
A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus

Effect of Prolactin on LymphocyteActivation from Systemic LupusErythematosus Patients
KARINA CHAVEZ-RUEDA,a VICTORIA MA. LEGORRETA-HAQUET,a
HERNANDO CERVERA-CASTILLO,b LOURDES SANCHEZ,c
LUIS JAVIER JARA,d EDGAR ZENTENO,e LUIS CHAVEZ-SANCHEZ,a
AND FRANCISCO BLANCO-FAVELAa
aImmunology Research Unit, Pediatric Hospital, Centro Medico Nacional “SigloXXI,” IMSS, Mexico, D.F.bRheumatology Department at the HGZ25, IMSS, Mexico, D.F.cRheumatology Department, Hospital de Especialidades Centro MedicoNacional “Siglo XXI,” IMSS, Mexico, D.F.dRheumatology Department, Hospital de Especialidades Centro MedicoNacional “la Raza,” IMSS, Mexico, D.F.eBiochemistry Department at the Medicine Faculty in the Autonomous NationalUniversity of Mexico, UNAM, Mexico, D.F.
ABSTRACT: The aim was to explore the role of prolactin (PRL) in thelymphocyte activation process in systemic lupus erythematosus (SLE) pa-tients in an in vitro model. Peripheral blood mononuclear cells (PBMCs)were isolated from SLE patients and healthy individuals. The mRNA forPRL and its receptor obtained by standard techniques, with an appro-priate primer, were subjected to polymerase chain reaction (PCR) andvisualized. The PBMCs were cultured with (a) medium alone as a nega-tive control, (b) unspecific mitogen as a positive control, (c) PRL alone, (d)mitogen plus PRL, (e) mitogen plus antibody anti-PRL, and (f ) mitogenplus a nonrelated antibody. Then CD69 and CD154 were determined byflow cytometry analysis. Twelve inactive and 15 active SLE patients werestudied. Twenty-five percent of the active patients displayed hyperpro-lactinemia. Under basal conditions CD69 expression was associated withdisease activity. The PBMCs activated in vitro were capable of producingand secreting PRL, measured by mRNA and Nb2 assay. In a similar way,the mRNA for the PRL receptor was visualized. Cells from SLE patientscultivated with PRL alone did not display increased CD69 and CD154expression. The addition of PRL to the unspecific stimulated culture doesnot have an additive effect. In contrast, the addition of antibodies against
Address for correspondence: Francisco Blanco-Favela, M.D., Ph.D., The Immunology ResearchUnit, Hospital de Pediatrıa, Centro Medico Nacional “Siglo XXI” Av. Cuahutemoc 330 Col. Doctores,PO Box: 73-032, CP: 03020 IMSS, Mexico, D.F. Voice/fax: (5) 56-27-69-43.
Ann. N.Y. Acad. Sci. 1108: 157–165 (2007). C© 2007 New York Academy of Sciences.doi: 10.1196/annals.1422.018
157

158 ANNALS OF THE NEW YORK ACADEMY OF SCIENCES
PRL in order to block the autocrine PRL resulted in a striking reductionof CD69 and CD154 expression.
KEYWORDS: prolactin; T cell activation; SLE
INTRODUCTION
Systemic lupus erythematosus (SLE) is an autoimmune rheumatic diseasecharacterized by widespread inflammation and most commonly affects women.Virtually every organ and/or system may be involved. The course of the dis-ease is characterized by remissions and exacerbation. The exacerbation ofthe disease has been linked to the activity of the immune system.1–4 The ac-tivation process of the immune system cells must be initiated by antigenicpeptides bound to class I or II MHC molecules by antigen-presenting cells(APC), which is recognized by the T cell receptor (TCR).5 Its signal deter-mines antigen specificity and plays a central role in initiating T cell activation.However, this interaction, by itself, is not sufficient to fully activate naıve Tcells. Thus, subsequent non-antigen-specific costimulatory signals are neces-sary to trigger cytokine gene expression. The best-known costimulatory signalfor T cells is provided by the interaction of CD28 on the T cell with membersof the B7 family (CD80 and CD86) on the APC. On the other hand, B cellsare also targets of costimulatory signals, mainly received through the CD40receptor after engagement by its ligand CD154 (CD40L) on activated T cells.A third type of signal with a crucial role in T and B cell activation is mediatedthrough the binding of soluble cytokines to their respective receptors.6,7 Theexpression of these molecules is a marker of activated cells8 together with theincreased expression of CD69 that is common its association in active SLEpatients.9–11
Another association described in active SLE patients is the high serum levelof prolactin (PRL).12 Although the clinical trials have yielded conflicting in-formation because some studies have found an association between serumlevels of PRL and disease activity12–15; however, others could not confirm thisrelationship.16–19 Inspite of this information other evidence supports the partic-ipation of PRL in the SLE disease activity through the immune system activitylike the hyperprolactinemia patients without autoimmune disease that showedan increased frequency of autoantibodies compared with healthy people20; orthe physiological hyperprolactinemia states such as pregnancy and lactancythat trigger lupus flares21; or the elevated frequencies of hyperprolactinemiain male SLE patients14,22; as well as the finding in the mouse model for SLEwhere implants of syngeneic pituitary glands, induce a hyperprolactinemicstate, resulting in accelerated autoimmunity and early mortality23,24 and thetreatment with bromocriptine improves the survival24; or the experiment wherePRL infusion induced a dramatic improvement in the survival of bromocriptine

CHAVEZ-RUEDA et al. 159
treatment mice after intraperitoneal injection of Listeria monocytogenes25 andthose studies in vitro where the aim was to show that the PRL has action as acomitogen for T, B, and NK cells from both humans26–28 and mice.29,30 More-over, recently, we describe that SLE patients with autoantibodies against PRLdisplay less activity when compared with those without these antibodies.31,32
Inspite of this information even with this very little knowledge about theparticipation of the PRL in the phenomenon of immune system activation, theaims of this research was to explore the participation of PRL in lymphocyteactivation, in cells from active or inactive SLE patients using an in vitro ap-proach and to determine if the PRL that participates in the immune responseis from pituitary or lymphoid origin.
METHODS
Peripheral blood mononuclear cells (PBMCs) were isolated from SLE pa-tients and healthy individuals. The mRNA for PRL and its receptor obtainedby standard techniques, with an appropriate primer, were subjected to poly-merase chain reaction (PCR) and visualized. The PBMCs were cultured with(a) medium alone as a negative control, (b) unspecific mitogen as a positivecontrol, (c) PRL alone, (d) mitogen plus PRL, (e) mitogen plus antibody anti-PRL, and (f ) mitogen plus a nonrelated antibody. Then CD69 and CD154 weredetermined by flow cytometry analysis.
RESULTS
The study population consisted of 27 SLE patients (mean ± SD age 36 ± 7years) and 12 healthy subjects with a mean age of 31 ± 10 years. The patientswere divided using SLEDAI into 12 inactive (42 ± 8 years) and 15 active (34 ±9 years) groups. The mean of serum levels of PRL in nonactive SLE patientswas 12 ± 1.9 ng/mL, similar to the mean in the healthy control group, 11.1 ±1.2 ng/mL. In contrast, the active SLE patients display a 16.8 ± 2.9 ng/mLlevel. Moreover, 25% of the active patients had hyperprolactinemia. The meanof SLEDAI in the group of active SLE patients was 14.5 compared to the zerofrom the nonactive patients.
We did not find statistically significant differences in the expressions ofCD154 among the active, inactive, and healthy groups. In contrast, CD69expressions were higher in CD4+ from patients with the disease activity (1 ±0.45%). It was statistically significantly different (P < 0.05) compared toCD4+ from inactive SLE patients (0.29 ± 0.09%) and healthy group (0.07 ±0.02%).
PBMCs from healthy people were activated in vitro using an unspecificmitogen. Then the supernatant was collected at time 0, 1, 2, and 3 h resulting

160 ANNALS OF THE NEW YORK ACADEMY OF SCIENCES
in concentrations of 0, 45.8, 26.2, and 63.4 pg/mL, respectively, measured bythe Nb2 assay. In contrast, we did not find Nb2 activation in cultures withoutstimulus and those in which anti-PRL antibody was added to the supernatant.In a similar way, the mRNA for PRL was obtained at 0, 1, 2, 3, 4, and 5 hafter the PBMCs were stimulated. Then RT-PCR and dot blotting were doneas can be seen in the FIGURE 1A. We found mRNA for PRL at time zero.In the measurement of time spent after cells activation an increased amountof mRNA was observed. In the same way, the mRNA for PRL receptor wasfound in activated PBMCs. The amount of mRNA for receptor appears withoutchanges throughout the time FIGURE 1B.
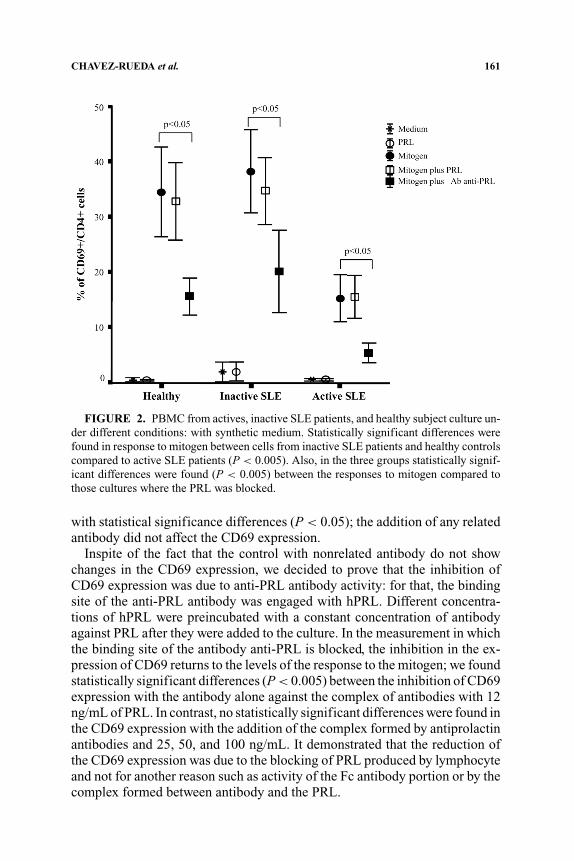
The cells culture conditions were a synthetic medium, nonsupplementedwith fetal bovine serum, in order not to have interference due to growth factors.The FIGURE 2 shows the behaviors of PBMCs from SLE patients in culture withhuman pituitary PRL (hPRL) or depleting the PRL with anti-PRL antibodies:The CD69 expression in the negative control was similar so that there wasCD69 expression in cells with only hPRL, meaning that hPRL alone is notcapable of activating cells from active or inactive SLE patients. The positivecontrol, PBMC, stimulated with unspecific mitogen shows high levels of CD69expression. In the healthy group, 30% of CD4+ display CD69 similar to the40% finding in the inactive SLE patients. Inexplicably, just 15% of cells fromactive patients display expression of CD69. In the experiments realized addinghPRL to the cells stimulated with unspecific mitogen, the expression of CD69does not change. In contrast, the addition of antibodies against PRL resulted ina striking reduction of CD69 expression in about 50% in patients and controls
FIGURE 1. (A). mRNA for PRL visualized by Dot Blotting; the expression of RNAwas found in cells without stimulus. The stimulus appears to increase the amount of mRNAfor PRL. (B). Displays the mRNA for PRL receptor. The amount of RNA did not increaseafter stimulus.
CHAVEZ-RUEDA et al. 161
FIGURE 2. PBMC from actives, inactive SLE patients, and healthy subject culture un-der different conditions: with synthetic medium. Statistically significant differences werefound in response to mitogen between cells from inactive SLE patients and healthy controlscompared to active SLE patients (P < 0.005). Also, in the three groups statistically signif-icant differences were found (P < 0.005) between the responses to mitogen compared tothose cultures where the PRL was blocked.
with statistical significance differences (P < 0.05); the addition of any relatedantibody did not affect the CD69 expression.
Inspite of the fact that the control with nonrelated antibody do not showchanges in the CD69 expression, we decided to prove that the inhibition ofCD69 expression was due to anti-PRL antibody activity: for that, the bindingsite of the anti-PRL antibody was engaged with hPRL. Different concentra-tions of hPRL were preincubated with a constant concentration of antibodyagainst PRL after they were added to the culture. In the measurement in whichthe binding site of the antibody anti-PRL is blocked, the inhibition in the ex-pression of CD69 returns to the levels of the response to the mitogen; we foundstatistically significant differences (P < 0.005) between the inhibition of CD69expression with the antibody alone against the complex of antibodies with 12ng/mL of PRL. In contrast, no statistically significant differences were found inthe CD69 expression with the addition of the complex formed by antiprolactinantibodies and 25, 50, and 100 ng/mL. It demonstrated that the reduction ofthe CD69 expression was due to the blocking of PRL produced by lymphocyteand not for another reason such as activity of the Fc antibody portion or by thecomplex formed between antibody and the PRL.

162 ANNALS OF THE NEW YORK ACADEMY OF SCIENCES
The hPRL alone was not capable of inducing CD154 expression in PBMCfrom SLE patients and healthy subjects. In contrast, phorbol myristate acetate(PMA) induced CD154 expression in about 50% of PBMC from SLE patientsand controls. The addition of hPRL to PMA-stimulated cells had no effect onCD154 expression. However, the supplement with anti-PRL antibodies resultedin a reduction of CD154 expression on PBMC in about 59%. The responses tounspecific stimulus display a similar pattern in both SLE patients and healthysubjects. However, the cells from SLE patients display less CD154 expressionthan the cells from healthy control. In fact, cells from active SLE patientsdisplay a weak response to unspecific mitogen, but display a higher reductionof the CD154 expression with anti-PRL antibodies compared to the inactivesand healthy control group. Similarly, in the three groups studied the inhibitoryactivity of anti-PRL antibody was ablated by absorption with PRL.
DISCUSSION
The information about the relationship between PRL and lupus activityin SLE patients has been inconsistent.12 The SLE patients in our researchshowed a similar pattern than did previous trials: 25% of active SLE patientsshowed hyperprolactinemia, which is similar with the ranges in the previousreports.12–14,31 The group of active SLE patients showed higher serum levels ofPRL compared to the nonactive patients and healthy group similar to previousresults from our laboratory.31 In fact, we did not find hyperprolactinemia inany inactive SLE patients supporting the idea that PRL is playing a role inthe disease activity. We found the association between CD69 expressions withdisease activity probably a reflection of immune system activity; it is similar tothe previous report.9,11The amount of active immune cells measured throughCD69 and CD154 expression in SLE patients is surprising.
Previous research suggests that the hyperprolactinemic state could activatethe immune system.23,24 In contrast, a low level of PRL has been associatedwith deficient immune response.33 This study was carried out using cells fromactive and inactive SLE patients in culture with or without PRL. The originalexpectation was that the hPRL could trigger the lymphocyte activation, espe-cially in inactive SLE patients or could well perpetuate immune cell activationin cells from active SLE patients. In contrast, we found that the hPRL by it-self is not capable of inducing activation in cells from either SLE patients orhealthy subjects. The PRL activity was found just after the trigger immuneresponse (unspecific mitogen) origin that the cells from the immune systemwere capable of producing and secreting PRL with autocrine and paracrineactivity similar to previous reports.28,29
Its results validate that the immune cells are capable to produce and secretePRL and this production is related to the amount of activation at the protein

CHAVEZ-RUEDA et al. 163
and mRNA levels. Moreover, the mRNA for PRL and its receptor were foundin cells without stimulus suggesting that it is constitutive.
The status of activation was measured through the expression of CD69 andCD154. Our results have shown that human PRL added to cells from SLEpatients (active or inactive) do not increase the expression of CD69 and CD154.Similarly, the lymphocytes from SLE patients and healthy subjects had a similarpattern of immune response. In contrast, the depletion of PRL produced bylymphoid with anti-PRL antibodies shows a decreased expression in the markedmolecules in about ∼50% patients, indicating that PRL has participation ina step between the trigger (antigen presentation) and costimulatory signal. Itcould be the explication why the SLE patients with autoantibodies againstPRL display less activity.31 This result is comparable to those experimentscarried out with tirocytos where PRL stimulus alone does not change theHLA-DR and CD40 expression.34 These findings are difficult to understandin the context of the literature because in knockout mouse for PRL not showsany evident abnormalities in the development of B and T cell and the innateresponse.35 However, adaptive immune response, where most of the effects ofPRL have been demonstrated including our results, was not studied. Moreover,we have found that the deletion of PRL with anti-PRL antibody also produces astriking reduction in the secretion of IL-2 and IFN-� by CD4+ supporting thisresult.36
Further studies are necessary in order to determine the steps and signalwhere the PRL trigger is in the action in T cell activation. This model displaysclearly that the hPRL do not trigger the immune response in resting humancells. However, after the initial stimulus the PRL appears to play a role in orderto continue the activation event, such as costimulatory molecule expressionand interleukin secretion necessary for complete response. In addition, thePRL, which takes effect in immune response, is produced by the lymphocytes,suggesting that the hyperprolactinemic states in lupus patients could be sec-ondary, in part by activity of the immune system. Moreover, the SLE patientswith autoantibodies against PRL that display a less clinical manifestation thanthose without antibodies, could be probably because its antibodies are blockingthe PRL in a manner similar to our in vitro model.31,32
ACKNOWLEDGMENT
This research was supported in part by Consejo Nacional de Ciencia y Tec-nologia (CONACYT): grant 34454-N. We would like to thank Dr. A.F. Par-low from the National Hormone & Pituitary Program of the Harbor-UCLAMedical Center for the donation of human PRL (hPRL) and polyclonal anti-body against PRL.

164 ANNALS OF THE NEW YORK ACADEMY OF SCIENCES
REFERENCES
1. TAN, E.M., A.S. COHEN & J.F. FRIES. 1982. The 1982 revised criteria for theclassification of systemic lupus erythematosus. Arthritis Rheum. 25: 1271–1277.
2. BONBARDIER, C., D. GLADMAN, M. UROWITZ, et al. 1992. The Committee on Prog-nosis Studies in SLE: derivation of the SLEDAI: a disease activity index forlupus patients. Arthritis Rheum. 35: 630–640.
3. BLANCO, F.J.K. & D.A. ISENBERG. 1991. Analysis of antibodies to RNA in patientswith systemic lupus erythematosus and other autoimmune rheumatic disease.Clin. Exp. Immunol. 86: 66–70.
4. BLANCO, F., J. KALSI, R. RAVIRAJAN, et al. 1992. IgG subclasses in systemic lupuserythematosus and other rheumatic disease. Lupus 1: 391–399.
5. KELSOE, G. 1996. The germinal center. A crucible for lymphocytes selection.Semin. Immunol. 8: 179–184.
6. DING, L., J.M. GREEN, C.B. THOMPSON & E.M. SHEVACH. 1995. B7/CD28 depen-dent and independent induction of CD40 ligand expression. J. Immunol. 155:5124–5132.
7. GREWAL, I.S. & R.A. FLAVELL. 1996. The role of CD40 ligand in co stimulationand T cell activation. Immunol. Rev. 153: 85–106.
8. TESTI, R., D. D’AMBROSIO, R. DE MARIA & A. SANTONI 1994. The CD69 receptor: amultipurpose cell-surface trigger for haematopoietic cells. Immunol. Today 15:479–483.
9. ATZENI, F., N. DEL PAPA, P. SARZI-PUTTINI, et al. 2004. CD69 expression on neu-trophils from patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 22: 331–334.
10. GONZALEZ-AMARO, R., D. PORTALES-PEREZ, L. BARANDA, et al. 1998. Role of IL-10in the abnormalities of early cell activation events of lymphocytes from patientswith systemic lupus erythematosus. J. Autoimmun. 11: 395–402.
11. CRISPIN, J.C., A. MARTINEZ, P. DE PABLO, et al. 1998. Participation of the CD69antigen in the T-cell activation process of patients with systemic lupus erythe-matosus. Scand. J. Immunol. 2: 196–200.
12. MOSZKORZOVA, L., Z. LACINOVA, J. MAREK, et al. 2002. Hyperprolactinaemia inpatients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 20: 807–812.
13. BLANCO-FAVELA, F., G. QUINTAL-ALVAREZ & A. LEANOS-MIRANDA. Associationbetween prolactin and disease activity in SLE. Influence of statistical power. J.Rheumatol. 26: 55–59.
14. LAVALLE, C., E. LOYO & R. PANIAGUA 1987. Correlation study between prolactinand androgens in male patients with systemic lupus erythematosus. J. Rheumatol.14: 268–272.
15. JARA, L.J., C. GOMEZ-SANCHEZ, L.H. SILVEIRA, et al. 1992. Hyperprolactinemiain systemic lupus erythematosus: association with disease activity. Am. J. Med.Sci. 303: 222–226.
16. BUSKILA, D., M. LORBER, L. NEUMANN, et al. 1996. No correlation between pro-lactin levels and clinical activity in patients with systemic lupus erythematosus.J. Rheumatol. 23: 629–632.
17. PAUZNER, R., M.B. UROWITZ, D.D. GLADMAN & J.M. GOUGH 1994. Prolactin insystemic lupus erythematosus. J. Rheumatol. 21: 2064–2067.
18. NEIDHART, M. 1996. Elevated serum prolactin or elevated prolactin/cortisol ratioare associated with autoimmune processes in systemic lupus erythematosus andother connective tissue diseases. J. Rheumatol. 23: 476–481.

CHAVEZ-RUEDA et al. 165
19. OSTENDORF, B., R. FISCHER, R. SANTEN, et al. 1996. Hyperprolactinemia in sys-temic lupus erythematosus? Scand. J. Rheumatol. 25: 97–102.
20. BUSKILA, D., M. BEREZIN, H. GUR, et al. 1995. Autoantibody profile in the sera ofwomen with hyperprolactinemia. J. Autoimmun. 8: 415–424.
21. RUIZ-IRASTORGA, G., F. LIMA, J. ALVES, et al. 1996. Increased rate of lupus flareduring pregnancy and the puerperium: A prospective study of 78 pregnancies.Br. J. Rheumatol. 35: 133–138.
22. FOLOMEEV, M., T. PROKAEVA & V. NASSONOVA. 1990. Prolactin levels in men withSLE and RA. J. Rheumatol. 17: 1569–1570.
23. MCMURRAY, R., D. KEISLER, K. KANUEKEL, et al. 1991. Prolactin influences au-toimmune disease activity in the female B/W mouse. J. Immunol. 147: 780–787.
24. MCMURRAY, R., D. KEISLER, S. IZUOI & S.E. WALKER. 1994 Hyperprolactinemia inmale NZB/NZW (B/W) F1 mice: accelerated autoimmune disease with normalcirculating testosterone. Clin. Immunol. Immunopathol. 71: 338–343.
25. BERTON, E.W., M.S. MELTZER & J.W. HOLADAY. 1988. Suppression of macrophageactivation and T lymphocyte function in hypoprolactinemic mice. Science 239:401–404.
26. CLEVENGER, C.V., D.O. FREIER & J.B. KLINE. 1998. Prolactin receptor signal trans-duction in cell of the immune system. J. Endocrinol. 157: 187–197.
27. RUSSEL, D.H., L. MATRISIAN, R. KIMBLER, et al. 1984. Prolactin receptor on humanlymphocytes and their modulation by cyclosporine. Biochem. Biophys. Res. Co.121: 899–906.
28. CLEVENGER, C.V., D.H. RUSSEL, P.M. APPASANY & M.B. PRYTOWKY. 1990. Regu-lation of IL-2 driven T lymphocyte proliferation by prolactin. Proc. Nat. Acad.Sci. USA. 87: 6460–6464.
29. HARTMANN, D.P., J.W. HOLODAY & E.W. BERTON. 1989. Inhibition of lymphocyteproliferation by antibodies to prolactin. FASEB 3: 2194–2202.
30. MATERA, L., A. CESANO, G. BELLONE & E. OBERHOLTZER. 1992. Modulatory ef-fect of prolactin on the resting and mitogen induced activity of T, B and NKlymphocyte. Brain Behav. Immun. 6: 409–417.
31. LEANOS-MIRANDA, A., D. PASCOE-LIRA, K. CHAVEZ-RUEDA & F. BLANCO-FAVELA.2001. Anti-prolactin antibodies in systemic lupus erythematosus patients; fre-quency and correlation with prolactinemia and disease activity. J. Rheumatol. 7:1546–1553.
32. BLANCO-FAVELA, F., M.A.G. QUINTAL, K. CHAVEZ-RUEDA, et al. 2001. Anti-prolactin autoantibodies in pediatric systemic lupus erithematosus patients. Lu-pus 11: 803–808.
33. BERCZI, I., E. NAGY, K. KOVACS & E. HORVATH. 1981. Regulation of humoralimmunity in rat by pituitary hormone. Acta Endocrinol. 98: 506–513.
34. LI, J., W. TENG & Z. SHAN. 2001. Effect of prolactin on HLA-DR and CD40expressions by human thyrocytes. Chin. Med. J. 114: 1151–1156.
35. HORSEMAN, N.D., W. ZHAO, E. MONTECINOS-RODRIGUEZ, et al. 1997. Defectivemammopoiesis, but normal haematopoiesis, in mice with a targeted disruptionof the prolactin gene. EMBO 16: 6926–6935.
36. CHAVEZ-RUEDA, K., J. HERNANDEZ, E. ZENTENO, et al. 2005. Identification ofprolactin as a novel immunomodulator on the expression of co-stimulatorymolecules and cytokine secretions on T and B human lymphocytes. Clin. Im-munol. 116: 182–191.
Copyright © 2022 FDOKUMEN




















