
Constitutive activation of AMP-activated protein kinase (AMPK) propel mitochondrial biogenesis
Upload
independentCategory
view
7download
0
Yu-Ting Xuan, Yiru Guo, Yanqing Zhu, Ou-Li Wang, Gregg Rokosh and Roberto BollipSer-Signal Transducers and Activators of Transcription1/3 Pathway−Kinase
p44/42 Mitogen-Activated Protein−εPreconditioning by Activating the Protein Kinase CEndothelial Nitric Oxide Synthase Plays an Obligatory Role in the Late Phase of Ischemic
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2007 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation doi: 10.1161/CIRCULATIONAHA.107.6894712007;116:535-544; originally published online July 2, 2007;Circulation.
http://circ.ahajournals.org/content/116/5/535World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2007/06/29/CIRCULATIONAHA.107.689471.DC2.html http://circ.ahajournals.org/content/suppl/2007/06/29/CIRCULATIONAHA.107.689471.DC1.html
Data Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialCirculationin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from

Endothelial Nitric Oxide Synthase Plays an Obligatory Rolein the Late Phase of Ischemic Preconditioning by Activating
the Protein Kinase C�–p44/42 Mitogen-Activated ProteinKinase–pSer-Signal Transducers and Activators of
Transcription1/3 PathwayYu-Ting Xuan, PhD; Yiru Guo, MD; Yanqing Zhu, MD; Ou-Li Wang, MD;
Gregg Rokosh, PhD; Roberto Bolli, MD
Background—The role of endothelial nitric oxide synthase (eNOS) in ischemic preconditioning (PC) and cardioprotectionis poorly understood. We addressed this issue using a genetic, rather than pharmacological, approach.
Methods and Results—In the nonpreconditioned state, eNOS�/� mice exhibited infarct sizes similar to those of wild-typemice. A sequence of six 4-minute coronary occlusion/4-minute reperfusion cycles (ischemic PC) induced late PC inwild-type mice; genetic deletion of eNOS abrogated the cardioprotection induced by late PC. In wild-type mice,ischemic PC induced membranous translocation of protein kinase C (PKC)� and an increase in pSer-MEK-1/2 andpTyr-p44/42 mitogen-activated protein kinase, nuclear pSer-signal transducers and activators of transcription (STAT)1and pSer-STAT3, and nuclear STAT1/3 DNA binding activity, followed by upregulation of cyclooxygenase-2 proteinand activity 24 hours later. All of these changes were abrogated in eNOS�/� mice. The NO donor diethylenetriamine/NOrecapitulated the effects of ischemic PC.
Conclusions—In contrast to previous reports, we found that basal eNOS activity does not modulate infarct size in thenonpreconditioned state. However, eNOS is obligatorily required for the development of the cardioprotective effects oflate PC and acts as the trigger of this process by activating the PKC�-MEK-1/2-p44/42 mitogen-activated protein kinasepathway, leading to Ser-727 phosphorylation of STAT1 and STAT3 and consequent upregulation of STAT-dependentgenes such as cyclooxygenase-2. The effects of eNOS-derived NO are reproduced by exogenous NO (NO donors),implying that nitrates can upregulate cardiac cyclooxygenase-2. (Circulation. 2007;116:535-544.)
Key Words: ischemia � myocardial infarction � occlusion � stress
The protective effects of ischemic preconditioning (PC)occur in 2 distinct phases: an early phase that develops
rapidly after the stimulus but dissipates within 2 to 3 hoursand a late phase that becomes apparent 12 to 24 hours laterand persists for �72 hours.1–12 Although nitric oxide (NO) isknown to be involved in both phases,3,4,7,8,13,14 the role ofendothelial NO synthase (eNOS; the major constitutivesource of NO in the heart) in the late phase of ischemic PCremains poorly understood. Specifically, 2 fundamental is-sues are unresolved.
Clinical Perspective p 544
The first issue is whether eNOS is responsible for trigger-ing this delayed adaptation of the heart to stress. The proposalput forth in previous investigations8,13,15 that eNOS-derived
NO triggers the late phase of ischemic PC is based on thefinding that pretreatment with L-nitro-arginine, a nonselectiveNOS inhibitor, blocks late PC8,15 and conversely that expo-sure to exogenous NO in the absence of ischemia induces adelayed protective effect.13 These studies, however, wereperformed using pharmacological inhibitors of NOS ratherthan genetic ablation of the protein. Furthermore, the use ofL-nitro-arginine does not enable one to identify the source ofthe increased NO formation that triggers late PC. Constitu-tively expressed NOS includes both eNOS and neuronalNOS; in principle, either or both of these isoforms couldcontribute to the burst of NO generation that triggers thedevelopment of late PC.
The second unresolved issue pertains to the mechanism bywhich eNOS triggers late PC. We have previously demon-
Received January 9, 2007; accepted May 29, 2007.From the Institute of Molecular Cardiology, University of Louisville, Louisville, Ky.The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.
107.689471/DC1.Correspondence to Roberto Bolli, MD, Division of Cardiology, University of Louisville, Louisville, KY 40292 (e-mail [email protected]); or
Yu-Ting Xuan, PhD, Division of Cardiology, University of Louisville, Louisville, KY 40292 (e-mail [email protected]).© 2007 American Heart Association, Inc.
Circulation is available at http://www.circulationaha.org DOI: 10.1161/CIRCULATIONAHA.107.689471
535 by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from

strated that the PC ischemia induces activation of Janustyrosine kinase-1 and -2, followed by tyrosine phosphoryla-tion and activation of signal transducers and activators oftranscription (STAT)1 and 3, which results in transactivationof STAT-responsive genes such as cyclooxygenase-2 (COX-2).16,17 Full transcriptional activation of STATs, however,requires not only tyrosine phosphorylation (Tyr-701 inSTAT1 and Tyr-705 in STAT3) but also serine phosphory-lation (Ser-727 in both STAT1 and STAT3).18–21 We haverecently found that Ser-727 phosphorylation of STAT1/3 ismodulated by the protein kinase C (PKC)�-Raf-1-MEK-1/2-p44/42 mitogen-activated protein kinase (MAPK) signalingcascade and is critical for the upregulation of COX-2 duringlate PC.17 The exact position of eNOS in this signalingscheme remains unclear. Because the promoter of the mouseCOX-2 gene contains the interferon-� activation site consen-sus sequence for the binding of STATs16,22,23 and becauseischemic PC is associated with increased constitutively ex-pressed NOS activity and NO generation,14 as well asactivation of PKC�6,9, STAT1, and STAT3,16,17 we hypothe-sized that rapid activation of eNOS by the initial PC ischemiais the early signal that generates NO, which in turn activatesa downstream PKC�-Raf-1-MEK-1/2-p44/42 MAPK-pSer-STAT1/3 pathway, leading to induction of COX-2 proteinand cardioprotection. However, it is unknown whether eNOSis necessary for serine phosphorylation of STATs and, if so,whether eNOS is proximal to the aforementioned pathway.
The objective of the present study was to determine therole of eNOS in ischemic PC using a genetic, rather than apharmacological, approach. The following specific questionswere addressed in a well-established murine model of myo-cardial infarction: Is eNOS necessary for the late phase ofischemic PC? If so, does eNOS trigger late PC through theactivation of the PKC�-Raf-1-MEK-1/2-p44/42 MAPK path-way, leading to Ser-727 phosphorylation of STAT1/3 duringthe initial PC ischemia and to subsequent upregulation ofCOX-2? Finally, can exogenous NO (NO donors) mimiceNOS-derived NO; ie, can it activate the PKC�-Raf-1-MEK-1/2-p44/42 MAPK-pSer-STAT1/3 pathway and induceCOX-2?
MethodsWe used eNOS�/� mice with deletion of the calmodulin bindingdomain.24 Wild-type (WT) control mice were C57BL/6 (The JacksonLaboratory, Bar Harbor, Me). Spontaneously hypertensive mice(high blood pressure mice [BPH]) were obtained from The JacksonLaboratory. All mice were maintained in sterile microisolator cagesunder pathogen-free conditions. The genotype of the eNOS�/� strainwas verified by polymerase chain reaction, as previously described,using DNA prepared from tail samples taken at the end of theexperiments.17 A total of 155 mice were used for the present study.
The murine model of late PC has previously been described indetail.16,25 Briefly, a nontraumatic balloon occluder was implantedaround the middle left anterior descending coronary artery inpentobarbital-anesthetized mice. To prevent hypotension, bloodfrom a donor mouse was given during surgery. Rectal temperaturewas maintained close to 37°C throughout the experiment.
Ischemic PC was elicited by a sequence of six 4-minute coronaryocclusion/4-minute reperfusion (O/R) cycles. Control mice under-went 1 hour of open-chest state (sham control), and sutures wereplaced as in the PC groups. Mice used for studies of myocardialinfarction underwent a 30-minute coronary occlusion, followed by
24 hours of reperfusion. At the conclusion of the study, theoccluded/reperfused vascular bed and the infarct were identified bypostmortem perfusion of the heart with phthalo blue dye andtriphenyltetrazolium chloride, respectively. Infarct size was calcu-lated as a percentage of the region at risk.16,25
The investigation consisted of 3 successive phases (A, B, and C).The objective of phase A was to determine whether eNOS isnecessary for the development of the cardioprotective effects of latePC. Mice were assigned to 6 groups (Figure 1A). On day 1, groups1 (WT acute myocardial infarction [AMI]) and 3 (eNOS�/� AMI)underwent 1 hour of open-chest state without ischemia, whereasgroups 2 (WT late PC [LPC]) and 4 (eNOS�/� LPC) underwent asequence of six 4-minute coronary O/R cycles. On day 2, all miceunderwent a 30-minute coronary occlusion. Groups 5 (BPH AMI)and 6 (BPH LPC) underwent a 30-minute occlusion on day 2 without(group 5) or with (group 6) six 4-minute coronary O/R cycles on day1. The purpose of studying these spontaneously hypertensive mice26
was to determine whether high blood pressure, in itself, could affectinfarct size.
The objective of phase B was to determine whether eNOS isrequired for the activation of PKC� and the MEK1/2-p44/42 MAPKpathway, the serine phosphorylation of STAT1/3, and the upregula-tion of COX-2 during late PC. Mice were assigned to 12 groups(Figure 1B). Groups 8 (WT PC–5 minutes), 10 (eNOS�/� PC–5minutes), 12 (WT PC–30 minutes), 14 (eNOS�/� PC–30 minutes), 16(WT PC–24 hours), and 18 (eNOS�/� PC–24 hours) underwent six4-minute coronary O/R cycles, whereas groups 7 (WT control–5minutes), 9 (eNOS�/� control–5 minutes), 11 (WT control–30 min-utes), 13 (eNOS�/� control–30 minutes), 15 (WT control–24 hours),and 17 (eNOS�/� control–24 hours) underwent 1 hour of open-cheststate without occlusion. Mice were euthanized at 5 minutes (groups7 through 10), 30 minutes (groups 11 through 14), or 24 hours(groups 15 through 18) after the open-chest state or the lastreperfusion. Myocardial samples were removed rapidly from theischemic-reperfused region or the left ventricle and frozen in liquidnitrogen until used.
The objective of phase C was to elucidate (by using the NO donordiethylenetriamine [DETA]/NO) whether exogenous NO is suffi-cient to activate PKC�, MEK-1/2, and p44/42 MAPKs and toincrease serine phosphorylation of STAT1/3 and expression ofCOX-2 protein. Mice were assigned to 6 groups (Figure 1C). Groups20, 22, and 24 received the NO donor DETA/NO (5 mg/kg IV every20 minutes 4 times), a dose that has previously been shown to elicita powerful late PC effect in mice,27 whereas control groups 19, 21,and 23 received the same volume of PBS. Mice were euthanized 5minutes (groups 19 and 20), 30 minutes (group 21 and 22), or 24hours (groups 23 and 24) after the end of the infusion of DETA/NOor PBS. Myocardial samples were removed rapidly from the leftventricle and frozen in liquid nitrogen until used.
Cytosolic, membranous, and nuclear fractions were prepared aspreviously described.16,17 Western immunoblotting analysis wasperformed using standard techniques16,17; the antibodies are speci-fied in Table I of the online Data Supplement. Equal loading wasconfirmed by staining with Ponceau-S.16,17 The DNA bindingactivity of STAT1/3 was measured with electrophoretic mobilityshift assays.16,17 A synthetic double-stranded probe with the se-quence 5�-GATCAGCTTCATTTCCCGTAAATCCCTA-3� (Gibco,Carlsbad, Calif) was end-labeled using [�-32P]ATP (3000 Ci/mmol,Amersham, Uppsala, Sweden) and T4 polynucleotide kinase. Thisoligonucleotide has the consensus sequence for interferon-� activa-tion site elements (italics).16,17 Prostaglandins (PGs) were extractedusing ODS-silica reverse-phase columns.11 The myocardial contentof PGE2, PGF2�, and 6-keto-PGF1� was determined by with enzymeimmunoassay kits (PGE2 and PGF2� enzyme immunoassay kits fromCayman Chemical, Ann Arbor, Mich; 6-keto-PGF1a kit from Amer-sham Life Science, Buckinghamshire, UK) and expressed as pico-grams per milligram of protein.11
Data are reported as mean�SEM, and sample sizes are stated inthe figures. Statistical comparisons were performed with 1-wayANOVA, followed by unpaired Student t test with Bonferronicorrection as appropriate. In all Western analyses, the content of the
536 Circulation July 31, 2007
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from

GROUP I(WT AMI)
GROUP II(WT LPC)
Day 2
Day 1 REPERFUSION24h
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
GROUP III(eNOS-/- AMI)
GROUP IV(eNOS-/- LPC)
Day 124h
Day 2
Day 2
Day 1 REPERFUSION24h
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Day 124h
Day 2 30 min occlusion REPERFUSION24h
Infarct size
30 min occlusion REPERFUSION24h
Infarct size
30 min occlusion REPERFUSION24h
Infarct size
30 min occlusion REPERFUSION24h
Infarct size
GROUP V(BPH AMI)
GROUP VI(BPH LPC)
Day 2
Day 1 REPERFUSION24h
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
30 min occlusion REPERFUSION24h
Infarct size
Day 124h
Day 2 30 min occlusion REPERFUSION24h
Infarct size
REPERFUSION1-h OPEN CHEST
REPERFUSION1-h OPEN CHEST
A
GROUP XV(WT control-24 h)
GROUP XVI(WT PC-24 h)
GROUP XVII(eNOS-/- control-24 h)
GROUP XVIII(eNOS-/- PC-24 h)
GROUP XI(WT control-30’)
GROUP XII(WT PC-30’)
GROUP XIII(eNOS-/- control-30’)
GROUP XIV(eNOS-/- PC-30’)
Day 1 REPERFUSION30’
1-h OPEN CHEST Tissue for MEK, p44/42, STAT1/3, DNA binding
Day 1 REPERFUSION
30’
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Day 1 REPERFUSION30’
1-h OPEN CHEST
Day 1 REPERFUSION
30’
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Tissue for MEK, p44/42, STAT1/3, DNA binding
Tissue for MEK, p44/42, STAT1/3, DNA binding
Tissue for MEK, p44/42, STAT1/3, DNA binding
Day 1 REPERFUSION24h
1-h OPEN CHEST
Day 2 Tissue for COX-2 protein and activity (prostaglandins)
Day 2
Day 1 REPERFUSION
24hTissue for COX-2 protein and activity (prostaglandins)
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Day 1 REPERFUSION24h
1-h OPEN CHEST
Day 2 Tissue for COX-2 protein and activity (prostaglandins)
Day 2
Day 1 REPERFUSION
24hTissue for COX-2 protein and activity (prostaglandins)
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
GROUP VII(WT control-5’)
GROUP VIII(WT PC-5’)
GROUP IX(eNOS-/- control-5’)
GROUP X(eNOS-/- PC-5’)
Day 1 REPERFUSION5’
1-h OPEN CHEST Tissue for PKCε translocation
Day 1 REPERFUSION
5’
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Day 1 REPERFUSION5’
1-h OPEN CHEST
Day 1 REPERFUSION5’
R1 R2 R3 R4 R5O2 O3 O4 O5 O6O1
Tissue for PKCε translocation
Tissue for PKCε translocation
Tissue for PKCε translocation
B
Figure 1. A, Experimental protocol for phase A. B, Experimental protocol for phase B. C, Experimental protocol for phase C.
Xuan et al eNOS and Preconditioning 537
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
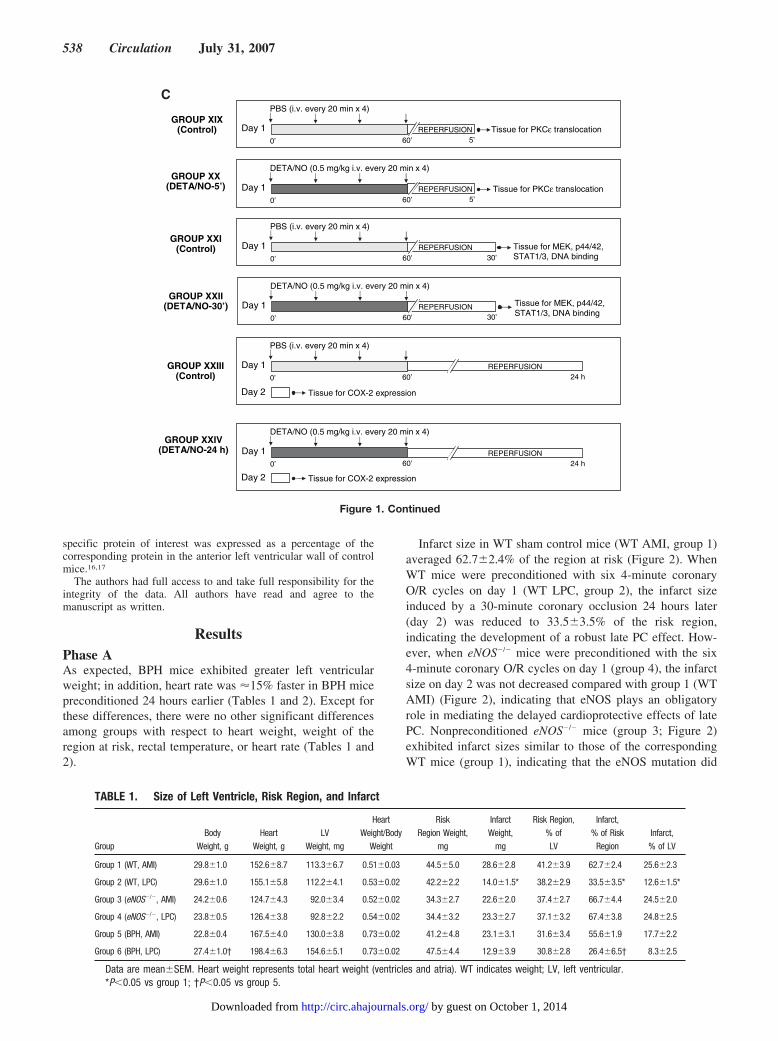
specific protein of interest was expressed as a percentage of thecorresponding protein in the anterior left ventricular wall of controlmice.16,17
The authors had full access to and take full responsibility for theintegrity of the data. All authors have read and agree to themanuscript as written.
ResultsPhase AAs expected, BPH mice exhibited greater left ventricularweight; in addition, heart rate was �15% faster in BPH micepreconditioned 24 hours earlier (Tables 1 and 2). Except forthese differences, there were no other significant differencesamong groups with respect to heart weight, weight of theregion at risk, rectal temperature, or heart rate (Tables 1 and2).
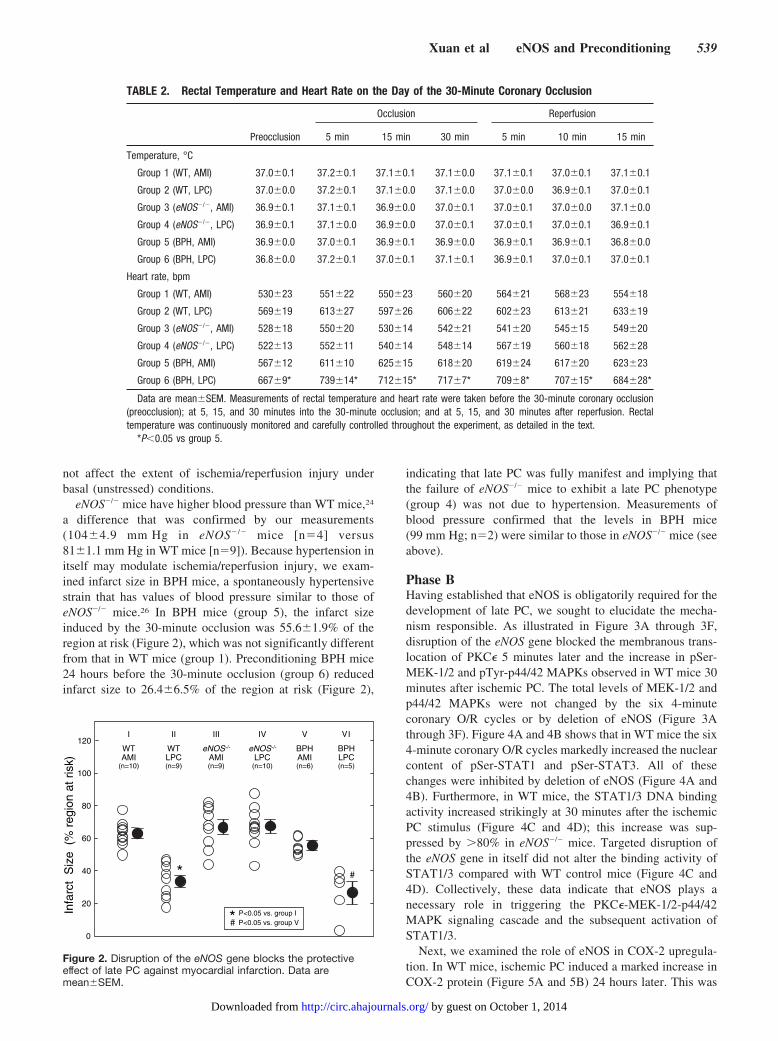
Infarct size in WT sham control mice (WT AMI, group 1)averaged 62.7�2.4% of the region at risk (Figure 2). WhenWT mice were preconditioned with six 4-minute coronaryO/R cycles on day 1 (WT LPC, group 2), the infarct sizeinduced by a 30-minute coronary occlusion 24 hours later(day 2) was reduced to 33.5�3.5% of the risk region,indicating the development of a robust late PC effect. How-ever, when eNOS�/� mice were preconditioned with the six4-minute coronary O/R cycles on day 1 (group 4), the infarctsize on day 2 was not decreased compared with group 1 (WTAMI) (Figure 2), indicating that eNOS plays an obligatoryrole in mediating the delayed cardioprotective effects of latePC. Nonpreconditioned eNOS�/� mice (group 3; Figure 2)exhibited infarct sizes similar to those of the correspondingWT mice (group 1), indicating that the eNOS mutation did
GROUP XIX(Control) Day 1 REPERFUSION
PBS (i.v. every 20 min x 4)
5’Tissue for PKCε translocation
0’ 60’
GROUP XX(DETA/NO-5’) Day 1 REPERFUSION
DETA/NO (0.5 mg/kg i.v. every 20 min x 4)
5’0’ 60’Tissue for PKCε translocation
GROUP XXI(Control) Day 1 REPERFUSION
PBS (i.v. every 20 min x 4)
30’
Tissue for MEK, p44/42, STAT1/3, DNA binding0’ 60’
GROUP XXII(DETA/NO-30’) Day 1 REPERFUSION
DETA/NO (0.5 mg/kg i.v. every 20 min x 4)
30’0’ 60’
Tissue for MEK, p44/42, STAT1/3, DNA binding
GROUP XXIII(Control)
Day 1
PBS (i.v. every 20 min x 4)
REPERFUSION24 h0’ 60’
GROUP XXIV(DETA/NO-24 h) Day 1
DETA/NO (0.5 mg/kg i.v. every 20 min x 4)
0’ 60’
Day 2 Tissue for COX-2 expression
REPERFUSION24 h
Day 2 Tissue for COX-2 expression
C
Figure 1. Continued
TABLE 1. Size of Left Ventricle, Risk Region, and Infarct
GroupBody
Weight, gHeart
Weight, gLV
Weight, mg
HeartWeight/Body
Weight
RiskRegion Weight,
mg
InfarctWeight,
mg
Risk Region,% ofLV
Infarct,% of Risk
RegionInfarct,% of LV
Group 1 (WT, AMI) 29.8�1.0 152.6�8.7 113.3�6.7 0.51�0.03 44.5�5.0 28.6�2.8 41.2�3.9 62.7�2.4 25.6�2.3
Group 2 (WT, LPC) 29.6�1.0 155.1�5.8 112.2�4.1 0.53�0.02 42.2�2.2 14.0�1.5* 38.2�2.9 33.5�3.5* 12.6�1.5*
Group 3 (eNOS�/�, AMI) 24.2�0.6 124.7�4.3 92.0�3.4 0.52�0.02 34.3�2.7 22.6�2.0 37.4�2.7 66.7�4.4 24.5�2.0
Group 4 (eNOS�/�, LPC) 23.8�0.5 126.4�3.8 92.8�2.2 0.54�0.02 34.4�3.2 23.3�2.7 37.1�3.2 67.4�3.8 24.8�2.5
Group 5 (BPH, AMI) 22.8�0.4 167.5�4.0 130.0�3.8 0.73�0.02 41.2�4.8 23.1�3.1 31.6�3.4 55.6�1.9 17.7�2.2
Group 6 (BPH, LPC) 27.4�1.0† 198.4�6.3 154.6�5.1 0.73�0.02 47.5�4.4 12.9�3.9 30.8�2.8 26.4�6.5† 8.3�2.5
Data are mean�SEM. Heart weight represents total heart weight (ventricles and atria). WT indicates weight; LV, left ventricular.*P�0.05 vs group 1; †P�0.05 vs group 5.
538 Circulation July 31, 2007
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
not affect the extent of ischemia/reperfusion injury underbasal (unstressed) conditions.
eNOS�/� mice have higher blood pressure than WT mice,24
a difference that was confirmed by our measurements(104�4.9 mm Hg in eNOS� /� mice [n�4] versus81�1.1 mm Hg in WT mice [n�9]). Because hypertension initself may modulate ischemia/reperfusion injury, we exam-ined infarct size in BPH mice, a spontaneously hypertensivestrain that has values of blood pressure similar to those ofeNOS�/� mice.26 In BPH mice (group 5), the infarct sizeinduced by the 30-minute occlusion was 55.6�1.9% of theregion at risk (Figure 2), which was not significantly differentfrom that in WT mice (group 1). Preconditioning BPH mice24 hours before the 30-minute occlusion (group 6) reducedinfarct size to 26.4�6.5% of the region at risk (Figure 2),
indicating that late PC was fully manifest and implying thatthe failure of eNOS�/� mice to exhibit a late PC phenotype(group 4) was not due to hypertension. Measurements ofblood pressure confirmed that the levels in BPH mice(99 mm Hg; n�2) were similar to those in eNOS�/� mice (seeabove).
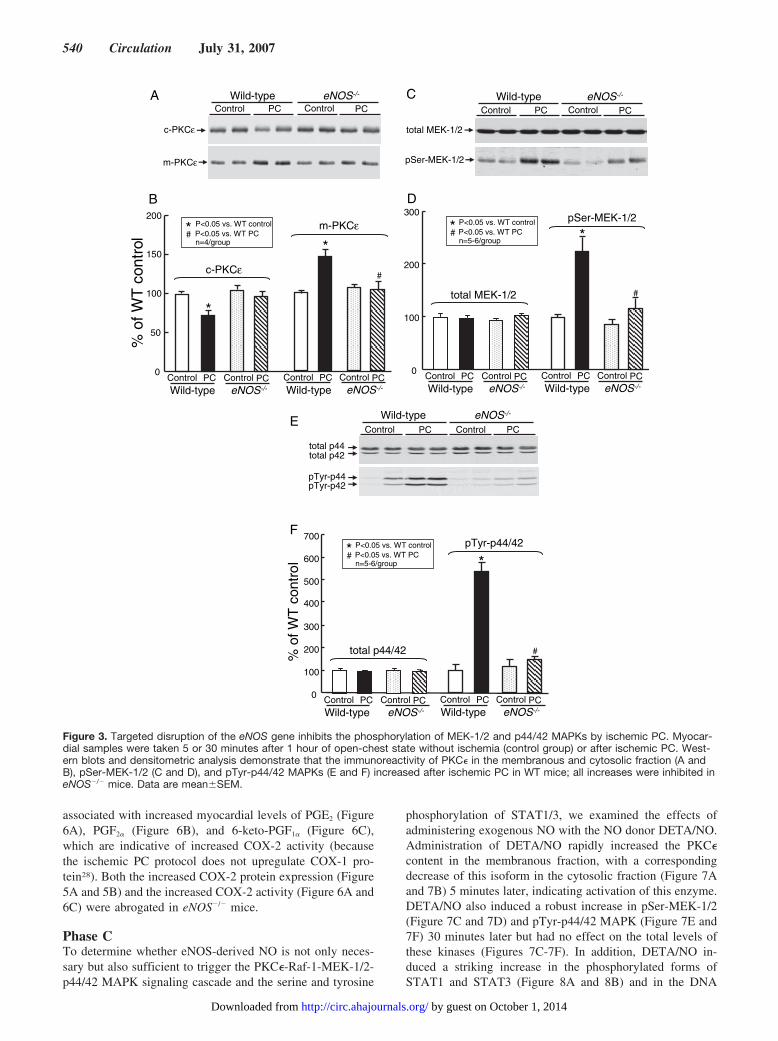
Phase BHaving established that eNOS is obligatorily required for thedevelopment of late PC, we sought to elucidate the mecha-nism responsible. As illustrated in Figure 3A through 3F,disruption of the eNOS gene blocked the membranous trans-location of PKC� 5 minutes later and the increase in pSer-MEK-1/2 and pTyr-p44/42 MAPKs observed in WT mice 30minutes after ischemic PC. The total levels of MEK-1/2 andp44/42 MAPKs were not changed by the six 4-minutecoronary O/R cycles or by deletion of eNOS (Figure 3Athrough 3F). Figure 4A and 4B shows that in WT mice the six4-minute coronary O/R cycles markedly increased the nuclearcontent of pSer-STAT1 and pSer-STAT3. All of thesechanges were inhibited by deletion of eNOS (Figure 4A and4B). Furthermore, in WT mice, the STAT1/3 DNA bindingactivity increased strikingly at 30 minutes after the ischemicPC stimulus (Figure 4C and 4D); this increase was sup-pressed by �80% in eNOS�/� mice. Targeted disruption ofthe eNOS gene in itself did not alter the binding activity ofSTAT1/3 compared with WT control mice (Figure 4C and4D). Collectively, these data indicate that eNOS plays anecessary role in triggering the PKC�-MEK-1/2-p44/42MAPK signaling cascade and the subsequent activation ofSTAT1/3.
Next, we examined the role of eNOS in COX-2 upregula-tion. In WT mice, ischemic PC induced a marked increase inCOX-2 protein (Figure 5A and 5B) 24 hours later. This was
TABLE 2. Rectal Temperature and Heart Rate on the Day of the 30-Minute Coronary Occlusion
Occlusion Reperfusion
Preocclusion 5 min 15 min 30 min 5 min 10 min 15 min
Temperature, °C
Group 1 (WT, AMI) 37.0�0.1 37.2�0.1 37.1�0.1 37.1�0.0 37.1�0.1 37.0�0.1 37.1�0.1
Group 2 (WT, LPC) 37.0�0.0 37.2�0.1 37.1�0.0 37.1�0.0 37.0�0.0 36.9�0.1 37.0�0.1
Group 3 (eNOS�/�, AMI) 36.9�0.1 37.1�0.1 36.9�0.0 37.0�0.1 37.0�0.1 37.0�0.0 37.1�0.0
Group 4 (eNOS�/�, LPC) 36.9�0.1 37.1�0.0 36.9�0.0 37.0�0.1 37.0�0.1 37.0�0.1 36.9�0.1
Group 5 (BPH, AMI) 36.9�0.0 37.0�0.1 36.9�0.1 36.9�0.0 36.9�0.1 36.9�0.1 36.8�0.0
Group 6 (BPH, LPC) 36.8�0.0 37.2�0.1 37.0�0.1 37.1�0.1 36.9�0.1 37.0�0.1 37.0�0.1
Heart rate, bpm
Group 1 (WT, AMI) 530�23 551�22 550�23 560�20 564�21 568�23 554�18
Group 2 (WT, LPC) 569�19 613�27 597�26 606�22 602�23 613�21 633�19
Group 3 (eNOS�/�, AMI) 528�18 550�20 530�14 542�21 541�20 545�15 549�20
Group 4 (eNOS�/�, LPC) 522�13 552�11 540�14 548�14 567�19 560�18 562�28
Group 5 (BPH, AMI) 567�12 611�10 625�15 618�20 619�24 617�20 623�23
Group 6 (BPH, LPC) 667�9* 739�14* 712�15* 717�7* 709�8* 707�15* 684�28*
Data are mean�SEM. Measurements of rectal temperature and heart rate were taken before the 30-minute coronary occlusion(preocclusion); at 5, 15, and 30 minutes into the 30-minute occlusion; and at 5, 15, and 30 minutes after reperfusion. Rectaltemperature was continuously monitored and carefully controlled throughout the experiment, as detailed in the text.
*P�0.05 vs group 5.
Infa
rct
Siz
e (
% r
egio
n at
ris
k)
0
20
40
60
80
100
120WTAMI
(n=10)
eNOS-/-
AMI(n=9)
BPHAMI(n=6)
eNOS-/-
LPC(n=10)
WTLPC(n=9)
BPHLPC(n=5)
I II III IV V I
* #
P<0.05 vs. group IP<0.05 vs. group V*#
V
Figure 2. Disruption of the eNOS gene blocks the protectiveeffect of late PC against myocardial infarction. Data aremean�SEM.
Xuan et al eNOS and Preconditioning 539
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
associated with increased myocardial levels of PGE2 (Figure6A), PGF2� (Figure 6B), and 6-keto-PGF1� (Figure 6C),which are indicative of increased COX-2 activity (becausethe ischemic PC protocol does not upregulate COX-1 pro-tein28). Both the increased COX-2 protein expression (Figure5A and 5B) and the increased COX-2 activity (Figure 6A and6C) were abrogated in eNOS�/� mice.
Phase CTo determine whether eNOS-derived NO is not only neces-sary but also sufficient to trigger the PKC�-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade and the serine and tyrosine
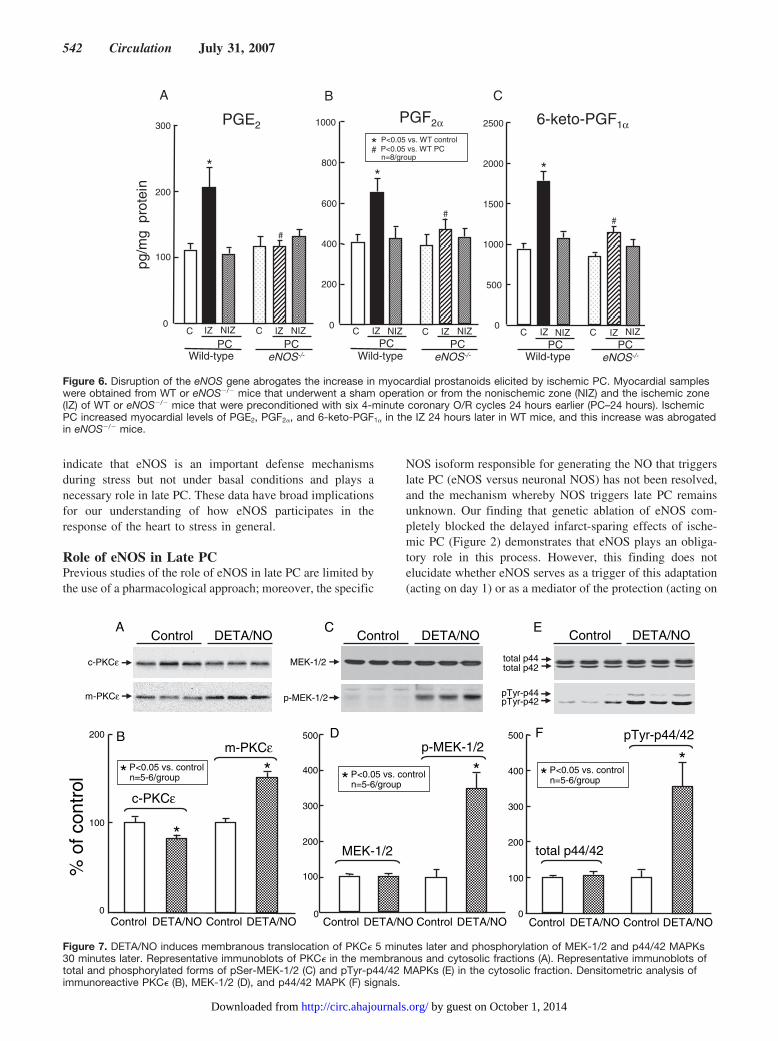
phosphorylation of STAT1/3, we examined the effects ofadministering exogenous NO with the NO donor DETA/NO.Administration of DETA/NO rapidly increased the PKC�content in the membranous fraction, with a correspondingdecrease of this isoform in the cytosolic fraction (Figure 7Aand 7B) 5 minutes later, indicating activation of this enzyme.DETA/NO also induced a robust increase in pSer-MEK-1/2(Figure 7C and 7D) and pTyr-p44/42 MAPK (Figure 7E and7F) 30 minutes later but had no effect on the total levels ofthese kinases (Figures 7C-7F). In addition, DETA/NO in-duced a striking increase in the phosphorylated forms ofSTAT1 and STAT3 (Figure 8A and 8B) and in the DNA
* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=5-6/group
D
0
100
200
300
total MEK-1/2
eNOS-/-Wild-typeControl PCControlPC
eNOS-/-Wild-typeControl PCControlPC
*
#
pSer-MEK-1/2
pSer-MEK-1/2
CControl PC
eNOS-/-Wild-typeControl PC
total MEK-1/2
* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=4/group
% o
f WT
con
trol
B
eNOS-/-Wild-typeControl PCControlPC
eNOS-/-Wild-typeControl PCControlPC
*
#
m-PKCε
AControl PC
eNOS-/-Wild-typeControl PC
c-PKCε
m-PKCε
0
50
100
150
200
c-PKCε
*%
of W
T c
ontr
ol
F
0
100
200
300
400
500
600
700
*
#
pTyr-p44/42
eNOS-/-Wild-typeControl PCControlPC
eNOS-/-Wild-typeControl PCControlPC
E
pTyr-p42pTyr-p44
total p44
Control PC
eNOS-/-Wild-typeControl PC
total p42
total p44/42
* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=5-6/group
Figure 3. Targeted disruption of the eNOS gene inhibits the phosphorylation of MEK-1/2 and p44/42 MAPKs by ischemic PC. Myocar-dial samples were taken 5 or 30 minutes after 1 hour of open-chest state without ischemia (control group) or after ischemic PC. West-ern blots and densitometric analysis demonstrate that the immunoreactivity of PKC� in the membranous and cytosolic fraction (A andB), pSer-MEK-1/2 (C and D), and pTyr-p44/42 MAPKs (E and F) increased after ischemic PC in WT mice; all increases were inhibited ineNOS�/� mice. Data are mean�SEM.
540 Circulation July 31, 2007
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from

binding activity of STAT1/3 (Figure 8C and 8D). Finally,administration of DETA/NO increased the expression ofCOX-2 protein 24 hours later (Figure 8E and 8F) but had noeffect on COX-1 expression.
DiscussionAlthough extensive evidence indicates a pivotal role of NO asa cardioprotectant (reviewed elsewhere15), the role of eNOSin myocardial ischemia/reperfusion injury and specifically inlate PC remains unclear. Studies of late PC have not resolvedeither the isoform involved or the mechanism whereby NOSinitiates late PC.14 The present study was conducted toaddress these unanswered questions.
The salient findings can be summarized as follows. First,eNOS is obligatorily required for the development of thecardioprotective effects of late PC. Second, eNOS activitytriggers late PC by activating the PKC�-Raf-1-MEK-1/2-p44/42 MAPK pathway, which leads to serine phosphoryla-tion of STAT1 and STAT3 and subsequent upregulation ofCOX-2. Third, administration of exogenous NO is sufficientto reproduce the effects of eNOS, ie, to activate PKC� and thedownstream MEK-1/2-p44/42 MAPK-pSer-STAT1/3 path-way with the consequent increase in COX-2 proteinexpression.
To the best of our knowledge, this is the first study todemonstrate that eNOS plays an obligatory role in thedelayed cardioprotection afforded by ischemic PC and that itdoes so by activating the PKC�-MEK-1/2-p44/42 MAPK-pSer-STAT1/3 pathway, leading to upregulation of STAT-dependent genes, including COX-2. This is also the firststudy to demonstrate that exogenous NO (NO donors) canreplicate the effects of eNOS activation, ie, that NO donorsrecruit the aforementioned pathway, resulting in increasedCOX-2 protein expression in the heart. In contrast, we foundno evidence that constitutive expression of eNOS modulatesmyocardial ischemia/reperfusion injury in the nonprecondi-tioned state in our mouse model. Collectively, the results
% o
f WT
con
trol
B
pSer-STAT3
pSer-STAT1
A Control PCControlPC
eNOS-/-Wild-type
0
100
200
300
400
500
*
#
eNOS-/-Wild-typeControl PCControlPC
pSer-STAT1*
#
eNOS-/-Wild-typeControl PCControlPC
pSer-STAT3* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=6/group
% o
f WT
con
trol
D
Control PCControlPCeNOS-/-Wild-type
C
STAT1/3
free
0
200
400
600
800
1000
eNOS-/-Wild-typeControl PCControlPC
*
#
STAT1/3-DNA Binding
* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=6/group
Figure 4. Disruption of the eNOS gene blocks the phosphorylation of STAT1/3 and the DNA binding activity by ischemic PC. Nuclearextracts were prepared from samples obtained as described in Figure 2. Western blots (A) and densitometric analysis (B) show that theimmunoreactivity of the serine phosphorylation of STAT1 and STAT3 increased markedly 30 minutes after ischemic PC in WT mice; thisincrease was blocked in eNOS�/� mice. C, Representative electrophoretic mobility shift assay shows that the STAT1/3–interferon-�activation site complex (arrow) increased markedly 30 minutes after ischemic PC in the nuclear extracts of WT mice and that thisincrease was abrogated in eNOS�/� mice. D, Densitometric analysis of STAT1/3 DNA binding activity.
COX-2
ControlA
ControlPC PCWild-type eNOS-/-
% o
f WT
con
trol
100
200
300
400
500
0
*
#
B
Control PC Control PCWild-type eNOS-/-
COX-2
*# P<0.05 vs. WT controlP<0.05 vs. WT PCn=6/group
Figure 5. Disruption of the eNOS gene prevents the upregula-tion of COX-2 by ischemic PC. Myocardial samples wereobtained from WT and eNOS�/� mice that underwent a shamoperation (WT control and eNOS�/� control, respectively) orfrom the ischemia-reperfused region of WT (WT PC-24 hour)and eNOS�/� (eNOS�/� PC-24 hour) mice. Representative immu-noblots (A) and densitometric analysis (B) show that ischemicPC increased COX-2 expression 24 hours later in WT mice andthat this increase was inhibited in eNOS�/� mice.
Xuan et al eNOS and Preconditioning 541
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
indicate that eNOS is an important defense mechanismsduring stress but not under basal conditions and plays anecessary role in late PC. These data have broad implicationsfor our understanding of how eNOS participates in theresponse of the heart to stress in general.
Role of eNOS in Late PCPrevious studies of the role of eNOS in late PC are limited bythe use of a pharmacological approach; moreover, the specific
NOS isoform responsible for generating the NO that triggerslate PC (eNOS versus neuronal NOS) has not been resolved,and the mechanism whereby NOS triggers late PC remainsunknown. Our finding that genetic ablation of eNOS com-pletely blocked the delayed infarct-sparing effects of ische-mic PC (Figure 2) demonstrates that eNOS plays an obliga-tory role in this process. However, this finding does notelucidate whether eNOS serves as a trigger of this adaptation(acting on day 1) or as a mediator of the protection (acting on
0
200
400
600
800
1000
*
#
PGF2α
C IZ NIZ C IZ NIZ
pg/m
g p
rote
in
0
100
200
300
*
#
C IZ NIZ C IZ NIZ
PGE2
#
0
500
1000
1500
2000
2500
*
6-keto-PGF1α
C IZ NIZ C IZ NIZ
Wild-type eNOS-/-PC PC
Wild-type eNOS-/-
PC PCWild-type eNOS-/-
PC PC
* P<0.05 vs. WT control# P<0.05 vs. WT PC
n=8/group
A B C
Figure 6. Disruption of the eNOS gene abrogates the increase in myocardial prostanoids elicited by ischemic PC. Myocardial sampleswere obtained from WT or eNOS�/� mice that underwent a sham operation or from the nonischemic zone (NIZ) and the ischemic zone(IZ) of WT or eNOS�/� mice that were preconditioned with six 4-minute coronary O/R cycles 24 hours earlier (PC–24 hours). IschemicPC increased myocardial levels of PGE2, PGF2�, and 6-keto-PGF1� in the IZ 24 hours later in WT mice, and this increase was abrogatedin eNOS�/� mice.
Control DETA/NO
c-PKCε
m-PKCε
% o
f con
trol
0
100
200
Control DETA/NO Control DETA/NO
c-PKCε
*
*
A
total p42
Control DETA/NO Control DETA/NO
*
total p44/42
pTyr-p44/42
0
100
200
300
400
500
total p44
pTyr-p44pTyr-p42
E
F
Control DETA/NOC
DB
MEK-1/2
p-MEK-1/2
p-MEK-1/2
Control DETA/NO Control DETA/NO0
100
200
300
400
500
MEK-1/2
n=5-6/groupP<0.05 vs. control**
DETA/NO
m-PKCε
Control
n=5-6/groupP<0.05 vs. control*n=5-6/group
P<0.05 vs. control*
Figure 7. DETA/NO induces membranous translocation of PKC� 5 minutes later and phosphorylation of MEK-1/2 and p44/42 MAPKs30 minutes later. Representative immunoblots of PKC� in the membranous and cytosolic fractions (A). Representative immunoblots oftotal and phosphorylated forms of pSer-MEK-1/2 (C) and pTyr-p44/42 MAPKs (E) in the cytosolic fraction. Densitometric analysis ofimmunoreactive PKC� (B), MEK-1/2 (D), and p44/42 MAPK (F) signals.
542 Circulation July 31, 2007
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
day 2). To distinguish between these 2 possibilities, weinvestigated whether deletion of eNOS interferes with theactivation of an early signaling cascade (the PKC�-MEK-1/2-p44/42 MAPK-pSer-STAT1/3 pathway) that we have pre-viously shown to be responsible for the initiation of the latePC adaptation on day 1.17 We reasoned that if eNOS acts asa trigger on day 1, this pathway will not be recruited ineNOS�/� mice; conversely, if eNOS acts as a mediator on day2, deletion of this protein should have no effect on therecruitment of the early signaling pathway on day 1. Ourfinding that targeted disruption of eNOS completely blockedor markedly reduced the increase in PKC� membranoustranslocation (Figure 3A and 3B), pSer-MEK-1/2 (Figure 3Cand 3D), pTyr-p44/42 MAPKs (Figure 3E and 3F), pSer-STAT1/3 (Figure 4A and 4B), and STAT1/3 DNA bindingactivity (Figure 4C and 4B) 30 minutes after ischemic PC, aswell as the subsequent upregulation of COX-2 protein (Figure5A and 5B) and activity (Figure 6A through 6C), demon-strates that this pathway is downstream of eNOS and isdependent on eNOS activity; ie, eNOS is an obligatory triggerof the adaptive response of late PC.
Effect of Exogenous NO on the PKC�-p44/42MAPK-pSer-STAT1/3 Pathway and on COX-2The results of phases A and B indicate that eNOS-dependentNO generation is necessary to trigger the late phase ofischemic PC. To determine whether increased NO availabilityin itself is sufficient to recruit the same signaling mechanismsthat are activated by eNOS during ischemic PC, we studiedthe effects of the NO donor DETA/NO given at the same dosepreviously shown to induce a delayed cardioprotective effect(NO-induced late PC)27 in mice. Our finding that DETA/NO,in the absence of ischemia, induced translocation of PKC� tothe particulate fraction (Figure 7A and 7B) and increased
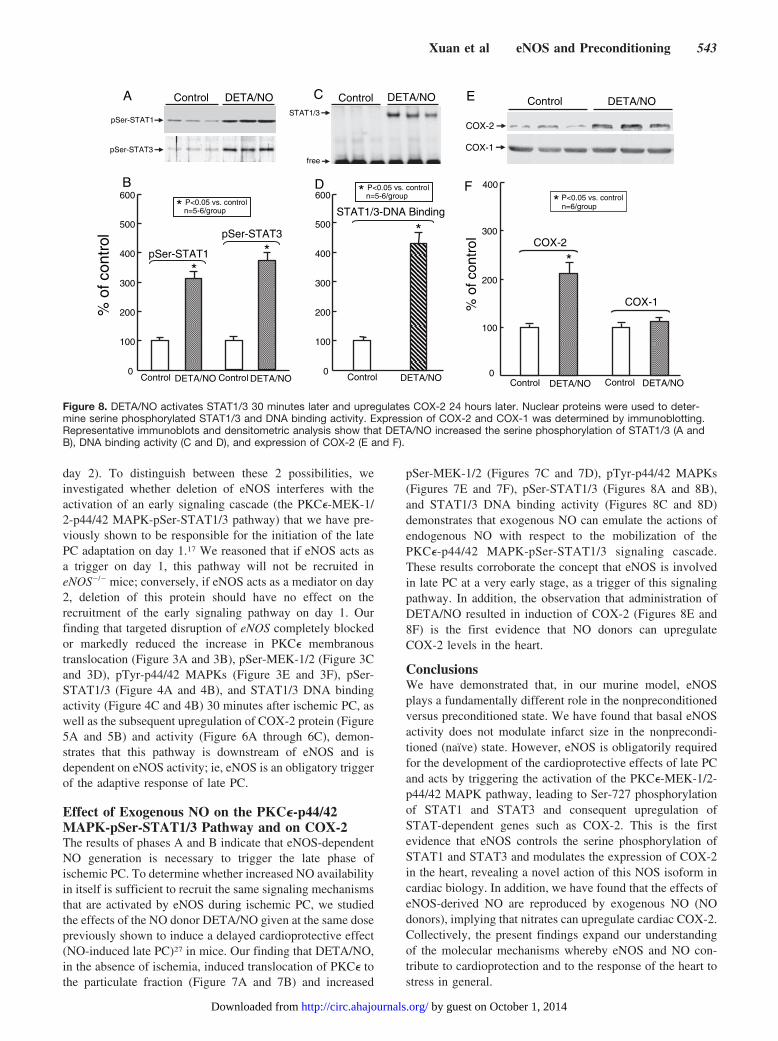
pSer-MEK-1/2 (Figures 7C and 7D), pTyr-p44/42 MAPKs(Figures 7E and 7F), pSer-STAT1/3 (Figures 8A and 8B),and STAT1/3 DNA binding activity (Figures 8C and 8D)demonstrates that exogenous NO can emulate the actions ofendogenous NO with respect to the mobilization of thePKC�-p44/42 MAPK-pSer-STAT1/3 signaling cascade.These results corroborate the concept that eNOS is involvedin late PC at a very early stage, as a trigger of this signalingpathway. In addition, the observation that administration ofDETA/NO resulted in induction of COX-2 (Figures 8E and8F) is the first evidence that NO donors can upregulateCOX-2 levels in the heart.
ConclusionsWe have demonstrated that, in our murine model, eNOSplays a fundamentally different role in the nonpreconditionedversus preconditioned state. We have found that basal eNOSactivity does not modulate infarct size in the nonprecondi-tioned (naïve) state. However, eNOS is obligatorily requiredfor the development of the cardioprotective effects of late PCand acts by triggering the activation of the PKC�-MEK-1/2-p44/42 MAPK pathway, leading to Ser-727 phosphorylationof STAT1 and STAT3 and consequent upregulation ofSTAT-dependent genes such as COX-2. This is the firstevidence that eNOS controls the serine phosphorylation ofSTAT1 and STAT3 and modulates the expression of COX-2in the heart, revealing a novel action of this NOS isoform incardiac biology. In addition, we have found that the effects ofeNOS-derived NO are reproduced by exogenous NO (NOdonors), implying that nitrates can upregulate cardiac COX-2.Collectively, the present findings expand our understandingof the molecular mechanisms whereby eNOS and NO con-tribute to cardioprotection and to the response of the heart tostress in general.
pSer-STAT3
pSer-STAT1
% o
f con
trol
0
100
200
300
400
500
600
Control DETA/NOControl DETA/NO
P<0.05 vs. control* n=5-6/group
*pSer-STAT3
*pSer-STAT1
A
B
Control DETA/NOSTAT1/3
free
Control DETA/NOC
D
Control DETA/NO
*STAT1/3-DNA Binding
0
100
200
300
400
500
600P<0.05 vs. control* n=5-6/group
E
F
% o
f con
trol
n=6/groupP<0.05 vs. control*
*
0
100
200
300
400
COX-2
COX-1
COX-2
COX-1
Control DETA/NO Control DETA/NO
Control DETA/NO
Figure 8. DETA/NO activates STAT1/3 30 minutes later and upregulates COX-2 24 hours later. Nuclear proteins were used to deter-mine serine phosphorylated STAT1/3 and DNA binding activity. Expression of COX-2 and COX-1 was determined by immunoblotting.Representative immunoblots and densitometric analysis show that DETA/NO increased the serine phosphorylation of STAT1/3 (A andB), DNA binding activity (C and D), and expression of COX-2 (E and F).
Xuan et al eNOS and Preconditioning 543
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from

Sources of FundingThe present study was supported in part by National Institutes ofHealth grants R01 HL-65660, HL-55757, HL-68088, HL-70897,HL-76794, and P01 HL-78825.
DisclosuresNone.
References1. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia.
Circulation. 1986;74:1124–1136.2. Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress
protein elevation 24 hours after brief ischemia or heat stress is associatedwith resistance to myocardial infarction. Circulation. 1993;88:1264–1272.
3. Bolli R. The late phase of preconditioning. Circ Res. 2000;87:972–983.4. Yellon DM, Downey JM. Preconditioning the myocardium: from cellular
physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151.5. Guo Y, Jones WK, Xuan YT, Bao W, Wu WJ, Han H, Laubach VE, Ping
P, Yang Z, Qiu Y, Bolli B. The late phase of ischemic preconditioning isabrogated by targeted disruption of the iNOS gene. Proc Natl Acad SciU S A. 1999;96:11507–11512.
6. Ping P, Zhang J, Qiu Y, Tang X-L, Manchikalapudi S, Cao X, Bolli R.Ischemic preconditioning induces selective translocation of protein kinaseC isoforms � and � in the heart of conscious rabbits. Circ Res. 1997;81:404–414.
7. Bolli R, Manchikalapudi S, Tang XL, Takano H, Qiu Y, Guo Y, ZhangQ, Jadoon AK. The protective effect of late preconditioning againstmyocardial stunning in conscious rabbits is mediated by nitric oxidesynthase. Circ Res. 1997;81:1094–1107.
8. Bolli R, Bhatti ZA, Tang XL, Qiu Y, Zhang Q, Guo Y, Jadoon AK.Evidence that late preconditioning against myocardial stunning in con-scious rabbits is triggered by the generation of nitric oxide. Circ Res.1997;81:42–52.
9. Qiu Y, Ping P, Tang XL, Manchikalapudi S, Rizvi A, Zhang J, Takano H,Wu WJ, Teschner S, Bolli R. Direct evidence that protein kinase C playsan essential role in the development of late preconditioning againstmyocardial stunning in conscious rabbits. J Clin Invest. 1998;101:2182–2198.
10. Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ,Bolli R. Nuclear factor-�B plays an essential role in the late phase ofischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–1109.
11. Shinmura K, Tang XL, Wang Y, Xuan YT, Liu SQ, Takano H, BhatnagarA, Bolli R. Cyclooxygenase-2 mediates the cardioprotective effects of thelate phase of ischemic preconditioning in conscious rabbits. Proc NatlAcad Sci U S A. 2000;97:10197–10202.
12. Guo Y, Bao W, Wu WJ, Shinmura K, Tang XL, Bolli R. Evidence for anessential role of cyclooxygenase-2 as a mediator of the late phase ofischemic preconditioning in mice. Basic Res Cardiol. 2000;95:479–484.
13. Takano H, Tang XL, Qiu Y, Guo Y, French BA, Bolli R. Nitric oxidedonors induce late preconditioning against myocardial stunning andinfarction in conscious rabbits via an antioxidant-sensitive mechanism.Circ Res. 1998;83:73–84.
14. Xuan YT, Tang XL, Qiu Y, Banerjee S, Takano H, Han H, Bolli R.Biphasic response of cardiac NO synthase isoforms to ischemic precon-ditioning in conscious rabbits. Am J Physiol. 2000;279:H2360–H2371.
15. Bolli R. Cardioprotective function of inducible nitric oxide synthase androle of nitric oxide in myocardial ischemia and preconditioning. J MolCell Cardiol. 2001;33:1897–1918.
16. Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of theJAK-STAT pathway in ischemic preconditioning. Proc Natl Acad SciU S A. 2001;98:9050–9055.
17. Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Messing RO, Bolli R.Role of the protein kinase C-�-Raf-1-MEK-1/2-p44/42 MAPK signalingcascade in the activation of signal transducers and activators of tran-scription 1 and 3 and induction of cyclooxygenase-2 after ischemicpreconditioning. Circulation. 2005;112:1971–1978.
18. Akira S. Roles of STAT3 defined by tissue-specific gene targeting.Oncogene. 2000;19:2607–2611.
19. Levy DE, Darnell JE Jr. Stats: transcriptional control and biologicalimpact. Nat Rev Mol Cell Biol. 2002;3:651–662.
20. Wen Z, Zhong Z, Darnell JE Jr. Maximal activation of transcription byStat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell.1995;82:241–250.
21. Shen Y, Schlessinger K, Zhu X, Meffre E, Quimby F, Levy DE, DarnellJE Jr. Essential role of STAT3 in postnatal survival and growth revealedby mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol.2004;24:407–419.
22. Horvath CM, Wen Z, Darnell JE. A Stat protein domain that determinesDNA sequence recognition suggests a novel DNA-binding domain.Genes Dev. 1995;9:984–994.
23. Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides witha major impact on cytokine-induced gene expression. J Interferon Cyto-kine Res. 1997;17:121–134.
24. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE,Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in micelacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A.1996;93:13176–13181.
25. Guo Y, Wu WJ, Qiu Y, Tang XL, Yang Z, Bolli R. Demonstration of anearly and a late phase of ischemic preconditioning in mice. Am J Physiol.1998;275:H1375–H1387.
26. Schlager G, Sides J. Characterization of hypertensive and hypotensiveinbred strains of mice. Lab Anim Sci. 1997;47:288–292.
27. Guo Y, Stein AB, Wu WJ, Zhu X, Tan W, Li Q, Bolli R. Late precon-ditioning induced by NO donors, adenosine A1 receptor agonists, and{delta}1-opioid receptor agonists is mediated by iNOS. Am J Physiol.2005;289:H2251–H2257.
28. Xuan YT, Guo Y, Zhu Y, Han H, Langenbach R, Dawn B, Bolli R.Mechanism of cyclooxygenase-2 upregulation in late preconditioning. JMol Cell Cardiol. 2003;35:525–537.
CLINICAL PERSPECTIVEPreconditioning (PC) is the most powerful and reproducible cardioprotective intervention identified to date, and mountingevidence suggests that it is effective in protecting human myocardium. Thus, PC represents an attractive strategy forinducing cardioprotection. PC consists of an early phase and a late phase (which occur 1 to 3 hours and 1 to 3 days afterthe PC stimulus, respectively). Although nitric oxide (NO) is known to be a key player in late PC, the role of endothelialNO synthase (eNOS) (the major constitutive source of NO in the heart) in late PC remains unclear. Here, we demonstratethat eNOS plays an obligatory role in triggering the development of the cardioprotective effects of late PC by activatinga downstream protein kinase C�-MEK-1/2-p44/42 mitogen-activated protein kinase signaling cascade that leads torecruitment of the signal transducers and activators of transcription (STAT)1 and STAT3 and the consequent upregulationof STAT-dependent genes such as cyclooxygenase-2. The effects of eNOS-derived NO are recapitulated by exogenous NO(NO donor administration), implying that nitrates can upregulate cardiac cyclooxygenase-2. Collectively, these resultsindicate that eNOS is an important defense mechanisms during stress and plays a necessary role in late PC. These dataexpand our understanding of the molecular mechanisms whereby eNOS and NO participate in the response of the heart tostress in general.
544 Circulation July 31, 2007
by guest on October 1, 2014http://circ.ahajournals.org/Downloaded from
Copyright © 2022 FDOKUMEN




















