
Edge-competing Pathological Liver Vessel Segmentation with ...

Effects of Competing Anions and Iron Bioreductionon Arsenic Desorption
Juscimar Silva & Jaime Wilson Vargas de Mello &
Massimo Gasparon &
Walter Antônio Pereira Abrahão
Received: 1 February 2012 /Accepted: 29 August 2012 /Published online: 18 September 2012# Springer Science+Business Media B.V. 2012
Abstract Dissimilatory iron-reducing bacteria play afundamental role in catalysing the redox transforma-tions that ultimately control the mobility of As inanoxic environments, a process also controlled bythe presence of competing anions. In this study, weinvestigated the decoupling of As from loaded Al andFe (hydr)oxides by competing anions in the presenceof iron-reducing bacteria. Hematite, goethite, ferrihy-drite, gibbsite and three aluminium-substituted goe-thites (AlGts) were synthesised and loaded witharsenate, followed by anaerobic incubation with dif-ferent phosphate or carbonate-containing media in thepresence of catalytic iron-reducing bacteria. SolubleAl, As, Fe and P contents were measured in aliquots
by inductively coupled plasma optical emission spec-trometry following periodical sampling. Shewanellaputrefaciens cells were able to utilise both non-crystalline and crystalline Fe (hydr)oxides as electronacceptors, releasing Fe and As into solution. Phos-phate and carbonate affected the Fe bioreduction,probably due to the precipitation of metastable mineralphases and also to phosphate-induced stabilisation onthe hydroxide surfaces. Phosphate precipitation actedas a sink for As, thus limiting its mobilisation. Thehighest fraction of desorbed As by phosphate wasobserved for gibbsite, followed by AlGts. Similarly,gibbsite showed significant amounts of arsenate dis-placed by carbonate. In spite of its low crystallinity,ferrihydrite was the most efficient compound in retain-ing arsenate, possibly due to As co-precipitation. Thisstudy provides new insight into the management ofAs-contaminated soils and sediments containing Al-goethites and gibbsite, where the Fe activity may betoo low to co-precipitate As-bearing vivianite. Thus,the dynamics of As(V) in flooded soils are significantin agriculture and environmental management.
Keywords Arsenic contamination . Al-substitutedgoethites . Soil . Sediments . Redox stability
1 Introduction
Coupling arsenic mobilisation with microbial reductivedissolution of Fe (hydr)oxide is one of the most
Water Air Soil Pollut (2012) 223:5707–5717DOI 10.1007/s11270-012-1308-0
J. Silva (*)Embrapa Vegetables,Rod. BR 060, km 09, Brasília/Anápolis, 70359-970,CP 218 Gama, Federal District, Brazile-mail: [email protected]
J. W. V. de Mello :W. A. P. AbrahãoSoil Department, Federal University of Viçosa,Av. P.H. Rolfs, s/n,Viçosa, Minas Gerais, Brazil 36570-000
M. GasparonSchool of Earth Sciences, The University of Queensland,St. Lucia,Brisbane, QLD 4072, Australia
J. Silva : J. W. V. de MelloNational Institute of Science and Technology, INCTAcqua,Belo Horizonte, Brazil

important aspects of the arsenic biogeochemistry. Thenatural input of arsenic in the environment is mainly dueto the weathering of As-bearing rocks and minerals.Anthropogenic sources, i.e. mining and smelting activi-ties, pesticides and wood preservative are potential addi-tions to the natural budget (Matschullat 2000; NHRMCand NRMMC 2004; Smedley and Kinniburgh 2002).An increase in the global cycling of As can be expecteddue to progressive industrialisation of developingnations (Leist et al. 2000) as well as to the increasedexploitation of groundwater resources.
Arsenic in nature can be found as inorganic andorganic compounds, in several valence states, i.e. −3,−1, 0, +3 and +5. In terrestrial environments, As occursmainly in inorganic forms as trivalent arsenite [As(III)](as H3AsO3) or pentavalent arsenate [As(V)] (asH2AsO4
− and HAsO42−). The lower is the valence state,
the higher is the toxicity of As compounds, with theinorganic forms being more toxic than the organic ones.Despite the different degrees of toxicity, there is nodistinction between As species in water quality stand-ards (Kim et al. 2002). Redox potential (Eh) and pHcontrol As distribution, and arsenite is expected to be thedominant species in anoxic conditions. However, due torelatively slow transformation following changes in re-dox conditions, both As(III) and As(V) can be oftenfound in either redox environment.
Aluminium and Fe (hydr)oxides are ubiquitous re-active constituents of soils, sediments and aquifers. Inhighly weathered tropical soils, these minerals arenaturally abundant, and due to their high reactivity,they play a fundamental role on the biogeochemicalcycle of many elements. The distribution of As underoxidising conditions is mostly controlled by theseminerals, and for this reason, soil materials have beenused as liners in remediation procedures at contami-nated sites. Ladeira and Ciminelli (2004) working withan oxisoil sample composed primarily of gibbsite,goethite and Al silicate minerals verified that thismaterial would be adequate as a protective liner inwaste dams due to its remarkable As immobilisationcapability.
In the absence of oxygen, however, Fe(III) mineralscan be used as electron acceptors during microbial res-piration. Therefore, based on the association betweenmany trace elements and Fe(III) (hydr)oxides, and thetendency of these minerals to be dissolved under suboxicconditions, biological Fe reduction can have a majorimpact on the persistence andmobility of toxic elements,
radionuclides and organic contaminants under such con-ditions (Cummings et al. 1999; Zachara et al. 2001).Cummings et al. (1999) investigated the biological re-duction of synthetic scorodite and noted that dissimila-tory Fe reduction led to the release of As(V) and Fe(II)into solution. Conversely, Al is rather stable under lowEh conditions and its intrusion into the Fe(III)(hydr)oxides structure by isomorphic substitutionreduces the rate of Fe reductive dissolution, as alreadyreported (Bousserrhine et al. 1999; Jeanroy et al. 1991;Schwertmann 1984).
Aluminium replacement of Fe under natural condi-tions is a very common process, and therefore most Feminerals found in soils, particularly from tropical cli-mates, show a certain degree of Al for Fe substitution.The use of such soils or precipitated mineral phasescombining the higher binding affinity of Fe(hydr)oxides for As, and the higher stability of Al underanoxic conditions, may be advantageous for As immo-bilisation. Very few studies have focused on As sorptionprocesses onto Al-substituted Fe (hydr)oxides or Asrelease from these compounds. Masue et al. (2007)reported a decrease in both As(III) and As(V) adsorptiononto Al/Fe hydroxides and an increase in As desorptionby phosphate as the Al/Fe molar ratio increased. Re-cently, Silva et al. (2010) reported that the presence ofAl substituting Fe in the goethite structure increasedboth the stability of goethite under low redox conditionsand its As adsorption capacity, being about three timesmore efficient than pure goethite.
Natural attenuation of As by sorption onto Al and Feminerals may be also limited by other anions competingwith arsenate for sorption sites (Hongshao and Stanforth2001; Sahai et al. 2007; Zhang et al. 2008). Due tosimilar acid dissociation constants, phosphate (pKa102.1, pKa207.2 and pKa3012.3) behaves much like arse-nate (pKa102.2, pKa206.9 and pKa3011.4). Besidesphosphate, carbonate may also, to a lesser extent, affectthe arsenate sorption reactions. The displacement ofadsorbed As by carbonate was theoretically examinedby Appelo et al. (2002) and this mechanism was pro-posed to be potentially one of the main reasons for highAs concentrations in groundwater.
Although investigations of competition betweenanions can provide insight into the reactions occurringon the mineral surface (Hongshao and Stanforth2001), most of the As desorption studies have evalu-ated those reactions individually. Nevertheless, Asmobilisation in natural environments may depend on
5708 Water Air Soil Pollut (2012) 223:5707–5717

the combination of different factors, such as the pres-ence of competing anions and bacterial catalytic activ-ities. Therefore, this work investigates the decouplingof As from Al-goethites and other synthetic Al and Fe(hydr)oxides by competing anions in the presence ofdissimilatory iron-reducing bacteria.
2 Materials and Methods
2.1 Synthesis of Al and Fe (hydr)oxides
Hematite (Hm), goethite (Gt) and two-line ferrihydrite(Fh) were synthesised according to Schwertmann andCornell (2000). Goethites with 13, 20 and 23 cmolmol−1
of Al, henceforth referred to as AlGt13, AlGt20 andAlGt23, respectively, were prepared by precipitation fol-lowing the reaction of ferrous and aluminium chloridesolution with a potassium hydroxide solution and set-tling in plastic bottle for 90 days. Slow oxidation of Fe2+
to Fe3+ and incorporation of Al3+ in the goethite struc-ture were achieved by opening the bottle daily andstirring the suspension vigorously for 5 min. To removethe excess of Al, the precipitates were washed twicewith a 0.01 molL−1 KOH solution. Precipitates of(hydr)oxides were washed several times with Milli-Qwater, centrifuged and air-dried at 50 °C in an oven,except for Fh, which was frieze-dried to prevent crys-tallisation. Gibbsite (Gb) was prepared after titrating anAl(NO3)3 solution with 4 molL−1 NaOH to a pH of 4.6±0.2 (Kyle et al. 1975). The gelatinous precipitate washeated for 2 h at 40 °C, then washed twice, dialysed
against Milli-Q water for 36 days and air-dried at 50 °C.The mineral phases were characterised by X-ray diffrac-tion, diffuse reflectance and Raman spectroscopy (datato be published elsewhere). Specific surface area wasdetermined by N2 adsorption using the multiple pointtechnique (Quantachrome model NOVA 1000, seeTable 1). Samples were degassed at 110 °C for 2 h undera continuous N2 stream prior to surface area determina-tion. These adsorbents were equilibrated with sufficientAs(V) to achieve their maximumAs adsorption capacity(Silva et al. 2010). Arsenate solutions were prepared bydissolving analytical reagent-grade disodium hydrogenarsenate heptahydrate (Na2HAsO4·7H2O; Ajax Fine-chem) in Milli-Q water. Solid samples (0.1000 g;<53 μm) along with 25 mL of arsenate solution (con-centrations ranging from 0.534 to 21.586 mmolL−1) atpH 5±0.2were placed into 50mL polypropylene conicalbase centrifuge tubes with screw caps and equilibratedfor 24 h on a rotary shaker. The pH was measured andadjusted during the first 4 h as necessary. The amount ofAs(V) adsorbed was estimated by the difference betweenthe initial and final As concentration in solution. Exper-imental tests with arsenic in solution but no adsorbentmaterial were carried out to evaluate the amount ofarsenic adsorbed by the walls of the reaction vessels.
2.2 Influence of Phosphate
Cultures of Shewanella putrefaciens obtained from theAustralian Collection ofMicroorganisms at The Univer-sity of Queensland (Strain ACM 4733(ATCC49138);incubation temperature/time (in days)037/1) were
Table 1 Specific surface area (SSA), adsorption maxima (b), binding constant (K) and correlation coefficient (R2) from the linear formof the Langmuir isotherm for arsenate adsorption onto different Fe and Al (hydr)oxides
Adsorbent SSA b K R2
gm−2 mmolg−1 Lmmol−1
Hm 34.8 0.193±0.006 2.562 0.9949
Gb 45.7 0.228±0.006 7.116 0.9964
Gt 20.6 0.101±0.002 10.188 0.9988
AlGt13 119.4 0.417±0.006 22.301 0.9988
AlGt20 124.7 0.395±0.004 35.401 0.9996
AlGt23 113.2 0.365±0.007 13.634 0.9975
Fh 260.4 1.258±0.034 2.041 0.9932
Experimental conditions: pH05±0.2, adsorbent mass 0.1000 g (<53 μm), ionic strength 10 mmolL−1 CaCl2, equilibration time 24 h.AlGt13, AlGt20 and AlGt23 are Al-goethites with 13, 20 and 23 cmolmol−1 of Al, respectively (Silva et al. 2010). (Mean values ±standard error, N03)
Hm hematite, Fh ferrihydrite, Gt goethite, Gb gibbsite
Water Air Soil Pollut (2012) 223:5707–5717 5709

grown aerobically in a Biosafety Cabinet Class II fol-lowing the specific procedures outlined in the AustralianCollection of Microorganism protocol. S. putrefacienswas selected for our experiments because of its well-documented ability to respire Fe(III), an anaerobic ter-minal electron acceptor (e.g. Cummings et al. 1999;Bonneville et al. 2006). The bacteria were grown for18 h to attain the late-log phase and routinely cultured at37 °C in a sterile medium containing 10 gL−1 peptone,5 gL−1 yeast and 5 gL−1 NaCl at pH 7.2. The compo-sition of the basal P-containing medium used in thisexperiment was (in millimoles per litre): NH4Cl (20),KCl (1.34), CaCl2 (1.0), MgCl2 (0.34), phosphate (asKH2PO4) (5.0) and sodium lactate (20) as the onlyelectron donor. The medium was extensively purgedwith high-purity N2 and autoclaved at 121 °C for20 min. After that, 0.2000 g of As-loaded adsorbentswas equilibrated with 96 mL of the sterile basal mediumand 4.0 mL of S. putrefaciens cell suspension in a 125-mL screw cap plastic bottle. Themixture was buffered atpH 7.0 by adding 10 mmolL−1 of 1,4-piperazinedietha-nesulfonic acid (PIPES). The suspensions were thenpurged with high-purity N2 and incubated anaerobicallyin a glove box for about 500 h. Aliquots of the equilib-rium solutions were periodically sampled to determineAs, Al, Fe and P concentrations. All aliquots weresyringe filtered using 0.22 μm membrane filters (Milli-pore Millex-GV, USA), and the filtered solutions wereacidified using high-purity concentrated HNO3 (2 %HNO3 in the final volume) and stored in a cold roomat ∼+2 °C until analysis. Sterility was maintainedthroughout the experiment as all reagents were keptinside a glove box vented using high-purity N2.
2.3 Influence of Carbonate
The decoupling of As by carbonate under anaerobicconditions was examined under conditions similar tothe experiment described above, except for the addi-tion of 30 mmolL−1 of carbonate (as NaHCO3) toreplace phosphate, and the lower concentration oflactate (10 mmolL−1) and As-loaded adsorbents(1.0 gL−1). In this experiment, NaHCO3 substitutedthe PIPES solution as the pH buffer.
2.4 Analytical Techniques
Soluble Al, As, Fe and P contents in the aliquots weremeasured by inductively coupled plasma optical
emission spectroscopy, using a Perkin Elmer Optima3300 DV. Scandium was used as the internal standardto correct for instrumental instabilities and matrixeffects. Solutions of this element were added to thealiquots to reach a final concentration of 44.5 μmolL−1. Typical detection limits (3σ) of 0.42 μmolL−1
were obtained for As.For both experiments, the total volume of the aliquots
did not exceed 10% of the initial volume. All chemicalsused were of reagent grade, and water was purifiedthrough a Milli-Q system. All cell additions, transferand culture samplings were performed with sterile sy-ringes or pipette tips previously flushed with high-purityN2. All assays were carried out in triplicates.
3 Results and Discussion
3.1 Influence of Phosphate as Competing Anion
The dynamics of P under anaerobic conditions at pH 7±0.2 and 25 °C showed that phosphate readily displacedarsenate and immediately reacted with other ions insolution and solid matrices (Fig. 1). In general, morethan 20% of the added phosphate was immobilised after4 h (first measurement) as a result of adsorption orprecipitation equilibria. Theoretical calculations usingVisual Minteq® (ver. 2.3) (Gustafsson 2000) confirmedthis observation, as they indicated saturation of calciumphosphate under our experimental conditions. The ad-sorption of phosphate followed byAs(V) desorption canalso be considered due to exchange reactions.
Initial P immobilisation was higher, almost 30 % inthe suspensions containing Fh or Al-goethites. Consid-ering that all adsorbents were submitted to similar ex-perimental conditions, i.e. equal concentrations of Caand P added to the medium, P immobilisation due to Ca-P precipitation should be similar. The additional amountof P immobilised for Fh and Al-goethites in relation tothe other (hydr)oxides can be attributed to adsorption ofP onto other sites not occupied by As(V). Consideringthat arsenic was loaded at near maximum adsorptioncapacity, and that P was supplied at concentrations(5 mmolL−1) sufficient to ensure a large excess withrespect to total adsorbedAs, the amounts of desorbedAs
Fig. 1 Fe(III) reduction, As release and P immobilisation fromdifferent Al and Fe (hydr)oxides suspensions in the presence ofS. putrefaciens
b
5710 Water Air Soil Pollut (2012) 223:5707–5717

0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
AsPFe
Hematite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2Goethite
2-line Ferrihydrite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
AlGt20
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
AlGt23
AlGt13
Time (h)
Time (h)
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Gibbsite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
As
rele
ased
, P im
mob
ilise
d (%
)
Fe
rele
ased
(m
mol
L-1
)
Water Air Soil Pollut (2012) 223:5707–5717 5711

does not explain the excess of P immobilised by thoseadsorbents. Previous investigations have shown thatsurface coverage in competitive adsorption experimentsis higher than the adsorption of the individual ions(Hongshao and Stanforth 2001; Zhang and Selim2008), suggesting that there are some specific sites foreach ion as well as other sites that can adsorb both(Manning and Goldberg 1996).
After 12 h, the fraction of P immobilised decreased,achieving a steady state by 144 h when no significantFe release was observed. This indicates remobilisationof P probably by precipitation of metastable phases, aspointed out previously. However, the late steady-stateequilibrium was not observed for Fh and Al-goethites(Fig. 1). On the contrary, there is a clear increase in thefraction of P immobilised by these minerals after144 h, and this coincides in general with the peak ofsoluble Fe, notably for Fh. This suggests further pre-cipitation of Fe(II)-P phases as a result of secondaryreactions, confirmed by thermodynamic calculations.For the conditions adopted in the experiment, suchcalculations revealed that vivianite [Fe3
(II)(PO4)2·8H2O]starts forming at Fe(II) concentration higher than 10−3
mmolL−1. Therefore, the formation of biogenic vivian-ite can account for the surprisingly low concentration ofsoluble Fe under anaerobic conditions (Fig. 1), as alsonoted by Burnol et al. (2007). Silva et al. (2010) carriedout a reduction experiment with a range of Fe-Al oxidesand hydroxides with and without adsorbed As(V), andreported higher concentrations of soluble Fe in the latter,with bioreduced Fe concentrations about 140 and 40times higher for Hm and Gt, respectively. In the pres-ence of adsorbed As(V), however, the effectiveness ofbacterial activity in reducing Fe was probably limited byelectrostatic repulsion between As-loaded (hydr)oxidesand bacteria cells. Borch et al. (2007) attributed thedecrease in ferrihydrite bioreduction to phosphate-induced stabilisation, a mechanism also proposed byEvangelou (2001) to stabilise iron (hydr)oxides filmsprecipitated on the surface of sulphide minerals, andtherefore to reduce oxidation of tailing materials. Ac-cordingly, the surface coverage restricting the associa-tion between (hydr)oxide minerals and S. putrefacienscells, and anion-induced stabilisation are expected to beeven higher in our experiment, since the surfaces of allminerals were loaded with As(V) and P.
Similar As desorption trends were observed for allmaterials. The fraction of mobilised As decreased inthe following order: Gb ≥ Al-goethites > Gt > Hm >
Fh. Arsenic release commenced readily after phos-phate addition (∼4 h), increasing quickly up to 48 hand decreasing slowly thereafter (Fig. 1). For example,nearly 70 % of the total As content was released fromGt within 48 h. Considering that no soluble Fe wasdetected during the initial interval (4–48 h), the Asdisplacement may be attributed exclusively to ligandexchange reactions.
More than 50 % of the As was released from Gband Al-goethites within the time frame of the experi-ment. For Gb, this can be attributed entirely to ex-change by phosphate on the Gb surface, but for Al-goethites, part of the As released resulted from Febioreduction. The fraction of As mobilised from Al-goethites increased with structural Al content (Fig. 1,AlGt13, AlGt20 and AlGt23), suggesting that the re-leased As was preferentially associated with Al sites.Such weak As retention to Al sites suggests the for-mation of unstable surface complexes likely related tosurface precipitation of As on these adsorbents. Thishypothesis is not fully supported by Ladeira et al.(2001), who showed that As forms an inner-spherebidentate binuclear complex on the surface of Al oxy-hydroxyl octahedra. Detailed spectroscopic investiga-tion is warranted to further elucidate the type ofsurface complexes formed between As and those ma-trices. Our data are in agreement with the report ofMasue et al. (2007) of increasing As(V) desorption byphosphate from poorly crystalline Al-Fe hydroxides asthe Al/Fe ratio increased, reaching almost total arse-nate desorption from pure Al hydroxide after 24 h.The much higher P/As (7500:1) ratio used by Masue etal. (2007) in their experiment may account for thefaster and higher amount of As desorbed.
The relatively low concentration of As desorbed byphosphate from crystalline Fe (hydr)oxides (26 % forFh and Hm and 37 % for Gt) is consistent with thefindings of Hongshao and Stanforth (2001), who sug-gested that arsenate is adsorbed mainly as a non-exchangeable ion, although a lesser amount is ex-changeable by phosphate (Fig. 1). Indeed, As(V) orphosphate adsorption kinetics are considered as a two-phase reaction, with a rapid initial step followed by amuch slower reaction (Hongshao and Stanforth 2001;Torrent et al. 1992; Zhang and Selim 2008).
A different behaviour was observed for Fh, where theamount of As released reached its maximum by 192 hand then suddenly dropped to almost 0. This depletionof soluble As coincided with P immobilisation, which is
5712 Water Air Soil Pollut (2012) 223:5707–5717

likely to be due to biogenic vivianite formation, as statedearlier. Unlike for P, there was no expectation for Asimmobilisation due to Fe(II)-As phase precipitation;however, arsenate can be associated with As-bearingvivianite by co-precipitation reactions, as arsenatebehaves much like phosphate. This hypothesis is realis-tic considering the structure of arsenate and phosphateminerals, where typical tetrahedral anions [XO4]
3− arebound to octahedrally coordinated transition metal ions.Variations and multiples of these bonding patterns tendto create relatively open structures that allow for exten-sive substitution of cations, anions, anions groups andwater (O'Day 2006). Consequently, the precipitation ofFe(II)-P phases could provide a sink for As, giving rise tovivianite-like minerals [Fe3
(II)[(As, P)O4]2·8H2O]. Theadsorption of As onto vivianite surface may to a lesserextent explain the depletion in soluble As noted byThinnappan et al. (2008), who showed that sorption ofarsenate onto natural well-crystallised vivianite in thepH range 3–11 varied between 25 and 40 % from astarting concentration of 100 μmolL−1. In addition, theadsorption was complete for lower As(V) concentra-tions (1 and 10 μmolL−1), and the sorption reactionswere little affected by pH variations. These observationshave significant environmental implications and provideuseful information on the fate of As in eutrophic-reducing aquatic systems.
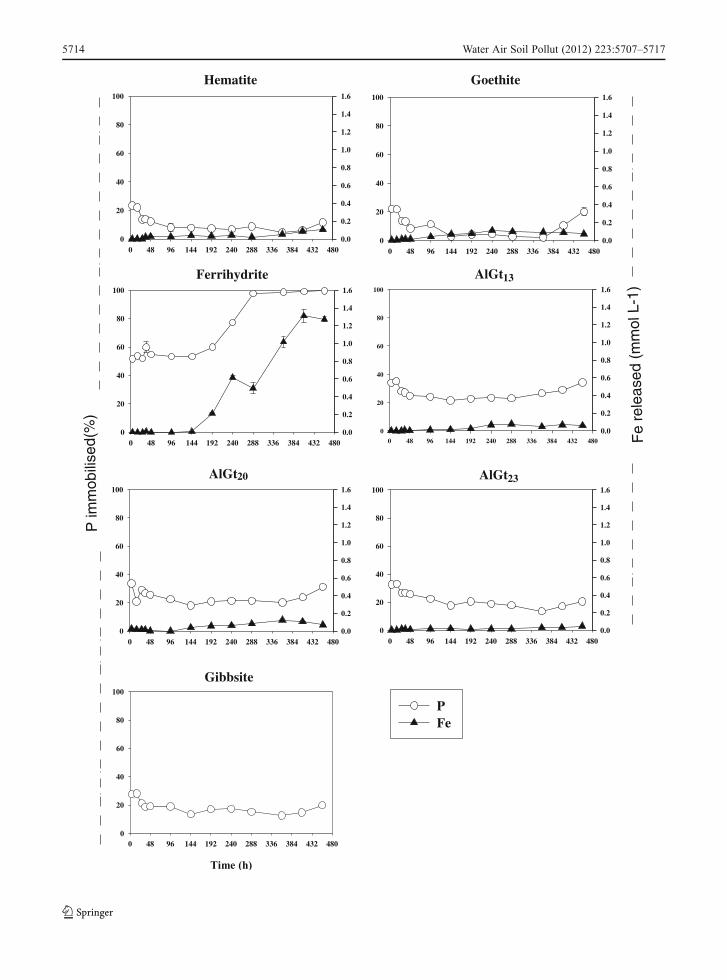
Adsorbents not loaded with As(V) showed similarP immobilisation patterns, with P precipitation/sorp-tion reactions taking place in the first hours, andequilibrium attained over the course of the experiment(Fig. 2). The amount of P immobilised was, however,greater than that observed in the As-loaded adsorbents,due mostly to the higher availability of adsorptivesites. The largest difference was noticed in Fh, whereP immobilisation varied from 30 to 50 % in the pres-ence and absence of adsorbed As(V), respectively. ForAl-goethites, the differences were smaller than for thewell-crystallised Gb, Gt and Hm and correspondedroughly to the amount of available adsorption sites,as measured by maximum As adsorption capacity(Table 1).
As previously addressed, the presence of adsorbedoxyanions decreased the efficiency of bacteria in usingFe as an electron acceptor due to phosphate coating.This effect is due to electrostatic repulsion betweennegatively charged mineral surface and bacteria cells,leading to phosphate-induced stabilisation. Similarresults were observed by Borch et al. (2007) for Fh,
where phosphate induced alterations in surface reac-tivity, by decreasing Fe(III) reduction nearly linearlyfor increasing P surface coverage. The amount ofbioreduced Fe was quite low and similar to that mea-sured in the presence of P and As(V), especially forAl-goethites that showed even lower Fe concentra-tions (<0.1 mmolL−1). Iron dissolution from Fh wascongruent with P immobilisation, which rapidly de-clined from 5 mmolL−1 in the first hour to near ana-lytical detection limits by 300 h. This pattern supportsthe hypothesis of the formation of an amorphousFe(II)-P phase, which was predicted by thermodynamiccalculations.
The reactions described here seem to mimic naturalsystems, where no Fe toxicity has been observed inphosphatic soils (Haldar and Mandal 1981; Wang etal. 1991), and changes in Fe forms in flooded soilshave a strong influence on P dynamics (Mello et al.1998; Chacon et al. 2006).
3.2 Influence of Carbonate as Competing Anion
Carbonate desorption of As from Al and Fe(hydr)oxides influenced by dissimilatory Fe reductionshowed different pattern from that induced by phos-phate (Fig. 3). Comparing the data from both systems,the content of soluble As was higher in Hm, Gt and Fhmedia and smaller in the AlGts ones when carbonatewas used as a competitor. Such pattern corroboratesthe hypotheses that, in phosphate systems, the precip-itation of Fe(II)-P phases could turn into an arsenicsink, forming a vivianite-like mineral.
Considering the smaller values of soluble As andthe fact that the fraction of As desorbed by carbonatefrom Al-goethites was almost the same in the presence(Fig. 3a) and absence (Fig. 3b) of Fe-reducing bacte-ria, it can be assumed that carbonate replaces arseniclinked to Al sites more efficiently than to Fe. This alsoindicates a weaker competitive effect of carbonatecompared with phosphate, as previously reported inthe literature (Arai and Sparks 2002; Goldberg andJohnston 2001; Radu et al. 2005). Radu et al. (2005),for instance, observed that carbonate decoupled less-adsorbed As(V) than phosphate even when present inmuch higher concentrations than phosphate. Gibbsitealso showed comparable fractions of As desorbed inboth presence and absence of S. putrefaciens cells andquite similar to those removed by phosphate. Thisresult contributes with the assumption that As(V)
Water Air Soil Pollut (2012) 223:5707–5717 5713
Hematite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
Goethite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
Ferrihydrite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
AlGt13
0 48 96 144 192 240 288 336 384 432 480
0
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
AlGt20
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
AlGt23
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
PFe
Gibbsite
0 48 96 144 192 240 288 336 384 432 4800
20
40
60
80
100
Fe
rele
ased
(m
mol
L-1
)
P im
mob
ilise
d(%
)
5714 Water Air Soil Pollut (2012) 223:5707–5717

adsorption onto the Gb surface is characterised by aweaker binding mechanism compared with other(hydr)oxides. It is also clear that As mobilisation fromGb and Al-goethites is primarily due to anion ex-change desorption, rather than to reductive dissolu-tion, as the fraction of Fe dissolved from theseadsorbents (Fig. 3c) was quite low (<2 %). On theother hand, Fh, Gt and to a lesser extent Hm showedhigher As release in the presence of bacteria, and thedifferences can be ascribed to the reductive dissolutionof Fe. The largest difference was observed for Fh,which showed less than 10 % of As released in theabsence of Fe-reducing bacteria, and roughly 50 % intheir presence.
In general, the concentrations of soluble Fe andAl were negligible in the absence of bacteria.
Significant fractions of Fe were solubilised in thepresence of bacteria only for Gt and Fh, and Asmobilisation was congruent with Fe bioreduction,reaching nearly 60 and 50 %, respectively. Al-though theoretical calculations indicated oversatu-ration with respect to siderite [Fe(II)CO3], unlikevivianite, this mineral cannot act as a sink forsoluble As.
Extensive spectroscopic evidence shows thatadsorbed As forms inner-sphere binuclear surfacecomplexes with Al and Fe (hydr)oxides (Fendorfet al. 1997; Goldberg and Johnston 2001; Ladeiraet al. 2001; Makris et al. 2007; Sherman andRandall 2003). Hence, these stable complexes canaccount for the generally low desorption of As bycarbonate. Nevertheless, our data suggest that thesecomplexes are to a certain extent more stable forFh, followed by Hm, compared to Gt and Gb.Prediction of As mobility, however, remains a verycomplex issue, and As sorption modelling requires
Time (h)
Time (h)
144 192 240 288 336 384 432 4800
20
40
60
80
0
20
40
60
80
Hm
Fh
Gt
AlGt13
AlGt20
AlGt23
Gb
0
2
4
6
8
10
12
14
Time (h)
0 48 96 144 192 240 288 336 384 432 4800 48 96
144 192 240 288 336 384 432 4800 48 96
a b
c
As
deso
rbed
(%
)D
isso
lved
Fe(
%)
Fig. 3 Fraction of As desorbed by carbonate and biologically reduced Fe(III) by S. putrefaciens cells. a Fraction of As mobilised in thepresence of bacteria; b fraction of As mobilised in the absence of bacteria; and c fraction of Fe solubilised by S. putrefaciens cells
Fig. 2 Fe(III) reduction and P immobilisation from different Aland Fe suspensions in the presence of S. putrefaciens. Data arerepresented as means ± standard error of the mean (n03); barsnot visible are smaller than symbol
R
Water Air Soil Pollut (2012) 223:5707–5717 5715

a more detailed understanding of the bondingmechanism and kinetics. Thus, investigations arewarranted to further elucidate the binding mecha-nisms that play a key role on As sorption on Al-substituted goethites surfaces.
4 Conclusions
This study has demonstrated the role of phosphateand carbonate on arsenate release from Al and Fehydroxides matrices, coupled with catalytic Fe-reducing bacteria. Despite the presence of adsorbedanions, the bacteria were able to use Fe as anelectron acceptor, thus releasing As into solution.Amorphous Fe hydroxides produced higheramounts of bioreduced Fe compared to their crys-talline counterparts. Both phosphate and carbonateremoved more efficiently As linked to matricescontaining Al, suggesting a weaker As-Al binding.Due to high concentrations of soluble Fe, phos-phate experienced secondary reactions resultingmost likely in the precipitation of vivianite, whichacted as an As sink by increasing the efficiency ofAs immobilisation. In natural systems, as observedin tropical regions, most soil goethites present acertain degree of Al for Fe substitution. Thisincreases their stability under low redox condi-tions and their efficiency in retaining potentialcontaminants.
This study provided useful information for themanagement of As-contaminated soils under anoxicconditions. Our data demonstrate that the presence ofsoluble phosphates and carbonates in waterloggedsoils can lead to an increase in the mobility and bio-availability of As, particularly in well-weathered trop-ical soils, if the Fe activity is not sufficient to co-precipitate As-bearing vivianite.
Acknowledgments The support from the Minas Gerais StateFoundation (FAPEMIG) and the Brazilian Research Council(CNPq) are gratefully acknowledged. Part of this study wascarried out while the first author was a visiting researcher atThe University of Queensland (UQ), with funding from CNPqand a UQ research grant. Laboratory work was carried out in theSchool of Earth Sciences at UQ (Australia) and in the SoilDepartment of the Federal University of Viçosa (Brazil). Theauthors are also grateful to Ms. Jenny Spratley (AustralianCollection of Microorganisms at UQ), who provided the S.putrefaciens cells used in this study.
References
Appelo, C. A. J., Van Der Weiden, M. J. J., Tournassat, C., &Charlet, L. (2002). Surface complexation of ferrous ironand carbonate on ferrihydrite and the mobilization of arse-nic. Environmental Science & Technology, 36, 3096–3103.
Arai, Y., & Sparks, D. L. (2002). Residence time effects onarsenate surface speciation at the aluminium oxide–waterinterface. Soil Science, 167, 303–314.
Bonneville, S., Behrends, T., Cappellen, P. V., Hyacinthe, C., &Roling, W. F. M. (2006). Reduction of Fe(III) colloids byShewanella putrefaciens: a kinetic model. Geochimica etCosmochimica Acta, 70, 5842–5854.
Borch, T., Masue, Y., Kukkadapu, R. K., & Fendorf, S. (2007).Phosphate imposed limitations on biological reduction andalteration of ferrihydrite. Environmental Science & Tech-nology, 41, 166–172.
Bousserrhine, N., Gasser, U. G., Jeanroy, E., & Berthelin, J.(1999). Bacterial and chemical reductive dissolution ofMn-, Co-, Cr-, and Al-substituted goethites. Geomicrobi-ology Journal, 16, 245–258.
Burnol, A., Garrido, F., Baranger, P., Joulian, C., Dictor, M. C.,Bodenan, F., Morin, G., & Charlet, L. (2007). Decouplingof arsenic and iron release from ferrihydrite suspensionunder reducing conditions: a biogeochemical model. Geo-chemical Transactions, 8, 12.
Chacon, N., Flores, S., & Ganzalez, A. (2006). Implication ofiron solubilization on soil phosphorus release in seasonallyflooded forest of the lower Orinoco River, Venezuela. SoilBiology and Biochemistry, 38, 1494–1499.
Cummings, D. E., Caccavo, J. F., Fendorf, S., & Rosenzweig, F.(1999). Arsenic mobilization by the dissimilatory Fe(III)-reducing bacterium Shewanella alga BrY. EnvironmentalScience & Technology, 33, 723–729.
Evangelou, V. P. (2001). Pyrite microencapsulation technolo-gies: principles and potential field application. EcologicalEngeneering, 17, 165–178.
Fendorf, S., Eick, M. J., Grossl, P., & Sparks, D. L. (1997).Arsenate and chromate retention mechanisms on goethite.1. Surface structure. Environmental Science & Technology,31, 315–320.
Goldberg, S., & Johnston, C. T. (2001). Mechanisms of arsenicadsorption on amorphous oxides evaluated using macro-scopic measurements, vibrational spectroscopy, and sur-face complexation modelling. Journal of Colloid andInterface Science, 234, 204–216.
Gustafsson, J. P. (2000). Visual Minteq 2.30: a free equilibriumspeciation model. Available at http://www2.lwr.kth.se/En-glish/OurSoftware/Vminteq/download.html. Accessed 20January 2008.
Haldar, M., & Mandal, L. N. (1981). Effect of Phosphorus andzinc on the growth and phosphorus, zinc, copper, iron andmanganese nutrition of rice. Plant and Soil, 59, 415–425.
Hongshao, Z., & Stanforth, R. (2001). Competitive adsorptionof phosphate and arsenate on goethite. Environmental Sci-ence & Technology, 35, 4753–4757.
Jeanroy, E., Rajot, J. L., Pillon, P., & Herbillon, A. J. (1991).Differential dissolution of hematite and goethite in dithion-ite and its implication on soil yellowing. Geoderma, 50,79–94.
5716 Water Air Soil Pollut (2012) 223:5707–5717

Kim, M. J., Nriagu, J., & Haack, S. (2002). Arsenic species andchemistry in groundwater of southeast Michigan. Environ-mental Pollution, 120, 379–390.
Kyle, J. H., Posner, A. M., & Quirk, J. P. (1975). Kinetics ofisotopic exchange of phosphate adsorbed on gibbsite. Jour-nal of Soil Science, 75, 34–43.
Ladeira, A. C. Q., & Ciminelli, V. S. T. (2004). Adsorption anddesorption of arsenic on an oxisol and its constituents.Water Research, 38, 2087–2094.
Ladeira, A. C. Q., Ciminelli, V. S. T., Duarte, H. A., Alves, M.C. M., & Ramos, A. Y. (2001). Mechanism of anionretention from EXAFS and density functional calculations:arsenic (V) adsorbed on gibbsite. Geochimica et Cosmo-chimica Acta, 65(8), 1211–1217.
Leist, M., Casey, R. J., & Caridi, D. (2000). The management ofarsenic wastes: problems and prospects. Journal of Haz-ardous Materials, 76(1), 125–138.
Makris, K. C., Sarkar, D., Parsons, J. G., Datta, R., & Gardea-Torresdey, J. L. (2007). Surface arsenic speciation of adrinking-water treatment residual using X-ray absorptionspectroscopy. Journal of Colloid and Interface Science,311, 544–550.
Manning, B. A., & Goldberg, S. (1996). Modelling competitiveadsorption of arsenate with phosphate and molybdate onoxide minerals. Soil Science Society of American Journal,60, 121–131.
Masue, Y., Loeppert, R. H., & Kramer, T. A. (2007). Arsenateand arsenite adsorption and desorption behaviour on copre-cipitated aluminium:iron hydroxides. Environmental Sci-ence & Technology, 41, 837–842.
Matschullat, J. (2000). Arsenic in the geosphere—a review. TheScience of the Total Environment, 249, 297–312.
Mello, J. W. V., Barrón, V., & Torrent, J. (1998). Phosphorusand iron mobilization in flooded soils from Brazil. SoilScience, 163, 122–132.
NHMRC, NRMMC (2004). Australian Drinking WaterGuidelines. In: National Water Quality ManagementStrategy Paper No 6. National Health and MedicalResearch Council and Natural Resource ManagementMinisterial Council. Canberra: Australian GovernmentPublishing Service.
O'Day, P. A. (2006). Chemistry and mineralogy of arsenic.Elements, 2(2), 77–83.
Radu, T., Subacz, J. L., Phillippi, J. M., & Barnett, M. O.(2005). Effects of dissolved carbonate on arsenic
adsorption and mobility. Environmental Science & Tech-nology, 39, 7875–7882.
Sahai, N., Lee, Y. J., Xu, H., Ciardelli, M., & Gaillard, J. F.(2007). Role of Fe(II) and phosphate in arsenic uptake bycoprecipitation. Geochimica et Cosmochimica Acta, 71(13), 3193–3210.
Schwertmann, U. (1984). The Influence of aluminum on iron-oxides. 9. Dissolution of Al-goethites in 6 M HCl. ClayMineralogy, 19, 9–19.
Schwertmann, U., & Cornell, R. M. (2000). Iron oxides in thelaboratory: preparation and characterization (2nd ed.).Weinheim: Wiley-VCH. Completely revised and extended.
Sherman, D. M., & Randall, S. R. (2003). Surface complexationof arsenic(V) to iron(III) (hydr)oxides: structural mecha-nism from ab initio molecular geometries and EXAFSspectroscopy. Geochimica et Cosmochimica Acta, 67(22),4223–4230.
Silva, J., Mello, J. W. V., Gasparon, M., Abrahão, W. A. P.,Ciminelli, V. S. T., & Jong, T. (2010). The role of As-goethites on arsenate mobility. Water Research, 44, 5684–5692.
Smedley, P. L., & Kinniburgh, D. G. (2002). A review of thesource, behaviour and distribution of arsenic in naturalwaters. Applied Geochemistry, 17, 517–568.
Thinnappan, V., Merrifield, C. M., Islam, F. S., Polya, D. A.,Wincott, P., & Wogelius, R. A. (2008). A combined exper-imental study of vivianite and As(V) reactivity in the pHrange 2–11. Applied Geochemistry, 23, 3187–3204.
Torrent, J., Schwertmann, U., & Barron, V. (1992). Fast andslow phosphate sorption by goethite-rich natural materials.Clays and Clay Mineralogy, 40, 14–21.
Wang, H. D., Harris, W. G., & Yuan, T. L. (1991). Relationbetween phosphorus and iron in Florida phosphatic soils.Soil Science Society of America Journal, 55, 554–560.
Zachara, J. M., Fredrickson, J. K., Smith, S. C., & Gassman, P.L. (2001). Solubilisation of Fe(III) oxide-bound trace met-als by a dissimilatory Fe(III) reducing bacterium. Geochi-mica et Cosmochimica Acta, 65(1), 75–93.
Zhang, H., & Selim, H. M. (2008). Competitive sorption–de-sorption kinetics of arsenate and phosphate in soils. SoilScience, 173, 3–12.
Zhang, J. S., Stanforth, R., & Pehkonen, S. O. (2008). Irrevers-ible adsorption of methyl arsenic, arsenate, and phosphateonto goethite in arsenic and phosphate binary systems.Journal of Colloid and Interface Science, 31, 35–43.
Water Air Soil Pollut (2012) 223:5707–5717 5717
Copyright © 2022 FDOKUMEN




















