
Draft Jamaican Standard Chemical Test Methods For ...
29
DJS 302: 2017 ICS: 91.100.10 Draft Jamaican Standard Chemical Test Methods For Hydraulic Cements BUREAU OF STANDARDS JAMAICA COMMENT DEADLINE: 30 AUGUST 2017- 28 OCTOBER 2017
-
Upload
khangminh22 -
Category
Documents
-
view
6 -
download
0
Transcript of Draft Jamaican Standard Chemical Test Methods For ...

DJS 302: 2017 ICS: 91.100.10
Draft
Jamaican Standard
Chemical Test
Methods
For
Hydraulic Cements
BUREAU OF STANDARDS JAMAICA
COMMENT DEADLINE: 30 AUGUST 2017- 28 OCTOBER 2017

IMPORTANT NOTICE
Jamaican Standards are subject to periodical review. The next amendment will be sent without
charge if you cut along the dotted line and return the self-addressed label. If we do not receive this
label we have no record that you wish to be kept up-to-date. Our address is:
Bureau of Standards Jamaica
6 Winchester Road P.O. Box 113
Kingston 10
Jamaica W.I. -----------------------------------------------------------(cut along the line)-------------------------------------------------------------------------------------------------------------------
DJS 302: 20XX
NAME OR DESIGNATION.......................................................................…..............................
ADDRESS..................................................................................…...............................................
............................................................................................................................…........
.............................................................................................….......................................

JBS CERTIFICATION MARK PROGRAMME
The general policies of the JBS Certification Mark Programme are as follows:
(a) The JBS provides certification services for manufacturers participating in the programme
and licensed to use the gazetted JBS Certification Marks to indicate conformity with
Jamaican Standards.
(b) Where feasible, programmes will be developed to meet special requirements of the
submitter.
(c) JBS certification is provided in the interest of maintaining agreed-upon standard
requirements. Where applicable, certification may form the basis for acceptance by
inspection authorities responsible for enforcement of regulations.
(d) In performing its functions in accordance with its policies, JBS does not assume or
undertake to discharge any responsibility of the manufacturer or any other party.
Participants in the programme should note that in the event of failure to resolve an issue arising
from interpretation of requirements, there is a formal appeal procedure.
Further information concerning the details of JBS Certification Mark Programme may be
obtained from the Bureau of Standards Jamaica, 6 Winchester Road, Kingston 10, Jamaica, W. I.
CERTIFICATION MARKS
Product Certification Marks Plant Certification Mark
Certification of Agricultural Produce
(CAP) Mark

DJS 171: 2017
ICS 67.100.01
Draft
Jamaican
Standard Chemical
Test Methods
For
Hydraulic Cements
Bureau of Standards Jamaica
6 Winchester Road
P.O. Box 113
Kingston 10
JAMAICA, W. I.
Tel: (876) 926 -3140-5/ 618 – 1534 / 632- 4275
Fax: (876) 929 -4736
Website: www.bsj.org.jm
E-mail: [email protected]
Month 201X
DJS 302: 20xx
© 20xx Bureau of Standards Jamaica
All rights reserved. Unless otherwise specified, no part of this publication may be reproduced or utilised in any form or
by any means, electronic or mechanical, including photocopying and microfilm, without permission in writing from the
Bureau of Standards Jamaica.
ISBN XXXXXXX Declared by the Bureau of Standards to be a standard specification pursuant to Section 7 of the Standards Act 1969.
First published November 2008
Second published xxxxxxxxxx.
Jamaican Standards establish requirements in relation to commodities, processes and practices, but do not purport to include all the necessary provisions of a contract.
The attention of those using this standard method of test is called to the necessity of complying with any relevant legislation.
Amendments
No.
Date of
issue
Remarks
Entered by
and date

iii
CONTENTS
Page
Foreword iv
Committee representation iv
Acknowledgement iv
Related documents iv
Note to User iv
Method of test
1. Scope 1
2. Reagents 1 3. Apparatus 1
4. Preparation of cement sample 1
5. Determination of loss on ignition 1 6. Determination of insoluble residue 2
7. Determination of main constituents 3 7.1 Principle 3
7.2 Decomposition with sodium peroxide 4
7.3 Precipitation and determination of silica (reference method) 5 7.4 Precipitation and determination of silica (alternative method) 6
7.5 Decomposition with hydrochloric acid and ammonium chloride and precipitation of silica (alternative method) 7
7.6 Determination of pure silica 8
7.7 Decomposition of the evaporation residue 8 7.8 Determination of soluble silica 9
7.9 Total silica 9 7.10 Determination of iron (iii) oxide 10
7.11 Determination aluminium oxide 11
7.12 Determination of calcium oxide (reference method) 11 7.13 Determination of magnesium oxide (reference method) 12
7.14 Determination of calcium oxide (alternative method) 13 7.15 Determination of magnesium oxide (alternative method) 14
7.16 Determination of the alkali content (reference method) 15
7.17 Determination of chloride content 19
7.18 Determination of SO3 20
Table
1. Volumes of solutions for preparation of calibration solutions and their
sodium oxide and potassium oxide concentration 17
Figure
1. Schematic diagram for chemical analysis of main constituents 5

iv
Foreword
The purpose of this standard is to provide the procedures for the chemical analysis of cement.
Committee representation
The preparation of this standard for the Standards Council established under the Standards Act of 1969,
was carried out under the supervision of the Cement Technical Committee, which at the time
comprised of the following members:
Mr. V. Douse, Chairman University of the West Indies, Mona
Mrs. M. Blankson, Vice Chairperson University of Technology, Jamaica
Mr. D. Young Jamaica Institution of Engineers
Mr. D. Allen Jentech Consultants Limited Mr. D. McCoy Jamaica Premix Limited
Mr. G. Blackwood Alumina de Caribe
Mr. E. Salkey Hardware Merchants Association
Mr. M. LynCook Caribbean Cement Company Limited
Mr. J. Gordon, 1st
Technical Secretary University of Technology, Jamaica
Mr. G. Hutchinson, 2nd
Technical Secretary Jentech Consultants Limited
Mr. S. Malcolm National Consumers League
Ms. J. Wright Consumer Affairs Commission Mr. J. Scott-Brown, Facilitator Bureau of Standards Jamaica
Mrs. E. Edwards, Recording Secretary Bureau of Standards Jamaica
Mrs. T. Clarke Morgan Bureau of Standards Jamaica
Mr. A. Blake Bureau of Standards Jamaica
Acknowledgement
Acknowledgement is made to the International Or g a n i za t i o n for Standardization (ISO), t h e
American Society for Testing and Materials (ASTM) and the British Standard Institution (BSI) for
granting permission to use parts of their standards as reference, and to the Caribbean Cement
Company Limited for their technical input into the development of this standard.
Special recognition is given to Mr. David Allen of Jentech Consultants Limited for his tremendous
contribution to the research involved in the development of this standard.
Related documents
This Standard makes reference to the following:
ISO 680 Cements – Test Methods – Chemical Analysis
BS EN 196 – 21 Determination of the Chloride, Carbon Dioxide and Alkali Content of Cement
ASTM C 114 Standard Test Methods for Chemical Analysis of Hydraulic Cement
Note to user
All bracketed clause references of the type”[ ]” relate to this document

DJS 302: 2008
1
Jamaican Standard chemical test methods for hydraulic cements
1.0 Scope
This Jamaican Standard describes the procedures for the chemical analysis of cement.
This standard describes the reference procedures and, in certain cases, an alternative method which
can be considered as giving equivalent results. In the case of dispute, only the reference procedures
are used.
2.0 Reagents
The general requirements for reagents used in this Standard shall be as described in ISO 680. The
degree of dilution is always given as a volumetric micrometre (µ m), for example: hydrochloric acid
1 + 2 means that volume 1 of concentrated hydrochloric acid is to be mixed with 2 volumes of
water.
3.0 Apparatus
Apparatus used shall be as described in ISO 680 and BS EN 196-21.
4.0 Preparation of cement sample
Before chemical analysis, the laboratory sample taken in accordance with EN 196-7 is treated to
give a sample for testing.
Take approximately 100 g of the sample by means of a sample divider or by quartering. Sieve this
portion on a 150 µ m or 125 µ m sieve until no more passes through. Remove with a magnet all the
metallic iron from the material retained on the sieve. Then grind the iron-free fraction of the
retained material so that it completely passes the 150 µ m or 125 µ m sieve. Transfer the sample to
a clean dry flask with an airtight closure and shake vigorously to mix it thoroughly.
The preceding operations are to be carried out as quickly as possible so that the sample is exposed to
ambient air for the minimum time.
Note. The sample is presented for analysis as marketed. If it contains particles of metallic iron, such as those that may be
introduced accidentally during grinding, the complete separation of these iron particles is carried out by means of a
magnetic stirrer in a suspension, for example in cyclohexane.
5.0 Determination of loss on ignition
5.1 Principle
The loss on ignition is determined in an oxidizing atmosphere (air). By igniting the air at (975 ±25) °C the carbon dioxide and water are driven off and any oxidisable elements present are oxidized. A
correction for the influence of this oxidation on the loss on ignition is described.
5.2 Procedure
Weigh (1± 0.05) g of cement (m7) into a crucible, which has been previously ignited and tared. Place
the covered crucible in the electric furnace controlled at (975± 25) °C. After heating for 5 minutes
slide the lid of the crucible slightly to one side without removing it to allow for the escape of gases and moisture and to prevent the loss of particles adhering to the lid and leave the crucible in the
furnace for a further 10 minutes. Cool the crucible to room temperature in the desiccator. Weigh
(m8). Determine constant mass in accordance with (3.5).
Note 2. For cement containing sulfides, a more precise determination of the loss on ignition can be obtained by determining the sulfate contents before and after ignition. The corrections applicable to these cements are given in 5.4.

DJS 302: 2008
2
5.3 Expression of results
The observed loss on ignition, in percentage, is calculated from the formula:
Observed loss on ignition = (m7 - m8) x 100 ……………1 (formula 9, ISO 680)
m7
Where
m7 is the mass of the test portion;
m8 is the mass of the ignited test portion.
5.4 Errors and corrections
Errors are caused either by the presence of carbon or by oxidation of sulfides of any remaining
metallic iron, of bivalent iron and of bivalent manganese. The error resulting from the presence of
these different elements may be corrected for, but only the correction due to oxidation of sulfides is
to be applied, the others usually being considered to be negligible.
In this case, the correction due to sulfide is 1.996 x (percentage of S
2-), and the corrected loss on
ignition = observed loss on ignition + 1.996 x (percentage of S2-)
For determination of sulfate present before and after ignition the relation is final percentage of SO3
initial percentage of SO3 = SO3 resulting from the oxidation of sulfides i.e. a correction of:
0.8 x (percentage of SO3 from oxidation of sulfite) = percentage of oxygen taken up, and corrected loss on ignition (%) = observed loss on ignition (%) + percentage of oxygen taken up.
Any corrections applied shall be indicated in the test report.
In the case of dispute, only the correction due to oxidation of sulfide shall be applied.
5.5 Repeatability and reproducibility
The standard deviation for repeatability is 0.04 %.
The standard deviation for reproducibility is 0.08 %.
6. Determination of insoluble residue - reference test method
6.1 Summary of test method:
In this test method, insoluble residue of cement is determined by digestion of the sample in
hydrochloric acid followed, after filtration, by further digestion in sodium hydroxide. The resulting
residue is ignited and weighed.
When this test method is used on blended cement, the decomposition in acid is considered to be
complete when the Portland-cement clinker is decomposed completely. An ammonium nitrate
solution is used in the final washing to prevent finely ground insoluble material from passing
through the filter paper.
6.2 Reagents:
(a) Ammonium Nitrate Solution (20 g NH4NO3/L).
(b) Sodium Hydroxide Solution (10 g NaOH/L).

DJS 302: 2008
3
6.3 Procedure: (a) To 1 g of the sample add 25 mL of cold water. Disperse the cement in the water and while
swirling the mixture, quickly add 5 mL of HCl. If necessary, warm the solution gently, and
grind the material with the flattened end of a glass rod for a few minutes until it is evident that
decomposition of the cement is complete.
(b) Dilute the solution to 50 mL with hot water (nearly boiling) and heat the covered mixture
rapidly to near boiling by means of a high-temperature hot plate. Then digest the covered
mixture for 15 minutes at a temperature just below boiling.
(c) Filter the solution through a medium-textured paper into a 400-mL beaker, wash the beaker,
paper, and residue thoroughly with hot water, and reserve the filtrate for the sulfur trioxide
determination, if desired.
(d) Transfer the filter paper and contents to the original beaker, add 100 mL of hot (near boiling)
NaOH solution (10 g/L), and digest at a temperature just below boiling for 15 minutes. During
the digestion, occasionally stir the mixture and macerate the filter paper.
(e) Acidify the solution with HCl using methyl red as the indicator and add an excess of 4 or 5
drops of HCl. Filter through medium-textured paper and wash the residue at least 14 times with hot NH4 NO3 solution (20 g/L) making certain to wash the entire filter paper and contents during
each washing.
(f) Ignite the residue in a weighed platinum crucible at 900 to 1000°C, cool in a desiccator, and
weigh.
6.4 Calculation— Calculate the percentage of the insoluble residue to the nearest 0.01 by
multiplying the weight in grams of the residue (corrected for the blank) by 100.
7. Determination of main constituents
7.1 Principle
The analysis is carried out after the sample is completely dissolved.
The cement is decomposed by sintering with sodium peroxide or by treatment with hydrochloric
acid in the presence of ammonium chloride. In the first case, after dissolution of the sintered solid
in hydrochloric acid, the major part of the silica is precipitated either by hydrochloric acid with
coagulation by polyethylene oxide or by double evaporation. In the second case, the major part of
the silica is separated by the treatment. The impure silica precipitated is treated with hydrochloric
acid and sulphuric acid to volatilize silica; the residue, treated with a mixture of sodium carbonate
and sodium chloride, is dissolved in hydrochloric acid and added to the silica filtrate.
In the case of the treatment with hydrochloric acid in the presence of ammonium chloride, if the
residue obtained after volatilization of impure silica by means of hydrofluoric acid and sulfuric acid
is greater than 0.5%, the method is not applicable.
The cement shall in this case be decomposed by sodium peroxide.
In the final solution made up to 500 mL the silica in solution is determined by photometric
determination. Iron (III) oxide, aluminium oxide, calcium oxide and magnesium oxide are
determined by complexometric methods.
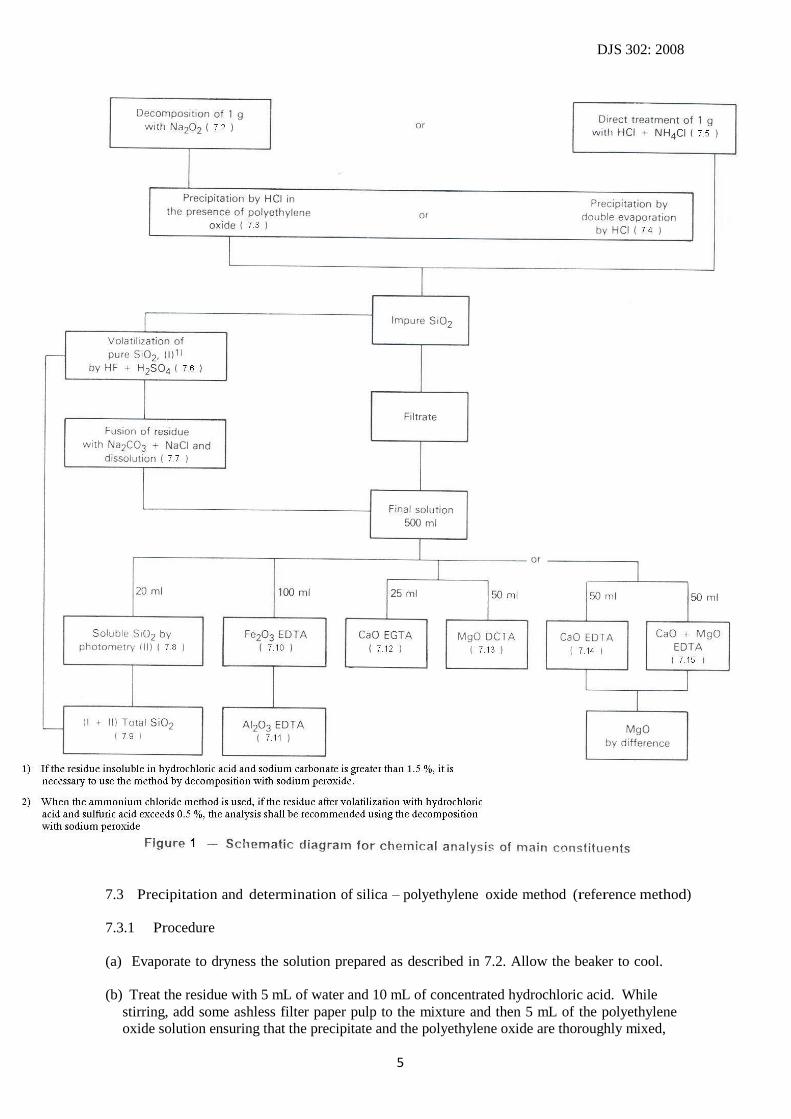
The schematic diagram of the chemical analysis is shown in Figure 1.

DJS 302: 2008
4
The scheme given in Figure 1 results, as far as the determination of silica is concerned; in the same
values for total silica whichever path is chosen.
7.2 Decomposition with sodium peroxide
(a) Weight (1 ± 0.05) g of cement (m17) and 1 g of sodium peroxide into a platinum crucible; mix
thoroughly with a spatula. Add to the mixture any particles adhering to the spatula. Cover the
mixture with 1 g of sodium peroxide. Carefully preheat the covered crucible for about 2 minutes at the opening of the furnace before placing it on its support in the heated zone
controlled at a uniform temperature of (500 ± 10)ºC.
(b) After 30 minutes, remove the crucible from the furnace and allow it to cool to room
temperature. The sintered mass should not stick to the sides of the crucible. If it does, then
repeat the decomposition at a temperature 10 º C lower than was first used.
(c) Transfer the sintered mass to a 400 mL beaker and rinse the crucible with 150 mL cold water.
(d) Cover the beaker with a watch glass and heat until the solid is completely dissolved. Then add
cautiously 50 mL of concentrated hydrochloric acid. The solution obtained shall be perfectly
clear. If not, reject it and repeat the decomposition by peroxide at a temperature increased by
10º C or for double the time in the furnace. Add 1 mL of sulfuric acid 1 + 1 to the solution.
Bring the solution to a boil and boil for 30 minutes.
(e) This solution is ready for use for the precipitation of silica in accordance with 7.3 or 7.4.
(See diagram – Figure 1 – Schematic diagram for chemical analysis of the main constituents)
DJS 302: 2008
5
7.3 Precipitation and determination of silica – polyethylene oxide method (reference method)
7.3.1 Procedure
(a) Evaporate to dryness the solution prepared as described in 7.2. Allow the beaker to cool.
(b) Treat the residue with 5 mL of water and 10 mL of concentrated hydrochloric acid. While
stirring, add some ashless filter paper pulp to the mixture and then 5 mL of the polyethylene
oxide solution ensuring that the precipitate and the polyethylene oxide are thoroughly mixed,

DJS 302: 2008
6
especially the precipitate adhering to the sides of the beaker. Stir the mixture thoroughly then
add 10 mL of water, stirring briefly and leave to stand for 5 minute. Then filter through a
medium filter paper into a 500 mL volumetric flask and rinse with hot hydrochloric acid 1 + 19.
Remove any precipitate adhering to the sides of the beaker using a rubber scraper. Wash the
filter and precipitate at least five times with hot hydrochloric acid 1 + 19, then rinse with hot
water, ensuring that the residue in the filter is broken up thoroughly during washing, until free
from C1- ions, tested by the silver nitrate test.
(c) Collect the washings in the same 500 mL volumetric flask.
(d) Ignite the filter and the precipitate in a platinum crucible at (1175 ± 25)ºC. Check for constant
mass. In general, an ignition period of 60 minutes is sufficient for obtaining constant mass (m18). Volatilize the decomposed residue as in 7.6. Add the decomposed residue [7.7] to the
filtrate and washings in the 500 mL volumetric flask. The combined solution is used for the calorimetric determination of silica remaining in solution [7.8] and also for the complexometric
determinations of iron (III) oxide [7.10], aluminium oxide [7.11], calcium oxide [7.12 or 7.14] and magnesium oxide, [7.13 or 7.15].
7.3.2 Expression of results
The impure silica content is calculated in percentage from the formula:
Impure SiO2 = m18 x 100 ………….. 3 (Formula 17, ISO 680)
m17
where
m17 is the mass of the test portion as in 7.2, in grams.
M18 is the mass determined in accordance with 7.3.1, in grams.
7.4 Precipitation and determination of silica double evaporation method (alternative method)
7.4.1 Procedure
(a) Evaporate to dryness the solution prepared as described in 7.2 on evaporation apparatus
controlled at (105 ± 3)ºC. Moisten with several drops of concentrated hydrochloric acid. Leave
for 1 h at this temperature.
(b) After cooling to room temperature, treat the residue with 10 mL of concentrated hydrochloric
acid. After a few minutes dilute with 50 mL of water, bring to the boil and filter the hot solution
through a medium filter paper into a 500 mL volumetric flask.
(c) Wash the filter and the residue three times with hot water. Evaporate the filtrate and washings in
the same way, treat with 10 mL of concentrated hydrochloric acid and dilute with 50 mL of
water. Boil, and then pass through the same filter into a 500 mL volumetric flask.
(d) Wash the filter and the residue with hot water until free from Cl- ions, tested by the silver nitrate
test. Collect the washings in the same 500 mL volumetric flask.
(e) Ignite the filter and precipitate in a platinum crucible at (1175 ± 25) º C. Check for constant mass
(m19). In general, an ignition period of 60 minutes is sufficient for obtaining constant mass.
Volatilize the decomposed residue as described in 7.6. Add the decomposed residue [7.7] to the filtrate and washings in the 500 mL volumetric flask.

DJS 302: 2008
7
(f) The combined solutions are used for the calorimetric determination of silica remaining in
solution [7.8] and for the complexometric determinations of iron (III) oxide [7.10], aluminium
oxide [7.11], calcium oxide [7.12 or 7.14] and magnesium oxide [7.13 or 7.15].
7.4.2 Expression of results
The impure silica content is calculated in percentage from the formula:
Impure SiO2 = m19 x 100 ……. 4 (Formula 18, ISO 680)
m17
where
m17 is the mass of the test portion as in 7.2, in grams;
m19 is the mass determined in accordance with 7.4.1, in grams.
7.5 Decomposition with hydrochloric acid and ammonium chloride and precipitation of
Silica (alternative method) 7.5.1 Procedure
(a) Weigh (1 ± 0.05) g of cement (m20) and place in a 100 mL beaker. Add about 1 g of ammonium
chloride and mix thoroughly with a glass-stirring rod. Cover the beaker with a watch glass and cautiously add 10 mL of concentrated hydrochloric acid taking care to let the acid run down the side of the beaker. When effervescence has stopped, add 10 drops of nitric acid and stir with a glass-stirring rod.
(b) Place the beaker and its watch glass on a boiling water bath and leave for 30 minutes. Filter
through a course filter paper into a 500 mL volumetric flask: transfer the gelatinous precipitate
to the filter as completely as possible without dilution, and allow the solution to drain through
the filter. Remove all precipitate adhering to the beaker by using a rubber scraper.
(c) Rinse the beaker and precipitate with hot hydrochloric acid 1 + 99. Then wash the precipitate
and filter 12 times with small amounts of hot water until free from Cl- ions, tested by the silver nitrate test. Collect the washings in the same 500 mL volumetric flask. This is used, together
with the decomposed residue, treated as described in 7.7. for the photometric determination of silica in solution in accordance with 7.8. Ignite the filter and the precipitate in a platinum
crucible at (1175 ± 25) º C. Check for constant mass (m21). In general, an ignition period of 60
minutes is sufficient for obtaining constant mass. Volatize the decomposed residues as
described in 7.6.
7.5.2 Expression of results
The impure silica content is calculated in percentage from the formula:
Impure SiO2 = m21 x 100 …………. 5 (Formula 19, ISO 680)
m20
where
m20 is the mass of the test portion as in 7.5.1, in grams;
m21 is the mass determined in accordance with 7.5.1, in grams.

DJS 302: 2008
8
7.6. Determination of pure silica 7.6.1 Procedure
(a) Moisten the residue obtained in accordance with 7.3.1 (m18 ) or 7.4.1 (m19) or 7.5.1 (m21) with
about 0.5 mL to 1 mL of water, add approximately 10 mL of hydrofluoric acid then two drops of sulfuric acid. Evaporate in a fume cupboard over a sand bath or hot plate then continue to heat until free from white sulfuric acid fumes.
(b) Ignite the crucible with the evaporation residue in an electric furnace (1175 ± 25) º C for 10
minutes, leave to cool to room temperature in a desiccator and weigh (m22).
(c) The evaporation residue is decomposed as described in 7.7. If the residue obtained by this
method exceeds 0.5%, the analysis shall be restarted and decomposition with sodium peroxide
used [7.2].
7.6.2 Expression of results
The pure silica content is calculated in percentage from the formula:
Pure SiO2 = m24 – m22 x 100 ………….. 6 (Formula 20, ISO 680)
m23
where
m22 is the mass determined in accordance with 7.6.1, in grams;
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams;
m24 is the mass determined in accordance with 7.3.1 (m18), 7.4.1 (m19) or 7.5.1
(m21), in grams. 7.7 Decomposition of the evaporation residue
To the evaporation residue, obtained in accordance with 7.6.1, add 2 g of the sodium carbonate and
sodium chloride mixture and fuse to a bright red heat using a gas burner. Swirl the melt frequently
until the residue is completely dissolved.
Check visually that no part of the residue remains at the base of the crucible. Allow the crucible and
its contents to cool, transfer to a 250 mL beaker, add about 100 mL water and acidify with a few
millimetres of concentrated hydrochloric acid.
When the decomposed mass is completely dissolved, remove the platinum crucible from the
solution and rinse it with water.
The solution shall be perfectly clear. If not, filter through a medium filter paper, wash, burn off the
paper, ignite and then repeat the decomposition as above. Transfer the solution to the 500 mL
volumetric flask containing the filtrate and washings from the precipitation of silica in accordance
with 7.3.1 or 7.4.1 or 7.5.1. Fill the flask up to the mark with water. After stirring, this solution is
ready for use. It is used in the photometric determination of the silica remaining in solution and also
in the complexometric determinations of iron (III) oxide, aluminium oxide, calcium oxide and
magnesium oxide.

9
DJS 302: 2008
7.8 Determination of soluble silica
7.8.1 Procedure
(a) Pipette 20 mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric
flask into a polyethylene beaker already containing a magnetic stirrer bar and add 20 mL
water. While stirring with the magnetic stirrer, add 15 drops of hydrofluoric acid 1 + 3. Stir
again for at least 1 minutes. Then pipette 15 mL of the boric acid solution.
(b) Adjust the pH of the solution to 1.15 ± 0.05 by adding, drop by drop, sodium hydroxide or
hydrochloric acid 1 + 2, using a pH meter calibrated with a buffer solution of similar pH
value (e.g. 1.40). Add from a pipette 5 mL of the ammonium molybdate solution to the
solution (time 0).
(c) Adjust the pH of the solution to 1.60 by adding, drop by drop, the sodium hydroxide solution
or hydrochloric acid 1 + 2. Transfer the solution to a 100 mL volumetric flask and rinse the
beaker with hydrochloric acid of pH 1.60. After 20 minutes add from a pipette 5 mL of the
citric acid solution, stir and leave to stand for 5 minutes, then add from a pipette 2 mL of the
reducing solution.
(d) Make up to volume with dilute hydrochloric acid of pH 1.60 and mix. At time 0 + 30
minutes measure the optical density with the photometer against a blank solution prepared
in a similar way and using the same wavelength of a cell of the same optical length as used
for the construction of the calibration graph. The silica concentration in mg SiO2 /100 mL is
read from the calibration graph.
7.8.2. Expression of results
The soluble silica content is calculated in percentage from the formula:
Soluble SiO2 = 500 x m25 x 100 ……………. 7 (Formula 21, ISO 680)
20 x 1000 x m23
Where
= 2.5 x m25
m23
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams;
m25 is the silica content of the solution in accordance with 7.8.1 in mg SiO2/100 mL.
7.9 Total silica
7.9.1 Expression of results
The total silica content is equal to the sum of the pure silica content and the soluble silica content.
7.9.2 Repeatability and reproducibility
The standard deviation for repeatability is 0.10 %.
The standard deviation for reproducibility is 0.25 %.

DJS 302: 2008
7.10 Determination of iron (III) oxide
7.10.1 Procedure
10
(a) Pipette 100 mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric
flask into a beaker compatible with the measuring apparatus. Then make up with water to a
volume suitable for the correct operation of the equipment.
(b) Add 0.5 g amino-acetic acid and 0.3 g to 0.4 g of sulfosalicylic acid indicator.
(c) Using a pH meter, adjust the pH of this solution to 1.5 ± 0.1 with the ammonium hydroxide 1 +
1 and 1 + 10.
(d) Heat to (47.5 ± 2.5)ºC. Place the beaker in the apparatus set a 520 nm and, while stirring the
solution, titrate with 0.03 mol/l Dehydrated disodium salt of ethylenediamine tetra-acetic acid
(EDTA – Na2; short EDTA) solution. In the vicinity of the indicator colour change, construct a
diagram of the optical density values as a function the volume of EDTA added. The volume V10
used is determined from the intersection of the line of greatest slope in the region of the colour change and the line of almost constant optical density after the colour change.
(e) During the titration, the temperature of the solution shall not exceed 50ºC. Otherwise the
determination shall be repeated.
This titrated solution is retained for the determination of aluminium oxide content in accordance with
7.11.1. The presence of titanium oxide (TiO2) can disturb the determination of iron oxide (Fe2O3) in
the presence of peroxide and the latter shall be completely decomposed.
NOTE. The presence of titanium affects the speed of the titration of iron by EDTA. This cause of error can be overcome
by proceeding slowly, for example with the help of an automatic burette. It is equally possible to mask the titanium by
adding 2 mL of sulfuric acid 1 + 1 to the solution before titration.
7.10.2 Expression of results
The iron (III) oxide content is calculated in percentage from the formula:
Fe2O3 = 0.03 x 159.692 x 500 x V10 x fD x 100
2 x 1000 x 100 x m23
= 1.1977 x V10 x fD ………… 8 (Formula 22, ISO 680)
m23
Where
V10 is the volume of 0.03 mol/l EDTA solution used for the titration, in millilitres;
fD is the factor of the 0.03 mol/l EDTA solution defined in 4.53.2;
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams.
7.10.3 Repeatability and reproducibility
The standard deviation for repeatability is 0.08 %.
The standard deviation for reproducibility is 0.15 %.

11
DJS 302: 2008
7.11 Determination of aluminium oxide
7.11.1 Procedure
(a) Cool the solution retained from 7.10.1 to room temperature. Then add 5 mL of acetic acid,
drop by drop to the ammonium acetate solution so as to obtain a pH of 3.05 ± 0.05. This
zone shall be strictly adhered to and controlled by means of a pH meter. Under no
circumstances is a pH of 3.1 to be exceeded.
(b) Bring to a boil; add three drops of copper complexonate solution and 10 drops of PAN
indicator (see ISO 680). During the titration, the solution shall be kept gently boiling (work
in a fume cupboard).
(c) Titrate with the 0.03 mol/l EDTA solution until the colour changes from violet-pink to pale
yellow. When the pink colour reappears, add the 0.03 mol/l EDTA drop by drop until the
yellow colour persists for at least 1 minute.
7.11.2 Expression of results
The aluminium oxide content is calculated in percentage from the formula:
A1203 = 0.03 x 101.961 x 500 x V11 x fD x 100
2 x 1000 x 100 x m23
= 0.7647 x V11 x fD …………… 9 (Formula 23, ISO 680)
m23
Where V11 is the volume of 0.03 mol/l EDTA solution used for the titration, in millilitres;
fD is the factor of the 0.03 mol/l EDTA solution defined in ISO 680;
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams.
7.11.3 Repeatability and reproducibility
The standard deviation for repeatability is 0.10 %.
The standard deviation for reproducibility is 0.25 %.
7.12 Determination of calcium oxide by ethylenebis (oxyethelenenitrilo) tetra acetic acid
(EGTA) (reference, method) 7.12.1 Procedure
(a) Pipette 25 mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric
flask into a beaker compatible with the measuring apparatus and make up to the same
volume as in with water and then add 25 mL of the triethanolamine solution 1 + 4 (4.45).
(b) Adjust the pH of this solution to 12.5 (using a pH meter) with a sodium hydroxide solution.
Add about 0.1 g of murexide or calcein indicator. Place the beaker in the apparatus set at
520 nm when using calcein or at 620 nm when using murexide and, while stirring, titrate
with the 0.03 mol/l EGTA solution. In the vicinity of the colour change, construct a diagram
of the readings from the measuring apparatus as a function of the volume of EGTA added.
The volume V12 used is determined from the intersection of the line of greatest slope in the
region of the colour change and the line of almost constant optical density after the colour
change.

12
DJS 302: 2008
7.12.2 Expression of results
The calcium oxide content is calculated in percentage from the formula:
CaO = 0.03 x 56.08 x 500 x V12 x fG x 100
1000 x 25 x m23
= 3.3648 x V12 x fG
m23
Where V12 is the volume of 0.03 mol/l EGTA solution used for titration, in millilitres;
fG is the factor of the 0.03 mol/l EGTA solution defined in ISO 680;
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20) in grams.
NOTE. Strontium oxide is determined and expressed as calcium oxide.
7.12.3 Repeatability and reproducibility
The standard deviation for repeatability is 0.18 %.
The standard deviation for reproducibility is 0.37 %.
7.13 Determination of magnesium oxide by DCTA (reference method)
7.13.1 Procedure
(a) Pipette 50 mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric flask
into a beaker compatible with the measuring apparatus, add 50 mL of triethanolamine solution 1 + 4 and a volume V13 required is calculated in millilitres from the formula:
V13 = 2 V12 + 1.5 ……………… 11 (Formula 25, ISO 680)
Where
V12 is the volume of the EGTA solution used for titration in accordance with
7.12.1, in millilitres;
V13 is the volume of the EGTA solution, in millilitres.
(b) After addition of the calculated volume of EGTA solution, dilute with water to a volume
suitable for the correct operation of the apparatus. Using a pH meter, adjust the pH of this
solution to 10.5 with concentrated ammonium hydroxide.
(c) Add about 0.1 g of methylthymol blue indicator.
(d) Place the beaker in the apparatus set at 620 nm and, while stirring the solution, titrate with the
0.01 mol/l DCTA solution. In the vicinity of the colour change of the indicator, plot a graph of the optical density values as a function of the volume of DCTA added. The volume V14 used is
determined from the intersection of the line of greatest slope in the region of the colour change and the line of almost constant optical density after the colour change.

13
DJS 302: 2008
7.13.2 Expression of results
The magnesium oxide content is calculated in percentage from the formula:
MgO = 0.01 x 40.311 x 500 x V14 x fc x100
1000 x 50 x m23
= 0.4031 x V14 x fc …………… 12 (Formula 26, ISO 680)
m23
Where
V14 is the volume of 0.01 mol/l DCTA solution used for the titration, in millilitres;
fc is the factor of the 0.01 mol/l solution as defined in ISO 680;
m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams.
7.13.3 Repeatability and reproducibility
The standard deviation for repeatability is 0.15 %.
The standard deviation for reproducibility is 0.15 %.
7.14 Determination of calcium oxide by EDTA (alternative method)
7.14.1 Restriction on the method
This method shall be preceded by the determination of manganese content [7.15.1].
7.14.2 Procedure
(a) Pipette 50mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric flask
into a beaker compatible with the measuring apparatus. Then dilute with water to a volume
suitable for the correct operation of the equipment. Add 50 mL of the triethanolamine solution
1 + 4.
(b) Using a pH meter, adjust the pH of this solution to 12.5 with the sodium hydroxide solution.
(c) Add about 0.1 g of murexide or calcein indicator. Place the beaker in the apparatus set at 620
mm when using murexide or at 520 nm when using calcein and, while stirring the solution,
titrate with the 0.03 mol/l EDTA solution. In the vicinity of the colour change of the indicator, plot a graph of the optical density values as a function of the volume EDTA added. The volume
V15 used is determined from the intersection of the line of greatest slope in the region of the
colour change and the line of almost constant optical density after the colour change.
7.14.3 Expression of results
The calcium oxide content is calculated in percentage from the formula:
CaO = 0.03 x 56.08 x 500 x V15 x fD x 100
1000 x 50 x m23
=1.6824 x V15 x fD ……………. 13 (Formula 27, ISO 680)
m23

14
DJS 302: 2008
Where
V15 is the volume of 0.03 mol/l EDTA solution used for the titration, in millilitres;
fD is the factor of the 0.03 mol/l EDTA solution defined in ISO 680; m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams.
NOTE: Strontium oxide is determined and expressed as calcium oxide.
7.14.4 Repeatability and reproducibility
The standard deviation for repeatability is 0.15 %.
The standard deviation for reproducibility is 0.43 %.
7.15 Determination of magnesium oxide by EDTA (alternative method)
7.15.1 Restriction on the method
For the rare particular instances where cements have a manganese oxide (Mn2O3) content greater
than 0.5 %, the method of determination of magnesium oxide by DCTA is applicable. Indeed in this case, the alternative method gives high results, or it requires a preliminary separation of hydroxides by precipitation.
7.15.2 Procedure
(a) Pipette 50 mL of the solution prepared in accordance with 7.7 from the 500 mL volumetric flask
into a beaker compatible with the measuring apparatus. Then dilute with water to a volume
suitable for the correct operation of the equipment. Add 50 mL of the triethanolamine solution
1 + 4.
(b) Using a pH meter, adjust the pH of this solution to 10.5 with dilute ammonium hydroxide 1 + 1.
(c) Using a burette, add the volume V15 of EDTA required for the titration of calcium oxide
previously determined in 7.14.2. (d) Then add about 0.1 g of methylthymol blue indicator.
(e) Place the beaker in the apparatus set at 620 nm and, while stirring the solution, titrate with the
0.03 mol/l EDTA solution. In the vicinity of the colour change of the indicator, plot a graph of the optical density values as a function of the volume of EDTA added. The volume V16 used is
determined from the intersection of the line of greatest slope in the region of the colour change and the line of almost constant optical density after the colour change.
7.15.3 Expression of results
The magnesium oxide content is calculated in percentage from the formula:
MgO = 0.03 x 40.311 x 500 x (V16 – V15) fD x 100
1000 x 50 x m23
=1.2093 x (V16V15) ………… 14 (Formula 28, ISO 680)
m23
Where
V15 is the volume of EDTA required for the determination of calcium oxide determined in
7.14.2, in millilitres;
V16 is the volume of EDTA required for the determination of calcium oxide and magnesium

15
oxide defined in 7.15.2, in millilitres;
fD is the factor of the 0.03 mol/lEDTA solution defined in ISO 680; m23 is the mass of the test portion as in 7.2 (m17) or 7.5.1 (m20), in grams.
7.15.4 Repeatability and reproducibility
The standard deviation for repeatability is 0.21 %.
The standard deviation for reproducibility is 0.25 %.
7.16 Determination of alkali content (reference method)
7.16.1 Principle
A butane or propane flame is used to excite the alkalis to emit their characteristic spectrum in the
visible range. The emission is proportional to the alkali content at low concentrations. The
influence of large quantities of calcium of the sodium determination is suppressed by means of
phosphoric acid.
7.16.2 Reagents
7.16.2.1 General requirements
References to water mean distilled water or water of the same degree of purity with an electrical
conductivity of approximately 2 µ S/cm. Use reagents of analytical quality; their alkali content shall
be tested by means of this method. If alkali content of a reagent exceeds 0.01 %, the batch
concerned is unsuitable and shall therefore be replaced by another, which shall be tested in the same
way.
(a) Concentrated hydrochloric acid (HC1).
(b) Dilute hydrochloric acid 1 + 19.
(c) Concentrated phosphoric acid (H3PO4).
(d) Dilute phosphoric acid 1 + 19; store this solution in a polyethylene flask.
(e) Concentrated nitric acid (HNO3).
(f) Concentrated perchloric acid (HCIO4).
(g) Concentrated hydrofluoric acid (HF).
(h) Sodium chloride (NaC1), dried at 105 °C to constant mass.
(i) Potassium chloride (KC1), dried at 105 °C to constant mass.
(j) Alkali Stock Solution. Weigh about 0.566 g of sodium chloride and about 0.475 g of potassium chloride, transfer them to a 1000 mL volumetric flask, Add 100 mL each of dilute hydrochloric acid 1 + 19 and dilute phosphoric acid 1 + 19, dissolve and make up to the mark with water. This solution contains approximately 0.300 g each of Na2O and K2O. The actual contents can
be determined from the original quantities from the following formulae:
K2O (in g/1) = 0.6138 x actual mass of potassium chloride in g ………..…...15
Na2O (in g/1) = 0.5303 x actual mass of sodium chloride in g ……………16

16
7.16.3 Apparatus
(a) Balance, capable of weighing to the nearest 0.0001g.
(b) Calibrated burettes.
(c) Flame photometer, with which the intensities of the sodium line at 589 nm.
(d) and the potassium line at 768 nm can be measured: a sufficiently stable
(e) apparatus shall be used.
(f) Platinum dish.
(g) Filter paper, medium pore size (pore diameter approximately 7 µ m).
(h) Platinum stirrer.
7.16.4 Preparation of calibration solutions and calibration graphs
Prepare the calibration solutions using the volumes of alkali stock solution, dilute hydrochloric acid
1 + 19 and dilute phosphoric acid.
1 + 19 listed in Table 1. The volumes listed in lines 1 to 8 shall be made up to 1000 mL with water.
Store these calibration solutions in polyethylene bottles.
Spray the calibration solutions into the flame of the flame photometer [7.16.3.3]. Spray the blank solution first and set the indication on the Apparatus to 0. Then spray the other calibration solutions in the order of increasing concentration (lines 2 to 8). Measure the intensities for Na2O at 589 nm
and for K2O at 768 nm.
Plot graphs of the measured intensities against the corresponding concentrations of sodium oxide
and potassium oxide and potassium oxide in the calibration solutions.
7.16.5 Dissolution of the test portion
7.16.5.1Cements completely soluble in acid (contents of insoluble residue < 3%)
(a) Weigh 0.1 g cement into a 50 mL beaker, make into a slurry with 10 mL of water and add
10 mL of dilute hydrochloric acid 1 + 19.
(b) Warm the mixture until the cement has dissolved, breaking any lumps down with a glass
rod. Filter the suspension through the filter paper [7.16.3.5] into a 100 mL volumetric flask
washing through with boiling water.
(c) Wash the filter paper and residue with boiling water until there is a quantity of
approximately 80 mL in the 100 mL volumetric flask. Then allow the filtrate and washing
water to cool to ambient temperature.
(d) Add 10 mL of dilute phosphoric acid 1 + 19 to the solution, make up to the mark and mix
thoroughly.
7.16.5.2Cements not completely soluble in acid (contents of insoluble residue > 3 %)
(a) Weigh 0.2 g of cement into a platinum dish and add 5 mL of concentrated nitric acid
[7.16.2.6]. Heat the mixture, for example, on a hot plate, and evaporate to dryness. Disperse
17
the residue from evaporation in 5 mL of water, add 2 mL of concentrated perchloric acid#
[7.16.2.7] and then add 10 mL of concentrated hydrofluoric acid [7.16.2.8].
(b) Heat the mixture and evaporate to dryness. Prevent overheating by frequent agitation by
means of the platinum stirrer [7.16.3.6]. Add 40 mL of water and 20 mL of dilute
hydrochloric acid 1 + 19 to the residue from evaporation and heat until the residue has
dissolved.
(c) Filter the suspension through the filter paper [7.16.3.5] into a 200 mL volumetric flask
washing through with hot water. Wash the filter paper and residue with hot water until the
200 mL volumetric flask contains a volume of approximately 150mL. Then allow the
filtrate and washing water to cool to ambient temperature.
(d) Add 20 mL of dilute phosphoric acid 1 + 19 to the solution make up to the mark with water
and mix thoroughly.
NOTE: Perchloric acid vapours form explosive mixture with organic materials. It is therefore necessary to take special
precautionary measures when working with perchloric acid: the use of fume cupboards flushed with water and a general
ban on the use of organic substances in the same fume cupboard.
7.16.6 Procedure
(a) Spray the measuring solution produced as described in 7.16.5.1 or 7.16.5.2 into the flame of
the flame photometer [7.16.3.3].
(b) Measure the intensity of the sodium in line at 589 nm and the potassium line at 768 nm.
Obtain the sodium oxide or potassium oxide concentration in the solution respectively by
means of a linear interpolation from the intensities and the associated concentrations of the
calibration solutions measured as described in 7.16.4.
Use the graphs plotted as in 7.16.4 to obtain the sodium oxide and potassium oxide concentrations
of the solution in mg/l or use the intensities and the associated concentrations of the calibration
solution with the next higher and next lower intensity for the calculation as indicated below
Table 1. Volumes of solutions for the preparation of calibration solutions and their sodium oxide and potassium oxide
concentrations.
Line Alkali stock solution
(7.16.2.11)
mL
Dilute hydrochloric
Acid 1 + 19
mL
Dilute phosphoric
acid 1 + 19 mL
Na2O and K2O
Concentrations mg/1
1
2
3 4
5 6
7
8
-
3.3
8.3 16.7
25.0 33.3
41.7
50.0
100.0
99.6
99.1 98.3
97.5 96.6
95.8
95.0
100.0
99.6
99.1 98.3
97.5 96.6
95.8
95.0
Blank solution
1.0
2.5 5.0
7.5 10.0
12.5
15.0
Calculate the sodium oxide (CNa2O) or potassium oxide (CK2O) concentration of the sample.
Form the intensities INa2O or IK2O) respectively using the following formulae:
CNa2O = CBn + (CBh – CBn) x INa2O – IBn ………………. 17
IBh - IBn

18
CK2O = CBn + (CBh – CBn) x IK2O - IBn ………………18
IBh - IBn
Where
CBn is the concentration of the sodium oxide or potassium oxide or potassium oxide respectively in
the calibration solution having a lower concentration than the measuring solution in mg/l;
CBh is the concentration of the sodium oxide or potassium oxide or potassium oxide respectively in
the calibration solution having a higher concentration than the measuring solution in mg/l;
IBn is the intensity of the calibration solution having a lower concentration than the measure
solution;
IBh is the intensity of the calibration solution having a higher concentration than the measuring
solution.
7.16.7 Expression of results
Calculate the contents of sodium oxide or potassium oxide as percentages in the cement from the
following equations using the corresponding concentrations as determined in accordance with
7.16.6.
Na2O = 0.1 CNa2O ………………………. 19
K2O = 0.1 CK2O …………………………20
Where
CNa2O is the sodium oxide concentration of the measuring solution as calculated by equation (17)
(in mg/l);
CK2O is the potassium oxide concentration of the measuring solution and calculated by formula (18)
(in mg/l).
The mean of the two results for each oxide shall be rounded to the nearest 0.01 %.
The total alkali content, A, is obtained by converting the potassium oxide content to equivalent
sodium oxide from the formula:
A = Na2O + 0.658 K2O …………… 21
7.16.8 Repeatability and reproducibility
The standard deviation of repeatability is:
0.01 % for the determination of Na2O; 0.02 % for the determination of K2O.
The standard deviation of reproducibility is:
0.02 % for the determination of Na2O;
0.03 % for the determination of K2O.

19
7.17 Determination of the chloride content 7.17.1 Principle
This method gives the total halogen content except for fluoride and expresses the result as Cl-. The
cement sample is decomposed with boiling dilute nitric acid. Sulphides are oxidized into sulphates
and do not interfere. The dissolved chloride is precipitated using a known volume of a standard
silver nitrate solution. After boiling, the precipitate is washed with dilute nitric acid and discarded.
The filtrate and washings are cooled to less than 25ºC and the residual silver nitrate is titrated with a
standard ammonium thiocyanate solution using an iron (III) salt as indicator.
7.17.2 Reagents
(a) Concentrated nitric acid (HNO3).
(b) Dilute nitric acid 1 + 2.
(c) Dilute nitric acid 1 + 100.
(d) Silver nitrate (AgNO3), dried at 150ºC.
(e) Silver nitrate solution, approximately 0.05 mol/l. Dissolve 8.494 g of silver nitrate in water in a
1000 mL volumetric flask and make up to the mark. Store the solution in a brown glass flask
and protect if from the light.
(f) Ammonium thiocyanate (NH4SCN).
(g) Ammonium thiocyanate solution, approximately 0.05 mol/l. Dissolve 3.8 g of ammonium thiocyanate in water and make up to 1000 mL.
(h) Ammonium iron (III) sulphate (NH4Fe(SO4)2. 12H2O).
(i) Indicator solution. Add 10 mL of dilute nitric acid 1 + 2 to 100 mL of a cold saturated solution
of ammonium iron (III) sulphate in water.
7.17.3 Apparatus
(a) Balance, capable of weighing to the nearest 0.0001 g.
(b) 10 mL burette, graduated to 0.1 mL.
(c) Desiccator, containing anhydrous magnesium perchlorate (Mg(C1O4)2).
(d) Filter paper, coarse (pore diameter approximately 20 µ m).
(e) 5 mL pipette
7.17.4 Procedure
(a) Weigh (5 ±0.05) g of cement and place in a 250 mL beaker, add 50 mL of water and, while
stirring with a glass rod, 50 mL of dilute nitric acid 1 + 2 [7.17.2.2]. Heat the mixture to
boiling, stirring occasionally, and boil for 1 minute.
(b) Add 5 mL of silver nitrate solution [7.17.2.5] by pipette [7.17.3.5] into the boiling solution.
Then boil for a maximum of 1 minute and filter through a filter paper [7.17.3.4] washed before use with dilute nitric acid 1 + 100 into a 500 mL flask. Wash the beaker, glass rod
and filter paper with dilute nitric acid 1 + 100 until the volume of the filtrate and the
washings is 200 mL. Cool the filtrate and washings to below 25ºC.

20
(c) Add 5 mL indicator solution [7.17.2.9] and titrate with the ammonium thiocyanate solution
[7.17.2.7] shaking vigorously until a drop of this solution produces a faint reddish-brown
coloration which no longer disappears on shaking. Record the volume V1.
If the chloride content of cement exceeds 0.17 %, it will be necessary to start the test again with a
smaller quantity of cement.
Carry out the same procedure with no cement sample and record the volume, V2 of ammonium
thiocyanate solution used in the blank titration.
7.17.5 Expression of results
Calculate the chloride content (in %) from the formula:
Cl-
= 1.773 x (V2 – V1) x 100 1000 m1
= 0.1773 x (V2 – V1) ……… 22
m1
Where
m1 is the mass of the cement test portion;
V1 is the volume of the ammonium thiocyanate solution used for the titration of the test
solution;
V2 is the volume of the ammonium thiocyanate solution used for the titration of the blank
solution.
The mean of the two results shall be rounded to the nearest 0.01 %.
7.17.6 Repeatability and reproducibility
The standard deviation of repeatability is 0.005 %.
The standard deviation of reproducibility is 0.010 %.
7.18 Determination of SO3 content
7.18.1 Reagents
(a) Concentrated hydrochloric acid
(b) 10 % Barium chloride
7.18.2 Procedure
(a) To 1 g of cement add 25 mL of cold water and while stirring vigorously add 5 mL of
concentrated hydrochloric acid. If necessary, heat the solution and break up the cement
with the flattened end of a glass rod until decomposition is completed. Dilute the solution
to 50 mL and heat it to just below boiling point for 15 minutes.
(b) Filter through a medium filter paper and wash the residue with hot water. Dilute the
solution to 250 mL and boil. Add 10 mL of hot solution of barium chloride drop-wise from
a pipette and boil until the precipitate is properly formed. Let the solution stand at just
below boiling point for 12 to 24 hours (may be placed in the oven set about 95˚C

21
overnight). To ensure that the volume of the solution remains between 225 and 260 mL
dilute the solution to about 280 mL.
(c) Filter the precipitate on a slow filter paper, wash it and place the filter paper and its contents
into a weighted porcelain or platinum crucible. Heat slowly and burn off the filter paper without inflaming. Ignite at 800 or 900˚C for 15 minutes, cool in a dessicator and weigh the
barium sulphate. Ignite at 15 minute intervals to constant weight.
Calculation:
%SO3 = wt of BaSO4ppt x 0.343 x 100 …………..23
Weight of sample

22
Standards Council
The Standards Council is the controlling body of the Bureau of Standards Jamaica and is responsible for the policy and
general administration of the Bureau.
The Council is appointed by the Minister in the manner provided for in the Standards Act, 1969. Using its powers in
the Standards Act, the Council appoints committees for specified purposes.
The Standard Act, 1969 sets out the duties of the Council and the steps to be followed for the formulation of a standard.
Preparation of standards documents
The following is an outline of the procedure, which must be followed in the preparation of documents:
1. The preparation of standards documents is undertaken upon the Standard Council’s authorisation.
This may arise out of representation from national organisations or existing Bureau of Standards Jamaica Committees of Bureau staff. If the project is approved it is referred to the appropriate sectional committee
or if none exists a new committee is formed, or the project is allotted to Bureau staff.
2. If necessary, when the final draft of a standard is ready, the Council authorises an approach to the Minister in
order to obtain the formal concurrence of any other Minister who may be responsible for any area, which the
standard may affect.
3. With the approval of the Standards Council, the draft document is made available for general public comment.
All interested parties, by means of a notice in the Press, are invited to comment. In addition, copies are
forwarded to those known to be interested in the subject.
4. The Committee considers all the comments received and recommends a final document to the Standards
Council.
5. The Standards Council recommends the document to the Minister for publication.
6. The Minister approves the recommendation of the Standards Council.
7. The declaration of the standard is gazetted and copies placed on sale.
8. On the recommendation of the Standards Council the Minister may declare a standard compulsory.
9. Amendments to and revisions of standards normally require the same procedure as is applied to the preparation
of the original standard.
Overseas standards documents
The Bureau of Standards Jamaica maintains a reference library, which includes the standards of many overseas
standards. These standards can be inspected upon request.
The Bureau can supply on demand copies of standards produced by some national standards bodies and is the agency for
the sale of standards produced by International Organisation for Standardization (ISO) members.
Application to use the reference library and to purchase Jamaican and other standards documents should be addresses
to:
Bureau of Standards Jamaica
6 Winchester Road
P.O. Box 113
Kingston 10
JAMAICA, W. I.




















