
ELANA CHERRY - Bibliothèque et Archives Canada
214
TRANS-DOMINANT NEGATIVE INHlBlTION OF HUMAN IMMUNODEFICtENCY VIRUS TVPE I REPLICATION BY EXPRESSION OF PROTEASE-REVERSE TRANSCRIPTASE FUSlON PROTEINS by ELANA CHERRY A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfillment of the requirements for the degree of Doctor of Philosophy ~eiartment of Microbiology and lmrnunology McGill Universityr Montreal, Canada January, 1999 O Elana Cherry. 1999
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of ELANA CHERRY - Bibliothèque et Archives Canada

TRANS-DOMINANT NEGATIVE INHlBlTION OF HUMAN IMMUNODEFICtENCY VIRUS TVPE I REPLICATION BY
EXPRESSION OF PROTEASE-REVERSE TRANSCRIPTASE FUSlON PROTEINS
by
ELANA CHERRY
A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfillment of the requirements for the degree of Doctor of Philosophy
~eiartment of Microbiology and lmrnunology
McGill Universityr Montreal, Canada January, 1999
O Elana Cherry. 1999

NaüonaC Libmy B * l of Canada Biblioth' ue nationaïe a du Cana a
Acquisitions and Acquisitions et BibIiogrâphgF Services seMces bibliographiques 395 Wellington Street 395. nre Wellington Ottawa ON KIA ON4 OrtewaON KTA ON4 Canada canada
The author has granted a non- exclusive licence dowiig the National Liôrary of Canada to reproduce, Ioan, distrr'bute or sen copies of this thesis m microforni, paper or eIectronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantid extracts fiom it may be piinted or otherwise reproduced without the author's permission.
L'auteur a accorde une licence non exclusive permettant à la Bibliothèque nationde du Canada de reproduire, prêter, distribuer ou vendre des copies de cette these sous la forme de microfiche/film, de reproduction sur papier ou sur formaî e1ectronique.
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni Ia thèse ni des extraits substantieIs de celIen ne doiveat être imprimés ou autrement reproduits sans son autorisation,

ABSTRACT
The molecular mechanisms involved in the regutation of protease (PR) activity
and human immunodeficiency virus type 1 (HIV-1) viral maturation are
incompletely elucidated. To better understand the importance of the cleavage
event between HIV-1 PR and reverse transcriptase (Rn, we have selectively
mutagenized specific residues at the junction between these genes to produce
a PR-FIT fusion protein Ri the conte& of a fuli-length proviral constnict. Mutant
viruses derived from COS-? cells transfected with this construct were analyzed
in regard to each of viral replication. maturation, and infectivity. l mmuno blot
analysis revealed that the mutation prevented cleavage between the PR and FlT
proteins and that both existed as a PR-RT fusion protein in each of cellular and
viral lysates. Interestingly, intracellular PR that existed within the PR-RT fusion
protein not only remained functionally active, but also processed HIV-1
precursor proteins with slightly increased efficiency as shown in time-course
experiments in transfected COS-? cells. In contrast, the RT component of the
fusion protein was active at wild-type levels in in vitro and endogenous RT
assays. Electron microscopy revealed that mutant viruses containing the
cleavage site mutation between PR and RT possessed wild-type morphology.
These viruses also displayed wild-type sensitivities to inhibitors of each of HIV-1
PR and FIT activities. However, viruses containing the PR-RT fusion protein
were 20-times less infectious than wild-type viruses. This defect was further
pronounced when mutated Gag-Pol proteins were overexpressed as a
consequence of an additional mutation that interfered with frameshifting. Thus,
unlike cleavage site mutations at the N-terminus of PR, a cleavage site mutation
between PR and K i did no€ prevent proteolytic processing and abolish
infectivity; rather, viruses containing PRRT fusion proteins were viable. in
agreement wth the notion that C-terminal liberation of PR is not as critical for its
activity as Kterminal processing. The diminished infectiousness of these
vinises likely results from cfysregulation of PR activity due to the fusion of the PR
and FiT components, leading to sfight premature cleavage of the precursor
proteins and hence reductions in infectivity of the released virus particles. In

addition. cotransfection experhents demoostrated that the mutant constmcts
inhibited replication of wild-type HLV-1 and diminished viral infectiousness in a dosedependent manner, suggesting a trans-dominant negative mode of action.
iii

Les mécanismes mol6culaires impliqu6s dans la regulation de i'activit6 de la prot6ase (PR) et de la maturation du virus d'immunodeficience de type 1 de l'humain (WH) ne sont pas compl6tement Blucides. Pour mieux comprendre l'importance du phhombne de la coupure entre la protbase (PR) et la
transcriptase inverse (RT) du VIH-1, nous avons fait des mutations s4lectives
sur des rdsidus spécifiques situes entre ces ghes pour produire une protéine
de fusion PR-RT en utilisant la longueur totale du provirus construit. La
r6plication, la maturation et I'infectiosite des virus mutants derivant des cellules
COS-7 transfectdes avec la construction, ont 8té analys6es. L'analyse
immunoblot a révélé que la mutation empBchait la coupure entre les protéines
PR et RT et que ces deux existaient comme une protéine de fusion PR-FITv tant
dans les lysats cellulaires que viraux. De façon intéressante, la prot6ine PR
intracellulaire qui existait dans la prot6ine de fusion PR-RT demeure non
seulement fonctionnellement active, mais aussi contribue à la maturation des
protéines pr6curseurs du VIH-1 avec une légère augmentation de l'efficacité
comme l'ont montre les expériences avec les cellules COS-7 transfectees. Par contre. la composante RT de la protéine de fusion était active in vitro et dans les
essais endogènes de KT comme dans le cadre du type sauvage. La
microscopie électronique a r6v616 que les virus ayant une mutation dans le site
de coupure entre PR et avaient la morphologie du type sauvage. Ces virus
prdsentaient Bgalement une sensibilité aux inhibiteurs des activités RT et PR
semblable a celle du VIH-1 sauvage. Cependant, les virus possédant la
protéine de fusion PR-RT dtaient vingt fois moins infectieux que le type
sauvage. Cette defaillance &ait plus prononcée quand des protéines ayant une
mutation Gag-Pol &aient surexprimees comme conséquence d'une mutation
additionnelle qui induirait un changement du cache de lecture. Ainsi,
contrairement aux mutations au site de coupure de rextrémite N-terminale de
PR. une mutation au site de coupure entre PR et FCT n'a pas empêché la
procession protéorytique et abolit i'infectiosite; les virus ayant les protéines de
fusion PR-RT étaient viables, en accord avec Pidée seion laquelle la lib6ration

de Itex€r6mit6 C-terminale de PR n'est pas aussi critique pour son activité que
I'est la digestion de L'extrémité N-terminale. La diminution de I'infectiosit8 de ces virus rdsulte probablement de la ddrgglernentation de I'actMt6 de PR, due
& la fusion des composantes PR et KL conduisant à une Idgère coupure pr6maturde des proteines pr6cuneurs et favorisant des r6ductions de
i'infectiosité des particules virales IibWes. Par ailleurs, des expériences de CO-
transfection ont d6montr6 que des mutants construits inhibaient la replication
du VIH-1 sauvage et diminuaient l'infectioçit& virale en fonctio" de la dose, suggdrant une mode d'action nhgative transdominante.

ACKNOWLEDGMENTS
First, l would Iike to express my deepest gratitude and appreciation to my
supervtsor. Dr. Mark WaÏnberg, for his excellent scientific guidance during the
course of my Ph.D. studies. On a personal level, I am particularly grateful for his
constant encouragement and support.
I also wish to thank the members of the laboratory for their assistance and
friendship. In particular I would like to acknowledge Mayla Hsu, Phil Inouye,
Nicolas Morin. Nathalie Richard. Chen Liang, Liwei Rong, Yudong Quan, Matthias Gotte, Horacio Salomon, Maureen Oliveira, and Xuguang Li. In
addition, I would like to thank Drs. Lawrence Kleiman and Ratph Germinario for
helpful and insightful discussions. Our lab coordinator, Bonnie Spira. has been
very helpful in providing materials for my research. I would also like to express
my gratitude to Marie-Pierre Aoun for her efficiency and expertise in preparation
of manuscripts for publication. And special thanks to Karidia Diallo for
translation of the abstract.
I am grateful to Health and Welfare Canada (National Health Research and
Development Program [NHRDP]). as well as tu the Province of Quebec (Fonds
pour la Formation de Chercheurs et l'Aide à la Recherche [FCARI) for financial
support during the course of my graduate training.
And finally. special thanks to rny parents, Ed and Evelyn, my brothers. Jamie
and Rob, and to Leslie for their unfaltering and tireless support.

In accordance with the Guidelines for Thesis Preparation from the Faculty of Graduate Studies and Research of McGill University. this Ph.D. thesis is written
t
in classical form. A general introduction and literature review are presented in
chapter 1. and are followed by a statement of the rationale and specific
objectives of the study. The materials and methods used in experiments
included in this thesis are described in chapter 2 The results are presented in
chapters 3 to S and appear in the following peer-reviewed publications:
1. Cherry, E., Morin, N.. and Wainberg, M.A 1998. Effects of HLV
constnicts containing protease-reverse transcriptase fusion proteins
on viral replication. AlDS l2:96%975.
2. Cherry, E., Liang, C., Rong, L, Quan, Y., Inouye, P., Li, X.. Morin,
N., Kotler, M., and Wainberg, M.A 1998. Characterization of human
immunodeficiency virus type4 (HIV-1) particles that express protease-reverse transcriptase fusion proteins. Journal of Moiecuîar
Biology 284:43-56.
The candidate was responsible for ail of the research described in chapters 3 to
5, with the following exceptions. Dr. Chen Liang and Liwei Rong provided
important assistance in the construction of the mutant clones; Dr. Yudong Quan
establislied and conducted experiments involving endogenous reverse
transcription reactions; Sharon Lemer was responsible for Northem blots;
electron microscopy analysis was perfomed by Robert Alain at the Institut
Armand-Frappier in Laval. Quebec.
The candidate was also involved in coflaborations with other researchers in the
faboratory which resulted in the following publications:
vii

3. Cherry, E., Slater, M., Salomon. He, Rud, El and Wainberg. M A
1997. Mutations at codon 184 in simian imrnunodeficiency virus
reverse transcriptase confer tesistance to the ') enantiomer of 2',3'-
dideoxy-3'-t h iacyt idine (3TC). Antimicrobial Agents end Chemo-
therapy 41 -2763-2765.
4. Morin. N., Cherry, E.. Li, X, and Wainberg, M A 1998.
Cotransfection of mutated foms of human immunodeficiency virus
type-1 gag-pol with wild-type constnicts can hterfere with processing
and viral replication. Journal of Human Virology 1~240-247.
5. Inouye, P., Cherry, E., Hsu, M.. Zolla-Pazner. S., and Wainberg.
M A 1998. Neutralizing antibodies directed against the V3 loop
select for different escape variants in a virus with mutated reverse
transcriptase (M 184V) than in wild-type human immunodeficiency
virus typa-1. AlDS Research and Human Retroviruses 1 4:?35-?40.
6. Liang. C., Rong, L, Morin, N., Cherry, E., Huang, Y., Kleiman, L..
and Wainberg, M.A. 1997. The roles of the human immunodeficiency
virus type4 pol protein and the primer binding site in the placement
of primer tRNAW onto viral genomic RNA Journal of VÎroIogy
71 ~9075-9086.
7. Hsu, M., Cherry, E.. Quan. Y., Richard, N., Kleiman, L, and
Wainberg, M.A 1998. Effects of mutations in NC7 on endogenous
reverse transcription in HIV-t . Leukemia (in press).
A general discussion is presented in chapter 6. and is followed by a description
of the candidate's 'contribution to original knowledgen. References cited in this
thesis are listed in chapter 7.
viii

TABLE OF CONTENTS
Page
CHAPTER 1:
INTRODUCTION AND LITERATURE REVIEW t-ttttto..ttttt~..-.l
1.2 The HIV4 genome and viral proteins .................. C..CC.C+.C..C.~~C~tL .*.-....6
1.21 The virion ........... . .....+. .CCCCC..-....t...+.....o...+..~-.,.,. ...+.. .. ........ -.... .......... . ..... ...-... 6 1.2.2 Genetic organization of HlV-1 ...................*..-.. .. L..L........* ttt.+*.J2
1 2 3 The gag gene ptoducts .........,.... ......... ..~.C.tt..CC .-.*..*............. ......12
1.2.3.7 The Gag precursor protein ............ 13
1 23.2 MA ..... ...... ......... ....- CCC-.I......--...-I ......... ........C.C..~CIC .... ......f 3
12.3.3 O-..... . ..... ....... -...-..-......-. .*-. C.....C......CCCCC* .*... . -.. 4
1.2.3.4 NG. ................ .~...~...-.~..~....t.Ct..................... .......+...............14
123.5 p6 .............................................. CCCCCCCCC...CC+C ..-............. . 1 5
1 23.6 p2 and p l ......................... ..-. -.-... .l....L~~CC~.C~C. ...-......... ~.L.~.~~........l 6

.............................................................. 12.4 The poi gene produ cts. ............. 17
.......................................... 1.24.1 The Gag-Pol precursor protein 7
1.2.4.2 Protease ..................................................................................... 18
.............................................................. 1 2-4-3 Reverse transcriptase 19
1 24.4 Integrase ., .... .. .... ............................. ........~................................. .1 9
..................................................................... 1 2.5 The env gene product s. ....20
12.6 Accessory proteins ................................................................................. 2 0
1 2.6.1 Ta t. ............................................................................................... 21
1 2.6.2 Rev .............................................................................................. Z 1.2.6.3 Nef ...............................................................*...........................*.* 23
1.2.6.4 Vpr ...........................................................~.............................. *.23 ........................................................................................... 126.5 Vl. .....24
1.2.6.6 Vpu .~.......................................~....................... A 5
.................................................................... 1.3 The replication cycle of HlV-1 25
1 .3.1 Virus entry and cellular tropism ....................................................*...... ..28 9 0 .... ................................... 1-32 Reverse transcription .... ....
1.3.3 Proviral DNA nuclear iocalization and integration ............................. 32 ............................................................. 1 .3.4 Expression of the viral genome 33
1.3.5 Viral packaging, assembfy. and budding ......................................... J 4
1.3.5.1 Other proteins incorporated into the vinon .............. ......... 36
1 .3.6 Maturation .................................................................................................. 38
.............................................. 1.4 Characterization of PR and RT activities 40 . . ...................................................... 1.4.1 Regulation of protease activity 2
1 4.1 . 1 Frameshaüng controls PR production ............. ~.L.....C.~~~..LC..~CL40
1 .4.1.2 Requirement for dimerkation ................................................ -42
1.4.1.3 Sequential cleavage of Gag and Gag-Pol ......................... .A4
1.4.1 -4 Reguiators of PR withÏn Gag and Gag-Pol ........................... 49 1 -42 The effect of PR cleavage site mutations ............................................. 52
1.421 N-termina1 mutations ................................................................ 52
.............................................. 1.4.22 C-teminaC mutations ..CCC..C..C.......S5

. 0
1 .4.3 Regulation of RT act~wty ................................................. ..-...*............... ..57
1 .4.4 The relationship between PR and RT .............................................~..... 57
1.4.4.1 The eKect of RT on PR acüvity ...............................*.*.............. 57 ............................................ 1 . 4.42 The effect of PR on RT actEQ ....59
1.4.4.2.1 RT activity in Gag-Pol ............ ... ....................*...... 59
1.4.422 RT activity in PR-defective vimses .......................... 59
1 . 5 Therapeuti c approaches for treatment of HIV-1 ................................ 60 1.5.1 Gene therapy strategie S. ......................................................................... 61
1.5.2 HlV-1 PR as a target for gene therapy .................................................. 62
RATIONALE AND SPECIFIC OBJECTIVES .......................*.......................... 65
CHAPTER 2:
MATERIALS AND METHODS .................................................................. 66
2-1 Construction of moiecular clones .......................................................~.. ..+67
2.2 Transfection of COS-7 tells.* ...................................~...............................~ ..?l
2.3 Purification of cellular and viral extracts from transfected
or infected cells .............................................................~.........................~~........ 71
2.4 Immunoblots ........................................................................................................ 72
2.5 Viral replication assays ........................................................................... C....C73
2.5. t In vitro RT and p24 assays ................................................................... ..73
2-52 Endogenous RT assays ....................~............................~............. J ................. ....................................................................... 25.3 Infections.. .-.. .....7 5
Ic . 25-4 TCID, determinations ~.........................................~..............~................. ..75
2.5.5 Assessrnent of vira[ sensiüvity to antf HlV drugs ................................ 76
2.6 Analysis of virai RNA .......................................................... 76
2.6.1 NorViem blotting ....~.~............................................................................... .76


packaging. .........---.-.----....-----.............................................................................. t 16
4.3 Eledron microscopy analysis of wild-type and mutant virus pactfclem ......*........................................................................~......~............ 120
...................................... 4.4 Thne course analysis of PR activity in vivo. 128
4.5 Sensitlvities of mutant viruses to antiviral drugs ........................... 132
CHAPTER 5:
PR-RT FUSlON PROTEINS INHIBIT HIV-1 REPLICATION
5.1 Cotiansfection of wild-type and mutant constiucts ..................................................................... decreases HIV-1 replication 1 35
5.2 Vlruses derived fiom cotiansfections of wlld-type and mutant constructs have diminished infectivity ........................ 1 38
CHAPTER 6:
GEN ER AL D 1 SCU S SION ......................................................................... 140
CONTRIBUTIONS TO ORIGINAL KNOWLEDGE---------LLt~-.~o~*c~c~c~~c~ccc*œ~.-~oœ15I
CHAPTER 7:
......................................................................................... R €FER EN C ES ....*153
xiii

LIST OF FIGURES AND TABLES
Page CHAPTER 1
............. .............. FIGURE 1-1. Schernatic representation of the HIV-1 virion ... 8
............................................................ FIGURE 1-2 Genetic organkation of HIV-1.- 1 O
FIGURE 1-3. Schematic representation of the HlV-I life cycle ............................. 26 FIGURE 1-4. The curent mode1 of retroviral reverse tmnscription ...........-Ct...tt..CC30
FIGURE 1-5. Schematic representation of cleavage sites for HIV-1
............................ protease within the Gag and Gag-Pol precursors 47
CHAPTER 2
.............. FIGURE 2-1. Description of HIV-1 mutant clones, derived from pBH1 O 69
CHAPTER 3
FIGURE 3-1. lmmunoblot of whole cet1 lysates harvested from COS-? ........................ transfected cells, using an anti-RT HIV-f IgGl mAb 82
FIGURE 3-2. lmmunoblot of purified concentrated viral lysates from COS-? ............. transfected cells. detected by an anti-RT HIV-1 lgGl mAb 84
FIGURE 3-3. lrnmunoblot of whole-cell lysates harvested from COS7
transfectioos, probed with an antCp24 HtV-1 lgGl mAb ................ .88
FIGURE 3-4. lmrnunoblot of purified concentrated viral lysates from COS-?
transfections. using paoled sera from HIV-1-infected . ........................................................................................... in&~Uuais..- ,,90
xiv

TABLE 3-1. Characterization of wild-type and mutant transfecüons of
............................................................................................. COS7 =Ils 95
FIGURE 3-5. The-course analysis of wild-type and mutant sarnples after
transfection of COS-7 cells ................................................ .*-.*-..---...--.*.96
FIGURE 3-6. PCR amplification of specific DNA transcripts produced from
.................................................................. endogenous RT reactions --99
CHAPTER 4
FIGURE 4-1. Replicative capacity of wild-type and mutant viruses derived
from transfected COS-7 cells in MF2 cells ..................................... 105
TABLE 4-1. Characterization of wild-type and mutant virus replicative
............................... a biiity and infect ivity in MT02 cells ............... ... 1 07
TABLE 4-2. Ability of wild-type and mutated viruses to infect different
................................................................................................ cell types 1 08
FlGURE 4-2. RNA and protein expression in MT-2 cells infected with
wild-type or mutant viruses derived from COS7 transfections ... 111
FIGURE 4-3. lmrnunoblot analysis of MT-2 cells infected with wild-type
or mutant viruses derived from COS-? transfections ..~..,~.C...C......~I 14
FIGURE 4-4. Genomic RNA packaging by wild-type and mutant
viruses.....,., ............................................................................................ 1 17
.......................................................... FIGURE 4-5. Electron m icroscopy anaIysis.. 1 22
....... .................. FIGURE 4-6. lntracellular PR actnnty assay ...--I.b.---.--.* LLL--.LoL.....oL.129
xu

CHAPTER 5
TABLE 5-1. TCID, determinations in MT-2 cells of culture fluids from COS-? cells derived from transfections or cotransfections with wild-type
and mutant HlV-1 constructs at various ratios.- +,.L+o *+w*.***** l 1 39

aa AIDS ASLV bp CA CPe dNTP Env Gag
amino acid acquired immunodef iciency syndrome avian sarcoma and leukosis vimses base pair capsid protein cytopathic effect deoxynucleoside triphosphate envelope protein gag protein
gP g Iycoprotein HLV human immunodeficiency virus [Cm 50% inhibitory concentration IN integrase protein kDa kilodalton Lm long terminal repeat mAb monoclonal antibody MA matrix protein NC nucleocapsid protein nt n ucleotide PCR polymerase chain reaction PCC preintegration corn plex Pol polymerase protein PR p rotease R repeat sequence RNaseH DNA/RNA-dependent ribonuclease RRE rewesponsive element RSV Rous sarcoma virus fT reverse transcriptase SlV simian immunodeficiency virus TAR tram-activation response elernent TCID, 50% tissue culture infective dose tRNA transfer RNA U3 3' unique region US 5' unique region
Note: Abbreviations are defmed in each chapter. The 'standardn one letter
symbolç for the a amino acid residues was the nomenclature used
throughout the text.

CHAPTER 1
lNTRODUCTlOff AND LITERATURE REVIEW

i .l .l Retroviruses Retrovifuses comprise a large and diverse family of enveloped RNA viruses,
and are characterized by two exceptional features. These include a replicative
strategy that invohres reverse transcription of genomic RNA into linear double-
stranded DNA and the subsequent integration of this DNA into the genome of
the cell. Second, viral maturation is completed outside of the host cell (Nemut
and Hockley, 1996; Vogt, 1997).
Retroviruses are divided into two broad categories. simple and complex,
based on considerations of genomic organization, mRNA splicing mechanisms,
and temporal regulaüon of gene expression (Culkn, 1992; Weiss, 1996). All
retroviruses contain three major coding domains, Le. gag, poi, and env, which
encode the structural, replicative, and envelope proteins, respectively. Simple
retroviruses usually contain only these three genes, whereas complex
retroviruses also encode additional regulatory proteins derived from multiply
spliced messages (Weiss, 1996). This alternative splicing mechanism is not
obsewed in simple retrovinises, which express only unspliced and singly
spliced viral mRNA transcripts. Finally, due to the fact that cornplex retroviruses
encode regulatory proteins, their gene expression can be divided into two
temporal phases, Le. an early, regulatory phase and a late, structural phase.
Simple retroviruses have not been shown to display this pattern of regulation of
gene expression (Cul[en, 1992).
Retroviruses are classified into seven genera according to evolutionary
sequence homology Veich, 1985). Formerly refened to as the oncoviruses due
to their oncogenic potential. this group is subdivided into five groups based on
virion morpho logy: avian sarcoma and leu kos is vimses (ASLV) , rn ammalian 8-
type viruses, murine leukemia-related viruses, human FcelI leokemia-bovine
leukemia vinises (HTLV-BLV), and D-type vimses (Vogt, 1997). Examples of
each group are: Rous sarcoma virus (RSV), mouse mammary tumor virus
(MhnlV), Moloney murine leukemia v h s (MoMLV). human T-ceII leukemia
virus (HTLV), and Mason-Pfizer monkey virus (MPMV), respectively. The other

two groups are the lentiviruses and the spumaviruses. Members of the former
inchde the human immunodeficiency viruses (HIV-1 and HIV-2), simian
immunodeficiency vinis (SIV), feline irnmunodeficiency virus (FIV). equine
infectious anemia virus (ElAV), and ovine maedi-visna virus (MW). An example
of spumaviruses is human foamy virus (HFV). AIl oncogenic viral groups except
the human T-ceIl virus-bovine leukemia vinis genus are simple retroviruses;
HTLV-BLV and the lentivfruses and spumaviruses are complex (Vogt, 1997).
Oncovinises occur in all classes of vertebrates, are highly pathogenic.
and usually cause malignancies. For example, human T-cell leukemia virus
type I (HTLV-1) can tesult in adult rceIl leukemia (Weiss. 1996). The
lentiviruses are 'slow' viruses that cause slow progressive degenerative
diseases in many vertebrates. This is a consequence of killing or impairing
specific celts and tissues. and frequentLy tesults in immunologie dysfunction and
neurological disorders (Levy, 1988; Weiss, 1996). Spumaviruses are less well
understood; they are not known to be pathogenic in vivo, despite the fact that in
vfim they are highly cytopathic, foming large. vacuolated syncytia with a
'foamy' appearance (Weiss. 1996).
The virion morphology of avian sarcorna and [eukosis viruses, murine
leukemia-re lated viruses, human T-cell leukernia-bovine leukernia viruses, and
lentiviruses is temed C-type. These virai partictes have a cent rally localized
spherical or conical core. and viral assembIy occurs at the cytoplasmic side of
the plasma membrane during budding (Nermut and Hockley, 1996). B-type
particles contain eccentric, spherical cores that are initially formed in the
cytoplasm, prÏor to migration towatds the plasma membrane. This type of
morphogenesk has been obsenred in mammalian B-type viruses, D-type
vinises, and spumavhses (Nemut and Hockley, 1996). P articles of A-type
morphology assemble wthin the cytoplasm or ai the endop[asmic reticulum
(ER) membrane. Such particles are immature and not infectious, and are most
probabIy aberrant forrns of other retroviruses (Nemut and Hockley, 1996).

1.1.2 Historical background of HlV-1 and AIDS Hurnan immunodeficiency virus type4 (HIV-1) was first isolated from a patient
in 1983, and rapidly became established as the causative agent of Acquired
lmmunodeficiency Syndrome (AIDS) (Mathews, 1982; Barre-Sinousi et al..
1983; Gallo et at., 1984; Popovic et al., 1984; Teich, 1985).
HIV-2 also causes AIDS, although it produces a lower virus load,
spreads much more slowly, and is less virulent than HIV-1 (Clavel et al., 1986;
Weiss, 1996). In fact, on the basis of serologic reactivity and nucleotide
sequence homology, HIV-2 is more closely related to SIV than to HIV-1 (Kanki
et al., 1985). It is thought that HIV-1 and HIV-2 are derived from ancestraI SIV
variants, and are not direct genetic descendants of each other (Smith et al., 1988; Gojobori et al., 1990). White HIV-2 is primarily located in Western Africa.
HIV-1 is found in North and South America, Europe. Central Africa, and Asia.
1.1.3 HIV-1 viral dynamics and AlDS pathogenesis HIV-1 infection is a dynamic process characterized by continuai new rounds of
viral infection and replication in susceptible cells (Ho et al.. 1995; Wei et al.,
1995). This process is ongoing throughout the course of HIV disease. even
during the prolongad asymptomatic phase between primary infection and the
devebpment of AIDS. There is high turnover of virus in the blood and of virus-
infected cells; the total number of virions produced, released into the
extracellular fluid. and cleared is a? ieast 10" particles per day (Ho et al., 1995;
Wei et al., 1995; Perelson et al., 1996). Furthemore, there is rapid turnover of
productively infected cells, whereby approximately 1 O9 new cells are infected
per day (Ho et al.. 1995; Wei et al., 1995; Perelson et al.. 1996; Cavert et al.,
1997).
Primary HIV-1 infection is characterized by extrernely high levels of
plasma virus. For example. values in excess of 106 copies of RNNml are
commonly seen (Perelson et al., 1996; Finzi and Silicano, 1998). As the viral-
specific immune response of the host develops, plasma virus ievels faII to lower
steady-state values. These Vary in dgierent individuals and are predictive of the
rate of disease progression (Mellors et al., 1996). In untreated asymptomatic

patients, the plasma HIV-1 RNA Ieveîs are typically in the range of 1 03-106
copieslml in blood (Finzi and Silicano, 1998).
HIV-1 principally infects activated CD& T lymphocytes and teninaity
differentiated cells of the monocyte-macrop hage lineage (McDoug al et al.,
1986; Maddon et al., 1986; Hwang et al., 1991; Emerman, 1996). Progressive
depletion of T lymphocytes is the defining feature of the immunodeficiency.
Infection of cells in the central nervous system, such as brain macrophages and
microglial cells, are responsible for AIDS dementia and other neurological
diseases (Nottet and Gendelman, 1995; Johnson, 1995). Infection of dendritic
and Langerhans cells of the macrophage Iineage are important for viral
transmission. These cells play a central role in the presentation of antigens to
CD4+ T lymphocytes, and thus infected macrophages likely transmit the
infection to T-cells during antigen presentation (Zhang et al.. 1993; Zhu et al.,
1993; Sotoramirez et al.. 1996).
The major sites of virus replication occur in the peripheral lymphoid
organs, such as the lymph nodes and the spleen, as well as in the mucosal
lymphoid tissue (Panteleo et al., 1991 and 1993; Embretson et al., 1993;
Frankel et al., 1996). It is estimated that at least 99% of the plasma virus is
produced by recently infected short-lived CD4+ T lymphocytes in the peripheral
lymphoid tissues (Ho et al., 1995; Wei et al.. 1995; Perelson et al., 1996 and
1997; Cavert et al., 1997; Chun et al., 1997; Finzi and Silicano, 1998). Long-
lived infected cells such as macrophages and latently infected T-celis make
only a minor contribution to plasma virernia in untreated patients (Ho et al.,
1995; Wei et al.. 1995; Perelson et al., 1996 and 1997; Cavert et al., 1997; Chun
et al.. 1997).
Despite the great deal of piogress that has been made in understanding
HIV-1 and AIDS, critical aspects of disease pathogenesis remain to be
elucidated. Primarily, it is still unclear how HIV-1 infection induces CD4+ T
lymphocyte depletion, the central pathophysiologic feature of the disease.
Numerous mechanisms have been proposed, including: (1) direct killing of HIV-
1-infected CD& T lymphocytes; (2) indirect effects on uninfected CD& and
CD& Fcells; (3) the failure of T-cell regeneration; (4) the disruption of the

lymphoid tissue architecture; and (5) multiple viral and host factors (Panteleo et
al.. 1993; Ho et al., 1995; Wei et al.. 1995; Perelson et al.. 1996; Fauci. 1996;
Hellerstein and McCune. 1997; Herbein et al., 1998; McMichael, 1988; Finzi
and Silicano. 1998). Second, the dominant host immunological factors involved
in controlling disease progression rernain unresolved. This is the case in spite
of the persistence of a vigorous HlV-specific immune response consisting of
cellular (including bath cytotoxic CD8+ T lymphocyte [CTL] and CD& helper
cell responses) and humoral mechanisms that effectively contain viral
replication for a time (Fauci, 1996; Rosenberg et al., 1997; 0gg et al.. 1998;
Finzi and Silicano. 1998).
Taken together. the uncertainty of how the virus persists in the presence
of host immune responses. how normal immune functions are compromised.
and which factors are involved in controlling andfor ttiggering the progression to
AlDS ciitically impair understanding of HIV disease, and therefore undemines
the search for a cure.
This chapter will concentrate primarily on HIV-1, with emphasis on viral
structure and genetic organization, biogenesis of viral proteins, and steps in
viral replication and maturation. Attention is directed to molecular mechan isms
involved in virus maturation and conditions to perturb that process. the focus of
this thesis.
1.2 THE HIV-1 GENOME AND VIRAL PROTEINS
1.2.1 The virion The HiV-1 virion is illustrated schematically in Figure 1-1. The viral envelope is fomed by a cell-derived lipid bilayer into which the viral glycosylated envefope
(Env) proteins have been inserted. These spiked projections consist of the
transrnembrane (TM. gp41) and the surface (SU, gp120) components linked
together noncovalently by disulfide bonds. The viral structural proteins are
iefened to as Gag proteins: in mature partides, the matrix protein (MA p17) is
located under the lipid bilayer, k i n g the inner surface of the membrane; the

capsid protein (CA, p24) fons the conical capsid shelI that encases the
genomic RNA and the nucleocapsid protein (NC, p7), which together fom part
of the ribonucleoprotein (RNP) core. Associated with the nucleocapsid and RNA
within the virion are the polymerase proteins, Le. reverse transcriptase (RT),
protease (PR), and integrase (IN). These are responsible for virus replication
(Gelderbloom et al., 1 987; Haseltine, 1991).

FIGURE 1-1. Schematic repiesentation of the HIV-1 virion.

(~32)
& P ~ V
su @piZO)
Upid Bilayer
NC (PT)
MA tp17)
primer tRNA
CA (~24)
RT @51/p66)
PR @Il)
Viral RNA

FIGURE 1-2. Genetfc organkation of HIV-1.


i 2.2 Genetic organization of HIV-1 Figure 1-2 is a schematic description of the HIV-I genome and the known
functions of its gene products. The genome consists of two functionally active
RNA molecules, each of which is 9.2 kb in length, single-stranded (ss),
nonsegmented, and of positive polarity (Popovic et al., 1984). Typical of al1
retroviruses, HIV-1 initially synthesizes the structural proteins and enzymes in
the fom of large precursor polyproteins, which are cleaved by the viral protease
to yield mature proteins. The HIV-1 genome consists of the three major retroviral
genes, gag, pol, and env, as well as six auxiliary genes. tat, rev, nef, vit vpr, and
vpu. (Cuilen, 1991).
HIV-1 also contains a large array of cis-acting elements that direct the
host cell machinery to function in viral gene expression from its proviral DNA
intermediate. Most of these elements reside in the long terminal repeats (LTRs)
which are located at each end of the provirus, and are divided into three
functionally distinct regions known as U3, R, and US. The U3 region that is
found upstrearn of the transcription start site contains most of the transcription
control elements, including the prornoter, multiple enhancer sequences, and
modulatory regions. RNA synthesis is initiated within the 5' LTR, at the junction
between the U3 and R regions. The 3' LTR contains control elements involved
in post-transcriptional processing of the 3' end of the RNA product. such as a
poly-A tail addition. The R and US regions fom part of the 5' untranslated leader
region, which contains multiple sequences critical for numerous aspects of viral
replication. For example, the leader region contains the TAR element, the
primer binding site (PBS), the dimeriration initiation signal (DIS), the packaging
signal (w), as well as the major splice donor (SD) site (Kingsman and
Kingsman, 1996; Rabson and Wills, 1997).
i.2.3 The gag gene products The gag gene encodes the PCSS*~ (Gag) polyprotein precursor. During virus
maturation, it is cleaved by the viral protease into several çmaller polypeptides
to yiefd the viral structural proteins, which each play critical roles in the virus Iife
cycb. The final products are: (1) MA (matrk pl?), which is located at the amino-

terminus of Gag and is essential for intracellular transport and membrane
association of precursor proteins du ring viral assem bly; (2) CA (capsid, pN),
which is derived from the central region of Gag and fons the viral core shell in mature particles; and (3) NCp (nucleocapsid, PIS), wtiich is located at the
carboxy-terminus of Gag and is further processed into four smaller fragments,
p2, p7 (nucleocapsid, NC), pl, and p6 Veronese et al., 1987; Kaplan and
Swanstrorn. 1991: Henderson et al.. 1992; Wondrak et al., 1993; Sheng and
Erickson-Viitanen, 1994).
1.2.3.1 The Gag piecuisor protein Gag is the only viral protein essential for virus assembly; it is sufficient to direct
the production and release of virus-like particles in the absence of any other
viral protein. including the env gene product (Gheysen et al., 1989; Karacostas
et al., 1989; Çhioda and Shibuta, 1990; Smith et al.. 1990; Gelderbloom, 1991 ;
Wills and Craven. 1991 ; Reicin et al.. 1995). Moreover, as the 'assembly
machine", Gag plays a critical role in orchestrating the assembly process. lt
contains within its structure all the functional elements required for: (1) targeting
and binding of Gag and Gag-Pol to the plasma membrane; (2) establishing
intemolecular interactions between Gag precursors as well as between Gag
and Gag-Pol polyproteins in the immature particle; (3) envelopment and release
of the particle; and (4) directing the incorporation of the viral genome and of
other molecules such as cyclophilin A and Vpr (Craven and Parent. 1996).
1=2,3.2 MA
The mat& protein (MA, p l 7) lies immediately undemeath the viral membrane
and forms the viral mat* (Gelderbloom et al., 1987). It is Kmyristyfated and is
responsible for targeting the Gag precursors to the plasma membrane, as well
as facilitating thek association with the membrane. These functions are
accomplished via the membrane-bhding (or M) domain that is located at its
amino-terminus (Bennett et al.. 1993; Facke et al.. 1993; Wang and Barklis,
1993; Speaman et al., 1994; Zhou et al., 1994a; Craven and Parent, 1996).
Mutations within MA have been shown €0 severely affect stable membrane

bhding, precursor transport. and particle assembIy (Wang and Barklis, 1993;
Yuan et al., 1993; Freed et al., 1994; Speaman et al., 1994; Wang et al., 1998).
The eff Ment incorporation of the envelo pe g tyco proteins into virions is also
dependent on the integrity of the MA domain (Yu et al.. 1992; Dohran et al.,
1994a). In addition, as part of the viral preintegration cornplex (PIC), MA is
critical for translocating the cornplex to the nucleus (Bukrinsky et al.. 1992 and
1993; Heinzinget et al., 1994; VonSchwedler et al.. 1994; Gallay et al., 1995).
Finally, the rnatrix protein has also been implicated in an early step in the virus
life cycle prior to the completion of reverse transcription (Parent et al., 1996;
Reicin et al., 1996; Casella et al., 1 997; Kieman et al., 1998).
1-2-3.3 CA
Capsid (CA, p24) is a hydrophobic protein that forrns a bullet-shaped core
which encases the nucleocapsid cornplex in the mature virion (Haseltine,
1991). CA plays essential roles earfy and late in infection. Its C-terminal
segment contains a highly conserved sequence, termed the major homology
region (MHR), that is essential for virion assernbly and release (VonPoblotzki et
al.. 1993; Dorfman et al., 1994b; Mammano et al.. 1994; McDermott et al.. 1996;
Zhang et al., 1996). This region is required for Gag oligomerization as well as
for CA dimerkation (Zhang et al.. 1996; Gamble et al.. 1997). Moreover, part of
this domain is thought to represent the minimal Gag region capable of particle
assembly and release (Wang et al.. 1998). In contrast, the N-terminal region of
CA appears to be dispensable for particle assembly, but is required for
formation of the mature core which is critical for viral infectivity (Wang and
Barklis, 1993; Dorfman et al., 1994b; Reicin et al.. 1995).
1,2-3.4 NC The mature nucleocapsid protein (NC, p7) is a small. basic protein containing
two zinc fingers of a characteristic CC-H-C motif that are associated with
nucleic acid binding proteins (Berg, 1986; South et al., 1990; Wondrak et al..
1993; Sheng and Erickson-Viitanen, 1 994). NC is essentiaI for virus infectivity,
and has numerous vital functions in the virus life cycle. These include: (1)

mediating Gag interactions - via the interaction (or I) dornain - which are critical
for the formation of mature, infectious cores (Gheysen et al., 1989; Gottlinger et
al., 1989; Wills and Craven. 1991; Bennett et al., 1993; Dorfman et al., 1993;
Chazal et al.. 1994; Freed et al., 1994; Mammano et al.. 1994; Ottman et al., 1995; Reich et al., 1995; Craven and Parent, 1996: Zhang et al., 1998); (2)
promoting dimeriration and maturation of viral RNA (Darlk et al, 1993; Fu and
Rein, 1993; Fu et al., 1994; Sheng and Erickson-Viitanen. 1994; You and
McHenry, 1994; Berkowitz et al., 1995; Feng et aL, 1996b; Sheng et al., 1997);
(3) facilitating the selective encapsidation of genornic RNA via interaction with
the y packaging site (Gorelick et al., 1988 and 1990; Linial and Miller, 1990;
Jowett et al,, 1992; Dannull et al., 1994; Berkowitz et al., 1995; Darlk et al.. 1995; ûttman et al.. 1995; Zhang and Barklis, 1995; Kaye and Lever, 1996;
Poon et al., 1996; DeGuman et al., 1998); (4) promoting primer tRNA
positioning and annealing to the primer binding site (PBS) (Prats et al., 1988;
DeRocquigny et al., 1992; Barat et al.. 1993; Mely et al., 1995); (5) unwinding
tRNA molecules (Kahn and Giedroc. 1 993); (6) stim u tating reverse transcription
by promoting the formation of a tripartite complex composed of the RNA
template, primer tRNA, and reverse transcriptase (RT) (Prats et al., 1988; Barat
et al., 1993; Mely et al., 1995). and by stimulating strand transfer (Darlix et al.,
1993; Allain et al.. 1994; You and McHenry, 1994); and (7) interacting with the
Vpr protein (Li et al., 1996).
1.2.3.5 p6
p6 (or pGg.O) is a peptide derived from the 3' region of the Gag precursor, and is
not present in Gag-Pol because of the tibosomal frameshifting event that occurs
upstream (Veronese et aL, 1987; Henderson et al., 1988). In the virion, it is
thought to be iocated between the core and envelope regions (Hoglund et al.,
1992). The role of p6 in the HIV-1 Iife cycle is not fully understood, but it
seemingly has numerous functions. Fint, it contains the L (or late) domain that
is required for efficient budding by facilitating the release of assemblecf viral
particles from the cell surface (Gheysen et al., 1989; Gottlinger et al., 1991;
Huang et aL, 1995; Yu et al., 1995; Craven and Parent, 1996; Schwartz et al.,

1996). Since p6 serves as a negative regulator of PR activity, its role in particle
morphogenesis is observed particularly in the presence of protease (Huang et
ai., 1995). However, accumulating data suggests that the p6 domain may not be
absolutely required for particle assembiy and release (Jowett et al., 1992;
Speaman et al., 1994; Yu et al.. 1995; Wang et al., 1 998). Second, p6 appears
to be critical for the incorporation of Vpr into particles (Lu et al., 1993 and 1995;
Paxton et al., 1993; Kondo et al., 1995). Third, this domain has recently been
show to play an important role in incorporating or retaining Pol proteins in the
assembling virus particles when protease is activated during budding (Yu et al.,
1998). Fourth, p6 was found to be the most important region critical for
detemining HIV-1 particle size (Garnier et al., 1998). Finally, sorne nuclear
targeting function was found to be directly or indirectly associated with this
domain (Chazal et al., t 994).
1.2.3.6 p2 and p l
Proteolytic processing of Gag also generates two small peptides, termed spacer
peptides 1 and 2 (pl and p2). which are located between NC and p6, and
between CA and NC, respectively (Mervis et al., 1988; Henderson et al., 1988
and 1992). Although they are poorly conserved in sequence and length, they
are conserved in location within Gag precursors of primate lentiviruses,
suggesting an important role in the retroviral life cycle (Henderson et al., 1988).
In fact, p2 functions transiently during sequential precursor processing, playing
a critical role in regulating the ordered particle assembly and core formation
which are crucial for normal virus maturation and infectivity (Gottlinger et al., 1989; Pettit et al., 1994; Krausslich et al., 1995; Reicin et al.. 1995; Accola et al., 1998). Deletion or alteration of p2 processing had drastic effects on intemal
core structure, resulting in aberrant virion morphology and loss of infectivity
(Gottlinger et al., 1989; Kaplan et al., 1993; Pettit et al., 1994; Krausslich et al,
1995; Reicin et al.. 1995). It is thought that p2 regulates the sequential
processing of Gag by serving as a negaüve regulator of cleavage at the CA-p2
site. In this way, it modulates the rate of processing and therefore regulates core
formation (Pettit et al., 1994). In addition, a novel function for p2 has recently

a been suggested in which it acts as a spherical shape deteninant of the Gag
particle during assernbly and maturation (Gay et al.. 1998). Although the
function of p l is unclear, this domain appears to play a roIe in RNA
encapsidation specificity (Zhang and Barklis, 1997).
1.2.4 The pol gene products The pd gene encodes the viral enzymes essential for replication, i.e. protease
(PR, p l 1 ), reverse transcriptase (RT, p511p66). and integrase (IN, p32),
positioned in that order from the 5' end of pot (Veronese et al., 1987; Goff,
1990). Protease is a homodimeric protein that is responsible for the proteolytic
processing and subsequent maturation of viral proteins (Graves et al.. 1988;
Mous et al., 1988, Roberts and Oroszlan, 1990). The heterodheric protein
reverse transcriptase converts viral genomic RNA into double-stranded (ds)
proviral DNA via reverse transcription (Gilboa et aL, 1979; Weiss et al.. 1 985).
The integrase protein catalyzes integration of dou ble-stranded provirat DN A
into the host chromosome (Bukrinsky et al.. 1992 and 1993).
1.2.4.1 The Gag-Pol polyprotein precursor
The translational reading frame of the pol gene is in a minus one position from
gag, and its expression occurs via a ribosomal frameshift to produce Pr160*Q901
polyproteins (Gag-Pol) (Dickson et al., 1984; Jacks et al., 1988; Wilson et al.. 1988). The frequency of the shift is about 5% and therefore the relative
abundance of the Gag and Gag-Pol precursor proteins is about 20:l.
respectively (Dickson et al., 1984; Jacks et al, 1988; Wilson et al., 1988).
Encoded in the region betuveen the frameshift site and protease itself is an
additionai peptide, the transframe region, designated p6' or p6P0'. This region
has not been ascribed a specific function, but k seems to play an important role
in regulating PR activity (Graves et al., 1988; Partin et al., 1991).
The Gag-Pol precursor is incotporated into virus particles and is
essential for infectivity (Popovic et al., 1984; Park and Motrow, t 991; Mergener
et al., 1992). It plays a central rote in the HIV-1 Iife cycle, not only due to the
structural and enzymatic proteins generated by its cleavage. but also because it

is a deteminant of the subsequent stability of the released virus particles (Park
and Morrow, 1993). The Gag portion of Gag-Pol is essential for targeting the
latter polyprotein into the virion, via multiple interactions between common
regions of Gag and Gag-Pol (Jones et al.. 1990; Weldon et al.. 1990; Wills and
Craven, 1991; Hunter, 1984; Craven and Parent. 1996; Huang and Martin,
1997). Furthemore, the balance between the levels of Gag and Gag-Pol is
critical in influencing the subsequent formation of virus particles (Felsenstein
and Goff. 1988; Quillent et al., 1996). In cootrast to Gag, the exclusive expression of Gag-Pol does not result in the release of virus-like particles
(Shioda and Shibuta, 1990; Park and Morrow, 1991; Mergener et al.. 1992).
This is due, in part, to the intracellular activation of the viral protease tesulting in
the premature and complete processing Gag-Pol .
1.2-4-2 Protease As with other retroviral proteinases. HIV-1 protease (PR) is absolutely required
for virion maturation and is therefore essential for viral infectivity (Kohl et al., 1988; Peng et al., 1989). Its inactivation, either by a single point mutation in the
catalytic active site (Asp-25) or by mutational amino acid insertions within
protease, has drastic effects on the assembly, stability, and infectivity of the
released virus particles (Gottlinger et al.. 1989; Peng et al., 1989 and 1991;
Park and Morrow, 1993).
HIV-1 PR is a member of the aspartic family of proteases. containing the
characteristic and conserved amino acid triplet Asp-Thr-Gly (DTG) in its catalytic
active site (Pearl and Taylor, 1987; Oroszlan and Luftig. 1990; Loeb et al.,
1989b). Mutagenic studies have shown the importance of the aspaw (D25)
and other conserved residues for activity (LeGtice et al., 1988b; Kohl et al.. 1988; Seelmeier et al-, 1988; Loeb et al.. 1989a.b). The active PR is an
obligatory dimerïc enzyme, consisting of two 1 1 kDa monomers which associate
symmetrically to fomi the substrate binding cleft. each monomer contributing
one catalytic aspartyl residue to the active site of the enzyme (Pearl and Taylor.
1987; Giam and Boros, 1988; Nutt et al.. 1988; Katoh et al., 1989; Lapatto et al..
1989; Miller et al.. 1989a.b; Navia et aL, 1989; Weber et aL, 1989; Wlodawer et

al.. 1989). The two monomers are heu together as a dimer by the amino- and
carboxy-termini of PR, which fom a fouFstranded antiparallel P-sheet
(Wlodawer et al., 1989; Weber et al.. 1990). Disruption of these interactions in
the dimer Riterface, for example by truncation due to self-digestion (Rose et al.,
1993) or by cornpetition by peptides (Zhang et al., 1991; Babe et al., 1992),
leads to loss of enzymatic activity.
1.2.4.3 Reverse transcriptase Reverse transcriptase (RT) is essential for viral replication, being responsible for
the conversion of single-stranded viral genomic RNA into do u ble-sttanded
proviral DNA by a process temed reverse transcription (Gilboa et al., 1979;
Weiss et ai., 1985). Functional RT molecules are heterodimers consisting of two
polypeptides, p66 and p51, which exist in approximately equal proportions in HIV-1 virions (Chandra et al., 1986; DiMano-Veronese et al., 1986; Lightfoote
et al., 1986; Lowe et al., 1988). While the two subunits share a common amino-
terminus, p51 is produced by proteolytic cleavage of p66 by PR near the
carboxy-terminus (DiMarzo-Veronese et al.. 1986; Lightfoote et al., 1986). In the
heterodimer, p66 is the subunit with reverse transcriptase activity, while its C-
terminal peptide (p15) exhibits RNaseH activity (Hansen et al., 1988, Lori et al.,
1988, Lowe et al., 1988, Stames et al., 1988; Tanese et al.. 1988; Hostomska et
al., 1 991). Reverse transcriptase has at least three enzymatic functions: RNA-
dependent DNA polyrnerase activity (RDDP), DNA-dependent DNA polymerase
activity (DDDP), and ribonuclease H (RNaseH) activity (Goff, 1990).
1.2.4.4 Integiase
lntegrase (IN) is a 32 kDa protein derived from the carboxy-terminus of the Gag-
Pol polyprotein by proteolytic cleavage during virus maturation. It is responsible
for the processing and joining teactions that nisert viral DNA into the host
genome in a process temed integration, an essential step in the virus
replication cycle (Bukrinsky et al., 1992 and 1993; Katz and Skalka. 1994). IN
functions as a multimer whose enzymatic activities require the presence of a
divalent metaI ion (Barsov et aL, 1896). lntegration is both site-specific,

involving U3 and U5 terminal sequences of the viral LTR. and nonspecific with
respect to the target site in host DNA (Katz and Skalka, 1994). Although
integrase is the only protein requîred for integration in vitro, additional viral and
host proteins participate during integration in vivo (Famet and Haseltine, 1991).
Initially, it was believed that the only function of integrase was to mediate the
integration of viral DNA. However, the structure of viral particles produced by
some IN-defective HIV-1 mutants is aberrant, suggesting that integrase could play a role in virion assembly and maturation (Shin et al., 1994; Engleman et
al., 1995).
1.2.5 The env gene products The envgene is translated from a spliced RNA into the precursor gp160, which
then enten the secretory pathway of the host cal1 (Freed and Martin, 1995).
During the process of transportation to the plasma membrane. the site at which
the envelope glycoproteins become incorporated into virions, gp160 undergoes
maturation to a bipartite complex composed of the N-terminal, externat subunit
(gp120, SU), and the transmembrane C-terminal protein (gp41, TM). In addition
to this proteolytic cleavage by cellular enzymes, the glycoproteins undeigo
additional modifications such as extensive glycosylation. disulfide bond
formation, and interactions with chaperon proteins which facilitate proper
folding (Earl et al., 1 991; Hallenberger et al., 1993; Einfeld, 1996). This results
in the formation of a noncovalently associated trimeric complex in the virion
membrane (Weiss et al.. 1990; Earl et al.. 1991; Hallenberger et al., 1993; Lu et
al.. 1995; Kwong et al., 1998; Wyatt et al., 1998). The gp120 subunits are
responsible for virus adsorption to receptors on host cells (McDougal et al.
1986), while gp41 anchors the complex in the viral membrane and mediates
cell fusion (Kowalski et al., 1987; Galiaher et al.. 1989).
1.2.6 Accessory proteins In addition to Gag, Pol, and Env present in al[ retrovinises, HIV-1 ako encodes
six accessory proteins, Le. Tat, Rev, Nef, Vif, Vpr, and Vpu (Haselthe, 1988).
These wn be classified into two groups based on the temporal regulation of

their expression. The Rev-independent proteins are produced at early times
after infection and include Tat, Rev, and Nef; the expression of Vif. Vpr, and Vpu
are Rev-dependent and thus occur fate in the viral life cycle (Cullen, 1992). Tat
and Rev are absolutely required foi virus growth. Tat is essential early in
replication with a primary rote in transcriptional activation of the viral promoter,
and Rev acts later to ensure the switch from the earIy. regulatory phase to the
late, structural phase of viral gene expression (ifono. 1995; Miller and Sarver,
1997). Although early evidence had suggested that Nef, Vif, Vpt. and Vpu were
dispensable for virus growth in many in vitro systems, ment studies have
show that they fulfill several critical functions in vivo, particularly with respect to
viral replication and pathogenesis (Trono, 1995; Miller and Sarver, 1997).
1.2.6,l Tat
Tat is a srnaIr protein encoded by a spliced mRNA derived from two exons
within the central region and env gene of the genome (Arya et al.. 1985;
Sodroski et al., 1905). Primarily located in the nucleus and nucleolus of infected
cells, Tat is a potent transactivator that enhances LTR-driven transcription of
viral genes by 10 to 1000-fold (Laspia et al., 1989; Sharp and Marciniak, 1989;
Kingsman and Kingsman, 1996). Tat binds to a stable stem-loop structure
iocated at the 5'-end of al1 RNAs, termed TAR Both Tat and TAR are essential
for HIV-1 replication and mutations that disrupt their interaction eliminate virus
production (Sharp and Marciniak, 1989; Kingsman and Kingsman, 1996;
Cullen. 1998). Binding of Tat to TAR RNA is thought to position Tat in proximity
to the RNA start site, allowing its transcriptional activation domains to interact
with the cellular transcription apparatus (Kingsman and Kingsman, 1996). Tat
activation of viral gene expression is believed to result from two modes of
action. FÏrst, it increases the processivity of RNA polyrnerase II, thereby
en hancing the efficiency of elongation of TAR-containing full-length RNA
transcripts. Second, the frequency of RNA initiation is increased by Tat (Laspia
et al.. 1989; Kingsman and Kingsman, 1996; Culfan, 1998).

L2.6.2 Rev
Rev is an essential viral protein encoded by a small, multipty spliced mRNA
synthesized at early times af€er infection. It is tocafed in the nucleus and
nucleolus of infected cells (Cullen, 1992). Rev acts posttranscriptionaliy, playing
a criticai role in regulating the temporal expression of viral proteins by driving
translocation of singly spliced and unspliced RNA out of the nucleus to the
cytoplasm (Sodroski et al., 1985; Hadzopolou-Cladares et ai., 1989; Malim et
al.. 1989a; Cullen, 1991; Greene, 1991). In the absence of Rev, only very low
levels of full-length RNAs and singly spliced mRNAs are found in the cytoplasm.
However, when a threshold level of Rev is produced, unspliced and singly
spliced RNAs begin to accumulate in the cytoplasm. This allows the switch from
the early, regulatory phase of viral gene expression to the late phase where
productive infection occurs (Rabson and Graves. 1997).
Rev functions through binding to a highly structured segment of HIV-1
RNA located within the env gene called the Rev response element (RRE)
(Hadzopolou-Cladares et al., 1989; Malim et al., l989a). The RRE is present in
al1 RNAs that are dependent on Rev for their cytoplasmic expression;
conversely, it is spliced out of Rev-nidependent RNAs.
There are a few possible models of Rev action. First, evidence strongly
suggests that the primary effect of Rev is in directLy mediating the nuclear export
of RRE-containing RNAs through interaction with a general nuclear export
pathway (Malim et al.. 1989a; Fisher et al., 1995; Wen et aL, 1995). Second,
Rev has atso been proposed to inhibit complete splicing of HIV-1 RNAs. This
results in the generation of a pool of unspllced and singly spliced RNAs that are
now available for nuclear export (Chang and Sharp, 1989). Finally, it has been
suggested that Rev rnay enhance the translation of unspliced and singly spticed
RNAs in the cytoplasm (Arrigo and Chen, 1991). Taken together, it appears that
Rev may have effects on several levers of RNA processing and function, acting
as a 'chaperone' through various stages of RNA transport (Rabson and Graves,
1997).

1.2-6-3 Nef
Encoded only by primate lentiviruses, Nef is produced at all stages of viral gene
expression. and is packaged in virions in low amounts. Nef is necessary for
high levels of viral replication and disease progression of SIV in vivo, as well as
for induction of AIDS (Kestler et al., 1991 ; Deacon et al., 1995; Kirchhoff et al.,
1995). White the mechanism of action of Nef in pathogenesis is unclear, it is
thought to reçult from the protein's capacity to alter several cellular functions.
First, Nef downregulates the surface expression of CD4 (Garcia and Miller,
1991 ; Aiken et al., 1994) and major histocompatibility complex class I molecules
(MHC 1) (Schwartz et al.. 1996; LeGall et al.. 1998). This is a consequence of
Nef triggering their rapid endocytosis and Iysosomal degradation, and results in
the inhibition of CL-mediated lysis of HIV-1 infected cells (Collins et al., 1998).
Second, virion-associated Nef enhances viral infectivity by promoting early
events after virus entry such as uncoating of the core and stimulating proviral
DNA synthesis (Miller et al.. 1994; Spina et al.. 1994; Aiken and Trono, 1995;
Bukovsky et al., 1997). In addition, Nef has been reported to alter T-ce11
activation pathways and increase viral transcription in vitro. but the relevance of
ffiese findings to HlV-1 pathogenesis in vivo has yet to be deterrnined (Trono,
1995; Miller and Sarver, 1997; Cullen. 1998).
1.2.6.4 Vpr
Vpr is a nuclear protein expressed from a singly spliced mRNk It is
incorporated hto budding virus particles in high amounts by a specific
mechanism. Le. by association with the p6 domain of Gag (Cohen et ai., 1990;
Lu et al.. 1993; Paxton et al., 1993; Kondo et al.. 1995). lts presence in mature
virions is relateci to its two principal functions. First, Vpr rnediates the active
transport of the preintegration cornplex to the nucleus of nondividing cells.
independent of MA'S rok in the same process (Heinzinger et al., 1994). The
second function of Vpr is to arrest dividing cells in the G, phase of the cell cycle,
by inhibiting cyclin-dependent kinase p34- activity (Jowett et ai., f 995; RogeI et al., 1995; Bartz et al., 1996). Cell-cycle anest has several important
consequences, such as: (1) maxirnknig virus production by delaying cells at the

point of the cell cycle where the vira! LTR is most active, before the infected celI
is eliminated by the host immune response (Emerrnan. 1996; Goh et al.. 1998);
(2) upregulating viral gene expression (Cohen et al.. 1990; Goh et al., 1998;
Yao et al., 1998); (3) preventing persistent infection in vitro (Rogel et al., 1995);
(4) causing terminal differentiation of some cells (Levy et al., 1993); and (5)
inducing HIV-mediated cell killing in vitro which rnay be associated with CD4+
%ceIl depletion during disease progression h vivo (Stewart et al., 1997; Yao et
al., 1998). Thus in wvo, it appears that the cytostatic Vpr-mediated O, arrest
plays a pivotal role in viral replication by providing the virus a selective
advantage, namely, maximizing virus production and perhaps mediating CD4+
T-cell depletion (Stewart et al., 1997; Goh et al., 1998; Yao et al., 1998).
Furthemore, recent evidence suggests that Vpr molecu les present in both
infectious and noninfectious viral particles are equally capable of arresting
CD4+ T-cells and may therefore contribute to immune dysfunction in vivo (Poon
et al., 1998). Interestingly, current antCHIV-1 drugs do not affect this virion-
associated function of Vpr (Poon et al.. 1998).
i.2.6.5 Vif
Vif is a cytoplasmic protein that is produced from a singly spliced mRNA late in
infection. it plays an important role in conferring infectivity on progeny virions.
rendering them competent for the early steps of infection (Gabuzda et al., 1992;
VonSchwedler et al., 1 993). This effect is probably indirect, because only trace
amounts are found in virions (Liu et al.. 1995; Camaur and Trono, 1996).
Moreover, the requirement for Vif is strïctly cell dependent. and is determined
solely by producer and not target cells (Sakai et al., 1993). Several
mechanisms for Vifs role in early events have been proposed. each relating to
infectivity enhancement and v i ~ o n association. First, Vif ensures the proper
packing of the nucleoprotein core (Hoglund et ai.. 1994). Second, it facilitates
the transport of virions through the microfiLament network that connects the
outer cell membrane to the nuclear membrane (Karczewski and Strebel. 1996).
Third, t stabilizes newly-synthesized DNA intemediates (Simon and Malim,
1996). Vi rnay also play a role in provirus formation; this function is believed to

be required during virus formation rather than during infection (Miller and
Sarver, 1997).
1.2.6.6 Vpu
Vpu is an integral membrane protein that is produced Iate in the replication
cycle from a singly spliced bicistronic mRNA that also contains the env open
reading frame. Two biological functions have been identified for Vpu. First, Vpu
interacts with the cytoplasmic domain of ER-retained CD4 molecules that are
complexed with gp160, and triggers their accelerated degradation (Willey et ai..
1992; Bou? et al., 1995; Margottin et al.. 1998). This not only enhances the
intracellular transport and maturation of Env. which is predicted to increase the
hfectivity of virions, but also reduces the density of CD4 at the cell surface. This
may preclude superinfection, and hence the premature destruction of the
infected cell (Miller and Sawer. 1997). Second, Vpu stimulates the release of
virions from the surface of infected cells (Klimkait et al., 1990; Gottlinger et al.,
1993). Although the mechanism for the release is unknown, it is thought to
result from the ability of Vpu to form ion channels in the cell membrane (Ewart et
al.. 1996; Schubert et al.. 1996: Lamb and Pinto, 1997).
1.3 THE REPLfCATlON CYCLE OF HlV-1
An ovewiew of the HIV-1 replication cycle is illustrated schematically in Figure
1-3.

FIGURE 1-3. Schemattc representation of the HlV-1 life cycle.


1.3.1 Virus entry and cellular tropism The process of HIV-1 infection begins with the vinis binding to a susceptible
target cell. Cellular entry requires bindhg of the viral gp120 envelope
glycoprotein both to the CD4 cell surface receptor (Maddon et al.. 1986; Stein et
al., 1987) and to one of the seven transrnembrane G-protein-coupled
chemokine receptors (GPCR) recently discovered to act as coreceptors and that
play an important rote in viral tropism (Alkhaüb et al., 1996; Deng et al.. 1996;
Dragic et al., 1996: Feng et al., 1996a). Most viruses that are able to infect
cultured T-cell Iines are temed T-tropic, or X4 viruses. These are syncytium
inducing (SI), are frequently found in late-stage HlV disease, and utilize the
chemokine receptor CXCR4. The majority of macrophage-tropic (M-tropic, or
R5) viruses are non-syncytium inducing (NSI) in T-cell lines, are found
throughout disease, and utiliza CCRS (Alkhatib et al., 1996; Berson et al., 1996;
Choe et al., 1996; Deng et al., 1996; Doranz et al.. 1996; Dragic et al., 1996;
Feng et al., 1996a; Littman. 1988). Although other members of the GPCR farnily
are involved in entry for various viral strains, CCRS andlor CXCR4 remain the
receptors used by a11 known strains of HIV-1 (Choe et al., 1996; Doranz et al..
1996; Littman, 1998). The natural ligands for these chemokine receptors (SDF-
1 for CXCR4; RANTES. MIP-la, and MIP-1 P for CCRS) inhibit the infection of
the particular HIV-1 variants that utilize these molecules for entry (Cocchi et al.,
1995; Bleui et al., 1996; Oberlin et al., 1996; Rapport et al., 1996; Sampson et
al., 1996).
1-cell tropic HIV-1 isolates can infect both resting and activated CD4+
cells. but replication does not occur in the former due to a block at a postentry
level (Bukrinsky et al., 1991 and 1992). Similarly, these viruses c m enter
macrophages efficiently by using CD4 and CXCR4 as coreceptors; however,
replication is restricted at the level of nuclear import of viral DNA
(Schmidtmayerova et al., 1990). Macrophage-tropic HIV-1 isolates can infect
activated CD4+ T-cells, but do not easiIy infect resting T-cells because of the
low expression of CCRS; productive infection of recently infected resting CD4+
T-cells requires antigen-driven activation (fack et al., 1990; Bukrinsky et al..
1991 and 1992; Spina et al., 1995).

The primary viral deteminant of cellular tropism is the third variable (V3)
loop of Env gp120 (Hwang et al.. 1991; Choe et al.. 1996). CD4 binding to
gp120 induces conformational changes in the gp120 glycoprotein, exposing or
creating high affinity binding sites for chemokine receptors on the V3 loop (Wu
et al., 1996; Kwong et al.. 1998; Rizzuto et al., 1998; Wyatt et al.. 1998).
Subsequently, gpf 20 interacts either CXCR4 or CCRS (Trkola et al.. 1996; Wu
et al.. 1996). Importantly. the affinity of gp120 for CCRS is greatly enhanced in the presence of CD4, emphasizing that CD4 not only provides a docking
surface for gp120. but also promotes exposure of a domain that interacts with
chemokine receptors (Trkola et al, 1996; Wu et al.. 1996). The binding site for
CCR5 on gp120 has been mapped to a fragment which contains the CD4
binding site and overlapping epitopes within the V3 loop (Wu et al., 1996); the
binding site of gp120 for CXCR4 is thought to lie within the highly consewed
stem of the VIN2 structure near the base of the V3 loop, and in other regions
folded into proximity (Kwong et al., 1998; Rinuto et al., 1998; Wyatt et al., 1998).
Chemokine-receptor binding is believed to trigger additional
conformational changes that lead to the exposure of the fusogenic peptide at
the amino-terminus of the gp41 glycoprotein (Kwong et al., 1998). This, in turn,
modifies gp41 into its fusion-active state, whereby it inserts into the tatget
membrane (Chan and Kim. 1998; Kwong et al., 1998). Subsequently, the viral
and plasma membranes fuse via a direct. pH-independent mechanism, thereby
releasing the viral core particle into the target cell cytoplasm to inliate
replication (Stein et al.. 1987). Furthermore, it is thought that in addition to their
rotes in viral entry, the chemokine recepton may also be involved in postentry
stages du ring virus replication (Chackerian et al.. 1997).
1.3.2 Reverse transcription Once the viral core enten the cytoplasm, reverse transcription converts viral
genomic RNA into double-stranded DNA (Gilboa et al.. 1979; Weiss et aL, 1985). The current mode1 for reverse transcription invohes nurnerous steps,
and is depicted in Figure 1-4.

FIGURE 1-4. The current modei of retroviral reverse transcription.

R U5 PBS U3 R
R US PBS
initiation of ') DNA synthesis RNAseH digestion Fis? strand tmsfer
U3 R
R U5 PBS
R U5 PBS
+ digestion
PPT U3 R
PBS' I U3t R' US' + Initiation of (+) DNA synthesis
I PPT U3 R US PBS s
U3 R U5 PBS Second strand transfer
U3' R' US' PBS'
U3 RU5 PBS
I Completion of DNA synthesis
U3' R' US'
PPT U3 R US
LTR
Legend: - genomic RNA r r r RNAseH digest& viral RNA - (9) DNA - (I) DNA ;). primertRNA

Reverse transcfiptase initiates minus-strand DNA synthesis by
elongating a partially unwound primer tRNA that is hybridized to the primer
binding site (PBS) in genomic RNA In HLV-1, tRNAw serves as the replication
primer (teis et ai., 1993). Synthesis continues to the 5' end of the genome,
generating minus-strand strong-stop DNA [(-)ssDNA]. As M reaches the end of
the template, its RNaseH activity degrades the RNA strand of the RNADNA
duplex (Champoux et al., 1984). This allows the first strand transfer to proceed
whereby (-)ssDNA is transferred to the 3' end of the genome, guided by the
repeat (R) sequences of the LTRs present on both ends of the RNA (Hu and
Temin, 1990; Luo and Taylor, 1990; Peliska and Benkovic, 1992). Minus-strand
DNA synthesis then resumes and is completed by RT, again accompanied by
RNaseH-mediated degradation of the template strand. Template digestion is
incomplete, and results in the generation of RNaseH-resistant oligoribo-
nucleotides rich in purines, called the polypurine tract (PPT) (Champoux, 1993).
Plus-strand DNA synthesis is Riitiated primarily at the PPT, and then proceeds
by copying minus-strand DNA to its 5' end (Pullen and Champoux. 1990;
Chameau and Clavel, 1991). RNaseH removal of the primer tRNA facilitates the
second strand transfer, in which complementary PBS segments in the plus-
strand DNA and in the minus-strand DNA anneal (Hu and Temin, 1 990; Peliska
and Benkovic, 1992). The plus- and minus-strand syntheses are then
completed, with each strand serving as a template for the other (Huber et al., 1989). This generates a linear dou ble-stranded DNA duplex. containing a
duplicated LTR at either end (Goff, 1990; Telesnitsky and Goff, 1997).
1.3.3 Proviral DNA nuclear localkation and integration Folfowing reverse transcription, the newly-synthesized double-stranded DNA is contained within a preintegration compfex (PIC), whose other components
include nT, IN, NC. MA. and Vpr (Famet and Haseltine, 1991; Bukrinsky et al*,
1993; Galfay et al., 1995). Entry of the PIC hto the nucleus is an essential step
in retrovirus re plicat ion and is required for su bsequent htegration of the proviral
genome into host DNA. and thus for vkus production (Bukrinsky et a!., 1993).
The oncovinises are dependent on cell proliferation for their replication,

because breakdown of the nuclear envelope at rnitosis allows the PIC to
interact with the host cell chromosomes. In contrast, HIV-1 and other lentiviruses
have evolved specific mechanisms for PIC nuclear import üiat are independent
of nuclear membrane breakdown, so that replication can occur in nondividing
cells. Consequently, HIV-1 can infect terminally differentiated and nondividing
cells such as macrophages and dendritic cells, a property important for viral
dissemination and transmission (Bukrinsky et al.. 1992 and 1993; Miller and
Sarver, 1997). Factors involved in HIV-1 PIC translocation include Vpr and MA
which independently, yet partially redundantly, permit the import of the viral
preintegration cornplex through the nuclear pore via distinct nuclear localkation
(NE) sequences (Heinzhger et al., 1994; VonSchwelder et al.. 1994; Freed et
al., 1995; Gallay et al., 1995; Yao et al., 1995).
Once inside the nucleus, the proviral DNA. which is a blunt-ended linear
molecule whose termini correspond to the boundaries of the LTRs, is integrated
into the host chromosome at non-specific target sites by the viral enzyme
integrase (IN) (Bukrinsky et al.. 1 992 and 1993). The first step (3'-processing) is
integrase-dependent cleavage of two nucleotides fmm the 3' end of each strand
of viral DNA; this occurs in the cytoplasm prior to nuclear entry of the PIC (Roth
et al., 1989). The next step (joining or DNA strand transfet) is a concerted
cleavage and ligation reaction, in which a staggered cut is generated in the
target DNA by nucleophilic attack involving the hydroxyl group present at the
recessed 3' ends of the viral DNA. This is foilowed by Iinkage of the viral DNA to
the 5' ends of the target DNA at the cleavage site (Engelman et al., 1991;
Pryciak and Varmus, 1992; Sherman et al.. 1992; Kulkosky and Skalka, 1994).
The resulting gaps in the target DNA are repaired, likeiy by host enzymes, to
create short direct repeats flanking the provirus, a hallmark of all retroviral
htegration reactions (Katz and Skal ka, 1994; Kulkosky and Skalka, 1994).
1.3.4 Expression of the viral genome Once integrated into the chromosome, HIV-1 utilizes the host cellular machinery
for transcription and translation of its genome. in addition to the viraliy encoded
Tat protein (Cullen, 1991; Haseltine, 1991). Host RNA polymerase II is

responsibb for synthesis of the primary RNA transcripts, which serve either as
mRNA for translation into viral proteins or as viral genomic RNA that will be
incorporated into progeny virions during assembly. The rate of initiation of viral
transcription depends on cellular transcription factors. particularly in the early
stages of viral gene expression prior to the production of fat Cellular activation
and proliferation signals cause the binding of transcription factors to the LTR and result in increased rates of transcription (Cullen, 1991; Kingsman and
Kingsrnan, 1996). The efficient transcription of the viral genome requires a series of complex mechanisms involving the viral regulatory proteins Tat and
Rev. as well as cellular transcription factors such as NF-KB, NFAT-1, AP-1, and
Spl (Kingsman and Kingsman, 1996).
Following the synthesis of full-iength RNA transcripts. a cornplex array of
alternativeIy spliced mRNAs are produced. The differential expression of distinct
species of viral mRNAs is controlled by the HIV-1 Rev protein (Cullen, 1998).
The Ievel of Rev present in an infected cell detemines the proportion of the
unspliced, singly spliced, and multiply spliced RNAs that are produced. This, in
tum, regulates the switch from the early phase of viral gene expression to the
late phase where productive infection occurs (Rabson and Graves, 1997;
Cullen, 1998). Because Rev, Tat, and Nef are encoded by fully spliced rnRNAs,
these gene products are expressed and function shortly after infection and are
therefore referred to as early gene products. In contrast. the Gag, Pol, Env, Vif.
Vpr. and Vpu proteins, as well as full-length genomic RNA, are all dependent on
Rev for the nucIeocytoplasrnic transport of their cagnate mRNAs and are
therefore expressed with delayed kinetics; they are referred to as the late
proteins (Hadzopolou-Cladares et al., 1989; Malim et al.. l989a; Cullen, 1 991
and 1998; Greene, 1991 ; Sodroski et al., 1985)-
1.3.5 Viral packaging, assembly, and budding HIV-1, like other lentivinrses and Ctype oncovinises, assembles at the plasma
membrane of the infected cells (Vogt, 1997). The formation of retroviral particles
is a self-assembly process requiring only the expression of the Gag precursor
polyprotein. Gag is responsible for orchestrathg the assembly process. as well

as recruiting viral proteins, viral genornic RN4 and host ceII-derived elements
into virus particles (Gheysen et ai.. 1989; Haselthe. 1991; Yu et al., 1992;
Luban et al., 1993a; Reicin et al.. 1995; Craven and Parent, 1996). However, it
is unclear whether the self-assernbly of Gag polyprotein monomers is initiated
in the cytoplasm or only after association of the molecules with the lipid bilayer
(Nermut and Hockley, 1996).
Assembly is thought to begin with the association of genomic RNA with
the Gag and Gag-Pol precursor proteins. Efficient encapsidation of the viral
genome requires the presence of cis-acting packaging sequences, termed
(psi), located upstream of Gag in the full-length vital genomic RNA (Linial and
Miller, 1990; Luban and Goff. 1991). Sequences in the NC domain of Gag are
necessary for specific genomic RNA recognition and packaging, particularly the
zinc finger motifs and the flanking basic reçidues (Linial and Miller, 1990; Luban
and Goff, 1991; Dannull et al., 1994; Berkowitz et al., 1995; Darlix et al., 1995;
Ottman et al., 1995; Kaye and Lever. 1996; Poon et al., 1996; DeGuman et al.,
1 998).
The Gag-Pol precursor is incorporated into virions via association with
Gag, presumably through interactions between the Gag domains of the two
polyproteins (Wills and Craven, 1991; Park and Morrow, 1992; Smith et al.,
1993; Hunter, 1994; Srinivasakumar et al.. 1995; Craven and Parent, 1996).
Although multiple regions throughout Gag are important in Gag interactions,
such as numerous domains within MA, CA. NC, and p2 (Chazal et al., 1994;
Carriere et al., 1995; Ottrnan et al., 1995; Reicin et al., 1995; Zhang et al., l998).
the critical determinants within Gag-Pol that mediate its entry into virions have
been mapped to the major homology region (MHR) and the adjacent C-terminal
sequences of CA (Park and Morrow. 1992; Smith et al., 1993; Mammano et ai.. 1994; Reicin et al., 1995; Srinivasakumar et al., 1995; Huang and Martin, 1997).
In addition. the RT and IN regions of GagPol may also play roles in Gag-Pol
incorporation b y faciiitating dimerkat ion of the po lyp roteh precurso rs
(Engelman et a[., 1995; Ansari-Lari et al.. 1996; AnsariLari and Gibbs. 1996;
Bukovslcy and Gottlinger, 1996).

Membrane targeting and binding of the precursors involves both the No
myrisüc acid that has been cotranslationally added to the amino-terminai
glycine residues of MA on both Gag and GagPol, as well as a cluster of basic
amino acids near the N-terminus of MA The myristate moiety is thought to
provide a hydrophobie anchor in the membrane while the basic charges
mediate electrostatic interactions that stabilize the association (Gottlinger et al..
1989; Bryant and Ratner, 1990; Park and Morrow, 1991 ; Speaman et al.. 1994;
Zhou et al., 1994a). In addition, it has recently been suggested that the
interaction of the membrane-binding domain of Gag with the plasma membrane
is facilitated by the interaction (or I) domain within NC (Sandefur et al., 1 998).
Myristylation of Gag is required for assembly and particle formation (Gheysen et
al., 1989; Gottlinger et al., 1989; Park and Morrow, 1992; Smith et al., 1993;
Charal et al., 1994). In contrast, myristylation of Gag-Pol is not necessary for
Gag-Pol assembly into virions; rather, the unmyristylated GagPol polyprotein is
recruited via interactions with Gag (Gottlinger et al., 1989; Bryant and Ratner,
1990; Park and Morrow, 1992; Smith et al.. 1993).
Budding is initiated following the association of genomic RNA/polyprotein
complexes with the Env glycoproteins at the cell membrane (Murphy and Goff,
1989; Barat et al., 1993; Berkowitr and Goff, 1993; Gottlinger et al., 1993;
Sakaguchi et al.. 1993; Lavallee et al., 1994; Mak et al., 1994). The envelope
proteins are incorporated into the virion through interactions with MA (Yu et al.,
1992; Einfeld, 1996; Freed and Martin, 1996; Lee et al., 1997). The efficient
release of virus particles is dependent upon a sequence elernent named the L
(or late) domain, located within the Cterminal p6 sequence of Gag (Gottlinger
et al., 1991; Wlts et ai., 1994; Craven and Parent, 1996).
1.3.5.1 Other proteins incorporated fnto the virion During virus assembly. additional molecules, i.e. other virus-encoded proteins
as well as cellular proteins and nucleic acids, are incorporated into virions mile
budding from infected cells. AIthough the mechanisrn by Mich some host-
derived proteins, such as MHC class l and class II, HU-DR. ~2-mÏcroglobulin,
and intercellular adhesion molecrile t ([CAM-1) are packaged is not dear. it

does appear that these host-derived proteins may have functional roles in the
virus life-cycle (Trernblay et al., 1998). Other proteins and RNA molecules,
which are criticai for virus replication and infectivity, are incorporated into
budding virions via specific mechanisms.
The Vpr protein iç found in large quantities in virions (Cohen et al., 1990).
and is recniited through interactions with the p6 domain of Gag (Lu et al., 1993;
Paxton et al., 1993; Kondo et a!., 1995). There remains some controversy
regarding the presence of Nef and Vif in mature particles, primarily because the
amounts of these proteins is low and the mechanisms for their encapsidation is
unclear; nevertheless, data supporting their presence is becoming stronger
(Trono, 1995; Miller and Sarver, 1997). Virion-associated Nef has been shown
to enhance the infectivity of the incoming virus, most likety by promoting
uncoating of the core and sümulating reverse transcription (Miller et al., 1994;
Spina et al., 1994; Aiken and Trono. 1995; Bukovsky et al., 1997). Although Vif
has been identified as a component of the virion which also enhances infectivity
(Liu et al., 1995; Camaur and Trono, 1996), recent data suggest that the
packaging of Vif is neither specific nor necessary for its function (Simon et al..
1 998).
The incorporation of the host cell chaperon cyclophilin A (CypA) into HIV-
1 virions is mediated by a specific interaction with the CA domain of Gag (Luban
et al., 1993a; Franke et al., 1994; Thali et al., 1994; Colgan et al., 1996).
Cyclophilin A is required for the formation of fully infectious virus particles
(Franke et al., 1994; Braaten et al., 1996; Franke and Luban, 1996). It is thought
that CypA binding to CA induces the correct Gag structure for proper assembly
and processing, and, after virus entry, facilitates destabilization of the viral core
and thereby pemits the initiation of reverse transcription (Braaten et al., 1996;
Franke and Luban, 1996; Luban, 1996; StrebIow et al., 1998).
In addition, viral particles contain numeruus cellular RNA species,
including primer tRNAw which is seiectively packaged into the virus via
specific RT sequences within Gag-Pol (Mak et al.. 1994 and 1997).

i .3.6 Maturation Newly-released viral particles are immature, conta ining uncleaved p recursor
proteîns which form a spherical protein shell closely apposed to the lipid
membrane, as well as an electron-lucent core (Gelderbloom, 1991; Hoglund et
al., 1992; Nemut and Hockley, 1996; Fuller et al., 1997). They are rendered
mature and therefore infectious by a process termed maturation. This involves
precursor protein processing whereby Gag cleavage induces a dramatic
reorganization of the intemal viral structure associated with condensation of the
core, yielding morphologically mature viruses (Vogt, 1996). Processing and
maturation are not required for particle assembly or release, but are essential
for viral infectivity (Kohl et al.. 1988; Peng et al., 1989).
Virion maturation involves cleavage of Gag by the virally encoded
protease, which is brought into the virion as a component of Gag-Pol (Crawford
and Goff, 1985; Kohl et al.. 1988; Gottlinger et al., 1989). Either at the
membrane of infected cells, or shortly after virion release, PR activity is initiated
by what is believed to be an autocatalytic process (Roberts and Oroszlan, 1990;
Kaplan et al., 1994). Protease activation only occurs after dimerkation of Gag-
Pol polyproteins at the time of virion assembly (Debouck et al., 1987; Luftig et
al., 1990). The active dimeric protease then processes the polyprotein
precursors into fonctional protein components. This causes the intemal
structure of the particle to rearrange and to condense into an electron-dense
core, generating mature, infectious viral particles (Dickson et al.. 1984; Oroszlan
and Lufiig, 1990; Roberts and Oroszlan. 1990; Craven and Parent. 1996; Vogt,
1996). Furthemore, protease activity in precursor processing iç also important
for maximizing the efficiency with which virions are released from the surface of
ïnfected cells (Kaplan et al., 1994).
Accurate and complete processing of the Gag precursor by PR is
essential for the formation of infectious, morphologically mature virions (Kaplan
et al., 1993 and 1994). Maturation and core condensation is a sequential
process that is regulated by the rate of cleavage at individual sites within Gag.
Three determinants are likely to controI the ordered proteolytic processing.
These are: (1) the sequence of the processing site; (2) the structural context of

the processing site; and (3) the accessibility of the site to protease (Parün et al.,
1990; Pettit et aL, 1994). The initial cleavage occurç at aie carboxy-terminus of p2 and separates an N-terminal MA-CA-p2 intenediate and a C-terminal NC-
plop6 intermediate (Mervis et al.. 1988; Gowda et al., 1989; Bennett et al., 1991 ;
Tritch et al.. 1991; Claven et al.. 1993). This rapid cleavage between CA and NC releases the RNA-binding NC protein and leads to condensation of the
ribonucleoprotein (RNP) core (Weigers et al., 1998). Subsequent cleavages
separating MA from CA-p2 and NC-pl from p6 occur at an approximately 10-
fold lower rate (Pettit et al., 1994). The release of MA ftom CA which allows CA
to separate from the membrane, plays a pivotal role in core condensation;
inhibition of processing at this site leads to noninfectious particles with
abnomal morphologies (Gottlinger et al., 1989; Partin et al., 1990; Kaplan et al.,
1993; Krausslich et al., 1995; Reicin et al., 1995; Vogt, 1996). The final cleavage
in the processing cascade teleases p2 from the carboxy-terminus of CA. This
iate event is slow and is thought to be mediated by cleavage at the p2-NC site,
which reduces the efficiency of cleavage at the upstrearn CA-p2 site (Pettit et al..
1994). In fact, C-terminally extended CA species have been observed in virions
or virus-like particles with reduced PR activity (Mergener et al., 1992; Rose et
al., t 995). This final CA cleavage is not needed for condensation of the RNP
core, but is essential for condensation of the capsid shell and is believed to
stabilize the viral core into its mature conformation (Weigers et al., 1998; Accola
et ai.. 1998).
lmplicit in the morphologicaI change precipîtated by the cleavage of Gag
during virion maturation is the condensation of RNA. The genomic RNA dimer is initially encapsidated in an exîended conformation, and later adopts a
condensed structure (Fu and Rein, 7993; Darlix et al.. 1995; Vogt, 1996). This
maturation requites the activity of PR, since Î t is the mature NC which induces
the conformational changes that stabilize the RNA dimer (Gelderbloom et a[..
1987; Oertle and Spahr. 1990; Stewart et al.. 1990; Fu and Rein, 1993; Fu et al.,
1994: Darlk et ai.. 1995; Berkowitz et al.. 1996; Feng et al., t W6b; Vogt, 1996).
Some Gag cleavages are dependent on the presence of RNA, and thus it is thought that events other than proteoIysis, in particular NGRNA interactions,

may be critical for proper morphological maturation (Sheng and Erickson-
Viitanen. 1994; Berkowitz et al., 1995; Sheng et al., 1997). Moreover, it has
been proposed that genomic RNA acts as a scaffold. organizing the assembly
process for condensation of a dense, infectious particle (Jowett et al., 1992;
Campell and Vogt, 1995; Craven and Parent, 1996; Fuller et al., 1997).
1.4.1 Regulation of protease activity The formation of infectious HIV-1 virions necessitates that intracellular cleavage
of the Gag and Gag-Pol precursors by the viral protease is limited. Proteolytic
activity of PR-containing precursors in the cytoplasm is suppressed primarily by
concentration and structural constraints. However, additional regulatory
mechanisms must be present during assembly and budding because Gag-Pol
precursois can undergo autoprocessing in the cytoplasm of some cells and in
some in vitro systems (Krausslich et al.. 1988; Smith et al., 1990; Kaplan and
Swanstrom, 1991). The mechanism of PR regulation is not fully understood,
although it is well-established that multiple factors play a role throughout the
virus life cycle. These control PR production and activation, as well as
sequential substrate processing. Tight regulation of PR activity is critical for
successful HIV-1 replication, as both the inhibition (Kohl et al.. 1988; Gottlinger
et al., 1989; Peng et al., 1989; Meek et al., 1990; Park and Morrow, 1993) and
the overproduction (Krausslich, 1991 ; Park and Morrow, 1991 ; Hoshikawa et al., 1991; Mergener et al., 1992; Karacostas et al.. 1993; Arrigo and Huffman, 1995)
of PR result in the generation of non-infectious particles.
t .4.l .t Frameshift lng controls PR production The balance between the levels of Gag and Gag-Pol is critical for efficient viral
particle assembly (Felsenstein and Goff, 1988; Park and Morrow, 1991; Dinman
and Wicknet, 1992; Karacostas et al., 1993; Hung et al., 1998). In contrast to
Gag, the exclusive expression of Gag-Pol does not resuIt in the release of virus-
Iike particles. This is primarily due to premature htracellular activation of the

viral protease, resu king in extensive degradat ion of polyprotein precu tsors
(Shioda and Shibuta, 1990; Park and Morrow, 1991; Mergener et al., 1992).
Gag-Pol is expressed via a ribosomal frameshift which occurs at a frequency of approximately 5%, such that the relative abundance of the Gag
and Gag-Pol precursoi proteins is about 20:1, respectively (Dickson et al., 1984;
Jacks et al.. 1988; Wilson et al., 1988). In this way, the amount of PR in the celI is regulated at the leveI of its production. Indeed, premature PR activation is
believed to be prevented by the limited concentration of Gag-Pol precursors in
the cytoplasrn. Consequently, only when there is a drarnatic increase in the
effective local concentration of PR monomers at the cell membrane during
assem bly, does protease become activated via dimerization of Gag-Pol
precursors (Navia and McKeever. 1990; Vogt, 1996).
Mutation of the frameshifting signal, so that gag and pol are in-frame and
Gag-Pol is produced 100% of the the, results in the ïnability to produce viral
particles (Park and Morrow, 1991; Karacostas et al., 1993). There are several
possible reasons for the absence of budding and particle formation, These
include structural andlor steric effects caused by the excess of Gag-Pol (Jacks
et al.. 1988; Hughes et al., 1993; Karacostas et al., 1993), as well as the
absence of p6 at the C-teminus of Gag which is important for vÎrion budding
(Gheysen et al.. 1989; Gottlinger et al., 1991 ; Mergener et al.. 1992). However,
the greatest contributor to the defect in particle production is that the
overproduction of Gag-Pol results in excess protease expressed intracellularly.
wusing its premature dimerization and activation (Kaplan and Swanstrom,
1991; Krausslich, 1991; Karacostas et aL, 1993; Luukkonen et al., 1995). This.
in tum. results in premature processing of the precursor proteins prior to their
transit to the plasma membrane. In this way. assembIy and particle formation
are abrogated as a consequence of dissociation of the virion wmponents from
the site of assembly (Hoshikawa et ai., 1 991; Kaplan and Swanstrom. 1991 ;
Ktausslicii, t 991; Park and Morrow, 1991; Mergener et al.. 1992; Karacostas et al., 1993; Arrîgo and Huffinan, f 995). This is supported by the finding that
inactivation of PR in the context of overproduced Gag-Pol padially restores

particte formation (Mergener et al., 1992; Hughes et al., 1993; Karacostas et al.,
1993).
In addition to the premature intracellular activation of PR damaging the
formation of progeny virions, it also causes increased cytotoxicity and cell death
in mammalian cells (Krausslich, 1991 and 1992; Kaplan and Swanstrom, 1991 ;
Mergener et al.. 1992; Krausslich et al.. 1993). These cytotoxic effects of PR
overexpression are thought to result from PR cleavage of cytoskeletal andor
other cellular proteins (Shoeman et al., 1990 and 1993; Oswald et al., 1991 ;
Riviere et al., 1991; Tomasselli et al., 1991a,b; Adams et al., 1992).
1 Al .2 Requirement for dimerization It is criticai that PR only becomes activated in budding particles during
assembly, since 8s cytoplasmic activation leads to premature cleavage of the
Gag and Gag-Pol precursors in the absence of virion production (Burstein et al.,
1991 ; Krausslich, 1991; Karacostas et al., 1993; Luukkonen et al., 1995). In addition to Iimiting the amount of PR produced via frameshifting, the
requirement for dimerization for protease activity is another level of control in
preventing its early activation and polyprotein cleavage prior to virion budding
(Krausslich and Wimmer, 1988).
Protease is initially generated as a monomer and must dirnerize in order to fomi an active enzyme (Erickson-Viitanen et al.. 1989; Lapatto et al., 1989;
Navia et al., 1989). Dimeriration of PR fotlows the dimerization of Gag-Pol
polyproteins during virion assembly (Debouck et al., 1987; Luftig et al., 1 990).
This is thought to be concentration-dependent insofar as it only occurs once
assembly has begun and Gag-Pol precursors are in close proximity (Krausslich
et al., 1988; Navia and McKeever, 1990). The dimerized PR then becomes
activated via an autocatalytic process, whereby it cleaves itself out from the
Gag-Pol precursor at the Phe-Pro cleavage sites on either side of the PR
domain. first at the amino- and then at ttie carboxyteminus of protease
(Debouck et ai.. 198ir; Giam and Boros. 1988; Oroszlan and Luftig, 1990;
Roberts and Oroszlan, 1990; Louis et aL, 1994; Wondrak et al., 1996). In w?m, it

has been shown that PR dimer formation must occur prior to the initiation of autocatalytic release of PR (Louis et al., 1994; Wondrak et al., 1996).
The crystal struqtures of HlV-I PR show that the extensive interface that
exists between the monomers is domhated by interactions between adjacent
amino- and carboxy-temini as well as those between twin amino acids at the
active site; the primary dimerization domain (about half of the intersubunit
contact area) is in the four-stranded antiparallel bsheet formed by the N- and
Gtmnini of the enzyme (Wlodawer et al., 1989; Weber et al., 1990). Alteration
of the dimerization interface interferes with PR'S dimerization potential and thus
its activity. For example, removal of several N-terminal amino acid residues of
PR, that are critical in foning the f3-sheet, reduces or aboiishes protease
activity (Pichova et al., 1992; Rose et al.. 1993; Schat. et al., 1997).
Furthermore, defective PR monomers lacking the N-terminal interface domain
form catalytically inactive heterodimers when mixed with wild-type monomers
via disruption of the normal dimer interface (Babe et al., 1991). Dimerization can
also be inhibited by peptides that mimic the N- and C-terminal $-strands of PR
These effectively block the dimerization interface and shin the monornerfdimer
equilibrium towards monomers by a mechanism temed 'dissociative inhibitionw
(Zhang et al., 1991; Babe et al., 1992). While both N- and C-terminal peptides
can bind to inactive PR protomers and prevent their association into the active
dimer, the C-terminal peptide is a more potent inhibitor of PR activation than the
N-terminal peptide (Zhang et al., 1991 ; Babe et al., t 992).
In addition, when the requirement for dimerization for PR activity is
eliminated, for example by forming a tethered dimer in which two PR monomers
are attached by a flexible Iinker, the intemiolecuhrly linked monomers fom a
more stable and enzymatically active enzyme. This dysregulation results in the
prem ature intracellular release of viral proteins instead of maturation wit hin
extracellu lar particles (Cheng et al., IWO; Burstein et al., 1991; Krausslich,
1991; Xiang et al.. 1997). Thus, both the promotion and the inhibition of PR
dime rizat ion affect its activity, em phasizing that PR'S de pendence on

dimerkation for activity is a regulatory mechanism to ensure that its activation is
control led.
1.4.1.3 Sequential cleavage of Gag and Gag-Pol
Another point of control in regulating processhg by PR is that the cleavage sites
in Gag and Gag-Pol are not functionally equivalent and thus are processed at
different rates. For example, the pBNC and p6*-PR sites are cleaved most
rapidly, while NCql and pl-p6 are the most slowly processed (Darke et al.,
1988; Krausslich et al., 1989; Loeb et al., 1989a; Tozser et al., 1991; Wondrak et
al., 1993). This ensures that the precursors are cleaved in a well-defined
sequential order, since accurate and complete processing of precunor proteins
is essential during maturation for the formation of mature cores in infectious
virions (Gottlinger et al., 1989; Kaplan et al., 1993 and 1994; Krausslich et al.,
1995; Reicin et al., 1995; Weigers et al., 1998; Zennou et al., 1998). Immature
HIV-1 particles formed in the presence of PR inhibitors are unable to achieve
maturity and infectivity upon removal of the inhibitor. presurnably as a
consequence of inappropriately timed and controllad precursor cleavages
(Lambert et al., 1992; Kageyama et al., 1994; Rayner et a[.. 1994). The
sequence and structural context of the cleavage site, as well as PR'S
accessibility to the site, together regulate the rate of cleavage at individual sites
within Gag and Gag-Pol, thereby controlling precursor processing (Krausslich et
al., 1989; Loeb et al., 1989a; Partin et ai., 1990; Pettit et al.. 1991 and 1994;
Tritch et al., 1991).
Protease substrate recognition is highly specific as only a few unique
sites in the Gag and Gag-Pol precursors are cleaved. Most of the specificity is
provided by four amino acids on either side of the scissile bonds that are
recognked by flaps present on each monomer of PR (Darke et al., 1988; Kotler
et al.. 1988; Loeb et ai., t989b; Miller et al.. 1989b; Weber et al.. 1989;
Tomasselli et al., 1990). However, residues in the flaps themselves also play a role in specificity (Moody et al., 1995). and distant residues significantly affect
the cleavage rate (Tritch et al., 1991).

Figure 1-5 is a schematic representation of protease cleavage sites in
HIV-1. Studies examining processing of Gag have shown that the initial
cleavage occurs between p2 and NC, generating MA-CA-p2 and NC-pl-p6.
This is followed by subsequent cleavages separating MA from CA-p2 and NC-
p l from p6, and the mal cleavage liberates p2 from CA (Gottlinger et al., 1989;
Ttitch et al., 1991 ; Pettit et al.. 1994; Krausslich et al., 1995; Reicin et al.. 1995;
Weigers et al., 1998). Processing studies in Gag-Pol have revealed that
protease autocatalysis absolutely requires the first cleavage to occur at the N-
terminus of PR, liberating it from p6'. This is followed by processing between PR
and RT, while the cleavage berneen FIT and IN is a rather late event during
Gag-Pol processing (Debouck et al.. 1 990; Louis et al.. 1991 and 7 994; Partin et
al., 1991; Pettit et al.. 1991; Poorman et al.. 1991; Lindhofer et al.. 1995;
Luukkonen et al., 1995).
DetaiIed analysis of multiple protease cleavage sites has revealed no
simple consensus sequence for PR, but common sequence motifs exist (Loeb et
al., 1989a,b; Partin et al., IWO; Pettit et al., 1991; Chou, 1993; Dunn et al.,
1994; Milder el al., 1994). The fact that the cleavage sites are not identical
renders some sites 'optimar and others 'su boptimal", whereby the cleavage
rates between sites h vitro can Vary by as much as 400-fold (Pettit et al.. 1994).
It is thought that the suboptimal sites have evolved because it is important that
they are cleaved later, ensuring that precursor processing is controlled and
follows a specifc order (Vogt, 1996). Sequential processing of Gag and Gag-Pol
results in discrete intenediates mat appear transiently before the final
products. The conformation of these intermediates likely regulate PR activity by
modifying the accessibility of the remaining cleavage sites (Pettit et al., 1994;
tindhofer et al., 1995; Wiegers et al., 1998). Furthemore, alteration of the order
of cleavage, either by preventing cleavage at one site or by replacing a
cleavage site sequence with that from another site. drastically affects virion
maturation and morphogenesis. presumably by interfering with the formation of an intermediate (Gottlinger et al.. 1989; Tritch et al., 1997; Pettit et al., 1994;
Krausslich et al.. 1995; Xiang et aL, 1997; Wiegers et al.. 1998). ln faa, mutation

at the CA-p2 region produced particles with decreased infectivity despite the presence of the proper final products (Pettit et al.. 1994; Krausslich et al., 1995).
Thus, the differential recognition of precursor cleavage sites by PR, based on primary sequence as well as structural context, appears to be
responsible for the different rates of processing observed for the specifk sites in
the Gag and GagPol precursors. This, in tum. regulates PR activity during
maturation (Erickson-Viitanen et al., 1989; Gowda et al., 1989. Krausslich et al.,
1989; Loeb at al., 1989a; Partin et al., 1990; Pettit et al., 1991 and 1994; Tritch
et al., 1991 ; Goobar-Larsson et al., 1995).

FIGURE 1-5. Schematlc representatlon of cleavage sites for HIV-1
protease withln the Gag and Gag-Pol precursors.

MA * CA SER GLN ASN TYR * PRO ILE VAL GLN CA * p2 ALA ARG VAL LEU * ALA GLU ALA MET p2 * NC ALA THR I L E MET * I L E GLN LYS GLY NC * pl ARG GLN ALA ASN * PHE LEU GLY ARG
PI * ~6 PRO GLY ASN PHE * LEU GLN SER ARG * PR SER PHE SER PHE * PRO GLN ILE THR
PR * RT THR LEU ASN PHE * PRO ILE SER PRO RT51* RNase H ALA GLU THR PHE * TYR VAL ASP GLY
RT * IN ARG LYS VAL LEU * PHE LEU ASP GLY

1.4.1.4 Regulators of PR within Gag and Gag-Pol The actMty of the PR domain, when embedded in Gag-Pol, is different than that
of the mature enzyme, in that its complete activation requires its Iiberation from aie precursor (Vogt, 1996). It will be telling to observe the conformation of PR
wîthin Gag-Pol once the crystal structure of the latter is detenined, and to map
structural elements in the precursor which enhance or constrain PR activity.
Although these conformational deteminants are undefined at this tirne, it is
clear that specific regions both upstream and downstream of PR in Gag and
Gag-Pol influence and reg ulate PR autoprocessing. and thus contribute to
budding of virions from the cell surface (Gottlinger et al., 1991; PaRin et al.,
1991 ; Pettit et al., 1994; Zybarth and Carter, 1995. Barrie et al., 1996; Bukovsky
and Gottlinger, 1996).
An important negative regulator of PR activity is the pokencoded p6'
domain, located proximal to the amino-terminus of PR. it is thought to regulate
PR activity in a manne? analogous to that of a zymogen conversion. Thus, p6'
may repress PR activity while in precursor form, presumably by looping outside
of the polyprotein molecular surface and reducing dimerkation of the PR
domain while embedded în Gag-Pol. Precursor dimerization during assernbly
alters the structural constraints and cleavage in the p6' region occurs. This, in
tum, removes the block limiting overall processing, and allows efficient PR
maturation followed by the cascade of proteolytic events (Zybarth et al., 1994).
This mechanisrn is supported by the observation that rernoval of the p6' region
leads to enhanced Gag polyprotein processing in an in vitro translation system.
and suggests that p 6 negatively regulates PR activation (Partin et al., 1991).
This domain has also been shown to inhibit PR activity h vivo, supporting the
notion that the proteins adjacent to andlot sunounding PR are crîtical for
maintabhg appropriate leveis of repression of PR activity in the cytoplasm
(Zybarth and Carter. 1995; Gatlin et al. 1998). Furthemore, the Gag p6 domain
also functions as a negative regulator of PR activity. and its removal from the C-
terminus of Gag results in reduced virion production (Gottbger et aL, 1991;
Huang et al. 1995).

Nucieocapsid has been shown to positively regulate protease activity in
ASLV and HIV-1, by promoting efncient PR dirnerization (Stewart and Vogt,
1993; Zybarth and Carter, 1995; Sellos-Moura and Vogt, 1996). Interestingly.
extended PR species contahing p 6 and a truncated NC domain failed to
dimerize and did not exhibit PR activity, suggesting an interplay between
negatively and positiveiy acting elements upstream of the PR domain (Zybarth
and Carter, 1 995).
Various regions in Gag have been implicated in mediating the
cytoplasrnic regulation of PR activity. For example, CA sequences were found to
repress the intracellulai activity of PR in trans, independent of carboxy-terminal
NC sequences (Gatlin et al., 1998). Other mutations in Gag that prevented
protein-protein interactions required for Gag-Pol dirnerization (particularly the I
domain) disrupted protease activation, and thus interfered with polyprotein
processing and virion maturation (Luban et al., 1993b; Xiang et al., 1997; Gatlin
et al., 1998). It is thought that the GaglGag-Pol interactions which are crucial for
PR regulation are dependent upon both specific sequences and appropriate
conformations of the precursor molecules.
Several pot domains within the Gag-Pol precursor are required for
optimal protease activation and partick maturation (Ross et al., 1991; Park and
Morrow, 1993; Quil lent et al., 1 993 and 1 996; Shin et al., 1 994; Engelman et al.,
1995; Bukovsky and Gottlinger. 1996). Reverse transcriptase is a positive
regulator of PR activity (Hu et al., 1990; Karacostas et al., 1993; Goobar-Lanson
et al., 1995; AnsariLari and Gibbs, 1996). It has been suggested that RT
promotes PR dimerkation and activation (Navia and McKeever, 1990; Zhang et
ai., 1991; Karacostas et al., 1993; Kumic, 1993; Darke et al.. 1994; Quillent et
al., 1996), while other studies have demonstrated that RT enhances PR activity
by increasing the catalytic efficiency of the PR dimer as well as its substrate
specificity (G~oba~Larsson et al., 1995 and 1996; Luukkonen et al., 1995).
Protease activity is suppressed by integrase, albeit only in the presence
of K i sequences (Shin et al., 1994; Engelman et al.. 1995; Bukovsky and
Gottlinger. 1996). Rernoval of the IN domain results in aberrant proteolysis by
PR and reductions in vira1 particle yield (Bukovsky and GottIinger, 1996). The

defects in particle production caused by deietion of integrase (or Gag p6) can be corrected by mutational inactivation of PR, indicating that IN is a negative
regulator of protease activity (Huang et al., 1995; Bukovsky and Gottlinger,
1 996).
In addition, regions within Gag-Pol modulate PR activity by providing an
appropriate conformational conte* For example, mutant HIV-1 particles lacking
R i or IN displayed global processing defects and were rnorphologically
immature, despite the presence of the entire PR domain (Quillent et al., 1893).
Similarly, virions containing various deletions in either Ri, RNaseH, or IN were
noninfectious and possessed aberrant morphology, consistent with defects in
precursor processing (Quillent et al., 1996). The severity of the alterations in
particle morphology were proportions[ to the extent of the deletion, illustrating
the important role of Gag-Pol structural detenninants in regulating PR activity
(Quillent et al., 1996). It is possible that these pol domain deletions alter
protease activity by changing the conformation of Gag-Pol, in effect modifying
the ability of PR to dimerize or cleave itself out of the polyptotein. Alternatively,
they could directly affect dimerization of the Gag-Pol precursor through the
removal of dimerization domains in F T or IN (Goff, 1990; Jones et aL, 1992;
Quillent et al., 1996). It has been suggested that the RT dornain of Gag-Pol,
because of its ske and ability to dimerize, may contribute to the formation of the
protease dimer and thus participate in the activation of PR (Navia and
McKeever, 1990). Taken together, several domains in polmust be kept intact for
optimal processing of Gag by PR,
Furthemiore, it has been reported that the sequences directly upstream
of the protease domain (eight amino acids on the 3' side of the N-terminal
cleavage site) postively regtilate the recovery of HIV-1 PR activity in vitro (Debouck et al.. 1990; Wan et al., t 996). In contrast. peptides that mimic both
the N- and C-terminal regions of PR were found to negatively regulate enzyme
activity, although the C-terminal peptide inhibited PR dimerization to a greater edent than the N-terminal peptide (ïhang et al.. 1991; Babe et al.. 1992).

1.4=2 The effects of PR cleavage site mutations 1.4.2.1 N-tetmf na1 mutations
The molecular rnechanisms leading to PR activation and autocataiytic release
are cumntly unknown. Although both the amino- and carbory- terminal amino
acids flanking protease are critical for its structure and function, previous work
analyzing the effect of cleavage site mutations on PR activity has focused
primarily at the N-terminus of protease.
It has been proposed that HIV-1 protease, while embedded in Gag-Pol,
is similar tu a rymogen in that proteoIytic removal of its N-terminal extensions
are required for activation of full enzymatic activity (Vogt, 1996). The 56
residues encoded by the pot open reading frarne upstream of the PR region
(p6') have not been ascribed a specific function and are in a position
corresponding to that of the prosegment observed in other aspartic proteases.
Based on this analogy, it has been suggested that p6+ may serve to regulate PR
activity in a manner analogous to that of a zymogen conversion and that
autocatalytic release of PR from p6* may be a triggering event in HIV
polyprotein processing (Partin et al., 1991; Phylip et al., 1992; Louis et al.. 1994;
Lindhofer et al., 1995).
lndeed, kinetic and mutational analysis have shown that the first and
most important cleavage during the autocatalysis of PR occurs at its N-terminus.
In HIV-1, this occun between p6' and PR in Gag-Pol (Debouck et al., 1990;
Pettit et al., 1991; Poorman et al.. 1991; Louis et al., 1991 and 1994; Luukkonen
et al.. 1995; Wondrak et al.. 1996; Tessmer and Krausslich, 1998). while in
avian retroviwses this occurs between NC and PR in the Gag precursor
(Burstein et al., 1992; Xiang et al.. 1997). Processing of a mode! precursor
protein ri, vitro revealed that cleavage at the N-terminus of PR is an
intramolecular reaction of the dimeric protein and follows fint-order kinetics,
whereas cleavage at its Gterminus is second order in protein concentration;
this provides a mechanistic explanation for PR autocatalysis (Louis et ai., 1994;
Wondrak et a!.. 1996).
Findings regarding the activity of protease in the cmtext of a precursor
form are controversial, and are highIy dependent on the system used. When

expressed in E. coli, the N-terminal release of HIV-1 PR is not a prerequisite for
its activity in most cases. That is, N-terminal cleavage site mutations which yield
pSPR, Gag-PR, or NC-PR fusion proteins are enzymatically active, and can
accurately process exogenous or natural substrates (Loeb et al., 1989a; Kotler
et al., 1992; Co et al.. 1994; Zybarth et al., 1994; Almog et al., 1996). Sirnilarly,
PR fused to nonviral proteins such as maltose binding protein (MBP) or p-
galactosidase @-gai) often remains active and can undergo autocatalysis
(Giam and Boros, 1988; Boutelje et al., 1990; Phylip et al.. 1992; Valverde et al., 1 992).
In contrast, other reports have found that some N-terminally extended PR
species produced in E. coli are enzymatically inactive in the absence of P R
rnediated cleavage. Hence, proteolytic activity was dependent on release of PR
from the fusion protein (Debouck et al., 1990; Loeb et al., 1989a; Louis et al.,
1991 and 1994). For example. PR fused to MBP was poorly active in an
exogenous proteolysis assay. but specific activity increased by over 100-fold
following self cleavage at its N-terminus (Louis et al., 1 994). This demonstrates
that N-terminal extensions of PR cause a decrease in autocatalytic release of
PR. Although some mutations at the N-temiinus of PR did not prevent
processing at other sites in Gag-Pol (Loeb et al., 1989a), others inhibited
cleavage at the C-terminus of PR and. in addition. significantly reduced global
PR activity (Loeb et al.. l989a, Debouck et al.. 1990; Louis et al., 1991).
Furthemore, other studies have shown that recovery of PR activity in bacterial
systems requires upstream sequences and the generation of a precise N-
terminus (Debouck et al.. 1990; Wan et al.. 1996). ln addition, although ASLV
NC-PR fusion proteins expressed in bacteria were devoid of detectable activity
(Sellos-Moura and Vogt, 1996). this fusion protein was active when genetated
as part of the Gag precursor (Arad et al., 1995). Interpretation of all results with
purifieci PRs expressed in bacteria are made difficuit by the need to denature
and renature- the protein, due to its insolubility or tendency to aggregate (Vogt,
1996). Thus, it is impossible to draw precise conclusions about the effect of N-
terminal PR extensions on enzyme activity fol1owing bacterial expression.

When expressed in an h vitro transcription-translation processing
systern, polyproteins consisting of only p6' and PR domains, or containing a
truncated NC region, exhibited no autoprocessing activity (Zybarth and Carter,
1995). Thus, in this systern, protease must fnst be released from its precursor
before it can initiate cleavage at other sites.
Similarly, virions €rom avian retroviruses that contain a cleavage site
mutation between NC and PR lacked protease activity and thus were unable to
process precursors (Oertle and Spahr, 1990; Buntein et al., 1992; Stewart and
Vogt, 1994; Schatz et al., 1997; Xiang et al., 1997). These particles did not
mature in COS cells and displayed 30-fold decreased RT activity compared to
wild-type (Stewart and Vogt, 1994). Interestingly, some of the mutant constructs
were not efficient at blocking cleavage even though they harbored strictly-
excluded pbranched amino acids at the P l site. Rather, precursor proteins in
these virions underwent m iscleavage at the N-terminus of protease, illustrating
the importance of cleavage at this site (Stewart and Vogt, 1994; Schatz et al.,
1997). These findings suggest that generation of a non-modified amino-
terminus of mature ASLV and RSV PR is required for protease activation in vivo,
and proteolytic liberation of the PR domain is essential for its activity (Burstein et
al., 1992; Stewart and Vogt, 1993 and 1994; Schatz et al., 1997; Xiang et al.,
1997 ).
In HIV-1. N-terminal cleavage of PR in vivo is absolutely required for
efficient Gag precursor processing and for viral infectivity (Zybarth et al., 1994;
Tessmer and Krausslich, 1998). Although one report suggested that p61PR
may be the predorninant form of active protease in mature HIV-1 particles
(Almog et al., 1996), other results indicated that this intermediate is not sufficient
to achieve complete proteolysis of precursor proteins during virion maturation
(Louis et ai., 1994; Zybarth et al., 1994; Lindhofer et ai.. 1995; Tessmer and
Krausslich. 1998). Interestingiy, while the p6-PR cleavage site mutation caused
a severe reduction in Gag polyprotein processing. cleavage at sites in the Pol
domain of the Gag-Pol polyprotein (i.e. the PRRT. RT-RNaseH, and RFIN
cleavage sites) were virtually unaffected messmer and Krausslich. 1998). The
fact that the N-terminal cleavage site mutation compteteIy prevented processing

at this site, yet did not affect cleavage at sites in the Pol domain of Gag-Pol, is in
agreement with findings in bacteria! systems in which some mutations at the N-
teminus of PR did not block creavage at other sites (although other N-teminal
mutations did inhibit cleavage at the Gteminus of PR as well as reduce global
PR activity) (Loeb et al., 1989a, Debouck et aL, 1990; Louis et al., 1991). Taken
together, it appears that N-terminal cleavage site mutations in vivo do not
completely prevent processing at other sites, but N-terminal release of PR is required for efficient Gag processing and for viral infectivity.
Not surprisingly, the N- and Co terminal regions of protease appear to be
interdependent, whereby sequences at one cleavage site can influence the
efficiency of cleavage at the other site (Louis et al., 1991). In this way, cleavage
at the first site (N-terminus) induces a conformational change in protease and
allows cleavage at the second site (C-terminus) to proceed; conversely, the lack
of cleavage at the first site fails to induce the conformational change in PR
thereby preventing processing at the other site (Louis et al., 1991 and 1994;
Wondrak et al.. 1996). This explains the demonstration that the N-terminal
cleavage of PR is required for subsequent Gag polyprotein processing and for
viral infectivity (Burstein et al., 1992; Stewart and Vogt. 1994; Zybarth et al.,
1994; Schatz et al.. 1997; Xiang et al., 1997; Tessmer and Krausslich, 1998).
1 A.2.2 C-terminal mutations Because cleavage at the N-terminus of PR occurs fint and is critical for
subsequent precursor processing, the effects of cleavage site mutations
between protease and reverse transcriptase have not been wett-studied. Until
now, HIV-1 PR-RT fusion proteins have only been analyzed in bacterial
systems. not in vivo (LeGrice et al., 1988b; Debouck et al., 1990; Louis et al.,
1991). Various mutations at and surroundhg the scissiie bond between PR and
RT have been studied to understand the sequence requirements that would
prevent processing at that site. The effects of the mutations ranged from being
ineffective, to allowing partial processing, and to cornplete[y blocking cleavage
at Viat site (Loeb et al., t989a; Debouck et al., $990; Louis et al., 1991). The
activity of the PR domain within the PR-FIT cleavage site mutants varied; in

some cases, processing at other sites in Gag or in vitro occuned efficiently,
m i le in other instances PR activity was diminished (Loeb et al., 1989a;
Debouck et al., 1990; Louis et al., 1991). Other mutations at the C-tennhus of
PR did not affect the enzyme's proteolytic capabilities. provided that the wild-
type site was preserved at the N-terminus (Debouck et al.. 1990; Louis et ai.,
1991). Interestingly, RT activity in these PR-Ki cleavage site mutants was not
assessed Ïn these reports (Loeb et al., 1989a; Debouck et al., 1990; Louis et al..
1991). A study analyzing the kinetics of PR autocatalysis demonstrated that the
addition of C-terminal flanking sequences to PR did not distort the active site of
the enzyme, and thus PR remained catalytically active (Wondrak et al.. 1996).
Analysis of a PR-RT cleavage site mutation in ASLV in vivo revealed that
the extended species retained their RT activity, yet the processing capabilities of
the PR domain was not studied (Stewart and Vogt. 1994). Other studies in RSV
have demonstrated that the C-teminus of PR must be unrestrained for optimal
activw in vivo, and that sequences fused to it are inhibitory to PR activity
(Bennett et al., 1991). However, HIV-1 differs from avian retroviruses in many
ways, particularly in that the PR domain in ASLV is encoded both at the C-
terminus of Gag as well as within Gag-Pot. The PR domain in Gag is sufficient
for polyprotein processing; however, ASLV PR is 10-fold less active than HIV-1
PR (due to differences in the enzymes' active sites), it is 20-fold more abundant
than HIV-1 PR. and it must be fully released from the precursor for activity
(Bennett et al., 1991 ; Craven et al., 199 1 ; Stewart and Vogt, 1991, 1993 and
1 994).
Taken together, it is difficult to draw conclusions regarding the effects of a
cleavage site mutation between PR and AT. ln the case of HIV-1. this problem
has onty been studied in bacterial systems and the results are inconclusive.
From extensive studies of N-terminal deavage site mutations, it is cfear that numerous discrepancies exist between reports that emplayed N, vitro venus in
vivo approaches. Furthemore, it is inappropriate to dkectly compare the effect
of PR-RT cleavage site mutations in ASLV and HIV-1. as their genetic
organization is different, particularIy in regard to protease expression and
regulation.

1.4.3 Regulation of RT activity The influence of the Gag and Gag-Po[ domains on reverse transcriptase activity
is corn ptex and not fut ly understood. In ASLV, it has been dernonstrated that RT
activity depends on the size of each of the Gag and Pol domains, and whether
or not PR is mutated (Stewart and Vogt, 1993). Gag domains inhibitory to RT
activity have not been fully elucidated, and tharefore, it is not well-understood
which cleavages are necessary for the activation of M (Stewart and Vogt,
1991). It has been shown, however, that integrase is a positive regulator of AT
activity. For example, the inhibition of RT activation by partial upstream Gag and
downstream Pol sequences can be alleviated by the presence of a cornplete
Pol domain (Stewart and Vogt, 1993; Shh et al.. 1994; Engleman et al., 1995).
1.4.4 The relationship between PR and RT Although both HlV-1 protease and reverse transcriptase are critical for
successful virus replication, their relationship is only partially understood. It is
clear that both proteins are important in optirniring the other's activity, however
characterization of the complex interactions between PR and RT is incomplete.
1.4.4.1 The effect of RT on PR activity Studies analyzing the effect of reverse transcriptase on protease activity
indicate that RT enhances PR proteolytic activity. The first evidence that
sequences downstream of protease affect its activity is that virions containing
deleted pol sequences outside the PR domain display reduced proteolytic
capabilities (Hu et ai., 1990; Karacostas et al., 1993; Ansari-Lari and Gibbs.
1996; Quillent et al., 1996). For example, protease within a Gag-PR precursor
processed Gag less efficiently than PR within Gag-PR-RT or Gag-Pol (Hu et al.,
1990), and various RT deletions result în diminished PR activity (Karacostas et al., 1993; AnsariLari and Gibbs, 1996; Quillent et al., 1996). Second, the block
in virus assembly caused by the overexpression of the Gag-Pol polyprotein,
resulting in increased intracelluiar protease activity, was partially overwme by
removal of KT (Karacostas et ai-, ? 993). Third. purified HIV-1 #r was found to

increase the activity of HIV-1 protease h vitro and in eokaryotic cells. in a dose-
dependent manner independent of pH or salt (Goobar-Larsson et al., 1995).
The fact that maximal enhancement was dependent on the
concentrations of both protease and reverse transcriptase suggests direct
protein-protein interactions between the two proteins (Goo bar-Larsson et al.,
1995). Direct interactions between PR and FIT was shown in one report, in
which purified PR was found to inhibit purified K i DNA synthesizing activty in
vitro (Bottcher and Grosse, 1 997).
A number of studies have proposed that pol sequences present in cis
enhance PR activity, and more specifically, that RT promotes protease
homodimerization (Navia and McKeever, 1990; Zhang et al., 1991 ; Karacostas
et al., 1993; Kuzmic, 1993; Darke et al., 1994; Goobar-Larsson et al., 1995;
Luukkonen et al., 1995; Quillent et al., 1996). However, other results
demonstrated that RT only marginally affects PR dimer formation andlor stability
in vitro (Goobar-Larsson et al., 1996). And, dimerization of a PR-RT fusion
protein does not appear to occur more efficiently than dimerization of PR alone
(Luukkonen et al., 1995).
lt has been proposed that the main effect of FIT on PR activity is not
facilitating PR dimerization, but increasing the catalytic efficiency of the PR
dimer and its affinity for the substrate (Goobar-Larsson et al., 1996). Indeed, RT has been shown to influence the cleavage efficiency of protease by improving
its substrate specificity in vitro and h vivo (Goobar-Larsson et al., 1995;
Luukkonen et al.. 1995). All testable cleavage sites were found to be more
eficientry processed in vitro in the presence of RT, indicating that interacts
with PR itself rather than with the substrate (Goobar-Larsson et al., 1995). The
presence of RT affected different peptide substrates to varying degrees, having
the greatest effect on the cleavage efkiency at the RT-IN junction, and the least
influence at PR-RT (Goobar-Lanson et al., 1995). In this way, it is thought that in VIVO, RT may regulate the activity of the PR dimer, rather than contribute to its activation by promoting dimer formation (Goobar-Larsson et al.. 1996). faken
together, however, it is likely that mature FtT enhances PR a c t M i by improving

activation by promoting dimer formation (GoobaFLarsson et al., 1906). Taken
toget her. however. it is likely that mature RT en hances PR activity by im p roving
its cataIytic efficiency, while RT ernbedded in Gag-Pol promotes precursor. and #us PR, dimerization.
1 A.4.2 The effect of PR on RT acüvity 1.4.4.2. r ~ n c t i v i t y in G B ~ P O I
Proteolytic maturation of the Gag-Pol precursor is not necessary for activation of
reverse transcriptase in mammalian systems. Gag-Pol possesses RT act ivity,
although the levels are considerably lower than for the mature RT (Peng et al.,
1990 and 1991; Hu and Kang, 1991; Stewart and Vogt, 1991 and 1993;
Mergener et al., 1992). Reverse transcriptase activity has been demonstrated in
unprocessed Gag-Pol in ASLV (Stewart et al., 1990; Stewart and Vogt, 1991
and 1993). Mo-MLV (Cmwford and Goff, 1985), and HIV-1 (Lori et al.. 1988; Hu
and Kang, 1991; Peng et ai.. 1991 ; Mergener et al., 1992).
In contrast, the Gag-Pol precursor protein expressad in E.coli has no
reverse transcriptase activity, suggesting that RT must be processed to acquire
enzymatic activity. (Leuthardt and LeGrice. 1988; LeGrice et al.. 1988b; Tanese
et al., 1988). Proteolytic removal of both the PR and IN domains are necessary
for full activation of RT in this system (Leuthardt and LeGrice, 1988; LeGrice et
al.. 1988b and 1989).
1.4.4.2.2 RT ectiviiy in PR-defective viruses In an E coli expression system, protease containhg an active-site mutation
(D25A) did not process Pol polyproteins, and these unprocessed precursors
displayed insignificant RT activity (LeGrice et a[-, 1988b). The same results
were shown in a PR-defective-RT fusion protein (LeGrice et ai., l988b).
However, wild-type protease supplied in tmns restored both processing and
reverse transcnptase activity in both constructs (LeGrice et al., 198813).
immature ASLV particles assembled from poIyprotein precursors with
defectke protease (0371) have redrrced but detecta ble RT act ivity (Oertle and
Spahr, 1990; Stewart et ai.. 1990; Crawn et ai., 1997; Stewart and Vogt. 1991

and 1993). Although the addition of wild-type protease in tram leads to a five-
fold increase in RT activity, the activation of reverse transcriptase is incomplete
and levels do not reach those of wild-type (Stewart et al.. 1990; Stewart and
Vogt, 1991 ). Unprocessed ASLV Gag-Po[ fusion proteins display RT activity
only when the protease domain is not mutated (Stewart and Vogt, 1993).
Furthemore, in both Mo-MLV and RSV, RT was enrymatically active only after
PR-mediated cleavage and release from the Gag-Pol polyprotein (Witte and
Baltimore, 1978; Craven et al., 199 1 ). Cleavage and reverse transcriptase
activation were restored by wild-type protease supplied in Pans (Craven et al.,
1991). These observations led to the hypothesis that RT is activated by its
release from the Gag-Pol precursot in avian retroviruses (Craven et al.. 1991 ;
Stewart and Vogt, 1993 and 1994).
In contrast, protease-defective mutant Mo-MLV and HIV-1 particles
display nearly normal levels of reverse transcriptase activity (Crawford and Goff,
1985; Katoh et al.. 1985; Peng et al., 1991). Transfection of HIV-1 proviral or subviral genomes containing inactive protease (D25A) resulted in the
production of noninfectious immature virus particles displaying low but
significant RT activity (Peng et al., 1991; Mergener et al., 1992). The levels of
RT activity in PR-defective particfes were found to be reduced by two- to ten-fold
(Gottlinger et al., 1989; Peng et al., 1991 ; Mergener et al., 1992; Park and Morrow, 1992).
1 .S THERAPEUTIC APPROACHES FOR TREATMEM OF HIV-1
Current antiretroviral therapies combining inhibitors of HIV-1 protease and
reverse transcriptase (highly active antiretroviral therapy [HAARTJ) are h ig h ly
efficient at diminishirtg viral replication and increasing CD& T-cell numbers.
resulting in a dramatic reduction in AlDS-related morbidity and mortality
(Markowitz et al.. 1995; Collier et al.. 1996; Harnmer et al., 1997; Palella et al.,
1998). However, viral eradication does not appear to be achievable with these
strategies, and long-term outcornes with respect to viral load. development of
multiply resistant virus. or disease status have not yet been fully ascertained.

1.5.1 Gene therapy strategies The challenges associated with traditional antiretroviral approaches have
caused attention to tum to the potential utility of genetic therapies for treatment
of HIV-1 infection. This is an attractive alternative. primarily because
antiretroviral therapy may need to be sustained for the life of the individual,
while gene therapy approaches might confer long-tenn antiviral effeds
(Poeschla et al.. 1 996). Gene therapy, or 'intracellular immunization', refers to
the genetic modification of cells - via transferring a Vierapeutic gene into target
cells - in order to render them resistant €0 HIV-1 replication. Infusion of cells
protected by in vitro gene therapy into HIV-1 -infected individuals should prevent
de novo infection of susceptible cells as well as suppress ongoing replication in
infected cells, thereby Iimiting virus spread and delaying disease progression
(Baltimore, 1988; Pomerantz and Trono, 1995).
Different inhibition strategies based on intracellular immunization include
both RNA-based and protein-based approaches. A variety of anti-HIV-1 genes
that have been developed and show to inhibit virus replication include
antisense RNA constructs (Rhodes and James, 1990; Sczakiel et al.. 1990;
Joshi et al., 1991; Meyer et al., 1993; Coli et al., 1994; Veres et al., 1996 and
1998). catalytic ribozymes (Sarver et aL, 1990; Ojwang et al., 1992; Zhou et al..
1994b and 1996), single-chain antibodies (Duan et al., 1994; Shaheen et al..
1 W6), HIV-1 -inducible cytokines (Viellard et al., 1994; Su et al., 1995) or toxins
(Curiel et al.. 1993), and RNA decoys which bind Tat (Çullenger et al., 1990) or
Rev (Lee et al., 1992 and 1994; Bahner et al., 1996).
A promising gene therapy approach to specificaliy compromise HIV-1
teplication appears to be the use of trans-dominant negative (or dominant
negative) mutants. These defective viral proteins. which can associate with and
inactivate the function of the wild-type gene products A vivo, have been
proposed for a number of HIV-1 proteins (Herskowk 1987; Baltimore. 1988;
Feinberg and Trono, 1992). Indeed, dominant-negative in hibition of virus
replication has been demonstrated for varÎants of Ta€ (Green et al.. 1989;
Pearson et al., 1990; Modesti et al.. 1991; Bahner et al.. 1993; Orsini and
Debouck, 1996; Ulich et al., 1 9Q6), Rev (Malim et al., 1 989b and f 992; Bevec et

al., 1992; Bahner et a1.. 1993; Escaich et ai., 1995). Gag (Trono et al., 1989;
Smyth et al., 1994; Lee and Linial, t995), and Env (Freed et al., 1992;
Buchschacher et al., 1995; Chen et al., 1996). Promising results have been
obtained with a number of these mutant proteins rii vivo. Moreover, early results
from a clinical trial in which patients received genetically modified peripheral
blood Iymphocytes (PBLs) carrying a trans-dominant Rev gene (RevM1 0) show
evidence of effectiveness, and also demonstrate that gene therapy for AIDS can
be safely approached in humans (Nabel et al., 1994; Woffendin et al., 1996;
Raoga et al., 1998).
1.5.2 HlV-1 PR as a target for gene therapy A gene therapy strategy for HIV-1 infection may be more effective at blocking a
step of the viral life cycle that involves a catalytic function such as proteolytic
processing. The obligate homodimeric nature of HIV-1 PR and its absolute
requirernent for viral maturation make it a good target for such an approach.
The use of defective PR monomers as inhibitors of protease function is
an altemate strategy to the use of small-molecule PR inhibitors. Active site-
directed protease in hibitors rel y on a few hig h-affinity interactions wth PR
Consequently, mutations within the PR gene that confer resistance to PR
inhibitors commonly arise (El-Fairash et al.. 1994; Ho et al., 1994; Wei et al., 1995). In contrast, interactions between the two protease su bunits involve many
residues. Thus, single mutations that preserve protease activity but prevent
dimerization with a trans-dominant defective monomer are unlikely to occur,
and trans-dominant inhibitors may be less prone to the emergence of
resistance-conferring mutations (Babe et al., 199 1 and 1995).
Inhibition of HIV-1 replication by intracellular expression of dominant
negative protease variants is an effective anti-HIV strategy. Constitutive
expression of trans-dominant PR inhibitors can efficiently suppress HIV-1
replication in vivo (Junker et al*, 1 996; McPhee et al., 1996). Trans-dom inant
inhibition of PR has been dernonstrated in two ways: (1) by inhibithg PR activity
via an active-site mutation which results in the formation of inactive PR
heterodimers (Atrigo and Huffman, 1995; Babe et aL. 1995; Junker et al., 1996;

McPhee et al.. 1996). and (2) by interfering with PR dimerization (Babe et al.,
1991 and 1992; Zhang et al., 1991). ln the former case, stable expression of
defective PR in cells reduced the yield of infectious particles by 90% following
transfection with wild-type proviral DNA (Babe et ai., 1995). In addition to the
inhibition of viral replication by dominant negative PR molecules provided in cis,
HIV-1 PR also functioned independently as an antiviral gene when expressed in tram (Amgo and Huffman, 1995).
The requirement for PR to dirnerize provides an alternative mechanism
for the inhibition of enzyme activity to active site-directed inactivation. Inhibition
of PR by the disruption of homodimer formation has been demonstrated in both
HIV-1 and HIV-2 (Babe et al., 1991 and 1992). In vitro incubation of wild-type
PR with a dimerization-defective PR mutant resulted in the production of
catalytically defective heterodimers which displayed 80% in hibition of
enzymatic activity (Babe et al., 1991). Dimerization can also be inhibited by
peptides that rnimic the N- and C-tennini of PR; these effectively block the
dimerization interface and prevent the formation of the active PR dimer (Zhang
et al., 199 1; Babe et al., 1992). Most notably, the C-terminal tetrapeptide of PR
was fomd to be an excellent dissociative inhibitor of its dimerization, and thus a
potent inhibitor of PR activation (Zhang et al., 1991 ; Babe et ai., 1992).
Thus, the PR dimer interface is an attractive site for antiproteolytic
intervention based on gene therapy approaches for several reasons. Mt. dimerization of the protease polypeptides is essential for activity and is also an
initial posttranslational step. Second, the extended interface created by the N-
and C-termini of PR is consenred and may be less vulnerable to mutational
escape than the active site or the substrate binding region of the protease.
Third, dissociative inhibitors bind to the protometic fom of PR that is present in
the polyprotein precursors. Accumulation of the poiyprotein-inhibitor complex in
the budding virion might concentrate the dissociative inhibitor at that location,
even before processing of the polyprotein can begin. ln contrast, inhibitors that
target the active site of the dimeric enzyme can onIy bind after that active site
has fomed by dimerization (Babe et al., 1991 and 1992; Zhang et al., 1991).

Based on the published literature, it would be niteresthg to detemine if the
cleavage site between HIV-t PR and FIT might be a target for a gene therapy
strategy against HIV-1 infection.

RATIONALE AND SPECIFIC OBJECTlVES
The global objective of this research was to characterire the relationship
between protease and reverse transcriptase in the context of full-length HIV-1 in
a relevant celCculture eukaryotic system. The first aim was to analyze the effects of a cleavage site mutation between PR and RTon both protease and reverse
transcriptase activities rii vivo. The second objective was to determine if and
how this PR-RT cieavage site mutation affected progeny virus particles with
respect to replication, assembly, maturation. and infectivity. The third goal was
to ascertain if the PR-RT fusion protein woutd possess a dominant negative
phenotype, and thereby interfere with normal virus function following
coexpresçion. This work demonstrated that white both HIV-1 PR and RT remain
enzymatically active when expressed as a fusion protein in vivo, viruses
containing the PR-RT fusion protein have diminished infectivity. Furthermore,
wild-type HIV-1 replication is suppressed by viruses containing the PR-RT
cleavage site mutation in a trans-dominant negative fashion. These studies
have identified novel HIV-1 dominant negative mutants which may have
usefulness when applied as a gene therapy approach for HIV-1 infection.

CHAPTER 2
MATERIALS AND METHOOS

The residue at the P l position in the cleavage site between PR and RT (Le. the
scissile bond is between the P l and Pl' amino acids) is always hydrophobic
and unbranched, and there is a notable exclusion of isoleucîne and valine at
this position. Additionally, a hydrophobic or uncharged polar residue always
occupies the P2' site, while charged amino acids, particularly lysine. are strictly
excfuded (Loeb et al., t989b; Debouck and Metcalf, 1990; Partin et al., 1990;
Pettit et al.. 1991: Dunn et al., 1994; Mildner et al., 1994). Based on these
considerations, we engineered our cleavage site mutation such that strictly
excluded residues replaced wild-type am ino acids, instead of more
consewative substitutions.
Figure 2-1 shows the construction of the mutant plasrnids. Briefly, the
Apal-Sall fragment (nucleotides 1 550 to 5366) from the infectious f ull-length
HIV-1 clone pBH1O was cloned into the eukaryotic expression vector pSVK3
(Pharmacia Biotech, Montreal, Canada). The resultant plasmid, pSVK3-
BH1 was then mutagenized using a Chameleon double-stranded site-
directed mutagenesis kit (Strategene, La Jolla, CA). To create the mutation
termed PR-RT, at the cleavage site between protease and reverse
transcriptase, the primer 5-GCACTTTAAATATTCCCAAGAGCCCTAmGAC-
3' (nucleotide positions 2160 to 2149) was used. The Pl and PZ' sites were
changed from Phe to IIe (F991) and from Ire to Lys (1101K), respectively, to
produce P 1F991P2'1 t 0 1 K. A mutation was introduced into the gag-pl
frameshift region to translocate the polgene into the gag reading frame in order
to improve the expression of the PR-RT fusion protein. The primer used to
create the frameshift readthrough mutant (FS) is S-GGCTAAllTiïTAAGGGA
AGATCTG-3' between nucleotides 1623 and 1646 (Park and Morrow, 1991 );
this resufîs in the overexpression of Gag-Pol in the context of full-length HIV-1.
FoIlowing site-directed mutagenesis and confnmation by double-strand
sequencing (Gibco BRL dsDNA Cycle Sequencing System, Life Technologies.
Mississauga. Canada), the mutant plasmids were digested with Apal and Sali
and cloned back into pBH 10 to yield two fulklength HIV-1 molecular clones

harboiing either the PR-nT cleavage site mutation alone @PR-Rq or in
combination with the fmeshift readthrough mutation (pFSIPR-RT). Large-scale proviral plasmid DNA was purifieci using Plasmid Maxi Kits (Qiagen tip-500,
Qiagen Inc. Chatsworth, CA) for use in othet studies.

FIGURE 2 Description of HIV-1 mutant cfones, derived from pBH10.
Construct pPR-RT contains amino acid substitutions (P1)F991 and (P231101 K at
the cleavage site between protease (PR) and reverse transcriptase (RT). Construct pFSPR-RT contains the same cleavage site mutations in combination
with a teadthrough insertion mutation at the frarneshift site (FS), in order to overexpress Gag-Pol in the context of fulklength HIV-1.

1 POL I
Frarneshift site Cleavage site
wild type Asn Phe Leu G l y
AAT TTT TTA GGG
PR-RT Cfeavaae site
Thr Leu A s n ~hei P r o Ile Ses P r o PBH~O 8
cleavage Asn Phe Leu G l y T h r Leu Asn 'I+ Pro Lsm Ser Pro pPR-Rf site
mutation AAT TTT TTA GGG ACT TTA AAT &TT CCC AGC CCT
-frarneshift Asn P h e Leu UQ Thr Leu Asn Ile Pro 4yL S e r Pro p ~ ~ / p ~ - ~ ~ + cfeavage AAT TTT TTA &X ACT TTA AAT &TT CCC A&& AGC CCT
site

COS-7 ceils, derived from simian virus (SV)4O-transformed African green
monkey kidney cells, were grown as described at 37°C under 5% CO2 as
monolayer cultures in Dulbecco's modified Eagle's medium (DMEM, containing
high glucose [4.5 g/L1 and with L-glutamine but without sodium pyruvate),
containhg 10% heat-inactivated fetal calf serum (FCS, Life Technologies,
Mississaoga, Canada), 100 U of penicillin G per ml. and 100 pg of streptomycin
per ml (Boulerice et al., 1990; Rooke et al.. 1990). Exponentially growing cells
were harvested by trypsinization and repiated at a density of 1 x ?O6 cells per
dish in 100 mm culture dishes, 24 hours before transfection. Wld-type and
mutant purified plasmid DNA (10 pg) were added to each dish using a standard
calcium phosphate (CaClJHepes-buffered saline (HBS) CO-precipitation
procedure (Maniatis et al., 1982), The cells were washed with phosphate-
buffered saline (PBS, pH 7.4) and replenished with fresh medium after
incubation for 16 hours at 37OC.
2.3 PUR~FICATION OF CELLULAR AND V ~ A L EXTRACTS FROM
TRANSFECTED OR [NFECTED CELLS
Cellular and viral extracts were harvested separately from transfected COS7
cells at approximately 48 hours after PBS wash and media replenishment (63
hours post-transfection, total time). Virus-containing culture supernatants were
clarified by centrifugation for 30 minutes at 4 O C at 3.000 rpm in a Beclanan OS-
6R centrifuge, then spun thtough a 20% (w/v) sucrose cushion for one hour at
4OC at 40,000 rpm in a Beckrnan SW41 rotor (Beckman Instruments, Inc., Palo
Alto, CA). Viral pellets were resuspended in one of two different ways: for
immunoblots (Western Mots), resuspension was in 100 pl NP-40-DOC lysis
buffer (20 mM Tris-HCI [pH 8-01, 120 mM NaCI, 2 mM EDTA, 0.5% deorycholate
[OOCJ, and 0.5% Nonidet P-40 [NP-401) containing 1.74 pglml
phenylmethylsuIfony1 f luoride (PMSF), 10 pghl apoprotein, and 10 Clglml

pepstatin A; for measurements of ih vitro cell-free W activity. resuspension was
in 100 pl PBS (Boulerice et al., 1990; Rooke et ai., 1990). With al1 samples, viral
capsid expression was detemined by p24 (capsid protein, CA) enzyme-linked
îmrnunosorption assay (ELISA; Abbott Laboratories, Abbott Park. IL) as
described (Boulerice et al., 1990; Rooke et al., 1990). Viral lysates were
aliquoted and stored at -20°C until use.
To prepare wholacell extracts. the transfected COS7 cells were washed
twice with icscold PBS, scraped from the culture dishes, and harvested by
centrifugation for five minutes at 1,000 rpm in a Beckman GS-6R rotor. The cell pellets were subjected to two rounds of washing and agitation. then
resuspended either in 100 pl PBS for detection of AT activity, or in 100 pl lysis
buffer for imrnunoblotting, in which case the lysed cells were kept for 30 minutes
on ice, followed by centrifugation of insoluble cellular material for 30 minutes at
12,500 rpm in a Beckman benchtop microfuge. Total protein content was
determined by the Bradford Protein Assay (Bio-Rad Laboratories Ltd.,
Mississauga, Canada) as previously described (Boulerice et al., 1990).
Cell lysates from MT02 infected cells were prepared as follows: MT-2
cells (1 x IO6) were infected with 100 ng of wild-type or 1000 ng of mutant COS-
? derived vinises. On day five post-infection, the infected cells were harvested
by centrifugation for five minutes at 1,000 rpm in a Beckman GS-GR rotor. After
two rounds of vigorous washing in PBS, the cell pellets were resuspended in
100 pl lysis buffer for immunoblotting.
Samples were standardized at 25 pg totai protein for cell lysates or 25 ng p24
for viral Iysates. These were fractionated on 12% (wlv) sodium dodecyf sulfate
(SDÇ)-polyacrylamide gels for 90 minutes at 125 volts, and transferred to
nitrocellulose filters for three hours at 200 mA and 90 volts, The filters were then
blocked with 5% (wh) skim miWO,OSX Tween-20IPBS at 4% for 16 hours.
Three different ptimary antibodies, diluted in 5% (w/v) skim milWBS, were

used an antCHlV p24 lgGl mAb, an antGHlV KT lgGl mAb, both used at
I:2.000 (ID h b s Inc, London, Canada), and a pool of sera from HlV-1
seropositive individuals at t :200 (Boulerice et al.. 1990). The filters were probed
with one of these primaiy antibodies for two hours at 37OC with constant
agitation, followed by extensive washing with 0.05% Tween-20/PBS,
Secondary antibodies were either sheep anti-mouse lgG, conjugated to
horseradish peroxidase (HRP; Amersham Life Science, Toronto, Canada) or
goat anti-human IgG-HRP (Bio-Rad Laboratones Ltd., Mississauga, Canada) at
12,000 (Boulerice et al., 1990). Proteins were visualized by the ECL chemi-
luminescence detection kit (Amersham Life Science, Toronto, Canada) and
exposed to Kodak X-Omat film (Eastman Kodak, Rochester. New York). In
certain experiments, purified recombinant nT, prepared as described (Quan et
al., 1996), served as a positive control.
2.5 VIRAL REPLICATION ASSAYS
2.5.1 In vitro RT and p24 assays The replication ability of the viruses produced from transfected or infected cells
was detemined by monitoring levels of in vitro celi-free RT activity and p24
capsid expression (Boulerice et al.. f9W; Rooke et al., 1990). For the FlT assay,
virus-containing culture fluids (50 pl) were added to 50 pl of polymerase
reaction mixture containing 50 mM Tris-HCI (pH 8.0). 150 mM KCI. 5 mM
dithiothreitol (DTT), 5 mM MgCk 0.3 rnM glutathione, 0.5 mM EDTA, 1% (v/v)
Triton X-100,50 pg of poly(rA)/oligo(dT),,, per ml, and 30 ci of [IHITTP per ml
(80 Cümmol). After incubation at 30°C for 22 hours. the reactions were stopped
by the addition of 2 ml of 10% (vlv) ice-cofd trkhloroacetic acid (TCA), incubated
for two hours at 4°C. and filtered onto Whatman GF/C filten. The filters, which
retained the precipitate containing incorporated pHllF. were then washed
mice with 2 ml of 1 0% (vfv) TCA, and counted for radioactivity in a Packard Tri-
Carb scintillation comte?. Viral p24 antigen expression was detemined by p24
enzyme-linked immunosorption assay (ELISA; Abbott Laboratories, Abbott
Park, IL).

2-59 Endogenous RT assays For endogenous RT reactions. clarified virus-containing culture fluids produced
from COS-? transfected cells were filtered Virough a 0.2 pm sterile membrane
prior to further purification by centrifugation for one hou? at 4OC at 35,000 rpm in
a Beckman SW41 rotor. Viral pellets were resuspended in 50 pl TNE (10 mM
Tris-HCI @H 7.41. 100 mM NaCI, 1 mM EDTA), and treated with 2 U of
Microccocal nuclease (Pharmacia Biotech, Montreal, Canada) for 20 minutes at
37OC. Endogenous FtT assays were modified from previously described
procedures (Quan et al., 1996) and were perfomed in a total volume of 50 pl
containing 50 mM Tris-HCI (pH 7.8). 5 mM MgC12, 60 mM KCI, 10 mM DIT, 10
mM NaCI, 1 mM ethylene glycol-bis (P-aminoethyl ether)-N, N, N; Wtetraacetic
acid (EGTA), 0.025% NP-40,0.4 mM each dATP, dGTP and dlTP, 10 pM dCTP,
10 pCi of [a-JaP]dCTP. purified virus particles normalized on the basis of p24
content (Le., 800 ng of p24, representing approximately 2 x 107 cpm of FtT
activity per ml per reaction), and variable concentrations of inhibitors. After 10
and 20 hours at 3g°C, reactions were terminated by addition of an equal
volume of stop buffer (1% SDS, 50 mM EDTA, 0.2 M NaCI). The reaction
mixture was digested with 20 pg of protease at 56OC for 30 minutes, and then
extracted with phenol-chloroform and chlorofon.
Following the endogenous K T reactions. products were then PCR-
amplified using specific primer pairs: the UPBS/AA5B1primer pair (153 bp) was
used to detect minus-strand Wang stop DNA (Li et ai., 1997); the PS/PA primer
pair (367 bp) was used for fulblength proviral DNA (Li et al., 1994). To ensure
that contaminating p lasmids from the transfection inocula were not PCR
amplified, COS-? cells were mock-transfected with pBHtO in the absence of
CaCI, and HBS. PCR assays were perfomed with 2 p1 of sarnple produced from
the endogenous FiT reactions, 50 mM Tris-HCI (pH 8.0). 50 mM KCI, 25 mM
MgC4, 2.5 U of Taq polymerase, 0.2 rnM deoxynucleoside triphosphates
(dNTPs). 10 pmol of "P-end-labeled sense primer, and 20 pmol of unlabeled
antisense primer. Reactions involved 26 cycles in which samples were

subjected to 94OC (1 minute), 60°C (1 minute), and 72OC (1 minute). Products
were separated on 4% nondenaturing polyacrylarnide gels, visualized by
autoradiography, and analyzed by molecular imaging in a phosphorimager
using a Bio-Rad 6s-250 32P-detection screen (Bio-Rad Laboratories Ltd.,
Mississauga, Canada).
2.5.3 Infections
For infectivity studies, the CD4+ MT-2. MT-4, and Jurkat T-lyrnphocytic cell lines
were grown in suspension culture (3-5 x lo5 cells per ml) in RPMI-1640 (Life
Technologies, Mississauga, Canada) supplemented with 10% heat-inactivated
FCS, 2 mM L-glutamine, 100 U of penicillin G per ml, and 100 pg of
streptomycin per rnl at 37OC under 5% CO, In some experiments,
phytohemagglutinin (PHA)-prestimulated primary cord blood mononuclear cells
(CBMCs), maintained at 5 x 10' cells per ml, were used.
A pellet of 0.5 - 1 x 1 o6 cells was resuspended in culture supernatants
from COS-7 transfections (standardized either by p24 levels [50 to 100 ng] or
TCID, units). Following a two hour incubation at 37OC with periodic tapping, the
cells were washed with serum-free medium, and resuspended in fresh serum-
containing medium. After five to seven days in culture (acute infections), these
cells were monitored for cytopathic effect (cpe) and levels of p24 and RT in
culture fluids. For long-term replication studies, infected cultures were passaged
twice weekly at which time 0.5 x 1 d fresh cells were added.
2.5.4 TCID, determinations
COS-7 cells were either transfected with 10 pg of wild-type or mutant DNA, or
were cotransfected at various ratios. For the latter, each of pPR-RT and pFS/PR-
RT was cotransfected with wild-type pBHt 0 at a 1 :I (5 pg of each DNA) and at
1:iO (1 pg pBHlO and I0 pg mutant DNA) wild-type to mutant ratios. As a
control, pBHlO was transfected using 1,5, or 10 pg of DNA in the presence of 9,
5, or 0 pg of negative controI pSVK3 DNA, respectively.

To detennine the 50% tissue culture infective dose (TCID,), dilutions of
clarified supematants produced h m COS7 transfected or cotransfected cells
were seeded in quadruplkate into 96-well culture plates containhg 10' MT-2
cells. The culture medium was changed after four days and infections were
scored after seven days for levels of p24. This method is more sensitive than
scoring by induction of cpe, and was necessary due to the diminished infectivity
of the mutant viruses. Samples were considered positive if the p24 levels in
infected cells were two-fold greater than those in equivalent mock-infected
cultures,
2.5.5 Assessrnent of viral sensitivity to anti-HIV drugs The susceptibility of these viruses to both anttPR drugs (saquinavir, indinavir,
and palinavir) and an anti-RT drug (zidovudine) was assessed as described
previously (Gu et al.. 1992). The antiretroviral drugs (saquinavir, indinavir,
palinavir, and zidovudine) were provided by Hoffman La Roche Inc., Basel,
Switzerland; Merck Inc, West Point, PA; Biomega Inc., Laval, Canada; and
Glaxo-Wellcome Inc., Research Triangle Park, NC, respectively.
MT-2 cells (0.5 x IO6) were infected with COS-7-derived wild-type and
mutant vinises at a multiplicity of infection (moi) of 200 TCID, units per IO6 cells.
lnfected cells were maintained in the presence of a range of drug
concentrations for five days. Cafcufations of 50% inhibitory concentration (IC,)
determinations were made on the basis of RT activity and p24 levels in culture
fluids (Gu et al.. 1992).
2.6 ANALYSB OF VIRAL RNA
2.6.1 Northern blotting MT-2 cells were infected with wild-type and mutant viruses derived from COS-?
transfected cells (standardized at 100 ng p24). After five days in culture. total
RNA was isolated from 10 x IO6 cells ushg TRlzol total RNA isolation kits (Life
Technologies. Mississauga. - Canada). The quaMy and intactness of RNA were verified by eth idiurn brom ide staining.

Samples of total RNA (5 pg for pBHlO and 10 pg for each of mock, pPR-
Ri, and pFS/PR-Rt) were electrophoresed onto 1.2% agarose-forrnaldehyde gels and then transfened onto nitrocellulose membranes (Amersham Life
Science, Toronto, Canada). RNA blots were prehybridized in 50% formamide, 5
X SSPE (1 X SSPE = 180 mM NaCl, 20 mM sodium phosphate, 1 mM EDTA,
pH 8.0), 5 X Denhardt's solution, 0.2% SDS and 100 Ciglml salmon sperm DNA
at 48OC for Iwo hours. ~~bkdizations were perfomied using HIV-1 and actin-
specific probes, which were PPI-labeled using a Nick Translation Labeling Kit
(Boehringer Mannheim, Germany). At the end of the hybridization periods, the
blots were washed up to a stringency of 0.1 x SSPE. 0.1% SDS at 48OC. The
blots were autoradiogaphed at -70°C using Kodak X-Omat film (Eastman
Kodak, Rochester, New York) and Ievels of RNA expression were quantifiad in a
phosphorimager using a Bio-Rad 6s-250 32P-detection screen.
2.6.2 Slot blottfng Analysis of vira! RNA expression in viruses produced by COS-? cells, transfected with wild-type and mutant DNA constructs. was perfomed by slot
blotting procedures described previously (Boulerice et al.. 1990). The efficiency
of transfection was routinely monitored by detection of viral p24. Viral RNA that
had been packaged into virions (purified by sucrose gradient centrifugation)
was purified with a commercial RNA extraction kit (Biotech, Houston, Texas).
The extracted RNA was treated with 100 U of DNase I, followed by phenoC
chlorofom extraction and ethano1 precipitation, to ensure removal of any
contaminating plasmids. The RNA pellets were resuspended in dieth y l
pyrocarbonate (DEPC)-treated double-distilled water. To rute out the possibility
that the samples tested also contained residual DNA, which might have been
hybridized by the radiolabeled DNA probe. RNase digestion of RNA extracted
from virions was performed with RNaseA (Life Technologies. Mississauga,
Canada) at a final concentration of 10 pglrnl at 37% for 30 minutes, following
which phenol-chlorofom extraction was performed.

To quantify viral RNA transcripts, total viral RNA (standardked by 024)
was immobilized on a Hybond-N nylon membrane (Amersham Life Science,
Toronto, Canada) using a slot blot apparatus (Bio-Rad Laborato ries LW.,
Mississauga, Canada). The blot was then subiected to UV irradiation, dried. and
hybridized with pBHlO viral DNA as a radiolabeled probe (Nick Translation
Labeling Kit; Boehringer Mannheim, Gemany) as described previously (Boulerice et ai., 1990). The membrane was exposed to Kodak X-Omat film
(Eastman Kodak, Rochester. New York) at -70°C, and the quantity of viral RNA
was detemiinad by molecular imaging analysis in a phosphorimager using a Bio-Rad GS-250 32P-detection screen,
2.7 ELECTRON MICROSCOPY
Negative staining of viral preparationç waç perforrned as previously described
(Alain et al., 1987). Samples (100 pl) of clarified, virus-containhg COS-?
derived culture fluîds were loaded into 240-pl nitrocellulose tubes into which an
electron microscopy grid had been inserted. The tubes were centrifuged at 120,000 rpm for five minutes, using a Beckrnan Airfuge (Rotor A-100); the grids
were then recovered, dried, and contrasted with phosphotungstic acid (3%; pH
6.0). Specimens were exarnined with a Philips 300 electron microscope.
For thin-section analysis, cells were centrifuged at 1,000 rpm and the
pellet was fked for one hour in 2% gluteraidehyde in PBS containing 3%
sucrose. Cell pellets were washed three times. The cells were then postfixed in 1% OsO, for 30 minutes, washed in buffer, dehydrated in graded acetone, and
embedded in Spun plastic. Observations were made with a Philips 300-electron
microscope.

2.8 [NTRACELLULAR PR ASSAY
Intracellular PR activity was studied by a C O W transfection t ime-course
analysis. COS-7 cells were transfected with either pBHlO or pPR-RT as
described above. Cell pellets were harvested at 12, 24, 36, 48, and 60 hours
post-transfection, and were lysed in 100 pl NP-40-DOC lysis buffer. Samples
(standardized at 25 pg total protein) were immunoblotted with anti-p24 m Ab.
Ratios of p24Ip55 at each time point were calculated based on densitometric
quantification of the corresponding bands in the immunoblots.
2.9 COTRANSFECTION OF J URKAT T-CELLS WlTH W ILD-TY P E
AND MUTANT CONSTRUCTS
CD4+ Jurkat T-lymphocytes were grown in suspension culture in RPMI-1640
(Life Technologies, Mississauga, Canada) supplemented with 1 0% heat-
inactivated FCS, 2 mM L-glutamhe, 100 U of penicillin G per ml, and 100 pg of
streptomycin per ml at 37OC under 5% COP Proviral DNA plasmids were
transfected into these cells by the diethylaminoethyl (DEAE)-Dextran Method
(Maniatis et al., 1982). Briefly, 5 x lo6 Jurkat cells were incubated with DNA and
100 pg/ml DEE-Dextran for 30 minutes at 37OC. Three different conditions
were employed for transfection of wild-type HlV-1 DNA and the mutant
constructs pPR-RT and pFS/PR-RE (1) wild-type pBHlO (1, 5 or, 10 pg) plus
addition of 9, 5 or, 0 pg, respectively, of negative control pSVK3 DNA; (2) 1 :1
wild-type to mutant ratio (5 pg of each DNA); and (3) 1:10 wild-type to mutant
ratio (1 pg pBH10/10 pg mutant DNA). The cells were washed with serum-free
medium, resuspended in I0 ml fresh serum-containing medium, and transferred
to a flask. CelI culture medium was changed at three- to four-day intervals.
Samples of calkfree culture supernatants were collected prior to passage, and
were analyzed for replicative ability by celbfree RT and p24 assays.

CHAPTER 3
HIV-1 PROTEASE AND REVERSE TRANSCRIPTASE REMAIN
ENNMATlCALLY ACTlVE WHEN EXPRESSED AS PR-RT
FUSION PROTEINS IN VIVO

3.1 THE PR-RT CLEAVAGE S E MUTATION PREVENTS CLEAVAGE
B ETWEEN THE TWO PROTEINS IN VIVO
lmrnunoblot anafysis was performed on cellular and viral lysates from COS-?
cells that had been transientty transfected with wiid-type and mutant purified
proviral DNA To confimi the presence of the PR-RT cleavage site mutation, cellular and viral lysates were probed with anti-RT mAbs (Figures 3-1 and 3-2, respectively). The expected pS1 and p66 subunits are clearly seen in wild-type
pBH1O (lane 2). as well as in the positive control of purified RT protein (lane 5)
for each of cellular and viral lysates (Figures 3-1 and 3-2; note that RT was
overloaded in the case of the control Ri lane 5, Figure 3-2). Wth the mutant
pPR-RT and pFWPR-RT constructs (lanes 3 and 4). the sites of the p51 and
p66 RT subunits are increased to 62 and ï7 kDa, respectively (Figures 3-1 and
3-2). These larger species represent the addition of the 1 1 kDa PR to the N-
terminus of RT. Thus, the cleavage site mutation between PR and RT resulted
Ri the expression of a novel PR-RT fusion protein ni transfected cells and viral
particles.

FIGURE 3-1. Immunoblot d whok cell lysates harvested from COS-7
tiansfected cells, using an anti-RT HlV-1 IgGf mAb.
Samples were standardized by total protein content, at 25 pg. Lanes 2. 3. and 4,
respectively, indicate transfections with wild-type pBHIO and the pFSIPR-RT
and pPR-RT constnicts. A mock transfection was a negative control (lane 1). As
a positive control. 50 ng of purified RT was used in lane 5. The p51 and p66
subunits of RT are indicated by anows on the right of the gel.


RGURE 3-2. lmmunoblot of purifiecf concentrated viral lysates frorn COS-? transfected cells. detected by an anti-nT HIV-1 lgG1 mAb.
Viral preparations were equalized at 25 ng p24. Mock transfection in lane 1 was
a negative control. The p51 and p66 subunits of purified RT (100 ng) are shown
in lane 5 and are indicated by arrows. Lanes 2, 3, and 4 respectively. indicate
transfections with wild-type pBHlO and the pFS/PR-RT and pPR-RT constructs.


3.2 PROTEASE WKHIN PR-RT Fusroff PROTEINS IS ENZYMATlCALLY
ACTIVE
The existence of a 62/77 kDa heterodimer in cellular and viral lysates indicated
that PR retained enzymatic activity as a fusion protein and coold process RT at
the RNaseH cleavage site. To extend these results and to determine if PR
within PR-RT fusion proteins could also process sites within Gag. cell lysates
harvested from COS-? transfected cells were immunoblotted with an anGp24
HIV-1 mAb (Figure 3-3). Studies with wild-type pBHlO (lane 2) show the
expected Gag proteins p55 (full-length Gag), ~39141 (CA-NC and MA-CA.
respectively), and p24 (CA); pl60 (Gag-Pol) is not readily detectable with this
mAb. The absence of p55 Gag in the pFSIPR-RT lysates is expected and
confimis the presence of the FS mutation (fane 3). While transfection of pFSIPR-
RT might have been expected to result in overexpression of the PR-RT fusion
protein, we instead observed a decrease in HIV protein expression in this
situation. This is in agreement with other reports of frameshift mutants (Park and
Morrow, 1991 ; Mergener et al., 1992; Smith et al.. 1993), and most Iikely results
from cellular toxicity arising from the overexpression of PR as previously
reported (Krausslich. 1991; Park and Morrow. 1991). The pPR-RT mutant (lane
4) expressed processed Gag proteins similar to wild-type, again indicating that
PR retained its enzymatic activity when expressed as a PR-RT fusion protein. In
fact. cells transfected with pPR-RT contained diminished amounts of the Gag
precursor proteins p55 and p39/41 as corn pared with wild-type pBHIO,
suggesting that PR activity was somewhat increased in PR-RT fusion ptoteins.
In addition. to more fully characterize protein expression in mutant viral
particles, lysates of porified virus were probed with pooled plasma from HIV-1
seropositive individuais (Figure 3-4). Analysis of wild-type pBHlO (lane 2)
clearly shows the presence of p l 60, gpt 20. p55, p39/4l, p24, p51, p66, and
p32; the 160 kDa protein likely represents a combination of Gag-Pol and the
envelope glycoprotein precursor gpf 60. As expected, pFS/PR-RT virus-like
particles (lane 3) did not express p55 Gag, had decreased ~39141 and p24
capsid Ievels, and overexpressed Gag-Pot in cornparison with pBHlO and pPR-

RT; these fmdings are consistent with previous reports of constnrcts containing
a frameshift mutation (Park and Monow, 1991; Mergener et al., 1992; Smith et
al.. 1993). The pPR-RT mutant (lane 4) expressed the same protein pattern as did wild-type pBH1O with the exception of the mutated 62177 kDa PR-RT fusion
protein, indicating that PR was enzymatically active and was able to process
HIV-1 precursor proteins to more mature forms. Taken together, the presence of mature, processed HIV-1 proteins in
both cellular extracts and viral particles illustrates that PR retained its catalytic
activity when expressed as PR-RT fusion proteins.

FIGURE 3-3. lrnmunoblot of wholacell lysates hanrested fiom COS-
7 transfections, probed with an antCp24 HIV-1 IgG1 mAb.
Samples were standardized at 25 pg total protein. Lanes 2. 3. and 4 indicate
transfections with pBH 10, pFS/PR-Rf, and pPR-RT, respectively. A mock
transfection was osed as a negative control (lane 1). Gag proteins p55. p39f41,
and p24 are indicated by arrows.


FIGURE 34. lmmunoblot of puiffled contentrated viral lysates from COS-? transfections. . using pooled sera from HIV-l -infected f ndfviduals.
Viral samples weie equalked at 25 ng p24. Anows on the left show the sites of
wild-type HIV-1 proteins. Mock transfection in lane 1 was a negative control. Lanes 2.3, and 4 depict transfections with wiId-type pBHlO and the pFSIPR-RT
and pPR-RT constructs, respectively.


3.3 DETERMINATlONS OF RT ACnVlTY AND p24 EXPRESSION
IN COS7 CELLS -
Table 3-1 shows the results of studies in which p24 and RT activity were measured in darified virus-containing culture fluids, whole-cell extracts, and
puriiied viral fractions derived from transfected COS7 cells. Levels of p24 and RT activity in each case were similar when cornparing pPR-RT to wild-type p ~ ~ l ~ , suggesting that the lack of cleavage between PR and RT did not affect
protein expression in transfected cells. Forthemore, the similar levels of p24 in
pPR-RT and wild-type demonstrates that normal levels of viral particles are
produced following transfection of pPR-€31. (The p24 EUSA assay also
measures p24 epitopes within p55; hence, this technique cannot detect more
rapid appearance of cleaved p24 in culture fluids).
Table 3-1 also shows k t culture fluids of COS-? cells transfected with
pFSPR-RT had a two-fold increase in K i activity compared to wild-type, while
whole-ceIl extracts had undetectable levels. This is in agreement with previous
reports of frameshifted HIV-1 Gag-Pol proteins, where cellular W activity was
found to be very low compared to levels of RT in cell culture fluids harvested 48
hours after transfection (Park and Morrow, 1991; Mergener et al., 1992); RT
activity in culture supematants from cells transfected with a frameshift mutant
was as much as two- to ten-fold higher than wild-type levels. depending on the
system used (Hoshikawa et al., 1991 ; Krausslich, 1991; Park and Morrow, 1991 ;
Mergener et al., 1992; Karacostas et al.. 1993: Arrigo and Huffman, 1995). This
most likely results from the overexpression of poI gene products, created by the
readthrough mutation at the frameshifî site; it fias been dernonstrated that the
overabundance of PR disrupts cellular integrity and leads to release of viral
proteins, since inactivation of PR restores the presence of cell-associated HIV
antigens (Krausslich, 1991 and 1992; Park and Morrow, 1991; Mergener et al., 1992; Luukkonen et al., 1995). Transfection with the double mutant also
resulted in decteased p24 levels in culture fluids and purified viral pellets
(Table S I ) , reflecting a seveie redtiction in budding and particle formation in
addition to an inability of the fiameshift mutants to produce infectious vidons,

consistent with previous reports (Shioda and Shibuta, 1990; Park and Morrow,
199 1, 1992, and 1993; Mergener et al., 1992; Hughes et al., 1993; Smith et al.,
1 993).
Sïmilar results were obtained in a time course analysis of FIT activity and
pz4 expression in clarified culture fluids, Mole-csll extracts. and viral pellets
harvested 24 and 48 hours after transfection of COS-? cells (Figure 3-5). When
corn paring wild-type pBHlO (lanes 1 and 2) and pPR-RT (lanes 5 and 6) at 24
and 48 hours post-transfection (solid and hatched ban, respectively, panels A-
F), simifar levels of M activity and p24 expression were observed in all
fractions. Small differences were obsewed in cellular extracts and viral pellets
in which FIT activity was slightly increased in pPR-RT samples compared to
wild-type at early times (24 hours; panels B and C. compare lanes 1 and 5), but
were similar at fater times (48 hours; panels 8 and C, lanes 2 and 6). Results
with pBH1O and pPR-RT are consistent with the findings reported in Table 3-1,
and further illustrate that transfections of pBHlO and pPR-RT resulted in a sim ilar pattern of viral expression and production.
In addition. analysis of pFS/PR-RT samples (Figure 3-5) indicate that p24
levels were diminished compared to wild-type in al1 cases, with only low levels
detected in culture fluids and cellular extracts (panels D and E, fanes 3 and 4).
and undetectable levels in purified viral pellets (panel F, lanes 3 and 4); these
results are in agreement with previotls reports in which pz4 expression in frameshifted mutants is severeIy reduced (Park and Monow, 1991; Mergener et
al., 1992). Interestingly, at early times after transfection of pFSPR-RT (24
hours), RT activity was elevated in wholscell extracts (panel B, lane 3). At later
times (48 hours), RT activity levels Ri celf extracts decreased significantly (panel
B. lane 4) while increasing in culture ffuids harvested a€ ais time (panel A, lane
4); this most likely refiects viral protein release from the cell. This pattern of
elevated RT activity in cellular extracts of frameshifted mutants collectecf at earIy
times. followed by undetectable levels in cells at later times, has been
dernonstrated previousiy, and is believed tu be a coosequence of pol
overexpression (Park and Morrow, 1991 ; Karawstas et al.. 1993).

Thus. M activity, p24 production, and viral expression are very similar when comparing wild-type HIV-1 and constnicts containing P R ! fusion proteins. Results from samples containing the cleavage site mutation in
combination with the readthiough mutation at aie frameshift site are in
agreement with previous reports of frarneshifted retroviral constnicts.

TABLE 91. Characteiizatlon of wildtype and mutant transtectlons of COS-7 cells.
Virus culture fluids cell pellets viral pellets culture fluids cell pellets viral pellets
RT activlty and p24 levels were monitored in culture supernatants, whole-cell extracts, and viral pellets as described. Data are means f standard deviation #rom four separate experirnents. Net RT activity after subtraction of values from mock-transfected cells,

FIGURE 3-5. nme-course analysils of wlld-type and mutant samples after transfectfon of COS-7 cells.
RT activity and p24 expression were monitored ni clarified culture fluids, whole-
cell extracts, and viral pellets at 24 and 48 hours post-transfection.
Transfections of pBH1O (lanes 1 and 2), pFS/PR-RT (lanes 3 and 4), and pPR-
F K (lanes 5 and 6). are indicated. Sarnples were harvested either at 24 (solid
bars; lanes 1, 3,s) or at 48 hours post-transfection (hatched bars; lanes 2. 4, 6).
respectively. Panels A, 6, C and D. E, F represent R i activity (cpm) and p24
levels (nglCI), respectively. in clarified culture fluids (A and D). whole-ceIl
extracts (6 and E), and viral pellets (C and F). FlT activity was determined after
subtraction of values from mock-transfected celk. Data are means t standard
deviations from three separate experiments.

a samples harvested at 24 hou= post-transfection samples hanresteci at 48 houn post-transfection

3.4 ENDOGENOUS REVERSE TRAN SCRlPTlON OCCU RS
EFFICIENTLY IN VIRUSES CONTAINING PR-RT FUSION PROTElNS
Use of an endogenous assay to study #r activity revealed similar levds of activity between wild-type pBHlO and pPR-RT virus particles (Figure 3-6). PCR
amplification of minus-strand strong stop [(-)ss] and fulklength DNA produw
indicates that reverse transcription in p8HlO and pPR-RT vinons was equally
efficient (Figure 3-64 compare lanes 2 and 3, and lanes 5 and 6). Similarly,
rnolecular imaging analysis showed no significant differences in FlT activity
between wild-type and mutant viruses (Figure 3-68). The jack of amplified DNA
products in viruses produced from COS7 celts mock-transfected with pBHlO in
the absence of CaCI, and HBS indicates that plasmids from the transfection
inocula were not contaminathg the assay.
Taken together, RT activity measured either in an in v#ro assay as well as
in endogenous reactions in virus particles is similar when comparing wild-type
and pPR-RT vinises. This demonstrates that RT remains enzyrnatically active at
wild-type levels when fused to PR, and that teverse transcription occurs
efficiently in vinises containing PR-RT fusion proteins.

FIGURE 3-6. PCR ampllfication of spaciflc DNA transcripts produced from endogenous RT reactions.
Purified viruses frorn COS-7 cells transfected with various molecular constructs
were studied in an endogenous RT assay for either 10 or 20 hours, and the fulC
length and minus-strand strong stop [(-)ss] DNA products were detected by
PCR. To ensure that contaminating plasmids from the transfection inocula were
not amplified, COS-? cells were mock-transfected with pBH10 Ri 1. absence of
CaCI, and HBS (lanes 1 and 4). Lanes 2 and 5, eodogenous M reactions of
wild-type pBH1 O for 10 and 20 hours, respectively; lanes 3 and 6, reactions of
mutant pPR-FIT virions for 10 and 20 hours, respectively. Lanes 7-1 1, several
dilutions of linearized pBHlO plasmid were used as a positive control (Le-, 10-
fold dilutions of plasrnids in ternis of copy number [O, 1 x 10'. 1 x 102, 1 xlo3,
and 1 x10*], respectively). (A) Samples were separated on a 4% nondenaturing
polyacrylamide gel and visualized by autoradiography. (B) Molecular imag ing analysis of band intensities relative to mock transfections. FulClength and minus
strong stop DNA products are show. Reactions were terminated after 10 (solid
bars) or 20 hours (hatched bars). Experiments were performed using three
replicate sampies; error bars represent standard deviations.

1 1 I C- c linearized ~ lasm id
full-length DNA (367 bp)
- (0) ss DNA - (153 bp)

fulblength DNA k) strong stop DNA

CHAPTER 4
DIMINISHED INFECTIVITY OF VIRUSES CONTAINlNG
PR-RT FUSION PROTEINS RESULTS FROM PROTEASE
DYSREGULATION

4.1 VlRUSES CONTAlNlNG PR-RT FUSION PROTEINS HAVE
REDUCED ~NFECTIVITY
Figure 4-1 graphically depicts the ability of wild-type and mutant viruses derived
from transfected COS-? cells to infect MT-2 cells. Studies involving acute
infections (panel A) as well as long-term infections (panel B) indicate that
viruses containing the PR-RT cleavage site mutation replicated much less
efficiently in the CD4+ T lymphocytic MT-2 cell line than did wild-type viruses-
These effects persisted over at least four weeks, during which tirne the infected
cells were maintained in culture (panel 8). As expected. pFSPR-RT viral
particles were noninfectious and did not replicate in these cells. Sirnilar results
were obtained when infections were monitored by p24 assay (not shown).
Table 4-1 characterizes the infectious capacity of COS7 derived wild-
type and mutant viruses Ri MT02 cells using various parameters. Vinrses
containing the PR-RT fusion protein yielded diminished levels of RT activity and
p24 antigen, as well as induced low levels of cpe. confirming that these viruses
had reduced infectivity- Other f indings revealed that viruses containing the
cleavage site mutation between PR and RT had titen of 1.89 x 1 os TCIDJml
compared with 3.61 x 16 TCID&nl for wild-type virus, representing a 20-fold
decrease in infectivity. As expected, pFSPR-RT virus particles did not induce
cpe and were virtually noninfectious. although the data suggest that a minor
degree of viral breakthrough might have occuned in some situations. Evidence
for infectiousness in these experiments is considered to be minimal, since
studies with heat-inactivated vinrses used as controls yielded shilar results, Le.
within limits of enor of the tests use&
The ability of these viruses to infect different ceII types is shown in Table
4-2. Sirnilar to data obtained in MT-2 cells in regard to levels of Ki activity
(Figure 4-1 and Table 4-1). pPR-RT viruses do not replicate as eficiently as
wifd-type viruses in MF4 and Jurkat T lymphocytes, or Ri cord blood
mononuclear cells (CBMCs). Variations in levels of M activity are consistent
witf~ differences observeci in viral replication assays perfoned in various cell types. As ni the case of infections of pFSIPR-RT in MT-2 cells, these viruses do

not readily infect CD4+ cells, and hence similar data in regard to levels of RT
activity were obtained in infections of each of CBMCs, MT4 and Jurkat cells.

FIGURE 4-1. Replicatlve capacity of wfld-type and mutant viruses derlved from transfected COS7 cells in MT92 cetCs.
Virus inocula were standardized at 100 ng p24. and used to infect 1 x 10' MT-2
cells. (A) Acute infection. Virus production from infected cells reached a
maximum after five days in culture, at which time extensive cell death was
obsewed in celis infected with wiId-type virus. (B) Long-terni infections. Cultures
were passaged twice weekly, at which time fresh cells (0.5 x Io6) were added.
RT activity was routinely monitored in culture fhids. A mock infection served as
a negative conttol. Data represent values o btained from four separate
experiments.

O CI C* m d YI IO
Days post-infect ion
a 0 8
Days post-infection

TABLE 4-1. Characteriration of wild-type and mutant virus replicatlve abillty and
intectivlty in MT-2 ceIlse.
Virus Relative RT Activlty ( ~ p m ) ~ @ p24 (ng/lrl)' Cytopathic TCIDJmld effect (c.p.e.)
a Cells were lnfected with culture fluids of COS-7 transfected cells, standardized at 100 ng p24. Infections were monitored aher five days in culture, Relative RT activlty was calculated by subtracting values in mock-infected cultures.
a Data are means f standard deviation #rom four separate experiments, TCID&nl values were calculated on the basis of p24 production in lnfected cells. Samples are considered positive il the p24 levels in infected cells are at least two-fold greater than those in virus alone. Data are means f standard deviation from three separate experiments. TCID,, 50% tissue culture infective doses,


The infectiousness of the mutant vituses was further confirmed by
analyzing v h I RNA and protein expression in MF2-infected cells (Figures 4-2A
and B. respecüvely). Northem blot analysis (Figure U A ) shows the expected 9.2.4.3, and 2.0 kb RNA species in cells infected with each of pBHlO and pPR-
these bands represent full-length and genomic viral RNA (9.2 kb), singly
spliced envmRNA (4.3 kb), and multiply spliced messages (2.0 kb). However,
levels of viral RNA are lower in cells infected with pPR-FIT compared with wild-
type virus (compare lanes 2 and 4). consistent with the diminished
infectiousness of the former. lmmunoblot analysis shows that m2 cells
infected with pPR-RT expressed lower levefs of Gag proteins than cells infected
with wild-type pBHlO viruses (Figure 4-28. compare lanes 2 and 4). These
results demonstrate that pPR-RT viruses are infectious, and that the matu ration
of Gag proteins was not affected in the case of this mutant. As expected, MT-2
cells infected with pFS/PR-RT contained no HIV-1 RNA or Gag proteins (Figures
4-2A and B. lane 3), indicating that these viruses are not infectious.
In addition. the expression of PR-RT fusion proteins in MT-2 cells infected
with the mutant viruses was confirmed by immunoblot analysis using anti-RT
mAbs (Figure 4-3). The expected p51 and p66 RT subunits are clearly seen in
wild-type pBHIO (lane 5). as well as in the positive control of purified RT protein
(lane 2). In cells infected with the mutant pPR-RT viruses (lane 3). the RT
subunits were increased to 62 and 77 kDa, respectively, confirrning the
expression of the PR-FtT fusibn protein in infected cells. Due to the fact that
pFSIPR-RT viruses are uninfectious. lysates from cells infected with this mutant
contain little HIV-specific protein (lane 4).
Taken together, viruses containing PR-FIT fusion proteins can infect and
replicate in CD& cells, as deterrnined by replication assays as well as by
detection of viml RNA and protein expression in infected cells; however. these
vinrses are 20-fold tess infectious corn pared to wild-ty pe when measu red b y
TCID, detemination. In agreement with previous reports of frameshifted mutants, pFSPRdT virus particles are not infectious, likety due to cellular
toxicity associated with the overexpression of PR (Shioda and Shibuta, 1990;

Krausslich, 1991; Park and Monow, 1991. 1992. and 1993; Mergener et ai..
1 992; Hughes et al., 1993; Smith et al.. 1993). I

FIGURE 4-2. RNA and protein expression In MT-2 cells lnfscted with wlld-type or mutant viruses derived from COS-? transfections.
Lanes 2, 3, and 4 indicate infections with pBH10, pFS/PR-RT, and pPR-RT,
respectively; a mock infection is shown in lane 1. (A) Northern Blot analysis.
Total RNA was isolated and hybridized with a [adioactively-labeled pBH1 O HIV-
1-specific probe, produced by ni&-translation. 10 pg was loaded in lanes 1, 3,
and 4, while 5 pg was used in lane 2. (B) lmmunoblot analysis. Samples were
standardized at 25 pg total protein, and probed with an anti-p24 HIV-1 mAb.



FIGURE 4-3. tmmunoblot anatysis of MT-2 cellr fnfected wlth wlld-
type or mutant viruses derived fiom COS7 tiansfections.
Cell lysates were standardized at 25 pg total protein. and proteins were
detected using an anti-RT HIV-1 mAb. Mock infection (lane 1) was a negative
control. The pS1 and p66 subunits of purified KT (50 ng) are shown in lane 2
and are indicated with arrows on the left of the gel. Lanes 3, 4. and 5.
respectively, indiçate infections with pPR-RT, pFS1PR-RT, and wild-type pBH10.


4.2 MUTANT VIRUSES HAVE NORMAL LEVELS OF GENOY~C RNA PACKAGtNG
To detemine if the PR-FtT cleavage site mutation affected packaging of
genomic RNA in virions. RNA dot blot analysis was perfomed (Figure 4-4).
Similar levels of viral RNA, based on RNNp24 ratios. were packaged into
viruses derived from COS7 cells that had been transfected with wild-type
pBHlO or pPR-RT (Figures 4-4A and B). When these RNA preparations were digested with RNaseA, little or no hybridizable material remained, indicating
that contarninating vira! DNA was not present in these preparations. Thus, the presence of PR-RT fusion proteins in virus particles does not
affect RNA packaging, and moreover, the diminished infectivity observed in pPR-RT vinises does not result from reduced efftciency of genomic RNA
packaging.

FIGURE 44. Genomic RNA packaging by wild-type and mutant v/ruses.
COS-? cells were transfected with either pBHlO or pPR-RT, and viruses in
culture fluids were purified by sucrose gradient ultracentrifugation. RNA was
extracted from equal quantities of viruses based on p24 content and quantified
by slot blot and molecular irnaging analysis. (A) Autoradiograph of RNA
packaging by slot Mot analysis. (B) Relative quantities of viral RNA packaged
into virions. Solid bars indicate pBHlO viral preparations; pPR-RT samples are
represented by hatched ban. Experiments were performed using three
replicate samples; error bars represent standard deviations. In some cases,
viral RNA was digested with RNaseA to eliminate hybridizable material. Results
are standardized to 100 for pBHlO (200 ng).

Viral p24 (ng) :- !
200 - i l
100 -, '

50 100 200 200 + RNaseA
Viral p24 (CA) (ng) in viral preparations

4.3 ELECTRON M ICROSCOPY ANALYSIS OF w ILD-TYPE AND
MüTANT VIRUS PARTICLES
The above results on RNA packaging and p24 levels. indicating few differences
between pBHlO and pPR-RT, showed that cells transfected with pPR-RT
produced normal levels of viral particles. To detemine the nature of the mutant
virus particles, electron microscopie analysis was performed. Negative staining
illustrates that morphologically mature viruses were present in culture fluids of
COS-? cells that had been transfected with either pBHlO or pPR-Rf; viral
structures in both cases displayed condensed conical cores, characteristic of
mature, infectious virions (Figures 4-5A and 0). ln addition, examination of thin
sections revealed that viral structures within the transfected cells contained
mature, electron-dense nucleoids within intracytop lasmic vacuoles in both
cases (Figures 4-5C and D). Thus, viruses containing PR-RT fusion proteins are
morphologically mature and indistinguishable from wild-type particles,
illustrating that the PR domain in this mutant remains capable of inducing and
mediating virus maturation.
Analysis of pFSPR-RT samples revealed no virus particles in the
negative stains, and thin section preparations contained very few virus-like
particles (compared to approximately 108 particleçlml in the case of pBHlO and
pPR-RT). This is consistent with results characteriring this mutant, presented
herein, as well as with previous reports of frameshifted mutants in which
budding and particle formation were severely diminished in these
circumstances, the formation of mature, infectious viral particies was completely
abrogated (Shioda and Shibuta. 1990; Park and Morrow, 199 1, 1992, and
1993; Mergener et al., 1992; Hughes et al., 1993; Karacostas et al.. 1993; Smith
et aL. 1993). Furthemore, the virus-like particles Viat were released from
transfected COS-? cells (Figure 4-5E) displayed a dense rayer of
ribonucleoprotein beneath the Iipid biiayer and no sign of nucleoid
condensation at the center of the particle, characteristic of immature particle
morphology as previously described (Gheysen et al.. 1989; Shioda and

Shibuta. 1990; Mergener et al., 1992; Hughes et al.. 1993; Smith et al.. 1993;
Quillent et al., 1996).

FIGURE 4-5. Electron microscopy analysis.
Negative stainîng of COS-7-derived wild-type pBHl0 (A) and pPR-RT (8). Also
shown aie thin section electron micrographs of COS-? cells tiansfected with pBH10 (C), pPRdf (D), and pFSPR-RT (E). Original magnification: Panels A
and B x 180,000; panels C and D x 90,000; panel E x 120,000.






4,4 TIME COURSE ANALYSIS OF PR ACTlVKY IN WVO
To compare intracellular PR activity between pPR-RT and wild-type h vivo,
processing was monitored over tirne in transfected COS-? celIs by immunoblot
with anti-pz4 mAbs (Figure 4-6A). For both wild-type pBHlO and mutant pPR-
RT, p55 Gag was processed into p24 CA. However, the rate of processing was increased in COS-? cells expressing PR-RT fusion proteins. After 12 houn, mature p24 was detected in lysates of cells transfected with pPR-RT but not
pBHlO (compare lanes 1 and 6). At later times, more p55 had been converted
into p24 in pPR-RT sarnples in cornparison with wild-type (compare lanes 2-5 to
7-10). Levels of processing are also presented through plots of p24/p55 ratios
(Figure 4-68), derived from densitometric quantification of the corresponding
bands in panel A. In this analysis. PR-RT clearly displays increased enzymatic
activity compared to wild-type.

FIGURE 4-6. lntracellular PR activity assay.
COS4 cells were transfected with wild-type or mutant constnicts. and whoie-
cell lysates were harvested at 12 hour intervals. (A) lmmunoblot of cell lysates
(standardized at 25 pg total protein) detected by a mAb directed against HlV-1
p24. Wild-type pBHlO (lanes 1-5) and mutant pPR-RT (lanes 6-10) were harvested at 12, 24, 36, 48, and 60 hours post-transfection. (8) Quantitation of
processing levels determined by densitometry of the immunobiot in panel A.
p24fp55 ratios are shown for pBHlO (solid bars) and pPR-RT (hatched bars) at
each time point. Data a n means I standard deviation from four separate
experiments.



4.5 SENSmVlTlES OF MUTANT VIRUSES TO ANTIVIRAL DRuGS
To assess whether vimses containing the PRnT fusion protein rnight have
possessed altered sensitivity to antiviral dnigs* the mutant viruses were tested
for sensitivity to zidovudine, an inhibitor of HIV-1 nT. as well as to each of the PR in hi bitors, saquinavir, indinavir, and palinavir. The res ults presented in Table 4-3 show that wild-type and pPRRT viruses possess similar 50%
inhibitory concentration (Cm) values in regard to each of the dnigs studied.
Thus, the fusion of PR and RT does not affect eiVier enzyme's senstivities to
specific inh ibitors.

TABLE 4-3. SensiUvltïes of wlld4ype and mutant vfruses to antlntrovlml drup.
Virus AZT Saqu inavir lndinavir Palinavir
a MT-2 cells were infected with culture fluids of COS-? transfected ceflç at a rnultiplicity of infection (moi) of 200 TCID, units per 1 d cells; p24 levels were detemrined after fve days and used to calculate IC, values. Data are means & standard deviation from three separate experiments.

CHAPTER 5
PR-RT FUSlON PROTEJNS tNHlBlT HIV-1 REPLlCATlON
BY A TRANS-DOMINANT NEGATIVE MECHANISM

5.1 COTRANSFECTiON OF WILD-TYPE AND MUTANT CONSTRUCTS D ECREASES HIV-1 REPLICATI ON
To determine whether the mutant constructs behaved in a trans-dominant
negative fashion, cotransfection experiments were performed using various
ratios of wild-type and mutant DNA. When pPR-RT was cotransfected in Jurkat
T-cells at a 1:l ratio with wild-type pBH 1 O. the resuit was a decrease in levels of
replication (Figure 5-1 A). This inhibition was even more pronounced when a
cotransfection ratio of 1:lO (wiid-type ta mutant) was used. In both cases, production of viral FIT activity was seen after 11 days in the case of pPR-RT in
contrast to 6 days with wild-type transfections. Viral replication in cotransfected
cells never reached the high levels obtained following transfection of wild-type
DNA alone. Sirnilar results were obtained in cotransfections of p8HlO with the
double mutant pF SPR-RT (Figure 5-1 6); in fact, these cotransfections resulted
in more pronounced inhibition of viral replication at both 1:1 and 1:10 ratios
when compared to results of pPR-RT and pBH IO cotransfections presented in
panel k Thus. both pPR-RT and pFS/PR-RT inhibited the replication of wild-
type vinises in a dose-dependent manne?. These experirnents were not
continued on a comparative basis beyond 11 days because of extensive ceil
death observed aftei this tirne in the case of control transfections.

FIGURE 5-1. Cotransfection analysis.
Jurkat cells were cotransfected with (A) pBHlO and pPR-RT or (6) @Hl0 and
pFS/PR-RT. 5 pg of each DNA was used to obtain a 1:1 cotransfection ratio. For
a 1:10 ratio. 1 pg of wild-type p6Hl O and 10 pg of mutant DNA was used. As
controls, 1.5. or 10 pg of pBHlO DNA was transfected in the presence of 9,5, or
O pg of negative control pSVK3 DNA, respectively. Culture fluids were
monitored for RT activity at various times. Data are means from four separate
experiments. and standard deviations are indicated.
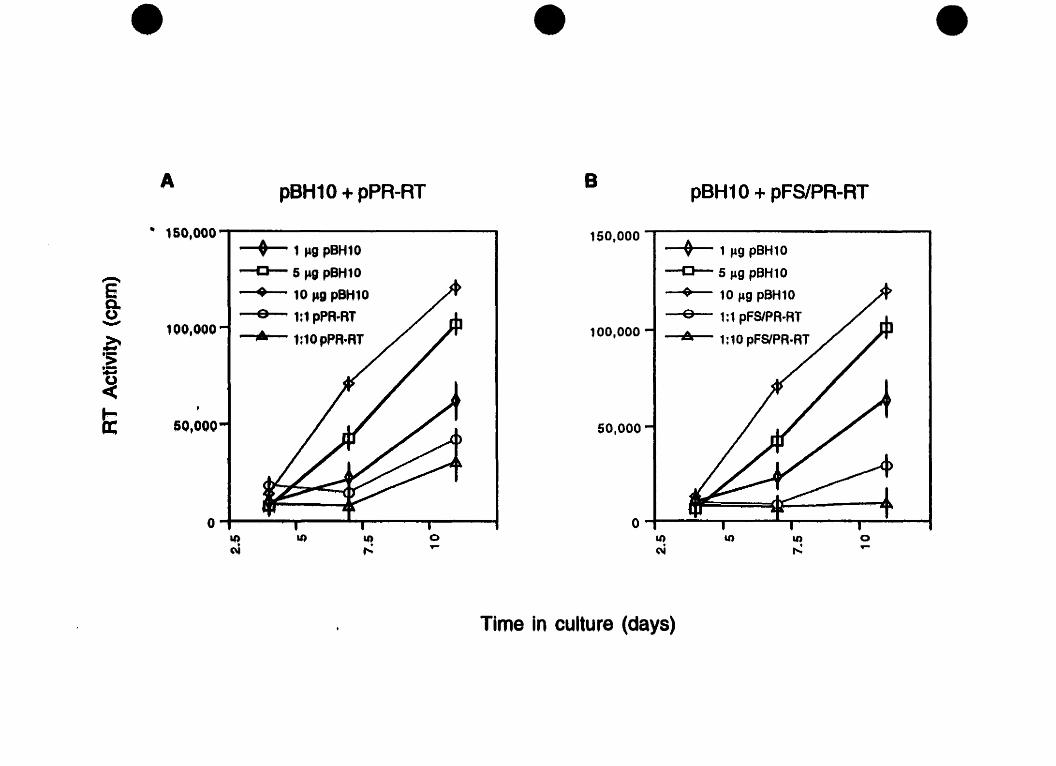
Time in culture (days)

5.2 VIRUSES DERlvED FROY COTRANSFECTIONS OF WILD-
TYPE AND MUTANT CONST RuCTS HAVE DIMINISHED
INFECT~VITY
Table 5-1 shows TCID, values in MT-2 cells of culture fluids derived €rom
cotransfections of COS7 cells. Transfection of 1.5, or 10 pg of wild-type pBHl O
alone resulted in values of 1.9 x 1o4, 9.7 x to4 and 2.2 x 10' TCIDJml.
respectively. At 1:l and 1:10 cotransfection ratios of wild-type pBHlO to mutant
pPR-RT, the TCID, values were decreased by 41% and 73%, respectively.
These diminutions were somewhat more pronounced with cotransfections of
pBHl O with mutant pFS/PR-RT (82% and 80% for 1:1 and 1 :IO cotransfection
ratios, respectively).
Therefore, in transient cotransfection experiments, both pPR-RT and
pFSPR-RT inhibited replication of wild-type HV-1 and diminished viral
infectiousness in a dose-dependent manner. suggesting a trans-dominant
negative mode of action.

TABLE 5-1. TCID, determinations in MT-2 cells of culture fluids f rom COS-7 cells derived from transfections or cotransfectlons with wild-type and mutant HIV-1 consfiucts at various ratios.
Amount of DNA transfected TCIDJmP
5 pg of wild-type pBHlO and either mutant DNA was used to obtain a 1:1 cotransfection ratio. For a 111 0 ratio, 1 pg of wild-type pBHt 0 and 10 pg of mutant DNA was used. TCIDSOI 50% tissue culture infective dose. Values were calculated on the basis of p24 production in infecteci celk and represent means from three separate experiments f standard daviation.

CHAPTER 6
GENERAL DiSCUSSlON

In retrovinises. mature protease (PR) and reverse transcriptase (RT) are
generated du ring virus maturation via proteoIytic processing of poly protein
precurson. The data presented herein demonstrate that a cleavage site mutation between the HlV-1 PR and RT genes prevenfed processhg between
the two proteins, resulting in a PRRT fusion protein. lmmunoblot analysis
showed that both cellular and viral lysates, derived from transfections of COS-?
cells, contained PR and RT fused as 62/77 kDa heterodimen. Although both PR
and RT enzyrnatic activities rernained intact, viruses containing the PR- fusion protein had diminished infectivity. These results are discussed in regard
to molecular detenninants which regulate PR activity, and to the development of
a novel gene therapy approach for HLV-1 infection.
Due to the importance of processing at the amino-terminus of PR for proteolytic activity, mutations at this cleavage site have been extensively
studied. It has been previously demonstrated in a vatiety of systems that
cleavage at the N-terminus of PR is essential for the initiation of protease activation, and that mutations which interfere with this event severely disrupt PR
activity (Kotler et al., 1988; Loeb et al. 1989a, Debouck et al.. 1990; Louis et al.,
1991 and 1994; Pettit et al., 1991; Poorman et al., 199 1 ; Burstein et al., 1992;
Zybarth et al., 1994; Wondrak et al., 1996; Xiang et al., 1997; Tessmer and
Krausslich. 1998). Processing at sites within the Gag poiyprotein were inhibited
or severely reduced by cleavage site mutations at Vie N-terminus of PR,
although processing at sites within the Pol domain of Gag-Pol occurred in some
cases (Kotler et al., 1988; Loeb et al., l989a; Louis et al., 1991; Tessmer and
Krausslich, 1998). These findings suggest that the N- and C- terminal regions of
protease are interdependent, whereby sequeoces at one cleavage site can
influence the efficiency of cleavages at others (Louis et al., 1991). Thus,
cleavage at the first site (at the N-terminus of PR) induces a conformational
change in protease, allowing cleavage at the second site (Gterminus) to
proceed. Taken together, this explains the demonstration mat the N-terminal
cleavage of PR is requ ired for sitbsequent Gag po lyprotein processing (6 urs tein
et aL, 1992; Zybarth et al.. 1994; Stewart and Vogt. 1994; Schatz et al., 1997;
Xiang et aL, 1997; Tessmer and Krausslidi, 1998). Furtherrnore, efficient Gag

processing has recently been shown to correlate with viral infectivity flessmer
and Krausslich, 1998). ln this report, a cleavage site mutation at the N-terminus
of HlV-1 PR in the context of a complete provirus resulted in a severe reduction
of Gag polyprotein processing and a complete abolition of virus infectivity, This
presumably resulted from a slowing of the rate of the initial cleavage event via
interference with PR dimerization vessmer and Krausslich, 1998).
In contrast, the effects of cleavage site mutations between protease and
reverse transcriptase have not been well-studied, and HIV-1 PR-RT fusion
proteins have not been previously analyzed in mammalian systems. Moreover,
such analyses have not been perfomed using complete proviral constnicts,
and thus, cleavage site mutations between PR and RT have not been ewmined
in virus particles. In Ecoli. certain cleavage site mutations at the carboxy-
teminus of PR had litt le effect on enzyme activity; they were tolerated, provided
that a wild-type site was presenred at the N-terminus. Yet other mutations at the
same cleavage site between PR and M rendered PR inactive (Loeb et al.,
1989a; Debouck et al., 1990; Louis et al*, 1991). However, the effect of such
mutations on RT activity was not analyzed in these studies.
We have now shown in a mammalian system that a cleavage site
mutation between HIV-1 PR and K i does not prevent PR dimerization and
activation, and that both protease and reverse transcriptase remain active when
expressed as PR-RT in both transfected cells and in punfied virus particles. Our
data is in agreement with the requirement for an hitial catalytic event to occur at
the N-terminus of PR (Debouck et al., 1990; Louis et al.. 1991; Pettit et al., 1991 ;
Poorrnan et al., 1991; Wan et al., 1996; Wondrak et al., 1996; Tessmer and
Krausslich, 1998). We found that preventing cleavage at the Cterminus of PR
did not block its proteolytic capacity, implying that this cleavage event follows N- terminal processing. Moreover, these determinations corroborate results
obtained from processing studies of a mode1 precursor protein in vitro, in which
PR species generated after the initial N-terminal cleavage are active when
tested on an exogenous substrafe provided h f m s (Wondrak et al., 1996). Out
findings also support the notion that generation of a non-rnodified N-terminus of
mature PR is required for protease activation in mammalian systems (Stewart

and Vogt, 1994; Zybarth et al., 1994; Wan et al., 1996). Furthemore, unlike
miscleavage that has been shown to occur following mutational disruption of
the N-terminal site, which illustrates the importance of cleavage at this site
(Phylip et al., 1992; Stewart and Vogt, 1994; Zybarai et al., 1994; Lindhofer et
aL, 1995; Schatz et al., 1997), we did not detect any aberrant foms of PR-RT
fusion proteins in either transfected cells or virus particles. Most notable.
however, is the fact that the infectiousness of vinises containing PR-nT fusion
proteins was diminished by only 20-fold, in contrast to the complete abolition of
infectivity reported in viruses containing p6*-PR fusion proteins (Tessrner and
Krausslich, 1998).
Taken together. we are the first to demonstrate in the context of a
complete provirus in vivo that cleavage at the C-terminus of PR is not as critical
for PR activation as processing at its N-terminus. Disruption of the C-terminal
cleavage site in our pPR-RT mutant did not result in severe viral defects such as
the abrogation of Gag precursor processing and virus infectivity. as reported in
vkuses in which cleavage at the N-terminus was blocked (Tesmer and
Krausslich, 1998). Rather. viruses containing PR-RT fusion proteins remained
viable and possessed both PR and RT enzymatic activities in vivo. These data
fuither contribute to the elucidation of mechanisms that reguiate PR activity in
infectious virions and to the molecular determinants underlying HIV-1 viral
maturation.
It is not surprising that RT activity within virus particles containing the PR-
RT fusion proteins remained at wild-type levels. It has been previously reported
that proteolytic maturation of the Gag-Pol precursor is not necessary for
activation of RT in mammalian systems; Gag-Pol possesses RT activity,
although at lower levels than those observed for mature RT (Peng et al., 1989.
1990, and 1991 ; Hu and Kang, 1991 ; Stewart and Vogt, 1991 and f 993;
Mergener et al., 1992). RT activity has been dernonstrated in unprocessed Gag-
Pol in ASLV (Stewart et al.. 1990; Stewart and Vogt, 1991 and 1993), Mo-MLV
(Ctawford and Goff. 1985), and HIV-1 (Lori et al., 1980; Hu and Kang, 1991;
Peng et al., 1991; Mergener et al., 1992). Protease-defective mutant HLV-1
particles also display signifiant M activity, Lee.. reductions of only two- to ten-

fold in cornparison with wild-type (Gottlinger et al., 1989; Peng et al.. 1991;
Mergener et al., 1992; Park and Morrow, 1992).
The PR-RT cleavage site mutation was combined with a readthrough
insertion mutation at the frameshift site in order to enhance the defects
associated with viruses containing the fusion protein. Indeed, cells transfected
with pFç/PR-RT produced noninfectious particles, in agreement with previous
reports analyzing the overexpression of Gag-Pol (Shioda and Shibuta, 1990;
Park and Morrow, 1991 and 1992; Mergener et al., 1992; Hughes et al., 1993;
Smlh et al., 1993). This is most likely a consequence of premature PR
activation and the subsequent proteolytic processing of polyprotein precursors.
An additional consideration is that a C-temiinal domain of the Gag p6 protein
may be required for efficient particle release and would not be expressed in
pFSPR-RT transfected cells (Gheysen et al., 1989; Gottlinger et al., 1991 ;
Huang et al., 1995; Yu et al.. 1995; Schwartz et al.. 1996). However, this would
not have affected our intracellufar results as demonstrated on the basis of the
immunoblots. Our findings that FtT levels in culture fluids from cells transfected
with a frameshift mutant are increased. are consistent with data of other groups
that have reported a range of increased values ranging from No- to ten-fold
(Hoshikawa et al., 1991 ; Krausslich. 1991; Park and Mo~~ow, 1991; Mergener et
al., 1992; Karacostas et al., 1993; Arrigo and Hufiman, 1995). When virus
particles from these transfections were centrifuged through sucrose, the virion-
associated FIT activity of the frameshift mutants was significantly diminished, in
accordance with previous observations (Gottlhger et al., 1989; Park and
Monow, 1991 ; Mergener et al., 1992; Hughes et al., 1993; Smith et al., 1993;
Arrigo and Huffman, 1995). Consistent findings were also obtained in regard to
p24 measurements.
Viruses containing the cleavage site mutation between PR and RT
replicated less efficiently in CD4+ Fcells and were about 20-times fess
infectious than wild-type vinises. The loss of infectivity did not result frorn a
restriction in DNA synthesis, RNA packaging, or virus production. Reverse
transcription of viral DNA products occuned with sim ilat effciency in wild-type
and mutant viruses when monitored by in vitro and endogenous reactions. Stot

blot analysis indicated that viral genomic RNA packaging was not disrupted in
mutant particles containing PR-RT fusion proteins. Nor was the decreased
infectiousness of PR-RT viruses due to aberrant virus production, as these
viruses expcessed normal levels of p24 capsid ptotein, indicating that mutant
particles were released from transfected cefls as efficiently as wild-type. In
addition, electron microscopy revealed that viruses containing the PR-RT fusion
protein possessed wild-type morphology. Finally, both types of viruses
displayed similat sensitivities to inhibitors of each of PR and RT, as measured in
vkus replication assays. However, the presence of the PR-FIT cleavage site
mutation must clearly be disadvantageous for virus replication.
Rather, we believe mat the diminished infectiousness of PR-RT viruses
may result from disruptioo of the normal tight regulation of protease activity. This
is a consequence of conformational changes at the PR dimerization interface
arÏsing from the fusion of PR and RT, and results in premature PR activation and
enhanced proteo[ytic activity as shown in our time-course experiments in
transfected COS-7 cells. Tight reg ulation of PR activity. both temporal and
spatial, are critical for successful HIV-1 replication. Protease production,
activation, and rate of precursor processing during maturation are controlled by
multiple mechanisms. such as the requirement for dimerization for enzyme
activation, and the numerous positive and negative regulators within Gag and
Gag-Pol. These mechanisms work together to prevent premature PR activation
and polyprotein cleavage prior to virion budding. Dysregulation of protease or
its overexpression has been shown to reduce the infectiousness of released
virus particles (Krausslich and Wimmer, 1988; Hoshikawa et al., 199 1 ;
Krausslich, 1991 and 1992; Park and Morrow, 1991; Mergener et al., 1992;
Karacostas et al.. 1993; Anigo and Huffman, 1995; Luukkonen et al., 1995).
Previous reports have also demonstratecf that RT c m enhance PR proteolytîc
activity in vitro and in eukaryotic cells in a dose-dependent manner, and that
mis increased intracellutar protease activity can be partially compensated by
rernoval of RT (Hu et al.. 1990; Karacostas et aL, 1993; Goobar-Larsson et al.,
1995; Luukkonen et al., 1995; Ansari-Lari and Gibbs, 1996; Quillent et al., 1996). Furthemore, RT has been shown to promote PR homodimerkation and

to improve its substrate speciiicity (Navia and McKeever, 1990; Zhang et al.,
1991; Karacostas et al., 1993; Kuzmic, 1993; Darke et al., 1994; Goobar-
Larsson et al., 1995 and 1996; Luukkonen et al., 1995; Quillent et al., 1996).
Our findings that PR activity is dysregulated via an enhancernent of
proteolysis when expressed as PR-RT fusion proteins in vivo are consistent with
these reports, and support the notion that RT enhances PR activity in transfected
ceils and virus particles. Our intracellular processing assay revealed that PR
fused to RT converted the Gag precursor to mature p24 slightly faster than wild-
type, Le. less than two-fold. We believe that this moderate increase in PR activity
within PR-RT fusion proteins is responsible for the diminution in infectivity, in
accordance with reports demonstrating that subtle changes in PR activity and
precursor processing resulted in drastic changes in infectivity (Kaplan et al.,
1993; Rose et al., 1995: Quillent et al.. 1996). Moreover, the increase in
proteolytic activity is small enough that it is not sufficient to disrupt DNA
synthesis or to affect virus production and morphology.
Conceivably. there are several possible mechanisms which could
explain the dysreg ulation of PR activity observed in viruses containing PR-RT
fusion proteins. Rst, FIT fused to PR enhances proteolytic activity in a manner
analogous to that whereby puriiied RT hcreases PR activity, when the two
proteins are expressed separately (Goobar-Larsson et al., 1995 and 1996). This
may be a consequence of the RT domain which promotes PR dimeriration and
improves catalytic efficiency. as previously reported (Hu et al., 1990; Karacostas
et al.. 1993; Darke et al.. 1994; Goobar-Larsson et al.. 1995 and 1996;
Luukkonen et al., 1995; AnsariLari and Gibbs, 1996; Quillent et al., 1996).
Second, regions at or surrounding the junction between PR and RT could
contain determinants that positively m odulate PR activity. These elements.
based on primary sequence andlor structural context, are p resumably
comparable to those located within Pol p 6 (Partin et al.. 1991; Zybarth and
Carter, 1995; Gatlin et al.. 1998) and Gag p6 (Gottlinger et al.. 1991; Huang et
al., 1995) which repress PR activity while in precursor form. A third
consideration is that PR must be completely processed into its mature form for
optimal activity. it has been previously demonstrated that release of N-terminal

PR extensions are required for optimal PR activity h vivo (Zybarth et a!., 1994;
Tessrner and Krausslich, 1998); our findings now illustrate that C-terminal
processing also ensures accurate PR activation in transfected cells and virus
particles. Finally, the absence of cleavage between PR and M could
dramatically alter the order of precursor processing such that PR activity is uncontrolled. Previous reports have show that Gag and Gag-Pol are cleaved
in a weli-defined sequential order, and that accurate and cornplete processing
of precursor proteins is essential for the formation of mature cores in infectious
virions (Gottlinger et ai., 1989; Kaplan et al., 1993 and 1994; Krausslich et al.,
1995; Reicin et al., 1995; Weigers et al.. 1998). Alteration of the order of
cleavage interferes with the formation of defined precursor intemediates.
whose structural conformations are important regulators of PR activity
(Gottlinger et al.. 1989; Tritch et aL, 1991; Pettit et al., 1994; Krausslich et al.,
1995; Lindhofer et al.. 1995; Xiang et al., 1997; Wiegers et al., 1998).
Presumably, these mechanisms all contribute to the dysregulation of PR activity
and to the enhancement of proteolytic processing observed in viruses
containing PR-RT fusion proteins.
We are currently planning to investigate the potential contributions of
these mechanisms in cell-free assays. In this context, we would like to express
and purify recombinant PR-RT fusion proteins to perforrn detailed kinetics
analyses of both enzyme activities. However, efforts to do so have thus far been
hindered by problems of cytotoxicity and autoproteolytic digestion in the & coii
expression system, similar to those reported by other investigators while
purifying recombinant PR (Debouck et aL, 1987; Giam and Boros, 1988;
McKeever et al., 1989; Rose et al., 1993; Stebbins and Debouck, 1994; Riuo
and Korant, 1994; Tomasselli et al., 1995; Wan et al., 1995).
Taken together, these findings advance the understanding of the
molecular determinants involved in HIV-1 PR regulation during virus maturation.
We have demonstrated that Gteminal cleavage of PR is less critical for
ptoteolytic actMty and virus maturation than N-terminal processing in vivo.
Moreover, ouf resuits have illustrated that the liberation of RT from PR is a novel
and important mechanism that regulates PR activation. In combination with

numerous additional rnodalities. C-terminal protease cleavage ensures that
premature enzyme activation and precursor processing do not occur. While N-
terminal cfeavage site mutations in hibit processing of Gag p recunors by PR
and abrogate virus infectivity (Tessmer and Krausslich. 1998), cleavage site
mutations at the C-terminus of PR enhance its activify, which also results in
diminished infectivity. In this way. multiple mechanisms regulate PR activity ln vivo, together ensuring that PR activation is appropriately timed and thus that
the rate of precursor processing is tightly controlled.
The characterization of our mutant constructs in these assays was a pre-
requisite for all additional studies. Our fhdings encouraged us to develop a
gene therapy approach to inhibit HIV-1 replication. in this regard. we have
shown that HIV-1 replication can be reduced by intracellular expression of
proviral trans-dominant negative PR-AT fusion proteins. using each of two
genetic constructs. In transient cotransfection experiments, both pPR-RT and
pFS/PR-RT inhibited replication of wild-type HIV-1 and dim inished viral
infectiousness in a dose-dependent manner.
lt is not surprising that pFS/PR-RT, which overexpresses the PR-FIT
fusion protein, inhibited viral replication to a greater extent than pPR-RT. It has
been previously reported that a more pronounced inhibition of HIV-1 replication
can result from increased intracellular expression levers of trans-dominant
protease inhibitors through manipulation of the viral frameshift (Junker et al.,
1 996).
Inhibition of HlV-1 replication by intracellular expression of dominant
negative protease variants has been established as an effective anti-HIV
strategy. Constitutive expression of trans-dominant PR inhibitors has been
shown to efficiently suppress HlV-1 replication N1 vivo (Junker et al., 1996;
McPhee et al., 1996). Trans-dominant inhibition of PR has been demonstrated
through mutations in the active site. either in c& or in tram. that blocked
enrymatic activity via the formation of inactive heterodirnen (Arrigo and
Huffmao, 1995; Babe et al.. 1995; Junker et al.. 1996; McPhee et al.. 1996).
Stable expression of such defective PR molecules in celis reduced the yieid of

infectious particles by 90% following transfection with wild-type proviral DNA
(Babe et al., 1995).
Another modality for inhibiting protease activity is through interference
with PR dimerization (Babe et al., 1991 and 1992; Zhang et al., 1991). Inhibition
of PR by the disruption of homodimer formation has been demonstrated in both
HIV-1 and HIV-2 (Babe et al., 1991 and 1992). In vitro incubation of wild-type
PR with a dimerkation-defective PR mutant resulted in the production of
catalytically defective heterodimen which disptayed 80% inhibition of
enzymatic activity (Babe et al., 3991).
In this study, we focused on the dimerization interface involving the N-
and C-temini of PR as a site for antiproteolytic intervention via a novel trans-
dominant negative strategy that blocked cleavage between PR and RT for
several reasons. First, dimeritafion of protease polypeptides is essential for
activity and is also an initial posttranslational step. Second, the extended
interface created by the N- and C-termini of PR is consewed and may be less
vulnerable to mutational escape than the active site or the substrate binding
region of PR itself. Third, dominant negative PR inhibitors rnight potentially bind
to the protomeric fom of PR that is present in polyprotein precursors.
Accumulation of such a polyprotein-inhibitor complex in the budding virion
might concentrate the inhibitor at that location, even before processing of the
polyprotein can begin. In contrast, inhibitors that target the active site of the
dimeric enzyme can only bind after that active site has been formed by
dimerization (Babe et al., 1991 and 1992; Zhang et al., 1991).
Although both the N- and C-termini of the HIV-1 PR are important in its
dimerization and activation, the C terminal tetrapeptide of PR was found to be
an excellent dissociative inhibitor of its dimerization (Zhang et al., 1991; Babe et
al., 1992). We have extended these findings by fusing the entire M protein to
PR. rather than fusing only a small tetrapeptide. Akhough out mutant wnstructs,
containing a cleavage site mutation between PR and K. did not inhibit
protease dimerization and activation, they seerningIy functioned as trans-
dominant negative inhibitors of HIV-1 replication, albel more modestly than
obsewed in other trans-dominant systems (Trono et al., 1989; Malim et al.,

1992; Arrigo and Huffman, 1995; Babe et al., 1995). Presumably, this occuned
by temporal and spatial dysregulation of PR activity through conformational
changes in the PR-RT fusion proteins, which were, in tum, imposed upon wild-
type protease in a dominant negative fashion. Consequently, wild-type PR
activity was affected, and viral replication and infectivity were diminished.
Although complete inhibition of HlV-1 replication in these studies was not
achieved, our research demonstrates the feasibility of a trans-dominant
approach that targets both the frameshift and the PR-RT cleavage sites. These novel dominant negative mutants may have usefulness when applied as a gene
therapy approach for HIV-1 infection. Furthemore, a similar strategy could
potentially be used in conjunction with currently available chernotherapeutic
agents that target the protease to synergistically reduce the viral load through
alternative modes of PR inhibition.

The present studies analyzing the effects of a cleavage site mutation between
HIV-1 protease (PR) and reverse transcriptase (RT) have contributed to a better
understanding of the molecular mechanisms involved in virus maturation and of
conditions that may pefturb that process. The candidate's major contributions to
original knowledge are the following:
1. For the first the. a cleavage site mutation was created between HIV-1
protease and reverse transcriptase in the context of a fuli-length viral genome.
Previous reports of a similar mutation did not involve full-iength HIV-1 constnicts
and were perfomed in bacterial systems. not in vivo (LeGrice et al.. 1988b;
Debouck et al.. 1990; Louis et al.. 1991). In these studies, the activity of the PR
domain within the PR-RT cleavage site mutants varied. Effects ranged from
being ineffective to allowing partial processing and to completely blocking
cleavage at aiat site (Loeb et al., 1989a; Debouck et al., 1990; Louis et al.,
1 991). Moreover, RT activity in samples containing these cleavage site
mutations was not assessed in previous reports (Loeb et al., 1989a; Debouck et
al., 1990; Louis et a!., 1991). Thus. the study presented herein is the first
analysis of PR-RT fusion proteins in HlV-1 viral particles.
2. While it has been previously demonstrated that C-teminally extended
HIV-1 PR species are enzymatically active (Loeb et al.. 1989a; Debouck et al.,
1990; Louis et al., 1991; Wondrak et al., 7996), this is the first report of such an
extension as studied in a mammalian system. PR-RT fusion proteins in transfected cells and viral particles retained Vieir ability to process other HIV
proteins. Moteovet, this is the fnst demonstration that C-terminally extended PR
species possess increased processing capabilities.
3. These data are the first to show that HIV-1 M is active when expressed
as a fusion protein in virus particles. F K activity has been previously
demonstrated in unprocessed Gag-Pol precunon in ASLV (Stewart et ai.,

1990; Stewart and Vogt, 1991 and 1993). Mo-MLV (Crawford and Go#, 1985).
and HIV-1 (Lori et al.. 1988; Hu and m g , 1991; Peng et al., 1991; Mergener et
al., 1992), as well as Ri protease-defective HIV-1 particles (Gottlinger et al., 1989; Peng et al.. 1991; Mergener et al.. 1992: Park and Morrow, 1992).
However, the results presented here indicate wild-type levels of RT activity in novel 6 W kDa heterodimers when measured by in vitro and endogenous
assays.
4. This is the fint determination that C-terminal cleavage of PR is less
critical for proteolytic activity and virus maturation than N-terminal processing in
vivo. Disruption of the Cterminal cleavage site in the pPR-RT mutant did not
result in severe viral defects such as the abrogation of Gag precursot
processing and virus infectivity, as reported in vinises in which cleavage at the
N-terminus was blocked (Tessmer and Krausslich, 1998). Rather, viruses
containing PR-RT fusion proteins rernained viable and displayed only a 20-fold
reduction in infectivity. Moreover, these results have illustrated that the
liberation of RT from PR is a novel and important regulatory mechanism for wild-
type PR activity, ensu ring that premature enzyme activation and precu rsor
processing do not occur. These findings contribute to the understanding of HIV-
1 PR regulation during the production of infectious virions.
5. NoveI trans-dominant negative viral constructs were identified in these
studies. They were found to inhibit the replication of wild-type HIV-1 and to
diminish viral infectiousness in a dose-dependen t manner. This research
demonstrates the potential utility of a gene therapy approach that targets both
the frameshift and the PR-RT cleavage sites in HIV-1 infection.

CHAPTER 7
REF ERENCES

Accola, M.A., S. Hoglund and H.G= Gottllnger. 1998. A putative a- helical structure which overfaps the capsid-p2 boundary in the human immunodeficiency type 1 Gag precursor is critical for viral particle assembly. J. ViroC. 72:2072-2078.
Adams, L.D., A.G. Tomasselli, P. Robbins, B. Moss and R.L. Heinrlkson. 1992. HIV-1 protease cleaves actin during acute infection of h m a n T4ymp hocytes. AlDS Res. Hum. Ret roviruses 8:2QI-295.
Aiken, Ce, J. Konner, N.R. Landau, M.E. Lenburg and D. Trono. 1994, Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasrnic domain. Cell76:853-864.
Aiken, C. and D. Trono. 1995. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J. Vkof. 69:5048-5056.
Alain, R., Fœ Nadon, C. Seguin, P. Payment and M. Trudel. 1987. Rapid virus subunit visualization by direct sedimentation of samples on electron microscope grids. J. Virol. Methods I6:209=2t 6.
Alkhatib, O., C. Combadiere, C C Broder, Y. Feng, P.€. Kennedy, P.M. Murphy and E.A. Berger. 1996. CC CKR5: a RANTES, MIP-la, and MIP-1 B receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272: 1 955-1 962,
AIIain, B., M. Lapadat-Tapolsky, C. Berlioz and Je-L. Darlix. 1994. Transactivation of the minus strand DNA transfer by nucleocapsid protein during reverse transcription of the retroviral genome. EMBO J. 132464-2472.
Almog, Na, R. RolIer, G. Arad, L. Passi-Even, M.A. Walnberg and M. Kotler. 1996. A pôPa~ptotease fusion protein is present in mature particles of human immunodeficiency virus type 1. J. Virol. 70:7228-7232.
Ansari-Lari, M.A., L.A. Donehower and RA. Gibbs. 1996. Analysis of human immunodeficiency type 1 integrase mutants. Virology 70:3870-3875.
AnsarbLari, M.A. and R.A. Gibbs. 1996. Expression of hurnan immunodeficiency virus type 1 reverse transcriptase in trans during virion release and after infection. J. Virol. 70:3870-3875.
Arrfgo, S.J. and [.S. Chan. 1991. Rev is necessary for translation but not cytoplasmic accumulation of HIV-i vif; vpr, and envfvpu 2 RNAs. Genes Dev. 5:808-819.
Arrlgo, S.J. and K. Hulfman. 1995. Potent inhibition of human immunodeficiency virus type 1 (HIV-1) replication by inducible expression of H IV-1 PR rnultimers. J. VkoL 695988-5994,

Arya, S.& C. Guo, S.J. Josephs and Fm Wong-Staal. 1985. fmns- Activator gene of human T-lymphotropic virus type III (HTLV-III). Science 229:69-73,
Babe, L.M., S. Pichuantes and C.S. Craik. 1991. Inhibition of HIV protease activity by heterodimer formation. Biochem. 30:106-111.
Babe, LM., J. Rose and C.S. Craik. 1992. Synthetic 'interfacem peptides alter dimeric assembly of the HlV 3 and 2 proteases. Protein Sci. 1 :l244-t 253.
Babe, LM., J. Rose and CS. Craik. 1995. Trans-dominant inhibitory human immunodeficiency virus type 1 protease monomers prevent protease activation and virion maturation. P roc. Natl. Acad. Sci. USA 92: 1 0069-1 0073,
Bahner, lm, CD Zhou, X . 4 Yu, C2.-L. Hao, J-C. Guatelli and D.B. Kohn. 1993. Cornparison of trans-dominant inhibitory mutant human immunodeficiency virus type 1 genes expressed by retroviral vectors in human T lymphocytes. J. Virol. 67:3199-3207.
Bahnei, ID, K. Kearns, Q.-L. Hao, €.M. Smogorzewska and D.6. Kohn. 1996. Transduction of human CD34+ hematopoietic progenitor cells by a retroviral vector expressing an RRE decoy inhibits human im munodeficiency virus type 1 replication in myelomonocytic cells produced in long-terni culture. J. Virol. 70:4352-4360.
Baltimore, D. 1988. lntracellular immunization. Nature 335:395-396.
Barat, C., O. Schatz, S.H.J. LeGrice and Je-L. Datlix. 1993. Analysis of the interactions of HIV-1 replication primer tRNA(Lys3) with nucleocapsid protein and reverse transcriptase. J. Molec. Biol. 231 :l85-l9O.
Barre-Sinoussi, F m , J.C. Cherman, Re Rey, MX. Nugeryte, S. Charnaret, J. Gruest, C. Dauget, C. Axler-Blin, F. Verinet-Brun, CD Rouzloux, W. Rosenbaum and L Montagnier. 1983. Isolation of a T- lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868-871.
Barrie, KDAmr E.E. Perez, S.L. Lamers, W.G. Farmerie, B.M. Dunn, J.W. Sleaseman and MM. Goodenow, 1996. Natural variation in HIV-I protease, Gag p7 and p6, and protease cfeavage sites within Gag-Pol polyproteins: amino acid substitutions in the absence of protease inhibitors in mothers and children infected by human immunodeficiency virus type 1. Virology 21 9:4O%4l6.
Barsov, E.V., W.€. Huber, J. Marcotrigiano, P.K. Clark, A.D. Clark, E. Arnold and S.H. Hughes. 1996. lnhibition of human immunodeficiency virus type t integrase by the Fab fragment of a specific monoclonal antibody suggests that different rnultirnetization states are requited for diierent enzymatic functions. J. Virof. 7014484-4494.

Bartz, S.R., M.E. Rogel and M. Emerman. 1996. Human immunodeficiency virus type I cell cycle controk Vpr is cyiostatic and mediates 62 accumulation by a mechanism which diffen from DNA damage checkpoint controf. 3. Virol. 70:2324-2331.
Bennett, R.P., S. Rhee, RE. Craven, E. Hunter and J-W. Wllls. 1991. Amino acids encoded downstream of Gag are not required by Rous sarcorna virus protease during Gag-mediated assembly. J. Virol. 65:272-280.
Bennett, R-P*, T.D. Nelk and LW. Wtlls. 1993. FundionaI chimeras of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol- 67:6487-6498,
Berg, J. 1986. Potential metai-binding domains in nucleic acid binding proteins. Science 25:485-487.
Berkowitz, RD. and S.P. Goff. 1993. Point mutations in Moloney murine leukemia virus envelope protein: effects on infectivity, virion association and superinfection-resistance. Virology I96:748-757.
Berkowitz, R.D., A. Ohagen, S. Hoglund and S.P. Goff. 1995. Retroviral nucleocapsid domains mediate the specific recognition of genomic viral RNAs by chimeric Gag polyproteins during RNA packaging N1 vivo. J. Virol. 69~644506456.
Berkowitz, R., J. Fisher and S.P. Goff. 1996. RNA packaging. Curr. Top. Microbioi. Crnmunol- 2W:I7? -21 8.
Berson, J.F., D. Long, G.J. Doranz. J. Rucker, F.R. Jirik and R.W. Doms. 1996. A seven-transmernbrane domain receptor involved in fusion and entry of Fcell-tropic human imm unodeficiency virus type 1 strains. J. Virol. 70:6288-6295 .
Bevee, O.,. M. Dobrovnik, J. Hauber and E. Bohnlein. 1992. Inhibition of human rmmuoodeficiency virus type 1 replication in human T cells by retroviral-mediated gene transfer of a dominant-negative Rev trans-activator. Froc. Natl. Acad, Sci. USA 89:9870-9874.
Bleul, C.C., M. Fartan, H. Choe, C. Paroln, I. Clark-Lewis, J. Sodroski and TA. Sprfnget. 1996. The lymphocyte chemoattractant SDF-1 is a ligand for LESTWfush and blocks HIV-t entry. Nature 382829-832.
Bottcher, M. and F. Grosse. 1997. HIV-I protease inhibits its homologous reverse transcriptase by protein-protein interaction. NucIeic Acids Res. 2S:I 709-1 71 4.
Boulerice, F., S. Baur, R. Geletfunas, A. Lvovich and Y.A. Wainbeig. 1990. High frequency of isoIation of defective human

immunodeficiency virus type 1 and heterogeneity of viral gene expression in clones of infected U-937 cells- S. Virol. 64= 1745-1755,
Bour, S.. U. Schubert and K. Strebef. 1995. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: implications for the mechanism of degradation. J. Virol. 69A510-1520.
Boutelje, JO, A.R. Karistrom, M.G.N. Harfmanis, E. Holmgrem, A. Sjogren and R.L. Levine. 1990. Human immunodeficiency viral protease is catalytically active as a fusion protein: characterization of the fusion and native enzymes produced in Escherichia coli. Arch. Biochem. Biophys. 283:141-149.
Braaten, D., L K * hanke and J. Luban. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. ?O:3551-3560.
Bryant, M. and L. Ratner. 1990. Myristylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 87523-527.
Buchschacher, G.L.J.! E.O. Freed and A.T. Panganiban. 1995. Effects of second-site mutations on dominant interference by a human immunodeficiency virus type 1 envelope glycoproteni mutant. J. Virol. 69:l344- 1348.
Bukovsky, A. and H. Gottlinger. 1996. Lack of integrase can markedly affect human immunodeficiency virus type 1 particle production in the presence of an active viral protease. J. Virol. ?0:6820-6825.
Bukovsky, A.A., T. Dorfman, A. Weimann and HG. Gottlinger. 1997. Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. J. Virol, 71 :IO 13-1 018.
Bukrinsky, M.I., T.L. Stanwick, M.$. Dempsey and M. Stevenson. 1991. Quiescent T lymphocytes as inducible virus reservoir in HIV-1 infection. Science 254:423-427.
Bukrinsky, M.!., N. Sharova, M.P, Dempsay, T.L. Stanwkk, A.G. Bukrins kaya, S. Haggerty and M. Stevenson. 1 992. Active nuclear import of HIV-1 preintegration complexes. Proc. Natl. Acad. Sci. USA 98:6580- 6584.
Bukrinsky, M . S. Haggerty, M.P. Dempsey, N. Sharova, A. Adzftubel, 1. Spk, Dm Goldfarb, Mm Emerman and M. Stevenson. 1993. A nuclear localkation signai4 mat& that govems infection of non- dividing cells. Nature 365:666-669.

Burstein, Hm, D. Bbub and AM. Skalka. 1991. Assembly and processing of avian retroviral Gag polyproteins containing Iinked protease dimers. J. ViroI. 65:6165-6172.
Burstein, H., D. Blzub, M. Kotlet, G. Schatr, V.M. Vogt and A.M. Skalka. 1992. Processirtg of avian retroviral Gag polyptotein precursors is blocked by a rn utation at the NC-PR cleavage site. J. Virol. 66: 1 78 1 -1 785.
Camaur, D. and D. Ttono. 1996. Characterization of human immunodeficiency virus type 1 Vif padicle incorporation. J. Virol. ? O S 1 06-61 1 1.
Casella, CR., L m J m Raffinf and A.T. Panganiban. 1997. Pleiotropic mutations in the HIV-1 rnattix protein that affect diverse steps in replication. Vitology 228~294-306.
Cavert, W., D.W. Notermans, K. Staskus, S.W. Wietgrefe, M. Zupancic, K. Gebhard, K. Henry, SrC. Zhang, R. Mills, H. Y cDade, C.M. Schuwirth, J. Goudsmit, S.A. Danner and AT. Haase. 1997. Kinetics of response in lymphoid tissue to antiretroviral therapy of HIV-1 infection. Science 276:960-964.
Chackerian, B., €.M. Long, P.A. Luclw and J. Overbaugh. 1997. Hurnan im rn unodeficiency virus type 1 coreceptors participate in postentry stages in the virus replication cycle and function in simian im munodeficiency virus infection. 3. Virol. 7k3932-3939.
Champoux, JJ., E. Gilboa and D. Baltimore. 1984. Mechanism of RNA primer removal by the RNaseH activity of avian myeloblastosis virus reverse transcriptase. J. Virol. 49~686-691.
Champoux, JJ. 1993. Roles of ribonucleaso H in reverse transcription. p. 103-1 17. In Skalka, AM. and S.P. Goff, (ed.). Reverse Transcriptase- Cold S p ring Harbor Labo rato ry P ress, Cold S p t ing Harbor, N.Y.
Chan, D.C. and P.S. Kim. 1998. HLV entry and its inhibition. Cell 93:681- 684.
Chandra, A., T. Gerber and P. Chandra. 1986. Biochernical heterogeneity of reverse transcriptase purifiecf from the AIDS virus, HTLV-III. FEBS Lett. 197:84-88.
Chang, D.D. and P.A. Sharp. 1989. Regufation by HIV Rev depends upon recognition of s pke sites. CelE59:769-795.
Chameau, P. and F. Cfavel. 1991. A single-sttanded gap in human immunodeficiency virus unintegrated Iinear DNA defined by a central copy of the po Lypu rine tract. J. Virol. 6S:24t 5-2421.

Chazal, N., C. Carrlere, B. Gay and P. Boulanger, 1994. Phenotypic characteriration of insertion mutants of the human immunodeficiency virus type 1 Gag precursor expressed in recombinant baculovirus-infected cells. J. Virol. 68:lll-l22.
Chen, S.S.-L., AA. Ferrante and EoF. Teiwilliger. 1996. Characterization of an enveIope mutant of HIV-I that interferes with viral infectivity. Virolog y 226:260-268.
Cheng, Y.-S.E., FoH* Yin, S. Foundling, D. Blomstrom and C.A. Kettner. 1 990. Stability and activity of human immunodef iciency virus protease: Cornparison of the natural dimer with a homologous, single-chain tethered dimer, Proc. Natl. Acad. Sci. USA 87:9660-9664,
Choe, H o , M. Fanan, Y. Sun, N. Sullivan, B. Rollins, P.D. Ponath, L. Wu, C.R. Mackay, G. LaRosa, W. Newman, N. Gerard, C. Gerard and J. Sodroski. 1996. The B-chernokine receptors CCR3 and CCRS facilitate infection by pnmary HIV-1 isolates. Cell 85:1 t 35-1 148.
Chou, J. J. 1993. A vectorized sequence-coding model for predicting HIV protease cleavage sites in proteins. J. Biol. Cham. 268: 1 6938-1 6948.
Chun, T.-W., L. Carruth, De Flnzi, X. Shen, J.A. DlGiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T.C. Quinn, Y.- H. Kuo, R. Brookmeyer, M.A. Zelger, P. Barditch-Crovo and R.F. Sillcano. 1997. Quantification of latent tissue reservoirs and total body viral toad in HIV-1 infection. Nature 387: 183-1 88.
Clavel, F.D., Do Guetard. F. Vezlnet-Brun, S. Chamaret, M.A. Rey, M.0. Santos-Ferreira, A.& LaurentF C. Dauguet, C. Katlama and C. Rouzioux. 1986. Isolation of a new human retrovirus from West African patients with AIDS. Science 233:343-346.
Co, E., O. Koelsch, Y. Lin, E. [do, J.A Hartsuck and L. Tang. 1994. Proteolytic processing mechanisms of a miniprecursor of the aspartic protease of human immunodeficiency virus type 1. Biochem. 33: tZ48-l2S4.
Cocchl, F., A. DeVico, A. Ganino-Demo, S. Arya, R. Gallo and P. Lusso. 1995. Identification of RANTES. MI?-la. and MIP-1 as the major HIV- 1 suppressive factors produced by CD8+ T celk Science 2?0:1811-1815.
Cohen, €.AA., G. Dehni, J.O. Sodroski and W.A. Haseltine. 1990. Human immunodeficiency virus Vpr product is a virion-associated regu latory protein. J. Virol. 64:3097-3099.
C~lbr inik~ Dy C Soskey and J. Lels. 1988. A retroviral RNA secondary structure required for the efficient inWon of reverse transcription. J. Virol. 64:3622-3630.

Colgan, J., H.E.H. Yuan, E.K. Franke and J. Luban. 1996. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerkation. J. Virol. iO:4299-431 O.
Coli, Hm, 8. Fan, RoL Joshl, A. Ramezanl, X. Li and S. Joshi. 1994. Inhibition of H1V-1 multiplication in a human CD& lymphocytic ceIl line expressing antisense and sense RNA molecules containing HIV-1 packaging signa1 and rev response elements. Antisense Res. Dev. 4:19-29.
Collier, A.C., A.W. Coombs, D.A. Schoenfeld, R.L Bassett, J. Tlmpone et al. 1998. Treatment of human immunodeficiency virus infection with saquinavif, zidovudine, and zalcitabine. N. Engl. J. Med. 334:lOll-lO?8.
Colllns, ICL., B.K. Chen, S.AD Kalams and B.D. Walker. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391 :397-401.
Craven, R.C., R.P. Bennett and J.W. WilIs. 1991. Role of avian retroviral protease in the activation of reverse transcriptase during virion assembly. J. VitoC. 656205621 7.
Ciaven, R.C., A.E. Leure-duPree, CR. Erdle. C.B. Wilson and J.W. Wills. 1993. Necessity of the spacer peptide between CA and NC in the Rous sarcoma virus Gag protein. J. Viro t. 67:6246-6252.
Craven, R.C. and LJ. Parent. 1996. Dynamic interactions of the Gag polyprotein. Curr. Top. Microbiol. lrnmunol. 214:65-94.
Qawford, S. and S.P. Goff. 1985. A deletion mutant in the 5' part of the pol gene of Moloney murine leukernia virus blocks proteolytic processing of the gag and PO/ polyproteins. J. Virol. 53:899-907.
Cullen, B.R. 1991. Regulation of HIV-1 gene expression. FASEB J. 52361- 2368,
CulIen, B.R. 1992. Mechanism of action of regulatory proteins encoded by corn plex ret roviruses. Micro biol. Rev. 56~375-394.
Cullen, B.R. 1998. HIV-1 auxiliary proteins: rnaking connections in a dying celL Cet[ 93S85-692.
Curiel, TA, DA- Cook. Y. Wang, B.H. Hahn. S.K. Ghosh and O.S. Harrison. 1993. Long-term inhibition of dinical and laboratory human imrnunodeficiency virus strains in human Tkell lines containing and HIV- regulated diphtheria toxh A chain gene. Hum. Gene Ther. 4:741-f47.

Dannull, J., A. Surovoy. G. Jung and K. Moelling. 1994. Specific binding of HIV-1 nucleocapsid protein to PSI RNA In vitru requires N-teminal zinc finger and fi anking basic amino acid residues. EMBO J. t 3: 1525-1533.
Darke, P L , R.F. Nutt, S.F Brady, V w M m Garsky, TOM. Ckcarone, C. Leu, P.K. Lumma, R.M. Freldinger, D.F. Veber and LS. SIgal. 1988. HlV-1 protease specificity of peptide cleavage is sufficient for processing of Gag and Pol polyproteins. Biochem. Biophys. Res. Commun. 156:29?-303.
Darke, POL, S.P. Jordan, D L Hall, JoA. Zugay, J.A. Shafer and L.C. KUO. 1994. Dissociation and association of the HIV-1 protease dimer subunits: equilibria and rates. Biochem. 33:98-105.
Darlix, Jœ-L., A. Vincent, C. Gabus, H. DeRocquigny and 8. Roques. 1993. Trans-activation of the 5' to 3' vira1 DNA strand transfer by nucleocapsid protein during reverse transcription of HIV-1 RNA. Life Sciences 316:763-771.
Dailix, JO-L, M. Lapadat-fapolsky, H m DeRocquigny and B.P. Roques. 1995. FirSt glimpses at structure-function relationships of the nucleocapsid protein of retroviruses. J. Molec. Biol. 254523437.
Deacon, N.J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D.J. Hooke?, D-A. McPhee, A.L. Greenway, A. Ellett, C. Chatfield, V.A. Lawson, S. Crowe, A. Maerz, S. Sonza, J. Learmont, J S . Sullivan, A. Cunningham, D. Dwyer, D. Dowton and J. Mllls. 1995. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 2?0:988-99 1 . Debouck, C., J.O. Gorniak, J. Strickler, T.D. Meek, B.W. Metcaif and M. Rosenberg. 1987. Human immunodeficiency virus protease expressed in Escherichia coli exhibits autoprocessing and specific maturation of the Gag precursor. Proc. Natl. Acad. Sci. USA 848903-8906.
Debouck, C., I.C. Deckman, SmK. Grant. RJ . Cralg and M.L. Moore. 1990. The HIV-1 aspartyl protease: maturation and substrate specificity, p. 9-1 7. In Pearl, LH., (ed.), Retrovrial Proteases: controi of maturation and mo~phogenesis. Stockton Press, N.Y.
Debouck, C. and B.W. Metcalf. 1990. Human immunodeficiency virus protease: a target for AIDS therapy. Drug Dev. Res. 21:l-17.
DeGuman, R.N., ZR. Wu, C.C. Stalllng, L. Pappalardo, P.N. Borer and M.F. Summers. 1998. Structure of the HV-1 nucleocapsid protein bound to the SL3 iy-RNA recognition element. Science 279:384-388.
Deng, H., R. Uu. W. EIlmeier, S. Choe, D. Unutmaz. M- Burkhart, P. DtManfo, R. Sutton, CM. Hill. C.B. Davis, S.C. Peiper, T.J. Schall, D.R. Llttman and N.R. Landau. 1996. Identification of a major coreceptor for primary isolates of HIV-1. Nature 38k661-666.

DeRocqulgny, Hm, Cm Gabes, A. Vincent, M.G. Formie-laluski, B. Roques and 3.4. Dadix. 1992 Viral RNA annealing activities of human immunodeficiency virus type I nucleocapsid protein requires only peptide domains outside of the zinc fingers. Proc. Natt Acad. Sci. USA 89:6472-6476.
Dlckson, Cm, R Elsenman, H. Fan, E. Hunter and N. Telch. 1984. Protein biosynthesis and assembly, p. 513-640. In Weiss. R, N. Teich, H. Varmus and J. Coffin, (ed.)), RNA tumor vifuses, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
DIMaizo-Veronese, F., T.D. Copeland, A.L. DeVico, R. Rahman, S. Orosdan, R.C. Gallo and M.G. Samgadharan. 1986. Characterization of highly imrnunogenic p66fp51 as reverse transcriptase of HTLV-IIVLAV. Science 231 : 1289-1 29 1 . Dinman, J.D. and R.B. Wicknet: 1992. Ribosomal frameshifting efficiency and gaa'gag-pol ratio are criticai for yeast M l double-stranded RNA virus propagation. J. Virol. 66:3669-3676.
Doranz, B.J., J. Rucker, Y. Yi, R.J. Smyth, M. Samson, S.C. Peiper, M. Parmentier, R.G. Collman and R.W. Doms. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the P-chernokirte receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149-1158.
Dorfman, Tm, J. Luban, S.P. Goff, W. HaseItine and HmG- Gottlinger. 1993. Mapping of functionally important residues of a cysteine-histidine box in the hurnan immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 6?:6 159-6 1 69.
Dorfman, TmT F. Mammano, W.A. Haseltine and H-G. GottIinger. 1994a Rote of the matrix protein in the virion association of the human imrnunodeficiency virus type 1 envelope glycoprotein- J. Virol, 68:1689-1696.
Dorfman, T., A. Bukovsky. A. Ohagen, S. Hoglund and H.G. Gottllnger. 1994b. FunctionaI domains of the capsid protein of hurnan imrnunodeficiency virus type 1. J. Virol. 68:81806187.
Dragic, T., V. Litwin, G.P. Allaway, S.R. Maitfn, Y. Huang, K.A. Nagashima, C. Cayanan, PJ* Maddon, RA. Koup, J.P. Moore and W.A. Paxton. 1996. HIV-1 entry into CD& cells is mediated by the chemokine receptor CC-CKR5. Nature 381 :667-673.
Duan, L., O. Bagasra, M-A. Laughltn, LW. Oakes and R.J. Pomerantr. 1994. Potent inhibition of human immunodeficiency virus type 1 replication by an intracelMar anti-Rev single chain antibody. Proc. Natl. Acad. Sci. USA 9 t 5075-5079.

Dunn, B.M., A. Gustchina, A. Wlodawer and J. Kay- 1994. Subsite pieferences of retroviral proteinases. Methods Enzymol. 241:254-278.
Earl, P L , B. Moss and R.W. Doms. 1991. Folding, interaction with GRP78-BiP. assembly and transport of human immunodeficiency virus type 1 envelope protein. J. Virol. 65:2047-2055.
Einfeld, D. 1996. Maturation and assembly of retroviral glycoproteins. Curr. Top. Microbiol. Immunol. 2M:t 33-176.
El-Farrash, M.A., M.J. Kuroda, T. Kitazakl, T. Masuda, K. Kato, M. Hatanaka and S. Harada. 1994. Generation and characterization of a human immunodeficiency virus type 1 (HIV-1) mutant resistant to an HIV-1 protease inhibitor. J. Virol. 68:233-239.
Ernbretson, J., M. Zupanclc, J.L. Ribas, A. Burke, P. Racz, K. Tenner-Racz and A.T. Haase. 1993. Massive covert infection of helper T lymphocytes and macrophages by HIV du ring the incubation period of AIDS. Nature 362~359-362.
Emerman, M. 1996. HIV-1, Vpr and the ceIl cycle. Curr. Biol. 6: 1 096-1 103.
Engelman, A., K. Mizuuchi and R. Craigie. 1991. HLV-1 DNA integration: mechanism of viral DNA cleavage and strand transfer. CeII 67: 12 1 1-1 221.
Engelman, A., G. Englund, J.M. Orenstein, M.A. Martin and RD Craigie. 1995. Multiple effectç of mutations in human immunodeficiency vinis type 1 integrase on viral replication. J. Virol. 69:2729-2736.
Erickson-Viltanen, S., J. Manfredi, P. Viitanen, D.E. Tribe, R. Tritch, C.A. Hutchison III, D.0. Loeb and R. Swanstrom. 1989. Cleavage of HIV-1 gag polyprotein synthesized in vitra sequential cleavage by the viral protease. AIDS Res. Hum. Retrovinises 5577-591.
Escafch, S., C. Kalfoglou, 1. Plavec, S. Kaushal, J.D. Mosca and E. Bohnlein. 1995. RevM 1 0-mediated inhibition of HIV-1 replication in chronically infected T cells. Hum. Gena Ther. 6:625-634.
Ewart, G.D., To Sutherland, P.W. Gage and G.B. Cox. 1996. The Vpu protein of human imrnunodeficiency vinis type 1 forms cation-selective ion channels- 3. Virol, 70:7108-7115.
Facke, Y., A. Janetzko, R.L, Shoeman and l4D-G- Krausslich. 1993. A large deletion in the matk domain of the human imrnunodeficiency virus gag gene redirects virus particle assembty from the plasma membrane ta the endoplasmic reticulum. J. Virol. 67:4972-4980.

Fainet, C.M. and WoA. Haselthe. 1991. Detemination of viral proteins present in the human immunodeficiency virus type 1 preintegration cornplex. J. Virol. 65:1910-1915,
Fauci, AS. 1996. Host factors and the pathogenesis of HIV-induced disease. Nature 384~529-534.
Felnberg, M.B. and Dm Trono. 1992. lntracellular immunkation: trans- dominant mutants of HIV gene products as tools for the study and interruption of viral replication. AIDS Res. Hum. Retroviruses 8:lO13-lO22.
Felsensteln, K.M. and S.P. Goff. 1988. Expression of the Gag-Pol fusion protein of Moloney murine leukemia virus without Gag protein does not induce virion formation or proteoIytic processing. JO Virol. 62:2179-2?82.
Feng, Y., CG. Broder, POE. Kennedy and E.A. Berger. 1996a. HIV-1 entry cofactor. functional cDNA cloning of a seven-transmem brane, G protein- coupled receptor. Science 272:872-877.
Feng, Y.-X., T.D. Copeland, LE. Henderson, RoJ. Gorelick, W.J. Bosche, J.G. Levin and A. Rein. 1996b HIV-1 nucleocapsid protein induces 'maturation' of dimeric retroviral RNA in vitro. Proc. Natl. Acad. Sci, USA 93~7577-7581-
Finzi, D. and R.F. Silicano. 1998. Viral dynamics in HIV-1 infection. Cell 93~665-671.
Fischer, U., Je Huber, W.C. Boelens, LW. Mattaj and R. Luhimann. 1995. The HIV-1 activation dornain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82~475-483.
Franke, E.K., H.E.H. Yuan and J. Luban. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 3?2:359-362.
Franke, E.K. and J. Luban. 1996. Inhibition of HIV-1 replication by cyclosporine A or related cornpounds correlates with the ability to disnipt the Gag-cyclophilin A interaction. Virology 222279-282,
Frankef, S.S., B.M. Wenlg, A.P. Burke, P. Mannan, L.D.R. Thompson, S.L. Abbondanzo, AM. Nelson, M. Pope and R.M. Steinman. 1996. Replication of HIV-1 in dendritic cell-derived syncytia at the mucosat surface of the adenoid. Science 2721 15-1 17.
Freed, E.O., E.L. Delwart, G.J. Bushschachei and Af.. Panganlban. 1992. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dorninantIy interferes with fusion and infectivity. Proc. NatL Acad. SCÎ. USA 89:tO-74.

Freed, €.O., J.M. Orenstein, A.J. Buckler-White and M.A. Martin. 1994. Single amino changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 68:53 t 1 -5320.
Freed, E.O., G. Englund and MA. Marün. 1995. Role of the basic domain of human immunodeficiency virus type t matrix in macrophage infection. J. Virol, 69:3949-3954,
Freed, E.O. and M.A. Martin. 1995. The role of human immunodeficiency virus type 1 envelope glycoproteins in virus infection. J. Biol. Chem. 2?0:23883-23886.
Freed, E.O. and M.A. Martin. 1996. Domains of the human immunodeficiency virus type 1 matrk and gp41 cytoplasrnic tail required for envelope incorporation into virions. J. Virol. ? O : W -351.
Fu, W. and A. Rein. 1993. Maturation of dimeric RNA of Moloney murine leukemia virus. 3. Virol. 6715443-5449.
Fu, W., R.J. Gorelick and A. Rein. 1994. Characterization of human imm unodeficiency virus type 1 dimeric RNA from wild-type and protease deficient virions- JI Virol. 68:SOf 3-50 18.
Fuller, S.D., T. Wilk, B.E. Gowen, H&. Krausslich and V.M. Vogt. 1997. Cryo-electron microscopy reveals ordered domains in the immature HlV particle. Curr. Biol. 7:729-738.
Gabuzda, D M , K. Lawrence, E. Langhoff, E. Tennilliger, 1. Dorfrnan and W.A. Haseltine. 1992. Role of vif in replication of human imm unodeficiency virus type 1 in CD4+ lymphocytes. J. Viro l. 66:6489-6495.
GaIlaher, W.R., J.M. Balf, R.F. Garry, M.C. Griffin and R.C. Montelaio. 1989. A general model for the transmembrane proteins of HIV and other retroviruses- AlDS Res. Hum. Retroviruses 5431 -440.
Gallay, P., S. Swlngler, CC. Aiken and 0. Trono. 1995. HlV-1 infection of non-dividing cells: C-terminal tyrosine phosphoqdation of the viral matrix protein is a key regulator. CelI 80:379-388.
GaIIo, R.C., S.Z. Salahuddln, M. Popovic, G.M. Shearer, Y. Kaplan, B.F. Haynes, T.J. Palker, R. Redffeld, 4. Oleske. B. Safai, G. White, P. Foster and P.D. Markham. 1984. Frequent detection and isolation of cytopathic retrovirus (HKV-III) from patients with AIDS and at risk for AIDS. Science 224:500-503.
Gamble, KR., S. Yoo, F.F. Vaidos, U.K. VonSchwedler, J.P. McCutcheon, W.I. Suncfquist and C.P. Hill. 1997. Structure of the carboxyterminal dimerkation domain of the HlV-1 capsid protein. Science 273:849-853.

Garda, JDVm and A.D. Miller. 1991. Serine phosphorylation-independent downregulation of celbsurface CD4 by nef. Nature 350:508-511.
Garnier, L., L. Ratner, B. Rovinskl, S.4. Cao and J.W. Wills. 1998. Particle size determinants in the human immunodeficiency virus type 1 Gag protein. J. Virol. 724667-4677.
Gatlln, J., S.J. Arfigo and M.G. Schmidt. 1998. Regulation of intracellular human imrnunodeficiency virus type-1 protease activity. Virology 244:87-96.
Gay, B., J. Tournier, N. Charal, C. Carilere and P. Boulanger. 1998. Morphopoietic determinants of HIV-1 Gag particles assembled in baculovirus- infected cells. Virology 247: 160-1 69.
Gelderbloom, H.R., E.H.S. Hausmann, M. Ozet. G. Pauli and M.A. Koch. 1987. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology 1 56: 1 7 1 -1 76.
Gelderbloom, H.R. 1991. Assembly and morphology of HIV: potential effect of structure on viral function. AIDS 5:617-638.
Gheysen, Dm, E. Jacobs, F. DeFoiesta, Cm Thirlart, M. hancotte, D m Thlnes and M. DeWilde. 1989. Assembly and release of HIV-1 precursor Pr55m virus-like particles from recombinant baculovirus-infected insect cels. Cell 59: 1 03-1 12.
Giam, C.Z. and 1. Boros. 1988. In vivo and in vitro autoprocessing of human immunodeficiency virus protease expressed in Escherichia coli J. Bio l. Chem. 263: 1 461 7- 14620.
Gilboa, E., S.W. Mitra, S.P. Goff and D. Baltimore. 1979. A detailed mode! of reverse transcription and tests for crucia! aspects. CeII 18:93-100.
Goff, S.?. 1990. Retroviral reverse transcriptase: Synthesiç, structure, and function, 3. AIDS 3:817-831.
Goh, W.C.. M.€. Roget, C.M. Kinsey, S.F. Michael, P.N. Fultz, M.A. Nowak, B.H. Hahn and M. Emerman. 1998. HIV-I Vpr increases viral expression by manipulating the cell cycle: a rnechanisrn for selection of Vpr in vivo, Nature Med- 4:65-71.
Gojobori, Tm. E. Moriyama, Y. [na, K. Ikeo, f. M b a , H. Tsujlmoto, M. Hayarni and S. Yokoyama. 1990. Evolutionary origin of human and simian irnmunodeficiency viruses. Proc. Nat[. Acad. Sci USA 87:4108-4111.
Goobar-Larsson, L.. BDGDM- Luukkonen, T. Unge, S. SchwartZ, G. Utter. B. Strandberg and B. Oberg. 1995. Enhancement of HIV-1 proteinase activity by HIV-I reverse transcriptase. ViroIogy 206:387-394.

Goobar-Larsson, L, P.T. Larsson. C. Debouck and €.M. Towler. 1996. HlV-1 RT enhances the activity of a tethered dimer of HlV-1 proteinase. Biochem. Biophys. Res. Commun. 220:203-207.
Gorelick, R.J., LE. Hendeison, J.F. Hanser and A Rein. 1988. Point mutants of the Moloney murine leukemia virus that fair to package viral RNA: evidence for specific RNA recognition by a 'zinc finger-liket protein sequence. Proc. Natt. Acad. Sci. USA 85:8420-8424.
Gorelick, R.J., S.M. Nigida, J.R. Bess, L.O. Arthur, LE. Henderson and A. Rein. 1990. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA J. Vïrol. 64:3207-3211.
Gottlinger, H.G., J.O. Sodroskl and W.A. Haseltine. 1989. Role of capsid precursor processing and myristyfation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 865781- 5785,
Gottllnger, H., T. Dorfman. J. Sodroskl and W. Haseltine. 1991. Effect of mutations affecting the p6 Gag protein on human immunodeficiency viral particle release. Proc. Natl. Acad. Sci. USA 88:3 1 95-3 1 99.
Gottlinger, H.G., 1. Dorfman. €.A. Cohen and W.A. Haseltine. 1993. Vpu protein of human immunodeficiency virus type 1 enhances the release of capsids produced by gag gene constructs of widely divergent retroviruses. Proc. Natl. Acad. Sci. USA 9O:?38 1 -7385.
Gowda, S.D., B.S. Stein and E.G. Engleman. 1989. Identification of protein intermediates in the processing of the p55 HIV-1 gag precursor in cells infected with recombinant vaccinia virus. J, Biol, Chernt 264:8459-8462.
Graves, M.C., J.J. Lim, €.P. Heimer and R.A. Krarner. 1988. An Il-kDa fom of human imm unodeficiency virus protease expressed in Escherichia coli is suffcient for enzymatk activity. Proc. Nat!. Acad- Sci. USA 852449-2453.
Green. M., M. lshlno and P. Loewenstein. 1989. Mutational analysis of HIV-1 Tat minimal domain peptides: identification of tramdominant mutants that suppress HIV-LTR-driven gene expression. CeII 58:ZlS-223.
Greene. W.C. 1991. The molecular bioIogy of human immunodeficiency virus type t infection. N. Engl. J. Med. 324:308-317.
Gu, Z., X. Gao, X. Li., M.A. Parniak and M.A. Wainberg. 1992. Novel mutation in the hum an imm unodeficiency virus type t reverse transcriptase that encodes cross-resistance to 2,Sdideoxyinosine and 2p3'-dideoxy~ytidine, J. VkoL 66:?1.28-7 1 35.

Hadzopolou-Cladares, Mw, B K Felber, C. Cladacos, A. Athanassopoulos, A. Tse and G o N e Pavlikis. 1989. The Rev (tdart) protein of human immunodeficiency virus type 1 affects viral mRNA and protein expression via a cisacting sequence in the env region. J. Virol. 63:1265-1274.
Hallenberger, S., V. Bosch, Hm Angliker, E. Shaw, H.-D. Klenk and W. Gartek 1992. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 360:358-362.
Hammei, S.M., KeE. Squires, M.D. Hughes, J.M. Grimes, L.M. Demeter et al. 1997. A controlled trial of hnro nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 pet cubic millimeter or less. N. Engl. J. Med. 337:725-733.
Hansen, J., T. Schulze, W. Mellert and K. MoeIIlng. 1988. Identification and characterization of H1V-specific RNase H by monoclonal antibody. EMBO J. 7:239-243.
Haseltine, W.A. 1988. Replication and pathology of the AIDS virus. J. Acquired 1 mmune Defic. Syndrome 1 :217-240.
Haseltine, W.A. 1991. Molecular biology of hurnan immunodeficiency virus type 1. FASEB J. 512349-2360.
Heinzfnger, N., M. Bukrinsky, S. Haggerty, Aœ Ragland. V. Kewal- Ramlnl, M. Lee, He Gendelman, M. Stevenson and M. Emerman. 1994. The HIV-1 Vpr protein influences nucfear targeting of viral nucleic acids in non-dividing ceIIs. Proc. Natl. Acad. Sci. USA 91 :73 1 1-73 15.
Hellerstein, M.K. and J.M. McCune. 1997. T cell turnover in HIV-1 disease. Imrnunity 2583-589.
Henderson. L.E., TeD. Copeland, R.C. Sowder, AmMe SchuItz and S. Orosdan. 1988. AnaIysis of proteins and peptides purified from sucrose gradient bonded HTLV-III, p. 135447. ln Bolognesi, D., (ed.), Human Retroviruses, Cancer. and AIDS: approaches to preventkm and rherepy. Alan R Liss, lm., N.Y.
Henderson, LE., MwAœ Bowers, R.C. Sowder, S.A. Serabyn, D.G. Johnson, J.W. Bess, L.O. Arthur, D.K. Bryant and C. Finselan. 1992. Gag proteins of the highly replicative LM strain of human immunodeficiency virus type 1: post-translationai modifications, proteolytîc processing and complete amino acid sequences. J. Virol- 66:1856-1865.
Herbeln, O., U. Mahlknecht, F. Batliwalle, P. Gregersen, T. Pappas, J. Butler, WoAœ O'Brien and E. Verdin. 1998. Apoptosis of CD8+ Tcelb is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 395: 189-1 94.

Heiskowitz, 1. 1987. Functional inactivation of genes by dominant negative mutations. Nature 329~219-222.
Ho, DeDos T. Toyoshlma, Hm Mo, D.J. Kempf, O. Norbeck, C.-M. Chen, E. Wldeburg, S.K. Buct, $.W. Eiickson and M.K. Singh. 1994. Characteritation of human immunodeficiency virus type 1 variants with increased resistance to a C2-syrnmetric protease inhibitor. J. Vkol. 68:2016- 2020-
Ho. D.D., A.U. Neumann, AS. Perelson, W. Chen, J.M. Leonard and M. Markowitz. 1995. Rapid turnover of piasma virions and CD4 lymphocytes in HIV-1 infection, Nature 373:123-126.
Hoglund, S., L-G. Ofverstedt, A, Nilsson, P. Lundquist, H. Gelderblom, M. Ozel and U. Skoglund. 1992. Spatial visualization of the maturing HIV-1 core and its Iinkage to the envelope. AlDS Res. Hum. Retroviruses 8:l-17.
Hoglund, S., A. Ohagen, K. Lawrence and D. Gabuzda. 1994. Role of vif during packing of the core of HIV-1. Virology 20t349-355.
Hope, T.J., N.P. Klein, Mo Elder end TmG. Parslow. 1992. Trans- dominant inhibition of human immunodeficiency virus type 1 Rev occurs through formation of inactive protein complexes. J. Virol. 66:1849-1855.
Hoshikawa, M.B., R.F. Jarrett, A Aidovlni, R.C. Gallo and S.F. Wong. 199 1. Role of the gag and pol genes of human immunodeficiency virus in the morphogenesis and maturation of retrovirus-like particles expressed by recombinant vaccinia virus: an ultrastructural study. J. Gen. Virol. 72:2509- 2517,
Hostomska, 2.. D.A. Matthews, J.F. Davies II, B.R. Nodes and 2. Hostomsky. 1991. Proteolytic release and crystallization of the RNaseH domain of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 266:f 4697-7 4702,
Hu, S.-L., B.M. Travis, J. Garriges, JoMe Zarllng, P. Siidhar, - T m Dykers, J.W. Eichberg and C. Alpers. 1990. Processing, assernbly and imm unogenicity of human immunodeficiency virus core antigens expressed b y recombinant vaccinia virus. Virology I79:3ZT -329.
Hu, W.-S. and H.M. Temin. 1990. Retroviral recombination and reverse transcription. Science 250: 1 227-1 233.
Hu, YPW. and C.Y. Kang. 1991, Enzyme activities in four different foms of human immunodeficiency virus 1 pol gene products. Proc. Nat[. Acad. Sci. USA 88~4596-4600.

Huang, M., J.M. Orenstein, M.A. Martin and E.O. Freed. 1995. p6vg is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J. Virol. 69:68l&6818.
Huang, M. and M.A. Martin. 1997. Incorporation of P r l G F C into virus particles requires the presence of both the major homorogy region and adjacent Gterminal capsid sequences within the Gag-Pd polyprotein. J. Virol. 7t :4472- 4478.
Huber, H.E., J.M. McCoy, J.S. Seehra and C.C. Richardson. 1989. Human imrnunodeficiency virus 1 reverse transcriptase: template binding, processivity, strand dis placement synthesis and tem plate switching. J. Biol. Chem. 264~4669-4678.
Hughes, B.P., T.F. Booth, AS. Belyaev, D. Mcllroy, J. Jowett and P. Roy. 1993. Morphogenic capabilities of human imrnunodeficiency virus type 1 gag and gag-pol proteins in insect cells. Virology 193:242-255.
Hung, M., P. Patel, S. Davis and S.R. Green. 1998. Importance of ribosomal frameshifting for human immunodeficiency virus type 1 particle assem bly and replication. J. Virol. 72:48 1 9-4824.
Hunter, E. 1994. Macromolecutar interactions in the assembiy of HIV-1 and other retroviruses, Semin- Virol- 5:71-83,
Hwang, S.S., T.J. Boyle, H.K. Lyetly and B.RD CulIen. 1991. Identification of the envelope V3 loop as the primary determinant of cal1 tropism in HIV-1, Science 2S3:f 1-74,
Jacks, T., M.D. Power, F.R. Masiarz, P.A. Luciw, P.J. Bari and H.E. Varmus. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280-283.
Johnson, R.T. 1995. The pathogenesis of HIV infections of the brain. Curr. Top. Microbiol. I mmunol. 2 O Z S I O .
Jones, KA., G. Blaug, M. Hansen and E. Barklis. 1990. Assembly of gag-beta-galactosidase proteins into retrovirus particles. J. Virol. 64:2265- 2279.
Jones, K.S., J. Cokman, J. Merkel, G.W. Laue and A.M. Skalka. 1992. Retroviral integrase functions as a rnultimer and can turn over catalytically. J. Biol. Chem. 267: 1 6037-1 6040.
Joshi, S., A. van Brunschot, S. Asad, I. van der Elst, S.E. Read and A. Bernstein. 1991. Inhibition of human bmunodeficiency virus type 1 multiplication by antisense and sense RNA expression. J. Virol, 655524-5530.

Jowett, J., D. Hockky, M.V. Nermut and LM. Jones. 1992. Distinct signals in human immunodeficiency virus type 1 Pr55 necessaiy for RNA binding and particle formation. J. Gan. Virol. 73:30?9-3086.
Jowett, AB., V. Planelles, B. Poon, N.F. Shah, M.L. Chen and I.S. Chen. 1995. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the O, + M phase of the cell cycle. J. Virol. 69:6304-6313.
Junker, U., S. Escaich. I. Plavic, J. Baker, F. McPhee, J.R. Rose, CS. Craik and E. Bohnlein. 1996. lntracellular expression of hurnan immunodeficiency virus type 1 (HIV-1) protease variants inhibits replication of wild-type and protease inhibitor-resistant HIV-I strahs in human T-cell fines. J. VitoL 70~7765-7772.
Kageyama, S., D-TB Hoekzema, Y. Muiakawa, E. Kojima, TB Shirasaka, D.J. Kempf, D.W. Norbeck, J. Eiickson and H. Mitsuya. 1994. A C2 symmetry-based HIV protease inhibitor. A77003, irreversibly inhibits infectivity of HlV-1 in vitro. AlDS Res. Hum. Retrovinises 10:735-743.
Kanki, P.J., M.F. McLans, N.W.J. King, N.L. Letvfn, R.D. Hunt, P. Sehgal, M.D. Daniel, R.C. Desrosiers and M. Essex. 1985. Serological identification and characterization of a macaque T-lymphotropic retrovirus closely related to HTLV-I II. Science 228: 1 199- 120 1 .
Kaplan, A.H. and R. Swanstrom. 1991. HIV-t Gag proteins are processed in two cellular compartments. Proc. Natl. Acad. Sci. USA 88:4528-4532.
Kaplan, A.H., J.A. Zack, M. Knigge, D. Paul, D.J. Kempf, L W , Notbeck and R. Swanstrom. 1993. Partiai inhibition of the hurnan immunodeficiency virus type 1 protease results in aberrant virus assembly and the formation of noninfectious particles. J. Virol. 624050-4055.
Kaplan, A.H., M. Manchester and R. Swanstrom. 1994. The activity of the protease of human irnmunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol. 68:6782-6786.
Karacostas, V., K. Nagashima, M.A. Gonda and B. Moss. 1989. Human immunodeficiency virus-like particles produced by a vaccinia virus expression vector. Proc. Natl. Acad. Sci. USA 86: 8964-8967.
Karacostas, V., E.J. WoMfe, K. Nagashlma, M.A. Gonda and B. Moss. 1 993. Overexpression of the HIV-1 Gag-Pol polyp rotein results in intracellular activation of HIV-1 protease and inhibition of assembly and budding of virus-like particles. Virology I93:66f -671.
Karczewski. M.K. and K. Strebel. 1996. Cytoskeleton association and virion incorporation of Vie human immunodeficiency virus type 1 Vif protein. J. Virol. 70:494-507.

Katoh, I., Y. Yoshinaka, A. Rein, M. Shibuya, T. Odaka and S. Oioszlan. 1985. Mu rine leukemia virus maturation: protease reg ion required for conversion from 'immaturea to 'mature' core form and for virus infectivity. Virology 145:280-292.
Katoh, L, Y. lkawa and Y. Yoshinaka. 1989. Retrovirus protease characterized as a dimeric aspartic protease. J. ViroL 63:2226-2232.
Katz, RDA. and A.M. Skalka. 1994. The retroviral enzymes. Annu. Rev. Biochem. 63: 133-1 73.
Kay, J. and B.M. Dunn. 1990. Viral proteinases: weakness in strength. Biochem. Biophys. Acta I040:l-18.
Kaye, J.F. and ML. Lever. 1996. Trans-acting proteins involved in RNA encapsidation and viral assembly in human immunodeficiency virus type 1. J. Virot. 70:880-886.
Kestler, H.W., D.J. Dingler, K. Mori. D.L. Pankali, P.K. Sehgal, M.0. Daniel and R.C. Desrosiers. 1991. Importance of the nef gene for maintenance of high virus loadç and for development of AIDS. Cell 65651-652.
Khan, R. and D.P. Giedroc. 1992. Recombinant human immunodeficiency virus type 1 nucleocapsid (NCp7) protein unwinds tRNA JO Biol. Chem. 267:6689-6695.
Klernan, R.E., A. Ono, G. Engtund and €.O. Freed. 1998. Role of rnatrix in an early postentry step in the human imrnunodeficiency type 1 life cycle. J. Virol, 72:4116-4126.
Kimkait, T., K. Strebel, M.D. Hoggan, M.A. Martin and J.M. Orenstein. 1990. The human immunodeficiency virus type 1-specific protein Vpu is required for efficient virus maturation and release. J. Virol. 64:62t -629.
Kingsman, S.M. and A.J. Kingsman. 1996. The regutation of human immunodeficiency virus type 1 gene expression. Eur. JO Biochem. 240:491-507.
Kirchhoff, F., T.C. Greenough, D.B. Btettler, J.L. Sullivan and R.C. Desrosiers, 1995. Absence of intact nef sequences in a long-terni sunrivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332228432.
Kohl. No€., €.A. Emini, W.A. Schleif, LJ. Davis, J-C. Helmbach, R.A.F. Dixon, E.M. ScoInIck and I.S. SigaI. 1988. Active human immunodeficiency virus protease is required for viral infectivity. P roc. Nat!. Acad. SC^, USA 85:4686-4690.

Kondo, E., F. Yammano, E.A. Cohen and KG. Gottlinger. 1995. The p6** domain of human immunodeficiency virus type 1 is sufficient for the incorporation of Vpr into heterologous viral particles. J. Virol. 692759-2764.
Kotler, M., R.A. Katz, W. Danho, J. Leis and A.M. Skalka. 1988. Synthetic peptides as substrates and inhibitors of a retroviral protease. Proc. Natf. Ac&, Sci. USA 85807-81 1,
Kotler, M., O. Arad and S.H. Hughes. 1992. Human immunodeficiency virus type 1 gapprotease fusion proteins are enzymatically active. J. Virol. 66:678 t -6783.
Kowalski, M., 4. Potz, L. Basiripour; T. Dorfman. W.C. Goh, E. Terwilliger, A. Dayton, C. Rosen, W. Haselthe and J. Sodroski. 1987. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 2W:T 351 -1 355.
Krausslich, H&., H m Schneider, O. Zybarth, C.A. Carter and E. Wimmer. 1988. Processing of NI vitresynthesized gag precursor proteins of human imrnunodeficiency virus (HIV) type 1 by HIV proteinase generated in Escherichia colt: 3, Virol. 62:4393-4397.
Krausslich, H.G. and E. Wimmer. 1988. Viral proteinases. Annu. Rev. Biochem. 54:701-754.
Krausslich, HG., R.H. Ingraham, M . Skoog, E. Wirnmer, P.V. Paltai and C.A. Carter. 1989. Activity of purified biosynthetic proteinase of human immunodeficiency virus on natural substrates and synthetic peptides. Proc. Natf. Acad. Sci. USA 86:807-8 1 1.
Krausslich, HG. 1991. Human immunodeficiency virus proteinase dimer as a component of the viral potyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA 88:3213-3217.
Krausslich, H.G. 1 992. Specific inhibitor of human immunodeficiency virus proteinase prevents the cytotoxic effects of a single-chain proteinase dimer and redores particle fomation. J. Virol. 665670572.
Krausslich, H-Gœ, C. Ochsenbauer, A.Y. ftaenckner, K. Mergener, M. Facke, H.R. Gelderblom and V. Bosch. 1993. Analysis of protein expression and virus-like particle formation in mammalian cell lines stably expressing HIV-1 gag and env gene products with or without active HIV proteinase. Virolcigy 192:605-6l?.
Kraussllch, H&.. M. Facke, APM. Heuset, J. Konvalinka and H. Zentgraf. 1995. The spacer peptide between human imrnunodeficiency virus capsid and nucIeocapsid proteins is essential for ordered assembly and viral infectivity. J. Virol. 69:34O7-34 19.

Kulkosky, J. and A M Skalka. 1994. Mokcular mechanism of retroviral DNA integration. P hannacol. Thet. 61 ~185-203.
Kuzmic, P. 1993. Kinetic assay for HlV proteinase subunit dissociation. Biochem. Biophys. Res. Commun. l9l:Q98-lOU3.
Kwong, P.De, R Wyatt, J. Robinson, R.WD Sweets, J. Sodtoski and W.A. Hendtickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659,
Lamb, RDA. and L.H. Phto. 1997. Do Vpu and Vpr of human immunodeficiency virus type 1 and NB of influenza B virus have ion channel activities in the viral Iife cycles? Virology 229:l-11.
Lambert, O-Mm, SDRe Petteway, C.E. McDanal, T.K. Hart, J.J. Leary, GoB. Dreyer, T.D. Meek, P.J. Bugelski, D.P. Botognesi, B.W. Metcalf and T.J. Matthews. 1992. Human immunodeficiency virus type 1 protease inhibitors ineversibly block infectivity of purified virions from chronically infected celis. Antimicrob. Agents Chernother. 36:982-988.
Lappatto, R., Tm Blundell, A. Hemmlngs, J. Overington, A. Wilderspin, S. Wood, R. Merson, P.J. Whittle, 0.E- Danley, K.F. Geoghegan, S.J. Hawiylik, S.E. Lee, K.G. Scheld and P.M. Hobart. 1989. X-ray analysis of HIV-1 proteinase at 2.7 A resolution confirms structural homology among retroviral enzymes. Nature 342:299-302.
Laspia, M.F., AmPm Rice and M.B. Matthews. 1989. HIV-1 Tat protein increases transcriptional activation and stabilizes elongation. Cell 59:283-292.
Lavallee, C., X.J. Yao, A. Ladha, H.G. Gottlinger, WmA Haseltine and E.A. Cohen. 1994. Requirement of Pr55@gprecursors for incorporation of the Vpr product into human immunodeficiency virus type 1 particles. J. Virol. 68:1926-l934.
Lee, P.P. and M.L. LiniaI. 1995. Inhibition of wild-type HIV-1 virus production be a matrix deficient Gag mutant. Virology 208:808-811.
Lee, S.W., B.A. Sullenger, H.F. Gallardo, G.E. Ungers and E. Gilboa. 1992. Overexpression of RRE-derived sequences inhibits HIV-1 replication in CEM cells. New Biol. 4:66-74.
Lee, S.W., H.F. GaHardo, E. Gilboa and C. Smith. 7994. Inhibition of human immunodeficiency virus type 1 human Fcells by a potent RRE decoy comprised of the 13-nucleotide minimal Rev-binding domain. J. Virol. 68:8254- 8264.
Lee, YtM., X.-B. Tang, LmM. Clmakasky, J . t K Hildreth and L F . Yu. 7997. Mutations in the m a t h protein of human immunodeficiency virus

type 1 inhibit surface expression and virion incorporation of viral envelope giycoproteins in CD4+ T lymphocytes. J. Virol. 71 :T443-1452.
LeGaII, S., L. Erdtmann, S. Benichou, C. BerIjoz-Torrent, L Liu, R. Benarous, JAR. Heard and 0. Schwartz. 1998. Nef hteracts with the p subunit of clatherin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. lmmunity 8:483-495.
LeGrice, S., R. Zehnle and J. Mous. t988a. A single 66-kilodalton peptide processed from the human immunodeficiency virus type 2 pol polyp rotein in Escherichia coli disp lays reverse transcriptase activity. J. Virol. 62~2525-2529.
LeGrice, S.F.J., J. Mills and J. Mous. 1988b. Active site mutagenesis of the AIDS virus protease and its alleviation by trans complementation. EMBO J. 7~2547-2553.
LeGrice, S.F.J., Re Ette, J. MilIs and J. Mous. 1989. Cornparison of the human immunodeficiency virus type 1 and 2 proteases by hybrid gene construction and tram-complementation. J. Biol. Chem. 264:14902-14908.
Lels, J., A. Aiyar and D. Cobrinik. 1993. Regulation of initiation of reverse transcription of retrovinises, p. 33-48. In Skalka, AM. and S.P. Goff, (ed.), Reverse Transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Leuthardt, A. and S.F.J. LeGrice. 1988. Biosynthesis and analysis of a genetically engineered HIV-1 reverse transcriptase/endonuclease polyp rotein in Escherichla coli Gene 68:35-42.
Levy, D.N., LE. Fernandes, W.V. Williams and 0.6. Weiner. 1993. Induction of cell differentiation by human immunodeficiency virus 1 vpr. Cell 72~547 -550,
Levy, JA. 1988. HIV and the pathogenesis of AIDÇ. JAMA 261:2887-3006.
Li. M., A.G. Garcia, U. Bhattacharyya, P. Mascagni, B.M. Austen and MœMœ Roberts. 1996. The Vpr ptotein of human immunodeficiency virus type 1 binds to nucleocapsid protein p7 in vitro. Biochem. Biophys. Res. Commun. 21 8~352-355.
Li, X, J. Mak, E.J. Arts, 2. Gu, L Kleiman and M.A. Wainberg. 1994. Effects of alterations of primer binding site sequences on hurnan immunodeficiency virus type I replication. J. ViroI. 68:6198-6206.
Li, Xe, C. Liang, Y. Quan, R. Chandok, M.A. bughrea, M.A. Parniak, L Klelman and A . Wainberg. 7997. Identification of seguences downstrearn of the primer binding site that are important for efficient repkation of human immunodeficiency virus me 1. J. Virol. ?¶:6OO3-6OlO.

Ughffoote, M.M., J.E. Coligan, T.M. Folks, AmS. Fauci, M.A. Martin and S. Venkatesan. 1986. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome retrovirus. J. Virol. 60:771-775.
Undhofer, H., K. Von de Helm and H. Nitschko. 1995. In vivo processing of Pr16QmHd from hurnan imrnunodeficiency vinis type 1 (HIV) in acutely infecteci, cultured T-lymphocytes. Virology 214:624-627.
Unial, M.L. and A.D. Miller. 1990. Retroviral RNA packaging: sequence requirements and implications. Cun. Top. Microbiol, Immunol. 1573 25-1 52.
Littman, DoRe 1998. Chemokine receptors: keys to AIDS pathogenesis? Cell 93:677-680.
Liu, H., X. Wu, M. Newman, G.M. Shaw, B.H. Hahn and J-C. Kappes. 1995. The Vif protein of human and simian hmunodeficiency viruses is packaged into virions and associates with viral core structures. J. Virol. 69~7630-7638-
Loeb, D.D., C.A. Hutchison III, M.H. Edgell, W.G. Farmeiie and R. Swanstrom. 1989a. Mutational analysis of hurnan immunodeficiency virus type 1 protease suggests functional homology with aspartic proteinases. J. Virol. 63:f 11-1 21.
Loeb, D-D., R. Swanstrom, L. Everitt, M. Manchester, S.E. Stamper and CA. Hutchison 111. 1989b. Complete mutagenesis of the HIV-1 protease. Nature 340:397-400.
Lori, F., A.!. Scovassi, O. tella, G. Achilll, E. Cattaneo, C. Casoli and U. Bertanoni. 1988. Enrymatically active foms of reverse transcriptase of the human imrnunodeficiency virus. AIDS Res. Hum. Retroviruses 5:393-398.
Louis, J.M., R A . McDonald, N.T. Nashed, E.M. Wondrak, D.M. Jerina, S. Oroszlan and P.T. Mora. 1991. Autoprocessing of the HIV-1 protease using purified wild-type and mutated fusion proteins expressed at high levels in EscherrCh;a col[ Eur, J. Biochem. ï99:361-369,
Louis, J.M., N.T. Nashed, K.D. Parris, A.R. Kimmel and DM. Jerina. 1994. Kinetics and mechanism of autoprocessing of human imrnunodeficiency vkus type 1 protease ftom an analog of the Gag-Pol polyprotein. Proc. Natl. Acadc Sci. USA 9t7970-7974.
Lowe, D.M., A. Aitken. C. Bradley, G.K Darby, B.A. Larder, K.L. Powell, D m J m M m Purtfoy, M. fistale and O.K. Stammers. 1988. HIV-1 reverse transcriptase: crystallization and ana[ysis of dornain structure by limited proteolysis. Biochern. 27:8884-8889.

Lu, M., S. Blackow and P. Kim. 1995. A tiimerïc structural domain of the HIV-t transmembrane glycoprotein. Nature Struct. Biol. 2:1075-1082.
Lu, Y.-L., P. Speaimsn and L. Ratner. 1993. Human immunodeficiency virus type 1 viral protein R localizes in infected cells and virions. J. Virol. 67:6542-6550.
Lu, Y.-L., R.P. Bennett, J.W. Wllls, R. Gorelick and L. Ratner. 1995. A leucine triplet repeat sequence (UO<), in ~6~ is important for Vpr incorporation into human immunodeficiency virus type 1 particles. J. Virol. 69:6873-6879.
Luban, J. and S.P. Goff. 1991. Binding of human immunodeficiency virus type 1 (HIV-1) RNA to recombinant HIV-1 Gag po1yprotein. J. Virol. 65~3203- 321 2.
Luban, J., K.L. Bossolt, E.K. Franke, G.V. KaIpana and S.P. Goff. 1993a. Human immunodeficiency virus type t Gag protein binds to cyclophilins A and B. Cet1 73:1067-1078.
Luban, J., C. Lee and S.P. Goff. 1993b. Effect of linker insertion mutations in the human imrnunodeficiency virus type 1 gag gene on activation of viral protease expressed in bacteria. J. Virol. 6?:3630-3634.
Luban, J. 1996. Absconding with the chaperone: essential cyclophilin-Gag interaction in HIV-1 virions. Cell 87: 1 157-1 159.
Luftig, R., 1. Kazuyoshi, M. Bu and P. Clakins. 1990. Terminal stages of retrovirus morp hogenesis in retroviral protease, p. 141 -1 48. In Pearl, L.H.. (ed.), Control of Maturation and Morphogenesjs. Macmillan. London.
Luo, G.X. and J. Taylor. 1990. Template switching by reverse transcriptase durhg DNA synthesis. J. Virol. 64:4321-4328.
Luukkonen, B.G.M.. E.M. Fenyo and S. Schwartz. 1995. Overexpression of human immunodeficiency virus type 1 protease increases cellular cleavage of Gag and reduces virus infectivity. Virdogy 206:854-865,
Maddon, P.J., A.J. Daîgîeish, J.S. McDougal, P.R. Clapharn, R.A. Weiss and Re Axel. 1986. The T4 gene encodes the AlDS virus nceptor and is expressed in the immune system and Vie brain. Cell47:333-348.
Mak, J., M. Jiang, M.A. Wainberg, M.L. Hammerskjold, D. Rekosh and L Kletman. 1994. Role of Pr160gag-pot in mediating the selective incorporation of tRNA(Lys) hto human immunodeficiency virus type 1 particles. 3- Virol- 68:2065-2072.
Mak, J., A. Khorchid, Q. Cao, Y. Huang, I. Lowy, M.A. Painiak, V.R. Prasad, M.A. Wainberg and t KIelman. 1997. Effects of mutations in

Pr160Mf upon tRNAm and Pr160wL incorporation into HIV-1. J. Molec. Biol. 265:419-43 1.
Mallm, M.H., J. Hauber, S.-Y. Le, J.V. Mafzeî and B.R. Cullen. 1989a The HIV-1 Rev trans-activator acts through a structural target sequence to activate nuclear export of unspliced viral mRNA. Nature 330254-257.
Malim, M.H., S. Bohnlein, J. Haubei and B.R. Cullen. 1989b. Funetionai dissection of the HIV-i Rev trans-activator - derivation of a trans- dominant repressor of Rev function. CeII 58205-214.
Malim, M.H., W.W. Feimuth, J. Lie, T.J. Boyle, H.K. Lyerly, B.R. Cullen and G.J. Nabel. 1992. Stable expression of transdominant Rev proteins in hurnan T cells inhibits human immunodeficiency virus replication. J. Exp. Med. l76:I lgM2OI .
Mammano, F., A. Ohagen, S. HogIund and HG. Gottlinger. 1994. Role of the major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J. Virol. 68~49274936.
Maniatis, T., E.F. Fritsch and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring ~arbor, N.Y.
Margottin, F., S.P. Bout, H. Durand, L. Sellg, S. Benichou, V. Richard. T. Thomas, K. Strebel and R. Benaious. 1998. A novel human WD protein. h-PTrCP. mat interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Moi. Cell 1565-574.
Maikowltz, M., M. Saag, W.G. Powderly, A.M. Hurley, A. Hsu, J.M. Valdes, D. Henry, Fm Sattler, A. LaMarca, LM. Leonard and D.D. Ho. 1995. A preliminary study of ritonavir, an inhibitor of HIV-1 protease to treat HIV- 1 infection. N. Engl. J. Med. 333:1534-1539.
Mathews, R.E.F. 1982. Classification and nomenclature of viruses. iVth Repart of the lntemational Cornmittee on Taxonomy of Vinrses. Karger, Basel.
McDermott, J., Le Farrell, R. Ross and E. Barklis. 1996. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions, using cysteinespecific teagents. J. Virol. 7051 06-51 14.
McDougaI, J.J., Y.S. Kenedy, J-M. Seigh. S.P. COR, A. Mawla and J.U. Nicholson. 1986. Binding of HTLV-IIIRAV to T4+ cells by a complex of the 1 1 0K viral protein and the T4 molecrile. Science 231 :382-385.
McKeeuer, B.M., M.A. Navla, P.M.D. Fitzgerald, J.P. Springer, C. Leu, AC. Heimbach, W.K. Hetber, LS. Slgal and P.L. Darke. 1989. Crystallization of the aspartyl protease from the hurnan im m unodeficiency virus, HlV-I - 3- BioI. Chern. 2w1919-f 921 .

McMichael, A 1998. T cell responses and viral escape. Cell 93:673-676.
McPhee, Fm, A.C. Good, I.D. Kuntz and C.S. Craik. 1996. Engineering human imrnunodeficiency virus 1 protease heterodimers as macromolecular inhibitors of viral maturation. Proc- NafC. Acad. Sci, USA 93:11477-11481.
Meek, T.D., DmMB Lambert, GoB. Dreyer, T.J. Carr, T.A. Tomaszek, MOL. Moore, J.E. Strickler, C. Debouck, L.J. Hyland, T.J. Mathews, B.W. Metcalf and S.R, Petteway. 1990. Inhibition of HIV-1 protease in infected T-lymphocytes by synthetic peptide analogues. Nature 343:90-92.
Mellors, LW., C.W. Rhaldo JI., P. Gupta, R.M. White, LA . Todd and L.A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272: 1 167-1 1 70.
Mely, Y., H. OeRocquigny, M. Sorinas-Jimeno, G. Keith, B.P. Roques, R. Marquet and D. Gerard. 1995. Binding of the HIV-1 nucleocapsid protein to the primer tRNA(3Lys), in vitro, is essentially not specific. J. BioL Chem. 270: 1650-1 656.
Mergener, K., M. Facke, R. Welker, V. Biinkmann, H.R. Gelderblom and H.G. Kiausslich. 1992. Analysis of HIV particle formation using transient expression of subviral constructs in mammalian cells. Virology 1 86:25-39.
Mervis, RJ., N. Ahmad, E.P. Lillehoj, MG. Raum, F.H.R. Salazar, H.W. Chan and S. Venkatesan. 1988. The gag gene products of hurnan immunodeficiency virus type 1: alîgnrnent within the gag open reading frame, identification of posttranslational modifications, and evidence for alternative gag precursors. J. Virol. 62:3993-4002,
Meyer, J., S. Nlck, T. Starnminger, Fm Grurnmt, G. Jahn and H.J. Lipps. 1996. Inhibition of HIV-1 replication by high-copy-number vector expressing antisense RNA for reverse transcriptase. Gene 129:263-268.
Mildner, A.M., D.J. Rothrock, J.W. Leone, COA. Bannow, J.M. Lull, I.M. Reardon, J.L. Sarcfch, W.J. Howe, Cg-S.C. Tomich, C.W. Smith, R.L Heinrikson and AG. TomasselII. 1994. The HIV-1 protease as enzyme and substrate: mutagenesis of autolysis sites and generation of a stable mutant with retained kinetic properties. Biochem. 33:9405-9413.
Mllrer, M., M. Jaskolski, J.K.M. Rao, J. Leis and A Wlodawer. 1989a- Ccystal structure of a retroviral protease proves relaüonship to aspartic protease fam ily. Nature 332576-579.
Miller, M.. B.K. Sathyanarayana, MN. Toth, OR. Marshall, L. Clawson, L Selk, J. Schneider. S.B.H. Kent and A. Wlodawer.

1989b Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3h resolution. Science 246:1149-1152.
Miller, M.D., M.T. Warmetdam, I. Gaston, W.C. Green and M.B. Feinberg. 1994. The human immunodeficiency virus4 nef gene product: a positive factor for viral infection and replication ni prhary lymphocytes and macrophages. J. Exp. Med. I79:lOMl3.
Mfller, RH. and N. Sarver. 1997. HIV accessory proteins as therapeutic targ ets. Nature Med. 3:389-394.
Modestf, N., J. Garcia, C. Debouck, M. Peterlln and R. Gaynor. 1991. Tians-dorninant tat mutants with alterations in the basic domain inhibits HIV-1 gene expression. New Biol. 3:759-5083.
Moody, MoDq SC. Pettit, W. Shao, L. Everitt, D.D. Loeb, C.A. Hutchlson III and Ro Swanstrom. 1995. A side chain at position 48 of the human imrnunodeficiency virus type4 protease flap provides an additional specificity deteminant, Virology 207:475-485.
Mous, Je, €.P. Heimer and SoFmJ* LeGrice. 1988. Processing protease and reverse transcriptase from human imm unodef iciency virus type 1 pol y p rotein in Escherichia coli. J. Virol. 62: 1 43 3- 1 436.
Murphy, J.E. and S.?. Goff. 1989. Construction and analysis of deletion mutations in the U5 region of Moloney murine leukemia virus: effects on RNA packaging and reverse transcription. J. Virol. 63:319-327.
Nabel, G.J., B.A. Fox, L. Post, C.B. Thompson and C. Woffendin. 1994. A molecular genetic intervention for AIDÇ - effects of a transdorninant negative f o n of Rev. Hum. Gene Ther. 5:79-92.
Navia, M.A., PeMoDo Fitzgerald, B.M. McKeever, C.-To Leu, J.C. Heimbach, W.K. Herbei, I.S. Sigal, P L Darke and J.P. Springer. t 989. Three-dimensional structure of aspartyl protease from human imm unodeficiency virus HIV-1. Nature 337:6? 5-620.
Navia, M.A and B.M. McKeever. 1990. A role for the aspartyl protease from the human immunodeficiency virus type 1 (HlV-1) in the orchestration of virus assembly. Ann. NY. Acad. Sci 616:73-85.
Netmut, M.V. and DoJ. Hockley. 1996. Comparative morphology and structural classification of retrovirusea Curr. Top. Microbiol. Irnmunol. 2W:f -24.
Nottet. H o S m and H*Eo Gendefman. 1995. Unraveling the neuroimmune mechanisms for the HIV-1-associated cogniüvelmotor cornplex. Immunol. Today t 6~441-448.

Nutt, R.F., S.F. Brady, PL. Darke, T.M. Ciccarone, E.M. Nutt, J.A. Rodkey, CmD. Bennett, L.H. Waxman and [.S. Sigal. 1988. Chernical synthesis and enzymatic activity of a 99-residue peptide with a sequence proposed for the human immunodeficiency virus protease. Proc. Natl. Acad. Sci. USA 85:7l29-7133.
Oberlln, E.. A. Amara, F. Bachelerie, C. Bessia, J.-L Virelizier, F. Arenzana-Seisdedos, O. Schwartz, Jœ-Mo Heard, 1. Clark-Lewis, D. F. Legler, M. Loetscher, M. Baggiolini and B. Mosei. 1996. The CXC chemokine SDFl is the ligand for LESTER/fusin and prevents infection by T- cell-line-adapted HIV-l. Nature 382833-835.
Oertle, S. and PPF* Spahr. 1990. Role of Gag polyprotein precursot in packaging and maturation of Rous sarcorna virus genomic RNA J. Virol. 64:5757-5763.
Ogg, O.S., X. Jln, S. Bonhoeffer, P.R. Dunbar, M.A. Nowak, S. Monard, J.F. Segal, Y. Cao, S.L. Rowland-Jones, V. Cerundolo, A. Hurley, M. Markowitz, D.D. Ho, D.F. Nixon and A.J. McMlchael. 1998. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279:2103-2106.
Ojwang, J.O., A. Hampel, 0.J. Looney, F. Wong-Staal and J. Rsppaport. 1992. Inhibition of human immunodeficiency virus type 1 by a hairpin ribozyme. Proc. Natl. Acad. Sci. USA 89:10802-10806.
Oroszlan, S. and R A Luftig. 1990. Retroviral Proteinases. Cun. Top. Microbiol. Cmmunot. l57:153-185.
Orsini, Mo and C.M. Debouck. 1996. Inhibition of human immunodeficiency virus type 1 and type 2 Tat function by transdominant Tat protein localited to both the nucleus and cytoplasm. J. Virol. 70:8055-8063.
Oswald, M. and K. Von der Hefrn. 1991. Fibronectin is a non-viral substrate for the HIV proteinase. FEBS L e t 292:298-300.
Ottman, M., C. Gabus and J.-L. Darlix. 1995. The centrai globular domain of the nucieocapsid protein of human immunodeficiency virus type 1 is critical for virion structure and infectivity. J. Virol. 69:ln8-I 784.
Palella, FeJq K.M. Delaney, AS. Moorman, M.0- Loveless, J, Fuhrer, G.A. Satten, D.J. Aschman and S.D. Holmberg. 1998. Declining rnorbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338:853-860.
Panteleo, O, C. Graziosi, L Butinl, RA- P i n o , S.M. Schnittman, D.P. Kotler and A.S. Fauci. 1991. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 88~9838-9842-

Panteleo. Ci., Cm Giazlosi, J.F. Demarest, L. Butin€, M. Montroni, C.H. Fox, JDMB Oienstein, D.P. Kotler and A.S. Fauci. 1993. HIV infection is active and progressive in Lymphoid tissue during the clinically latent stage of disease. Nature 362:355-358.
Parent, LJq C.B. Wilson, M.D. Resh and J.W. WilIs. 1996. Evidence for a second function of the MA sequence in the Rous sarcoma virus Gag protein. J. Virol, 7O:I O1 6-1 026.
Park, J. and CD- Moiiow. 1991. Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteoIytic processing in the absence of virion production. J. Virol. 6551 11- 51 17,
Park, J. and C.D. Morrow. 1992. The nonmyristylated P r 1 6 0 ~ ~ ~ polyprotein of human immunodeficiency virus type 1 interacts with Pr5!jPg and is incorporated into virus-like particles. J. Virol. 66:6304-6313.
Park, J. and C.D. Morrow. 1993, Mutations in the protease gene of human immunodeficiency virus type 1 affect release and stabiiity of virus particles. Virology 194:843-850.
Partin, K., HtG. Krausslich, L. Ehrlich, E. Wimmer and C. Carter. 1990. Mutational analysis of a native substrate of the human immunodeficiency virus type 1 proteinase. J. Virol. 64:3938-3947.
Partin, K., G. Zybaith, GD Ehrlich, Lm Decrombrugghe, E. Wimmer and Cm Carter. 1991. Deletion of sequences upstrearn of the proteinase improves the proteolytic processing of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 88:47ï6-4780,
Paxton, W., RD[- Connor and N.R. Landau. 1993. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J. Virol. 6?:7229-7237.
Pearl, L.H. and W.R. Taylor. 1987. A structural model for the retroviral proteases. Nature 329:351-354.
Pearson. L., J. Garcia, F. WU, N. Modesti, J. Nelson and R. Gaynor. 1990. A transdominant taf mutant that inhibits tat-induced gene expression from the human immunodeficiency virus long terminal repeat. Proc. Natl. Acad. Sei. USA 825079-5083.
Pellska, J.A. and S.J. Benkovic. 1992. Mechanisrn of DNA strand transfer reactions catalyzed by HIV-l reverse transcriptase. Science 258: t 1 t 2-1 1 18.

Peng, CD, B.K. Ho, T.W. Chang and N.T. Chang. 1989. Role of human immunodeficiency virus type t -specific protease in core protein maturation and virai infectivity. J. Virol. 63:2550-2556.
Peng, CD, K Moelllng, N.T. Chang and T.W. Chang. 1990. Functional characterization of HIV-1 gag-pol fusion protein, p. 55-62. In Pearl, LH.. (ed.), Retro viral Proteases: con trol of maturation and morphogenesis. Stockton Press, N.Y.
Peng, C., NiTe Chang and T.W. Chang. 1991. Identification and characterization of human immunodeficiency virus type 1 gag-pol fusion protein in transfected mammalian cells. 3- Virol. 65:275f-2756-
Perelson, A.SD, AoU- Neumann, M. Mackowitt, J.M. Leonard and DmDo Ho. 1 996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life- span, viral generation the. Science 271 :1582-1586.
Perelson, AS., P. Essunger, Y. Cao, M. Vesanen, A. Huiley, K. Saksela, M. Markowitz and D.D. Ho. 1997, Decay characteristics of HIV- 1 -infected compartments during combination therapy. Nature 3879 88-1 91.
Pettit, S.C., J. Simsic, D.D. Loeb, L. Eveiitt, C.A. Hutchlson III and Re Swanstrom. 1991, Analysis of retroviral protease cleavage sites reveals two types of cleavage sites and the structural requirements of the P l amino acid. 3. Biot. Chern. 266:14539-14547.
Pettit, S.C., M.D. Moody, RS- Wehbie, A.H. Kaplan, P.V. Nantermet, C.A. Klein and R. Swanstrom. 1994, The ~2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virus. J. Virol. 68:8017-8027.
Phylip, LHm, J.S. Mills, B.F. Parten, B.M. Dunn and J. Kay. 1992. Intrinsic activity of precursor foms of HIV-1 proteinase. FEBS L e t 31 4:449- 454.
Pichova, lm, P. Strop, J. Sedlacek, F. Kapralek, V. Benes, M. fravnlcek, L. Pavlfckova, M. Soujcek, V. Kostka and S. Foundling. 1992. lsolation and biochernical characterization and crystallization of the p15Gag proteinase of myeloblastosis associated virus expressed in E coli. lnt- 3- Biochem. 24:235-242.
Poeschla, E., P. Corbeau and F. Wong-Staal. 1996. Development of HIV vectors for anttHIV gene therapy. Roc. Natl. Acad. Sci. USA 93: 11395- 11399.
Pomerantz, RJ. and D. Trono. 1995. Genetic therapies for HIV infections: promise for the future. AlDS 9:985-993.

Poon, B., K. Grovit-Ferbas, S.A. Stewart and 1.S.Y- Chen. 1998. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science 281 ~266-269.
Poon, DmTm, J. Wu and A. Aldovlni. 1996. Charged amino acid residues of hurnan immunodeficiency virus type I nucleocapsid p7 protein involved in RNA packag ing and infectivity. J. Virol. ?0:6607-6616.
Pooiman, RmAq A.O. Tomasselli, R.L. Heinriksan and FmJ- Kezdy. 1991. A cumulative specificity mode1 for proteases from human irnmunodeficiency virus types 1 and 2, inferred from statfstical analysis of an extended su bstrate data base. J. Biol. Chem. 266: 14554-1456 1.
Popovic, M., M.G. Sarngadharan, E. Read and R.C. Gallo. 1984. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) €rom patients with AIDS and pre-AIDS. Science 224:497-500.
Prats, A.-Cm, L. Sarlh, C. Gabus, S. Litvak, G. Keith and J.-L. Darllx. 1988. Small finger protein of avian and murine retroviruses has nucleic acid annealing activity and positions the replication primer tRNA ont0 genomic RNA EMBO 3.7: 1777-1783.
Pryciak, P*M- and HmE- Varmus. 1992. Nucleosomes, DNA-binding proteins. and DNA sequence modulate retroviral integration target site selection. Celt 69~769-780,
Pullen, K.A. and J.J. Champoux. 1990. Plus-strand origin for human immunodeficiency virus type 1: implications for integration. J. Virol. 64:6274- 6277,
Quan, Y., 2. Gu, X. Li, CD Mortow and M.A. Wainberg. 1996. Endogenous reverse transcriptase assays reveal high-level resistance to the triphosphate of (-)2'-dideoxy-3'-thiacytidine by mutated Ml 84V human immunodeficiency virus type 1. J. Virol. 70:5642.
Ouillent, C., N. Dumey, CD Dauguet and F. Clavel. 1993. Reversion of a polymerase-defective integrated HIV-1 genome. AIDS Res. Hum. Retroviruses 9:1031-1037.
QuiIlent, C., A.M. Borrnan, S. Paulous, CD Dauguet and F. Clavel. 1996. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle formation. Virology 21 928-36.
Rabson, A-B. and J.W. Wills. 1997. Synthesis and ptocessing of viral RNA, p. 205-262. In Coffin. J.M., S.H. Hughes and H.E. Varmus. (ed-), Rstroviiuses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.

Ranga, U., C. Woffentin, S. Verma, L. Xu, C.H. June, D m K m BIshop and G.J. Nabel. 1998. Enhanced T celt engraftment after retroviral delivery of an antiviral gene in HIV-infected individuals. Proc. Natl. Acad. Sci. USA 95:lZOl -1 206.
Rapport, C.J., J. Gosllng, V.L. Schweickart, P.W. Gray and I.F. Charo. 1996. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCRS) for RANTES, MIP-If3, and MIP-la J. Biol. Chern. 2fi:l ï l6l-l?I 66.
Rayner, M., B.C. Cordova, RœPm Meade, P.€. Aldrkh, P.K. Jadhav, Y. Ru and P.Y. Lam. 1994. DMP 323, a nonpeptide cyclic urea inhibitor of human immunodeficiency virus (HIV) protease. specifically and persistently blocks intracellular processing of HIV Gag polyprotein. Anthicrob. Agents Chemother. 38:l635-t 640.
Reeves, R., C. Gorman and B. Howard. 1985. Minichromosome assembly of non-integrated plasrnid DNA transfected into mammalian cells. Nucleic Acids Res. 1 3:3599-56 1 S.
Reicin, AS., S. Paik, R.D. Beikowitz, J. Luban, 1. Lowy and S.P. Goff. 1995. Linker insertion mutations in the human immunodeficiency virus type 1 gag gene: effects on virion particle assembly, release, and infectivity. J. Virol. 69:642-650.
Relcln, A.S., A. Ohagen, L. Yin, S. Hoglund and S.P. Goff. 1996. The toîe of Gag in human immunodeficiency virus type 1 virion morphogenesis and early steps of the viral life cycle. J. Virol. 70:8645-8652.
Rhodes, A. and W. James. 1990. Inhibition of human immunodeficiency virus replication in cell culture by endogenously synthesized antisense RNA. J. Gen, ViroC. 71 : 1 965- 1 974,
Riviere, Y., V. Blank, P. Kourilsky and A. Israel. 1991. Processing of the precursor of NF-kappa 6 by the HIV-1 protease during acute infection. Nature 350~625-626.
Riuo, CmJ* and B.D. Korant. 1994. Genetic approaches designed to rninimize cytotoxicity of retroviral protease. Methods Enzyrnol. 241 :16-29.
Riuuto, CD.. R. Wyatt, NN. Hernandez-Ramos, Y. Sun, P.D. Kwong, W.A. Hendrickson and J. Sodroski. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280: 19494 953-
Roberts, M.M. and S. Oroszlan. 1990. The action of retrovirai protease in various phases of virus replication, p. 131-139. ?n Pearl, LH.. (ed.), Retroviral Pmteases: Confrol of Maturation and MorphogenesrS. Stockton Press, N.Y.

Rogel, M.E., L.I. Wu and M. Emerman. 1995. The human immunodeficiency virus type 1 vpr gene prevents ceil proliferation during chronic infection. J. Virol. 69:882-888.
Rooke, R., M. Tremblay and M.A. Wainbeig. 1990. AZT (zidovudine) may act postintegrationally to inhibit generation of HIV-1 progeny virus in chronically infected cells. Virology 176:205-215.
Rose, J.R., R. Salto and CS. Craik. 1993. Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem. 268: 1 t 939-1 1945
Rose, J.R., LM. Baba and C.S. Craik. 1995. Defining the level of human immunodeficiency virus type 1 (HIV-1) protease activity required for HIV-1 particle maturation and infectivity. J. Virol. 69:275 1-2758.
Rosenberg, E.S., J.M. Billfngsley, AmMo Caliendo, SOL Boswell, P*Eo Sax, SA. Kalams and B.D. Walker. 1997. Vigorous HIV-1-specific CD4+ T ceIl responses associated with control of viremia. Science 2?8:I44?-145O.
Ross, E.K., T.R. Fuerst, J.M. Orenstein, Te O'Neill, M.A. Martin and S. Venkatesan. 1991. Maturation of human immunodeficiency virus particles assembled from the Gag precursor protein requires in sfiu proceçsing by Gag- Pol protease. AIDS Res. Hum. Retrovinises 7 :475-483.
Roth, M.J., P.L. Schwartzberg and S.P. Goff. 1989. Structure of the termini of DNA intermediates in the integration of retroviral DNA: dependence on IN function and terminal DNA sequence. Cell58:47-54.
Saikai, H., R. Shlbata, J.4.. Sakuragi, S. Sakuiagi, M. Kawanura and A. Adachi. 1993. Cell-dependant requirement of human Rnrnunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 67: 1663- 1666.
Sakaguchi, K., N. Zambrano, E X Baldwin, B.A. Shaplro, J.W. Erickson, J-G. Ornichinski, G.M. Clore, J.W. Gronenbon and E. Appella. 1993. Identification of a binding site for hurnan imrnunodeficiency virus type 1 nucleocapsid protein. Proc. Natl. Acad. Sci. USA 905219-5223.
Sampson, M., 0. Labbe, C. Yollereau, O. Vassart and M. Parmentier. 1996. Molecular cloning and functional expression of a new CC- chernome receptor gene. Biochern. 353362-3367.
Sandefur, S., V. Varthakavi and P. Spearman. 1998. The I domain is required for efficient plasma membrane binding of hurnan immunodeficiency virus type 1 Pr55-O. J. Virol. 72:2723-2732.

Sacver, No, E-M. Canth, P.S. Chang. LA. Zaia, A. Ladne, L A . Stephens and J.J. Rossi. 1990. Ribozymes as potential anttHIV-1 therapeutic agents. Science 24?:I222-l225.
Schatz, G.. 1. Ptchova and V.Y. Vogt. 1997. Analysis of cleavage site mutations between the NC and PR Gag domains of Rous sarcoma virus. J. Vkol. 71 ~444-450.
Schmidtmayerova, Ho, M. Alfano, G. Nuovo and M. Bukrinsky. 1998. Human immunodeficiency virus type 1 T-iymphotropic strains enter macrophages via a CD4- and CXCR4-mediated pathway: replication is restricted at a postentry level. J. Virol. 72:4633-4642.
Schubert, U., A.V. Ferrer-Montief, M. Oblatt-Montal, P. Henklein. K. Strebel and M. Montal. 1996. Identification of an ion channe1 activity of the Vpu transrnembrane domain and its Vivolvement in the regulation of virus release from HIV-1 infected celk FEBS L e t 398:l2-18.
Schwartz, O., V. MaiechaI, S. Le Gall, F. Lemonnier and J.-M. Heard. 1996. Endocytosis of major histocompatibility complex class 1 molecules is induced by HHIV-1 Nef protein. Nature Med. 2338-342.
Sczakiel, GD, M- Pawlfta and A. Kleinheinz. 1990. Specific inhibition of human immunodeficiency virus type 1 replication by RNA transcribed in sense and antisense orientation from the 5'leader-gag region. Biochem. Biophys. Res. Commun. i 69:643-65 1.
Seelmeier, S., H. Schmidt, V. Turk and K. von der Helm. 1988. Human imrnunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proc. Natl. Acad. Sci. USA 8S:66t 2-661 6.
Sellos-Moura, M. and VDM. Vogt. 1996. Proteolytic activity of purified avian sarcoma and leukemia virus NC-PR protein expressed in Escherichia coli. Virology 221 :335-345.
Shaheen, F., L. Duan, M. Zhu, O. Bagasra and RoJ. Pomerantz. 1996. Targeting human immunodeficiency virus type 1 reverse transcriptase by intracellular expression of single-chain variabLe fragments to inhibit early stages of the viral iife cycle. J. Virol. 70:3392-3400.
Sharp, P.A. and RA. Marciniak. 198% HIV TAR an RNA enhancer? Ceil 59:229-230.
Sheng, N., SC. Pettit. R.J. Tritch, DoH- Odurk, MM. Rayner, R. Swanstrom and Se ErlcksonlViitanen, 1997. Determinants of the human immunodeficiency virus type 1 pl5NC-RNA interaction that affect enhanced cleavage by the vira1 protease. J. Virol. 715723-5732.

Sheng, S. and S. Ericksoo-Viltanen. 1994. Cleavage of p l5 in vitro by human immunodeficiency vkus type 1 protease is RNA dependant. J. Virol. 68:6207-6214,
Sherman, P.A., MoL Dickson and JwA- Fyfe. 1992. Hurnan imrnunodeficiency virus type 1 integration protein: DNA sequence requirements for cleaving and joining reactions. J. Virol. 66:3593-3601.
Shin, C.-G., Bo Taddeo, WmA. Haseltine and CmMo Farnet. 1994. GeneNc analysis of the human irnmunodeficiency virus type 1 integrase protein. J. Virol. 68A 633-1 642.
Shioda, T. and H. Shi buta. 1990. Production of human immunodeficiency virus (HIV)-like particles ftom cells infected with recombinant vaccinia viruses carrying the gag gene of HlV. Virology t75:I 39-1 48.
Shoeman, RoLw, B. Honer, TwJ. Stoller, C. Kesselrneier, M.C. Meidel, P. Traub and MoC. Graves. 1990. Hriman immunodeficiency virus type 1 protease cleaves the intermediate filament proteins vimentin, desrnin and glial fibrillary acidic protein. Proc. Nat!. Acad. Sci. USA 87:6336-6340.
Shoeman, R.L., C. Sachese, B. Honer, E. Mothes, M. Kaufman and P. Ttaub. 1993. Cleavage of human and mouse cytoskeletal and sarcomeric protehs by human immunodeficiency virus type 1 protease. Am. J. Pathol. I42:22t -230.
Simon, J.H.M. and M.H. Malim. 1996. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 70:5297-5305.
Simon, JoHwM., D.L. MIIIer, R-A.M. Fouchier and M.H. Malim. 1998. Virion incorporation of human immunodeficiency virus type-1 Vif is deterrnined by intracellular expression level and may not be necessary for function. Virology 248~182-187,
Skalka, A.M. 1989. Retroviral proteases: first glimpses at the anatomy of a processing machine. Cell 56:9 1 1 -9 13.
Smith, A.J., M.-l. Cho, MrL. Hammerskjold and D. Rekosh. 1990. Human immunodeficiency virus type 1 Pr55@'= and Pr160flPPOi expressed frorn a simian virus 40 late replacement vector are efficiently processed and assembled into virus-like particles. J. Virol. 642743-2750.
Smith, AJ., N. Siinivasakumar, MPL. Hammerskjold and D. Rekosh. 1993. Requkements for incorporation of P r 1 6 F f from human immunodeficiency virus type 1 into virus-like particles. J. Virol. 67:2266-2275.
Smythe, J.A., D. Sun, M. Thomson, P-Do Markham, M.S. Reitz Jr., R.C. Gallo and J. Lisziewics. 1994. A Rev-inducible mutant gag gene

stably transferred into T lymphocytes: an approach to gene therapy against human immunodeficiency vkus type 1 infection. Proc. Natl. Acad. Sci. USA 91 :3657-3661.
Sodioski, J., R. Pataica, Cm Rosen, F. Wong-Staal and W. Haselthe. 1985. Location of the tran~~activating region on the genome of human T-ceII lymphotropic virus type III. Science 229:74-77.
Sotorarniiez, L-E., B. Renjlfo, M.F. McLane, RD Marlink. C. Ohara, R. Sutthent, C. Wasi, P. Vithayasai, V. Vithayasai and C. Apichartplyakul. 1996. HIV-I Langerhans cell tropism associated with heterosexual transmission of HlV. Science 271 A29 1-1 293.
South, TL, B. Kim, D.R. Hare and M.F. Summers. 1990. Zinc fingers and molecular recognition. Biochem. Pharmacol. 40:123-129.
Spearman, P., J m J m Wang, N*Vm Heydon and L Ratner. 1994. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 68:3232-3242.
Spina, C.A., T J m Kwoh, M X Chowers, J.C. Guatelli and DmDo Rlchman. 1994. The importance of nef in the induction of human immunodeficiency virus type 1 replication from primary quiescent CD4 lymphocytes. J, Exp. Med. 179:IlS-123.
Spina, C.A.! J.C. Guatelll and D.D. Richman, 1995. Establishment of a stable, inducible fomi of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 69~2977-2988.
Srinivasakumar, N., M.-J. Hammerskjold and D. Rekosh. 1995. Characteriration of deletion mutants in the capsid region of human immunodeficiency virus type t that affect particle formation and Gag-Pol precursor incorporation. J. Virol. 69:6106-6114.
Starnes, M.C., W. Gao, RrnYmCm Ting and Y&. Chang. 1988. Enzyme activity gel analysis of human immunodeficiency virus reverse transcriptase. J. Biol. Chem. 26352-54,
Stebblns, J. and Cm Debouck. 1994. Expression system of retroviral proteases. Methods Enzymol. 241 :3-16.
Stein, B.S., S.D. Gowda, J.D. Lifson, R.C. Penhallow, K.O. Bensch and E.G. Engelman. 1907. pH-independent HlV entry into CD4positive T cells via virus envelope fusion to the plasma membrane. Celt 49:659-668.
Stewart, L, O. Schafz and V.M. Vogt. 1990. Propetües of aviao retrovirus particles defective in vira! protease. J. Virol. 645076-5092.

Stewart, L and V.M. Vogt. 1991. trans-acting vital protease is necessary and sufficient for activation of avian leukosis virus reverse transcriptase. J. Virol. 64:6218-623 1 . Stewart, L. and VM. Vogt. 1993, Reverse transcriptase and protease activities of avian leu kosis virus Gag-Pol fusion proteins exp ressed in insect cells. 3. Virol. 67~7582-7596.
Stewart, L. and V.M. Vogt. 1994. Proteolytic cleavage at the Gag-Pol junction in avian leukosis virus: differences in vitro and in vivo. Virology 204:45- 59-
Stewart, S.A., B. Poon, J.B.M. Jowett and 1.S. Chen. 1997. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 71 :433 1-4338.
Streblow, D.N., M. Kitabwalla, M. Malkovsky and CD. Pauza. 1998. Cyclophilin A modulates processing of human immunodeficiency virus type 1 ~ 5 5 ~ . mechanism for antiviral effects of cyclosporin A. Virology 24S:I9?-202.
Su, Y., W. Popik and P.M. Pitha. 1995. Inhibition of human immunodeficiency virus type 1 replication by a Tat-activated, transduced interferon gene: targeted expression to human irnmunodeficiency type 1- infected cells. 3. Viro t. 69: t 10-121.
Sullenger, B.A., H.F. Gallardo, G.E. Ungers and E. Gllboa. 1990. Overexpression of TAR sequences renders ce1 ls resistant to h uman irnmunodeficiency virus replication. Cell 63:601-608.
Tanese, N., V.R. Piasad and S.P. Goff. 1988. Structural requirernents for bacterial expression of stable, enzymatically active fusion proteins containing human immunodeficiency vims reverse transcrÎptase. DNA 7:407-4? 6.
Teich, N. 1985. Taxonomy of retroviruses, p. 1-16, In Weiss, R.A. N. Teich, H. Varmus and J. Coffin, (ed.), RNA Timor Vinses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N-Y.
Telesnitsky, A. and S.P. Goff. 1997. Reverse transcription and the generation of retroviral DNA, p. 721-160. In Coffin, LM., S.H. Hughes and H.E. Varmus, (ed.), Retrovinrses. Cold Spring Harbor Laboratory Press. Cold Sprhg Harbor, N.Y.
Tessmer, U. and HA. Kiausslich. 1998. Cleavage of human immunodeficiency virus type t proteinase from the N-terminally adjacent p6' protein is essential for effkient Gag polyprotein processing and viral infectivity. 3. ViroC. 72:3459-3463,

Thall, M., A Bukovsiû, E. Kondo, B. Rosenwirth, C X Walsh, J. Sodroski and H.G. Gottllngei. 1994. Functional association of cyciop hilin A with HIV-1 virions, Nature 372~363-365.
Tomasselli, A.G., J.O. Hu€, T.K= Sawyer, D.J. Staples, C.A. Bannow, I.M. Reardon, V.K. Chaudhary, C.M. Fryllng, 1. Pastan. D.J. Fitzgerald and R.L. Heinrikson. 1990, Proteases from human immunodeficiency virus and avian myeloblastosis virus show distinct specificities in hydrolysis of multidomain protein substrates. J. Virol. 64:3157- 3161.
Tomasselli, ADGD, W.J. Howe, J.O. Hui, T.K. Sawyer, 1-MD Reardon, C-D. De, CS. Craik and R.L. HeInrikson. 1991a. Calcium-free calmodulin is a substrate of proteases from human immunodeficiency viruses 1 and 2. Protein Struc. Funct, Genet, t0:l-9.
Tomasselli, A.G., J.O. Hui, L Adams, J. Chosay, D. Loweiy, B. Greenberg, A. Yem, M.R. Deibel, N.H. Zurcher and R.L. Heinrikson. 1991b. Actin, troponin C, Alzheimer amyloid precursor protein and prointerleukin 1 beta as substrates of the protease from human immunodeficiency virus. J . Biol. Chem. 266: 1 4548-1 4553.
Tomasselli, A.G., A.M. Mildner, D.J. Rothrock, J L Sarclch, J. Lull, J. Leone and R.L. Heinrickson. 1995. Mutants of HlV-1 protease with enhanced stability to autodegradation, p. 387-398. In Takahashi, Ki, (ed.), Aspaltic Proteinases: Structure, Function, Biologfi and Biomedical Implcatiuns. Plenum Press, New York.
Tozser, J., 1. Blaha, T.D. Copeland, E.M. Wondiak and S. Oroszlan. 199 1. Cornparison of the HlV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol poIyproteins. FEBS L e t 281 :77-80.
Tremblay, M.J., &Fe Fortin and R. Cantin. 1998. The acquisition of host- encoded proteins by nascent HIV-1. Immunol. Today l9:346-35?.
Tritch, R.J., Y.-S. Cheng, EH. Yin and S. Eiickson-Viitanen. 1991. Mutagenesis of protease cleavage sites in the human immunodeficiency virus type 1 Gag polyprotein. J. Virol. 65:922-930.
Trkola, A., T. Dragic, J. Arthoi, J.M. Binley, W.C. Olson, C. Chen- Meyer, J. Robinson, P.J. Maddon and J.P. Moore. 1996. CD4- dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5- Nature 384:184-18?.
Trono* Dm, M.B. Feinberg and 0. Baltimore. 1989. HIV-1 Gag mutants can dominantIy interfere with the replication of the wild-type virus. Cell 59: 1 1 3- 120.

Trono, D. 1995. HlU accessory proteins: feading roles for the supporting cast CeH 82: 189-1 92.
Ullch, C., D. Harrich, P. Estes and R.B. Gaynor. 1996. Inhibition of human immunodeficiency virus type 1 replication is enhanced by a combination of transdominant Tat and Rev proteins. J. Virol. 70:48714876.
Valverde, V., P. Lemay, J.M. Masson, B. Gay and P. Boulanger. 1992. Autoprocessing of the human imrnunodeficiency virus type 1 protease precursor expressed in Escherichia coli from a synthetic gene. J. Gen. Virol. 73~639-651.
Veres, O., S. Escaich, J. Baker, C. Barske, C. Kalfoglou, H. Ilves, H. Kaneshima and E. Bohnlein. 1996. Intracellular expression of RNA ttanscripts complementary to the human irnmunodeficiency virus type 1 gag gene inhibits viral replication in human CD4+ lymphocytes. J. Virol. 70:8792- 8800.
Veies, G., U. Junker, J. Baker, C. Barske, C. Kalfoglou, H. Ilves, S. Escalch, H m Kaneshima and E. BohnIein. 1998. Comparative analysis of intracellularly expressed antisense RNAs as inhibitors of human immunodeficiency virus type 1 replication. J. Virol. 72:1894-19Ol.
Veronese, F.D., R. Rahman, T.D. Copeland, S. Oroszlan and R.C. Gallo. 1987. Immunological and clinical analysis of p6, the carboxy-terminal fragment of HIV p l 5 AIDS Res. Hum. Retroviruses 3:253-264.
Viellard, V:, E. Lauret, V. Rousseau and E. DeMaeyet. 1994. Blocking of retroviral infection at a step prior to reverse transcription in cells transformed to constitutively express interferon beta Proc. Natl. Acad. Sci. USA 9112689- 2693.
Vogt, P.K. 1997. Historical introduction to the general properties of retroviruses, p. 1-27. In Coffin, LM., S.H. Hughes and HE. Varmus, (ed.), Retrovfruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Vogt, V.M. 1996. Proteolytic processing and particle maturation. Curr. Top. Microbiol. lmmunol, 214:95-132.
VonPoblotzki, A., R. Wanger, M. Neidrlg, O. Wannet, H. Wolf and S. Modrow. 1993. ldentification of a region in the PrS5gag polyprotein essential for HIV-1 particle formation. Virology l93:981-985.
VonSchwedler, U., J. Song, C, Aiken and 0. Trono. 1993. Vif is crucial for human ~munodeficiency virus type 1 proviral DNA synthesis in infected cells. J. ViroI. 6?:4945-4955.
VonSchwedIer, U., R.S. Kornbluth and O. Trono. 1994. The nuclear Iocalization signal of the rnatrk protein of HIV-1 allows the establishment of

infection in macrophages and quiescent T lymphocytes. Proc. Natl. Acad. Sci. USA 91 :6992-6996.
Wan. M., M. Takagf, B A . Loh and T. Imanaka. 1995. Comparison of HIV-1 protease expression in different fusion forms. Biachemistry and Molecular Biology International 36:411-419.
Wan, M., M. Takagi, B.-ND Loh, X d . Xu and T. Imanaka. 1996. Autoprocessing: an essential step for the activation of HIV-1 protease. Biochem. J. 316:569-573.
Wang, C.-T. and E. Barktls. 1993. Assembly, processing, and infectivity of human immunodeficiency virus type 1 Gag mutants. J. Virol. 67:4264-4273.
Wang, C.-T., H.Y. Lai and J.4. LI. 1998. Analysis of minimal human immunodeficiency type 1 gag coding sequences capable of virus-like particle assembly and release. J. Virol. 72:7950-7959.
Weber. I.T., M, Miller, M. Jaskolski, J. Leis, A.M. Skalka and A. Wlodawer. 1989. Molecular modefing of the HIV-1 protease and its substrate binding site. Science 243:928-93 1.
Weber, 1.T. 1990. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J. BioL Chem. 265A 0492-1 0496,
Wei, Xe, S.K. Ghosh, M.E. Taylor, V.A. Johnson, EDAe Emini, P. Deutsch, J.D. Lifson, S. Bonhoeffer, M.A. Norwak, B.H. Hahn, M.S. Saag and G.M. Shaw. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373: 1 17-1 22.
Weiss, C.D., J.A. Levy and J.M. White. 1990. Oligorneric organization of gpt20 on infectious human imrnunodeficiency type 1 particles. J. Virol. 64:5674-5677.
Weiss, R., N. Teich, H. Varmus and J. Coffin. 1985. RNA Tumor Viruses. Cold Spring Harbor Laboratory Press, Cold Spn'ng Harbor, N.Y.
Weiss, RA. 1 996. Retrovirus classification and ceIl interactions. Journal of Antimicrobial Chemotherapy 37 (Suppl. 8):l-11.
Welson, R.A., C.R. Erdie, M.G. Oliver and LW. Wltb. 1990. incorporation of chimeric Gag protein inm retroviral particles. J. Virol. 64:4169- 41 79,
Wen, W., +Lm Meinkoth. R.Y. Tskn and S.S. Taylor. 1995. Identification of a signal for rapid export of proteins from the nucleus. Cell 82:463-473.

Wlegers, K., O. Rutter, H. Kottler, U. Tessrnet. K Hohenberg and H.-O. Krausslich. 1998. SequentiaI steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavag e sites. J. Virol. 72:2846-2854.
Willey, R.L., F. Maldrelli, M.A. Martin and K. Strebel. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 66:?193-7200-
Wills, LW. and R.C. Craven. 1991. Fom, function, and use of retroviral Gag proteins. AIDS 5639-654.
Wills, J.W., C.E. Cameion, C.B. Wilson, Y. Xiang, R.P. Bennett and J. Leis. 1994. An assernbly domain of the Rous satcoma vkus Gag protein required late in budding. J. Virol. 68:6605-6618.
Wilson, W., M. Braddock, S.E. Adams, P.D. Rathjen, S.M. Kingsman and A.J. Kingsman. 1988. HIV expression strategies: ribosomaI frameshifting is directed by short sequence in both mammalian and yeast systems. Cell 553 159-1 1 69.
Witte, O.N. and D. Baltimore. 1 978. Relationship of retrovirus polyprotein cleavages to virion maturation studied with temperature-sensitive murine leukemia virus mutants. 3. ViroL 26:750-761.
Wlodawer, Am, M. Miller, M. Jaskolski, B.K. Sathyanaiayana, E. Baldwin, 1.T. Weber, L.M. Selk, L Clawson, J. Schneider and S.B.H. Kent. 1989. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 26:6 16-621.
Woffendin, C., U. Ranga, 2. Yang, L Xu and G.J. Nabel. 1996. Expression of a protective gene prolongs survival of T cells in human immunodeficiency virus infected patients. Proc. Natl. Acad. Sci. USA 93:2889- 2894.
Wondrak, EMm, JmM- Louis, H. DeRocqufgny, J.C. Chermann and B.P. Roques. 1993. The Gag precursor contains a specific HIV-1 protease cleavage site between the NC (p7) and p l proteins. FEBS Lett. 333:21-24.
Wondrak, E.M., N.T. Nashed. M I , Haber, LM. Jerina and JmM. Louis. 1996. A transient precursor of the HLV-1 protease. J. Biol. Chern. 271 :44??-448l,
Wu, L., N.P. Gerard. R. Wyatt, H- Choe, C. Parolln, N. Ruffing, A. BoisettI, A.A. Cardoso, E. Desiardin, W. Newman, C. Gerard and J. Sodroski. 1996. CDGinduced interaction of primary HIV-I gp120 glycoproteins with the chemokine teceptor CCR-5. Nature 384: 179-1 83.

Wyatt, R., P.D. Kwong, E. Desjardins, R.W. Sweet, J. Robinson, W.A. Hendrickson and J.O. Sodroski. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705-711.
Xiang, Y:, T.W. Ridky, N-K. Krishna and J. Lets. 1997. Altered Rous sarcoma vrrus Gag polyprotein processing and its effects on particle formation. J. Virol. 71 :2083-209 1,
Yao, 4 . AmJ. Mouland, R.A. Subbramanian, J. Forget, ND Rougeau, Dm Bergeron and E.A. Cohen. 1998. Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1- infected dividing Jurkat T cells. J. Virol. 72:4686-4693.
You, J.C. and C.S. McHenry. 1994. Human immunodeficiency virus type 1 nucleocapsid protein accelerateç Strand transfer of the terminally redundant sequences involved in reverse transcription. J. Biol. Chem. 269:31491-31495.
Yu, X., X- Yuan, 2. Matsuda, T.H. Lee and M. Essex. 1992. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J. Virol. 66:4966-4971,
Yu, XPF., 2. Matsuda, Q.-C. Yu, TmHm Lee and M. Essex. 1995. Role of the C-terminus Gag protein in human irnmunodeficiency virus type 1 assembly and maturation, 3. Gen, Virol. 76331 71 -31 79.
Yu, X.F-, L. Dawson, C.-J. Tian, C. Flexner and M. Dettenhofer. 1998. Mutations of the human immunodeficiency virus type 1 pGWg domain result in reduced retention of Pol proteins during -assernbly. J. ~irol . 72:3412- 3417.
Zack, JA., S.J. Arrlgo, S.R. Weitsman, AS. Go, A. Haislip and LS.Y. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61 :213-222.
Zennou, V., F. Mammano, S. Paulous, O. Mathez and F. Clavel. 1998. Loss of viral fitness associated with multiple Gag and Gag-Pol defects in human immunodeficiency virus type 1 variants sefected for resistance to protease inhibitors in vivo. J. Virol. 72:3300-3306.
Zhang, L-Q, P. Mackenzie, A. Cleland, EX. Holmes, A.J. Brown and P. Simmonds. 1993. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon prhary infection. J. Viral- 67:3345-3356.
Zhang, W.-H., D.J* Hockley, MDVm Nerrnut, Y. Morikawa and I.M. Jones. 1996. Gag-Gag interactions in the C-terminal domain of human immunodeficiency virus type 1 p24 capsid antigen are essential for Gag particle assembly. J. Gen. Virol. P.743-751.

Zhang, Y. and E. Barklis. 1995. Nucleocapsid protein effects on the specificity of retrovirus RNA encapsidation. J. Virol. 69:5716-5722.
Zhang, Y., H. Qian, 2. Love and E. Bariûis. 1998. AnaIysis of the assembly function of the human imrnunodeficiency virus type 1 Gag protein nucleocapsid domain. J. VRoL 72:1782-1789.
Zhang, Z X , RDA- Poorman, LL. Maggtora, R.L. Helnrikson and F.J. Kezdy. 1991. Dissociathe inhibition of dimeric enzymes, kinetic characterization of the inhibition of HlV-1 protease by its COOH-teminal tetrapeptide. J. Biol. Chem. 266: 1559 1 -1 5594.
Zhou, W., LJ. Parent, J.W. WilIs and M.D. Resh. 1994a. Identification of membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556-2569.
Zhou, CD, I.C. Bahner, G.P. Lacson, J.A. Zaia, J.J. Rossi and D.B. Kohn. 1994b. Inhibition of HIV-1 in hurnan T4ymphocytes by retrovirally transduced anti-tat and rev hammerhead ribozymes. Gene i49:33-39.
Zhou, C., 1. Bahner, J.J. Rossi and D.B. Kohn. 1996. Expression of hamrnerhead ribozymes by retroviral vectors to inhibit HIV-1 replication: corn parison of RNA Ievels and viral inhibition. Antisense Res. Dev. 6: 11-24.
Zhu, T., H. Mo, N. Wang, D.S. Man, Y. Cao, R.A. Koup and D.D. Ho. 1993. Genotypic and phenotypic characterization of HIV-1 patients with prirnary infection. Science 261 : 1 1 79-1 1 81.
Zybarth, O, H.G. Krausslich, K. Partin and C. Carter. 1994. Proteolytic activiry of novel human immunodeficiency virus type 1 proteinase proteins from a precursor wHh a blocking mutation at the N-terminus of the PR domain. J. Virol. 68:240-2SO.
Zybarai, G. and C. Carter. 1995. Domains upstream of the protease (PR) in human imrnunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J. Virol. 69:3878-3884.




















