
Elevations in cytosolic free Ca2+ are not required to trigger apoptosis in human leukaemia cells
7
Clin. exp. Immunol. (1992) 87, 465-471 Elevations in cytosolic free Ca2+ are not required to trigger apoptosis in human leukaemia cells S. V. LENNON, S. A. KILFEATHER*, M. B. HALLETTt, A. K. CAMPBELLt & T. G. COTTER Immunology Unit, Department of Biology, St Patrick's College, Maynooth, Co. Kildare, Republic of Ireland, *Department of Pharmacology, Royal College of Surgeons in Ireland, Dublin, Republic of Ireland, tDepartment of Surgery and $Medical Biochemistry, University of Wales College of Medicine, Cardiff, UK (Acceptedfor publication 6 November 1991) SUMMARY Previous studies have indicated that Ca2+ is a trigger for apoptosis (programmed cell death) in thymocytes and related cell lines. Recently we have shown that levels of apoptosis in leukaemic cells are diminished in Ca2+-deficient conditions, indicating that Ca2+ may be important in the mechanism of apoptosis in these cells. In the present study we investigated the possibility that Ca2+ serves as a trigger for apoptosis in the human leukaemic cell line, HL-60. Using fura-2 to measure cytosolic free Ca2+ concentrations, [Ca2+], in cell suspensions, and by using ratio imaging of fura-2 in single cells, we did not observe an early significant increase in [Ca2+]i in HL-60 cells undergoing apoptosis. The latter stages of apoptosis were, however, accompanied by increasing [Ca2+]1; these increases were apparently a result of, rather than a cause of, apoptosis. Furthermore, apoptosis could be induced in HL-60 cells under conditions of vastly reduced [Ca2+]i achieved by loading these cells with fura-2 in the presence of EGTA. These results indicate that elevation of [Ca2+]j is not a prerequisite for apoptosis in HL-60 cells and that apoptosis can occur in these cells in the presence of low [Ca2+]. Keywords apoptosis programmed cell death calcium fura-2 INTRODUCTION Apoptosis or programmed cell death is the mode of cell death observed under physiological or mildly pathological conditions [1]. Apoptosis takes the form of a series of well-ordered stages [1,2], beginning with plasma membrane blebbing, and ending with the packaging of the cell's contents into vesicles termed apoptotic bodies which are subsequently phagocytosed by neighbouring cells in vivo [3,4] or undergo secondary necrosis in vitro [5]. The hallmarks of this important death process include nuclear fragmentation and endonuclease activation leading to fragmentation of the cell's nuclear DNA into oligonucleosome- length fragments [6]; processes which occur while membrane integrity is maintained. Examples of this important death process include death of tumour cells killed by cytotoxic T cells [7], receptor-mediated death in T cell hybridomas [8] and death of autoreactive thymocytes [9]. Possible roles of Ca2+ in apoptosis have been well docu- mented. In thymocytes, a sustained increase in [Ca2 +] occurs Correspondence: Dr T. G. Cotter, Immunology Unit, St Patrick's College, Maynooth, Co. Kildare, Republic of Ireland. immediately after addition of several apoptosis-inducing sti- muli. Inhibition of this Ca2+ rise blocks DNA fragmentation and subsequent cell death [10-12]. Thus, there is considerable evidence for a role of Ca2+ in initiation of apoptosis in these cells. The importance of Ca2+ in apoptosis in other cell lines has also been demonstrated. For example, receptor-mediated apo- ptosis in T cell hybridomas [8] and pore-former-induced apoptosis in tumour target cells [13] are also dependent on extracellular Ca2 . Also, rat liver nuclei contain a Ca 2+- dependent endonuclease which, when activated results in DNA fragmentation typical of apoptosis [14]. Recently we have shown that apoptosis is induced in a range of leukaemic cell lines by many different stimuli, when applied at experimentally determined low levels [15]. The levels of apopto- sis were greatly diminished in Ca2+-deficient medium [15,16] and we have also demonstrated the presence of a Ca2+_ dependent, zinc-inhibitable endonuclease in HL-60 cells capable of inducing DNA fragmentation typical of apoptosis [17]. Furthermore HL-60 and other cell lines underwent apoptosis when cultured in zinc-deficient medium [18], and UV-induced apoptosis in HL-60 cells is zinc-inhibitable [16]. These findings 465
Transcript of Elevations in cytosolic free Ca2+ are not required to trigger apoptosis in human leukaemia cells

Clin. exp. Immunol. (1992) 87, 465-471
Elevations in cytosolic free Ca2+ are not required to trigger apoptosisin human leukaemia cells
S. V. LENNON, S. A. KILFEATHER*, M. B. HALLETTt, A. K. CAMPBELLt & T. G. COTTERImmunology Unit, Department of Biology, St Patrick's College, Maynooth, Co. Kildare, Republic of Ireland,
*Department of Pharmacology, Royal College of Surgeons in Ireland, Dublin, Republic of Ireland, tDepartment of Surgery and
$Medical Biochemistry, University of Wales College of Medicine, Cardiff, UK
(Acceptedfor publication 6 November 1991)
SUMMARY
Previous studies have indicated that Ca2+ is a trigger for apoptosis (programmed cell death) inthymocytes and related cell lines. Recently we have shown that levels of apoptosis in leukaemic cellsare diminished in Ca2+-deficient conditions, indicating that Ca2+ may be important in themechanism of apoptosis in these cells. In the present study we investigated the possibility that Ca2+serves as a trigger for apoptosis in the human leukaemic cell line, HL-60. Using fura-2 to measurecytosolic free Ca2+ concentrations, [Ca2+], in cell suspensions, and by using ratio imaging of fura-2in single cells, we did not observe an early significant increase in [Ca2+]i in HL-60 cells undergoingapoptosis. The latter stages of apoptosis were, however, accompanied by increasing [Ca2+]1; theseincreases were apparently a result of, rather than a cause of, apoptosis. Furthermore, apoptosiscould be induced in HL-60 cells under conditions of vastly reduced [Ca2+]i achieved by loading thesecells with fura-2 in the presence of EGTA. These results indicate that elevation of [Ca2+]j is not aprerequisite for apoptosis in HL-60 cells and that apoptosis can occur in these cells in the presenceof low [Ca2+].
Keywords apoptosis programmed cell death calcium fura-2
INTRODUCTION
Apoptosis or programmed cell death is the mode of cell deathobserved under physiological or mildly pathological conditions[1]. Apoptosis takes the form of a series of well-ordered stages[1,2], beginning with plasma membrane blebbing, and endingwith the packaging of the cell's contents into vesicles termedapoptotic bodies which are subsequently phagocytosed byneighbouring cells in vivo [3,4] or undergo secondary necrosis invitro [5]. The hallmarks of this important death process includenuclear fragmentation and endonuclease activation leading tofragmentation of the cell's nuclear DNA into oligonucleosome-length fragments [6]; processes which occur while membraneintegrity is maintained. Examples of this important deathprocess include death of tumour cells killed by cytotoxic T cells[7], receptor-mediated death in T cell hybridomas [8] and deathof autoreactive thymocytes [9].
Possible roles of Ca2+ in apoptosis have been well docu-mented. In thymocytes, a sustained increase in [Ca2+] occurs
Correspondence: Dr T. G. Cotter, Immunology Unit, St Patrick'sCollege, Maynooth, Co. Kildare, Republic of Ireland.
immediately after addition of several apoptosis-inducing sti-muli. Inhibition of this Ca2+ rise blocks DNA fragmentationand subsequent cell death [10-12]. Thus, there is considerableevidence for a role of Ca2+ in initiation of apoptosis in thesecells. The importance of Ca2+ in apoptosis in other cell lines hasalso been demonstrated. For example, receptor-mediated apo-ptosis in T cell hybridomas [8] and pore-former-inducedapoptosis in tumour target cells [13] are also dependent onextracellular Ca2 . Also, rat liver nuclei contain a Ca2+-dependent endonuclease which, when activated results in DNAfragmentation typical of apoptosis [14].
Recently we have shown that apoptosis is induced in a rangeofleukaemic cell lines by many different stimuli, when applied atexperimentally determined low levels [15]. The levels of apopto-sis were greatly diminished in Ca2+-deficient medium [15,16]and we have also demonstrated the presence of a Ca2+_dependent, zinc-inhibitable endonuclease in HL-60 cells capableof inducing DNA fragmentation typical of apoptosis [17].Furthermore HL-60 and other cell lines underwent apoptosiswhen cultured in zinc-deficient medium [18], and UV-inducedapoptosis in HL-60 cells is zinc-inhibitable [16]. These findings
465

S. V. Lennon et al.
suggest that activation of a Ca2+-dependent/zinc-inhibitableendonuclease present in these cells may be the trigger forapoptosis in HL-60 cells.
In the present study, we investigated the possibility thatincreasing [Ca2+]i serves as a triggering signal for apoptosis inHL-60 cells. This was achieved using the fluorescent calciumindicator, fura-2, to observe changes in [Ca2+]i both in cellsuspensions and in single cells. Surprisingly, we found that cellsundergo blebbing and DNA fragmentation with no apparentearly increase in [Ca2+],. Increases in [Ca2+], only occurred afterDNA fragmentation and before secondary necrosis of thesecells, i.e. in the final stages of apoptosis. Furthermore, apopto-sis could be induced in cells following clamping of [Ca2+]- andremoval of extracellular Ca2+. These results suggest that Ca2+is not a trigger for apoptosis in HL-60 cells, and that apoptosiscan proceed in these cells even in the presence of very low[Ca2+].
MATERIALS AND METHODS
MaterialsRPMI 1640 medium was obtained from GIBCO (Hertfordshire,U.K.). Fetal calf serum (FCS) was obtained from FlowLaboratories (Middlesex, UK). Hydrogen peroxide (H202), andphenol were obtained from BDH Chemicals (Poole, UK).Proteinase K was obtained from Boehringer Mannheim (Ger-many). Ethanol was obtained from Rathburn Chemicals (UK).Fura-2-AM was obtained from Sigma (Poole, UK) and fromMolecular Probes (OR, USA). Calcium ionophore (A23187),Percoll and all other chemicals (except where otherwise stated)were obtained from Sigma.
Cells and culture conditionsHL-60, a promyelocytic leukaemia line [19], was cultured inRPMI 1640 medium supplemented with 10%/0 FCS. Cells weremaintained at 37 C in a humidified 5% CO2 atmosphere andwere passaged three times weekly. Exponentially growing cellswere seeded atS x 1 05/ml for all experiments. When loading cellswith fura-2-AM, either HBSS or Krebs medium was used.HBSS consisted of: 140 mm NaCl, 5 mm KCl, 0-3 mm Na2HPO4,0 4 mM KH2PO4, I mM CaCl, 0 5mM MgCI2, 05mM MgSO4, 6mm glucose and 25 mm HEPES, pH 7-2. Krebs mediumconsisted of 120 mm NaCl, 4-8 mm KCI, 1-2 mm KH2PO4, 1-2mMMgSO4, 1-3 mm CaCl2 and 25 mm HEPES, pH 7-2. For Ca2+-deficient conditions, Ca2+ was replaced with 1 mm EGTA(ethyleneglycol-bis-[,B-amino-ethyl ether] NN'-tetra-aceticacid) in the above buffers.
Induction of apoptosisApoptosis was induced by exposing exponentially growingcultures to either 100 pM H202, 55% ethanol or by UVirradiation. UV irradiation was carried out by exposing cells inpolystyrene culture flasks (Nunc) from below to a 302 nm UVtransilluminator source at a distance of 1 5 cm for 10 min andirradiated cells were subsequently incubated under standardculture conditions.
Cell viability and morphologyCell viability was determined using the trypan blue exclusiontest. Cell morphology was evaluated on Rapi Diff II stainedcytocentrifuge samples. Apoptotic cells were identified by the
condensed and fragmented state of their nucleus [1,20]. Furtherverification of apoptosis in cultures was achieved by electro-phoresis of DNA from cell cultures containing a high level ofapoptosis or from purified populations of apoptotic cells,showing cleavage of the DNA into nucleosome size fragments.Apoptotic cells maintained the ability to exclude vital dyes aspreviously described [5,15,21].
Cytosolic Ca2+ measurementsCell suspensions. Cells were exposed to various apoptotic-
inducing stimuli (as described above). At various time intervals,5 x 106 cells were taken from culture, washed once in culturemedium, resuspended in1 ml of culture medium containing 2-5yM fura-2-AM, and incubated for 20-30 min at 37 C in the dark.The cells were then washed three times in HBSS. Cells were thenresuspended at 2 x 106/ml in this buffer and transferred to acuvette in a Perkin-Elmer (IOOOM) Fluorimeter with the cuvetteholder maintained at 37 C. Cell viability in the cuvette wasalways >90%. Excitation and emission wavelengths were 336nm and 536 nm respectively. [Ca2+]j values were calculated aspreviously described [22]. Briefly, following recording of thefluorescence corresponding to basal [Ca2+]j, Triton X-100 (finalconcentration 0-1% (v/v)) was added to lyse the cells anddetermine maximum fluorescence on release of dye into Ca2+>1 mm. Minimum fluorescence was achieved by addition ofEGTA (final concentration 10 mM) in 4M Tris. [Ca2+], was alsomeasured in control cells for each time point. Changes in theautofluorescence of unloaded cells caused by the variousadditions were taken into account in the calculation of [Ca2+],.[Ca2+ ], was determined using a value of 224 nm as the effective Kdof the probe [22].The ability of HL-60 cells to take up and hydrolyse Fura-2-
AM to fura-2 free acid was determined by manganese sensitivityoffluorescence observed after lysing fura-2-AM loaded cells. Nocompartmentalization of fura-2 was demonstrable, using digito-nin to selectively permeabilize the plasma membrane [23].
Measurement of[Ca2+]I in single cells. Cells were loaded with2-5-5 yM Fura-2-AM for 20-30 min at 37°C in the dark. Thecells were then washed twice in Krebs medium, and resuspendedin this buffer. The cells were then allowed to adhere to 0-1 mg/mlpoly-L-lysine-treated coverslips for 15 min and placed onto amicroperfusion chamber [24] and thermostatically controlledstage heater [25]. Fluorescence was excited at 350 nm and 380nm by coupling the output from a Spex Fluorolog dualwavelength fluorometer (Glen Spectra, UK) to a Zeiss photo-microscope III fitted with an Omega Optical dichroic mirrorwith 50% transmission at approximately 400 nm (Glen Spectra,UK). The emitted images were detected using an ISIS intensifiedCCD camera (Photonic Science, Tunbridge, UK) and ratioimages acquired using a Spex IM201 analysis system. Theoffsets were set for background subtractions and accuratedetermination of 350/380 nm ratios. To minimize exposure to340 nm light, images were acquired at 15 min intervalsthroughout the experiments, the cells being unilluminated forthe remainder of the time. The intensity of the fura-2 signals wassufficiently high compared to the autofluorescence signal forcalculation of ratio images to be uncontaminated by autofluor-escence artefact. The Ca2+ concentration was calculated asdescribed by Grynkiewicz et al. [22] from knowledge of themaximum and minimum ratio values, using a Kd of 224 nM.Whilst acquiring ratio images of the field of cells, either 100 gM
466

Apoptosis in human leukaemia cells
H202 or 5-5% ethanol (both in Krebs medium containing 0-5 ,ug/ml acridine orange) was added and the subsequent ratio imageswere recorded for analysis. Nuclear staining by acridine orangefluorescence was observed by excitation at 475 nm and nuclearimages were recorded immediately after addition of stimulusand at various timepoints thereafter.
Purification of apoptotic cellsApoptotic cells were isolated from normal cells as previouslydescribed [17]. Briefly, cells were washed twice in 0 01 M
phosphate-buffered saline, pH 7 2 and resuspended in I ml of1 06 g/ml Percoll solution. The cells were then layered onto a
discontinuous Percoll gradient consisting of sequentially-layered Percoll solutions with densities of 108 g/ml (I ml),1075 g/ml (2 ml) and 107 g/ml (2 ml). Cells were thencentrifuged in a swing-out rotor at 400 g for 30 min. The pelletthus formed contained a purified population of apoptotic cells,which were washed free of Percoll with PBS and resuspended inthis buffer.
DNA isolationCells, 106, were resuspended in 50 p1 of lysis buffer (10 mMEDTA, 50 mm Tris, pH 8-0, 0-5% sodium lauryl sarcosine, 0-5mg/ml proteinase K) and incubated at 50 C for I h. RNAse Awas then added to a final concentration of 0 5 mg/ml andincubation was continued at 50'C for 1 h. Samples were thenextracted twice with equal volumes of phenol followed by twoextractions with chloroform: isoamylalcohol (24:1). Sampleswere then pelleted at 13 000 g for 15 min in order to separateintact from fragmented chromatin. The supernatants were
placed in separate tubes and precipitated overnight in twovolumes of ice-cold ethanol at -70°C.
Electrophoresis ofDNASupernatant DNA was spun-down at 13 000 g for 15 min, air-dried at room temperature for 5 min, and resuspended in 50 piof TE buffer (0 01 M Tris, pH 8-0 containing 1 mm EDTA).Loading buffer (10 mm EDTA, 0-25% bromophenol blue, 50%glycerol) was then added at a 1:5 ratio. Samples were thenheated to 65°C for 10 min in a water-bath and were thenplunged into ice. Samples were then loaded onto a 1% agarose
gel, and electrophoresis was carried out at 6 V per cm of gel inTBE buffer (2 mm EDTA, pH 8 0, 89 mm Tris, 89 mM boricacid). Molecular weight markers of 23 5, 9 6, 6-6, 4 3, 2-2, 22-1and 0 5 kbp respectively were provided by an Hind III digest ofA-DNA. After electrophoresis, DNA was visualized by soakingthe gel in TBE containing 1 pg/ml ethidium bromide.
RESULTS
Changes in [Ca2+ , in HL-60 cell cultures undergoing apoptosisApoptosis was initiated in HL-60 cells by three different stimulias described in Materials and Methods. At certain time intervalsafter addition of stimulus, cells were removed from culture,loaded with the acetoxymethyl ester of fura-2, and [Ca2+],measured (as described in Materials and Methods). [Ca2+], ofcontrol cells was also determined at each time point. Simulta-neously, the percentage of apoptotic cells in both treated andcontrol cultures was monitored on Rapi Diff II stained cytocen-trifuge preparations.
Table 1. Asynchronous increases in [Ca2+]i and apoptosis in HL-60 cellsfollowing exposure to ethanol or H202
% Apoptosis [Ca2+], (nM)Time(hours) n Control Treated P Control Treated P
3 5 1 5 17 <0005 51 58 NS6 5 2 50 <0 0001 50 124 <0 001
Cells were treated with 5 5% ethanol and 100 PM H202 and their[Ca2+1] measured at 3 and 6 h timepoints. Simultaneous measurementsof the levels of apoptosis were performed. Results are given as the meanof five different experiments. Statistical significance was evaluated byusing Student's (-test.
Levels of apoptosis and [Ca2+]- measured at 3 and 6 hfollowing exposure to ethanol or H202 showed an asynchronousdevelopment of apoptosis and [Ca2+], increase (Table 1). Thus,while apoptosis was significant at 3 h, [Ca2+], was not signifi-cantly different from control cells at this time point. Figure lashows a representative experiment for each of the agents used inthis study. While the basal levels of [Ca2+], in control cells variedbetween 25 and 55 nm, there was a gradual rise in the [Ca2+1] oftreated cells. Concomitant with this increase in [Ca2+], was anincrease in the number of apoptotic cells in culture. [Ca2+], intreated cells rose to between 115 nm (ethanol-treated cells) and175 nM (H202-treated cells) over the time frame of the experi-ment. The morphological characteristics of HL-60 cells under-going apoptosis after exposure to UV irradiation are shown inFig. lb. Similar morphological patterns were observed afterexposure to H202 or ethanol. Electrophoresis of the nuclearDNA from purified populations of apoptotic cells indicatesDNA cleavage into fragments which represent multiples ofapproximately 200 bp typical of apoptosis in HL-60 cells afterexposure to H202, ethanol or UV irradiation (Fig. lc).
A23187-induced increases in [Ca2+], and related apoptosisWe then went on to investigate whether the increases in [Ca2+]observed in HL-60 cells undergoing apoptosis represented atrigger for apoptosis in these cells. This was conducted bydetermining the concentration of the calcium ionophoreA23187 which would induce increases in [Ca2+1] similar to thosedetermined in apoptotic HL-60 cells and by investigatingwhether such concentrations of A23 187 could induce apoptosisin HL-60 cells.
As shown in Fig. 2a, A23 187 at a concentration of 1 x 10 -7Minduced an increase in [Ca2+], from 50 nm to above 175 nM.Figure 2b shows however that this concentration of A23187failed to induce significant apoptosis in HL-60 cells over the timeframe used for H202, ethanol or UV irradiation. Furthermore,higher concentrations of A23187 such as 3 x 10-6 M wereobserved to induce relatively high levels ofapoptosis (40%) onlyafter 6 h, whereas at 5 h post-UV irradiation, levels of apoptosiswere 75%. This indicated that the [Ca2+], observed in culturesundergoing apoptosis may not, by itself, provide a signal fortriggering apoptosis in HL-60 cells.
The fact that concentrations of A23 187 which induce Ca2+-influx into the cell may also induce apoptosis in these cellsindicates that influxing Ca2+ is not inhibitory to the apoptotic
467

S. V. Lennon et al.
w(b)
1501
Time ( h )
100
-
a)C-
I
o
-C
0
iii
ona-0z
CLI
Time ( h )
150I ,'i
40
80
40 0 tQa0
CL
20 't
n
_'. a. And}-..
(c ) 2 3 4 5
Kbp
-23-5-9-6-6-6-4.3
-2-2-2-1
- 0-5
2 4 6 8
Time (h )
Fig. I (a). [Ca' in HL-60 suspensions and corresponding levels of apoptosis following exposure to three different stimuli. Cells wereeither untreated (open symbols) or treated with various apoptotic-inducing stimuli (closed symbols), and their [Ca]2+ andcorresponding levels of apoptosis determined at various timepoints. Stimulus: (top) 10 min u.v-irradiation, (middle) 100 PM H202,(bottom) 5-5', ethanol. The levels of apoptosis are expressed as the mean 0 of apoptotic cells present in five randomly selected fields( > 500 cells) ± s.e.m. (b). Morphological features of apoptosis in HL-60 cells. Indicates nuclear fragmentation typical of apoptosis in
cells treated with UV irradiation for 10 min and subsequently cultured under standard conditions for 3 h. 'N' indicates normal cells. (c).Electrophoresis of supernatant DNA extracted from purified populations of apoptotic cells. This indicates DNA fragmentation typicalofapoptosis in HL-60 cells exposed to 10 min UV irradiation (lane 1), 100 pM H202 (lane 2), 5.50,, ethanol (lane 3). Lane 4 shows that noDNA fragmentation is associated with control cells. Lane 5 is a Hind III digest of .-DNA providing molecular size markers of 23-5, 9-6,6t6, 4-3, 2 2, 2-1, and 0-5 kbp.
process. However, we have recently shown that A23187 can
induce significant apoptosis in HL-60 cells cultured in Ca>+-deficient medium [15]. It is apparent therefore that A23187 can
also induce apoptosis by a mechanism independent of Ca>+influx.
Imaging of Ca>+ in individual cellsDue to the possibility of transient and asynchronous increases in
[Ca2+], in individual cells undergoing apoptosis, effects whichmay have been undetectable in cell populations, the [Ca2+],distribution in individual cells was determined by ratio imagingof fura-2 signals.
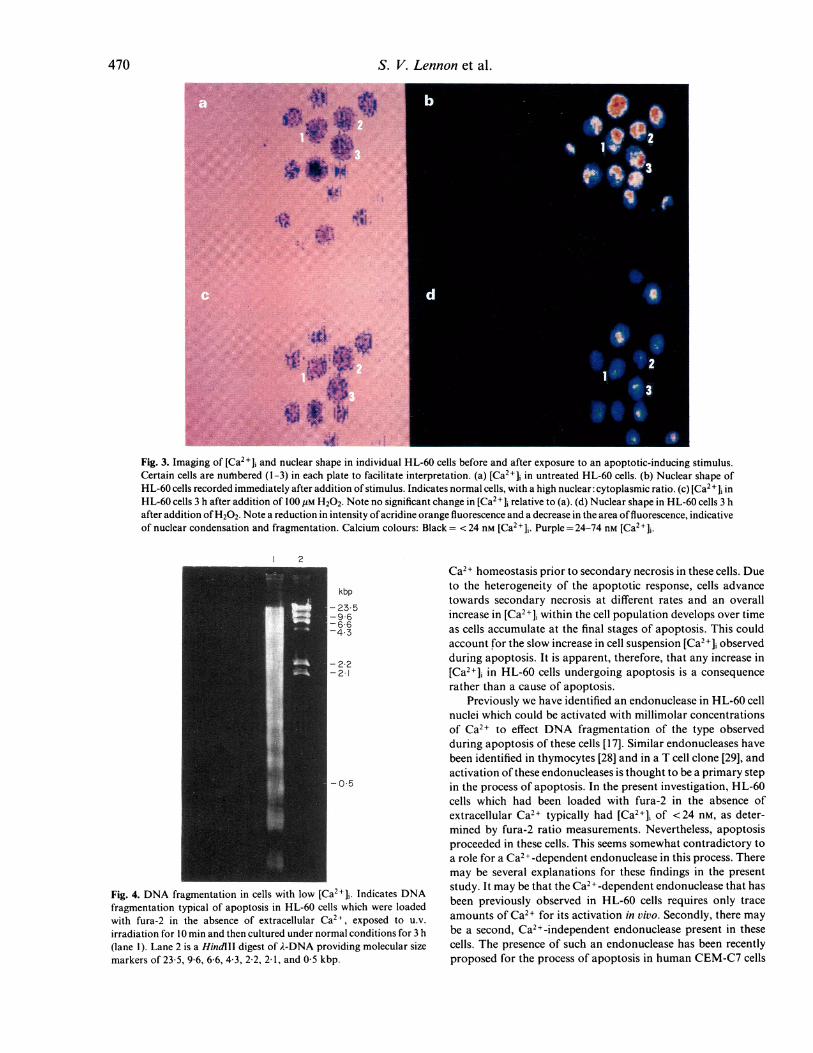
Whilst ratio images were being acquired of a field of HL-60cells (Fig. 3b), H202 was added at a concentration of 100 pM andsubsequent ratio images of the cells observed. No significantchanges in [Ca2+], occurred in HL-60 cells immediately afteraddition of stimulus, or during the formation of blebs on the cellsurface (which occurred approximately 20-30 min after addi-tion of stimulus).
Simultaneous determinations of nuclear morphology were
made using acridine orange fluorescence. Images recordedimmediately after addition of the stimulus showed for themajority of cells a high intensity fluorescence from most of thecell body, indicative of a high nuclear: cytoplasmic size ratio
468
(a )
180
-iYEciE
.2
C
Ln
0
0
0>1en
YE
0 120E
029
0.y 60o 1>, 30
.u
-f
b. =- -4- -t%- - - + - -1- - I - - -o

Apoptosis in human leukaemia cells
700(a )
2 600r_
E.,, 500
u400.9
° 300
w 200
_00
0
*0
T1
A23187 (nM)
a
a)
0
CL0CLCl
0 3 6 9 12Time (h)
Fig. 2. Dose response of HL-60 [Ca>+]i to the calcium ionophore A23 187and induction of apoptosis by this agent. (a) Cells were treated withvarious concentrations of A23187 and their [Ca2+]i was determinedapproximately 2 min later. Results are expressed as triplicate determina-tions from a representative experiment (mean of triplicate + s.e.m.). (b)Time course of appearance of apoptotic cells in HL-60 cultures treatedwith various concentrations of ionophore (M). Results from a represen-
tative experiment are shown (mean of triplicate + s.e.m.).
typical of HL-60 cells (Fig. 3b). Images recorded at various
timepoints after addition of stimulus indicated a progressiveweakening of the intensity of acridine orange fluorescence, andreduction of the area of fluorescence indicative of nuclearcondensation and fragmentation (Fig. 3d). Similar changes in
acridine orange fluorescence were also observed in cells treatedwith ethanol or UV-irradiation. In control cells, no changeswere observed in acridine orange fluorescence over the timescale of the experiment (data not shown).
No significant changes in [Ca2+], was observed in HL-60 cellsup to and including the time when nuclear condensation andfragmentation were apparent from acridine orange fluorescence(Fig. 3c). Hence, the HL-60 cells under investigation were ableto undergo the main characteristics of apoptosis, namelynuclear condensation and fragmentation, without any signifi-cant increases in [Ca2+],. Similar results were also observed forethanol-induced apoptosis (data not shown).
Using the same experimental system as described above,ratio images of fura-2 fluorescence were obtained for cells which
had previously been UV-irradiated. Again, similar results were
observed as before, namely that nuclear condensation andfragmentation can proceed without any increase in [Ca>].Hence, 3 h post UV-irradiation, when cytospin preparations ofthese cells showed that > 90% were undergoing apoptosis, theaverage [Ca2+], measured in individual cells was 40+8 nM
(n= 10). However, towards the latter stages of apoptosis,namely 5 h post UV irradiation, when cells would be progressingtowards secondary necrosis, [Ca2+], in cells was observed toincrease (data not shown). This supports the data recorded for[Ca2+]i determined during apoptosis in cell suspensions, namely,that an increase in [Ca2+], could be a result, rather than a causeof, apoptosis.
Depletion of [Ca2]+[Ca2] was depleted by loading the cells with fura-2 in theabsence of extracellular Ca2+. This would be expected toseverely deplete cells of their [Ca2+]j. This was verified byquantifying the [Ca2] in individual HL-60 cells loaded withfura-2 in this way. Under these conditions, HL-60 cells typicallyhad [Ca2+]- of < 24 nm. However, such cells were still capable ofundergoing apoptosis (Fig. 4).
DISCUSSION
The concept of Ca2+ playing a pivotal role in apoptotic celldeath was provided mainly from work conducted on thymo-cytes [10-12] and related cell lines [8] where a sustained Ca2+influx occurs following induction of apoptosis by differentstimuli. Inhibition of Ca2+ elevation in these cells blocks DNAfragmentation and cell death. Ca>2+ influx may be enhanced by a
cytosolic factor which is newly synthesized after treatment ofthese cells with an apoptosis-inducing stimulus [10,11]. Hence,by blocking protein or RNA synthesis, the synthesis of thisfactor and the increase in [Ca2+]i and subsequent cell death are
blocked. There is evidence, however, that Ca2+ increases are nota universal requirement for apoptosis in all cells. A recent reporthas indicated that apoptosis induced by glucocorticoid andnovobiocin in human CEM-C7 cells involves activation of a
constitutive Ca2 + -independent endonuclease [26]. However,other work, using 45Ca2+ to measure Ca2+ uptake in these cellsundergoing glucocorticoid-induced apoptosis could not entirelyrule out the involvement ofCa2+ in the initiation of apoptosis or
release of Ca2+ from internal stores during apoptosis [27].In the present investigation, we have shown that apoptosis
can be initiated in HL-60 cells without any increase in [Ca2+],.Furthermore, apoptosis could still be induced in the HL-60 cellsin conditions of exceedingly low [Ca2+] (< 24 nM) achieved byusing fura-2 to buffer [Ca2+]i and EGTA to deplete theextracellular Ca>.
In cell suspensions, a gradual increase in [Ca2 + ], was
observed during the development of apoptosis in HL-60 cells.However, A23 187 at a concentration which was found to mimicthis increase in [Ca2+]- did not induce apoptosis in these cells,indicating that this Ca2+ increase alone was not a signal forinitiation of apoptosis, but possibly a secondary event. Determi-nation of [Ca2+], in individual cells by using ratio measurementsof fura-2 indicated that an increase in [Ca2+], occurred only afternuclear fragmentation had occurred. This latter increase in[Ca>]2 is probably indicative of a progressive breakdown in
469
S. V. Lennon et al.
tt
*so
Fig. 3. Imaging of [Ca2+]i and nuclear shape in individual HL-60 cells before and after exposure to an apoptotic-inducing stimulus.
Certain cells are nuflbered (1-3) in each plate to facilitate interpretation. (a) [Ca2+] in untreated HL-60 cells. (b) Nuclear shape of
HL-60 cells recorded immediately after addition of stimulus. Indicates normal cells, with a high nuclear: cytoplasmic ratio. (c) [Ca2 + ]i inHL-60 cells 3 h after addition of 100 gM H202. Note no significant change in [Ca2+]j relative to (a). (d) Nuclear shape in HL-60 cells 3 h
after addition ofH202. Note a reduction in intensity ofacridine orange fluorescence and a decrease in the area offluorescence, indicativeof nuclear condensation and fragmentation. Calcium colours: Black = < 24 nm [Ca2+11. Purple = 24-74 nm [Ca2+],.
2
kbp
- 23-5-96
6 6-4.3
- 22-21
O
Fig. 4. DNA fragmentation in cells with low [Ca2+],. Indicates DNAfragmentation typical of apoptosis in HL-60 cells which were loadedwith fura-2 in the absence of extracellular Ca2 , exposed to u.v.
irradiation for 10 min and then cultured under normal conditions for 3 h(lane 1). Lane 2 is a HindIII digest of A-DNA providing molecular sizemarkers of 23-5, 9 6, 6 6, 4-3, 2-2, 21 , and 05 kbp.
Ca2+ homeostasis prior to secondary necrosis in these cells. Dueto the heterogeneity of the apoptotic response, cells advancetowards secondary necrosis at different rates and an overallincrease in [Ca2+]i within the cell population develops over timeas cells accumulate at the final stages of apoptosis. This couldaccount for the slow increase in cell suspension [Ca2+]j observedduring apoptosis. It is apparent, therefore, that any increase in[Ca2+]i in HL-60 cells undergoing apoptosis is a consequencerather than a cause of apoptosis.
Previously we have identified an endonuclease in HL-60 cellnuclei which could be activated with millimolar concentrationsof Ca2+ to effect DNA fragmentation of the type observedduring apoptosis of these cells [17]. Similar endonucleases havebeen identified in thymocytes [28] and in a T cell clone [29], andactivation of these endonucleases is thought to be a primary stepin the process of apoptosis. In the present investigation, HL-60cells which had been loaded with fura-2 in the absence ofextracellular Ca2+ typically had [Ca2+], of <24 nm, as deter-mined by fura-2 ratio measurements. Nevertheless, apoptosisproceeded in these cells. This seems somewhat contradictory toa role for a Ca2+-dependent endonuclease in this process. Theremay be several explanations for these findings in the presentstudy. It may be that the Ca2+-dependent endonuclease that hasbeen previously observed in HL-60 cells requires only traceamounts of Ca2+ for its activation in vivo. Secondly, there maybe a second, Ca2+-independent endonuclease present in thesecells. The presence of such an endonuclease has been recentlyproposed for the process of apoptosis in human CEM-C7 cells
470

Apoptosis in human leukaemia cells 471
[26]. Alternatively, it is possible that Ca2+ activation of anendonuclease in vitro has minimal relevance to the in vivostimulation of the endonuclease. Thus, the endonuclease pre-viously isolated from HL-60 cells may not require Ca2+ forinitiation of action in vivo.
We have recently shown that the levels of apoptosis inducedin HL-60 cells were diminished, but not abolished when thesecells were cultured in Ca2+-deficient conditions [15,16]. Thisindicated a role for Ca2+ in the mechanisms ofHL-60 apoptosis.From the data presented in the present paper it appears thatrather than acting as a signal for apoptosis, Ca2+ may act merelyto speed up this death process in HL-60 cells.
In conclusion, in this study we have shown that HL-60 cellscan undergo apoptosis without any early increase in [Ca2+]-. Anincrease in [Ca2+], occurs in the latter stages of apoptosis, and isapparently a consequence, rather than a cause of, apoptosis.Furthermore, HL-60 cells could be induced to undergo apopto-sis in conditions of very low [Ca2+]i. These observations indicatethat elevation of [Ca2+]i does not serve as a signal for triggeringapoptosis in HL-60 cells and that apoptosis may occur in thesecells in conditions of very low [Ca2+],.
ACKNOWLEDGMENTS
The authors are grateful to the Children's Leukaemia Research Projectand Eolas-the Irish Science and Technology Agency, the Royal IrishAcademy/Royal Society and FEBS for generous financial support.
REFERENCES
I Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biologicalphenomenon with wide ranging implications in tissue kinetics. Brit JCan 1972; 26:239-57.
2 Searle J, Kerr JFR, Bishop CJ. Necrosis and apoptosis: distinctmodes of cell death with fundamentally different significance. PatholAnnu 1982; 17:229-59.
3 Duvall E, Wyllie AH, Morris RG. Macrophage recognition of cellsundergoing programmed cell death. Immunology 1985; 56:351-9.
4 Savill JSA, Wyllie AH, Henson JE, Walport MJ, Henson PE,Haslett C. Macrophage phagocytosis of aging neutrophils ininflammation: programmed cell death in the neutrophil leads to itsrecognition by macrophages. J Clin Invest 1989; 83:865-75.
5 Wyllie AH, Duvall E, Blow JJ. In: Davies I, Sigee DC, eds. Cellageing and cell death. Cambridge: Cambridge University Press,1984:269-94.
6 Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is asso-ciated with endogenous endonuclease activation. Nature 1980;284:555-6.
7 Duke RC, Chervenak R, Cohen JJ. Endogenous endonuclease-induced DNA fragmentation: an early event in cell-mediatedcytolysis. Proc Natl Acad Sci 1983; 80:6361-5.
8 Odaka C, Kizaki H, Tadakuma T. T cell receptor-mediated DNAfragmentation and cell death in T cell hybridomas. J Immunol 1990;144:2096-101.
9 MacDonald HR, Lees RK. Programmed death of autoreactivethymocytes. Nature 1990; 343:642-4.
10 McConkey DJ, Hartzell P. Duddy SK, Hakansson H, Orrenius S.2,3,7,8-Tetrachlorodibenzo-p-dioxin kills immature thymocytes byCa2+-mediated endonuclease activation. Science 1988; 242:256-9.
11 McConkey DJ, Nicotera P. Hartzell P. Bellomo G, Wyllie AH,Orrenius S. Glucocorticoids activate a suicide process in thymocytesthrough an elevation of cytosolic Ca2+ concentration. Arch Bio-chem Biophys 1989; 269:365-70.
12 McConkey DJ, Hartzell P. Amador-Perez JF, Orrenius S, Jondal M.Calcium-dependent killing of immature thymocytes by stimulus viathe CD3/T cell receptor complex. J Immunol 1989; 143:1801-6.
13 Hameed A, Olsen KJ, Lee MK, Lichtenheld MG, Podak ER.Cytolysis by Ca2 + -permeable transmembrane channels: pore forma-tion causes extensive DNA degradation and cell lysis. J Exp Med1989; 169:765-77.
14 Jones DP, McConkey DJ, Nicotera P, Orrenius S. Calcium-activated DNA fragmentation in rat liver nuclei. J Biol Chem 1989;264:6398-403.
15 Lennon SV, Martin SJ, Cotter TG. Dose-dependent induction ofapoptosis in human tumour cell lines by widely diverging stimuli.Cell Prolif 1991; 24:203-14.
16 Martin SJ, Cotter TG. Ultraviolet B irradiation ofhuman leukaemiaHL-60 cells in vitro induces a suicidal cell response (apoptosis). Int JRadiat Biol 1991; 59:1001-16.
17 Martin SJ, Lennon SV, Bonham AB, Cotter TG. Induction ofapoptosis (programmed cell death) in human leukaemic HL-60 cellsby inhibition of RNA or protein synthesis. J Immunol 1990;145:1859-67.
18 Martin SJ, Mazdai G, Strain JJ, Cotter TG, Hannigan BM.Programmed cell death (apoptosis) in lymphoid and myeloid celllines during zinc deficiency. Clin Exp Immunol 1991; 83:338-43.
19 Collins SJ, Gallo RC, Gallagher RE. Continuous growth anddifferentiation ofhuman myeloid cells in suspension culture. Nature1977; 270:347-9.
20 Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance ofapoptosis. Int Rev Cytol 1980; 68:251-306.
21 Newman SL, Henson JE, Henson PM. Phagocytosis of senescentneutrophils by human-derived macrophages and rabbit inflamma-tory macrophages. J Exp Med 1982; 156:430-42.
22 Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J BiolChem 1985; 260:3440-50.
23 Roe MW, Lemasters JJ, Herman B. Assessment of Fura-2for measurements of cytosolic free calcium. Cell Calcium 1990;11:63-73.
24 Hallett MB. Quantal secretion and response lag demonstrated insingle rat neutrophils. Biochem Biophys Acta 1985; 847:15-19.
25 Lowndes RH, Hallett MB. A versatile light microscope stage heaterfor biological temperatures. J Microsc 1985; 142:371-4.
26 Alnemri ES, Litwack G. Activation of internucleosomal DNAcleavage in human CEM lymphocytes by glucocorticoid andnovobiocin. J Biol Chem 1990; 265:17323-35.
27 Bansal N, Houle AG, Melnyovych G. Dexamethasone-inducedkilling of neoplastic cells of lymphoid derivation: lack of earlycalcium involvement. J Cell Physiol 1990; 143:105-9.
28 Cohen JJ, Duke RC. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. JImmunol 1984; 132:38-42.
29 Nieto MA, Lopez-Rivas A. IL-2 protects T lymphocytes fromglucocorticoid-induced DNA fragmentation and cell death. J Immu-nol 1989; 143:4166-70.




















