
Siamese-Twin Porphyrin: A Pyrazole-Based Expanded Porphyrin Providing a Bimetallic Cavity
Upload
eastangliaCategory
view
2download
0
Electrochemical studies of porphyrin-appended dendrimers
Conor F. Hogan,aAlexander R. Harris,
bAlan M. Bond,*
bJoseph Sly
cand
Maxwell J. Crossley*c
Received 16th November 2005, Accepted 23rd February 2006
First published as an Advance Article on the web 24th March 2006
DOI: 10.1039/b516281e
The electrochemical properties of porphyrin-appended dendrimers containing 2-, 4-, 8-, 16-, 32-
and 64-porphyrin macrocycles in their free-base and zinc(II) forms have been investigated. Both
series gave diffusional based voltammetric responses in dichloromethane. There was minimal
effect of dendrimer generation on the redox potentials. Multiple p-cation and anion radicals as
well as dications and dianions were formed on the surface of the dendrimers on oxidation or
reduction as appropriate, with each cyclic voltammetric wave representing electron transfer to or
from multiple non-interacting porphyrin sites. Electrostatic interactions in the higher generation
dendrimers result in kinetic effects being observed for the highly charged species generated when
each porphyrin unit is doubly or triply oxidised. The number of electrons transferred on
reduction or oxidation of the dendrimers was evaluated using steady-state microelectrode
voltammetry. For the lower generations of species a good correlation was observed between
numbers of electrons transferred and number of porphyrin entities per molecule; for the
dendrimers containing 32 and 64 units, however, slight negative deviations were observed,
possibly due to electrostatic interactions as the porphyrins become closer packed.
Introduction
There has been considerable interest in construction of synthetic
analogues of the light-harvesting and redox-active pigment
arrays involved in photosynthesis. Structural studies of the
natural systems have shown that porphinoid pigments are used
as the photo- and electro-active chromophores.1 Arrays of these
pigments are arranged in well-defined geometry usually on or
near the surface of a 3-D shape. The light-harvesting pigments
are relatively close packed but their number is organism
dependent. In the case of photosystems I and II, the pigments
lie in the outer regions of ellipsoid molecular spaces that
encapsulate the photosynthetic reaction centre.2,3 The photo-
system I complex of the cyanobacterium Synechococcus elonga-
tus is trimeric and has 96 chlorophyll pigments so arranged in
each sub-unit.2 The light-harvesting system of green sulfur
bacteria has only 7 (bacteriochlorophyll) pigments per subunit
but has similar molecular architecture.4,5 Purple photosynthetic
bacteria have light-harvesting units with 27- and 32-porphinoid
pigments arranged on the outer periphery of a toroid.6,7
The redox properties and substantially large extinction
coefficients of systems containing multiple porphyrin units
make them particularly useful models of natural photosyn-
thetic antennas and may also underpin developments in
molecular photonic devices such as sensors, nanoscale optical
sources and solar collectors. The stability of the p-cation
radicals and p-anion radicals that are formed upon oxidation
or reduction of multiple porphyrin arrays provide the basis for
nanoscale hole/electron storage reservoirs.
The unique hyperbranched polymeric structure and well de-
fined 3-D architecture of dendrimers affords precise control of
molecule size,8 as well as the positioning of desired functionalities
at the core,9 within the dendric branches10 or on the exterior
surfaces.11 In addition, each branch in the dendrimer structure
allows the attachment of a greater number of external substitu-
ents. In an attempt to mimic the properties of light-harvesting
arrays, we constructed oligoporphyrin systems based on dendri-
mers, the porphyrins being appended to the outer surface of the
dendrimers thereby mimicking an important feature of the
natural systems. We have prepared poly(propylene) imine den-
drimers with 4-, 8- 16-, 32-, and 64-porphyrins attached to the
perimeter, thereby approximating the number of pigments from
the smaller to the larger natural light-harvesting systems.
In previous work we have thoroughly investigated the
dynamics of energy transfer using time-resolved fluorescence
anisotropy, in the first, third and fifth generation of the
dendrimers functionalised with free-base porphyrins,12 and
the first and second generation compounds have been used
in supramolecular photovoltaic cells.13–15 We have also studied
energy transfer and conformational dynamics,16 and singlet–
singlet annihilation kinetics17 in the corresponding zinc(II)
porphyrin dendrimers. In this study we have systematically
evaluated the electrochemistry of free-base (2-H2–7-H2) and
metalated (2-Zn–7-Zn) porphyrin functionalized dendrimers
containing 2-, 4-, 8-, 16-, 32- and 64-porphyrin functionalities
and compared these properties with those of the corresponding
monoporphyrin model species 1-H2 and 1-Zn, respectively,
which both have a single branch of the dendrimer as a
substituent. Electrochemical and photophysical properties of
aDepartment of Chemistry, La Trobe University, Bundoora, Victoria,3086, Australia
b School of Chemistry, Monash University, Clayton, Victoria, 3800,Australia. E-mail: [email protected]; Fax: +613 99054597; Tel: +613 9905 1338
c School of Chemistry, The University of Sydney, NSW 2006,Australia. E-mail: [email protected]
2058 | Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 This journal is �c the Owner Societies 2006
PAPER www.rsc.org/pccp | Physical Chemistry Chemical Physics

these porphyrin-appended dendrimers are of particular inter-
est because of their relevance to natural systems.
Experimental
Materials
The synthesis of the free-base (2-H2–7-H2) and metalated
(2-Zn–7-Zn) porphyrin-appended dendrimers as well as the
monoporphyrin model compounds, 1-H2 and 1-Zn, will be
described elsewhere.18 Free-base and zinc 5,10,15,20-tetra-
kis(3,5-di-tert-butylphenyl)porphyrin, TDBPP-H2 and
TDBPP-Zn, and the corresponding 5,10,15,20-tetraphenyl-
porphyrins, TPP-H2 and TPP-Zn, were prepared by literature
methods.19
For UV-Vis absorption measurements, spectroscopic grade
dichloromethane (Aldrich) was used. Electrochemical mea-
surements were carried out in AR grade dichloromethane
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 | 2059

(Aldrich). The solvent was stored over molecular sieves prior
to use and basic alumina was used to remove any residual
hydrochloric acid. The electrolyte, [Bu4N][PF6], (GFS) was
recrystallised twice from ethanol/water before use and all
solutions were deoxygenated using solvent saturated nitrogen
prior to the electrochemical experiments. All measurements
were carried out at ambient temperature (20 � 2 1C).
Absorption measurements, to evaluate the concentration of
the dendrimers in the electrochemical solutions, were made
using a Varian Carey 5 spectrophotometer. Large molar
absorptivity values of the dendrimer species necessitated using
a small path length (1 mm) cell and in some cases dilution was
necessary.
Apparatus
Voltammetric measurements were carried out with an Autolab
PGSTAT100 (ECO-Chemie, Utrecht, Netherlands), or a Mac
lab/4e (ADInstruments, NSW, Australia). A Faraday cage
was used which, in addition to isolating the cell from electrical
noise, maintained the solutions in darkened conditions. A
three-electrode assembly was used with a small volume (0.5
cm3) electrochemical cell (Cypress Systems). The working
electrode consisted of a 1 or 1.5 mm diameter glassy carbon
disk shrouded in polyether ethylketone (Cypress Systems) or a
10 mm diameter Au microelectrode shrouded in glass (Cypress
Systems), while a Pt wire served as the auxiliary electrode. The
reference electrode was an Ag wire that was separated from
the test solution by a Vycor frit. All potentials are quoted
relative to the ferrocene/ferrocenium couple (Fc0/+) measured
in situ. The supporting electrolyte was 0.1 M [Bu4N][PF6] for
all measurements. IUPAC conventions are used for presenta-
tion of all voltammograms.
Results and discussion
Free-base porphyrin dendrimers
Fig. 1 shows the cyclic voltammetric responses at a 1 mm
diameter glassy carbon electrode for solutions of free-base
mono-porphyrin 1-H2 and the second generation dendritic
octakis-porphyrin 4-H2, dissolved in dichloromethane con-
taining 0.1 M [Bu4N][PF6] at a scan rate of 1 V s�1. The
concentrations are approximately 1 and 0.1 mM for the two
compounds, respectively. In general, the electrochemical be-
haviour exhibited in Fig. 1 is mirrored in each successive
generation of dendrimer; that is, two reversible reductions
and one reversible oxidation process are observed in each case.
For each compound the voltammetry exhibited in this solvent
is characteristically diffusional, with no significant evidence of
adsorption occurring, even for the higher molecular weight
species.
The first oxidation process in each case shows an Ioxp /Iredp
ratio (Ioxp = oxidation peak current, Iredp = reduction peak
current), greater than unity when scan rates below 1 V s�1 are
used, but approaches unity and hence becomes reversible at
higher scan rates. This chemical irreversibility is due to a
secondary homogeneous reaction of the electrogenerated por-
phyrin cation-radical and gives rise to an unidentified product
that is reduced in the region of �0.3 V vs. Fc0/+. Also, over
time (>4 h), with exposure to ambient light, photolysis of the
dendrimers occurs, which is signalled by a colour change from
2060 | Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 This journal is �c the Owner Societies 2006

red to green, giving rise to a new quasi-reversible redox process
centred at�0.98 V vs. Fc0/+. For some of the lower generation
species, additional irreversible oxidation peaks could also be
discerned close to the edge of the solvent limit.
The formal potential (E0f ) data for the mono- and bis-
porphyrin species and five generations of porphyrin-appended
dendrimers are compared with the corresponding data for
model compounds, TPP-H2 and TDBPP-H2 in Table 1 and
displayed graphically in Fig. 2. E0f values have been calculated
as the average of the oxidation (Eoxp ) and reduction (Ered
p ) peak
potentials obtained at a scan rate of 1 V s�1. As expected, the
TDBPP-H2 oxidation is more facile (and reduction more
difficult) than the un-substituted TPP-H2 (by 20–40 mV),
due to the electron-donating effect of the tert-butyl groups.
As seen in Table 1 and Fig. 2, compounds 1-H2 to 7-H2 show
no significant variation in oxidation and reduction potentials
as the number of porphyrin units is increased. Moreover, the
potentials are the same within experimental error as those of
the model species TDBPP-H2, the amide linker group see-
mingly having little effect on the electron density at the
porphyrin macrocycles.
The uniformity of the redox potentials with dendrimer gen-
eration is in contrast with results reported for dendrimers with
electroactive groups located within their cores, where a strong
correlation between redox potential and degree of dendric
branching is found.20 This implies that the porphyrin entities in
the compounds investigated in this study experience similar
micro-environments regardless of dendrimer generation, as ex-
pected, given their peripheral location. The constancy in the
redox potentials is mirrored in the spectroscopic data presented
in Table 1, i.e., the position of the intense Soret band at 425 nm
does not change with generation, though the intensity of the
bands increases proportionally with increasing numbers of por-
phyrins. The positions of the Q-bands at 488, 521, 555, 593 and
649 nm are similarly invariant with increasing number of por-
phyrin units and show the same trend with respect to intensity.
An interesting feature of the data presented in Fig. 2 is the
constancy, for all dendrimers and model porphyrin com-
pounds, of the difference in potential between the first oxida-
tion and first reduction processes (2.20 � 0.03 V). The so-
called electrochemical HOMO–LUMO gap (DH�L) has often
Fig. 1 Cyclic volammograms of the monoporphyrin, 1-H2 and the
dendrimer containing eight free-base porphyrins, 4-H2 dissolved in
CH2Cl2 (0.1 M [Bu4N][PF6]) at a 1 mm diameter glassy carbon
electrode. The scan rate is 1 V s�1 in both cases.
Table 1 Electrochemical and spectroscopic data for free-base dendrimers and other compounds. Values in italics represent peak potential (Eoxp )
data for irreversible processes
p E0f /V (vs. Fc0/+) DH�L/V lSoret/nm Log (eSoret)/M
�1 cm�1
TPP-H2 1 �1.98 �1.67 0.52 0.82 — 2.19 419 5.69TDBPP-H2 1 �2.02 �1.69 0.48 0.78 — 2.17 422 5.661-H2 1 �2.03 �1.70 0.48 0.76 1.10 2.21 425 5.552-H2 2 �2.05 �1.74 0.45 0.86 1.10 2.19 425 5.803-H2 4 �2.10 �1.78 0.41 0.72 1.10 2.19 425 6.114-H2 8 �2.13 �1.77 0.44 0.76 — 2.21 425 6.385-H2 16 �2.19 �1.79 0.44 0.76 — 2.23 424.5 6.756-H2 32 �2.13 �1.80 0.41 — — 2.21 424.5 6.957-H2 64 �2.11 �1.78 0.43 — — 2.20 424 7.3
Fig. 2 Redox potential data for the free-base dendrimers and model
compounds. Open circles represent peak potential (Ep,ox) data for
irreversible processes. The dashed lines represent the limits for solvent
decomposition.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 | 2061
been used as a diagnostic criterion for assigning the site of
electron transfer to the metal or the macrocycle. Its value has
been given as 2.25 � 0.15 V for reactions at the conjugated
p-ring system of metalloporphyrins containing tetraphenyl-
porphyrin (TPP-M) macrocycles.21 It is also noteworthy that
the difference between E1
f’s for the first and second reductions
is also constant (Dox1,ox2 = 0.34 � 0.03 V). This constancy is
also frequently taken as evidence for ring-centred electron
transfer reactions.22
As is known to be the case for 5,10,15,20-tetraphenylpor-
phyrin23 (TPP-H2), stepwise electrooxidation of the mono-
porphyrins TDBPP-H2, and 1-H2 is presumed to yield p-cation radicals and p-dications while the stepwise reduction
yields p-anion radicals and p-dianions according to eqn (1).
P2þÐe�
P�þÐe�
PÐe�
P��Ðe�
P2� ð1Þ
The third irreversible oxidation process observed for free-
base compounds 1-H2, 2-H2 and 3-H2 in the vicinity of the
anodic solvent limit is assigned to the amide linker groups as it
is absent in the voltammetric responses of TPP-H2 and
TDBPP-H2.
The almost identical redox behaviour observed for all free-
base multi-porphyrin species and mono-porphyrin model spe-
cies as well as the insensitivity of the absorption maxima to the
number of porphyrins per molecule, indicates a lack of
electronic communication between individual sites.24 There-
fore, the waves observed for compounds 2-H2 to 7-H2 must
represent the simultaneous oxidation and reduction of multi-
ple non-interacting units. If it is assumed that each wave
results from the oxidation/reduction of all the porphyrin units
in each species, i.e., that n= p, then their redox behaviour can
be represented as follows in eqn (2).
P2nþn Ð
ne�
Pnð�þÞn Ð
ne�
PnÐne�
Pnð��Þn Ð
ne�
P2n�n ð2Þ
In order to test this, the number of electrons transferred (n) in
the first reduction wave for several of the compounds was
determined by measuring the steady state voltammetric re-
sponse at a 10 mm diameter gold microelectrode and using the
formula:
Ilim = 4nFrDc (3)
where Ilim is the limiting current, r the electrode radius, D the
diffusion coefficient and c the concentration of electroactive
species. The concentration c was determined from absorption
measurements at the wavelength of the Soret band or one of
the Q-bands, while D was evaluated using the Stokes–Einstein
relationship:
D ¼ kT
6prZð4Þ
where k is the Boltzmann constant, T is the temperature, r is
the radius of the molecule and Z is the viscosity of the solvent.
Because of the bulky porphyrin end-groups, back-folding in
these molecules is not possible, therefore, the dendrimers are
assumed to adopt either a disc or spherical shape depending on
generation12 and r has been estimated by taking the sum of the
radius of the porphyrin molecule and the radius of gyration of
the dendrimers determined from a molecular dynamics
study.25 The values of D calculated in this way were found
to vary from 5.6 � 10�6 to 2.3 � 10�6 cm2 s�1 on going from
compound 1-H2 to 7-H2.
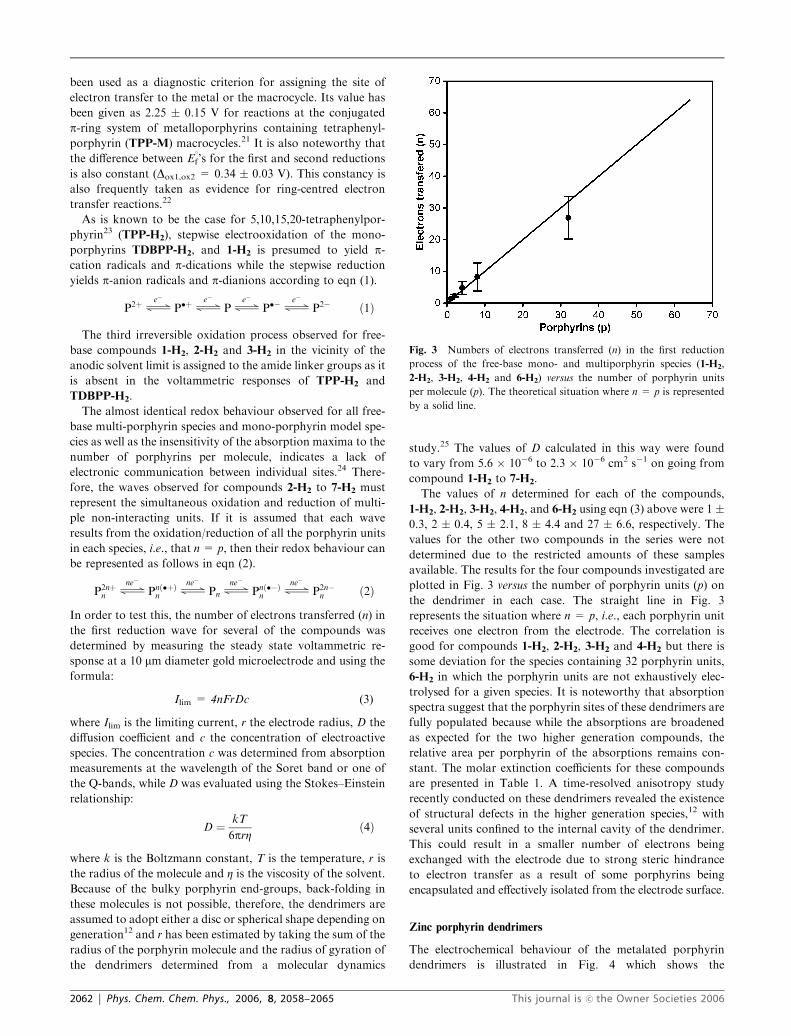
The values of n determined for each of the compounds,
1-H2, 2-H2, 3-H2, 4-H2, and 6-H2 using eqn (3) above were 1 �0.3, 2 � 0.4, 5 � 2.1, 8 � 4.4 and 27 � 6.6, respectively. The
values for the other two compounds in the series were not
determined due to the restricted amounts of these samples
available. The results for the four compounds investigated are
plotted in Fig. 3 versus the number of porphyrin units (p) on
the dendrimer in each case. The straight line in Fig. 3
represents the situation where n = p, i.e., each porphyrin unit
receives one electron from the electrode. The correlation is
good for compounds 1-H2, 2-H2, 3-H2 and 4-H2 but there is
some deviation for the species containing 32 porphyrin units,
6-H2 in which the porphyrin units are not exhaustively elec-
trolysed for a given species. It is noteworthy that absorption
spectra suggest that the porphyrin sites of these dendrimers are
fully populated because while the absorptions are broadened
as expected for the two higher generation compounds, the
relative area per porphyrin of the absorptions remains con-
stant. The molar extinction coefficients for these compounds
are presented in Table 1. A time-resolved anisotropy study
recently conducted on these dendrimers revealed the existence
of structural defects in the higher generation species,12 with
several units confined to the internal cavity of the dendrimer.
This could result in a smaller number of electrons being
exchanged with the electrode due to strong steric hindrance
to electron transfer as a result of some porphyrins being
encapsulated and effectively isolated from the electrode surface.
Zinc porphyrin dendrimers
The electrochemical behaviour of the metalated porphyrin
dendrimers is illustrated in Fig. 4 which shows the
Fig. 3 Numbers of electrons transferred (n) in the first reduction
process of the free-base mono- and multiporphyrin species (1-H2,
2-H2, 3-H2, 4-H2 and 6-H2) versus the number of porphyrin units
per molecule (p). The theoretical situation where n = p is represented
by a solid line.
2062 | Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 This journal is �c the Owner Societies 2006

voltammetric responses at a 1 mm diameter glassy carbon
electrode, solutions of the Zn2+ monoporphyrins TDBPP-Zn
and 1-Zn, and the corresponding fourth generation dendrimer
containing 32 porphyrin entities (6-Zn), dissolved in dichlor-
omethane containing 0.1 M [Bu4N][PF6]. The formal potential
(E0f ) data for the mono- and bis-porphyrin species and the five
generations of porphyrin-appended dendrimers are compared
with the corresponding data for two model compounds TPP-
Zn and TDBPP-Zn in Table 2 and Fig. 5.
As observed for the corresponding free-base compounds,
the potentials for TDBPP-Zn are shifted negative relative to
un-substituted TPP-Zn and essentially the same redox pattern
is observed for each compound in the series. The third oxida-
tion process, ascribed to the amide linkers, is once again
absent for the two model species. The redox processes for
the metalated species are observed at more negative potentials
than the free-base compounds, due to the increased electron
density on the porphyrin macrocycles provided by the zinc.
Both the similarity in oxidation potential between successive
generations and the negative shift on co-ordination with zinc
are mirrored in the absorption spectra. The spectroscopic data
presented in Table 2 show that the position of the Soret band
does not vary between the metalated compounds and has a
value that is shifted to longer wavelengths by 2 to 5 nm relative
to the free-base compounds.
In general, for the metalated compounds, only a single
reduction wave is detected, while all three oxidation processes
are now observed in each case. As a result of the negative shift
in potentials on metalation with zinc(II), the second and third
oxidation processes for these compounds are now more clearly
defined, not being complicated by proximity to the oxidative
solvent limit, while the second reduction for most of the Zn
compounds is now completely obscured by the negative limit
where the solvent is reduced.
As can be seen from Table 2, in common with the free-base
compounds, the HOMO–LUMO gap,(DH�L) for the metal-
lated species does not vary appreciably with dendrimer gen-
eration (2.12 � 0.04 V) although it does decrease slightly on
metalation. The constancy of the gap between reduction
processes observed for the free-base compounds in Table 1 is
Fig. 4 Cyclic voltammograms at a 1 mm diameter glassy carbon
electrode, for the Zn2+ mono porphyrins TDBPP-Zn (4 mM) and 1-
Zn (1.5 mM) and the fourth generation dendrimer containing 32
metalated porphyrin entities, 6-Zn (50 mM) dissolved in dichloro-
methane (0.1 M [Bu4N][PF6]). The scan rate is 0.1 V s�1 in each case.
Table 2 Electrochemical and spectroscopic data for zinc dendrimers and other compounds. Values in italics represent peak potential data forirreversible processes
p E0f /V (vs. Fc0/+) DH�L/V lSoret/nm Log (eSoret)/M
�1 cm�1
TPP-Zn 1 �2.17 �1.79 0.42 0.71 — 2.21 420 5.45TDBPP-Zn 1 — �1.89 0.30 0.62 — 2.19 423 5.711-Zn 1 �2.16 �1.86 0.30 0.56 0.95 2.16 426.5 5.772-Zn 2 — �1.85 0.30 0.59 0.94 2.15 426.5 6.073-Zn 4 — �1.84 0.30 0.70 1.00 2.14 427 6.344-Zn 8 — �1.80 0.30 0.63 0.90 2.10 427 6.625-Zn 16 �2.18 �1.80 0.29 0.63 0.90 2.09 427 6.896-Zn 32 — �1.79 0.31 0.64 0.91 2.10 427 7.227-Zn 64 — �1.80 0.30 0.76 1.09 2.10 426 7.49
Fig. 5 Redox potential data for the zinc dendrimers and model
compounds. Open circles represent peak potential (Ep,ox) data for
irreversible processes. The dashed lines represent the limits for solvent
decomposition.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 | 2063

mirrored in Table 2 for the gap between adjacent oxidation
processes (Dox1, ox2 = 0.33 � 0.06 V).
The Zn(II) porphyrins displayed more stability toward
ambient light than the free-base compounds, with no evidence
of new voltammetric processes developing over time as ob-
served for the unmetalated species. A homogeneous reaction
was observed, following oxidation to the dication, which gave
rise to a reduction peak at ca. �0.5 V vs. Fc0/+, which can be
seen in the uppermost curve of Fig. 4. Doubly oxidised zinc
porphyrin compounds are known to be susceptible to nucleo-
philic attack at both the meso and b-positions resulting in the
formation of substituted products;23,26 the coupled homo-
geneous reaction observed here, therefore, may be the result
of trace levels of nucleophiles in the electrochemical medium.
As evidenced by Fig. 4, a trend toward chemical and
electrochemical irreversibility is observed as the generation
number increases. This trend is also observed for the free-base
compounds but is most apparent for the second and third
oxidations of the metalated dendrimers. With increasing num-
ber of porphyrins per molecule, the peak current per unit
concentration times the number of porphyrins decreases and
additionally a significant increase in DEp accompanied by a
broadening of the voltammetric waves is observed. For the
third oxidative process, at ca. 1.0 V vs. Fc0/+, which is
assigned to the amide functional groups, these changes could
be regarded as being consistent with a strong steric inhibition
to electron transfer as these groups are buried ever deeper
within the porphyrin core for each successive generation.
Moreover, with increasing dendrimer generation, access to
the electrode surface will be increasingly restricted as the
porphyrin surface becomes more crowded. For the first two
oxidations, however, which are assigned to the formation of
p-cation radicals and p-dications on the peripheral porphyrin
rings, significant steric crowding is unlikely. Another possible
explanation for the observed kinetic effects, therefore, may be
electrostatic interactions contributing to an increasingly large
barrier to electron transfer as ever-larger numbers of electrons
are transferred with increasing dendrimer generation. Finally
it is noted that a higher probability of kinetic and/or thermo-
dynamic dispersion is likely for higher generation dendrimers
and that these factors will lead to broadening of voltammo-
grams.
Fig. 6 shows the results for the determination of n, the
number of electrons transferred, for the series of zinc porphyr-
in compounds 1-Zn to 7-Zn. The n values determined for the
seven compounds were 1 � 0.3, 2 � 0.5, 4 � 0.4, 7 � 1.5 17 �2.3, 26 � 8.1 and 58 � 6.6. The trend observed for the
unmetalated species is mirrored here, with n increasing almost
proportionately with p, the negative deviations once again
being observed for the two highest generations. The absorp-
tivity data in Table 2 shows that, as with the unmetalated
species, the maximum intensities of the Soret bands (e) showthe same trend as for the electrons transferred (n), with a
negative divergence from simple proportionality of e to p for
the largest dendrimers. Broadening of the electronic spectra
occurs as the dendrimer generation increases, however, and
use of area rather than maximum values of absorption
significantly improves the level of linearity.
Conclusions
Cyclic voltammetry of both free-base and zinc metalated
dendrimers show diffusional responses when dissolved in
dichloromethane, with no significant evidence of adsorption
phenomena. For the free-base dendrimers and free-base
monoporphyrin models, generally two porphyrin-ring based
reversible reductions are observed, corresponding to the for-
mation of single or multiple p-anion radicals and p-dianionsand one reversible oxidation process resulting in single or
multiple p-cation radicals. For the zinc porphyrin compounds,
being more difficult to reduce, the second reduction is ob-
scured by the solvent limit, but one or two additional oxida-
tion processes are observed. The second oxidation in this case
is due to the formation of single or multiple porphyrin
p-dications, while the third oxidation process is assigned to
the amide linker groups, as it is absent in the voltammetric
response of a model compound which is not substituted at the
b-pyrrole position.
For the higher generation dendrimers, kinetic or thermo-
dynamic dispersion effects are evident, with increased DEp
values and broadening of the voltammetric waves. The effects
are most pronounced for the doubly- and triply-oxidised zinc
porphyrin arrays, which suggests they may be a consequence
of the heterogeneous kinetics being perturbed by electrostatic
interactions, though strong steric inhibition of electron trans-
fer may also be a contributing factor in the case of the
oxidation of the amide linkers, which are located at the
dendrimer core.
The positions of the redox waves are very similar for each
successive generation free-base dendrimer and also similar to
the corresponding model species. The same applies to the
series of metalated compounds but the potentials are shifted
negative relative to the free-base species, due to the increased
electron density at the porphyrin rings provided by the metal.
The electrochemical behaviour is mirrored in the spectroscopic
Fig. 6 Numbers of electrons transferred (n) in the first reduction
process of the zinc mono- and multiporphyrin species (1-Zn, to 7-Zn)
versus the number of porphyrin units per molecule (p). The theoretical
situation where n = p is represented by a solid line.
2064 | Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 This journal is �c the Owner Societies 2006

response with the maximum positions of the Soret bands in the
visible absorption spectra remaining constant with increasing
generation and shifting to longer wavelengths on metalation
with zinc(II). The absence of a dendrimer effect implies that
most or all of the porphyrins experience essentially the same
microenvironment regardless of generation. Other features of
the voltammetric response such as the HOMO–LUMO gap
and the gap between adjacent redox processes were also
invariant with generation but the magnitude of the
HOMO–LUMO gap decreased by 50–100 mV when the
porphyrin macrocycles were metalated.
The uniformity in redox potentials within each series is
consistent with each electron transfer process representing
the almost simultaneous oxidation of multiple non-interacting
centres. In order to verify this, the number of electrons
transferred (n) for each dendrimer was evaluated from limiting
current data using steady state microelectrode voltammetry,
with the diffusion coefficients in each case being estimated
from the Stokes–Einstein equation. For both free-base and
zinc metalated porphyrins, values of n determined were rea-
sonably consistent with the number of porphyrin macrocycles
(p) contained in each dendrimer. Slight deviations were ob-
served for the higher generation species in each case, which
may be a consequence of structural defects, with one dendron
of the molecule confined to the dendrimer core due to re-
stricted growth. More likely, the slight deviation might be due
to electrostatic interactions of the porphyrins as these redox-
active chromophores become closer packed in each successive
higher generation. Calculations indicate that the porphyrins
are on average about 1.8 nm apart in the tetrakis-porphyrins
3-H2 and 3-Zn but are only about 0.5 nm apart in the 64-mers
7-H2 and 7-Zn. It is interesting that in the absorptivity values
there is also a slight divergence from simple proportionality of
e with p in the fourth and fifth generation species and the
absorptions are broadened as expected while the relative area
of the absorptions remains constant.
The light harvesting potential of these molecules is clearly
demonstrated by the increased absorption properties with
array size. From an electrochemical viewpoint, dendrimers
are very interesting species because the electron transfer
properties of potentially electroactive units may be modulated
by placing them in the branches or the core, or a large number
of topologically equivalent units may be linked to the surface,
giving rise to simultaneous exchange of a large and pre-
determined number of electrons.27 Electrochemistry is also a
powerful technique for the evaluation of the degree of electro-
nic interaction among the electroactive units.28
Moreover, oxidation or reduction of the Zn porphyrin
dendrimer species results in ‘‘supercharged’’ molecules that
are stable on the cyclic voltammetric time-scale and fully
soluble in dichloromethane. This suggests that these com-
pounds may form the basis for efficient hole/electron storage
devices.
Acknowledgements
We thank the Australian Research Council for a Discovery
Research Grant (DP0208776) to M.J.C.
References
1 R. E. Blankenship, Molecular Mechanisms of Photosynthesis,Blackwell Science, Oxford, 2002.
2 P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Seanger and N.Krauss, Nature, 2001, 411, 909–917.
3 K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S.Iwata, Science, 2004, 303, 1831–1838.
4 A. Camara-Artigas, R. E. Blankenship and J. P. Allen, Photosynth.Res., 2003, 75, 49–55.
5 D. E. Tronrud and B. W. Matthews, Photosynthetic ReactionCentre, 1993, 1, 13–21.
6 M. Z. Papiz, S. M. Prince, T. Howard, R. J. Cogdell and N. W.Isaacs, J. Mol. Biol., 2003, 326, 1523–1538.
7 A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. J.Law, N. W. Isaacs and R. J. Cogdell, Science, 2003, 302,1969–1972.
8 G. R. Newkome, C. N. Moorefield and F. Vogtle, Dendrimers andDendrons, Wiley-VCH, Weinheim, 2001.
9 N. Tomioka, D. Takasu, T. Takahashi and T. Aida, Angew.Chem., Int. Ed., 1998, 37, 1531–1534.
10 G. R. Newkome, E. He and L. A. Godinez,Macromolecules, 1998,31, 4382–4386.
11 C.-F. Shu and H. M. Shen, J. Mater. Chem., 1997, 7, 47–51.12 E. K. L. Yeow, K. P. Ghiggino, J. N. H. Reek, M. J. Crossley, A.
W. Bosman, A. P. H. J. Schenning and E. W. Meijer, J. Phys.Chem. B, 2000, 104, 2596.
13 T. Hasobe, Y. Kashiwagi, M. A. Absalom, J. Sly, K. Hosomizu,M. J. Crossley, H. Imahori, P. V. Kamat and S. Fukuzumi, Adv.Mater., 2004, 16, 975–979.
14 S. Fukuzumi, T. Hasobe, K. Ohkubo, M. J. Crossley, P. V. Kamatand H. Imahori, J. Porphyrins Phthalocyanines, 2004, 8, 191–200.
15 T. Hasobe, P. V. Kamat, M. A. Absalom, Y. Kashiwagi, J. Sly, M.J. Crossley, K. Hosomizu, H. Imahori and S. Fukuzumi, J. Phys.Chem. B, 2004, 12865–12872.
16 J. Larsen, J. Andersson, T. Polıvka, J. Sly, M. J. Crossley, V.Sundstrom and E. Akesson, Chem. Phys. Lett., 2005, 403, 205–210.
17 J. Larsen, B. Bruggemann, T. Polıvka, V. Sundstrom, E. Akesson,J. Sly and M. J. Crossley, J. Phys. Chem. A, 2005, 109,10654–10662.
18 J. N. H. Reek, J. Sly, A. W. Bosman, M. A. Absalom, T. Khouryand M. J. Crossley, in preparation.
19 G. H. Barnett, M. F. Hudson and K. M. Smith, J. Chem. Soc.,Perkin Trans. 1, 1975, 1401–1403.
20 P. Weyermann, J.-P. Gisselbrecht, C. Boudon, F. Diedrich and M.Gross, Angew. Chem., Int. Ed., 1999, 38, 3215–3219.
21 J.-H. Fuhrhop, K. M. Kadish and K. M. Davis, J. Am. Chem. Soc.,1973, 95, 5140–5147.
22 D. W. Clack and N. S. Hush, J. Am. Chem. Soc., 1965, 87,4238–4242.
23 K. M. Kadish, L. R. Shiue, R. K. Rhodes and L. A. Bottomley,Inorg. Chem., 1981, 20, 1274–1277.
24 A. Tsuda and A. Osuka, Science, 2001, 293, 79–82.25 L. Cavallo and F. Fraternalli, Chem.-Eur. J., 1998, 4, 927–934.26 G. H. Barnett and K. M. Smith, J. Chem. Soc., Chem. Commun.,
1974, 772–773.27 C. M. Casado, B. Gonzalez, I. Cuadrado, B. Alonso, M. Moran
and J. Losada, Angew. Chem., Int. Ed., 2000, 39, 2135–2138.28 B. Garcia, C. M. Casado, I. Cuadrado, B. Alonso, M. Moran and
J. Losada, Organometallics, 1999, 18, 2349–2356.
This journal is �c the Owner Societies 2006 Phys. Chem. Chem. Phys., 2006, 8, 2058–2065 | 2065
Copyright © 2022 FDOKUMEN




















