
Domains of Estrogen Receptor (ER) Required for ER/Sp1-Mediated Activation of GC-Rich Promoters by...
14
Domains of Estrogen Receptor (ER) Required for ER/Sp1-Mediated Activation of GC-Rich Promoters by Estrogens and Antiestrogens in Breast Cancer Cells KYOUNGHYUN KIM, NGUYEN THU, BRAD SAVILLE, AND STEPHEN SAFE Department of Veterinary Physiology & Pharmacology (K.K., N.T., S.S.) and Department of Biochemistry & Biophysics (B.S.), Texas A&M University, College Station, Texas 77843-4466 Estrogen receptor (ER)/Sp1 activation of GC- rich gene promoters in breast cancer cells is dependent, in part, on activation function 1 (AF1) of ER, and this study investigates contributions of the DNA binding domain (C) and AF2 (DEF) regions of ER on activation of ER/Sp1. 17- Estradiol (E2) and the antiestrogens 4-hydroxyta- moxifen and ICI 182,780 induced reporter gene activity in MCF-7 and MDA-MB-231 cells co- transfected with human or mouse ER (hER or MOR), but not ER and GC-rich constructs con- taining three tandem Sp1 binding sites (pSp1 3 ) or other E2-responsive GC-rich promoters. Estro- gen and antiestrogen activation of hER/Sp1 was dependent on overlapping and different re- gions of the C, D, E, and F domains of ER. Antiestrogen-induced activation of hER/Sp1 was lost using hER mutants deleted in zinc fin- ger 1 [amino acids (aa) 185–205], zinc finger 2 (aa 218–245), and the hinge/helix 1 (aa 265–330) do- mains. In contrast with antiestrogens, E2-depen- dent activation of hER/Sp1 required the C- terminal F domain (aa 579–595), which contains a -strand structural motif. Moreover, in peptide competition experiments overexpression of a C- terminal (aa 575–595) F domain peptide specifi- cally blocked E2-dependent activation of hER/ Sp1, suggesting that F domain interactions with nuclear cofactors are required for ER/Sp1 action. (Molecular Endocrinology 17: 804–817, 2003) T HE NUCLEAR RECEPTOR superfamily of tran- scription factors includes the ligand-activated steroid and thyroid hormone, vitamin D and retinoid receptors, and several orphan receptors with un- known endogenous ligands (1–6). Transcriptional activation induced by steroid hormone receptors requires initial ligand binding and formation of a nuclear hormone receptor dimer that interacts with cognate hormone response elements in promoter regions of responsive genes. DNA-bound nuclear hormone receptors activate the basal transcription machinery through interactions with a growing num- ber of coregulatory proteins including coactivators, p300, cAMP response element binding protein- associated factor, and TATA binding protein-asso- ciated factors (7–13). Functional and physical inter- actions of these factors with several steroid hormone receptors have been reported; however, the precise assembly of interacting proteins has not been de- fined and depends on the specific receptor, cell, and promoter context (14, 15). Ligand-activated estrogen receptor (ER) and ER signaling pathways have been extensively in- vestigated and involve interactions of ER homo- or heterodimers with estrogen responsive elements (EREs) in 17-estradiol (E2) responsive gene pro- moters (1–6, 15–19). Several studies have also de- scribed ligand-activated ER action through activator protein 1 (AP1) elements in which transcriptional enhancement is associated with interactions be- tween ER and the c-jun component of the AP1 com- plex (20–25). Estrogens and antiestrogens activate ER/AP1; however, this response is ligand structure and cell context dependent, as antiestrogens exhibit minimal agonist activity in breast cancer cells. In contrast, ER/AP1 is preferentially activated by antiestrogens but not E2 or diethylstilbestrol (20). Research in this laboratory has identified GC-rich motifs in promoters of several E2-responsive genes that are activated by ER/Sp1 and, like ER/AP1, this hormone-dependent response does not require di- rect binding of ER to promoter elements and is activated by HE11, a DNA-binding domain (DBD) deletion mutant of ER (26–36). Human ER (hER)/ Sp1 is activated by estrogens and antiestrogens in MCF-7 and MDA-MB-231 breast cancer and LnCaP prostate cancer cells but not in HeLa cells. In con- Abbreviations: aa, Amino acids; ADA, adenosine deaminase; AF, activation function; AP1, activator protein 1; CHO, Chinese hamster ovary; DBD, DNA-binding domain; DMSO, dimethyl- sulfoxide; DPBS, Dulbecco’s PBS; E2, 17-estradiol; ER, es- trogen receptor; ERE, estrogen responsive element; FBS, fetal bovine serum; hER, human ER; MOR, mouse ER; 4-OHT, 4-hydroxytamoxifen; RAR, retinoic acid receptor; SERM, selec- tive ER modulator; SRC, steroid receptor coactivator. 0888-8809/03/$15.00/0 Molecular Endocrinology 17(5):804–817 Printed in U.S.A. Copyright © 2003 by The Endocrine Society doi: 10.1210/me.2002-0406 804
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Domains of Estrogen Receptor (ER) Required for ER/Sp1-Mediated Activation of GC-Rich Promoters by...

Domains of Estrogen Receptor � (ER�) Required forER�/Sp1-Mediated Activation of GC-Rich Promotersby Estrogens and Antiestrogens in BreastCancer Cells
KYOUNGHYUN KIM, NGUYEN THU, BRAD SAVILLE, AND STEPHEN SAFE
Department of Veterinary Physiology & Pharmacology (K.K., N.T., S.S.) and Department ofBiochemistry & Biophysics (B.S.), Texas A&M University, College Station, Texas 77843-4466
Estrogen receptor � (ER�)/Sp1 activation of GC-rich gene promoters in breast cancer cells isdependent, in part, on activation function 1 (AF1)of ER�, and this study investigates contributionsof the DNA binding domain (C) and AF2 (DEF)regions of ER� on activation of ER�/Sp1. 17�-Estradiol (E2) and the antiestrogens 4-hydroxyta-moxifen and ICI 182,780 induced reporter geneactivity in MCF-7 and MDA-MB-231 cells co-transfected with human or mouse ER� (hER� orMOR), but not ER� and GC-rich constructs con-taining three tandem Sp1 binding sites (pSp13) orother E2-responsive GC-rich promoters. Estro-gen and antiestrogen activation of hER�/Sp1was dependent on overlapping and different re-gions of the C, D, E, and F domains of ER�.
Antiestrogen-induced activation of hER�/Sp1was lost using hER� mutants deleted in zinc fin-ger 1 [amino acids (aa) 185–205], zinc finger 2 (aa218–245), and the hinge/helix 1 (aa 265–330) do-mains. In contrast with antiestrogens, E2-depen-dent activation of hER�/Sp1 required the C-terminal F domain (aa 579–595), which contains a�-strand structural motif. Moreover, in peptidecompetition experiments overexpression of a C-terminal (aa 575–595) F domain peptide specifi-cally blocked E2-dependent activation of hER�/Sp1, suggesting that F domain interactions withnuclear cofactors are required for ER�/Sp1action. (Molecular Endocrinology 17: 804–817,2003)
THE NUCLEAR RECEPTOR superfamily of tran-scription factors includes the ligand-activated
steroid and thyroid hormone, vitamin D and retinoidreceptors, and several orphan receptors with un-known endogenous ligands (1–6). Transcriptionalactivation induced by steroid hormone receptorsrequires initial ligand binding and formation of anuclear hormone receptor dimer that interacts withcognate hormone response elements in promoterregions of responsive genes. DNA-bound nuclearhormone receptors activate the basal transcriptionmachinery through interactions with a growing num-ber of coregulatory proteins including coactivators,p300, cAMP response element binding protein-associated factor, and TATA binding protein-asso-ciated factors (7–13). Functional and physical inter-actions of these factors with several steroid hormonereceptors have been reported; however, the preciseassembly of interacting proteins has not been de-
fined and depends on the specific receptor, cell, andpromoter context (14, 15).
Ligand-activated estrogen receptor � (ER�) andER� signaling pathways have been extensively in-vestigated and involve interactions of ER homo- orheterodimers with estrogen responsive elements(EREs) in 17�-estradiol (E2) responsive gene pro-moters (1–6, 15–19). Several studies have also de-scribed ligand-activated ER action through activatorprotein 1 (AP1) elements in which transcriptionalenhancement is associated with interactions be-tween ER and the c-jun component of the AP1 com-plex (20–25). Estrogens and antiestrogens activateER�/AP1; however, this response is ligand structureand cell context dependent, as antiestrogens exhibitminimal agonist activity in breast cancer cells. Incontrast, ER�/AP1 is preferentially activated byantiestrogens but not E2 or diethylstilbestrol (20).Research in this laboratory has identified GC-richmotifs in promoters of several E2-responsive genesthat are activated by ER�/Sp1 and, like ER/AP1, thishormone-dependent response does not require di-rect binding of ER to promoter elements and isactivated by HE11, a DNA-binding domain (DBD)deletion mutant of ER� (26–36). Human ER� (hER�)/Sp1 is activated by estrogens and antiestrogens inMCF-7 and MDA-MB-231 breast cancer and LnCaPprostate cancer cells but not in HeLa cells. In con-
Abbreviations: aa, Amino acids; ADA, adenosine deaminase;AF, activation function; AP1, activator protein 1; CHO, Chinesehamster ovary; DBD, DNA-binding domain; DMSO, dimethyl-sulfoxide; DPBS, Dulbecco’s PBS; E2, 17�-estradiol; ER, es-trogen receptor; ERE, estrogen responsive element; FBS, fetalbovine serum; hER, human ER; MOR, mouse ER�; 4-OHT,4-hydroxytamoxifen; RAR, retinoic acid receptor; SERM, selec-tive ER modulator; SRC, steroid receptor coactivator.
0888-8809/03/$15.00/0 Molecular Endocrinology 17(5):804–817Printed in U.S.A. Copyright © 2003 by The Endocrine Society
doi: 10.1210/me.2002-0406
804

trast, hER�/Sp1 exhibits minimal activity in all celllines examined (35). Studies with wild-type hER�,various hER� chimeras, and mutants demonstratedthat the activation function 1 (AF1) region of hER�was required for hER�/Sp1-mediated transactiva-tion in MCF-7 and MDA-MB-231 breast cancer cells;however, this did not exclude a role for AF2 in thisresponse (35).
This study further investigates estrogen and anties-trogen activation of hER�/Sp1 action in MCF-7 andMDA-MB-231 breast cancer cells and AF1-indepen-dent domains of ER� required for ligand-dependenttransactivation. Estrogen activation of hER�/Sp1 isobserved in cells transfected with wild-type hER� anddeletions of one or both zinc fingers and point muta-tions in helix 12 (D538N, E542Q, and D545N) thatblock interactions with AF2-interacting coactivators.Activation of hER�/Sp1 by E2 is not observed in hER�mutants with deletion of the hinge region [amino acids(aa) 271–300] or helix 12 and the F domain (aa 538–595), whereas these latter mutants are activated by theantiestrogens 4-hydroxytamoxifen (4-OHT) and ICI182,780. In contrast, antiestrogen activation of hER�/Sp1 is not observed with zinc finger 1 or zinc finger 2deletion mutants. Thus, hER�/Sp1 activation by estro-gens and antiestrogens is complex and requires mul-tiple domains of hER�.
RESULTS
Role of Zinc Fingers 1 and 2 of ER� and ER� inHormonal Activation of GC-Rich Promoters
Previous studies in this laboratory showed thathormone-induced activation of hER�/Sp1 in breastcancer cells required the AF1 domain of hER� (35),and activation by E2 was also observed in cells co-transfected with the DBD deletion mutant (aa 185–251)hER11 (26–36). The role of other domains of hER� onestrogen and antiestrogen activation of hER�/Sp1 hasnot been defined and is investigated in this study.Although hER�11/Sp1 is activated by E2, deletion ofthe entire DBD resulted in loss of antiestrogen-induced transactivation (35, 37), and therefore initialstudies determined the role of zinc fingers 1 and 2deletion mutants on estrogen/antiestrogen activationof ER�/Sp1.
Wild-type and zinc finger deletion mutants for hER(� and �) and mouse ER� (MOR) (Table 1) werecloned into pcDNA3 and translated in vitro using[35S]methionine, and the radiolabeled proteins wereseparated by SDS-PAGE (Fig. 1A). The results showthat in vitro translated proteins gave distinct bandswith comparable intensities and the expected mo-lecular weights, indicating that the �ZF1 or �ZF2
Table 1. Summary of Primers Used for Mutagenesis of the DBDs of hER� and MOR
ER Deletion Mutant PCR Primer Sets (A1/B1, A2/B2) for Mutagenesisa Deletion
hER��ZF1
A1: 5� GCAAATGGGCGGTAGGCGTGTA 3� 21-aadeletion
(185 aa–205 aa)B1: 5� CTTGAAGAAGGCCTTGTAGCGAGTCTCCTTGG 3�A2: 5� AAGGAGACTCGCTACAAGGCCTTCTTCAAGAG 3�B2: 5� GAGACCAATCATCAGGA
hER��ZF2
A1: 5� GCAAATGGGCGGTAGGCGTGTA 3� 28-aadeletion
(218 aa–245 aa)B1: 5� CATTCCCACTTCGTAGTTATGTCCTTGAATACT 3�A2: 5� GTATTCAAGGACATAACTACGAAGTGGGAATGAT 3�B2: 5� GAGACCAATCATCAGGA 3�
hER��ZF1
A1: 5� GACGTCAATGGGAGT 3� 21-aadeletion
(96 aa–111 aa)B1: 5� CTTTTAAAAAAGGCCTTGAAGTGAGCATCCCTCTTC 3�A2: 5� GGATGCTCACTTCAAGGCCTTTTTTAAAAG 3�B2: 5� GAGACCAATCATCAGGA
hER��ZF2
A1: 5� GTGTACGGTGGGAG 3� 28-aadeletion
(124 aa–131 aa)B1: 5� CCATTCCCACTTCGTAATTATGTCCTTGAATGCTTC 3�A2: 5� CAAGGACATAATTACGAAGTGGGAATGG 3�B2: 5� AACTCTCGAAACCTTGAA 3�
MOR��ZF1
A1: 5� GCATCGCCTACGG 3� 21-aadeletion
(189 aa–209 aa)B1: 5� CTTAAAGAAAGCCTTGTAGCGAGTCTCCTTGGC 3�A2: 5� GGAGACTCGCTACAAGGCTTTCTTTAAGAGAAGC 3�B2: 5� GGTCAATAAGCCCATCA 3�
MOR�ZF2
A1: 5� GCATCGCCTACGG 3� 28-aadeletion
(222 aa–249 aa)B1: 5� CATGCCCACTTCGTAATTGTGTCCTTGAATGCT 3�A2: 5� TCAAGGACACAATTACGAAGTGGGCATGATG 3�B2: 5� GGTCAATAAGCCCATCA 3�
a Overlapping regions are noted in bold type.
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 805
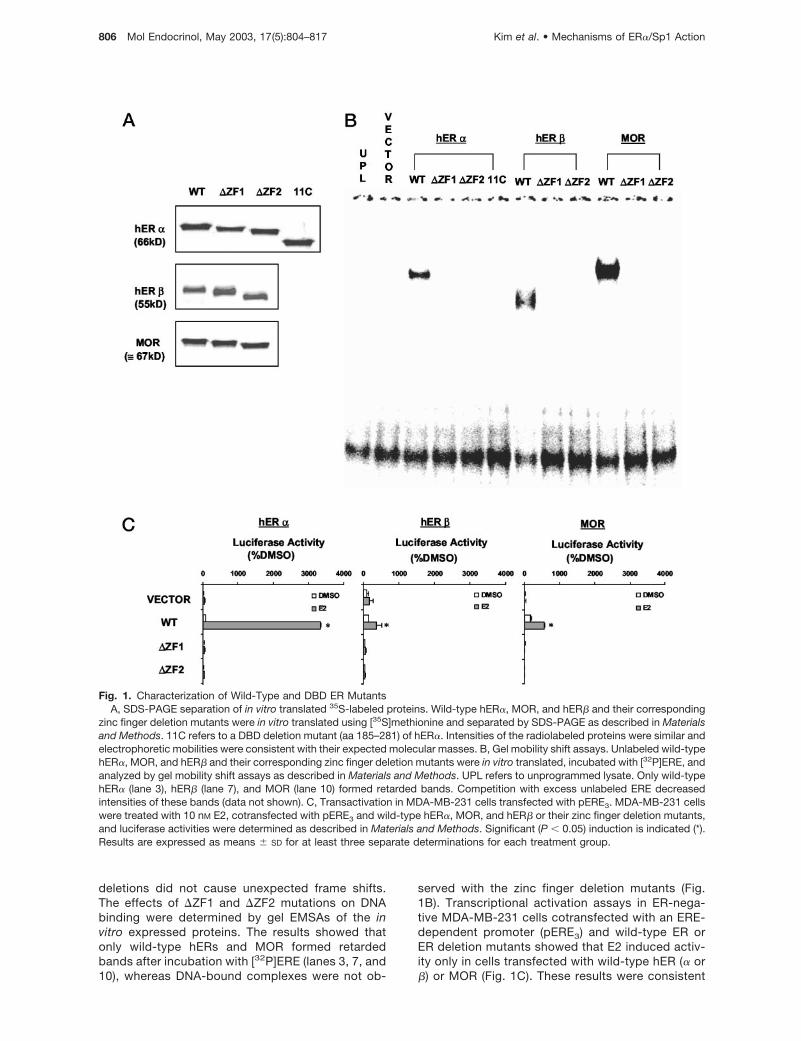
deletions did not cause unexpected frame shifts.The effects of �ZF1 and �ZF2 mutations on DNAbinding were determined by gel EMSAs of the invitro expressed proteins. The results showed thatonly wild-type hERs and MOR formed retardedbands after incubation with [32P]ERE (lanes 3, 7, and10), whereas DNA-bound complexes were not ob-
served with the zinc finger deletion mutants (Fig.1B). Transcriptional activation assays in ER-nega-tive MDA-MB-231 cells cotransfected with an ERE-dependent promoter (pERE3) and wild-type ER orER deletion mutants showed that E2 induced activ-ity only in cells transfected with wild-type hER (� or�) or MOR (Fig. 1C). These results were consistent
Fig. 1. Characterization of Wild-Type and DBD ER MutantsA, SDS-PAGE separation of in vitro translated 35S-labeled proteins. Wild-type hER�, MOR, and hER� and their corresponding
zinc finger deletion mutants were in vitro translated using [35S]methionine and separated by SDS-PAGE as described in Materialsand Methods. 11C refers to a DBD deletion mutant (aa 185–281) of hER�. Intensities of the radiolabeled proteins were similar andelectrophoretic mobilities were consistent with their expected molecular masses. B, Gel mobility shift assays. Unlabeled wild-typehER�, MOR, and hER� and their corresponding zinc finger deletion mutants were in vitro translated, incubated with [32P]ERE, andanalyzed by gel mobility shift assays as described in Materials and Methods. UPL refers to unprogrammed lysate. Only wild-typehER� (lane 3), hER� (lane 7), and MOR (lane 10) formed retarded bands. Competition with excess unlabeled ERE decreasedintensities of these bands (data not shown). C, Transactivation in MDA-MB-231 cells transfected with pERE3. MDA-MB-231 cellswere treated with 10 nM E2, cotransfected with pERE3 and wild-type hER�, MOR, and hER� or their zinc finger deletion mutants,and luciferase activities were determined as described in Materials and Methods. Significant (P � 0.05) induction is indicated (*).Results are expressed as means � SD for at least three separate determinations for each treatment group.
806 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action
with the gel mobility shift assays showing that thezinc finger mutants do not bind EREs (Fig. 1B).
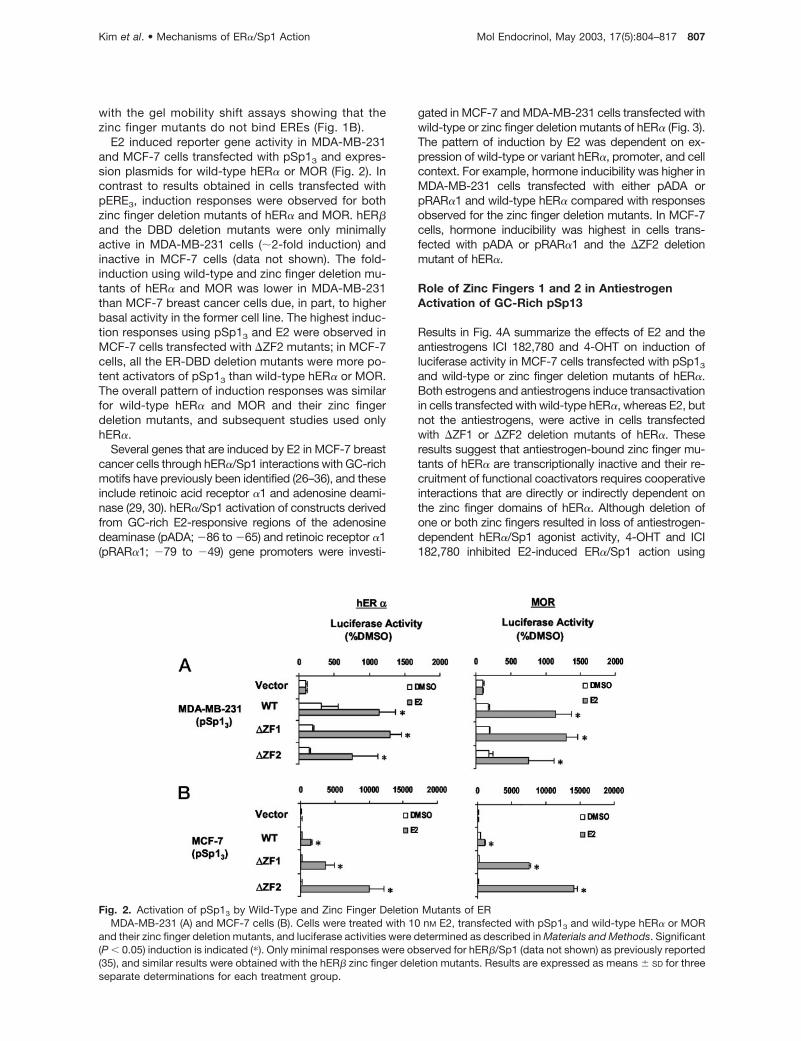
E2 induced reporter gene activity in MDA-MB-231and MCF-7 cells transfected with pSp13 and expres-sion plasmids for wild-type hER� or MOR (Fig. 2). Incontrast to results obtained in cells transfected withpERE3, induction responses were observed for bothzinc finger deletion mutants of hER� and MOR. hER�and the DBD deletion mutants were only minimallyactive in MDA-MB-231 cells (�2-fold induction) andinactive in MCF-7 cells (data not shown). The fold-induction using wild-type and zinc finger deletion mu-tants of hER� and MOR was lower in MDA-MB-231than MCF-7 breast cancer cells due, in part, to higherbasal activity in the former cell line. The highest induc-tion responses using pSp13 and E2 were observed inMCF-7 cells transfected with �ZF2 mutants; in MCF-7cells, all the ER-DBD deletion mutants were more po-tent activators of pSp13 than wild-type hER� or MOR.The overall pattern of induction responses was similarfor wild-type hER� and MOR and their zinc fingerdeletion mutants, and subsequent studies used onlyhER�.
Several genes that are induced by E2 in MCF-7 breastcancer cells through hER�/Sp1 interactions with GC-richmotifs have previously been identified (26–36), and theseinclude retinoic acid receptor �1 and adenosine deami-nase (29, 30). hER�/Sp1 activation of constructs derivedfrom GC-rich E2-responsive regions of the adenosinedeaminase (pADA; �86 to �65) and retinoic receptor �1(pRAR�1; �79 to �49) gene promoters were investi-
gated in MCF-7 and MDA-MB-231 cells transfected withwild-type or zinc finger deletion mutants of hER� (Fig. 3).The pattern of induction by E2 was dependent on ex-pression of wild-type or variant hER�, promoter, and cellcontext. For example, hormone inducibility was higher inMDA-MB-231 cells transfected with either pADA orpRAR�1 and wild-type hER� compared with responsesobserved for the zinc finger deletion mutants. In MCF-7cells, hormone inducibility was highest in cells trans-fected with pADA or pRAR�1 and the �ZF2 deletionmutant of hER�.
Role of Zinc Fingers 1 and 2 in AntiestrogenActivation of GC-Rich pSp13
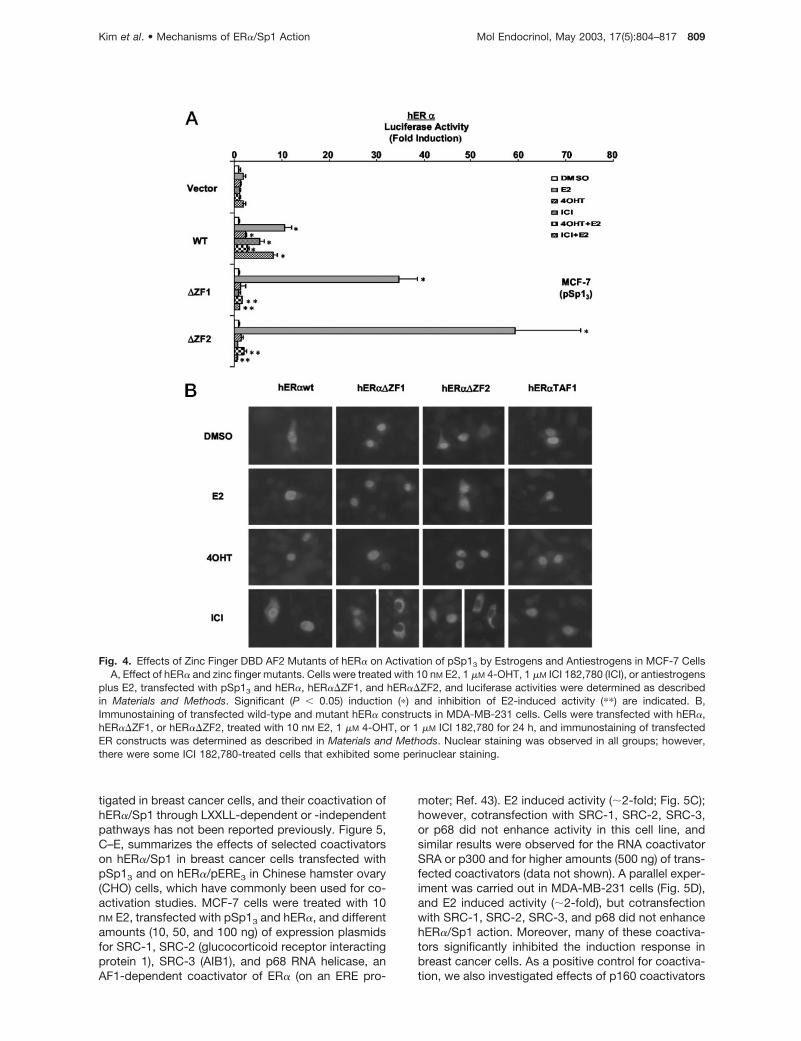
Results in Fig. 4A summarize the effects of E2 and theantiestrogens ICI 182,780 and 4-OHT on induction ofluciferase activity in MCF-7 cells transfected with pSp13
and wild-type or zinc finger deletion mutants of hER�.Both estrogens and antiestrogens induce transactivationin cells transfected with wild-type hER�, whereas E2, butnot the antiestrogens, were active in cells transfectedwith �ZF1 or �ZF2 deletion mutants of hER�. Theseresults suggest that antiestrogen-bound zinc finger mu-tants of hER� are transcriptionally inactive and their re-cruitment of functional coactivators requires cooperativeinteractions that are directly or indirectly dependent onthe zinc finger domains of hER�. Although deletion ofone or both zinc fingers resulted in loss of antiestrogen-dependent hER�/Sp1 agonist activity, 4-OHT and ICI182,780 inhibited E2-induced ER�/Sp1 action using
Fig. 2. Activation of pSp13 by Wild-Type and Zinc Finger Deletion Mutants of ERMDA-MB-231 (A) and MCF-7 cells (B). Cells were treated with 10 nM E2, transfected with pSp13 and wild-type hER� or MOR
and their zinc finger deletion mutants, and luciferase activities were determined as described in Materials and Methods. Significant(P � 0.05) induction is indicated (�). Only minimal responses were observed for hER�/Sp1 (data not shown) as previously reported(35), and similar results were obtained with the hER� zinc finger deletion mutants. Results are expressed as means � SD for threeseparate determinations for each treatment group.
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 807

these DBD deletion constructs, suggesting that theantiestrogen-mediated responses are intact in cells ex-pressing zinc finger mutants of hER�. We also investi-gated the possibility that the failure to observe antiestro-gen activation of zinc finger mutants of hER�/Sp1 maybe due to failure of the transfected constructs to accu-mulate in nuclei of breast cancer cells. However, resultsof immunofluorescent studies in MDA-MB-231 cellstransfected with hER� or the zinc finger mutantsshowed that wild-type and mutant constructs were pri-marily nuclear in cells treated with dimethylsulfoxide(DMSO) or E2/antiestrogens, although some perinuclearstaining was observed with hER��ZF1 using ICI 182,780(Fig. 4B).
Activation of hER�/Sp1 by E2, 4-OHT, and ICI182,780 Does Not Require AF2-Helix 12-Coactivator Interactions
Saville et al. (35) previously reported that activation ofhER�/Sp1 by E2 was lost after deletion of aa 51–117;however, this did not exclude a role for AF2 alone or asa modifier of AF1-dependent hER�/Sp1 action.hER�TAF1 contains three aa mutations (D538N,E542Q, and D545N) that do not affect ligand bindingbut inactivate AF2 by selectively blocking interactionswith AF2-dependent coactivators (38–41). In MCF-7cells transfected with pSp13 and hER�TAF1, E2 sig-nificantly induced reporter gene activity, and ICI182,780 and 4-OHT also slightly increased this re-sponse (Fig. 5A). Hormone-mediated transactivationwas also observed in MCF-7 cells transfected with
pSp13 and hER�TAF1 containing deletions of zincfinger 1 or zinc finger 2 in the DBD; in contrast, theantiestrogens ICI 182,780 and 4-OHT did not activateluciferase activity using the hER�TAF1 DBD mutants,and similar results were observed for hER��ZF1 andhER��ZF2 (Fig. 4A). Increased hormone-inducedtransactivation was observed in MCF-7 cells trans-fected with pSp13 and hER��ZF1 or hER��ZF2 com-pared with wild-type hER� (Figs. 2B and 4A). In con-trast, deletion of zinc fingers 1 or 2 in hER�TAF1 didnot result in increased hormone responsiveness inMCF-7 cells transfected with the zinc finger deletionmutants compared with hER�TAF1 (Fig. 5A). This sug-gests that helix 12 may contribute to E2-inducedhER��ZF1/Sp1 and hER��ZF2/Sp1 action. hER�19and hER�null were also inactive, and this was consis-tent with previous studies showing the importance ofAF1 for hER�/Sp1 action (35). The results in Fig. 5Bshow that expression of the LXXLL peptide 2XF6 (38)significantly decreased hormone-induced transactiva-tion in MDA-MB-231 cells transfected with pERE3, andinhibition was not observed in cells transfected withpSp13.
These data suggest that interactions of hER� withprototypical steroid receptor coactivators containingLXXLL motifs may not be critical for hER�/Sp1 action.Coactivators of hER� and other nuclear receptorshave been extensively investigated, and these includeAF2-dependent steroid receptor coactivators (SRCs)and AF1-dependent p68 RNA helicase (42, 43). How-ever, many of these coactivators have not been inves-
Fig. 3. Activation of GC-Rich E2-Responsive pRAR�1 and pADA Promoters by E2MDA-MB-231 (A) and MCF-7 (B) cells transfected with pRAR�1 and pADA. Cells were treated with E2 or DMSO, transfected
with pADA or pRAR�1 constructs, wild-type hER�, or zinc finger deletion mutants of hER�, and luciferase activities weredetermined as described in Materials and Methods. Significant (P � 0.05) induction by E2 is indicated (�). Results are presentedas means � SD for three separate determinations for each treatment group.
808 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action
tigated in breast cancer cells, and their coactivation ofhER�/Sp1 through LXXLL-dependent or -independentpathways has not been reported previously. Figure 5,C–E, summarizes the effects of selected coactivatorson hER�/Sp1 in breast cancer cells transfected withpSp13 and on hER�/pERE3 in Chinese hamster ovary(CHO) cells, which have commonly been used for co-activation studies. MCF-7 cells were treated with 10nM E2, transfected with pSp13 and hER�, and differentamounts (10, 50, and 100 ng) of expression plasmidsfor SRC-1, SRC-2 (glucocorticoid receptor interactingprotein 1), SRC-3 (AIB1), and p68 RNA helicase, anAF1-dependent coactivator of ER� (on an ERE pro-
moter; Ref. 43). E2 induced activity (�2-fold; Fig. 5C);however, cotransfection with SRC-1, SRC-2, SRC-3,or p68 did not enhance activity in this cell line, andsimilar results were observed for the RNA coactivatorSRA or p300 and for higher amounts (500 ng) of trans-fected coactivators (data not shown). A parallel exper-iment was carried out in MDA-MB-231 cells (Fig. 5D),and E2 induced activity (�2-fold), but cotransfectionwith SRC-1, SRC-2, SRC-3, and p68 did not enhancehER�/Sp1 action. Moreover, many of these coactiva-tors significantly inhibited the induction response inbreast cancer cells. As a positive control for coactiva-tion, we also investigated effects of p160 coactivators
Fig. 4. Effects of Zinc Finger DBD AF2 Mutants of hER� on Activation of pSp13 by Estrogens and Antiestrogens in MCF-7 CellsA, Effect of hER� and zinc finger mutants. Cells were treated with 10 nM E2, 1 �M 4-OHT, 1 �M ICI 182,780 (ICI), or antiestrogens
plus E2, transfected with pSp13 and hER�, hER��ZF1, and hER��ZF2, and luciferase activities were determined as describedin Materials and Methods. Significant (P � 0.05) induction (�) and inhibition of E2-induced activity (��) are indicated. B,Immunostaining of transfected wild-type and mutant hER� constructs in MDA-MB-231 cells. Cells were transfected with hER�,hER��ZF1, or hER��ZF2, treated with 10 nM E2, 1 �M 4-OHT, or 1 �M ICI 182,780 for 24 h, and immunostaining of transfectedER constructs was determined as described in Materials and Methods. Nuclear staining was observed in all groups; however,there were some ICI 182,780-treated cells that exhibited some perinuclear staining.
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 809
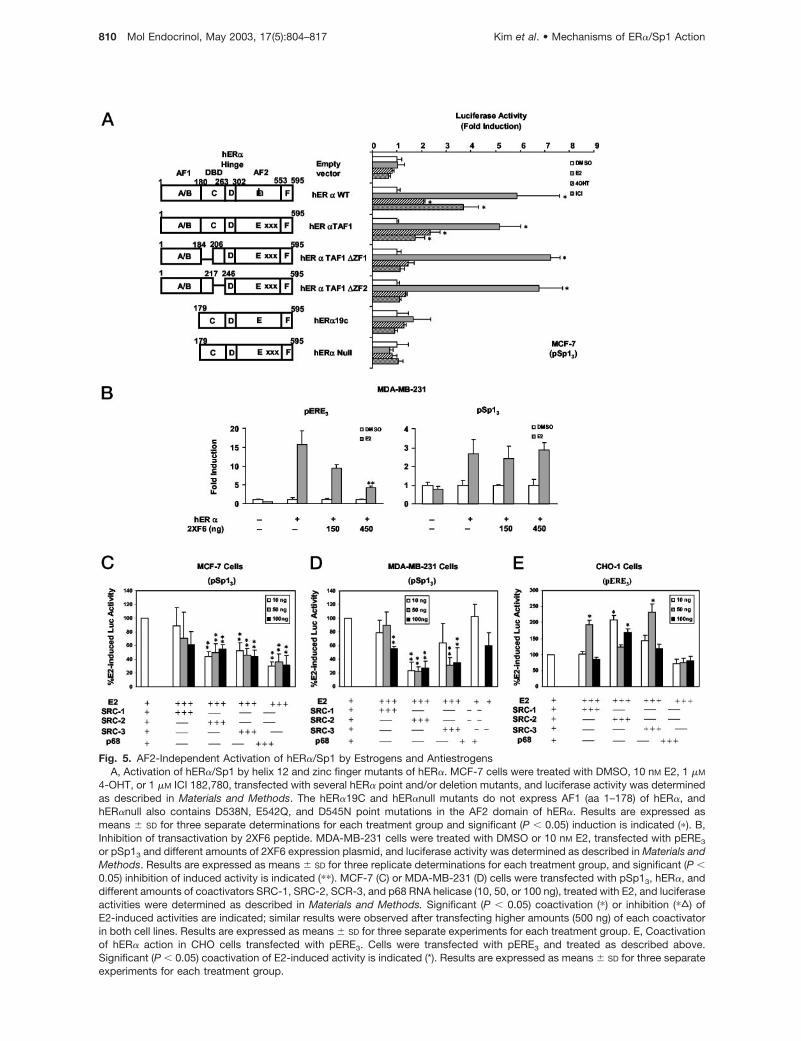
Fig. 5. AF2-Independent Activation of hER�/Sp1 by Estrogens and AntiestrogensA, Activation of hER�/Sp1 by helix 12 and zinc finger mutants of hER�. MCF-7 cells were treated with DMSO, 10 nM E2, 1 �M
4-OHT, or 1 �M ICI 182,780, transfected with several hER� point and/or deletion mutants, and luciferase activity was determinedas described in Materials and Methods. The hER�19C and hER�null mutants do not express AF1 (aa 1–178) of hER�, andhER�null also contains D538N, E542Q, and D545N point mutations in the AF2 domain of hER�. Results are expressed asmeans � SD for three separate determinations for each treatment group and significant (P � 0.05) induction is indicated (�). B,Inhibition of transactivation by 2XF6 peptide. MDA-MB-231 cells were treated with DMSO or 10 nM E2, transfected with pERE3
or pSp13 and different amounts of 2XF6 expression plasmid, and luciferase activity was determined as described in Materials andMethods. Results are expressed as means � SD for three replicate determinations for each treatment group, and significant (P �0.05) inhibition of induced activity is indicated (��). MCF-7 (C) or MDA-MB-231 (D) cells were transfected with pSp13, hER�, anddifferent amounts of coactivators SRC-1, SRC-2, SCR-3, and p68 RNA helicase (10, 50, or 100 ng), treated with E2, and luciferaseactivities were determined as described in Materials and Methods. Significant (P � 0.05) coactivation (�) or inhibition (�„) ofE2-induced activities are indicated; similar results were observed after transfecting higher amounts (500 ng) of each coactivatorin both cell lines. Results are expressed as means � SD for three separate experiments for each treatment group. E, Coactivationof hER� action in CHO cells transfected with pERE3. Cells were transfected with pERE3 and treated as described above.Significant (P � 0.05) coactivation of E2-induced activity is indicated (*). Results are expressed as means � SD for three separateexperiments for each treatment group.
810 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action

and p68 in CHO cells treated with 10 nM E2 andtransfected with pERE3 and hER�. This cell line hasfrequently been used to demonstrate coactivation ofhER� using ERE-dependent promoter-reporter con-structs. E2 induced luciferase activity (8- to 15-fold)and SRC-1, SRC-2, and SRC-3 (but not p68) en-hanced the induction response (Fig. 5E). Coactivationof hER�/Sp1 by SRCs was not observed in ER-posi-tive or negative breast cancer cell lines; this was con-sistent with the importance of AF1 for hER�/Sp1-mediated transactivation (35). Surprisingly, we did notobserve coactivation of hER� or hER�/Sp1 by theAF1-interacting coactivator p68 in ER-negative or-positive cell lines, suggesting that cell context mod-ulates the effects of p68 as a coactivator.
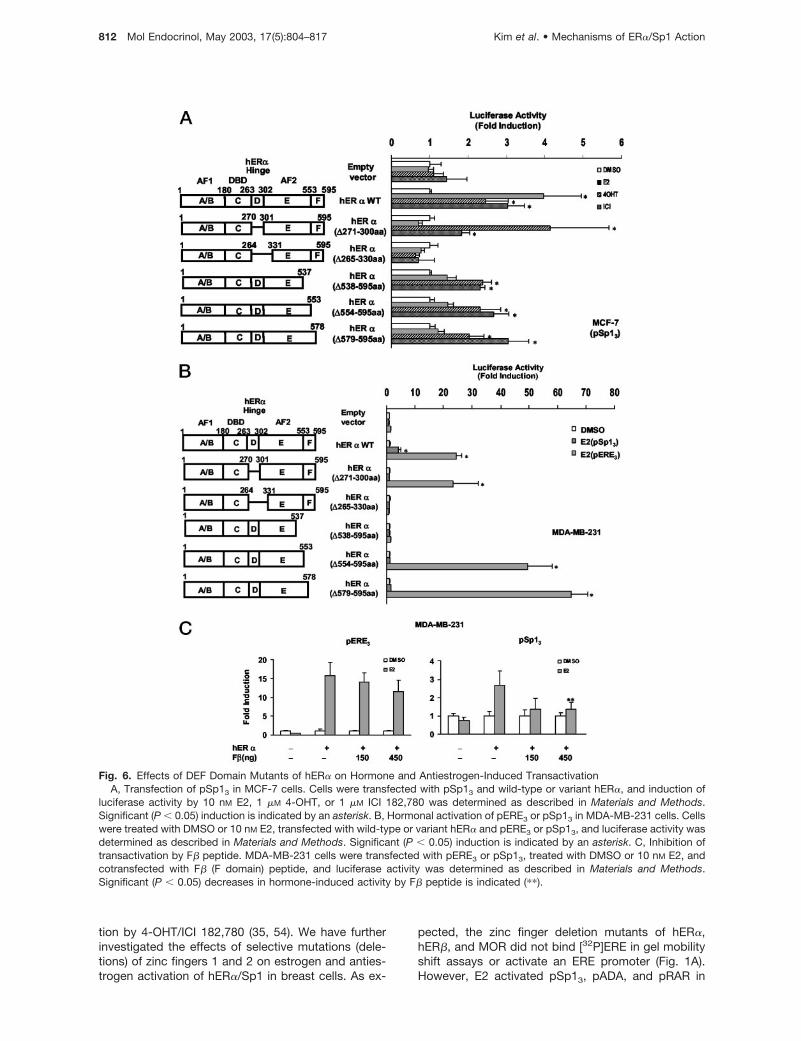
Role of AF2/Hinge (DEF) Region for Activation ofhER�/Sp1 by Estrogen and Antiestrogens
We further investigated requirements for other regionswithin the DEF domains for activation of hER�/Sp1 byestrogens and antiestrogens in MCF-7 cells. E2,4-OHT, and ICI 182,780 did not induce luciferase ac-tivity in MCF-7 cells transfected with pSp13, whereasa 2.5- to 4.5-fold induction was observed by all threecompounds in cells cotransfected with hER� (Fig. 6A).E2 did not induce activity in MCF-7 cells transfectedwith hER�(�271–300) or hER�(�265–330) which con-tain deletions of the hinge (D) or hinge (D) plus helix 1of the E domain. ICI 182,780 and 4-OHT were alsoinactive in cells transfected with hER�(�265–330),whereas induction by the antiestrogens was observedin cells transfected with hER�(�271–300). Estrogen/antiestrogen-dependent activation of hER�/Sp1 wasalso investigated in MCF-7 cells transfected with aseries of C-terminal deletion mutants, namelyhER�(�538–595), hER�(�554–595), and hER�(�579–595). These mutants contain deletions of helix 12 (E)and the C-terminal F domain (538–595), the F domain(554–595) alone, and the �-strand region of the Fdomain (579–595). In MCF-7 cells, both 4-OHT and ICI182,780 induced luciferase activity in cells transfectedwith these hER� deletion mutants, whereas E2 wasinactive. The failure of E2 to induce transactivation incells transfected with pSp13 and hER�(�579–595)suggests that the C-terminal aa 579–595, which con-tains a QKYYIT �-strand motif (44), may be critical fortranscriptional activation by E2 but not 4-OHT or ICI182,780. We further confirmed the F domain require-ment for hormonal activation of hER�/Sp1 by exam-ining a similar series of hER� deletion mutants in MDA-MB-231 cells cotransfected with pSp13 or pERE3. E2induced transactivation in cells cotransfected withpERE3 and hER�(�271–300) or hER�(�554–595), con-firming results of previous studies in other cell linesshowing that the hinge region and F domain are notnecessary for hormonal activation of ER�/pERE (42,44–48). E2 did not induce transactivation in MCF-7 orMDA-MB-231 cells cotransfected with pSp13 andhER�(�554–595). Thus, hormonal activation of hER�/
Sp1 by E2 was dependent on the hinge (D) and Fdomains of hER�, whereas these same regions ofhER� were not required for activation of pERE3.
Peptides targeted to different regions of hER� blockhormone-induced transactivation of ERE-dependentpromoters/genes (38, 39, 48). This has been exten-sively investigated with peptides containing LXXLLmotifs that block coactivator interactions with ER� (38,39) and inhibit hormone-induced activation in cellstransfected with pERE3 (Fig. 5B). Hormone-inducedtransactivation in MDA-MB-231 cells transfected withpERE3 was not significantly decreased after cotrans-fection with the F-� strand peptide containing aa 575–595 from the F domain of hER� fused to the DBD ofthe yeast GAL4 protein. In contrast, the F domainpeptide blocked hormone-induced transactivation inMDA-MB-231 cells transfected with pSp13, whereasthe 2XF6 peptide was inactive, and similar results wereobtained with other peptides containing LXXLL se-quences (data not shown). These results are consis-tent with the activity of wild-type and variant hER�constructs and confirm that the F domain of ER� isalso essential for E2-dependent activation ofhER�/Sp1.
DISCUSSION
Development of selective ER modulators (SERMs) fortreatment of breast cancer and other hormone-relatedproblems is dependent on their tissue-specific activa-tion or inhibition of ER-mediated genes/responses(49–53). There are an increasing number of factors thatregulate cell context-dependent ER action, and theseinclude relative expression of ER subtypes and a com-plex network of nuclear proteins that uniquely interactwith specific surfaces or domains of ER�, ER�, andother coregulatory proteins (7–13). The classicalmechanism of ER activation involves ligand-depen-dent formation of ER dimers that bind consensus ornonconsensus EREs and recruit SRCs and other nu-clear proteins that facilitate interactions with basaltranscription factors (1–13). In contrast, nonclassicalpathways that involve ligand activation of ER/Sp1 andER/AP1 do not require interactions of ER with pro-moter DNA but with other DNA-bound transcriptionfactors, namely Sp1 and c-jun, respectively. Researchin this laboratory has identified a number of E2-responsive genes regulated by ER�/Sp1 (26) in breastcancer cells (27–36), suggesting that the nonclassicalpathways for activation of ER� may play a significantrole in cell context-dependent regulation of genes byE2 and SERMs.
Previous studies showed that both estrogens andantiestrogens activated a construct containing a GC-rich promoter (pSp1) in breast cancer cells and thisresponse was AF1 dependent (35). Moreover, the DBDof hER� was not required for activation by E2, whereasdeletion of this region resulted in loss of transactiva-
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 811
tion by 4-OHT/ICI 182,780 (35, 54). We have furtherinvestigated the effects of selective mutations (dele-tions) of zinc fingers 1 and 2 on estrogen and anties-trogen activation of hER�/Sp1 in breast cells. As ex-
pected, the zinc finger deletion mutants of hER�,hER�, and MOR did not bind [32P]ERE in gel mobilityshift assays or activate an ERE promoter (Fig. 1A).However, E2 activated pSp13, pADA, and pRAR in
Fig. 6. Effects of DEF Domain Mutants of hER� on Hormone and Antiestrogen-Induced TransactivationA, Transfection of pSp13 in MCF-7 cells. Cells were transfected with pSp13 and wild-type or variant hER�, and induction of
luciferase activity by 10 nM E2, 1 �M 4-OHT, or 1 �M ICI 182,780 was determined as described in Materials and Methods.Significant (P � 0.05) induction is indicated by an asterisk. B, Hormonal activation of pERE3 or pSp13 in MDA-MB-231 cells. Cellswere treated with DMSO or 10 nM E2, transfected with wild-type or variant hER� and pERE3 or pSp13, and luciferase activity wasdetermined as described in Materials and Methods. Significant (P � 0.05) induction is indicated by an asterisk. C, Inhibition oftransactivation by F� peptide. MDA-MB-231 cells were transfected with pERE3 or pSp13, treated with DMSO or 10 nM E2, andcotransfected with F� (F domain) peptide, and luciferase activity was determined as described in Materials and Methods.Significant (P � 0.05) decreases in hormone-induced activity by F� peptide is indicated (��).
812 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action

MCF-7 and MDA-MB-231 cells transfected with wild-type hER� (and MOR) and both zinc finger mutants(Figs. 2 and 3), and similar results were obtained inMCF-7 cells transfected with wild-type and zinc fingermutants of hER�TAF1 (Fig. 5A). In contrast, minimalresponses were observed for hER�/Sp1 (data notshown) as previously reported (35). Zinc fingers 1 and2 are important for DNA binding, and the D box regionof zinc finger 2 plays a role in ER� homodimerization(42, 55). The DBD of ER� is also an important deter-minant for antiestrogen activation of ER�/AP1 andER�/Sp1. Point mutations in zinc finger 1 either de-creased (E207G/G208S), eliminated (K201A), or didnot affect (E207A/G208A) ICI 182,780 activation ofER�/AP1 in TSA cells, whereas an A227T mutation inzinc finger 2 resulted in loss of ICI 182,780 inducibilitythrough an AP1 element (25). E2 decreased activationof ER�/AP1 in MCF-7 and TSA cells, and this was alsoobserved in all but one (K210a) of the DBD pointmutants (25). We also investigated activation of hER�/Sp1 by the zinc finger point mutants (25) in breastcancer cells, and minimal transactivation was ob-served after treatment with E2, 4-OHT, or ICI 182,780(data not shown). In contrast, deletion of the DBD ofhER� did not affect activation of hER�/Sp1 by E2 inbreast cancer cells (35); however, our results showthat both zinc fingers of hER� and hER�TAF1 arerequired for the activity of antiestrogens (Fig. 5). Thus,there are significant differences between hER�/Sp1and ER�/AP1 and their requirements for regions withinthe DBD for activation by E2 and SERMs, suggestingthat cell context-specific interactions of nuclear pro-teins with the DBD region of hER� may be importantfor ligand-dependent activation of hER�/Sp1. DBD-interacting proteins that coactivate hER� and otherhormone receptors have been reported (56–58), andcurrent studies are investigating coactivation of hER�/Sp1 by SNURF, a small RING finger protein initiallyidentified as an androgen receptor DBD-interactingprotein.
Previous studies with AF1 deletion mutants of hER�showed that aa 51–117 were required for ER�/Sp1-mediated responses (35); however, contributions ofthe DEF domains have not been determined. The AF2domain of hER� and other nuclear receptors is re-quired for ligand-dependent activation of hER�through classical DNA-dependent pathways, and thisactivation process involves recruitment of AF2-inter-acting coactivators (7–13). NR box (LXXLL) motifs inSRCs and other coactivators specifically interact withhelix 12 of hER�. D538N, E542Q, and D545N muta-tions in helix 12 give hER�TAF1 abrogate interactionswith most AF2-interacting coactivators and decreasetransactivation from ERE promoters (38–41). MaximalER�/AP1 activation by E2 requires intact activationsurfaces of both AF1 and AF2, and AF2-dependent responses require helix 12 and the corre-sponding NR box interacting sites. Moreover, the AF1domain of ER� inhibits antiestrogen-induced ER�/AP1action (21). In contrast, both estrogens and antiestro-
gens activated hER� and hER�TAF1/Sp1, and over-expression of the NR box peptide 2XF6 (38) derivedfrom SRC-2 did not affect activation of hER�/Sp1 butinhibited ER� on an ERE promoter (Fig. 5B). Theseresults imply that regions of AF2 that interact withcoactivators through their NR boxes are not necessaryfor hER�/Sp1 action, and this is supported by studiesshowing that prototypical AF2-interacting SRCs didnot enhance hER�/Sp1-mediated transactivation inbreast cancer cells transfected with pSp13 (Fig. 5).Interestingly, p68, an AF1-interacting protein, also didnot enhance hER�/Sp1 activation of a GC-rich pro-moter in breast cancer cells, indicating that AF1-dependent p68 coactivation of hER� previously ob-served in COS-1 and HeLa cells transfected with anERE promoter is also dependent on cell context (43).Results obtained with hER�TAF1, the SRCs, and pep-tide competition experiments clearly define that somemechanistic differences between hormone-dependentactivation of hER�/Sp1 and hER� are due, in part, tohelix 12 of hER�.
We further investigated other regions within the DEFdomains required for ligand-dependent activation ofhER�/Sp1 (Fig. 6). The antiestrogens 4-OHT and ICI182,780 activated hER�/Sp1 in MCF-7 cells trans-fected with hER�(�271–300), a hinge region deletionmutant (Fig. 6A), whereas E2 was inactive. In contrast,E2 activated an ERE promoter in MDA-MB-231 cellstransfected with hER�(�271–300) (Fig. 6B), and thiswas consistent with previous reports showing that thehinge region was not required for activation of ERE-dependent constructs (42). Deletion of the hinge re-gion and helix 1 [i.e. hER�(�265–330)] resulted in lossof E2 and antiestrogen activation of hER� and hER�/Sp1 (Fig. 6, A and B), and the importance of helix 1within the E domain for activation of ERE-dependentpromoters has previously been reported (42). Pissiosand co-workers (59) recently showed that E2, 4-OHT,and ICI 182,780 induced interactions of a helix 1-GAL4chimeric protein with the ligand binding domain (LBD)or ER� in a mammalian two-hybrid assay. Thus, helix1 may stabilize ligand interactions with the LBD, andthis process may be functional for both DNA-depen-dent and -independent mechanisms of ER� action.However, the importance of helices 1 and 2 as inter-acting domains for other nuclear factors has not beendetermined.
Activation of hER�/Sp1 by estrogens was also de-pendent on the C-terminal region of hER� (aa 538–595), which encompassed part of helix 12 within the Edomain (aa 538–553) and the F domain (aa 554–595),which potentially contains helix 13 and �-strand motifsbased on secondary structure calculations (42, 58).Helix 12 is required for E2-dependent activation ofER� in cells transfected with an ERE promoter (42)(Fig. 6B), and similar results were observed for activa-tion of hER�/Sp1 by E2 (Fig. 6A). In contrast, both4-OHT and ICI 182,780 activated hER�(�538–595)/Sp1, and this result coupled with antiestrogen activa-tion of hER�TAF1/Sp1 confirms that helix 12 is not
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 813

required for this induction response by antiestrogens.The failure of E2 to activate hER�(�538–595)/Sp1was not due to the requirement for helix 12 becauseactivation was also not observed in cells transfectedwith hER�(�554–595) (i.e. F domain deletion) orhER�(�579–595) in which only the C-terminal �-strandregion of the F domain has been deleted. F domaindeletions can modulate activation of ERE promotersby antiestrogens but have minimal effects on E2-mediated transactivation (45). However, the results inFig. 6A clearly demonstrate that F domain aa 579–595are required for activation of hER�/Sp1 by E2. Thiswas also confirmed in selective NR box (2XF6) and Fdomain (aa 575–595) peptide competition studies,which demonstrate preferential inhibition of hER�/Sp1action in cells transfected with the F� expression plas-mid (Fig. 6C). These results demonstrate that E2- andSERM-mediated activation of hER�/Sp1 in breastcancer cells is complex and dependent on multipleoverlapping and distinct regions of hER� (Fig. 7).These domains of hER� may impart unique structuralfeatures required for hER�/Sp1 action and may alsoserve as binding sites for essential interacting nuclearcoregulatory proteins. Decreased hER�/Sp1-medi-ated transactivation in cells transfected with the Fdomain peptide (Fig. 6B) suggests that this region ofhER� may interact with other nuclear coregulatoryproteins, and current studies are focused on identify-ing F domain-interacting factors and their function inbreast cancer cells.
MATERIALS AND METHODS
Chemicals and Biochemicals
DMEM nutrient mixture F-12 (DME/F12) without phenol red,PBS, E2, 4-OHT, BSA (Fraction V), and 100� antibiotic/antimycotic solution were purchased from Sigma (St Louis,MO). Fetal bovine serum (FBS) was obtained from JRH Bio-sciences (Lenexa, KS). ICI 182,780 was kindly provided byDr. Alan Wakeling (Astra USA, Inc.-Zeneca Pharmaceuticals,Macclesfield, UK). [�-32P]ATP (3000 Ci/mmol) was purchasedfrom NEN Life Science Products (Boston, MA). Polydeoxy-(inosinic-cytidylic)acid, and T4-polynucleotide kinase werepurchased from Roche Molecular Biochemicals (Indianapo-lis, IN). All the restriction enzymes and modifying enzymes (T4DNA ligase, calf intestinal alkaline phosphatase) used in thisstudy were purchased from Promega Corp. (Madison, WI) orRoche Molecular Biochemicals. Plasmid preparation kitswere purchased from QIAGEN (Valencia, CA), and 40% poly-
acrylamide was obtained from National Diagnostics (Atlanta,GA). All other chemicals were obtained from commercialsources at the highest quality available.
Cell Maintenance and Transient Transfection Assay
MCF-7, CHO, and MDA-MB-231 cells were obtained fromthe American Type Culture Collection (ATCC, Manassas, VA).MCF-7, MDA-MB-231, and CHO cells were grown in DME/F12 (Sigma) supplemented with 2.2 g/liter sodium bicarbon-ate, 5% FBS, BSA (Sigma), and 10 ml/liter antibiotic/antimy-cotic solution (Sigma). Cells were cultured and maintained in150-cm2 tissue culture dishes in a 37 C in 5% CO2-95% air.For transient transfection assays, cells were seeded ontosix-well tissue culture plates in DME/F12 without phenol redsupplemented with 2.2 g/liter sodium bicarbonate, 5% dex-tran-coated charcoal-stripped FBS, BSA, and 10 ml/literantibiotic/antimycotic solution (Sigma). After 24 h, cells weretransfected with the calcium phosphate method with 2 �g ofluciferase reporter construct (pSp13, pERE3, pADA, andpRAR�1), 250 ng pcDNA3/His/lacZ (Invitrogen, Carlsbad,CA) as a standard reference for transfection efficiency, and 1�g or 100 ng (for cotransfection with pERE3) of the appro-priate ER expression plasmid. In studies where variableamounts of coactivators were also used, the amount of DNAtransfected was kept at a constant value by adding sufficientamount of empty vector. After 5–6 h, the media were re-moved and cells were shocked with 20% glycerol in PBS (pH7.4) for 1 min. Cells were rinsed twice with 1 ml of PBS andtreated with 5% charcoal-stripped DME/F12 either contain-ing DMSO, E2 (10 nM), 4-OHT (1 �M), or ICI 182,780 (1 �M) for36–40 h. After harvesting cells by scraping in 1� reporterlysis buffer (Promega Corp.), luciferase activity of aliquots ofthis extract were determined using the luciferase assay sys-tem (Promega Corp.). �-Galactosidase activity was per-formed using Tropix Galacto-Light Plus assay system(Tropix, Bedford, MA). Light emission was detected on alumicount micro-well plate reader (Packard Instruments, Me-riden, CT), and luciferase reporter gene activity was correctedby normalizing against �-galactosidase activity obtainedfrom the same sample. Results are expressed as means � SD
with at least three determinations for each treatment group.
Oligonucleotides and Plasmids
hER� expression plasmid was kindly provided by Dr. Ming-jerTsai (Baylor College of Medicine, Houston, TX); ER-null andhER�TAF1 and 2XF6 NR box peptide (fused to the yeastGAL4 DBD) were obtained from Dr. D. McDonnell (DukeUniversity, Durham, NC). The hER deletion constructshER11C, hER�(�265–330) (HE15), and hER�(�271–300)(HE12) were originally obtained from Dr. Pierre Chambon(Institut de Genetique et de Biologie Moleculaire et Cellulaire,Illkirch, France). MORs were generously provided by Dr. Mal-com G. Parker (Imperial Cancer Research Fund, London,UK), and hER� was supplied by Dr. J. A. Gustafsson (Karo-linska Institute, Huddinge, Sweden). Our experiments werecarried out using a shorter variant form of ER�; however, inpreliminary experiments using a longer form of ER� (providedby Dr. S. Mosselman, N.V. Organon, Oss, The Netherlands),minimal induction of ER�/Sp1 was also observed. SRC-1,SRC-2 (glucocorticoid receptor interacting protein 1), SRC-3(AIB1), and p68 RNA helicase were graciously provided byDrs. B. O’Malley (Baylor College of Medicine), M. R. Stallcup(University of Southern California, Los Angeles, CA), P. Melt-zer (National Cancer Institute, Bethesda, MD), and S. Kato(University of Tokyo, Tokyo, Japan), respectively. The hERcDNAs and the MOR cDNA were inserted into vectorspcDNA3 or pcDNA3.1/His C (Invitrogen, Carlsbad, CA) in thislaboratory for in vitro translation and for expression in mam-malian cells in transient transfection assays. For gel mobilityshift assays, a consensus estrogen response element (ERE);
Fig. 7. Summary of Domains of hER� Required for Activa-tion of hER�/Sp1 by E2 and Antiestrogens 4-OHT and ICI182,780.
814 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action

5�-GTC CAA AGT CAG GTC ACA GTG ACC TGA TCA AAGTT-3� (sense) was used and obtained from the Gene Tech-nologies Laboratories, Texas A&M University (College Sta-tion, TX). The DNA oligonucleotides used for construction ofplasmids were also synthesized by the Gene TechnologiesLaboratories (Texas A&M University). pXP1 luciferase re-porter construct was obtained from ATCC, and the minimalTATA sequences were inserted into pXP1 in this laboratory.The following promoter sequences were cloned into HindIIIand BamHI sites of pXP1 TATA-luciferase reporter construct:three consensus GC-rich Sp1 binding sites for pSp13 (5�-GCT TAT TCG ATC G)GG GCG GGG CGA GCA TTC GATCGG GGC GGG GCG AGC ATT GAT CGG GGC GGG GGCGAG CG-3� (sense)]; and three consensus EREs for pERE3(5�-AGC TTT CCG GAT CTA)GGT CAC TGT GAC CCG GGATCC TAG GTC ACT GTG ACC CGG GAT CCT AGG TCA CTGTGA CCT GAT CAA AGT G-3� (sense)]. The GC-rich genomicpromoter sequence from RAR�1 gene (pRAR�1; �79 to �49)(5�-AGC TTG ATT GGT CGG T)GG GCG GGC AGG GGCGGG CCT-3� (sense)] and the GC-rich genomic promotersequence from the pADA gene (�86 to �65) (5�-AGC TTGGCG AGA G)GG CGG GCC CCG GGA-3� (sense)] were alsocloned into HindIII and BamHI sites of pXP1 luciferase re-porter construct. The GC-rich and ERE motifs are underlined.
Generation of ER Deletion Mutant Constructs
ER DBD deletion constructs were prepared by site-directedmutagenesis by overlap extension using the PCR as previ-ously described (60). For example, hER��ZF1 in pcDNA3was constructed by carrying out the following procedures.One set of primers (A1/B1) from the HindIII site (A1) in themultiple cloning site in pcDNA3 to the site before the firstamino acid in the region to be deleted was amplified by PCR;the latter primer (B1) has an overlapping region of approxi-mately 15 to 20 bp that starts at the next amino acid in thedeletion construct. In addition, another set of primers begin-ning just after the last amino acid to be deleted to the HindIIIsite in hER� cDNA sequence were also used and amplified byPCR. This second set of primers (A2/B2) contained a 15- to20-bp overlapping region complementary to the last 15- to20-bp DNA sequence in the first PCR product. Both PCRreaction products have their own regions of overlap, andthese were coincubated to anneal the overlapping regions;this was followed by PCR amplification with the primer (A1)that starts at the multiple cloning site and the primer (B2) thatstarts at the hER� cDNA sequence. The resulting PCR prod-uct containing the desired deletion and a unique restrictionsite at both ends (HindIII) was purified, digested with HindIII,and finally cloned into pcDNA3 construct to give the appro-priate expression plasmid for electrophoretic mobility shiftand transient transfection assays.
The primers used for the mutagenesis assays are summa-rized in Table 1. hER��ZF2 in pcDNA3 was also generated byusing a unique HindIII restriction site for cloning into thisvector. Generation of hER��ZF1 and hER��ZF2 used theunique NheI and EcoRI sites of previously modified hER� inpcDNA3.1 (35). MOR cDNA was inserted into EcoRI site ofpMT2 mammalian expression vector that contains uniqueNotI and XbaI sites in MOR cDNA sequence suitable forcloning the PCR-amplified insert containing deletion of onezinc finger domain, into pMT2. The EcoRI fragment contain-ing the desired deletion from pMT2 MOR vector was clonedinto EcoRI site of pcDNA3.1 for in vitro translation. ThehER�TAF1 construct (in pcDNA3) has three point mutations(D538N, E542Q, and D545N) in AF2 (40, 41). hER�TAF1�ZF1and hER�TAF1�ZF2 constructs were created by cloning theXbaI fragment (� 0.7 kb) from hER�TAF1 (in pcDNA3) intopcDNA3.1, and zinc finger mutants were prepared as de-scribed above. hER�(�538–595) was made by PCR amplifi-cation of wild-type hER� using primer sets: (F1) 5�-TGC TAGCAT GAC CAT GAC CCT CCA CAC C-3� and (R1) 5�-GACTCG AGT CAA GTG GGC GCA TGT AGG CGG TG-3�.
hER�(�554–595) and hER�(�579–595) were also amplifiedwith the same F1 primer above and (R2) 5�-GAC TCG AGTCAA GTG GGC GCA TGT AGG CGG TG-3� or (R3) 5�-TACTCG AGT CAC AAG GAA TGC GAT GAA GTA GAG-3�, re-spectively. The amplified fragments were digested and in-serted into NheI and XhoI sites of pcDNA3.0 or 3.1 expres-sion vector. GAL4-DBD fusion F� peptide [21 amino acidsfrom C-terminal end of hER� (aa 575–595)] was made byusing BamHI and HindIII sites within the multiple cloningregion of pM vector (CLONTECH Laboratories, Inc., PaloAlto, CA) with (F) 5�-GAT CCG TTCA TCG CAT TCC TTG CAAAAG TAT TAC ATC ACG GGG GAG GCA GAG GGT TTC CCTGCC ACA GTC TGA A-3� and (R) 5�-AGC TTT CAG ACT GTGGCA GGA AAC CCT CTG CCT CCC CCG TGA TGT AAT ACTTTT GCA AGG AAT GCG ATG AAC G-3�. All constructs weremapped by restriction enzymes and sequenced to confirmthat proper deletion or insertions were introduced into thetarget cDNA.
In Vitro Translation and Detection of theTranslated Proteins
Wild-type ER and ER deletion mutants were synthesized invitro using TNT T7 quick coupled transcription/translationSystem (Promega Corp.) in the presence or absence of[35S]methionine for EMSAs and separation by 10%SDS-PAGE.
EMSAs
The consensus [32P]ERE oligonucleotide was annealed andend labeled using T4-polynucleotide kinase and [�-32P]ATP.To characterize DNA binding of wild-type ER and corre-sponding zinc finger deletion mutants, 0.5 �l of in vitro trans-lated protein or 0.5 �l of unprogrammed lysate was incu-bated in 1� binding buffer (25% glycerol, 0.5 mM EDTA, 0.5mM dithiothreitol, 50 mM potassium chloride, 10 mM HEPESat pH 8.0) for 5 min at 4 C. Radiolabeled consensus EREoligonucleotide (60,000 cpm) was added to the reaction, andthe reaction mixture was incubated at 25 C for 15 min.Samples were then applied to the gel and separated bypolyacrylamide gel electrophoresis at 120 V in 0.9 mM Tris,0.9 M borate, 2 mM EDTA (pH 8.0) for 2–3 h. Protein-DNAcomplexes were visualized by autoradiography using X-Omatfilm (Eastman Kodak Co., Rochester, NY).
Fluorescence Immunocytochemistry
MDA-MB-231 cells were subcultured in four-well Lab-Tekchambered slides (Nunc Inc., Naperville, IL) in DME/F12 me-dium without phenol red 5% FBS stripped with dextran-coated charcoal. After 24 h, cells were transiently transfectedwith 500 ng of hER� or hER� mutant expression plasmids.For transient transfection studies, cells were incubated withFuGENE Transfection Reagent (Roche) at 37 C for 5 h, fol-lowed by 24 h of recovery in DME/F12. Before fixation, slideswere washed three times in Dulbecco’s PBS (DPBS) and thenfixed for 10 min at �20 C at 100% methanol. For nuclearlocalization of ER, the rat monoclonal antibody raised againstthe N-terminal domain of the hER� (H184; Santa Cruz Bio-technology, Inc., Santa Cruz, CA) was diluted to a final con-centration of 3 �g/ml in DPBS containing 0.5% BSA, 0.1%goat serum, and 0.3% Tween 20. Rat IgG at the same con-centration was used as a control. Tween 20 (0.3%) wasincluded in all antibody, blocking steps, and washes for nu-clear localization of ER. Followed by incubation with H184antibody for 16 h, cells were washed with DPBS (three times),then incubated for 1 h in a 1:200 dilution of fluoresceinisothiocyanate-conjugated goat-antirat IgG (62–9511; ZymedLaboratories, Inc., South San Francisco, CA) in DPBS con-taining 0.1% goat serum. Cells were then washed (four times)
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 815

over a period of 2 h and transferred to DPBS before coverslipmounting with ProLong Antifade mounting reagent (Molecu-lar Probes, Inc., Eugene, OR). For each treatment, represen-tative fluorescence images were recorded using an Axioplanmicroscope (Carl Zeiss, Thornwood, NY) equipped with aHamamatsu chilled three charge-coupled device color cam-era (Hamamatsu, Japan) using Adobe Photoshop 5.0 (AdobeSystems, Seattle, WA) image capture software. Images fromall treatment groups were captured at the same time usingidentical image capture parameters. Slides were subse-quently washed three times in DPBS followed by a 1-h block-ing step in 3% normal goat serum (G-9023; Sigma).
Statistics
For transient transfection studies, results are expressed asmeans � SD for at least three separate experiments for eachtreatment group. Statistical differences (P � 0.05) betweencontrol (DMSO) and treatment groups or between E2-induced responses and treatment groups (coactivator exper-iments) were determined by ANOVA and Scheffe’s post hoctest.
Acknowledgments
Received December 5, 2002. Accepted January 27, 2003.Address all correspondence and requests for reprints to:
Stephen H. Safe, Department of Veterinary Physiology &Pharmacology, Texas A&M University, 4466 TAMU, CollegeStation, Texas 77843-4466. E-mail: [email protected].
This work was supported in part by NIH Grant ES-09106and the Texas Agricultural Experiment Station. S.S. is a SidKyle Professor of Toxicology.
REFERENCES
1. Tsai MJ, O’Malley BW 1994 Molecular mechanisms ofaction of steroid/thyroid receptor superfamily members.Annu Rev Biochem 63:451–486
2. Beato M, Herrlich P, Schutz G 1995 Steroid hormonereceptors: many actors in search of a plot. Cell 83:851–857
3. Mangelsdorf DJ, Thummel C, Beato M, Herrlich P,Schutz G, Umesono K, Blumberg B, Kastner P, Mark M,Chambon P, Evans RM 1995 The nuclear receptorsuperfamily: the second decade. Cell 83:835–839
4. Enmark E, Gustafsson JA 1996 Orphan nuclear recep-tors—the first eight years. Mol Endocrinol 10:1293–1307
5. Perlmann T, Evans RM 1997 Nuclear receptors in Sicily:all in the famiglia. Cancer Res Cell 90:391–397
6. Kliewer SA, Lehmann JM, Willson TM 1999 Orphan nu-clear receptors: shifting endocrinology into reverse. Sci-ence 284:757–760
7. Horwitz KB, Jackson TA, Bain DL, Richer JK, TakimotoGS, Tung L 1996 Nuclear receptor coactivators core-pressors. Mol Endocrinol 10:1167–1177
8. Glass CK, Rose DW, Rosenfeld MG 1997 Nuclear recep-tor coactivators. Curr Opin Cell Biol 9:222–232
9. Edwards DP 1999 Coregulatory proteins in nuclear hor-mone receptor action. Vitam Horm 55:165–218
10. McKenna NJ, Xu J, Nawaz Z, Tsai SY, Tsai MJ, O’MalleyBW 1999 Nuclear receptor coactivators: multiple en-zymes, multiple complexes, multiple functions. J SteroidBiochem Mol Biol 69:3–12
11. Robyr D, Wolffe AP, Wahli W 2000 Nuclear hormonereceptor coregulators in action: diversity for sharedtasks. Mol Endocrinol 14:329–347
12. Lemon BD, Freedman LP 1999 Nuclear receptor cofac-tors as chromatin remodelers. Curr Opin Genet Dev9:499–504
13. Klinge CM 2000 Estrogen receptor interactions with co-activators and corepressors. Steroids 65:227–251
14. Chen JD 2000 Steroid/nuclear receptor coactivators. Vi-tam Horm 58:391–448
15. Katzenellenbogen JA, Katzenellenbogen BS 1996 Nu-clear hormone receptors: ligand-activated regulators oftranscription and diverse cell responses. Chem Biol3:529–536
16. Cowley SM, Hoare S, Mosselman S, Parker MG 1997Estrogen receptors � and � form heterodimers on DNA.J Biol Chem 272:19858–19862
17. Barkhem T, Carlsson B, Nilsson Y, Enmark E, GustafssonJ, Nilsson S 1998 Differential response of estrogen re-ceptor � and estrogen receptor � to partial estrogenagonists/antagonists. Mol Pharmacol 54:105–112
18. Hyder SM, Chiappetta C, Stancel GM 1999 Interaction ofhuman estrogen receptors � and � with the same natu-rally occurring estrogen response elements. BiochemPharmacol 57:597–601
19. Hall JM, McDonnell DP 1999 The estrogen receptor�-isoform (ER�) of the human estrogen receptor modu-lates ER� transcriptional activity and is a key regulator ofthe cellular response to estrogens and antiestrogens.Endocrinology 140:5566–5578
20. Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J,Kushner PJ, Scanlan TS 1997 Differential ligand activa-tion of estrogen receptors ER� and ER� at AP1 sites.Science 277:1508–1510
21. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR,McInerney E, Katzenellenbogen BS, Enmark E, Gustafs-son J-Å, Nilsson S, Kushner PJ 1999 The estrogen re-ceptor enhances AP-1 activity by two distinct mecha-nisms with different requirements for receptortransactivation functions. Mol Endocrinol 13:1672–1685
22. Webb P, Lopez GN, Uht RM, Kushner PJ 1995 Tamox-ifen activation of the estrogen receptor/AP-1 pathway:potential origin for the cell-specific estrogen-like effectsof antiestrogens. Mol Endocrinol 9:443–456
23. Uht RM, Anderson CM, Webb P, Kushner PJ 1997 Tran-scriptional activities of estrogen and glucocorticoid re-ceptors are functionally integrated at the AP-1 responseelement. Endocrinology 138:2900–2908
24. Weatherman RV, Scanlan TS 2001 Unique protein deter-minants of the subtype-selective ligand responses of theestrogen receptors (ER� and ER�) at AP-2 sites. J BiolChem 276:3827–3832
25. Jakacka M, Ito M, Weiss J, Chien P-Y, Gehm BD, Jame-son JL 2001 Estrogen receptor binding to DNA is notrequired for its activity through the nonclassical AP1pathway. J Biol Chem 276:13615–13621
26. Porter W, Saville B, Hoivik D, Safe S 1997 Functionalsynergy between the transcription factor Sp1 and theestrogen receptor. Mol Endocrinol 11:1569–1580
27. Wang F, Hoivik D, Pollenz R, Safe S 1998 Functional andphysical interactions between the estrogen receptor-Sp1and the nuclear aryl hydrocarbon receptor complexes.Nucleic Acids Res 26:3044–3052
28. Wang W, Dong L, Saville B, Safe S 1999 Transcriptionalactivation of E2F1 gene expression by 17�-estradiol inMCF-7 cells is regulated by NF-Y-Sp1/estrogen receptorinteractions. Mol Endocrinol 13:1373–1387
29. Xie W, Duan R, Safe S 1999 Estrogen induces adenosinedeaminase gene expression in MCF-7 human breastcancer cells: role of estrogen receptor-Sp1 interactions.Endocrinology 140:219–227
30. Sun G, Porter W, Safe S 1998 Estrogen-induced retinoicacid receptor �1 gene expression: role of estrogenreceptor-Sp1 complex. Mol Endocrinol 12:882–890
31. Qin C, Singh P, Safe S 1999 Transcriptional activation ofinsulin-like growth factor binding protein 4 by 17�-estra-diol in MCF-7 cells: role of estrogen receptor-Sp1 com-plexes. Endocrinology 140:2501–2508
816 Mol Endocrinol, May 2003, 17(5):804–817 Kim et al. • Mechanisms of ER�/Sp1 Action

32. Duan R, Porter W, Safe S 1998 Estrogen-induced c-fosprotooncogene expression in MCF-7 human breast can-cer cells: role of estrogen receptor Sp1 complex forma-tion. Endocrinology 139:1981–1990
33. Dong L, Wang W, Wang F, Stoner M, Reed JC, Harigai M,Kladde M, Vyhlidal C, Safe S 1999 Mechanisms of tran-scriptional activation of bcl-2 gene expression by 17�-estradiol in breast cancer cells. J Biol Chem 174:32099–32107
34. Samudio I, Vyhlidal C, Wang F, Stoner M, Chen I, KladdeM, Barhoumi R, Burghardt R, Safe S 2001 Transcriptionalactivation of DNA polymerase � gene expression inMCF-7 cells by 17�-estradiol. Endocrinology 142:1000–1008
35. Saville B, Wormke M, Wang F, Nguyen T, Enmark E,Kuiper G, Gustafsson J-A, Safe S 2000 Ligand-, cell- andestrogen receptor subtype (�/�)-dependent activation atGC-rich (Sp1) promoter elements. J Biol Chem 275:5379–5387
36. Xie W, Duan R, Chen I, Samudio I, Safe S 2000 Tran-scriptional activation of thymidylate synthase by 17�-estradiol in MCF-7 human breast cancer cells. Endocri-nology 141:2439–2449
37. Duan R, Porter W, Samudio I, Vyhlidal C, Kladde M, SafeS 1999 Transcriptional activation of c-fos protooncogeneby 17�-estradiol: mechanism of aryl hydrocarbon recep-tor-mediated inhibition. Mol Endocrinol 13:1511–1521
38. Chang C, Norris JD, Gron H, Paige LA, Hamilton PT,Kenan DJ, Fowlkes D, McDonnell DP 1999 Dissection ofthe LXXLL nuclear receptor-coactivator interaction motifusing combinatorial peptide libraries: discovery of pep-tide antagonists of estrogen receptors � and �. Mol CellBiol 19:8226–8239
39. Schaufele F, Chang CY, Liu WQ, Baxter JD, Nordeen SK,Wan YH, Day RN, McDonnell DP 2000 Temporally dis-tinct and ligand-specific recruitment of nuclear receptor-interacting peptides and cofactors to subnuclear do-mains containing the estrogen receptor. Mol Endocrinol14:2024–2039
40. Tzukerman MT, Esty A, Santiso-Mere D, Danielian P,Parker MG, Stein RG, Pike JW, McDonnell DP 1994Human estrogen receptor transactivational capacity isdetermined by both cellular and promoter context andmediated by two functionally distinct intramolecular re-gions. Mol Endocrinol 8:21–30
41. McDonnell DP, Clemm DL, Hermann T, Goldman ME,Pike JW 1995 Analysis of estrogen receptor function invitro reveals three distinct classes of antiestrogens. MolEndocrinol 9:659–669
42. Kumar V, Green S, Stack G, Berry M, Jin J-R, ChambonP 1987 Functional domains of the human estrogen re-ceptor. Cell 51:941–951
43. Endoh H, Maruyama K, Masuhiro Y, Kobayashi Y, GotoM, Tai H, Yanagisawa J, Metzger D, Hashimoto S, KatoS 1999 Purification and identification of p68 RNA heli-case acting as a transcriptional coactivator specific forthe activation function 1 of human estrogen receptor �.Mol Cell Biol 19:5363–5372
44. Schwartz JA, Zhong L, Deighton-Collins S, Zhao C, Ska-far DF 2002 Mutations targeted to a predicted helix in theextreme carboxyl-terminal region of the human estrogen
receptor-� alter its response to estradiol and 4-hydroxytamoxifen. J Biol Chem 277:13202–13209
45. Montano MM, Muller V, Trobaugh A, KatzenellenbogenBS 1995 The carboxy-terminal F domain of the humanestrogen receptor: role in the transcriptional activity ofthe receptor and the effectiveness of antiestrogens asestrogen antagonists. Mol Endocrinol 9:814–825
46. Nichols M, Rientjes JM, Stewart AF 1998 Different posi-tioning of the ligand-binding domain helix 12 and the Fdomain of the estrogen receptor accounts for functionaldifferences between agonists and antagonists. EMBO J17:765–773
47. Peters GA, Khan SA 1999 Estrogen receptor domains Eand F: role in dimerization and interaction with coactiva-tor RIP-140. Mol Endocrinol 13:286–296
48. Koide A, Abbatiello S, Rothgery L, Koide S 2002 Probingprotein conformational changes in living cells by usingdesigner binding proteins: application to the estrogenreceptor. Proc Natl Acad Sci USA 99:1253–1258
49. McDonnell DP 1999 The molecular pharmacology ofSERMs. Trends Endocrinol Metab 10:301–311
50. Smith CL, O’Malley BW 1999 Evolving concepts of se-lective estrogen receptor action: from basic science toclinical applications. Trends Endocrinol Metab 10:299–300
51. Jordan VC 1999 Targeted antiestrogens to preventbreast cancer. Trends Endocrinol Metab 10:312–317
52. Fuqua SA, Russo J, Shackney SE, Stearns ME 2000Estrogen, estrogen receptors and selective estrogen re-ceptor modulators in human breast cancer. J Women’sCancer 2:21–32
53. Krishnan V, Heath H, Bryant HU 2001 Mechanism ofaction of estrogens and selective estrogen receptormodulators. Vitam Horm 60:123–147
54. Porter W, Wang F, Duan R, Qin C, Castro-Rivera E, SafeS 2001 Transcriptional activation of heat shock protein27 gene expression by 17�-estradiol and modulation byantiestrogens and aryl hydrocarbon receptor agonists:estrogenic activity of ICI 164,384. J Mol Endocrinol 26:31–42
55. Schwabe JW, Neuhaus D, Rhodes D 1990 Solutionstructure of the DNA-binding domain of the oestrogenreceptor. Nature 348:458–461
56. Moilanen AM, Poukka H, Karvonen U, Hakli, Janne OA,Palvimo JJ 1998 Identification of a novel RING fingerprotein as a coregulator in steroid receptor-mediatedgene transcription. Mol Cell Biol 18:5128–5139
57. Saville B, Poukka H, Wormke M, Janne OA, Palvimo JJ,Stoner M, Samudio I, Safe S 2002 Cooperative coacti-vation of estrogen receptor � in ZR-75 human breastcancer cells by SNURF and TATA-binding protein. J BiolChem 277:2485–2497
58. Ko L, Cardona GRH, Chin WW 2002 Identification andcharacterization of a tissue-specific coactivator, GT198,that interacts with the DNA-binding domains of nuclearreceptors. Mol Cell Biol 22:357–369
59. Pissios P, Tzameli I, Kushner P, Moore DD 2000 Dynamicstabilization of nuclear receptor ligand binding domainsby hormone or corepressor binding. Mol Cell 6:245–253
60. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR 1989Site-directed mutagenesis by overlap extension usingthe polymerase chain reaction. Gene 77:51–59
Kim et al. • Mechanisms of ER�/Sp1 Action Mol Endocrinol, May 2003, 17(5):804–817 817





![[sp1] oocyte collection from superstimulated disease-free ...](https://static.fdokumen.com/doc/165x107/631dd3361aedb9cd850f788f/sp1-oocyte-collection-from-superstimulated-disease-free-.jpg)




![Saugu[!] Asmundar, er kalladur er Kappabani :](https://static.fdokumen.com/doc/165x107/63264a17051fac18490dae0e/saugu-asmundar-er-kalladur-er-kappabani-.jpg)









