
DOCTOR AT THFS1S Aspects of Drinking Water Supply in ...
119
LULEA I UNIVERSITY »JL^ t OF TECHNOLOGY 2002:27 DOCTOR AT THFS1S Aspects of Drinking Water Supply i n Areas o f Humic Water TALIS JUHNA Department of Environmental Engineering Division of Sanitary Engineering 2002:27 • ISSN: 1402 - 1544 • 1SRN: LTU - D T - - 02/27 - - SE
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of DOCTOR AT THFS1S Aspects of Drinking Water Supply in ...

LULEA I UNIVERSITY » J L ^ t
OF TECHNOLOGY
2 0 0 2 : 2 7
D O C T O R AT THFS1S
Aspects of Drinking Water
Supply in Areas of Humic Water
TALIS JUHNA
Department o f Environmental Engineering
Division o f Sanitary Engineering
2002:27 • ISSN: 1402 - 1544 • 1SRN: L T U - D T - - 02/27 - - SE

Aspects of Drinking Water Supply in Areas of Humic Water
T a l i s J u h n a
Department of Environmental Engineering Division of Sanitary Engineering Luleå University of Technology
SE-971 87 Luleå, Sweden
Akademisk avhandling som med vederbörligt tillstånd från Tekniska Fakultetsnämnden vid Luleå tekniska universitet för avläggande av teknisk doktorsexamen kommer att offentligt försvaras vid Luleå tekniska universitet i sal D 531, fredagen den 13 september 2002, kl . 10.00. Fakultetsopponent: Professor Torsten Hedberg, Chalmers tekniska högskola. Ordförande: Professor Jörgen Hanaeus, Luleå tekniska universitet.
Doctoral Thesis 2002:27 ISSN: 1402:1544 ISRN: LTU-DT-02/27-SE

A S P E C T S OF DRINKING WATER S U P P L Y IN A R E A S OF HUMIC WATER
Talis Juhna
Division of Sanitary Engineering
Department of Environmental Engineering
Luleå University of Technology SE-971 87 Luleå
Sweden
2002
Academic dissertation

Preface
Drinking water supply from naturally coloured waters, sometimes called humic waters, has often been associated with aesthetic problems and high chlorine demand during water
disinfection. Growing evidence suggests that, due to high amounts of natural organic matter, humic waters may also produce cancer-causing products and increase hygienic risks to consumers. Development of new methods for the removal of natural organic matter, including the main constituent humic substances, has been a challenge for water treatment engineers
and scientists in many countries over the last decades. However, traditionally used water treatment methods should not be disregarded and their potential and limitation in removing organic matter has to be understood. This thesis studies the removal of natural organic matter
and its effects on bacteria growth during water distribution in a system supplied with humic
water treated by two traditional methods, namely artificial groundwater recharge and chemical precipitation.
As there is no singular composition of humic substances, their properties may vary f rom place to place. Hence, results obtained f rom a single humic water supply cannot always be applicable directly for another. Thus, any generalisation of the results presented in this thesis
should be done with precaution.
The thesis is a result of cooperation between the Div . of Sanitary Engineering at Luleå University of Technology in Sweden and the Div . o f Water Supply and Sewerage at Riga
Technical University in Latvia. In Latvia, experiments and f ie ld studies were carried out. whereas most of the academic and pedagogic work needed for obtaining the doctoral degree
was accomplished in Sweden during annual visits.
A Swedish doctoral thesis is produced either as a monograph or as a collection of papers. In
the latter case, the introductory part constitutes the formal thesis, which summarizes the accompanying papers. The papers have either already been published or are manuscripts at
various stages (in press, submitted or in ms). This thesis is based on six papers that wi l l be referred to by their roman numerals in the text o f t h e introductory part.

Abstract
Due to high amount of humic substances (HS) drinking water supply from naturally coloured waters (humic waters) is associated with aesthetic and hygienic problems. HS produce carcinogenic compounds during water disinfection. Humic waters may also contain microbially assimilable organic carbon (AOC). which together with other nutrients such as phosphorus and nitrogen, supports bacteria growth, thus promoting biofilm formation in distribution networks. This thesis investigates different aspects of HS and AOC removal during water treatment and its influence on bacteria growth during water distribution. The studies were carried out in a drinking water supply system that was fed with humic waters (from a lake and river) treated with traditional methods - chemical precipitation or artificial recharge of groundwater.
The important mechanism of HS removal during artificial recharge of groundwater - sorption on aquifer material - was studied in batch equilibrium experiments. The main sorption mechanisms and influences of several factors that determine sorption efficiency were evaluated. Sorption increased with an increase of clay content, a decrease of loosely bound organic matter in the aquifer material, or a decrease of pH of water. Larger molecular weight, more aromatic and hydrophobic fractions of HS (humic acids) were more efficiently sorbed than acidic fractions (fulvic acids). The result suggested that due to the low amount of binding sites in the material, physical sorption (mainly hydrophobic attraction) was more important than chemical sorption. The testing of different sorbents revealed that fractions of HS (mainly fulvic acids), which were not retained during artificial groundwater recharge, were effectively removed with an additional treatment by macroreticular weak anion exchange resin.
To investigate the effect of humic lake blooming on organic matter biodegradation during artificial recharge of groundwater, total bacterial numbers and the number of cyanobacteria were measured in the aquifer material below infiltration basins used for artificial recharge of groundwater. Analyses showed that cyanobacteria were present below the basins; the total number of bacteria was high and positively correlated with the cyanobacteria number, thus indirectly indicating that blooming did not have a significantly negative impact on the biodegradation of organic matter.
Bacterial growth potential (biological stability) of drinking water from the studied water supply system was investigated by measuring the concentration of AOC, microbially available phosphorus (MAP) (potentially limiting nutrient in humic waters), and the biofilms formation density. Chemical precipitation of humic water significantly removed MAP whereas reduction of AOC was moderate. As a result, phosphorus became a limiting nutrient for bacteria growth. However, MAP levels were sufficiently high to support bacterial biofilm formation in the distribution networks. Due to insufficient removal through the aquifer, the MAP concentration was higher in the recharged groundwater than in chemically treated surface water, and did not always limit bacteria growth. Concentrations of chlorine or phosphorus influenced biofilm formation in distribution net. Due to higher MAP levels in the groundwater bacteria formed a denser biofilm. The addition of chlorine in drinking water did not stop the biofilm formation unless high concentration of residual chorine was maintained.
In addition to a better understanding of water supply from humic water the thesis has supplied material regarding physicochemical forms (species) of iron in humic groundwater. By using a metal speciation method (based on macroreticular weak anion exchange resin), it was shown that nearly half of the iron in the recharged groundwater was bound to HS and that these fractions were even greater in groundwater affected by drainage of peat deposits or a swamp. This finding has important practical implications at the selection of iron removal methods from humic groundwater.

Sammanfattning
Höga halter humusämnen i dricksvatten är orsak till flera problem. Humusämnen kan bilda cancerogena ämnen vid desinfektion av vattnet. Humusrika vatten kan också innehålla mikrobiellt assimilerbart organiskt kol (AOC), vilket tillsammans med andra näringsämnen såsom fosfor och kväve gynnar bakterietillväxten som bidrar till biofilmtillväxt i distributionsnätet. Föreliggande avhandling undersöker olika aspekter på avlägsnande av humusämnen och AOC i vattenbehandlingen och därav följande påverkan på bakterietillväxten under distributionen. Studierna ägde rum på ett dricksvattensystem med humusrikt vatten (från en sjö och en flod) som behandlats med traditionella metoder - kemisk fällning eller artificiell grundvattenbildning.
Den viktiga mekanismen för avlägsnande av humusämnen vid produktion av artificiellt grundvatten -sorption på akvifermaterialet - studerades i satsvisa jämviktsexperiment. Påverkan av flera faktorer som bestämmer sorptionseffektiviteten och dess mekanismer utvärderades. Sorptionen ökade med ökat lerinnehåll, med en minskning av löst bundet organiskt material i akvifermaterialet och med en minskning av vattnets pH. Större molekylvikt, fler aromatiska och hydrofoba fraktioner av humusämnen (humussyror) absorberades effektivare än syra fraktioner (fulvosyror). Resultatet föreslår att pga de få bindningsmöjligheterna i materialet var (är) den fysiska Sorptionen (huvudsakligen hydrofob attraktion) viktigare än den kemiska. Försöken med de olika sorbenterna visade att den fraktion av humusämnen (huvudsakligen fulvosyror) som inte avskiljdes under infiltrationen avlägsnades effektivt vid behandling i svaga anjonbytare.
För att undersöka effekten av algblomning på nedbrytningen av organiskt material under konstgjord infiltration mättes totala antalet bakterier samt antalet cyanobakterier i akvifermaterialet under infiltrationsbassängerna för artificiellt infiltrerat grundvatten. Analyserna visade att det fanns cyanobakterier under bassängema. Det totala antalet bakterier var högt och positivt korrelerat med antalet cyanobakterier. således indirekt påvisande att algblomning inte har en signifikant negativ påverkan på biologisk nedbrytning av organiskt material.
Det studerade dricksvattnets bakterietillväxtpotential (biologisk stabilitet) undersöktes genom att mäta AOC-koncentrationen. mikrobiellt tillgängligt fosfor (MAP) (potentiellt begränsande näringsämne i humusrika vatten) och den bildade biofilmens densitet. Kemisk fällning av humusrika vatten avlägsnade MAP i stor utsträckning medan separationen av AOC var måttlig . Fosfor blev därigenom det begränsande ämnet för bakterietillväxt. Pga otillräcklig avskiljning genom akviferen var MAP-koncentrationen högre i det artificiella grundvattnet än i det kemiskt behandlade ytvattnet och begränsade inte alltid bakterietillväxten. Pga högre halter MAP i grundvattnet tillväxte bakterierna snabbare och bildade en tätare biofilm. Tillsatsen av låga klordoser i dricksvattnet stoppade inte biofilmtillväxten utan denna begränsades först då en hög klorresidual kunde hållas.
Förutom en bättre förståelse av vattenförsörjningen med humusrika vatten, har avhandlingen bidragit till en förståelse av järnets fysikalisk-kemiska förekomstformer i humusrika grundvatten. Genom användning av en metod för metallspeciering (baserad på svag anjonbytare) kunde visas att nästan hälften av järnet i det konstgjort infiltrerade grundvattnet var bundet t i l l humusämnen och att dessa fraktioner var ännu större i grundvatten som påverkats av dränering av torvfyndighet eller våtmark. Dessa observationer ger viktiga praktiska slutsatser vid valet av avjärningsmetod för humusrika grundvatten.

Kopsavilkums
Dzeramä üdens apgäde no üdens tilpnem, kuras satur augstu humlnvielu (HV) koncentraciju. ir saistTta ar vairäkäm Odens kvalitätes problemäm. Odeni hloréjot, HV veido kancerogenas vielas. Sädas üdens tilpnes var saturet mikrobiologiski viegli assimilejamu organisko oglekli (assimilable organic carbon, AOC). kas kopä ar citäm mikroorganismu barlbas vieläm, tädäm kä fosfors un släpeklis, veicina bakteriju vairosanos un biologisko apaugumu (biopleves) veidosanos dzeramä Odens apgädes sistémäs. Sajä disertäcijä petita dzeramä Odens attlrisana no HV un AOC, un täs ietekme uz bakteriju vairosanos un biopleves veidosanos dzeramä üdens sadales tlklä. Petljumi veikti Odens apgädes sistémä, kura iegüst üdeni no tilpnem ar augstu HV koncentraciju (upe un ezers), izmantojot tradicionälas Odens attTrTsanas metodes: ^Tmisko koaguläciju vai intensTvo gruntsüdens papildinäsanu.
Intensivas gruntsüdens papildinäsanas procesä Odens attlräs no HV, sim vieläm sorbejoties uz ieza materiäla. Laboratorijas apstäklos pétlti fizikäli-klmiskie faktori, kuri ietekme HV sorbciju. Konstatets. ka HV sorbcija pieaug palielinoties mäla Ipatsvaram. samazinoties nestabilo organisko vielu koncenträcijai iezl vai Odenim paskäbinoties. HV ar lielu molekuläru masu. aromätisku un hidrofobu struktüru (humlnskäbes) sorbejäs labäk kä HV ar augstu karbonskäbju koncentraciju (fulvoskäbes). Rezultäti parädlja. ka intensivas papildinäsanas procesä HV fizikälä sorbcija (galvenokärt. hidrofobä iedarblba) ir svarlgäka par kimisko sorbciju. Pärbaudot dazädus sorbentus konstatets. ka HV daju, kas nesorbejas intenslväs papildinäsanas procesä (galvenokärt, fulvoskäbes), no gruntsüdens var izdallt. izmantojot anjonu apmainas makroporainus svekus.
Analizejot kopejo bakteriju skaitu un zilajgu skaitu iezl zem infilträcijas baseina. petits, vai zilajgu ziedésana eitrofä ezerä bötiski ietekme organisko vielu biodegradäciju intenslväs papildinäsanas procesä. Konstatets, ka neskatoties uz zilajgu klätbutni. kopejais bakteriju skaits iezl bija liels un pozitlvi koreleja ar zilajgu koncenträciju. Tas netiesi norädlja. ka zilajgu ziedésana bütiski negatlvi neietekmeja organisko vielu noärdlsanos infilträcijas procesä.
Odens ir biologiski stabils, ja tajä nevairojäs bakterijas. Dzeramä Odens biologiska stabilitäte noteikta analizejot AOC. mikrobiologiski izmantojamä fosfora (microbially available phosphorus, MAP) koncenträcijas un biopleves veidosanäs ätrumu. Klmiskä koaguläcija ievérojami samazinäja MAP un nedaudz AOC koncenträciju. Tä rezultätä. nevis ogleklis. bet fosfors limiteja bakteriju vairosanos dzeramä udenl. Biopleves veidosanäs bija atkarlga no MAP vai hlora konceträcijas. Papildinätais gruntsüdens satureja augstäku MAP koncenträciju. täpec tajä biopléve veidojäs intenslväk nekä klmiski attlrltä upes OdenT. Bija nepieciesamas lielas hlora dozas lai pilnlgi apturetu biopleves veidosanos.
IegOts jauns materials par dzeramä Odens apgädi no üdens tilpnem ar augstu HV koncenträciju un par dzelzs atrasanäs formäm gruntsüdenl. Izmantojot anjonu apmainas (makroporains anjonits) metodi. konstatets, ka papildinätä gruntsüdenl apmeram puse no dzelzs ir saistlta kompleksä ar HV un sis Tpatsvars ir lieläks gruntsüdenos. kuri drene kudras laukus vai purvus. Metodi var izmantot izveloties gruntsüdenu atdzelzosanas tehnologijas.

C O N T E N T S
1. Abbreviations
2. List of publications
3. INTRODUCTION
4. A I M OF THE THESIS
5. B A C K G R O U N D
5.1. D O M in waters 5.2. Removal of D O M f rom surface water during artificial
recharge of groundwater 5.3. Removal of HS f rom artificially recharged groundwater using sorbents 5.4. Effect of B O M on drinking water during its distribution in networks..
6. STUDY SITE A N D E X P E R I M E N T A L METHODS 6.1. Description of study sites and water quality ' 4
6.2. Analyses of bacterial growth potential in water ' 8 6.3. Determination of HS concentrations and composition in water 20
6.4. Measuring b iof i lm formation 21 6.5. Determination of bacterial number in water and in b io f i lm 21 6.6. Sorption experiments with HS on aquifer material and sorbents 21 6.7. Determination of iron-humic complexes in humic groundwater 22
7. MAJOR RESULTS A N D DISCUSSION 7.1. Sorption of HS on aquifer material during artificial recharge of groundwater 23
7.2. Effect of blooming on the biodegradation of B O M during artificial recharge of humic water 25
7.3. Removal of HS f rom artificially recharges groundwater using sorbents 26
7.4. Bacterial growth potential in drinking waters prepared f rom humic surface water 26
7.5. B i o f i l m formation in drinking water distribution networks of humic waters 29
7.6. Iron species in humic groundwaters ™
8. CONCLUSIONS 3 2
9. Future research 3 3
10. Acknowledgements 34
11. List of references ^5

1. A B B R E V I A T I O N S
AOCnative assimilable organic carbon without addition of inorganic nutrients
AOC potential assimilable organic carbon with addition of inorganic nutrients ARG artificial recharge of groundwater BDOC biologically degradable organic carbon (measurement-specific) B O M biodegradable organic matter (measurement-unspecific) CFU bacterial colony forming unit
COD chemical oxygen demand CP chemical precipitation DEAE diethylaminoethyl groups
DOC dissolved organic carbon (< 0.45 p m by fil tration) D O M dissolved organic matter FA fulvic acid, fraction of HS that is soluble at any pH GAC granular activated carbon HA humic acid, fraction of HS that is insoluble below pH 1-2 HGR heterotrophic growth response bioassay HPC heterotrophic plate count HS humic substances M A P microbially available phosphorus N O M natural organic matter T B N total bacterial number THMs trihalomethanes TOC total organic carbon X A D non-ionic acrylic ester polymer resin
1

2. L I S T O F P U B L I C A T I O N S
I . Juhna, T., Klavins, M . and Eglite, L . Sorption of humic substances to aquifer material at artificial recharge of groundwater. Chemosphere, 2002, submitted.
I I . Springe, G., Druvietis, I . and Juhna, T. (2001) Development o f potentially toxic
cyanobacteria and bacteria during artificial recharge o f groundwater. In: Proceedings of International Conference on Harmful Algal blooms. Ninth Conference, Tasmania 2000.
I I I . Klavins, M . , Juhna, T. and Eglite, L. (2000) Removal o f humic substances during treatment
of drinking water using sorbents. Vatten, 56, 79-83.
I V . Juhna, T.. Nikolajeva, V. , Juhna, V . and Hanaeus. J. Microbially available phosphorus and
assimilable organic carbon in a drinking water supply system. Water Research, 2002,
submitted.
V. Juhna, T. (2002) Effect of phosphorus removal f rom humus-rich drinking water on b iof i lm formation. In: Proceedings of International Specialized Conference on B io f i lm Monitoring,
Porto, Portugal 2002.
V I . Juhna, T., Gulbe, B. and Klavins, M . (2002) Speciation o f iron in groundwater f rom areas of humic waters by ion-exchange method. Vatten, 58 (3), in press.
2

3. I N T R O D U C T I O N
In cold and temperate climate areas that are abundant in wetlands or soils rich in organic matter natural waters frequently contain high amounts o f dissolved organic matter (DOM). The bulk o f DOM usually consists of humic substances - yellow to black, relatively recalcitrant, naturally derived organic material with unspecified structures and largely colloidal properties. Drinking
water supply f rom these sources (humic waters) is related to several hygienic and aesthetic water quality problems. Humic substances impart colour and turbidity to water, consume disinfectant
power, and form carcinogenic compounds such as trihalomethanes (THMs) during drinking water chlorination. Humic substances interfere with most water treatment processes including removal of particles, xenobiotics, heavy metals, and pathogenic protozoa (Dai and Hozalski, 2002; L u and
Speitel, 1991).
In addition, drinking water prepared from humic waters may contain biodegradable organic matter (BOM) originating from the raw water source or produced during the treatment process. Even at
very low concentrations, B O M serves as nutrients and energy source for bacteria l iving in water
distribution networks, thus promoting their growth in drinking water (Ellis et al. 2000). Bacteria in the network colonize the surface of the pipes while forming a slimy b io f i lm (Flemming et al. 2002; Block et al. 1993). The biofi lms deteriorate the aesthetic water quality, accelerate pipe corrosion, consume chlorine, and potentially increase the risk o f pathogenic bacteria occurrence in tap water
(Franzmann et al. 2001; L u et al. 1999; Geldreich, 1996; LeChevallier et al. 1996; Astier et al.
1995; Rice et al. 1991).
Even though there are some positive effects of D O M in drinking water, including a decrease of iron pipe corrosion rates (Bro et al. 1999), longer inhibition o f bacterial growth in water after
ultraviolet light disinfection (Lund and Hongve, 1994), and a decrease in the toxicity of heavy metals, the overall impacts are probably detrimental. Therefore, knowledge about removal of
DOM, especially humic substances and B O M , is important for managing a water supply system
that utilises humic waters.
Traditional drinking water treatment methods such as chemical precipitation (CP) or artificial recharge of groundwater (ARG) are primarily designed for removal of turbidity and colour. The
processes involved in removal of D O M , at least in the case of A R G systems, are not wel l understood. In these systems concentrations o f D O M decrease during surface water infiltration and
movement through an aquifer to groundwater. Several physical, chemical (e.g. straining, interception, sorption), and microbiological processes (e.g. biodegradation) are involved in
retention of D O M during A R G . Humic substances are mainly retained by sorption on the aquifer material (Juhna et al. 1998a; Marmonier et al. 1995; McCarthy et al. 1993) as a result of different
mechanisms (e.g. ligand exchange, hydrophobic and electrostatic attraction) similar to those occurring in soils (Jacks and Frycklund, 1995). The chemical and structural heterogeneities
associated wi th the complex nature of aquifer material and humic substances complicate evaluation of the influence of these mechanisms. Many physical and chemical factors are
determining the sorption efficiency including composition o f aquifer material and the properties of
humic substances and water chemistry. Increased knowledge about the influence of these factors would allow to better understanding the sorption mechanisms. This information in turn can be used
to optimise the artificial recharge systems to better remove humic substances.
3

In the process of water passage f rom surface water to groundwater B O M is mainly removed as a result o f biodegradation by bacteria l iving attached to the surfaces of aquifer material (Marmonier
et al. 1995). Bacteria are sensitive to different environmental stress that may affect their metabolism. The development o f cyanobacteria blooms is common in eutrophicated waters (Wei et
al. 2001). Blooming may induce changes in phylogenetic composition and the number of microbes including free l iving and attached bacteria (Rohrlach et al. 2001; Riemann et al. 2000), which in
turn may influence the biodegradation process of B O M . Many studies have shown several negative effects o f algae blooming on drinking water including a decrease of aesthetic and hygienic quality and an increase in the occurrence risk of toxic and carcinogenic compounds in drinking water
(Bruce et al. 2002; Rapala et al. 2002; Graham et al. 1998; Schmidt et al. 1998). However, the influence of blooming on bacterial ability to degrade B O M has not received considerable attention.
This is important because an inhibition o f the biodegradation process may increase B O M concentration in drinking water, thereby increasing bacterial growth potential in the distribution
networks.
During A R G , the total amount as well as the composition of humic substances may be changed
(Miettinen et al. 1997b), e.g. molecular weight, charge density, polarity, and biodegradability. The composition of HS is important in the selection o f an optimal method for further treatment (post-
treatment) o f artificially recharged groundwater. Different sorbents including granular activated carbon (GAC) and anion exchange resins have been proposed for removal o f humic substances
f r o m surface water (Ødegaard, et al 1999). However, properties of HS in surface water and artificially recharged groundwater differ, possibly resulting in a different affini ty towards the
sorbents. Thus, more information about the possibilities of applying sorbents for the removal of D O M , and especially HS f rom artificially recharged groundwater, is of interest.
Microbial growth and formation of biofilms in drinking water distribution networks are usually
reduced by adding disinfectant, most frequently chlorine, to water. The chlorine doses for humic waters should be carefully selected given the high formation potential of THMs in these waters.
Another approach is to reduce concentration of B O M or other nutrients essential for bacteria growth in water. Usually B O M is removed because carbon regulates bacteria growth in waters
wi th low levels of D O M (LeChevallier et al. 1991; van der Koo i j , 1992). However, inorganic nutrients in humic waters, most frequently phosphorus, can l imit bacterial growth (Miettinen et al.
1997a). More knowledge about reducing B O M and inorganic nutrients by traditional methods and its effect on bacterial growth in water and b io f i lm in distribution networks is needed for managing
drinking water supply systems in areas of humic waters.
Humic substances influence removal of heavy metals f r om drinking water. Iron removal is o f
special concern in humic groundwater. When water contains humic substances, iron removal with commonly used methods such as aeration is diff icul t . This is further complicated as the actual
portion o f iron bound to humic substances is usually unknown in common drinking water treatment. A t the moment, the degree of iron complexation is determined indirectly and is based on
analyses of other parameters. Different, advanced methods such as anodic stripping voltammetry and fluorescence spectroscopy have been proposed for determination of iron-humic complexes.
However, most of them are laborious and require expensive equipment. Thus, a simple method for speciation o f different iron forms in humic groundwater could be of great help for sanitary
engineers.
4

4. A I M O F T H E T H E S I S
The overall objective of this thesis was to gain more knowledge about the removal of D O M by ARG and CP, and about the bacterial growth potential in humic waters through the fol lowing
items:
• to identify and evaluate the influence of main factors determining the sorption of humic
substances during ARG;
• to test the effectiveness of different sorbents for removal of humic substances, with an emphasis on humic substances removal f r om artificially recharged groundwater:
• to investigate i f algae blooms significantly impair biodegradation of D O M during ARG;
• to evaluate the bacterial growth potential o f drinking water prepared f rom humic waters
by CP or ARG;
• to measure formation of b iof i lm in drinking water prepared by CP and A R G from humic
water and test which chorine doses are needed to reduce b io f i lm formation;
• to develop a simple method to measure the concentration of iron-humic complexes in
humic groundwaters.
5

5. B A C K G R O U N D
5.1. D O M in waters
D O M concentration, measured as dissolved organic carbon (DOC), varies with the type of water f rom about 0.5 mg/1 in groundwater to 100 mg/1 in coloured bog water. Aquatic D O M can be
produced by soils and vegetation of terrestrial watersheds or f rom algae and bacterial growth, and leaching of vegetation within the water body (Aiken and Cotsaris, 1995; Thurman, 1985; Wetzel,
1983). D O M constitutes about 90% of natural organic matter (NOM) of most natural waters (Aiken and Cotsaris, 1995; Thurman, 1985) whereas the rest is particulate organic matter. The source material, hydrological pathways, physical and geochemical characteristics of the watershed,
temperature, sunlight, trophic state, and other factors influence the proportions of each class of D O M and their properties in natural water (Wilkinson et al. 1997; Will iamson et al. 1999).
D O M can be arbitrarily subdivided into two large categories: humic and non-humic substances.
Humic substances (HS) account for approximately 30 to 50% of the DOC in clear waters and 50 to 90% in humic waters (e.g. Kullberg, 1994). Usually, lake DOC contains less HS than in rivers (Imai et al. 2001); their HS properties often being different. HS are operationally divided further
into humic acids (HA), fu lvic acids (FA), and sometimes humin. H A are components that precipitate when a water sample is acidified between pH 1 and 2, and FA remains soluble whereas
humin is not soluble at any pH. FA are more acidic than H A ; with an intermediate molecular
weight (600 to 1000 Da), they may constitute 20 to 90% of the HS (Aiken and Cotsaris, 1995). H A have higher molecular weight (1500-5000 Da), are more hydrophobic, have more heterogeneous
molecular weights, more colloid alike, and are commonly in lower concentrations than FA (Aiken
and Cotsaris, 1995; Thurman, 1985).
HS are formed f rom either products o f microbial degradation or metabolism products o f plants and microorganisms that are modified by humification, i.e. reactions including polymerization,
condensation, and oxidation that produce highly structurally complex macromolecules. Cold
climate HS has a tendency to accumulate in natural waters; however, many other factors such as humidity, season, vegetation type, composition of soil, hydrology, anthropogenic pollution, and
disturbances of the surrounding watershed may render high HS levels in water (Imai et al. 2001; Williamson et al. 1999; Aiken and Cotsaris, 1995). HS consist of carbon, oxygen, hydrogen, and occasionally small amounts of nitrogen or phosphorus and sulphur. The structure of HS is i l l -defined despite many decades of research and numerous tentative structures having been proposed
(Figure 1). In broadest terms, HS structures can be described as assemblies of covalently linked
aromatic and aliphatic residues carrying carboxyl, phenolic, and alkoxy groups, and possibly
sulphate esters, semiquinone, phoshate esters, and hydroquinone moieties (Jones and Bryan, 1998 and reference within).
Non-humic substances are mainly by-products of microbial activity including zooplankton grazing,
viral lysis of bacteria, and algal exudation. They are generally less hydrophobic and are comprised
of compounds with specific chemical structures including hydrophilic acids, proteins, amino acids,
and carbohydrates. Analytically HS can be distinguished f rom non-humic substances by adsorption of the former substances on either hydrophobic (e.g. X A D ) resins at acidic pH or weak base anion
exchanger (e.g. DEAE) at neutral p H (Martin-Mousset et al. 1997).
6

Figure 1. Model of humic macromolecule structure as proposed by Schulten and Schnitzer (1993).
B O M is mainly composed of non-humic substances, though lately it was proven that 10 to 20% of river and lake HS are biodegradable and thus also contribute to B O M (Hunt et al. 1999). In the
drinking water industry, two measurement-specific B O M subsets are widely used. Biologically
degradable organic carbon (BDOC) is a portion (10-20%) of the DOC that can be mineralised by heterotrophic bacteria (Servais et al. 1987), whereas assimilable organic carbon (AOC) is the portion of DOC (1-10%) that can be converted to cell mass by either a single organism or a
consortium of bacteria (van der Kooi j et al. 1982).
Physicochemical characteristics of HS
Although the composition o f HS is very heterogeneous and varies among waters, some properties are similar. In principle, the polydispersity of molar masses and chemical structure comprising HS
are responsible for their physicochemical characteristics in raw and drinking water. HS impart colour to water due to the presence of chromophores, i.e. simple or condensed aromatic compounds with hydroxylic and carboxylic substituents that have a strong absorbance in an
ultraviolet and visible light spectrum. This property is commonly used to determine the
concentration o f HS in water (Hautala et al. 2000; Peuravuori and Pihlaja, 1997). Aromatic groups in HS, e.g. meta-dihydroxybenzene, generate potentially harmful disinfection by-products such as
THMs and haloacetic acids during chlorination (Pomes et al. 1999).
HS contain carboxylic and phenolic functional groups that are deprotonated at neutral pH levels
(Jones and Bryan, 1998). The resulting negative charge of the molecule accounts for many HS
characteristics including aqueous solubility, binding capacity for metals, and buffering capacity.
Functional groups also participate in a specific sorption process (ligand exchange) to oxides surfaces (Edwards et al. 1996). Because of the net negative charge. HS are anions in water and can thus be removed by anion exchange resins (Fu and Symon. 1990)
7

Hydrophobie moieties, such as the long alkyl side chains of fatty acid residues, provide an amphiphilic character for humic molecules. Because o f their amphiphilic nature, they tend to
organize spontaneously in an aqueous solution, forming micelle-like structures with hydrophobic groups inside and acidic dissociated functional groups outside the molecule. Due to the
hydrophobic interaction, HS accumulate on surfaces and sequester solution-borne hydrophobic species. In part, HS have colloidal properties because of high density the functional groups
(Simpson et al. 2002). Unlike non-humic substances, e.g. oligosaccharides and polysaccharides that promote aggregation o f colloids, HS stabilize mineral particles (Wilkinson et al. 1997; Zhou et al. 1994), rendering their removal during water treatment processes more dif f icul t .
HS behave like flexible anionic polyelectrolytes at sorption to surfaces (Vermeer et al. 1998, De
Wit t et al. 1993; Schnitzer, 1991). A t an acidic p H and high salt concentration, the polyelectrolytes have a coiled confirmation. Thus, a substantial fraction of the sorbed HS is not in direct contact
with the surface, resulting in a relatively high sorbed amount in a thick layer. A t a basic pH where the polymer is highly negative (due to dissociated phenol groups), it assumes an extended "stretched out" shape in the solution as a result o f intermolecular electrostatic repulsive
interactions. When adsorbed at the surface under these conditions, they assume a flat
configuration; thus the sorption is low (Vermeer et al. 1998).
Because HS are end products of earlier biological activity, they were assumed to be biologically
inactive. However, recent research has shown that HS not only participate as electron acceptors for microbial respiration in anoxic habitats (Lovely et al. 1996), but can also serve as energy and
carbon sources for bacteria (Bano et al. 1997; Grøn et al. 1992; Tranvik, 1990). High molecular weight HS may be even more biodegradable than low molecular weight HS (Tranvik, 1990),
possibly because the former are relatively younger and generally contain more carbohydrate
moieties (Haiber et al. 2001) that are readily utilised by bacteria.
5.2. Removal of D O M from surface water during artificial recharge of groundwater
Art i f i c ia l recharge of groundwater (ARG) is achieved by putting surface water into basins, furrows, ditches, or other facilities where it infiltrates into the soil and moves downward to
recharge aquifers (Bower, 2002). Another approach is to induce surface water to f low directly through the banks of a river or a lake to groundwater (Kuehn and Mueller, 2000). This simple and
economical method, as a single water treatment step or in combination with a pre- or post-treatment, has been used for more than a century in Europe and about 50 years in the United States (Ray et al. 2002). During surface water passage to groundwater many raw water impurities are
attenuated f rom a combination o f processes such as fi l tration, microbial degradation, sorption on aquifer material, and dilution with natural groundwater (Ray et al. 2002; Kuehn and Mueller, 2000;
von Gunter and Zobrist, 1993; Huisman, and Olsthoorn, 1983). The porous media of the aquifer
used for A R G efficiently trap fine particles, colloids, iron, and microorganisms (Ray et al. 2002; Frycklund and Jacks, 1997; Miettinen et al. 1997b). D O M concentration decreases due to physical, chemical, and microbial processes that take place in both the infiltration basins and the subsurface
during water filtration to groundwater. Due to heterogeneity of the aquifer material and D O M , as
well as varying operational practice, the removal of HS and B O M can differ between plants.
Frycklund et al. (1995) reported that during favourable summer conditions, about 50% of total
organic carbon (TOC) were retained in the top 20 cm of the aquifer material in a Swedish A R G plant. In Germany, along the Rhine River, bank fil tration removed 75% of D O M present in the
river (Sontheimer, 1980). In Finland, about 60% of N O M was removed f rom humic lake water
8

after its passage through an esker (Miettinen et al. 1994). A similar or slightly lower reduction was reported f rom several plants in United States (Ray et al. 2002, see references within). However, a Swedish study that employed pilot columns to evaluate the possibility of using A R G for Stockholm's water supply showed less than 15% reduction o f D O M (Hanson, 2000), which is
about the usual efficiency achieved by slow sand fi l trat ion (Collins et al. 1992).
Removal efficiency of HS during ARG
Since only a few studies have done direct measurements of HS, it is always di f f icul t to evaluate the
removal efficiency of HS during ARG. In Denmark, Alborzfar et al. (2001) showed that when humic rich groundwater was sprinkled onto soil, HS were already reduced about tenfold in the
upper layer. Results obtained f rom studies by Marmonier et al (1995) showed that refractory D O M , mostly HS, are effectively removed in the first metre of water passage f rom the Rhone River to groundwater. Miettinen et al. (1994) showed that during bank filtration a high molecular weight fraction o f D O M were eliminated by 87%, whereas concentration of low molecular weight
fraction was not significantly effected. Myllykangas et al. (2002) also observed the greater reduction o f a higher molecular weight fraction compared to a smaller molecular weight fraction.
HS represent the bulk of high molecular weight D O M , meaning that HS are fractions of D O M that are effectively reduced during ARG. It has also been reported that the amount o f halogented
organic by-products in chlorinated recharged groundwater is significantly reduced (Miettinen et al. 1998). HS are major precursors of these products, thus the reduction of HS during A R G appears to
be effective.
Mechanisms for removal of HS during ARG
Mechanisms involved in the removal of HS during A R G include straining and interception,
sorption and biodegradation. Dominant fractions o f HS are recalcitrant or slowly degradable, whereas sorption on surfaces makes HS more readily available for b io f i lm bacteria (Camper,
2002). Hence, sorption to aquifer material most likely precedes biodegradation; accordingly, sorption is the l imiting step for HS removal during A R G . Given the colloidal properties of HS it could be assumed that some HS are removed by interception and straining in porous media as
discrete humus colloids, or humus adsorbed on inorganic colloids (Davis et al. 2002). Physicochemical forces, such as van der Waals and electrostatic attraction, along with
hydrodynamic forces govern colloidal removal by interception. Whi le developing the concept of HS colloidal behaviour in porous media, Gerlach and Gimbel (1999) proposed a colloids transport theory application for removal of HS during ARG. The straining of colloids occurs when the pores
are most l ikely clogged, thereby forming an organic mat (also called "'schmutzdecke"). Some HS
may be removed by flocculation with multivalent cations within the organic mat or in the porous media f rom a similar process as it occurs during chemical coagulation treatment (Jacks and Frycklund, 1995). The relative infuence of the above mentioned mechanisms for removal of HS
during A R G is not well known. This thesis mainly focuses on one o f the most important processes
of HS removal - sorption to aquifer material.
Sorption of HS on aquifer material
Sorption on mineral surfaces, dissolution, and precipitation o f humic materials under changing
solution conditions are common processes in soil and natural waters (Tombåcz et al. 2000). When
9

HS in surface water percolates into porous underground environment, the system strives to achieve chemical equilibrium (Weber et al. 1991) leading to sorption of HS on surfaces of aquifer material.
There is no coherent theory that exactly describes sorption o f HS on aquifer material, though major
mechanisms are identified (Jacks and Frycklund. 1995). They appear to be similar to sorption mechanisms on mineral phases in soils. The sorption of HS on mineral phases occurs in two steps -initial sorption is followed by rearrangement of the HS structure in the sorbed layer due to re-
conformations and their polyelectrolytic properties (Aven and Koopal, 1999; Vermeer et al. 1998). For initial sorption to occur HS should be transported f rom bulk water to the water/surface interface and then attached to the surface. Either of these steps can l imit the entire retention
process (Aven and Koopal, 1999). When HS are transported close enough to the surface, they are
attached as a result of different forces. In general, sorption occurs due to physical sorption (including electrostatic attraction, London-van der Waals force, hydrogen bounding, and hydrophobic attraction) and chemisorption (mainly ligand exchange). The most important forces
for removal o f HS on aquifer material are electrostatic and hydrophobic attractions (with the assistance of London-van der Waals force and hydrogen bounding) and ligand exchange
(Vermmer et al. 1998; Jacks and Frycklund, 1995). The polyelectrolytic type of sorption is also important - at low pH and high salinity HS tend to agglomerate and precipitate in well-packed
segments on the surface, while remaining in the vicinity o f the surface, but not in direct contact
(Vermeer e ta l . 1998).
Factors affecting the sorption of HS
Composition o f minerals, properties of HS. and solution properties such as pH, ionic strength, and
temperature are the most important factors in the sorption process (Zhou et al. 1994). Aluminium
and iron oxide, common constituents of aquifer materials, provide sorption sites for HS. Depending on pH, ligand exchange between the surfaces and functional groups in HS or electrostatic interaction in between them may occur (Tombåcz et al. 2000; Edwards et al. 1996;
Tipping, 1981). Thus, higher iron and aluminium concentrations in the aquifer material increase
sorption of HS, especially FA, because of their higher carboxyl group content that can be used for ligand exchange. Aluminium may also act as a bridging cation for the binding o f HS (Tombåcz et
al. 2000), which thereby increases the agglomeration of HS and favours their removal. Silica oxides (as quartz) at neutral pH is either neutral or negatively charged, hence electrostatic
attraction of negatively charged carboxyl groups of HS is not likely. However, some ligand exchange between them may occur (Filip and Alberts, 1994). For surfaces wi th few binding sites hydrophobic interaction appears to be most important process for sorption of organic substances
(Stumm, 1992). The aquifer material texture of is also important. Due to their small size, silt and clay particles have high specific surface areas available for sorption, they consequently actively
participate in sorption processes (Schwarzenbach et al. 1983 reference within). Clay increases sorption due to the presence of aluminium oxides. Clay also sorbs about tenfolds more HS than
quartz (Filip and Alberts, 1994) probably because of these properties. Different types of clay in terms of density, origin of charge, and expansibility have different abilities to sorb HS.
Organic matter that covers aquifer material grains can either favour or decrease a removal of HS
(Marmonier et al. 1995: Jardine et al. 1989: Hrubec et al. 1986). Metals in water favour sorption
(and interception) of HS by aquifer material (Juhna and Klavins, 1999) probably due to a more compressed structure of metal-humic complexes as compared to a single HS and a smaller lateral
electrostatic repulsion between the sorbed humic acid molecules (Vermeer et a). 1998).
10

Decreasing the pH and increasing the ion strength in most cases favours sorption of HS due to its polyelectolytes behaviour in the vicinity o f the surfaces (see section 5.1). In addition, at lower pH surfaces become more positively charged whereas the charge of HS decreases, thus allowing for
charge neutralization. Also, the hydrodynamic radius of HS decreases, thereby favouring their diffusion towards surfaces and sorption (Aven and Koopal, 1999).
Biodegradation of DOM during ARG
During surface water passage from infiltration basins to production wells, most aquatic
microorganisms (algae, bacteria, viruses, and protozoa) become trapped in the pores of the aquifer
material or adsorbed on surfaces, thus forming biofilms (Burns, 1983). When attached to the aquifer material, bacteria may use more energy for self-nourishment (anabolism) rather than for motility, and are more protected f rom abiotic (e.g. toxic substances) and biotic (e.g. grazing) stresses (Ghiorse and Wilson, 1988). As a result aquifer material is covered with a high number of
active bacteria that are able to utilize B O M and a part of HS as energy or carbon source.
Most bacteria live in the upper zone below the infiltration basins; however, degradation may also
take place in deeper layers. The conditions of groundwater f low are conducive to biodegradation in two respects. Low velocities assure long residence times whereas a porous medium provides a
surface for attachment o f bacterial f i lms. The long residence times of a substrate in the aquifer that result f r o m sorptive retention provide opportunities for significant biodegradation to occur, even i f very slow (Roberts et al. 1982). More important, given the long residence times, substantial degradation may occur under either aerobic or anaerobic conditions, implying that removal o f
B O M during ARG is efficient in most cases. HS substances can also be partially biodegraded, but
their degradation occurs after enzymatic hydrolysis (Laurent et al. 1999), which is more efficient when both bacteria and HS are sorbed on the surface (Grossart and Simon, 1998). As wel l , the structure of sorbed HS changes and becomes more available to bacteria (Camper, 2002). Easily
degradable substrates such as lactate and benzoate stimulate the degradation rate of HS (De Haan, 1983), whereas HS enhance biodegradation of hydrophobic organic compounds (Ortega-Calvo and
Saiz-Jimenez, 1998).
Cyanobacteria (blue-green algae) and green algal blooms are common phenomena in eutrophicated waters (Wei et al. 2001). Blooming in raw water is of concern to the drinking water industry primeraly because during and after blooms, l iving or dead cyanobacteria release toxins (e.g. microcystins) (Long et al. 2001; Karner et al. 2001), produce earthy and musty odours (Bruce et al.
2002) and act as THMs precursors (Graham et al. 1998). In addition, cyanobacteria blooming may infuence drinking water quality indirectly by inhibiting bacteria that carry our biodegradation
during A R G . During the bloom, algae induce changes in phylogenetic composition and number of
bacteria. (Riemann et al. 2000). Algae produce organic compounds that are antagonistic to the growth o f bacteria (Boualam et al. 2002; Metting and Pyne, 1986) or inhibit feeding activity o f
bacterial grazers (e.g. Daphnia spp.) (Rohrlack et al. 2001). This may influence the ability o f bacteria l iv ing in the subsurface to degrade nutrients including B O M . Because degradation is a
major process in the removal o f nutrients f r om raw water during ARG, algal blooms, though a secondary mechanism (inhibition of bacteria growth), may increase the potential of bacteria
growth during water distribution.
11

5.3. Removal of H S from artificially recharged groundwater using sorbents
Several methods available to remove HS f rom water include CP, sorption, membrane fil tration, or oxidation-biofiltration. Sorption is an attractive method of removing HS because inherent
properties allow them to accumulate on different sorbent surfaces. Due to hydrophobic moieties HS can be sorbed on nonpolar sorbents such as GAC, whereas through acid groups (carboxyl and
phenol) they can be removed by anion exchange resins. Both GAC and anion exchange resins are used to remove colour f rom humic surface waters (Ødegaard, et al 1999). Because of its large
surface area (1000-1400 m7g) (Fettig, 1999) GAC effectively sorbs small organic molecules. However, since about 90% of GAC surface area is located in the pore system (Pelekani and Snoeynik, 1999, references within), the large HS that slowly diffuse into the pores interior may
block them, thus reducing GAC sorption capacity (Pelekani and Snoeyink, 1999). Properties o f HS
in surface water and artificial groundwater differ. Large and intermediate humic fractions are the dominant fractions in surface water, whereas intermediate and small fractions dominate in natural and artificially recharged groundwaters (Nissinen et al. 2001). In groundwater HS are often
composed of acidic fractions such as FA (Petersen. 1992). Anion exchange resins are ineffective in removing large and hydrophobic fractions (Hongve et al. 1999; Croué et al. 1999), but are efficient in removing those that are acidic (Fu and Symons, 1990). Hence, GAC and anion exchange resins
can potentially be used to remove HS f rom artificial groundwater.
The efficiency of an anion exchanger to remove HS is dependent on its properties, including the type o f functional groups (strong or weak bases) and the composition of the matrix (gel-type or
macroreticular). Because functional groups of strong base anion exchangers dissociate over a
broad range of pH values, HS are effectively sorbed through the formation o f salts between the f ixed ions of the anion exchanger and carboxylic groups o f HS (Fu and Symons, 1990).
Dissociation of those groups is suppressed in weak base anion exchangers at neutral and subalkaline pH (Mamchenko et al. 1997), possibly hampering ion exchange. Therefore, i t was
suggested that HS by weak anion exchanger can be removed due to hydrophobic sorption at the surface of the resin (Fu and Symons, 1990), though this mechanism has been questioned lately (Mamchenko et al. 1997). Unlike gel-type resins the matrix of a macroreticular anion-exchanger
has a true pore phase in addition to the gel phase with an internal surface area that is easily
accessible by HS (Fu and Symons, 1990), i.e. its capacity for removal of HS may be higher than
that of a gel-type matrix.
5.4. Effect of B O M on drinking water quality during its distribution in networks
The main problem associated with B O M in drinking water distribution networks is its ability to provide energy or nutrients needed for bacteria to grow. The distribution network used to supply
public water is not a sterile environment. Bacteria, including pathogens, can enter the network as a result of insufficient treatment, through pipeline breakages (Craun et al. 2001), or during pipe
installation (Haas et al. 1999). Meaning that when B O M is not sufficiently removed or becomes produced during water treatment it can potentially support the proliferation o f bacteria along the
distribution system. High bacteria numbers, in turn, may contribute to the production of a musty taste (Astier et al. 1995) or swampy odour (Franzmann et al. 2001) o f the water, stimulate the
development of a trophic food web (Gatel et al. 2000), accelerate the corrosion rates of pipes (Rompé et al. 2000), and may increase the number of incidences of bacterial diseases (Geldreich,
1996; LeChevallier et al. 1996; Rice et al. 1991). The cause of these problems largely stems from
the ability of bacteria to form biofilms on the inner surfaces of pipes. The b io f i lm, also known as "Aufwuchs", "slime", or "biological deposits", results f rom adhesion and growth of
microorganisms and consists of microbial cells embedded in a matrix of extracellular polymeric
12

substances. The pathogens and opportunistic pathogens that invade the distribution network may hide f rom a disinfectant effect and even slowly multiply in the biofi lms (Camper et al. 1996).
In Europe, an average o f about 6,000 reported cases o f gastrointernal diseases are caused annually
by tap water consumption (Kramer et al. 2001). In the USA most waterborne disease outbreaks in community drinking water systems are caused by distribution networks contaminations rather than
by the raw water source (Craun and Calderon, 2001). Studies in Northern Canada showed that the number of gastrointernal symptoms such as nausea, vomiting, diarrhoea, sore throat, cold, and f l u were about 20% higher in a population that consumed drinking water delivered through
distribution networks than in a population drinking bottled water (Payment et al. 1997). These problems are at least partly attributable to b iof i lm formation caused by nutrients such as B O M in
water. Several studies have shown that the occurrence of faecal pollution indicators - col iform bacteria - in the distribution networks correlated positively with B O M levels in drinking waters (LeChevallier, 1996; Rice et al. 1991). Falkinham et al (2001) showed that number of opportunistic human pathogens in drinking water such as Mycobacterium avium and Mycobacterium intracellulare increased with the increase of B O M (both AOC and BDOC)
concentration. I t is therefore, even though epidemiological surveys hitherto were not able to show that communities consuming drinking water with high B O M have higher rates o f gastrointernal
illnesses (Payment et al. 1997). nutrients such as B O M are still considered as an important factor compromising drinking water quality.
Limiting nutrient for bacterial growth
Bacterial growth in drinking water is regulated mainly by temperature, disinfectant residual, and
nutrients concentrations (Zhang and DiGiano. 2002). A trophic food web was observed in some distribution systems where predators such as protozoa were also partly controlling the bacterial
population (Gatel et al. 2002; Sibille et al. 1998). Maintaining a high residual concentration of chlorine requires either boosting chlorination or the addition of high chlorine doses at the plant, considered undesirable in humic waters (Koivusalo, 1998). At moderate and low doses chlorine
reacts with pipes and organic matter while rapidly loosing it disinfectant power (Haas et al. 2002; Frateur et al. 1999). A t this point bacterial growth is mainly dependent on the nutrient that is in
critical concentration for bacterial cell requirements (limiting nutrient). In drinking water prepared from water with low D O M organic carbon is limiting bacterial growth (LeChevallier et al. 1991).
Therefore, in these waters, either the AOC or BDOC methods is usually utilised to evaluate the bacterial growth potential. Another situation may be present in waters with a high content of B O M such as humic water. Recently, it was reported that in some humus-rich drinking waters, bacterial
growth was limited by phosphorus (Miettinen et al. 1997a). Therefore, to correctly evaluate bacterial growth in drinking water that is produced f rom humic water, the nutrient that limits
bacterial growth should be determined.
13

6. S T U D Y S I T E AND E X P E R I M E N T A L M E T H O D S
6.1. Description of study sites and water quality
Field experiments were carried out using either groundwater f rom the vicinity of Riga City, or raw and drinking water f rom the city water supply system, Figure 2. Riga, the capital of Latvia, is supplied with drinking water (ca. 200,000 m 3/day) produced f rom surface water (50%) and
groundwater (50%). The surface water, taken f rom the Daugava River, was pre-chlorinated (3 to 5 mg CL/ l ) and treated with chemical coagulation (alum), fol lowed by precipitation and rapid sand
filtration at the Daugava water treatment plant (WTP). A chlorine dose of 2 to 3 mg/1 is used for final water disinfection at the plant. During the period studied, Daugava WTP was rebuilt and the
treatment train was upgraded with a two-stage ozonation unit fol lowed by a biologically active carbon treatment (BAC). Primary chlorination was substituted with pre-ozonation, and the final chorine dose was lowered three to four times.
The groundwater is abstracted at several naturally and artificially recharged groundwater
abstraction sites (Figure 2). During the A R G process, surface water f r o m Lake Mazais Baltezers is
pumped into the infiltration basins where it percolates down to the groundwater. After underground transport (retention time 30-180 days) the recharged groundwater is abstracted by a siphon system (production wells are connected to vacuumed siphon-pipes), disinfected by adding 1 mg of Ch per litre, and pumped to the city water distribution.
In the network the groundwater, including artifically recharged groundwater, is mixed with treated surface water in the central part of the city. The part of city situated to the east of the Daugava was
supplied only with groundwater while the west side only wi th treated surface water, Figure 2. This
enabled to study the changes in water quality of groundwater and surface water in the distribution network separately.
Raw water quality
The water chemistry and biology of inland waters of Latvia are generally determined by its physico-geographic location: a wet and temperate climate, f lat surface topography, dominance of Quaternary glacial and ancient sea sediments (parent soil materials are moraine loam and sand),
and the dominance of humic podsol soils (Klavins et al. 2000). Calcium and hydrogen carbonate dominate inorganic water ions; in general, waters are moderately hard with a high buffering capacity though there are several lakes with soft water.
Latvian surface waters mostly contain high concentrations of N O M , which is highest in peatland and swamp drainage areas (Apsite and Klavins, 1998). Due to a relatively cold climate and an abundance o f soils rich in organic carbon the concentration o f HS in Latvia is higher than in many
European and Scandinavian countries (Klavins, 1993a). The trophic state of lakes in Latvia varies f rom dystrophic to eutrophic (Klavins and Apsite, 1997). Local water sources are polluted with
nutrients, though overall surface water quality in Latvia has been improving over the last decade (Juhna and Klavins, 2001; Klavins et al. 2000).
14

Figure 2. The main study site. Raw water sources, the drinking water distribution network of Riga City with surface water treatment plant Daugava (S) and groundwater sources ( • ) , including an artificially recharge groundwater abstraction plant (G) with infiltration basins.
The watershed of the Daugava is largely covered with swamps (7%) and forests (38%) (DRBP, 2000). The concentration of DOC in the river varies from 8.5 to 23.6 mg/1 (Klavins, 1993b) with the lowest values in the autumn and winter. The highest DOC concentrations are found during the
summer, the season with highest rain intensity in the river watershed (DRBP, 2000). For comparison purposes, the average value o f DOC in rivers and streams on Earth is 5.75 mg/1 (Meybeck, 1982), but in the humus-rich waters f rom the boreal climate of Finland this value is 12
mg/1 (Kortelainen, 1993). Hence, the DOC concentration in the Daugava can be regarded as high. The absorbance of ultraviolet light by water (Abs254) is also high (Table 1) and occasionally
correlated with DOC (DOC (mg/1) = 19.5 A b s 2 5 4 ( c m 1 ) + 3.42; r = 1) (RWB, 2002). This was similar to that found in Norwegian humic waters (Vik et al. 1995). Assuming that HS are composed of 5067r carbon (see also Table 2) and that determination of HS concentration is correct,
as is shown below in Table 1, it can be calculated that HS constituted more than 90% of DOC in
the Daugava water.
According to COD analyses, concentration or organic matter in Lake Mazais Baltezers is lower
than in the Daugava. The ration between AbS254 and COD were similar in both waters, indicating similar percentages of HS in DOC as well (Table 1). Usually, HS in lakes comprise about 50%) of
DOC (Imai et al. 2001). The higher level o f HS in Lake Mazais Baltezers could be due to the inflow of waters from the Daugava during the autumn and winter (see hydrological networks in
Figure 2).
Both the river and lake are polluted with nutrients (Table 1), resulting in a high number of algae that are occasionally accompanied by blue-green algae blooms in the summer and autumn (Druvietis, 1997). Based on HS concentration (measured as colour) and content o f nutrients both
sources can be classified as eutrophic humic waters.
15

Composition of HS in the Daugava River and Lake Mazais Baltezers
Because HS have unspecific structures information about their chemical composition is important. Composition and properties that determine HS removal during drinking water treatment (e.g.
aromaticity, acidity, molecular weight, polydispersity, biodegradability) are dependent on the source material of HS and biochemical processes occurring within the water reservoir.
Percentages of main elements composing HS, their atomic ration, and contents of carboxyl and phenolic groups can be used to identify the source material. HS derived f r o m vegetation (vascular
plants) in watersheds are highly aromatic (H/C < 1), with high phenolic (>0.5 mmol/g) and low nitrogen (<1%) contents (Pomes et al. 1999). HS derived f rom algal and cyanobacterial biomass
within the water reservoir are abundant in nitrogen (>1%) and carboxylic groups (6 mmol/g). The N level can also be high for soil-derived HS (Pomes et al. 1999) as for carboxylic groups f rom
swamp-derived HS. FA f r o m the Daugava and Lake Mazais Baltezers were moderately aromatic with high phenolic contents and moderate N and carboxylic contents. Accordingly, vegetation is a dominant source material; however, algal biomass and soil drainage are also important.
In addition to source materials, biochemical processes involving carbon cycling (e.g. humification)
within terrestrial and aquatic systems influence the chemical composition o f HS (Aiken and Cotsaris, 1995). The degree o f humification can be determined f rom a weight average molecular
weight ( M w ) , a number average molecular weight (M„), and the polydispersity of HS (the closer to unity, the more homogeneous is HS). Even assuming possible discrepancies in results due to the
application of different methods for D O M fractionation (Müller and Frimmel, 2002), the M w of FA f rom the Daugava River and Lake Mazais Baltezers appears to be higher than in Finnish and
Swedish (1850-2650) rivers (Pettersson, 1992).
This could be because both Latvian waters were more eutrophic (molecular weight increase with the increase of trophic state) (Klavins and Apsite, 1997) and because their FA were less degraded
(humification decrease the M w ) (Petterson, 1992). Polydisperisty of Lake Mazais Baltezers FA was similar to that o f Nordic rivers (1.5) (Petterson, 1992). The Daugava FA were more heterogeneous and contained more carboxyl groups, indicating lower humification (in summer) than in Lake
Mazais Baltezers. In the autumn and winter, the amount of carboxyl groups and polydispersity
decreased in HS f rom the Daugava, indicating more intense humification during these seasons
(Klavins et al. 1997).
The 1 3 C nuclear magnetic resonance spectra analyses agreed well wi th elemental analyses; HS in the Daugava and Lake Mazais Baltezers were similar and displayed a moderate aromatic and
aliphatic character and a high O-alkyl character, indicative o f carbohydrates (Klavins, 2002). A high concentration of carbohydrates reflects that HS are not subjected to long microbial and
chemical degradation, thus they are potentially bioavailable. A moderate N concentration (Table 2) also indicated relative bioavailability of HS from the Daugava and Lake Mazais Baltezers, since
biodegradability is at a low level of N in HS (Hunt et al. 1999). To summarise, HS f rom the Daugava River and Lake Baltezers were moderately aromatic and acidic with slightly increased
molecular weights and were, hypothetically, rather biodegradable.
16

Table 1. Chemical analyses o f important parameters for the study of raw and drinking water quality of Riga water supply a
Water source Abs254nm H S e
C O D M n Phosphate b Nitrate b
cm" mg HS/1 mg O2/I PO4-P ug/1 NO3 mg/1
-N
River Daugava 0.570 + 0.146 27.70 16.1 1 ? ± 4 . 4 8 b 70 ± 5 1 0.81 ± 0.37 CP drinking water 0.127 + 0.012 5.76 4.55 ± 0 . 4 7 < 2 0 c 0.75 ± 0 . 3 4 CPBAC drinking water d 0.072 + 0.01 3.03 3.24 ± 0 . 4 4 17 + 19 1.03 ± 0 . 5 4 Lake Mazais Baltezers 0.325 + 0.146° 15.56 9.70 ± 1.56 b 68 ± 7 3 0.46 ± 1.09 Artif icial groundwater 0.161 ± 0.016 c 7.44 3.80 ± 0 . 1 0 < 2 0 c 0.26 ± 0 . 0 4
a values are annual arithmetic mean f rom monthly average ± standard deviation f rom mean;
symbols: HS, humic substances; CODMn, chemical ( K M n 0 4 ) oxygen demand; CP - drinking water produced from the Daugava using chemical precipitation; CPBAC - drinking water produced f rom
the Daugava River using chemical precipitation upgraded with two step ozonation and biologically active carbon filtration; b values for year 2000; c values are averages f rom scattered sampling occasions during the last 10 years; d values f rom September 2001 to Apr i l 2002; e determined
spectrophotometrically f rom the calibration graph for Aldrich humic acid: HS = (Abs2 5 4 -0.016V0.0202 ( r = 0.999)
Table 2. Elemental and functional composition and molecular weight distribution of fulvic acids
(FA) f rom the Daugava River (DR), Lake Mazais Baltezers ( L M B ) , compared with Suwannee River (SR) f, the river that originates f r o m a swamp and is strongly influenced by terrestrial
Humic ' " C S " " " • 1 s ^ -* I
C O O H d A r O H e M n
f M w
g Mw/ substance C,% H , % N , % 0 ,% H/C c C /N mmol/g M „ h
FA-DR a 51.42 4.48 0.97 40.21 1.05 61.85 4.35 1.11 1800 3100 1.72 FA- L M B 52.37 4.11 0.91 42.61 0.94 67.14 3.98 1.17 2200 3300 1.50 FA-SR b 52.65 3.71 0.47
b» 39.28 0.81 135.66
/ 1 n o n \ . C .
6.00 1.5 n.d. n.d. n.d.
content of reactive phenolic groups; f a number average molecular weight; s weight average
molecular weight; h polydispers
Substance Society (IHSS, 1981).
molecular weight; h polydispersity; n.d., no data; f reference FA of the International Humic
Studied drinking water quality
The drinking water quality of Riga complies with the current European Council directive (Council
of European Union, 1998), wi th exception o f the parameters COD (5 mg 0 2 / l ) and THMs (100 ug/1) that occasionally exceeded maximum allowed values both in ARG and CP treated water.
After upgrading Daugava WTP with B A C , the THMs at the plant effluent were reduced to low levels. However, COD remained close to the guideline level (RWB, 2002). Because of the high
concentration of organic substances, chlorine consumption is high, residual chlorine concentration
in the networks disappears rapidly especially in summer, and T H M s formation potential is still above 100 ug/1. Removal level of COD and colour during A R G at Baltezers plant is about 70%.
Iron and manganese both exceed the concentration 0.2 mg/1, which is a problem for artificial groundwater at Baltezers plant.
17

6.2. Analysis of the bacterial growth potential in water
Determination of bacterial growth-limiting nutrient
Gibson (1971) postulated that i f the addition of a nutrient does not result in an increase o f the
biomass, then that nutrient is l imiting. This principle is applied in enrichment tests to determine the l imit ing nutrient in pelagic (see review by Beardall et al. 2001) and soil (Aldén et al. 2001) ecosystems. Recently, a similar test showed that not only organic carbon, but also phosphorus
could l imi t bacterial growth in drinking water (Miettinen et al. 1997a; Sathasivan and Ohgaki, 1999). In these tests planktonic bacteria isolated f rom drinking water were usually used. I t is recognized that most bacterial biomass in drinking water develops in the biofilms (e.g. Flemming
et al. 2002), whereas plankton bacteria have little impact on b iof i lm composition (Norton and
LeChevallier, 2000). Thus, in this thesis (paper I V ) , b iof i lm bacteria were used to determine the bacterial growth-limiting nutrient. The b io f i lm bacteria were sampled f r o m the distribution network pipes of Riga. The bacterial biomass was inoculated in two aliquots of the water sample.
Enough phosphorus was added in the first aliquot, but no addition was done to the second one. I f biomasses increased by more than double in the aliquot in which phosphorus was added, phosphorus was assumed to l imi t bacterial growth in the sample. The test did not exclude the
predators and the bacteriovores that might be present in the sample. Thus, the absolute number o f bacteria water has been underestimated.
Analyses of BOM
In principle it is possible to measure some constituents of B O M (e.g. acetic, formic, amino acids,
and carbohydrates) using chemical methods (Peldszus et al. 1998; Jahnel and Frimmel, 1995; Mopper et al. 1992). However, because B O M is composed of many different complex species at
very low concentrations (van der Kooi j et al. 1982), chemical analysis does not provide a complete picture of B O M . Therefore, microbial methods (bioassays) are used instead, which measure B O M
based on two principles: (1) the amount o f B O M is calculated f rom the increase in the number o f pure strain (Pseudomonas fluorescence and Aquaspirillum sp.) of standardized bacteria in a sterile
sample, or (2) by measuring the decrease o f DOC in a water sample after it has been in contact
with a biomass. The fraction of carbon calculated f rom an increase o f t he biomass is A O C (van der Kooi j et al. 1982), whereas a fraction of carbon calculated f rom a decrease of DOC is BDOC (Servais et al. 1987).
Several modifications for either A O C or BDOC methods exist (see review by LeChevallier, 1999). Both methods have their advantages and disadvantages. When both methods were used by Escobar
and Randall (2001) or LeChevallier (1999), no correlation between BDOC and A O C was found,
probably because the methods identify different fractions of the B O M . BDOC represent larger molecules that are mostly responsible for chlorine consumption, whereas AOC represent smaller molecules (<1000 Da) that are most rapidly assimilated in the biomass by bacteria, though larger
molecules can also be included in AOC (Hem and Efraimsen, 2001). AOC test was used in this study. To confirm that AOC really represents the bacterial growth potential of water, heterotrophic
growth response bioassay (HGR) and b i o f i l m formation in the water were also estimated. Because other nutrients can restrict bacterial growth in humic water, analyses of AOC should be done wi th
(AOCpotentiai) and without the addition (AOC„ a tive) of nutrients (Miettinen et al. 1999). A O C n a , l v e
shows the amount of AOC consumed by bacteria at prevailing conditions, whereas AOCpotenuai shows the amount of potentially available AOC.
18

Determination of biologically available phosphorus
As there is a high probability that carbon does not l imi t bacterial growth in humic waters the
concentration of biologically available phosphorus was measured. In drinking water (unless it is treated wi th phosphate containing corrosion inhibitors), phosphorus (P) concentration is generally below the sensitivity levels of standard spectrometric methods (Miettinen et al. 1997a; Appenzeller
et al. 2001; Chandy and Angles, 2001). There are relatively sensitive methods available for the analysis o f P such as a magnesium-induced co-precipitation procedure that measures down to 31 ng of P per liter (Karl and Tien, 1992). However, the bioavailability of P (similar as with DOC) in
water is not only related to its concentration, but also to its distribution over different species, including free orthophosphate (mainly H2PO4 and HPO~ 4), organic phosphorus, condensed
phosphates, and colloidal complexes. Free orthophosphate is most likely to be rapidly utilized by bacteria, though there are several difficulties with using orthophosphate as a measurement for bacterial growth potential. First, orthophosphate analyses in P limiting waters using common chemical (e.g. molybdenum-blue reagent) spectrophotometeric and radiochemical techniques (e.g.
Rigler radiobioassay) (Hudson and Taylor, 1996) are d i f f icul t to accomplish. Generally, the orthophosphate concentration in water is severely overestimated (Selig et al. 2002; Hudson et al. 2000; Baldwin, 1998). Secondly, bacteria under certain conditions catabolize other species of P,
including both natural and synthetic organic P (Lemke et al. 1995; Bujacz et al. 1995; Langowska, 1982). The amount of bioavailble P is not only dependent on the form of P in water, but also on the
properties of the bacteria (e.g. phosphates activity, size and hydrophobicity of the cell surface) living in the studied water (Lemke et al. 1995). For example, mainly bacteria wi th hydrophobic surfaces are able to utilize organic phosphates released f rom cytoplasm and outer membranes of other bacteria (Lemke et al. 1995).
Bioassays, in which the productions of biomass are used to detect biodegradable P, have been used for P analyses in purified wastewater (Ekholm and Krogerus, 1998) and freshwater (Hudson and Taylor, 1996). In this thesis (Paper V , I V ) microbially available phosphorus (MAP) was analysed
using a bioassay recently suggested for drinking water by Lehtola et al. (1999). According to the proposed methodology the maximum growth of test bacteria in a pre-sterilized water sample is related to the P concentration. Inorganic salts (except P) and carbon (sodium acetate) are added to
the water to ensure that only P limits the growth of the test bacteria. Pseudomonas fluorescence
P17, the bacterium strain used in the AOC test, was selected because i t is common in drinking water (Ribas et al. 2000), it is versatile in using organic carbon compounds, and it has the
phosphatase activity needed for hydrolyses of organic phosphorus. M A P is likely to underestimate the total biologically available P concentration because only one bacteria species is used. Results from papers I V and V showed that during P l imit ing conditions native bacteria l iving in the
drinking water assimilated P more efficiently than Pseudomonas fluorescence, meaning that that true amount of P assimilated by bacteria was higher than M A P . However, the M A P method, in
combination with AOC bioassay and HGR, provided relevant information about the bacterial growth potential in water.
Heterotrophic bacteria growth response bioassay
AOC and M A P are indirect measurements of bacterial growth potential. Pure cultures were used in these tests. However, indigenous bacteria l iving in the drinking water distribution network may
utilize nutrients at different rates due the involvement of a complex interaction between species
and other factors discussed by Servais et al. (1987). Unlike AOC or M A P analyses in HGR bioassay, the bacteria are subject to bacteriophage and pathogenic pressure. By incubating samples
19

with indigenous bacteria it is possible to obtain information about the maximum number o f bacteria that potentially can grow in the sample under conditions more common to those occurring
in the net. In short, the procedure was as follows. The sample in which HGR was to be measured was dechlorinated and, with indigenous bacteria present in the sample, incubated in a batch for two
weeks at 20°C in darkness. Increase of bacteria (mesophilic and psychrophilic) numbers (as HPC) was monitored over the whole period, where the maximum number was assumed to show bacterial
growth potential in water.
6.3. Determination of HS concentrations and composition in water
Assuming that HS constitute the bulk of DOC, their concentration can be approximately calculated by measuring DOC or chemical oxygen demand (COD). However, it is only a rough estimate and
does not allow, for example, fractions of DOC responsible for bacterial growth and fractions responsible for THMs formation to be distinguished. Despite applying powerful analytical methods, molecular structure identification in the classical sense has not been successful and is not
anticipated in the near future (Frimmel, 1998). Hence, because there is no unique molecular structure, the concentration of HS can be determined either by measuring surrogate (unspecific)
parameters or by first isolating them and then determining the concentration from measurements o f
isolated mass. Common properties of HS that can be measured to calculate their concentration are: yellow colour, intensive ultraviolet light absorbance, polydispersity, high molecular mass, polyfunctionality, THMs formation potential, as wel l as other more advanced methods such as
enzyme-linked immunoassay or chemiluminescence (Hu et al. 1994). The concentration of HS in water samples f rom laboratory experiments in papers I , I I I , and V I was determined
spectrophotometrically by measuring the colour at a wavelength of 410 nm. The concentration of
HS was calculated from a calibration graph. This method is rapid and accurate, however, the disadvantage is that iron, manganese, nitrate, and turbidity affect the colour of HS, and that absorbance differs with pH, aromaticity, carbon content, and molecular weight of HS (Hautala et
al. 2000).
Elemental (C,0 ,H, and N) and functional composition (carboxyl and phenolic groupos) of HS were determined by standard analyses; this information was used to evaluate some properties of
HS. The (0+N)/C atomic ratio was used to establish (polar/nonpolar)
hydrophilicity/hydrophobicity o f HS. The H/O ratio was used to indicate the humification degree of humic substances (De Paolis and Kukkonen. 1997).
HS (FA and H A ) was used for experiments in papers I , I I I , and V I . Here, HS were isolated and fractioned into FA and HA. Several techniques of isolating HS are available, including freeze
concentration, chemical precipitation, solvent extraction, reverse osmosis, or ultrafiltration (Maurice et al. 2002). Nowadays, sorbents such as non-ionic macroporous resins (e.g. acrylic-
ester X A D - 8 ) (Aiken, 1985) or a weak basic anionic exchanger (e.g. DEAE) (Thurman and Malcolm, 1981) are most frequently used. For isolation of HS with X A D , samples are acidified,
meaning that the natural properties of HS are changed (Peuravuori and Pihlaja, 1998). Therefore, in this thesis HS isolation on D E A E using a method proposed by Thurman and Malcolm (1981)
was preferred.
20

6.4. Measuring biofilm formation
Measuring b iof i lm formation is another approach to estimating bacterial growth potential in water (van der Kooi j et al. 1995). A first step in understanding biof i lm accumulation processes in such a
complex system, as a distribution network, is to measure biomass accumulation on surfaces in an experimental setup under defined conditions (van der Kooi j et al. 1995). In paper V the modified methods earlier described by van der Kooi j (1995) were used. According to this method, the water, f rom where biof i lm formation is to be measured, was pumped through a b io f i lm collector - a glass
column f i l led with plastic cylinders. The cylinders were occasionally removed to measure the number of bacteria (as HPC) that had accumulated on their surfaces. Pipe material might be important, at least in the initial step of bacteria sorption (Niquette et al. 2000; Schwartz et al.
1998). Here, the materials used were inert against corrosion, presumably did not release B O M , and did not consume chlorine. Bacteria were removed f rom the surface of the cylinders with a sterile swab and then pooled in sterile water. This technique is simple; however, i t presumably did not remove all bacteria (Gagnon and Slawson, 1999). The biof i lm collectors were installed in several
places along the distribution system. Unlike AOC and HGR analyses the b iof i lm formation was subjected to actual conditions regarding chlorine and temperature.
6.5. Determination of bacterial number in water and biofilm
A direct count method using a bright f ield microscope was used to determine the total bacterial
number (TBN) after staining the sample with erythrosine. Viable heterotrophic bacterial number was determined with HPC on low nutrient agar R 2 A after seven-day incubation at 20°C (Reasöner and Geldreich, 1985). Agar R?A was chosen because i t shows higher numbers of heterotrophic
bacteria growing in drinking water (i.e. high accuracy) compared to the traditional, high nutrient media (e.g. 5% sheep blood agar) (Carter et al. 2000).
6.5. Sorption experiments with HS on aquifer material and sorbents
Sorption of HS was investigated in dynamic laboratory scale column tests (Paper I I I ) and in equilibrium experiments in a batch system (Papers I , I I I ) . In the column test, a small glass column
was f i l led with the respective sorbent. Water with a known amount o f HS was then pumped through the column. Breakthrough curves o f HS through the sorbent were constructed according to
the measurements of HS concentrations at the outlet of the column. In batch sorption experiments, water with either a known amount of HS and sorbents (Paper I I I ) or aquifer material (Paper I ) was agitated until maximal sorption occurred. The sorbed amount was estimated f rom differences in concentrations of HS in the water before and after contact with the sorbent. These kinds of
experiments allowed studying the effects o f different variables such as HS type, composition of sorbent, pH, and ion strength on the sorption o f HS. Straining and interception may be involved in
removal of HS in the column tests because o f t he involvement o f hydrodynamic forces (in addition to physicochemical forces). However, as hydrodynamic forces are less important in a batch system, the sorption was assumed to be a major mechanism for HS removal in this batch mode.
Aquifer material f rom infiltration basins (Paper I) contained organic matter that was most likely formed f rom humic water infiltration though this material. Due to the release of previously
retained D O M from the aquifer material the results of the sorption experiments could not been analysed by Freundlich or Langmuir isotherms. Hence, the initial mass approach used for sorption
studies on soil was used instead (Nodvin et al. 1986). Release of organic matter f r om aquifer
21

material was considered when calculating the amounts of sorbed HS. The concentration of a substance, retained or released (normalized to sorbent mass), was plotted as a function of the initial concentration of the substances (also normalized to sorbent mass).
Analyses of aquifer materials
Grain size distribution and chemical composition o f minerals influence their ability to adsorb HS. Aquifer material for the sorption experiment was sampled f rom infiltration basins used for ARG
for more than 50 years at Baltezers plant. Grain size distribution was estimated by dry sieve analysis. Clay content was determined by applying the sedimentation-hydrometer method. Chemical composition of aquifer material was determined by standard methods after its digestion
with strong acid. Mineralogy was determined wi th x-ray diffraction, and the content of organic carbon using wet digestion wi th potassium dichromate.
6.6. Determination of iron-humic complexes in humic groundwater
In groundwater at neutral pH dissolved-free iron dominates as a positively charged ion (cation). When iron bounds to HS the produced iron-humic complex acquires a negative charge (anion). Hence, free iron can be separated f rom water by cation exchange resin whereas iron-humic
complex by anion exchange resins (Hiraide et al. 1988). This principle has been used in several studies for speciation o f iron in surface water (Appelblad et al. 1999; Hiraide, 1992; Petterson et al.
1992) and for speciation of other heavy metals (Cu, Cd) in groundwater (Christensen and Christensen, 1999). Applying this approach for iron speciation in groundwater is d i f f icul t because
by fol lowing the procedure proposed for surface water, part of the iron becomes oxidised during the sampling, and the actual speciation o f iron in groundwater is altered. To avoid this problem a
special water sampler that allows taking a sample without contact with ambient air was proposed
(Paper V I ) . First, one sampler was f i l led wi th D E A E anion exchange resin (fibrous cellulose type weak base exchanger with diethylamino groups) and another with cation exchanger resin (gel-type strong acid exchanger with styrene divinylbenzene groups). The former resin removed iron-humic
complexes whereas the latter removes free iron ions. The difference between the initial
concentration and amount removed allowed the determination o f the respective species o f iron in groundwater. Analyses were performed only for dissolved (truly dissolved and colloids) species
that are defined as a fraction of the total iron that passes through 0.45 pm membrane filters. The fraction of total iron retained by the fi l ter was defined as particulate iron. According to this methodology the speciation analyses done for A R G at Baltezers plant and for some humic groundwaters in the vicinity of Riga (Paper V I ) .
22

7. M A J O R R E S U L T S AND D I S C U S S I O N
7.1. Sorption of HS on aquifer material during artificial recharge of groundwater
Experiments in a batch system (Paper I) were conducted to evaluate the influence of physical and chemical factors that determine the sorption of HS on aquifer material. The influence of aquifer
material composition was evaluated by measuring the sorption of HS to the material sampled f rom
below the infiltration basin and its major representative sorbing phases: aluminium and iron oxide, different clays, and organic matter (lake sediments). The influences of HS properties were tested by comparing the sorption of H A , isolated f r o m soil (soil H A ) , w i th the sorption of FA, isolated from a river (aquatic FA). The soil H A was of higher molecular weight and more aromatic,
hydrophobic, and with a lower amount of carboxyl groups than aquatic FA. Wel l defined, relatively homogenous, commercially available H A (Aldrich H A ) were also used for comparison. For the sorption study the aquifer material was taken f rom below of the infiltration basin, dried,
and equilibrated with different concentrations of various HS in a batch system. The analyses of aquifer material revealed a composition mainly of silica (90-95 wt % as quartz), aluminium, iron
(0.06-0.07 wt % ) , and organic carbon (0.3-0.6 wt % ) . The texture o f the aquifer material ranged from clay (1-22%) to fine sand.
The batch sorption tests showed that sorption o f Aldrich H A increased with an increase in the
percentage of clay, and decreased wi th an increase of organic carbon in the aquifer material,
whereas iron did not contribute significantly to sorption. Sorption isotherms revealed that soil H A and Aldrich HS were better sorbed than aquatic FA on aquifer material wi th a low percentage of clay, Figure 3. Therefore, sorption increased with an increase o f molecular weight and aromaticity
and decreased with an increase of carboxylic groups. Aquifer material wi th a higher amount o f clay sorbed slightly higher amounts of FA; however, H A were generally removed better than FA. A l l relevant phases of aquifer material, including aluminium oxide, clay, and organic rich
sediments, showed similar or better sorption of H A compared to FA wi th the exception of iron
oxide, which showed a higher aff ini ty for FA. No increase of HS removal by aquifer material with an increase of its iron content can possibly be explained as fol lows. Since the tested aquifer material had been used for ARG for a long time, positively charged iron had become coated wi th
organic matter, resulting in negatively charged surfaces (Day et al. 1994). Consequently, ligand exchange or electrostatic interaction between carboxyl groups o f HS and iron hydroxyl groups was hampered, decreasing the overall sorption of HS.
23

O 10 20
Humic substances added (mg/g)
Figure 3. Sorption of different humic substances on upper layer of aquifer materials wi th similar properties, but with clay content of 1% (—) and 3.3% (— — ) (sample size 500 mg; pH 6.5; sorption time 16 h).
Sorption of HS on aquifer material fol lowed the general trends for sorption characteristics of HS on minerals (Marshall et al. 1998), viz. sorption increases with decreasing pH, increasing aromaticity, and decreasing polarity of the humic substances. I t is known that FA are effectively
sorbed on pure mineral surfaces rich in hydroxides as a result of ligand exchange or electrostatic interaction (Gu et al. 1993). Aquifer materials f r o m artificial recharge aquifers contained a low
amount of oxides and were covered wi th organic matter f rom the infiltrated surface water. As a result the hydrophobic HS were preferably sorbed compared to more acid fraction o f HS most
likely as a result of hydrophobic interactions. The strength of the bonds between aquifer material and organic coverage as well as polyelectrolytic properties largely controls the sorption process of
HS. HS are macromolecules behave like polyelectrolytes; thus their sorption is determined not only by the availability of sites, but also by lateral interaction between themselves and the steric
arrangements of the macromolecule (Vermeer et al. 1998). Due to a high amount of functional
groups, HS stretches (due to lateral repelling between the functional groups) and when becomes sorbed on the surfaces occupies a large area that makes overall sorption efficiency o f FA lower than o f HA. Results f rom present study, that showed preferable removal of larger molecular
weight, more hydrophobic fractions of HS in batch mode, are consistent with earlier results f r o m f ie ld studies at ARG plants and slow sand filtration (McCarty et al. 1993; Collins et al. 1992; Schwardzenbach et al. 1983). Preferable sorption o f more aromatic, hydrophobic H A over acidic, hydrophilic FA was shown also in soil (Kaiser and Zech, 1997; Jardine at al. 1989) and other
geological material (Meiere ta l . 1999).
Overall, sorption efficiency o f HS during humic water artificial recharge to groundwater was
highly dependent on their properties and composition of aquifer material. The removal of HS f r o m
humic water with a high amount of FA using A R G would not be highly efficient unless aquifer material is rich in iron or aluminium hydroxides. In aquifer material that is poor with oxide
surfaces and clay, FA are conservative and would appear as a major constituent of D O M in recharged groundwater.
24

depth (cm)
Figure 4. Changes in total bacterial number (TBN) and abundance o f cyanobacteria with the depth
in soil o f the borehole under on of the infiltration basins at Baltezers plant
7.2. Effect of blooming on the biodegradation of B O M during humic water artificial recharge
The biodegradation rate o f B O M in porous media is dependent on the bacterial biomass in the
biof i lm (Carlson and Amy, 1998: Collins et al. 1992), where most bacterial activity takes place (Lehman and O'Connell, 2002). Hence, a significant decrease in the number of bacteria can be assumed to indicate lower biodegradation efficiency. It was examined in Paper I I whether the
total bacterial number below an infiltration basin used for ARG was significantly affected by a possible increase of the cyanobacteria number in the subsurface; this resulting f rom algal bloom in
the recharging surface water (Lake Mazais Baltezers).
Increase in concentration of toxins in water due to cyanobacteria growth is common in Lake
Mazais Baltezers (Eynard et al. 2000; Druvietis. 1997). Soon after one of the blooms, the number of cyanobacteria and total number of bacteria below the basins were analysed. Cyanobacteria were
found down to a depth o f 200 cm whereas other bacteria proceeded deeper and were found in concentrations almost a mil l ion times higher, Figure 4. The cyanobacterial numbers were
positively correlated wi th the total bacteria number possibly indicating that produced toxins or other mechanisms that may impede bacterial metabolism did not have a significantly negative
effect on the total bacterial number in the b iof i lm and subsequently did not inhibit biodegradation of B O M . Moreover, the total number of bacteria was even higher during and after bloom than in
the period when blooms did not occur (Juhna and Springe, 1998b). Thus, the biodegradation of B O M in artificially recharged groundwater was not considerably influenced by the cyanobacteria blooms. This also agrees with earlier findings that blooms through toxins and others blooming
exudes (e.g. amino acids, peptides, fatty acids, carbohydrates) can rather stimulate the growth of bacteria (e.g Shingomonas sp.) by serving as a carbon or nitrogen source (Boualam et al. 2002;
Acea et al. 2001), and that bacteria are able to degrade toxins at a reasonable level during ARG
(Miller and Fallowfield, 2001).
25

7.3. Removal of HS from artificially recharged groundwater using sorbents
In paper I I I different sorbents (anthracite, GAC, other nonpolar sorbent) and ion exchange resins (weak base anion exchange resin, strong base anion exchange resin, cation exchange resins) were
tested for removal of different HS (FA and H A ) in batch sorption tests and in a laboratory scale column. Results generally showed that macroreticular weak anion resins (matrix of methacrylic
acid derivatives) were the most efficient sorbent for the majority o f HS. Despite the lower carboxyl group density (acidity) HA were better removed than FA. Nevertheless, it was observed that
removal of acidic, low aromatic FA by anion exchange resin was significantly higher than by GAC. Results f rom Paper I suggest that FA could be found as a dominant fraction of HS in some artificially recharged groundwaters. Hence, macroreticular weak anion resins appear to be well
suited for use in additional treatments of such waters. Despite the lower concentrations of
functional groups that can be used for the exchange, macroreticular, weak base anion exchange resins were more efficient in removing HS than a gel-type, strong base anion exchanger, which indicates that an availability of surfaces is important to the sorption o f HS. However, the results also showed that (1) H A were also partly retained by a cation exchange resin and nonpolar sorbent,
and (2) sorption at acidic pH was more pronounced for a nonpolar sorbent. These results agreed with earlier findings that HS are partly removed by a hydrophobic attraction to a nonpolar matrix
of anion exchange resin (Croué et al. 1999).
In pilot scale columns, the removal of D O M f rom artificially recharged groundwater (at Baltezers plant) using a strong base anion exchange resin or activated carbon was studied. The anion
exchange resins were more efficient than GAC, though the difference in retention capacity was lower than that observed for removal of FA in the batch mode. This may be because artificially
recharged groundwaters, besides FA, also contained D O M fractions, presumably low molecular
substances, which were better removed by GAC than by an anion exchanger resin. These fractions contributed to B O M , creating a problem in applying anion exchange resins for post-treatment of
A R G as it may be insufficient in eliminating B O M . Another constraint in applying anion exchange resin could be the high regeneration expense of the resin, and its decrease of treatment effectiveness due to the presence of iron or sulphate ( K i m and Symons, 1991), thereby increasing
the expense o f these methods. Thus, before practical application o f anion exchange resin for post-treatment of artificially recharged groundwaters the economical and engineering aspects should be
evaluated.
7.4. Bacterial growth potential in drinking waters prepared from humic surface water
Results in Paper I V showed (Figure 5) that B O M levels, measured as AOC, in the eutrophic humic Lake Mazais Baltezers and Daugava River were similar or higher than values reported f rom
other humic waters (Miettinen et al. 1999). The concentration of A O C in a water sample, measured
after adding a mixture o f inorganic nutrients (AOCpotentiaiX was significantly higher than AOC in a sample to which inorganic nutrients were not added (AOC n ative) (Figure 5), indicating that inorganic nutrients, such as phosporus or nitrogen, limited bacterial growth in the river on all sampling occasions f rom summer to winter. This difference was lower in the Mazais Baltezers and was absent in the autumn, showing that inorganic nutrients were not always restricting the bacterial
growth humic lake.
26

ü o
1000
900
800
700
600
500
400
300
200
100
0
fl r i , fl I
il k
11
J L , J L
R Snet Gnet
20 13 AOC
18 D AOCp 0 l e n t i a ]
16 B M A P
14
12 a:
10 < o BX
3.
Figure 5. Mean concentrations of assimilable organic carbon (AOC) and microbially available phosphorus (MAP) in water f r om different sampling sites: R, raw water f rom Daugava River; S, chemically precipitated surface water at the plant effluent; Snet, treated surface water in the
distribution net; L, raw water of Lake Mazais Baltezers; G, artificially recharged groundwater; Gnet, groundwater in the distribution net over a nine-month period.
The finding that organic carbon was not always l imit ing bacteria development in surface water is not unique. The role of phosphorus and nitrogen in l imit ing bacteria growth has been recognized
for both humic and fresh waters (Olsen et al. 2002; Jansson et al. 1996). Phosphorus limitation is expected more in humic water because phosphate is readily bound to iron-humus complexes and may not be biologically available to bacteria (Shaw et al. 2000; Jones, 1988; Tipping, 1981).
Chemical treatment removed 20% of AOCpotentiai f rom water of the Daugava River, whereas more than 95% of M A P was removed (Figure 5). As a result, phosphorus became l imit ing for bacterial growth in drinking water, but no other nutrients, which was confirmed by nutrient enrichment experiments using indigenous b iof i lm bacteria f rom the distribution network, Figure 6. Due to a
deficiency of phosphorus, only 40% of AOCpo tentiai was assimilable (as AOC n ative) for bacteria in drinking water. The bacterial formation potential had decreased mainly because o f phosphorus reduction during chemical water treatment, and only moderately due to the reduction o f organic
matter. Although microbiologically available phosphorus (MAP) was effectively eliminated during chemical treatment, a significant part of phosphorus was still present since AOC n ative remained rather high (278 pg AOC-C/1) compared to the usual concentration (10-50 pg AOC-C/1) for
biologically stable water (LeChevallier, 1991; van der Koo i j , 1992). High bacteria growth potential was also confirmed when measuring maximum growth of indegenious bacteria (HGR test). According to the tests the drinking water could potentially support the growth o f as many as 10 s-109 CFU/L of a natural microbial population.
27

5 10 15
incubation time (d)
Figure 6. Growth of a b iof i lm bacteria consortium (Pseudomonas aeruginosa and Sphingomonas
paucimobilis) in chemically treated surface water. The bacteria consortiums were removed f rom
the walls of distribution pipes and incubated in water samples before (LO), after the addition of
100 pg/L of phosphorus (LP), or after the addition of a mixture o f inorganic nutrients ((NHihSO.^,
K H 2 P 0 4 , M g S 0 4 x 7 H 2 0 , CaCl 2 x2H 2 0 , NaCl) containing no phosphorus (LN-P). The number o f
bacteria was determined by heterotrophic plate counts (HPC) on R2A agar after 7 days at 20 ± 2°C.
Points represent average values obtained f rom two dilutions of two replicates.
During water filtration f rom Lake Mazais Baltezers to groundwater, about 60% of M A P was
removed, whereas the removal of AOC p o t entiai was about 30%, Figure 5. Since M A P was not effectively reduced during water infiltration to groundwater, phosphorus did not always l imit the growth of bacteria in the artificially recharged groundwater at the plant effluent and nearly all
A O C (AOCnative) was available (as AOCpotentiai). The ratio between AOCpotentiai and M A P was close to the critical requirements for bacterial growth (C:P = 1:100), meaning that any of these two nutrients may easily become limiting in drinking water.
The role of phosphorus in regulating bacterial growth was observed earlier in water supply systems
in Finland and Japan (Miettinen et al. 1997a; Sathasivan and Ohgaki, 1999). Here was found that the water treatment process largely determine nutrient that is l imiting bacterial growth in drinking water. Phosphorus is likely to become limiting during humic water treatment with aluminium
coagulant. This is probably because phosphorus becomes bound to aluminium during coagulation more effectively than organic carbon (Nishijima et al. 1997) and nitrogen.
Although groundwater at the plant contained higher levels of M A P and recieved lower chlorine
doses (see section 6.1) than chemically treated water, the increase of bacteria number in water during groundwater distribution was not greater (Figure 7). The lack of positive correlation
between M A P and the number of suspended bacteria could be because most of bacterial growth in distribution networks occurs in biofilms rather than in water.
28

10 20
water distribution time (h)
30 10 20 30
water distribution time (h)
Figure 7. Increase of bacteria number during distribution of (A) chemically treated surface water
and (B) groundwater in distribution networks. The number of bacteria was determined by
heterotrophic plate counts (HPC) on R 2 A agar after 7 days at 20 ± 2°C.
7.5. Biofilm formation in drinking water distribution networks of humic waters
Measurement of b io f i lm formation (Paper V ) in chemically treated water ( f rom days 0 to 80) showed significantly lower levels than in artificially recharged groundwater at the plant, Figure 8.
After introducing an additional treatment step (BAC) at the chemical treatment plant (day 83), the MAP concentration decreased further to limits of method detection (<0.08 pg P/l). However, this
treatment resulted in rapid b iof i lm formation that reached levels similar to artificially recharged groundwater. This is because high chlorine doses (residual C l 2 >1.2 mg/1) were previously used at
the chemical treatment plant whereas after start-up of B A C chlorine doses were decreased to levels used for the recharged groundwater (residual C l 2 <0.4 mg/1). This shows that the chlorine concentration had a significant effect on regulating b io f i lm formation in drinking water prepared
from humic waters. However, this effect was observed through the start-up stage of B A C treatment
during the summer season, and should be tested after start-up stages and at different seasons before general conclusion are drawn.
Although removal of M A P did not decrease b io f i lm formation at the plant the effect of decreasing MAP concentration on b io f i lm formation was observed in the distribution network where residual
chlorine was absent. In distributed, chemically treated surface water, which contained low levels of MAP, b iof i lm growth was on average almost ten-fold lower than in distributed groundwater which
contained significantly higher levels o f M A P , Figure 8. Hence, an increase o f M A P concentration increased the b iof i lm formation rate, which is in agreement with other studies that showed
phosphorus influence on bacteria growth in b io f i lm (Lehtola et al. 2002).
29

7
Biofilm formation time (d)
Figure 8. Heterotropic plate counts (HPC) in biofi lms formed f rom chemically treated surface
water at the plant ( • ) and in the distribution network ( • ) , and f rom groundwater at the plant ( • ) and in the distribution network (o).
7.6. Iron species in humic groundwaters
Figure 9, adapted f rom paper V I , shows the distribution of different iron species as determined by
a proposed ion-exchange speciation method in groundwater f rom areas of humic waters. This
simple method allowed amounts of iron-humic complexes, free iron, and particulate iron to be rapidly measured. The obtained results were compared with analyses of iron species by the
computer model WinHumicV (Gustafsson, 1999), and revealed that with the ion-exchange speciation method the amount of iron-humic complexes in studied groundwaters was about 30%
higher than determined by the computer model. The difference was explained after measuring the size of iron-humic complexes. The DEAE resins used for isolation o f iron-humic complexes retained not only truly dissolved (< 10 kDa), but also colloidal species, whereas the latter species
were not included in the the computer model calculation.
0,00 0,50 1,00
Fe, mg/1
1,50 2,00
Figure 9. Concentrations of iron-humic complexes (FCNOM), free iron cations (Fepc), and particulate iron (Fe PM), as determined with an ion-exchange method, in artificially recharged (G2) and natural ( G l ) groundwater at Baltezers plant, in artesian groundwater ( D l ) and in groundwaters
draining peat deposits areas (P) or swamp (B).
30

About 40% o f iron in the recharged groundwater at Baltezers plant was bound to HS. forming
either truly dissolved or colloidal species, Figure 9. Note that this was in groundwater where the concentrations o f organic matter (see section 6.1) did not significantly exceed the maximum allowable levels in drinking water. In groundwater affected by drainage f rom peat deposits or
swamps, the amount of organic matter was higher than in the recharged groundwater. This resulted in higher amounts o f iron-humic complexes as well as of total iron. This agrees with earlier studies that showed iron concentration increases with increases of HS (Olivié-Lauquet et al. 1999). In groundwater, an increase in the amount of iron with an increase of HS is l ikely. The proposed
method allows to identify the amount of iron bound to HS, which may be an important criteria at the selection o f methods for iron removal f rom humic groundwater
31

8. C O N C L U S I O N S
The fo l lowing main findings can be drawn f rom this study.
• Sorption o f humic substances to aquifer material during artificial recharge o f groundwater was
significantly affected by the amount of clay, the amount o f organic matter in the aquifer
material, the elemental, functional composition and molecular weight o f the humic substances,
and pH of water. Sorption increased with (a) clay content, (b) a decrease o f loosely bound
organic matter in the aquifer material, and (c) a decrease in the p H of water. Larger molecular
weight, more aromatic and hydrophobic fractions of HS (humic acids) were more efficiently
sorbed than acidic fractions (fulvic acids). Physical sorption (mainly hydrophobic attraction)
appears to be more important mechanisms than chemical sorption in removal of humic
substances by aquifer material during artificial recharge of groundwater.
• Macroreticular anion resin and granular activated carbon were effective sorbents for removal
of humic substances f rom artificially recharged groundwater.
• Algal blooms in a humic lake appear to not significantly affect the biodegradation of D O M
below the infiltration basins during the lake water artificial recharge to groundwater
• During chemical treatment, bacterial growth potential mainly decreased due to removal of
phosphorus, rendering this element l imiting for bacterial growth in drinking water. Because of
a lower phosphorus removal, artificially recharged groundwater produced f rom a humic lake
had a higher bacterial growth potential (i.e. concentration of l imit ing nutrient) than water
produced f rom a river using chemical precipitation. Even though concentrations of the limiting
nutrient in chemically treated water were lower than in groundwater the increase of bacterial
numbers in both waters during their distribution in the net was not significantly different.
• Concentrations of chlorine or phosphorus influenced b io f i lm formation in distribution net.
Because of lower phosporus concentration, chemically treated water produced lower b iof i lm
growth than groundwater in parts of the network where chlorine was absent. Residual chlorine
concentration higher than 1.2 mg/1 was needed to signicantly reduce b i o f i l m formation.
• It is possible to determine the amount of the iron-humic complexes (truly dissolved and in
colloidal form) in humic groundwater using ion-exchange resins. Groundwater affected by
humic water, including natural and artificially recharged groundwater, contains significant
amounts of iron-humic complexes.
32

9. Future research
In this thesis sorption of humic substances on aquifer material used for artificial recharge of groundwater was investigated at equilibrium conditions in a static mode (batch sorption tests).
However, in real systems, equilibrium is not likely to be attained. In addition, in real systems, due to the colloidal properties of HS and water f low through the aquifer material, straining and
interception of HS in the porous part of the aquifer material are likely to be important mechanisms for removal of HS. Thus, future studies about removal of HS should consider its colloidal properties during water transport in the porous media.
We observed that the bacterial strain used in M A P bioassays exhibited lower growth response than
indigenous bacteria l iving in the drinking water. Therefore, to evaluate bacterial growth more completely in water where phosphorus is l imit ing, M A P analyses should be improved by adding
more bacterial species.
The study concluded from previous findings that phosphorus rather than carbon is l imi t ing bacterial growth in chemically treated humic waters. In theory, phosphorus rather than carbon can
continuously be accumulated in the distributing network and bacteria can recycle it rapidly as it
occurs in natural waters (Hudson and Taylor, 1996), thus they survive long after phosphorus is depleted (Vadstein, 2000). Hence, studies about the possibilities to produce biologically stable water (no bacteria regrowth) by removing phosphorus while considering its possible recycling
should be continued. The influence of phosphorus removal on the survival of pathogens in b io f i lm should also be studied.
Even though chemical coagulation effectively removes phosphorus and increases the biological
stability of humic waters, removal of carbon is moderate, resulting in a high amount o f organic matter in drinking water. Organic carbon, though not available for bacteria, consumes chlorine and
increases the by-product levels in drinking water. The usual biological treatment that utilises bacteria is not very effective in removing this carbon because of phosphorus shortages for the
bacteria. Thus, methods to remove carbon under conditions of phosphorus limitation should be
investigated.
33

10. Acknowledgements
During my doctoral studies numerous individuals and several organizations have assisted me.
Firstly, I thank Jörgen Hanæus , Professor at Division of Sanitary Engineering at Luleå University of Technology, for giving me the opportunity to study under his guidance and for helping me with
all kinds of scientific, academic, and administrative matters.
I am indebted to docent Janis Sprogis, my supervisor at Riga Technical University. His scientific
reasoning, amazing natural buoyancy, and human warmth have been an example and
encouragement to me throughout these years.
I acknowledge the financial support o f Riga Municipal Enterprise Riga Water. Without our cooperation in several projects about the water supply of Riga, the completion of this thesis would
not have been possible. I am indebted to all the helpful staff at the company including Una Zilbere, Silvija Pastare and Benite Naudzune for providing me with data and assistance in water sampling.
Thanks are due to the co-author of several articles, Professor Maris Klavins, for leading me into the mysterious world of humic substances, helping with good advice during the study, and for
providing me with data and useful comments on the thesis. Thanks to all my colleagues at his laboratory including Mara, Linda, Ilga, Baiba, and Kristine for assisting me in the many laborious
humus sorption experiments.
I am indebted to all-former and present doctoral students, and staff including Anneli , Annelie,
Daniel, Erica, Elisabeth, Helena, Magnus, Maria, Kerstin, Roger, and the ever-changing secretaries f rom our division at Luleå University of Technology. Although you may not have noticed, you
have been my family during my visits to Sweden and made my life more fun .
I thank my co-workers Vizma, Zaiga, and Daina f rom Microbial Strain Collection of Latvia for carrying out microbiological analyses and always being so responsive to my spontaneous ideas.
I thank all f r om the Laboratory of Hydrobiology at Institute of Biology in Salaspils for their cooperation. We proved that a natural scientist and sanitary engineers could also possibly do
research together.
I acknowledge Professor Pertti Martikainen and researchers Markku Lehtola and I lkka Miettinen
f rom the National Public Health Institute in Finland. The research carried out by these intellectuals
encouraged me to study biological stability of drinking water in Latvia.
The thesis was partly financed by a grant received from the Latvian Academy of Science for which
this organization is acknowledged.
Wayne Chan is acknowledged for proofreading the English.
I acknowledge all the kind Swedish people. The Nordic climate seemed not that cold in an
atmosphere of human warmth and helpfulness.
I am thankful to the most important people in my l ife: my mother, father, sister, wife Linda, and
son Matiss for continually loving me and believing in me. Without a doubt, this doctoral thesis has been a test for all of us!
34

11. List of references
Acea, M.J., Diz, N . and Prieto-Fernåndez, A . (2001) Microbial populations in heated soils inoculated wi th cyanobacteria. Biol. Fertil. Soils, 33, 118-125.
Aiken, G. (1985) Isolation and concentration techniques for aquatic humic substances. In: Humic substances in soil, sediments, and water. Wiley. New York.
Aiken, G. and Cotsaris, E. (1995) Soil and hydrology: their effect on N O M . Journ. Amer. Water
Works Assoc., 87(1), 36-45. Alborzfar, M . , Villumsen, A . and Gron, C. (2001) Ar t i f ic ia l recharge of humic ground water. J.
Environ. Qual, 30(1), 200-209.
Aldén, L . , Demoling, F. and Bååth, E. (2001) Rapid method of determining factors l imit ing
bacterial growth in soil. Appl. Environm. Microb., 67(4), 1830-1838. Appelblad, P.K., Baxter, D.C. and Thunberg, J.O. (1999) Determination of metal-humic
complexes, free metal ions and total concentrations in natural waters. J. Environ. Monit., 1, 211-217.
Appenzeller, B . M . , Batté, M „ Mathieu, L . , Block, J.C., Lahousseine, V. , Cavard, J. and Gatle, D. (2001) Effect of adding phosphate to drinking water on bacterial growth in slightly and highly corroded pipes. Wat. Res., 35(4), 1100-1105.
Apsite, E. and Klavins, M . (1998) Assessment o f the changes of COD and colour in rivers of
Latvia during the last twenty years. Environment International, 24 (5/6), 637-643. Astier, F., Paquin, J.L., Mathieu, L . , Morlot , M . and Hartemann, P. (1995) Study of development
of musty taste in water according to its aging process in pilot plant. Environ. Technol., 16, 955-965.
Aven, M.J . and Koopal, L . K . (1999) Kinetics of humic acid adsorption at solid-water interfaces. Environ. Sei. Technol., 33, 2739-2744.
Averett, R.C., Leenheer, J.A., McKnight , D . M . and Thorn, K . A . (1989) Humic substances in the
Suwannee river, Georgia: Interactions, Properties, and proposed structures. United States Geological Survey Open-File Report 87-557; Denver, Colorado.
Baldwin, D.S. (1998) Reactive "organic" phosphorus revised. Wat. Res., 32 (8), 2265-2270. Bano, N . , Morän M.A . , and Hodson R.E. (1997) Bacterial utilization of dissolved humic
substances f rom a freshwater swamp. Aquatic Microbial Ecology, 12, 233-238.
Beardall, J., Young, E. and Roberts, S. (2001) Approaches for determining phytoplankton nutrient limitation. Aquatic Sciences, 63, 44-69
Block, J.C., Haudidier, K. , Paquin, J.L., Miazga, J. and Levi , Y . (1993) B io f i lm accumulation in drinking water distribution systems. Biofouling, 6, 333-343.
Boualam, M . , Mathieu, L . , Fass, S., Cavard, J. and Gatel, D . (2002) Relationship between coliform culturability and organic matter in low nutritive waters. Wat. Res., 36(10), 2618-2826.
Bower, H . (2002) Ar t i f ic ia l recharge o f groundwater: hydrology and engineering. Hydrology
Journal, 10, 121-142.
Bro, A.E. , Berghult, B. and Hedberg, T. (1999) Drinking water distribution - the effect of natural organic matter (NOM) on corrosion o f iron and copper. Wat. Sei. Tech., 40 (9), 17-24.
Bruce, D., Westerhoff, P. and Brawley-Chesworth, A . (2002) Removal of 2-mefhylisoborneol and geosmin in surface water treatment plants in Arizona. AQUA, 51(4), 183-197.
Bujacz, B. , Wieczorek, P., Krzysko-Lupicka, T., Golab, Z., Lejczak B., and Kavfarski, P. (1995) Organophoshonate utilization by the wild-type strain of Penicillium notatum. Appl. Envir.
Microbiol., 61(8), 2905-2910.
Burns, R . G (1983) Extracellular enzyme-substrate interaction in soil. I n : Microbes in Their
Natural Environments. Editors: Slater J. H . , Whittenderg R. and Winpenny J. W . T. Publisher: Cambridge University Press.
35

Camper, A . K . (2002) Involvement of humic substances in regrowth. 423-438. pp. In: Proceeding f rom NSF International/World Health Organization Symposium on HPC Bacteria in Drinking Water. Apr i l 22-24, 2002. Geneva. Switzerland.
Camper, A .K . , Jones, W . L . and Hayes, J.T. (1996) Effect of growth conditions and substratum
composition on the persistence of coliforms in mixed-population b iof i lm. Appl. Emir.
Microbiol, 62(11), 4014-4018. Carlson, K. and Amy, G.L. (1998) B O M removal during biofiltration. Journ. Amer. Water Works
Assoc., 90(12), 42-52.
Carter, J.T., Rice, E.W., Buchberger, S.G. and Lee, Y . (2000) Relationships between levels o f
heterotrophic bacteria and water quality parameters in a drinking water distribution system. Wat. Res., 34(5), 1495-1502.
Chandy, J.P. and Angles, M . L . (2001) Determination of nutrient l imit ing b io f i lm formation and the subsequent impact on disinfectant decay. Wat. Res., 35(11), 2677-2682.
Christensen, J.B. and Christensen, T .H . (1999) Complexation of Cd, N i , and Zn by DOC in
polluted groundwater: a comparison of approaches using resin exchange, aquifer material sorption and computer speciation models ( W H A M and MINTEQ2) . Environ. Sc. Technol., 33, 3857-3863.
Collins, M.R., Eighmy, T.T., Fenstermacher, J.M. Jr. and Spanos, S.K. (1992) Removing natural
organic matter by convention slow sand filtration. Journ. Amer. Water Works Assoc., 5(84), 80-90.
Council of European Union (1998) Council Directive 98/83/EC of 3 November 1998 on the quality of water indented for human consumption. Official Journal of the European
Communities, L33, 32-54.
Crandall, C A . , Katz, B.G. and Hirten, J.J. (1999) Hydrochemical evidence for mixing of river water and groundwater during high-flow conditions, lower Suwannee river basin, Florida,
USA. Hydrogeology Journal, 1, 454-467.
Craun, G.F. and Calderon, R.L. (2001) Waterborne disease outbreaks caused by distribution system deficiencies. Amer. Water Works Assoc., 93(9) 64-74.
Croué, J.P., Violleau, D., Bodaire, C. and Legube, B. (1999) Removal o f hydrophobic and hydrophilic constituents by anion exchange resin. Water Science and Technology, 40(9), 207-215.
Dai, X . and Hozalski, R . M . (2002). The effect of N O M on removal of Cryprosporidium parvum
oocysts in rapid filters. Wat. Res., in press.
Davis, C.J., Eschenazi, E. and Papadopoulos, K .D. (2002) Combined effect o f Ca 2 + and humic acid and colloid transport though porous media. Colloid. Polym Sei, 280, 52-58.
Day, G.M., Hart, B.T., McKelvie , I .D. and Beckett, R. (1994) Adsorption o f natural organic matter onto goethite. Colloid and Swface A, 89, 1-13
De Haan, H . (1983) Use of ultraviolet spectroscopy, gel fi l tration, pyrolysis/mass spectrometry and number of benzoate-metobolizing bacteria in the study of humification and degradation o f
aquatic humic matter. In: Aquatic and Humic Material. Editors: R.F. Christman and E.T. Gjessing. Publisher: Ann Arbor Science.
De Paolis, F. and Kukkonen, J. (1997) Binding of organic pollutants to humic and fulvic acids: influence of pH and the structure of humic material. Chemosphere, 34(8), 1693-1704.
De Wit t , J.C.M. and Van Riemsdijk, W . H . and Koopal L .K. (1993) Proton binding to humic
substance. 1. Electrostatic effects. Environ. Sei. Technolo., 27, 2005-2010. DRBP (2000) Reports f rom Daugava River Basin Project, www.daugava.lv
Drikas, M . , Chow, C.W.K., House, J. and Burch, M . D . (2001) Using coagulation settling to remove toxic cyanobacteria. Journ. Amer. Water Works Assoc., 93(2), 101 -111.
Druvietis, I . (1997) Algae as criterion of environmental state of Latvia inland water bodies. Doctoral thesis. University of Latvia, Institute of Biology. Riga. 102 pp.
36

Edwards, M , Benjamin, M . M . , and Ryan, J.N. (1996) Role of organic acidity in sorption of natural organic matter ( N O M ) to oxide surfaces. Colloids and Surfaces A, 107, 297-307.
Ekholm, P. and Krogerus, K. (1998) Bioavailability of phosphorus in purified municipal wastewater Wat. Res., 32(2), 343-351
Ellis, B.D., Butterfield, P., Jones, W . L . , McFeters, G.A. and Camper. A . K . (2000) Effects of carbon source, carbon concentration, and chlorination on growth related parameters of heterotrophic b io f i lm bacteria. Microb. Ecoi, 38, 330-347.
Escobar, I . and Randall, A . A . (2001) Ozonation and distribution system biostability. Journ. Amer.
Water Works Assoc., 93(10) 77-89 Eynard, F., Mez, K. and Walther J.-L. (2000) Risk of cyanobacterial toxins in Riga waters, Latvia
Wat. Res., 34(11), 2979-2988 Falkinham, J.O.III, Norton, C D . and LeChevallier, M . W . (2001) Factors influencing number of
Mycobacterium avium, Mycobacterium intracellular, and other mycobacteria in drinking water distribution systems. Appl. Envir. Microbiol, 67(3) 1225-1231.
Fettig, J. (1999) Removal of humic substances by adsorption/ion exchange. Water Science &
Technology, 40(9), 173-183 Filip, Z. and Alberts, J.J. (1994) Adsorption and transformation of salt march related humic acids
by quartz and clay minerals. The Science ofthe Total Environment, 153, 141-150.
Flemming, H . -C , Percival, S.L. and Walker, J.T. (2002) Contamination potential o f biofi lms in water distribution systems. Water Supply, 2(1), 271-280.
Franzmann, P.D., Heitz, A. , Zappia, L.R, Wajon, J.E and Xanthis, K. (2001) The formation of malodorous dimethyl oligosulphides in treated groundwater: the role of biofilms and potential precursors. Wat. Res., 35(7), 1730-1738.
Frateur, I . , Deslous, C , Keine, L . , Levi , Y. , and Tribollet, B . (1999) Free chlorine consumption
induced by cast iron corrosion in drinking water distribution systems. Wat. Res., 33(8), 1781-
1790. Frimmel, F.H. (1998) Characterization of natural organic matter as major constituents in aquatic
systems. Journal of Contaminant Hydrology, 35, 201-216.
Frycklund, C. (1995) Total organic carbon retention by filtersand in an infiltration ponds for artificial groundwater recharge. Aqua Fennica, (25), 5-14.
Frycklund, C. and Jacks, G. (1997) Iron and artificial recharge of groundwater. Boreal
Environmental Research, 2, 171-181. Fu, P.L.K, and Symons, J .M. (1990) Removing aquatic organic substances by anion exchange
resins. Journ. Amer. Water Works Assoc., 82(10), 10-11.
Gagnon, G.A. and Slawson, R .M. (1999) An efficient b iof i lm removal methods for bacterial cell exposed to drinking water. Journal of Microbiological Methods, 34. 203-214.
Gatel, D., Servais, P., Block, J.C, Bonne, P. and Cavard, J. (2000) Microbiological water quality management in the Paris suburbs distribution system. AQUA, 49(5), 231-241.
Geldreich, E.E. (1996) Microbial quality of water supply in distribution system. Lewis Publisher CRC Press, Boca Raton, New York, London, Tokyo.
Gerlach, M . and Gimbel, R. (1999) Influence of humic substance alteration during soil passage on their behaviour. Water Science and Technology, 40(9), 231-241.
Ghiorse, W . C and Wilson, J.T. (1988) Microbial ecology of the terrestrial subsurface. Adv. Appl
Microbiol, 33, 107-172. Gibson, C E . (1971) Nutrient limitation. Journal of Water Pollution Control Federation, 43, 2436-
2440. Graham, N.J.D., Wardlaw, V.E. , Perry, R. and Jiang, J.-Q. (1998) The significance of algae as
trihalomethane precursors. Wat. Sei. Tech., 37(2), 83-89. Grossart, H.-P. and Simon, M . (1998) Bacterial colonization and microbial decomposition of
limnetic organic aggregates (lake snow). Aquat. Microb. Ecol., 15, 127-140.
37

Grøn, C , Tørsløv, J., Albrechtsen, H.-J. and Jensen, H . M . (1992) Biodegradability o f dissolved
organic carbon in groundwater f rom an unconfined aquifer. The Science of the Total
Environment, 117/118, 241-251. Gu, B., Schmitt, J., Chen, Z., Lian, L . and McCarthy, J.F. (1993) Adsorption and desorption of
natural organic matter iron oxide: mechanisms and models. Environmental Science &
Technology, 28(1), 38-46.
Gustafsson, LP. (1999) http://amov.ce.kth.se/people/gustafjp/winhumicv.htm. Haas, C.N., Gupta, M . , Chirluru, R. and Burlingame, G. (2002). Chlorine demand in disinfecting
water mains. Journ. Amer. Water Works Assoc., 94(1) 97-102.
Haas, N.C., Gupta, M „ Burlingame, G A . , Chitluru, R.B. and Pipes, W.O. (1999) Bacterial levels of new mains. Journ. Amer. Water Works Assoc., 91(5), 78-84.
Haiber, S., Herzog, H. , Burba, P., Gosciniak, B. and Lambert, J. (2001) Quantification of
carbohydrate structures in size fractionated aquatic humic substances two-dimensional nuclear magnetic resonance. Fresenius J. Anal. Chem. 369, 457-460.
Hanson, G. (2000) Art i f ic ia l groundwater recharge - A method used in Swedish drinking water supply for 100 years. VA-Forsk report no. 2000-05. 204 pp.
Hautala, K., Peuravuori, J. and Pihlaja, K. (2000) Measurement of aquatic humus content by spectroscopic analyses. Wat. Res., 34 (1), 246-258.
Hem, L.J and Efraimsen, H . (2001) Assimilable organic carbon in molecular weight fractions of natural organic matter. Wat. Res., 35(4), 1106-1110.
Hiraide. M . (1992) Heavy metal complexed with humic substances in fresh water. Analytical
Science, 8, 454-459.
Hiraide, M . , Ishii , M . and Mizuike, A . (1988) Speciation of iron in river water. Anal. Sei., 4, 605-609.
Hongve, Y., Baann, J., Becher, G. and Beckmann, O.A. (1999) Experience f rom operation and
regeneration of an anionic exchanger for natural organic matter ( N O M ) removal. Water
Science and Technology, 40(9), 215-223.
Hrubec, J., Luijten, J.A., Luijten, W . C . M . M . and Piet, G.J. (1986) A pilot plant study on water quality changes during groundwater recharge. Wat. Res., 20(9), 110-1127.
Hu, X . , Kitano, M . , Takenaka, N . , Ban§dow, H , Meada, Y . and Zhang, D. (1994) Sensitive determination of humic acid in natural water by chemiluminiscence-flow-injection method. Bunseki Kagaku, 43(12), 1077-1082.
Hudson, J.J. and Taylor, W . D . (1996) Measuring regeneration of dissolved phosphorus in
planktonic communities. Limnol. Oceanogr., 41(7), 1560-1565. Hudson, J.J., Taylor, W . D . and Schindler, D.W. (2000) Phosphate concentration in lakes. Nature,
406 (6), 54-56.
Huisman, L . and Olsthoorn, T .N . (1983) Artificial Groundwater Recharges. Pitman Books Limited. Boston.
Hunt, A.P., Parry, J.D. and Hamilton-Taylor, J. (1999) Further evidence o f elemental composition
as an indicator of the bioavailability of humic substances to bacteria. Limnol. Oceanogr., 45(1), 237-241.
IHSS (1982) International Humic Substance Society, http://www.ihss.gatech.edu/
Imai, A. , Fukushima, T., Mutsushige, K . and K i m , Y . H . (2001) Fractionation and characterization of dissolved organic matter in a shallow eutrophic lakes, its inf lowing rivers, and other organic matter sources. Wat. Res., 35(17), 4019-4028.
Jacks, G. and Frycklund, C. (1995) Removal of organic matter during groundwater formation. Vatten 51 , 66-69. (In Swedish).
Jahnel, J.B. and Frimmel, F.H. (1995) Enzymatic release of amino acids f rom different humic substances, Acta hydrochimica et hydrobiologica, 23, 31-35.
38

Jansson, M . , Blomqvist, P., Jonsson, A. and Berström, A . K . (1996) Nutrient limitation of bacterioplankton, autotrophic and mixotrophic phytoplankton, and heterotrophic nanoflagellates in Lake Örträsket. Limnol. Oceanogr. 41(7), 1552-1559.
Jardine, P.M., Weber, N . L . and McCarthy, J.F. (1989) Mechanisms of dissolved organic carbon adsorption on soil. Soil Science Society of America Journal, 53, 1378-1385.
Jones, M . N . and Bryan, N . D . (1998) Colloidal properties of humic substances. Advances in
Colloidal and Interface Science, 78, 1-48. Jones, R. I . , Salonen, K. and DeHaan, H . (1988) Phosphorus transformations in the epilimnion of
humic lakes: Abiotic interaction between dissolved humic material and phosphate. Freshwater
Biol., 19,357-369. Juhna. T., Klavins. M . and Sprogis. J. (1998)a Retention of humic substances on filtersand f rom
artificial groundwater recharge infiltration basins. Latvian Journal of Chemistry, 4, 87-93. Juhna, T. and Springe, G. (1998)b Distribution of microorganisms in the course of artificial
recharge of groundwater at Baltezers waterworks, Riga. Vatten, 54, 259-264.
Juhna, T. and Klavins, M . (1999) Retention of metal-humic complexes during artificial recharge of groundwater. Latvian Journal of Chemistry, 2, 94-101.
Juhna, T. and Klavins, M . (2001) Water-quality changes in Latvia and Riga 1980-2000: Possibilities and Problems. AMBIO, 30 (4-5) 306-314.
Kaiser, K . and Zech, W . (1997) Competitive sorption of dissolved organic matter fraction to soil and related mineral phase. Soil. Sei. Soc. Am. J., 61 , 64-69.
Karl, D . M . and Tien, G. (1992) M A G I C : a sensitive and precise methods for measuring dissolved phosphorus in aquatic environment. Limnol. Oceanogr., 37, 105-116.
Karner, D.A., Standridge, J.H., Harrington, G.W. and Barnum, R.P. (2001) Microcystin algal
toxins in source and finished drinking water. Journ. Amer. Water Works., 93(8), 72-81. Kim, P.H.S. and Symon, J .M. (1991) Using anion exchange resins to remove T H M precursors
Journ. Amer. Water Works., 12(83), 61-68. Klavins, M . (1993)a Humic substances of Latvia, (in Latvian) Habil. PhD Thesis. University o f
Latvia, pp 43. Klavins, M . (1993)b Immobilization of aquatic humic matter. Latvian Journal of Chemistry, 1, 91-
103. Klavins, M . and Apsite, E. (1997) Sedimentary humic substances f rom lakes in Latvia.
Environment International, 23(6), 783-790.
Klavins, M . , Springe, G„ Rodinov, V. . Druvietis. I . , Parele, E. and Briede. A. (2000) Water quality changes in Latvia in relation to changing loading levels. Vatten, 56, 39-47.
Klavins, M . (2002) personal communication. Koivusalo, M . (1998) Drinking water mutagenicity and cancer. Doctoral Thesis. National Public
Health Institute, Division of Environmental Health, Kuopio, Finland.
Kortelainen, P. (1993) Content of total organic carbon in Finnish Lakes and its relationship to catchment characteristics. Can. J. Fish. Aquat. Sei., 50, 1477-1483.
Kramer, M . H . . Quade, G.. Hartenmann. P. and Exner. M . (2001) Waterborne diseases in Europe 1986-1996. Journ. Amer. Water Works., 93(l),48-53.
Kuehn. W . and Mueller, U . (2000) Riverbank filtration. A n overview. Journ. Amer. Water Works.,
92(12), 60-69. Kullberg, A . (1994) Mobilisation and transport of dissolved organic matter and its biological effect
in acidified humic freshwaters. PhD thesis. Department of Ecology, Lund University, Sweden.
Langowska, I . (1982) Utilization of organic phosphates by selected bacteria and algae. Wat. Res.,
16. 167-167. Laurent, P., Prévost, M . , Cigana, J., Niquette, P. and Servais, P. (1999) Biodegradable organic
matter removal in biological filters: evaluation of the C H A B R O L model. Wat. Res., (33)6,
1387-1398.
39

LeChevallier, M.W. , Shulz, W. and Lee, R.G. (1991). Bacterial nutrients in drinking water Appl.
Environ. Microbiol., 57(3), 857-862.
LeChevallier, M.W. , Welch, N.J. and Smith, D.B. (1996) Full-scale studies o f factors related to col i form regrowth in drinking water. Appl. Environ. Microbiol., 62(7), 2201-2221.
LeChevallier, M . W . (1999) The case for maintaining a disinfectant residual. Amer. Water Works
Assoc., 91(1) 86-94.
Lehman, R . M . and O'Connell, S.P. (2002) Comparison of extracellular enzyme activities and community composition of attached and free-living bacteria in porous medium columns. Appl.
Environ. Microbiol., 68 (4), 1569-1575.
Lehtola, J .M., Miettinen, LT. , Vartinainen, T. and Martikainen, P.J. (1999) A new sensitive bioassay for determination of microbially available phosphorus in water. Appl. Environ.
Microbiol., 65(5), 2032-2034.
Lehtola, M.J. , Miettinen, I .T. and Martikainen, P.J. (2002) B i o f i l m formation in drinking water
affected by low concentration of phosphorus. Can. J. Microbiol., 48, 1-6. Lemke, J .M., Churchill, P.F. and Wetzel, R.G. (1995) Effect of substrate and cell surface
hydrophobicity on phosphate utilization in bacteria. Appl. Environ. Microbiol., 61(3), 913-919. Long, B . M . , Jones, G.J. and Orr, P.T. (2001) Cellular microcystin content in N-limited Microcystis
aerigonosa can be predicted f rom growth rate. Appl. Environ. Microbiol., 67(1), 278-283.
Lovely, D. , Coates, J.D., Blunt-Harris, E.L., Philips, E.J.P. and Woodward, J.C. (1996) Humic substances as electron acceptors for microbial respiration. Nature, 383(1), 445-448.
Lu, C.-J. and Speitel Jr., G.E. (1991) Effect of natural organic matter on biodegradation of recalcitrant synthetic organic chemical. Journ. Amer. Water Works., 83(2), 55-61.
Lu, W. , Kiéné, L . and Lévi , Y . (1999) Chlorine demand of b io f i lm in water distribution systems. Wat. Res., 33(3), 827-835.
Lund, V . and Hongve, D . (1994) Ultraviolet irradiated water containing humic substances inhibit bacterial metabolism. Wat. Res., 28(5), 1111-1116.
Mamchenko, A .V . , Vaynman, A . B . , Philipanko, I .V . , Novozhenyuk, M.S., Oshurkova, M . V . and Rhyzhikova, N .V . (1997) Journal of Water Chemistry and Technology, 9 (19) 17-20.
Marmonier, P., Fontveille, D., Gibert. J. and Vanek.V. (1995) Distribution o f dissolved organic carbon and bacteria at the interface between the Rhone River and its alluvial aquifer. J. N. Am.
Benthoi, 14(3), 382-391.
Marshall, S.J., House, W.A. , Russell, N.J. and White, G.F. (1998) Comparative adsorption of
natural and commercially available humic acids to river sediments. Physicochemical and
Engineering Aspects, 144, 127-137. Martin-Mousset, B. , Croue, LP., Lefebvre, E. and Legube, B . (1997). Distribution and
characterization of dissolved organic matter of surface waters. Wat. Res., 31(3), 542-553. ( in French)
Maurice, P.A., Pullin, M.J. , Cabiniss, S.E., Zhou, Q., Namjesnik-Dejanovic, K„ Aiken, G.R. (2002) A comparison o f surface water natural organic matter in raw filtered water samples,
X A D and reverse osmosis isolates. Wat. Res., 36(9), 2357-2371. McCarthy, J.F., Will iams, T . M . , Liang, L . , Jardine, P., Jolley, L .W. , Taylor, D.L. , Palumo, A . V . ,
Cooper, L .W. (1993) Mobi l i ty of natural organic matter in a sandy aquifer. Environ. Sei.
Technol. 27, 667-676.
Meier, M . , Namjesnik-Dejanovic, K. , Maurice, P.A., Chin,Y.-P. and Aiken, G.R. (1999) Fraction
of aquatic natural organic matter upon sorption to geothite and kaolinite. Chemical Geology
(157)275-284.
Metting, B . and Pyne J.W. (1986) Biologically active compounds f rom microalgae. Enzyme
Microb Technol (8), 386-394.
Meybeck, M . (1982) Carbon, nitrogen and phosphorus transport by worlds rivers. Am. J. Sei. 401-450.
40

Miettinen, LT. , Martikainen, P.J. and Vartiainen, T. (1994) Humus transformation at the bank fi l trat ion water plant. Wat. Sei. Tech., 30(10), 179-187.
Miettinen LT. , Vartiainen T.K. and Martikainen P.J. (1997)a Phosphorus and bacterial growth in drinking water. Appl. Environ. Microbiol., 63 (8), 3242-3245.
Miettinen, LT. , Vartiainenm T. and Martikainen, P. J. (1997)b Changes in water microbial quality during bank filtration of lake water. Can. J. Microbiol., 43, 1126-1132.
Miettinen, LT., Martikainen, P.J. and Vartiainen, T. (1998) Mutogenicity and amount of chloroform after chlorination of bank filtered water. The Science of the Total Environment,
215, 9-17. Miettinen I .T. , Vartiainen T. and Martikainen P.J. (1999) Determination of assimilable organic
carbon in humus-rich drinking waters. Wat. Res., 33(10), 2277-2282.
Miller, M.J . and Fallowfield, H.J. (2001) Degradation o f cyanobacterial hepatotoxins in batch experiments. Water Science and Technology, 43(12), 229-232.
Mopper, K. , Schultz, C A . , Chevolot, L . , Germain, C , Revueita, R. and Dawson, R. (1992) Determination o f sugars in unconcentrated seawater and other natural waters by liquid chromatography and pulsed aperometric detection. Environ. Sei. Technol., 26, 133-138.
Müller, M . B and Frimmel. F.H. (2002) A new concept for the fractionation of D O M as a basis f ro
its combined chemical and biological characterization. Wat. Res., 36(10), 2643-2655.
Myllykangas, T., Nissinen, T.K., Rantakokko, P., Martikainen, P.J. and Vartiainen, T. (2002) Molecular size fractions of treated aquatic humus. Wat. Res., 36(12), 3045-3053.
Niquette, P., Servais, P. and Savoir, R. (2000) Impact o f pipe material on densities of f ixed
bacterial biomass in a drinking water distribution system. Wat. Res., 34(6), 1952-1956. Nishijima, W., Shoto, E. and Okada, M . (1997) Improvement of biodegradation of organic
substances by addition of phosphorus in biological activated carbon. Water Science and
Technology, 36, 251-257. Nissinen, T.K., Miettinen, LT. , Martikainen, P.J. and Vartiainen, T. (2001) Molecular size
distribution of natural organic matter in raw and drinking waters. Chemosphere, 45, 865-873. Nodvin, S.C, Driscoll, C T . , Likens, G.E., (1986) Simple partitioning o f anions and dissolved
organic carbon in a forest soil. Soil Science, 142(1), 27-35.
Norton, C D . and LeChevallier. M . W . (2000) A pilot study o f bacteriological population changes though potable water treatment and distribution. Appl. Envir. Microbiol., 66(1), 268-276.
Olivié-Lauquet, Q., Allard, T., Benedetti, M . and Muller, J.-P. (1999) Chemical distribution of
trivalent iron in riverine material f rom a tropical ecosystem: a quantitative EPR study. Wat.
Res., 33.(11), 2726-2734. Olsen, L . M . , Reinertsen, H. and Vadstein, O. (2002) Can phosphorus limitation inhibit dissolved
organic carbon consumption in aquatic microbial food webs? A study of three food web structures in microcosms. Microbial Ecology, 43(3), 353-366.
Ortega-Calvo, J-J. and Saiz-Jimenez, C. (1998) Effect of humic fractions and clay on
biodegradation of phenanthrene by a Pseudomonas fluorescens strain isolated f rom soil Appl.
Envir. Microbiol., 64(8), 3123-3226. Payment, P., Siemiatycki, J., Richardson, L . , Renoud, G., Franco, E. and Prévost, M . (1997) A
prospective epidemiological study of gastrointestinal health effects due to the consumption of
drinking water. International Journal of Environmental Health Research, 7, 5-31. Peldszus, S., Huch, P.M. and Andrews, S.S. (1998) Quantitative determination of oxalate and other
organic acids in drinking water at low pg/1 concentrations. Journal of Chromatography A, 793,
198-203. Pelekani, C. and Snoeyink, L . V . (1999) Competitive adsorption in natural water: role of activated
carbon pore size. Wat. Res., 33(5), 1209-1219. Pettersson, C. (1992) Properties of Humic Substances from Groundwater and Surface Water.
Doctoral Thesis. Faculty of Arts and Sciences at Linköping University.
41

Peuravuori, J. and Pihlaja, K. (1997) Molecular size distribution and spectroscopic properties of aquatic humic substances. Analytica Chimica Acta, 337, 133-149.
Peuravuori, J. and Pihlaja, K. (1998) Multi-method characterization of lake aquatic matter isolated
with two different sorbing solids. Analytica Chimica Acta, 363, 235-247.
Pomes, M . L . , Green, W.R., Thurman, E .M. , Orem, W.H. and Lerch, H.E. (1999) DBP formation of aquatic humic substances. Journ. Amer. Water Works., 91(3), 103-115.
Rapala, J.. Lahti, K., Räsänen, L .A . , Esala, A . - L . , Niemelä, S.I. and Sivonen, K . (2002) Endotoxins
associated with cyanobacteria and their removal during drinking water treatment. Wat. Res.,
36(10), 2627-2635. Ray, C., Grischek, T., Schubert, J., Wang, J.Z. and Speth, T.F. (2002) A perspective of riverbank
fil tration. Jour. Amer. Water Works Assoc., 94(4), 149-160.
Reasöner. D.J. and Geldreich. E.E. (1985) A new medium for the enumeration and subculture of bacteria f rom potable water. Appl. Environm. Microbiol., 49, 1-7.
Ribas, F., Perramon, J., Terradillos, A. , Frias, J. and Lucena, F. (2000) The Pseudomonas group as
an indicator of potential regrowth in water distribution system. Journal of Applied
Microbiology, 88. 704-710. Rice, E.W., Scarpino, P.V., Reasöner, D.J., Logsdon, G.S. and W i l d , D.K. (1991) Correlation of
col iform growth response with other water quality parameters. Journ. Amer. Water Works
Assoc., 83,98-102. Riemann, L. , Steward, G.F. and Azam, F. (2000) Dynamics of bacterial community composition
and activity during a mesocosm diatom bloom. Appl. Envir. Microbiol., 66(2), 578-587.
Roberts, P.V., Schreiner, J. and Hopkins. G.D. (1982) Field study of organic water quality changes
during groundwater recharge in the Palo Alto Baylands. Wat. Res., 16(6), 1025-1035. Rohrlack, T., Dittmann, E., Börner, T. and Christoffersenl, K. (2001) Effects of cell-bound
microcystins on survival and feeding of Daphnia spp. Appl. Envir. Microbiol., 67 (8), 3523-
3529. Rompé , A„ Prévost, M „ Coallier, J., Brisebois, P. and Lavoie, J. (2000) Impact of implementing a
corrosion control strategy on b io f i lm growth. Water Science & Technology. 41(4-5), 287-294
R W B Consulting Engineers and SIA FIDIC S A (2002) Daugava WTP Follow-up of water quality.
Intermediate report (01E28) for Riga Municipal Enterprise RigaWater and Swiss Federal
Office of Foreign Economical Affai rs . 16 pp. Sathasivan, A. and Ohgahi, S. (1999) Application of new bacterial regrowth potential methods for
water distribution system - clear evidence pf phosporus limitation. Wat. Res., 33(1), 137-144. Sathasivan, A. and Ohgaki, S. (1999) Application of new bacterial regrowth potential method for
water distribution system - a clear evidence of phosphorus limitation. Wat. Res., 33, 251-257. Schmidt, W., Hambsch. B. and Petzoldt, H. (1998) Classification of algogenic organic matter
concerning its contribution to the bacterial regrowth potential and by-products formation. Wat.
Sei. Tech., 37(2), 91-96. Schnitzer, M . (1991) Soil organic mat te r - the next 75 years. Soil Science, 151(1), 41-58.
Schulten. H.R. and Schnitzer, M . (1993) A state of art structural concept for humic substances. Naturwissenschaften, 80, 29-30
Schwartz, T., Hoffmann. S. and Obst, U . (1998) Formation and bacterial composition of young. natural b iof i lm obtained f rom public bank-filtered drinking water system. Wat. Res., 32(9),
2787-2797. Schwarzenbach, R.P., Giger, W. , Hoehn, E. and Schneider, J.K. (1983) Behaviour of organic
compounds during infiltration of river water to groundwater. Field studied. Environ. Sei.
Technol., 17,472-479. Selig, U . . Hübener. T. and Michalik, M . (2002) Dissolved and particulate phosphorus forms in a
eutrophic shallow lake. Aquatic Sciences, 64. 97-105.
42

Servais, P., Billen, G. and Hascoet, M.C. (1987) Determination of the biodegradable fraction of dissolved organic matter. Wat. Res.. 21 , 445-450.
Shaw, P.J., Jones, R . I . and De Haan, H . (2000) The infuence of humic substances on the molecular wieght distribution of phosphate and iron in epilimnetic lakes water. Freshwater Biology, 45,
383-393. Sibille, I . , Sime-Ngando, T., Mathieu, L . and Block, J.C. (1998) Protozoan bacterivory and
Escherichia coli survival in drinking water distribution systems. Appl. Envir. Microbiol., 64(1), 197-202.
Simpson, A.J., Kingery, W . L . , Hayes, M.H.B. , Spraul, M . , Humpfer, E., Dvortsak, P., Kerssebaum, R., Godejohann, M . and Hofmann, M . (2002) Molecular structures and
associations of humic substances in the terrestrial environment Naturwissenschaften, 89, 84-88. Sontheimer, H . (1980) Experience with riverbank filtration along the Rhine River. Journ. Amer.
Water Works Assoc., 72(7), 368-390. Stumm, W . (1992) Chemistry ofthe solid-water interfaces. John Wiley & Sons. Inc. USA. Thurman, E .M. (1985) Organic geochemistry of natural waters. Martinus Ni jhof f . Dr. W . Junk
Publishers. Wageningen. Thurman, E .M. and Malcolm, R.L. (1981) Preparative isolation of aquatic humic substances.
Environmental Science and Technology, 15(4) 463-466.
Tipping, E. (1981) The adsorption of aquatic humic substances by iron oxides. Geochimica et
Cosmochimica Acta, (45)191-199. Tombåcz, E., Dobos, A , Szekeres, M . , Narres, H.D., Klumpp, E., Dékäny, I . (2000) Effect of pH
and ion strength on the interaction of humic acid with aluminium. Colloid. Polym. Sei., 278, 337.345.
Tranvik, L.J. (1990) Bacterioplankton growth on fractions of dissolved organic carbon of different molecular weights f rom humic and clear lakes. Appl. Envir. Microbiol., 56(6), 1672-1677.
Vadstein, O. (2000) Heterotrophic, planktonic bacteria and cycling of phosphorus. Phosphorus requirements, competitive ability and food wed interactions, pp 115-167. In: Advances in
Microbial Ecology, 16. Kluwer Academic/Plenum Publishers, New York, van der Koo i j , D., Visser, A . and Hijnen, W . A . M . (1982) Determining the concentration of easily
assimilable organic carbon in drinking water. Journ. Amer. Water Works Assoc., 74, 540-545. van der Koo i j , D. (1992) Assimilable organic carbon as an indicator of bacterial regrowth. Journ.
Amer. Water Works Assoc., 84(2), 57-66.
van der Koo i j , D., Veenendaal, H.R., Baars-Lorist, C , van der K l i f t , D.W. and Drost, C.Y. (1995) B i o f i l m formation on surfaces of glass and Teflon exposed to treated water. Wat. Res., 29(7),
1655-1662. Weber Jr, W.J., McGinley, P .M. and Katz, L.E. (1991) Sorption phenomena in subsurface systems:
concepts, models and effects on contaminant fate and transport. Wat. Res., 25(5) 499-528.
Wei, B. , Sugiura, N . and Maekawa, T. (2001) Use of artificial neural network in the prediction of algal blooms. Wat. Res., 35 (8), 2022-2028.
Vermeer, A.W.P., van Riemsdijk, W.H . and Koopal L . K . (1998) Adsorption of humic acid to mineral particles. 1. Specific and electrostatic interactions. Langmuir, 14, 2810-2819.
Wetzel, R.G. (1983) Limnology. 2nd ed. Sounders College. Philadelphia. Vik, E.A., Carlson, D A . , Eikum, A.S. and Gjessing, E.T. (1985) Removing aquatic humus f rom
Norwegian lakes. Journ. Amer. Water Works Assoc., 77 (3), 58-66.
Wilkinson, K.J., Négre , J .C, Buff le , J. (1997) Coagulation of colloidal material in surface water: the role of natural organic matter. Journal of Contaminant Hydrology, 26, 229-243.
Williamson, C.E., Morris , D.P., Pace, M . and Olson, O.G. (1999) Dissolved organic carbon and
nutrients as regulators o f lake ecosystem: Resurrection of a more integrated paradigm. Limnol.
Oceanogr., 44(3/2), 795-803.
43

von Gunter, U . and Zobrist, J. (1993) Biogeochemical changes in groundwater-infiltration systems:
Column studies. Geochimica et Cosmoshimica Acta, 57, 3895-3906. Zhang, W . and DiGiano, F.A. (2002) Comparison of bacterial regrowth in distribution systems
using free chlorine and chloramine: a statistical study a causative factors. Wat. Res., 36(6),
1469-1482. Zhou, J.L., Rowland, S., Mantoura. R.F.C. and Braven, J. (1994) The formation of humic coatings
on mineral particles under simulated estuarine condition - a mechanistic study. Wat. Res.,
28(3), 571-579. Ødegaard, H . , Eikebrokk, B . and Storhaug, R. (1999) Process for removal of humic substances
f rom water-an overview based on Norwegian experiences. Water Science & Technology,
40(9), 37-47.
44

P a p e r I
Sorption of humic substances to aquifer material at artificial recharge of groundwater.
Chemosphere, 2002, submitted.

S O R P T I O N O F H U M I C S U B S T A N C E S ON A Q U I F E R M A T E R I A L A T A R T I F I C I A L R E C H A R G E O F G R O U N D W A T E R
TALIS J U H N A 1 2 ) , *MÄRIS K L A V I N S 3 ) , L I N D A EGLlTE 3 )
' Department of Environmental Engineering, Luleå University of Technology, SE 97187, Luleå, Sweden
"'Department o f Civi l Engineering, Riga Technical University, Azenes Street 16, L V 1048, Riga, Latvia
3 1 Department of Environmental Science, University of Latvia, Raina blvd. 19, L V 1586, Riga, Latvia
Abstract
Experiments in batch equilibrium system were carried out to evaluate the importance of physical and chemical factors determining the sorption efficiency o f humic substances (HS) on aquifer material, which has been used for artificial recharge of groundwater in drinking water production. Results showed that an increase of the
amount of clay in the aquifer material and a decrease of pH in water increased the
sorption efficiency. The sorption of higher molecular weight, more hydrophobic and aromatic HS (Aldrich and forest soil humic acids) were greater than the sorption of acidic HS (river fulvic acids), either on the aquifer material or to its representative sorbing phases, clay and organic matter. The sorption on the aquifer material was
largely due to physical sorption (hydrophobic attractions). This study showed the importance of HS composition on their removal during artificial recharge of groundwater and contributed to an understanding o f the HS sorption mechanisms in this process.
Keywords: humic substances, sorption, artificial recharge o f groundwater, aquifer material, drinking water.
1. Introduction
Arti f ic ia l recharge o f groundwater (ARG) is achieved by putting surface water in basins or other facilities where it infiltrates into the ground and moves through
porous media downwards to recharge aquifers (Bower, 2002). In this process, without adding any reagents, suspended particles and human pathogens are effectively
removed f rom the water (Huisman and Olsthoorn, 1983). Therefore, in areas where permeable aquifers are available, A R G has been used for drinking water production
for more than 150 years. Nowadays, it is recognized that removing humic substances (HS) (humic acids and fulvic acids) f rom drinking water is important because they
impart a yellow colour to water and may lead to the formation o f carcinogenic byproducts during disinfections (Pomes et al. 1999) as well as undesirable bacterial
b iof i lm growth (Ellis et al. 2000) in distribution systems. The removal efficiently of
HS during A R G varies f rom place to place; the causes of these variations are often not understood. Thus, more knowledge about the mechanisms involved in removing HS is of interest.
1

Due to partially colloidal properties (Jones and Bryan, 1998) and its ability to associate with inorganic colloids (Burba et al. 2001), straining and interception in porous media both in solid-water and gas-water interface may be involved in HS retention during the ARG. However, sorption to aquifer material (hereafter filtersand)
(McCarthy et al. 1993) followed by microbial degradation are the most important mechanisms. Because HS are relatively resistant to degradation whereas attachment to
surfaces may increase their availability for bacteria (Camper, 2002), sorption is most likely a l imit ing step for HS removal during ARG. The mechanisms of HS sorption to
filtersand have not been widely studied. However, given the similarity in composition of material, these mechanisms are assumed to be similar to those occurring in soils
(Jacks and Frycklund, 1995), river sediments, and natural aquifer materials. Among the different mechanisms proposed, ligand exchange (chemisorption), hydrophobic attraction, and electrostatic attraction (physical sorption) are considered the most
important for removal of HS by surfaces (Stumm, 1992) because HS has both acidic functional groups (carboxyl and phenol) and hydrophobic moieties. Due to the
carboxyl groups, HS may participate in ligand exchange or electrostatic interaction with oxide surfaces, whereas hydrophobic properties allow HS to accumulate on surfaces because of their incompatibility with water, which is more polar than the
surface. The importance of these mechanisms can be evaluated by comparing the sorption of fulvic acid (FA) (i.e. more acid fraction of HS) with humic acids (HA) (i.e.
more hydrophobic fraction of HS). In addition to the type of HS, the physical-chemical properties of sorbing material (e.g. texture, surface areas, amount of oxides
groups) and of water (e.g. pH, ion strength, concentration of divalent metals) govern
the sorption mechanisms. In the present study, the sorption of FA and H A on the filtersand and its major
composing phases participating in the sorption (e.g. iron and aluminium oxides,
organic matter) were evaluated in laboratory batch equilibrium systems. The
objectives were to investigate how composition and properties of HS influence their sorption efficiency on the filtersand used for A R G and evaluate the influence of
sorption mechanisms involved in removing HS during ARG.
Materials and Methods
2.7. Isolation and analyses of humic substances
H A and FA f rom typical forest soil (podzolic) in Latvia and filtersand f rom the infiltration basin used for drinking water production at Baltezers ARG plant in Riga, Latvia were isolated by extraction with 0.1M NaOH (Thurman, 1985). H A and FA
f rom water of Lake Baltezers (used for ARG) and the Salaca River were isolated by
the Malcolm and Thurman method, which was modified by KJavins and Cinis (1990). Commercial (Aldrich) H A was used for comparison. The elemental composition of
isolated HS was determined on a Perkin Elmer 240 B C H N analyser. Ash content was determined by ignition of samples at 500 C for 5 h. The content of carboxyl groups
was determined using Ba(OH)i titration, and the content of hydroxyl groups was done after acetylation (APHA 1988). Aromaticity of HS was determined based on
measurement of their ultraviolet light absorption and calculated according to Chin et
al. (1994). Properties o f the HS used are summarised in Table 1. Concentrations of HS in water were measured colorimetrically at 410 nm wavelengths using a H A C H DR
2

2000 spectrophotometer and calculated from calibration graphs initially prepared for
each HS.
2.2. Sampling, preparation and analysis of the filtersand
Filtersand was sampled at the Baltezers plant where groundwater is artificially recharged by infi l trat ion of water f r om Lake Baltezers through the infiltration basins. The basins were excavated in quaternary deposits at the end of the 1950s. In 1997, samples were collected f rom the bottoms of three infiltration ponds numbered accordingly 3, 4, and 8. Samples f rom each basin were taken close to the periphery
( A ) , the centre (C) of the basins and in between these sampling points (B). The surficial filtersand layer ( 0 - 4 cm) was sampled by Ekman drag, and deeper layers (4
- 24 cm) were taken using a core sampler and dissected into segments of 4 cm. The samples were air dried, sieved (<2 mm), and subsequently used for analyses and sorption experiments.
Particle-size was estimated by dry sieve analysis or by the hydrometer method. BET surface area for minerals was determined by Micrometric FlowSorb I I 2300 (30% H i , 70% He) or Coulter SA 3100. The mineral composition was estimated using x-ray diffraction (diffractometer DRON-3). The filtersand chemical composition was
determined by standard methods (APHA 1988). For chemical analyses iron was
extracted f rom filtersand samples (~ 10 g) with 25 ml cone. HNO3, together wi th 5 ml 60% HCIO4. The content of organic carbon (OC) in the filtersand was determined wi th the Walkley and Black method.
2.3. Filtersand with organic matter removed
The organic matter in filtersand samples was removed by oxidation in acidic
media: 500 g o f the filtersand treated with a mixture of 375 ml 15% H2O2 and 10% CH3COOH (55ml) at 75°C for 4 hours, after which a new portion (100 ml) of 30% H 2 0 2 was added wi th heat for an additional 2 h. Next, the samples were washed with 0.01 M NaCl to remove the entrained salts, then by distilled water, and dried at 105°C
(24 h).
2.4. Oxides, clays and lake sediments
To determine more specifically what phases contributing to HS sorption on
aquifer material, phases representing those found in aquifer material were obtained for
the study. Reanal supplied Alumina (a-ALOs). Goethite (a-FeOOH) was synthesised
accordingly (Lattimer and Jones, 1992). Clays were collected in Latvia; their
properties are summarised in Table 4. Surficial sediments layer (67% OC, BET of
1.51 n r /g , 7.37 mg/g Fe) were collected by Ekman drag f rom the bottom of Lake
Baltezers and analysed as per the methods cited above.
2.5. Sorption experiments
Sorption studies for HS on different phases were conducted as batch experiments performed in 50 ml sealed glass bottles on a rotary shaker table for 16 h
at 20°C. In 50 ml glassware, 5, 10, 25, 50. 75. 100, and 250 mg/L solutions of HS
were equilibrated with 0.5 g of sorbent until equilibrium occurred. After separation of
3

the sorbent by filtration through filter paper, the concentration o f HS in the filtrate was analysed. The quantity of HS sorbed to different phases was determined f rom the difference between the initial aqueous phase concentration and the concentration in
solution at equilibrium. Due to the release of organic matter f r om the filtersand and lake sediments, the results of the sorption experiments could not be interpreted by
Freundlich or Langmuir isotherms. When initially sorbed (background) organic matter needs to be considered, the initial mass approach (Nodvin et al. 1986) is useful. This
approach was also used in this study. In the initial mass approach, the concentration o f a substance retained or released (normalized to sorbent mass) is plotted as a function
of the initial concentration of the substances (also normalized to sorbent mass).
3. Results
3.1 Composition of the filtersand
Mineralogical analysis using x-ray diffraction spectroscopy revealed that the filtersand, regardless the sampling location and depth, was dominantly composed of SiGs (quartz) wi th some admixture of aluminium silicate (labradorite plagioclase). A n
absence of iron oxide minerals (e.g. celadonite and glauconite) indicated that iron,
detected by chemical analyses, (Table 2) presumably infiltrated in via surface water. The texture of the aquifer material ranged f rom clay to fine sand. Typical
concentrations of clay in filtersand ranged between 1-3% (basins no. 8 and 3), whereas it was as higher in basin no. 4 close to that found in soils, see Table 2. The amount of organic matter (OC), which was mainly composed of HS, was about 0.5%.
3.2. Composition of humic substances
Soil H A and commercially available (Aldrich) H A , were more aromatic and contained less f rom the carboxyl groups than river FA, Table 1. The (0+N)/C atomic
ratio was used to establish hydrophilicity/hydrophobicity of HS as suggested by De Paolis and Kukkonen, (1997). The ratios for Aldrich H A and soil H A were significantly lower than for river FA (Table 1) and indicated that both HA:s were
more hydrophobic than the FA. The molecular mass of the aquatic FA was lower
(500-1000 Da) than for both HA:s (500-3000 Da).
3.3. HS sorption on filtersand and related phases
Sorption of Aldrich HS increased with increased amounts of clay and
decreased wi th increased amount of OC in the filtersand, Fig. 1 and Fig. 2. Sorption was heterogeneous among samples, but generally increased wi th the depth below the
basins. Fig. 2. Correlations between OC and clay and the sorption of HS for all filtersand samples were presented in a previous article (Juhna et al. 1998). Sorption
experiments of different HS on clays revealed that HS sorption exceeded the FA sorption, while it was better on smectite (the expandable clays) compared to koalinite,
Table 3. Sorption to the filtersand from which organic matter was chemically removed was low or negative. A t the same pH, Aldrich and soil H A were sorbed better than
river FA to typical filtersand (basins no. 3 and 8) f rom Baltezers plant, Fig. 3.
Regardless o f the filtersand composition, the lowering o f pH significantly increased the sorption efficiently of HS, Fig. 4. To understand the influence o f different phases
4

composing the filtersand, the sorption o f H A and FA to iron, aluminium oxides, and organic rich sediments were tested. H A were always better or similarly sorbed to all filtersand components except iron oxide, which instead better sorbed FA (Fig. 5- Fig.
7).
4. Discussion
Shen (1999) suggested that chemisorption (ligand exchange) is the dominant mechanism for sorption of HS to soil. Jardine et al. (1989), after observing
temperature independency of HS sorption to soil, suggested that physical sorption driven by entropy changes (hydrophobic attraction) is more important. Gu et al. (1994) supported this view and proposed that the ligand exchange is unlikely to be a
dominant interaction in the soil system because of negatively charged clay surfaces; hence, the lack of ligand exchange sites, except at a few broken edge surfaces in such systems. The composition o f the filtersand used for ARG systems was similar to that
of soils but with lower amounts of clay and iron oxide (Table 2), the surfaces bearing major binding sites for sorption of HS. A relatively high amount of organic matter
(OC) (Table 2) indicated that a large proportion of these sites had already been occupied with organic matter making their surfaces negatively charged (Day et al.
1994), and thus unavailable for ligand exchange. The quartz was a major constituent of the filtersand. Ligand exchange of HS with quartz surfaces is minor, as proven in experiments by Spark et al. (1997), in which no proton consumption was observed in
the interaction between the quartz and HS. Thus, under conditions where filtersand has a limited number of binding sites, ligand exchange is probably not the dominant process for removing HS.
The results showed that sorption o f HS increases with an increase the fraction
of clay in the filtersand; explanations for such behaviour could vary. Due to small size
the clay particles have a large specific surface area that, depending o f chemical and physical properties of clay (not identified herein), can be conductive for either physical or chemical sorption to occur. However, physical interaction rather than
ligand exchange between HS and the filtersand was supported by observations where more aromatic and higher molecular mass fractions of HS ( H A ) were removed instead
of those more acidic (FA) that contained higher amounts of functional groups, Fig. 3.
This explanation, however, should be carefully considered since lower sorption efficiency of HS wi th a higher amount of functional groups (e.g. FA) is not always indicative of a lack of coordinative interaction (ligand exchange). HS are
macromolecules behave like polyelectrolytes; thus their sorption is determined not only by the availability of sites, but also by lateral interaction between themselves and the steric arrangements of the macromolecule (Vermeer et al. 1998; Kaiser and Zech,
1997 reference within). Due to a high amount of functional groups, HS stretches (due
to lateral repelling between the functional groups) and when becomes sorbed on the surfaces occupies a large area that makes overall sorption efficiency of FA lower than of H A . This is also demonstrated by the finding that HS, with high amounts of
aromatic groups and lower amounts o f carboxyl groups, can be preferably sorbed to
the surface with high amounts of binding sites such as iron or aluminium oxides (Meier et al. 1999; Evanko and Dzombak, 1999). This occurs even though ligand
exchange is the dominant mechanism of interactions of HS with those surfaces (Tombåcz et al. 2000; Edwards et al. 1996; Day et al. 1994; Gu et al. 1994; Tipping
1981).
5

In the sorption process HS replaced nearly all previously retained HS before any sorption could occur, Fig. 3. This indicates that HS were weakly sorbed on the
bare filtersand, perhaps as a result of physical interaction. Elemental analyses showed an insignificant increases of oxygen and carboxyl groups in the filtersand HS
compared to HS of the water used for recharge. Thus, chemisorption, which generally contributes to higher level o f theses moieties in the sorbed HS, was not significant.
A change of pH affected both HS and filtersand properties. A decrease of pH should increase the positive charge o f oxides, thereby making an electrostatic attraction with negatively charged HS molecules possible. However, at acidic pH HS
become more hydrophobic, thus increasing the possibility for hydrophobic attraction. Since the filtersand contained low amounts of iron and clay, the strong positive effect
of sorption (Fig. 4) that was due to a lowering of pH, was perhaps also due to a hydrophobic effect rather than an increase of electrostatic attraction. Therefore, the
sorption may be dominated not so much by the forces between the filtersand surface and HS, but rather by the intermolecular association forces between HS and its incompatibility with water.
However, we observed that background organic matter also plays an important role. HS is better attracted to organic rich sediments than to organic filtersand, which
had a relatively lower amount o f OC. Elemental analyses revealed that filtersand showed significantly lower H/O ratio compared to sediments. Table 1. The H/O ratio
indicates the humification degree of humic substances (De Paolis and Kukkonen, 1997). Thus, the higher ratio indicated that HS in sediments were more humified than
the filtersand HS, which apparently also increases the ability to remove HS from water.
From this discussion, we conclude that due to low amounts of binding sites in
filtersand, a hydrophobic fraction such as H A is preferably removed over more acidic
fraction such as FA. This may lead to a fractionation of HS during A R G where large
hydrophobic and aromatic molecules are removed in the earlier in the process, though FA are conserved unless the aquifer is rich with iron oxides or clay. The conclusion reached f rom this laboratory scale study is consistent with earlier findings f rom fu l l -
scale studies presented by McCarthy et al. (1993) and Schwarzenbach et al. (1983). The aromatic fractions o f HS are a major source of by-products and colour,
thus A R G decreases the risk o f carcinogenicity and improves the aesthetic quality of surface water. I f filtersand, through which ARG is accomplished, is poor in iron oxide
minerals or clay, the removal of FA would be low even after long infiltration distances.
5. Conclusion
Sorption experiments showed that clay content increased whereas organic matter decreased the sorption o f HS to filtersand at A R G . Organic matter was released
because it was not chemically bound and completely stabilized. More aromatic, hydrophobic fractions of HS (HA) were to a higher extend sorbed than acidic
fractions of HS (FA). Chemisorption appears to be less important than physical sorption (hydrophobic sorption enhanced by polyelectrolytic effect) for HS removal during ARG.
6

6. Acknowledgments
We acknowledge Prof. Jörgen Hanæus, Luleå University of Technology, for useful comments on the manuscript and Wayne Chan for proofreading the English.
References
APHA, 1988. Standard methods for analysis waters and wastewaters. APHA, N . Y .
Bower, H. , 2002. Art i f ic ial recharge of groundwater: hydrology and engineering. Hydrogeology Journal 19, 121-141.
Burba, P., van den Bergh, L , Klockow, D., 2001. On-site characterization of humic-
rich hydrocolloids and their metal loading by means of mobile size-fractionation and exchange techniques. Fresenius J. Anal . Chem. 371, 660-669.
Camper, A .K . , 2002. Involvement of humic substances in regrowth. Presented at the
NSF international Wor ld Health Organization Symposium on HPC Bacteria in Drinking Water, Apr i l 22-24, 2002, Geneva, Switzerland.
Chin, Y-P., Aiken, G., O'Loughlin, E., 1994. Molecular weight, polydispersity, and
spectroscopic properties o f aquatic humic substances. Environ. Sei. Technol. 28(11), 1853-1858.
Day, G.M., Hart, B.T., McKelvie, I .D. , Beckett, R., 1994. Adsorption of natural organic matter onto goethite. Colloid and Surface A 89, 1-13.
De Paolis, F., Kukkonen, J., 1997. Binding o f organic pollutants to humic and fulvic
acids: influence of pH and the structure of humic material. Chemosphere 34(8), 1693-1704.
Edwards, M . , Benjamin, M . M . , Ryan, J.N., 1996. Role of organic acidity in sorption of natural organic matter ( N O M ) to oxide surfaces. Colloids and Surfaces A 107,
297-307. Ellis, B.D., Butterfield, P., Jones, W.L. , McFeters, G.A., Camper A . K . , 2000. Effects
of carbon source, carbon concentration, and chlorination on growth related
parameters of heterotrophic b iof i lm bacteria. Microb. Ecol. 38, 330-347 Evanko, CR. , Dzombak, D.A. , 1999. Surface complexation modelling of organic
acids sorption to goethite. J. Colloid Interface Sei. 214(2), 189-206. Gu, B. , Schmitt, J., Chen., Z., Liang, L.,McCarty, J.F., 1994. Adsorption and
desorption of natural organic matter on iron oxide: mechanisms and models. Environ. Sei. Technol. 28(1), 38-46.
Huisman, L. , Olsthoorn, T.N. , 1983. Art i f ic ia l Groundwater Recharges. Pitman Books
Limited, Boston. Jacks, G., Frycklund, C , 1995. Removal o f organic matter during groundwater
formation. Vatten 51, 66-69.
Jardine, P.M, Weber, N . L . , McCarty, F .M. , 1989. Mechanisms of dissolved organic carbon adsorption on soil. Soil. Soc. A m . J., 53, 1378-1385.
Jones, M . N . , Bryan, N.D. , 1998. Colloidal properties of humic substances. Advances in Colloidal and Interface Science 78, 1 -48.
Juhna, T., Klavins, M . , Sprogis, J., 1998. Retention of humic substances on fil ter sand from artificial groundwater recharge infiltration basins. Latvian Journal of
Chemistry 4, 87-93.
Kaiser, K., Zech, W., 1997. Competitive sorption of dissolved organic matter to soils and related mineral phases. Soil. Sei. A m . J. 61, 64-69.
7

Klavins, M . , Cinis, I L , 1990 Isolation of humic substances f rom surface waters. Izv. A N Latv. SSR, Khim.Ser. 3, 360-364. (in Russian).
Lattimer, L . V . , Jones V.P., 1992. Sorption of cadmium and copper onto iron oxides. Inorg. Chem. 23, 326-332.
McCarthy, J.F., Williams, T . M . , Liang, L . , Jardine, P, Jolley, L.W. , Taylor, D .L . ,
Palumo, A . V . , Cooper, L .W. , 1993. Mobi l i ty of natural organic matter in a sandy aquifer. Environ. Sei. Technol. 27, 667-676.
Meier, M . , Namjesni-Dejanovic, K. , Maurice, P.A., Chin, Y.-P., Aiken, G.R., 1999. Fractionation of aquitic natural organic matter upon sorption to goethite and kaolinite. Chemical Geology 157, 275-284.
Nodvin, S.C., Driscoll, C.T., Likens, G.E., 1986. Simple partitioning of anions and
dissolved organic carbon in a forest soil. Soil Science 142(1), 27-35. Pomes, M . L . , Green, W.R., Thurman, E .M. , Orem, W.H. , Lerch, H.E.. 1999. DBP
formation potential of aquatic humic substances. Journ. Amer. Water Works Assoc. 91(3), 103-115.
Shen, Y . - H . , 1999. Sorption o f natural dissolved organic matter on soil. Chemosphere 38(7), 1505-1515.
Spark, M . K . , Wells, J.D., Johnson, B.B., 1997. Characteristics of the sorption of humic acids by soil minerals. Aust. J. Soil. Res. 35, 103-112.
Schwarzenbach, R.P., Giger, W „ Hoehn, E. and Schneider, J.K., 1983. Behaviour of
organic compounds during infiltration of river water to groundwater. Field studied. Environ. Sei. Technol. 17, 472-479.
Stumm, W . 1992. Chemistry of the solid-water interfaces. John Wiley & Sons. Inc. U.S.A.
Thurman, E .M. , 1985. Organic geochemistry of natural waters. Martinus N i jho f f , Dr. W . Junk Publishers, Wageningen.
Tipping, E., 1981. The adsorption of aquatic humic substances by iron oxide. Geochimica et Cosmochimica Acta 45, 191-199.
Tombåcz , E., Dobos, Å., Szekeres, M „ Narres, H.D, Klumpp, E., Dékåny, L , 2000. Effect of p H and ion strengh on the interaction of humic acid with aluminium oxide. Colloid. Polym. Sei. 278, 337-345.
Vermeer, A.W.P. , Riemsdijk, W . H . , Koopal, L .K. , 1998. Adsorption of humic acid to
mineral particles. 1. Specific and electrostatic interactions. Langmuir 14, 2810-2819.

Table 1. Elemental and functional composition of humic acid (HA) f rom filtersand of infiltration basins at Baltezers plant (Sand H A ) ; H A f rom water of Lake Baltezers (Lake H A ) ; H A f rom deposits of Lake Baltezers (Sediment H A ) ; fulvic acid (FA) from Salaca River (River FA) ; podzolic soil H A (Soil H A ) and Aldrich H A .
Humic Elemental composition, % Atomic ratio aC-COOH ^AiOH carom.
substance C H N O H/O (N+0) /C mmol/g % Sand H A 54.60 3.70 0.93 40.80 1.45 0.57 4.88 1.18 32.5 Lake H A 57.21 3.94 1.03 40.21 1.57 0.63 4.15 1.42 19.3 Sediment H A 52.14 6.04 4.30 37.52 2.58 0.61 2.43 - -
River FA 46.28 3.72 2.90 47.10 1.26 0.82 4.45 0.98 16.3 Soil H A 49.85 4.73 2.11 43.31 1.75 0.69 2.21 - 65.3 Aldrich H A 49.89 4.76 2.30 43.06 1.76 0.69 2.15 - 53.5
a carboxylic groups; phenolic hydroxyl groups; c aromaticity
Table 2. Physical and chemical characteristics of aquifer material (filtersand) recovered f rom surficial layers (0-4 cm) of infiltration ponds used for artificial groundwater recharged f rom Lake Baltezers compared to composition of soil analysed by Jardine et al. (1989).
Basin no, sample position BET, OC, Particle size analyses, Fe, m 2 /g dry wt dry wt% mg/g
% Sand Silt Clay
8B n.d. 0.5 77.0 22.0 1.0 0.637 3A 0.49 0.6 65.7 31.0 3.3 0.715 4A 0.85 0.5 40.8 37.2 22.0 0.695 Reference soi l a n.d. 0.2 32.2 44.5 23.6 16.82b
n.d., not determined; BET, surface area, OC, percent of organic matter, a Jardine et al. (1989); b dithionite-citrate-bicarbonate extractable Fe
Table 3. Sorption (mg/g) of different humic acids (HA) and fulvic acids (FA) of
different origins on clay (pH 3.4, 100 mg of clay). Characterization of clay given in Table 4.
Humic substance Clay sample no. 1 Clay sample no. 2 Clay sample no. 3
Aldrich HA 27.1 29.2 16.0
Soil H A 26.3 28.4 13.5 River FA 12.1 14.2 6.4
Table 4. Characterization of studied samples of clay
Sample Age, origin Characteristics Mineralogy of clay no. fraction
1 T l Inda Stage, Nemuna Greenish gray, Smectite dominates Formation T l Inda calcareous clay
2 Stage, Nemuna Reddish brown fat clay Smectite dominates Formation J3,
3 Callovian Stage Black, lean clay Kaolinite dominates

"Si
•3 ii
o c ss
-C 3
3
8A 8C 3 A 3B 4 A
S a m p l i n g position
Fig. 1. Sorption of Aldrich humic acid (100 mg/1) on filtersand samples f rom different infiltration basins (sample size 500 mg; pH 6.5; sorption time 16 h). A l l samples were taken f rom the uppermost layer, 0 - 4 cm, but at different positions (A, B, C) in the basin. See composition of filtersand in Table 2.
O
5£
>> u
•a
TS
0,6
0,2
0-4 4-8 8-12 12-16
Depth in basin, cm
16-20
et "Bfc E
•6
A 0,4 2
u C es *J ir.
JS 3 u
i 3
X
Fig. 2. Relationship between sorption o f Aldrich humic acid (100 mg/1) to filtersand
and the different depths below infiltration basin no. 8A (sample size 500 mg; pH 6.5;
sorption time 16 h). Legend: lefty-axis, OC, organic carbon (white bars); clay (black bars); right y-axis, adsorbed humic substances (open squares).

O 10 20
Humic substances added, mg/g
Fig. 3. Sorption of different humic substances on the upper layer o f filtersand f rom basin 8A (—) and 3A (— — ) (sample size 500 mg; p H 6.5; sorption time 16 h).
1.2
3 4 5 6 7 8 9 10
p H
Fig. 4. Relationship between the sorption of humic substances and pH of filtersand
samples f rom the infiltration basins. Each data point represents mean values of
filtersand collected f rom all basins at different depths and positions. Vertical error bars represent the standard deviation f rom mean values.

Fig. 5. Sorption (sample size 500 mg; pH 6.5; sorption time 16 h) o f fu lvic acid (FA) and different humic acids (HA) on crystalline Alumina (AI2O3).
12
0 5 10 15 20 25 30
Humic substances added, mg/g
Fig. 6. Sorption (sample size 500 mg; pH 6.5; sorption time 16 h) o f fulvic acid (FA)
and different humic acids (HA) on crystalline goethite (a-FeOOH).

'(DO %£'£!) sjazaireg s^q UIOJJ sjuauiipss oj (VH) sppe oiuinq jusjajjip puu (yg) ppe oiAjnj jo (q 9j auiij uoqdjos i5-9 j-fd ;3ui OOS
3zis qdures) jo uoqdjos 7,
8/SUI 'pappe saauejsqns aiuinjj
0€ SS 06 SI 01 S 0
I 1 *-
ft

Paper II
Development of potentially toxic cyanobacteria and bacteria during artificial recharge o f groundwater.
In : Proceedings of International Conference on Harmful Algal blooms. Ninth Conference, Tasmania 2000.

DEVELOPMENT O F POTENTIALLY TOXIC CYANOBACTERIA AND BACTERIA DURING ARTIFICIAL R E C H A R G E OF GROUNDWATER
Gunta Springe*. Ivars Druvietis* & Talis Juhna**
institute of Biology. University of Latvia. 3 Miera St.. Salaspils LV-2169. Latvia Fax: 371-2-944986; e-mail: [email protected] **Riga Technical University. Div. of Water Supply and Sewage. 16 Azenes St.. Riga LV-1048. Latvia e-mail: [email protected] & Luleå University of Technology. Div. of Sanitary Engineering, Luleå SE-97187. Sweden
ABSTRACT
The occurrence of cyanobacterial blooms is typical for the Lake Mazais Baltezers, the water source for the artificial groundwater recharge plant supplying up to 25% of drinking water for Riga City, Latvia. In 1997-98, investigations of algae and bacteria were carried out in the lake water as well as in water and sediments from one infiltration basin and in sand from bore-holes between infiltration basin and a siphon-pipe, i.e.. in the course of artificial recharge of groundwater.
In 1997, cyanobacteria appeared in water of the lake and the infiltration basin in July (0.381 and 0.847 mg L ' J , correspondingly) with Microcystis spp. dominating, and in August (0.847 and 1.925 mg L" 1. correspondingly) with Aphanizomenon flos-aquae (L.) Ralfs. The development of algae including cyanobacteria as well as bacteria (measured as aerobic heterotrophic oligocarbophillous bacteria and total bacterial number) in water of the infiltration basin was substantially higher than in the lake indicating favourable conditions for development of biota. Cyanobacteria were observed also in sediments to a depth of 3 cm below the bottom's surface of the infiltration basin. In contrast to water, cyanobacteria in the sediments were detected not only in late summer, but also during other seasons. The investigations of sand taken from 10 bore-holes between infiltration basin and siphon-pipe to a depth of 900 cm showed that cyanobacteria developed to a depth of 200 cm in the bore-hole under the infiltration pond while bacteria were found a depth of up to 900 cm in all bore-holes. A positive correlation was observed between cyanobacteria and total bacterial number in sediments from the bottom's surface of the infiltration basin (r=0.82; n=41 and in sand from the bore-hole under the infiltration basin (r=0.87; n=8. a=0.01), and further investigations are needed to find the explanation of this phenomenon. A direct link between development of cyanobacteria and heterotrophic bacteria was neither established in water nor in the sediments or sand of the infiltration system.
Consequently, this study revealed that at low quantities of cyanobacteria interaction between them and heterotrophic
bacteria was not found in the course of artificial recharge of groundwater and thus the influence of cyanobacteria on the biodegradation of organic matter was not established.
INTRODUCTION
Artificial recharge of groundwater is being used for drinking water production (about 25% of total) in Riga. Latvia. In the process of infiltration, surface water from Lake Mazais Baltezers (M. Baltezers) is pumped to the infiltration basins from where it percolates down to the groundwater. After underground transport (retention time 30-180 days) the recharged groundwater is abstracted by a siphon system (production wells are connected to vacuumed siphon-pipes), disinfected and pumped to the city water distribution.
Lake M. Baltezers is located in the vicinity of Riga City. Its trophic state is eutrophic. In comparison with the lake in the beginning of the century concentrations of organic matter and biogenic substances in water as well as in sediments has increased [1], In 1996. mineral nitrogen and phosphorus concentrations in lake water varied from 1.0 to 1.83 mg L ' 1 , and from 0.015 to 0.055 mg L"'. respectively. The major pari of phosphorus (64-97 % of total phosphorus) in the lake sediments was found bound to organic substances [2]. Nitrogen is more abundant than phosphorus in the lake, and the N:P ratio exceed 12 [3], suggesting that the algal production was limited by phosphorus. While the development of Aphanizomenon flos-aquae (L.) Ralfs, and A. scheremetievii Elenkin was scanty in 1951, potentially toxic blue-greens Microcystis aeruginosa Kiitz.. Anahaena flos-aquae (Lyngb.) Breb. and A. scheremetievii Elenkin were found in very high amounts in the 1990's [4. 5]. Microcystis aeruginosa Kiitz. has previously been found to be dominant in phytoplankton from eutrophic and hypereutrophic lakes in Latvia with minor amounts of Anahaena spp.. diatoms and green algae [6].
With increasing eutrophication toxic cyanobacterial blooms can be expected to cause a risk for drinking water [7]. In 1995. toxic cyanobacterial blooms occurred in the lake M. Baltezers at the end of the summer and microcystin-LR was found in the algal biomass [5].
Further filtration through sediments and sand results in a rather efficient removal of cyanobacterial toxins and cells except when abundant cyanobacterial blooms occurred [8]. Toxins from water are removed mainly as a result of microbial degradation or photolysis in the presence of humus [9,10]. Besides the fact that toxins excreted by cyanobacteria in the basin directly threaten the health of drinking water consumers, they may influent metabolism of biota in the water body [11]. and subsequently the degradation processes of organic matter in the recharged groundwater.
Organic substances in drinking water may form potentially carcinogenic compounds upon chlorination [12]. and serve as a substrate for bacterial re-growth in the water distribution system [13]. In the process of artificial groundwater recharge, organic substances are mostly removed during water passage

through the subsurface from the infiltration basins to the production wells. Adsorption on sand and biodegradation by heterotrophic bacteria living in the subsurface are the two major processes responsible for this reduction [14].
The groundwater quality largely depends upon the input from Lake M. Baltezers and sediment biology. Potentially toxic cyanobacteria and bacteria living in the infiltration ponds and in the sand between the infiltration ponds and drinking water production wells influence self-purification process of the recharged groundwater. It is therefore, the knowledge about development of cyanobacteria and bacteria, and interactions between them are important. This study attempted to find whether the cyanobacteria significantly influenced the development of bacteria in the course of artificial recharge of groundwater.
MATERIALS AND METHODS
The samples for bacteriological and algological analyses were collected from water with a Ruttner water sampler and from sediments with microbenthometer from Lake M. Baltezers (M. Baltezers) and from one infiltration basin (May. July. September 1997). Sand cores were taken with a special vibrator-drill sampler from 10 sampling bore-holes between
the infiltration basin and the siphon-pipe (October 1998) (Fig. 1). In samples from water, sediment and sand the total bacterial number (TBN) was determined by direct count method. The number of aerobic oligocarbophillous heterotrophic bacteria was determined by the pour-plate method with specific nutrient medium [15. 16]. Algal analyses were conducted according to Standard Methods [16].
RESULTS AND DISCUSSION
During the period of investigation (May-September 1997) cyanobacteria occured in the lake at the input sites for artificial groundwater in July (0.381 mg L"1) when Microcystis spp. was dominant, and in early September (0.847 mg L~') when Aphanizomenon flos-aquae (L.) Ralfs, was dominant. In 1997. contrary to previous years [4. 5] a development of cyanobacteria was less pronounced. In 1997. TBN in lake water was high, although low mesophillous saprophytes and the coli index characterized the water as clean [17].
In the water of the infiltration basin, the development of cyanobacteria and bacteria was substantially higher than in the lake (Table 1).
Fig. 1 Map of the study site and a principal scheme of water passage during anificial recharge of groundwater - Lake M. Baltezers (1), infiltration basin (2), sand sampling bore-holes (3). and production wells connected to a siphon pipe (4).

Table 1. Abundance of heterotrophic oligocarbophilous bacteria (HB). total bacterial number (TBN) and cyanobacteria in the water of Lake M . Baltezers and one infiltration basin. May-September 1997
The overall composition of algae in the infiltration basin was similar to that of the lake, but the phytobenthic species Oscillaloria lemtis (AG.) Kiitz. apparently washed in from the lake was overgrown the bottom of infiltration basin. Bacillariophyceans dominated the upper sediment layer (to 10 cm) known to reduce the infiltration rate of water by clogging pores between sediment particles [18]. At the end of the vegetation period (September), diatoms together with decaying cyanobacteria (Microcystis spp.. Aphanizomenon flos-aquae (L.) Ralfs.) formed a crust on the surface of the sediment, further reducing the infiltration process. Cyanobacteria were observed to a depth of 3 cm and in contrast to the overlying water they were detected not only in the late summer but also during other seasons. A similar trend of development of cyanobacteria and bacteria on the surface of sediments, and a correlation was found between numbers of cyanobacteria and TBN (r=0.819: n=4). Correlations between numbers of cyanobacteria and oligocarbophillous heterotrophic bacteria and TBN were neither established in the water nor in deeper layers of sediment.
In the sand taken from 10 bore-holes between the infiltration basin and the siphon-pipe (depth down to 900 cm), the TBN was related to the concentration of NOV (r=0.862; n=7. a=0.05) and to organic matter (r=0.987: n=7, ot=0.01). In October 1998. algal cells were found to 700 cm only in the bore-hole below the infiltration basin, dominated by Bacillariophyceae. The Cyanobacteria Oscillaloria tenuis (Ag.) Kiitz were found to a depth of 200 cm. and also O. princeps Vauch.,. typical for autumn, as well as Lyngbia sp. occurred (Table 2).
Table 2 Distribution of cyanobacteria in sand from bore-hole below an infiltration basin to a depth of 900 cm
Date HB. cells mL - ' TBN. cells 10" mL-'
Cyanobacteria. cells L 1
Date
lake inf. bas.
lak e
inf. bas.
lake inf. bas.
7.05. 2 100 2 930 5.6 8.4 0 0 26.05. 150 790 3.5 9.3 0 7 740 15.07. 120 1 500 6.1 9.2 304 000 2 348 800 17.09. 111) 490 8.0 8.0 888 200 16 836 850
Depth, cm
Cyanophvta Depth,
cm Oscillaloria
princeps Oscillaloria
tenuis Lyngbia sp.
0 + + + 10 + + + 20 + + + 30 + 0 40 + + 0 50 + 0 0 100 + 0 0 200 + 0 0 300 0 0 0 400 0 0 0 500 0 0 0 600 0 0 0 700 0 0 0 900 0 0 0
In the sand from bore-holes a positive correlation between the concentration of cyanobacteria and TBN was found: r=0.87 (n=8. cx=0.01) (Fig.2) but correlations between cyanobacteriacea and heterotrophic bacteria were not observed.
400.0
350.0
300.0 -
250.0 -
ffl 200.0 -H 150.0 -
100.0 -y =
5 0 . 0 - R- = 0.8391
0.0 1 1—I—h
G sO rg) ^ $> ^
y = -21.414x +377.13
R2 = 0.8602
2500
2000
I 1500
-j - 1000
\ 500
0
depth (cm)
• - - Total bacterial number (TBN). million cells/g
• Abundance of cyanobacteria. cells/a
Fig. 2 Changes in total bacterial number (TBN) and abundance of cyanobacteria with the depth in sand of the bore-hole below the infiltration basin

In general, the presence of cyanobacteria in the course of artificial recharge of groundwater appears to be determined by their development in Lake Mazais Baltezers. The numbers and the biomass of cyanobacteria as well as the numbers of total bacteria increased substantially in the infiltration basin indicating favourable conditions for development of both algae and bacteria. A correlation was found between the concentration of cyanobacteria and the total bacterial number in the sediments from the infiltration basin and in the sand from bore-hole below this basin. At the same time significant correlations between cyanobacteria that occurred in 1997 at low quantities and heterotrophic bacteria in the infiltration basin and in the sand from bore-holes were not found, indicating that there was no significant interaction between algae and heterotrophic bacteria. Thus the biodegradation of organic matter in artificially recharged groundwater was not considerably influenced by the cyanobacteria.
ACKNOWLEDGEMENTS
Authors are grateful to municipal enterprises Riga Water for financial support and to Jörgen Hanaeus, Professor at the Division of Sanitary Engineering. Luleå University of Technology. Sweden, for academic support.
R E F E R E N C E S
Amsterdam, Netherlands, publisher A.A Balkema. (Rotterdam), p. 211 -216 (1998).
9. G. J. Jones. D.G. Bourne. R.L. Blakely. H. Doelle. Nat. Toxins, 2, 228-235 (1994).
10. M. Welker. C. Steinnberg. Environ. Sei. Technol, 34, 3415-3419 (2000).
11. W.W Carmichael. A Status Report on Planktonic Cyanobacteria (Blue-Green Algae) and Their Toxins. (Environmental monitoring systems laboratory office of research and development U.S. Environmental Protection Agency Cincinnati. Ohio 45268). 141 p.. (1992).
12. A. Liimatainen. T. Grummt. Bull. Environ. Contam. Toxicol., 41,712-718 (1988).
13. P.M. Huch. Jour. AWWA. 82.(7), 78-86 (1990). 14. R.P. Schwarzenbach, W. Giger, E. Hoehn. J.K.
Schneider. Environ. Sc. Technology. 17. 472-479 (1983).
15. V . l . Romanenko. S.I. Kuznetsov. (Nauka, Leningrad), (in Russ.), 194 p. (1974).
16. Standard Methods for the examination of Water and Wastewater. A.E. Greenberg. L.S. Clesceri. A.D. Eaton, eds.. 18,h edition. (APHA, AWWA. WPCF). 1268 p. (1992).
17. T. Juhna. G. Springe. Vatten, 54. 259-264 (1998). 18. B.R. Jackes. in: Proceedings of the Groundwater
Recharge Conference, Townswille. Australia. July, 1980. 119-131 (1980).
1. A. Bnede. G. Springe, in: Sustainable Lake Management. Book of Abstracts "8 International Conference Lake 99". Denmark. I I , S8.A-7 (1999).
2. A. Briede. M. Kjavirjs. V. Rodinov. Proc.Latvian Acad.Sci., Section B, 53,48-53 (1999).
3. C. Forsberg. S.-O. Ryding. A. Claeson. A. Forsberg. Mitt.Imernat.Verein.Limnol.,21, 352-363 (1978).
4. G. Spripge. A. Briede. I . Druvietis, E. Parele. V. Rodinovs. L. Urtäne. in: Hydrobiological Research in the Baltic Countries. Part I . Rivers and Lakes. R. Volskis. ed.. (Institute of Ecology. Lithuania, Vilnius), pp. 184-324(1999).
5. F. Eynard, K. Mez. J.-L. Walther, Wat. Res.. 34. 2979-2988 (2000).
6. I . Druvietis. in: Harmful Algae. B.Reguera. J.Blanco. M.L.Fernandez and T.Wyatt. eds., (Xunta de Galicia and Intergovernmental Oceanographic Commission of UNESCO), p. 65 (1998).
7. J.R. Falconer, in: Harmful Algae. B.Reguera. J.Blanco. M.L.Fernandez and T.Wyatt. eds.. (Xunta de Galicia and Intergovernmental Oceanographic Commission of UNESCO), p. 37-38 (1998).
8. K. Lahti. J.Vaitomaa, A.-L. Kivimäki, in: Proceedings of The Third International Symposium on Artificial Recharge of Groundwater - T1SAR 98,

Paper III
Removal of humic substances during treatment of drinking water using sorbents.
Vatten, 56, 79-83 , 2000

VATTEN 56:79-83. Lund 2000
REMOVAL OF HUMIC SUBSTANCES DURING TREATMENT OF DRINKING WATER USING SORBENTS
by MARIS KLAVINS, TALIS JUHNA1'2, LINDA EGLITE
Department of Environmental Sciences, University of Latvia, Raina blvd. 19, LV-1586, Riga, Latvia
e-mail: [email protected]
1 Department of Civil Engineering, Riga Technical University, LV-1658, Riga, Latvia
2 Luleå University of Technology, Division of Sanitary Engineering, S-971 87, Luleå, Sweden
Abstract The removal of humic substances during drinking water preparation by anificial recharge of groundwaters using synthetic sorbents, activated carbon and anthracite was assessed. Humic (HA) and fulvic (FA) acids isolated from soil, peat, water, filters, from the infiltration basin for artificial groundwater recharge, as well as commercial humic acid, were used in study. I t was found that adsotption is determined by the sorbent propenies, and the nature of the humic substance. The sorption process is slow, controlled by p H and ionic strength (sorption increases in acidic and high ion strength conditions), and depends on the porosity of the used sorbents and the character of their functional groups. The tested sorbents proved their efficiency in pilot scale experiments.
Key words — humic substances, sorption, drinking water, anificial groundwater recharge.
Introduction
Water treatment aimed at drinking water preparation is the removal of undesired water ingredients. Numerous techniques can be used for these purposes, including coagulation, filtration and sorption approaches (Water Quality and Treatment... 1990). Humic substances are among the different groups of substances present in natural waters which adversely influence the quality of drinking water (Thurman 1985, Klavins 1998). These high molecular weight polycationites form carcinogenic and mutagenic organochlorines upon chlorination (Loper 1980, Kronberg and Vartiainen 1988). Humic substances also form stable complexes with metal ions and persistent organic xenobiotics, and thus are responsible for the transport of many contaminants during the drinking water preparation process. The presence of humic substances also gives an unpleasant odour, colour and taste to drinking water. Substantial efforts have been made to develop drinking water treatment technologies to reduce the concentration of humic substances during drinking water treatment process (Eikebrokk 1989). The sorption of humic substances from the aqueous phase on solid surfaces occurs as a result of attractive interactions due to Van der Waals forces, hydrogen bonding and electrostatic forces (Klavins 1998). As the molecular mass of humic substances can reach up to several thousands Daltons, high porosity of the sorbents (Thurman and Malcolm 1981) is of importance. More effort should be paid to the development of efficient
sorbent systems for preparation of drinking water. The main problems related to removal of humic substances are comparatively high concentrations in surface waters and major differences in their properties depending on their origin. In surface waters, use of sorbents for removal of HS may be economically unacceptable, but their concentrations in groundwater are much lower and use of sorbents may be practical. There is a need to develop techniques for removal of organic substances after artificial recharge of groundwater — a sustainable, energy saving method used in water supply in Riga (Juhna et al. 1999). This process consists of filtration of lake waters (Lake Baltezers) through the surface sediment layer mostly consisting from loose quaternary sediments, composed from silica, clays, minor amounts of organic sediments and iron oxides, thus allowing to remove bacteria, fungi, algae, particulate matter from treated water and to reduce the concentration of organic substances (from 15 mg 0 2 H C O D l M n in infiltration ponds, to 7 mg 0 2 1 _ 1
in groundwater). However, the achieved organic matter concentration can be considered as too high for further use without purification but too low to efficiently use the common water treatment methods (coagulation etc.)
The objectives of the study were to assess in respect to humic substances the sorption properties of different sorbents and to evaluate the possibilities to use sorbents for drinking water treatment with additional cleaning of water after artificial recharge.
V A T T E N • 2 • 00 79

Table 1. Elemental and functional composition, of humic substances used for adsorption experiments.
Humic substance Elemental composition, %
C H N
- C O O H
mmol g - 1
A r O H Aromaticity
%
Aquatic FA from Lake Islienas (FA-I) 56.41 3.85 0.87 3.87 1.43 22.4 Commercial H A (Aldrich) (HA-A) 49.89 4.76 2.30 2.15 1.17 53.5 Soil FA (FA-So) 46.13 4.09 2.48 3.10 1.65 28.7 Soil HA (HA-So) 54.05 5.21 3.71 2.10 1.56 34.3 Peat FLA (Sphagnum peat) (HA-Sp) 51.39 4.08 2.25 — — — Peat H A (pine peat) (HA-P) 52.34 4.28 3.86 1.84 0.96 65.3
Materials and methods
Isolation and analyses of humic substances
Humic (HA) and fulvic (FA) acids were isolated from soil and peat by extraction with 0.1M NaOH (Methods of Soil Analysis, 1989), but from water of Lake Islienas by a method described by Klavins and Cinis (1990). The elemental composition of isolated humic substances was determined on a Perkin Elmer 240 B C H N analyzer. The content of carboxyl groups was determined using Ba(OH) 2 titration, and the content of hydroxyl groups after acetylation (Methods of Soil Analysis, 1989). Aromaticity of the humic matter was calculated using the following equation: aromaticity = 0.05 3 + 6.74 where: 3- molar absorptivity (at 280 nm) of humic substances (l(mol cm) - 1 ) (Chin et al. 1994). Commercial (Aldrich) humic substances were used for comparison. Properties of used humic substances are summarised in Table 1. Concentrations of humic substances in water were determined spectrophotometrically (410 nm) using a H A C H DR 2000 spectrophotometer.
Adsorption Studies
Sorbents used in this study were used as obtained from Dow Chemicals, Enola Ltd., Lachema, Biolat, Chem-viron Carbon, Reachim, Rohm&Haas (Table 2). Before use the sorbents were conditioned and washed. Adsorption studies of humic substances on different sorbents were conducted as batch experiments performed in 100 ml sealed glass bottles on a rotary shaker table 24 h at 20°C. 100 ml of either 5, 10, 25, 50, 75, 100, 250 mg 1 _ 1 solutions of humic substances with 1 g of sorbent were shaken until equilibrium occurred (24 hrs.). After separation of phases by filtration, the concentrations of humic matter in the supernatant phase were determined spectrophotometrically (HACH 2000, glass cell with path length 2.5 cm) as absorption at 410 nm (values obtained from initially prepared calibration graph). The quantity of humic substances adsorbed to different phases was determined from the difference of the initial aqueous phase concentration and the amount in solution at equilibrium. The type of humic substance, pH of the solution, and sorption time were varied. The sorp-
Table 2. Properties of sorbents used for testing of sorption of humic substances.
Name Type* M a t r i x " Porosity*** Exchange capacity ' meq g"'
Activated carbon (GAC) Hydrophobic Carbon M R — Amberlite IRA-405 Strong anion* St-Dvb M R 4.2 AB-17-8 Strong anion* St-Dvb Gel-type 4.4 Spheron DEAE Weak anion Ma M R 0.34 Amberlite IR-120 Strong cation St-Dvb Gel-type 4.3 Amino propyl silica Weak anion Silica M R 0.18 DiaionPA-17-8 Weak anion St-Dvb M R — Amberlite IRA-95 Weak anion St-Dvb M R 4.7 Amberlite IR-124 Strong cation St-Dvb M R 4.3 Anthracite Hydrophobic Carbon — — Polisorb-1 Hydrophobic St-Dvb M R —
* Strong anion — strong anion exchange resin (commonly containing functional groups such as - N ( C H 3 ) 3 ' ' ; Weak anion - weak anion exchange resin (commonly containing functional groups such as - N H j or - N ( C 2 H ; ) 2 ; Strong cation - strong cation exchange resin (contain functional groups -S0 3 H~) .
** St-Dvb — styrene-divinylbenzene; Ma - Methacrylic acid derivatives. ** M R - macroreticular.
80 VATTEN -2-00

C, mgl"1
Figure 1. Sorption of humic acid (Aldrich) onto ion exchange resins.
tion test was performed in a laboratory scale column (30x250 mm) filled with sorbents (GAC, Polysorb-1, Amberlite IRA-95). A humic acid solution (Aldrich, 25 mg l"1) was percolated through the column with a velocity 6 ml min - 1 , sampling fractions of 10 ml with a "Pharmacia Fine Chemicals" fraction collector and analysing concentrations of the remaining humic acids in solution. The sorption test in a pilot scale column was performed taking groundwater from a depth of 15 m at the artificial recharge plant "Baltezers". A pilot scale column (820 x 200 mm; volume - 25 dm 3) was filled with GAC or anion exchange resin AB-17-8 and tested (flow rate 100 1 h - 1 , volume of sorbent 5 liters, C O D M n
of ingoing water 8.5 m g 0 2 H ) . In the outlet, C O D M n , TDS, N t o t , and PTOt, were estimated for 2 months, taking weekly samples.
Results and discussion
Since humic substances are anionic polyelectrolytes, macroporous anion exchange resins can be considered to be well suited for their removal. However, the preference of the resin type (strong base resins vs. weak base resins) and the rigidity of the resin matrix are highly controversial (Fu and Symons 1990, Hand et al. 1994). In real processes the interaction with other ions in treated water should be considered (Eikebrokk 1989). Also carbonaceous resins have been recommended for drinking water treatment (Hand et al. 1994), which seems yet more prospective as such polymers are used for isolation of aquatic humic substances (XAD-8). Other resins comparatively widely studied for treatment include granulated activated carbon (GAC), obtained from wood and cleaned for use in water purification, and more recently, anthracite (Removal of humic substances... 1999). Sorption of humic substances on carbon-based sorbents proceeds due to Van der Waals interaction and depends on the pore size of the sorbent. In the case of GAC, macropores (- 40 % of rotal pore volume) and micropores (> 1 nm) dominate, but the specific surface area is 1000-1400 m 2 g" 1. In comparison, the specific surface
• • • - • Amberlite IRA-405
0 50 100 150 200
Cmgr 1
Figure 2. Sorption of humic acid (Aldrich) onto ion exchange resins.
area of anthracite is - 5 m 2 g _ 1 and it does not contain micro and mesopores, but only a relatively increased (eroded) surface (Removal of humic substances... 1999)
Humic substances present in the groundwater after its artificial recharge can originate from aquatic, soil, peat and sedimentary HS. As the properties of HS depend on their origin, we have separately studied the sorptive behaviour of these substances, using as a surrogate commercially available humic acid (Table 1). The properties of the studied HS fall into the range common for humic substances (Orlov 1990, Klavins, 1998). The humic substances differ in their aromaticity, since the dominant structures of humic substances are benzene- and phenolecarboxylic acids (Klavins 1998) (aromatic structural units), and residues of carbohydrates and mono-and dicarboxvlic acids (aliphatic structural units). The aromaticity is highest for peat and soil humic substances, and lowest for aquatic humic substances (Klavins et al. 1999).
The sorptive properties of studied humic substances were determined in batchwise test conditions, in a laboratory scale column and in a pilot scale column. Among the studied sorbents (Figures 1-3), the macroreticular weak anion exchange resins (DEAE Spheron, Amino propyl silica, Diaion PA-408) were the most efficient. Porosity appeared to prevail over functional group con-
0 L « _ ^ , , 1
0 50 100 150
C mg r'
Figure 3. Sorption of humic acid (Aldrich) onto GAC, anthracite and Polysorb-1.
VATTEN -2-00 9 81

centration and type in determining sorption efficiency: weak anion exchangers with a low concentration of functional groups were more efficient than strong anion exchange resins with nearly ten times higher concentrations of functional groups.
Sorption on anthracite led to lower equilibrium concentrations (< 90 mg l* 1), which is to be expected in water treatment, and the sorption was improved over that on GAC which is more commonly used for water purification. These differences show that the micropores in GAC are not easily accessible to humic substances. The sorption efficiency was high on carbonaceous (hydrophobic) resins (Polysorb-1) and moderate on cation exchange resins, implying that there are several mechanisms involved in retention of humic substance. The observed differences in sorption, depending on the sorbent properties, shows the importance of optimisation of sorbent selection or even a need to design special resin types for removal of humic substances.
The sorption of humic substances on the sorbents (Figure 4) is highest at low pH of the humic matter solution. At pH that is more common in the real environ-
I 4
I I at • GAC
rjAmberlite IRA-95
ment, sorption of humic matter is much lower. The dependence on pH is higher for hydrophobic sorbents, but less expressed for ion exchangers. However, the sorption is low at pH where the humic substances as polycation-ites are thermodynamically less stable and tend to precipitate, which means that the sotption is dominated not so much by the forces of adsorption of humic matter onto sorbents, but rather by the intermolecular association forces between humic matter.
Study of sorption kinetics (Figure 5) shows that the sorption process is slow (especially for GAC and Anthracite). The observed pore size values for nitrogen adsorption (BET method) were much higher than size of humic macromolecule. The sorption kinetics indicate that the sorption process is diffusion controlled. The sorption of humic substances much depends also on the type and properties of humic substances (Figure 6). Depending on their humic substance origin, the sorption efficiency could be optimal on hydrophobic sorbents (GAC) or ion exchangers (Amberlite IRA-95). Therefore, the optimal sorbent for each case, depending on the dominant sources of humic substances can differ.
To study the potential for practical groundwater purification, the most perspective sorbents were tested first in laborator)' scale columns (Figure 7), but afterwards in a pilot scale system (Figure 8).
Amberlite IRA-95 Polysorb-1 GAC
400 600
V, ml
1000
Figure 6. Sorption depending on type of humic substances. Figure 7. Breakthrough curves for three types of resins in labo Wry scale column.
82 VATTEN -2-00

1,0
0,8
o 0,6 II
D ~ 0.4 < >J
f -O-GAC / -&-AB-17-8
0,2
0,0 '
O 20 40 60 80
V , m 3
Figure 8. Breakthrough curves for two types of resins in pilot scale column.
The use of the tested sorbents increased the quality of
tteated water (Figure 7, 8). Anion exchange resins and
GAC proved to be more efficient for removal of organic
mattet. The sorprion curves show comparatively rapid
saturation of the used sorbents and achievement of equi
librium with organic matter in aqueous phase. A possi
ble explanation for this may be that humic substances
comprise up to 60-80% of the total dissolved organic
matter in groundwater (and this is in general agreement
with data from literature) (Klavins 1998). To confirm
this suggestion, biologically degradable organic carbon
(BDOC) was estimated (Volk et al. 1997). In water of
lake Baltezers, BDOC comprised 3 0 ^ 0 % of the total
DOC, compared to 15-25 % of total DOC in ground
water The experimental pilot scale experiments showed
that the major part of HS are removed during the test
period, while low molecular weight organic substances
are nor retained, but those may be efficiently removed
using known biodegradation methods or other ap
proaches.
Conclusions
Humic substances may be efficiendy removed from
groundwater using different types of sorbents. Macro-
reticulat anion exchange resins and GAC were found to
be the most efficient. The sorption depends also on the
properties of HS.
References Chin, Y.-P., Aiken, G., O'Loughlin, E. (1994) Molecular
weight, polydispersity, and spectroscopic properties of aquatic humic substances. Environ. Sei. Technol., 28(11), 1853-1858.
EikebrokkB. (1989) Fjerningavhumus. Vatten,45, 297-315. Fu, P. L. K., Symons J. M . (1990) Removing aquatic organic
substances by anion exchange resins. J.Amer .Water Works Assoc., 82, 10, 70-77.
Hand D. W., Herlevich, J. A , Perram, D. L , Crittenden, J. C. (1994) Synthetic adsorbent versus GAC for TCE removal. J.Amer. Water Works Assoc., 86, 64-72.
Juhna T. M . Klavins, J. Sprogis. (1998) Retention of humic substances by fiker-sand from infiltration ponds for artificial groundwater recharge. Latv. Chem.J., 4, 87-93.
Klavins, M . , Apsite, E., Parele, E. (1997) Humic substances in surface waters of Latvia. Proc. Latv. Acad. Set., Ser. S, 51 (3/4), 143-153.
Klavins, M . (1998) Aquatic humic substances: Characterisation, Structure and Genesis. Riga: LU, 286 pp.
Klavins, M . , Cinis U . (1990) Isolation of humic substances from surface waters. Izv. A N Latv. SSR, Khim.Ser., 3, pp. 360-364 (in Russian).
Klavins M . , Seriäne, J., Supe, A. (1999) Properties of soil and peat humic substances from Latvia. Proc. Latv. Acad. Sei, ser B, 53(5), 249-256.
Kronberg, L , Vartiainen, T. (1988) Ames mutagenicity and concentration of the strong mutagen 3-chloro-4-(di-chloromethyl)-4-oxo-butenoic acid in chlorine-treated tap waters. Mutation Res., 206, 177-182.
Loper, J. C. (1980) Mutagenic effects of organic compounds in drinking water. Mutation Res., 76, 241—268.
Methods of Soil Analysis. (1989) American Society of Agronomy Inc. Publisher, Madison, Wise. pp. 1247.
Orlov, D.S. (1990) Soil humic acids and general humification theory. Moscow:MGU, 324 p. (in Russian).
Removal of humic substances from water. (1999) (Ed. H . Odegaard) International IAWQ-IWSA Conference, Norges forskningsrad., Trondheim, Norway, 280 p.
Thurman, E. M . (1985) Organic Geochemistry of Natural Waters. Martinus Nijhoff /Dr. W. Junk Publishers, Wa-geningen, 234 pp.
Thurman, E. M . , Malcolm, R. L. (1981) Preparative isolation of aquatic humic substances. Environ. Sei. Technol., 15, 463-466.
Volk, C. J., Volk, K. D., Kaplan, L. A. (1997) Chemical composition of biodegradable dissolved organic matter in streamwater. Limnol. Oceanogr., 42(1), 39—44.
Water Quality and Treatment: A Handbook of Community Water Supply (1990) American Waterworks Association, Ed. F. W. Pontius, McGrawHill: N.Y., 1193 p.
VATTEN -2-00 83

Paper IV
Microbially available phosphorus and assimilable organic carbon in a drinking water supply system.
Water Research, 2002, submitted.

M I C R O B I A L L Y A V A I L A B L E P H O S P H O R U S AND A S S I M I L A B L E O R G A N I C C A R B O N IN A D R I N K I N G W A T E R S U P P L Y S Y S T E M
TALIS J U H N A 1 ' 2 ) , V I Z M A N I K O L A J E V A 3 ' , VIKTORS J U H N A 4 ) , J Ö R G E N H A N A E U S 1 ' "Department o f Environmental Engineering, Luleå University of Technology, SE 97187,
Luleå, Sweden "'Department of Civ i l Engineering, Riga Technical University, L V 1048, 16, Azenes Street,
L V 1048, Riga, Latvia 3 ) Microbial Strain Collection of Latvia, University of Latvia, 4, Kronvalda boulevards, L V
1586, Riga, Latvia 4 ) Riga Municipal Enterprise Riga Water, 1, Basteja boulevards, L V 1495, Riga, Latvia
Corresponding author: Talis Juhna telephone: + 371 9226441
telefax: + 371 7089253 e-mail: [email protected]
Running title: M A P and AOC in humus waters
Keywords: drinking water, humus water, assimilable organic carbon, microbially available
phosphorus, microbial growth, l imit ing nutrient.
Abstract
It was recently shown that bacterial growth in some humus-rich drinking waters is regulated by phosphorus rather than by organic carbon. In this study, to determine the bacterial growth
potential of drinking water, prepared by chemical precipitation or groundwater that has been
artificially recharged f rom humic surface waters, microbially available phosphorus (MAP) and assimilable organic carbon (AOC) were analysed with the use of bioassays along the water supply system of Riga, Latvia. Results showed M A P was effectively removed (95%) by
chemical treatment, but only moderately (60%) in the process of artificial recharge of groundwater, whereas removal of potential AOC was low (<30%) in both water treatment processes. As a result phosphorus became growth limiting for bacteria in the former, but not in the latter water. In chemically treated drinking water, the potentially available AOC content
was high (672 p.g/1), but only 40% of it could be used by bacteria due to a deficit of M A P , which was as low as 0.43 pg/1 in this water. Contrarily, in recharged groundwater, the
potentials of AOC, 584 pg/1, and M A P , 5.41 pg/1, were high. Thereby there were no shortages of phosphorus and the whole amount of potential AOC was used for bacterial growth. The
phosphorus limitation was confirmed by a nutrient enrichment test using a consortium of
bacteria sampled from deposits along pipe walls in the distribution net. Although the M A P concentration was ten times lower in the chemically treated surface water than compared to the artificially recharged groundwater, bacterial growth in distributed water was only 40%
lower in the former. This was because at phosphorus growth limiting conditions, the bacterial
strain used in M A P bioassays exhibited lower growth response than indigenous bacteria l iving in the drinking water. It was concluded that in humus-rich drinking water prepared by
chemical coagulation phosphorus may not only regulate the bacterial number, but also the
distribution among the different bacterial species.
1

I N T R O D U C T I O N
Growth of heterotropic bacteria in drinking water distribution systems may lead to unaesthetic and unsafe water for consumers. Microbial growth in drinking water usually is regulated by
organic carbon (LeChevallier et al., 1991). Assimilable organic carbon (AOC) is commonly used to measure bacterial growth potential (van der Kooij et al., 1992). Recently, it was reported that in some humus-rich drinking waters, bacterial growth was limited by
phosphorus (Miettinen et al., 1997; Sathasivan et ai, 1997; Sathasivan and Ohgaki, 1999). To evaluate the potential of bacteria to grow in these types of waters, the use of a newly
developed bioassay of microbially available phosphorus (MAP) (Lehtola et al., 1999) has
been suggested (Lehtola et al., 2001). This study aims, by analysing M A P and A O C concentrations, to evaluate the bacterial
growth potential of drinking water f r o m a water supply system fed with humus-rich raw waters. The tasks were to determine (1) changes of M A P and AOC concentrations after the
raw water chemical treatment or infiltration to groundwater (artificial recharge of groundwater), and during drinking water distribution in network; (2) the effects o f M A P and
AOC concentrations on heterotrophic plate counts (HPC) of bacteria and maximum bacterial
growth numbers ( H P C m a x ) in distributed drinking water; and (3) which nutrient is l imiting bacterial growth along a particular water supply system. The present paper reports a nine-month full-scale study at the water supply system of Riga City, Latvia.
M A T E R I A L S AND M E T H O D S
Study site
Riga, the capital o f Latvia, is supplied with drinking water (-200 000 m /day) produced f rom surface water (50%) and groundwater (50%). The surface water, taken f rom the
Daugava River (S r a w ) (Fig. 1), is pre-chlorinated and treated with chemical coagulation-
flocculation (alum) followed by sedimentation and rapid filtration. A chlorine dose of 2-3
mg/1 is used for final water disinfection at the plant (Spiant). The groundwater is abstracted at several naturally ( N p | a m ) and artificially recharged groundwater abstraction sites ( A p i a n i , Gpian,).
In this study, the plant G p i a n i was investigated in particular (Fig. 1). At this plant abstracted groundwater is artificially recharged (without pre-treatment) f rom the eutrophic (Eynard et
ai, 2000) humic lake Mazais Baltezers (Lraw)- The lake water is purified while it flows f rom the infiltration basins through the sandy (Quaternary) aquifer. The retention time in the
subsurface was about one month. The water is then treated only by an addition of a chlorine dose of about 1 mg/1 before being pumped to the city network. The drinking water distribution network is more than 50 years old wi th a total length of about 1,275 km. Cast iron dominates
as pipe material. The corroded network generates plenty of soft deposits in the net. A part (S n e l ) of the network is supplied wi th only chemically treated surface water, another part (G n e i )
with groundwater, and one part wi th mixed waters (Fig. 1). About half of the distributed water
in the part G n e i is artificially recharged groundwater, mainly f rom plant G P ] a m , whereas the rest is natural groundwater.
Water sampling
During the study period of nine months (April-December) in 2000 (plus in Apr i l of 2001),
a total of 66 water samples were collected f rom the Riga water supply system on 19 sampling
occasions ( - two per month). The samples originated f rom the raw water sources S, a w (/? = 3)
and (n = 3) (sampling depth - 0.5 m), at the effluents of the drinking water plants S p i a n t (« = 6) and G p i a m (n = 7), f rom 16 points in the distribution network (no. 1-16) in part G n e t (/; =
2

19), f rom 13 points (no. 23-35) in part S n e t (« = 20), and 6 points (no. 17-22) in the mixture zone (SGnet) (« = 8) (Fig. 1). Most of the drinking water samples in the network were taken from hydrants after flushing them for 15 minutes, while a few (points no. 14, 16, 24, and 30) were f rom apartment water taps in the morning hours. The samples were collected in 1.000 ml sterile borosilicate glass bottles (Pobel, Deltalab, Spain), delivered to the laboratory, and
analysed within six hours.
Biofilm sampling
On two occasions the b iof i lm was removed f rom the transition main of the networks G n e t
and Snet- About 500 g of loose deposit layer (thickness varied f r o m 0.2 cm to 3 cm) was removed using a sterile scalpel f r om the inner side of the large diameter pipes immediately after opening the pipe joints. The sample was placed into a sterile plastic bag (Whirl-Pak Nasco, Inc.) and delivered to the laboratory within 30 minutes. The pipes f rom where the samples were taken have been used to supply water for more then 50 years. X-ray diffraction analyses revealed that the deposits were mainly fine grains of various forms of iron
(hydr)oxides.
Preparation of glassware
A l l glassware (Pobel, Deltalab, Spain; Simax. Czech Republic) and plastic pipettes (Elkay Products, Inc.), were first washed with detergent, immersed in 2% HCl solution for 2 hours, and then rinsed several times wi th deionised distilled water before being used for analyses. Finally, borosilicate glassware was muffled at + 250° C for 8 hours.
Mineral salts, thiosulfate and acetate stock solutions
Analyses and test procedures required several stock solutions. For AOC p o t entiai analysis (see below) and nutrient enrichment test, a nutrient solution (LNP) was prepared by dissolving 4.55 g (NH4) 2 S0 4 , 0.2 g K H 2 P 0 4 , 0.1 g M g S 0 4 x 7 H 2 0 , 0.1 g C a C l 2 x 2 H 2 0 , and 0.2 g NaCl in 1,000 ml deionised water. For M A P analyses (see below) and nutrient enrichment test, a
nutrient solution without phosphorus (LN-P) was prepared by dissolving 0.83 g NH4NO3,
0.10 g M g S 0 4 x 7 H 2 0 , 0.10 g C a C l 2 x 2 H 2 0 , 0.10 g KCl , and 0.10 g NaCl in 1,000 ml of deionised water. Dechlorination of drinking water samples required the preparation of a sodium thiosulfate solution by dissolving 30.0 g Na 2 S 2 03 in 1,000 m l of deionised water. For
bacterial stock culture and M A P analyses a sodium acetate stock solution was made by dissolving 2.267 g CH3COONax3H 2 0 in 1,000 ml of deionised water. A l l reagents used were
of analytical grade purity and purchased f rom Lachema Ltd. , Czech Republic. A l l solutions were sterilized (autoclaved) 121 °C for 20 minutes and stored at 4 °C in darkness for use
within no more than six months.
Determination of AOCnalive and AOCp(rtmaa\
The concentration of AOC was measured by a modification (Miettinen et al., 1999) of the
standard method (Van de Kooi j et al., 1982) suggested for humus-rich waters. According to
the modification, AOC was measured with (AOCpotentiai) and without (AOCnative) adding a
mixture (40 u.1 of LNP solution to 40 ml sample) o f inorganic nutrients. A bioassay is based
on measuring the growth of the bacteria Pseudomonas fluorescens P17 (ATCC 49642) and
Aquaspirillum sp. N O X (ATCC 49643) in water samples. The strains were maintained in liquid nitrogen at -196 °C in 10% glycerin and were cultivated by spread plating on R2A agar
(Merck KGaA, Darmstad, Germany) at 28 °C. The stock cultures for the AOC test were grown in commercially available, sterile Zakumuiza spring water (Markol Ltd. , Latvia) by
adding 100 ug/1 of sodium acetate stock solution. Stock cultures were incubated at 20 ± 2 °C until the maximum count, i.e. a stationary phase, was reached. Stock cultures were stored at 4
3

C and used for analyses within six months after preparation. Two 40 ml replicates of the
sample were poured into 100 ml borosilicon vials. Prior to inoculation, chlorinated water
samples were neutralized by adding 100 pi sodium thiosulfate solution to 40 ml samples. The water samples were pasteurised by heating the bottles for 30 minutes at 70 °C in a water-bath, and then cooled at room temperature. About 500 colony-forming units (CFU)/ml each of P.
fluorescens PI7 and Aquaspirillum sp. N O X were inoculated using carbon-free pipettes. The
bottles were capped tightly and incubated at 20 ± 2 °C in darkness. On incubation days seven, eight, and nine, 0.1 ml of the sample was withdrawn with a sterile pipette and in three dilutions surface-plated over R2A agar plates. CFU were counted after three days incubation at 20 C. AOC was calculated f rom a three-day average of CFU (Swanson et al., 1992) using
the previously derived empirical yield values of 4.1 x 10 6 CFU P. fluorescence!'pg acetate-C and 2.9 x 10' CFU Aquaspirillum sp./pg oxalate-C (APHA, 1995). AOC analyses were done
for two replicates. For simplicity all AOC values are given as p.g AOC-C/1, with the understanding that the values are acetate (for P. fluorescence) for and oxalate (for
Aquaspirillum sp.) carbon equivalents per litre.
Determination of microbially available phosphorus (MAP)
M A P concentration in water was determined using a bioassay. where the maximum
growth of P. fluorescens in pasteurised water samples is related to phosphorus concentration
(Lehtola el ai, 1999). Briefly, 40 p.1 of an inorganic salts (without phosphorus) stock solution
(LN-P) and 200 jal of a sodium acetate stock solution (as organic carbon source) were added
to 40 ml of investigated water, to ensure that neither o f the nutrients except phosphorus
restricted bacterial growth. Inoculation was carried out using P. fluorescens PI7 (ATCC
49642). Water samples were incubated at 20 ± 2 °C in darkness. On incubation days four,
f ive, and six, 0.1 ml of sample was withdrawn daily with a sterile pipette and surface-plated
over R2A agar plates in three dilutions. CFU were counted after three days of incubation at 20
°C. To calculate the concentration o f M A P the yield factor 3.73 x 10 s CFU/u.g o f PO4-P, as
suggested by Lehtola et al. (1999), was used. M A P analyses were done for two replicates.
Heterotrophic bacterial number and bacterial growth
Heterotrophic plate count (HPC) were enumerated by a spread-plate procedure using R2A
low nutrient agar media (Reasöner and Geldreich, 1985) and incubated at 20 ± 2 °C for 7
days. Bacterial growth in water was measured by incubating water samples at 20 + 2 °C in 40
ml glass flasks for 14 days in darkness fol lowing the methodology by Miettinen et al. (1997).
Prior, 100 pd of thiosulfate solution in 40 ml of sampled water was applied to remove
chlorine. After 2, 3, 5, 7, 9, and 14 days of incubation, water samples were withdrawn and
HPC bacteria were enumerated by spread-plate technique on R2A media. The highest value
(mean of two replicates) was defined as the maximum heterotrophic bacterial number
( H P C m a x ) that can grow in a water sample.
Determination of limiting nutrient for bacterial growth (nutrients enrichment experiment)
This procedure consisted of four steps: b io f i lm sampling from the pipe (see above), inoculums preparation, and inoculation and measurement of bacterial number. Since
microbial activities in a drinking water distribution network take place mostly on surfaces (e.g. pipe, soft deposits, suspended particles) (Gagnon et al., 2000), and the bacterial
population of the b iof i lm and water column can be very different (Norton and LeChevallier. 2000), the inoculums were prepared f rom b iof i lm rather then suspended bacteria. Separate
inoculums were made f rom G n e t and Sn et parts. Each soft deposit was placed in a clean beaker and 50 ml of sterile drinking water was poured over it . The suspension was shaken on a rotary table (180 rpm) for 15 minutes and left to settle for 10 minutes. Then 100 pi of the suspension
4

was inoculated into 40 ml dechlorinated (100 u.1 sodium thiosulfate solution/40 ml) pasteurised (heated for 30 minutes at 70 °C) water f rom both groundwater and surface water.
The samples were incubated at 20 ± 2 °C for 7 days until maximum HPC was reached. This procedure ensured that the l imiting nutrient of the inoculated sample was reduced to a minimum and that the bacteria were acclimatized to the sample. The acclimatized bacterial inoculum was added to triplicate flasks containing 40 ml of water samples to give an initial concentration of 500 CFU/ml. The growth of heterotrophic bacteria in the water sample was
measured without any nutrients added (LO), in samples enriched with a mixture of nutrients (LNP), and in samples enriched with only 100 pg/1 phosphorus solution (LP). To distinguish
from co-limitation (more than one nutrient is limiting), some samples were enriched with a mixture of nutrients excluding phosphorus (LN-P). Samples were incubated for two weeks in darkness at room temperature on an inclined rotary table (180 rpm). After days 2, 5, 7, 9, 12, and 14 o f incubation, 0.1 ml of aliquots was withdrawn each day. HPC was performed in two
replicates f rom three dilutions. The averages of the maximum HPC numbers for the two
replicates were used to compare the effects o f the nutrient enrichment experiments. In nutrients enrichment tests, microbial growth was considered to be strongly limited by a mineral nutrient i f the addition of the nutrient at least doubled (LP or LNP/LO >2) the maximum HPC, and barely limited i f HPC increased between 1 .3 -2 times. Dominant species
of bacteria were recovered f rom R2A medium on the basis of colony morphology, and
isolated and identified.
Identification of bacteria
Isolates f rom each type of bacterial colony were tested with Becton Dickinson Microbiology Systems reagents/stain droppers for oxidase reaction and indole production. Gram-reaction o f the bacterial strains was determined with a non-staining K O H technique
(Powers, 1995). The bacteria were identified applying B B L C R Y S T A L ™ Enteric/Nonfermenter and Gram-Positive Identification Systems (Becton Dickinson and
Company, USA) and Bergey's Manual of Determinative Bacteriology (Holt et ai, 1994).
Statistical analyses and detection of water retention times
To determine the significance of the differences between AOCnative and AOC p o t e n t i a i , Student's paired (two tailed) f-test was performed. The differences were considered as significant at P < 0.05 and very significant at P < 0.01. Pearson correlation coefficients (r)
were used to evaluate the correlations between AOC, M A P , and H P C m a x . Water retention time from the average daily f low in the distribution networks was calculated with a software program (Licwater V I 4 0 2 , Dekho, Denmark). The computer network model was calibrated
from water pressure measurements in the f ield.
R E S U L T S
Arithmetic mean values over the study period of AOCpotentiai, AOC„ati v e, and M A P were determined in samples taken f rom the lake (Law), river ( S r a w ) , and f rom effluent of plants
(Gpiant and S p i a n t) (Table 2-3). The AOC and M A P concentrations of water sampled f rom the distribution net were related to water retention time (RT) in the net. Because no significant correlations with RT were found neither for groundwater nor for surface water, arithmetic means over the study period of all the water samples taken f rom points (1-16), points (23-35),
and points (17-22), (Fig. 1), were estimated (Table 4-6). These values are assumed to be representative for mean water RT in the networks that were about 10 hours for groundwater
(Table 4) and 14 hours for surface water (Table 5).
5

MAP
M A P concentrations were similar in the lake and river (Fig. 2). M A P concentration was very low in a sample taken during the July month algae bloom (Table 2). Chemical treatment
of the river water reduced M A P to very low concentrations, whereas the lake-recharged groundwater contained relatively high M A P . M A P concentration in the chemically treated
water did not change significantly in the distribution net (S n et), though it increased during groundwater transportation in the net (Gnet) (Fig. 2). The highest values were found in the
mixture zone (SG n e i) (Table 6).
AOCnative Olid AOCpotentiai
AOCpotentiai concentrations were similar in the lake and river (Fig. 2). The difference between AOCpotentiai and AOCnative during sampling occasions (Fig. 2) was significant in the river, but not in the lake, indicating that inorganic nutrients limited bacterial growth in the former, but not in the latter raw water. The reduction of AOC p ot entiai was greater during groundwater recharge than during chemical treatment (Fig. 2). The difference between AOCpotentiai and AOCnative increased significantly in chemically treated water and decreased in artificially recharged groundwater. There were no significant AOC n ative changes during distribution of either water in the net.
AOC yield was calculated f rom the growth of Pseudomonas fluorescens PI7 and
Aquaspirillum sp. NOX. A O C n a , j v e yield was about 70-80% in groundwater (G p i a n t , G n e t) whereas between 10-15% in surface water ( S p i a m , Snet) due to growth of Pseudomonas
fluorescens P I 7 (Fig. 2). AOCp ot entiai yield was between 60-80% in both waters due to growth of Pseudomonas fluorescens P17.
Ratios between AOC and MAP
The ratio of AOCpotentiai to M A P in the river and lake was about 55 ( f ig AOC-C/p.g M A P -P). During water treatment by chemical coagulation the ratio increased about 60 times, but only two times during groundwater infiltration. The ratio between AOC n atwe. w'hen yield was calculated only f rom Pseudomonas fluorescens P17 growth, and M A P was 90 in treated surface water.
HPC and bacteria growth
HPC of bacteria increased during both treated surface water and groundwater transport in
the network (Fig. 3). It was possible to relate HPC of bacteria to water retention time in the net. Exponential increase of HPC in both groundwater (r2 = 0.34) and treated surface water (r2
= 0.61) was found (Fig. 3). The relations showed that HPC increased faster in surface water
than in groundwater.
A maximum number of heterotrophic bacteria, H P C m a x , at the existing levels of nutrients in the water sample, was analysed during 14 days of batch incubation. In chemically treated water, a mean H P C m a x of 279,700 ± 55,150 CFU/ml was reached after 14 days, and in
groundwater ( G p I a m plus G n e t ) 441,100 ± 45,630 CFU/ml after 12 days o f incubation (Fig. 4). Thus, groundwater sustained approximately 40% more (P = 0.047, unpaired Student's f-test)
bacteria than treated surface water. The bacterial number was low, though H P C m a x was very
high in the part o f the net SG n e t where groundwater was mixed with surface water (Table 6).
Bacterial growth limiting nutrient
The population of bacteria, sampled from the biofi lms of the pipes at a single occasion,
was used as inoculums in enrichment experiments. Different species of bacteria were identified in biofilms sampled f rom the S n e t and G n e t parts of network. Sphingomonas
6

paucimobilis and Pseudomonas aeruginosa were the dominant bacteria in biofi lms formed from surface water; Brevundimonas vesicularis, Burkholderia cepacia, and Acinetobacter
hvoffii were the dominant bacteria in groundwater. The maximum bacterial number in samples enriched with phosphorus only (LP) or with a
mixture of nutrients (LNP) was compared with the maximum bacterial number in samples to which no nutrients were added (LO). The LP to LO ratio was close to one in the lake water
(0.88). as well as in the recharged groundwater plant effluent (1.02). There was no significant increase of bacterial number in the river water (22/11/00) when enriched with phosphorus
only (LP/LO = 0.8), whereas the addition of other nutrients increased the bacterial number moderately (LNP/LO = 1.5). In chemically treated surface water bacterial numbers increased significantly when a mixture of inorganic nutrients (LNP/LO = 3.6) or phosphorus only (LP/LO = 3.9) were added (Fig. 5 A ) . When in chemical treated water taken in other sampling
occasion all inorganic nutrients except phosphorus were added, no significant increase of the bacterial numbers was observed (Fig 5 B, LN-P). The addition of phosphorus alone increased the production of bacteria by up to 12 times (Fig 5 B, LP).
D I S C U S S I O N
Nutrients removal during water treatment
Raw water. Due to a comparatively cold climate and plenty o f humic podsol soils in the
drainage areas many surface waters of Latvia contain relatively high concentrations of natural organic matter largely composed of humic substances. Both main water sources used for
Riga's drinking water supply were also humus-rich in terms o f CODMn (Table 1) and
contained relatively high amounts of labile organic carbon, measured as AOCpotentiai (Fig. 2). This agrees with recent findings arguing that humic substances can serve as significant organic carbon sources for bacteria in addition to photosynthetic exudates and cell breakdown products (Hunt et al, 2000). Thus, humic waters can support a higher bacterial biomass than clear water aquatic systems (Tranvik, 1988).
Both raw waters sources studied also contained relatively high amounts of inorganic
nutrients (nitrogen and phosphorus) (Table 1). However, a significantly higher AOC p o, entiai than AOCnative level indicated that inorganic nutrients were l imiting bacterial growth in the
river, which is common for humic waters (Jansson et al, 1998). The closeness of AOC p o I entiai to M A P ratio to critical nutrient requirements for bacteria (100:1-2) as well as results f rom enrichment experiments showed that inorganic nutrients other than phosphorus are more likely to restrict bacterial growth in the river during the period of the study. However, it was noted that the role o f inorganic nutrients for bacterial growth was not as pronounced in the
lake.
Chemical treatment of river water. The chemical treatment removed only 20% of AOC p o I entiai, whereas M A P was removed by more than 95%. As a result, phosphorus became limit ing for bacterial growth in drinking water (Fig. 2). While no other nutrients were l imit ing (or co-
limiting) in chemically treated surface water, phosphorus was confirmed by nutrient enrichment experiments using indigenous b io f i lm bacteria f rom the distribution network (Fig.
5). Due to a deficiency of phosphorus, only 40% of AOCpotentiai was assimilable (as AOC n a tive) for bacteria in drinking water. Therefore, the bacterial formation potential was decreased mainly because of phosphorus reduction during chemical water treatment, and only moderately because of the reduction of organic matter. The role o f alum treatment in
phosphorus inactivation has been earlier recognised for drinking water (Nishijima et al.
7

1997) and for natural aquatic systems (Rydin et al, 2000). Although M A P was effectively eliminated during chemical treatment, a significant part of phosphorus was still present since
AOCnative remained rather high (278 pg AOC-C/1) compared to the usual concentration (10-50 pg AOC-C/1) for biologically stable water (LeChevallier, 1991; van der Kooi j , 1992).
The results in this study showed the importance o f carrying out AOCpotemiai and M A P
analyses in humus-rich waters. I f only AOC n ative or, for example, biologically degradable organic carbon (BDOC) would be analysed the decrease of bacterial formation potential would be wrongly attributed to a removal of organic matter only.
Artificial groundwater recharge using lake water. During water filtration f rom the lake to
groundwater, about 60% of M A P was removed, whereas the removal of AOC p o, entiai was about 30% (Fig. 2). Since M A P was not effectively reduced during water infiltration to groundwater phosphorus did not l imit the growth of bacteria in the artificially recharged groundwater at the
plant effluent and thus nearly all AOCpotentiai was available as AOCnative- Notably, in some samples, the addition of inorganic nutrients decreased the AOC yield. Such a phenomenon has been found earlier (Miettinen et al., 1999), but an explanation for the growth decrease is
not known. The ratio between AOC p o t e n t ia i and M A P was close to the critical requirements for bacterial growth, meaning that any of these two nutrients may easily become limiting. During groundwater distribution, possibly due to a contribution f rom the natural groundwater that
supplied the same part of the distribution net (Gnet), the M A P concentration increased f rom
about 5 to 12 ug P/l, thereby doubling (Fig. 2). As a result the AOC p o , e n t iai /MAP ratio decreased, making carbon l imi t bacterial growth. The significant positive correlation (P =
0.021, r = 0.61; n = 14) between AOCnative and the maximum number of bacteria that water could support (Fig. 6) indicated that organic carbon largely regulated the growth of bacteria in
the distributed groundwater. Earlier, it was shown that phosphorus is very effectively eliminated in the course of artificial recharge (Miettinen et ai, 1996), and that M A P can be
reduced to a concentration below 1 pg P/l (Lehtola et ai, 1999). Why M A P removal was not that great during water filtration f rom lake to groundwater in this study is not known. As natural groundwaters may contain high amounts of M A P (Lehtola et al., 1999) and AOC
(Noble et ai, 1996) the nutrification of artificial groundwater aquifers by natural groundwater
did probably occur in this study.
Nutrients effect on bacterial growth HPC in water distribution net. Although groundwater at the plant effluent contained higher
levels of M A P (Fig. 2) and received lower chlorine doses than chemically treated water, the
increase of HPC during groundwater distribution did not appear to be greater (Fig. 3).
Moreover, despite the high content of M A P in some samples in the part of network (SG n et) where groundwater and chemically treated water were mixed (mixture zone), HPC in the water was relatively low (Table 6). The lack o f positive correlation between higher
phosphorus concentrations in water and increases in the number of suspended bacteria could be attributed both to the difference in water temperature among different water sampling places and to the fact that major biological activity occurs on the surfaces of pipes in b io f i lm
rather than in the bulk water (Gagnon et al., 2000). Bacteria enters bulk water largely through
the release of attached bacteria f rom drinking water b iof i lm (Chandy and Angles, 2001,
reference therein); this process, besides the phosphorus concentration, is also regulated by residual chlorine concentration, water velocity in the pipes, and other factors that were not
taken into account in this study.
Maximum bacterial growth. Distributed groundwater contained about 2-fold AOC n ative and 10-fold greater M A P concentrations compared to chemically treated surface water (Fig. 2).
8

The incubation of indigenous bacteria in the respective waters showed that groundwater supported a 40% higher growth of the bacterial number than surface water (Fig. 4). This
shows that AOCnative was reasonably related whereas the M A P concentration underestimated the growth of bacteria potential in water (since phosphorus was l imiting for surface water and close to critical nutrients requirements in groundwater). The yield analyses of different species used in the A O C and M A P bioassays offered an explanation to the cause o f these discrepancies. In the chemically treated surface water the AOC n a ,j Ve yield was only 10%, while 70% in groundwater due to Pseudomonas fluorescens PI 7 growth (the rest was a result of
NOX growth) (Fig. 2). Since a single Pseudomonas fluorescens Pil strain was used in the MAP bioassay, the low growth of these bacteria in chemically treated water resulted in relatively low M A P values. In the chemically treated water, the indigenous bacteria with better growth responses to low phosphorus concentrations than Pseudomonas fluorescens P I 7
apparently dominated. These bacteria metabolise a part of phosphorus that was not
assimilated by Pseudomonas fluorescens PI 7 in the AOCnative and M A P bioassays. The addition of inorganic nutrients to samples of chemically treated water increased the yield of
AOCpotentiai mainly due to the growth of Pseudomonas fluorescens P17 (Fig. 2). Thus, a low growth o f Pseudomonas fluorescens PI 7 could not be attributed to factors such as the dominance of organic substances not assimilated by this bacterial strain (e.g. oxalic acid) (Huck et al., 1991 reference within) or the presence of a specific inhibitor (e.g. aluminium) in chemically treated water. The results presented in this study a priori suggest that in drinking water distribution systems with low phosphorus concentrations, bacteria with better growth
response to low phosphorus levels wi l l dominate. This finding may be important in the
selection of bacterial species for bioassays to be used in the growth o f bacteria potential evaluation.
C O N C L U S I O N S
(1) Chemical (alum) surface water treatment was 10-fold more effective in the removal
of M A P than artificial recharge of groundwater. However, removal of AOCpotentiai was about 10% higher during the latter water treatment process. Due to an effective M A P
removal, only 40% of AOCp ot entiai was microbially available (as AOC n ative) in the chemically treated surface water. During distribution of the surface water M A P did not change, but increased during groundwater distribution. Neither AOC p o , e n t iai nor AOCnative concentrations changed significantly during drinking water distribution.
(2) Although chemically treated water contained 40% less AOCnative and 10 times less M A P than groundwater, HPC's were similar after distribution, wi th a higher tendency
to increase in the chemically treated water. Distributed groundwater supported about
a 40% higher ( H P C m a x ) growth o f bacteria than distributed surface water. (3) In chemically treated drinking water microbial growth was limited by phosphorus,
whereas in artificially recharged groundwater the phosphorus content was close to
critical requirements for bacteria. However, because of an increase of M A P during distribution, perhaps as a result of input f rom other groundwater sources, organic
carbon became limiting. (4) When interpreting the results from M A P bioassays it is important to consider the
differences in growth rates between the bioassay bacteria and the bacteria of the
actual distribution-pipe environment.
9

Acknowledgements
The study was supported by Riga Municipal Enterprise Riga Water f rom which the director
Mr . Uldis Bambe is especially acknowledged. Part of the financing was granted by the Latvian Academy of Science (grant no. 01.0599). We thank Prof. Janis Sprogis for his useful discussions and we acknowledge the staff of Riga Water for help in sampling deposits f rom
the distribution net. We are also grateful to Una Zilbere for providing chemical data and we thank Zaiga Petri a and Dainu Ezi f rom the Microbial Strain Collection of Latvia for carrying
out the bioassays. Mr . Wayne Chan is acknowledged for proofreading the English of the manuscript.
R E F E R E N C E S
A P H A (1995) Standard methods for the examination of water and wastewater, 19 t h ed., Eaton
A . D . , Clesceri L.S. and Greenberg A.E. , American Public Health Association, Washington, DC.
Chandy J.P. and Angles M . L . (2001) Determination of nutrients l imiting b io f i lm formation
and the subsequent impact on disinfectant decay. Water Res. 35 (11), 2682-2682. Eynard F., Mez K . and Walther J.-L. (2000) Risk of cyanobacterial toxins in Riga waters
(Latvia). Water Res. 34 (11), 2979-2988.
Gagnon G.A., Slawson R . M . and Huck. P.M. (2000) Effect o f easily biodegradable organic compounds on bacterial growth in a bench-scale drinking water distribution system. Can.
J. Civ. Eng. 27, 412-420.
Hol t J .G, Krieg N.R., Sneath P.H., Staley A.J.T. and Williams S.T. (1994) Bergey's Manual
of Determinative Bacteriology 9 I h ed. pp. 787. Williams & Wilkins. Baltimore, Philadelphia, Hong Kong, London, Munich, Sydney, Tokyo.
Huck P.M., Fedorak P.M. and Anderson W.B. (1991) Formation and removal of assimilable organic carbon during biological treatment. J. Am. Water Works Assoc. 83 (12), 69-80.
Hunt A.P., Parry J.D. and Hamilton-Taylor J. (2000) Further evidence of elemental composition as an indicator of bioavailability of humic substances to bacteria. Limnol.
Oceanogr., 45 (1), 237-241. Jansson M . , Blomqvist P., Jonsson A. and Bergström A . - K . (1996) Nutrient limitation of
bacterioplankton, autotrophic and mixotrophic phytoplankton, and heterotrophic nanotlagellates in Lake Örträsket. Limnol. Oceanogr., 41 (7), 1552-1559.
LeChevallier M . W . , Schulz W. and Lee R.G. (1991) Bacterial nutrients in drinking water. Appl. Environ. Microbiol. 57, 857-862.
Lehtola J.M., Miettinen LT. , Vartiainen T., Myllykangas T. and Martikainen. P.J. (2001)
Microbially available organic carbon, phosphorus, and microbial growth in ozonated drinking water. Water Res. 35 (7), 1635-1640.
Lehtola J.M., Miettinen LT. , Vartinainen T. and Martikainen P.J. (1999) A new sensitive bioassay for determination of microbially available phosphorus in water. Appl. Environ.
Microbiol. 65 (5), 2032-2034.
Miettinen LT. , Vartiainen T. and Martikainen P. (1996) Bacterial enzyme activities in ground
water bank fi l trat ion of filtration of lake water. Water Res. 30 (10), 2495-2501. Miettinen LT. , Vartiainen T. and Martikainen P.J. (1999) Determination of assimilable
organic carbon in humus-rich drinking waters. Water Res. 33 (10), 2277-2282.
Miettinen I .T. , Vartiainen T .K. and Martikainen P.J. (1997) Phosphorus and bacterial growth in drinking water. Appl. Environ. Microbiol. 63 (8), 3242-3245.
10

Nishijima W., Shoto E. and Okada M . (1997) Improvement of biodegradation of organic substances by addition o f phosphorus in biological activated carbon. Water Sei. Techn. 36 (12), 251-257.
Noble P.A., Clark D.L . and Olson. B .H . (1996) Biological stability of groundwater. J. Am.
Water works Assoc. 88 (5). 87-96. Norton C D . and LeChevallier M . W . (2000) A pilot study of bacteriological population
changes through potable water treatment and distribution. Appl. Environ. Microbiol., 66 (1), 268-276.
Powers E .M. (1995) Efficacy o f t h e Ryu nonstaining K O H technique for rapidly determining Gram reactions o f foodborne and waterborne bacteria and yeasts. Appl. Environm.
Microbiol. 61, 3756-3758. Reasöner D.J. and Geldreich E E . (1985) A new medium for the enumeration and subculture
of bacteria f rom potable water. Appl. Environm. Microbiol. 49, 1-7.
Rydin E., Huser. B. and Welch E.B. (2000) Amount of phosphorus inactivated by alum treatments in Washington lakes. Limnol. Oceanogr. 45 (1). 226-230.
Sathasivan A. and Ohgaki S. (1999) Application of new bacterial regrowth potential method
for water distribution system - a clear evidence of phosphorus limitation. Water Res. 33 (1), 137-144.
Sathasivan A. , Ohgahi S., Yamamoto K. and Kamiko N . (1997) Role of inorganic phosphorus in controlling regrowth in water distribution system. Water Sei. Tech. 35 (8), 37-44.
Swanson K.M.J . . Bust F.F., Peterson E.H. and Johnson M.G. (1992) Colony count methods. In Compendium of methods for the microbial examination of foods. 3rd edn, Vanderzant C.
and Splittstoesser D.F. (eds) pp. 78-88. American Public Health Association, Washington. Tranvik L.J. (1988) Availabili ty of dissolved organic carbon for planktonic bacteria in
oligotrophic lakes o f differ ing humic content. Microb. Ecol. 16, 311-322. Van der Kooi j D., Hijnen W . A . M . and Visser A.J. (1982) Determining the concentration of
easily assimilable organic carbon in drinking water. J. Am. Water Works Assoc. 74 (10),
540-545. Van der Kooij D. (1992) Assimilable organic carbon as an indicator of bacterial regrowth. J.
Am. Water Works Assoc. 84 (2), 57-66.
11

Table 1. Chemical analyses o f raw and drinking water o f Riga water supply in 2000 ;
Water source C O D M n
b Phosphateb Nitrate b
mg 0 2 / l P0 4-P ug/1 NO3-N mg/1
S raw 16.19 ± 4 . 4 8 7 0 ± 51 0.81 ± 0 . 3 7
Lraw 9.70 ± 1.56 68 ± 7 3 0.46 ± 1.09
Spiam effluent 4.55 ± 0.47 < 2 0 0.75 ± 0.34
Gpiant effluent 3 . 8 0 ± 0 . 1 0 < 2 0 0.26 ± 0.04 a Symbols: C O D m h , chemical (KMnCu) oxygen demand; S r a w , Daugava River L r a w , lake Mazais Baltezers; S p i a m , chemical precipitation plant;
Gpiam, artificially recharged groundwater abstraction plant. b Annual arithmetic mean from monthly average ± standard deviation f rom mean.
Table 2. Results from analyses o f AOC, based on growth o f P. fluorescens PI7 (AOC n ative
PI7) and Aquaspirillum sp. N O X without (AOC n a tive) and with (AOCpotentiai) inorganic nutrients addition, microbially available phosphorus (MAP) , heterotrophic plate count (HPC) and maximum HPC ( H P C m a x ) after 14 days o f incubation in raw water f rom Daugava River
(Sraw) and lake Mazais Baltezers (Lraw) used for drinking water production.
Date Raw water AOC, native AOC potential AOCnative HPC HPCmax M A P AOCpotentiai source P17 : M A P
d/m/y rig AOC-C/1 % CFU/ml pg P/l
19/7/00 Sraw 443 861 27 5400 184000 1.28a 673 a
18/10/00 Sraw 604 856 45 190 180000 14.34 60
22/11/00 Sraw 728 928 79 43000 330000 18.50 50
Mean 592 882 50 16197 231333 16.42 55
± S . D . b ±143 +40 ±26 +2.94
19/7/00 Lraw 988 1305 64 2500 516000 15.30 85
25/10/00 Lraw 789 650 75 3300 80000 22.12 29
15/11/00 Lraw 716 735 83 2320 112000 13.80 53
Mean 831 897 74 2 707 236 000 17.07 56
± S . D . b +141 ±356 ±10 ±4.43 a Value excluded from calculation o f mean. b S.D., standard deviation o f the mean.

Table 3. Results f rom analyses o f A O C , based on growth o f P. fluorescens P I 7 (AOC n a tive PI7)
and Aquaspirillum sp. N O X without (AOCnative) and wi th (AOCpotentiai) inorganic nutrients addition, microbially available phosphorus (MAP) , heterotrophic plate count (HPC) and maximum HPC ( H P C m a x ) after 14 days o f incubation in drinking water samples taken from the chemical precipitation plant (S p i a n t ) and the artificial recharged groundwater abstraction ( G p i a n t ) plant. b
Date Plant AOCnative AOCpotentiai AOCnative HPC HPCmax M A P AOCpotentiai • effluent P17 M A P
d/m/y pg AOC-C/1 % CFU/ml pg P/l lig/ug 19/7/00 Gplant 295 397 76 300 n.d 4.40 67
Gplant 227 269 71 2300 271 000 2.97 76 25/10/00 Gplant 798 651 81 900 445 000 3.67 220
Gplant 533 686 78 120 500 000 5.08 105 15/11/00 Gplant 804 643 85 12000 410 000 9.70 83
Gplant 771 858 78 20 620 000 6.70 115 5/04/01 Gplant 820 730 76 10 n.d. 5.20 158
Mean 571 584 78 2235 449 000 5.41 111
±S.D. + 262 ± 2 1 3 ± 5 ± 2 . 6 4
19/07/00 e a
gplant 1 1 33 240 384 000 0.01 n.d 18/10/00 c a gplant 269 993 3 1 420 000 1.57 633 8/11/00 c a
gplant 487 751 15 10 227 000 n.d n.d 6/12/00 Spiant 299 671 11 10 n.d 0.17 3941 11/12/00 Spiant 343 1029 15 60 n.d 0.33 3095 5/04/01 Spiant 263 589 7 10 n.d 0.07 8808
Mean 278 672 14 55 343 667 0.43 3305
±S.D. ± 158 ± 3 7 3 ±10 ± 0 . 6 5 a Samples were taken before chlorination o f drinking water started b Symbols: S.D., standard deviation o f the mean; n.d., no data available.

Table 4. Results from analyses o f AOC, based on growth o f P. fluorescens PI7 (AOCn ative PI7) and
Aquaspirillum sp. NOX without (AOCnative) and wi th (AOCpo tentiai) inorganic nutrients addition, microbially available phosphorus (MAP) , heterotrophic plate count (HPC) and maximum HPC (HPCmax) after 14 days o f incubation in drinking water samples taken from sampling points (1-16) in the part the net (G n e t ) supplied with groundwater after a certain water retention time (RT) down f low from the Gpiantplant.b
Sampling ! AOCnative AOCpotentiai A O C native HPC HPCmax M A P AOCpotentiai • RT Date point P-17 M A P
d/m/y no. ug AOC-C/1 % CFU/ml ug P/l M-g/ug h
5/4/00 1 574 512 73 20 n.d. c 3.60 142 0.04 2 458 512 76 2000 n.d. 18.20 28 0.05
3 453 340 80 20 n.d. 1.70 200 0.6 4 499 508 73 2120 n.d. 2.10 242 2.3 5 412 421 72 2500 n.d. 18.80 22 3.85
19/4/00 6 631 715 70 30 450 000 0.10 7150 a 5.58 7 550 705 67 60 300 000 0.10 7050 a 6.27
8 420 644 62 2140 150 000 22.10 29 7 15 633 487 87 1350 300 000 8.50 57 28
10 1068 733 89 1470 600 000 16.60 44 12.5 5/5/00 11 483 1299 80 220 270 000 24.90 52 15.12
21/6/00 12 775 410 83 2000 780 000 17.00 24 18.16 13 433 492 69 27000 540 000 16.40 30 24.19
9 674 669 65 7000 540 000 18.20 37 10.4
27/9/00 14 256 286 64 n.d. n.d. 11.89 24 n.d.
16 480 563 85 n.d. n.d. 9.92 57 n.d.
6 491 445 65 n.d. n.d. 12.87 35 5.58 7 403 391 61 n.d. n.d. 13.37 29 6.27
15 403 506 76 n.d. n.d. 19.54 26 28
Mean 531 560 73 3424 436 667 12.42 63 10.23
± S . D . ± 176 ± 2 2 0 ± 9 ± 7 . 7 7 a Value excluded from calculation o f mean b Symbols: S.D. , standard deviation o f the mean; n.d., no data available.

Table 5. Results f rom analyses o f AOC, based on growth o f P. fluorescens P I 7 (AOCnative P I 7) and
Aquaspirillum sp. N O X without (AOCn ative) and with (AOCpotentiai) inorganic nutrients addition, microbially available phosphorus (MAP) , heterotrophic plate count (HPC) and maximum HPC (HPCmax) after 14 days o f incubation in drinking water samples taken from sampling points (27-35) in the part o f the net (S n et) supplied wi th chemically treated surface water after a certain water retention
time (RT) downflow from the S pi a nt plant.'
Date Sampling AOC n a tive AOCpotentiai AOC native HPC HPCmax M A P AOCpotentiai RT
point P-17 : M A P
d/m/y no. pg AOC-C/1 % CFU/ml ugP/1 ug/pg h
3/5/00 29 242 1005 1.57 10 820 000 0.30 3350 6.40 29 358 801 3.75 50000 300 000 0.70 1144 18.00 31 299 982 0.88 6400 2 100 000 0.20 4910 18.00
5/5/00 27 320 1814 2.31 50 130 000 0.60 3023 7.50 26 352 865 36.52 10000 960 000 0.70 1236 12.13
23 390 980 0.45 2000 130 000 0.70 1400 16.04
33 290 1199 1.14 13000 650 000 0.50 2398 15.22
6/9/00 30 24 590 3.80 680 49 000 0.15 3932 n.d.
29 586 697 33.12 70 35 000 0.59 1181 6.40
28 319 736 4.24 890 61 000 1.15 640 18.00
32 462 925 3.64 132000 22 000 1.97 470 16.02
11/10/00 25 254 548 19.28 7000 114 000 0.60 914 n.d.
24 567 741 6.56 2000 246 000 0.63 1176 n.d. 27 175 345 0.95 1340 66 000 0.48 718 7.50
23 212 347 0.35 1480 183 000 0.07 4957 16.04
26 217 327 2.86 n.d 540 000 0.10 3274 12.13
8/11/00 31 264 706 8 680 300 000 0.32 2200 18.00
33 235 588 10.39 1320 119 000 0.23 2579 15.22 34 215 741 33.30 2830 107 000 0.31 2398 18.00 35 169 972 15.42 1270 290 000 0.53 1849 24.15
Mean 297 797 9.45 12 264 361 100 0.54 2187 14.39
±S.D. ±133 ±337 ±11.8 ±0.42
Symbols: S.D., standard deviation o f t he mean; n.d., no data available.

Table 6. Results from analyses o f AOC, based on growth o f P. fluorescens P17 (AOC n a Iive P17) and
Aquaspirillum sp. N O X without (AOCn ative) and with (AOCpotenciai) inorganic nutrients addition, microbially available phosphorus (MAP), heterotrophic plate count (HPC) and maximum HPC (HPCmax) after 14 days o f incubation in drinking water samples taken f rom sampling points (17-21) in the part o f the net (SGnet) supplied with a mixture o f groundwater and treated surface water.3
Datum Sampling AOCriatjve AOCpotentiai AOC native HPC HPCmax M A P AOCpotentiai • point P-17 M A P
d/m/y no. pg AOC-C/1 % CFU/ml l ig P/l Hg/ug
19/4/00 17 483 1299 80 220 600 000 24.90 52
2/8/00 20 249 1569 80 370 1 790 000 3.00 523
21 861 1330 61 520 3 660 000 19.41 69
22 1730 5826 87 n.d. n.d. 3.71 1570
19 2064 4738 87 600 770 000 2.39 1982
18 1270 3400 83 n.d. n.d. 1.11 3063
29/11/00 20 1558 1326 86 2450 620 000 39.81 33
19 891 1938 54 2000 890 000 4.37 443
Mean 1071 2455 76 1 027 1 388 333 12.29 967
±S.D. ±621 ±1788 ±12 ±14.20 a Symbols: S.D., standard deviation o f the mean; n.d., no data available.

R i g a B a y
Mazais Baltezers L a k e L r
Sampling points 1-16 supplied by groundwater Sampling points 17-22 mixed supply Sampling points 23-35 supplied by surface water
5 k m S Daugava
raw ^ ° River
Fig. 1. Layout o f the Riga drinking water supply including raw water sources (S n
and Lraw), surface water chemical treatment plant (Spiant), artificially recharged
groundwater (G pia n t, Aplm), and natural groundwater abstraction plants (Npiant)-Transition mains and major water flows are shown.

1000
900
800
700
600
ü 500 c < SU
400 a.
300
200
100
** *** 20
D A o r j g r v w v-nauve
Q A O C p 0 t e n t j a |
16 • M A P
14
12
10 1 6
4
\ 2
0
^raw Sp] a n t S n e t Gnet G p i a n t L > r a w
Fig. 2. Mean concentration of assimilable organic carbon (AOC) and microbially
available phosphorous (MAP) in water f rom different sampling sites: S r a w , raw water of Daugava River; S p i a n ! , chemically treated surface water at the plant effluent; S n e t . treated surface water in the distribution net; L , a w , raw water of Lake Mazais Baltezers;
Gpiant, artificially recharged groundwater; G n e t , groundwater in the distribution net. The line across the bars shows the yield of AOC from Pseudomonas fluorescens PI 7
(height of the bar below the line) and the yield f rom Aquaspirillum sp. N O X (height
of the bar above the line); whiskers represent standard error. Statistical significances
of difference between AOC n ative and AOCp 0 , e n t i a i were tested with Student's f-test: P<0.05:*, P<0.01 :**. and /°<0.001 :***.

to 20 30
CL,
y u OH X
i ooo ooo
100 000 b
10 000
1 000
10 20
water retenton time (h)
30
Fig. 3. Increase of heterotrophic plate counts (on R2A agar after 7 days at 20 ± 2 °C)
in (A) treated surface water and (B) in groundwater during water transport in the network. Data points are f rom samples taken at different locations away f rom water
works in the network during the study period o f nine months.

Fig. 4. Growth of autochthonous heterotrophic bacteria in drinking water.
Dechlorinated water samples were incubated at 20 ± 2 °C in the dark. Bacterial
number was found by plate counting on R2A agar after 7 days at 20 ± 2 °C. The data
points represent average values f rom groundwater (n = 14 f rom three sampling
occasions) and treated surface water (n = 22 f rom five sampling occasions) sampled
both at the plant and at the distribution network. The arrows show the maximum
number of bacteria ( H P C m a x ) that growth in respective water can support.
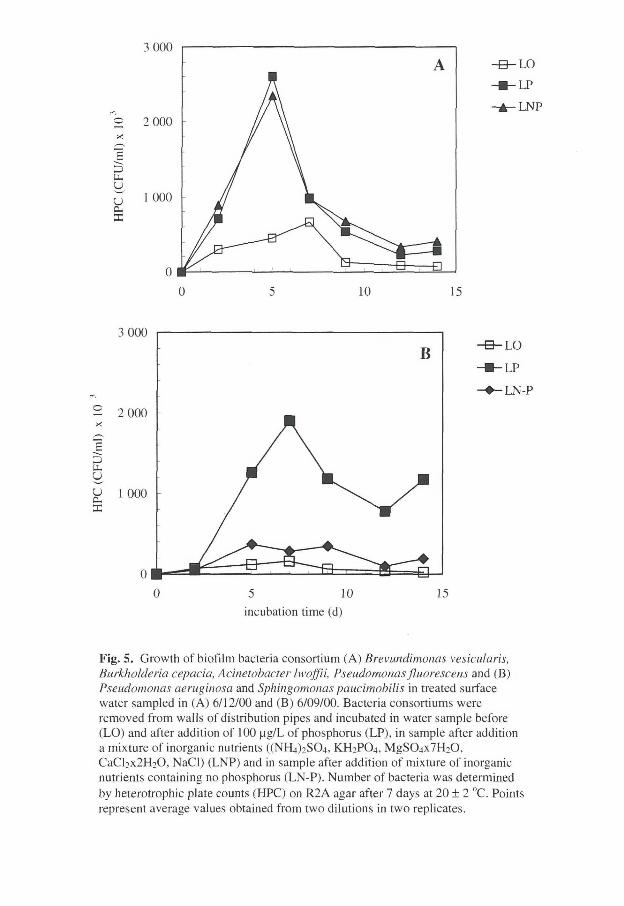
E
—
y u
3 000
2 000
000
- B - L O
- A - L N P
10
3 000
2 2 000
D —
U U 000
LO
LP
LN-P
5 10
incubation time (d)
15
Fig. 5. Growth of biof i lm bacteria consortium (A) Brevundimonas vesicularis,
Burkholderia cepacia, Acinetobacter Iwoffii, Pseudomonas fluorescens and (B)
Pseudomonas aeruginosa and Sphingomonas paucimobilis in treated surface
water sampled in (A) 6/12/00 and (B) 6/09/00. Bacteria consortiums were
removed f rom walls of distribution pipes and incubated in water sample before
(LO) and after addition of 100 pg/L of phosphorus (LP), in sample after addition
a mixture of inorganic nutrients ((NLLihSOd, KH2PO4, MgSOjxTI-LO,
CaCLx2H20, NaCl) (LNP) and in sample after addition of mixture of inorganic
nutrients containing no phosphorus (LN-P). Number of bacteria was determined
by heterotrophic plate counts (HPC) on R2A agar after 7 days at 20 ± 2 °C. Points
represent average values obtained f rom two dilutions in two replicates.

Fig. 6. Correlation between native assimilable organic carbon (AOCn ative)
(Pseudomonas fluorescens PI 7 and Aquaspirillum sp. N O X ) and maximum growth o f
autochthonous heterotrophic bacteria ( H P C m a x ) in distributed groundwater. For
detection of H P C m a x dechlorinated water samples were incubated at 20 + 2 °C in the
dark until maximum growth occurred. Bacterial numbers were found by plate
counting on R2A agar after 7 days at 20 ± 2 °C.

Paper V
Effect o f phosphorus removal f rom humus-rich drinking water on b io f i lm formation.
In : Proceedings of International Specialized Conference on B i o f i l m Monitoring, Portugal 2002.

Effect of phosphorus removal from humus-rich drinking water on biofilm formation
T.Juhna
Div. of Sanitary Engineering, Luleå University o f Technology, SE 97187 Luleå, Sweden Div. of Water Supply and Sewerge, Riga Technical Univesity, LV 1048 Riga, Latvia
Abstract The effect of reducing the concentrations of microbially available phosphorus (MAP) and residual chlorine on bacterial biofilm formation in humus-rich drinking water was studied. Heterotrophic plate counts (HPC) of microbes in biofilms, formed from drinking water containing low (chemically treated surface water) or high MAP (artificial recharged ground water) concentrations, were compared during a four months period in a water supply system. In the chemically (alum) treated surface water, where bacterial growth was limited by phosphorus, with a MAP concentration < 0.1 ug P/L and a total residual chlorine concentration of 1.2 mg/L, HPCs of the biofilm were very low (< 102 cfu/cm 2), but increased to 105 cfu/cm 2
when the chlorine concentration was reduced to 0.4 mg/L. HPCs were high (> 106 cfu/cm") in biofilm formed from artificially recharged ground water, containing 3 pg P/L of MAP and 0.4 mg/L of residual chlorine. In the networks where both waters contained no chlorine HPCs in biofilms formed from chemically treated surface water was slightly lower (by 0.9 log) compared to ground water biofilm. The study showed that biofilm formation in humus-rich drinking water could be effectively controlled by combining a reduction of MAP with a not too low residual chlorine concentration. Keywords AOC; biofilm; drinking water; MAP; phosphorus.
Introduction Bacterial formation o f biofilms in drinking water supply systems causes economical and hygienic problems. Besides temperature and properties o f pipe material, perhaps concentrations o f nutrients and disinfectant are the important factors that regulate biofilm formation in the drinking water distribution networks. In humus-rich drinking water phosphorus rather than organic carbon is bacterial growth limiting nutrient (Miettinen et al., 1997). Thus, a reduction o f concentration o f this element is expected to decrease levels of b iof i lm formation. However, knowledge to what extent removal of nutrients is sufficient to significantly reduce the b iof i lm formation and whether such a reduction allows to apply lower chlorine doses are not conclusive. The objective of this full-scale study was to investigate whether (1) the reduction o f microbially available phosphorus (MAP) may decrease the b iof i lm formation in humus-rich drinking water and (2) how the concentration o f residual chlorine affects this process. During a four months period the b iof i lm formation in biof i lm collectors fed with drinking water, taken from waterworks and from distribution networks o f Riga (Latvia) water supply system, was measured. Heterotrophic plate counts (HPC) o f biofilms, formed from the water containing either high or low levels of MAP (ground water or chemically treated surface water, respectively) in presence o f low (< 0.4 mg/L) or moderate concentration (1-2 mg/L) o f residual chlorine, respectively, were compared. In addition, the effect o f onset o f biological treatment (ozonation followed by biologically active carbon filtration) on biofilm formation from chemically treated surface water (hereafter surface water) was evaluated.
Materials and Methods Biofi lm collector: Vertically placed glass columns (length 62 cm; inner diameter 2.8 cm) were filled with about 30 pieces of hard polystyrene (Giacotherm) cylinders (length 1.8 cm; inner diameter 1.5 cm) one on top of each other. Water was pumped upwards through the column wi th a f low rate o f 50 ml/min. The column was wrapped in fo i l and placed in the dark to avoid growth of phototropic organisms. Installation o f biofilm collectors: two collectors were fed with chemically (alum) treated drinking water taken from waterworks after chlorination and from a point in the part o f distribution network supplied only with this type of water, two other collectors were fed with (artificially recharged) ground water taken from the waterworks after chlorination and from a point in the part o f network supplied with ground water. The retention time of water f rom the waterworks t i l l the network points was about one day. Biofi lm and water sampling and analyses: Biofilms were sampled at 14 occasions during a four months period (spring-summer). At each sampling occasion the two upper cylinders were taken from the column and placed in 40 ml of autoclaved tap water in 100 ml vials. To determine the number and composition o f biofilm bacteria the cylinders were scoured with sterile swabs (Celsis) in water. From the suspension
1 I International Specialised Conference on Biofilm Monitonng - Porto 2002

achieved, samples were taken to determine HPCs and identify the bacteria. Water samples were collected (before the columns), at conditions required for bacteriological analyses, in order to determine assimilable organic carbon (AOC), M A P (four occasions), and HPCs (14 occasions). Biof i lm formation: HPC bacteria were enumerated by a spread-plate procedure using R2A low nutrient
agar media incubated at 20 ± 2 °C for 7 days. Identification o f dominant b iof i lm bacteria: Colonies were selected based on morphology. The bacteria were identified applying B B L CRYSTAL™ Enteric / Nonfermenter and Gram-Positive
Identification Systems (Becton Dickinson and Company, USA) and Bergey's Manual o f Determinative Bacteriology (Holt et al, 1994). At each sampling occasion five to 10 dominant colonies in two replicates were identified. Determination o f AOC, MAP and chlorine: The AOC-concentration was determined by a modification (Miettinen et al, 1999) o f t h e standard methods (van der Kooij et al, 1982) using two bacteria strains (Pseudomonas fluorescens PI7 and Aquispirülum sp. NOX). According to the modification the AOC
yield was measured before (AOCnative) and after addition of inorganic nutrients (AOCpotentiai)- The MAP concentration in water was determined by a bioassay (Lehtola el al., 1999), where maximum growth of Pseudomonas fluorescens in sterilized water samples, and enriched in excess with acetate and mixture of inorganic nutrients (except phosphorus), was related to the phosphorus concentration. Total residual chlorine was measured by the standard AyV-diethyl-1,4-phenylenediamine method. Statistical analyses: To compare biof i lm densities under different nutrient conditions one-way variance test assuming null hypotheses (95% confidence intervals) was used.
Results and Discussion MAP was removed during chemical water treatment to a very low concentration (0.11 pg P/L). As a result phosphorus became limiting for bacterial growth in the surface water, while in the ground water, for which no treatment was applied, phosphorus was close to the critical nutrient requirements for bacteria (Juhna et al., in preparation), with a MAP concentration some 30 times higher than in the surface
water. Although potentially assimilable carbon (AOCpotentiai) was high in both waters, only a portion o f AOCpotentiai could be used, as native assimilable organic carbon (AOC n a tive), by bacteria in the surface water due to shortage of phosphorus. In spite that nearly all MAP was eliminated in the surface water, the AOCnative concentration was still rather high (261 pg AOC-C/L). A n explanation is as follows; because MAP represents a part o f phosphorus that is consumed by P. fluorescens, whereas the high content of AOCnative was mainly due to growth of Aquspirillum sp., another bacteria used in this bioassay. In the distribution network where chlorine was absent, the density of b iof i lm formed f rom surface water was high ( lO 3 cfu/cm 2 ). Although, due to a much lower MAP concentration in the surface water the biofi lm density was by one order o f magnitude (0.9 log) lower then in ground water (P < 0.05), Fig. I B .
Table 1. Water quality parameters in ground water and in surface water. Drinking water was sampled at the respective waterworks and at locations in the distribution network. Surface water samples were taken after and before onset of biological treatment at the waterworks. Average values over the study period are
shown.
Ground water Chemically treated surface water
Parameters Waterworks Network Waterworks Network
Before After Before After biotreat. biotreat. biotreat. biotreat.
MAP, pg P/L 3.00 8.65 0.11 <0.08 O.08 <0.08
AOCnative, fig AOC-C/L 606 583 261 57 118 69
AOCpotentiai , Ug AOC-C/L 554 432 799 810 612 94
Total chlorine residual, mg/L 0.4 <0.1 1.2 0.4 <0.1 <0.1 PH 7.6 7.7 6.8 7.0 7.0 7.1
Temperature, °C 9 9-14 5-14 6-15 18 17
Aluminium, mg/L <0.02 O.02 0.07 0.02 0.05 0.02
2 I International Specialised Conference on Btofilm Monitoring - Porto 2002

At the waterworks in the surface water wi th moderate chlorine residual concentration (1.2 mg/L) number of attached bacteria (< 10 2 cfu/cm 2) and free-floating bacteria (10 cfu/mL) was very low, Fig. 1A, I B . After the implementation o f biological treatment at the surface water treatment plant (on day 83 after start of the biof i lm experiments) the MAP concentration in the surface water was reduced even more (< 0.08 pgP/L) and AOCnative was decreased to 25% of the initial value. Nevertheless, the bacterial number both in the biofi lm and in the bulk water increased to levels similar those found in the distribution networks (103 cfu/cm 2 and 10 4 cfu/mL, respectively), where chlorine was absent, Fig. 1. It is because, at implementation o f the biological treatment pre-chlorination was substituted by pre-ozonation and the final disinfection dosages were lowered and the residual chlorine content decreased from 1.2 to 0.4 mg/L. In summary, this study showed that removal of MAP to very low concentrations decreased the biofi lm density but marginally, however increased chlorine doses had much greater effect on restricting biofi lm formation. Notably in surface water before the chlorine dosage was decreased, the biofi lm density was very low; yet AOCn aiive was still rather high (> 250 pg AOC-C/L). This is an encouraging finding, since earlier it was shown that significant biof i lm formation occurs even in presence of moderate concentrations o f chlorine (Norton and LeChevallier, 2000; Volk and LeChevallier, 1999), unless AOC (as AOCnaiive) is removed to extremely low (< 10 ug AOC-C/L) concentrations (Van der Kooj ei al., 1999).
0 20 40 60 80 100 120 Days
Fig. 1. Heterotropic plate counts in water column (A) and in biofilms (B) formed from chemically treated surface water at the waterworks ( • ) and in the network (o) and from ground water at the waterworks ( • ) and in distribution network (o) .
Other species o f bacteria were isolated from ground water b iof i lm and compared to the biof i lm formed in chemically treated surface water in the beginning o f the experiments. In the ground water, Brevudinamonas vesicularis was dominant (68% of HPC) over the study period, while in the surface water, Sphingomonas paucimobilis and Stenotrophomonas maltophilia were the two major species during the first two months (Fig.2). However later, possibly as a result of changes in temperature, or in chlorine doses, or from the periodical ozonation testing (before start o f the biological treatment), B. vesicularis and Pantoea agglomerans were two significant biof i lm bacteria. In chemically treated water with a low content o f M A P the bacterial population was more heterogeneous than in M A P rich ground water. Most bacteria in the biofilms were gram-negative except for very young ground water b iof i lm (Micrococcus luteus) or in b iof i lm formed soon after start up of the biological treatment (Corynebacterium sp), when some gram-positive bacteria were present.
3 I International Specialised Conference on Biofilm Monitonng - Pono 2002

Fig. 2. Composition o f biof i lm bacteria formed from ground water (A) and chemically treated surface water (B) in the distribution networks. Percentage of HPC vs. time o f b iof i lm formation. \-Brevudimonas vesicularis; 2-Sphingomonas paucimobilis; 3-Burkholderia cepacia; ^-Micrococcus luteus; 5-P.agglomerans; 6-Acinetobacter Iwoffii; 1-Stenotrophomonas maltophilia; S-Bacillus subtilis; 9-Corynebacterium sp; 10- unidentified.
Conclusions Reduction solely of MAP to very low concentrations decreased b io f i lm formation moderately in humus-rich drinking water. In water with low MAP concentration and moderate content o f residual chlorine biof i lm formation was reduced significantly during the three months period.
Acknowledgements I acknowledge Prof. Jörgen Hanæus, Luleå University o f Technology, and Prof. Jänis Sprogis, Riga Technical University for academic support; Mr. Wayne Chan for proofreading the English; Ms. Benita Naudzune, for chemical analyses data; Dr. Vizma Nikolajeva, Ms. Daina Eze, Ms. Zaiga Petrina for doing microbiological analyses. Riga Municipal Enterprise Riga Water, and Latvian Academy o f Science financed this study.
References Holt J.G., Krieg N.R., Sneath P.H., Staley A.J.T. and Williams S.T. (1994). In: Bergey's Manual of
Determinative Bacteriology, 9th edn, Williams & Wilkins, Baltimore et al., pp. 787. Juhna T., Nikolajeva V., Juhna V. and Hanæus J. Microbially available phosphorous and assimilable organic carbon in a drinking water supply system (in preparation). Lehtola M.J., Miettinen LT., Vartinainen T. and Martikainen P.J. (1999). A new sensitive bioassay for determination o f microbially available phosphorus in water. Appl. Environ. Microbiol., 65(5), 2032-2034. Miettinen I.T., Vartiainen T. and Martikainen P.J. (1999). Determination o f assimilable organic carbon in humus-rich drinking waters. Water Res., 33(10), 2277-2282. Miettinen I.T., Vartiainen T. and Martikainen P.J. (1997). Phosphorus and bacterial growth in drinking water. Appl. Environ. Microbiol., 63(8), 3242-3245. Norton C D . and LeChevallier M.W. (2000). A pilot study o f bacteriological population changes through potable water treatment and distribution. Appl. Environ. Microbiol., 66(1), 268-276. Van der Kooij D., Hijnen W . A . M . and Visser A.J. (1982). Determining the concentration o f easily assimilable organic carbon in drinking water. J. Am. Water Works Assoc., 74(10), 540-545. Van der Kooj D., van Lieverloo J.H.M., Schellart J.A. and Hiemstra P. (1999). Distributing drinking water without disinfectant: highest achievement or height o f folly? J. Water S R. 77. Aqua, 48(1), 31-37. Volk J.C. and LeChevallier M . (1999). Impact o f the reduction o f nutrient level on bacterial water quality in distribution systems. Appl. Environ. Microbiol., 65(11), 4957-4966.
4 I International Specialised Conference on Biofilm Monitonng - Porto 2002

Paper VI
Speciation of iron in groundwater from areas of humic waters by ion-exchange method.
Vatten, 58, 2002, in press.

VATTEN 58:00-00. Lund 2002
SPECIATION OF IRON IN GROUNDWATER FROM AREAS OF HUMIC WATERS BY AN ION-EXCHANGE METHOD
by TALIS JUHNA,'- 2 BAIBA GULBE? MARIS KIA VINS3
1 Department of Environmental Engineering, Luleå University of Technology, SE-97187, Luleå, Sweden
2 Department of Civil Engineering Riga Technical University, Azenes Street 16, LV-1048, Riga, Latvia
e-mail: [email protected]
3 Department of Environmental Sciences, University of Latvia, Raina blvd. 19, LV-1586, Riga, Latvia
Abstract The removal efficiency of iron from groundwater for drinking water production is related to iron physico-chemical forms (species). An ion-exchange method was suggested for iron speciation analysis in groundwater. This method is based on the sorption of negatively charged iron species, mainly organically bound iron, on to DEAE cellulose anion-exchange resin, and positively charged species, mainly hydrated iron ions, on to Amberlite IRA 120 cation-exchange resin. The method was designed for taking water directly into a sampler filled with the respective resin, thereby largely preserving the natural conditions of the aquifer. The analytical results obtained were compared with simulation results using the WinHumicV computer speciation model. The results showed that this method could be used equally efficient for speciation of iron in groundwater with high (peat land and bog drained) and moderate (artificially recharged) concentrations of humic substances.
Key words — iron-humic complexes, speciation analysis, groundwater.
Introduction
Among the natural ingredients in groundwater, iron
appears to be a major burden for its use in drinking
water production (Deutsch, 1997). In addition to causing turbidity, unpleasant taste, and discolouration in
drinking water, iron can also facilitate bacterial growth
and corrosion in distribution networks. Thus, iron re
moval is needed. Iron exists in groundwater in various
physicochemical forms (species) including hydrated ions, hydroxo complexes, inorganic and organic com
plexes, colloids, or suspended particles (Stumm and
Morgan, 1998). In areas rich in organic soils, humic substances (HS) are common ingredients of groundwater.
HS is a general category of naturally occurring heterogeneous, high molecular weight, refractory macromole
cules that are formed through the breakdown of plant and animal tissues by chemical and biological processes,
and constitute 50-90% of organic matter in natural
waters (Thurman, 1985). Carboxylic and phenolic functional groups in humic substances provide a high bind
ing affinity for metals induding iron (Livens, 1991).
Iron is one of the metals that form the most stable com
plexes with organic matter (Stumm and Morgan, 1998) and its concentration frequendy increases in natural
waters with increasing H S concentration (Olivié-
Lauquet et al. 1999; Kortelainen et al. 1986). Several physicochemical properties of iron are altered as a result
of this complexation, including oxidation kinetics of
ferrous iron (Theis and Singer, 1974; Jobin and Ghosh, 1972; Barry et al. 1994), iron sorption on solid phases,
hydrolysis, and polymerisation reactions (Rose et al.
1998), as well as the molecular and colloid size of iron
species, and participation in various other processes
(Stumm and Morgan, 1998). Since iron removal tech
nologies such as oxidation with liquid—solid separation,
cation exchange, membrane filtration, and biological re
moval largely rely on one or several of these properties,
the selection of a water purification method is depen
dent on the predominating iron species. Most iron speciation studies have dealt with surface waters (Pullin and
Cabaniss, 2001; Appelblad et al. 1999; Kawakubo et al.
1999; Olivié-Lauquet et al. 1999; Rose et al. 1998; Petterson et al. 1992; Ephraim and Marinsky, 1990; Hiraide et al. 1988). However, in drinking water pro
duction from groundwater only ferrous Fe(II) and ferric
Fe(III) species of iron are normally identified. Thus, further research on the iron speciation in groundwater
along with the development of a simple and operative
method for the determination of major iron species including organically bound iron is of interest. The sorp
tion of iron species on ion exchangers has proven to be efficient for the determination of iron species in surface water (Hiraide et al. 1988; Hiraide, 1992; Petterson et
al. 1992; Appelblad et al. 1999). As groundwater is mostly an anaerobic environment, sampling that avoids
contact with aerobic atmosphere is preferable to prevent
changes of speciation forms due to oxidation. This in addition to other reported problems (Hiraide, 1992)
makes the application of other commonly used metal speciation methods such as anodic stripping voltamme-
V A T T E N - 3 - 0 2 01

cry and fluorescence spectrometry difficult to use for
iron speciation analysis in groundwater.
Theoretical speciation models such as the W H A M
(Tipping, 1994) and che M I N T E Q A 2 (Gustafsson,
1999) have shown co be efficient cools for speciacion
studies. These models contain useful databases for the
study of metal speciacion processes in aquacic syscems,
and have recendy been applied in che study of mecal speciacion in groundwater (Chriscensen and Chrisrensen,
1999; Jensenetal. 1999). The overall objective of chis study was to investigate
whether the ion-exchange mechod (IEM) can be used for iron speciacion in groundwacer. The study aimed to dis
tinguish between dissolved anionic (mainly organically-
bound iron complexes), cacionic (free and hydraced ions,
hydroxo complexes), and particle-relaced species of iron.
The following methodology was used in this study.
(1) In batch equilibrium experiments the appropriate
ion-exchange resin for I E M was selected and the
methodology of sampling, which allowed caking che
groundwacer sample withouc significancly alcering the
condicions in the aquifer, was developed. (2) I E M was
tested at different iron and organic matter concentrations in groundwater by means of spiking the ground
water sample with different amounts of artificially
prepared iron-humic complexes solution; (3) The ob
tained results were compared with the predictions of the computer speciation model WinHumicV (improved
W H A M ) containing default databases. (4) By using
the developed methodology, we were able to identify iron species in groundwater samples taken from aquifers which were affected by peat land, bog, or humic surface water in the vicinity of Riga City (Latvia).
Materials and methods
Materials
Unless stated otherwise, all chemicals were of analytical
grade and used without any further purification. De
ionised and distilled water were used throughout.
Sorbencs for iron speciacion used in chis scudy were ob
tained from the Dow Chemicals, Enola, Lachema, and
Rohm&Haas companies. In all laboratory experiments
with ferrous iron, prepared from analytical quality NH 4 (Fe(S04) 2 ) , water was purged with high purity
N 2 gas.
Methods
Unless stated otherwise, all ingredients in the water
were analysed according to Standard Methods (1999).
Ferrous iron concentration was determined speccropho-
tometrically using 2,2'-bipyrid.il (Water Analysis, 1988). Total iron concentrations (Fe I 0 t ) were determined by
flame atomic absorption spectrometry (Perkin Elmer
403) or spectrophotometrically with the FerroZine®
mechod ( H A C H ) (adapted from Stookey, 1970) as sug
gested for organically bound iron (Viollier ec al. 2000).
The elemental composition of isolated humic substances
was determined on a Perkin Elmer 240 B C H N ana
lyzer. For spectrometric analysis a spectrophotometer
H A C H DR/2000, SF-46 was used. In che field. p H,
redox (Pt eleccrode), temperature, and dissolved oxygen
were measured using a W T W Multiline P4; a H A C H
T D S measured electro-conductivity.
Isolation and analyses of humic substances
For laboratory experiments humic (HA) and fulvic (FA)
acids were isolated from groundwater of Lake Mazais
Baltezers (Latvia) using the Thurman and Malcolm
method (1981). The isolated HS had the following com-
position (FA from Lake Mazais Baltezers): C 54.60%,
H 3.70%, N 0.93%, O 39.43%, - C O O H 4.88 mmol
g _ l . Commercial H A (Aldrich): C 49.89%, H 4.76%,
N 2.30%, O 42.03%. - C O O H 2.15 mmol g - 1 . Con
centrations of HA andFA in synthetic and natural waters
were determined spectrophotometrically (Water Analysis,
1988) at a wavelength of 410 nm as suggested by
Hautala et al. (2000), using a H A C H D R 2000
spectrophotometer. Solutions containing 0.5, 1.0. 2.0,
5.0, 10.0, 20.0, and 50 mg l _ l of Aldrich H A or Lake
Mazais Baltezers FA were used for the calibration.
Size fractionation
Before size fractionation of iron, the ultrafiltration
system (Sarcorius) was carefully washed with 500 ml deionised water, 500 ml 0.1 N H N 0 3 , and 1,000 ml of
deionised water. The groundwater samples under a
N 2 stream were passed through a series of membrane filters differing in nominal pore size: 0.46 pm (200 ml),
0.05 pm (150 ml), molecular size exclusion limit of
100 kDa (100 ml), and 10 kDa (50 ml). F e t o , was deter
mined in the respective filtrate. The fraction retained ac 0.46 pm was defined as suspended particular macter
( F e p M ) . The fraction passing the 10 kDa membrane was
defined as truly dissolved, whilst the rest was defined as
colloidal.
Sorption of iron-humic complexes onto sorbents
The solutions of iron-humic acid complexes were pre
pared by mixing 5 mg 1 of Fe(III) with 25 mg 1"' H A (Aldrich) ac pH 6, followed by dialysis of che obcained
solution against distilled water. Before being used, the
sorbents were conditioned and washed. Adsorption
studies of che different sorbencs were conducted as batch
02 V A T T E N - 3 - 0 2

experiments performed in 100 ml sealed glass botdes on
a rotary shaker table (24 h at 20°C): 100 ml of iron-
humic complex with 100 mg of sorbent until equilib
rium had occurred. After the phases were separated by
passing them through filter paper, the concentrations of
iron in the supernatant phase were determined spec
trophotometrically (see above).
Effect of sample spiking with artificially prepared iron-humic solution
In the laboratory iron-humic acid (.Aldrich) complexes
were prepared as follows. Deionised water was purged
with nitrogen gas for - 30 min to remove dissolved
oxygen. The stock solution of Aldrich H A (100 mg l"1)
was then added until the concentration of H A in oxygen free solution was 2 mg 1_ I. Purging proceeded for ap
proximately another 30 min, then the stock solution of
Fe(II) (25 mg 1" ) was added until the concentration of
Fe(II) in solution was 2 mg 1"'. The obtained iron-
humic solutions were allowed to equilibrate under ni
trogen atmosphere until redox conditions became stable
(after about four hours). The prepared solution was
added to natural groundwater (G2) so that total concen
tration of iron in the spiked groundwater solutions
ranged from 0.45 to 1.83 mg l - ' , while HS ranged from
2.7 to 8.6 mg H (with FA 21 to 65% of H A ) . This dlowed different iron to humic substances ratios in the complexes to be formed. Spiked samples were then
shaken for two hours in darkness to avoid possible iron reduction by light (Fukushima and Tatsumi, 1999). Iron species in the solution were estimated by I E M (see below) and verified by a computer model (see below).
Iron speciation analysis with IEM
At sampling water was immediately added, after passing
through the 0.46 pm membrane filter, into the acid
washed 50 ml sampling device (a plastic syringe) (Figure
1) filled with a suspension (1 g) of D E A E cellulose (weak
anion exchanger - fibrous cellulose with diethylamino-
groups) or Amberlite IRA 120 (strong acidic - S 0 3 H styrene divinylbenzene gel type cation-exchange resin).
Prior to sampling, the resins were rinsed with deionised water and purged with nitrogen gas for about 45 min to
remove oxygen traces. Pressure produced by the pumped
water displaced the piston in the syringe allowing for the
collection of water without contact with atmospheric oxygen. The samples were collected and instantly deliv
ered to the laboratory for water quality analyses. Samples
were taken in triplicates. Additional samples using the same procedure, except for the addition of sorbent in the
similar device, were taken for analyses of iron that passed
through the 0.46 pm filter.
The amount of iron retained by the ion exchanger was
calculated as the differences between the concentrations
in aqueous phases before and after equilibration with resins (mass balance relations). Preliminary results
showed that there is statistically no significant difference
between the amount of iron recovered from a resin with
4 M H N 0 3 and the amount of iron obtained by mass balance relations. Thus, for simplicity the latter method
was used. The iron species studied were defined as
follows, see Figure 2: (1) a part of iron that passed 0.46
pm filter - colloidal and truly dissolved ( F e p C D ) ; (2) the
difference between Fe I O [ and Fe p cn — particulate iron and iron bound co particulate matter ( F e p M ) ; (3) a part
retained on the anion-exchanger D E A E (calculated as a
difference between F e p C D and the concentration of iron
in filtrate from che D E A E cellulose) - anionic, mainly
organically-bound iron (FeNof»t)i (4) a pare recained on cation-exchanger IRA-120 - cacionic iron (Fe p C ) (calcu
lated as the difference between FepcD a n t l t ^ l c concentration of iron in the filtrate from the IRA.-120 resin). The binding capacicy was assumed to be independent of temperature (Shaker et al. 2000). No distinction
between Fe(II) and Fe(III) was made due to the com
plexity of these analyses in waters with high concentra
tions of organic matter (e.g. Pullin and Cabaniss, 2001).
Study sites
Baltezers well fields were chosen for the study due to its
importance for supplying water to Riga Cicy (Latvia)
and for the heterogeneity of factors that influence iron speciation forms in the groundwater of this locality (e.g.
geology and influence of surface water). The ground
water at this site is artificially replenished with surface
water from Lake Mazais Bakezers through series of infiltrations basins. These fields are used to produce drink
ing water for the city of Riga, providing about 30%
(50,000 m 3 /h) of the total water supply. Two areas ( G l . G2 , Figure 3) that potentially differed regarding the
sources of iron were selected, and groundwater samples
were taken from each target area. The groundwater at
site G 2 was artificially recharged while at site G l it was largely of natural origin. Wells in both areas were located
in an unconfined aquifer in Quaternary deposits. In addition, samples were taken from a confined aquifer in
Devonian deposits ( D l ) , and from wells supplied with
organic-rich groundwater that drains either peat land
(PI) or bogs ( B l , B2), (Figure 3). Three replicates of samples from each of the areas were taken at two sam
pling occasions. Sampling at the sites G2 , G l , and D l
were carried out on July 21, 2000, and on October 9,
2000 for the sites P I , B1, and B2.
Sampling procedure
The sampling procedure was designed to ensure that the
samples reflected the in situ conditions with respect to
V A T T E N - 3 - 0 2 03

p H and redox of the groundwater. Redox, p H , Fe(II),
oxygen, and conductivity were monitored during the
pumping process. Samples were taken (after about 45
minutes) when all these indicator parameters had
reached steady values (pumping rate 9 1 min ). Water
was extracted direcdy from the water production wells.
A sampling hose attached to a submergible electric
turbine pump was either inserted through the well head
opening into the interior of the well (G 1, G2) or taken
from sampling tap at the well ( D l , B l , B2, P I ) . For iron
speciation analyses water was directly (without contact
with atmosphere) filled into a sampling device (Figure
1). For general water quality analyses, the samples
were collected in plastic acid-washed containers
(500 ml) and delivered immediately to the laboratory in
an icebox.
Computer model
A modified Windermere Humic Aqueous Model
( W H A M ) computer code (Tipping, 1994), speciation
model WinHumicV (Gustafsson, 1999) with default
database was used to determine the concentration of
organically bound iron. The model describes the bind
ing of metal ions to humic substances by a discrete
binding sites model in which binding is modified by
electrostatic interactions. There is an empirical relation
between net humic charge and an electrostatic interac
tion factor. The model also takes into account the accu
mulation of counterions in the diffuse layer using a
Donnan-type expression. Two types of sites (types A and
B) represent the discrete binding sites, where within each
site are four different sites presented in equal amounts.
Both intrinsic proton binding constant (pKA and pK%)
and spreads of the values ( A p K A and A p K B ) are de
scribed within each of the two sites (Table 1). Metal
binding occurs at single proton binding sites or by bide-
nate complexation between pairs of sites depending on a
proximity factor (f p r ) that defines if pairs of proton bind
ing groups are close enough to form bidenate complexes.
The two types of sites (A and B) have separate intrinsic
exchange constants i p K M H A
a n < 1 P-^MHB) (Table 1). The
default model parameters contained in the database orig
inate from published data describing proton and metal
binding to isolated humic and fulvic acids (Tipping,
1992). The model permits the binding of the first
hydrolysis product (e.g. in case of ferric ion; Fe(OH)*)
as well as the parent species. In calculating the amount
of iron bound to humic substances, a default database in
aqueous speciation mode was used. All determined
chemical parameters of water (Table 2) were introduced.
When samples were amended with an artificially pre
pared iron-humic complexes solution (see above), the
chemical composition to be used in the model was cal
culated as that of completely mixed solutions. After run
ning the model, the amount of organically bound iron
was calculated as the difference between F e t o t and sum of
free Fe(II) and Fe(III) in solution as determined by the
model. Iron within the diffused double layer ( D D L ) of
H S or associated with H S were assumed to be organi
cally bound. Even though WinHumicV is a predictive
semi-empirical model, Bryan et al. (2000) showed that
results of metal speciation analyses by this model agrees
considerably with physicochemical models based on the
understanding of mechanisms of metal humic interac
tions.
Accuracy of speciation analyses
Care has been taken to achieve relevant accuracy of the
speciation analysis procedure. Iron speciation analyses
were made using triplicates of each sample. T o avoid and
to check for possible air contact with the samples after
the sampling process, redox values and oxygen concen
trations were thoroughly controlled.
Results and discussion
Selection of the ton-exchange resin for separation of iron species
Iron-humic complexes, as well as HS , carry both non-
polar (hydrophobic) and negatively charged moieties,
whereas an iron ion that is not bound to H S (free iron)
is positively charged. For this reason iron-humic com
plexes can be separated from a water solution using
either a hydrophobic sorbent or an anion-exchange resin
(or sorbent) (Hiraida et al. 1988), which leaves free iron
in solution. Another approach is to separate free iron
with cation-exchange resins leaving iron-humic com
plexes in solution (Christensen and Christensen, 1999).
In this study both approaches were tested and compared
against each other.
A hydrophobic macro reticular styrene-divinylben-
zene copolymer, such as Amberlite X A D - 2 , has pre
viously been used for separation of iron-humic com
plexes (Hiraida et al. 1988). This sorbent removes iron
via low-energy physical interaction, depending on the
aromaticity and size of the hydrophobic groups of humic
substance. However, to use this sorbent application a
sample acidification is needed, which is difficult to do
at in situ analyses. As well, X A D removes significant
amounts of inorganic metal species due to polar impuri
ties in the resin unless specially prepared (by indium
treatment) (Hiraida et al. 1988). A strong-base anion
exchanger (e.g Dowex 1X8) has also been used for metal
speciation and found to be less appropriate than weak
anion exchangers (Appelblad et al. 1999). Thus, in this
study weak-base anionic resins were chosen for the sepa
ration of iron-humic complexes. T o select a weak an
ionic exchanger, several common commercially available
04 V A T T E N - 3 - 0 2

resins were compared in batchwise sorption experiments
regarding their sorption capacity for artificially prepared
iron-humic complexes. The results showed that weakly
basic D E A E cellulose (diethylaminoethylcellulose, macro-
porous structure, hydrophilic matrix to which secondary
amines were attached) had the highest sorption capacity,
Table 3. The effectiveness of D E A E cellulose for metal
including iron speciation has been earlier shown
(Hiraide, 1992; Petterson et al. 1992; Appelblad ec al.
1999). The high sorption capacity is attributed to bind
ing efficiency and a highly developed surface of D E A E
cellulose. Some cation species (e.g amide) may con
tribute to the pool of organically bound iron in ground
water. However, these species were neglected in the
present study due to their low concentrations in natural
waters (Niessner et al. 1998; Pandeya and Sing, 1997);
thus, the amount of iron removed by D E A E resin was
defined as organically bound iron ( F e N 0 M ) .
For separation of free iron ion species (Fe p c), che
cation-exchange resin Amberlite IRA 120 was used due
co its frequent application in water purification prac
tices, e.g. iron removal and softening. This resin removes
inorganic iron cations (e.g., F e O H * 2 , Fe* 2, Fe* 3, and
FeOH*), positively charged inorganic colloids, and
readily dissociable complexes. No comparison cests with
ocher cation-exchange resins were done in che present
scudy.
The iron speciacion was done for samples chat were
filtered through a 0.46 urn pore size membrane. The
part of iron that was retained on the membrane was
arbicrarily denoced as particulace matter ( F e p M ) , while
the species in the filtrate were assumed to include coarse
to fine colloids and truly dissolved fractions of iron or its
complexes.
Importance of anaerobic conditions at sampling
The resulcs showed chat redox conditions at the sam
pling were important in iron speciacion analyses. When
an artificially prepared solution of iron-humic com
plexes was aerated, the amounc of F e N 0 M (detected in an
unfiltered sample by adsorption on a D E A E resin)
increased along wich an increase of F e p M , Fig. 4. The
Fe(II) concencracion decreased due to oxidation to
Fe(III). The oxidation rate was rapid in the beginning,
but decreased significantly after about 7 min. The de
crease of the oxidation rate is perhaps due to the onset
reduction of Fe(III) to Fe(II) by H S (Theis and Singer,
1974; Jobin and Ghosh, 1972; Barry et al. 1994).
However, the rate of Fe(II) oxidation was faster than the
rate of Fe(III) reduction, thus different aggregates of
Fe(III) were formed. These were most likely Fe(III)-HS
colloids and suspended solids covered with humic sub
stances (Rose et al. 1998; Olivié-Lauquet ec al. 1999)
wich the size and surface properties suicable co be re-
cained on 0.46 pm membrane and in D E A E resin. Thus,
co obtain samples that are representative of a ground
wacer environment, sampling should be carried ouc ac
anaerobic condicions (e.g. in nicrogen acmosphere). The
device presenced in Figure 1 was developed for this pur
pose.
Comparison of the ion-exchange method with the computer model WinHumicV
Results f rom iron speciation analyses with the IEM
(using DEAE resin) were compared to results obtained
f rom the WinHumicV computer model. Different
amounts o f artificially prepared iron-humic complexes
were added to groundwater (G2) samples, and
concentrations o f iron-humic complexes ( F e S 0 M ) in the
amended samples were measured using both speciation
methods. Results revealed that according to I E M , the
F e N 0 M concentration was about 30% higher than
forecasted by the computer model, Fig. 5. This
discrepancy could be because the WinHumicV computer
model calculates only concentrations of truly dissolved
organically bound iron (Tipping. 1994), whereas the
anion-exchange resin in addition to truly dissolved iron-
humic complexes may also retain negatively charged
iron colloids.
Iron colloids are common in organic rich
groundwater (Hiraide 1992; Jensen ci al. 1999),
Negative charged colloids are formed after iron oxides
are coated with humic substances (Hiraide, 1992) or
inorganic ligands such as silicate (H 4 SiOj ) and
phosphorus (e.g. H : P0 4 ") are included into a structure o f
hydrated iron aggregates (Deng, 1996). In this study, to
investigate how much iron was truly dissolved or
colloidal, a size fractionation of iron in the groundwater
samples (G2) was carried out after adding high amount
of humic substances. The water samples were passed
through several membrane filters with decreasing pore
size. The results showed that about 30 to 40% o f iron
was of colloidal size (lOkDa - 0.46 urn). Figure 6.
Therefore, we believe that when F e N 0 M was determined
by I E M , it included not only dissolved iron-humic
complexes, but also colloidal species of iron.
V A T T E N - 3 - 0 2

Speciation of iron at field conditions
Groundwater samples for iron speciation analyses were
taken from natural aquifers ( D l ) , anificially recharged
aquifers with low ( G l ) and high (G2) surface water in
fluence, and artificially recharged aquifers affected by
peatland (PI) and bog drainages affected areas ( B l , B2),
Fig. 3. Sites D l , G 2 , and G l supplied drinking water to
Riga City, Latvia. The chemical composition of the
groundwater samples was different (Table 2); compared
to natural (D l ) or anificially recharged groundwater
( G l , G2) , groundwater from P I and B2 were more
acidic, contained concentrations of organic matter
almost five times higher (measured as C O D M n and HS) ,
and had higher iron concentrations.
Iron speciation analyses showed that in peat land and bog affected areas, iron exists largely as particulate matter (Fe p M ) , while only a small fraction of F e p M was found in artificial groundwater and in groundwater from confined aquifer, Fig. 7. Most of iron was organically bound in peat-affected areas followed bogs drainages, artificially recharged groundwater and natural groundwaters. Field sampling showed that at concentration typical for drinking water in Riga, more that 40% of the iron was in form of F e N 0 M - This is in agreement with Kawakubo et al. (1999), who by using catalytic spectrophotometery found that in drinking water even with low content of HA (1.2 mg 1*') iron largely exists as humic complexes.
Comparisons of cation and anion-exchange resins
showed that either of the sorbents could be used. The iron concentration calculated from sum of the indi
vidual species ( F e ^ , F e N 0 M and F e p M ) was 5 to 15 % higher than the F e I 0 t concentration measure in a sample
before the speciation analyses, Fig. 7. This difference
could be partially attributed to inaccuracy of the results
as well as to species that were retained by both resins.
Prospects and limitations of the IEM method
Although interaction of iron and humic substances has
been widely investigated in natural sciences (Ohzeki et
al. 1991 references therein) and several methods were developed for humic-iron speciation, they have not
frequendy been adapted for use in the water production industry. The proposed I E M method is simple, rapid in
its procedure, gives reliable results in the range of tested
conditions that are typical for drinking water in humic areas, and could potentially be used in the design of a drinking water treatment process.
However, the I E M method distinguishes between dif
ferent iron species based purely on the sorption proper
ties, whereas humic complexes having similar sorbtion
properties may have different other properties relevant for drinking water treatment. Knocke et al. (1992)
showed that depending on the molecular size of actual
humic substances, removal of iron-humic complex by
oxidation or by coagulation should be preferred. Thus,
knowledge about iron-humic complexes per se cannot
always be enough in the design of iron removal
processes. Further research about the role of iron-humic
complexes in water treatment is needed, and the method
presented here can be helpful in these endeavours.
Conclusions
A method for iron speciation analysis in groundwater
from areas of humic waters is proposed. Negatively
charged, dominantly organically bound, positively
charged, dominantly iron ions, and particulate matter
species of iron can be quantified with this method. The
sampling was found to have a significant influence on
the distribution of iron species. D E A E separates not
only dissolved but also partially colloidal species. T h e
method can be used in the design of iron removal facili
ties for drinking water production. In this study it was
found that the substantial part of the total iron in
groundwater, including artificially recharged ground
water, existed in the form of iron-humic complexes.
Acknowledgment
Thanks are due to Professor Jörgen Hanæus at Div.
Sanitary Engineering, Luleå University of Technology
for reviewing the article.
References
Appelblad, P. K... Baxter, D. C. and Thunberg, j . O. (1999) Determination of metal-humic complexes, free metal ions and coca! concentrations in natural waters. / Environ. Moni!.. 1. 211-217.
Barry, R. C., Schnoor, J. L , Sulzerbergcr, B., Sigg, L. and Stumm, W. (1994) Iron oxidation kinetics in an acidic Alpine lake. Wat. Res.. 28 (2), 323-333.
Bryan, N . D., Jones, D. M . , Appleton, M . , Livens, F. R.. Jones, M . N . , Warwick. P.. King, S. and Hall, A. (2000) A physicochemical model of metal-humate interactions. Phys. Chem. Chem. Phys., 2, 1291-1300.
Christensen, J. B. and Christensen, T. H . (1999) Complexa-tion of Cd, Ni , and Zn by DOC in polluted groundwater: a comparison of approaches using resin exchange, aquifer material sorption and computer speciation models ( W H A M and MINTEQ2). Environ. Sc. TcchnoL, 33. 3857-3863.
Deng, Y. (1996) Formation of iron (III) hydroxides from homogeneous solutions. Wat. Res., 31 (6), 1347-1354.
Deutsch, W. J. (1997) Groundwater geochemistry. Lewis Publ., Boca Raton, 221 pp.
Ephraim, J. and Marinsky, J. A. (1990) Ultrafikration as a technique for studying metal-humate interactions: study with iron and copper. Analytica Chimica Acta, 232,
06 V A T T E N - 3 - 0 2

171-180. Fukushima, M. andTatsumi, K. (1999) Light acceleration of
iron(III) reduction by humic acid in the aqueous solution. Coll. Surf. A: Physicochem. Eng. Asp., 155, 249-258.
Gustafsson, J. P. (1999) http://amov.ce.kth.se/people/gustafjp/winhumicv.htm.
Hautala, K. Peuravuori, J. and Pihlaja, K. (2000) Measurement of aquatic humus content by spectroscopic analyses. Wat. Res., 34 (1), 246-258.
Hiraide, M. (1992) Heavy metal complexed with humic substances in fresh water. AnalyticalScienc, 8, 454—459.
Hiraide, M., Ishii, M. and Mizuike, A. (1988) Speciation of iron in river water. Anal Sei., 4, 605-609.
Jensen, D. L. , Ledin, A. and Christensen, T .H. (1999) Speciation of heavy metals in landfill-leachate polluted groundwater. Wat. Res., 33 ( I I ) , 2642-2650.
Jobin, R. and Ghosh, M. M. (1972) Effect of buffer intensity and organic matter on oxygentation of ferrous iron. Jour. Amer. Wat. Works Ass., 64, 590-595.
Kawakubo, S., Hagihara, Y., Honda, Y., and Iwatsuki, M. (1999) Speciation of iron in river and tap waters by catalytic spectrophotometry using oxidation of ophenylene-diamine with peroxide. Analytica Chimica Acta, 388, 35-43.
Knocke, W. R , Conley I_, and Van Benschoten, J. E . (1992) Impact of dissolved organic carbon on the removal of iron during water treatment. Wat. Res., 26 (11), 1515-1522.
Kortelainen, P., Mannio, J., and Pcnnanen, V. (1986) Characteristics of the allochthonous organic matter in Finnish forest lakes and reservoirs. Publications of the Water Research Institute, National Board of Waters, Finland, 65, 88-97.
Livens, F. R. (1991) Chemical reactions of metals with humic material. Environmental Pollution, 70, 183-208.
Niessner, G., Buchberger, W. and Bonn, K. G. (1998) Anion exchange solid phase extraction of humic substances for the determination of complexed heavy metals in natural waters with high dissolved organic carbon contents. Monatsh. Chem., 129, 597-605.
Ohzeki, K , Tatehana, M., Nukatsuka, I. and Ishida, R (1991) Determination of humic acid and iron(III) buy solid-state spectrophotometry to study their interactions. Analyst., 116, 199-205.
Olivié-Lauquet, Q., Allard, T. , Benedetti, M. and Muller, J.-P. (1999) Chemical distribution of trivalent iron in riverine material from a tropical ecosystem: a quantitative EPR study. Wat. Res., 33 (11), 2726-2734.
Pandeya, S. B. and Sing, A. K (1997) Discontinuous spectro-colorimetric titration method for determining stability constants of fulvic acid-iron complexes. Aust. J. Soil Res., 35, 1279-1290.
Pettersson C , Håkansson K., Karlsson S, and Allard B. (1992) Metal speciation in a humic surface water system polluted by acidic leachates from a mine deposits in Sweden. Wat. Res., 27 (5), 863-871.
Pullin, M. J. and Cabaniss, S. E. (2001) Colorimetric flow-injection analysis of dissolved iron in high D O C waters. Wat. Res., 35 (2), 363-372.
Rose, }., Vilge, A., Olivié-Lauquet, G., Masion, A., Frechou, C. and Bottero, J. (1998) Iron speciation in natural organic matter colloids. Coll Surf. A:Physicochemic. Eng. Asp., 136, 11-19.
Shaker, M., Ghabbour, E. A., Davies, G. , EI-Toukhy, A. A. and Abid, I. M. (2000) Temperature independent site occupation in the tight binding of iron (III) by a solid-derived humic acid. Proceedings (Vol. 1): 10th International Meeting of IHSS, 24-28 July 2000 - Toulouse, France 688 pp.
Standard Method (1999) Standard methods for the examination of water and wastewater. Washington, APHA.
Stookey, L L. (1970) Ferrozine — a new spectrophotometric reagent for iron. Analyt. Chem., 42, 770-781.
Stumm, W. and Morgan, J. J . (1998) Aquatic Chemistry. Wiley Interscience, N.Y., 583 pp.
Theis, T . L. and Singer, P. C . (1974) Complexation of iron (II) by organic matter and its effect on iron (II) oxygenation. Envir. Sei. Technol. 8, 569-572.
Thurman, E . M. and Malcolm R L. (1981) Preparative isolation of aquatic humic substances. Environ. Set. Technol, 15,463-466.
Thurman, E . M. (1985) In: Organic geochemistry of natural waters. Martinus NijhofF/Dr.W.Junk Publishers: Wage-ningen.
Tipping, E. and Hurley, M. A. (1992) A unifying model of cation binding by humic substances. Geochim. Cosmochim. Acta, 56, 3627-3641.
Tipping, E . (1994) WHAM - A chemical equilibrium model and computer code for waters, sediments, and soils incorporating a discrete site/electrostatic model of ion-binding by humic substance. Comput. Geosci., 20,973-1023.
Viollier, E. , Inglett P. W., Hunter, K., Roychoudhury, A. N. and Van Cappellen, P. (2000) The ferrozine method revised: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem., 15,785-790.
Water Analysis (1988) Fresenius. W., Quentin, K. E. and Schneider W. (eds.). Springer-Verlag: Berlin, Heidelberg, N.Y., 804 pp.
V A T T E N - 3 - 0 2 07

Table 1. Default parameters used in the WinHumic V model
Parameter Humic acid Fulvic acid
type A sites, mol g 1
P*A
P*B
APK a
ApK B
Plog I
molecular radius (nm)
molecular weight
P^MHA FeöD püT M H A Fe(III)
Double layer overlap factor
K Z , charge factor
0.00329 0.00473
4.02 3.26
8.55 9.64
1.78 3.34
3.43 5.52
-374 -103
0.5 0.4
1.72 0.8
15000 1500
2.1 1.3
0.8 -0.2
0.25
1000
Table 2. Chemical composition of groundwater from different sampling sites (Gl, G2, Dl, PI, Bl, and B2). Average values of two replicates taken at a single sampling occasion. All chemical paramers value are shown as mg 1" except for chemical oxygen demand (COD M „), which is shown as mg0 21 .
Parameter G l G2 D l PI Bl B2
PH 7.23 7.56 7.64 6.10 7.13 6.52
C O D M n 3.75 3.78 3.40 24.06 11.72 14.23
HA 2.91 2.57 1.54 11.65 11.88 14.74
FA 5.73 5.67 5.73 5.73 5.45 7.67
Ca* 2 49 67 49 24 78 69
H C O j - 180 239 219 75 312 330
N C y 1.1 0.8 1.5 2.6 2 8.9 F e,o, 0.65 0.28 0.71 1.32 1.57 1.24
Table 3. Sorption capacity of different weak anion sorbents for iron-humic acid complexes (P'NQM) in batch experiments performed in 100 ml sealed glass bottles on a rotary shaker table (24 h at 20°C). Iron-humic complexes were prepared by mixing 5 mg l" 1 of Fe(III) with 25 mg 1"' HA (Aldrich) at pH 6.
Sorbent Porosity Functional group Matrix F E NOM m gg~'
D E A E cellulose Fibrous - C H 2 C H 2 N ( C 2 H 5 ) 2 Cellulose 2.14
DEAE SephadexA25 Gel-type - C H 2 C H 1 N ( C j H 5 ) J Dextran 1.15
DEAESpheron L C 1000 MR* - C H 2 C H 2 N ( C 2 H 5 ) 2 Poly(oxymethylacrylate) 1.10
Aminopropylsilica MR- - C H 2 C H , C H 2 N H 2 Silica 0.63
Diaion PA308 MR" - C H 2 N ( C H J ) J * ar Styrene-divinylbenzene 1.90
* MR - macroreticular
08 V A T T E N - 3 - 0 2

^ 50mm
ion-exchange resin
0.46um filter
pump pressurized water
Sample Fe«,,
Filtration 0.46 um FepcD
_^ FepM
Anionic resin DEAE cellulose
FeNOM
Cationic resin IRA 120
Fepc
Figure 2. /ron speciation analysis scheme.
Figure 1. A device for water iron species sampling using an ion-exchange method
V A T T E N - 3 - 0 2 09

Figure 3. Map shotting the study sites Gl, G2, Dl, Bl, B2, PI (black dots), lakes, river and a part of the Baltic Sea (grey areas), peat land and bog areas (checkered areas).
0.13 0.14 0.02 * G2 E F e N 0 M
• Fepc 0.27 0.47 0.02 *** E F e N 0 M
• Fepc G l • Fep„
OJ 0.44 0.03 ** • Fep„
D l
0.65 0.16 0.52 * PI ]
0.65 0.52 0.41 * B l
0.75 0.38 0.23 B2
( ) 0.5 1 1.5
Fe,mgr'
2
Figure 7. Iron speciation (FeN0M, FepC
FefM) applying an ion-exchange method for groundwater abstracted from different sites (G2, GI, Dl, PI, Bl, B2, see Fig. 3). Bars represent the arithmetic mean of three replicates; the deviation from means were < 10% and are not shown. The arithmetical differences between the sum of individual species
( F e N O M - F e p C > F c p M > s e e F i S - 2) a n d
the total iron (Fe t o I, see table 2) concentration were < 15% :***;< 10% : ** or < 5 % : *.
010 V A T T E N - 3 - 0 2

Figure 4. Effect of aeration time on the distribution of iron species in groundwater. In batchwise experiments iron-humic complexes were prepared from 4 mgl"1 of humic acid (Aldrich) and 4 mgl"1 of iron in N 2 atmosphere (contact time 0.5 hr). Next, the samples was aerated by purging them with air while withdrawn and organically bound iron ( F e N O M ) were determined in the unfiltered sample by separation on DEAE resin, particular iron (Fe p M) by membrane (0.46um pore size) filtration and ferrous iron (Fe(II)) spectrophotometrically using 2,2'-bipyridil.
S
>0.46 (im
0.05 - 0.46 um |
100 kDa-0.05 um
10-100 kDa
<10kDa
0.00 0.10 0.20 0.30
Fe, mg!-1
Figure 6. Size fractionation of iron in groundwater samples (G2) after adding artificially prepared iron-humic complexes (Fe„„ 0.58 mgf. CODs*, 12.3 mg02r').
100
90
80
70
60 I 50 -
I
40 S
30
20
10
0
0 0.5 1 1.5 2
Feto,, mg r1
Figure 5. Determination comparison of the amount of organically bound iron FeN0M (% Fem) by an ion-exchange method (IEM) and by a WinHumicV computer speciation model at different iron and humus concentrations in groundwater (G2). This was amended with different concentrations of artificially prepared iron humic acid complexes (Aldrich). Error bars represent the standard error of the mean.
V A T T E N - 3 - 0 2 011

NR: 2002:27 ISSN: 1402-1544 ISRN: LTU-DT -02/27-SE
U N I V E R S I T E T
Education
Doctoral Thesis
Department
Environmental Engineering
Edition
Division
Sanitary Engineering
Title Aspects o f drinking water supply in areas of humic water
Date
2002-08-13
Author
Talis Juhna
Language
English
Summary
The thesis investigated several aspects that are important for drinking water supply f r o m
waters wi th high amounts of humic substance (humic water).
The results showed that the composition o f humic substances is important in their
sorption to aquifer material during surface water ar t i f ic ia l recharge through inf i l t ra t ion
basins to groundwater. The more acidic fract ion o f humic substances was less effectively
sorbed than the more hydrophobic fraction. The removal o f the former fraction f r o m the
recharged groundwater was effective using weak base anion exchange resin.
A high total number of bacteria in presence o f cyanobacteria in the subsurface below the
inf i l t ra t ion indicated that significant decrease o f biodegradation o f organic matter, during
water passage f r o m blooming humic lake to groundwater, is not l ikely.
Because o f high amounts o f microbially assimilable organic carbon in humic water and its
low reduction during chemical treatment, bacterial growth in drinking water distribution
systems was dependent on the phosphorus concentration. In the distributed water bacteria
were forming b i o f i l m on the pipe surfaces. To reduce the b i o f i l m growth fa i r ly high doses
o f chlorine were needed.
The anion exchange resin was successfully used to determine concentrations of
iron-humic complexes in humic groundwater. According to this analysis a significant part
of iron in drinking water, produced by ar t i f ic ial (cont.)
Examiner/Supervisor
prof. Jörgen Hanaeus. docent Janis Sprogis
URL: http://epubl.luth.se/1402-1344/2002/27





















