EO. 1) WRITE(C5,2020} PUBDAT(1,Y,J) FORMAT(' **** H =',I2,' ****') IF (SNP .EO. 2) WRITE(C5,2021l PUBDAT(1,Y,J) FORMAT(' **** K =',I2,' ****~) IF (SNP .EO. 3) WRITE(C5,2022\ PUBDAT(1,Y,J) FORMAT(' **** L =',I2,' ****~) CONTINUE
~~IJ~uBJg~~tB6~{1~uBgRfcP~gg~z~1>~~N~~ ;1 ~~4) FORMAT I3l1X,IJ,1X,I4,1X,I4J GO TO 00::> JT6 = JT6 + 1 IF (JT6 .EQ. 3) GO TO 1115 WRITE(C6,1I56) FORMAT ( 1X I I ) IF (JT6 .EQ. 4) JT6 = 0 GO TO 2005 IF (SNP .EO. 1) WRITE(C6,2023) PUBDAT(1,Z,J) FORMAT(' **** H =',I2 I****') IF (SNP .EO. 2) WRITE(C6,2024} PUBDAT(1,Z,J) FORMAT(' **** K =',I2 I ****') IF (SNP .EO. 3) WRITE(C6,2025} PUBDAT(1,Z,J) FORMAT ( I * * * * L =I , I 2 , I * * * * I ) CONTINUE WRITE (6,999) C1,C2tC3~C4 1 C5lC6 FORMAT ( '+ 'I, 3X, 3A6 , ::> ( 2x, 3A6) J CONTINUE CONTINUE CONTINUE WRITE(6,1203) FORMAT(////,1X,6('.ALIGNMENT. ',9X) ,'.ALIGNMENT.') CONTINUE STOP END
178

PROGRAM
c BONDSTAT
******************** c c c c c c c c c c c c c c c c c c
c c c c c c
READS TITLE,NUMBER OF BONDS
~~t£sc5&§~fN~~~~~~D0~Y~~~~A~B~E~A~~~~~~s'~NgA~~~G~2cH) REQUIRES ASYMMETRIC UNIT TO BE A CONNECTED CHEMICAL UNIT
WRITES OUT CALCULATED BOND LENGTHS
CALCULATES STANDARD DEVIATIONS AND MEANS OF THE SPECIFIED TYPES AND PRINTS THEM OUT ********************
INPUT ***** TITLE 13A6,A2 NUMBER OF BOND TYPESt MAXIMUM CELL CONSTANTS oF10.6
BOND LENGTH I5,Fl0.6 {ANGLES)
BOND TYPES A2,3X,A2 ATOM PARAMETERS AS
1 PER CARD,NBONDT OF THEM FOR CUCLS 2 CARDS PER ATOM
REAL NAME, NNAME DIMENSION TITLE(l4) DIMENSION TAB1{6)~TAB2{6) DIMENSION DELTAX{~) DIMENSION CELL~6) DIMENSION NAME 150(6) ,NNAME{2,500) ,X{3,150) ,SUM{lOO) ,DIST{500) ,BON
*D{l00,2) ,SUMSQ 1001 ,NUM{lOO)
READS TITLE,NUMBER OF BONDS READS TITLE,NUMBER OF BOND TYPES,MAXIMUM BOND LENGTH, NUMBER OF SYMBOLS REQUIRED FROM ATOM LABEL CELL CONSTANTS,BOND TYPES,ATOM PARAMETERS{2 CARDS EACH)
LIM=500 READA5~.10000l TITLE
lOOOO ~g~E1J!rft861f)TITLE 10001 FORMAT{~OX,l4A6)
READ(5,50)NBONDTtNOID,NOID2,D,DMIN 50 FORMAT{3I5 2Fl0.o)
c
WRITE{6 47~ NBONDT,D,NOID,NOID2 47 FORMAT{iX,'THE NUMBER OF BOND TYPES EXAMINED IS ',I4,' FOR ATOMS
*LESS THAN ',F5.l,'A APART. NOID = ',Il,' NOID2 = ',Il) DD=D**2 DDMIN=DMIN**2 READ{5,5llTABl READ{5,51 TAB2
51 FORMAT { 6I ) READ{5,49) CELL
49 FORMATJ6Fl0.6)
45 ~~~ft<f~~~c~ftLCONSTANTS ARE ',6Fl0.6) DO 10 K=l NBONDT READ{5,.60~BOND(K,l) ,BOND{K,2)
60 FORMAT(A6,4X,A6) 10 CONTINUE
DO 100 J=l,l50 READ{5,.500,END=200) {NAME{J,JJ) ,JJ=l,6), {X{II,J) ,II=l,3)
500 FORMAT(6Al,24X,3Fl0.6) X~3,J1=Xj3,J~*CELL{3)+X(l,J)*COS{CELL{5)*ATAN{l.0)/45.0)*CELL{l) X 2,J =X 2,J *CELL{2) X l,J =X llJ *SIN{CELL{5)*ATAN{l.0)/45.0)*CELL{l) READ{ ,5 1 DUMMY
501 FORMAT(F6. ) 100 CONTINUE 200 N=J-1
179

C CALCULATES BOND LENGTHS FOR ALL BONDS IN STRUCTURE C REQUIRES ASYMMETRIC UNIT TO BE A CONNECTED CHEMICAL UNIT c
c
DO 2000 JJ=1,N-1 DO 300 JK=JJ+1,N DIST2=0 DO 400 I=1 3 DIST2=DIST2+(X(I
6JJ)-X(I,JK))**2
IF(I.LT.3) GOT 400 IF(DIST2.GT.DD) GO TO 300
IF(DIST2.LT.DDMIN) GO TO 300 NN=NN+1 IF(NN.GT.LIM) GO TO 600 DIST(NN)=DIST2 DO 297 JE=1 2
297 NNAME(JE,NNf=· I
DO 298 L=1 NOID NNAME(1,NNf=CONCAT(NNAME(1,NN) ,NAME(JJ,TAB1(L)) ,(7-L)*8-1,47,8)
298 CONTINUE DO 296 L=1 NOID2 NNAME(2LNNf=CONCAT(NNAME(2,NN) ,NAME(JK,TAB2(L)) ,(7-L)*8-1,47,8)
296 CONTINU.t!i WRITE(6,301) (NAME(JJ,LN) ,LN=1,6) ,(NAME(JK,LM) ,LM=1,6) ,NN,SQRT(DIST
*2) 301 FORMAT(1X,2(6A1,2X) ,I5,F10.6) 400 CONTINUE 300 CONTINUE
2000 CONTINUE GO TO 700
C WRITES OUT CALCULATED BOND LENGTHS c
c
600 WRITE(6 800)D JJ JK 800 FORMA~~iXt'506 DfSTANCES LESS THAN',F5.3,' HAVE BEEN CALCULATED AN
*D JJ= ,I~,' JK= ',I3) NN=NN-1
700 CONTINUE
C CALCULATES STANDARD DEVIATIONS AND MEANS OF THE SPECIFIED C TYPES AND PRINTS THEM OUT c
DO 900 JR=1,NN DO 910 NB=1,NBONDT IFCNNAME(1,JR}.IS.BOND(NB,1).AND.NNAME(2,JR} .IS.BOND(NB,2)) GO TO
*920 910 CONTINUE
GO TO 900 920 CONTINUE
SUM(NB)=SUM(NB?+SORT(DIST(JR)) SUMSO(NB)=SUMS (NB)+DIST(JR) NUM(NB}=NUM(NB +1
900 CONTINUE
~g=§g~T~C1s~~~~9~)-SUM(M)**2/NUM(M))/CNUM(M)-1) ) WRITE(6i930) NUM(M),BOND(M~1),BOND(M,2),SD,SUM(M)/NUM~M)
930 FORMAT( Xt'THE STANDARD DEviATION OF THE',I4f' BONDS ,A6,'-',A6,' *IS',F6.4, A. THE MEAN BOND LENGTH IS ',F6.4, A.')
980 CONTINUE STOP END
180

8 1235 125 32.508 AS 1 ZN 1 AS 2 ZN 2 AS 3 ZN 3 AS 4 ZN 4
SAMPLE INPUT
BOND LENGTH STATISTICS FOR AS S ZN COMPOUND 4 3 3.0 1.5
18.489 s 1 s 1 s 2 s 2 s 3 s 3 s 4 s 4
30.589 90.0 95.699
181
90.0

PROGRAM SCALE
FILE l=BITRI/IN,UNIT=DISKLRECORD=11fBLOCKING=90 6SAVE=7~AREA=900*8 FILE 2=BITRI/OUT,UNIT=DISK,RECORD=1 6BLOCKING=9 ,SAVE=t,AREA=900*8 DIMENSION INPUT~20)jPROCEZ(40,2 )
c c c c
c c c
8lM~~~i8~ g~~l~3d 1
6 ao?~DAT2(30~8ol 6couNT1(30,80) DIMENSION SCAL1(3 ,80} ,SCAL2(~0,8 ) INT=1 SIG=2 H=7 K=8 L=9 NREF=11 N=11 NUM=5040
READS REFLECTION DATA FROM FILE SORTS OUT HKO AND -HKO REFLECTIONS
100 CONTINUE IVXX=1 READ(IVXX,END=99) (INPUT(J) ,J=1,N)
99 CONTINUE I~~~~~J~Nlru~:~~~~F~>boG~6 ~H~> Go To 4oo IF INPUT H .LT.O GO TO 200 IF INPUT H .GT.O GO TO 300 GO TO 10
200 CONTINUE INPUT{K)=INPUT(K)+1 DAT1{ABSCINPUT(H)) ,INPUT(K))=INPUT(INT} GO TO 100
300 CONTINUE DATA INPUT(K}=INPUT(K)+1 DAT2(ABS{INPUT(H)) ,INPUT(K))=INPUT(INT) GO TO 100
400 CONTINUE REWIND IVXX
500 CONTINUE READ(IVXX,END=199) (INPUT(J) ,J=1,N)
199 CONTINUE IVYY=2 IF(ABS(INPUT(NREF)).GT.7308) GO TO 1500 IF(ABS(INPUT(NREF)).GT.NUM.AND.ABS(INPUT(NREF)) .LE.7308) GO TO 40
*00 IF(ABS(INPUT(NREF)).GT.2303.AND.ABS(INPUT(NREF)).LT.2851} GO TO 40
*00 GO TO 4001
4000 CONTINUE WRITE(IVYY) (INPUT(J} ,J=1,N) GO TO 500
4001 CONTINUE INPUT(K)=INPUT(K)+1 IF(INPUT(L).EQ.O) GO TO 600
ESTABLISHES SCALE FACTORS FOR HKO REFLECTIONS
GO TO 700 600 CONTINUE
IF(COUNT.GT.O) GO TO 800 IF(DAT1(ABS(INPUT(H)) ,INPUT(K)).LE.O) GO TO 1700 SCAL1CABS(INPUT(H}) ,INPUT(K})=(DAT2(ABS(INPUT(H)) ,INPUT(K))/DAT1 *J~B~~1:~~Ha(H)) ,1:NPUT(K))+l)*0.5
1700 CONTINUE SCAL1(ABS(INPUT(H)) ,INPUT(K})=1
182

c c c
c c c
1800 CONTINUE COUNT=O
700 CONTINUE COUNT = COUNT+l DO 900 JJJ=l N PROCEZ(COUNT:JJJ)=INPUT(JJJ)
900 CONTINUE GO TO 500
800 CONTINUE IF(DATl(ABS(INPUT(H)) ,INPUT(K)) .LE.O) GO TO 1900 SCAL2(ABS(INPUT(H)) ,INPUT(K))=(DAT2(ABS(INPUT(H)) ,INPUT(K))/DATl
*(ABS(INPUT(H)) ,INPUT(K))+l)*0.5 GO TO 2000
1900 CONTINUE SCAL2(ABS(INPUT(H)) ,INPUT(K))=l
2000 CONTINUE DROCEZ(l,H)=PROCEZ~l,H)
~~?~~Kl~~~~s1¥~~~~~~ff~INPUT(K)) .EQ.0.5) SCAL2(ABS(INPUT(H)) ,I *NPUT(K))=l.O
3000
106
1000
1200 1100
1300
1500
IF(SCALl(ABS(DROCEZ(l6H)) ,DROCEZ(l,K)).EQ.0.5) SCALl(ABS(DROCEZ(l, *H)) ,DROC:E:Z (l,K) b = 1.
*B:~I~i~aJ¥~oJ12.b*~~~kt~t~~~1~&gfif~h~f 1
1~&gfif~k~>:0.5>>> IF(DROCEZ(l H) .GT.O) SCALl(ABS(DROCEZ( H)),DROCEZ(l K)) = *0. 5 * ( 1. 0+1. 6; ( 2. 0 * ( SCALl (ABS ( DROCEZ ( 1, Hf) , DROCEZ ( 1, Kf) -0. 5) ) )
SCALES H~L REFLECTIONS
DO 1000 JJ=l,COUNT JJ= JJ-1 SCALE=((COUNT-JJ)/COUNT)*SCALl(ABS(DROCEZ(l,H)) ,DROCEZ(l,K))
*+JJ*SCAL2(ABS(INPUT(H)) ,INPUT(K))/COUNT JJ= JJ+l PROCEZlJJ,K)=PROCEZ(JJ,K)-1 PROCEZ JJ,INT)=PROCEZ(JJ,INT)*SCALE PROCEZ JJ,SIG)=PROCEZ(JJ,SIG)*SCALE CONTIN E WRITE(6fl06) (PROCEZ(JJ,M) ,M=l,N) FORMAT( X,llF12.1) WRITE(IVYY) (PROCEZ(JJ,M) ,M=l,N) CONTINUE IF(ABS(INPUT(NREF)) .EQ.NUM) GO TO 1300 DO 1100 JK=l,COUNT DO 1200 JL=l N PROCEZ(JK,JLf=O CONTINUE CONTINUE COUNT=O GO TO 600 CONTINUE PROCEZ!1,INT~=INPUT(INT)*SCAL2(ABS(INPUT(H)) ,INPUT(K)) PROCEZ 1,SIG =INPUT(SIG)*SCAL2(ABS(INPUT(H)) ,INPUT(K)) PROCEZ 1,NRE )=5040 PROCEZ 1,K)=PROCEZ(1,K)+1
WRITES SCALED REFLECTION DATA TO PRINTER AND FILE
WRITE(6,106) (PROCEZ(l,MK) ,MK=l,11) WRITE(IVYY) (PROCEZ(l,MN) ,MN=1,N) GO TO 500 CONTINUE
PROCEZ 1,K = 99 PROCEZ~l,H! = 99 PROCEZ 1,L = 99 WRITE(IVYY (PROCEZ(1,J) ,J=1,N) LOCK IVYY STOP
183




















