
Electronic and optical properties of dibenzothiophen-S,S-dioxide and EDOT based conducting polymers
8
Synthetic Metals 161 (2011) 1898–1905 Contents lists available at ScienceDirect Synthetic Metals j o ur nal homep ag e: www.elsevier.com/locate/synmet Electronic and optical properties of dibenzothiophen-S,S-dioxide and EDOT based conducting polymers Pinar Camurlu a,∗ , Tarık Durak a , Abidin Balan b,1 , Levent Toppare b a Akdeniz University, Department of Chemistry, 07058 Antalya, Turkey b Middle East Technical University, Department of Chemistry, 06531 Ankara, Turkey a r t i c l e i n f o Article history: Received 15 January 2011 Received in revised form 16 June 2011 Accepted 21 June 2011 Available online 22 July 2011 Keywords: Dibenzothiophen-S,S-dioxide EDOT Donor–acceptor polymers Electrochromism Electrochromic device Electropolymerization a b s t r a c t Among various techniques, utilization of donor–acceptor–donor (DAD) approach is known to be powerful in tuning the color of the conducting polymers. For this purpose, a new DAD type monomer 3,7- di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl) dibenzothiophen-S,S-dioxide (EDOTSO 2 ) was developed through Stille cross coupling between tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)tin and 3,7- dibromodibenzothiophen-S,S-dioxide. EDOTSO 2 was polymerized electrochemically and the resultant polymer displayed color alternation from yellow to purple upon oxidation. Furthermore, EDOTSO 2 was subjected to electrochemical copolymerization in the presence of 3,4-ethylenedioxythiophene (EDOT) under both potentiodynamic and potentiostatic techniques. Spectroelectrochemical analysis demon- strated that the neutral copolymers possess reveal a shift in their dominant wavelength of absorbance upon increasing applied potential. Both homopolymer and the copolymers were characterized by means of cyclic voltammetry, FTIR, spectroelectrochemistry, kinetic properties and electrochromic device per- formance of EDOTSO 2 based systems were highlighted. © 2011 Elsevier B.V. All rights reserved. 1. Introduction Electrochromic materials based on conducting polymers have become a recent focus of research due to their high electroactivity and high degree of color tailorability. Among various techniques, utilization of donor–acceptor–donor (DAD) approach is known to be powerful in tuning the color of the conducting polymers. Enclo- sure of electron-rich donor (D) and electron-deficient acceptor (A) groups in an alternating, conjugated manner generally results in a conjugated polymer with a reduced band gap having various colors depending on judicious selection of D–A units hence, controlling the HOMO and LUMO energy levels [1]. Incorporation of electroni- cally different D and A units yields fine control of band gap, redox and electrochromic behavior of the polymers. Owing to its facile electrochemical/chemical polymerization and electronic proper- ties, 3,4-ethylenedioxythiophene (EDOT) and its derivatives have been subject of a growing interest. Especially, within last decade numerous papers have been published utilizing EDOT as the donor group in D–A systems [2,3]. Up to date, many electron-accepting moieties, including quinoxaline derivatives [4,5], benzotriazole ∗ Corresponding author. Tel.: +90 242 3102308; fax: +90 242 2278911. E-mail address: [email protected] (P. Camurlu). 1 Present address: Department of Chemical Engineering and Chemistry, Eind- hoven University of Technology, P.O. Box 513 5600 MB, Netherlands. [6,7], pyridine [8,9], pyridopyrazine [10,11], fluorenone [12] and benzothiadiazole [13–15], have been used in -conjugated D–A–D type polymers. Correspondingly, dibenzothiophen-S,S-dioxide is a promising acceptor group for D–A type polymers since it reveals strong electron-withdrawing effect due to presence of sulpho- nyl group in its structure. In fact, since 2005 extensive effort has been devoted on the synthesis and utilization of dibenzothiophene- S,S-dioxide together with poly(p-phenylenevinylene) [16] and polyfluorene [17–21] in the field of light emitting diodes (PLEDs). Studies [19,20,22] have shown that inclusion of dibenzothiophene- S,S-dioxide unit within the backbone of the conjugated polymers generally enhance the electron injection characteristics and device performances. The underlying reason was considered to be related to the changes in the frontier orbital energy levels and increase in the electron affinity of these materials due to strong electron- withdrawing ability of dibenzothiophene-S,S-dioxide unit [21]. Though dibenzothiophene-S,S-dioxide-containing polymers have been studied in PLEDs, to our knowledge, there were no reports on their electrochromic behavior and use in electrochromic devices. Hence, in this study a new DAD type monomer 3,7- di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl) dibenzothiophen- S,S-dioxide (EDOTSO 2 ) was synthesized through Stille coupling between tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)tin and 3,7-dibromodibenzothiophen-S,S-dioxide (Scheme 1). EDOTSO 2 was electrochemically homopolymerized and copolymerized in the presence of 3,4-ethylenedioxythiophene in tetrabutylam- 0379-6779/$ – see front matter © 2011 Elsevier B.V. All rights reserved. doi:10.1016/j.synthmet.2011.06.033
Transcript of Electronic and optical properties of dibenzothiophen-S,S-dioxide and EDOT based conducting polymers

Eb
Pa
b
a
ARRAA
KDEDEEE
1
baubsgcdtcaetbngm
h
0d
Synthetic Metals 161 (2011) 1898– 1905
Contents lists available at ScienceDirect
Synthetic Metals
j o ur nal homep ag e: www.elsev ier .com/ locate /synmet
lectronic and optical properties of dibenzothiophen-S,S-dioxide and EDOTased conducting polymers
inar Camurlua,∗, Tarık Duraka, Abidin Balanb,1, Levent Toppareb
Akdeniz University, Department of Chemistry, 07058 Antalya, TurkeyMiddle East Technical University, Department of Chemistry, 06531 Ankara, Turkey
r t i c l e i n f o
rticle history:eceived 15 January 2011eceived in revised form 16 June 2011ccepted 21 June 2011vailable online 22 July 2011
eywords:
a b s t r a c t
Among various techniques, utilization of donor–acceptor–donor (DAD) approach is known to be powerfulin tuning the color of the conducting polymers. For this purpose, a new DAD type monomer 3,7-di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl) dibenzothiophen-S,S-dioxide (EDOTSO2) was developedthrough Stille cross coupling between tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)tin and 3,7-dibromodibenzothiophen-S,S-dioxide. EDOTSO2 was polymerized electrochemically and the resultantpolymer displayed color alternation from yellow to purple upon oxidation. Furthermore, EDOTSO2 was
ibenzothiophen-S,S-dioxideDOTonor–acceptor polymerslectrochromismlectrochromic devicelectropolymerization
subjected to electrochemical copolymerization in the presence of 3,4-ethylenedioxythiophene (EDOT)under both potentiodynamic and potentiostatic techniques. Spectroelectrochemical analysis demon-strated that the neutral copolymers possess reveal a shift in their dominant wavelength of absorbanceupon increasing applied potential. Both homopolymer and the copolymers were characterized by meansof cyclic voltammetry, FTIR, spectroelectrochemistry, kinetic properties and electrochromic device per-formance of EDOTSO2 based systems were highlighted.
© 2011 Elsevier B.V. All rights reserved.
. Introduction
Electrochromic materials based on conducting polymers haveecome a recent focus of research due to their high electroactivitynd high degree of color tailorability. Among various techniques,tilization of donor–acceptor–donor (DAD) approach is known toe powerful in tuning the color of the conducting polymers. Enclo-ure of electron-rich donor (D) and electron-deficient acceptor (A)roups in an alternating, conjugated manner generally results in aonjugated polymer with a reduced band gap having various colorsepending on judicious selection of D–A units hence, controllinghe HOMO and LUMO energy levels [1]. Incorporation of electroni-ally different D and A units yields fine control of band gap, redoxnd electrochromic behavior of the polymers. Owing to its facilelectrochemical/chemical polymerization and electronic proper-ies, 3,4-ethylenedioxythiophene (EDOT) and its derivatives haveeen subject of a growing interest. Especially, within last decade
umerous papers have been published utilizing EDOT as the donorroup in D–A systems [2,3]. Up to date, many electron-acceptingoieties, including quinoxaline derivatives [4,5], benzotriazole∗ Corresponding author. Tel.: +90 242 3102308; fax: +90 242 2278911.E-mail address: [email protected] (P. Camurlu).
1 Present address: Department of Chemical Engineering and Chemistry, Eind-oven University of Technology, P.O. Box 513 5600 MB, Netherlands.
379-6779/$ – see front matter © 2011 Elsevier B.V. All rights reserved.oi:10.1016/j.synthmet.2011.06.033
[6,7], pyridine [8,9], pyridopyrazine [10,11], fluorenone [12] andbenzothiadiazole [13–15], have been used in �-conjugated D–A–Dtype polymers. Correspondingly, dibenzothiophen-S,S-dioxide is apromising acceptor group for D–A type polymers since it revealsstrong electron-withdrawing effect due to presence of sulpho-nyl group in its structure. In fact, since 2005 extensive effort hasbeen devoted on the synthesis and utilization of dibenzothiophene-S,S-dioxide together with poly(p-phenylenevinylene) [16] andpolyfluorene [17–21] in the field of light emitting diodes (PLEDs).Studies [19,20,22] have shown that inclusion of dibenzothiophene-S,S-dioxide unit within the backbone of the conjugated polymersgenerally enhance the electron injection characteristics and deviceperformances. The underlying reason was considered to be relatedto the changes in the frontier orbital energy levels and increasein the electron affinity of these materials due to strong electron-withdrawing ability of dibenzothiophene-S,S-dioxide unit [21].Though dibenzothiophene-S,S-dioxide-containing polymers havebeen studied in PLEDs, to our knowledge, there were no reports ontheir electrochromic behavior and use in electrochromic devices.
Hence, in this study a new DAD type monomer 3,7-di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl) dibenzothiophen-S,S-dioxide (EDOTSO2) was synthesized through Stille coupling
between tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)tin and3,7-dibromodibenzothiophen-S,S-dioxide (Scheme 1). EDOTSO2was electrochemically homopolymerized and copolymerized inthe presence of 3,4-ethylenedioxythiophene in tetrabutylam-
P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905 1899
SOO
S SOO
BrBr
SOO
OO
S
OO
SSOO
BrBr
OO
S
OO
SSnBu3
n-BuLi
SnBu3Cl
Pd(PPH3)2Cl 2
THF
OO
SSnBu3
MCPBA
CH2Cl 2
NBS, H2SO 4
+
EDOTSO2
oute o
mrmkea
2
2
pNdDc(Tw
2
Ds(n5wwpaisavc
Scheme 1. Synthetic r
onium hexafluorophosphate (TBAPF6)/dichloromethane. Theesulting homopolymer and copolymers were characterized byeans of cyclic voltammetry, FTIR, spectroelectrochemistry and
inetic studies. The effect of synthesis conditions on the spectro-lectrochemical properties of polymer was also investigated andn electrochromic device was constructed.
. Experimental
.1. Materials
All chemicals (except the supporting electrolyte) wereurchased from Aldrich, Merck Chemical as analytical grade.-bromosuccinimide was recrystallized from hot water. Tetrahy-rofuran (THF) was distilled over Na/benzophenone prior to use.imethyl formamide (DMF) was distilled under vacuum overalcium hydride (CaH2). Acetonitrile (ACN), dichloromethaneDCM) were distilled over CaH2 and kept on 4 A molecular sieves.ributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)stannane [23]as prepared according to previously reported method.
.2. Equipments
NMR spectra were recorded with Bruker Spectrospin AvancePX-400 Spectrometer at 400 MHz (1H NMR) and Bruker Ultra-
hield at 300 MHz (1H NMR), 75 MHz (13C NMR). Chemical shiftsı) were given relative to tetramethylsilane (TMS) as the inter-al standard. Mass spectroscopy (MS) was performed on DP-MS973 HP quadruple MS. High resolution mass spectroscopy (HRMS)as performed on Waters SYNAPT MS System. The FTIR spectraere recorded on a Brucker Tensor 27 spectrometer. Gamry 600otentiostat/galvanostat was used for electrochemical synthesisnd cyclic voltammetry. Spectroelectrochemical and kinetic stud-es of the polymers were performed on Varian Cary 100 UV–vis
pectrophotometer. Colorimetry measurements were recorded onMinolta CS-100A Chroma Meter with a 0/0 (normal/normal)iewing geometry as recommended by CIE. Elemental analyses ofopolymers were performed on LECO, CHNS-932
f monomer EDOTSO2.
2.3. Monomer synthesis
2.3.1. Synthesis of dibenzothiophene-S,S-dioxide0.11 mol of 77% m-chloroperbenzoic acid (MCPBA) (in 50 mL
chloroform) and 0.05 mol of dibenzothiophene (in 80 mL chloro-form) were slowly mixed and refluxed for 7 h. After the mixture wascooled, it was washed with saturated NaHCO3, dried over MgSO4,and recrystallized from ethanol to give the title compound in 70%yield [21]. 1H NMR (400 MHz, CDCl3): ı: 7.45–7.49 (t, 1H), 7.56–7.60(t, 1H), 7.72–7.77 (m, 2H).
2.3.2. Synthesis of 3,7-dibromodibenzothiophene-S,S-dioxideDibenzothiophene-S,S-dioxide (0.046 mol) was dissolved
in concentrated H2SO4 (300 mL). N-bromosuccinimide (NBS)(0.092 mol) was added to this solution in several portions and themixture was stirred at room temperature for 24 h. The white solidwas filtered off, washed with H2SO4 and water (44%) [21]. 1H NMR(400 MHz, CDCl3): ı: 7.58 (d, 1H), 7.72 (d, 1H), 7.87 (s, 1H).
2.3.3. Synthesis of3,7-di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)dibenzothiophen-S,S-dioxide (EDOTSO2)
A mixture of 3,7-dibromo-dibenzothiophene S,S-dioxide(0.224 mmol), tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)stannane (0.89 mmol), PdCl2(PPh3)2 (50 mg), and THF (15 mL)were refluxed for 24 h. The solvent was evaporated the crude prod-uct was purified by flash chromatography and the title compoundas a yellow solid (45% yield) was achieved (Scheme 1, Fig. 1a andb). 1H NMR (300 MHz, CDCl3) ı 8.33 (s, 1H), 7.8 (dd, 2H), 6.41 (s,1H), 4.49–4.15 (m, 4H),13C NMR (75 MHz, DMSO) ı 142.80, 140.76,138.21, 135.54, 130.78, 128.55, 123.69, 117.94, 114.16, 100.84,65.61, 64.56. MS calcd. for C24H16O6S3 (m/z): 496.5, found: 496.1;
HRMS (MH+) calcd: 497.0187, found: 497.0164 �absmax : 263–227 nm,344–387 nm; �ems
max (excitation at 390 nm): 459 nm; FTIR (in KBrdisk, cm−1): 3112, 2925, 2871,1504 (s), 1432, 1359, 1299, 1162,1069 (s),942, 906, 828 (w), 706 (w), 592.

1900 P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905
1 ) 13C
2
tsaeed
Fig. 1. (a) H NMR (in d-chloroform), (b
.4. Synthesis of homopolymer (PEDOTSO2)
PEDOTSO2 films were prepared potentiodynamically on indiumin oxide coated glass slides (ITO), using 0.01 M monomer in aolution containing TBAPF6 and DCM. A platinum wire was used
s the counter electrode against a Ag wire pseudo referencelectrode (Scheme 2). Electrochromic measurements; spectro-lectrochemistry, and switching studies of the polymer filmseposited on ITO electrode were performed using a UV-cuvetteNMR (in DMSO) spectrum of EDOTSO2.
with three-electrodes placed in the sample compartment of a spec-trophotometer.
2.5. Synthesis of copolymers (P(EDOTSO2-EDOT))
EDOT was used as the comonomer for the synthesis of conduct-ing copolymer of EDOTSO2. EDOTSO2 (50 mg) was dissolved in 5 mlof DCM and 10 �L of EDOT was introduced into the electrolysiscell containing TBAPF6 (Scheme 2). The films were either prepared

P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905 1901
SOO
OO
S
OO
S
SOO
OO
S
OO
S
S
OO
S
OO
S
OO
S
Electrochemical
polymerization
Electrochemical
copolymerization
n
n
OO
S m
hemic
p1
2
pgpttep
3
3
DiwowTSyctwHh
3
tiaAam(fvc
Scheme 2. Electroc
otentiodynamically scanning the potential between −0.5 V and.5 V or potentiostatically at 1.1–1.4 V on ITO electrodes.
.6. Construction of electrochromic devices (ECDs)
Both anodically (copolymer) and cathodically (PEDOT) coloringolymers were electrochemically deposited onto the ITO-coatedlass from a 0.1 M TBAPF6/ACN at 1.1 V. The redox sites of theseolymer films were matched by stepping the potentials betweenheir extreme states. ECDs were built by arranging two elec-rochromic polymer films (one doped, the other neutral) facingach other and separated by gel electrolyte. The gel electrolyte wasrepared according to literature [24].
. Results and discussion
.1. Monomer synthesis
The synthetic route to the monomer is revealed in Scheme 1.ibenzothiophene was treated with m-chloroperbenzoic acid
n chloroform to yield benzothiophene-S,S-dioxide. Later, itas brominated with n-bromosuccinimide in the presence
f concentrated H2SO4 in high yields. Stannylation of EDOTas achieved according to previously reported method [23].
he Stille coupling reaction of 3,7-dibromo-dibenzothiophene,S-dioxide with tributyl(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-l)stannane was performed in dry THF with Pd(PPh3)2Cl2 as theatalyst. The reaction proceeded with moderate yield to afford theitle compound EDOTSO2. The structural investigation of monomeras performed with 1H NMR, 13C NMR FTIR, Floresence, MS andRMS analyses. The monomer is solid in room temperature andas yellow color.
.2. Electrochemical polymerization
EDOTSO2 was examined via cyclic voltammetry (CV) on indiumin oxide coated glass slides (ITO) in 0.1 M TBAPF6/DCM support-ng electrolyte solution in order to investigate its redox behaviornd to synthesize the electroactive homopolymer (PEDOTSO2).s seen in Fig. 2a EDOTSO2 revealed an irreversible oxidationt around 1.3 V, signifying the formation of radical cation of theonomer through its EDOT moieties and new redox processes
quasi reversible) appeared at lower potentials, indicating theormation of polymer. Such behavior is in accordance with the pre-ious studies based on Bis EDOT derivatives in literature. Uponontinuous cycling a homogeneous PEDOTSO2 film was synthe-
OO
al polymerizations.
sized. FTIR spectrum of PEDOTSO2 revealed 2958–2853 cm-1(C–Haliphatic), 1649–1682 cm−1 (polyconjugation), 1503–1490 cm−1
(C C, C–C stretching), 1400, 1303, 1164 cm−1 (SO2), 1238 cm-1
(C–O–C asymmetric stretching), 1050 cm−1 (C–O–C symmetricstretching), 850 cm−1 (dopant anion), 763–620 cm−1 (C–H out ofplane bending). Hence, upon the homopolymerization most of thetypical peaks of EDOTSO2, especially those related to SO2, remainedunperturbed indicating that the electrochemical polymerizationwas achieved [25].
Fig. 2c displays the cyclic voltammogram of EDOT the undersame conditions where EDOT’s oxidation peak was observed ataround 1.2 V. These results imply that both EDOT and EDOTSO2 areoxidized within the similar potential window and radical cations ofboth monomers might form almost simultaneously on the workingelectrode. Hence, these two monomers could be utilized in copoly-mer synthesis since their radical cations can react with each otherand form a true copolymer [26]. Fig. 2b shows the CV of a systemcontaining both monomers in 0.1 M TBAPF6/DCM supporting elec-trolyte solution. As seen, in the first cycle there is a steady increasein the current around 1.2 V (indicating the irreversible oxidation ofEDOT) and a well defined peak (1.39 V) corresponding to the oxida-tion of EDOTSO2. Upon repetitive scanning, evolution of new peakson both anodic (0.83 V) and cathodic (−0.26 V) sides were observeddue to formation of the copolymer which is significantly differ-ent than the pristine EDOT and EDOTSO2 systems. Fig. 2d showsthe scan rate dependence of the copolymer film in monomer freesupporting electrolyte system where a linear increase of the peakcurrent was observed upon increase in the scan rate. In literature,such observation is attributed to non-diffusion limited redox pro-cesses and formation of well adhered electroactive polymer film onthe electrode [27].
3.3. Spectroelectrochemistry
Spectroelectrochemistry experiments are performed to inves-tigate the optical changes upon doping process and band gap ofthe polymer [28]. The spectral behavior of the PEDOTSO2 wasrecorded on a UV–vis spectrophotometer in a monomer free elec-trolyte system while incrementally increasing the applied potentialfrom −1.5 V to 2.0 V. In neutral form the polymer showed a distinctabsorption centered at 357 nm that is attributed to �–�* transition(Fig. 3). Stepwise oxidation of the polymer resulted in a gradual
decline in the intensity of this transition and simultaneously, for-mation of new bands (centered at 537 nm) was observed due tocharge carrier band formation. The polymer displayed distinct colorchange from yellow (Y: 730, x: 0.322, y: 0.310) to purple (Y: 518,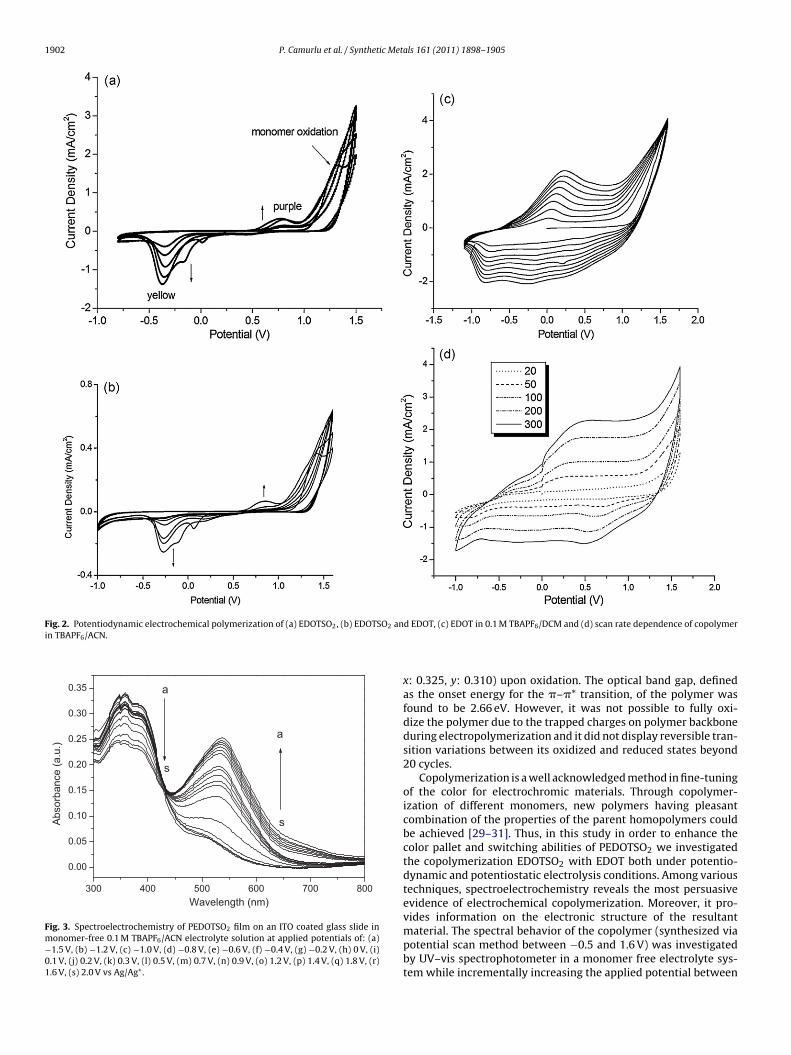
1902 P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905
Fig. 2. Potentiodynamic electrochemical polymerization of (a) EDOTSO2, (b) EDOTSO2 anin TBAPF6/ACN.
800700600500400300
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
a
s
s
a
Abso
rban
ce (a
.u.)
Wavelength (nm)
Fig. 3. Spectroelectrochemistry of PEDOTSO2 film on an ITO coated glass slide inmonomer-free 0.1 M TBAPF6/ACN electrolyte solution at applied potentials of: (a)−1.5 V, (b) −1.2 V, (c) −1.0 V, (d) −0.8 V, (e) −0.6 V, (f) −0.4 V, (g) −0.2 V, (h) 0 V, (i)0.1 V, (j) 0.2 V, (k) 0.3 V, (l) 0.5 V, (m) 0.7 V, (n) 0.9 V, (o) 1.2 V, (p) 1.4 V, (q) 1.8 V, (r)1.6 V, (s) 2.0 V vs Ag/Ag+.
d EDOT, (c) EDOT in 0.1 M TBAPF6/DCM and (d) scan rate dependence of copolymer
x: 0.325, y: 0.310) upon oxidation. The optical band gap, definedas the onset energy for the �–�* transition, of the polymer wasfound to be 2.66 eV. However, it was not possible to fully oxi-dize the polymer due to the trapped charges on polymer backboneduring electropolymerization and it did not display reversible tran-sition variations between its oxidized and reduced states beyond20 cycles.
Copolymerization is a well acknowledged method in fine-tuningof the color for electrochromic materials. Through copolymer-ization of different monomers, new polymers having pleasantcombination of the properties of the parent homopolymers couldbe achieved [29–31]. Thus, in this study in order to enhance thecolor pallet and switching abilities of PEDOTSO2 we investigatedthe copolymerization EDOTSO2 with EDOT both under potentio-dynamic and potentiostatic electrolysis conditions. Among varioustechniques, spectroelectrochemistry reveals the most persuasiveevidence of electrochemical copolymerization. Moreover, it pro-vides information on the electronic structure of the resultant
material. The spectral behavior of the copolymer (synthesized viapotential scan method between −0.5 and 1.6 V) was investigatedby UV–vis spectrophotometer in a monomer free electrolyte sys-tem while incrementally increasing the applied potential between
P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905 1903
8007006005004000.10
0.15
0.20
0.25
0.30
0.35
o
a
Abs
orba
nce
Wavelength (nm)
Fig. 4. Spectroelectrochemistry of copolymer film (synthesized on an ITO via poten-tiodynamic cycling) in 0.1 M TBAPF6/DCM electrolyte solution at applied potentialso−A
−amtca
tFcfictntcttgp
Fs
Fig. 6. Images of (a) PEDOTSO2 in neutral and oxidized states, (b) P(EDOTSO2-
f: (a) −1.3 V, (b) −1.2 V, (c) −1.1 V, (d) −1.0 V, (e) −0.9 V, (f) −0.8 V, (g) −0.7 V, (h)0.6 V, (i) −0.5 V, (j) −0.3 V, (k) −0.2 V, (l) 0.2 V, (m) 0.7 V, (n) 1.0 V, (o) 1.3 V vsg/Ag+.
1.3 V and 1.3 V. In neutral form copolymer exhibited a broadbsorption at 568 nm due �–�* transition (Fig. 4). The copoly-er’s band gap was calculated as 1.7 eV and it was in between the
wo parent polymers (PEDOT: 1.6 eV and PEDOTSO2: 2.66 eV). Theopolymer revealed indigo and transparent gray color in its neutralnd oxidized states, respectively.
Moreover, the effect of electrochemical synthesis conditions onhe electrochromic properties of the copolymer was investigated.or this purpose, copolymers were synthesized by using the sameomonomer feed ratio and solvent–electrolyte couple under dif-erent (1.1, 1.2, 1.3, 1.4 V) applied potentials. This simple approachs based on one of the unique properties (selectivity) of electro-hemical polymerization. The resultant polymers were subjectedo spectroelectrochemistry studies. Figs. 5 and 6 represent theormalized absorption spectra of copolymers in neutral state andhe colors of each polymer, respectively. As seen in Fig. 6 all theopolymer films were homogeneous and well adhered on ITO elec-
rode. Contrary to potentiodynamically synthesized copolymer,hese copolymers displayed two well defined transitions showingradual shift and a broadening upon increase of polymerizationotential from 1.1 V to 1.4 V. It is important to note that the tran-8006004000.0
0.5
1.0
1.5
2.0
1.1 V
1.2 V
1.3 V
1.4 V
Nor
mal
ize
Abs
orba
nce
Wavelength (nm)
ig. 5. Normalized UV–vis spectrum of neutral P(EDOTSO2–EDOT)s which wereynthesized at different polymerization potentials.
EDOT) synthesized under constant potentials (given below) in neutral state,(c) P(EDOTSO2-EDOT)/PEDOT absorptive–transmissive type electrochromic device(ECD) at ultimate coloration conditions.
sitions at higher wavelengths increased in intensity relative tothe transitions at lower wavelengths. Among those, the former isconsidered to be related with the EDOT and the latter with theEDOTSO2 rich domains. This situation is also reflected on the col-ors of the copolymers where a direct relation between the colors inneutral state and applied potential during copolymerization wasobserved. The color of the copolymers varied from light purpleto blue color. Such distinct behavior could be related to presenceof longer segments of EDOT units in copolymers. Electrochemicalpolymerization of thiophene and its derivatives are known [32] toproceed through ECE mechanism which involves formation of rad-ical cation, coupling of the two radicals to produce dihydro dimercation and loss of two protons, respectively. Since EDOT has a loweroxidation potential than EDOTSO2 monomer, the number of radi-cal cations available within the vicinity of the electrode throughthis unit could be more than the EDOTSO2 radical cations. Thus,incorporation of more EDOT units within the copolymer backboneis expected. Moreover, the difference in the rate of electrochemicaloxidation of both monomers is valid by the CV studies, where undersimilar conditions EDOTSO2 revealed a well defined peak contraryto EDOT (Fig. 2a and c) during oxidation. This is perhaps related to
relatively slow oxidation of EDOTSO2, leading to a decrease of elec-troactive species within the diffusion layer. Assuming similar rateof diffusion, we could infer that EDOT is comparatively more reac-65
70
75
80
85
90
3210
Tran
smitt
ance
( T
%)
Time (min.)
Fig. 7. Electrochromic switching, optical absorbance change of copolymer depositedon ITO electrode monitored at 540 nm in 0.1 M TBAPF6/ACN.
1904 P. Camurlu et al. / Synthetic Metals 161 (2011) 1898– 1905
D at ap
tilprltmE
3
ossceamocctia
3
m
TM
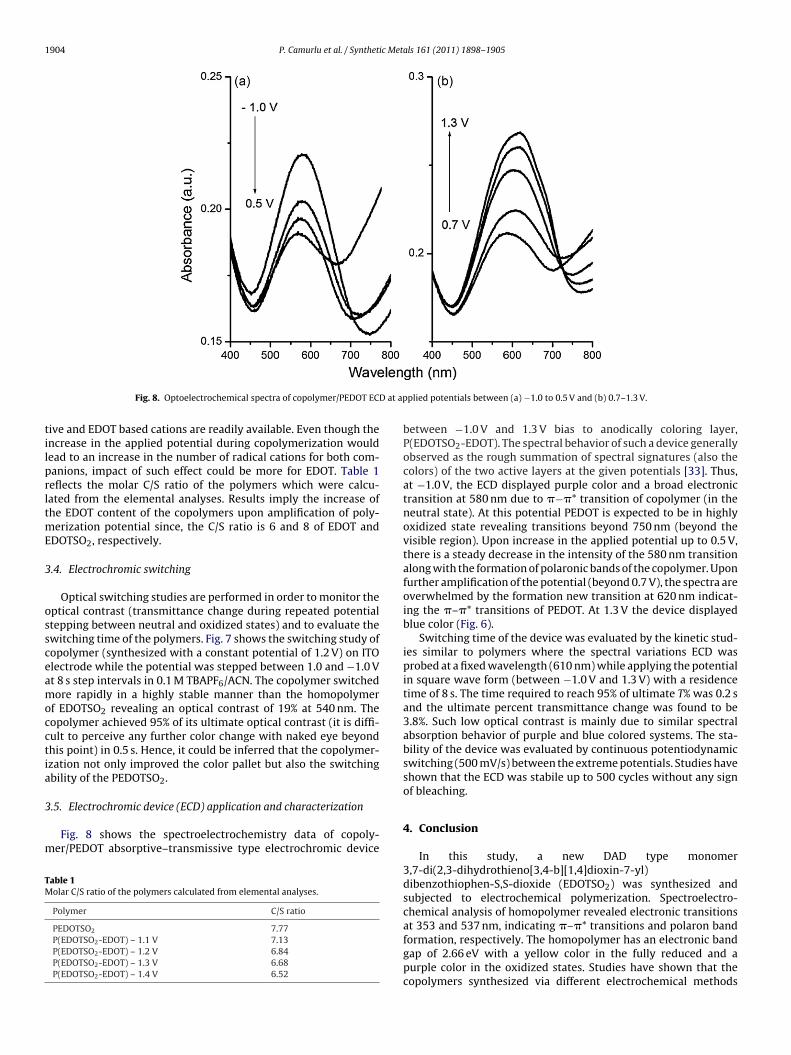
Fig. 8. Optoelectrochemical spectra of copolymer/PEDOT EC
ive and EDOT based cations are readily available. Even though thencrease in the applied potential during copolymerization wouldead to an increase in the number of radical cations for both com-anions, impact of such effect could be more for EDOT. Table 1eflects the molar C/S ratio of the polymers which were calcu-ated from the elemental analyses. Results imply the increase ofhe EDOT content of the copolymers upon amplification of poly-
erization potential since, the C/S ratio is 6 and 8 of EDOT andDOTSO2, respectively.
.4. Electrochromic switching
Optical switching studies are performed in order to monitor theptical contrast (transmittance change during repeated potentialtepping between neutral and oxidized states) and to evaluate thewitching time of the polymers. Fig. 7 shows the switching study ofopolymer (synthesized with a constant potential of 1.2 V) on ITOlectrode while the potential was stepped between 1.0 and −1.0 Vt 8 s step intervals in 0.1 M TBAPF6/ACN. The copolymer switchedore rapidly in a highly stable manner than the homopolymer
f EDOTSO2 revealing an optical contrast of 19% at 540 nm. Theopolymer achieved 95% of its ultimate optical contrast (it is diffi-ult to perceive any further color change with naked eye beyondhis point) in 0.5 s. Hence, it could be inferred that the copolymer-zation not only improved the color pallet but also the switchingbility of the PEDOTSO2.
.5. Electrochromic device (ECD) application and characterization
Fig. 8 shows the spectroelectrochemistry data of copoly-er/PEDOT absorptive–transmissive type electrochromic device
able 1olar C/S ratio of the polymers calculated from elemental analyses.
Polymer C/S ratio
PEDOTSO2 7.77P(EDOTSO2-EDOT) – 1.1 V 7.13P(EDOTSO2-EDOT) – 1.2 V 6.84P(EDOTSO2-EDOT) – 1.3 V 6.68P(EDOTSO2-EDOT) – 1.4 V 6.52
plied potentials between (a) −1.0 to 0.5 V and (b) 0.7–1.3 V.
between −1.0 V and 1.3 V bias to anodically coloring layer,P(EDOTSO2-EDOT). The spectral behavior of such a device generallyobserved as the rough summation of spectral signatures (also thecolors) of the two active layers at the given potentials [33]. Thus,at −1.0 V, the ECD displayed purple color and a broad electronictransition at 580 nm due to �−�* transition of copolymer (in theneutral state). At this potential PEDOT is expected to be in highlyoxidized state revealing transitions beyond 750 nm (beyond thevisible region). Upon increase in the applied potential up to 0.5 V,there is a steady decrease in the intensity of the 580 nm transitionalong with the formation of polaronic bands of the copolymer. Uponfurther amplification of the potential (beyond 0.7 V), the spectra areoverwhelmed by the formation new transition at 620 nm indicat-ing the �–�* transitions of PEDOT. At 1.3 V the device displayedblue color (Fig. 6).
Switching time of the device was evaluated by the kinetic stud-ies similar to polymers where the spectral variations ECD wasprobed at a fixed wavelength (610 nm) while applying the potentialin square wave form (between −1.0 V and 1.3 V) with a residencetime of 8 s. The time required to reach 95% of ultimate T% was 0.2 sand the ultimate percent transmittance change was found to be3.8%. Such low optical contrast is mainly due to similar spectralabsorption behavior of purple and blue colored systems. The sta-bility of the device was evaluated by continuous potentiodynamicswitching (500 mV/s) between the extreme potentials. Studies haveshown that the ECD was stabile up to 500 cycles without any signof bleaching.
4. Conclusion
In this study, a new DAD type monomer3,7-di(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)dibenzothiophen-S,S-dioxide (EDOTSO2) was synthesized andsubjected to electrochemical polymerization. Spectroelectro-chemical analysis of homopolymer revealed electronic transitionsat 353 and 537 nm, indicating �–�* transitions and polaron band
formation, respectively. The homopolymer has an electronic bandgap of 2.66 eV with a yellow color in the fully reduced and apurple color in the oxidized states. Studies have shown that thecopolymers synthesized via different electrochemical methods
c Meta
rctoavclb
A
i2
R
[
[[
[[[
[
[
[[
[
[
[
[[
[[[
[
[
[
P. Camurlu et al. / Syntheti
evealed diverse spectroelectrochemical behavior, band gap andoloration. All the copolymers can be switched easily betweenheir neutral states to a common blue oxidized state with moderateptical contrast. Through the careful selection of comonomersnd by varying the applied potential (that is far more easier thanariation of comonomer concentration) the spectral behavior andolor of copolymers in neutral state ranging from purple to blue,ed to augmentation of color pallet of the electrochromic polymersased on dibenzothiophen-S,S-dioxide.
cknowledgements
We are grateful to “2008 L’Oreal Turkey for Young Womenn Science” fellowship, Akdeniz University (Project no:008.01.0105.009) and METU for the support of this study.
eferences
[1] H.A.M. van Mullekom, J.A.J.M. Vekemans, E.E. Havinga, E.W. Meijer, Mater. Sci.Eng. 32 (2001) 1.
[2] P.M. Beaujuge, J.R. Reynolds, Chem. Rev. 110 (2010) 268.[3] J. Roncali, Macromol. Rapid Commun. 28 (2007) 1761.[4] A. Tsami, T.W. Bunnagel, T. Farrell, M. Scharber, S.A. Choulis, C.J. Brabec, U.
Scherf, J. Mater. Chem. 17 (2007) 1353.[5] G.E. Gunbas, P. Camurlu, I.M. Akhmedov, C. Tanyeli, A. Onal, L. Toppare, J. Elec-
troanal. Chem. 615 (2008) 75.[6] A. Balan, D. Baran, G. Gunbas, A. Durmus, F. Ozyurt, L. Toppare, Chem. Commun.
(2009) 6768.
[7] D. Baran, A. Balan, S. Celebi, E.B. Meana, H. Neugebauer, S.N. Sariciftci, L. Top-pare, Chem. Mater. 22 (2010) 2978.[8] C.J. DuBois Jr., F. Larmat, J.R. Reynolds, Adv. Mater. 14 (2002) 1844.[9] D.J. Irvin, C.J. DuBois Jr., J.R. Reynolds, Chem. Commun. (1999) 2121.10] C.J. DuBois Jr., F. Larmat, D.J. Irvin, J.R. Reynolds, Synth. Met. 123 (2001) 425.
[
[[
ls 161 (2011) 1898– 1905 1905
11] B.L. Lee, T. Yamamoto, Macromolecules 32 (1999) 1375.12] R. Demadrille, N. Delbosc, Y. Kervella, M. Firon, R. De Bettignies, M. Billon, P.
Rannou, A. Pron, J. Mater. Chem. 17 (2007) 4661.13] A. Durmus, G.E. Günbas , P. Camurlu, L. Toppare, Chem. Commun. (2007) 3246.14] P.M. Beaujuge, S. Ellinger, J.R. Reynolds, Adv. Mater. 20 (2008) 2772.15] M. Sendur, A. Balan, D. Baran, B. Karabay, L. Toppare, Org. Electron. 11 (2010)
1877.16] B. Wang, J. Yina, M. Xue, J. Wang, G. Zhong, X. Ding, Thin Solid Films 424 (2003)
186.17] K.T. Kamtekar, H.L. Vaughan, B.P. Lyons, A.P. Monkman, S.U. Pandya, M.R. Bryce,
Macromolecules 43 (2010) 4481.18] Y. Li, H. Wu, J. Zou, L. Ying, W. Yang, Y. Cao, Org. Electron. 10 (2009) 901.19] S.M. King, I.I. Perepichka, I.F. Perepichka, F.B. Dias, M.R. Bryce, A.P. Monkman,
Adv. Funct. Mater. 19 (2009) 586.20] F.B. Dias, S. King, A.P. Monkman, I.I. Perepichka, M.A. Kryuchkov, I.F. Perepichka,
M.R. Bryce, J. Phys. Chem. B 112 (2008) 112, 6557.21] I.I. Perepichka, I.F. Perepichka, M.R. Bryce, L. Palsson, Chem. Commun. (2005)
3397.22] R. Grisorio, C. Piliego, P. Cosma, P. Fini, P. Mastrorilli, G. Gigli, G.P. Suranna, C.F.
Nobile, J. Polym. Sci. Polym. Chem. 47 (2009) 2093.23] C. Edder, J.M.J. Frechet, Org. Lett. 5 (2003) 1879.24] E. Sahin, P. Camurlu, L. Toppare, V.M. Mercore, I. Cianga, Y. Yagci, Polym. Int. 54
(2005) 1599.25] B. Lu, Y. Li, J. Xu, J. Electroanal. Chem. 643 (2010) 67.26] C.L. Gaupp, J.R. Reynolds, Macromolecules 36 (2003) 6305.27] A. Kumar, D.M. Welsh, M.C. Morvant, F. Piroux, K.A. Abboud, J.R. Reynolds,
Chem. Mater. 10 (1998) 896.28] S. Beyazyildirim, P. Camurlu, D. Yilmaz, M. Gullu, L. Toppare, J. Electroanal.
Chem. 587 (2006) 235.29] P. Camurlu, S. Tarkuc , E. S ahmetlioglu, I.M. Akhmedov, C. Tanyeli, L. Toppare,
Sol. Energy Mater. Sol. Cells 92 (2008) 154.30] P. Camurlu, E. S ahmetlioglu, E. Sahin, I.M. Akhmedov, C. Tanyeli, L. Toppare,
Thin Solid Films 516 (2008) 4139.
31] E. Yildiz, P. Camurlu, C. Tanyeli, I.M. Akhmedov, L. Toppare, J. Electroanal. Chem.612 (2008) 247.32] J. Roncali, Chem. Rev. 92 (1992) 711.33] L.B. Groenendaal, G. Zotti, P.H. Aubert, S.M. Waybright, J.R. Reynolds, Adv.
Mater. 15 (2003) 855.




















