
Electronic Structure and Bonding Acids and Bases
713
Electronic Structure and Bonding Acids and Bases
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Electronic Structure and Bonding Acids and Bases

Electronic Structure and
Bonding
Acids and Bases

• Organic compounds are compounds containing carbon
• Carbon neither readily gives up nor readily accepts electrons
• Carbon shares electrons with other carbon atoms as well as with several different kinds of atoms
Organic Chemistry

The Structure of an Atom
• An atom consists of electrons, positively charged protons,and neutral neutrons
• Electrons form chemical bonds
• Atomic number: numbers of protons in its nucleus
• Mass number: the sum of the protons and neutrons of an atom
• Isotopes have the same atomic number but different mass numbers
• The atomic weight: the average weighted mass of its atoms
• Molecular weight: the sum of the atomic weights of all the atomsin the molecule

The Distribution of Electrons in an Atom
• Quantum mechanics uses the mathematical equation of wavemotions to characterize the motion of an electron around a nucleus
• Wave functions or orbitals tell us the energy of the electron and the volume of space around the nucleus where an electron ismost likely to be found
• The atomic orbital closer to the nucleus has the lowest energy
• Degenerate orbitals have the same energy

Table 1.1


• The Aufbau principle: electrons occupy the orbitals with the lowest energy first
• The Pauli exclusion principle: only two electrons can occupy one atomic orbital and the two electrons have opposite spin
• Hund’s rule: electrons will occupy empty degenerated orbitals before pairing up in the same orbital

• Ionic compounds are formed when an electropositive element transfers electron(s) to an electronegative element

Covalent Compounds• Equal sharing of electrons: nonpolar covalent bond (e.g., H2)
• Sharing of electrons between atoms of different electronegativities: polar covalent bond (e.g., HF)

Electrostatic Potential Maps

• A polar bond has a negative end and a positive end
dipole moment (D) = µ = e x d
(e) : magnitude of the charge on the atom(d) : distance between the two charges
A Dipole

Lewis Structure
Formal charge = number of valence electrons –(number of lone pair electrons +1/2 number of bonding electrons)


Important Bond Numbers
H F ICl Brone bond
Otwo bonds
Nthree bonds
Cfour bonds

The s Orbitals

The p Orbitals

Molecular Orbitals
• Molecular orbitals belong to the whole molecule
• σ bond: formed by overlapping of two s orbitals
• Bond strength/bond dissociation: energy required to break a bond or energy released to form a bond


In-phase overlap forms a bonding MO; out-of-phase overlap forms an antibonding MO

Sigma bond (σ) is formed by end-on overlap of two p orbitals
A σ bond is stronger than a π bond

Pi bond (π) is formed by sideways overlap of two parallel p orbitals

Bonding in Methane and Ethane: Single Bonds
Hybridization of orbitals:

The orbitals used in bond formation determine the bond angles
• Tetrahedral bond angle: 109.5°
• Electron pairs spread themselves into space as far from each other as possible

Hybrid Orbitals of Ethane

Bonding in Ethene: A Double Bond

• The bond angle in the sp2 carbon is 120°
• The sp2 carbon is the trigonal planar carbon
An sp-Hybridized Carbon

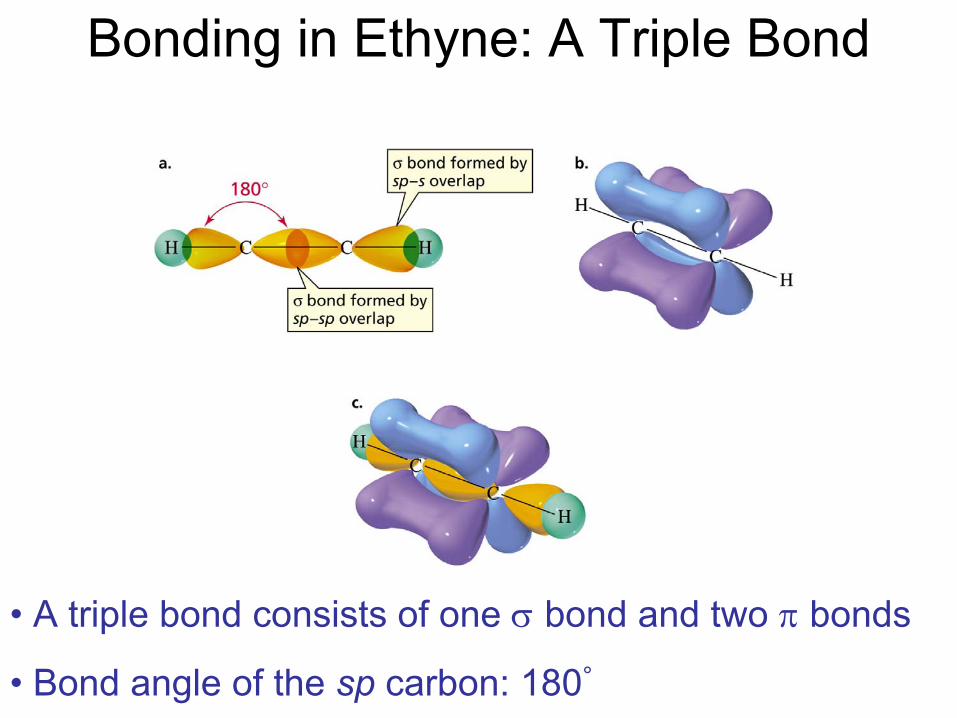
Bonding in Ethyne: A Triple Bond
• Bond angle of the sp carbon: 180°
• A triple bond consists of one σ bond and two π bonds

Bonding in the Methyl Cation

Bonding in the Methyl Radical

Bonding in the Methyl Anion

Bonding in Water

Bonding in Ammonia and in the Ammonium Ion

Bonding in Hydrogen Halides

Summary
• A π bond is weaker than a σ bond
• The greater the electron density in the region of orbital overlap, the stronger is the bond
• The more s character, the shorter and stronger is the bond
• The more s character, the larger is the bond angle

The vector sum of the magnitude and the direction of the individual bond dipole determines the overall dipole moment of a molecule
Molecular Dipole Moment

Brønsted–Lowry Acids and Bases• Acid donates a proton
• Base accepts a proton
• Strong reacts to give weak• The weaker the base, the stronger is its conjugate acid• Stable bases are weak bases

An Acid/Base Equilibrium
Ka: The acid dissociation constant
H2O + HA H3O+ + A-
[H3O+][A-]Ka = [H2O][HA]
pKa = -log Ka

The Henderson–Hasselbalch Equation
• A compound will exist primarily in its acidic form at a pH< its pKa
• A compound will exist primarily in its basic form at a pH > its pKa
• A buffer solution maintains a nearly constant pH upon addition of small amount of acid or base
[ ][ ]−+=AHAlogpHp aK
The pH indicates the concentration of hydrogen ions (H+)

• When atoms are very different in size, the stronger acid will have its proton attached to the largest atom

• When atoms are similar in size, the stronger acid will have its proton attached to the more electronegative atom
• Inductive electron withdrawal increases the acidity of a conjugate acid

Acetic acid is more acidic than ethanol
CH3COHO
CH3CH2OH
pKa = 4.76 pKa = 15.9acetic acid ethanol
The delocalized electrons in acetic acid are shared by more than two atoms, thereby stabilizing the conjugated base
CH3CO-
OCH3CO-
O

• Lewis acid: non-proton-donating acid; will accept two electrons
• Lewis base: electron pair donors
Lewis Acids and Bases

Electronic Structure and
Bonding
Acids and Bases

• Organic compounds are compounds containing carbon
• Carbon neither readily gives up nor readily accepts electrons
• Carbon shares electrons with other carbon atoms as well as with several different kinds of atoms
Organic Chemistry

The Structure of an Atom
• An atom consists of electrons, positively charged protons,and neutral neutrons
• Electrons form chemical bonds
• Atomic number: numbers of protons in its nucleus
• Mass number: the sum of the protons and neutrons of an atom
• Isotopes have the same atomic number but different mass numbers
• The atomic weight: the average weighted mass of its atoms
• Molecular weight: the sum of the atomic weights of all the atomsin the molecule

The Distribution of Electrons in an Atom
• Quantum mechanics uses the mathematical equation of wavemotions to characterize the motion of an electron around a nucleus
• Wave functions or orbitals tell us the energy of the electron and the volume of space around the nucleus where an electron ismost likely to be found
• The atomic orbital closer to the nucleus has the lowest energy
• Degenerate orbitals have the same energy

Table 1.1


• The Aufbau principle: electrons occupy the orbitals with the lowest energy first
• The Pauli exclusion principle: only two electrons can occupy one atomic orbital and the two electrons have opposite spin
• Hund’s rule: electrons will occupy empty degenerated orbitals before pairing up in the same orbital

• Ionic compounds are formed when an electropositive element transfers electron(s) to an electronegative element

Covalent Compounds• Equal sharing of electrons: nonpolar covalent bond (e.g., H2)
• Sharing of electrons between atoms of different electronegativities: polar covalent bond (e.g., HF)

Electrostatic Potential Maps

• A polar bond has a negative end and a positive end
dipole moment (D) = µ = e x d
(e) : magnitude of the charge on the atom(d) : distance between the two charges
A Dipole

Lewis Structure
Formal charge = number of valence electrons –(number of lone pair electrons +1/2 number of bonding electrons)


Important Bond Numbers
H F ICl Brone bond
Otwo bonds
Nthree bonds
Cfour bonds

The s Orbitals

The p Orbitals

Molecular Orbitals
• Molecular orbitals belong to the whole molecule
• σ bond: formed by overlapping of two s orbitals
• Bond strength/bond dissociation: energy required to break a bond or energy released to form a bond


In-phase overlap forms a bonding MO; out-of-phase overlap forms an antibonding MO

Sigma bond (σ) is formed by end-on overlap of two p orbitals
A σ bond is stronger than a π bond

Pi bond (π) is formed by sideways overlap of two parallel p orbitals

Bonding in Methane and Ethane: Single Bonds
Hybridization of orbitals:

The orbitals used in bond formation determine the bond angles
• Tetrahedral bond angle: 109.5°
• Electron pairs spread themselves into space as far from each other as possible

Hybrid Orbitals of Ethane

Bonding in Ethene: A Double Bond

• The bond angle in the sp2 carbon is 120°
• The sp2 carbon is the trigonal planar carbon
An sp-Hybridized Carbon
Bonding in Ethyne: A Triple Bond
• Bond angle of the sp carbon: 180°
• A triple bond consists of one σ bond and two π bonds

Bonding in the Methyl Cation

Bonding in the Methyl Radical

Bonding in the Methyl Anion

Bonding in Water

Bonding in Ammonia and in the Ammonium Ion

Bonding in Hydrogen Halides

Summary
• A π bond is weaker than a σ bond
• The greater the electron density in the region of orbital overlap, the stronger is the bond
• The more s character, the shorter and stronger is the bond
• The more s character, the larger is the bond angle

The vector sum of the magnitude and the direction of the individual bond dipole determines the overall dipole moment of a molecule
Molecular Dipole Moment

Brønsted–Lowry Acids and Bases• Acid donates a proton
• Base accepts a proton
• Strong reacts to give weak• The weaker the base, the stronger is its conjugate acid• Stable bases are weak bases

An Acid/Base Equilibrium
Ka: The acid dissociation constant
H2O + HA H3O+ + A-
[H3O+][A-]Ka = [H2O][HA]
pKa = -log Ka

The Henderson–Hasselbalch Equation
• A compound will exist primarily in its acidic form at a pH< its pKa
• A compound will exist primarily in its basic form at a pH > its pKa
• A buffer solution maintains a nearly constant pH upon addition of small amount of acid or base
[ ][ ]−+=AHAlogpHp aK
The pH indicates the concentration of hydrogen ions (H+)

• When atoms are very different in size, the stronger acid will have its proton attached to the largest atom

• When atoms are similar in size, the stronger acid will have its proton attached to the more electronegative atom
• Inductive electron withdrawal increases the acidity of a conjugate acid

Acetic acid is more acidic than ethanol
CH3COHO
CH3CH2OH
pKa = 4.76 pKa = 15.9acetic acid ethanol
The delocalized electrons in acetic acid are shared by more than two atoms, thereby stabilizing the conjugated base
CH3CO-
OCH3CO-
O

• Lewis acid: non-proton-donating acid; will accept two electrons
• Lewis base: electron pair donors
Lewis Acids and Bases





















































5-1
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Introduction to Introduction to Organic Organic
ChemistryChemistry2 ed2 ed
William H. BrownWilliam H. Brown

5-2
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alkenes Alkenes & &
AlkynesAlkynesChapter 5Chapter 5

5-3
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Unsaturated HydrocarbonsUnsaturated Hydrocarbons•• Unsaturated hydrocarbonUnsaturated hydrocarbon: contains one or more
carbon-carbon double or triple bonds•• AlkeneAlkene: contains a carbon-carbon double bond
and has the general formula CnH2n
Ethene(an alkene)
HC C
H
H H

5-4
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Unsaturated HydrocarbonsUnsaturated Hydrocarbons•• AlkyneAlkyne: contains a carbon-carbon triple bond and
has the general formula CnH2n-2
Ethyne(an alkyne)
H- C C- H

5-5
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Unsaturated HydrocarbonsUnsaturated Hydrocarbons•• ArenesArenes: benzene and its derivatives (Ch 9)
CC
CC
C
C
H
H
H
HH
H
Benzene(an arene)

5-6
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Benzene & Phenyl GroupBenzene & Phenyl Group• We do not study benzene and its derivatives until
Chapter 9• But, we show structural formulas of compounds
containing the phenyl group before that time• the phenyl group is not reactive under any of the
conditions we describe in Ch 3-8
Benzene Alternative representations for the phenyl group
C6 H5 - Ph-

5-7
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure of AlkenesStructure of Alkenes• The two carbon atoms of a double bond and the
four atoms attached to them lie in a plane, with bond angles of approximately 120°
• According to the orbital overlap model, a double bond consists of
• one sigma bond formed by overlap of sp2 hybridorbitals
• one pi bond formed by overlap of parallel 2porbitals

5-8
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure of AlkenesStructure of Alkenes• Length of C-C bonds: single > double > triple• Strength of C-C bonds:triple > double > single
sp-sp, two 2p- 2p 1.21 200
1461.34sp2 -sp 2 , 2p- 2p
MoleculeC-C OrbitalOverlap Å kcal/mol
sp3 -sp 3 1.54 90ethane
ethylene
acetylene

5-9
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CisCis--Trans IsomerismTrans Isomerism• Because of restricted rotation about a C-C double
bond, groups on adjacent carbons are either cisor trans to each other
cis-2-Butenemp -139°C, bp 4°C
trans -2-Butenemp -106°C, bp 1°C
C
H3 C
C
H
CH3
C
HH
C
CH3
HH3 C

5-10
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CisCis,Trans Isomerism,Trans Isomerism• Trans alkenes are more stable than cis alkenes
because of nonbonded interaction strain between alkyl substituents of the same side of the double bond

5-11
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure of AlkynesStructure of Alkynes• The functional group of an
alkyne is a carbon-carbon triple bond
• A triple bond consists of • one sigma bond formed by
the overlap of sp hybridorbitals
• two pi bonds formed by the overlap of sets of parallel 2porbitals

5-12
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkenesAlkenes• Use the infix -enen- to show the presence of a
carbon-carbon double bond• Number the parent chain to give the 1st carbon of
the double bond the lower number• Follow IUPAC rules for numbering and naming
substituents• For a cycloalkene, the double bond must be
numbered 1,2

5-13
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkenesAlkenes
6 5 4 3 2 1
4-Methyl-1-hexeneCH3 CH2 CHCH2 CH= CH2
CH3124 3
2-Ethyl-3-methyl-1-pentene
CH3 CH2 CHC= CH25
CH3
CH2 CH3
1 2
34
5
3-Methylcyclo-pentene
CH3
1,6-Dimethylcyclo-hexene
CH3
CH31
65
4
32

5-14
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkenesAlkenes
allyl chloride
vinyl chloride
allyl
vinyl
ExampleCommonName
Alkenyl Group
CH2 = CH-
CH2 = CHCH2 -
CH2 = CHCl
CH2 = CHCH2 Cl

5-15
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkenesAlkenes• Some alkenes, particularly low-molecular-weight
ones, are known almost exclusively by their common names
IsobutylenePropyleneEthyleneCommon:
IUPAC: 2-MethylpropenePropeneEthene
CH2 = CH2 CH3 CH= CH 2 CH3 C= CH2
CH3

5-16
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkynesAlkynes• IUPAC: use the infix -ynyn- to show the presence of
a carbon-carbon triple bond
1
3-Methyl-1-butyne 6,6-Dimethyl-3-heptyne
234 1 2 3 4 5 6 7CH3 CH2 C CCH2 CCH3
CH3
CH3CH3
CH3 CHC CH

5-17
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlkynesAlkynes• Common names: prefix the substituents on the
triple bond to the name “acetylene”
Methylacetylene D imethylacetylene VinylacetylenePropyne 2-Butyne 1-Buten-3-yne
CH3 C CH CH3 C CCH 3 CH2 =CHC CHIUPAC:
Common:

5-18
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Configuration Configuration -- ciscis, trans, trans•• TheThe ciscis--trans systemtrans system: configuration is determined
by the orientation of atoms of the main chain
cis -3,4-Dimethyl-2-pentene
1
2 3
4C
H
C
CH3
CH( CH3 ) 2H3 Ctrans -3-Hexene
C
H
C
CH2 CH3
HCH3 CH2

5-19
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Configuration Configuration -- E,ZE,Z• The E,Z system uses the priority rules of the R,S
system to assign to the groups on each carbon of a carbon-carbon double bond1. Each atom bonded to the C-C double bond is assigned
a priority2. If groups of higher priority are on the same side,
configuration is Z (German, zusammen)3. If groups of higher priority are on opposite sides,
configuration is E (German, entgegen)

5-20
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Configuration Configuration -- E,ZE,Z•• ExampleExample: name each alkene and specify its
configuration by the E,Z system
(a) (b)C
H3 C CH( CH3 ) 2
C
CH3H
C
Cl
C
H
CH2 CH3H3 C
(d)(c) C
Br CH3
C
HCl
C
ClCH2
C
CH3
CH2 CH3H3 C

5-21
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CisCis--Trans inTrans in CycloalkenesCycloalkenes• Configuration of the double bond in
cyclopropene through cycloheptene must be cis

5-22
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CisCis--Trans inTrans in CycloalkenesCycloalkenes• trans-Cyclooctene is the smallest trans-
cycloalkene stable at 25°C • the cis isomer is 9.1 kcal/mol more stable than the
trans isomer.

5-23
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical PropertiesPhysical Properties• Alkenes are nonpolar compounds• The only attractive forces between their
molecules are dispersion forces• The physical properties of alkenes are similar to
those of alkanes

5-24
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
TerpenesTerpenes•• TerpeneTerpene: a compound whose carbon skeleton
can be divided into two or more units identical with the carbon skeleton of isoprene
CH2 =C-CH= CH2
CH3
2-Methyl-1,3-butadiene(Isoprene)
C-C-C-C1 2 3 4head tail
Isoprene unit
C

5-25
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
TerpenesTerpenes• Myrcene, C10H16, a
component of bayberry wax and oils of bay and verbena
12
34
12
3
4
two isoprene units joined here

5-26
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
TerpenesTerpenes• Menthol, from peppermint
OH
5-27
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
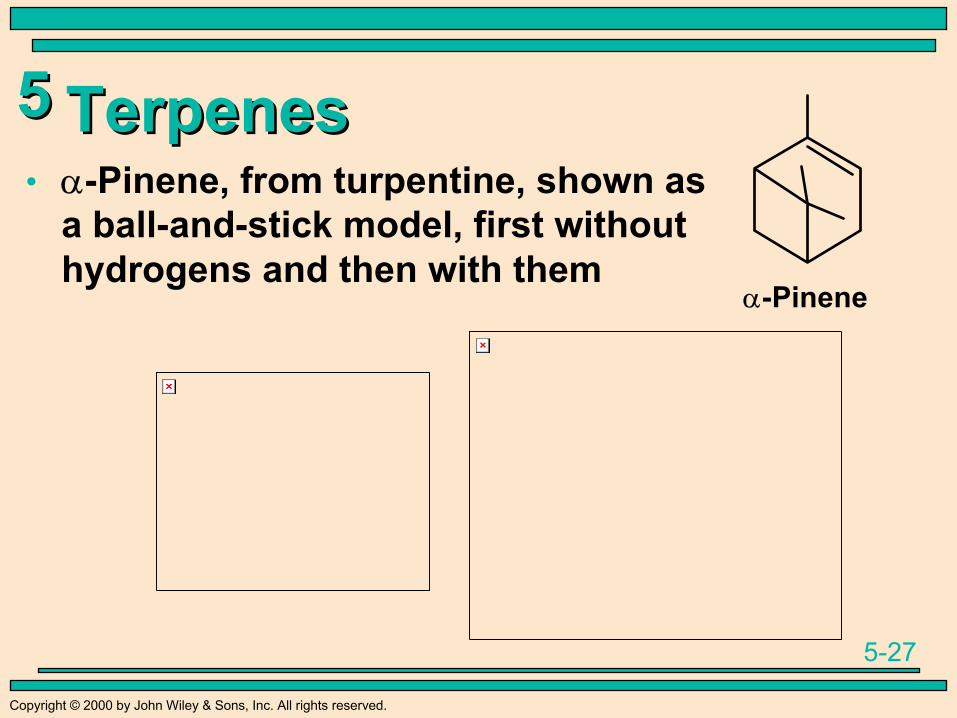
TerpenesTerpenes• α-Pinene, from turpentine, shown as
a ball-and-stick model, first withouthydrogens and then with them
α-Pinene
5-28
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
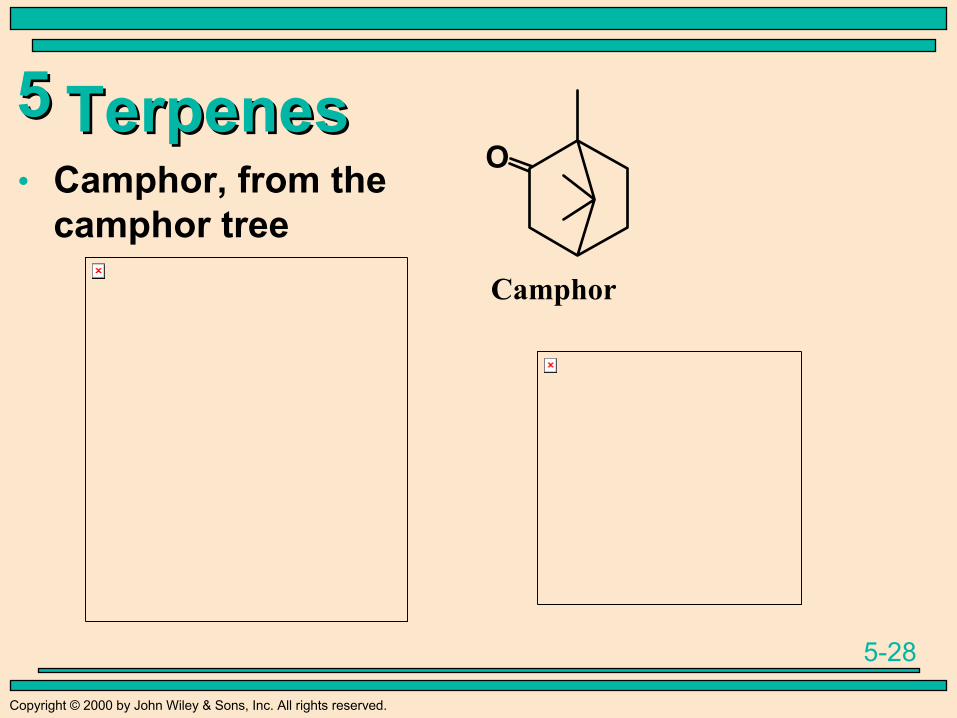
TerpenesTerpenes• Camphor, from the
camphor tree
O
Camphor

5-29
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Vitamin A (Vitamin A (RetinolRetinol))How many stereoisomers are possible for vitamin A?
H3 C CH3
CH3
CH3 CH3
CH2 OH

5-30
55
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alkenes & AlkynesAlkenes & AlkynesEnd Chapter 5End Chapter 5

6-1
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Introduction to Introduction to Organic Organic
ChemistryChemistry2 ed2 ed
William H. BrownWilliam H. Brown

6-2
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reactions of Reactions of AlkenesAlkenes
Chapter 6Chapter 6

6-3
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Characteristic ReactionsCharacteristic Reactions
+
Hydrochlorination (hydrohalogenation)
CC C CH Cl
HCl
+
Hydration
C C C CH OH
H2 O

6-4
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Characteristic ReactionsCharacteristic Reactions
+
Bromination (halogenation)
C C C CBr Br
Br 2
+
Hydroxylation (oxidation)
C C C CHO OH
OsO4

6-5
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Characteristic ReactionsCharacteristic Reactions
Polymerization
CC C Cinitiator
nn
+
Hydrogenation (reduction)
CC C CHH
H2

6-6
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction MechanismsReaction Mechanisms• A reaction mechanism describes how a reaction
occurs• which bonds are broken and which new ones are
formed• the order in which bond-breaking and bond-forming
steps take place• the role of the catalyst (if any is present)• the energy of the entire system during the reaction

6-7
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
PE DiagramsPE Diagrams• Potential energy (PE)
diagram: a graph showing the changes in energy that occur during a chemical reaction
• Reaction coordinate: a measure of the change in position of atoms during a reaction
Reaction coordinate
Pote
ntia
l en
ergy

6-8
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
PE DiagramsPE Diagrams
Ea
∆H
transition state
A + B
C + D
Reaction Coordinate
Pote
ntia
l ene
rgy

6-9
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
PE DiagramsPE Diagrams• Heat of reaction, ∆H: the difference in potential
energy between reactants and products• exothermic: products are lower in energy than
reactants; heat is released• endothermic: products are higher in energy than
reactants; heat is absorbed• Transition state: an energy maximum on a PE
diagram• represents an unstable species of maximum PE
formed during the course of a reaction

6-10
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Activation EnergyActivation Energy• Activation Energy, Ea: the difference in
potential energy between reactants and the transition state• determines the rate of reaction• if activation energy is large, only a few
molecular collisions occur with sufficient energy to reach the transition state, and the reaction is slow
• if activation energy is small, many collisions generate sufficient energy to reach the transition state, and the reaction is fast

6-11
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Developing a MechanismDeveloping a Mechanism• Design experiments to reveal details of a particular
chemical reaction• Propose a set or sets of steps that might account for the
overall transformation• A mechanism becomes established when it is shown to
be consistent with every test that can be devised• This does mean that the mechanism is correct, only that
it is the best explanation we are able to devise

6-12
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Why Mechanisms?Why Mechanisms?• A framework within which to organize descriptive
chemistry• The intellectual satisfaction derived from
constructing models that accurately reflect the behavior of chemical systems
• A tool with which to search for new information and new understanding

6-13
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ElectrophilicElectrophilic AdditionsAdditions• Hydrohalogenation using HCl, HBr, HI• Hydration using H2O, H2SO4
• Halogenation using Cl2, Br2

6-14
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of HXAddition of HX• Carried out with pure reagents or in a polar
solvent such as acetic acid• Addition is regioselective • Regioselective reaction: a reaction in which one
direction of bond-forming or bond-breaking occurs in preference to all other directions

6-15
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of HXAddition of HX• Addition is regioselective
•• MarkovnikovMarkovnikov’’ss rulerule: in the addition of HX or H2O to an alkene, H adds to the carbon of the double bond that has the greater number of hydrogensbonded to it
1-Chloropropane (not observed)
2-Chloropropane
Propene
+
+
ClCl
CH3 CH= CH2 HCl
CH3 CH-CH2 CH3 CH- CH2
H H

6-16
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
HClHCl + 2+ 2--ButeneButene• A two-step mechanism
• Step 1: formation of sec-butyl cation, a 2° carbocationintermediate
-+
+δ-δ+
H- ClCH3 CH= CHCH3
Clsec-Butyl cation
slow, rate-limiting step
+H
CH3 CH- CHCH3

6-17
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
HClHCl + 2+ 2--ButeneButene• Step 2: reaction of the sec-butyl cation with chloride
ion
- +
sec-Butyl cation (a Lewis acid)
+
ClCl CH3 CHCH2CH3CH3 CHCH2CH3
Chloride ion(a Lewis base)
fast

6-18
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CarbocationsCarbocations•• CarbocationCarbocation: a species containing a positively
charged carbon• Carbocations are
• classified as 1°, 2°, or 3° depending on the number of carbons bonded to the carbon bearing the positive charge
• electrophiles; that is, they are “electron-lovers”

6-19
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CarbocationCarbocation StructureStructure• Bond angles about the
positively charged carbon are 120°
• Carbon uses sp2 hybrid orbitalsto form sigma bonds to the three attached groups
• The unhybridized 2p orbital lies perpendicular to the sigma bond framework and contains no electrons
R CR
R+

6-20
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CarbocationCarbocation StabilityStability• A 3° carbocation is more stable (requires a lower
activation energy for its formation) than a 2°carbocation
• A 2° carbocation is, in turn, more stable (requires a lower activation energy for its formation) than a 1° carbocation
• 1° and methyl cations is so unstable that they are rarely if ever observed in solution

6-21
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CarbocationCarbocation StabilityStability• Relative stability
Methyl cation
(methyl)
Ethyl cation
(1°)
Isopropyl cation
(2°)
tert -Butyl cation
(3°)
Increasing carbocation stability
+ + + +C
H
H
CH3 CCH3
CH3
HC
CH3
CH3
CH3CHH
H

6-22
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CarbocationCarbocation StabilityStability• Carbocations are stabilized by the electron-
withdrawing inductive effect of the positively-charged carbon• according to molecular orbital calculations:
HH
CH
H3 CCH3
CCH3
+0.83
+0.06
+0.60
+0.13

6-23
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of HAddition of H22OO• Addition of water is called hydration• Acid-catalyzed hydration of an alkene is
regioselective - hydrogen adds to the less substituted carbon of the double bond
Propene 2-Propanol+
OH
CH3 CH= CH2 H2 OH2 SO4 CH3 CH- CH2
H

6-24
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of HAddition of H22OO• Step 1: proton transfer from solvent to the alkene
+
++
intermediateA 2o carbocation
+HO
H
HOH
H
CH3 CH= CH2
CH3 CHCH 3
slow, rate-limiting step

6-25
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of HAddition of H22OO• Step 2: reaction of the carbocation intermediate with
H2O to form an oxonium ion
• Step 3: proton transfer to solvent
+
++
An oxonium ion
H OHH
CH3 CHCH3 O-H CH3 CHCH 3fast
++
+OH HOH
H
HH O HCH3 CHCH 3 CH3 CHCH 3
OH
fast

6-26
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of ClAddition of Cl22 and Brand Br22• Carried out with either the pure reagents or in an
inert solvent such as CCl4 or CH2Cl2
2,3-Dibromobutane2-Butene+
Br BrBr2CH3 CH= CHCH3 CH3 CH- CHCH3CCl4

6-27
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of ClAddition of Cl22 and Brand Br22• Addition is stereoselective
•• StereoselectiveStereoselective reactionreaction: a reaction in which one stereoisomer is formed or destroyed in preference to all others than might be formed or destroyed
trans -1,2-Dibromo-cyclohexane
Cyclohexene
+ Br2 CCl4Br
Br

6-28
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of ClAddition of Cl22 and Brand Br22• Addition involves a two-step mechanism
• Step 1: formation of a bridged bromonium ion intermediate
C CBr
C C
BrBr
Br -
A bridged bromonium ion intermediate
+

6-29
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of ClAddition of Cl22 and Brand Br22• Step 2: Attack of halide ion from the opposite side of
the three-membered ring
Anti addition-
C C
Br
C C
Br
Br
Br
+

6-30
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Addition of ClAddition of Cl22 and Brand Br22• For a cyclohexene, anti coplanar addition
corresponds to trans-diaxial addition
trans-Diaxial (less stable)
trans-Diequatorial (more stable)
Br
Br
BrBrBr2

6-31
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation/ReductionOxidation/Reduction• Oxidation: the loss of electrons• Reduction: the gain of electrons• Recognize using a balanced half-reaction
• write a half-reaction showing one reactant and its product(s)
• complete a material balance. Use H2O and H+ in acid solution; use H2O and OH- in basic solution
• complete a charge balance using electrons, e-

6-32
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation with OsOOxidation with OsO44• Oxidation by OsO4 converts an alkene to a
glycol, a compound with -OH groups on two adjacent carbons• oxidation is syn stereoselective
H
HO
H
OH
+ ROOHOsO4
cis-Cyclopentanediol(a cis glycol)
Cyclopentene

6-33
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation with OsOOxidation with OsO44• Intermediate is a cyclic osmic ester containing a
five-membered ring
H
O
H
OOs
O OA cyclic osmic ester
O OOs
O O
H
HO
H
OH

6-34
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reduction of AlkenesReduction of Alkenes• Most alkenes react with H2 in the presence of a
transition metal catalyst to give alkanes• commonly used catalysts are Pt, Pd, Ru, and Ni
• The process is called catalytic reduction or, alternatively, catalytic hydrogenation
+ H2Pt
Cyclohexene Cyclohexane

6-35
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reduction of AlkenesReduction of Alkenes• the most common pattern is syn stereoselectivity
70% to 85%cis-1,2-Dimethyl-
cyclohexane
1,2-Dimethyl- cyclohexene
+
CH3
CH3
CH3
CH3
CH3
CH3
H2 / Pt
30% to 15%trans -1,2-Dimethyl-
cyclohexane

6-36
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reduction of AlkenesReduction of Alkenes• Mechanism of catalytic hydrogenation
• H2 is absorbed on the metal surface with formation of metal-hydrogen bonds
• the alkene is also absorbed with formation of metal-carbon bonds
• a hydrogen atom is transferred to the alkene forming one new C-H bond
• a second hydrogen atom is transferred forming the second C-H bond

6-37
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction StereochemistryReaction Stereochemistry• Consider the addition of bromine to 2-butene?
• three stereoisomers are possible for 2,3-dibromobutane; one pair of enantiomers
CH3 CH= CH2 CH3 CH3 CH- CHCH3
2-Butene 2,3-Dibromobutane
Br2Br Br

6-38
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction StereochemistryReaction Stereochemistry• how many stereoisomers are formed in the addition of
bromine to cis-2-butene?
C C HHH3 C CH3cis- -2-Butene
C CH
HH3 C
CH3Br
BrC C
H
HH3 C
CH3Br
Br
+
A pair of enantiomers(a racemic mixture)
Br2

6-39
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction StereochemistryReaction Stereochemistry• Consider the oxidation of 2-butene by OsO4
• three stereoisomers are possible for 2,3-butanediol; one pair of enantiomers and one meso compound
• which are formed in this oxidation?
CH3 CH= CH2 CH3 CH3 CH- CHCH3
2-Butene 2,3-Butanediol
OsO4OH OH

6-40
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction StereochemistryReaction Stereochemistry• syn hydroxylation of cis-2-butene produces only the
meso compound
identical; a meso compound
(2S,3R)-2,3-Butanediol
(2R,3S)-2,3-Butanediol
2
2
3
3C
HO
HO
CH
C
OH
HCH3
H3 C
C
OH
H3 CH
CH3H

6-41
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction StereochemistryReaction Stereochemistry• Enantiomerically pure products can never be
formed from achiral starting materials and achiralreagents
• An enantiomerically pure product can be generated in a reaction if at least one of the reactants is enantiomerically pure, or if the reaction is carried out in an achiral environment

6-42
66
Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reactions of Reactions of AlkenesAlkenes
End Chapter 6End Chapter 6



















































Chapter 15
AromaticityReactions of
Benzene
Organic Chemistry4th Edition
Paula Yurkanis Bruice
Irene LeeCase Western Reserve University
Cleveland, OH©2004, Prentice Hall

Criteria for Aromaticity1. A compound must have uninterrupted cyclic cloud of
π electrons above and below the plane of the molecule
2. The π cloud must contain an odd number of pairs of π electrons

Hückel’s Rule
For a planar, cyclic compound to be aromatic, its uninterrupted π cloud must contain (4n + 2) π electrons,where n is any whole number

Monocyclic hydrocarbons with alternating single and double bonds are called annulenes
Cyclobutadiene is not aromatic because it has an even number of electron pairs
Cyclooctatetraene is not aromatic because it is nonplanar

Neither cyclopropene nor the cyclopropenyl anion isaromatic
The cyclopropenecation is aromatic

sp3 sp3
cycloheptatriene cyclopentadiene
interrupted π cloudnot aromatic
2 pairs of π electronsnot aromatic

These compounds are aromatic

Aromatic Heterocyclic Compounds
A heterocyclic compound is a cyclic compound in whichone or more of the ring atoms is an atom other than carbon

Pyridine Is Aromatic

Pyrrole Is Aromatic

Furan Is Aromatic

Examples of Heterocyclic Aromatic Compounds

The Effect of Aromaticity on the pKaValues of Some Compounds


Aromaticity influences chemical reactivity

A compound is antiaromatic if it is a planar, cyclic compound with an uninterrupted ring of π cloud,but it contains even number of pairs of π electrons

A Molecular Orbital Description of Aromaticity and Antiaromaticity

Benzene is a nucleophile that reacts with an electrophile

Reaction Coordinate Diagrams for the Two Benzene Reactions

General Mechanism for ElectrophilicAromatic Substitution of Benzene
Y+
+
H
Y
H
Y
H
Y
Y
B:
slow
fast

Halogenation of Benzene



Nitration of Benzene

Sulfonation of Benzene



Friedel–Crafts acylation must be carried out with more than one equivalent of AlCl3


The carbocation will rearrange to a more stable species



alkylation of benzene by an alkene

It is not possible to obtain a good yield of an alkylbenzene containing a straight-chain group via Friedel–Crafts alkylation

However, a Friedel–Crafts acylation–reduction works well
This method avoids using a large excess of benzene in the reaction

Methodologies Used for the Reduction Step

One needs to consider an alternative if there is another functional group present in the compound

TARGET="display">












































77
7-1Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Introduction to Introduction to Organic Organic
ChemistryChemistry2 ed2 ed
William H. BrownWilliam H. Brown

77
7-2Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alkyl Alkyl HalidesHalides
Chapter 7Chapter 7

77
7-3Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
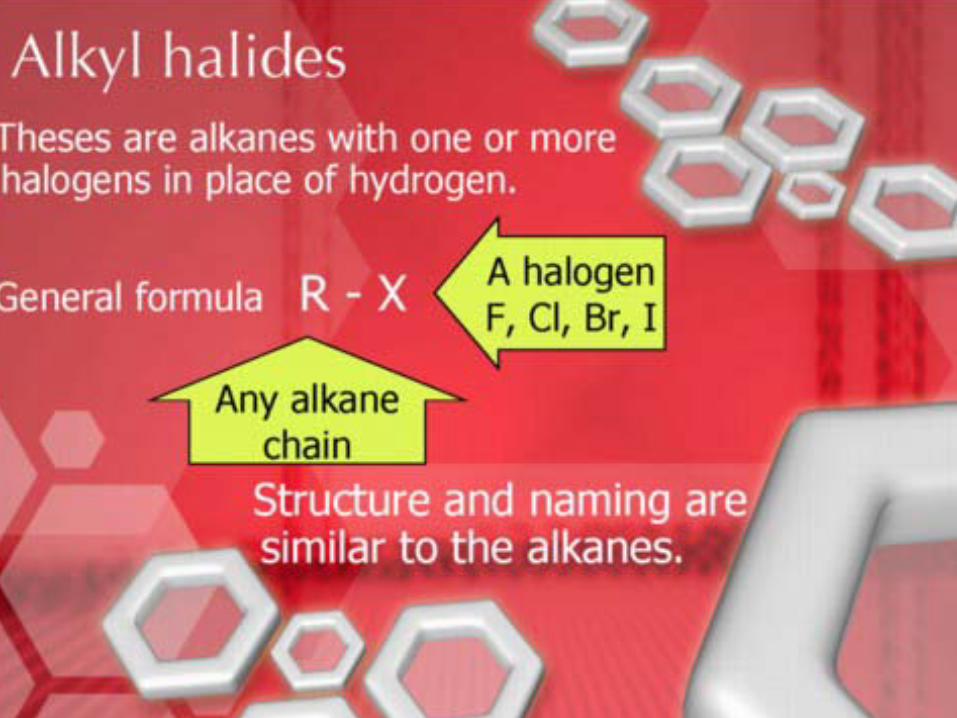
StructureStructure•• Alkyl halideAlkyl halide: : a compound containing a halogen
atom covalently bonded to an sp3 hybridized carbon atom• given the symbol RX
A haloalkane (an alkyl halide)
R X

77
7-4Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
VinylicVinylic and Aryl Halidesand Aryl Halides• if the halogen is bonded to an sp2 hybridized carbon,
the compound is called a vinylic halide• if the halogen is bonded to a benzene ring, it is called
an aryl halide, given the symbol Ar-X
• we do not study vinylic and aryl halides in this chapter
A haloalkene (a vinylic halide)
C CR
R
X
RX
A haloarene (an aryl halide)

77
7-5Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NomenclatureNomenclature• locate the parent alkane• number the parent chain to give the substituent
encountered first, be it halogen or an alkyl group, the lower number
• halogen substituents are indicated by the prefixesfluoro-, chloro-, bromo-, and iodo- and listed in alphabetical order with other substituents
3-Bromo-2-methylpentane 2,3-Dichlorobutane
Br CH3 Cl Cl
CH3 CHCH CH3CH3 CH2 CHCHCH31 12 2 3345 4

77
7-6Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NomenclatureNomenclature• for haloalkenes, numbering is determined by the
location of the C-C double bond
4-Bromo-cyclohexene
Br
1
23
4CH3 CCH= CH2
CH3
Cl3-Chloro-3-
methyl-1-butene
1234
56

77
7-7Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NomenclatureNomenclature• common names - name the alkyl group followed by the
name of the halide
3-Chloropropene (Allyl chloride)
Chloroethene (Vinyl chloride)
Chloroethane (Ethyl chloride)
CH3 CH2 Cl CH2 =CHCl CH2 =CHCH 2 Cl

77
7-8Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NomenclatureNomenclature• several polyhaloalkanes are common solvents and are
generally referred to by their common or trivial names
Dichloromethane (Methylene chloride)
Trichloromethane (Chloroform)
Trichloroethylene (Trichlor)
1,1,1-Trichloroethane (Methyl chloroform)
CHCl 3CH2 Cl2
CCl 2 =CHClCH3 CCl 3

77
7-9Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
FreonsFreons & Their Alternatives& Their Alternatives• The Freons are chlorofluorocarbons (CFCs).
Among the most widely used are/were
• Nonozone-depleting alternatives arehydrofluorocarbons (HFCs) andhydrochlorofluorocarbons (HCFCs) including
CCl3 FTrichlorofluoromethane
(Freon-11)
CCl2 F2Dichlorodifluoromethane
(Freon-12)
CH2 FCF3HFC-134a
CHCl2 CF3HCFC-123

77
7-10Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NucleophilicNucleophilic SubstitutionSubstitution
•• NucleophilicNucleophilic substitutionsubstitution: any reaction in which one nucleophile is substituted for another
•• NucleophileNucleophile: a molecule or ion that donates a pair of electrons to another molecule or ion to form a new covalent bond: a Lewis base
nucleophilic substitution +
•••• + C NuC XNu - X-

77
7-11Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NucleophilicNucleophilic SubstitutionSubstitution• One of the most important reactions of alkyl
halides
a thiol (a mercaptan)HS - CH3 SH
an ether
an alcohol
Reaction:+ -+CH3 NuCH3 Br Br
RO -
HO -
Nu
CH3 OH
CH3 OR

77
7-12Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NucleophilicNucleophilic SubstitutionSubstitution
+an alcohol (after proton transfer)
an alkylammonium ion
an alkyl iodide
H
I -
NH 3
HOH
CH3 I
CH3 NH3+
CH3 O-H
a sulfide (a thioether)RS - CH3 SR

77
7-13Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
MechanismsMechanisms• Chemists propose two limiting mechanisms for
nucleophilic aliphatic substitution• a fundamental difference between them is the timing of
bond-breaking and bond-forming steps• they are designated SN1 and SN2

77
7-14Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN22• Bond breaking and bond forming occur
simultaneously• S = substitution
N = nucleophilic2 = bimolecular
•• Bimolecular reactionBimolecular reaction: a reaction in which two reactants are involved in the transition state of the rate-limiting step

77
7-15Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN22• Simultaneous backside attack of the nucleophile
and departure of the leaving group
C Br
H
HH
HO- +
C
H
H H
HO Brδ- δ-
Transition state with simultaneous bond breaking and bond forming
C
H
HH
HO + Br -

77
7-16Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN11• Bond breaking is complete before bond forming
begins• S = substitution
N = nucleophilic1 = unimolecular
•• UnimolecularUnimolecular reactionreaction: a reaction in which only one species is involved in the transition state of the rate-limiting step

77
7-17Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN11• An SN1 mechanism is illustrated by the reaction
of 2-bromo-2-methylpropane (tert-butyl bromide) and methanol
CH3 C- BrCH3
+ CH3 OH CH3 C- OCH3
CH3
+ HBr
2-Bromo-2-methylpropane
Methanol tert -Butylmethyl ether
methanolCH3 CH3

77
7-18Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN11• Step 1: ionization of the C-X bond to form a
carbocation intermediate
C
CH3
CH3H3 C
+C
H3 C
H3 CBr
H3 C
slow, rate-limiting step
A carbocation intermediate; carbon is trigonal planar
+ Br -

77
7-19Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN11• Step 2: reaction of the carbocation (a Lewis acid) with
methanol (a Lewis base) to form an oxonium ion
C
CH3
CH3H3 CC
CH3
CH3CH3
O
H3 C
HC
H3 C
H3 C
O
H3 CH
CH3
+ CH3 OH
++ +
+

77
7-20Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism Mechanism -- SSNN11• Step 3: proton transfer to solvent completes the
reaction
••
••
•• + ••+
+
••••+ fastC
H3 CH3 C
O
H3 C
OCH3
H
OHOCH3
CH3
H
H
CH3
H3 C
CH3 C
H3 C

77
7-21Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Stereochemistry of SStereochemistry of SNN11• For an SN1 reaction at a stereocenter, the product
is a racemic mixture
(R)-Enantiomer Planar carbocation (achiral)
CH
Cl
C6 H5
Cl
C+
C6 H5
H
Cl
CH3 OH-Cl -
-H +
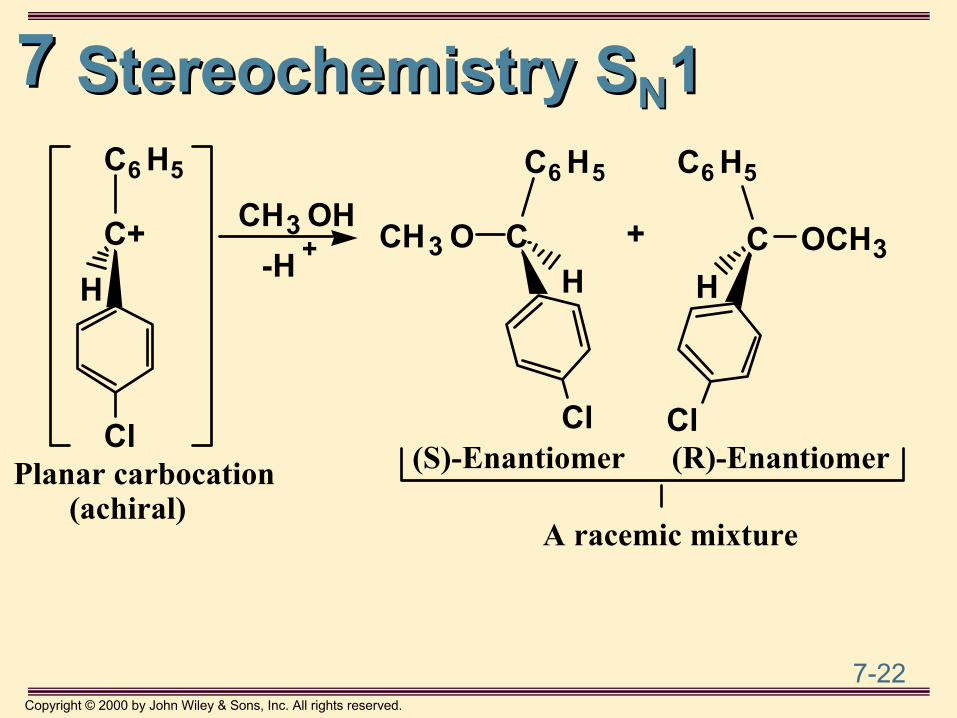
77
7-22Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Stereochemistry SStereochemistry SNN11
+
A racemic mixture
Cl
C6 H5 C6 H5
C OCH3H
CH3 O CH
Cl(R)-Enantiomer(S)-EnantiomerPlanar carbocation
(achiral)
C+
C6 H5
H
Cl
CH3 OH-H +

77
7-23Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Evidence of SEvidence of SNN reactionsreactions• What effect of does
• the structure of the nucleophile have on rate?• the structure of alkyl halide have on rate?• the structure of the leaving group have on rate?• the solvent have on the reaction mechanism?

77
7-24Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NucleophilicityNucleophilicity•• NucleophilicityNucleophilicity: a kinetic property measured by
the rate at which a Nu attacks a reference compound under a standard set of experimental conditions• for example, the rate at which a set of nucleophiles
displaces bromide ion from bromoethane in ethanol at 25°C
• Table 7.2 shows common nucleophiles and their relative nucleophilicities

77
7-25Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NucleophilicityNucleophilicity
good
poor
Br - , I-
HO- , CH3 O- , RO-
CH3 S- , RS-
CH3 CO2- , RCO2
-
H2 OCH3 OH, ROHCH3 CO2 H, RCO2 H
NH3 , RNH2 , R2 NH, R3 NCH3 SH, RSH, R2 S
Effectiveness Nucleophile
moderate

77
7-26Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure of RXStructure of RX• SN1 reactions are governed by electronic factorselectronic factors,
namely the relative stabilities of carbocationintermediates• 3° > 2° > 1° > methyl
• SN2 reactions are governed by stericsteric factorsfactors, namely the relative ease of approach of thenucleophile to the site of reaction• methyl > 1° > 2° > 3°

77
7-27Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure of RXStructure of RX• Reactivities for SN1 and SN2 are in opposite
directions
R3 CX R2 CHX RCH 2 X CH 3 X(3°) (2°) (methyl)(1°)
Increasing stability of cation intermediate
Increasing ease of access to the reaction site
SN1
SN2

77
7-28Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
The Leaving GroupThe Leaving Group• The more stable the anion, the better the leaving
ability• the most stable anions are the conjugate bases of
strong acids
Increasing leaving ability
I- > Br - > Cl - >> F - > CH 3 CO2- > HO - > NH 2
-
Stability of anion; strength of conjugate acid

77
7-29Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CommonCommon ProticProtic SolventsSolvents•• ProticProtic solventsolvent: a solvent that contains an -OH
group • These solvents favor SN1 reactions; the greater the
polarity of the solvent, the easier it is to formcarbocations in it
Solvent Structurewaterformic acidmethanolethanol
H2 OHCO2 HCH3 OHCH3 CH2 OH
acetic acid CH3 CO2 H
Solvent Polarity

77
7-30Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
CommonCommon AproticAprotic SolventsSolvents•• AproticAprotic solventsolvent:does not contain an -OH group
• it is more difficult to form carbocations in aproticsolvents
• these solvents favor SN2 reactions
diethyl etherdichloromethane
Aprotic Solvent Structure
dimethyl sulfoxideacetone
CH2 Cl2(CH 3 CH2 ) 2 O
(CH 3 ) 2 S=O
Solvent Polarity
(CH 3 ) 2 C=O

77
7-31Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Summary of SSummary of SNN RexnsRexnsAlkyl Halide
CH3Xmethyl
RCH 2Xprimary
R2CHXsecondary
SN 2 SN 1
SN 2 favored SN 1 does not occur; the methyl cation too unstable
SN 1 rarely occurs; 1° cations rarely observed in solution
SN 1 favored in protic solvents with poor nucleophiles
SN 2 favored in aprotic solvents with good nucleophiles
SN 2 favored

77
7-32Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Summary of SSummary of SNN RexnsRexns
R3CXtertiary
Substitution at stereocenter
SN 2 does not occur; steric hindrance
SN 1 favored; ease of formation of 3° carbocations
Inversion of configuration
Racemization
Alkyl Halide SN 2 SN 1

77
7-33Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
SSNN1/S1/SNN2 Problems 2 Problems -- 11• Predict the type of mechanism for this reaction,
and the stereochemistry of the product
+
+
Cl
OCH3
CH3 CHCH2 CH3
CH3 CHCH2 CH3
CH3 OH
HCl(R)-enantiomer

77
7-34Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
SSNN1/S1/SNN2 Problems 2 Problems -- 22• Predict the mechanism of this reaction
+
+
DMSO
CH3
CH3CH3 CHCH2 I
CH3 CHCH2 Br Na+ I-
Na+ Br -

77
7-35Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
SSNN1/S1/SNN2 Problems 2 Problems -- 33• Predict the mechanism and the configuration of
product
+
+
acetone
Br
SCH3
CH3 CHCH2 CH3 CH3 S- Na +
CH3 CHCH2 CH3 Na+ Br -
(S configuration)

77
7-36Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ββ--EliminationElimination•• ββ--EliminationElimination: removal of atoms or groups of
atoms from adjacent carbons to form a carbon-carbon double bond
β α
C C +
An alkyl halide
Base
An alkene
CH3 CH2 O-Na+CH3 CH2 OH
CH3 CH2 OH Na+ X -+
+C CH X

77
7-37Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ββ--EliminationElimination• Example
CH3 ( CH2 ) 7 CH2CH2 Br
CH3 ( CH2 ) 7 CH= CH2
αβ( CH3 ) 3 CO- K+
1-Bromodecane Potassiumtert-butoxide
( CH3 ) 3 COH + K+ Br -
1-Decene+
+

77
7-38Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ββ--EliminationElimination•• ZaitsevZaitsev rulerule: the major product of a β-elimination
is the more highly substituted alkene
+
Br
CH3 CH= CCH3 CH3 CH2 C= CH2
CH3 CH2 CCH 3
2-Methyl-2-butene(major product)
CH3 CH2 O-Na+
CH3 CH2 OHCH32-Bromo-2-
methylbutaneCH3 CH3
2-Methyl-1-butene

77
7-39Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ββ--EliminationElimination• Example (follows Zaitsev rule)
+
CH3
CH3 CH2
1-Methyl- cyclopentene
(major product)
CH3 O- Na +
CH3 OH1-Bromo-1-methyl-
cyclopentane
Br
Methylene- cyclopentane

77
7-40Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ββ--EliminationElimination• There are two limiting mechanisms for β-
elimination reactions•• E1 mechanismE1 mechanism: breaking of the R-X bond is complete
before reaction with base to break the C-H bond begins. Only R-X is involved in the rate-limiting step.
•• E2 mechanismE2 mechanism: breaking of the R-X and C-H bonds is concerted. Both R-X and base are involved in the rate-limiting step.

77
7-41Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
E1 MechanismE1 Mechanism• Step 1: rate-limiting ionization of the C-X bond to form
a carbocation intermediate
slow, rate limiting
+(a 3° carbocation
intermediate)
CH3 -C- CH3
CH3
Br
CH3 -C- CH3
CH3
+ Br -

77
7-42Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
E1 MechanismE1 Mechanism• Step 2: proton transfer from the carbocation
intermediate to the base (in this case, the solvent)
fast+
OH
H3 C
CH2 = C- CH3
CH3
OH
H3 CH+ +
H- CH2 - C- CH3
CH3

77
7-43Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
E2 MechanismE2 Mechanism• A one-step mechanism; all bond-breaking and
bond-forming steps are concerted
+ H-CH-CH2-Br
CH3
CH3 CH2 OH CH3 CH= CH2+ + Br -
CH3 CH2 O-

77
7-44Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Summary of E1 versus E2Summary of E1 versus E2
E2
Primary cations are rarely formed in solution and, therefore, E1reactions of primary halides are rarely observed.
Halide Reaction Comments
primary
( RCH2 X)E1
Main reaction with strong bases such as HO- and RO-

77
7-45Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Summary of E1 versus E2Summary of E1 versus E2E2
E2
E1
secondary
tertiary
Main reaction with strong bases such as HO- and RO-
( R3 C- X)
( R2 CH- X)
Main reaction with strong bases such as HO- and RO-
Main reaction with weak bases such as CH3CO2
- and ROH
E1 Common in reactions with weak bases such as CH 3CO2
-

77
7-46Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alkyl Alkyl HalidesHalides
End Chapter 7End Chapter 7

Chapter 8
Nucleophilic Substitution Reactions

Reactivity of Alkyl halides
Structure– sp3
– permanent dipole– polar
Alkyl halides react in substitution reactions.Vinyl halides or aryl halides do NOT react
δ−δ+
HH C Cl
H
ClBr

Nucleophilic Substitution
Reaction:
Nu_
R X+ R Nu + X-
nucleophile electrophile
leaving group

Substitution Reaction
Nucleophilic Substitution:– nucleophile can be an anion:
– or a neutral molecule with lone pair of electrons:
Nu_
R X+ R Nu + X-
X-+R Nu+ R XNu+

Different nucleophiles produce different products:
Nu +R Nu+R X X
+R X OH + XR OH alcohol
+R X O CH3 + XO CH3R ether
+R X SH R SH + X thiol
+R X SCH3 R SCH3 + X thioether

Nu +R Nu+R X X
Br+R X
R X + I
+R X C N
C C CH3R X +
R Br + X alkyl bromide
R I X+ alkyl iodide
R C N + X nitrile
X+R C C CH3

Substitution of One Halide for Another
(Lab Experiment 8.1)
– NaCl and NaBr are insoluble in acetone:
– Hard to make alkyl fluorides: need lots of heat
Br + NaII + NaBr (ppt)acetone
Cl + NaIacetone I + NaCl (ppt)
Br + KF
ethyleneglycol
(b.p. 120)F + KBr

Neutral nucleophiles (amines):
Form alkyl ammonium salts
+R X NH3
R X + CH3N H
H
+ XR NH3+
R NCH3
H
H
X++

Predict the products:
CH3CH2CH2CH2Cl OH+
Br
+ I
CH2CH2CH2 I + CN
CH3CH2CH2CH2OH + Cl
I
Br+
CH2CH2CH2 CN + I

Predict the products:
Br + CH3C
OO-
I +
Br +
NaN3
CH3SNa
O CCH3
O
N3
SCH3
+ Br-
+ NaI
+ NaBr

Nucleophilic SubstitutionRelative Reactivity of Halide Leaving Groups
RF <<<<< RCl < RBr < RI
fluorides are very, very slow to reactfluorides are worst “leaving group”– leaving group ability related to basicity: – weaker the base, better the leaving group
fluoride is strongest base, worse leaving groupiodide is weakest base, best leaving group
Nu_
R X+ R Nu + X-
nucleophile electrophile
leaving group

Rate Law for SN2 Reaction:Bimolecular: two species involved in rate-determining step (second order reaction)– both nucleophile and electrophile (RX) are
important to rate of reaction; increasing concentrations of either increase rate of reaction.
– rate = k[nucleophile][RX]Energy diagram: one step = one hump– Ea, TS, ∆H

Mechanism of Substitution Reactions: SN2
SN2 (substitution, nucleophilic, bimolecular)– concerted (one-step) reaction:– nucleophile attacks back-side the carbon with
the leaving group
C
H
H
H
BrH O C
H
H
H
H O + Br
C BrH
HHH O +
C
H
HO Br
HH
δ−δ−
transition state
H O CH
+ BrH
H

Stereochemistry: inversion of configuration:
Backside attack on a stereogenic center:
– configuration changes: inversion
C BrH
+C
H
HO Brδ−δ−
transition state
CH3 CH2CH3
H OCH2CH3
CH3 CH2CH3
H O CH
CH3
(S) (R)
H Br H
(R)-2-bromobutane (S)-2-iodobutane
I- I

Predict the Products
Show stereochemistry, where appropriate
CH2CH3
HCH3
Br NaI
acetone
IH
NaN3
DMF
I
HCH3
CH2CH3
N3H

Steric Effects in the SN2 Mechanism:
Backside attack: easier it is for nucleophile to get close to the carbon, the faster the reaction:
C BrH
HHH O + methyl halide
C BrH
C
H
H
H C HHH
C BrC
C
H
H
H
HH
H
C HHH
C BrH
C
H
H
H H
primary halide secondary halide tertiary halide
methyl > primary > secondary >>>> tertiary(doesn’t go at all)

Steric Effects in the SN2 Mechanism:
β-branching also affects rate of SN2 reaction
– methyl bromide >>> ethyl bromide
– 1-bromopropane >>> 1-bromo-2-methylpropane
– neopentyl halides??? Primary halide, but very sterically hindered….lots of β-branching

Nucleophiles and Nucleophilicity
Nucleophilicity increases from right to left across a row (opposite trend to electronegativity)
nucleophilicity
RCH2- RNH- RO- F> > > nucleophilicity
_

Periodic Trends:Nucleophilicity increases going down a column
nucleophilicity
I- Br- Cl- F-
strongestnucleophile
weakestnucleophile

Periodic Trends down a column:Polarizability:
– polarizability and size increase going down a column
– polarizability increases nucleophilicity
nucleophilicity
size

Effect of Charge
Anions are stronger nucleophiles than their neutral conjugate acids:
Stronger nucleophile
weaker nucleophile
H O
HH O >
_
>R O R O
H
_
> NH3NH2

Some Common Nucleophiles:
I. Strong nucleophiles– Very good nucleophiles I-, HS-, RS-– Good nucleophiles Br-, OH-, RO-, CN-, N3
-
II. Fair Nucleophiles: NH3, Cl-, F-, RCO2-
III. Poor Nucleophiles– Weak nucleophiles: H2O, ROH– Very weak nucleophiles: RCO2H

Practice problems:
Identify the stronger nucleophile:
H2Sa) HS
PH3 NH3 AsH3b)
c) CH3Te CH3S CH3O
d) CH3O CH3NCH3CH3CCH3
CH3

Practice problems:
Identify the stronger base:
H2Sa) HS
PH3 NH3 AsH3b)
c) CH3Te CH3S CH3O
d) CH3O CH3NCH3CH3CCH3
CH3

tertiary halides can’t do SN2 (steric hindrance)neutral molecules are poor nucleophiles - generally don’t do SN2But this reaction works, so there must be another way to do nucleophilic substitution!!
C
CH3
CH3
CH3
Br + C
CH3
CH3
CH3
OCH3 + H BrCH3OH
MTBE
SN1 Mechanism

Mechanism of SN1 Reaction:
SN1- substitution nucleophilic unimolecular
– rate depends only upon the concentration of the electrophile (substrate, RX), not the nucleophile
– first order reaction
– rate = k [RX]

Mechanism:Step 1: loss of the leaving group to generate carbocation (slow step = rds)
Step 2: addition of nucleophile to carbocation
(Step 3: loss of proton to generate neutral product)
C
CH3
CH3
CH3
Br C
CH3
CH3
CH3
+ + Br_
CH3OHC
CH3
CH3
CH3
++ +C
CH3
CH3
CH3
O CH3
H
+C
CH3
CH3
CH3
O CH3
H
C
CH3
CH3
CH3
O CH3H+
+

Implications of SN1 Mechanism:
Intermediate is carbocation:– the more stable the carbocation, the lower
energy it has and the easier it is to form• tertiary > secondary >>> primary >>>>> methyl
Energy diagram:– 2 main steps= 2 humps– 2 transition states, 1 intermediate– formation of carbocation is rate determining
step:

Stereochemistry: stereorandom reaction
Carbocations are sp2 hybridized --> planar
nucleophile can attack backside or frontside -->CH3CH2CH2
CH3 C CH2CH3
CH3CH2CH2
CH3 C CH2CH3
OH
C CH2CH3CH3
CH3CH2CH2
OHH
OHH
OH
C CH2CH3
CH3
CH3CH2CH2
Can get a mixture of enantiomers with SN1 reaction

Example:
Predict the products:
– not totally racemization• carbocation-halide ion pair• preferential attack on other side of
carbocation– rearrangement
CCH3CH2
Br
CH3
HH2O∆
CCH3CH2
OH
HCH3
CCH3CH2
OHCH3
H
+
racemic mixture

Solvent Effects
Solvent can effect rate of substitution reaction
Rate of SN1 reaction increases in polar solvents (solvents with high dielectric constant)
Dielectric: ability to support separated positive and negative charge species
Polar solvent with high dielectric constant helps stabilize transition state– help charge separation

Solvent Effects in SN1
Comparison of tert-butyl chloride with various solvents:
Rate Solvent Dielectric1 acetic acid 64 methanol 335,000 formic acid 58
150,000 water 78

Solvent Effects in SN2
Solvent must be polar enough to dissolve nucleophile (ionic)Protic vs aprotic solvents– protic solvents have OH group (water, alcohol,
etc.); solvate nucleophiles– aprotic solvents lack OH group, can’t solvate
nucleophiles, so are more reactiveCommon polar, aprotic solvents:– acetone, DMF, DMSO, acetonitrile

Solvent Effects in SN2
Rate of Reaction of 1-bromobutane with azide ion
Rate Solvent Dielectric Type1 methanol 32.6 polar protic7 water 78.5 polar protic
1,300 DMSO 48.9 polar aprotic2,800 DMF 36.7 polar aprotic5,000 CH3CN 37.5 polar aprotic

Solvents:
Polar, protic solvents favor SN1
Polar, aprotic solvents favor SN2:
CH3OHH2O CH3CH2OH CH3C
O
OH
CH3CCH3
ON
CH3
CH3C
O
HS
CH3
CH3
O
acetone dimethylformamide (DMF)
dimethylsulfoxide (DMSO)

SN2 vs SN1 Reaction:Effect of substrate (RX)– tertiary halides only do SN1– primary and methyl halides only do SN2– secondary halide - can do either SN2 or SN1
Effect of nucleophile– good nucleophiles (anions) do SN2 if they can– poor nucleophiles (neutral species) do SN1
Effect of solvent: – polar protic solvents (having OH) favor SN1– polar, aprotic solvents (without OH) favor SN2

SN2 or SN1?
Substitution: SN2 or SN1
SN2 only
Methyl or primary
SN2apolar protic solvent
Strong nucleophile(polarizable anion)
SN1polar protic solvent
Poor nucleophile(neutral species)
Secondary
SN1 only
Tertiary
Electrophile/Substrate

Elimination Reactions:
Alkyl halides can also undergo elimination reactions:
– elimination is favored by:• strong base• tertiary > secondary > primary (never methyl)
Nu+C C Br
H
H
H
H
H
C CH
H
H
H+ Br+Nu H

E2 Mechanism (dehydrohalogenation)
Elimination, bimolecular:– concerted one-step reaction– both base and alkyl halide are involved in the
rate determining step
– rate = k[base][alkyl halide]– proton and leaving group must be anti to each
other.
C CH
XHH
HH
B
C CH
H
HH
X
H
B
C CH
H
H
HH B+
X+

Predict the products of E2 elimination
Forms alkene between carbon containing the leaving group and an adjacent carbon having a proton
forms most stable alkene (Saytzeff’s Rule)
Br OH-
12 3 4
major minor
1 2 34
IOH-

Factors affecting Elimination:
Strong bases (OH-, NH2-, CH3O- or other
alkoxides) favor elimination reactions. (E2)
Weak bases (halide ions, NH3, PH3) favor substitution
With SN1 reactions, usually get some E1 too (common intermediate)

Summary:Species that are Good Nucleophiles but Weak Bases promote SN2 reactions
I-, Br-, Cl-, HS-, NH3, PH3,
Species that are Good Nucleophiles but Strong Bases promote both SN2 and E2 (depends on substrate)
OH-, RO-, H2N-
Species that are Poor Nucleophiles and Weak Bases promote SN1/(E1) reactions
H2O, ROH

Comparison of Reactions:Substrate
(RX)Good Nuc,Weak Base
Good Nuc,StrongBase
Poor NucWeak Base
I- Br- HS-
NH3 PH3
OH- RO-
NH2-
H2O ROH
methyl SN2 SN2 No Rxn
primary SN2 MostlySN2 (some
E2)
No Rxn
secondary SN2 E2 SN1
tertiary No Rxn E2 SN1

Sulfonate Esters as SubstratesSulfonate esters are excellent leaving groups:
– very weak bases, since conjugate bases of very strong acids
– abbreviation:
CH3 S
O
O
O H
methanesulfonic acid
S
O
O
O HCH3
p-toluenesulfonic acid
O S
O
CH3
O
O S
O
O
CH3
OTs
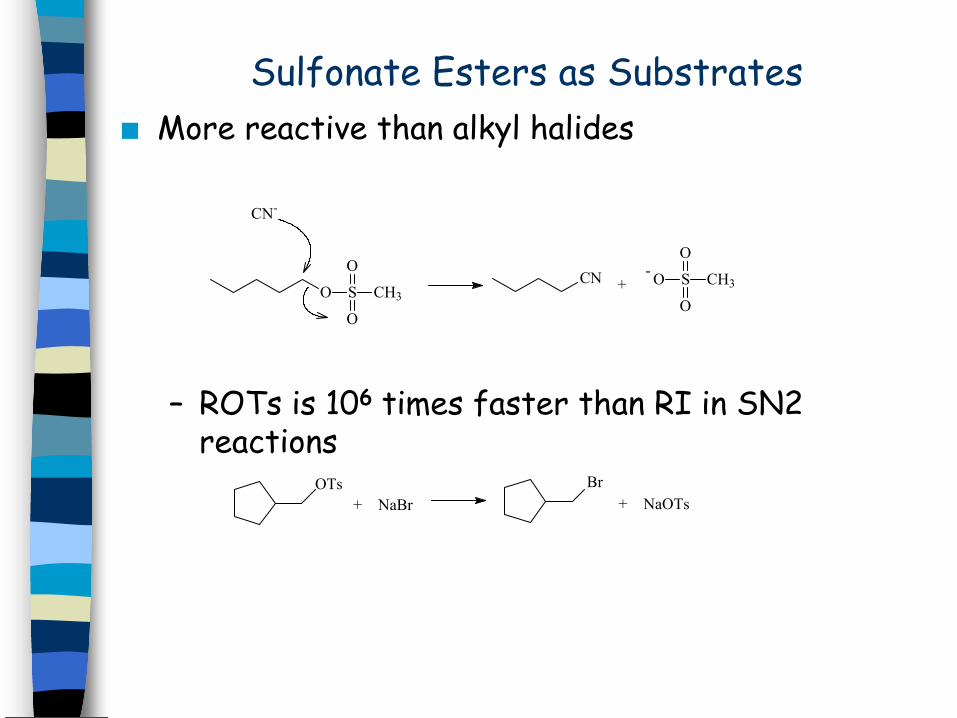
Sulfonate Esters as SubstratesMore reactive than alkyl halides
– ROTs is 106 times faster than RI in SN2 reactions
O S
O
CH3
O
CN-
CN + O S
O
CH3
O
-
OTs+ NaBr
Br+ NaOTs

Sulfonate Ester StereochemistryPreparing tosylate:
– stereochemistry retained (no bond breaking to chiral center)
SN2 reaction: inversion ofconfiguration
OHH
(S)
OH
S
O
O
(S)
CH3p-tosyl chloride
pyridine
OH
S
O
O
(S)
CH3
NaCN
DMFH
CN
(S)

Predict the Products:
Br CN-
acetone
Cl H I-
DMF
I H2O
Cl H OH-
a)
b)
d)
CN (SN2)
H I
(SN2)
(E2)
OH+
(SN1) (E1)

Predict the Products:
I OH-
ClCH3O-
Cl CH3OH
HBr
CH3OHh)
e)
f)
g)
(E2)
OCH3 (SN2)
No Reaction
OCH3 OCH3H H
racemic mixture
(SN1) (E1)

88
8-1Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Introduction to Introduction to Organic Organic
ChemistryChemistry2 ed2 ed
William H. BrownWilliam H. Brown

88
8-2Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alcohols, Alcohols, Ethers, andEthers, andThiolsThiols
Chapter 8Chapter 8

88
8-3Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure Structure -- AlcoholsAlcohols• The functional group of an alcohol
is an -OH group bonded to an sp3
hybridized carbon• bond angles about the hydroxyl
oxygen atom are approximately 109.5°• Oxygen is also sp3 hybridized
• two sp3 hybrid orbitals form sigma bonds to carbon and hydrogen
• the remaining two sp3 hybrid orbitalseach contain an unshared pair of electrons

88
8-4Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure Structure -- EthersEthers• The functional group of an ether is an oxygen
atom bonded to two carbon atoms• Oxygen is sp3 hybridized with bond angles of
approximately 109.5°. In dimethyl ether, the C-O-C bond angle is 110.3°
H
H O
H
C H
H
H
C••
••

88
8-5Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Structure Structure -- ThiolsThiols• The functional group of a thiol is an
-SH (sulfhydryl) group bonded to an sp3 hybridized carbon
• The bond angle about sulfur inmethanethiol is 100.3°, which indicates that there is considerably more p character to the bondingorbitals of divalent sulfur than there is to oxygen

88
8-6Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
NomenclatureNomenclature--AlcoholsAlcohols• IUPAC names
• the longest chain that contains the -OH group is taken as the parent.
• the parent chain is numbered to give the -OH group the lowest possible number
• the suffix -ee is changed to -olol• Common names
• the alkyl group bonded to oxygen is named followed by the word alcoholalcohol

88
8-7Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlcoholsAlcohols
1-Propanol(Propyl alcohol)
CH3 CH2 CH2 OH
2-Propanol(Isopropyl alcohol)
OH
CH3 CHCH3
1-Butanol(Butyl alcohol)
CH3 CH2 CH2 CH2 OH
2-Butanol(sec-Butyl alcohol)
OH
CH3 CH2 CHCH3
2-Methyl-1-propanol (Isobutyl alcohol)
2-Methyl-2-propanol (tert-Butyl alcohol)
CH3
CH3
CH3CH3 CHCH2 OH
CH3 COH

88
8-8Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlcoholsAlcohols• Problem: write IUPAC names for these alcohols
(a) (b)
(c)
CH3 OH
CH3
CH3 CHCH 2 CHCH3
OH
(d) C
CH2 OH
H3 CHCH2 CH3
CH3 ( CH2 ) 6CH 2OH

88
8-9Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlcoholsAlcohols• Compounds containing more than one -OH group
are named diols, triols, etc.
1,2-Ethanediol(Ethylene glycol)
1,2-Propanediol(Propylene glycol)
1,2,3-Propanetriol(Glycerol, Glycerin)
HO OH
HO OH HO HO OH
CH3 CHCH 2
CH2 CH2
CH2 CHCH2

88
8-10Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- AlcoholsAlcohols• Unsaturated alcohols
• the double bond is shown by the infix -enen-• the hydroxyl group is shown by the suffix -olol• number the chain to give OH the lower number
HOCH2 CH2
C CCH2 CH3
H H
4
5 6
trans -3-Hexen-1-ol
3
21

88
8-11Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- EthersEthers• IUPAC: the longest carbon chain is the parent. Name the
OR group as an alkoxy substituent• Common names: name the groups attached to oxygen
followed by the word etherether
Ethoxyethane(Diethyl ether)
2-Methoxy-2-methylpropane(methyl tert -butyl ether, MTBE)
CH3
CH3
CH3 CH2 OCH 2 CH3
CH3 OCCH3
trans -2-Ethoxycyclohexanol
OCH2 CH3
OH

88
8-12Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- EthersEthers• Although cyclic ethers have IUPAC names, their
common names are more widely used
Ethylene oxide
Tetrahydro- furan, THF
Tetrahydro- pyran
1,4-DioxaneO O
O
OO

88
8-13Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Nomenclature Nomenclature -- ThiolsThiols• IUPAC names:
• the parent is the longest chain that contains the -SH group
• change the suffix -ee to -thiolthiol• Common names:
• name the alkyl group bonded to sulfur followed by the word mercaptanmercaptan
1-Butanethiol(Butyl mercaptan)
CH3 CH2 CH2 CH2 SH2-Butanethiol
(sec-Butyl mercaptan)
SH
CH3 CH2 CHCH3

88
8-14Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop Physical Prop -- AlcoholsAlcohols• Alcohols are polar compounds
• Alcohols are associated in the liquid state by hydrogen bonding
δ-
δ+
δ+O
HH
H
C
H

88
8-15Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop Physical Prop -- AlcoholsAlcohols•• Hydrogen bondingHydrogen bonding: the attractive force between a
partial positive charge on hydrogen and a partial negative charge on a nearby oxygen, nitrogen, or fluorine atom
• the strength of hydrogen bonding in water is approximately 5 kcal/mol• hydrogen bonds are considerably weaker than
covalent bonds• nonetheless, they can have a significant effect on
physical properties

88
8-16Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop Physical Prop -- AlcoholsAlcohols• Ethanol and dimethyl ether are constitutional
isomers. • Their boiling points are dramatically different
• ethanol forms intermolecular hydrogen bonds which increases attractive forces between its molecules, which results in a higher boiling point
bp -24°CEthanolbp 78°C
Dimethyl etherCH3 CH2 OH CH3 OCH3

88
8-17Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- AlcoholsAlcohols• In relation to alkanes of comparable size and
molecular weight, alcohols• have higher boiling points• are more soluble in water
• The presence of additional -OH groups in a molecule further increases boiling points and solubility in water

88
8-18Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- AlcoholsAlcoholsFormula Name MW
bp (°C)
Solubility in Water
methanol 32 65 infiniteethane 30 -89 insoluble
ethanol 46 78 infinitepropane 44 -42 insoluble
1-propanol 60 97 infinitebutane 58 0 insoluble
1-pentanol 88 138 2.3 g/100 g1,4-butanediol 90 230 infinite
hexane 86 69 insoluble
CH 3 CH 2 CH 2 OHCH 3 CH 2 CH 2 CH 3
CH 3 OHCH 3 CH 3
CH 3 CH 2 OHCH 3 CH 2 CH 3
CH 3 ( CH 2 ) 4 OHHO ( CH 2 ) 4 OH
CH 3 ( CH 2 ) 4 CH 3

88
8-19Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- EthersEthers• Ethers are polar molecules;
• the difference in electronegativitybetween oxygen (3.5) and carbon (2.5) is 1.0
• each C-O bond is polar covalent• oxygen bears a partial negative
charge and each carbon a partial positive charge

88
8-20Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- EthersEthers• Ethers are polar molecules, but because of steric
hindrance, only weak attractive forces exist between their molecules in the pure liquid state
• Boiling points of ethers are • lower than alcohols of comparable MW and • close to those of hydrocarbons of comparable MW
• Ethers hydrogen bond with H2O and are more soluble in H2O than are hydrocarbons

88
8-21Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- ThiolsThiols• Low-molecular-weight thiols have a STENCHSTENCH
• the scent of skunks is due primarily to these two thiols
2-Butene-1-thiol3-Methyl-1-butanethiol
CH3 CH= CHCH 2 SHCH3 CHCH 2 CH2 SH
CH3

88
8-22Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Physical Prop. Physical Prop. -- ThiolsThiols• The difference in electronegativity between S
(2.5) and H (2.1) is 0.4. Because of the low polarity of the S-H bond, thiols• show little association by hydrogen bonding• have lower boiling points and are less soluble in water
than alcohols of comparable MW
1177865
1-butanolethanolmethanol
9835
6
1-butanethiolethanethiolmethanethiol
bp (°C)Alcoholbp (°C)Thiol

88
8-23Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Acidity of AlcoholsAcidity of Alcohols• In dilute aqueous solution, alcohols are weakly
acidic
HHOH
[CH 3 O-] [H 3 O+ ][CH 3 OH]
CH3 O-H O H
H
CH3 O -+
Ka =
+ +
= 15.5

88
8-24Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Acidity of AlcoholsAcidity of AlcoholsCompound pKa
-7
15.5
15.7
15.9
17
18
4.8
CH3 OH
H2 O
CH3 CH2 OH
(CH 3 ) 2 CHOH
(CH 3 ) 3 COH
CH3 CO2 HHClhydrogen chloride
acetic acid
methanol
water
ethanol
2-propanol
2-methyl-2-propanol
FormulaStronger acid
Weaker acid

88
8-25Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
BasicityBasicity of Alcoholsof Alcohols• In the presence of strong acids, the oxygen atom
of an alcohol behaves as a weak base • proton transfer from the strong acid forms an oxonium
ion
••
••
•• ••
An acidA base
An oxonium ion
+
++••
••+
H
HCH3 H
HCH3
H
H
H
H
OO
O
H2 SO4
O

88
8-26Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction with MetalsReaction with Metals• Alcohols react with Li, Na, K, and other active
metals to liberate hydrogen gas and form metalalkoxides
2 CH3 OH + 2 Na 2 CH3 O- Na + H2Sodium
methoxide
+
Methanol

88
8-27Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Conversion of ROH to RXConversion of ROH to RX• Conversion of an alcohol to an alkyl halide
involves substitution of halogen for -OH at a saturated carbon • the most common reagents for this purpose are the
halogen acids, HX, and thionyl chloride, SOCl2

88
8-28Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction with HXReaction with HX• Water-soluble 3° alcohols react very rapidly with
HCl, HBr, and HI. Low-molecular-weight 1° and 2° alcohols are unreactive under these conditions
CH3 COH 25°C
2-Methyl-2-propanol
2-Chloro-2-methylpropane
+ HCl CH3 CCl + H2 OCH3 COH 25°C
2-Methyl-2-propanol
2-Chloro-2-methylpropane
+ HCl + H2 OCH3 CH3
CH3CH3

88
8-29Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction with HXReaction with HX• Water-insoluble 3° alcohols react by bubbling
gaseous HX through a solution of the alcohol dissolved in diethyl ether or THF
CH3
OHHCl
CH3
ClH2 O
1-Chloro-1-methyl- cyclohexane
1-Methylcyclo- hexanol
0 oCether
+ +

88
8-30Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction with HXReaction with HX• 1° and 2° alcohols require concentrated HBr and
HI to form alkyl bromides and iodides
CH3 CH2 CH2 CH2 OH HBr
CH3 CH2 CH2 CH2 Br H2 O
H2 Oreflux
1-Bromobutane
1-Butanol+
+

88
8-31Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism: 3Mechanism: 3°° ROH +ROH + HClHCl• An SN1 reaction
• Step 1: rapid, reversible acid-base reaction that transfers a proton to the OH group
2-Methyl-2-propanol(tert-Butyl alcohol)
••••
••
An oxonium ion
+
rapid andreversible
+••••
••+
+
CH3
CH3
O- H
CH3
CH3
OH
H
H
H
H
H HCH3 - C
CH3 - C O
O

88
8-32Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism: 3Mechanism: 3°° ROH +ROH + HClHCl• Step 2: loss of H2O to give a carbocation intermediate
••
An oxonium ion
+rate-limiting
step+
••••
CH3
CH3
OH
H
H
H
CH3 -C O
CH3
CH3
CH3 -C +
A 3° carbocation intermediate

88
8-33Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism: 3Mechanism: 3°° ROH +ROH + HClHCl• Step 3: reaction of the carbocation intermediate (a
Lewis acid) with halide ion (a Lewis base)
2-Chloro-2-methylpropane (tert-Butyl chloride)
••
••
••
•• •••• ••rapidCl+
CH3
CH3
CH3
CH3
CH3 - C-ClCH3 - C+

88
8-34Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism: 1Mechanism: 1°°ROH +ROH + HBrHBr• An SN2 reaction
• Step 1: rapid and reversible proton transfer••
••••
rapid and reversible
+H
HCH3 CH2 CH2 CH2 -
CH3 CH2 CH2 CH2 -O-H
O
An oxonium ion
H O HH
+••
+
••O HH
••+

88
8-35Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanism: 1Mechanism: 1°° ROH + HXROH + HX• Step 2: displacement of HOH by halide ion
•• ••
••
••
••
••
••
rate-limiting step
Br
Br ++ ••
H
H
CH3 CH2 CH2 CH2 -O
CH3 CH2 CH2 CH2 - +
••
H
H
O
••
SN2

88
8-36Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Mechanisms: SMechanisms: SNN11 vsvs SSNN22• Reactivities of alcohols for SN1 and SN2 are in
opposite directions
3° alcohol 2° alcohol 1° alcohol
Increasing stability of cation intermediate
Increasing ease of access to the reaction site
SN 1
SN 2

88
8-37Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reaction with SOClReaction with SOCl22• Thionyl chloride is the most widely used reagent
for the conversion of 1° and 2° alcohols to alkyl chlorides• a base, most commonly pyridine or triethylamine, is
added to neutralize the HCl
Thionyl chloride
1-Heptanol
1-ChloroheptaneCH3 (CH 2 ) 5 CH2 Cl + SO 2 + HCl
CH3 (CH 2 ) 5 CH2 OH + SOCl 2pyridine

88
8-38Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• An alcohol can be converted to an alkene by
elimination of H and OH from adjacent carbons (a β-elimination)• 1° alcohols must be heated at high temperature in the
presence of an acid catalyst, such as H2SO4 or H3PO4
• 2° alcohols undergo dehydration at somewhat lower temperatures
• 3° alcohols often require temperatures only at or slightly above room temperature

88
8-39Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH
140oCCyclohexanol Cyclohexene
OH+ H 2 OH2 SO4
180oCCH3 CH2 OH
H2 SO4CH2 =CH 2 + H 2 O
+ H2 OCH3 COH
CH3
CH350oC
H2 SO4 CH3 C= CH2
CH3
2-Methylpropene (Isobutylene)

88
8-40Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• Where isomeric alkenes are possible, the alkene
having the greater number of substituents on the double bond generally predominates (Zaitsevrule)
1-Butene (20%)
2-Butene (80%)
2-Butanol+
heat
OH85% H3PO4
CH3 CH= CHCH3
CH3 CH2 CHCH3
CH3 CH2 CH= CH2

88
8-41Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• A three-step mechanism
• Step 1: proton transfer from H3O+ to the -OH group to form an oxonium ion
+••••
An oxonium ion
rapid andreversible
+
••••
••
•• ++
HO
OH H
H H
H
CH3 CHCH2 CH3 O
CH3 CHCH 2 CH3 O
H
H

88
8-42Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• Step 2: the C-O bond is broken and water is lost,
giving a carbocation intermediate
+A 2o carbocation
+
slow and rate limiting
+
••
••
••
OHH
CH3 CHCH 2 CH3
CH3 CHCH2 CH3 H2 O

88
8-43Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• Step 3: proton transfer of H+ from a carbon adjacent to
the positively charged carbon to water. The sigma electrons of the C-H bond become the pi electrons of the carbon-carbon double bond
rapid++
••••
+••
H
H
HH
H
CH3 -CH -CH- CH3
CH3 -CH = CH -CH3
O
O
H
+

88
8-44Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Dehydration of ROHDehydration of ROH• Acid-catalyzed alcohol dehydration and alkene
hydration are competing processes
• large amounts of water favor alcohol formation• scarcity of water or experimental conditions where
water is removed favor alkene formation
An alkene An alcohol
C C C C
H OH
+ H2 O
acidcatalyst

88
8-45Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation: 1Oxidation: 1°° ROHROH• A primary alcohol can be oxidized to an aldehyde
or a carboxylic acid, depending on the oxidizing agent and experimental conditions
OH[O]
CH3 -CH 2 CH3 -C- H CH3 -C- OHO O
[O]
A primary alcohol
An aldehyde A carboxylic acid

88
8-46Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation: chromic acidOxidation: chromic acid• Chromic acid is prepared by dissolving
chromium(VI) oxide or potassium dichromate in aqueous sulfuric acid
Potassium dichromate
Chromic acidK2 Cr 2 O7
H2 SO4 H2 Cr 2 O7H2 O
2 H2 CrO 4
+Chromic acidChromium(VI)
oxide
CrO 3 H2 O H2 CrO 4H2 SO4

88
8-47Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation: 1Oxidation: 1°° ROHROH• Oxidation of 1-octanol by chromic acid gives
octanoic acid• the aldehyde intermediate is not isolated
Octanal(not isolated)
Octanoic acid
1-Octanol
O O
CH3 ( CH2 ) 6 CH2 OHCrO3
H2 SO4 , H2 O
CH3 ( CH2 ) 6 CH CH3 ( CH2 ) 6 COH

88
8-48Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
PCCPCC•• Pyridinium chlorochromatePyridinium chlorochromate (PCC)(PCC): a form of
Cr(VI) prepared by dissolving CrO3 in aqueousHCl and adding pyridine to precipitate PCC
• PCC is selective for the oxidation of 1° alcohols toaldehydes; it does not oxidize aldehydes further to carboxylic acids
NH
+CrO 3 Cl-

88
8-49Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation: 1Oxidation: 1°° ROHROH• PCC oxidation of a 1° alcohol to an aldehyde
CH2 OH CH
O
PCC
Geraniol Geranial

88
8-50Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Oxidation: 2Oxidation: 2°° ROHROH• 2° alcohols are oxidized to ketones by both
chromic acid and and PCC
2-Isopropyl-5-methyl- cyclohexanone (Menthone)
2-Isopropyl-5-methyl- cyclohexanol (Menthol)
acetone
CH3
CH(CH 3 ) 2
OH O
CH(CH 3 ) 2
CH3
H2 CrO 4

88
8-51Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Reactions of ethersReactions of ethers• Ethers, R-O-R, resemble hydrocarbons in their
resistance to chemical reaction• they do not react with strong oxidizing agents such as
chromic acid, H2CrO4
• they are not affected by most acids and bases at moderate temperatures
• Because of their good solvent properties and general inertness to chemical reaction, ethers are excellent solvents in which to carry out organic reactions

88
8-52Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
EpoxidesEpoxides•• EpoxideEpoxide: a cyclic ether in which oxygen is one
atom of a three-membered ring• Common names are derived from the name of the
alkene from which the epoxide is formally derived
Ethylene oxide
CH2CH2
O
CHCH 3CH2
OPropylene oxide

88
8-53Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Synthesis ofSynthesis of EpoxidesEpoxides--11• Ethylene oxide, one of the few epoxides
manufactured on an industrial scale, is prepared by air oxidation of ethylene
Oxirane (Ethylene oxide)
CH2CH2O
2 CH2 =CH 2 + O 2Ag 2

88
8-54Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Synthesis ofSynthesis of EpoxidesEpoxides--22• The most common laboratory method for the
synthesis of epoxides is oxidation of an alkeneusing a peroxycarboxylic acid (a peracid) such as peroxyacetic acid
CH3 COOHPeroxyacetic acid (Peracetic acid)
O

88
8-55Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Synthesis ofSynthesis of EpoxidesEpoxides--22• Epoxidation of cyclohexene
A carboxylic acid
1,2-Epoxycyclohexane (Cyclohexene oxide)
A p eroxy-carboxylic
acid Cyclohexene
+
+
OH
H
O
O
RCOH
CH2 Cl2RCOOH

88
8-56Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• In the presence of an acid catalyst, an epoxide is
hydrolyzed to a glycol
+
Ethylene oxide 1,2-Ethanediol(Ethylene glycol)
CH2O
CH2 HOCH2 CH2 OHH2 O H+

88
8-57Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• Step 1: proton transfer to the epoxide to form a
bridged oxonium ion intermediate
H2 C CH2
OOH HH
H2 C CH2
O
H++
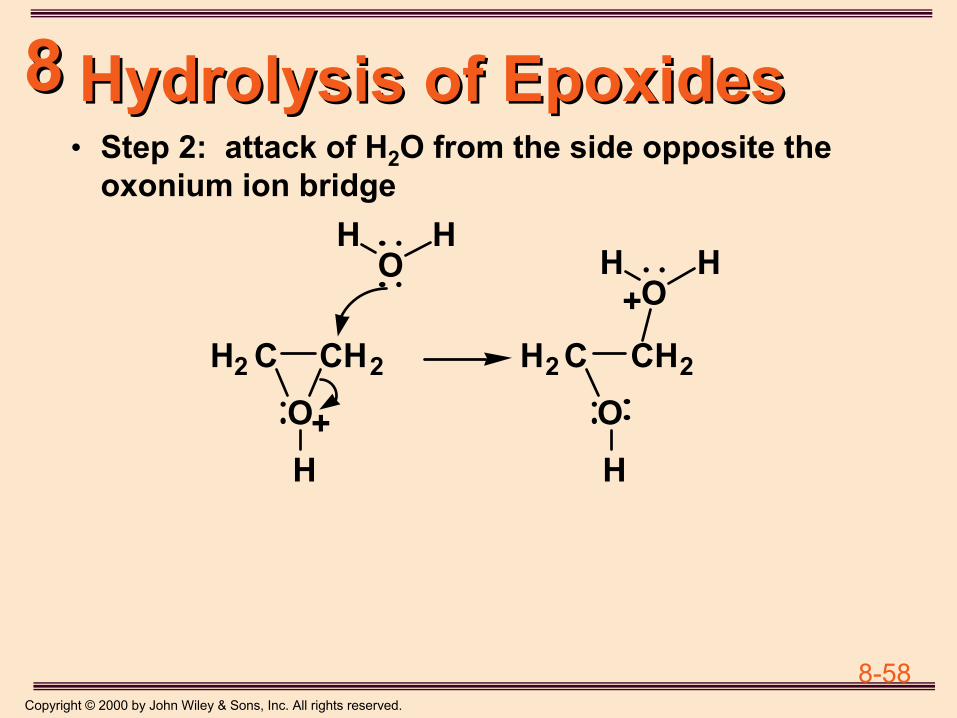
88
8-58Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• Step 2: attack of H2O from the side opposite the
oxonium ion bridge
OH H
H2 C CH2
OH
+
OH H
H2 C CH2
OH
+

88
8-59Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• Step 3: proton transfer to solvent to complete the
hydrolysis
OH H
H2 C CH2
O
H
+
OH H
OH
H2 C CH2
O
H
+ OH HH
+

88
8-60Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• Attack of the nucleophile on the protonated
epoxide shows anti stereoselectivity• hydrolysis of an epoxycycloalkane gives a trans-diol
H
H
O + H2 OH+
OH
OH1,2-Epoxycyclopentane (Cyclopentene oxide)
trans -1,2-Cyclopentanediol

88
8-61Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Hydrolysis ofHydrolysis of EpoxidesEpoxides• Compare the stereochemistry of the glycols
formed by these two methods
trans -1,2-Cyclopentanediol
cis-1,2-Cyclopentanediol
O
H
H
OH
OH
OH
OH
H2 OH+RCO3 H
OsO4
ROOH

88
8-62Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ThiolsThiols• Thiols are stronger acids than alcohols
pKa = 8.5CH3 CH2 SH CH3 CH2 S- + H3 O++ H2 O
pKa = 15.9CH3 CH2 OH CH3 CH2 O- + H3 O++ H2 O

88
8-63Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ThiolsThiols• When dissolved an aqueous NaOH, they are
converted completely to alkylsulfide salts+
+Stronger
acidStronger
base
Weaker base Weaker acid
pKa = 8.5
pKa = 15.7
CH3 CH2 SH Na +OH-
CH3 CH2 S- Na + H2 O

88
8-64Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
ThiolsThiols• Thiols are oxidized to disulfides by a variety of
oxidizing agents, including O2. • they are so susceptible to this oxidation that they must
be protected from air during storage
• The most common reaction of thiols in biological systems in interconversion between thiols and disulfides, -S-S-
A thiol A disulfide2
+ 1 +2 RSH O2 RSSR H2 O

88
8-65Copyright © 2000 by John Wiley & Sons, Inc. All rights reserved.
Alcohols,Alcohols,Ethers, andEthers, andThiolsThiols
End of Chapter 8End of Chapter 8

Carboxylic acids:R-COOH, R-CO2H,
Common names:
HCO2H formic acid L. formica ant
CH3CO2H acetic acid L. acetum vinegar
CH3CH2CO2H propionic acid G. “first salt”
CH3CH2CH2CO2H butyric acid L. butyrum butter
CH3CH2CH2CH2CO2H valeric acid L. valerans
R COH
O

Carboxylic acids, common names:
…
CH3(CH2)4CO2H caproic acid L. caper goat
CH3(CH2)5CO2H ---
CH3(CH2)6CO2H caprylic acid
CH3(CH2)7CO2H ---
CH3(CH2)8CO2H capric acid
CH3(CH2)9CO2H ---
CH3(CH2)10CO2H lauric acid oil of lauryl

5 4 3 2 1C—C—C—C—C=Oδ γ β α used in common names
CH3CH2CH2CHCOOHBr
CH3CHCH2COOHCH3
α− bromovaleric acid β -methylbutyric acid
isovaleric acid

COOH
COOH COOH COOHCH3
CH3CH3
benzoic acid
o-toluic acid m-toluic acid p-toluic acid
special names

IUPAC nomenclature for carboxylic acids:
parent chain = longest, continuous carbon chain that contains the carboxyl group alkane, drop –e, add –oic acid
HCOOH methanoic acid
CH3CO2H ethanoic acid
CH3CH2CO2H propanoic acid
CH3CH3CHCOOH 2-methylpropanoic acid
BrCH3CH2CHCO2H 2-bromobutanoic acid

dicarboxylic acids:
HOOC-COOH oxalic acid
HO2C-CH2-CO2H malonic acid
HO2C-CH2CH2-CO2H succinic acid
HO2C-CH2CH2CH2-CO2H glutaric acid
HOOC-(CH2)4-COOH adipic acid
HOOC-(CH2)5-COOH pimelic acid
Oh, my! Such good apple pie!

CO2H
CO2H
CO2H
CO2H
CO2H
CO2Hphthalic acid isophthalic acid terephthalic acid
CCOOHH
CCOOHH
CCOOHH
CHHOOC
maleic acid fumaric acid

salts of carboxylic acids:
name of cation + name of acid: drop –ic acid, add –ate
CH3CO2Na sodium acetate or sodium ethanoate
CH3CH2CH2CO2NH4 ammonium butyrate
ammonium butanoate
(CH3CH2COO)2Mg magnesium propionate
magnesium propanoate

HOC
OH
O
NaOC
ONa
O
HOC
ONa
O
carbonic acid sodium bicarbonatesodium hydrogen carbonate
sodium carbonate
NaHCO3
Na2CO3

physical properties:
polar + hydrogen bond relatively high mp/bp
water insoluble
exceptions: four carbons or less
acidic turn blue litmus red
soluble in 5% NaOH
RCO2H + NaOH RCO2-Na+ + H2O
stronger stronger weaker weakeracid base base acid

RCO2H RCO2-
covalent ionicwater insoluble water soluble
Carboxylic acids are insoluble in water, but soluble in 5% NaOH.
1. Identification.
2. Separation of carboxylic acids from basic/neutral organic compounds.
The carboxylic acid can be extracted with aq. NaOH and then regenerated by the addition of strong acid.

Carboxylic acids, syntheses:
1. oxidation of primary alcohols
RCH2OH + K2Cr2O7 RCOOH
2. oxidation of arenes
ArR + KMnO4, heat ArCOOH
3. carbonation of Grignard reagents
RMgX + CO2 RCO2MgX + H+
RCOOH
4. hydrolysis of nitriles
RCN + H2O, H+, heat RCOOH

1. oxidation of 1o alcohols:
CH3CH2CH2CH2-OH + CrO3 CH3CH2CH2CO2Hn-butyl alcohol butyric acid1-butanol butanoic acid
CH3 CH3CH3CHCH2-OH + KMnO4 CH3CHCOOHisobutyl alcohol isobutyric acid
2-methyl-1-propanol` 2-methylpropanoic acid

2. oxidation of arenes:
CH3
CH3
H3C
CH2CH3
KMnO4, heat
KMnO4, heat
KMnO4, heat
COOH
COOH
HOOC
COOH
toluene benzoic acid
p-xylene terephthalic acid
ethylbenzene benzoic acid
+ CO2
note: aromatic acids only!

3. carbonation of Grignard reagent:
R-X RMgX RCO2MgX RCOOH
Increases the carbon chain by one carbon.
Mg CO2 H+
CH3CH2CH2-Br CH3CH2CH2MgBr CH3CH2CH2COOHn-propyl bromide butyric acid
Mg CO2 H+
CO
O
RMgX + R CO
O-+ +MgX
H+
R CO
OH

CH3
Br
MgCH3
MgBr
CO2 H+CH3
COOH
p-toluic acid
CH3
Br2, hvCH2Br
MgCH2MgBr
CO2
H+
CH2 COOH
phenylacetic acid

4. Hydrolysis of a nitrile:
H2O, H+
R-C≡N R-CO2Hheat
H2O, OH-
R-C≡N R-CO2- + H+ R-CO2H
heat
R-X + NaCN R-CN + H+, H2O, heat RCOOH1o alkyl halide
Adds one more carbon to the chain.R-X must be 1o or CH3!

CH3Br2, hv
CH2BrNaCN
CH2 CN
H2O, H+, heat
CH2 COOH
CH3CH2CH2CH2CH2CH2-BrKCN
CH3CH2CH2CH2CH2CH2-CN
H2O, H+, heat
CH3CH2CH2CH2CH2CH2-COOH
1-bromohexane
heptanoic acid
toluene
phenylacetic acid

CO2H
CH2OH
CH3
Br
C N
MgBr
KMnO4, heat
KMnO4
Mg CO2; then H+
H2O, H+, heat

carboxylic acids, reactions:
1. as acids
2. conversion into functional derivatives
a) acid chlorides
b) esters
c) amides
3. reduction
4. alpha-halogenation
5. EAS

as acids:
a) with active metals
RCO2H + Na RCO2-Na+ + H2(g)
b) with bases
RCO2H + NaOH RCO2-Na+ + H2O
c) relative acid strength?
CH4 < NH3 < HC≡CH < ROH < HOH < H2CO3 < RCO2H < HF
d) quantitative
HA + H2O H3O+ + A- ionization in water
Ka = [H3O+] [A-] / [HA]

Ka for carboxylic acids ≈ 10-5
Why are carboxylic acids more acidic than alcohols?
ROH + H2O H3O+ + RO-
RCOOH + H2O H3O+ + RCOO-
∆Go = -2.303 R T log Keq
The position of the equilibrium is determined by the free energychange, ∆Go.
∆Go = ∆H - T∆S
∆Go ≈∆H ∴Ka is inversely related to ∆H, the potential energy difference between the acid and its conjugate base. The smaller the ∆H, the larger the Ka and the stronger the acid.

HA + H2O
H3O+ + A-po
tent
ial e
nerg
y
ionization
∆H
The smaller the ∆H, the more the equilibrium lies to the right, giving a larger Ka ( a stronger acid ).

R CO-
OR C
O-
O
R CO
O
Resonance stabilization of the carboxylate ion decreases the ∆H, shifts the ionization in water to the right, increases the Ka, and results in carboxylic acids being stronger acids.

Effect of substituent groups on acid strength?
CH3COOH 1.75 x 10-5
ClCH2COOH 136 x 10-5
Cl2CHCOOH 5,530 x 10-5
Cl3CCOOH 23,200 x 10-5
-Cl is electron withdrawing and delocalizes the negative charge on the carboxylate ion, lowering the PE, decreasing the ∆H, shifting the ionization to the right and increasing acid strength.

Effect of substituent groups on acid strength of benzoic acids?
Electron withdrawing groups will stabilize the anion, decrease the ∆H, shift the ionization to the right, increasing the Ka, increasing acid strength.
Electron donating groups will destabilize the anion, increase the ∆H, shift the ionization in water to the left, decreasing the Ka, decreasing acid strength.
COO-
G
COO-
G

-NH2, -NHR, -NR2-OH-OR electron donating-NHCOCH3 -C6H5-R-H-X-CHO, -COR-SO3H-COOH, -COOR electron withdrawing-CN-NR3
+
-NO2

Relative acid strength?
Ka
p-aminobenzoic acid 1.4 x 10-5
p-hydroxybenzoic acid 2.6 x 10-5
p-methoxybenzoic acid 3.3 x 10-5
p-toluic acid 4.2 x 10-5
benzoic acid 6.3 x 10-5
p-chlorobenzoic acid 10.3 x 10-5
p-nitrobenzoic acid 36 x 10-5
2. Conversion into functional derivatives:
a) acid chlorides
R COH
O SOCl2
or PCl3orPCl5
R CCl
O
CO2H + SOCl2 COCl
CH3CH2CH2 CO
OH
PCl3CH3CH2CH2 C
O
Cl

b) esters
“direct” esterification: H+
RCOOH + R´OH RCO2R´ + H2O
-reversible and often does not favor the ester
-use an excess of the alcohol or acid to shift equilibrium
-or remove the products to shift equilibrium to completion
“indirect” esterification:
RCOOH + PCl3 RCOCl + R´OH RCO2R´
-convert the acid into the acid chloride first; not reversible

C CO
O
CH3
+ H2O
SOCl2
CCH3OH
O
OH+ CH3OH
O
Cl
H+

c) amides
“indirect” only!
RCOOH + SOCl2 RCOCl + NH3 RCONH2amide
Directly reacting ammonia with a carboxylic acid results in an ammonium salt:
RCOOH + NH3 RCOO-NH4+
acid base
OH
O
3-Methylbutanoic acid
PCl3Cl
O NH3
NH2
O

CO
OH
PCl3C
O
ClC
O
NH2
NH3
NH3
amide
CO
O NH4ammonium salt

3. Reduction:
RCO2H + LiAlH4; then H+ RCH2OH1o alcohol
Carboxylic acids resist catalytic reduction under normal conditions.
RCOOH + H2, Ni NR
CH3CH2CH2CH2CH2CH2CH2COOH
Octanoic acid(Caprylic acid)
LiAlH4 H+
CH3CH2CH2CH2CH2CH2CH2CH2OH
1-Octanol

CH2 CO
OH
H2, PtNR
LiAlH4
H+
CH2CH2OH

4. Alpha-halogenation: (Hell-Volhard-Zelinsky reaction)
RCH2COOH + X2, P RCHCOOH + HXXα-haloacid
X2 = Cl2, Br2
COOH
Br2,PNR (no alpha H)
CH3CH2CH2CH2COOH + Br2,P CH3CH2CH2CHCOOHBrpentanoic acid
2-bromopentanoic acid

RCH2COOH + Br2,P RCHCOOH + HBrBr
NH2OH
RCHCOOH RCHCOOH
RCH=CHCOOHRCH2CHCOOHBr
aminoacid
NaOH;then H+
NH3
KOH(alc)
then H+

5. EAS: (-COOH is deactivating and meta- directing)
CO2H
CO2H
NO2
CO2H
SO3H
CO2H
Br
NR
HNO3,H2SO4
H2SO4,SO3
Br2,Fe
CH3Cl,AlCl3
benzoic acid

spectroscopy:
IR: -COOH O—H stretch 2500 – 3000 cm-1 (b)
C=O stretch 1680 – 1725 (s)
nmr: -COOH 10.5 – 12 ppm

p-toluic acid
-COO—H stretch C=O

COOH
CH3 a
b
c
c b a

Carboxylic acids, syntheses:
1. oxidation of primary alcohols
RCH2OH + K2Cr2O7 RCOOH
2. oxidation of arenes
ArR + KMnO4, heat ArCOOH
3. carbonation of Grignard reagents
RMgX + CO2 RCO2MgX + H+
RCOOH
4. hydrolysis of nitriles
RCN + H2O, H+, heat RCOOH

carboxylic acids, reactions:
1. as acids
2. conversion into functional derivatives
a) acid chlorides
b) esters
c) amides
3. reduction
4. alpha-halogenation
5. EAS





































Functional Derivatives of Carboxylic Acids
R CNH2
O
R CO
O
R CCl
O
R COR'
O
CO
R
acid chlorideanhydride
amide ester
R may be H or Ar

Nomenclature: the functional derivatives’ names are derived from the common or IUPAC names of the corresponding carboxylic acids.
Acid chlorides: change –ic acid to –yl chloride
Anhydrides: change acid to anhydride
CCl
OCH3CH2CH2C
O
Cl
butanoyl chloridebutyryl chloride
benzoyl chloride
H3C CO
H3C CO
O
O
O
O
O
O
O
ethanoic anhydrideacetic anhydride
phthalic anhydride maleic anhydride

Amides: change –ic acid (common name) to –amide
-oic acid (IUPAC) to –amide
Esters: change –ic acid to –ate preceded by the name of the alcohol group
CNH2
OCH3CH2CH2C
O
NH2
butanamidebutyramide
benzamide
CO CH2CH3
O
ethyl benzoate
CH3CH2CH2CO
O CH3
methyl butanoatemethyl butyrate

Nucleophilic acyl substitution:
R CZ
OR C
W
O+ :Z R C W
O
Z+ :W
-W = -OH, -Cl, -OOCR, -NH2, -OR'
R CO
= "acyl" group

R CZ
O
R CW
O+ :Z R C W
O
Z
+ :WR C WO
Z
RDS
Mechanism: Nucleophilic Acyl Substitution
1)
2)

R CZ
O
R CW
OH+ :ZH R C W
OH
ZH
+ HW + H+R C WOH
ZH
RDS
R CW
OHR C
W
O+ H+
Mechanism: nucleophilic acyl substitution, acid catalyzed
1)
2)
3)

nucleophilic acyl substitution vs nucleophilic addition to carbonyl
aldehydes & ketones – nucleophilic addition
functional deriv. of carboxylic acids – nucleophilic acyl substitution
R CZ
OR C
W
O+ :Z R C W
O
Z+ :W
-W = -OH, -Cl, -OOCR, -NH2, -OR'
R CR'
O+ :Z R C R'
O
Z
YR C R'
OY
Z

Acid Chlorides
Syntheses:SOCl2
RCOOH + PCl3 RCOClPCl5
COH
O+ SOCl2 C
Cl
O
benzoic acid benzoyl chloride
OH
O+ PCl3
Cl
O
3-methylbutanoic acidisovaleric acid
3-methylbutanoyl chlorideisovaleryl chloride

Acid chlorides, reactions:
1. Conversion into acids and derivatives:
a) hydrolysis
b) ammonolysis
c) alcoholysis
2. Friedel-Crafts acylation
3. Coupling with lithium dialkylcopper
4. Reduction

acid chlorides: conversion into acids and other derivatives
Cl
O H2O
OH
OHydrolysis
isovaleryl chloride3-methylbutanoyl chloride
isovaleric acid3-methylbutanoic acid
Ammonolysis CH3CH2 CCl
O NH3CH3CH2 C
NH2
O
propionyl chloridepropanoyl chloride
propionamidepropanamide
AlcoholysisC
O
Cl
CH3CH2OHC
O
OCH2CH3
benzoyl chloride ethyl benzoate

Schotten-Baumann technique – aromatic acid chlorides are less reactive than aliphatic acid chlorides. In order to speed up the reactions of aromatic acid chlorides, bases such as NaOH or pyridine are often added to the reaction mixture.
O2N
O2N
COClpyridine
CH3CH2CH2OHO2N
O2N
CO
OCH2CH2CH3
3,5-dinitrobenzoyl chloride n-propyl-3,5-dinitrobenzoate

acid chlorides: Friedel-Crafts acylation
R CO
Cl+ ArH
AlCl3R C Ar
O+ HCl
phenone
CH3CH2CH2CO
Cl CH3+
toluene
butyryl chloride
AlCl3CH3CH2CH2C
OCH3 + ortho-
p-methylbutyrophenone
CH3CH2CH2CO
Cl
butyryl chloride
+ NO2
AlCl3No reacton

acid chlorides: coupling with lithium dialkylcopper
R CO
Cl+ R'2CuLi R C R'
O
ketone
CO
Cl+ (CH3CH2CH2)2CuLi C CH2CH2CH3
O
benzoyl chloride lithium di-n-propylcopper butyrophenone
CCl
O+ 2CuLi
O
2,4-dimethyl-3-pentanoneisobutyryl chloride lithium diisopropylcopper

acid chlorides: reduction to aldehydes
R CCl
O LiAlH(t-BuO)3R C
H
O
CO
ClC
O
H
LiAlH(t-BuO)3
mechanism, nucleophilic acyl substitution by hydride :H-
R CCl
O1) + :H R C Cl
O
HRDS
2) R C ClO
HR C
H
O+ Cl

Anhydrides, syntheses:
Buy the ones you want!
Anhydrides, reactions:
1) Conversion into carboxylic acids and derivatives.
a) hydrolysis
b) ammonolysis
c) alcoholysis
2) Friedel-Crafts acylation

O
O
Ophthalic anhydride
+ H2OCOOH
COOH
(CH3CO)2O + NH3 CH3 CNH2
OCH3 C
ONH4
O+
acetic anhydride
phthalic acid
acetamide
O
O
O
+ CH3CH2OH
succinic anhydride
CH2COCH2CH3
O
CH2COHO
ethyl hydrogen succinate
ammonium acetate

2) anhydrides, Friedel-Crafts acylation.
(RCO)2O + ArHAlCl3
R COH
O+R C Ar
O
phenone
(CH3CO)2O + CH3AlCl3 H3C C
OCH3 + CH3CO2H
acetic anhydridetoluene p-methylacetophenone
O
O
O
phthalic anhydride
+AlCl3
CO
CO
OH
o-benzoylbenzoic acid

Amides, synthesis:
Indirectly via acid chlorides.
R COH
O SOCl2R C
Cl
O NH3R C
NH2
O
[ carboxylic acids form ammonium salts when reacted directly with ammonia ]
CH3CH2CH2CO2H CH3CH2CH2CO
Cl
PCl3 NH3CH3CH2CH2C
O
NH2butyric acid butyryl chloride butyramide
COOHPCl5
CCl
O NH3C
NH2
O
benzoic acid benzoyl chloride benzamide

Amides, reactions.
1) Hydrolysis.
R CNH2
O H2O, H+ or OH-
heatR C
OH
O
CH3CHCH2CCH3
NH2
O
isovaleramide
+ H2OH+
heatCH3CHCH2C
CH3
OH
O
isovaleric acid

HN CHCO
R
HN CHC
R
O HN CHC
R
O HN CHC
R
O HN CHC
R
O HN CHC
R
O
proteins are polyaminoacids
H2N CHCR
OHO
aminoacids
"peptide bond"hydrolysis

Wool, hair, silk, spider web: fibrous proteins.
Silk is an extremely strong, thin, lightweight fiber, perfect for making sheer stockings for women as well as parachutes. It is made by the silkworm, a domesticated moth larva raised in Japan and China. During World War II a substitute material was needed and developed by DuPont – Nylon-66, a synthetic polyamide of adipic acid and hexamethylenediamine:
C(CH2)4CO O
Cladipoyl chloride
+ H2N (CH2)6 NH2hexamethylenediamine
C(CH2)4CO O
NH (CH2)6 NHC(CH2)4CO O
NH (CH2)6 NH
Nylon-66
Cl

Esters, syntheses:
1) From acids
RCO2H + R’OH, H+ RCO2R’ + H2O
2) From acid chlorides and anhydrides
RCOCl + R’OH RCO2R’ + HCl
3) From esters (transesterification)
RCO2R’ + R”OH, H+ RCO2R” + R’OH
RCO2R’ + R”ONa RCO2R” + R’ONa

Esters often have “fruity” or “floral” odors:
isopentyl acetate banana oil
n-pentyl butyrate apricot
isopentyl isovalerate apple
ethyl butyrate peach
ethyl heptanoate cognac
ethyl nonate flower bouquet
ethyl laurate tuberose
methyl butyrate pineapple
octyl acetate orange

CO
OH
isovaleric acid
+ CH3CH2OH
ethyl alcohol
H+
CO
O
ethyl isovalerate
+ H2O
SOCl2
CO
Cl
isovaleryl chloride
+ CH3CH2OH
ethyl alcohol
CO
O
ethyl isovalerate
+ HCl
“Direct” esterification is reversible and requires use of LeChatelier’s principle to shift the equilibrium towards the products. “Indirect” is non-reversible.

In transesterification, an ester is made from another ester by exchanging the alcohol function.
CH3CH2CH2CO
OCH3
methyl butanoate
+
isopropyl alcohol
H+CH3CH2CH2C
O
O
isopropyl butanoate
+ CH3OHCHCH3
CH3CHCH3HOCH3
CH3CH2CH2CO
OCH3
methyl butanoate
+
CH2ONa
benzyl alcoholCH3CH2CH2C
O
O+
CH2
CH3ONa
benzyl butanoate

Esters, reactions:
1) Conversion into acids and derivatives
a) hydrolysis
b) ammonolysis
c) alcoholysis
2) Reaction with Grignard reagents
3) Reduction
a) catalytic
b) chemical
4) Claisen condensation

COCH2CH3
O H2O; H+ or OH-
heatC
OH
O+ CH3CH2OH
ethyl benzoate
CH3CHCCH3 O
O CH3
methyl isobutyrate
NH3CH3CHC
CH3 O
NH2
+ CH3OH
CH3CO
OCH2CH3+ OH
H+
CH3CO
O + CH3CH2OH
ethyl acetate cyclopentyl acetate

R C18O R'
O OH-
H2O, heatH+
R COH
O+ R'18OH
OH-
R CO-
18OOH
R
Tracer studies confirm that the mechanism is nucleophilic acyl substitution:

H2CHC
OOCROOCR'
H2C OOCR''
triglycerides, fats/oilstriesters of glycerol
NaOH, H2O
heat
H2CHC
OHOH
H2C OH
glycerol
+
RCOO-Na+
R'COO-Na+
R''COO-Na+
"SOAP"
Hydrolysis of a fat or oil is also called "saponification

Esters, reaction with Grignard reagents
R CO
O R''+ R'MgX
H2OR C R'
OH
R'+ R''OH
3o alcoholnucleophilicacyl substitution
R C R'O
ketone
+ R'MgX
nucleophilicaddition

CH3CH2CH2CO
OCH3
methyl butanoate
+ MgBr
phenyl magnesium bromide
H2O
CH3CH2CH2COH
1,1-diphenyl-1-butanol

Esters, reduction
a) catalytic
b) chemical
RO R'
O+ H2, Ni NR
RO R'
O H2, CuO, CuCr2O4
150o, 5000 psiRCH2OH + R'OH
RO R'
O LiAlH4 H+
RCH2OH + R'OH

O
O
isopropyl isobutyrate
H2, CuO, CuCr2O4
150o, 5000 psi
OH
OH
+
isobutyl alcohol isopropyl alcohol
CH3CH2CO
O
phenyl propanoate
1. LiAlH42. H+ CH3CH2CH2OH +
OH
n-propyl alcohol phenol

Spectroscopy:
Infrared: strong absorbance ~ 1700 cm-1 for C=O
RCO2R 1740 ArCO2R 1715-1730 RCO2Ar 1770
Esters also show a strong C—O stretch at 1050-1300
Amides show N—H stretch at 3050 –3550 and N—H bend in the 1600-1640 region.
Nmr: NB in esters the protons on the alcohol side of the functional group resonate at lower field than the ones on the acid side.
RCOO—C—H 3.7 – 4.1 ppm
H—C—COOR 2 – 2.2 ppm

methyl propionate
C=O
C--O

butyramide
N—H
C=O
N—H bend

Ethyl acetate
CH3CO2CH2CH3b c a
Note which hydrogens are upfield.
c b a

Methyl propionate
CH3CH2CO2CH3a b c
Note which hydrogens are upfield.
c b a

Aldehydes and Ketones

Before you can learn about aldehydes and ketones, you must first know something about the nomenclature of carboxylic acids since many of the names of aldehydes and ketones are derived from the names of the corresponding carboxylic acids.

Carboxylic acids:
R-COOH, R-CO2H,
Common names:
HCO2H formic acid L. formica ant
CH3CO2H acetic acid L. acetum vinegar
CH3CH2CO2H propionic acid G. “first salt”
CH3CH2CH2CO2H butyric acid L. butyrum butter
CH3CH2CH2CH2CO2H valeric acid L. valerans
R COH
O

Carboxylic acids, common names:
…
CH3(CH2)4CO2H caproic acid L. caper goat
CH3(CH2)5CO2H ---
CH3(CH2)6CO2H caprylic acid
CH3(CH2)7CO2H ---
CH3(CH2)8CO2H capric acid
CH3(CH2)9CO2H ---
CH3(CH2)10CO2H lauric acid oil of lauryl

5 4 3 2 1C—C—C—C—C=Oδ γ β α used in common names
CH3CH2CH2CHCOOHBr
CH3CHCH2COOHCH3
α− bromovaleric acid β -methylbutyric acid
isovaleric acid

COOH
COOH COOH COOHCH3
CH3CH3
benzoic acid
o-toluic acid m-toluic acid p-toluic acid
Special names!

ALDEHYDES AND KETONES
“carbonyl” functional group:
Aldehydes Ketones
HC
H
O
RC
H
O
RC
R'
O
R can be Ar
CO

Nomenclature:
Aldehydes, common names:
Derived from the common names of carboxylic acids;
drop –ic acid suffix and add –aldehyde.
CH3CH3CH2CH2CH=O CH3CHCH=O
butyraldehyde isobutyraldehyde(α-methylpropionaldehyde)

CHO
benzaldehyde
CHOCH3
o-tolualdehyde
HC
H
O
formaldehyde
CH2CH=O
phenylacetaldehyde

Aldehydes, IUPAC nomenclature:
Parent chain = longest continuous carbon chain containing the carbonyl group; alkane, drop –e, add –al. (note: no locant, -CH=O is carbon #1.)
CH3CH3CH2CH2CH=O CH3CHCH=O
butanal 2-methylpropanal
H2C=O CH3CH=O
methanal ethanal

Ketones, common names:
Special name: acetone
“alkyl alkyl ketone” or “dialkyl ketone”
H3CC
CH3
O
CH3CH2CCH3
OCH3CH2CCH2CH3
O
ethyl methyl ketone diethyl ketone
CH3CCH2CH2CH3
O
methyl n-propyl ketone

(o)phenones:
Derived from common name of carboxylic acid, drop –ic acid, add –(o)phenone.
CRO
CO
H3CCO
benzophenone acetophenone

Ketones: IUPAC nomenclature:
Parent = longest continuous carbon chain containing the carbonyl group. Alkane, drop –e, add –one. Prefix a locant for the position of the carbonyl using the principle of lower number.
CH3CH2CCH3
OCH3CH2CCH2CH3
O
2-butanone 3-pentanone
CH3CCH2CH2CH3
O
2-pentanone

Physical properties:
polar, no hydrogen bonding
mp/bp are relatively moderate for covalent substances
water insoluble
(except: four-carbons or less)
C O sp2 120o
C O C O
π

Spectroscopy:
IR: C=O stretch, strong ~1700 cm-1
RCHO 1725 ArCHO 1700
R2CO 1710 ArCOR 1690
C—H stretch for aldehydes 2720
nmr: -CHO 9-10 ppm

C=Ostretch
acetophenone

valeraldehyde
CHOC—Hstretch2720 cm-1
C=O stretch
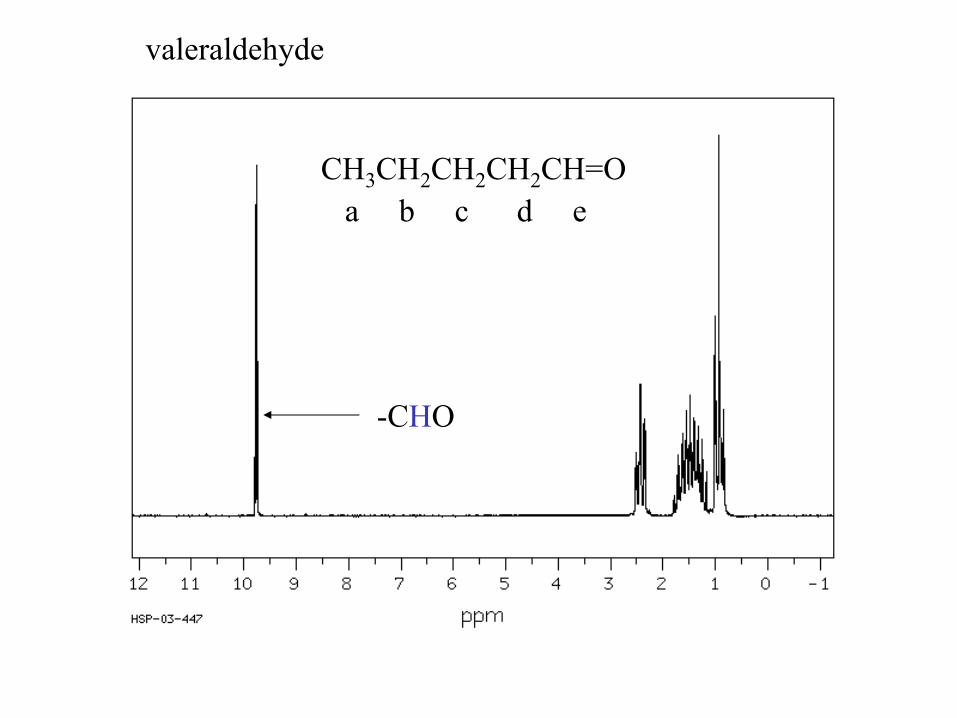
valeraldehyde
CH3CH2CH2CH2CH=Oa b c d e
-CHO

Oxidation/Reduction:
oxidation numbers:
oxidation
-4 -2 0 +2 +4 CH4 CH3OH H2C=O HCO2H CO2
alkane alcohol aldehyde carboxylic acid
reduction

Aldehydes, syntheses:
1. Oxidation of 1o alcohols
2. Oxidation of methylaromatics
3. Reduction of acid chlorides
Ketones, syntheses:
1. Oxidation of 2o alcohols
2. Friedel-Crafts acylation
3. Coupling of R2CuLi with acid chloride

Aldehydes synthesis 1) oxidation of primary alcohols:
RCH2-OH + K2Cr2O7, special conditions RCH=O
RCH2-OH + C5H5NHCrO3Cl RCH=O
(pyridinium chlorochromate)
[With other oxidizing agents, primary alcohols RCOOH]

CH3CH2CH2CH2CH2OH
+ K2Cr2O7 CH3CH2CH2CH2CO2H
1-pentanol
pentanoic acid
K2Cr2O7, special conditions!CH3CH2CH2CH2CH=O
pentanalvaleraldehyde
CH2OHC5H5NHCrO3Cl
pyridinium chlorochromateCH=O
benzaldehydebenzyl alcohol
CH3CH2CH2CH2CH2OH
1-pentanol

Aldehyde synthesis: 2) oxidation of methylaromatics:
+ CrO3, (CH3CO)2O
geminal diacetate
H2O, H+
CH3
BrBr
CH OOC C
H3C
OO
H3C
Br
CHO
p-bromobenzaldehydeAromatic aldehydes only!

CH3
CH3O CH3
CrO3
(CH3CO)2O
CrO3
(CH3CO)2O
H2O
H2O
CHO
CH3O CH=O
2-methylnaphthalene 2-naphthaldehyde
p-methylanisole p-anisaldehyde

Aldehyde synthesis: 3) reduction of acid chloride
LiAlH(O-t-Bu)3
lithium aluminum hydride tri-tert-butoxide
O
Clisovaleryl chloride
O
Hisovaleraldehyde
RCO
ClLiAlH(O-t-Bu)3
RCO
H

CO
Cl
LiAlH(O-t-Bu)3C
O
H
LiAlH(O-t-Bu)3
benzoyl chloride benzaldehyde
CH3CHCH2CO
Cl
CH3
CH3CHCH2CO
H
CH3
isovaleryl chloride isovaleraldehyde

Ketone synthesis: 1) oxidation of secondary alcohols
NaOCl
cyclohexanol cyclohexanone
isopropyl alcohol acetone
K2Cr2O7
H OH O
H3CC
CH3
OCH3CHCH3
OH

Ketone synthesis: 2) Friedel-Crafts acylation
RCOCl, AlCl3 + ArH + HClAlCl3
ArCRO
Aromatic ketones (phenones) only!
CH3CH2CH2CO
Cl+
AlCl3CH3CH2CH2C
O
butyrophenone

+AlCl3
m-nitrobenzophenone
O2N
C ClO
CO
O2N
+AlCl3C Cl
O
NO2
NR
Friedel Crafts acylation does not work on deactivated rings.

Mechanism for Friedel-Crafts acylation EAS
RC
Cl
O+ AlCl3 RC=O + AlCl4
+ RC=ORDS
H
CRO
H
CRO
+ AlCl4 C RO
+ HCl + AlCl3

Ketone synthesis: 3) coupling of RCOCl and R2CuLi
RCOCl + R'2CuLiR
C
O
R'
Cl
O
+ (CH3CH2)2CuLi
O
Isobutyryl chloride 2-Methyl-3-pentanonelithium diethylcuprate

CuLi2
+ CHCH2CH2CH3O
ClCCH2CH2CH3
O
butyrophenone
CH3CH2CH2CO
Cl+ CH3CH
CH3
2CuLi CH3CH2CH2CCHCH3
O
CH3
2-methyl-3-hexanone

Aldehydes, syntheses:
1. Oxidation of 1o alcohols
2. Oxidation of methylaromatics aromatic only
3. Reduction of acid chlorides
Ketones, syntheses:
1. Oxidation of 2o alcohols
2. Friedel-Crafts acylation aromatic only
3. Coupling of R2CuLi with acid chloride

aldehyde
1o alcohol
Ar-CH3
acid chloride
CrO3 H2O
(AcO)2O
LiAlH(O-t-Bu)3
K2Cr2O7, special cond.
or C5H5NHCrO3Cl

ketone
2o alcohol
acid chloride + ArH
acid chloride + R2CuLi
NaOCl, etc.
AlCl3

1. outline three different syntheses for benzaldehyde
2. outline three different syntheses for benzophenone
3. outline a different synthesis for each of the following compounds:
cyclohexanone, 4-bromobenzaldehyde, 2-pentanone, valeraldehyde, acetophenone, isobutyraldehyde,

CH2OHK2Cr2O7
special conditions
CH3CrO3
(CH3CO)2O
CH(OOCCH3)2
H2O
CO
ClLiAlH(O-t-Bu)3
CH=O
benzaldehyde
Synthesize benzaldehyde three different ways.

CHOH NaOCl
CO
Cl+
AlCl3
CO
Cl+ CuLi
2
CO
Synthesize benzophenone three different ways.

cyclohexanone, 4-bromobenzaldehyde, 2-pentanone,valeraldehyde, acetophenone, isobutyraldehyde, using a different method for each one.
OBr CHO
CH3CH2CH2CCH3
OCH3CH2CH2CH2CHO
CH3CO
CH3CHCHOCH3
oxidation of 2o alcohol oxidation of Ar-CH3
R2CuLi + R'COCl
Friedel-Crafts acylation
oxidation of 1o alcohol
reduction of acid chloride

O
CH3CH2CH2CCH3
O
CH3CO
OHH
K2Cr2O7
CH3 CCl
O+
AlCl3
(CH3CH2CH2)CuLi + CH3 CCl
O

Br CHO
CH3CH2CH2CH2CHO
CH3CHCHOCH3
Br CH3
CH3CH2CH2CH2CH2-OHK2Cr2O7
special conditions
CrO3
(CH3CO)2O
H2O
CH3CHCCH3 O
Cl
LiAlH(O-t-bu)3
The methods could be reversed for the last two syntheses.

Reactions of aldehydes and ketones:
oxidation
reduction
nucleophilic addition
1) Aldehydes are easily oxidized, ketones are not.
2) Aldehydes are more reactive in nucleophilic additions than ketones.

alkane alcohol
aldehydeketone
carboxylic acid
oxidation
reductionreduction
additionproduct
nucleophilicaddition

nucleophilic addition to carbonyl:
CO
+ Y Z CZ
OYδ
δ

Mechanism: nucleophilic addition to carbonyl
CO
+ ZRDS
CO
Z
CO
Z+ Y C
OY
Z
1)
2)

Mechanism: nucleophilic addition to carbonyl, acid catalyzed
CO
+ H COH
COH
+ HZRDS
COH
ZH
COH
ZHCOH
Z+ H
1)
2)
3)

Aldehydes & ketones, reactions:
1) Oxidation
2) Reduction
3) Addition of cyanide
4) Addition of derivatives of ammonia
5) Addition of alcohols
6) Cannizzaro reaction
7) Addition of Grignard reagents
8) (Alpha-halogenation of ketones)
9) (Addition of carbanions)

1) Oxidation
a) Aldehydes (very easily oxidized!)
CH3CH2CH2CH=O + KMnO4, etc. CH3CH2CH2COOHcarboxylic acid
CH3CH2CH2CH=O + Ag+ CH3CH2CH2COO- + Ag
Tollen’s test for easily oxidized compounds like aldehydes.
(AgNO3, NH4OH(aq))
Silver mirror

Ketones only oxidize under vigorous conditions via the enol.
O
+ KMnO4 NR
O
Cyclohexanone
+ KMnO4, heat HOOCCH2CH2CH2CH2COOH
adipic acid
OH
enol

b) Methyl ketones:
RC
CH3
O+ OI-
RC
O-
O+ CHI3
iodoform
test for methyl ketonesYellow ppt
CH3CH2CH2CCH3 + (xs) NaOI CH3CH2CH2CO2- + CHI3
O
2-pentanone

2) Reduction:
a) To alcohols
H2, Ni
NaBH4 or LiAlH4
then H+
CO
COH
H

H2, Pt
1. NaBH4
2. H+
O
cyclopentanone
OHcyclopentanol
C CH3
OCHCH3
OH
acetophenone 1-phenylethanol
H

CO
H
H2, PtCH2OH
CH3CHCH=OCH3 LiAlH4 H+
CH3CHCH2OHCH3
benzaldehyde benzyl alcohol
isobutyraldehyde isobutyl alcohol

RDS1)
2)
mechanism: nucleophilic addition; nucleophile = hydridehydride reduction
CO
+ H: CH
O
CH
O+ Al H C O Al
Al Al+
Then + H+ alcohol

Reduction
b) To hydrocarbons
NH2NH2, OH-
Zn(Hg), HCl
Clemmensen
Wolff-KishnerCO
CO
CH2
CH2

+ AlCl3
Zn(Hg), HCl
n-pentylbenzene
cannot be made by Friedel-Crafts alkylation due to rearrangement of carbocation
Cl
O O

3) Addition of cyanide
CO 1. CN-
2. H+CCN
OH
cyanohydrin
O + NaCN; then H+OH
CN

CO
mechanism for addition of cyanidenucleophilic addition
RDSCO
CN
CO
CN
+ Na+ CONa
CN
+ C N
then + H+
1)
2)

Cyanohydrins have two functional groups plus one additional carbon. Nitriles can be hydrolyzed to carboxylic acids in acid or base:
CH2CHOH
C NH2O, OH-
heat CH2CHOH
COO-
H2O, H+
heatCH
CH
COOHCH2CHOH
C N

4) Addition of derivatives of ammonia
O+
N+ H2OH2N G
(H+)
G
HN
phenylhydrazine
H2N NH2
hydrazine
H2N OH
hydroxylamine
HN NO2
O2N
2,4-dinitrophenylhydrazine
H2N NH
O
NH2
semicarbazide
H2NH2N

CO
+ H+COH
COH
+ H2N G
acid catalyzed nucleophilic addition mechanism followed by dehydration
CNH2
OH
G
CNH2
OH
G
CN
G
+ H2O + H+
RDS
1)
2)
3)

CH2 CHO
phenylacetaldehyde
+ H2NOH CH2 CH NOH
an oxime
O + H2NHNCNH2
O H+
NHNCNH2
O
a semicarbazonecyclohexanone
CH3CH2CH2CH2CHO + NHNH2
phenylhydrazine
hydroxylamine
semicarbazide
pentanalCH3CH2CH2CH2CH N NH
a phenylhydrazone

melting points of derivativesketones bp semi- 2,4-dinitro- oxime
carbazone phenylhydrazone
2-nonanone 195 119 56
acetophenone 202 199 240 60
menthone 209 189 146 59
2-methylacetophenone 214 205 159 61
1-phenyl-2-propanone 216 200 156 70
propiophenone 220 174 191 54
3-methylacetophenone 220 198 207 55
isobutyrophenone 222 181 163 94

5) Addition of alcohols
CO
+ ROH, H+COR
OR acetal
COH
OR hemiacetal

Mechanism = nucleophilic addition, acid catalyzed
1) CO
+ H COH
2)COH2 + ROH C
OH
HOR
3) COH
HORCOH
OR+ H
RDS

CH2CHO(xs) EtOH, H+
CH2 CHOEt
OEt
O (xs) CH3OH, dry HClOCH3
OCH3
acetal
ketal

CHOOHHHHOOHHOHH
CH2OH
OH
HO
H
HO
H
OHOHH H
OHO
H
HO
H
HO
H
HOHH OH
OH
nucleophilic addition of -OH on carbon 5 to the aldehyde functional group
CHOOHHHHOOHHOHH
CH2OH
CHOHHHHOOHHHHOH2C
OH
OH
HO
H
HO
H
OHOHH H
OH
O
OH
HO
H
HO
H
HOHH OH
OH
α
βrotate C-5 OH to rear

6) Cannizzaro reaction. (self oxidation/reduction)
a reaction of aldehydes without α-hydrogens
CHO
Br
conc. NaOH
CH2OH COO-
Br Br+
CH3OH + HCOO-H2C=Oconc. NaOH

Formaldehyde is the most easily oxidized aldehyde. When mixed with another aldehyde that doesn’t have any alpha-hydrogens and conc. NaOH, all of the formaldehyde is oxidized and all of the other aldehyde is reduced.
Crossed Cannizzaro:
CH=O
OCH3OH
vanillin
+ H2C=Oconc. NaOH
CH2OH
OCH3OH
+ HCOO-

7) Addition of Grignard reagents.
CO
+ RMgX CO
R
MgBr
CO
R
MgBr+ H2O C
OH
R
+ Mg(OH)Br
larger alcohol
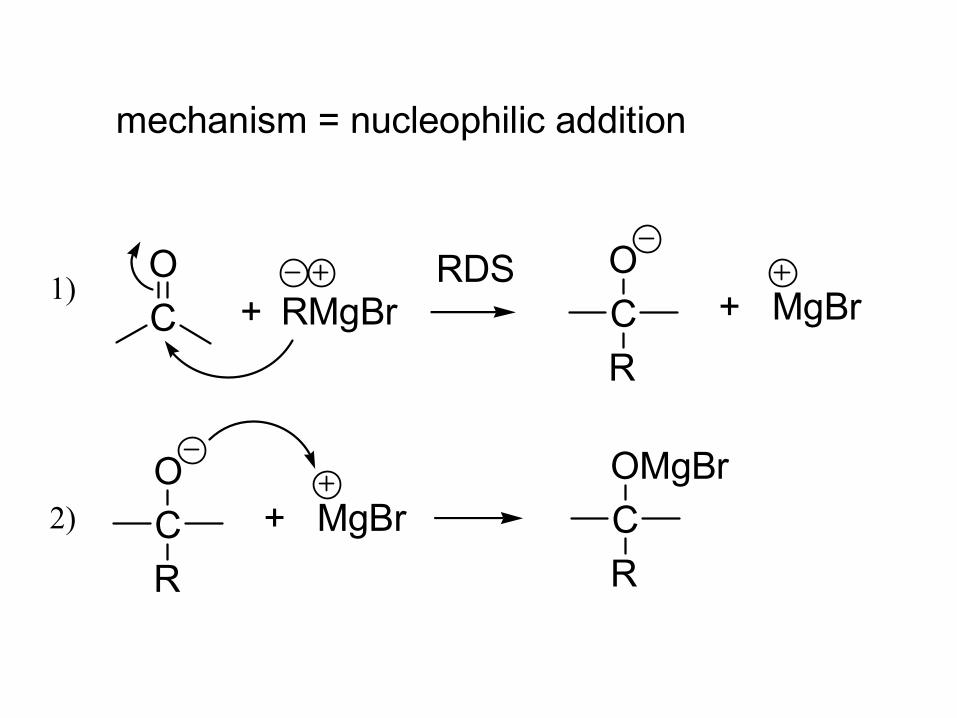
CO
RMgBr+RDS
CO
R+ MgBr
CO
R+ MgBr C
OMgBr
R
mechanism = nucleophilic addition
1)
2)

#3 synthesis of alcohols. Used to build larger molecules from smaller organic compounds.
RMgX +H
CH
ORCH2OMgX
H+RCH2OH
formaldehyde 1o alcohol + 1 C
RMgX +R'
CH
OR'CHOMgX
R
H+R'CHOH
Rother aldehydes 2o alcohol + X C's

R-MgX +R'
CR"
OR-COMgX
R'
R"
H+
R-COH
ketone 3o alcohol + X C's
RMgX +H2C CH2
ORCH2CH2OMgX
H+
RCH2CH2OH
ethylene oxide 1o alcohol + 2 C's
R'
R"

Aldehydes & ketones, reactions:
1) Oxidation
2) Reduction
3) Addition of cyanide
4) Addition of derivatives of ammonia
5) Addition of alcohols
6) Cannizzaro reaction
7) Addition of Grignard reagents
8) (Alpha-halogenation of ketones)
9) (Addition of carbanions)

Planning a Grignard synthesis of an alcohol:
a) The alcohol carbon comes from the carbonyl compound.
b) The new carbon-carbon bond is to the alcohol carbon.
CO
+ RMgX H+COH
R
New carbon-carbon bond

“The Grignard Song” (sung to the tune of “America the Beautiful”)
Harry Wasserman
The carbonyl is polarized,the carbon end is plus.A nucleophile will thus attackthe carbon nucleus.
The Grignard yields an alcoholof types there are but three.It makes a bond that correspondsfrom “C” to shining “C.”

CH3CH2CH2CH2 C CH3
CH3
OH2-Methyl-2-hexanol
CH3CH2CH2CH2MgBr + CH3CCH3
OH2O
CH3CH2CH2CH2 C CH3
CH3
OH2-Methyl-2-hexanol
CH3CH2CH2CH2CCH3 + CH3MgBrH2O
O
or

ROH RX
-C=O
RMgX
R´OH
HX Mg
ox.
H2O largeralcohol

Stockroom:
alcohols of four-carbons or less:
(methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, 2-methyl-2-propanol, 2-methyl-1-propanol.)
benzene
cyclohexanol
any needed inorganic reagents or solvents.

Grignard synthesis of 4-methyl-2-pentanol from alcohols of four-carbons or less:
Step one: determine the carbonyl compound and Grignard reagent that you would use:
CH3CH3CHCH2CHCH3
OH
H2OCH3CH3CHCH2MgBr + CH3CH=O
Step two: show the syntheses of the Grignard reagent and the carbonyl compound from alcohols…

CH3 HBr CH3 Mg CH3CH3CHCH2OH CH3CHCH2Br CH3CHCH2MgBr
H+
K2Cr2O7 CH3CH3CH2OH CH3CH=O CH3CHCH2CHCH3
special cond. OH
4-methyl-2-pentanol

Br2,FeBr
MgMgBr
CH3CHCH3
OH CrO3CH3CCH3
O
H2OC CH3CH3
OH
2-phenyl-2-propanol
2-phenyl-2-propanol

H3C OH
1-Methylcyclohexanol
OHH
Cyclohexanol
NaOCl
O
Cyclohexanone
CH3OHHBr
CH3BrMg CH3MgBr
H2O
1-methylcyclohexanol

CH2OHH
Cyclohexylmethanol
OHH BrH MgBrH
CH3OH H2C=O
HBr Mg
K2Cr2O7
special cond.
H2O
cyclohexylmethanol

aldehyde RCOOHketone
ROR
alkyne
alkene
RH
RX
ROH
Alcohols are central to organic syntheses

ROH RX
-C=O
RMgX
R´OH
HX Mg
ox.
H2O largeralcohol

Using the Grignard synthesis of alcohols we can make any alcohol that we need from a few simple alcohols. From those alcohols we can synthesize alkanes, alkenes, alkynes, alkyl halides, ethers, aldehydes, ketones, carboxylic acids…
eg. Outline all steps in a possible laboratory synthesis of 3-methyl-1-butene from alcohols of four carbons or less.
CH3CH3CHCH=CH2

Retrosynthesis:
alkenes, syntheses:
1. Dehydrohalogenation of an alkyl halide
2. Dehydration of an alcohol
3. Dehalogenation of a vicinal dihalide
4. Reduction of an alkyne
Methods 3 & 4 start with compounds that are in turn made from alkenes.

Dehydration of an alcohol?
CH3 H+
CH3CHCHCH3 yields a mixture of alkenesOH
CH3 H+
CH3CHCH2CH2-OH yields a mixture of alkenes
E1 mechanism via carbocation!

Dehydrohalogenation of an alkyl halide?
CH3 KOH(alc)CH3CHCHCH3 yields a mixture of alkenes
Br
CH3 KOH(alc) CH3CH3CHCH2CH2-Br CH3CHCH=CH2
only product ☺
E2 mechanism, no carbocation, no rearrangement

CH3 HBr CH3CH3CHCH2CH2-OH CH3CHCH2CH2-Br
1o alcohol, SN2 mechanism, no rearrangement!
CH3 KOH(alc) CH3CH3CHCH2CH2-Br CH3CHCH=CH2
Use the Grignard synthesis to synthesize the intermediate alcohol from the starting materials.

CH3 PBr3 CH3 Mg CH3CH3CHCH2-OH CH3CHCH2Br CH3CHCH2MgBr
K2Cr2O7CH3OH H2C=O
special cond. H2O
CH3CH3CHCH2CH2-OH
HBr
CH3 KOH(alco) CH3CH3CHCH=CH2 CH3CHCH2CH2-Br

Notes on Exam I:
1. Given the structures, be able to name aldehydes/ketones using either common or IUPAC names.
2. Be able to outline all steps in the mechanisms: nucleophilic addition & acid catalyzed nucleophilic addition.
3. Be able to draw structures for the organic products of the reactions of aldehydes/ketones.
4. Be able to outline possible laboratory syntheses of aldehydes/ketones and Grignard syntheses of alcohols, etc.
5. Be able to match structures with their IR spectra.
6. Be able to predict the 1H-nmr spectrum, given the structure.
7. Given the 1H-nmr spectrum and formula, be able to draw the structure of the compound.




















