
Orthodontic Appliance Preferences of Children and Adolescents

Epithelioid Sarcoma in Children and AdolescentsA Report from the Italian Soft Tissue Sarcoma Committee
Michela Casanova, M.D.1
Andrea Ferrari, M.D.1
Paola Collini, M.D.2
Gianni Bisogno, M.D.3
Rita Alaggio, M.D.4
Giovanni Cecchetto, M.D.5
Alessandro Gronchi, M.D.6
Cristina Meazza, M.D.1
Alberto Garaventa, M.D.7
Andrea Di Cataldo, M.D.8
Modesto Carli, M.D.3
1 Pediatric Oncology Unit, Istituto Nazionale Tu-mori, Milan, Italy.
2 Pathology Department, Istituto Nazionale Tumori,Milan, Italy.
3 Division of Hematology-Oncology, Department ofPediatrics, University Hospital of Padova, Padova,Italy.
4 Department of Pathology, University Hospital ofPadova, Padova, Italy.
5 Department of Pediatric Surgery, University Hos-pital of Padova, Padova, Italy.
6Melanoma Sarcoma Surgical Unit, Istituto Nazio-nale Tumori, Milan, Italy.
7 Division of Pediatric Oncology, Istituto G. GasliniGenova, Genova, Italy.
8 Division of Pediatric Oncology, Universita di Ca-tania, Catania, Italy.
Supported in part by Fondazione Citta della Spe-ranza and by MURST ex 60%.
Address for reprints: Andrea Ferrari, M.D., Pediat-ric Oncology Unit, Istituto Nazionale Tumori, Via G.Venezian, 1–20133 Milan, MI, Italy; Fax: (011) 39 0223902648; E-mail: [email protected]
Received July 11, 2005; revision received August22, 2005; accepted August 23, 2005.
BACKGROUND. Epithelioid sarcoma (ES) is an uncommon malignant soft tissue
tumor. To the authors’ knowledge, little information is available to date concerning
its clinical features and management in children and adolescents, particularly with
regard to the recently described proximal-type variant. The current study concerns
30 patients age � 18 years who were enrolled in the Italian Soft Tissue Sarcoma
Committee protocols.
METHODS. Histopathologic specimens, clinical data, and treatment modalities
were reviewed for the current analysis. Nineteen patients had classic ES and 11 had
proximal-type ES. Surgery was the mainstay of treatment; radiotherapy was given
to six patients considered to be at risk of local disease recurrence due to incom-
plete resection, and chemotherapy was administered to eight patients.
RESULTS. With a median follow-up of 66 months, the 5-year event-free survival
(EFS) and overall survival (OS) rates were 61.7% and 92.4%, respectively, but the OS
rate dropped to 86.9% and 72.4%, respectively, at 10 years and 15 years. Local
disease recurrence was the major cause of treatment failure. The most significant
finding influencing both EFS and OS was tumor site, with a tumor location in the
extremities predicting a favorable outcome. Initially unfavorable clinical findings
and a worse outcome were associated with the proximal-type variant of ES. A
response to chemotherapy was noted in three of the seven patients with measur-
able disease.
CONCLUSIONS. The current study confirms some typical features of ES (i.e., the
peculiar superficial distal location [i.e., the hand, fingers], indolent growth, and a
tendency to recur locally). The current study data do not clearly confirm the strong
tendency for the lymph node involvement described in adult ES patients. Further
studies are needed to better define the clinical behavior and biology of the prox-
imal-type variant of ES. Cancer 2006;106:708 –17.
© 2005 American Cancer Society.
KEYWORDS: epithelioid sarcoma, pediatric soft tissue sarcomas, proximal-typeepithelioid sarcoma, lymph node metastases, nonrhabdomyosarcoma soft tissuesarcoma.
Epithelioid sarcoma (ES) was first described by Enzinger in 1970.1 Itis an uncommon, malignant soft tissue tumor that generally pre-
sents in fascial planes, aponeuroses, and tendon sheaths of the ex-tremities, particularly in the hand (the fingers) and foot. The tumor ismost prevalent in young adults ages 20 – 40 years. It rarely is found inchildren and older people, but no age group is exempt.1–3
Pediatric oncologists usually classify ES within the heterogeneousgroup of nonrhabdomyosarcoma soft tissue sarcomas (NRSTS); inparticular, ES recently has been included in the subset of so-calledadult-type NRSTS.4 In what to our knowledge are two of the largestreported series on NRSTS, ES is reported to represent 5-8% of cases.4,5
708
© 2005 American Cancer SocietyDOI 10.1002/cncr.21630Published online 13 December 2005 in Wiley InterScience (www.interscience.wiley.com).

Histologically, in its classic form, the tumor has adistinctive nodular, granuloma-like pattern, with spin-dle and epithelioid cells circumscribing areas of cen-tral degeneration and necrosis.1,6,7 A subtype charac-terized by a more aggressive behavior and worseoutcome recently was described in rare cases, mostlyoccurring as a deep-seated soft tissue mass at proxi-mal body sites, hence the definition of proximal-typeES.8,9 It is characterized histologically by sheets oflarge, atypical, epithelioid cells with vesicular nucleiand prominent nucleoli, with a rhabdoid phenotype.Despite the differences in clinical presentation andbehavior, classic ES and proximal-type ES share asimilar immunophenotypic profile (although peculiaralterations of chromosome 22 with deletions of theSMARCB1/INI1 gene recently were described in theproximal-type variant10), suggesting that they repre-sent different variants of a distinctive tumor entity.8,11
To our knowledge, few reports have been pub-lished to date and little information currently is avail-able regarding the clinical features and managementof ES in children and adolescents,6,12–14 and no data atall are available concerning the clinical history ofproximal-type ES in pediatric patients.
To contribute information regarding the naturalhistory and management of ES in children and ado-lescents, the Italian Soft Tissue Sarcoma Committee(which is affiliated with the Associazione Italiana Ema-tologia Oncologia Pediatrica– [AIEOP]) reviewed theirexperience with children and adolescents diagnosedwith ES.
MATERIALS AND METHODSBetween January 1978 and December 2004, 30 consec-utive, previously untreated patients age � 18 yearswith a diagnosis of ES were registered at AIEOP cen-ters. Half of these patients were enrolled prospectivelyin the RMS’88 and RMS’96 AIEOP cooperative proto-cols for soft tissue sarcomas, whereas the other halfwere treated at the Istituto Nazionale Tumori of Milan(INT), which only joined the national protocols in1996. The 30 patients represented approximately 7%of all NRSTS cases registered during the study period(n � 404 cases). Complete information regarding clin-ical data, treatment modalities, and outcome wasavailable for all patients and was reviewed for thecurrent analysis. Fourteen patients treated at the INTwere included in the recently reported single-institu-tional series of adult-type NRSTS.4 Informed consentwas provided by all patients or their guardians, ac-cording to the rules established by the study institu-tions.
Histopathologic ReviewThe histopathologic specimens and slides were re-viewed for all patients by two of the authors (P.C. andR.A.) to confirm the diagnosis (using the revised WorldHealth Organization [WHO] criteria),15 and in partic-ular to identify cases of proximal-type ES. The criteriafor the diagnosis of the proximal-type variant was thepresence of large, epithelioid cells; marked nuclearatypia; frequently rhabdoid features; and the lack of agranulomatous-like pattern.8 Immunoreactivity forcytokeratins, epithelial membrane antigen, and CD34helped to confirm the diagnosis. The grading systemof the French Federation of Cancer Centers SarcomaGroup (FNCLCC)16 was tentatively applied to assign ahistologic grade, although this practice is not recom-mended by the latest WHO classification.15 We de-cided to tentatively evaluate tumor grade because it isone of main factors used to predict patient prognosisand to determine the indication for adjuvant therapiesin patients with NRSTS. The FNCLCC is a 3-gradesystem that defines a score in relation to tumor differ-entiation (for ES, a value of 3 is given by default),mitotic index (score of 1–3, according to the numberof mitoses present in 10 high-power fields), and tumornecrosis (score of 0 –2).16 This system was chosen forits great reproducibility and because it enables a validcomparison with adult patient series.
Clinical GroupingAt the time of diagnosis, local tumor extent was as-sessed using computed tomography (CT) and/or mag-netic resonance imaging (MRI); pretreatment investi-gations included chest X-ray and/or chest CT scan,abdominal ultrasound, and whole-body bone scan inthe majority of patients. Regional lymph nodes wereassessed radiologically (by ultrasound) in the majorityof patients (e.g., axillary ultrasound for tumors of theupper extremity). Lymph nodes were assessed surgi-cally in two patients with no clinical signs of lymphnode spread (both biopsies proved negative); one pa-tient with clinically involved lymph nodes underwentlymphadenectomy.
Disease stage was assessed according to both theclinical TNM pretreatment staging system17 and theIntergroup Rhabdomyosarcoma Study (IRS) postsur-gical grouping system.18 The TNM definition of T1refers to tumors that are confined to the organ ortissue of origin, whereas T2 lesions invade contiguousstructures; the T1 and T2 tumor groups are classifiedfurther as “a” or “b” according to whether they have agreatest tumor dimension of � 5 cm or � 5 cm,respectively. Regional lymph node involvement is des-ignated as N1 (no lymph node involvement– [N0]) and
Childhood Epithelioid Sarcoma/Ferrari et al. 709

distant metastases at the time of disease onset is clas-sified as M1 (no metastases– [M0]).17 The IRS systemcategorizes patients into four groups based on theamount and extent of residual tumor after initial sur-gery: Group I includes patients with completely ex-cised tumors with negative microscopic margins;Group II indicates patients with macroscopically re-sected tumors with microscopic residual diseaseand/or resected involved regional lymph nodes;Group III patients have macroscopic residual diseaseafter incomplete surgical resection or biopsy; andGroup IV patients have metastases at the baseline.18
TreatmentPatients were treated using multimodality approachesincluding surgery, chemotherapy, and radiotherapy,based on the ongoing protocols at the time of diagno-sis. Overall treatment strategies did not change sub-stantially over the years. Surgery was the mainstay oftreatment. In principle, primary excision was at-tempted when complete, nonmutilating resection wasconsidered feasible; otherwise a biopsy was performedand chemotherapy was administered to shrink thetumor and make it more amenable to subsequentsurgery. Primary reexcision was recommended beforeany other treatment when microscopic residual dis-ease was suspected, particularly in those patients whohad undergone inadequate surgery initially based onthe clinical assumption of a benign lesion. Surgery wasdefined as complete (IRS Group I) when histologicmargins were free of disease, and therefore includedablative surgery, compartment resections (en bloc re-section of the tumor and the entire compartment oforigin in which the tumor was anatomically confined),and wide excisions (en bloc excisions through normaltissue, beyond the reactive zone but within the mus-cular compartment, removing the tumor together withits pseudocapsule). Marginal surgical resection (IRSGroup II) was a resection performed just outside thepseudocapsule, with suspected microscopic residualdisease (marginal resection according to Enneking’scriteria), and cases of microscopically infiltrated mar-gins (considered by Enneking to be intralesional).19
Radiotherapy was given to patients considered tobe at risk of local disease recurrence because of mi-croscopically or macroscopically incomplete surgicalresection. It was delivered using megavoltage photonor electron beam energies, using conventional frac-tionation (200 centigrays [cGy] daily for 5 days perweek) or hyperfractionated accelerated radiotherapy(HART) (2 daily fractions of 160 cGy, with a 6 – 8-hrinterval), according to protocols in use at the time.
Different chemotherapeutic regimens wereadopted over the years, and were the same as those
used to treat rhabdomyosarcoma. The VACA regimenwas comprised of vincristine at a dose of 1.5 mg/m2
given at Weeks 1, 4, and 7; doxorubicin at a dose of 30mg/m2/day given for 2 days at Weeks 1 and 7; cyclo-phosphamide at a dose of 1200 mg/m2 given at Weeks1, 4, and 7; and actinomycin D at a dose of 0.5 mg/m2/day given for 3 days at Week 4, for a total of either37 weeks or 46 weeks. Ifosfamide replaced cyclophos-phamide in the VAIA regimen (vincristine at a dose of1.5 mg/m2 on Weeks 1–7; actinomycin D at a dose of1.5 mg/m2 at Weeks 1 and 7; ifosfamide at a dose of 3g/m2/day for 2 days at Weeks 1, 4, and 7; and doxo-rubicin at a dose of 40 mg/m2/day given for 2 days atWeek 4, for a total of 27 weeks). Carboplatin andetoposide were included in the CEVAIE schedule,which was comprised of carboplatin at a dose of 500mg/m2 given at Week 1; epirubicin at a dose of 150mg/m2 given at Week 1; vincristine at a dose of 1.5mg/m2 given at Weeks 1–7; actinomycin D at a dose of1.5 mg/m2 given at Week 4; ifosfamide at a dose of 3g/m2/day given for 3 days at Weeks 4 and 7; andetoposide at a dose of 150 mg/m2/day given for 3 daysat Week 7, for a total of 27 weeks (in all regimens, themaximum dose of vincristine and actinomycin D ad-ministered was 2 mg). One patient with metastases atthe time of disease onset received high-dose chemo-therapy (comprised of thiotepa, cyclophosphamide,and melphalan) with stem cell rescue. In those pa-tients who received primary chemotherapy, responsewas evaluated after 3 cycles (at Week 9 or 10) and wasbased on a reduction in the sum of the products of theperpendicular dimensions of all measurable lesions,defined as follows: a complete response (CR) indicateda complete disappearance of tumor, a partial response(PR) indicated a tumor reduction � 50%; and a minorresponse (MR) indicated a reduction of � 25%. Stabledisease or a reduction in tumor of � 25% was recordedas no response (NR), whereas an increase in tumorsize or the detection of new lesions was considered tobe progressive disease.
Isolated limb perfusion (with the administrationof doxorubicin) was performed in one patient.
Statistical MethodsEvent-free survival (EFS) and overall survival (OS)were estimated according to the Kaplan–Meier meth-od.20 Patients were evaluated from the date of histo-logic diagnosis until the most recent uneventful fol-low-up or disease progression, recurrence, or deathfrom any cause (whichever occurred first) for EFS, anduntil to death for OS. The local recurrence-free sur-vival (LRFS) rate was calculated from the time of di-agnosis until local disease progression or recurrence.Patients who died of their tumor after distant failure
710 CANCER February 1, 2006 / Volume 106 / Number 3

occurring before any local disease progression/recur-rence were censored at the time of death in the anal-ysis of LRFS. The metastasis-free survival (MFS) ratewas calculated from the time of diagnosis until theonset of distant metastases.
The log-rank test was used to compare the sur-vival curves for the different subgroups of patients toestablish the potential value of prognostic factors.21
The duration of patient follow-up (as of April 2005)ranged from 3–180 months (median, 66 mos).
RESULTSClinical CharacteristicsThe ages of the patients ranged from 4 –18 years (me-dian, 12 yrs); 17 patients were male and 13 were fe-male. The time elapsing between the first symptoms(generally a swelling or a lump) and the correct diag-nosis was 1–36 months (median latency period of 10mos; the latency period was � 12 mos in 13 patients).
Some of the clinical features of the 30 patients arepresented in Table 1, comparing classic ES patients(19 patients) with patients with proximal-type ES (11patients). Overall, 6 patients were classified as havingFNCLCC Grade 3 tumors, and 24 patients were clas-sified as having FNCLCC Grade 2 tumors. In the ma-
jority of cases, the primary tumor sites were in theextremities, and were distal in several patients (5 casesof a tumor arising in a finger). The median tumor sizewas 3 cm, and the majority of tumors measured � 5cm, with no local invasion reported.
Three patients had regional lymph node involve-ment and two patients had distant metastases (onepatient in the lung and one patient in soft tissue).
With regard to the 11 patients with the proximal-type ES variant, 6 tumors arose at a proximal site (4 inthe trunk, 1 in the head and neck area, and there was1 case of intraabdominal disease). High-grade (FN-CLCC Grade 3 disease), large (measuring � 5 cm), andunresectable tumors were more common amongthose patients with proximal-type ES compared withthe patients with classic ES (the difference was notfound to be statistically significant, but it should benoted that the number of patients in each group wassmall).
TreatmentComplete surgical resection was the only treatment inthe 18 IRS Group I patients (no adjuvant treatmentswere given); of these 18 patients, 11 achieved histo-logically free surgical margins after undergoing a pri-
TABLE 1Clinical Features
Histology Entire series (n � 30 patients) Classic ES (n � 19 patients)Proximal-type ES (n � 11patients)
FNCLCC grade Grade 2 (24 patients)Grade 3 (6 patients)
Grade 3 (2 patients) Grade 3 (4 patients)
Primary tumor site Extremities (23 patients)Upper (16 patients [arm in 2 patients, forearm in 6 patients,
hand in 3 patients, and finger in 5 patients])Lower (7 patients [gluteus/thigh in 4 patients, leg in 2
patients, and foot in 1 patient])Head and neck (3 patients [head in 1 patient, oral cavity in
1 patient, and tongue in 1 patient])Proximal site (nonextremities)
(2 patients)Proximal site (nonextremities)
(6 patients)Trunk (3 patients)Intraabdomen (omentum) (1 patient)
Tumor size 1-11 cm (median, 3 cm)� 5 cm (22 patients)) � 5 cm (3 patients) � 5 cm (5 patients)� 5 cm (8 patients
TNM classification T1a (20 patients) T2b (1 patient) T2b (4 patients)T1b (3 patients)T2a (2 patients)T2b (5 patients_N1 (3 patients) N1 (2 patients) N1 (1 patient)M1 (2 patients) M1 (1 patient) M1 (1 patient)
IRS grouping Group I (18 patients) Group III (2 patients) Group III (4 patients)Group II (4 patients)Group III (6 patients)Group IV (2 patients)
ES: epithelioid sarcoma; FNCLCC: French Federation of Cancer Centers Sarcoma Group; IRS: Intergroup Rhabdomyosarcoma Study.
Childhood Epithelioid Sarcoma/Ferrari et al. 711

mary reexcision performed within 8 weeks of initialinadequate surgery. In other words, 11 patients whowere classified as Group II were downstaged to GroupI after primary reexcision, thereby avoiding radiother-apy. Fingers were amputated in three patients.
Four patients were classified as IRS Group II be-cause of surgical margin excisions (not amenable toresurgery) in three patients and regional lymph nodeinvolvement in one patient (both the primary forearmtumor and the enlarged axillary lymph nodes werecompletely resected).
With regard to the eight patients with advanceddisease (six of whom were in IRS Group III and two ofwhom were classified as Group IV), the first surgicalapproach was a biopsy in six patients and a macro-scopically incomplete resection in two patients. Six ofthese patients subsequently underwent delayed sur-gery (achieving histologically clear surgical margins inthree patients). One patient was treated with radio-therapy and chemotherapy (with no further surgeryperformed). One 18-year-old patient with a large tu-mor of the upper limb received isolated limb perfu-sion, but experienced severe local side effects (cuta-neous and soft tissue necrosis) that subsequentlymade emergency amputation necessary.
Overall, radiotherapy was administered to 6 pa-tients (2 patients in IRS Group II, 3 patients in IRSGroup III, and 1 patient in IRS Group IV), with con-ventional fractionation used in 1 patient (a dose of 70grays [Gy]) and HART administered to 5 patients(doses ranged from 40 –50 Gy).
Chemotherapy was given to eight patients (theone patient in IRS Group II with N1 disease, five of thesix patients in IRS Group III, and the two patients inIRS Group IV). The chemotherapy regimen used wasVACA in two patients, VAIA in three patients, CEVAIEin two patients, and CEVAIE followed by high-dosechemotherapy with stem cell rescue in one patient.The response to primary chemotherapy was evaluablein seven patients and was a CR in two patients, a PR inone patient, and NR in four patients (for an overallresponse rate 43%). With regard to the regimen ad-ministered, responses were observed in one of the twopatients treated with VAIA (the other patient was notevaluable), two of the three patients treated withCEVAIE, and neither of the two patients treated withVACA. All three patients who responded to chemo-therapy had received regimens including ifosfamideand anthracyclines.
OutcomeAt the time of last follow-up, the EFS for the entireseries of patients was 61.7% at 5 years, and did notchange significantly at either 10 years or 15 years. The
OS was 92.4% at 5 years, but decreased to 86.9% and72.4%, respectively, at 10 years and 15 years (1 patientdied of tumor 132 mos after diagnosis).
Seventeen patients were alive in first completedisease remission and 1 patient was alive with stabledisease. Twelve patients developed a disease recur-rence 3–112 months after diagnosis (median time todisease recurrence was 11 mos). The first disease re-currence was local in 9 patients, in the regional lymphnodes in 1 patient, and at distant sites (lung) in 2patients (associated with local recurrence in 1 pa-tient), for 5-year LRFS and MFS rates of 65.0% and82.1%, respectively. Among the 12 patients with recur-rent disease, 4 died of their tumor and 3 were alivewith progressive disease at the time of last follow-up.Five patients (who developed local recurrence) wereable to achieve further complete disease remissionthanks to salvage treatments (mutilating surgery in 3patients) and were alive without evidence of disease ata range of 54 –120 months (median of 100 mos) aftertheir last disease recurrence (2 patients experiencedonly 1 disease recurrence, and 3 patients developedseveral subsequent disease recurrences, with featuresresembling in-transit metastases noted in 2 of thesepatients).
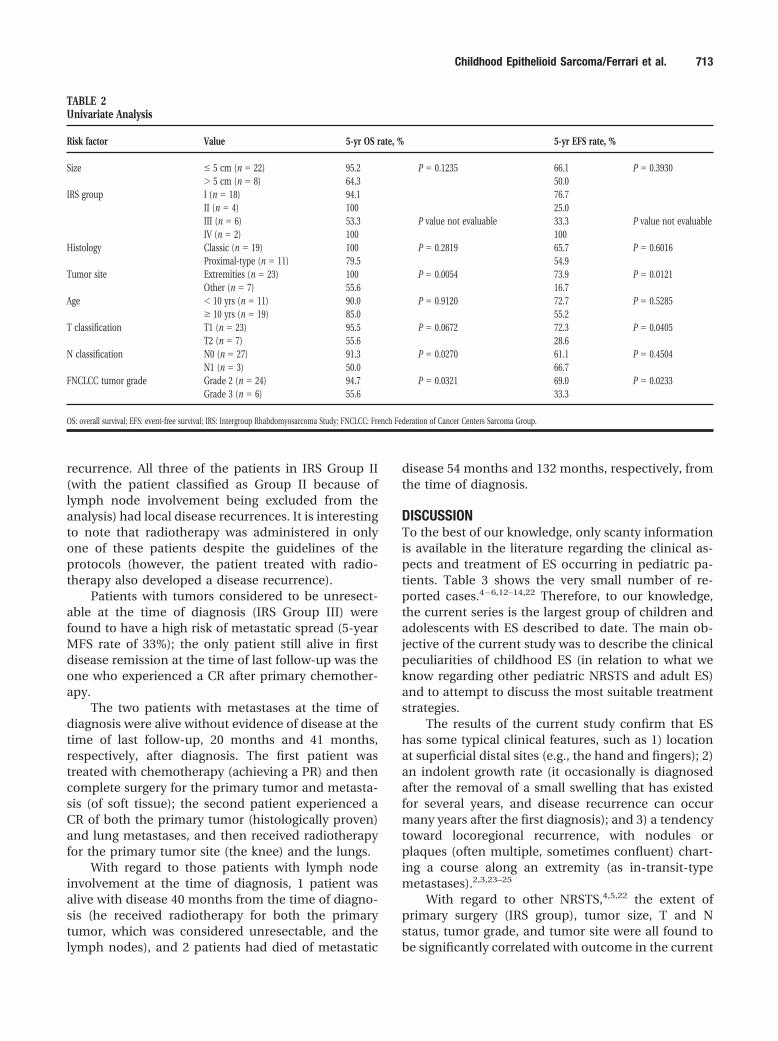
Table 2 shows the results of univariate analysis(considering the variables usually significant forNRSTS). The most significant factor found to influenceboth EFS and OS was the site of the tumor, with alocation in the extremities predicting a favorable out-come. Tumor size (considering the usual cutoff valueof 5 cm) appeared to be less significant. However,because the median tumor size in this series was 3 cm,the analysis also was performed considering a cutoffvalue of 3 cm; the 5-year EFS was reported to be 88.9%in patients with tumors measuring � 3 cm and 47.4%in those patients with tumors measuring � 3 cm. Thisdifference was statistically significant (P � 0.0140).
The proximal-type variant of ES was associatedwith unfavorable survival curves (P value was not sig-nificant): It is interesting to note that the combinationof proximal-type variant and high grade (FNCLCCGrade 3 disease) was found to be associated with apoor prognosis (four patients with a total of threecases of disease recurrence and two deaths).
With regard to the treatment administered as vari-ables, it is difficult to establish any significant corre-lation with outcome. As expected, initial completeresection with histologically free surgical margins wasassociated with a good EFS; among the 18 patients inIRS Group I (all of whom were treated with surgeryalone), 4 developed disease recurrence (all 4 locally,and 1 patient also had distant spread). Microscopicallyinfiltrated surgical margins were associated with local
712 CANCER February 1, 2006 / Volume 106 / Number 3
recurrence. All three of the patients in IRS Group II(with the patient classified as Group II because oflymph node involvement being excluded from theanalysis) had local disease recurrences. It is interestingto note that radiotherapy was administered in onlyone of these patients despite the guidelines of theprotocols (however, the patient treated with radio-therapy also developed a disease recurrence).
Patients with tumors considered to be unresect-able at the time of diagnosis (IRS Group III) werefound to have a high risk of metastatic spread (5-yearMFS rate of 33%); the only patient still alive in firstdisease remission at the time of last follow-up was theone who experienced a CR after primary chemother-apy.
The two patients with metastases at the time ofdiagnosis were alive without evidence of disease at thetime of last follow-up, 20 months and 41 months,respectively, after diagnosis. The first patient wastreated with chemotherapy (achieving a PR) and thencomplete surgery for the primary tumor and metasta-sis (of soft tissue); the second patient experienced aCR of both the primary tumor (histologically proven)and lung metastases, and then received radiotherapyfor the primary tumor site (the knee) and the lungs.
With regard to those patients with lymph nodeinvolvement at the time of diagnosis, 1 patient wasalive with disease 40 months from the time of diagno-sis (he received radiotherapy for both the primarytumor, which was considered unresectable, and thelymph nodes), and 2 patients had died of metastatic
disease 54 months and 132 months, respectively, fromthe time of diagnosis.
DISCUSSIONTo the best of our knowledge, only scanty informationis available in the literature regarding the clinical as-pects and treatment of ES occurring in pediatric pa-tients. Table 3 shows the very small number of re-ported cases.4 – 6,12–14,22 Therefore, to our knowledge,the current series is the largest group of children andadolescents with ES described to date. The main ob-jective of the current study was to describe the clinicalpeculiarities of childhood ES (in relation to what weknow regarding other pediatric NRSTS and adult ES)and to attempt to discuss the most suitable treatmentstrategies.
The results of the current study confirm that EShas some typical clinical features, such as 1) locationat superficial distal sites (e.g., the hand and fingers); 2)an indolent growth rate (it occasionally is diagnosedafter the removal of a small swelling that has existedfor several years, and disease recurrence can occurmany years after the first diagnosis); and 3) a tendencytoward locoregional recurrence, with nodules orplaques (often multiple, sometimes confluent) chart-ing a course along an extremity (as in-transit-typemetastases).2,3,23–25
With regard to other NRSTS,4,5,22 the extent ofprimary surgery (IRS group), tumor size, T and Nstatus, tumor grade, and tumor site were all found tobe significantly correlated with outcome in the current
TABLE 2Univariate Analysis
Risk factor Value 5-yr OS rate, % 5-yr EFS rate, %
Size � 5 cm (n � 22) 95.2 P � 0.1235 66.1 P � 0.3930� 5 cm (n � 8) 64.3 50.0
IRS group I (n � 18) 94.1 76.7II (n � 4) 100 25.0III (n � 6) 53.3 P value not evaluable 33.3 P value not evaluableIV (n � 2) 100 100
Histology Classic (n � 19) 100 P � 0.2819 65.7 P � 0.6016Proximal-type (n � 11) 79.5 54.9
Tumor site Extremities (n � 23) 100 P � 0.0054 73.9 P � 0.0121Other (n � 7) 55.6 16.7
Age � 10 yrs (n � 11) 90.0 P � 0.9120 72.7 P � 0.5285� 10 yrs (n � 19) 85.0 55.2
T classification T1 (n � 23) 95.5 P � 0.0672 72.3 P � 0.0405T2 (n � 7) 55.6 28.6
N classification N0 (n � 27) 91.3 P � 0.0270 61.1 P � 0.4504N1 (n � 3) 50.0 66.7
FNCLCC tumor grade Grade 2 (n � 24) 94.7 P � 0.0321 69.0 P � 0.0233Grade 3 (n � 6) 55.6 33.3
OS: overall survival; EFS: event-free survival; IRS: Intergroup Rhabdomyosarcoma Study; FNCLCC: French Federation of Cancer Centers Sarcoma Group.
Childhood Epithelioid Sarcoma/Ferrari et al. 713

series, although the small number of patients in eachsubgroup severely limited the statistical significance ofthe analysis. The tumor’s site of origin was found to beone of the most significant prognostic variables. ESarising in the extremities was found to have a morefavorable prognosis than ES originating in the trunkand head and neck region. When tentatively applied topediatric ES patients, the FNCLCC grading system ap-peared to be a statistically significant prognostic vari-able for both EFS and OS on univariate analysis.
A specific comment concerning the role of tumorsize in predicting outcome in ES patients may bewarranted. With regard to the group of NRSTS patientsas a whole, tumor size is one of the most significantprognostic factors, as confirmed by the recently re-ported series from the INT, in which the usual dimen-sion of 5 cm was considered as a cutoff value withwhich to compare patient subsets (the 5-year EFS ratewas 88% in patients with tumors measuring � 5 cmand 41% in those patients with tumors measuring � 5cm) and it is worth noting that the median tumordimension was 6 cm.4 Conversely, in the current seriesof ES patients, tumor size appeared to be less impor-tant in predicting outcome when the cutoff value of 5cm was considered, but it became significant if a cut-off of 3 cm was used (5-year EFS rate of 89% forpatients with tumors measuring � 3 cm and 47% forpatients with tumors measuring � 3 cm; P � 0.0140).The 3-cm latter cutoff appeared to be more suitable(and therefore significant), considering that the me-
dian size of ES tumors in the current series was 3 cm(ES lesions at onset are usually smaller in size thanother NRSTS).
The results of the current study suggest that theoverall outcome for children and adolescents with ESis comparable to that in patients with other pediatricNRSTS (the 5-year EFS rate was 64% for the 182 pa-tients at the INT).4 In other words, the ES histotypedoes not appear to have a worse outcome than otherhistotypes. Conversely, in the University of TexasM. D. Anderson Cancer Center study of a large seriesof 1225 adult patients with localized soft tissue sarco-mas (including 29 cases of ES [2%]), Zagars et al.reported that, together with malignant fibrous histio-cytoma and neurogenic sarcoma, the ES histotype wasone of the factors found to be predictive of local andmetastatic disease recurrence.26 The authors sug-gested that ES might be correlated with unfavorablesurvival rates because this histotype is “lymphoge-nous” (i.e., characterized by a tendency toward lymphnode spread).26 In fact, adult patients with ES havebeen recognized as demonstrating lymph node in-volvement more frequently than patients with othersoft tissue sarcomas.1,27–30 This tendency, describedlargely in adult ES patients, appears to be less evidentin pediatric patients; although de Vries et al.14 re-ported that all four ES patients in their study devel-oped lymph node metastases,14 none of the eight pa-tients in the St. Jude series13 were found to have lymphnode spread, and none of the three patients in the
TABLE 3Cases of Children and Adolescents with ES Reported in the Literature
Published series regardingchildhood ES No. of cases Tumor site Outcome
Schmidt and Harms, 19876 6 1 in the extremities5 at axial sitesa
5 alive with NED (at a follow-upof 6-14 mos)1 DOD
De Vries et al., 198914 4 4 in the extremities 2 alive with NED (after regionaldisease recurrence)2 DOD
Kodet et al. 199412 11 7 in the extremities4 at axial sites
6 alive with NED (at a follow-upof 0-7 yrs)1 AWD3 DOD
Gross et al., 199613 8 4 in the extremities4 at axial sites
6 alive with NED (at a follow-upof 12-126 mos)2 DOD
Major published series regarding NRSTSPratt et al., 199922 3 ES/81 NRSTS (4%)Spunt et al., 19995 6 ES/121 NRSTS (5%)Ferrari et al., 20054 14 ES/179 NRSTS (8%)
ES: epithelioid sarcoma; NED: no evidence of disease; DOD: dead of disease; AWD: alive with disease; NRSTS: nonrhabdomyosarcoma soft tissue sarcomas.a Axial extremities are considered to be the head and neck, trunk, and pelvis.
714 CANCER February 1, 2006 / Volume 106 / Number 3

same study who underwent lymph node dissection forstaging purposes were found to have histologicallypositive lymph nodes.13 In the current series, 3 of 30patients had lymph node involvement at the time ofdiagnosis, a percentage (10%) that is lower than thatnoted in adult series (up to 45%),1,27–30 but higher thanhas been reported for pediatric NRSTS patients as awhole (approximately 5%).4,5,31 It is possible that therate of lymph node involvement might actually behigher than we found because only two patients weresampled. However, other findings in the current seriesappear to suggest a weaker propensity for lymph nodespread in pediatric ES patients compared with adults.In fact, surgical lymph node staging was performed intwo cases, both of which were negative, and only onepatient developed disease recurrence with lymphnode metastases. It is difficult to speculate as to whychildren and adolescents with ES appear to be lesslikely to develop lymph node involvement comparedwith adults. Most likely, a combination of differentfactors (e.g., patient age, treatment, and tumor biol-ogy) might be hypothesized.
A further issue for debate concerns the recentlydescribed proximal-type ES entity. This variant ischaracterized by large, epithelioid cells; marked nu-clear atypia; frequently rhabdoid features; a lack of agranulomatous-like pattern; and a preference forproximal and axial regions (e.g., the pelvis and trunk)rather than the usual extremities.8 To our knowledge,it is not yet clear whether proximal-type ES is a differ-ent variant of ES (its immunophenotypic profile isquite similar to that of classic ES)8,11,32 or whether itshould be considered to be a form of extrarenal rhab-doid tumor.9 Recently, studies have appeared thathighlight a difference in the mechanisms of the alter-ation of the SMARCB1/INI1 gene between proximal-type ES and rhabdoid tumor because deletions werefound in the former and inactivating mutations in thelatter.10 This different genetic background, coupledwith a morphologic heterogeneity described in proxi-mal-type ES compared with rhabdoid tumor, suggeststhat they are two distinct entities.10 This issue goesbeyond the objective of the current study, but thelimited information available in the adult literaturesuggests a worse outcome for patients with this vari-ant.8 –11 It is difficult to state conclusively whether thisunfavorable prognosis has to do with different bio-logic features, or simply with the well known classicprognostic factors (proximal location in particular, butalso tumor size and depth) that affect this variant. Toour knowledge, the current study is the first to de-scribe the clinical features and outcome of proximalES diagnosed in childhood. Far from being able tospeculate on the biologic and clinical behavior of
proximal-type ES in pediatric patients, we can onlydescribe the clinical findings and outcome in the cur-rent series. Among the 11 patients with histologic fea-tures that were similar to those described as beingpeculiar to this variant, 6 tumors were in fact locatedat proximal sites (the trunk, head and neck, and ab-domen). It should be noted that the characteristicsthat usually identify the clinically more aggressive sar-comas (i.e., high grade, large size, unresectability, andlocally advanced disease) were represented morestrongly in the 11 patients with proximal-type ES thanin the other 19 patients with classic ES. Survival anal-ysis demonstrated a worse outcome for the patientswith proximal-type ES (5-year EFS and OS rates of54.9% and 79.5%, respectively, vs. 65.7% and 100%,respectively), although the survival rates were found tocorrelate more with the proximal tumor site than theproximal-type histotype. It also must be said thatthese survival rates are distinctly higher than thosereported for patients with extracranial rhabdoid tu-mors,33 suggesting that the clinical behavior of proxi-mal-type ES (at least in the current series of pediatricpatients) differs considerably from that of rhabdoidtumors.
The current study describes the clinical behaviorafter therapy of ES diagnosed in pediatric patients.With regard to the optimal treatment of ES, the smallnumber of patients in the current study limits thepossibility of reaching definite conclusions. Surgicalresection clearly remains the mainstay of treatment,with clinicians striving to obtain histologically dis-ease-free margins (primary reexcision was found to behelpful in 11 patients in the current study). Becausedistal sites often are affected, and the fingers in par-ticular, finger amputation should be considered as anoption, especially after a first local disease recurrence.Little could be inferred from the current series withregard to the role of adjuvant therapies. Patients whoundergo marginal surgical resection, with microscop-ically infiltrated margins, are at a high risk of localdisease recurrence and should, in principle, be givenradiotherapy, but the data from the current study didnot allow us to establish a clear role for radiotherapy.The possible benefit of chemotherapy remains to bedetermined, but it is worth emphasizing that threepatients in the current study (of seven patients withmeasurable disease) responded well to chemotherapy(two of whom achieved CRs), and all had receivedchemotherapy regimens that included ifosfamide andanthracyclines.
ES is a rare subtype of soft tissue sarcoma withpeculiar features, and to our knowledge little is knownregarding its clinical behavior and management whenit occurs in children. Although the large group of soft
Childhood Epithelioid Sarcoma/Ferrari et al. 715

tissue sarcomas clearly includes an assortment of dif-ferent diseases, the rarity of each histotype preventsthe performance of clinical trials on a single tumortype; therefore, soft tissue sarcomas usually are ana-lyzed as a group. The new European protocol forNRSTS (developed by the new European pediatric SoftTissue Sarcoma Study Group [EpSSG]) includes ESwithin the group of “adult-type” soft tissue sarcomas(together with adult-type fibrosarcoma, malignant pe-ripheral nerve sheath tumor, leiomyosarcoma, clearcell sarcoma of soft tissue, liposarcoma, and others).34
In the EpSSG NRSTS protocol, chemotherapy with thecombination of ifosfamide and doxorubicin, followedby a local treatment strategy (surgery and/or radio-therapy), is recommended in the case of tumors con-sidered to be unresectable at the time of diagnosis.Adjuvant therapies are recommended according to therisk of local and distant disease recurrence, as sug-gested by the presence of residual disease after initialsurgery, tumor size, and tumor grade. ES patients con-sequently will receive adjuvant chemotherapy in thecase of FNCLCC Grade 3 histology and a tumor sizemeasuring �than 5 cm, whereas postoperative radio-therapy will be given to patients with completely re-sected ES measuring � 5 cm, and to patients whounderwent an initial marginal resection.33 The needfor surgical lymph nodes assessment remains an openissue; because experience in pediatric patients did notentirely confirm the high propensity toward lymphnode spread suggested by the adult literature, lymphnode biopsy is not required for ES patients in theEpSSG protocol.
REFERENCES1. Enzinger FM. Epithelioid sarcoma: a sarcoma simulating a
granuloma or carcinoma. Cancer. 1970;26:1029 –1041.2. Chase DR, Enzinger FM. Epithelioid sarcoma: diagnosis,
prognostic indicators, and treatment. Am J Surg Pathol.1985;9:241–261.
3. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: theclinicopathological complexities of this rare soft tissue sar-coma. Ann Surg Oncol. 2000;7:218 –225.
4. Ferrari A, Casanova M, Meazza C, et al. Adult-type soft tissuesarcomas in pediatric age: experience at the Istituto Nazio-nale Tumori in Milan. J Clin Oncol. 2005;23:4021– 4030.
5. Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors forchildren and adolescents with surgically resected nonrhab-domyosarcoma soft tissue sarcoma: an analysis of 121 pa-tients treated at St Jude Children’s Research Hospital. J ClinOncol. 1999;17:3697–3705.
6. Schmidt D, Harms D. Epithelioid sarcoma in children andadolescents: an immunohistochemical study. Virchows ArchA Pathol Anat Histopathol. 1987;410:423– 431.
7. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithe-lioid sarcoma: an immunohistochemical analysis of 11 clas-sical and variant cases and a discussion of the differentialdiagnosis. Hum Pathol. 1999;30:934 –942.
8. Guillou L, Wadden C, Coindre JM. “Proximal-type” epithe-lioid sarcoma: a distinctive aggressive neoplasm showingrhabdoid features. Clinicopathologic, immunohistochemi-cal, and ultrastructural study of a series. Am J Surg Pathol.1997;21:130 –146.:
9. Hasegawa T, Matsuno T, Shimoda T, et al. Proximal-typeepithelioid sarcoma: a clinicopathologic study of 20 cases.Mod Pathol. 2001;14:655– 663.
10. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1tumor suppressor gene is frequently inactivated in epithe-lioid sarcomas. Cancer Res. 2005;65:4012– 4019.
11. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecularcytogenetic characterization of proximal-type epithelioidsarcoma. Genes Chromosomes Cancer. 2004;41:283–290.
12. Kodet R, Smelhaus V, Newton WA Jr., et al. Epithelioidsarcoma in childhood: an immunohistochemical, electronmicroscopic, and clinicopathologic study of 11 cases under15 years of age and review of the literature. Pediatr Pathol.1994;14:433– 451.
13. Gross E, Rao NB, Pappo A, et al. Epithelioid sarcoma inchildren. J Pediatr Surg. 1996;31:1663–1665.
14. de Vries J, Hoekstra HJ, Oosterhuis JW, et al. Epithelioidsarcoma in children and adolescents: a report of four cases.J Pediatr Surg. 1989;24:186 –188.
15. [authors]. Tumours of soft tissue and bone. In: FletcherCDM, Unni KK, Mertens F, editors.World Health Organiza-tion classification of tumours. Pathology and genetics. Lyon,France: IARC Press, 2002.
16. Trojani M, Contesso G, Coindre JM, et al. Soft tissue sarco-mas of adults: study of pathological prognostic variables anddefinitions of a histopathological grading system. Int J Can-cer. 1984;33:37– 42.
17. Harmer MH. TNM classification of pediatric tumors. Ge-neva: International Union Against Cancer, 1982:23–28.
18. Maurer HM, Beltangady M, Gehan EA, et al. The IntergroupRhabdomyosarcoma Study I: a final report. Cancer. 1988;61:209 –220.
19. Enneking WF, Spanier SS, Goodman MD. A system for thesurgical staging of musculoskeletal sarcoma. Clin OrthopRelat Res. 1990;153:106 –120.
20. Kaplan EL, Meier P. Non-parametric estimation from in-complete observation. J Am Stat Assoc. 1958;53:457– 481.
21. Conover WJ. Practical nonparametric statistics. New York:Wiley, 1980153–169.:
22. Pratt CB, Pappo AS, Gieser P, et al. Role of adjuvantchemotherapy in the treatment of surgically resected pe-diatric nonrhabdomyosarcomatous soft tissue sarcomas:a Pediatric Oncology Group Study. J Clin Oncol. 1999;17:1219 –1226.
23. Bos GD, Pritchard DJ, Reiman HM. Epithelioid sarcoma: ananalysis of fifty-one cases. J Bone Joint Surg Am. 1988;70:862– 870.
24. Halling AC, Wollan PC, Pritchard DJ, et al. Epithelioid sar-coma: a clinicopathologic review of 55 cases. Mayo ClinProc. 1996;71:636 – 642.
25. Callister MD, Ballo MT, Pisters PW, et al. Epithelioid sar-coma: results of conservative surgery and radiotherapy. IntJ Radiat Oncol Biol Phys. 2001;51:384 –391.
26. Zagars GK, Ballo MT, Pisters PWT, et al. Prognostic factorsfor patients with localized soft-tissue sarcoma treated withconservative surgery and radiation therapy: an analysis of1225 patients. Cancer. 2003;97:2530 –2543.
716 CANCER February 1, 2006 / Volume 106 / Number 3

27. Prat J, Woodruff JM, Marcove RC. Epithelioid sarcoma: ananalysis of 22 cases indicating the prognostic significance ofvascular invasion and regional lymph node metastasis. Can-cer. 1978;41:1472–1478.
28. Mazeron JJ, Suit HD. Lymph nodes as sites of metasta-ses from sarcomas of soft tissue. Cancer. 1987;60:1800 –1808.
29. Riad S, Griffin AM, Liberman B, et al. Lymph node metas-tasis in soft tissue sarcoma in an extremity. Clin OrthopRelat Res. 2004;426:129 –134.
30. Ross HM, Lewis JJ, Woodruff JM, Brennan MF. Epithelioidsarcoma: clinical behaviour and prognostic factors of sur-vival. Ann Surg Oncol. 1997;4:491– 495.
31. Spunt SL, Ashley Hill D, Motosue AM, et al. Clinical features
and outcome of initially unresected nonmetastatic pediatricnonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol.2002;20:3225–3235.
32. Rakheja D, Wilson KS, Meehan J, et al. “Proximal-type” andclassic epithelioid sarcomas represent a clinicopathologiccontinuum: a case report. Pediatr Dev Pathol. 2005;8:105–114.
33. Kodet R, Newton WA Jr., Sachs N, et al. Rhabdoid tumors ofsoft tissues: a clinicopathologic study of 26 cases enrolled onthe Intergroup Rhabdomyosarcoma Study. Hum Pathol.1991;22:674 – 684.
34. Ferrari A, Casanova M. New concepts for the treatment ofpediatric non-rhabdomyosarcoma soft tissue sarcomas. Ex-pert Rev Anticancer Ther. 2005;5:307–318.
Childhood Epithelioid Sarcoma/Ferrari et al. 717
Copyright © 2022 FDOKUMEN




















