
DISSERTATION Erlangung des akademischen Grades eines ...
267
DISSERTATION ZUR Erlangung des akademischen Grades eines Doktors der Naturwissenschaften der Naturwissenschaftlichen Fakultät der Karl-Franzens-Universität Graz Institut für Chemie, Organische und Bioorganische Chemie Thema: Synthesen und Reaktionen von Heterocyclen mit Fluoreszenz-optischen Eigenschaften Vorgelegt von Mag. Guy Crépin ENOUA Im November 2010 Graz-Österreich
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of DISSERTATION Erlangung des akademischen Grades eines ...

DISSERTATION
ZUR
Erlangung des akademischen Grades eines Doktors der Naturwissenschaften
der
Naturwissenschaftlichen Fakultät der Karl-Franzens-Universität Graz
Institut für Chemie, Organische und Bioorganische Chemie
Thema:
Synthesen und Reaktionen von Heterocyclen mit Fluoreszenz-optischen Eigenschaften
Vorgelegt von Mag. Guy Crépin ENOUA
Im November 2010
Graz-Österreich

DOCTORAL THESIS
SYNTHESES AND REACTIVITY OF
HETEROCYCLES WITH
FLUORESCENCE-OPTICAL PROPERTIES
ENOUA Guy Crépin, Mag.
Univ. Prof. Dr. Wolfgang STALDBAUER, Dissertation Supervisor
A Thesis presented for the degree of Doctorate in Natural Sciences, Option: Organic Chemistry Examiners committee Chairman : Univ. Pr. Dr. Georg URAY Examiners: Univ. Pr. Dr. Wolfgang KROUTIL Univ. Pr. Dr. Wolfgang STADLBAUER
Institute of Chemistry / Organic and Bioorganic Chemistry
Karl-Franzens University of Graz, Austria November, 2010

First of all, I would like to express my praises to God for His
guidance, protection and all the blessings He has bestowed upon
me. He granted me the strength to fulfill my doctoral studies
during these 3 years. To Him be the glory, honor and thanksgiving.

Dedications
I am dedicating this thesis to my mother KOBI Julienne, my father
ENOUANSOULOU Okira Felix, my grandmother NDIMI Catherine and am
thankful for their selfless sacrifices.
I am also writing this thesis in remembrance of my deceased brother Gaétan
Ndzila Okougou ENOUA, grandfathers Henri OKOU, Pierre NGOUMBA, and
uncles Isidore ANDA-MBELE and Firmin EFFENGUET.
This thesis is also dedicated to my brothers, my sisters, nephews, nieces,
cousins, and uncles who cannot partake in its defense because of the
geographical distance between the Republic of Congo and the Republic of
Austria.

Acknowledgements
The work presented in this thesis was conducted at the Institute of Chemistry,
Division of Organic and Bioorganic Chemistry, at the Karl-Franzens University
of Graz from December 2007 to November 2010.
I would like to express my deepest gratitude to Univ. Prof. Dr. Wolfgang
STADLBAUER for having accepted me in his laboratory and for his exceptional
supervision. As advisor he provided me with his full professional support in
dealing with this Ph.D research project. His constant encouragement helped
and challenged me throughout my studies.
I am also thankful to Univ. Prof. Dr. Georg URAY for his helpful contribution,
especially regarding the aspects of separation, purification, fluorescence
measurements of compounds and his invested time and valuable input to this
thesis. He greatly encouraged, advised and supported me during the writing of
this thesis. I feel much honored to have Professor URAY as second supervisor.
Both Professors STADLBAUER and URAY treated me like fathers would tutor a
son, they were very patient with my ``unforgivable mistakes`` in chemistry.
They are well respected due to their wide range of knowledge, good
interpersonal skills and humility which are worthy to emulate. What I learned
from both of them during these three years makes up a very important
foundation for my future research and teaching career.

I would also like to express my sincere thankfulness to Univ. Prof. Dr. Wolfgang
KROUTIL for examining this thesis and to Univ. Prof. Dr. Nadia Mösch-Zanetti,
head of Institute of Chemistry Karl-Franzens University, I am grateful to Univ.
Prof. Dr. Ulrike WAGNER, Institute of Biomolecular Sciences, Karl-Franzens
University of Graz for her help with the X-ray analyses. Univ. Prof. Dr. Klaus
ZANGGER was a helpful partner for various discussions about the
interpretation of NMR spectral analyses. His and Mr. Bernhard WERNER’s
assistance for recording numerous NMR and infrared spectra is greatly
appreciated as well as, Dr. Claudia REIDLINGER’s support for recording LC-
Mass spectral analyses.
My sincere thanks also go to Prof. Jean Boukari LEGMA, Prof. Adama SABA,
and Dr. Honorat Charles Roger NEBIE from University of Ouagadougou
(Burkina Faso) as well as Dr. Auguste BOUSSOUKOU from University Marien
NGOUBI of Brazzaville (Republic of Congo) for their teaching in
Electrochemistry and Organic Chemistry.
I am also thankful for Dr. Clement Bienvenu LOUBAKI’s efforts in talking with
Professor STADLBAUER for the provision of a place in Prof. STADLBAUER’s
laboratory for me.
I am grateful to Dr. Toma N. GLASNOV for reading my thesis. He and Dr.
Tahseen RAZZAQ accompanied me with their wisdom and encouragements.
The cooperation with them and also with my co-workers Ms. Nathalie
LACKNER and Mr. Günther LAHM made a real difference in my studies and
elaboration of this doctoral thesis.
My deep gratitude also goes to Mrs. Gritschi KERL and Mrs. Mag. Petra
RADESCHNIG in Vienna/Austria, both worked for OeAD in Burkina Faso, for

their generosity. Mrs. KERL especially played the role of a mother to me during
my stay in Austria. The friendship with M.Sc. Raymond OUEDRAOGO and
M.Sc. Albert SOUDRE also provided great comfort to me.
The “Church of Christ” in Graz/Austria became my Christian family while
staying in Austria. My special thanks go to Ms. Yasna VUČKOVIĆ from
Slovenia/Maribor, and GM Mag. Petra LANG and DI. Thomas LANG for their
generosity.
I gratefully acknowledge the financial support administered by the Austrian
Academic Exchange Service OeAD (Österreichischer Austauschdienst) through
a scholarship granted under the North-South-Dialog (Doctorate) scheme,
particularly Mrs Katharina ENGEL and Mrs. Mag. Christina DÜSS for their
generosity.
Finally, I feel deeply indebted to my fiancée Aicha Moussokoro BASSOLE for
being such a great source of encouragement to me. Her patience,
understanding and constant positive stimulation helped me to overcome the
stress related to my studies and the fact of being separated from for three
years, not to forget our baby.

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
LIST OF PRESENTATIONS AND CONFERENCE PROCEEDINGS List of Poster Presentations
1. Synthesis and Fluorescence Properties of 4-Cyano-6-methoxyquinolones
Enoua G. C., Uray G. and Stadlbauer W.
16th European Symposium on Organic Chemistry (16th ESOC), Prague (Czech
Republic), 12 - 16 July 2009 (Book of Abstracts p.197, poster P1.130).
2. 6-Methoxy- and 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile: syntheses and fluorescence properties
Enoua G. C., Uray G. and Stadlbauer W.
XXIVth European Colloquium on Heterocyclic Chemistry (24th ECHC), at the
Vienna University of Technology, Vienna (Austria), August 23 – 27, 2010 (Book
of Abstracts, PO-4)
List of Publications online
1. 4-Cyano-6-methoxyquinolones: Syntheses and Luminescence Properties
Enoua G. C., Uray G. and Stadlbauer W.
ECSOC-13, The Thirteenth International Electronic Conference on Synthetic
Organic Chemistry, http://www.usc.es/congresos/ecsoc/13/index.htm,
November 1-30, 2009 (a012)

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2. Fluorescence Properties of 6-Methoxy- and 6,7-Dimethoxyquinoline-3,4-
dicarbonitriles
Enoua G. C., Uray G., Stadlbauer W.,
ECSOC-14, The Fourteenth International Electronic Conference on Synthetic
Organic Chemistry, http://www.usc.es/congresos/ecsoc/14/index.htm,
November 1-30, 2010 (a###); submitted.
List of Conference Proceedings
1. 4-Cyano-6-methoxyquinolones: Syntheses and Luminescence Properties
Enoua G. C., Uray G., Stadlbauer W.
In Proceedings of ECSOC-13, The Thirteenth International Electronic
Conference on Synthetic Organic Chemistry,
http://www.usc.es/congresos/ecsoc/13/index.htm, November 1-30, 2009
(a012); J. A. Seijas, Shu-Kun Lin, M. P. Vázquez Tato (Eds). CD-ROM edition,
ISBN 3-906980-23-5, Published in 2009 by MDPI, Basel, Switzerland.
2. Fluorescence Properties of 6-Methoxy- and 6,7-Dimethoxyquinoline-
dicarbonitriles
Enoua G. C., Uray G., Stadlbauer W.
In Proceedings of ECSOC-14, The Fourteenth International Electronic
Conference on Synthetic Organic Chemistry,
http://www.usc.es/congresos/ecsoc/14/index.htm, November 1-30, 2010; J.
A. Seijas, Shu-Kun Lin, M. P. Vázquez Tato (Eds). CD-ROM edition, ISBN 3-
906980-##-#, To be published in 2010 by MDPI, Basel, Switzerland; in
preparation.

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CONTENTS
ABSTRACT……………………………………………………………………. 1
GENERAL INRODUCTION……………….…………………………………2
A. INTRODUCTION………………………………………………………..……………….2
B. METHODS FOR THE SYNTHESIS OF 4-HYDROXY-
QUINOLONE DERIVATIVES…………………………………………...................5
B.1. Cyclization of malondianilides with methane
sulfonic acid……………………………..…………………………………………..…5
B.2. Reaction of anilines with Meldrum’s acid…………………………………….....6
B.3. Reaction between aniline and carbon suboxide………………………………..6
B.4. Reactions of carbanions with isatoic anhydrides………………………………7
B.5. Cyclization of N-acetylanthranilic acid……………………………………………8
C. METHODS FOR THE SYNTHESIS OF QUINOLINES…………………………..8
C.1. Skraup quinoline synthesis……………………………………….………………..9
C.2. Döbner-von Miller reaction………………………………………………………….9
C.3. Döbner reaction……………………………………………………………………….10
C.4. Combes quinoline synthesis……………………………………………………….10
C.5. Friedländer synthesis…………………………………………………… …………11
C.6. Synthesis of quinolines by the Pfitzinger reaction………………… ………..11
D. SYNTHETIC ROUTES FOR THE PREPARATION OF 4-
HYDROXYQUINOLONES AND QUINOLINES USED IN THIS
WORK………………………………………………………………………..…..….……12
E. FLUORESCENCE PROPERTIES OF CARBOSTYRILS…………….….………16
F. APPLICATION OF FLUORESCENCE RESONANCE ENERGY
TRANSFER (FRET)………………………………………………………………..…..18
G. OTHER IMPORTANT FIELDS OF FLUORESCENT DYES………………...…20
REFERENCES FOR INTRODUCTION…………………………………………...……24

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CHAPTER 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY- 6-
AND 7-METHOXYQUINOLIN-2(1H)-ONES…….…..…...39
1.1. INTRODUCTION……………………………………………………….…………....39
1.2. RESULTS AND DISCUSSION…………………………………………….…….…39
1.2.1. Cyclocondensation of p-anisidin 1 and malonic acid 2 to
4-hydroxy-6-methoxyquinolin-2(1H)-one 3 as precursor…………...……39
1.2.1.1. Optimization of the reaction…………………………………………...……….41
1.2.1.2. Proposed mechanism of the formation of 4-hydroxy-6-
methoxyquinolin-2(1H)-one 3…………..……………………………………...42
1.2.1.3. Proposed mechanism of the formation of dianilide 4……………………..43
1.2.1.4. Structural elucidation : IR and 1H-NMR spectroscopy
study of compounds 3 and 4………………...………………………………..43
1.2.2. Chlorination of 4-hydroxyquinolone 3 to 2,4-dichloroquinoline 5
and 4-chloroquinolone 6…………………………………………………………44
1.2.2.1. Study of Infrared and 1H-NMR Spectra of 5 and 6………………………..45
1.2.2.2. 13C-NMR spectrum of 4-chloro-6-methoxyquinolin-
2(1H)-one 6………………………………………………………………………...46
1.2.3. Introduction of cyano substituents into 4-chloro-6-
methoxyquinolin-2(1H)-one 6…………………………………………………..46
1.2.4. Synthesis of 4-sulfinyloxyquinolin-2(1H)-one 8…………………………….49
1.2.4.1.Structure elucidation of 4-sulfinyloxyquinolin-2(1H)-one 8……………….50
1.2.4.2. 13C-NMR spectrum of 4-sulfinyloxyquinolin-2(1H)-one 8………………..51
1.2.5. Experiment to show the reaction pathway…………………………………..51
1.2.5.1. Cyanation of 4-sulfinyloxyquinolin-2(1H)-one 8 to 4-
cyanoquinolone 9 and 3,4-dicyanoquinolone 7…………..………………..52
1.2.5.2. One-pot synthesis of 3,4-dicyanoquinolone from 4-
sulfinyloxyquinolone 8…………………………………………………………..54
1.2.6. One-pot reaction to 4-cyano-3-unsubstituted quinolone 9……………...54
1.2.7. Chlorination reaction with sulfuryl chloride………………………………...55
1.2.8. Reduction reaction of 3,3-dichloroquinolin-2,4-dione 10 to
3-chloro-4-hydroxy-6-methoxyquinolin-2(1H)-one 11…………………….56
1.2.9. Bischlorination reaction of 3-chloro-4-hydroxyquinolone 11
with phosphoryl chloride…….………………………………………………..…57

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.10. Hydrolysis reaction of 2,3,4-trichloroquinoline 12………….……………57
1.2.11. Introduction of cyano substituents into 3,4-dichloro-
6-methoxyquinolin-2(1H)-one 13………………………………………….….58
1.2.12. Synthesis of 4-chloro-6-methoxy-3-nitroquinolin-2(1H)-
one 16……………………………..………………………………………………..59
1.2.12.1. Nitration reaction of 4-hydroxy-6-methoxyquinolin-2(1H)-one 3…..…..59
1.2.12.2. Bischlorination of 4-hydroxy-6-methoxy-
3-nitroquinolin-2(1H)-one 14…………………………………………………..60
1.2.12.3. Regioselective hydrolysis of 2,4-dichloro-6-methoxy-
3-nitroquinolin-2(1H)-one 15……………………………………………….....62
1.2.13. Synthesis of N-(4-chloro-6-methoxy-2-oxo-1,2-
dihydroquinolin-3-yl)acetamide 18………………………………………....63
1.2.13.1 Reduction of 4-hydroxy-6-methoxy-3-nitroquinolone 14 into
acetylaminoquinolone 17……………………………………………………….63
1.2.13.2. Regioselective chlorination of N-(4-hydroxy-6-
methoxy-2-oxo-1,2-dihydroquinolin-3-yl)acetamide 17………………….64
1.2.14. Amination of 3,3-dichloroquinoline-2,4-dione 10………………………..66
1.2.15. Reduction of 6-methoxy-3,3-di(piperidin-1-yl)quinoline-
2,4(1H,3H)-dione 19.……………………………………………………………66
1.2.16. Synthesis of 4-chloro-6-methoxy-3-(piperidin-1-
yl)quinolin-2(1H)-one 22………..…………………………….………….……67
1.2.17. Synthesis of 3,4-dicyanoquinolones 7 from
3-substituted 4-chloroquinolones…..…………..……………….…………..68
1.2.18. About an “isomer” of 6-methoxyquinoline-dicarbonitrile
7, compound 23a…….…………………………………………………….……70
1.2.19. Cyclocondensation of m-anisidin 27 and malonic acid 2
to 4-hydroxy-7-methoxy-2(1H)-one 28………………………………….…..75
1.2.20. Chlorination of 4-hydroxyquinolin-2(1H)-one 28 to
2,4-dichloroquinoline 30 and 4-chloroquinolin-2(1H)-one 31.……….76
1.2.21. Synthesis of 7-methoxyquinoline-carbonitriles 32 and 33…………....77
1.2.22. Synthesis of 6-methoxy 4-trifluoromethylquinolin-
2(1H)-one 38………………………………………………………………….…..80

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.23. Synthesis of 4-methoxy-N-methylaniline 41……………………….……...82
1.2.23.1. Selective monoalkylation of p-anisidine 1 to
N-(4-methoxyphenyl)-N-methylformamide 40……………………….…... 82
1.2.23.2. Acidic hydrolysis of N-(4-methoxyphenyl)-N-methylformamide 40…...83
1.2.24. Cyclocondensation of 4-methoxy-N-methylaniline 41 and malonic
acid 2 to 4-hydroxy-6-methoxy-1-methylquinolin-2(1H)-one 42……..83
1.2.25. Chlorination of 4-hydroxy-6-methoxy-1-methylquinolin-
2(1H)-one 42 with phosphoryl chloride …………………………………….84
1.2.26. Introduction of a cyano substituent into 4-chloro-6-methoxy-
1-methylquinolin-2(1H)-one 43…………………………………………….....85
1.2.27. N-Methylation of 6-methoxy-2-oxo-1,2-dihydroquinoline
-4-carbonitrile 9……………………………………………………………………85
1.2.28. N-Alkylation of 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile 7……………………………………………………………....86
1.2.28.1. N-Alkylation with iodomethane…………………………….………………..86
1.2.28.2. N-Alkylation reaction with ethyl bromoacetate………… ………….......87
1.2.29. O-Alkylation of 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile 7…………………………….…………………………………88
1.2.30. N-Methylation of 4-chloro-6-methoxyquinolin-2(1H)-one 6…………….89
1.2.31. N-Methylation of 3,4-dichloro-6-methoxyquinolin-2(1H)-one 13…......89
1.2.32. Nitration reaction 4-chloro-6-methoxyquinolin-2(1H)-one 6……………90
1.3 CONCLUSION……………………………………………………………………….92
CHAPTER 2. SYNTHESIS OF 4-CYANO-3-SUBSTITUTED
QUINOLIN-2(1H)-ONES….……………………………….94
2.1. INTRODUCTION……………………………………………………………………..94
2.2. RESULTS AND DISCUSSION………………………………………................94
2.2.1. Esterification reaction of arylacetic acid 52…………………………………94
2.2.2. Condensation reaction of arylethyl ester 53 with diethyl
carbonate 54…………………………………………………………………….…..95
2.2.3. Thermal cyclization of aryl malonates 55………….................................96
2.2.4. Bischlorination of 4-hydroxy-3-substituted quinolin-2(1H)-ones 57…..98

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.5. Regioselective hydrolysis of 2,4-dichloro-3-substituted
quinolines 58……………………………………………………………………100
2.2.6. Introduction of the cyano group into 4-chloro-6-methoxy-
3- substituted quinolin-2(1H)-ones 59…………………………………….101
2.2.7. Synthesis of 2-chloro-6-methoxy-3-phenylquinoline-4-
carbonitrile (61)………………………………………………………...103
2.2.8. Synthesis of ethyl 3-[(4-methylphenyl)amino]-
2-[(4-methoxyphenyl)carbamoyl]-3-oxopropanoate 64…………………103
2.2.9. Synthesis of ethyl 4-hydroxy-2-oxo-1,2-dihydroquinoline-
3-carboxylate (67)……………………………………………………………....105
2.2.10. Bischlorination of ethyl 4-hydroxy-2-oxo-1,2-dihydro
quinoline-3-carboxylate (67) with phosphoryl chloride………………107
2.2.11. Synthesis of ethyl 4-chloro-2-oxo-1,2-dihydroquinoline-
3-carboxylate (70)…………………………………………………………......108
2.2.12. Synthesis of 4-hydroxyquinolin-2(1H)-one 69………………………….110
2.2.13. N-Alkylation of 4-chloro-3-(4-chlorophenyl)-6- methoxy
quinolin-2(1H)-one 59b with ethyl bromoacetate……………..………111
2.3. CONCLUSION……………………………………………………………………..112
CHAPTER 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-
DIHYDROQUINOLINE-3,4-DICARBONITRILE…….114
3.1. INTRODUCTION…………………………………………………………..……..114
3.2. RESULTS AND DISCUSSION…………………………………………..….....114
3.2.1. Synthesis of 4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one
(74) as precursor………………………………………………………………...114
3.2.2. Bischlorination of 4-hydroxy-6,7-dimethoxyquinolin-
2(1H)-one (74) with phosphoryl chloride……………………………………115
3.2.3. Regioselective hydrolysis of 2,4-dichloro-
6,7-dimethoxyquinoline (75)………………………………………………….116
3.2.4. Chlorination of 4-hydroxy-6,7-dimethoxyquinolin-
2(1H)-one (74) with sulfuryl chloride……………………………………….117

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
3.2.5. Reduction of 3,3-dichloro 6,7-dimethoxyquinolin-
2,4(1H,3H)-dione (77) to 3-chloro-4-hydroxyquinolin-
2(1H)-one 78…………………………………………………………………......117
3.2.6. Bischlorination of 3-chloro-4-hydroxy-6,7-
dimethoxyquinolin-2(1H)-one 78……………………………………………..118
3.2.7. Regioselective hydrolysis of 2,3,4-trichloro-6,7-
dimethoxyquinoline 79………………………………………………………….119
3.2.8. Introduction of the cyano groups into 4-chloro
quinolin-2(1H)-ones 76 and 80………………………………………………119
3.2.9. Alkylation of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile (82)…………………………………………………………...121
3.2.9.1. N-Methylation of 6,7-dimethoxy-2-oxo-1,2-
dihydroquinoline-3,4-dicarbonitrile (82)……………………..……………..121
3.2.9.2. Alkylation of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline
-3,4-dicarbonitrile (82) with ethyl bromoacetate……..……………………122
3.2.10. N-Alkylation of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline
3,4-dicarbonitrile (82) with ethyl bromoacetate…………………..........123
3.3. CONCLUSION………………………………………………………..……………..124
CHAPTER 4: SYSTEMATIC INVESTIGATION OF
SUBSTITUENT EFFECTS ON FLUORESCENCE
AND PHOTOPHYSICAL PROPERTIES OF
4-CYANOCARBOSTYRIL DERIVATIVES………….126
4.1. INTRODUCTION………………………..…………………………………………126
4.2. RESULTS AND DISCUSSION…………………………………………..……….128
4.2.1. Influence of methyl, trifluoromethyl and cyano groups
on the fluorescence properties on quinolones core……………………….128
4.2.2. The effects of methoxy groups in positions 6 and/or 7………………….132
4.3. ELECTRONIC SPECTRA OF THE NEW CARBOSTYRILS...................135
4.3.1. Known 4-cyano-3-H-6,7-dimethoxy-carbostyrils …… ..……….135
4.3.2. O- versus N-Alkylation…………………………………………………………..141
4.3.3. Influence of differently substituted aryl groups
in position 3 on the fluorescence properties………………………………..141

PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4.3.4. Stacking cyanocarbostyrils……………………………………………..…...144
4.3.5. Fluorescence properties of 4-sulfinyloxycarbostyrils…………………..146
4.4. CONCLUSION……………..………………………………………………........147
GENERAL CONCLUSION……………………………………………….148
EXPERIMENTAL PART FOR CHAPTERS 1, 2 AND 3………….150
EXPERIMENTAL PART FOR CHAPTER 4…………………………214
REFERENCES OF THEORETICAL PART
FOR CHAPTERS 1, 2 AND 3………………………………………….218
REFERENCES OF THEORETICAL PART
FOR CHAPTER 4……………………………………………….……..…234
REFERENCES FOR EXPERIMENTAL PART………………………242

ABSTRACT 1
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
ABSTRACT
Carbostyrils have found in the last decade great interest as stable
fluorophors with excellent photophysical properties. In this thesis a number of
4-cyanocarbostyrils were investigated which show very interesting fluorescence
properties and offer the introduction of linkers to label biological material,
which make them particularly interesting as probes in biological, biochemistry
and medicine applications.
The syntheses started from methoxyanilines which gave with malonic acid or
arylmalonates either 3-unsubstituted or 3-aryl-4-hydroxycarbostyrils.
Functionalization of the 3-H derivatives at position 3 by bis-chlorination and
reduction gave 3-chloro derivatives. Amination of the dichloro derivatives with
piperidine and reduction gave 3-piperidinocarbostyrils. Nitration of the 3-H
derivatives gave 3-nitro derivatives; reduction afforded 3-
acetylaminocarbostyrils.
All these 4-hydroxycarbostyrils were chlorinated at position 2 and 4 and
hydrolyzed in acidic media regioselectively at position 2 to 4-chlorocarbostyrils.
The introduction of the cyano group in the 4-chloro-6-methoxycarbostyril
series gave 3,4-dicyanocarbostyrils. 7-Methoxy- and 3-aryl analogues gave
under the same conditions 4-cyanocarbostyrils. Alkylation of 3,4-dicyano-
carbostyrils led to the expected 1-alkyl derivatives.
Fluorescence spectra of 3,4-dicyano-6-methoxycarbostyril show λmaxexc at 470
nm, λmaxem at 560 nm, and a fluorescence quantum yield of ~15%. The
fluorescence of the 6,7-dimethoxycarbostyril analogue suffered a small blue-
shift, but exhibits an excellent quantum yield of ~50 %. The introduction of
alkyl groups at N-1 did not affect the fluorescence properties. Excitation and
emission maxima of 3-aryl-4-cyanocarbostyrils and 7-methoxycarbostyrils are
both ~50 nm blue-shifted, compared with dicyano analogues; the quantum
yield is ~25%.

GENERAL INTRODUCTION 2
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
GENERAL INTRODUCTION
A. INTRODUCTION
4-Cyanoquinolin-2(1H)-ones (4-cyanocarbostyrils) of type I (R2 = CN)
which synthesis and chemical modifications described in this thesis, are
heterocyclic compounds with two fused rings containing a benzene nucleus (II)
and pyridin-2(1H)-one moiety (III).
N O
H
R1
R2
R3
R4
R5
R6
R4
R3
R5
R6
N
R2
R1
O
H
I II III
Figure 1: Structure of quinolin-2(1H)-one I, benzene II and pyridine-2-(1H)-
one III
Quinolin-2(1H)-ones are also subunits of the pyrido[3,2,1-jk]carbazol-6-
ones VII and their tetrahydroderivatives VIII, which possess biologically
interesting combination of pharmacologically relevant core structures [1].
Pyridocarbazolones have gained attention in medicinal chemistry as blood
coagulation inhibitors, or antiallergics, analgesics and antipyretics. Few of
them can be used for the treatment of certain mental disorders [2].
Pyridocarbazolone derivatives are also useful in dye chemistry [3] as
fluorescent whiteners as greenish-yellow azodyes for polyester fibers.

GENERAL INTRODUCTION 3
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
OOH
R1
R3
R2
VII
N
OOH
R1
R3
R2
VIII
Pyrido[3,2,1-jk]carbazol-6-one 8,9,10,11-Tetrahydropyrido[3,2,1-jk]
carbazol-6-one
Figure 2: Structures of compounds VII and VIII.
Electron-donating groups such as methoxy groups on the quinolin-
2(1H)-one moiety have been chosen in this work, potentially serving as a
handle for further functionalization [4, 5, 6]. On the other side, methoxy
groups are common for many quinoline alkaloids [7a].
Quinolin-2(1H)-one, particularly the 4-hydroxyquinolin-2(1H)-one moiety
is a basic structure found in many natural products or is synthesized and used
in industries. It plays an important role as precursor in various reaction such
as transformation into quinoline nucleus, which is a very important class of
heterocyclic compound. The quinoline core is present in many alkaloids [7],
such as Swietinidine A [8], Daurine, Folidine [9], Glycolone A [10],
Glycocitridine [11], Flindersine [12, 13], or Almein [14].
Moreover, substituted quinolines are known to display a wide range of
pharmacological activities such as anti-inflammatory [15-19], antibacterial [18-
25], antiprotozoan [26-28], antimalarial [18, 19, 29-32], antiasthmatic [18, 19,
33], antituberculosis [34, 35 ], anti-Alzheimer [36], antihypertensive [18, 19,
37], anthelmintic [38], anti-HIV [39-41], anticancer [42-45], anti-platelet [18,
19] activity and tyro-kinase PDGF-RTK inhibiting agents.

GENERAL INTRODUCTION 4
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Hydroxyquinolin-2(1H)-ones unsubstituted at the fused benzene ring
such as 4-hydroxyquinolin-2(1H)-one IX are considered as glycosyl acceptor
[46]. Glycosides are largely distributed in nature and play important roles in
living organisms.
N O
H
OH
IX
Figure 3: Structure of compound IX
Quinoline glycoalkaloides, in particular (β-D-glucopyranosyloxy)quinolines and
their quinolin-2(1H)-one analogues, were isolated from different natural
sources [46]. These include larvae, pupae and adults of some wasps (Vespa
levisi) and honey bees (Apis mellifera and others), [47], corn kernels [48] and
Chinese medicinal plant Echinops gmelinii (Compositae) [49]. Plants of the
Haplophyllum (Rutaceae) genus are especially rich sources of quinoline
alkaloids including glycoalkaloides of furanoquinoline structure [49c, 50].
Quinoline and quinolinone derivatives are metabolized in animals to
hydroxyl derivatives which then become conjugated with D-glucuronic acid
prior to their excretion [46]. β-D-glucopyranosiduronic acid derivatives of
quinoline, quinolin-2(1H)-one and quinolin-4(1H)-one were isolated from urine
of rabbits [51], rats [52], and mice [52b].
Several (β-D-glucopyranosyloxy)quinolines and their quinolin-2(1H)-ones were
prepared for treatments of chloroquinone-resistant malaria [53], tuberculosis
[54] and as anti-allergic agents [55].
A literature survey showed that quinolin-2-(1H)-ones could be obtained
in different pathways.

GENERAL INTRODUCTION 5
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
B. METHODS FOR THE SYNTHESIS OF 4-HYDROXYQUINOLONE
DERIVATIVES
Several methods for the preparation of quinolin-2(1H)-ones of type I have
been developed. Among them, some have been established for a wide range of
derivatives of 4-hydroxyquinolin-2(1H)-ones such as: Cyclization of
malondianilides with methane sulfonic acid, reaction of anilines with
Meldrum’s acid, reaction between aniline and carbon suboxide, reactions of
carbanions with isatoic anhydrides, cyclization of N-acetylanthranilic acid,
condensation of anilines or alkyl anthranilates with malonic acid
and/ormalonic acid esters.
B.1. Cyclization of malondianilides with methane sulfonic acid
Malondianilides X were treated with aluminium trichloride [56] or
polyphosphoric acid [57] to form by intramolecular cyclization the
corresponding 4-hydroxyquinolin-2(1H)-one XI in poor to moderate yields and
in some cases the reaction failed. However, the cyclization of X in the presence
of methanesulfonic acid containing 10 % of phosphoric pentoxide as catalyst
[58] afforded the corresponding 4-hydroxyquinolin-2(1H)-one XI in excellent
yield of 90 % after a simple work-up.
R3
R4
R5
R2
NH
CH
R5
R4
R3
R2
NH
O
O
R1
N
R4
R2
R5
R3
H
O
R1
OH
CH3SO
3H / P
2O
5
X XI
Scheme 1

GENERAL INTRODUCTION 6
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
B.2. Reaction of anilines with Meldrum’s acid
Quinolin-2(1H)-ones XV can readily be synthesized from anilines XII and
Meldrum’s acid XIII [59]. The reaction involves an intermediate Malonic acid
monoaryl ester XIV, which treated with cyclization reagent such as Eaton’s
reagent afforded quinolin-2(1H)-ones XV. The crucial problem in this
procedure is to reduce or eliminate the decarboxylation as the authors state.
Thus, Eaton’s reagent is used for the cyclization of the intermediate under mild
reaction conditions.
N
R3
R1
R2
H
O
OH
R3
R2
R1
NH2
O
O
O
O
CH3
CH3
- (CH3)2CO R
3
R2
R1
NH
O
COOH
- H2O
+
XII XIII XIV
Scheme 2
B.3. Reaction between aniline and carbon suboxide
The reaction between aniline XVI and carbon suboxide XVII usually
gives malonanilide [60] quantitatively. It was found that the reaction of aniline
XVI with carbon suboxide (C3O2) in the presence of aluminium chloride
produced in the first step an N-acetylated intermediate 3-oxo-N-
phenylacrylamide XVIII in good yield, which upon cyclization in benzene at

GENERAL INTRODUCTION 7
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
80 °C reflux conditions gave 4-hydroxycarbostyril IX in only 17 % yield [61].
N
H
O
OH
NH2
N
H
C
O
C O
AlCl3
Et2O C
6H
6
O C C C O
XVI XVII XVIII
IX
++
Scheme 3
B.4. Reactions of carbanions with isatoic anhydrides
A literature survey showed that some works have addressed on the
synthesis of quinolin-2(1H)-ones starting from isatoic anhydrides [62-67].
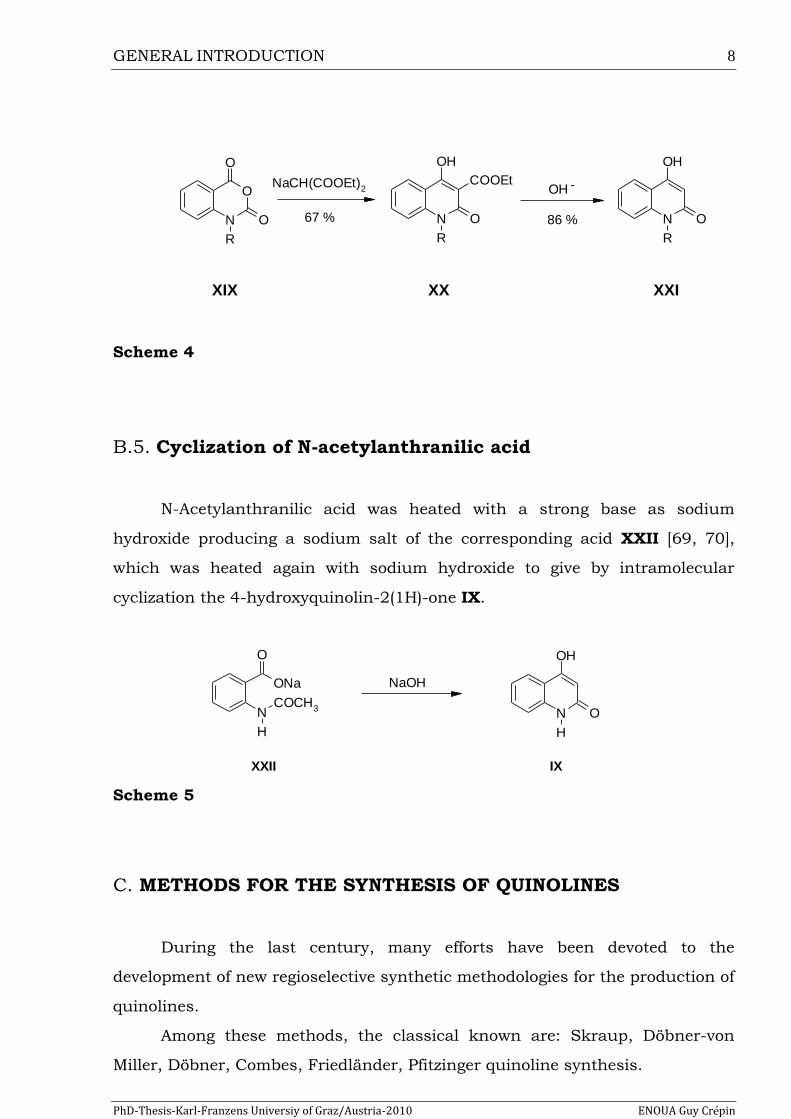
G. M. Coppola et al. [68] reported the synthesis of quinolin-2(1H)-ones XXI by
reaction of the sodium salt of diethylmalonate with isatoic anhydride XIX,
allowing the isolation of the intermediate ethyl 1-alkyl-4-hydroxy-2-oxo-1,2-
dihydroquinoline-3-carboxylate XX, which was then subsequently hydrolyzed
in alkaline media followed by decarboxylation to give the corresponding
quinolin-2(1H)-ones XXI.
GENERAL INTRODUCTION 8
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
R
O
OH
N
O
O
O
R
NaCH(COOEt)2
N O
OH
COOEt
R
OH -
86 %67 %
XIX XX XXI
Scheme 4
B.5. Cyclization of N-acetylanthranilic acid
N-Acetylanthranilic acid was heated with a strong base as sodium
hydroxide producing a sodium salt of the corresponding acid XXII [69, 70],
which was heated again with sodium hydroxide to give by intramolecular
cyclization the 4-hydroxyquinolin-2(1H)-one IX.
N
H
O
OH
N
H
ONa
O
COCH3
NaOH
XXII IX
Scheme 5
C. METHODS FOR THE SYNTHESIS OF QUINOLINES
During the last century, many efforts have been devoted to the
development of new regioselective synthetic methodologies for the production of
quinolines.
Among these methods, the classical known are: Skraup, Döbner-von
Miller, Döbner, Combes, Friedländer, Pfitzinger quinoline synthesis.

GENERAL INTRODUCTION 9
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
C.1. Skraup quinoline synthesis
The Skraup synthesis is a chemical reaction used to synthesize
quinolines. It is named after the Czech chemist Zdenko Hans Skraup (1850-
1910) and was developed at the chemistry institute of Graz. In the archetypal
Skraup reaction, aniline is heated with sulfuric acid, glycerol, and an oxidizing
agent, such as nitrobenzene to yield quinoline [71].
NNH2
OH
OH
OH
H2SO
4
PhNO2
XVI XXIII
Scheme 6
In this example, nitrobenzene serves as both the solvent and the oxidizing
agent. The reaction, which otherwise has a reputation for being violent ("the
Chemical Inquisition"), is typically conducted in the presence of ferrous sulfate
[72]. Arsenic acid may be used instead of nitrobenzene and the former is better
since the reaction is less violent [73].
C.2. Döbner-von Miller reaction
The Döbner-Miller reaction is the organic reaction of an aniline XVI with
α, β-unsaturated carbonyl compounds XXIV to form quinolines XXV [74].
N RNH2
H
O
R
XVI XXV
XXIV
Scheme 7

GENERAL INTRODUCTION 10
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
This reaction is also known as the Skraup-Döbner-Von Miller quinoline
synthesis, and is named after the Czech chemist Zdenko Hans Skraup (1850-
1910), and the German chemists Oscar Döbner (Doebner) (1850-1907) and
Wilhelm von Miller (1848-1899). When the β-unsaturated carbonyl
compound is prepared in situ from two carbonyl compounds (via an Aldol
condensation), the reaction is known as the Beyer method for quinolines. The
reaction is catalyzed by lewis acids such as tin tetrachloride, scandium (III)
triflate or Brönsted acids such as p-toluenesulfonic acid, perchloric acid,
amberlite and iodine.
C.3. Döbner reaction
The Döbner reaction is the chemical reaction of an aniline XVI with an
aldehyde XXVII and pyruvic acid XXVI to form quinoline-4-carboxylic acids
XXVIII [71a, 71b].
N R
OHO
NH2
CH3
OH
O
O
R H
O
XVI XXVIII
XXVI
XXVII
Scheme 8
C.4. Combes quinoline synthesis
The Combes quinoline synthesis is a chemical reaction involving the
condensation of unsubstituted anilines (XVI) with β-diketones (XXIX) to form
substituted quinolines (XXXI) after an acid-catalyzed ring closure of an
intermediate Schiff base (XXX) [75].

GENERAL INTRODUCTION 11
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N R1
R2
NH2
O
R2
O
R1
R1
O
R2 H
2SO
4
- H2O- H
2O
XVI XXX XXXI
XXIX
Scheme 9
C.5. Friedländer synthesis
The starting materials for this quinoline synthesis are o-aminoaryl
aldehydes or ketones XXXII and a ket -methylene group
XXXIII. After an initial amino-ketone condensation, the intermediate
undergoes base- or acid-catalyzed cyclocondensation to produce a quinoline
derivative XXXIV. It is named after the German chemist Paul Friedländer
(1857-1923) [76, 77].
N
R2
R3
R4
R2
O
NH2
R1
R1
R3
O
R4+
XXXII XXXIII XXXIV
Scheme 10
C.6. Synthesis of quinolines by the Pfitzinger reaction
The Pfitzinger reaction (also known as the Pfitzinger-Borsche reaction) is
the chemical reaction of isatin XXXV with a base and a carbonyl compound
XXXVI to yield substituted quinoline-4-carboxylic acids XXXVII [78].

GENERAL INTRODUCTION 12
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
R2
R1
OHO
N
O
O
H
R1
O
R2
KOH
XXXVI
XXXV XXXVII
Scheme 11
D. SYNTHETIC ROUTES FOR THE PREPARATION OF 4-
HYDROXYQUINOLONES AND QUINOLINES USED IN THIS
WORK
Our synthetic routes for the preparation of 4-hydroxyquinolin-2(1H)-ones
were based on the cyclocondensation of methoxyanilines XXXVIII with malonic
acid XXXIX, heated in phosphoryl chloride to form the 4-hydroxyquinolin-
2(1H)-ones XL.
R3
R2
NH
R1
N
R3
R2
R1
O
OH
COOH
COOH
POCl3
XXXIX
XXXVIII XL
Scheme 12
Another approach to 4-hydroxyquinolin-2(1H)-ones was the synthesis of
3-aryl-4-hydroxyquinolin-2(1H)-ones XLV having substituents in the phenyl
moiety, which are connected via the conjugated bonds to the 2-quinolone dye
system.

GENERAL INTRODUCTION 13
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The synthesis started from suitable substituted aryl acetates XLI which
were treated with diethyl carbonate XLII to give aryl malonates XLIII.
Thermal cyclization of aryl malonates XLIII with anilines XLIV gave the
corresponding 3-aryl-4-hydroxyquinolin-2(1H)-ones XLV.
O
OEt
R
O
EtO
EtO
O
OEt
O
OEtR
NaH / THF
+
XLI XLII XLIII
R3
NH2 N
R3
O
OH
H
RO
OEt
O
OEtR
Ph2O
+
XLIV XLIII XLV
> 250 °C
Scheme 13
On the other hand, 3-ethyloxycarbonyl-4-hydroxyquinolin-2(1H)-one XLVIII
was obtained from methyl anthranilate XLVI by treatment with
diethylmalonate XLVII in sodium ethoxide.
N O
OH
H
COOEt
NH2
O
OMe COOEt
COOEt
EtONa / EtOH
+
XLVI XLVII XLVIII
Scheme 14
GENERAL INTRODUCTION 14
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
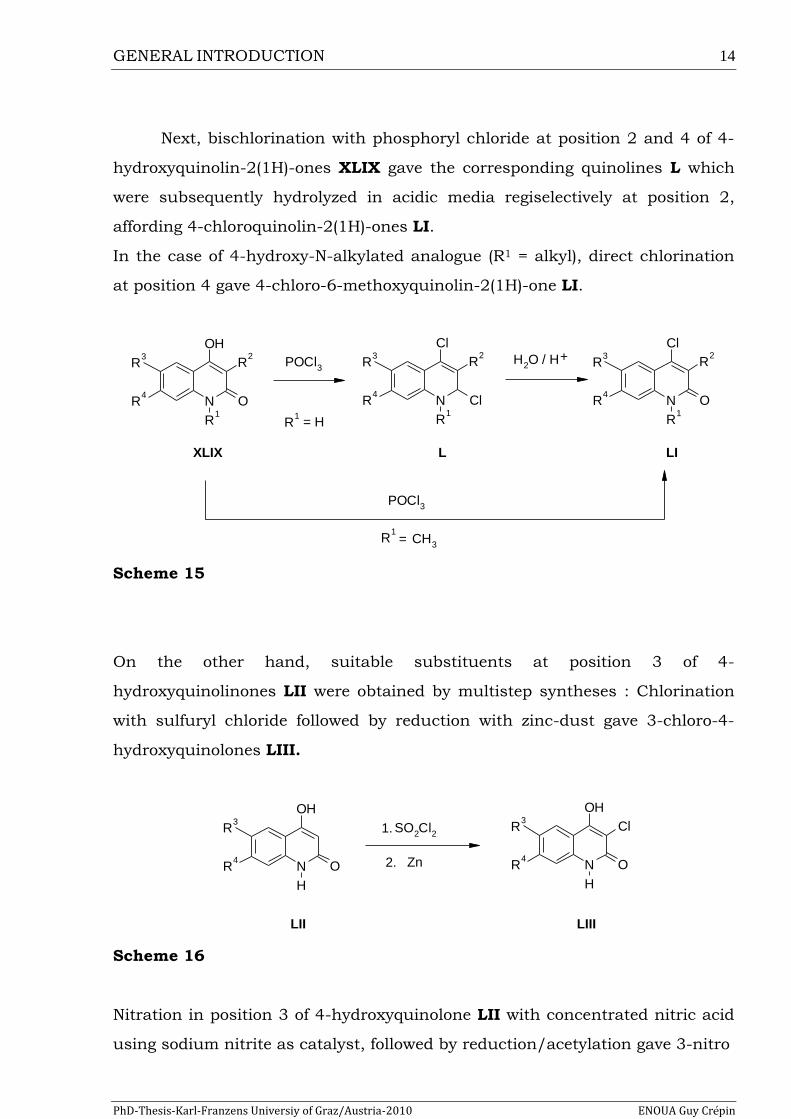
Next, bischlorination with phosphoryl chloride at position 2 and 4 of 4-
hydroxyquinolin-2(1H)-ones XLIX gave the corresponding quinolines L which
were subsequently hydrolyzed in acidic media regiselectively at position 2,
affording 4-chloroquinolin-2(1H)-ones LI.
In the case of 4-hydroxy-N-alkylated analogue (R1 = alkyl), direct chlorination
at position 4 gave 4-chloro-6-methoxyquinolin-2(1H)-one LI.
N O
OH
R1
R2
R3
R4
N
R2
R3
R4
Cl
Cl
R1
N O
R2
R3
R4
Cl
R1
POCl3
H2O / H
R1
CH3
POCl3
R1
+
XLIX L LI
=
= H
Scheme 15
On the other hand, suitable substituents at position 3 of 4-
hydroxyquinolinones LII were obtained by multistep syntheses : Chlorination
with sulfuryl chloride followed by reduction with zinc-dust gave 3-chloro-4-
hydroxyquinolones LIII.
N O
OH
R3
R4
H
N O
R3
R4
Cl
OH
H
SO2Cl
2
Zn
LII LIII
1.
2.
Scheme 16
Nitration in position 3 of 4-hydroxyquinolone LII with concentrated nitric acid
using sodium nitrite as catalyst, followed by reduction/acetylation gave 3-nitro

GENERAL INTRODUCTION 15
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
and 3-acetylaminoquinolones LIV and LV respectively.
N O
OH
R3
R4
H
N
R3
R4
O
H
OH
NO2
N O
R3
R4
OH
H
NHCOCH3
HNO3 / NaNO
2 Zn/ Ac2O
LII LIV LV
Scheme 17
Amination of 3,3-dichloroquinolin-2,4-dione LVI with piperidine and
reduction of the resulting intermediate dipiperidine LVII with sodium dithionite
in ethanol-water mixture gave 4-hydroxy-3-piperidinoquinolone LVIII.
N O
H
OCl
ClR
3
R4
N
N O
H
OH
R4
R3
N
N
N O
H
O
R4
R3
Piperidine Na2S
2O
4
LVI LVII LVIII
Scheme 18
All these 4-hydroxyquinolones derivatives were chlorinated with
phosphoryl chloride at position 2 and 4, and subsequently hydrolyzed in acidic
media regioselectively at position 2 to give the 4-chloroquinolone derivatives. In
the case of 3-acetylaminoquinolones LV the chlorination reaction with
phosphoryl chloride gave directly 4-chloroquinolone.
The introduction of the cyano group upon 4-chloroquinolone derivatives
at position 4 and/or 3 was carried out by reaction with potassium cyanide in
the presence of sodium p-toluenesulfinate.
Interesting chromophores having the cyano groups at position 3 and 4

GENERAL INTRODUCTION 16
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
were alkylated with methyl iodide or ethyl bromoacetate at position N-1.
Also, 6-methoxy-4-trifluoromethylquinolin-2(1H)-one was synthesized
from p-anisidine and 4,4,4-trifluoroacetoacetate in the presence of sulfuric
acid in order to compare its fluorescence properties with those of 6-methoxy-2-
oxo-1,2-dihydroquinoline-4-carbonitrile.
E FLUORESCENCE PROPERTIES OF CARBOSTYRILS
Fluorescence is generally defined as a luminescence emission that is
caused by the flow of some form of energy into the emitting body, this emission
ceasing abruptly when the exciting energy is shut off.
In the literature of organic luminescence, the term fluorescence is used
exclusively to denote a luminescence which occurs when a molecule makes an
allowed optical transition.
The fluorescence properties of carbostyrils [quinolin-2(1H)-ones] were
first reported by O. A. Ponomarev. [79] Compared with similar coumarin
fluorophors, luminescence properties of most quinolin-2(1H)-ones have the
disadvantage of shorter absorption and emission wavelengths [80, 81]. In
contrast, the advantages of carbosytril systems are high stability against
chemicals, thermal and photochemical stress, for instance an aqueous solution
pH 6 of the europium-complex was kept in the dark at room temperature and
its luminescence was found to be unchanged after more than one year [58],
insensitivity to oxygen quenching, independence of luminescence in a broad pH
region, for instance an aqueous
Recently, our group investigated on the vastly improved luminescence
properties of a big number of carbostyril [quinolin-2(1H)-one) systems [82].
These properties we achieved by suitable substituents, e.g. acceptor
groups such as trifluoromethyl in position 4 (Acc) and donor groups such as
methoxy or amino in position 6 and 7 (Don1 and Don2). The molecules with
methoxy groups gave excitation/emission maxima at ~370/430 nm, together

GENERAL INTRODUCTION 17
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
with large Stoke' s shifts and sufficient quantum yields [4, 5, 83]. These
fluorescence properties are comparable with umbelliferon (7-
hydroxycoumarin), but the molecules have much better stability.
O OOH
Figure 4: Structure of umbelliferon
6,7-Amino groups show longer wavelengths and better quantum yields, but
have the big disadvantage of pH sensitivity [83b]. 4-Cyano substituents
improved the fluorescence properties with longer wavelengths and better
quantum yields [61].
N O
Acc
Don2
Don1
Linker
R
Donor: MeO, NHR, NR2
Acceptor: CF3, CN
Absorption: 360-390 nm
fluorescence: 420-450 nm;
Quantum yield up to 0.5
Figure 5
Fluorescence has many practical applications, including mineralogy,
gemology, chemical sensors, fluorescent labeling, dyes, biological detectors and
most commonly fluorescent lamps.

GENERAL INTRODUCTION 18
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Fluorescence is used in the life sciences generally as a non-destructive
way of tracking or analysing biological molecules by means of fluorescence.
Some protein or small molecules in cells are naturally fluorescent, this is called
intrinsic or autofluorescence (such as NADH, tryptophan or endogenous
Chlorophyll, Phycoerythrin or green fluorescent protein), alternatively specific
or general proteins, nucleic acids, lipids or small molecules can be labeled with
an extrinsic fluorophore, a fluorescent dye which can be a small molecule,
protein or quantum dot.
F. APPLICATION OF FLUORESCENCE RESONANCE ENERGY
TRANSFER (FRET)
Förster resonance energy transfer (abbreviated FRET), also known as
fluorescence resonance energy transfer, resonance energy transfer (RET) or
electronic energy transfer (EET), is a mechanism describing energy transfer
between two chromophores.
Several techniques exist, often exploiting additional properties, such as
fluorescence resonance energy transfer, where the energy is passed non-
radiatively to a particular neighbouring dye, allowing proximity to be detected;
another is the change in properties, such as intensity, of certain dyes
depending on their environment allowing their use in structural studies [84].
Luminescence chromophores as entities of fluorescence resonance
energy transfer systems are important tools to study supramolecular
interactions with a special emphasis in the real of biomolecules like DNA, RNA,
and proteins [85]. FRET Systems allow monitoring distance-dependent
interactions on the molecular level, and in real-time mode. For instance the
group of Bannwarth [86] already used our fluorescent carbostyrils,
incorporated in peptides, in a Fluorescence-Resonance-Energy Transfer
(FRET)-system for distance determinations.

GENERAL INTRODUCTION 19
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
R
O
MeO
MeO
O
H-His-Ala-Lys-Tyr-His-Lys-Gly-NH2
Ru (II)-bathophenanthroline R = CF3, CN
Figure 6: FRET carbostyril with peptides
The FRET technology is also based on the non-emissive transfer of
energy between a donor and acceptor fluorophore, it decreases with r-6, r being
the distance the donor and the acceptor [87]. Thus, FRET molecules are used
for displaying distances or approaches by changing of fluorescence, the
distance markers between dyes and fluorescence of the molecule depend of the
cation (K+) complexation in the molecule [88]. Another field of application for
FRET is in diagnostics and drug research, where tools like molecular beacons
[89], TaqMan probes [90] or fluorescent protease substrates, for instance, use
the same principle [91].

GENERAL INTRODUCTION 20
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Figure 7: Synthetic Valinomycin-Carbostyril, FRET with rhodamin after K+
Complexation
G. OTHER IMPORTANT FIELDS OF FLUORESCENT DYES
Another important field that has registered development is the
application of fluorescent labels for many compounds with potential biological
activity [92, 93].
Among them, the fluorescent peptides [94-98] have a big number of
applications in biochemistry and biology, mainly in studies of protein
interactions and conformational analysis. Fluorescent markers are also
investigated for in vivo imaging studies, such as in Alzheimer disease [99]. The
most used fluorescent markers for peptides are rhodamine, fluorescein,
coumarin and their derivatives [97, 100].
The fluorescence dyes of the coumarin type which are of interest not only
because of their pharmacological activity [101] but also because of their

GENERAL INTRODUCTION 21
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
applications as laser dyes [102], fluorescent labels [103, 104] (e.g. in biological
applications), emission layers in organic light-emitting diodes (OLED) [105],
and as optical brighteners [106].
Sugars have also been used in the development of fluorescent reagents
because they confer water solubility to organic fluorophores with no significant
change in absorption and fluorescence properties [107].
Molecules containing europium antenna chelates or europium complex
change the fluorescence properties by addition of small molecules like water or
carbon dioxide (CO2) [108].
Structure of Europium complex
N
O
O
CH3
CH3
CF3
N
O
H
Eu-chelate
O-
Structure of europium antenna chelate
Figure 8
Eu
O
N
NN
O
O
HN
O
NH
O
CF3
H3CO
H3CO
HO
OO
OO

GENERAL INTRODUCTION 22
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Figure 9
Luminescence lanthanide (terbium and europium chelates) have many
useful applications, including as alternatives to standard fluorescent dyes
especially when there is significant autofluorescence [109] or as donors in
energy transfer experiments to measure both static and time-varying distance
[110]. These applications arise because of the chelates’ unsual spectral
characteristics, including millisecond lifetime, spiked emission (< 10 nm full
width at half-maximum), large Stoke’s shifts (> 150 nm) and excellent solubility
in aqueous solvents [111].
The new carbostyril chromophores 7 and 82 presenting a second
maximum above 300 nm have the possibility to be excited both in UV and
visible regions that make them to promising candidates for the construction of
FRET molecules, and might be useful as alternatives to established probes for
biological investigations.
N
O
CH3
O
H
CN
CN
7
N
O
CH3
O
H
CN
CN
O
CH3
82
Figure 10. Structures of compounds 7 and 82

REFERENCES FOR INTRODUCTION 23
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES FOR INTRODUCTION

REFERENCES FOR INTRODUCTION 24
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES FOR INTRODUCTION
[1] V. H. Dang, Ph.D. thesis, 2006, p. 1, University of Graz, Austria.
[2] a) E. Ziegler, U. Rossmann, F. Litvan , H. Meier, Monatsh. Chem. 1962,
93, 26; b) E. Ziegler, F. Litvan (Geigy Chemical Corp.), US Patent, 1959,
3 052 678, Chem. Abstr., 1963, 58, 3437e; c) Geigy Chemcal Corp., Brit.
Patent, 1960, 912 289; Chem. Abstr. 1963, 59, 645; d) M. Harfenist, E.
Magnien, J. Org. Chem., 1963, 28, 538; e) M. Harfenist, J. Org. Chem.,
1962, 27, 4326-31.
[3] a) O. S. Wolfbeis, E. Ziegler, A. Knierzinger, H. Wipfler, I. Trummer,
Monatsh. Chem., 1980, 111, 93; b) U. Zirngibl (Sandoz Ltd.), Ger. Offen.
DE 2 142 334, 19 720 302 (1972); DE 71-2 142 334-19710824 (1971).
[4] N. S. Badgujar, M. Pazicky, P. Traar, A. Terec, G. Uray, W. Stadlbauer,
Eur. J. Org. Chem. 2006, 2715–2722.
[5] A. B. Avhale, H. Prokopcová, J. Šefčovičová, W. Steinschifter, A. E.
Täubl, G. Uray, W. Stadlbauer. Eur. J. Org. Chem., 2008, 563-71.
[6] G. Uray, N. S. Badgujar, S. Kováčková, W. Stadlbauer, J. Heterocycl.
Chem., 2008, 45, 165-172.
[7] a) P. Michael, Nat. Prod. Rep., 2007, 24, 223-246; b) J. P. Michael, Nat.
Prod. Rep., 2005, 22, 627; c) J. P. Michael, Nat. Prod. Rep., 2004, 21,
650.

REFERENCES FOR INTRODUCTION 25
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[8] G. M. Coppola, J. heterocycl. Chem. 1985, 22, 1087.
[9] J. Reisch, G. M. K. B. Gunaherath, Monatsh. Chem. 1988, 119, 1169.
[10] P. Bhattacharya, B. K. Chwdhury, Phytochemistry, 1985, 24, 634.
[11] a) Y. Tagawa, T. Kawaoka, Y. Goto, J. Heterocycl. Chem. 1997, 34, 1677;
b) T. S. Wu, F. C. Chang, P. L. Wu, Phytochemistry, 1995, 39, 1453.
[12] a) J. A. Bosson, M. Rasmussen, E. Ritchie, A. V. Robertson, W. C. Taylor,
Australian J. Chem., 1963, 16, 480; b) G. Brader, G. Wurz, H. Greger, O.
Hofer, Liebigs Ann. Chem., 1993, 355-358; c) R. F. C. Brown, J. J.
Hobbs, G. K. Hughes, E. Ritchie, Australian J. Chem., 1954, 7, 348.
[13] a) M. F. Grudon, V. N. Ramachandran, B. M. Sloan, Tetrahedron Lett.,
1981, 22, 3105; b) S. Mitaku, A. L. Skaltsounis, F. Tillequin, M. Koch, J.
G. Chauvier, J. Nat. Prod., 1985, 48, 772; c) A. Ulubelen, Photochemistry,
1985, 24, 372.
[14] J. Reisch, M. Iding, Monatsh. Chem., 1989, 120, 363.
[15] P. R. Kym, M. E. Kort, M. J. Coghlan, J. L. Moore, R. Tang, J. D.
Ratajczyk, D. P. Larson, S. W. Elmore, J. K Pratt, M. A. Stashko, H. D.
Falls, C. W. Lin, M. Nakane, L. Miller, C. M. Tyree, J. N. Miner, P. B.
Jacobson, D. M. Wilcox, P. Nguyen, B. C. Lane, J. Med. Chem. 2003, 46,
1016-1030;
[16] S. W. Elmore, M. J. Coghlan, D. D. Anderson, J. K. Pratt, B. E. Green, A.
X. Wang, M. A. Stashko, C. W. Lin, C. M. Tyree, J. N. Miner, P. B.
Jacobson, D. M. Wilcox, B. C. Lane, J. Med. Chem. 2001, 44, 4481-
4491.

REFERENCES FOR INTRODUCTION 26
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[17] a) L. Savini, L. Chiasserini, C. Pellerano, W. Filippelli, G. Falcone,
Farmaco 2001, 56, 939-945; b) S. Falk, R. Goggel, U. Heydasch, F.
Brasch, K. M. Muller, A. Wendel, S. Uhlig, Am. J. Resp. Crit. Care 1999,
160, 1734-1742.
[18] a) Y. L. Chen, K. C. Fang, K. J.-Y. Sheu, S. L. Hsu, C. J. Tzeng, Med.
Chem. 2001, 44, 2374–2377; b) G. Roma, M. D. Braccio, G. Grossi, F.
Mattioli, M. Ghia, Eur. J. Med. Chem. 2000, 35, 1021–1035.
[19] a) M. P. Maguire, K. R. Sheets, K. McVety, A. P. Spada, A. Zilberstein, J.
Med. Chem. 1994, 37, 2129–2137; b) O. Bilker, V. Lindo, M. Panico, A.
E. Etiene, T. Paxton, A. Dell, M. Rogers, R. E. Sinden, H. R. Morris,
Nature 1998, 289–292.
[20] a) P. Narender, U. Srinivas, M. Ravinder, B. A. Rao, C. Ramesh, K.
Harakishore, B. Gangadasu, U. S. N. Murthy, V. Rao, V. J. Bioorg. Med.
Chem. 2006, 14, 4600-4609.
[21] a) B. S. Holla, K. N. Poojary, B. Poojary, K. S. Bhat, N. S. Kumari, Indian
J. Chem. B 2005, 44, 2114-2119; b) A. K. Sadana, Y. Mirza, K. R. Aneja,
O. Prakash, Eur. J. Med. Chem. 2003, 38, 533-536.
[22] a) R. N. Kumar, T. Suresh, P. S. Mohan, Indian J. Chem. B 2003, 42,
688-689; b) M. Kidwai, K. R. Bhushan, P. Sapra, R. K. Saxena, R. Gupta,
R. Bioorg. Med. Chem. 2000, 8, 69-72.
[23) a) M. Fujita, K. Chiba, Y. Tominaga, K. Hino, Chem. Pharm. Bull. 1998,
46, 787-796; b) M. G. Kayirere, A. Mahamoud, J. Chevalier, J. C. Soyfer,
A. Cremieux, J. Barbe, Eur. J. Med. Chem. 1998, 33, 55-63.
[24] a) P. Narender, U. Srinivas, M. Ravinder, B. A. Rao, C. Ramesh, K.
Harakishore, B. Gangadasu, U. S. N. Murthy, V. J. Rao, Bioorg. Med.

REFERENCES FOR INTRODUCTION 27
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Chem. 2006, 14, 4600; b) B. S. Holla, K. N. Poojary, B. Poojary, K. S.
Bhat, N. S. Kumari, Indian J. Chem. B 2005, 44, 2114;
[25] a) A. K. Sadana, Y. Mirza, K. R. Aneja, O. Prakash, Eur. J. Med. Chem.
2003, 38, 533; b) R. N. Kumar, T. Suresh, P. S. Mohan, Indian J. Chem.
B 2003, 42, 688.
[26] a) A. G. Tempone, A. C. M. P. da Silva, C. A. Brandt, F. S. Martinez, S. E.
T. Borborema, M. A. B. da Silveira, H. F. de Andrade, Antimicrob. Agents
Chemother. 2005, 49, 1076-1080; b) X. Franck, A. Fournet, E. Prina, R.
Mahieux, R. Hocquemiller, B. Figadere, Bioorg. Med. Chem. Lett. 2004,
14, 3635-3638.
[27] N. P. Sahu, C. Pal, N. B. Mandal, S. Banerjee, M. Raha, A. P. Kundu, A.
Basu, M. Ghosh, K. Roy, S. Bandyopadhyay, Bioorg. Med. Chem. 2002,
10, 1687-1694;
[28] a) E. Chiari, A. B. Oliveira, M. A. F. Prado, R. J. Alves, L. M. C. Galvão, F.
G. Araujo, Antimicrob. Agents Chemother. 1996, 40, 613-615, b) A.
Fournet, A. A. Barrios, V. Munoz, R. Hocquemiller, A. Cave, J. Bruneton,
Antimicrob. Agents Chemother. 1993, 37, 859-863.
[29] a) A. A.Joshi, C. L. Viswanathan, Bioorg. Med. Chem. Lett. 2006, 16,
2613-2617; b) A. A. Joshi, S. S. Narkhede, C. L. Viswanathan, Bioorg.
Med. Chem. Lett. 2005, 15, 73-76; c) C. Portela, C. M. M. Alfonso, M. M.
M. Pinta, M. J. Ramos, Bioorg. Med. Chem. 2004, 12, 3313.
[30] a) G. S. Dow, M. L. Koenig, L. Wolf, L. Gerena, M. Lopez-Sanchez, T. H.
Hudson, A. K. Bhattacharjee, Antimicrob. Agents Chemother. 2004, 48,
2624-2632.

REFERENCES FOR INTRODUCTION 28
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[31] a) J. Ziegler, R. Linck, D. W. Wright, Curr. Med. Chem. 2001, 8, 171-189;
b) O. Billker, V. Lindo, M. Panico, A. E. Etienne, T. Paxton, A. Dell, M.
Rogers, R. E. Sinden, H. R. Morris, Nature 1998, 392, 289-292.
[32] a) F. M. D. Ismail, M. J. Dascombe, P. Carr, S. A. M. Merette, P.
Rouault, J. Pharm. Pharmacol. 1998, 50, 483-492; b) M. L. Go, T. L.
Ngiam, A. L. C. Tan, K. Kuaha, P. Wilairat, Eur. J. Pharm. Sci. 1998, 6,
19-26.
[33] a) H. Heitsch, Curr. Med. Chem. 2002, 9, 913-928; b) C. Buccellati, F.
Fumagalli, S. Viappiani, G. Folco, G. Farmaco 2002, 57, 235-242; c) D.
Dube´, M. Blouin, C. Brideau, C. C.Chan, S. Desmarais, D. Ethier, J. P.
Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P.
Tagari, R. N. Young, Bioorg. Med. Chem. Lett. 1998, 8, 1255-1260.
[34] a) A. Nayyar, A. Malde, R. Jain, E. Coutinho, Bioorg. Med. Chem. 2006,
14, 847; b) A. Nayyar, R. Jain, Curr. Med. Chem. 2005, 12, 1873; c) V.
Monga, A. Nayyar, B. Vaitilingam, P. B. Palde, P. B. Jhamb, S. S. Kaur,
P. P. Singh, R. Jain, Bioorg. Med. Chem. 2004, 12, 6465.
[35] a) S. Vangapamdu, M. Jain, R. Jain, S. Kaur, P. P. Singh, Bioorg. Med.
Chem. 2004, 12, 2501-2508; b) R. Jain, B. Vaitilingam, A. Nayyar, P. B.
Palde, Bioorg. Med. Chem. Lett. 2003, 1051-1054.
[36] (a) P. Camps, E. Gómez, D. Muňoz-Torrero, A. Badia, N. M. Vivas, X.
Barril, M. Orozco, F. J. Luque, J. Med. Chem. 2001, 44, 4733-4736; b) P.
Camps, R. El Achab, J. Morral, D. Muňoz-Torrero, A. Badia, J. E. Baňos,
N. M. Vivas, X. Barril, M. Orozco, F. J. Luque, J. Med. Chem. 2000, 43,
4657-4666; c) T. Suzuki, T. Usui, M. Oka, T. Suzuki, T. Kataoka, Chem.
Pharm. Bull. 1998, 46, 1265-1273.
[37] a) N. Muruganantham, R. Sivakumar, N. Anbalagan, V. Gunasekaran, J.
T. Leonard, Biol. Pharm. Bull. 2004, 27, 1683-1687, b) R. H. Bradbury,

REFERENCES FOR INTRODUCTION 29
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
C. P. Allott, M. Dennis, J. A Girdwood, P. W. Kenny, J. S. Major, A. A.
Oldham, A. H. Ratcliffe, J. E. Rivett, D. A. Roberts, P. J. Robins, J. Med.
Chem. 1993, 36, 1245-1254.
[38] a) S. Rossiter, J. M. Peron, P. J. Whitfield, K. Jones, Bioorg. Med. Chem.
Lett. 2005, 15, 4806-4808; b) B. Kalluraya, S. Sreenivasa, Farmaco
1998, 53, 399-404.
[39] a) X. Franck, A. Fournet, E. Prina, R. Mahieux, R. Hocquemiller, B.
Figadere, Bioorg. Med. Chem. Lett. 2004, 14, 3635; b) C. Bénard, F.
Zouhiri, M. Normand-Bayle, M. Danet, D. Desmaële, H. Leh, J. F.
Mouscadet, G. Mbemba, C. M. Thomas, S. Bonnenfant, M. Le Bret, J.
d’Angelo, Bioorg. Med. Chem. Lett. 2004, 14, 2473.
[40] a) M. A. Fakhfakh, A. Fournet, E. Prina, J. F. Mouscadet, X. Franck, R.
Hocquemiller, B. Figadere, Bioorg. Med. Chem. 2003, 11, 5013-5023.
[41] a) A. Fournet, R. Mahieux, M. A. Fakhfakh, X. Franck, R. Hocquemiller,
B. Figadere, Bioorg. Med. Chem. Lett. 2003, 13, 891-894; b) F. Zouhiri,
D. Desmaele, J. d'Angelo, M. Ourevitch, J. F. Mouscadet, H. Leh, M. Le
Bret, Tetrahedron Lett. 2001, 42, 8189-8192.
[42] a) A. Tsotinis, M. Vlachou, S. Zouroudis, A. Jeney, F. Timar, D. E.
Thurston, C. Roussakis, Lett. Drug Des. Discov. 2005, 2, 189-192; b) A.
R. Martirosyan, R. Rahim-Bata, A. B. Freeman, C. D. Clarke, R. L.
Howard, J. S. Strobl, Biochem. Pharmacol. 2004, 68, 1729-1738.
[43] a) A. Perzyna, F. Klupsch, R. Houssin, N. Pommery, A. Lemoine, J. P.
Hénichart, Bioorg. Med. Chem. Lett. 2004, 14, 2363-2365; b) A. H. Abadi,
R. Brun, Arzneimittel-Forsch. 2003, 53, 655-663; c) J. Charris, P.
Martinez, J. Dominguez, S. Lopez, J. Angel, G. Espinoza, Heterocycl.
Commun. 2003, 9, 251-256.

REFERENCES FOR INTRODUCTION 30
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[44] a) C. Lamazzi, S. Leonce, B. Pfeiffer, P. Renard, G. Guillaumet, C. W.
Rees, T. Besson, Bioorg. Med. Chem. Lett. 2000, 10, 2183-2185; b) J.
Osiadacz, L. Kaczmarek, A. Opolski, J. Wietrzyk, E. Marcinkowska, K.
Biernacka, C. Radzikowski, M. Jon, W. Peczynska-Czoch, Anticancer Res.
1999, 19, 3333-3342.
[45] a) L. Kaczmarek, W. Peczynska-Czoch, J. Osiadacz, M. Mordarski, W. A.
Sokalski, J. Boratynski, E. Marcinkowska, H. Glazman-Kusnierczyk, C.
Radzikowski, Bioorg. Med. Chem. Lett. 1999, 7, 2457-2464; b) W.
Peczynskaczoch, F. Pognan, L. Kaczmarek, J. Boratynski, J. Med. Chem.
1994, 37, 3503-3510.
[46] R. Kimmel, S. Kafka, J. Košmrlj, Carbohydrate Research, 2010, 345,
768-779.
[47] I. Ishiguro, T. Ikeno, H. Matsubara, Yakugaku Zasshi, 1974, 94, 116–
123; Chem. Abstr. 1974, 80, 118417.
[48] a) K. Tateishi, H. Shibata, Y. Matsushima, T. Iijima, Agric. Biol. Chem.
1987, 51, 3445–3447; b) K. Tateishi, Y. Matsushima, H. Shibata, Agric.
Biol. Chem. 1989 53, 2545–2551; c) H. Shibata, T. Nanbu, K. Tateishi, S.
Shimizu, Agric. Biol. Chem. 1989, 53, 849–850.
[49] a) Y.–F. Su, Y. Luo, C.-Y. Guo, D.-A. Guo, J. Asian Nat. Prod. Res. 2004,
6, 223–227; b) J. P. Michael, Nat. Prod. Rep. 2007, 24, 223–246; c) V. M.
Dembitsky, Lipids 2005, 40, 1081–1105.
[50] a) K. A. Rasulova, I. A. Bessonova, M. R. Yagudaev, S. Y. Yunusov, Khim.
Prirod. Soed. 1987, 6, 876–879; b) V. I. Akhmedzhanova, K. A. Rasulova,
I. A. Bessonova, A. S. Shashkov, N. D. Abdullaev, L. Angenot, Chem. Nat.
Compd. 2005, 41, 60–64.

REFERENCES FOR INTRODUCTION 31
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[51] J. N. Smith, Biochem. J. 1953, 55, 156–160; b) J. N. Smith, R. T.
Williams, Biochem. J. 1954, 56, 325–329; c) J. N. Smith, R. T. Williams,
Biochem. J. 1955, 60, 284–290.
[52] a) C. Baglioni, P. Fasella, C. Turano, N. Siliprandi, Biochem. J. 1960, 74,
521–525, b) L. T. Burka, J. M. Sanders, H. B. Matthews, Xenobiotica
1996, 26, 597–611.
[53] a) H. Suzuki, H. Igarashi, I. M. Igarashi, I. Tanaka, N. Suzuki, Jpn. Kokai
Tokyo Koho JP 2006056872, 2006; Chem. Abstr. 2006, 144, 205733; b)
H. Suzuki, N. S. M. Aly, Y. Wataya, H.-S. Kim, I. Tamai, M. Kita, D.
Uemura, Chem. Pharm. Bull. 2007, 55, 821–824.
[54] T. Nógrádi, L. Vargha, G. Ivánovics, I. Koczka, Acta Chim. Acad. Sci.
Hung. 1955, 6, 287–293.
[55] H. Takagaki, S. Nakanishi N. Kimura, S. Yamaguchi, Y. Aoki, Eur. Pat.
Appl. EP 1999, 933378, Chem. Abstr. 1999, 131, 116454. 18.
Hirayama, B. A.; Diez-Sampedro, A.; Wright, E.
[56] a) E. Ziegler, H. Junek, Monatsh. Chem. 1956, 87, 503; b) E. Ziegler, R.
Wolf, T. Kappe, Monatsh. Chem. 1965, 96, 418.
[57] C. M. Mehta, G. H. Patel, J. Ind. Res.(India), 1959, 18B, 391.
[58] T. Kappe, A. S. Karem, W. Stadlbauer, J. Heterocycl. Chem., 1988, 25,
857-862.
[59] W.-T. Gao, W.-D. Hou, M.-R. Zheng, L.-J. Tang, Synth. Commun. 2010,
40, 732-738.
[60] L. H. Reyrson, K. Kobe, Chem. Rev., 1930, 7, 479.

REFERENCES FOR INTRODUCTION 32
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[61] A. Omori, N. Sonoda, S. Tsutsumi, Bull. Chem. Soc. Jpn., 1970, 43,
1135-1138.
[62] G. M. Coppola, J. Heterocycl. Chem., 1985, 22, 1087.
[63] G. M. Coppola, G. E. Hardtmann, J. Heterocyl. Chem., 1979, 16, 1605.
[64] a) L. A. Mitscher, H. E. Gracey, G. W. Clark, T Suziki, J. Med. Chem.,
1978, 21, 485; b) L. A. Mitscher, D. L. Flynn, H. E. Gracey, S. D. Drake,
J. Med. Chem., 1979, 22, 1354.
[65] M. R. Bell, A. W. Zalay, R. Oesterlin, P. Shane, G. O. Potts, J. Med.
Chem., 1970, 13, 664.
[66] D. R. Shridar, C. V. Reddy Sastry, A. K. Mehrotra, c. Seshagiri Rao, V.
Taneja, India J. Chem., 1979, 17b, 488.
[67] K. Tsuji, G. W. Spears, K. Nakamura, T. Tojo, N. Seki, A. Sugiyama, M.
Matsuo, Bioorg. Med. Chem. Lett., 2002, 12, 85-88.
[68] G. M. Coppola, G. E. Hardtmann, O. R. Pfister, J. Org. Chem., 1976, 41,
825-831.
[69] Farbenfabrik Hoechst, D. R. P. 102, 894; Chem. Zblt., 1899, I, 462.
[70] Badische Anilin- und Sodafabrik, D. R. P. 117, 167; Chem. Zblt., 1901, I,
236.
[71] a) Z. H. Skraup, Monatsh. Chem., 1880, 1, 316; b) R. H. F. Manske,
Chem. Rev., 1942, 30, 113.
[72] H. T. Clarke, A. W. Davis, Org. Synth. 1941, 1, 478.

REFERENCES FOR INTRODUCTION 33
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[73] ]I. L. Finar, Org. Chem., 1973, 1, 857.
[74] a) O. Doebner, O.; W. v. Miller, Ber. Dtsch. Chem. Ges 1881, 14, 2812.,
b) O. Doebner, O.; W. v. Miller, Ber. Dtsch. Chem. Ges 1883, 16, 1664 & 2464;
c) O. Doebner, O.; W. v. Miller, Ber. Dtsch. Chem. Ges.,1884, 17, 1712; d F. W.
Bergström, Chem. Rev. 1944, 35, 153. (Review).
[75] a) A. Combes, Bull. Chim. Soc. France 1888, 49, 89; b) F. W. Bergstrom
Chem. Rev., 1944, 35, 156 (Review).
[76] a) C.-S. Jia, Z. Zhang, S.-J. Tu, G.-W. Wang, Org. Biomol. Chem., 2006,
4, 104-110; b) J. Wu, H.-G. Xia, K. Gao, Org. Biomol. Chem., 2006, 4,
126-129; c) R. Martínez, D. J. Ramón, M. Yus, J. Org. Chem., 2008, 73,
9778-9780; d) J. S. Yadav, B. V. S. Reddy, P. Sreedhar, R. S. Rao, K.
Nagaiah, Synthesis, 2004, 2381-2385.
[77] a) R. Varala, R. Enugala, S. R. Adapa, Synthesis, 2006, 3825-3830; b) S.
S. Palimkar, S. A. Siddiqui, T. Daniel, R. J. Lahoti, K. V. Srinivasan, J.
Org. Chem., 2003, 68, 9371-9378; c) P. G. Dormer, K. K. Eng, R. N. Farr,
G. H. Humphrey, J. C. McWilliams, P. J. Reider, J. W. Sager, R. P.
Volante, J. Org. Chem., 2003, 68, 467-477.
[78] W. Pfitzinger, W. J. Prakt. Chem. 1886, 33, 100; b) W. Pfitzinger, J. Prakt.
Chem. 1888, 38, 582.
[79] O. A. Ponomarev, E. R. Vasina, V. G. Mitina, A. A. Sukhorukov, Zh. Fiz.
Khim., 1990, 64, 974.

REFERENCES FOR INTRODUCTION 34
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[80] a) J. Chen, P. R. Selvin, J. Photobiol. 2000, 135A, 27-32; b) C. Marzano,
A. Chilin, A. Guitto, F. Baccichetti, F. Carlassare, F. Bordin, Farmaco
2000, 55, 650-658; c) G. Saroja, N. B. Samanta, Chem. Phys. Lett. 1996,
249, 392-398; d) M. Li, P. R. Selvin, J. Am. Chem. Soc. 1995, 117, 8132-
8138; e) R. Nakagaki, N. Kitamura, I. Aoyama, H. Ohtsubo, J. Photochem.
Photobiol. 1994, A80, 113-119.
[81] a) O. A Ponomarev, Yu F. Pedash, O. V. Prezhdo, Zh. Fiz. Khim. 1991, 65,
1846-1850; b) O. A. Ponomarev, E. R. Vasina, S. N. Zarmolenko, V. G.
Mitina, N. S. Pivnenko, Zhurn. Obshch. Khim. 1990, 60, 1161-1170; J
Gen. Chem. USSR. (Transl. Plenum Pub. Corp.) 1990, 60, 1035-1042.
[82] A. Lobnik, N. Majcen, K. Niederreiter, G. Uray, Sensors and Actuatrors B
2001, 74, 200-2056.
[83] a) W. M. F. Fabian, K. S. Niederreiter, G. Uray, W. Stadlbauer J. Mol.
Structure, 1999, 477, 209-220; b) G. A. Strohmeier, W. M. F. Fabian, G.
Uray, Helv. Chim. Acta, 2004, 87, 215-226; c) G. Uray, K. H. Niederreiter,
F. Belaj, W. M. F. Fabian, Helv. Chim. Acta, 1999, 82, 1408-1417.
[84] a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy 3 rd Ed. 2006,
New York: Springer, XXVI, 954 p;
b) http://www.invitrogen.com/site/us/en/home/References/Molecular-
Probes-The-Handbook/Introduction-to-Fluorescence-Techniques.html.
[85] K. E. Sapsford, L. Berti, I. L. Medintz, Angew. Chem., Int. Ed. 2006, 45,
4562.
[86] R. A. Kramer, R. Flehr, M. Lay, M. U. Kumbe, W. Bannwarth, Helv. Chim.
Acta., 2009, 92, 1933-1943.
[87] T. Förster, Ann., Phys., 1948, 2, 55.

REFERENCES FOR INTRODUCTION 35
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[88] G. Uray, Sallegger, unpublished results.
[89] S. Tyagi, F. R. Kramer, Nat. Biotechnol., 1996, 14, 303-308.
[90] P. M. Holland, R. D. Abramson, R. Waston, D. H. Gelfand, Proc. Nat.
Acad. Sci. U. S. A., 1991, 88, 7276-7280.
[91[ E. K. Kainmüller, E. P. Ollé, W. Bannwarth, Chem. Commun., 2005,
5459-5461.
[92] A. Cuppoletti, C. Younjin, P. Jin-Seong, C. Strässler, E. T. Kool,
Bioconjugate Chem., 2005, 16, 528-534.
[93] B. Wetlz, M. Gruber, B. Oswald, A. Dürkop, B. Weidgans, M. Probst, O.
Wolfbeis, J. Chromatogr. B 2003, 793, 83-92.
[94] S. J. Danishefsky, J. R. Allen, Angew. Chem., Int. Ed. 2000, 39, 836-863.
[95] J. Fernandez-Carteado, M. J. Kogan, S. Castel, E. Giralt, Angew. Chem.,
Int. Ed. 2004, 43, 1811-1814.
[96] R. David, Z. Machova, A. G. Beck-Sickinger, Biol. Chem., 2003, 384,
1619-1639.
[97] M.-P. Faure, P. Gaudreau, I. Shaw, N. R. Cashman, A. Beaudet, J.
Histochem. Cytochem., 1994, 42, 755-763.
[98] S. Fuchs, H. Otto, S. Jehle, P. Henklein, A. D. Schüter, Chem. Commun.
2005, 1830-1832.
[99] V. J. Mayo, J. Prabhakaran, J. J. Mann, J. S. D. Kumar, Tetrahedron
Lett. 2003, 44, 8535-8537.

REFERENCES FOR INTRODUCTION 36
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[100] F. A. Bennett, D. J. Barlow, A. N. O. Dodoo, R. C. Hider, A. B. Lansley,
M. J. Lawrence, C. Marriott, S. S. Bansal, Anal. Biochem., 1999, 270,
15-23.
[101] a) K. R. Romines, J. K. Morris, W. J. Howe, P. K. Tomich, M.-M. Horng,
K.-T. Chong, R. R. Hinshaw, D. J. Anderson, J. W. Strohbach, S.-R.
Turner, S. A. Miszak, J. Med. Chem. 1996, 39, 4125-4130; b)
Coumarins–Biology, Applications and Mode of Action (Eds.: R.
O×Kennedy, R. D. Thornes), Wiley, Chichester, 1997.
[102] a) R. Raue in Ullmannns Encyclopedia of Industrial Chemistry, Vol. A15,
5thed. (Eds.: B. Elvers, S. Hawkins, G. Schulz), VCH, Weinheim, 1990,
pp. 155-157; b) R. S. Koefod, K. R. Mann, Inorg. Chem. 1989, 28, 2285-
2290.
[103] a) P. D. Edwards, R. C. Mauger, K. M. Cottrell, F. X. Morris, K. K. Pine,
M. A. Sylvester, C. W. Scott, S. T. Furlong, Bioorg. Med. Chem. Lett.
2000, 10, 2291-2294; b) M. Adamczyk, M. Cornwell, J. Huff, S. Rege, T.
V. S. Rao, Bioorg. Med. Chem. Lett. 1997, 7, 1985-1988; c) C. A. M.
Seidel, A. Schulz, M. H. M. Sauer, J. Phys. Chem. 1996, 100, 5541-
5553.
[104] a) A. Adronov, S. L. Gilat, J. M. Fre¬ chet, K. Ohta, F. V. R. Neuwahl, G.
R. Fleming, J. Am. Chem. Soc. 2000, 122, 1175-1185; b) K. H.
Shaughnessy, P. Kim, J. F. Hartwig, J. Am. Chem. Soc. 1999, 121, 2123-
2132.
[105] a) J. Kido, Y. Lizumi, Appl. Phys. Lett. 1998, 73, 2721-2723; b) A. Niko,
S. Tasch, F.Meghdadi, C. Brandstätter,G. Leising, J. Appl. Phys. 1997,
82, 4177-4182; c) S. Tasch, C. Brandstätter, F. Meghdadi, G. Leising, G.
Froyer, L. Athouel, Adv. Mater. 1997, 9, 33-36.

REFERENCES FOR INTRODUCTION 37
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[106] A. E. Siegrist, H. Hefti, H. R. Meyer, E. Schmidt, Rev. Prog. Coloration
1987, 17, 39-55.
[107] M. J. V. Reddington, J. Chem. Soc., Perkin Trans. 1, 1998, 143-147.
[108] a) J. Hynes, T. C. O'Riordan, A. V. Zhdanov, G. Uray, Y. Will, D. B.
Papkovsky, Analyt. Biochemistry, 2009, 390, 21-28, b) Ribitsch; Uray,
unpublished results.
[109] a) L. Seveus, M. Vaisala, S. Syrjanen, M. Sandberg, A. Kuusito, R. Harju,
J. Salo, I. Hmmila, H. Kojola, E. Soini, Cytometry, 1992, 13, 329-335; b)
L. Seveus, M. Vaisala, I. Hemmilä, H. Kojola, E. Soini, Microsc. Res.
Technol. 1994, 28, 149-153.
[110] P. R. Selvin, T. M. Rana, J. E. Hearst, J. Am. Chem. Soc., 1994, 116,
6029-6032.
[111] A. Lobnik, N. Majcen, K. Niederreiter, G. Uray, Sens. and Actuators B,
2001, 74, 200-206.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 38
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CHAPTER 1
SYNTHESIS AND REACTIVITY OF 4-HYDROXY-
6- AND 7-METHOXYQUINOLIN-2(1H)-ONES

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 39
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-
6- AND 7-METHOXYQUINOLIN-2(1H)-ONES
1.1. INTRODUCTION
4-Hydroxyquinolin-2(1H)-one and 4-hydroxycoumarin derivatives are
naturally occurring biologically active compounds [1, 2]. The key step in our
overall synthetic scheme is methoxyquinolin-2(1H)-ones of type 3 having the
hydroxide group in position 4. These heterocycles are useful intermediates for
many industrial products such as dyes [3, 4], herbicides [5, 6] and anticancer
agents [7].
On the other hand the 4-hydroxy group of the 2-quinolone moiety can
easily be substituted by various nucleophiles and converted to reactive
derivatives such as 4-chloroquinolines 6, or 4-azido-2-quinolones [8], which
are suitable precursors for further syntheses. For instance, exchange of the 4-
hydroxy group in 4-hydroxyquinolin-2(1H)-one against the arylthio moiety
leads to compounds with potent HIV-1 reverse transcriptase inhibitory
properties [9]. 4-Hydroxyquinolone derivatives of type 3 are important
biosynthetic [10] and synthetic [11] precursors of quinoline alkaloids.
1.2. RESULTS AND DISCUSSION
1.2.1 . Cyclocondensation of p-anisidin 1 and malonic acid 2 to
4-hydroxy-6-methoxyquinolin-2(1H)-one 3 as precursor.
The synthesis of 4-hydroxyquinolones was first described in 1923
starting from quinoline by heating with potassium hydroxyde to 225 °C under
anhydrous conditions [12].

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 40
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Recently, Gao et al. [13] reported a one-pot formation of 4-hydroxy-6-
methoxyquinolin-2(1H)-one from Meldrum’s acid and p-anisidin using
phosphoric anhydride/methanesulfonic acid as cyclization agent. However,
this method gave a moderate yield of 61 %. Recently, it could be shown [14,
15] that 3-acyl-4-hydroxy-2-quinolones could be readily converted to 3-acyl-4-
azido-2-quinolones and further cyclized to isoxazoles.
The presented synthetic procedure using phosphoryl chloride as both
reagent and solvent is a convenient, simple and fast alternative for
synthesizing 4-hydroxyquinolin-2(1H)-one derivatives.
NH2
O
CH3
COOH
COOH
N
OH
O
H
O
CH3
N
N
O
O H
H
OCH
3
OCH
3
POCl3
+
1 2
95 °C, 90 min.
+
73 % 19 %
4
Scheme 1
Malonic acid 2 is not reactive enough to react with dinucleophile 1 [16].
However, conversion of malonic acid in situ with phosphoryl chloride [17a]
gave the corresponding reactive acid chloride, which was treated with p-
anisidine similar as described in ref. [17b] to give 3 in 63 % yield besides the
by-product 4 in 32 %.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 41
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.1.1. Optimization of the reaction
In order to optimize the reaction, several methods were attempted:
increasing of the equivalents of p-anisidine or malonic acid, longer reaction
time, direct dissolution of the raw product in aqueous sodium hydroxide; in all
cases the overall yield of the compound 3 was between 41-63 %. It was found
that, large excess of phosphoryl chloride should be avoid in order to prevent
subsequent reaction between phosphoryl chloride with the 4-hydroxy group
and the 2-oxo group of the formed 4-hydroxyquinolone 3 [18].
Best results were obtained at temperatures in the range of 90-95 °C for
1.5 hour and following reaction work-up with ice/water. Dissolving the crude
product into large excess of sodium hydroxide (1 M, 1000 mL) allowed the
isolation of product 3 in 73 % yield as well as 19 % of the dianilide by-product
4.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 42
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.1.2. Proposed mechanism of the formation of 4-hydroxy-6-
methoxyquinolin-2(1H)-one 3
Scheme 2

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 43
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.1.3. Proposed mechanism of the formation of dianilide 4
Scheme 3
1.2.1.4. Structural elucidation : IR and 1H-NMR spectroscopy
study of compounds 3 and 4
Compounds 3 and 4 were characterized by infrared and 1H-NMR spectra.
The infrared spectrum of compound 3 showed a medium band in the region of
3434-3290 cm-1, ascribable to the vibration of NH and a carbonyl band at 1663
cm-1 confirming the presence the lactam function. The 1H-NMR spectrum of 3
revealed a singlet at 5.79 ppm for C-3 proton. It should be noted that the
corresponding NH group in amines, amides and/or lactams also exhibit
hydrogen bonding NMR shifts although to a lesser degree. Furthermore, OH
and NH groups can undergo rapid proton exchange with each signal at an
average chemical shift and often only one of them could be observed. The
sharp singlet at 11.08 ppm was assigned to NH.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 44
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Infrared spectrum of dianilide 4 exhibited the absorption bands at 1655
cm-1 and 1620 cm-1 ascribable to carbonyls groups and a broad NH absorption
at 3435 cm-1. The 1H-NMR spectrum of 4 showed two doublets at 6.88 ppm
and 7.45 ppm typical of the para-substituted patterns of benzene ring, a
singlet at 3.49 ppm was due to the methylene proton.
1.2.2. Chlorination of 4-hydroxyquinolone 3 to 2,4-
dichloroquinoline 5 and 4-chloroquinolone 6
Survey of the literature showed that the quinoline moiety is an important
class of various pharmacologically active compounds and is present in many
biologically active natural products, particularly in alkaloids [20a-c] as outlined
in the general introduction. The quinoline scaffold display an important role as
intermediate in the synthesis of fluorescence compounds such as 2-
aminoquinolines and 2-amino-arylpeptides quinoline [21].
In order to prepare 4-chloro-6-methoxyquinolone 6, 4-hydroxy-6-
methoxyquinolone 3 was refluxed in excess amount of phosphoryl chloride for
8 hours to provide 83 % yield of 2,4-dichloroquinoline 5. The latter was then
regioselectively converted into 4-chloro-2-oxo-quinoline 6 in excellent yield of
95 % by reaction with 70 % methanesulfonic acid in ethanol; selective
monochlorination in position 4 of the N-1 unsubstituted quinolin-2(1H)-one 3
was not possible.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 45
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
OH
O
H
O
CH3
POCl3
N
Cl
Cl
O
CH3
N
Cl
O
H
O
CH3CH
3SO
3H / EtOH
73 %
3
reflux, 8 h83 %
reflux, 28 h
95 %
5 6
Scheme 4
1.2.2.1. Study of Infrared and 1H-NMR Spectra of 5 and 6
Comparison of spectral data of compound 5 and 6 showed that the
infrared spectrum of 5 did not exhibit the carbonyl band, which was present in
the compound 6 at 1623 cm-1. In the 1H-NMR spectrum of 5, the signal of NH
proton was not observed, which is present in the compound 6. However, the
signal of C-3 proton appeared at 7.92 ppm for the compound 5 due to the
strong electronegativity of an additional chlorine in position 2, and in
compound 6 this signal was shifted up field at 6.82 ppm.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 46
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.2.2. 13C-NMR spectrum of 4-chloro-6-methoxyquinolin-
2(1H)-one 6
The 13C-NMR spectrum of compound 6 was simple. The methoxy carbon
appeared at 56.0 ppm.
The carbon of the lactam function appeared at 160.4 ppm, those of
phenyl nuclei undergo different electronic effects of pyridine ring. Thus,
carbons 9-C and 10-C recognizable as carbon 2-C of lactam function by
their small size appeared at 133,6 ppm and 1178.2 ppm respectively.
The complete assignment of 4-chloro-6-methoxyquinolini-2(1H)-one 6 is
given below.
N
O
Cl
O
H
CH3
56.0
121.2
117.8133.6
160.4
106.3
143.8118.2121.7
155.1
Figure 1: 13C-NMR values of compound 6
The mass spectral and elemental analysis were in accordance to the
corresponding structure.
1.2.3. Introduction of cyano substituents into 4-chloro-6-
methoxyquinolin-2(1H)-one 6
The synthesis of 2-chloroquinolin-4-carbonitrile and 4-chloroquinolin-2-
carbonitrile is described in the literature, however not via direct chloro
exchange [22]. So far, it is still a challenge to substitute a halide group of

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 47
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
benzene, pyridones, quinolones moieties or related compounds by nucleophilic
substitution against nitrile. Whereas C-C coupling in aromatic chemistry
follows in most cases the mechanism of electrophilic substitution, in
heterocyclic chemistry the nucleophilic substitution is preferred for the large
-deficient heterocycles such as pyridine and its related heterocycles
[23].
N O
Cl
H
O
CH3
N O
CN
H
CNO
CH3
6
KCN/ p-tol.sulfinate Na, DMF
140 °C, 43 h
64 %
6 7
(Pathway A)
Scheme 5
Survey of the literature shows that the introduction of a cyano group into
the similar 4-chloro-N-substituted quinolones of type 6 using cyanide anion,
cyanide or crown ether failed [23]. In early experiments [24], attempts to
introduce the cyano by using Rosenmund-Braun aromatic cyanation with
copper (I) cyanide in high-boiling solvents failed.
The use of p-toluenesulfinate as catalyst [25] or via heavy metal cyanides
such as zinc or copper cyanides [26] is described for the cyanide introduction
into chloro heterocycles. The reaction of similar compounds of type 6 with zinc
cyanide gave the corresponding 4-cyanoquinolones which were however
cumbersome to purify [23].
The introduction of the cyano group in position 3 and/or 4 of 4-
chloroquinolin-2(1H)-one 6 was carried out by reaction with dry potassium
cyanide using dry sodium p-toluenesulfinate in dry dimethylformamide as
recently described in our group [18] and [19], in continuation of early work on
the synthesis of highly fluorescent carbostyrils.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 48
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
We wanted to prepare 4-cyanoquinolone from one equivalent of 4-
chloroquinolone 6 with 2.1 equivalents of dry potassium cyanide and 2.5
equivalents of dry sodium p-toluenesulfinate as a catalyst in dry
dimethylformamide as solvent. It should be noted that the reaction requires
strict anhydrous conditions. The mixture was heated to 110 °C, after 25 hours
reaction time, we obtained a mixture of two UV-active compounds, but very
difficult to separate and purify. This means, at 110 °C during 25 hours the
reaction involves an intermediate sulfinate and a cyano compound in the same
time. Thus, when a longer reaction time (43 hours) at 140 °C was used, a
single yellow-green fluorescence cyano carbostyril appeared, which could be
isolated. The structure was identified by spectroscopic and analytical methods.
According to the spectral data, infrared spectrum of compound 7 showed
the signal of cyano group at 2228 cm-1 and another signal at 1650 cm-1
ascribable to lactam carbonyl. Surprisingly, the assignment of 1H-NMR showed
that the C-3 proton was missing; it was also exchanged against the cyano
group visible from the mass spectrum and elemental analysis data. The NH
proton appeared at 13.08 ppm. The assignment of 13C-NMR showed the main
relevant carbons such as 2-C for the lactam carbonyl at 157.0 ppm, the 6-C
bonded to the methoxy group was observed at 156.1 ppm, and at 118.8 ppm,
almost the same value as described in the literature [20d] with benzonitrile,
probably two cyano carbons are in the same position because the intensity of
the signals is twice the usual size.
Crystallization of compound 7 from acetone gave single crystals that
were suitable for X-ray analysis, which confirmed the structure of 7. One can
think that the mechanism from 4-chloro-6-methoxyquinolin-2(1H)-one 6 to 6-
methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 7 proceeds via a
Meisenheimer type sulfinyl addidtion of p-toluene sulfinate in position 3,
followed by elimination of the 3-substituent.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 49
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Figure 2: Molecular structure of 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile 7 (ORTEP presentation).
1.2.4. Synthesis of 4-sulfinyloxyquinolin-2(1H)-one 8
The reaction of 4-chloroquinolone 6 into the reactive 4-
sulfinyloxyquinolone 8 was achieved according to procedure used in our group
[18] by treatment of 6 with sodium p-toluenesulfinate in dimethylformamide as
solvent. The reaction allowed the nucleophilic substitution of the sulfinate as
more reactive good leaving group in position 4 in good yields of 69 %.
N
Cl
H
O
O
CH3
N
H
O
OS
O
CH3
O
CH3
6
8
sodium p-toluene sulfinate, DMF 120 °C, 20 h
69 %
(Pathway A)
Scheme 6

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 50
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.4.1.Structure elucidation of 4-sulfinyloxyquinolin-2(1H)-one
8
The structural elucidation of 4-sulfinyloxyquinolone 8 was based on
spectral analyses. In the infrared spectrum, the lactam carbonyl stretching was
observed at 1667 cm-1. Assignment of all signals and H-H coupling constants
in 1H-NMR measurements encountered some difficulties due to the roof effect
[27] and also to the overlap between the carbon components, because of the
big number of aromatic rings in compound 8.
There is an overlap from 7.23 to 7.26 ppm between 3-H and 7-H, which
was not easy to distinguish the signal from each proton. Gratifyingly, the roof
effect technique allowed a conclusive assignment to be made. Thus, the signal
of 7-H appeared in singlet at 7.23 ppm, the signal of 3-H appeared in doublet
of doublets at 7.26 ppm (J = 9.0 Hz). At 7.33 ppm a doublet is accounted for 8-
H (J = 9.17 Hz). From 7.45 to 7.50 ppm, there was also another overlap
between 5-H and protons from aromatic substituent patterns HBB’ again with
the roof effect we could distinguished each of them. The signal of 5-H proton
appeared in doublet at 7.46 ppm (J = 2.8 Hz), the two protons HB and HB’ which
are equal were observed in doublet at 7.48 ppm (J = 8.2 Hz) and the doublet (J
= 8.2 Hz) corresponding to the protons HA’ and HA’ was shifted down at 7.96
ppm due to the influence of SO2 group, and the signal of NH-proton was
observed at 12.31 ppm.
N
O
OCH
3
O
H
SO
H7
H8
H5
H3
HB'
HA' HA
HB
CH3
Scheme 3: Structure of compound 8

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 51
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.4.2. 13C-NMR spectrum of 4-sulfinyloxyquinolin-2(1H)-one 8
The 13C-NMR of compound 8 exhibited aromatic peaks between 106.7
and 154.6 ppm and a peak at 159.8 ppm for the lactam carbon. The peaks of
two carbons CBB’ and for carbon 2-C appeared in the same position at 130.9
ppm, and the peaks of two aryl carbons CAA’ appeared also in the same position
at 136.2 ppm. However the intensities of the signals are 3 times and two times
respectively the usual size.
Mass spectrum showed with APCI negative method the base peak with
100 %. However, using ESI (electrospray ionization) mass spectral method,
additional signals could be observed which could be assigned as adducts of
compound 8. It showed only very few peaks: the ratio of intensities varies is
dependent on the fragmentor voltage: At 50 V, a base peak with the high mass
681 could be identified as the combination between 2 molecules carbostyril
and one sodium ion, [2M + Na+]. At 100V this peak is less intense but can still
easily identified (56 %) and the base peak is 352, identified as MNa+. MH+ is
typically of minor intensity, 11 % at 50 V and 15 % at 100 V fragmentor
voltage. Further the mass 368 can be identified as MK+, 7 % with fragmentor
50 V and 3 % with 100 V. In case of 4-sulfinyloxyquinolone 8, also an
additional cluster peak (mass 703) was found. We interpreted it as a double
molecule containing two sodium ions having lost a proton. The formation of
such adducts with alkali metal ions in ESI mass spectra were investigated
recently by Takayama [28].
1.2.5. Experiment to show the reaction pathway
The reaction of one equivalent of 4-sulfinyloxyquinolin-2(1H)-one 8 with
1.5 equivalent of dry potassium cyanide in dry dimethylformamide was carried
out by heating to 130 °C for 30 hours.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 52
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
After worked up, the thin layer chromatography (TLC) analysis showed a
mixture of two compounds, which could not separated. Thus, we though there
could be two pathways to form 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile 7 from 4-sulfinyloxyquinolone 8, since the experiment from
compound 9 to compound 7 worked at about the same rate as experiment from
compound 8 to compound 7, most probably the rate determining step was
experiment from 8 to 9.
N
H
O
OS
O
CH3
O
CH3
N
H
O
CN
O
CH3
N
H
O
CN
CNO
CH3
8
97
KCN / DMF 70 °C, 5 h 67 %
KCN / DMF140 °C, 72 h58 %
Sodium p-toluene sulfinate, DMF 140 °C, 65 h
72 %
(Pathway B)(Pathway B)
(Pathway C)
KCN
Scheme 7
1.2.5.1. Cyanation of 4-sulfinyloxyquinolin-2(1H)-one 8 to 4-
cyanoquinolone 9 and 3,4-dicyanoquinolone 7
For the conversion into the intermediate 4-cyanoquinolone 9, one
equivalent of 4-sulfinyloxyquinolone 8 was treated with 3 equivalents of dry
potassium cyanide at 140 °C in dry dimethylformamide as solvent. The
exchange of sulfinate as reactive good leaving group against cyano at position 4

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 53
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
was complete after 5 hours reaction time, leading the compound 9, which was
then isolated, purified and characterized by analysis and spectroscopic
methods.
Infrared spectrum exhibited the most relevant signal for cyano at 2227
cm-1 and the signal corresponding for lactam carbonyl was observed at 1655
cm-1. The number of proton signals observed in the 1H-NMR spectrum and
their chemical shifts also supported the proposed structure; the most relevant
signals being a singlet at 8.68 ppm ascribable to C-3 proton and the singlet at
12.40 ppm for NH proton. In the 13C-NMR spectrum, the signals of aromatic
carbons were observed between 106.7 and 158.7 ppm, the most relevant were
observed at 118.7 ppm for cyano and at 158.7 ppm for lactam carbonyl.
The mass spectrum analysis showed with APCI ionization-negative method, the
base peak with 100 %, a weak peak (M-15) with 11 % intensity corresponding
to the loss of methyl group and another weak peak (M+1) with only 13 %,
which supported the proposed structure 9. The purification was found to be
difficult.
Comparison of 1H-NMR data of compounds 3, 6 and 9 having
respectively hydroxide, chloro and cyano at position 4 indicate that the
chemical shift of C-3 proton of compound 9 is more shifted down fields caused
by the greater electronegativity of cyano group position 4.
Then, one equivalent of the resulting 6-methoxy-2-oxo-1,2-
dihydroquinoline-4-carbonitrile 9 was subsequently converted into 6-methoxy-
2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 7 by reaction with 2.4
equivalents of dry sodium p-toluenesulfinate as catalyst and 3 equivalent of
dry potassium cyanide in dry dimethylformamide as solvent with 72 % of yield.
The substitution of hydrogen in position 3 of compound 9 was assumed.
The findings were confirmed by analytical and spectroscopic data (IR, 1H
and 13C-NMR, MS, and elemental analysis) similar to those obtained in chapter
1.2.3.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 54
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.5.2. One-pot synthesis of 3,4-dicyanoquinolone from 4-
sulfinyloxyquinolone 8
To react the 4-sulfinyloxyquinolone 8 with dry potassium cyanide in this
way, 1:3 molar ratio of 8 and potassium cyanide, in dry dimethylformamide
was heated to 140 °C to give 3,4-dicyanoquinolone 7 after 72 hours reaction
time in moderate yield of 58 %. The structure was confirmed by spectroscopic
data.
1.2.6. One-pot reaction to 4-cyano-3-unsubstituted quinolone 9
The one-pot synthesis of 4-cyanoquinolone 9 could be achieved by
treatment of 4-chloroquinolone 6 with dry potassium cyanide using dry sodium
p-toluenesulfinate as catalyst. The reaction gave a mixture of 4-cyanoquinolone
9 and 4-sulfinyloxyquinolone 8 after 27 hours reaction time. The separation of
8 and 9 could be accomplished by simple recrystallization from freshly distilled
dioxane.
N
Cl
O
H
O
CH3
N O
H
CN
O
CH3
N O
H
O
O
CH3
SO
CH3
KCN / p-tol.sulfinate Na DMF, 130 °C, 27 h
85 %
6 9 8
+
(Pathway B)
Scheme 8

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 55
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.7. Chlorination reaction with sulfuryl chloride
Survey of the literature showed that 3,3-disubstituted quinoline-2,4-
dione systems have found interest because of their biological activity (e.g. 1-
substituted 3,3-diazido-quinolinediones as platelet aggregation inhibitors [29,
30], 3-hydroxy-3-alkylquinoline-2,4-diones as contents of bacteria with
antibiotic activity [31-33].
Electrophilic substitution of the hydrogen atom at position 3 of 4-
hydroquinolone 3 by chlorine was easily achieved by substitution reaction with
sulfuryl chloride as chlorination agent, which is known as Cl+ source for
electrophilic ionic reactions at these conditions. A radical process can be ruled
out because all requirements for this reaction type such as high temperature,
radical starters, light irradiation or catalyst were missing. Furthermore, it is
described in the literature [34] that reactive hydroxypyridones and quinolones
react already at room temperature or need cooling which is again a strong hint
for an electrophilic mechanism.
A simple reaction uses sulfuryl chloride as source of chloronium ions
[30, 31, 34c, 35]. Another chlorination method was found by reaction of t-
butyloxy chloride with carbocyclic 1,3-dicarbonyl compounds [36].
The chlorination reaction was performed with sulfuryl chloride, because the
chlorination with chlorine gas is not easy to handle [37].
The chlorination of 4-hydroxyquinolone 3 to 3,3-dichloroquinolin-2,4-
dione 10 proceeds in 73 % yield. The temperature should not exceed 60 °C,
otherwise many unwanted by-products might be formed, which are very
difficult to separate. This matter can be explained by the fact that also the
benzene nucleus can be chlorinated in an electrophilic aromatic substitution
by sulfuryl chloride at high temperature [34a].

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 56
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
OH
O
H
O
CH3
N O
H
OCl
ClO
CH3SO
2Cl
2 / dioxane
60 °C
73 %
3 10
Scheme 8
The infrared spectrum of 10 exhibited the absorption bands at 1688 cm-1
and at 1622 cm-1 ascribable to lactam carbonyl and another band at 1725 cm-1
for carbonyl at C-4. The band at 3434 cm-1 was assigned to NH absorption.
1H-NMR spectrum showed the proton signal of NH lactam at 12.27 ppm.
1.2.8. Reduction reaction of 3,3-dichloroquinolin-2,4-dione 10
to 3-chloro-4-hydroxy-6-methoxyquinolin-2(1H)-one 11
By reacting 3,3-dichloroquinolin-2,4-dione 10 in ethanol/acetic acid
mixture using zinc-dust as reduction agent, the reaction allowed to remove one
atom of chlorine of 3,3-dichloroquinolin-2,4-dione 10 and so to obtain 3-
chloro-4-hydroxyquinolin-2(1H)-one 11 in excellent yield of 89 %. The end of
the reaction step could be easily observed by the changing of the color from
yellow to greenish. Attention must be paid for the amount of zinc-dust to be
used, otherwise, both chlorine atoms might be removed and the reaction
recovers the starting material 4-hydroxyquinolone 3 if more zinc-dust is used
or the reaction can not take place if less zinc-dust is used.
Spectral data were consistent with the structure. Comparison of infrared
data of 10 and 11 indicates the absence of additional carbonyl band at C-4 in
compound 11, caused by the formation of hydroxide at this position.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 57
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N O
H
OCl
ClO
CH3
N O
H
OH
ClO
CH3Zn / AcOH, EtOH
reflux
89 %
10 11
Scheme 10
1.2.9. Bischlorination reaction of 3-chloro-4-hydroxyquinolone
11 with phosphoryl chloride
The nucleophilic displacement of the 4-hydroxy group and conversion of
2-oxo of 3-chloro-4-hydroxyquinolone 11 by chlorine was easily achieved by
treatment with phosphoryl chloride to give 2,3,4-trichloroquinoline 12.
Comparison of spectral data of compounds 11 and 12 showed that in the
infrared spectrum of 12 the carbonyl band was not observed, and in the 1H-
NMR spectrum of 12, the signal of NH proton disappeared. The elemental
analysis was in good agreement with the proposed structure.
N O
H
OH
ClO
CH3
POCl3
N
Cl
Cl
Cl
O
CH3
11
110 °C, 8 h
90 %
12
Scheme 11
1.2.10. Hydrolysis reaction of 2,3,4-trichloroquinoline 12
2,3-Dichloroquinolin-2(1H)-one 13 was obtained by regioselective
hydrolysis of compound 12 at position 2 using 70 % methanesulfonic acid in

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 58
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
n-butanol, as described in chapter 1.2.2, to form 13 in excellent yield of 88 %.
Survey of the literature showed that alkaline hydrolysis gave significantly lower
yield [18].
Compound 13 was characterized by analytical and spectroscopic
methods. Infrared spectrum clearly showed the presence of lactam carbonyl
band at 1657 cm-1 and an absorption at 3467 cm-1 corresponding to NH. The
1H-NMR showed again the signal of NH protons at 12.47 ppm. The elemental
analysis confirmed the structure.
CH3SO
3H / n-BuOH
N
Cl
Cl
O
H
O
CH3
N
Cl
Cl
Cl
O
CH3
reflux, 45 h
88 %
1312
Scheme 12
1.2.11. Introduction of cyano substituents into 3,4-dichloro-6-
methoxyquinolin-2(1H)-one 13
In order to check this unusual reaction pathway (replacing of 3-H by
cyano in position 3 of 4-chloro-6-methoxyquinolone 6) and to install properly
the cyano groups in both position 3 and 4 of the quinolone core, we prepared
independently 3,4-dichloroquinolone 13 starting from the known 4-hydroxy-6-
methoxyquinolone 3 via dichloro derivative 10, subsequently followed by
reduction, bischlorination at position 2 and 4 of 11 and hydrolysation in acidic
media regioselectively at position 2 to give 13, which was found to be highly
reactive for the introduction of dicyano substituents.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 59
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
Cl
Cl
O
H
O
CH3
N O
H
O
CH3
CN
CN
13 7
72 %
(Pathway D)
KCN / sodium p-toluene sulfinate, DMF 140 °C, 46 h
Scheme 13
Treatment of 13 with dry potassium cyanide in the presence of dry
sodium p-toluenesulfinate as catalyst in dry dimethylformamide at 140 °C
leaded to a clean 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 7.
Spectroscopic and analytical investigations are similar to those of compound 7
obtained in chapter 1.2.7.
Moreover, using ESI (electrospray) ionization method, some higher mass
fragments from alkali metal complexes were observed. A base peak with the
high mass 248 could be identified as the combination between one molecule
carbostyril and one sodium ion [MNa]+, the peaks corresponding to [MH]+,
[MK]+ and [2M + Na]+ complexes are respectively less intense 8, 17, and 21 %.
1.2.12. Synthesis of 4-chloro-6-methoxy-3-nitroquinolin-2(1H)-
one 16
1.2.12.1. Nitration reaction of 4-hydroxy-6-methoxyquinolin-
2(1H)-one 3
Survey of the literature showed that 4-hydroxy-3-nitroquinolin-2(1H)-
ones were selective glycine site antagonists related to several central nervous
system disorders including stroke, epilepsy, schizophrenia, Parkinson disease,
and Alzheimer disease [13, 38-43].

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 60
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The 2-quinolone 3 was nitrated by reaction of concentrated nitric acid in
acetic acid according to ref. [8, 44]. The electrophilic substitution at position 3
of 4-hydroxy-6-methoxyquinolin-2(1H)-one 3 was catalyzed by sodium nitrite
to accelerate its velocity and allow to give at room temperature the expected 4-
hydroxy-6-methoxy-3-nitroquinolin-2(1H)-one 14 in good yield of 76 %.
The catalytic effect of sodium nitrite could be explained by an initial nitrosation
in position 3 of compound 3 and subsequent oxidation of the nitroso to the
desired nitro group [44]. The nitration reaction of the similar quinolone
systems using nitric acid in boiling acetic acid [45] should lead to a direct
oxidation of the C-H acid in position 3 of 4-hydroxy-6-methoxyquinolin-2(1H)-
one 3 by the oxidative agents present in the mixture such as nitrous and nitric
acid or by exchange of the already formed nitro group against the hydroxy
group [37], or again leads to the nitration of the benzo part of the 2-quinolone
3. The similar substituted 3-nitro and 3-bromo-4-hydroquinolin-2(1H)-ones
showed biological activities such as antituberculosis activity [46].
1H-NMR spectrum of compound 14 clearly showed the absence of C-3 proton
and the signal of NH proton was observed at 11.89 ppm. The infrared spectrum
exhibited the lactam carbonyl band at 1688 cm-1, the signal at 1530 cm-1 was
assigned to the nitro group.
N
OH
O
H
O
CH3
N
OH
O
H
NO2O
CH3HNO
3 / NaNO
2, AcOH
r.t., 30 min.
76 %
3 14
Scheme 14
1.2.12.2. Bischlorination of 4-hydroxy-6-methoxy-
3-nitroquinolin-2(1H)-one 14
The chlorination of the similar 4-hydroxy-3-nitro compounds with
phosphoryl chloride or phosphoryl chloride/phosphorous pentachloride gave

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 61
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
impure products in poor yields [47]. Recently, it was shown in our work group
that the addition of triethylamine as a basic catalyst to the mixture of
phosphoryl chloride and and similar carbostyrils accelerated the exchange of
the hydroxyl against chloro group and allowed to obtain the desired product in
excellent yields of 92-94 % after 1 hour reaction time [8].
Treatment of one equivalent of 4-hydroxy-6-methoxy-3-nitroquinolin-
2(1H)-one 14 with 1.2 equivalent of triethylamine in phosphoryl chloride
afforded the expected 2,4-dichloro-6-methoxy-3-nitroquinoline 15 in excellent
yield of 91 %.
The addition of triethylamine could be explained in terms of destruction
of the hydrogen bonds between the 3-nitro and the 4-hydroxy group, which
prevented or retarded the attack of phosphoryl chloride and accelerated the
reaction speed [8, 48]. A similar hindrance was found in the chlorination of 3-
amido-4-hydroxyquinolone and also in the chlorination of 3-acyl-4-
hydroxyquinolones [15a], which will be described in the chapter 2.2.10.
The structure of compound 15 was confirmed by infrared and 1H-NMR
spectroscopic data.
N
OH
O
H
NO2O
CH3
N
NO2
Cl
Cl
O
CH3POCl
3 / Et
3N
91 %
14
reflux, 1 h
15
Scheme 15

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 62
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.12.3. Regioselective hydrolysis of 2,4-dichloro-6-methoxy-3-
nitroquinolin-2(1H)-one 15
4-Chloro-3-nitroquinolone derivatives of type 16 and the corresponding
2-pyridones are found to be useful as intermediates of synthetic routes
because of their reactivity [8, 23, 48-50].
N
NO2
Cl
Cl
O
CH3 CH
3SO
3H / n-BuOH
N
Cl
O
H
NO2O
CH3
110 °C, 45 h
67 %
15 16
Scheme 16
Hydrolysis of 2,4-dichloro-6-methoxy-3-nitroquinolin-2(1H)-one 15 was
attempted in the presence of 70 % methanesulfonic acid using various
conditions (solvents, temperatures, and reaction time) as listed in the table 1.
In all these cases, a mixture of two compounds was obtained which were found
to be difficult to resolve.
Table 1. Attempts reactions condition from 15 to 16
Solvent Temperature (C) Time (h)
Ethanol
78 35
n-butanol
110 35
AcOH/H2O 115 35
1-dodecanol
150 30
C6H5Br
150 30
DMF 150 44
DMF
150 64
1-dodecanol 210 40
N-methyl-2-pyrrolidone 200 48

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 63
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Finally, the best conditions for the regioselective hydrolysis at position 2
of compound 15 in the presence of 70 % methanesulfonic acid were obtained
by heating the mixture to 110 °C in n-butanol for 45 hours. A careful column
chromatography using chloroform/acetone 3:7 as eluent afforded the expected
4-chloro-6-methoxy-3-nitroquinolin-2(1H)-one 16 in 76 % yield.
Structural investigations showed the most relevant band in the infrared
spectrum at 1663 cm-1 ascribable as lactam carbonyl and the absorption band
in the range of 3437-2916 cm-1 was assigned to the NH. 1H-NMR spectrum
showed a singlet at 12.99 ppm due to the presence of NH proton.
The mass spectrum showed with APCI negative and positive methods the
base peak with 100 % intensity. The proposed structure was supported by the
elemental analysis.
1.2.13. Synthesis of N-(4-chloro-6-methoxy-2-oxo-1,2-
dihydroquinolin-3-yl)acetamide 18
1.2.13.1 Reduction of 4-hydroxy-6-methoxy-3-nitroquinolone
14 into acetylaminoquinolone 17
Survey of the literature showed that reduction of compounds of type 14
using sodium dithionit failed [14].
4-Hydroxy-6-methoxy-3-nitroquinolone was treated with zinc-dust in
acetic acid in the presence of acetic anhydride according to ref. [51], to avoid
the isolation of the sensitive free amino derivatives. The corresponding
acetylaminoquinolone 17 was obtained after 30 minutes reaction time.
Similar reactions heated for a long time leaded to the formation of oxazolo
[5,4:4,5]pyrido [2,2-d] pyrimidinetriones [52], and oxazolo [4,5-c]quinolines or
oxazoloquinolones [14].
Comparison 1H-NMR data of 14 and 17 indicated the appearance of the
additional NH proton signal at 9.74 ppm and a methyl signal at 2.24 ppm

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 64
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
caused by the formation of the amide group at position 3, which was formed by
reduction of nitro (NO2) to amide (-NHCOCH3). The signal at 11.73 ppm is
accounted for NH proton from lactam, and the signal at 11.98 ppm is assigned
to OH proton. Infrared also showed an additional amide carbonyl band at 1645
cm-1, the lactam carbonyl band was observed at 1640 cm-1. Elemental analysis
was in accordance with the proposed structure.
N
OH
O
H
NO2O
CH3
Zn / Ac2O, AcOH
N
H
O
OH
O
CH3
N CH3
O
H
reflux, 30 min.
68 %
1417
Scheme 17
1.2.13.2. Regioselective chlorination of N-(4-hydroxy-6-
methoxy-2-oxo-1,2-dihydroquinolin-3-yl)acetamide 17
The chlorination reaction of N-(4-hydroxy-6-methoxy-2-oxo-1,2-
dihydroquinolin-3-yl)acetamide 17 to the 4-chloroquinolone 18 was performed
similar as described for compound 15. To our surprise, analytical and
spectroscopic investigations revealed that only the 4-hydroxide of compound
17 was exchanged against the chloro atom to give directly compound 18 with
excellent yield of 90 %.
N
H
O
Cl
O
CH3
N CH3
O
POCl3 / Et
3N
N
H
O
OH
O
CH3
N CH3
O
HH
1718
reflux, 1 h
90 %
Scheme 18

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 65
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Infrared spectrum of 18 showed two carbonyl bands assigned for the
lactam carbonyl at 1620 cm-1 and for amide at 1651 cm-1 as already observed
with compound 17. 1H-NMR spectrum also exhibited the main NH proton
signals at 9.60 ppm for NH-amide and at 12.18 ppm ascribable for the NH-
lactam; moreover the signal of OH proton observed at 11.98 ppm in 17
disappeared, caused by the exchange of hydroxyl group against the chloro
atom. These findings clearly confirm that the 2-oxo function of compound 17
did not react with phosphoryl chloride.
Since the carbonyl group in position 2 of the quinolone ring participates only in
the formation of significantly less stable intramolecular hydrogen bonds (IMHB)
with the hydrogen from 3-amide, this lactam carbonyl becomes surrounded;
thus, the chlorination at position 2 was hampered, probably by steric reasons.
The elemental analysis showed a small deviation of 0.09 % from the theoretical
percentage for the carbon component. Mass spectrum with APCI ionization
showed the base peak with 100 %, and exhibited a weak peak (M-43) with only
8 % intensity corresponding to the loss of acylium radical CH3CO+ (m/e = 43)
as shown below.
N O
H
Cl
NH
MeO CH3
ON O
H
Cl
NH
MeO CH3
O
N O
H
Cl
MeO NH
CH3 O
e -
+
.
.
+ +
M-43
m/e = 43
Scheme 19Fragmentation process of M-43

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 66
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.14. Amination of 3,3-dichloroquinoline-2,4-dione 10
3,3-Dipiperidinylquinolinedione 19 was prepared similar as described in
ref. [53] by treatment of 3,3-dichloroquinolinedione 10 with piperidine in
dimethylformamide at room temperature. The nucleophilic exchange of both
chlorine atoms against secondary aliphatic amine such as piperidine leads to
19 in 78 % yield. Infrared spectrum confirm the carbonyl bands of ketone at
1668 cm-1 and for lactam at 1657 cm-1. In the 1H-NMR spectrum, the signal of
NH proton was observed at 8.31 ppm.
N O
H
OCl
ClO
CH3
N
N
N O
H
O
O
CH3piperidin / DMF
10 19
78 %
0 °C, 30 min.
Scheme 20
1.2.15. Reduction of 6-methoxy-3,3-di(piperidin-1-yl)quinoline-
2,4(1H,3H)-dione 19
For the reduction of compound 19 to compound 20 we have used the
procedure described in ref. [54]. 3,3-Dipiperidinylquinolinedione 19 was
treated with sodium dithionite as a reduction agent in a mixture of
ethanol/water (1:1) as solvent to give 4-hydroxy-6-methoxy-3-(piperidin-1-
yl)quinolin-2(1H)-one 20 with an excellent yield of 96 % in pure form. The
structure was confirmed by analytical and spectroscopic methods (IR, 1H-NMR,
MS, and elemental analysis). Infrared showed only one carbonyl band at 1638
cm-1 assigned to –HN-C=O lactam.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 67
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
N O
H
OH
O
CH3
N
N
N O
H
O
O
CH3 Na
2S
2O
4, EtOH / H
2O
reflux, 30 min.
96 %
2019
Scheme 21
1.2.16. Synthesis of 4-chloro-6-methoxy-3-(piperidin-1-
yl)quinolin-2(1H)-one 22
4-Chloro-3-piperidinylquinolone 22 was first prepared from 20 in a two
pot synthesis, similar as described in chapters 1.2.9 and 1.2.10.
Bischlorination of one equivalent of 4-hydroxy-3-piperidinylquinolone 20 with
excess amount of phosphoryl chloride at positions 2 and 4 formed 2,4-
dichloro-6-methoxy-3-(piperidin-1-yl)quinoline 21 in moderate yield of 47 %.
Following hydrolysis in acidic media regioselectively at position 2 of 21 allowed
the conversion of 2-chloro into 2-oxo in 53 %. In this way, the overall yield was
low, the step reactions took longer time and the compound 22 was not easy to
isolate. It was next developed a one-pot synthesis of 4-chloroquinolone 22
directly from the corresponding hydroxy compound 20, using less phosphoryl
chloride and shorter reaction time. A careful treatment of compound 20 with
phosphoryl chloride at 80-90 °C allowed as expected the nucleophilic
substitution of 4-hydroxy to 4-chloro and gave 22 in good yield of 82 %. It
should be noted that in this chlorination step the exchange of the 4-hydroxy
group by chlorine occurs rapidly. Even after 15 minutes the starting 4-
hydroxy-3-piperidinylquinolone 20 was not found in the reaction mixture and
the single reaction product was 22 obtained after 25 minutes of reaction time.
The structure was confirmed the spectral data.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 68
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
N
OH
O
H
O
CH3
N
N Cl
Cl
O
CH3
N
N O
H
Cl
O
CH3
POCl3
POCl3
CH3SO
3H / n-BuOH
20 21
22
110 °C, 8 h
80-90 °C, 25 min.
82 %
47 %
reflux, 48 h53 %
Scheme 22
1.2.17. Synthesis of 3,4-dicyanoquinolones 7 from 3-
substituted 4-chloroquinolones
It was shown that the reaction of nucleophiles with 4-chloroquinolin-
2(1H)-ones usually affords the corresponding 4-substituted quinolin-2(1H)-
ones. The reaction rate was found to be strongly dependent on the electronic
effects of substituents in position 3 of the quinolone moiety. For instance,
electron-withdrawing groups facilitate the substitution, whereas electron-
donating groups impair or prevent the reaction [8].
According to these findings, the 4-chloro atom of compound 16 should
be exchanged easily against nucleophiles by the influence of the nitro group in
position 3. It was shown that in 4-chloro-3-nitrocoumarin [55], both the 4-
chloro and 3-nitro group could be exchanged depending on the nature of the
nucleophiles, and these results should be extended to the 2-quinolone

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 69
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
nucleus. Thus, in the reaction of 16 with dry potassium cyanide in the
presence of sodium p-toluenesulfinate as catalyst in dry dimethylformamide,
the C-Cl bond in position 4 on the quinolone moiety could be activated by the
strong electron-withdrawing effect of the neighboring nitro group, thus
enabling the substitution by p-sulfinate, which is a good leaving group and
allowed easily the introduction of the cyano groups in positions 3 and 4, to give
7 in excellent yield of 93 % after 2.5 hours reactions times (Pathway E).
N
Cl
H
O
NO2O
CH3
N
Cl
H
O
N CH3
O
O
CH3
N
N
Cl
H
O
O
CH3
N
H
O
CN
CN
O
CH3
H
16
18
22
KCN / p-tol.sulfinate Na, DMF 140 °C, 2.5 h
KCN / p-tol.sulfinate Na, DMF 70-140 °C, 17-45 h
KCN / p-tol.sulfinate Na, DMF 140 °C, 5 days
65-86 %
93 %
7
(Pathway E)
(Pathway F)
(Pathway G)
Scheme 23
Attempts to react compound 18 having a 4-chloro substituent and amide
in position 3 even in short reaction time and at low temperature in order to
exchange only the 4-chloro atom and to favor a kinetically controlled reaction
at C-4 position of compound 18, resulted again in the formation of 7 with
overall yield of 65-86 % (Pathway F).
Spectroscopic and analytical investigations of compounds 7 obtained
from 16 and 18 were similar with those obtained previously.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 70
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
However, in the case of 22, the presence of amine in position 3 gave less
reactivity after 5 days (Pathway G). The pure product was obtained by HPLC-
chromatography. The structural investigations of this compound could not be
done, but its fluorescence investigations were very interesting, with an
absorption maximum at 450 nm, an emission maximum at 570 nm in the
mixture of n-heptane/dioxane/DCM 45, and an excellent quantum yield of
0.786.
1.2.18. About an “isomer” of 6-methoxyquinoline-dicarbonitrile
7, compound 23a
In order to investigate the formation of dicyano compound 7 using a
short reaction time, 3,4-dichloro-6-methoxyquinolone 13 was treated with
potassium cyanide in presence of sodium p-toluenesulfinate as catalyst at 140
°C, surprisingly we isolated an obvious isomer of 7, compound 23a.
UV and fluorescence investigations of the isolated intermediate 23a showed
remarkable fluorescence properties. An absorption wavelength up to 497 nm
and an emission maximum of about 570 nm in acetonitrile, never observed
previously with carbostyrils. The structure elucidation by means of
spectroscopic methods (IR, 1H and 13C-NMR, MS) showed that the most
relevant signal of carbonyl lactam in the infrared spectrum was not observed.
However, a stronger peak of cyano group was observed at 2213 cm-1. In the 1H-
NMR spectrum, the signal of NH proton was not observed, but the number of
proton signals in the benzo part and their chemical shifts also support this
part of carbostyril. The most relevant signal being a doublet at 6.84 ppm (J =
1.9 Hz) corresponding to C-5 proton, a multiplet at 7.17-7.21 ppm assigned to
two protons of C-7 and C-8.
13C-NMR spectrum showed the main carbons such as 2-C at 153.9 ppm,
6-C at 148.5 ppm and probably two cyano carbons in the same position at
118.0 ppm because of the intensity of the signal is twice the usual size

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 71
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
O
CH3
Cl
Cl
O
H
CN
N
O
CH3
O
H
CN
CN
O
CH3
N CN
OHKCN / Na p-toluene sulfinate, DMF 140 °C, 3 h
76 %
13 23A
7
soution acetic acid
Scheme24
Mass spectrum analysis showed with APCI-negative the base peak of 23a
corresponding to the molecular ion with 100 %. Efforts to elucidate the
structure by X-ray crystal analysis failed, because it was not possible to get a
single crystal. However, after a second recrystallization of compound 23a in
glacial acetic acid, the 1H-NMR spectrum showed a completely expected
spectrum of 7 with a signal of NH proton at 13.08 ppm, and in the infrared
spectrum, the band of lactam carbonyl was observed at 1655 cm-1 and the
usual band of cyano observed at 2233 cm-1.
The question arises is, where this hydrogen of NH come from ? Whereas
it was not observed in the same sample before the recrystallization from glacial
acetic acid. We attempted to understand this phenomenon.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 72
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
To test if this unknown compound 23a was the sodium salt (compound
24), several experiments have been done.
N
O
ONa
CH3
CN
CN
24
Figure 4
We prepared a 10-2 M stock solution in aqueous concentrated ammonia
and diluted 10 L respectively with 1 mL water (pH = 9), sodium hydroxide 5 M
(pH = 10 and pH = 12); In all cases the UV spectra were almost similar to the
reference compound 23a.
We next added HCl (pH = 1) to the stock solution, we observed a 10 % intensity
increasing spectrum, but the wavelength maximum was still the same. HClO4
(6 M) was added to the stock solution, we just observed a small red-shift of
wavelength value. This means, protonated and deprotonated of compound 23a,
the UV spectra were very similar. The results of the different tests showed that
the compound 23a was not the sodium salt.
We also considered that compound 23a might have the structure 25
resulting from a rearrangement between 3-cyano and oxygen at position 2, but
in this case the formation of structure 7 after recrystallization from glacial
acetic acid could not be explained.
N
O
CH3
OH
CN
CN
25
Figure 5

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 73
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
We also envisaged the possibility of 4-pyridon structure 26 based on the
strong peak at 1583 cm-1 in the infrared spectrum. A literature survey showed
that 4-pyridon derivatives are known to possess carbonyl signals below 1600
cm-1 [56] similar to 26, in this case the signal of the NH, which should also be
seem in the tautomer 26a should be observed in the 1H-NMR spectrum.
N
CNO
CH3
H
O
CN N
CNO
CH3
CN
OH
26 26aaa
Scheme 25
One more probable hypothesis of compound 23a is 2-hydroxy-6-
methoxyquinoline-3,4-dicarbonitrile 7a, which could be explained by the fact
that the stronger signal of cyano observed at 2213 cm-1 is caused not by the
two cyano groups, but because the cyano groups are not close to 2-hydroxide
as observed in the previous cases. In this case, the cyano and 2-hydroxide are
coplanar, that increase strongly the intensity of the band ascribable to cyano
group [57] and a broad-strong band observed in the range of 3600-3200 cm-1 is
due to the presence of OH.
N
CNO
CH3
OH
CN
7a
Figure 6
Since it is speculated that, a tautomeric equilibrium between lactam
form 7 and lactim form 7a could be more easily shifted

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 74
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
O
CH3
O
H
CN
CN
N
O
CH3
CN
CN
OH
7 7a
Scheme 26
and knowing that the lactim form 7a is usually less stable than the lactam
form 7, the molecule could then be stacking in the lactim form (23b), the
tautomeric equilibrium could not be shifted towards the lactam form 7.
N
O
OH
CN
CN
N
O
OH
CN
NC
23b
(2 motives)
Figure 7
The lack of the lactam carbonyl band in the infrared was then caused by
the presence of the bridged 2-hydroxy group, and the absence of the signal OH
proton in the 1H-NMR spectrum is probably due to the quick exchange of this
OH against heavy water (D2O) which is not visible in 1H-NMR.
However, when 23a was dissolved in glacial acetic acid, the stacking form (23b)
which clearly confirms the presence of a OH group either at the position 2 was
broken and the tautomeric equilibrium shifted to the predominate lactam form.
This is a convincing argument to proof that this stacking molecule was the
lactim form of carbostyril.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 75
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.19. Cyclocondensation of m-anisidin 27 and malonic acid 2
to 4-hydroxy-7-methoxy-2(1H)-one 28
4-Hydroxy-7-methoxyquinolin-2(1H)-one 28 was prepared according to
the previously reported procedure described in chapter 1.2.1, by heating at 95
°C m-anisidin 27 with malonic acid 2 using phosphoryl chloride as solvent and
condensation agent. The reaction afforded mainly 28 in 72 % besides the
isomer 4-hydroxy-5-methoxyquinolin-2(1H)-one 29 in 6 % yield and 2,4-
dichloro-7-methoxyquinoline 30 in 16 % yield.
NH2
OCH
3
COOH
COOH
N
OH
H
OOCH
3 N O
OH
H
OCH
3
N
Cl
ClOCH
3
POCl3
+
27 2
28 2930
95 °C, 90 min.
16 % 72 % 6 %
+ +
Scheme 27
The formation of the isomer 29 could be explained by the fact that the
electron-donating group methoxy is ortho- and para-directing (Hollemann’s
rules), therefore in the last step of the formation of the 2-quinolone ring as
described in chapter 1.2.1.2, the attack of carbonyl took place both in position
6 of m-anisidin moiety to form 28 and in position 2 to form the isomer 29.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 76
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The lower yield of 4-hydroxy-5-methoxyquinolin-2(1H)-one 29 in this
reaction is due to the fact that para-electrophilic substitution on the benzene
ring having donor substituent is more favored to give preferentially compound
28.
The formation of 2,4-dichloro-7-methoxyquinoline 30 is due as described
in chapter 1.2.1, to the excess amounts of phosphoryl chloride, which reacted
with the 4-hydroxy group and the 2-oxo group of the formed 4-
hydroxyquinolone 28 to form 30.
Comparison of 1H-NMR data of 28 and 29 indicated that the methoxy
group in position 7 (compound 28) or in position 5 (compound 29) did not
influence the signal of C-3 proton which appeared at 5.60 and 5.61 ppm
respectively, in contrast NH was influenced, the signal was observed at 11.02
ppm for compound 28 and at 10.00 ppm for compound 29.
1.2.20. Chlorination of 4-hydroxyquinolin-2(1H)-one 28 to 2,4-
dichloroquinoline 30 and 4-chloroquinolin-2(1H)-one
31
4-Hydroxyquinolin-2(1H)-one 28 was brought to reaction with
phosphoryl chloride for 28 hours to form 2,4-dichloroquinoline 30 in 81 %
yield. The product was pure enough for further use.
In the infrared spectrum, the characteristic band of carbonyl disappeared and
in the 1H-NMR spectrum, the signal of NH proton was not observed. Infrared
and 1H-NMR data confirmed the structure of compound 30.
Next, the resulting 2,4-dichloroquinoline 30 was then regioselectively
hydrolyzed in position 2 by reaction with 70 % methanesulfonic acid in n-
butanol to give the desired 4-chloro-7-methoxyquinolin-2(1H)-one 31 in
excellent yield of 96 %.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 77
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
Cl
ClOCH
3N
OH
H
OOCH
3
POCl3
N
H
O
Cl
OCH
3
CH3SO
3H / n-BuOH
3028
reflux, 8 h
81 %
31
110 °C, 28 h 96 %
Scheme 28
Compound 31 was characterized by infrared and 1H-NMR spectra. In
infrared, the main carbonyl lactam stretching was observed at 1669 cm-1 and
in the 1H-NMR the signal of NH proton appeared at 11.87 ppm.
1.2.21. Synthesis of 7-methoxyquinoline-carbonitriles 32 and
33
In order to influence the fluorescence properties of 4-cyanocarbostyrils
with methoxy groups in position 6 and/ or 7-methoxy-2-oxo-1,2-
dihydroquinoline-4-carbonitrile 33 was planed to be synthesized.
Reaction of 4-chloro-7-methoxyquinolone 31 with dry potassium cyanide in the
presence of dry sodium p-toluenesulfinate as catalyst at 120 °C during 30
hours, afforded compound 32.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 78
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
H
O
Cl
OCH
3 NOCH
3
CN
OH
3132
KCN / sodium p-tol.sulfinate, DMF 120 °C, 30 h
79 %
Scheme 29
Structural investigations revealed the absence of lactam carbonyl in the
infrared spectrum, and the presence of a strong peak at 2220 cm-1 ascribable
to cyano group, in the 1H-NMR spectrum the signal of C-3 proton clearly
appeared at 7.98 ppm. The number of protons in the benzo part and their
chemical shifts supported this part of structure with a doublet of doublets at
6.46 ppm (J = 8.7 + 2.5 Hz) corresponding to C-6 proton, a doublet at 6.50
ppm (J = 2.50 Hz) for C-8 proton, another doublet at 7.28 ppm (J = 8.7 Hz)
assigned to C-5 proton, and the protons of methoxy group appeared at 3.76
ppm. However, the signal of NH proton was not observed.
Mass spectal analysis showed with APCI-negative and positive methods, very
clean spectra with only one peak with 100 % with 199 and 201 respectively
corresponding to the base peak for each of them.
Here also, we considered that compound 32 might have the 4-pyridonic
structure 34 because of the strong peak at 1614 cm-1 in the infrared spectrum.
However, the lack of NH signal in the 1H-NMR spectrum and the stronger peak
at 2220 cm-1 in the infrared spectrum corresponding to the cyano group could
not confirmed the 4-pyridonic structure 34.
Figure 8
N
H
O
MeO CN
34

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 79
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Survey of the literature revealed that K. Kim et al. achieved since 2000 in
a short communication the synthesis of 2-cyano-4-quinolone 34 [58], which
was obtained by intramolecular cyclocondensation of 2-(2,2-dimethyl-4,6-
dioxo-1,3-dioxane-5-ylidene)-3-(3-methoxyphenyl)propanenitrile A, heated in
diphenyl ether as solvent or without a solvent at 200 to 220 °C for 3 to 5
minutes. However, there is no spectroscopical proof of the data.
H
O O
O
O
CN
OMe
N
H
O
MeO CN
Ph2O, reflux
or neat 200-220°C 3-5 min
63 %
34A
Scheme 30
Through a similar approach as described in chapter 1.2.18, we assume
that the structure of compound 32 might be the stacking form of compound
35.
N OH
CN
OCH
3
35
Figure 9
When 4-chloro-7-methoxyquinolin-2(1H)-one 31 was treated with dry
potassium cyanide in dry sodium p-toluenesulfinate at 140 °C for 46 hours,
the expected compound 7-methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile
33 was isolated and characterized by spectroscopic methods. The infrared
spectrum exhibited the relevant peak at 2220 cm-1 assigned to cyano and the

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 80
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
peak at 1661 cm-1 was accounted for lactam carbonyl band. 1H-NMR spectrum
showed the signal of C-3 proton at 8.66 ppm as already observed with 6-
methoxy analogue (compound 9 ) in chapter 1.2.5.1.
N
H
O
Cl
OCH
3 N O
H
CN
OCH
3
31 33
KCN / sodium p-tol.sulfinate, DMF 140 °C, 46 h
87 %
Scheme 31
Mass spectrum showed with APCI-positive and negative methods the
base peak with mass 201 and 199. However, using ESI (electrospray) mass
spectral method, four peaks were observed, these additional signals observed
could be assigned as adducts of compound 33 with sodium and potassium
cations.
A base peak with the high mass 423 corresponds to the complex [2M +
Na] +, and the complex [M + Na]+ is also much stable with an intensity of 95 %,
the peaks of the complexes [M +K] + and [M + H] + are less stable with 11 % and
23 % intensities respective.
It should be noted that, the substitution of 3-H against cyano in position
3 was not assumed in the case of 4-chloro-7-methoxyquinolin-2(1H)-one 31, as
observed with 4-chloro-6-methoxyquinolin-2(1H)-one 6.
1.2.22. Synthesis of 6-methoxy 4-trifluoromethylquinolin-
2(1H)-one 38
2-Quinolone moieties play an important role in organic chemistry as
outlined in the introduction. The introduction of trifluoromethyl group into
bioactive molecules such as 2-quinolones often increases their therapeutic

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 81
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
efficiency due to an increasing in their lipophilicity [59].
For comparison purposes with 4-cyanoquinolone derivatives on the
fluorescence properties, compound 38 was also synthesized.
The cyclocondensation of p-anisidine 1 with ethyl 4,4,4-trifluoroacetoacetate
36 to 6-methoxy-4-trifluoromethylquinolin-2(1H)-one 38 was performed as
described in ref. [60, 61] in one-pot reaction via an intermediate 4,4,4-trifluoro-
N-(4-methoxyphenyl)-3-oxobutanamide 37, which was cyclized without
isolation by treatment with 76 % sulfuric acid as catalyst to form only the
expected compound 38 in 71 % yield.
The structural investigations using analytical and spectroscopic data (IR,
1H-and 13C-NMR, MS and elemental analysis) confirmed the proposed
structure.
In the infrared spectrum, the lactam carbonyl band was observed at
1672 cm-1, and the absorption band of NH at 3432 cm-1, in the 1H-NMR
spectrum the signal of NH proton appeared at 12.24 ppm, a singlet observed at
6.98 ppm was assigned to C-3 proton, a doublet at 7.05 ppm ascribable to C-5
proton was not well resolved, it seemed to get a roof effect.
NH2
O
CH3
O CF3
OO
N O
CF3
H
O
CH3
H2SO
4
N
CF3
O
O
H
O
CH3
+
90 °C, 12 h71 %
136
38
1) 130 °C, 1 h
2) 76 %
37
Scheme 32

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 82
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Comparison of 1H-NMR data of compounds 9 and 38 indicated that the
chemical shift of C-3 proton of compound 9 is more shifted down field caused
by the greater electronegativity of cyano group in position 4.
N
CN
O
O
CH3
H
9
N O
O
CH3
H
CF3
38
Figure 20
1.2.23. Synthesis of 4-methoxy-N-methylaniline 41
1.2.23.1. Selective monoalkylation of p-anisidine 1 to N-(4-
methoxyphenyl)-N-methylformamide 40
Treatment of p-anisidine 1 with the corresponding orthoformate 39 in
the presence of sulfuric acid as catalyst gave via a Chapman rearrangement
[62] N-(4-methoxyphenyl)-N-methylformamide 40. Other monoalkylation
approaches described in the literature did not show any advantages [63-65].
The structural study by 1H-NMR showed a singlet at 8.33 ppm assigned to the
formyl proton (-CH=O), the signal of the methoxy group was observed at 3.81
ppm, the additional signal observed at 3.26 ppm ascribable to N-CH3 was
caused by selective monoalkylation. At 6.92 and 7.12 ppm two doublets
accounted for the benzene substituent patterns.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 83
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NH2
O
CH3
N
CH3
CHO
O
CH3
H2SO
4 conc.
OO
OCH
3
CH3
CH3
+
103 °C, 3.5 h
70 %
40391
Scheme 33
1.2.23.2. Acidic hydrolysis of N-(4-methoxyphenyl)-
N-methylformamide 40
The reaction of compound 40 in 10 % hydrochloric acid was heated
under reflux for 90 minutes similar as described in a text-book procedure [66],
and afforded 4-methoxy-N-methylaniline 41 in 74 % yield.
Structural assignment by 1H-NMR spectrum showed a doublet of N-CH3 at
2.62 ppm and a quadruplet at 5. 14 ppm assigned to N-H proton.
N
CH3
CHO
O
CH3
N
CH3
H
O
CH3
4041
10 % HClreflux, 90 min.
74 %
Scheme 34
1.2.24. Cyclocondensation of 4-methoxy-N-methylaniline 41
and malonic acid 2 to 4-hydroxy-6-methoxy-
1-methylquinolin-2(1H)-one 42
4-Hydroxy-6-methoxy-1-methylquinolin-2(1H)-one 42 was prepared in
moderate yield of 47 %, similar as described in chapter 1.2.29 by reaction of

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 84
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N-methylaniline 41 with dry malonic acid 2 using phosphoryl chloride as
solvent and condensation agent.
The formation of compound 42 was supported in the 1H-NMR spectrum by the
observation of peaks in the expected shifts of N-CH3 and O-CH3 protons as
singlets at 3.52 and 3.81 ppm respectively. The singlet at 5.93 ppm was due to
C-3 proton and at 11.46 ppm another singlet for OH proton. In the infrared
spectrum, the amide carbonyl band was observed at 1643 cm-1.
NH
CH3
O
CH3
COOH
COOH N O
CH3
OH
O
CH3POCl
3
+95 °C, 90 min.
47 %
41 2 42
Scheme 35
1.2.25. Chlorination of 4-hydroxy-6-methoxy-1-methylquinolin-
2(1H)-one 42 with phosphoryl chloride
The nucleophilic displacement of the 4-hydroxy group of compound 42
by chlorine was easily achieved by treatment with phosphoryl chloride, to form
4-chloro-6-methoxy-1-methylquinolin-2(1H)-one 43 in 82 % yield. It should be
noted that 2-oxo function of compound 42 could not be chlorinated, hampered
by the N-methyl substituent. The structure was characterized by infrared and
1H-NMR spectroscopic data. Infrared clearly showed the amide carbonyl band
at 1642 cm-1.
N O
CH3
OH
O
CH3
N O
CH3
Cl
O
CH3POCl
3
4243
reflux, 8 h
82 %
(Method A)
Scheme 36

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 85
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.2.26. Introduction of a cyano substituent into 4-chloro-6-
methoxy-1-methylquinolin-2(1H)-one 43
4-Chloro-6-methoxy-1-methylquinolin-2(1H)-one 43 was treated at 120
°C with dry potassium cyanide in the presence of dry sodium p-
toluenesulfinate in dry dimethylformamide. The reaction was monitored by thin
layer chromatography (TLC). By stirring the reaction mixture at this
temperature for 28 hours, the starting material totally disappeared. However,
the purity of the resulting 6-methoxy-1-methyl-2-oxo-1,2-dihydroquinoline-4-
carbonitrile 44 was not sufficient. Then we decided to prepare compound 44 by
N-alkylation from the previous 6-methoxy-2-oxo-1,2-dihydroquinoline-4-
carbonitrile 9 synthesized in chapter 1.2.5.1.
N O
CH3
Cl
O
CH3
N O
CH3
CN
O
CH3
43 44
KCN/ p-tol.sulfinate Na, DMF
120 °C, 28 h
(Method A)
84 %
Scheme 37
1.2.27. N-Methylation of 6-methoxy-2-oxo-1,2-dihydroquinoline
-4-carbonitrile 9
The methylation reaction of 2-quinolones using NaOH/CH2Cl2 or
K2CO3/CH3CN heated to 20-50 °C for 3-5 hours afforded a mixture of N- and
O-methylated products in the ratio 4:1 respectively [18].
The reaction of 4-cyanoquinolone 9 with methyl iodide (CH3I) was performed
using dry sodium carbonate as base in dry dimethylformamide at 90 °C for 15
minutes. A simple recrystallization from ethanol afforded the expected 6-
methoxy-1-methyl-2-oxo-1,2-dihydroquinoline-4-carbonitrile 44 in 82 %.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 86
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
It should be noted that methyl iodide is an excellent reagent for
methylation, but there are some disadvantages to its use. It has a high
equivalent weight: one mole of methyl iodide weights almost three times as
much as one mole of methyl chloride. Moreover, iodides are generally expensive
to the more common chlorides and bromides, though methyl iodide is
reasonably affordable.
1H-NMR spectrum revealed an additional methyl group at 3.64 ppm
caused by the substitution of a hydrogen by methyl at N-1 position. Infrared
spectrum showed the relevant peak of amide carbonyl at 1644 cm-1, the peak
at 2229 cm-1 was assigned to cyano group. The 13C-NMR supported the
proposed structure.
N
O
O
H
CH3
CN
N
O
O
CH3
CH3
CNCH3I / Na
2CO
3, DMF
90 °C, 15 min.
9 44
82 %
(Method B)
Scheme 38
1.2.28. N-Alkylation of 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile 7
1.2.28.1 N-Alkylation with iodomethane
The reaction of compound 7 with iodomethane to produce N-methylated
compound 45 was performed similar as described in chapter 1.2.27. It involves
in the first step the attack of hydrogen at N-1 by the base on the compound 7,
then followed by electrophilic substitution to give 6-methoxy-1-methyl-2-oxo-
1,2-dihydroquinoline-3,4-dicarbonitrile 45 in 65 % yield. The structure was
confirmed by infrared and 1H-NMR data.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 87
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Mass spectral using ESI (electrospray ionization) method showed very
few peaks. The high mass observed could be identified as the combination
between one and 2 molecules carbostyrils and alkaline metal ions such as Na+
and K+.
N O
H
CN
CNO
CH3
N O
CN
CN
CH3
O
CH3
CH3I /Na
2CO
3, DMF
120 °C, 25 min.
65 %
7
45
Scheme 39
1.2.28.2 N-Alkylation reaction with ethyl bromoacetate
To attach a reactive linker group at the N-1 position, compound 7 was
brought to reaction with ethyl bromoacetate and dry sodium potassium in dry
dimethylformamide similar as described in chapter 1.2.27, to give the expected
ethyl [3,4-dicyano-6-methoxy-2-oxoquinolin-1(2H)-yl]acetate 46 in 84 % yield.
In addition to the simple reaction conditions, this procedure has the
advantages of very short reaction time, easy experimental and work-up.
procedures.
N O
H
CN
CNO
CH3
N O
CN
CNO
CH3
O
O
CH3
BrCH2COOEt / Na
2CO
3, DMF
7
46
90 °C, 15 min.
84 %
Scheme 40

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 88
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Infrared spectrum showed the amide carbonyl band at 1656 cm-1 and
the ester carbonyl band at 1746 cm-1. The signal of cyano was observed at
2230 cm-1. The 1H-NMR spectrum showed a triplet at 1.22 ppm assigned to
CH3 protons, caused by the formation of N-ester, a quadruplet at 4.17 ppm
assigned to O-CH2 and the singlet corresponding to N-CH2 was observed at
5.20 ppm. 13C-NMR showed the relevant signals of cyano at 117.4 and 118.6
ppm, the signal at 156.6 ppm was ascribable for lactam carbonyl (C-2) and at
167.6 ppm a signal accounted for ester carbonyl. Elemental analysis was
consistent with the proposed structure.
Furthermore, mass spectrum using ESI method at 50 V showed a base
peak with high mass 334, which could be identified as the combination
between one molecule carbostyril 46 and one sodium ion [M + Na]+. The peaks
corresponding to the complexes [M + H]+ and [M + K]+ are respectively 62 %
and 51 % intensities.
The peak corresponding to the combination between 2 molecules
carbostyril 46 and one sodium ion was not observed.
1.2.29. O-Alkylation of 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile 7
The reaction of 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile
7 with halide such as benzyl chloride 47 in dry dimethylformamide using dry
potassium carbonate as base was performed similar as described in ref. [60a].
Surprisingly, after worked-up the analysis of the reaction mixture by thin layer
chromatography (TLC) revealed only the presence of one compound, which was
isolated, purified by dry flash chromatography using toluene-dichloromethane
(3:1) as eluent and characterized by spectroscopic analyses, to give 2-
benzyloxy-6-methoxyquinoline-3,4-dicarbonitrile 48. Infrared spectrum did not
show the lactam carbonyl band and in the 1H-NMR spectrum, the signal of N-H
proton was not observed confirming the O-alkylation. ESI-MS confirmed the
proposed structure. No N-alkylated product was obtained by this reaction.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 89
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N O
H
CN
CN
O
CH3
N
O
CH3
O
CN
CNCl K
2CO
3, DMF
7
+
1) 80 °C, 3 h2) 110 °C, 2 h
47 48
61 %
Scheme 41
1.2.30. N-Methylation of 4-chloro-6-methoxyquinolin-2(1H)-one
6
4-Chloro-6-methoxyquinolin-2(1H)-one 6 was treated at 120 °C with
iodomethane and dry sodium carbonate in dry dimethylformamide as
described in chapter 1.2.27, to give the desired 4-chloro-6-methoxy-1-
methylquinolin-2(1H)-one 43 in 81 %.
The spectroscopic data were consistent with those obtained in chapter 1.2.25.
N O
H
O
CH3
Cl
N O
CH3
O
CH3
ClCH3I / Na
2CO
3, DMF
6 43
120 °C, 25 min.
81 %
(Method B)
Scheme 42
1.2.31. N-Methylation of 3,4-dichloro-6-methoxyquinolin-2(1H)-
one 13
The reaction of 3,4-dichloro-6-methoxyquinolin-2(1H)-one 13 with
iodomethane and dry sodium carbonate in dry dimethylformamide at 120 °C
afforded the desired 3,4-dichloro-6-methoxy-1-methylquinolin-2(1H)-one 49 in

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 90
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
80 % yield.
1H-NMR spectrum showed the additional signal of N-CH3 at 3.67 ppm
caused by substitution of hydrogen against methyl group. The elemental
analysis supported the proposed structure.
N O
H
O
CH3
Cl
Cl
N O
CH3
O
CH3
Cl
Cl
CH3I / Na
2CO
3, DMF
13 49
120 °C, 25 min.
80 %
Scheme 43
1.2.32. Nitration reaction 4-chloro-6-methoxyquinolin-2(1H)-
one 6
The one-pot synthesis of 4-chloro-6-methoxy-7-nitroquinolin-2(1H)-one
50 was achieved by reaction of 4-chloro-6-methoxyquinolin-2(1H)-one 6 with
concentrated nitric acid in acetic acid and sodium nitrite at 110 °C.
The ortho directing substitution leaded by methoxy in position 6 was applied
successfully for the electrophilic substitution of hydrogen against nitro in
position 7 of 4-chloro-6-methoxyquinolin-2(1H)-one 6, to form this rare
chemical compound 50 in excellent yield of 82 %. The reaction showed
excellent selectivity in the synthesis of compound 50 because the attack of
nitro in position 5 of compound 6 is very low.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 91
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N O
H
O
CH3
Cl
N
O
CH3
Cl
O
H
O2N
HNO3 / AcOH, NaNO
2
N
O
CH3
O
H
O2N
CN
110 °C, 3 h
6
82 °C
50
KCN / p-toluene sulfinate Na, DMF 100-140 °C, 20-72 h
51
Scheme 44
The stretching frequency in infrared spectrum at 1534 cm-1 supported
the nitro group and the lactam carbonyl band was observed at 1659 cm-1. The
1H-NMR spectrum showed the signal of C-3 at 6.95 ppm and at 12.32 ppm a
singlet corresponding to N-H proton. The C-5 proton appeared as a singlet at
7.56 ppm and at 7.78 ppm another singlet assigned to C-8 proton, its chemical
shift is inflenced by the nitro group at position 7. However, the signal of C-7
proton has disappeared, caused by the replacing of C-7 hydrogen against nitro
group.
Mass spectrum using the ESI (electrospray) gave the correct mass of
base peak, with some additional signals assigned to adducts with alkali metal
ions.
However, attempts to introduce the cyano in position 4 of the obtained 4-
chloro-6-methoxy-7-nitroquinolin-2(1H)-one 50 failed.

Chapter 1: SYNTHESIS AND REACTIVITY OF 4-HYDROXY-6- AND 7-METHOXYQUINOLIN-2(1H)-ONES 92
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1.3 CONCLUSION
In conclusion, we have developed efficient and convenient methods for the
preparation of 3,4-dicyanocarbostyrils. The results of these syntheses showed
that the presence of chloro, nitro or acetylamino group in position 3 of 4-
chloroquinolin-2(1H)-ones in the reaction to 3,4-dicyanocarbostyrils leads to
excellent yields. However, the reactive intermediate 4-chloro-3-nitroquinolin-
2(1H)-one 16 was found to be very difficult to purify. The structure of 6-
methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 7 has been confirmed
by X-ray diffraction analysis. 3,4-Dicyanocarbostyril derivatives 7 showed
excellent fluorescence properties, absorption wavelengths and large Stoke’s
shifts. 4-Sulfinyloxycarbostyril 8 also showed encouraging UV absorption and
fluorescence spectra.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 93
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CHAPTER 2
SYNTHESIS OF 4-CYANO-3-SUBSTITUTED
QUINOLIN-2(1H)-ONES

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 94
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED
QUINOLIN-2(1H)-ONES
2.1. INTRODUCTION
3–Substituted quinolin-2(1H)-ones are an important class of heterocyclic
compounds having interesting biological activities [67-72], and particularly 3-
aryl-4-hydroxyquinolin-2(1H)-ones [69c] recently described as novel and potent
tyrosine kinase (RTK) receptor
Survey of the literature show that 4-hydroxyquinolin-2(1H)-ones are
selective glycine site antagonists [73-75], related to several central nervous
systems disorders including stroke, epilepsy, schizophrenia, Parkinson disease,
Alzheimer disease [76-81], and serotonin (5-HT3) receptor antagonists [82].
Moreover, 3-aryl-4-hydroxyquinolin-2(1H)-ones have been found to serve as
key intermediates in the synthesis of non-peptide GnRH (Gonadotropin
releasing hormone) receptor antagonists [83]. Such compounds are of interest
for the treatment of sex hormone related conditions [84].
In this chapter the synthesis, isolation and analysis of various 4-cyano-
3-alkyl or aryl substituted quinolin-2(1H)-ones is described.
2.2. RESULTS AND DISCUSSION
2.2.1. Esterification reaction of arylacetic acid 52
Esterification of arylacetic acid 52a-d with primary alcohol such as ethanol in
the presence of catalytic amounts of concentrated sulfuric acid in chloroform
leads to the formation of the corresponding arylacetic acid ethyl ester 53a-d

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 95
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
in moderate to excellent yields. The water formed during the reaction was
removed by the terms of Dean-Stark apparatus. The reaction was finished
upon ceased water formation.
Infrared spectra of compound 53a-d contained strong ester bands in the range
of 1730–1736 cm-1. The ethyl group appeared clearly in the 1H-NMR spectra as
triplet and quadruplet. The singlets at 3.57-3.87 ppm correspond to a
methylene group (CH2).
COOH
O
O
R REtOH
H2SO
4 / CHCl
3
reflux, 5 h
52 53
+
p-NO2
52, 53 R
a
b
c
d
p-MeO
p-Cl
m-Cl
Yield (%)
76
75
87
56
Scheme 1
2.2.2. Condensation reaction of arylethyl ester 53 with diethyl
carbonate 54
The condensation reactions between arylacetate 53a,b,d and diethyl carbonate
54 were promoted by a strong base such as 60 % sodium hydride in dry
tetrahydrofuran and were achieved as described in ref. [85]. The reaction
mechanism involves a Claisen condensation in which the enolizable -hydrogen
at the arylacetate reacts with diethyl carbonate to give the expected
arylmalonates 55a,b,d. The yields were within 48-83 %.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 96
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Infrared spectra showed ester carbonyl bands at 1733-1736 cm-1 and
additionally at 1752-1754 cm-1 corresponding to the presence of second ester.
In the 1H-NMR spectra, the signal of methyne (-CH-) protons appeared at 4.86-
5.03 ppm.
O
O
R OOEt
OEt
NaH / THFO
O
OEt
OEt
R
53
+reflux
54 55
53, 55 R
a
b
d
p-MeO
p-Cl
m-Cl
Yield (%)time (h)
72 69 %
2 83 %
7 48 %
Scheme 2
2.2.3. Thermal cyclization of aryl malonates 55
The reaction of p-anisidine 1 with diethyl malonates 55a-g was performed
similar as described in ref. [9, 86]. In our hands 2-aryl malonates of type 55
proved as good starting materials at reaction temperatures above 250 °C for
the simple and quick cyclization reaction [16, 86], probably via intermediates
arylketene ester 56 [87]. Moreover, the only disadvantage of this reaction
sequence is the high reaction temperature, which prevents its use for sensitive
substituents [16]. The ketene mechanism is also supported by observations
obtained during the reaction [16]: at temperature below 200 °C, the first mole
of alcohol is observed to be liberated when the open chain ester or amide is

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 97
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
formed, then it needs a temperature of more than 250 °C to liberate the second
mole of alcohol to form the ketene intermediates 56.
Previously we have also performed [87] the thermal investigations of similar
reaction between phenol and malonate. Differential scanning calorimetry (DSC)
showed that, at about 160 °C a first exothermic reaction starts immediately
after the endothermic boiling point of phenol, then a weak exothermic reaction
begins at about 240 °C followed by a strong exothermic reaction (onset 260 °C).
Thus, the thermal reactions of p-anisidine 1 with diethyl malonates 55a-g in
diphenyl ether as solvent were performed similarly as described in ref. [88-89]
to give the corresponding 4-hydroxy-3-substituted quinolin-2(1H)-ones 57a-g
in moderate to excellent yields of 43-98 %. It should be noted that diphenyl
ether had to be used as solvent in the case of 55f-g having boiling points below
200 °C (respectively 198-199 °C, 75-77 °C) in order to raise the reaction
temperature of about 250 °C and to liberate the second molecule of ethanol,
otherwise no reaction took place. However, in the case of 55a,b,d,e having
boiling points above 250 °C the solvent diphenyl ether had to be used to
maintain the reaction temperature stable at about 250 °C, and to obtain the
corresponding 4-hydroxy-3-substituted quinolones 57a,b,d in pure form,
without such a temperature control, the reaction temperature reaches more
than 300 °C and the by-product dianilide could be formed besides the desired
products 57a,b,d.
Each reaction was heated until the distillation of ethanol ceased, indicating
completion of the reaction.
Spectroscopic investigations showed in the infrared spectra the lactam
carbonyl bands at 1644-1648 cm-1 for all compounds 57a-g, and in the 1H-
NMR spectra the signals of N-H protons were observed at 10.00-10.30 ppm and
at 11.22-11.44 ppm the singlets assigned to OH protons for compounds 57a-f.
In the case of the known compound 57g, only the signal of N-H was observed
at 11.27 ppm, but the 13C-NMR of compound 57g was consistent with the
structure.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 98
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NH2
O
CH3
N
OH
R1
O
H
O
CH3
Ph2O
R1
O O
O
O
Et
Et
CR1
O O Et
O
- EtOH
- EtOH
250 °C, 3 h
1
55a-g
57a-g
>
56a-g
CH3
R1
CH3CH
2m-Clp-Cl
a b d e f
R p-MeO
Yield (%) 98 80 73 97 94 43
g
Scheme 3
2.2.4. Bischlorination of 4-hydroxy-3-substituted
quinolin-2(1H)-ones 57
Transformation of 4-hydroxy-3-substituted quinolin-2(1H)-ones 57a-g to the
reactive 4-chloro-3-substituted analogues 59a-g were started by reaction of
57a-g with an excess amount of phosphoryl chloride under reflux, which
allowed the conversion of 4-hydroxy and 2-oxo of compounds 57a-g into

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 99
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2,4-dichloro-3-substituted quinolines 58a-g in good to excellent yields of 79-97
%. The presence of meta substituent on the phenyl moiety gave rise to the high
reaction yields (compound 58d compared with compound 58a), and when a
short reaction time was used, the additional para substituent on the phenyl
moiety gave rise to low reaction yields (compound 58b compared with
compound 58e), also a long alkyl chain in position 3 of compounds 57
decreases the reaction yields (compound 58f compare with compound 58g).
Finally, the reactions are more sluggish with alkyl in position 3 than aryl in the
same position.
N
OH
R1
O
H
O
CH3
N
R1
Cl
Cl
O
CH3POCl
3
57 58
reflux
CH3 CH
3CH
2R
1p-Cl
a b d e f
p-MeO m-Cl
g
79 82 97 97 92 84
time (h)12 8 12 8 24 24
Yield (%)
Scheme 4
All structures were confirmed by spectroscopic and analytical data (IR, 1H-and
13C-NMR, and elemental analyses). In the 1H-NMR spectrum of compound 58g,
the meta coupling between H-5 and H-7 was not well resolved, thus the signal
of C-5 proton appeared as singlet at 7.35 ppm and the signal C-7 proton
coupled with C-8 appeared as doublet at 7.47 ppm (J = 8.8 Hz).

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 100
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The signal of C-8 proton observed at 7.86 ppm also appeared as doublet (J =
8.8 Hz). In contrast, in the case of compound 58b, the 1H-NMR spectrum
showed the overlap correlation signals between H-7 and two protons from aryl
ring at 7.57-7.64 ppm, on the other hand between H-5 and two others protons
from aryl ring at 7.45-7.48 ppm. Thus, the conclusive assignment could be
possible with the roof effect technique as described below:
A doublet at 7.45 ppm (J = 1.8 Hz) corresponding to C-5 proton.
A doublet at 7.47 ppm (J = 8.2 Hz) corresponding to two protons aryl
patterns (HBB’).
A doublet at 7.59 ppm (J = 9.2 + 2.0 Hz) corresponding to C-7 proton.
A doublet at 7.63 ppm (J = 8.2 Hz) corresponding to two protons aryl
patterns (HAA’).
A doublet at 7.99 ppm (J = 9.2 Hz) corresponding to C-8 proton.
2.2.5. Regioselective hydrolysis of 2,4-dichloro-3-substituted
quinolines 58
Reaction of 2,4-dichloro-3-substituted quinolines 58a-g in the presence of 70
% methanesulfonic acid in n-butanol under reflux allowed the regioselective
conversion of 2-chloro substituted compounds 58a-g into the 2-oxo derivatives
59a-g in excellent yields of 80-94 %.
All structures were characterized by spectroscopic and analytical data, with the
presence of the lactam carbonyl bands at 1640-1666 cm-1 in infrared spectra,
and the signals of N-H protons in the 1H-NMR were observed at 11.95-12.21
ppm. The complete assignment of compounds 59a and 59e in the 1H-NMR
spectra could not obtained because of many overlaps between the aromatic
rings.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 101
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
R1
Cl
Cl
O
CH3
N
R1
Cl
O
H
O
CH3CH
3SO
3H / n-BuOH
58 59
reflux
CH3
CH3CH
2R1
p-Cl
58, 59 a b d e f
p-MeO m-Cl
g
Yield (%)
time (h) 84 45 45 30 48 48
93 94 90 89 92 80
Scheme 5
2.2.6. Introduction of the cyano group into 4-chloro-6-methoxy-
3- substituted quinolin-2(1H)-ones 59
The introduction of the cyano group in position 4 of 4-chloro-6-methoxy-3-
substituted quinolin-2(1H)-ones 59a-g was carried out by reaction with dry
potassium cyanide mediated by sodium p-toluenesulfinate, similar as
described in chapter 1.2.11. The substitution of 4-chloro of 3-aryl-4-
chloroquinolin-2(1H)-ones 59a,b,d,e with p-methoxy, p-chloro, m-chloro and
hydrogen as substituents at the 3-phenyl moiety worked cleanly, the phenyl
ring and its substituents remained unaffected and the yields of 96-98 %
obtained were excellent. The substituents on the phenyl moiety and the
reaction time did not influence the yields.
However, the substitution of 4-chloro against cyano in the case of compounds
59f, g having either methyl or ethyl in position 3 did not proceed, probably due

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 102
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
to the low reactivity of electron-donating group such as ethyl or methyl in
position 3 of compounds 59f, g, the starting materials were recovered.
The compounds 60a,b,d,e were characterized by spectroscopic and analytical
methods. The most relevant signals of lactam carbonyl in the infrared spectra
corresponded to an intense absorption bands at 1658-1670 cm-1, and weak
peaks at 2229-2236 cm-1 assigned to nitrile. In the 13C-NMR spectrum of
compound 60a, the two carbons CAA’ and two carbons CBB’ of pattern benzene
ring appeared at 113.8 and 132.1 ppm respectively and their intensities are
higher. In the case of compound 60e, the 13C-NMR spectrum also exhibited two
higher peaks at 129.8 and 130.2 ppm respectively corresponding to two
carbons CAA’ and two carbons CBB’ of pattern benzene ring. Mass spectra of
compounds 60a,d,e using APCI and/or ESI methods showed the base peaks
with 100 %.
N
R1
Cl
O
O
CH3
N
R1
O
H
CN
O
CH3KCN / p-toluenesulfinate Na, DMF
120-130 °C
59 60
R1
CH3
CH3CH
2m-Clp-MeO p-Cl
a b d e f
Yield (%)
g59, 60
time (h) 72 63 43 45 48 48 72
96 98 97 96
Scheme 6

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 103
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.7. Synthesis of 2-chloro-6-methoxy-3-phenylquinoline-4-
carbonitrile (61)
The conversion of the 2-oxo compound 60e to the reactive 2-chloro moiety in
compound 61 could be performed by reaction with phosphoryl chloride under
reflux at 110 °C to give 2-chloroquinoline-4-carbonitrile 61 in excellent yield of
95 %.
Similar results were obtained [21] by reacting 6,7-dimethoxy-4-
(trifluoromethyl)quinolin-2(1H)-one in phosphoryl chloride using conventional
heating.
The infrared spectrum of the obtained 2-chloro-4-carbonitrile did not show the
carbonyl band and in the 1H-NMR spectrum the signal of NH proton
disappeared, compared with the starting material 6-methoxy-2-oxo-3-
phenylquinoline-4-carbonitrile 60e.
N O
H
O
CH3
CN
N
O
CH3
Cl
CNPOCl3
110 °C, 12 h
95 %
60e 61
Scheme 7
2.2.8. Synthesis of ethyl 3-[(4-methylphenyl)amino]-
2-[(4-methoxyphenyl)carbamoyl]-3-oxopropanoate 64
Attempts to prepare ethyl 4-hydroxy-6-methoxy-2-oxo-1,2-dihydroquinoline-
carboxylate 63 from primary arylamine such as p-anisidine 1 and triethyl
methanetricarboxylate 62 by conventional heating to 160-220 °C without any
solvent similar as described in ref. [90] using secondary benzo amine such as
tetrahydro-indole or 1,2,3,4-tetrahydroquinoline failed.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 104
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
When diphenyl ether was added as solvent to the mixture of compounds 1 and
62 under a nitrogen atmosphere for 3 hours, the starting material was
consumed, however the product was not isolated. The reaction of 1 and 62 was
also unsuccessfully attempted in N-methyl-2-pyrrolidone (NMP) at 210 °C
during 5 days.
In the best case, the reaction of 1 with 62 in bromobenzene under reflux gave
an unexpected dianilide 64 in 84 % yield. The structure was confirmed by
infrared and 1H-NMR data.
A. Kutyrev and T. Kappe [91] have obtained the compounds of type B (R 2-
hydroxy-4-oxo-1,9a-dihydro-4H-pyrido[1,2-a]pyrimidine-3-carboxylate), C
(methyl 4-oxo-4H-pyrimido[1,2-a]pyrimidine-3-carboxylate), and D (methyl 7-
hydroxy-5-oxo-8,8a-dihydro-5H-[1,3]thiazolo[3,2-a]pyrimidine-6-carboxylate)
by reacting respectively pyridine-2-amine, pyrimidin-2-amine or 1,3-thiazol-2-
amine as primary amines with trialkyl methanetricarboxylate under
conventional heating.
N
N
O
OH
O
O
R
B
N
N
N
O
OH
O
O
CH3
C
N
NS
O
OCH
3
O
OH
D
Figure 1
J. Jampilek et al. [92] have reported the synthesis of ethyl -4-hydroxy-2-oxo-
1,2-dihydroquinoline-3-carboxylate 67 (Scheme 27) in 50 % yield using aniline
and triethyl methanetricarboxylate under microwave conditions.
According to these findings, it was speculated that no quinolin-2(1H)-ones can
be obtained from primary aniline derivatives using conventional heating.
Moreover, the methods of the preparation of 3-ethoxycarbonylquinolin-2(1H)-
ones can greatly depend on the character of their substituents at the nitrogen
atom or in the benzene part of the molecule.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 105
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NH
O
CH3
O
NHO
OCH
3
O
O
CH3
O
NH2
CH3
EtOOC
EtOOC
COOEtO
CH3
NH
O
OH
COOEt
64
PhBrreflux, 6 h
86 %
+
1 62 63
Scheme 8
2.2.9. Synthesis of ethyl 4-hydroxy-2-oxo-1,2-
dihydroquinoline-3-carboxylate (67)
Compounds of type 67 are known as powerful acylating agents [93]. They
readily react with primary and many secondary aliphatic, aromatic and
heterocyclic amines to give the corresponding amides in high yields [94-97].
They also are good synthons for the synthesis of thiosemicarbazones which
present a broad spectrum of pharmacological activities such as
antiproliferative [98] and antiamebal [99] activity. Thiosemicarbazones are
effective in the fight against malaria [100], the herpes simplex virus [101],
carcinoma of the prostate gland [102], hormone dependent breast cancer [103],
and other types of malignat neoplasms [104, 105]. Also, the large use of the
thiosemicarbazones is found in the treatment of various microbial infections
[106-113].

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 106
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
In the literature [114] a rather complicated procedure is used, and in the
literature [115] no exact reaction conditions are described for the preparation
of compounds of type 67. Compound 67 was obtained according the procedure
described in ref. [116], starting from the commercially available methyl
anthranilate 65 which was treated with diethylmalonate and sodium ethoxide.
During the reaction, the formed ethanol was distillated off and the solid
mixture heated for 15 hours at about 140-150 °C until no further ethanol was
liberated.
O
O
CH3
NH2 N
OH
O
H
COOEt
O
O
OEt
OEt
EtONa / EtOH, HCl
+140-150 °C, 15 h
77 %
65 66 67
Scheme 9
The compound was isolated, purified and characterized by spectroscopic
analyses. Infrared spectrum showed the lactam carbonyl signal at 1638 cm-1
and at 1669 cm-1 the absorption band for the ester carbonyl. The low frequency
for an ester carbonyl group is well known [117] and is caused by strong
intramolecular hydrogen bonding [118]. In the 1H-NMR spectrum, the signal of
OH proton was observed at 13.40 ppm and the signal of NH proton at 1.50
ppm. The presence of the ester in the 1H-NMR was confirmed by a triplet at
1.31 ppm and a quadruplet at 4.34 ppm assigned respectively to methyl and to
methylene of the ethyl group.

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 107
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.10. Bischlorination of ethyl 4-hydroxy-2-oxo-1,2-
dihydroquinoline-3-carboxylate (67) with phosphoryl
chloride
Bischlorination at positions 2 and 4 of ethyl 4-hydroxy-2-oxo-1,2-
dihydroquinoline-3-carboxylate (67) in phosphoryl chloride using triethyl
amine as catalyst in order to prevent slow and incomplete reactions caused by
hydrogen bonding between hydrogen group and the ester carbonyl group,
occurs readily and the expected ethyl 2,4-dichloroquinoline-3-carboxylate 68
could be isolated in excellent yield of 98 %.
By reacting compound 67 in phosphoryl chloride at 100 °C without the use of
triethyl amine, I. V. Ukrainetes et al. [119] obtained 68 in only 12 % yield
besides a side compound ethyl 2-chloro-4-hydroxyquinoline-3-carboxylate E in
69 % yield.
N
OH
O CH3
O
Cl
E
Figure:2
The presented synthetic procedure using triethyl amine as catalyst is a simple,
convenient and practical route to transform compound 67 to compound 68 as
a single product and in an excellent yield.
The infrared spectrum of compound 68 showed only the ester carbonyl band at
1728 cm-1, the signal of lactam carbonyl disappeared, and in the 1H-NMR
spectrum the signal of NH proton was not observed.
N
OH
O
H
COOEt
N
COOEt
Cl
ClPOCl3 / Et
3N
67 68
60 °C, 2 h
98 %
Scheme 10

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 108
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.11. Synthesis of ethyl 4-chloro-2-oxo-1,2-dihydroquinoline-
3-carboxylate (70)
The reaction of one equivalent of ethyl 2,4-dichloroquinoline-3-carboxylate 68
in the presence of 3.1 equivalent of 70 % methanesulfonic acid was performed
in different solvents such as methanol, ethanol or n-butanol, under reflux for
20-24 hours. In all cases the unexpected reaction product 4-hydroxyquinolin-
(1H)-one 69 was isolated in which not only the chloro atoms in position 2 and
4 of ethyl 2,4-dichloroquinoline-3-carboxylate 68 was exchanged against the
hydroxy group but surprisingly also the ester group in position 3 was cleaved.
The use of a lower reaction temperature and/or a shorter reaction time in order
to prevent the cleavage of ester in position 3 of 68 were unsuccessful. The
cleavage of 3-ester might be attributed to the use of excess amount of stronger
Brönsted acid such as methanesulfonic acid.
Infrared spectrum of the obtained compound 69 showed only the lactam
carbonyl band at 1653 cm-1 and a broad signal in the range of 3093-2560 cm-1
was ascribable to NH. The 1H-NMR spectrum clearly showed that the 3-ester
group was cleaved, it rather showed a singlet at 5.72 ppm assigned to C-3
proton. The signal of NH proton appeared at 11.29 ppm. The reported melting
point (mp > 350 °C) was in accordance with the literature [120-128].

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 109
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
COOEt
Cl
Cl
N O
H
OH
N O
H
COOEt
Cl
CH3SO
3H
CH3SO
3H
68
69 70
3.1 equiv.
MeOHor n-BuOHor EtOH
in
71-77 %
1.54 equiv.
in
EtOH67 %
reflux, 24 hreflux, 48 h
Scheme 11
Finally, the best reactions conditions were found by using one equivalent of 68
in the presence of 1.54 equivalent of 70 % methanesulfonic acid in n-butanol.
The reaction mixture was refluxed for 48 hours to form the desired 4-chloro-2-
oxo-1,2-dihydroquinoline-3-carboxylate (70) in 67 % yield.
The structure elucidation was based on the spectroscopic analyses. Infrared
spectrum of compound 70 showed the lactam carbonyl band at 1649 cm-1 and
a strong signal of ester carbonyl clearly appeared at 1736 cm-1. In the 1H-NMR,
the ethyl group of ester appeared as a singlet at 1.31 ppm corresponding to
methyl (CH3) and at 4.34 ppm a quadruplet corresponding to methylene (CH2).
The infrared and 1H-NMR spectra of compound 70 were in accordance with the
literature [116].

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 110
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.12. Synthesis of 4-hydroxyquinolin-2(1H)-one 69
Cyanation attempts upon 4-chloro-2-oxo-1,2-dihydroquinoline-3-carboxylate
70 with dry potassium cyanide in the presence of sodium p-toluenesulfinate as
catalyst in dry dimethylformamide were unsuccessful. Surprisingly the infrared
spectrum of the isolated compound 69 did not show the signal of ester
carbonyl. The signal of the lactam carbonyl was observed at 1653 cm-1 In the
1H-NMR spectrum a broad signal was observed at 11.29 ppm assigned to OH
proton, caused by the replacement of chloro by the hydroxy in position 4 of 70
and an additional singlet observed at 5.72 ppm due the presence of C-3 proton
indicating the cleavage of 3-ester group, which confirmed the structure of
compound 69, similar as already obtained in chapter 2.2.11.
N
Cl
O
H
COOEt
N O
H
COOEt
CN
N
OH
O
H
70
7169
KCN / p-toluenesulfinate Na, DMF 130 °C, 24 h
97 %
Scheme 12

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 111
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.2.13. N-Alkylation of 4-chloro-3-(4-chlorophenyl)-6-
methoxyquinolin-2(1H)-one 59b with ethyl
bromoacetate
Treatment of 3-aryl-4-chloroquinolin-2(1H)-one 59b with ethyl bromoacetate in
the presence of dry sodium carbonate gave as expected ethyl [4-chloro-3-(4-
chlorophenyl)-6-methoxy-2-oxoquinolin-1(2H)-yl]acetate (72) in excellent yield
of 95 % but it was found to be very difficult to purify.
Comparison of infrared data of 59b and 72 indicated the appearance of an
ester carbonyl band at 1737 cm-1 in compound 72, caused by the formation of
the N-ester at position N-1, the amide carbonyl band was observed at 1638 cm-
1. In the 1H-NMR spectrum, the NH proton disappeared, however, the presence
of a singlet at 5.14 ppm was observed corresponding to N-CH2 proton, at 4.16
ppm a quadruplet corresponding to O-CH2 proton, the methyl group of ester
function appeared as triplet at 1.21 ppm.
N O
H
O
CH3
ClCl
N
O
CH3
Cl
O
O
O
CH3
ClBrCH
2COOEt / Na
2CO
3, DMF
120 °C, 25 min.
95 °C
59b
72
Scheme 13

Chapter 2: SYNTHESIS OF 4-CYANO-3-SUBSTITUTED QUINOLIN-2(1H)-ONES 112
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2.3. CONCLUSION
3-Aryl-4-cyanoquinolones are readily obtained in excellent yields from the
starting 4-chloro analogues after easy work-up. The phenyl ring and its
substituents remained unaffected. These compounds showed encouraging
emission spectra and may prove to be potential candidates for FRET
experiments due to their long and favorable emission wavelength values (λmax
emis. = ~510 nm). But in the case of compounds 60b and 60d having chlorine
on the aryl moiety, the purification was very difficult. However, in the case of
ethyl 4-chloro-2-oxo-1,2-dihydroquinoline-3-carboxylate 70, the reaction to 4-
cyano analogue gave an unwanted product.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 113
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CHAPTER 3
SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-
DIHYDROQUINOLINE-3,4-DICARBONITRILE

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 114
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-
DIHYDROQUINOLINE-3,4-DICARBONITRILE
3.1. INTRODUCTION
It was shown previously that 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile derivatives exhibited excellent spectroscopic properties but have
the disadvantage of the low quantum yield of fluorescence (see chapter 4).
Knowing that an additional methoxy group in position 7 is required to
improved the quantum yield, we envisioned to synthesized 3,4-
dicyanocarbostyrils in order to increase the quantum yield of 3,4-
dicyanocarbostyrils compounds.
On the other hand, the advantages of 6,7-dimethylcarbostyrils were
shown in their use as fluorescence energy transfer systems [129, 130a], in the
study of luminescence resonance transfer techniques [129a], incorporated in a
time resolved pH-sensor as covalently attached europium complex [129b], for
the use in sensor and luminescence devices [130b-e].
3.2. RESULTAS AND DISCUSSION
3.2.1. Synthesis of 4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one
(74) as precursor
4-Hydroxyquinolin-2(1H)-one moiety is the basic structure found in
many natural products such as flavipucine, the long known and highly toxic
ricinime and the yellow pigments bassianin and tellenin [37], it plays also an

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 115
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
important role as precursor in various reactions such as transformation into
quinoline nucleus, which is a framework of many pharmacologically active
compounds with antiasthmatic [131], antibacterial [132], antifungal [133],
antimalarial [134], anti-viral [135], and anti-inflammatory [136] activities.
Commercially available 4-aminoveratrol (73) was treated with dry
malonic acid 2 in phosphoryl chloride as condensing agent similar as described
in ref. [18] to form 4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one (74) in 84 %.
Infrared spectrum of 74 showed the lactam carbonyl band at 1682 cm-1. 1H-
NMR spectrum revealed the signal of NH proton as singlet at 11.06 ppm and
the singlet of C-3 proton was observed at 5.64 ppm.
O
CH3
O
CH3
NH2 NO
CH3
O
CH3
H
OH
O
COOH
COOH
POCl3
1) 50 °C, 3 h2) 90 °c, 30 min.
+84 %
73 2 74
Scheme 1
3.2.2. Bischlorination of 4-hydroxy-6,7-dimethoxyquinolin-
2(1H)-one (74) with phosphoryl chloride
The nucleophilic displacement of the 4-hydroxy group and the
conversion of 2-oxo of compound 74 by chlorine was easily performed by
reaction with phosphoryl chloride to form 2,4-dichloro-6,7-dimethoxyquinoline
(75) in 84 %.
Comparison of spectral data of compounds 74 and 75 clearly showed the
absence of lactam carbonyl signal in the infrared spectrum of 75, and in the
1H-NMR spectrum of 75, the signal of NH proton disappeared indicating the
conversion of the 2-oxo into 2-chloro group.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 116
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NO
CH3
O
CH3
H
OH
O NO
CH3
O
CH3
Cl
Cl
POCl3
7475
reflux, 8 h
84 %
Scheme 2
3.2.3. Regioselective hydrolysis of 2,4-dichloro-6,7-
dimethoxyquinoline (75)
2,4-Dichloroquinoline 75 in n-butanol, was hydrolyzed regioselectively in
the presence of 70 % methanesulfonic acid at position 2, to give 4-chloro-6,7-
dimethoxyquinolin-2(1H)-one (76) in excellent yield of 96 %.
The structure was confirmed by infrared and 1H-NMR spectra. Infrared showed
again a strong lactam carbonyl signal at 1656 cm-1, the absorption band at
3418 cm-1 was assigned to NH. In the 1H-NMR spectrum the signal of NH
proton appeared at 11.84 ppm.
Comparison of the signal of C-3 proton of compounds 75 and 76 showed
that the chemical shift of C-3 proton shifted down field with the
electronegativity of the neighboring group.
NO
CH3
O
CH3
Cl
Cl NO
CH3
O
CH3
H
O
ClCH3SO
3H / n-BuOH
75 76
reflux, 48 h
96 %
Scheme 3

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 117
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
3.2.4. Chlorination of 4-hydroxy-6,7-dimethoxyquinolin-
2(1H)-one (74) with sulfuryl chloride
Electrophilic substitution of the hydrogen atom at position 3 of 4-
hydroxy-6,7-dimethoxyquinolin-2(1H)-one 74 by chlorine was easily achieved
by substitution reaction with sulfuryl chloride as chlorination agent, which is
well known as Cl+ generator for electrophilic ionic reactions at these conditions,
to give 3,3-dichloroquinolin-2,4-dione 77 in 82 % yield. The reaction was
performed at a temperature below 60 °C to avoid many unwanted by-products,
which are very difficult to separate.
Infrared spectrum showed a strong lactam carbonyl band at 1673 cm-1 and at
1709 cm-1 a ketone carbonyl band. In the 1H-NMR spectrum the signal of NH
proton was observed at 11.29 ppm. The elemental analysis was consistent with
the proposed structure.
NO
CH3
O
CH3
H
OH
O NO
CH3
O
CH3
H
O
Cl
Cl
OSO2Cl
2 / dioxane
74 77
40-60 °C
82 %
Scheme 4
3.2.5. Reduction of 3,3-dichloro 6,7-dimethoxyquinolin-
2,4(1H,3H)-dione (77) to 3-chloro-4-hydroxyquinolin-
2(1H)-one 78
The reduction reaction of 3,3-dichloroquinolin-2,4-dione 77 was
achieved by treatment with ethanol/acetic acid mixture using zinc-dust as
reduction agent under reflux, similar as described in chapter 1.2.8, to form 78
in 87 %.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 118
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The assignment of 3-chloro-4-hydroxyquinolin-2(1H)-one 78 was confirmed by
1H-NMR, IR, and elemental analysis. Infrared showed the absence of ketone
carbonyl at C-4 position.
NO
CH3
O
CH3
H
O
Cl
Cl
O
NO
CH3
O
CH3
H
OH
O
ClZn / EtOH, AcOH
77 78
reflux
87 %
Scheme 5
3.2.6. Bischlorination of 3-chloro-4-hydroxy-6,7-
dimethoxyquinolin-2(1H)-one 78
It was shown in chapter 1.2.11 that 3,4-dichloroquinolin-2(1H)-one is
suitable reactive for the introduction of dicyano groups. The follow-up step in
the introduction of 3,4-dicyano groups in the 2-quinolone core was the
chlorination of 78 at positions 2 and 4 with phosphoryl chloride to give 2,3,4-
trichloroquinoline 79 in excellent yield of 92 %.
The structure was characterized by spectroscopic and analytical data (IR, 1H-
NMR, and elemental analysis). Comparison of 1H-NMR data of 78 and 79
revealed the absence of NH proton in 79 and in the infrared spectrum, the
lactam carbonyl band was not observed.
NO
CH3
O
CH3
H
OH
O
Cl
NO
CH3
O
CH3
Cl
Cl
ClPOCl
3
78 79
reflux, 8 h
92 %
Scheme 6

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 119
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
3.2.7. Regioselective hydrolysis of 2,3,4-trichloro-6,7-
dimethoxyquinoline 79
2,3,4-Trichloroquinoline 79 was hydrolyzed regioselectively at position 2
by reaction with 70 % methanesulfonic acid in n-butanol to form 3,4-
dichloroquinolone 80 in 73 % yield. Compound 80 was found to be highly
reactive for the introduction of the dicyano groups.
In the infrared the signal of lactam carbonyl appeared at 1671 cm-1 and in the
1H-NMR spectrum the singlet of NH proton was observed at 12.37 ppm. The
elemental analysis confirmed the proposed structure.
NO
CH3
O
CH3
Cl
Cl
Cl
NO
CH3
O
CH3
Cl
Cl
O
H
CH3SO
3H / n-BuOH
79 80
110 °C, 24 h
73 %
Scheme 7
3.2.8. Introduction of the cyano groups into
4-chloroquinolin-2(1H)-ones 76 and 80
The introduction of the cyano at positions 3 and 4 of compounds 76 and
80 were achieved by nucleophilic substitution of chlorine, mediated by sodium
p-toluenesulfinate as catalyst.
In the case of 4-chloro-3-unsubstituted quinolone 76 the purity and the
yield of the obtained product are not sufficient, the reaction needs longer time
and involves probably the intermediate 81a (R = H), which is less reactive in
the formation of 3,4-dicyanocarbostyril 82. However, with the highly reactive
3,4-dichloroquinolin-2(1H)-one 80, the clean 3,4-dicyanocarbostyril 82 could
be easily obtained in 80 % just after 5 hours reaction time.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 120
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NO
CH3
O
CH3
H
O
R
Cl
NO
CH3
O
CH3
O
CN
CN
H
KCN / Na p-toluenesulfinate, DMF
NO
CH3
O
CH3
O
CN
R
H
H Cl
76, 80
82
130 °C
81a, b
R
82
Yield (%)
time (h) 86 5
57 80
76: R = H80: R = Cl
Scheme 8
In the infrared spectrum, an intensive absorption band observed at 1665 cm-1
was assigned to the lactam carbonyl and the signal of cyano appeared at 2230
cm-1. 1H-NMR spectrum showed the signal of NH proton at 12.94 ppm. The
elemental analysis found was in accordance with the theoretical percentages.
Mass spectrum using APCI negative method showed only one peak with 100 %
corresponding to the base peak with the mass 254. However, using ESI positive
method with the fragmentor voltage 50 V, mass spectral analysis showed
additional signals which could be assigned as adducts of compound 82. The
base peak with the mass 278 could be identified as the combination between
one molecule of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile
82 and one sodium ion [M + Na]+. Furthermore, the mass 533 can be identified
as the combination between two molecules of 82 and one molecule of sodium
ion with 49 % intensity, the mass 294 and 256 of the complex [M + K]+ and

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 121
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[M + H]+ could be observed with 39 and 36 % intensities respectively. The mass
of 209 corresponding to the loss of a methyl radical was also observed; its
intensity was weak (24 %) because the resulting radical is less stable.
Additionally, the dimeric material was observed, which means this compound
is very interesting for the study of its reactivity towards polymers for instance.
3.2.9. Alkylation of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile (82)
3.2.9.1. N-Methylation of 6,7-dimethoxy-2-oxo-1,2-
dihydroquinoline-3,4-dicarbonitrile (82)
Methylation of 82 with methyl iodide as alkylation agent in the presence
of dry sodium carbonate as the base was achieved similar as described in
chapter 1.2.27, to give the expected 6,7-dimethoxy-1-methyl-2-oxo-1,2-
dihydroquinoline-3,4-dicarbonitrile (83) in excellent yield of 88 %.
Infrared spectrum of 83 exhibited a strong absorption signal at 1637 cm-1
ascribable to amide carbonyl, the 1H-NMR spectrum showed an additional
singlet at 4.06 ppm assigned to N-CH3.
NO
CH3
O
CH3
O
CN
CN
H
NO
CH3
O
CH3
O
CN
CN
CH3
CH3I / Na
2CO
3, DMF
8283
90-100 °C, 15 min.
88 %
Scheme 9

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 122
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
3.2.9.2. Alkylation of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline
-3,4-dicarbonitrile (82) with ethyl bromoacetate
When compound 82 was treated with ethyl bromoacetate as alkylation
agent and dry sodium carbonate as the base, thin layer chromatography (TLC)
analysis showed a mixture of two products, which were separated by
recrystallization from toluene-acetone (9:1), to give the ethyl [3,4-dicyano-6,7-
dimethoxy-2-oxoquinolin-1(2H)-yl]acetate 84 in 56 % yield besides the isomer
O-alkylated product 85 in 29 %.
Infrared spectrum of compound 84 showed the an intensive absorption
band at 1659 cm-1 ascribable to amide carbonyl and another strong band at
1732 cm-1 assigned to ester carbonyl. Infrared spectrum of compound 85 did
not show the amide carbonyl, however, the ester carbonyl was observed 1761
cm-1. 1H-NMR spectrum of compound 84 showed a triplet at 1.23 ppm
corresponding to CH3 and a quadruplet at 4.19 ppm corresponding to CH2 of
the ethyl group. A singlet observed at 5.26 ppm corresponds to CH2 of acetate.
1H-NMR spectrum of compound 85 exhibited the signals almost at the same
positions, with a triplet a 1.21 ppm corresponding to CH3 of ethyl, a quadruplet
at 4.19 ppm corresponding to CH2 of ethyl and a singlet at 5.22 ppm
corresponding to CH2 of the acetate. It should be noted that the similar
chemical shift values for N-ester and O-ester in the 1H-NMR spectra have been
already obtained in our group with similar compounds [18, 60a].
In the 13C-NMR spectrum of compound 84, the most relevant signals
were observed at 167.6 ppm corresponding to the ester-C=O, at 157.4 ppm the
signal of the lactam-C=O, at 114.7 ppm the signal of CN at position 4 and at
113.5 ppm the signal of CN at position 3. The structure was confirmed by the
elemental analysis.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 123
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NO
CH3
O
CH3
O
CN
CN
H
NO
CH3
O
CH3
O
CN
CN
O
CH3
O
BrCH2COOEt / Na
2CO
3, DMF
ONO
CH3
O
CH3
CN
CN
O
CH3
O
84
80 °C, 15 min.
85
+
56 %
29 %
82
Scheme 10
3.2.10. N-Alkylation of 6,7-dimethoxy-2-oxo-1,2-
dihydroquinoline 3,4-dicarbonitrile (82) with ethyl
bromoacetate
In order to prevent the side O-alkylation reaction in the reaction of 6,7-
dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 82 with ethyl
bromoacetate, the modified procedure described by W. J. Bannwarth et al.
[130a] was used appliying lithium diisopropylamide (LDA) as base for the
deprotonation at N-1 position instead of sodium carbonate. The lithium
diisopropylamide led to a quantitative deprotonation of compound 82 under
the mild conditions followed by the attack of ester to give 84 in 65 % yield. This
way avoid at the same time the formation of the O-alkylated product. The
structural investigations were similar to those obtained previously.

Chapter 3: SYNTHESIS OF 6,7-DIMETHOXY-2-OXO-1,2-DIHYDROQUINOLINE-3,4-DICARBONITRILE 124
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
NO
CH3
O
CH3
O
CN
CN
H
NO
CH3
O
CH3
O
CN
CN
O
CH3
O
LDA / THF
BrCH2COOEt
82
84
0 °C, 30 min.
3) r.t., 15 h
2)
1) 0 °C, 1 h
65 %
Scheme 11
3.3. CONCLUSION
As previously reported in chapter 1, an additional methoxy group in
position 7 of 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 7 was
necessary to improve the quantum yield of fluorescence and the resulting
compound 82 and its N-alkylated analogues exhibited interesting fluorescence
properties.
It was observed that in the reaction of the N-alkylation of 6,7-dimethoxy-
2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile 82, with ethyl bromo acetate,
using sodium carbonate as base, a mixture of N- and O-alkylated product was
obtained. To avoid such a side O-alkylated by-product, lithium
diisopropylamine (LDA) has been used instead of sodium carbonate.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties125
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
CHAPTER 4
SYSTEMATIC INVESTIGATION OF SUBSTITUENT
EFFECTS ON FLUORESCENCE AND
PHOTOPHYSICAL PROPERTIES OF
4-CYANOCARBOSTYRIL DERIVATIVES

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties126
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Chapter 4: SYSTEMATIC INVESTIGATION OF SUBSTITUENT
EFFECTS ON FLUORESCENCE AND
PHOTOPHYSICAL PROPERTIES OF
4-CYANOCARBOSTYRIL DERIVATIVES
4.1. INTRODUCTION
Coumarins both naturally occurring as well as from syntheses are used
in a great number of fluorescence applications [1-7], such as fluorescent dyes
[8], photosensitizers [9], laser dyes [10-14] or pH indicators in biochemistry
and medicine [15-16].
Survey of the literature showed that only a few studies have been paid to
their aza-analogues quinolin-2(1H)-ones (carbostyrils) [10, 17-26], probably
because of their lower extinction coefficients, shorter absorption and emission
wavelengths, and more-cumbersome tuning of photophysical properties [27-
30]. However, the important advantages of carbostyrils are their high stability
against chemicals compared with coumarins [6b, 6c], and to many other
fluorescent dyestuffs [38] such as fluorescein, thermal and photochemical
stress (e.g. compared with azodyes), and they are not sensitive to oxygen
quenching (compared with e.g. 1,10-phenanthroline complexes). All these
properties make them very useful as for instance fluorescence markers in e.g.
natural polymers [32-36].
O. A. Ponomarev and coworkers published their efforts on the
photochemical data of a reasonably number of analytes [18-22].
Recently, our group has shown that by systematic investigation substituent
effects using the push-pull concept [37] with two electron donating group
(EDG) such as methoxy or amino in the position 6 and/or 7, and an electron

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties127
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
withdrawing group (EWG) such as trifluoromethyl (CF3) in the position 4 of
carbostyrils, we could improve their absorption, emission and fluorescence
quantum yields comparable to those of coumarins [37, 38]. About 140
structures were calculated and synthesized, and we found out absorption
maxima in a range of 350-380 nm, emission maxima at 410-425 nm and
fluorescence quantum yields up to 0.41, almost the same fluorescence
properties like umbelliferone (7-hydroxycoumarin).
More recently [31], our group investigated on the effect of a strong
electron withdrawing cyano group in the position 4 combined with methoxy as
electron donor groups in the positions 6 and 7 of the carbostyril derivatives.
The spectra revealed a broad double maximum at 385 nm and 410 nm in polar
and apolar solvents, independent of the pH value (pH 1-11), an emission range
of 430-440 nm in all solvents, and fluorescence quantum yields up to 0.5 were
obtained. This means, 4-cyanocarbostyril derivatives were useful new
fluorophores and well suited as fluorescence standards [31].
The construction of Fluorescence Resonance Energy Transfer (FRET)
carbostyril complexes such as Valinomycin-carbostyrils, FRET with rhodamin
[39] or FRET peptides with [RuII(bathophenanthroline)] complex [40] is suited
for the characterization of biochemical events both in vitro and in vivo, and also
it involves binding of ligands to their pertinent protein receptors [41-42], DNA-
protein complexation [43-45], and RNA-folding and catalysis [46-47].
In this chapter, we describe the fluorescence of 3,4-dicyano carbostyril
derivatives and compare their photophysical properties with others carbostyrils
previously investigated, differently substituted, and also with some dimeric
carbostyrils [bisquinolin-2(1H)-ones]. In addition, fluorescence properties of 4-
sulfinyloxycarbostyrils 8 and U are also described.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties128
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4.2. RESULTS AND DISCUSSION
4.2.1. Influence of methyl, trifluoromethyl and cyano groups on
the fluorescence properties on quinolones core
Coumarins with electron donating group such as methoxy in position 7
(e.g. coumarin A1) compared with simple coumarin A, show blue fluorescence
of 415 nm wavelengths and good quantum yield up to 0.6. But in alkaline
solution, coumarins suffer ring opening, this losing fluorescence properties.
O
CH3
OMeO O
CH3
O
N
CH3
O
H
A1 A B 7-methoxy-4-methylcoumarin 4-methylcoumarin 4-methylcarbostyril
Figure 1
Table 1. Photophysical data for absorption and emission of compounds A1, A and B
A1 A B
Absorption (nm) 320 330 330
Emission (nm) 415 380 375
Quantum yield 0.6 0.002 0.02
In comparison with coumarins analogues, methoxy group in position 7 of
4-methyquinolone C (Table 2) lowers fluorescence properties, but shows good
pH-stability, almost no solvent dependency; the quantum yield of 0.052 is very
low, therefore not suitable for fluorescence applications. Compared with 6-
methoxyquinolone D (Table 2), 7-methoxyquinolone C has a high extinction
coefficient (ε =10000) and the emission maximum of 342 nm.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties129
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Table 2. Photophysical data for published carbostyril derivatives (letter
assigned in order to be distinguished from the new Compounds)
Cpd
Structure
Solvent
λmax 1
UV-Vis (nm)
2
λmax 2
UV-Vis
(nm)
2
λmax
em. (nm)
Φ
C N
CH3
O
H
MeO
DMSO
340
10000
-
-
370
0.052
D N
CH3
O
H
MeO
DMSO
-
6550
-
-
353
0.033
E
N
CH3
O
H
MeO
MeO
DMSO
346
9000
-
-
392
0.08
F
N
CF3
O
H
MeO
MeO
DMSO
H2O
368
365
9400
10400
-
-
-
438
428
0.470
0.450
G
N O
H
MeO
MeO
CN
DMSO
397
11300
385
11200
440
0.308
H
N
CF3
O
MeO
CH3
MeO
DMSO
H2O
365
365
9700
10100
-
-
435
428
0.340
0.160
I
N O
MeO
CH3
MeO
CN
DMSO
H2O
390
380
12400
11000
-
-
-
-
436
432
0.610
0.500
J
N
Cl
O
MeO
CH3
MeO
DMSO
*
358
372
-
-
-
412
0.017
* Double maximum
(continued on next page)

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties130
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Table 2 (continued)
Cpd
Structure
Solvent
λmax 1
UV-
Vis (nm)
2
λmax 2
UV-
Vis
(nm)
2
λmax
em. (nm)
Φ
K NNH
2
CF3
O
H
MeO
DMSO
373
438
L N
CF3
O
H
NH2
MeO
DMSO
404
7350
533
0.380
M N
CF3
O
H
MeO
(CH3)2N
DMSO
384
8470
557
0.280
N N O
CN
MeO
O
O
CH3
MeO
DMSO
EtOH
H2O
390
385
380
12400
13100
11000
-
-
-
-
-
-
436
432
432
O N O
MeO
O
O
CH3
MeO
CF3
DMSO
368
8000
-
-
430
0.269
P N O
MeO
MeO
CF3
O
O
CH3
DMSO
350
9280
337
8300
384
0.143
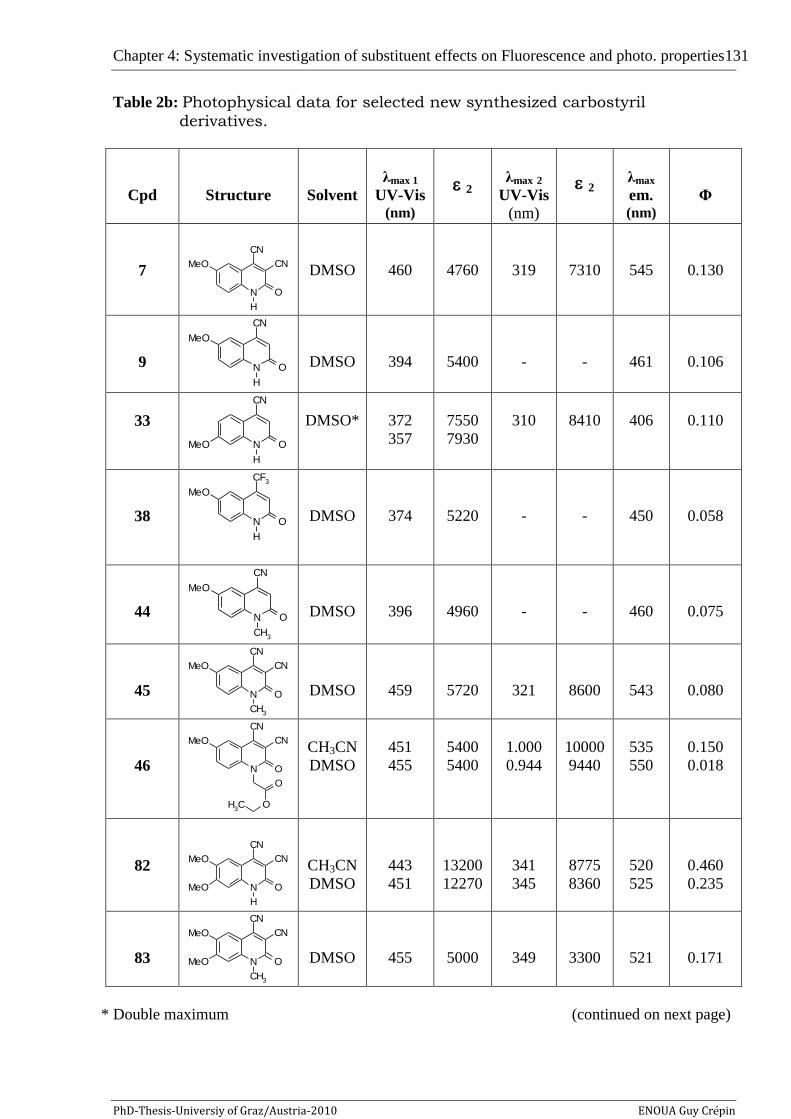
Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties131
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Table 2b: Photophysical data for selected new synthesized carbostyril
derivatives.
Cpd
Structure
Solvent
λmax 1
UV-Vis (nm)
2
λmax 2
UV-Vis
(nm)
2
λmax
em. (nm)
Φ
7
N O
H
CN
CN
MeO
DMSO
460
4760
319
7310
545
0.130
9 N O
MeO
H
CN
DMSO
394
5400
-
-
461
0.106
33
N O
H
MeO
CN
DMSO*
372
357
7550
7930
310
8410
406
0.110
38 N O
MeO
H
CF3
DMSO
374
5220
-
-
450
0.058
44 N O
MeO
CH3
CN
DMSO
396
4960
-
-
460
0.075
45 N O
CH3
CN
CN
MeO
DMSO
459
5720
321
8600
543
0.080
46 N O
CN
CN
MeO
O
O
CH3
CH3CN
DMSO
451
455
5400
5400
1.000
0.944
10000
9440
535
550
0.150
0.018
82
N O
H
CN
CN
MeO
MeO
CH3CN
DMSO
443
451
13200
12270
341
345
8775
8360
520
525
0.460
0.235
83 N O
CH3
CN
CN
MeO
MeO
DMSO
455
5000
349
3300
521
0.171
* Double maximum (continued on next page)

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties132
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Table 2b (continued)
84 N O
CN
CN
MeO
O
O
CH3
MeO
CH3CN
DMSO
CH3OH
H2O
442
451
440
440
9250
10850
9320
9520
344
350
344
347
6440
7540
6160
6190
520
520
520
520
0.300
0.088
0.359
0.078
85 N O
CN
CN
MeO
MeO
O
O
CH3
DMSO
406
10000
353
7000
472
*
* The compound was not completely free of N-alkylated carbostyril isomer 84.
4.2.2. The effects of methoxy groups in positions 6 and/or 7
Spectral data of 7-methoxy and 6-methoxy 4-methylquinolone C and D (Table
2) show compared with those of 6,7-dimethoxy-4-methylquinolone E (392 nm,
Table 2) an 20 respectively 40 nm redshifted absorption maximum. The
fluorescence quantum yield of E (Φ = 0.08) is only slightly higher than the
monomethoxy 4-methylanalogues. Extinction coefficient of C is with about
10000 about 25% higher than that of D and E. When the electron donating 4-
methyl group was replaced by an electron-withdrawing 4-trifluoromethyl
group, 6,7-dimethoxyquinolone F (Table 2) exhibited a red shifted emission at
438 nm in DMSO. The fluorescence quantum yield (Φ = 0.47) is 83 % higher
than for the 6,7-dimethoxyquinolone E, the absorption maximum is about 20
nm red shifted, whereas the extinction coefficient is rather similar.
N
CH3
O
H
MeO
N
CH3
O
H
MeO
N
CH3
O
H
MeO
MeO
N O
H
MeO
MeO
CF3
C D E F
Figure 2

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties133
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Trifluoromethyl-6,7-dimethoxyquinolone F and 6-methoxy-4-
trifluoromethylquinolone 38 (Table 2b) show that the removal of the methoxy
0.47 vs 0.06) and 50 % reduction of the extinction coefficient. However, note
the slightly blue shifted absorption and emission maxima of F.
N O
H
MeO
MeO
CF3
N O
H
MeO
CF3
N O
H
MeO
CF3
NH2
N O
H
MeO
CF3
NH2
F 38 K L
Figure 3
Comparison of 6,7-dimethoxy-4-trifluoromethylquinolone F with 7-
amino-6-methoxy analogue K (Table 2) [48] shows that absorption and
emission maxima are very similar. One can conclude that the amino group in
position 7 of the 2-quinolone has the same effect on the fluorescence properties
as a methoxy group at the same position.
In contrast, by comparing the data of 4-trifluoromethylquinolone F with
those of 6-amino-7-methoxy-4-trifluoromethylquinolin-2(1H)-one L (Table 2)
[37a], there were not the same effects observed. A bathochromic shift was
found for the latter with an absorption maximum of 403 nm and an emission
maximum of 533 nm, whereas the fluorescence quantum yield of 0.38 is about
20 % lower than F.
N O
CH3
MeO
MeO
CF3
H M
Figure 4
N
CF3
O
H
MeO
(CH3)2N

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties134
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The N-methylation of 6,7-dimethoxy-4-trifluoromethylquinolone F
yielded 6,7-dimethoxy-1-methyl-4-trifluoromethylquinolone H (Table 2), which
presents almost the same absorption and emission maxima, and extinction
coefficient in DMSO and in water. However, the fluorescence quantum yield is
much less in water for N-methylderivative H. When F is compared with 6-
dimethylamino-7-methoxy-4-trifluoromethylquinolin-2(1H)-one M (Table 2)
[37a], replacement of the 6-methoxy by the 6-dimethylamino group leads to the
highest fluorescence wavelength of 557 nm, which is 120 nm red shifted
compared with F. This increasing of fluorescence can be deduced from the
strongly electron delivering dimethylamino group, whereas the quantum yield
of 0.28 is 2 times less than for F.
Similarly, 6,7-dimethoxy-1-methyl-4-trifluoromethylquinolone H and 4-
chloro-6,7-dimethoxy-1-methylquinolone J (Table 2) bearing respectively
trifluoromethyl and a chloro group in position 4 show that, their absorption
maxima do not differ significantly, but emission maxima of H (λmax em. = 435
nm) having a strong acceptor group trifluoromethyl (CF3) in position 4 is only
23 nm longer wavelength compared with compound J, and the chloro
substituent in position 4 (compound J,
fluorescence quantum yield compared with compounds H I
0.61). As also can be seen in Table 2, more strongly fluorescing 4-cyano
derivative I absorbs also with 390 nm about 20 nm blueshifted compared with
trifluoromethyl analogue H). Also the fluorescence quantum yield in water is 68
% better.
N O
CH3
MeO
MeO
CF3
N O
H
MeO
CF3
(CH3)2N
N
Cl
CH3
O
MeO
MeO
N O
CH3
MeO
MeO
CN
H M J I
Figure 5

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties135
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N O
R
MeO
CN
N O
MeO
CN
MeO
R
N O
R
MeO
CN
4.3. ELECTRONIC SPECTRA OF THE NEW CARBOSTYRILS
4.3.1. Known 4-cyano-3-H-6,7-dimethoxy-carbostyrils
Figure 6
6- And 7-methoxy substituted 4-cyanoquinolones 9 and 33 (Table 2b)
showed that methoxy in position 7 shifted in the absorption and emission
maxima to shorter wavelength, but have a somewhat better extinction
coefficient (ε = 7550), versus 5400 for compound 9 having the methoxy in
position 6. This confirms once again that only a methoxy in position 7
increases the extinction coefficient.
3,4-Dicyanocarbostyril 7 (Table 2b) is absorbing and emitting at higher
wavelength than 3-H-4-cyano analogue 9 (Table 2b) and 3-arylsubstituyed-4-
cyanoanalogue 60a-e. In contrast, 3-aryl substituted-4-cyanoanalogue 60a-d
show no change in the absorption wavelength compared with 3-H-4-cyano
analogue 9. However, emission of 60a-d is red shifted by about 50 nm, the
quantum yield is about 54 % higher than 9. For compound 60e which contains
only a phenyl in position 3, the fluorescence quantum yield is about 50 %
lower compared with 9.
N
MeO
CN
O
H
CN
N
MeO
CN
O
H
R
7 60a-e
Figure 7

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties136
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Trifluoromethylquinolone F (Table 2) [38] and 4-cyanoquinolone G
(Table 2) [31] which are different only at the acceptor group in position 4, and
dicyanocarbostyril 7 (Table 2b) show that the fluorescence spectra of these
three dyes F, G and 7 are significantly red-shift in more polar solvent (DMSO).
Emission maxima of F and G are nearly similar due to the fact that the main
electron withdrawing group cyano in F has almost the same electro negativity
effect as that of trifluoromethyl in G. However, in the similar 4-trifluoromethyl
H (Table 2) and 4-cyano-6,7-dimethoxycarbostyrils I (Table 2) containing an
additional methyl group at N-1, we observed an excellent fluorescence
quantum yield for I ( = 0.61 in DMSO). In both cases, the absorption intensity
is higher for G and I having cyano in position 4.
N O
H
MeO
MeO
CF3
N
MeO
CN
O
H
MeO
N O
CH3
MeO
MeO
CN
N O
CH3
MeO
MeO
CF3
F G I H
Figure 8
Until this work was done, compounds F and G were the most push-pull
efficiently substituted known carbostyrils. To our surprise, the effect of an
additional 3-cyano substituted (compound 7) showed a dramatic increment of
absorption maximum to 460 nm at pH = 1, and emission maximum to 545 nm
but this absorption is less bright at pH = 9.2 (λmax. = 322 nm), with a large
Stoke’ s shift of 85 nm compared with 4-cyanocarbostyrils 9 (Table 2b) and
60a-e (Table 3), this is due to the presence of the additional cyano group in
position 3 of 4-cyanocarbostyril 7. However, the lack of a methoxy group in
position 7 of 4-cyanocarbostyril 7 lowered the fluorescence quantum yield and
extinction coefficient. These disadvantages (low fluorescence quantum yield
and extinction coefficient) were overcome with the introduction of an additional
methoxy group in position 7, compound 82 which will be described later.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties137
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
In the case of coumarins, it has been observed that the presence of a
strong electron withdrawing group, such as cyano or ester group in position 4
on the parent chromophore leads to enhanced non-radiative rates for
coumarins [49-55] and in differents solvents.
Rettig and El-Kemary [56] have shown that the cyano group in position 4
of coumarins (coumpounds Q and R) exhibited a strong red shift compared to
the corresponding primary coumarin C153 [57], indicating that the effective
size of the -system is larger for coumarins Q and R than for compound C153.
ON O
S
NCN
ON O
O
NCN
Cl
Q R
ON
CF3
O
Coumarin 153 (C153)
Figure 9
However, different from our carbostyrils, they exhibit a strong
dependence on solvent polarity, being most effective in non polar solvents. In
Acetonitrile and especially in DMSO, quantum yields are lowered by 50 % and
80 % respectively. Data in water were not published.
These findings are however also in contrast to 4-trifluorometyl coumarin
C153, which does not show this solvent polarity dependence.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties138
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
By comparing the data of 6,7-dimethoxy-1-methyl-2-oxo-1,2-
dihydroquinoline-4-carbonitrile I (Table 2) with those of 6-methoxy-1-methyl-2-
oxo-1,2-dihydroquinoline-4-carbonitrile 44 (Table 2b), one can confirm once
again that methoxy in position 7 combined with N-methyl (compound I) have a
significant increment on the fluorescence quantum yield and absorption
I
44 in DMSO). The highest increasing of fluorescence quantum yield is
especially due to the presence of a strong acceptor group in position 4 and a
strong donor group in position 7 at the carbostyril. In contrast, with electron
donating group such as methyl in position 4, this increasing of fluorescence
quantum yield is not substantial even if carbostyrils contain methoxy groups
in positions 6 and 7 (compound E Table 2). However, in the case of 3,4-
dicyanoquinolones 82 and 83 (Table 2b) the effects of N-methyl are
exceptionally reversed. The reasons of this quenching are not clear, since we
did not observe it in the case of 4-cyanoquinolones G versus I.
N
MeO
CN
O
H
MeO
N O
CH3
MeO
MeO
CN
N
CH3
O
H
MeO
MeO
N
MeO
CN
O
CH3
G I E 44
N
MeO
CN
O
H
CN
MeO
N
MeO
CN
O
CH3
CN
MeO
82 83
Figure 10

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties139
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The shape and wavelength of the absorption and emission bands are not
affected by the three 4-cyanocarbostyril derivatives 7, 45 and 46 (Table 2b)
differently substituted at position N-1; however their fluorescence quantum
yields are affected. The fluorescence quantum yield of 0.08 for the N-
methylquinolone 45 is about 38 % less when compared to compound 7 having
NH at position 1. This weak fluorescence quantum yield is more pronounced
when compound 7 -1 to give N-methylquinolone 46
= 0.018).
N
MeO
CN
CN
O
H
N
MeO
CN
O
CH3
CN
N
MeO
CN
O
CN
O
O
CH3
7 45 46
Figure 11
In order to optimize the low fluorescence quantum yield of 0.13 obtained
with 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (compound 7,
Table 2b), 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile
(compound 82, Table 2b) was prepared; the UV-visible absorption spectrum of
compound 82 shows absorption maximum at 443 nm and epsilon coefficient of
ε = 13200 in acetonitrile, and absorption maximum at 451 nm, epsilon
coefficient of ε = 12270 in DMSO, while for compound 7 the absorption
maximum was observed at 460 nm and extinction coefficient was only ε = 4760
in DMSO. The emission maximum of 525 nm for compound 82 in DMSO is
only 20 nm blue-shifted compared to compound 7. However, compound 82
shows large Stoke’s shift of about 80 nm with an excellent fluorescence
quantum yield of about 50 %, which is 20 % more than fluorescein at pH 3.3 in
acetonitrile, due to the replacement of hydrogen against the methoxy group at
position 7. The new chomophore (compound 82) which showed intense
luminescence, exhibited a double maximum and can be therefore excited at a
wavelength range of 350-430 nm, is a promising probe for further

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties140
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
investigations as an optical sensor for pH measuring, oxygen or environmental
important gases such as ammonia.
0
50
100
320 370 420 470 520 570 620
Figure 12: Excitation (dotted line) and emission (continuous line) spectra for
carbostyrils 7 (red curves), 82 (blue curves) and fluorescein (black
curves).
Comparison of 3,4-dicyanocarbostyrils 82, 83 (Table 2b) and 84 (Table
2c) leads to the same observations as for 3,4-dicyanocarbostyrils 7, 45, and 46
(Table 2b), with the only difference that the absorption coefficient is about 50
% less for 83 compared to 82 and 84. However, in all solvents compound 84
shows similar absorption and emission spectra with the main peaks around
450 nm and 520 nm respectively, which means that the spectra show virtually
no solvent dependence, but there is an increasing in the fluorescence quantum
yields along the series H2O < DMSO < CH3CN < CH3OH.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties141
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N
MeO
CN
O
H
CN
MeO
N
MeO
CN
O
CH3
CN
MeO
N
MeO
CN
O
CN
O
O
CH3
MeO
82 83 84
Figure 13
4.3.2. O- versus N-Alkylation
A side product of N- alkylation of 82 was at position 2 O-linked 3,4-
dicyanoquinoline 85 (Table 2b), which causes a small blue shift of absorption
and emission maxima compared to N-alkylated 3,4-dicyanoquinolone 84 (Table
2b), The fluorescence data of N-alkylated-4-cyanocarbostyril N are almost
solvent insensitive for the absorption and emission wavelengths.
O
O
CH3
N
MeO
CN
OMeO
O
O
CH3
N
MeO
CN
O
CN
MeO
84 85
Figure 14
4.3.3. Influence of differently substituted aryl groups in
position 3 on the fluorescence properties
3-Aryl substituted-6-methoxyquinolone derivatives have not been
described previously. We investigated substituent effects in position 3 on the

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties142
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
fluorescence properties. 3-Arylquinolone derivatives (table 3) have been
synthesized and 3-aryl-4-hydroxy (57) and 3-aryl-4-cyanoquinolone (60)
derivatives were selected for fluorescence investigations.
Luminescence spectra of these 3-aryl-4-cyanoquinolones show the
similar absorption maxima of about 410 nm and emission maxima of about
510 nm, which are respectively 20-40 nm and 50-70 nm shifted to the longer
wavelengths compared to the simple 4-cyano or 4-trifluoromethylquinolones
derivatives such as compounds F, G (Table 2), 9, 33 and 38 (Table 2b).
However the fluorescence quantum yields are bellow to 10 %.
The influence of substituents at the aryl moiety is weak and has almost
no effect on the fluorescence, while one can see some differences regarding the
quantum yield of fluorescence depending of the meta or para substituent at the
aryl moiety (compounds 60b, 60d, Table 3).

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties143
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Table 3. Photophysical data of selected 3-arylcarbostyril derivatives.
Cpd
Structure
Solvent
λmax 1
UV-
Vis (nm)
2
λmax 2
UV-
Vis
(nm)
2
λmax
em. (nm)
Φ
S
N
CN
O
MeO
CH3
MeO
DMSO
-
-
485
0.500
T N O
MeO
H
MeO
CF3
OMe
DMSO
368
20800
-
-
435
0.210
57b
N O
MeO
H
OHCl
DMSO
352
8720
-
-
416
0.034
57e N O
MeO
H
OH
DMSO
-
8070
-
-
351
0.040
60a
N
CN
O
MeO
H
OMe
DMSO
405
10370
341
8490
503
0.198
60b N
CN
O
MeO
H
Cl
DMSO
406
6840
311
9270
510
0.228
60d N
CN
O
MeO
H
Cl
DMSO
408
5500
310
7080
510
0.113
60e N
CN
O
MeO
H
DMSO
H2O
4850
5100
-
-
-
-
505
500
0.057
0.050

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties144
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4.3.4. Stacking cyanocarbostyrils
Table 4. Photophysical data of stacking cyanocarbostyrils.
Cpd
Concentration
Solvent
λmax 1
UV-Vis (nm)
2
λmax 2
UV-Vis
(nm)
2
λmax
em. (nm)
23
2 mm cell
4.00E-05 CH3CN 497 5025 264 5025 567
0.081
DMSO 470 4320 320 5200 557
0.102
H2O 454 5400 322 6450 553
0.130
4.00E-06 CHCl3 452 5250 320 10500 522
0.203
N
O
O
CN
CN
N
O
O
CN
NC
N
CN
O
MeO
H
CN
Concentration dependence in DMSO only
1.00E-05 DMSO 460
8.00E-05 DMSO 479 4320 320 5200 557 0.102
1.00E-04 DMSO 483 4640 319
2.20E-04 DMSO 490 4668 316
4.30E-04 DMSO 497
3.00E-03 DMSO 507
32
NMeO
CN
OH
DMSO
425
1100
-
-
500
0.019

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties145
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
These stacking cyanocarbostyrils 23 and 32 (table 4) were prepared
under the same conditions as for 4-cyanocarbostyril derivatives 7 and 33 (table
2b) respectively, using a short reaction time. To our surprise, compound 23,
which is the lactim form of compound 7 exhibits remarkable fluorescence
properties with an absorption maximum at 497 nm and an emission maximum
of about 570 nm in acetonitrile, ever observed so far with carbostyrils. In
DMSO and in water, compound 23 is absorbing in the range of 454-470 nm,
emitting at about 560 nm, and shows the fluorescence quantum yield of about
10-13 %, with an excellent Stoke’ s shift of about 100 nm in water. The
fluorescence quantum yield is 2-times higher than those of the best
biscarbostyrils published [49]. This means that the new 3,4-dicyanocarbostyril
derivatives far outclass all biscarbostyrils in terms of absorption, emission
maxima, fluorescence quantum yields and Stoke’ s shifts data.
N
O
OH
CN
CN
N
O
OH
CN
NC
NMeO
CN
OH
23 32
Figure 15
It should be noticed that the fluorescence data of 23 are nearly the same
as those of its isomer compound 7 in DMSO with the only difference for the
extinction coefficient. The values of Stoke’s shifts increase with increasing
solvent polarity.
The absorption maxima of 23 are concentration and solvent dependence,
which means the absorption maxima increase with increasing of concentration.
However, spectral data of 23 in chloroform at 4.10-6 M. lowered slightly the
absorption and emission maxima, but exhibited a high extinction coefficient (ε
= 10500) and a better fluorescence quant F = 0.203). Interestingly,
when compound 23 was more diluted in DMSO to 3.10-3 M. in 2 mm cell the
absorption maximum moved up to 520 nm.

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties146
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
In contrast, in the case of 7-methoxy-cyanocarbostyril 32, the absorption
maximum of 425 nm and the emission maximum of 500 nm are respectively
53 nm and 94 nm shifted to longer wavelength regions compared with its
lactam form 33 (table 2b), whereas the fluorescence quantum yield and
extinction coefficient are much lower.
4.3.5. Fluorescence properties of 4-sulfinyloxycarbostyrils
Table 5. Photophysical data of 4-sulfinyloxycarbostyrils.
Cpd
Concentration
Solvent
λmax 1
UV-
Vis (nm)
2
λmax 2
UV-
Vis
(nm)
2
λmax
em. (nm)
Φ
8 N O
MeO
H
OS
O
CH3
DMSO
385
7750
-
-
490
0.180
U
N O
MeO
H
OS
O
CH3
MeO
DMSO
378
9180
-
-
486
0.270
The absorption maximum of 4-toluenesulfinyloxycarbostyril 8 (Table 5)
does not differ significantly from that of 4-cyanoquinolone 9 (Table 2b). To our
surprise, the emission maximum is 490 nm, 30 nm red-shifted compared with
9 higher than
the quantum yield observed for 4-cyanoquinolone 9, the epsilon coefficient (ε =
7700) is higher than 4-cyanoquinolone 9 (ε = 5400). We conclude that p-
toluenesulfinate at position 4 of carbostyrils is stronger electron-withdrawing
than a cyano group. These findings prompted us to measure also the
absorption and emission data of the already previously published 6.7-
dimethoxy analogue U. As hoped, the presence of sulfinate in position 4 and

Chapter 4: Systematic investigation of substituent effects on Fluorescence and photo. properties147
PhD-Thesis-Universiy of Graz/Austria-2010 ENOUA Guy Crépin
two electron delivering groups in pos 6 and 7 revealed a strongly redshifted
emission maximum at 486 nm, which is compared with F even 10 nm and
about 50 nm shifted to longer wavelength regions. Not so exciting is the
fluorescence quantum yield because it is about 50 % of F. The absorption
maximum is 386 nm hence we observed in case U a 100 nm Stoke´s shift.
4.4. CONCLUSION
Compared with published 4-cyano-6,7-dimethoxy push-pull substituted
carbostyrils, the novel 3,4-dicyano analogues show vastly improved absorption
and emission properties. For example, 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile 7 shows a red shift of 66 nm compared with 6-methoxy-2-
oxo-1,2-dihydroquinoline-4-dicarbonitrile 9 and 6,7-dimethoxy analogue 82
absorbed with 451 nm, a red shift of 57 nm compared with 9.
6-Methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7) shows excellent
spectroscopic properties. In addition, a methoxy group in position 7 is
required to improve the fluorescence quantum yield. The new carbostyril
chromophores 7 and 82 presenting a second maximum above 300 nm have
the possibility to be excited both in UV and visible regions that make them to
promising candidates for the construction of FRET molecules, and might be
useful as alternatives to established probes for biological investigations.

GENERAL CONCLUSION 148
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
GENERAL CONCLUSION
In conclusion, the current study demonstrates that the synthesis of 3,4-
dicyanoquinolones via the reactive 3-acetylamino-, 3-chloro- or 3-nitro-4-
chloroquinlones derivatives in presence of sodium p-toluenesulfinate and
potassium cyanide is an efficient and convenient method for the introduction of
the cyano groups into the heterocyclic moiety. The higher and interesting
spectroscopic properties of compounds 7 and 82 as well as their N-linkers
make them the probes of potential interest for attachment to biomolecules
such as peptides, proteins, carbohydrates.
The synthesis of 3,4-dicyanoquinolones via 3-H-4-chloroquinolones were
found to be very difficult to obtain. In this way the purity and the yields were
not sufficient. The syntheses of 3-aryl-4-cyanoquinolones are characterized by
an easier work-up and good yields. Absorption and emission of N-H
carbostyrils and their N-alkylated are very similar.
In view of possible applications of the new dyes (e.g. as fluorescent labels
in biological systems), compounds 7, 82 as well as their N-linkers have
greatest interest since these dyes can be linked to biomolecule through N-1
position and are well suited as fluorescence standards for FRET applications.

EXPERIMENTAL PART 149
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
EXPERIMENTAL PART
FOR
CHAPTERS 1, 2 AND 3

EXPERIMENTAL PART 150
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
EXPERIMENTAL PART
General Remarks:
Melting points (Mp) were determined with a Stuart SMP3 melting point
apparatus in open capillary tubes.
1H and 13C NMR spectra were recorded with a Bruker AMX 360 instrument
(360 or 90 MHz) or a Bruker Avance III instrument (300 MHz), or a Bruker
Avance DRX 500 instrument (500 or 125 MHz). The operator gives a locator
number called the Chemical Shift having units of parts per million (ppm) and
designated by the symbol , from the internal TMS standard. Data are reported
as follows: (recorded as: s, singlet; d, doublet; dd, doublet of doublets; t, triplet;
q, quadruplet and m, multiplet), coupling constants (J in Hertz, Hz),
integration, and assignment (phenyl, Ph; aryl, Ar).
Infrared spectra (IR) were recorded with a Mattson Galaxy Series FTIR 7020
instrument with potassium bromide discs or a Bruker Alpha –P, with
Attennated Total Reflectance (ATR) measurement, using a reflexion method and
are reported in terms of frequency of absorption (, cm-1), end expressed as: s,
strong; m, medium; w, weak; br, broad.
Elemental analyses were performed at the Microanalytical laboratory of the
University of Vienna, Austria.
Mass spectra (MS) were obtained with a HP 1100 LC/MSD mass spectral
instrument (positive or negative APCI ion source, 50–200 V, nitrogen) or
(positive or negative ESI ion source, 50–200 V, nitrogen). Mass spectral data
are reported as m/z.

EXPERIMENTAL PART 151
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
UV/Vis spectra were recorded with a Shimadzu UV/Vis scanning
spectrophotometer UV-2101 PC; concentration: 1x10–4 M.
Fluorescence data: Excitation and emission spectra were recorded with a
Perkin-Elmer LS50B luminescence spectrometer.
Determination of quantum yields: emission signals were set in relation to the
known area of the emission signal of quinine sulfate at pH 1. Corrections were
made for other solvents by using the factor (nwater/nsolvent) [ex-1].
Analytical HPLC was performed with a Shimadzu LC 20 system equipped with
a diode array detector (215 and 254 nm) with a Pathfinder AS reversed phase
(4.6150 mm, 5 µm) column, running an acetonitrile/water gradient (30–100 %
acetonitrile).
All reactions were monitored by thin-layer chromatography (TLC) on 0.2-mm
silica gel F-254 (Merck) plates by using UV light (254 and 366 nm) for
detection, and are expressed in reference frontal (Rf).
Dry column flash chromatography [ex-2] was carried out on silica gel 60 H
(5-40 µm) (Merck, Darmstadt, Germany). Common reagent-grade chemicals
were either commercially available and were used without further purification
or prepared by standard literature procedures. All optical measurements were
performed by using analytical-grade solvents.
X-Ray structures were solved by Prof. Dr. U. WAGNER at the Karl Franzens
University of Graz, Austria. The structure determination was performed on an
automatic four-circle Siemens P3/PC diffractometer, using graphite
monochromatized MoK radiation (0.71069 ) at 293 K, /2 scanning, 2
max.50°. The structure was solved by direct methods using the SHELX97
program package [83].

EXPERIMENTAL PART 152
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Hydroxy-6-methoxyquinolin-2(1H)-one (3)
A mixture of p-anisidin (1: 18.90 g, 154.0 mmol) and dry malonic acid (2:
23.40 g, 225.0 mmol) in phosphoryl chloride (24.0 mL, 40.08 g, 261.0 mmol)
was heated in an open flask to 95 °C for 90 minutes under stirring with a
thermometer, then cooled to 20 °C, poured onto ice/water (500 mL) and filtered
by suction. The precipitate was dissolved in aqueous sodium hydroxide (1000
mL, 1 M) under warming to 60 °C. The remaining insoluble solid [mainly N,N'-
bis (4-methoxyphenyl)malonamide (4)] was filtered.
To the alkaline filtrate, concentrated hydrochloric acid was added until pH = 1–
2 was reached, the precipitate filtered by suction, washed with water and dried
at 40 °C under reduced pressure, which afforded 4-hydroxy-6-
methoxyquinolin-2(1H)-one (3). The yield was 21.290 g (73 %), yellow prisms,
mp 324-328 °C (methanol), lit. mp 298–320 °C [ex-3, ex-4].
Rf = 0.23 (chloroform/aceton 3:7).
IR (ATR-measurement): 3434 (m), 3290 (s), 1663 (w), 1642 (s), 1616 (w), 1602
(w), 1539 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO-d6): = 3.77 (s, 3 H, CH3O), 5.79 (s, 1 H, 3-H),
7.14 (dd, J = 8.9 + 2.7 Hz, 1 H, 7-H), 7.20 (d, J = 8.8 Hz, 1 H, 8-H), 7.21 (d, J =
2.8 Hz, 1 H, 5-H), 11.08 (s, 1 H, NH).
C10H9NO3 (191.19).
N,N'-Bis(4-methoxyphenyl)malonamide (4)
This compound was obtained as insoluble solid during the work-up of
compound 3. The solid was purified by recystallization from acetic acid-
methanol (1:3). The yield was 9.167 g (19 %), grey prisms, mp 228.1–231.1 °C
(acetic acid-methanol), lit. mp 226–240 °C [ex-3a-c, ex-5, ex-6, ex-7, ex-8].
Rf = 0.87 (chloroform/aceton 3:7).
IR (KBr): 3435 (m), 1655 (w), 1620 (s), 1588 (s) cm -1.

EXPERIMENTAL PART 153
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (360 MHz, CDCl3): = 3.49 (s, 2 H, CH2), 3.81 (s, 6 H, 2 CH3O), 6.88
(d, J = 8.8 Hz, 4 H, Ar-H), 7.45 (d, J = 8.8 Hz, 4 H, Ar-H), 8.64 (s, 2 H, NH).
C17H18N2O4 (314.34).
2,4-Dichloro-6-methoxyquinoline (5)
A solution of 4-hydroxy-6-methoxyquinolin-2(1H)-one (3: 3.710 g, 19.42 mmol)
in phosphoroxychloride (45.0 mL) was heated under reflux for 8 hours. The
excess amount of phosphoroxychloride was removed under reduced pressure.
The residue was poured onto ice/water (300 mL) and brought to pH = 4–6 with
aqueous sodium hydroxide (5 M), filtered by suction, and washed with water.
The solid was dried at 40 °C under reduced pressure, to give 2,4-dichloro-6-
methoxyquinoline (5). The yield was 3.733 g (83 %), brown prisms, mp 175–
177 °C (ethanol), lit. mp: 130 °C [ex-9].
Rf = 0.90 (chloroform/aceton 3:7).
IR (KBr): 3439 (s), 1622 (m), 1564 (m) cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 3.95 (s, 3 H, CH3O), 7.43 (d, J = 2.6 Hz, 1
H, 5-H), 7.57 (dd, J = 9.1 + 2.7 Hz, 1 H, 7-H), 7.92 (s, 1 H, 3-H), 7.95 (d, J =
9.2 Hz, 1 H, 8-H).
C10H7Cl2NO (228.08).
4-Chloro-6-methoxyquinolin-2(1H)-one (6)
A solution of 2,4-dichloro-6-methoxyquinoline (5: 13.30 g, 58.33 mmol) and 70
% methanesulfonic acid (37.0 g, 25 mL, 385.42 mmol) in ethanol (300.0 mL)
was heated under reflux for 28 hours. The mixture was cooled to 20 °C and
poured into ice/water (100 mL), brought to pH = 4–6 with sodium hydroxide (2
M), filtered by suction, washed with water and dried at 40 °C under reduced
pressure, to give 4-chloro-6-methoxyquinolin-2(1H)-one (6). The yield was
11.552 g (94 %), brownish prisms, mp 296–300 °C (toluene).

EXPERIMENTAL PART 154
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Rf = 0.72 (chloroform/aceton 3:7).
IR (KBr): 1645 (s), 1623 (m), 1576 (w), 1569 (w), 1558 (w), 1541 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 (s, 3 H, CH3O), 6.82 (s, 1 H, 3-H), 7.27
(dd, J = 7.6 + 2.5 Hz, 1 H, 7-H), 7.31 (d, J = 2.6 Hz, 1 H, 5-H), 7.34 (d, J = 8.5
Hz, 1 H, 8-H), 11.95 (s, 1 H, NH).
13H-NMR (360 MHz, [D6]DMSO): = 56.0 (CH3O), 106.3 (3-C), 117.8 (8-C),
118.2 (10-C), 121.7 (5-C), 112.1 (7-C), 133.6 (9-C), 143.8 (4-C), 155.1 (6-C),
160.4 (lactam-C=O).
MS (APCI pos): m/z (%) = 212 (30, M + 2), 211 (13, M + 1), 210 (100, M).
MS (APCI neg): m/z (%) = 210 (35, M +2), 209 (10, M + 1), 208 (100, M), 193 (7,
M - 15).
Anal. for C10H8ClNO2 (209.63).
Calcd: C 57.30, H 3.85, N 6.68
Found: C 57.21, H 3.70, N 6.62
6-Methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7)
Pathway A
A mixture of 4-chloro-6-methoxyquinolin-2(1H)-one (6: 0.600 g, 2.86 mmol),
sodium p-toluenesulfinate (1.068 g, 6.0 mmol), and potassium cyanide (0.465
g, 7.15 mmol) in dry dimethylformamide (17.0 mL) was heated to 140 °C for 43
hours with vigorous stirring. The solution was cooled to 20 °C, poured into
ice/water (100 mL) and acidified with concentrated hydrochloric acid to pH =
1–2. Then it was filtered by suction, washed with water and dried at 40 °C
under reduced pressure, to afford 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile (7). The yield was 0.41g (64 %), yellow prisms, mp 303–305 °C
(acetonitrile).
Rf = 0.81 (chloroform/aceton 3:7).

EXPERIMENTAL PART 155
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Pathway B
A mixture of compound 6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl 4-
methylbenzenesulfinate (8: 1.00 g, 3.04 mmol) and potassium cyanide (0.600
g, 9.12 mmol) in dry dimethylformamide (20.0 mL) was heated to 140 °C for 72
hours, and worked up as described for the pathway A, to give 6-methoxy-2-
oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7). The yield was 0.396 g (58 %),
yellow prisms, mp 307–310 °C (acetic acid).
Rf = 0.82 (chloroform/aceton 3:7).
Pathway C
A mixture of 6-methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (9: 1.00 g,
5.0 mmol), sodium p-toluenesulfinate (2.140 g, 12.02 mmol) and potassium
cyanide (0.975 g, 15 mmol), in dry dimethylformamide (30.0 mL) was heated to
140 °C for 65 hours with vigorous stirring and worked-up using the procedure
described for the pathway A, to give 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile (7). The yield was 0.812 g (72 %), red prisms, mp 309–312 °C
(acetone).
Rf = 0.78 (chloroform/aceton 3:7).
Pathway D
A mixture of 3,4-dichloro-6-methoxyquinolin-2(1H)-one (13: 0.620 g, 2.54
mmol), sodium p-toluenesulfinate (0.950 g, 5.34 mmol) and potassium cyanide
(0.413 g, 6.35 mmol), in dry dimethylformamide (15.0 mL) was heated to 140
°C for 46 hours with vigorous stirring and worked-up using the procedure
described for the pathway A, to give 6-methoxy-2-oxo-1,2-dihydroquinoline-
3,4-dicarbonitrile (7). The yield was 0.408 g (72 %), red prisms, mp 302–306 °C
(acetone).
Rf = 0.82 (chloroform/aceton 3:7).
Pathway E
A mixture of 4-chloro-6-methoxy-3-nitro-quinolin-2(1H)-one (16: 32.0 mg, 0.13
mmol), potassium cyanide (30.0 mg, 0.46 mmol) and sodium p-
toluenesulfinate (50.0 mg, 0.27 mmol) in dry dimethylformamide (1.0 mL), was

EXPERIMENTAL PART 156
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
heated to 140 °C for 2.5 hours and worked-up using the procedure described
for the pathway A, to give 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile (7). The yield was 0.026 g (93 %), red prisms, mp 301–304 °C
(acetonitrile).
Rf = 0.79 (chloroform/aceton 3:7).
Pathway F
A mixture of N-(4-chloro-6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)acetamide
(18: 1.670 g, 6.27 mmol), potassium cyanide (1.018 g, 15.67 mmol) and
sodium p-toluenesulfinate (2.342 g, 13.16 mmol) in dry dimethylformamide
(38.0 mL), was heated to 140 °C for 45 hours with vigorous stirring and worked
up using the procedure described for the pathway A, to give 6-methoxy-2-oxo-
1,2-dihydroquinoline-3,4-dicarbonitrile (7). The yield was 1.373 g (86 %), red
prisms, mp 300–302 °C (acetonitrile).
Rf = 0.79 (chloroform/aceton 3:7).
Pathway G
A mixture of 4-chloro-6-methoxy-3-piperidin-1-ylquinolin-2(1H)-one (22:
0.338 g, 1.16 mmol), sodium p-toluenesulfinate (0.440 g, 2.47 mmol) and
potassium cyanide (0.200 g, 2.90 mmol), in dry dimethylformamide (7.0 mL)
was heated to 140 °C for 5 days with vigorous stirring and worked up using the
procedure described for the pathway A, to give 6-methoxy-2-oxo-1,2-
dihydroquinoline-3,4-dicarbonitrile (7). The yield was (66 %), red-green prisms.
Rf = 0.80 (chloroform/aceton 3:7).
IR (KBr): 2972–2742 (w, br), 2228 (w), 1650 (s), 1615 (w), 1555 (w), 1498 (m)
cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.88 (s, 3 H, CH3O), 7.12 (d, J = 2.7 Hz, 1
H, 5-H), 7.42 (d, J = 9.2 Hz, 1 H, 8-H), 7.52 (dd, J = 9.2 + 2.7 Hz, 1 H, 7-H),
13.08 (s, 1 H, NH).
13C-NMR (360 MHz, [D6]DMSO): = 56.3 (CH3O), 106.5 (3-C), 113.4 (8-C),
114.6 (10-C), 116.7 (5-C), 118.8 (CN), 126.4 (7-C), 129.5 (9-C), 135.8 (4-C),
156.1 (6-C), 157.0 (lactam-C=O).

EXPERIMENTAL PART 157
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
MS (API-ESI neg): m/z (%) = 225 (15, M + 1), 224 (100, M), 209 (6, M – 15).
Anal. For C12H7N3O2 (225.21).
Calcd: C 64.00, H 3.13, N 18.66
Found: C 63.70, H 3.00, N 18.33
6-Methoxy-2-oxo-1,2-dihydroquinolin-4-yl
4-methylbenzenesulfinate (8)
Pathway A
A mixture of 3-chloro-6-methoxyquinolin-2(1H)-one (6: 5.400 g, 25.77 mmol),
and sodium p-toluenesulfinate (6.880 g, 38.66 mmol) in dry
dimethylformamide (150.0 mL) was heated to 120 °C for 20 hours. The
resulting solution was cooled to 20 °C and poured into ice/water (500 mL). The
obtained solid was filtered by suction, washed with water and dried at 40 °C
under reduced pressure to afford 6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl 4-
methylbenzenesulfinate (8). The yield was 5.836 g (69 %), yellow prisms, mp
255–258 °C (dioxane).
Rf = 0.77 (chloroform/aceton 3:7).
Pathway B
This compound was obtained during the work-up of the compound 9 from 3-
chloro-6-methoxyquinolin-2(1H)-one (6) by crystallization from dioxane and the
separated solid was recrystallized from dioxane to yield 8, 1.256 g (32 %),
yellow prisms, mp 256–259 °C (dioxane).
Rf = 0.79 (chloroform/aceton 3:7).
IR (ATR-measurement): 3146 (w), 2955 (w), 2839 (w), 1667 (s), 1622 (w), 1596
(w), 1559 (w), 1503 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 2.38 (s, 3 H, CH3), 3.73 (s, 3 H, CH3O),
7.24 (s, 1 H, 3-H), 7.26 (dd, J = 9.0 + 2.7 Hz, 1 H, 7-H), 7.33 (d, J = 9.1 Hz, 1
H, 8-H), 7.46 (d, J = 2.8 Hz, 1 H, 5-H), 7.48 (d, J = 8.2 Hz, 2 HBB´ Ar-H), 7.96 (d,
J = 8.4 Hz, 2 HAA´ Ar-H), 12.31 (s, 1 H, NH).

EXPERIMENTAL PART 158
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
13C-NMR (300 MHz, [D6]DMSO): = 21.6 (CH3), 66.8 (CH3O), 106.7 (3-C),
118.3 (Aromatic-C), 121.4 (Aromatic-C), 125.7 (Aromatic-C), 128.4 (Aryl-C),
130.9 (9-C + 2 x Aryl-CBB´), 136.2 (2 x Aryl-CAA´), 146.1 (Aryl-C), 147.5 (4-C),
154.6 (6-C), 159.8 (lactam-C=O).
MS (API-ESI neg): m/z (%) = 329 (21, M + 1), 328 (100, M).
Anal. for C17H15NO4S (329.38).
Calcd: C 61.99, H 4.59, N 4.25 O 19.43
Found: C 61.83, H 4.38, N 4.50 O 19.67
6-Methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (9)
Methode A
A mixture of compound 6-methoxy-2-oxo-1,2-dihydroquinolin-4-yl 4-
methylbenzenesulfinate (8: 1.00 g, 3.04 mmol) and potassium cyanide (0.30 g,
4.62 mmol) in dry dimethylformamide (150.0 mL) was heated to 70 °C for 5
hours. The solution was cooled to room temperature, poured into ice/water
and acidified with concentrated hydrochloric acid to pH = 1–2. The resulting
solid was filtered by suction, washed with water and dried at 40 °C under
reduced pressure to afford 6-methoxy-2-oxo-1,2-dihydroquinoline-4-
carbonitrile (9). The yield was 0.455 g (67 %), brown prisms, mp 290–293 °C
(acetone).
Rf = 0.81 (chloroform/aceton 3:7).
Methode B
A mixture of 4-chloro-6-methoxyquinolin-2(1H)-one (6: 2.50 g, 11.93 mmol),
sodium p-toluenesulfinate (4.460 g, 25.05 mmol), and potassium cyanide
(1.940 g, 29.83 mmol) in dry dimethylformamide (71.0 mL) was heated to 130
°C for 27 hours with vigorous stirring and worked up as described for 7,
pathway A. TLC analysis showed two products: 6-methoxy-2-oxo-1,2-
dihydroquinolin-4-yl 4-methylbenzenesulfinate (8) and 6-methoxy-2-oxo-1,2-
dihydroquinoline-4-carbonitrile (9).

EXPERIMENTAL PART 159
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The yield of the mixture was 2.027 g (85 %). The 2 products were separated by
recrystallization from dioxane to give first compound 9 and secondly compound
8 in 0.87 g (32 % yield), respectively.
6-Methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (9) was recrystallized
from methanol, to give 1.068 (49 %), gold prisms, mp 293–295 °C (methanol).
Rf = 0.83 (chloroform/aceton 3:7).
IR (ATR-measurement): 3156 (w), 3082 (w), 2991 (w), 2914 (w), 2825 (w), 2227
(w), 1655 (s), 1622 (m), 1571 (w), 1499 (m) cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 3.80 (s, 3 H, CH3O), 7.29 (d, J = 2.4 Hz, 1
H, 5-H), 7.31 (d, J = 8.4 Hz, 1 H, 8-H), 7.36 (dd, J = 9.0 + 2.6 Hz, 1 H, 7-H),
8.68 (s, 1 H, 3-H), 12.40 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 56.0 (CH3O), 106.7 (10-C), 110.1 (5-C),
116.5 (7-C), 117.6 (8-C), 118.7 (CN), 124.3 (4-C), 135.3 (9-C), 149.4 (3-C),
155.1 (16-C), 158.7 (lactam-C=O).
MS (API-ESI neg): m/z (%) = 200(13, M + 1), 199 (100, M), 184 (11, M – 15).
Anal. for C11H8N2O2 (200.20).
Calcd: C 66.00, H 4.03, N 13.99
Found: C 65.16, H 3.85, N 13.59
The elemental analysis showed a small deviation of 0.84 % for carbon
component due to the fact that the compound 9 was not well soluble in almost
all organic solvents.
3,3-Dichloro-6-methoxyquinoline-2,4(1H,3H)-dione (10)
A suspension of 4-hydroxy-6-methoxyquinolin-2(1H)-one (3: 35.00 g, 183.25
mmol) in dioxane (400.0 mL) was warmed to 40–50 °C, and then under
vigorous stirring, keeping the temperature between 50–60 °C, sulfuryl chloride
(35.00 mL, 439.80 mmol ) was added dropwise. As soon as the temperature
reached 60 °C, the reaction was stopped and cooled to 20 °C. The mixture was
filtered and the filtrate was poured into ice/water (1500 mL) under stirring,
and then filtered by suction, washed with water and dried at 40 °C under
reduced pressure, to yield 3,3-dichloro-6-methoxyquinoline-2,4(1H, 3H)-dione

EXPERIMENTAL PART 160
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
(10). The product was pure for further reactions. The yield was 34.983 g (73
%), yellow prisms, mp 219–223 °C, lit. mp 215–216 °C [ex-3a].
Rf = 0.89 (chloroform/aceton 3:7).
IR (KBr): 3434 (m), 3198 (m), 3118 (w), 3076 (m), 2976 (m), 2915 (m), 2839 (w),
1725 (s), 1688 (s), 1622 (m), 1500 (s) cm-1.
1H-NMR (360 MHz, [D6] DMSO): = 3.79 (s, 3 H, CH3O), 7.07 (d, J = 8.5 Hz, 1
H, 8-HH), 7.28 (d, J = 2.8 Hz, 1 H, 5-H), 7.30 (dd, J = 8.6 + 2.9 Hz, 7-H), 11.27 (s,
1 H, NH).
C10H7Cl2NO3 (260.08).
3-Chloro-4-hydroxy-6-methoxyquinolin-2(1H)-one (11)
To a solution of 3,3-dichloro-6-methoxyquinoline-2,4(1H,3H)-dione (10: 5.250
g, 20.20 mmol), in ethanol (52.0 mL), and acetic acid (26.0 mL), zinc-dust
(5.210 g, 79.66 mmol) was added in small portions, the solution was kept to
boiling. The yellow solution got decolorized (from yellow to grey-greenish),
which indicated the end of reaction. The solution was cooled to room
temperature and filtered from zinc. To the filtrate, ice/water (500 mL) was
added. The colorless precipitate was filtered by suction, washed with water and
dried at 40 °C under reduced pressure to give 3-chloro-4-hydroxy-6-
methoxyquinolin-2(1H)-one (11). The yield was 4.023 g (89 %), colorless
powder, mp 270–273 °C (ethanol), lit. mp not given [ex-10].
Rf = 0.33 (chloroform/aceton 3:7).
IR (KBr): 3432 (m), 3281 (w), 2960 (w), 2835 (w), 1633 (w), 1586 (s), 1537 (m)
cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 3.68 (s, 3 H, CH3O), 7.00 (dd, J = 8.7 + 2.6
Hz, 1 H, 7-H), 7.12 (d, J = 8.8 Hz, 1 H, 8-H), 7.41 (d, J = 2.5 Hz, 1 H, 5-H ),
10.84 (s, 1 H, NH).
C10H8ClNO3 (225.63).

EXPERIMENTAL PART 161
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2,3,4-Trichloro-6-methoxyquinoline (12)
A solution of 3-chloro-4-hydroxy-6-methoxyquinolin-2(1H)-one (11: 6.00 g,
28.64 mmol) in phosphoroxychloride (31.50 mL) was brought to the reaction
and worked up according to the method described for 5. The yield was 6.244 g
(90 %), beige powder, mp 276–279 °C (toluene).
Rf = 0.92 (chloroform/aceton 3:7).
IR (ATR-measurement): 3012 (w), 2944 (w), 2359 (w), 2339 (w), 1619 (m), 1546
(m), 1486 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.97 (s, 1 H, CH3O), 7.40 (d, J = 2.8 Hz, 1
H, 5-H ), 7.57 (dd, J = 9.2 + 2.7 Hz, 1 H, 7-H ), 7.96 (d, J = 9.2 Hz, 1 H, 8-H).
Anal. for C10H6Cl3NO (262.52).
Calcd: C 45.75, H 2.30, N 5.34
Found: C 45.71, H 2.44, N 5.27
3,4-Dichloro-6-methoxyquinolin-2(1H)-one (13)
A solution of 2,3,4-trichloro-6-methoxyquinoline (12: 6.500 g, 24.76 mmol),
and 70 % methanesulfonic acid (6.50 mL) in n-butanol (130.0 mL) was heated
under reflux for 45 hours. The mixture was cooled to 20 °C and poured into ice
/ water (50 mL), brought to pH = 4–6 with sodium hydroxide (2 M), filtered by
suction, washed with water and dried at 40 °C under reduced pressure to give
3,4-dichloro-6-methoxyquinolin-2(1H)-one (13). The yield was 13.439 g (88 %),
beige prisms, mp 264–265 °C (xylen).
Rf = 0.82 (chloroform/aceton 3:7).
IR (KBr): 3467 (m), 2840 (m), 2720 (w), 1657 (s), 1599 (s) cm-1.
1H-NMR (360 MHz, [D6] DMSO): = 3.83 (s, 3 H, CH3O), 7.25 (d, J = 2.2 Hz, 1
H, 5-H), 7.30 (dd, J = 8.9 + 2.4 Hz, 1 H, 7-H), 7.36 (d, J = 8.9 Hz, 1 H, 8-H),
12.47 (s, 1 H, NH).
Anal. for C10H7Cl2NO2 (244.08).
Calcd: C 49.21, H 2.89, N 5.74
Found: C 49.24, H 2.83, N 5.68

EXPERIMENTAL PART 162
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Hydroxy-6-methoxy-3-nitroquinolin-2(1H)-one (14)
A solution of 4-hydroxy-6-methoxyquinolin-2(1H)-one (3: 13.60 g, 71.20 mmol)
in glacial acetic acid (140.0 mL) was treated with concentrated nitric acid (14.0
mL) and with sodium nitrite (0.60 g, 8.70 mmol) to start a strongly exothermic
reaction. The starting material dissolved, followed immediately by precipitation
of the product. The mixture was stirred for 30 minutes and poured onto
ice/water (1500 mL), stirred, filtered by suction and washed with water. The
solid was dried at 40 °C under reduced pressure to give 4-hydroxy-3-nitro-7-
methoxyquinolin-2(1H)-one (14). The yield was 12.731g (76 %), yellow prisms,
mp 253.5–256.8 °C (methanol), lit. mp. not given [ex-11].
Rf = 0.33 (chloroform/aceton 3:7).
IR (KBr): 3435 (w), 3001 (m), 2896 (m), 2847 (m), 2785 (w), 2839 (w), 1668 (s),
1636 (w), 1609 (s), 1530 (s) cm-1.
1H-NMR (360 MHz, [D6] DMSO): = 3.81 (s, 3 H, CH3O), 7.28 (d, J = 8.9 Hz, 1
H, 8-H), 7.31 (dd, J = 9.1 + 2.5 Hz, 1 H, 7-H), 7.49 (d, J = 2.2 Hz, 1 H, 5-H),
11.89 (s, 1 H, NH).
C10H8N2O5 (236.19).
2,4-Dichloro-6-methoxy-3-nitroquinoline (15)
A solution of 4-hydroxy-6-methoxy-3-nitroquinolin-2(1H)-one (14: 0.862 g,
3.65 mmol) and dry triethylamine (0.60 mL, 4.40 mmol) in phosphor-
oxychloride (4.50 mL) was refluxed for 1 hour. The excess amount of phosphor-
oxychloride was removed under reduced pressure. The residue was poured
onto ice/water (100 mL) and the solution was brought to pH = 4–6 with
aqueous sodium hydroxide (5 M), filtered by suction, and washed with water.
The solid was dried at 40 °C under reduced pressure. The yield was 0.905 g (91
%), brown prisms, mp 195–196 °C (dioxane), lit. mp: not given [ex-12].
Rf = 0.95 (chloroform/aceton 3:7).
IR (KBr): 3437 (s), 2970 (w), 1617 (s), 1566 (m), 1547 (s) cm-1.

EXPERIMENTAL PART 163
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (360 MHz, [D6] DMSO): = 4.01 (s, 3 H, CH3O), 7.52 (d, J = 2.7 Hz, 1
H, 5-H), 7.75 (dd, J = 9.3 + 2.7 Hz, 1 H, 7-H), 8.10 (d, J = 9.3 Hz, 1 H, 8-H).
C10H6Cl2N2O3 (273.08).
4-Chloro-6-methoxy-3-nitroquinolin-2(1H)-one (16)
A solution of 2,4-dichloro-6-methoxy-3-nitroquinoline (15: 2.00 g, 7.33 mmol),
and 70 % methanesulfonic acid (6.0 mL) in n-butanol (40.0 mL ) was heated to
110 °C for 45 hours. The mixture was cooled to room temperature and poured
into ice/water (100 mL), the solution was brought to pH = 4–6 with sodium
hydroxide (2 M), filtered by suction, washed with water and dried at 40 °C
under reduced pressure. The purification was performed by dry flash
chromatography using chloroform-acetone (3:7) as eluent, which afforded 4-
chloro-6-methoxy-3-nitroquinolin-2(1H)-one (16). The yield was 1.244 g (67 %),
mp 274–276 °C (chloroform/acetone 3:7), yellow-green prisms.
Rf = 0.75 (chloroform/aceton 3:7).
IR (KBr): 3437 (m), 2997 (w), 2916 (w), 2866 (m), 2761 (w), 1663 (s), 1626 (w),
1596 (w), 1543 (s) cm-1.
1H-NMR (360 MHz, [D6] DMSO): = 3.87 (s, 3 H, CH3O), 7.30-7.39 (m, 1 H, 7-
H), 7.42-7.49 (m, 2 H, 5-H and 8-H), 12.99 (s, 1 H, NH).
MS (APCI pos): m/z (%) = 257 (30, M + 2), 256 (10, M + 1), 255 (100, M).
MS (APCI neg): m/z (%) = 255 (34, M + 2), 254 (10, M + 1), 253 (100, M).
Anal. For C10H7ClN2O4 (254.63).
Calcd: C 47.17, H 2.77, N 11.00
Found: C 47.26, H 2.62, N 10.87

EXPERIMENTAL PART 164
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
N-(4-Hydroxy-6-methoxy-2-oxo-1,2-dihyroquinolin-3-
yl)acetamide (17)
A solution of 4-hydroxy-6-methoxy-3-nitroquinolin-2(1H)-one (14: 10.750 g,
45.55 mmol) in acetic acetic (350.0 mL) and acetic anhydride (236.0 mL) was
heated to reflux. Then, zinc-dust (about 20 g, 300 mmol) was added until the
yellow color was decolorized, then the mixture was refluxed 30 minutes. The
mixture was filtered from zinc salt, the filtrate was taken to dryness at reduced
pressure. The residue was dissolved in aqueous sodium hydroxide (600 mL, 1
M), kept for 10 minutes at 20 °C and then under stirring, acidified with
concentrated hydrochloric acid to pH = 1. The precipitate was filtered by
suction, washed with water and dried at 40 °C under reduced pressure to
afford N-(4-hydroxy-6-methoxy-2-oxo-1,2-dihyroquinolin-3-yl)acetamide (17).
The yield was 7.702 g (68 %), beige powder, mp 300–303 °C (toluene).
Rf = 0.70 (chloroform/aceton 3:7).
IR (ATR-measurement): 3434 (m), 3326 (m), 2964 (m), 2913 (m), 2873 (w),
2837 (m), 1654 (w), 1640 (w), 1615 (s), 1530 (s), 1502 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 2.24 (s, 3 H, CH3), 3.79 (s, 3 H, CH3O),
7.15 (dd, J = 8.8 + 2.3 Hz, 1 H, 7-H), 7.22 (d, J = 8.9 Hz, 1 H, 8-H), 7.27 (d, J =
2.1 Hz, 1 H, 5-H), 9.74 (s, 1 H, NHAc), 11.73 (s, 1 H, NH), 11.98 (s, 1 H, OH).
Anal. for C12H12N2O4 (248.24).
Calcd: C 58.06, H 4.87, N 11.28
Found: C 57.92, H 4.62, N 11.01
N-(4-Chloro-6-methoxy-2-oxo-1,2-dihydroquinolin-3-
yl)acetamide (18)
To a solution of N-(4-hydroxy-6-methoxy-2-oxo-1,2-dihyroquinolin-3-
yl)acetamide (17: 5.50 g, 22.20 mmol) in phosphoroxychloride (26.50 mL), dry
triethylamine was added (3.70 mL, 26.60 mmol), then the solution was
refluxed for 1 hour. The excess amount of phosphoroxychloride was removed

EXPERIMENTAL PART 165
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
under reduced pressure. The residue was poured onto ice/water (400 mL) and
brought to pH = 4–6 with sodium hydroxide (2 M). The solution was filtered by
suction, washed with water, and dried at 40 °C under reduced pressure to
afford N-(4-chloro-6-methoxy-2-oxo-1,2-dihydroquinolin-3-yl)acetamide (18).
The yield was 5.248 g (90 %), dark-brown powder, mp 283–285 °C (dioxane).
Rf = 0.42 (chloroform/aceton 3:7).
IR (ATR-measurement): 3435 (m), 3225 (m), 3169 (w), 3094 (w), 3010 (w), 2937
(w), 2874 (w), 1651 (s), 1620 (m), 1518 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 2.05 (s, 3 H, CH3), 3.84 (s, 3 H, CH3O),
7.25 (d, J = 2.4 Hz, 1 H, 5-H), 7.31-7.35 (m, 2 H, 7-H and 8-H), 9.60 (s, 1 H,
NHAc), 12.18 (s, 1 H, NH).
MS (APCI pos): m/z (%) = 269 (30, M + 2), 268 (12, M + 1), 267 (100, M), 225
(8, M – 43),
Anal. for C12H11ClN2O3 (266.69).
Calcd: C 54.05, H 4.16, N 10.50
Found: C 54.30, H 3.97, N 10.46
6-Methoxy-3,3-di(piperidin-1-yl)quinoline-2,4(1H,3H)-dione (19)
A solution of 3,3-dichloro-6-methoxyquinolin-2,4(1H,3H)-dione (10: 4.60 g,
17.70 mmol) was dissolved in dry dimethylformamide (20.0 mL), and cooled to
0 °C, then piperidine (12 mL, 10.32 g, 121.41 mmol) was added, which gave a
dark color of the mixture, the temperature raised to 35 °C, and piperidine
hydrochloride precipitated. The mixture was stirred for 30 minutes, cooled to 0
°C, and water (80 mL) added dropwise. The solution was filtered by suction,
washed with water and dried at 40 °C under reduced pressure, to afford 6-
methoxy-3,3-di(piperidin-1-yl)quinoline-2,4(1H,3H)-dione (19). The yield was
4.917 g (78 %), yellow-green prisms, mp 162–164 °C (ethanol).
Rf = 0.69 (chloroform/aceton 3:7).
IR (KBr): 3435 (s), 3202 (m), 3088 (w), 3059 (w), 2988 (w), 2932 (s), 2851 (m),
2796 (w), 1688 (s), 1657 (s), 1616 (w), 1502 (s) cm-1.

EXPERIMENTAL PART 166
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (360 MHz, [D6]CDCl3): = 1.47-1.54 (m, 12 H, piperidin-H), 2.62-2.64
(m, 8 H, NCH2 of piperidin), 3.86 (s, 3 H, CH3O), 6.82 (d, J = 8.7 Hz, 1 H, 8-H ),
7.11 (dd, J = 8.7 + 2.7 Hz, 1 H 7-H), 7.39 (d, J = 2.7 Hz, 1 H, 5-H), 8.36 (s, 1 H,
NH).
C20H27N3O3 (357.46).
The elemental analysis of 19 could not be obtained because of the easy
decomposition, but elemental analysis for its follow-up compound is correct.
4-Hydroxy-6-methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (20)
6-Methoxy-3,3-di(piperidin-1-yl)quinoline-2,4(1H,3H)-dione (19: 2.00 g, 5.60
mmol) were dissolved in water/ethanol (1:1, 32.0 mL), the solution heated to
reflux and sodium dithionite (2.00 g, 13.0 mmol) added The mixture was
stirred for 30 minutes under reflux, cooled to 5 °C and kept it 2 hours at this
temperature. The product precipitated, was filtered by suction, washed with
water and dried at 40 °C under reduced pressure, to give 4-Hydroxy-6-
methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (20). The product was sufficiently
pure for further reactions. The yield was 1.44 g (96 %), beige prisms, mp 228–
231°C.
Rf = 0.44 (chloroform/aceton 3:7).
IR (KBr): 3439 (m), 2991 (m), 2952 (m), 2851 (m), 2831 (w), 1638 (s), 1597 (s),
1544 (s) cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 1.50-1.52 (m, 2 H, piperidin-H), 1.70-1.80
(m, 4 H, piperidin-H), 3.39 (s, 3 H, CH3O), 3.73-3.76 (m, 4 H, piperidin-NH),
7.04 (dd, J = 8.8 + 2.8 Hz, 1 H, 7-H), 7.10 (d, J = 8.8 Hz, 1 H, 8-H), 7.28 (d, J =
2.7 Hz, 1 H, 5-H), 10.66 (s, 1 H, NH).
MS (APCI neg): m/z (%) = 274 (5, M + 1), 273 (100, M).
MS (APCI pos): m/z (%) = 276 (18, M + 1), 275 (100, M).
Anal. for C15H18N2O3 (274.32).
Calcd: C 65.68, H 6.61, N 10.21
Found: C 65.67, H 6.37, N 10.22

EXPERIMENTAL PART 167
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2,4-Dichloro-6-methoxy-3-(piperidin-1-yl)quinoline (21)
A solution of 4-hydroxy-6-methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (20:
0.90 g, 3.30 mmol) in phosphoroxychloride (10.0 mL) was brought to the
reaction and worked up as described for 5, to give 2,4-Dichloro-6-methoxy-3-
(piperidin-1-yl)quinoline (21). The yield was 0.482 g (47 %), brown prisms, mp
182–185 °C (methanol).
Rf = 0.94 (chloroform/aceton 3:7).
IR (KBr): 3433 (m), 2932 (m), 2847 (w), 1620 (s), 1556 (w) cm-1.
1H-NMR (360 MHz, [D3] CDCl3): = 1.66-1.81 (m, 6 H, piperidin-H), 3.23-3.25
(m, 4 H, piperidin-NH), 3.97 (s, 3 H, CH3O), 7.30 (dd, J = 9.1 + 2.6 Hz, 1 H, 7-
H), 7.39 (d, J = 2.4 Hz, 1 H, 5-H), 7.86 (d, J = 9.2 Hz, 1 H, 8-H).
C15H16Cl2N2O (311.21).
4-Chloro-6-methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (22)
Method A
A solution of 2,4-dichloro-6-methoxy-3-(piperidin-1-yl)quinoline (21: 2.00 g,
6.43 mmol) and 70 % methanesulfonic acid (1.50 mL) in n-butanol (15.0 mL)
was refluxed for 48 hours and worked up as described for 6 to give 4-chloro-6-
methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (22). The yield was 0.997 g (53
%), pale green prisms, mp 210–214 °C (ethanol).
Rf = 0.88 (chloroform/aceton 3:7).
Method B
A solution of 4-hydroxy-6-methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one
(20:1.00 g, 3.65 mmol) in phosphoroxychloride (8.0 mL) was heated to 80 °C
for 25 minutes and worked up as described for 5 to form directly 4-chloro-6-
methoxy-3-(piperidin-1-yl)quinolin-2(1H)-one (22). The yield was 0.875 g (82
%), pale green prisms, mp 213–215.9 °C (ethanol).
Rf = 0.85 (chloroform/aceton 3:7).

EXPERIMENTAL PART 168
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
IR (KBr): 3454 (s), 2991 (w), 2924 (m), 2840 (m), 1643 (s), 1599 (w), 1554 (w)
cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 1.56-1.60 (m, 6 H, piperidin-H), 3.12-3.14
(m, 4 H, piperidin-NH), 3.82 (s, 3 H, CH3O), 7.11 (dd, J = 8.8 + 2.7 Hz, 1 H, 7-
H), 7.22 (d, J = 8.9 Hz, 1 H, 8-H), 7.26 (d, J = 2.7 Hz, 1 H, 5-H), 11.83 (s, 1 H,
NH).
MS (APCI pos): m/z (%) = 295 (33, M + 2), 294 (20, M + 1), 293 (100, M).
C15H17ClN2O2 (292.77).
Dicyano-6-methoxyquinoline (23)
A mixture of 3,4-dichloro-6-methoxyquinolin-2(1H)-one (13: 3.690 g, 15.12
mmol), sodium p-toluenesulfinate (1.50 g, 8.43 mmol) and potassium cyanide
(2.52 g, 38.77 mmol), in dry dimethylformamide (50.0 mL) was heated to 140
°C for 3 hours with vigorous stirring and worked up using the procedure
described for 7 (pathway A), to give the compound 23. The yield was 31.747 g
(76 %), red prisms, mp > 350 °C (acetonitrile).
Rf = 0.80 (chloroform/aceton 3:7).
IR (KBr): 3433 (m), 2213 (s), 1657 (w), 1628 (w), 1583 (s), 1550 (s), 1500 (s) cm-
1.
1H-NMR (360 MHz, [D6]DMSO): = 3.80 (s, 3 H, 6-CH3O), 6.84 (d, J = 1.9 Hz, 1
H, 5-H), 7.17-7.21 (m, 2 H, 7-H and 8-H).
13C-NMR (300 MHz, [D6]DMSO): = 55.7 (CH3O), 102, 115.0, 116.7, 118.0
(CN), 122.5, 125.0, 127.0, 148.5, 153.8 .
MS (APCI neg): m/z (%) = 225 (8, M + 1), 224 (100, M), 209 (6, M – 15).
Anal. For C12H7N3O2 (225.21).
Calcd: C 64.00, H 3.13, N 18.66 O 14.21
Found: C 63.87, H 3.00, N 18.27 O 14.47

EXPERIMENTAL PART 169
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Hydroxy-7-methoxyquinolin-2-(1H)-one (28)
A mixture of m-anisidin (27: 18.950 g, 17.30 mL, 154.06 mmol) and dry
malonic acid (2: 23.40 g, 225.0 mmol) in phosphoryl chloride (24.0 mL) was
heated in an open flask to 95 °C for 90 minutes under stirring with a
thermometer, then cooled to 20 °C, poured onto ice/water (500 mL) and filtered
by suction. The precipitate was dissolved in aqueous sodium hydroxide (450
mL, 1 M) under warming to 60 °C. The remaining insoluble solid (mainly 2,4-
dichloro-7-methoxyquinoline, 30) was filtered.
To the alkaline filtrate, concentrated hydrochloric acid was added until pH = 1–
2 was reached, the precipitate filtered by suction, washed with water (150 mL)
and dried at 40 °C under reduced pressure.
The solid was separated by recrystallization from methanol which afforded as
first crop 4-hydroxy-5- methoxyquinolin-2(1H)-one (29) and as second crop the
main compound, 4-hydroxy-7-methoxyquinolin-2(1H)-one (28). The yield was
19.348 g (72 %), brown prisms, mp 347–348.8 °C (methanol), lit. mp 318-332
°C [ex-4b, ex-13].
Rf = 0.21 (chloroform/aceton 3:7).
IR (KBr): 3427 (w), 3132 (w), 3075 (w), 3009 (w), 2966 (w), 1638 (s), 1607 (w),
1565 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.78 (s, 3 H, CH3O), 5.60 (s, 1 H, 3-H), 6.72
(d, J = 2.4 Hz, 1 H, 8-H), 6.75 (dd, J = 9.2 + 2.4 Hz, 1 H, 6-H), 7.67 (d, J = 9.1
Hz, 1 H, 5-H), 11.02 (s, 1 H, NH), 11.15 (s, 1 H, OH).
C10H9NO3 (191.19).
4-Hydroxy-5-methoxyquinolin-2-(1H)-one (29)
This compound was obtained during the recristallization of compound 28 from
methanol. The yield was 1.765 g (6 %), yellow prisms, mp 255–258 °C
(methanol), lit. mp 255–256 °C [ex-14].
Rf = 0.43 (chloroform/aceton 3:7).

EXPERIMENTAL PART 170
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
IR (KBr): 3430 (w), 3317 (m), 2973 (w), 2941 (w), 2840 (w), 1657 (s), 1619 (w),
1567 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.91 (s, 3 H, CH3O), 5.61 (s, 1 H, 3-H), 6.73
(d, J = 8.1 Hz, 1 H, 6-H), 6.89 (d, J = 8.3 Hz, 1 H, 8-H), 7.40 (t, J = 8.3 Hz, 1 H,
7-H), 10.00 (s, 1 H, NH), 11.24 (s, 1 H, OH).
C10H9NO3 (191.19).
2,4-Dichloro-7-methoxyquinoline (30)
Method A
This compound was obtained as insoluble solid during the work-up of
compound 28. The solid was purified from methanol. The yield was 5.62 g (16
%), grey prisms, mp 139–142 °C (methanol), lit. mp 120–156 °C [ex-9, ex-15].
Rf = 0.90 (chloroform/aceton 3:7).
Method B
A solution of 4-hydroxxy-7-methoxyquinolin-2(1H)-one (28: 16.450 g, 86.13
mmol) in phosphoroxychloride (103.0 mL) was heated under reflux for 8 hours
and worked up using the procedure described for 5, which afforded 2,4-
dichloro-7-methoxyquinoline (30), brown prisms, pure enough for further use.
The yield was 15.974 g (81 %), brown prisms, mp 135 -138 °C (methanol), lit.
mp 120–156 °C [ex-9, ex-15].
Rf = 0.94 (chloroform/aceton 3:7).
IR (KBr): 3437 (m), 3090 (w), 1624 (m), 1574 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.94 (s, 3 H, CH3O), 7.40 (d, J = 2.4 Hz, 1
H, 8-H), 7.44 (d, J = 8.4 Hz, 1 H, 5-H), 7.77 (s, 1 H, 3-H), 8.10 (dd, J = 8.4 +
2.6 Hz, 1 H, 6-H).
C10H7Cl2NO (228.08).

EXPERIMENTAL PART 171
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Chloro-7-methoxyquinolin-2-(1H)-one (31)
A solution of 2,4-dichloro-7-methoxyquinoline (30: 7.00 g, 30.70 mmol) and 70
% methanesulfonic acid (5.0 mL) in n-butanol (30.0 mL) was reacted according
to the method described for 6, to give 4-chloro-7-methoxyquinolin-2-(1H)-one
(31). The yield was 6.138 g (96 %), grey prisms, mp 244–246 °C (methanol), lit.
mp 252 °C [ex-15a].
Rf = 0.74 (chloroform/aceton 3:7).
IR (KBr): 3416 (w), 2965 (w), 2924 (w), 2851 (w), 1669 (s), 1626 (m), 1553 (w),
1507 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 (s, 3 H, CH3O), 6.60 (s, 1 H, 3-H), 6.86
(d, J = 2.4 Hz, 1 H, 8-H), 6.91 (dd, J = 8.9 + 2.5 Hz, 1 H, 6-H), 7.74 (d, J = 8.9
Hz, 1 H, 5-H), 11.87 (s, 1 H, NH).
C10H8ClNO2 (209.63).
Cyano-7-methoxyquinoline (32)
A mixture of 4-chloro-7-methoxyquinolin-2(1H)-one (31: 1.10 g, 5.25 mmol),
sodium p-toluenesulfinate (2.00 g, 11.24 mmol), and potassium cyanide
(1.058 g, 16.28 mmol) in dry dimethylformamide (31.50 mL) was heated to 120
°C for 30 hours with vigorous stirring, and worked up as described in the
procedure for compound 7 (pathway A), to furnish the compound 32. The yield
was 0.829 g (79 %), green prisms, mp 328–331 °C (acetonitrile).
Rf = 0.81 (chloroform/aceton 3:7).
IR (KBr): 3433 (m), 2939 (w), 2220 (m), 1614 (s), 1537 (m), 1517 (w), 1499 (w)
cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.76 (s, 3 H, CH3O), 6.46 (dd, J = 8.7 + 2.5
Hz, 1 H, 6-H), 6.50 (d, J = 2.5 Hz, 1 H, 8-H), 7.28 (d, J = 8.7 Hz, 1 H, 5-H),
7.98 (s, 1 H, 3-H).
MS (APCI pos): m/z (%) = 201 (100, M).
MS (APCI neg): m/z (%) = 199 (100, M).
C11H8N2O2 (200.20 ).

EXPERIMENTAL PART 172
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
7-Methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (33)
A mixture of 4-chloro-7-methoxyquinolin-2(1H)-one (31: 0.630 g, 3.15 mmol),
sodium p-toluenesulfinate (1.121 g, 6.30 mmol), and potassium cyanide (0.540
g, 8.31 mmol) in dry dimethylformamide (18.0 mL) was heated to 140 °C for 46
hours with vigorous stirring, and worked up as described in the procedure for
compound 7 (pathway A), to form 7-methoxy-2-oxo-1,2-dihydroquinoline-4-
carbonitrile (33). The yield was 0.525 g (87 %), pale green prisms, mp 304–308
°C (acetonitrile).
Rf = 0.77 (chloroform/aceton 3:7).
IR (ATR-measurement): 2985 (w), 2944 (w), 2878 (w), 2848 (w), 2816 (w), 2220
(w), 1661 (s), 1623 (s), 1562 (m), 1506 (m), 1489 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.85 (s, 3 H, CH3O), 6.81 (d, J = 1.9 Hz, 1
H, 8-H), 6.92 (dd, J = 8.9 + 2.1 Hz, 1 H, 6-H), 7.70 (d, J = 8.8 Hz, 1 H, 5-H),
8.66 (s, 1 H, 3-H), 12.31 (s, 1 H, NH).
MS (APCI pos): m/z (%) = 202 (15, M + 1), 201 (100, M).
MS (APCI neg): m/z (%) = 200 (13, M + 1), 199 (100, M).
Anal. For C11H8N2O2 (200.20).
Calcd: C 66.00, H 4.03, N 13.99 O 15.98
Found: C 65.86, H 3.82, N 13.81 O 16.17
4-Trifluoromethyl-6-methoxyquinolin-2(1H)-one (38)
To ethyl 4,4,4-trifluoroacetoacetate (36: 2.99 g, 16.25 mmol) at 130 °C in an
open flask p-anisidin (1.00 g, 8.13 mmol) was added dropwise under stirring,
the reaction mixture was kept for 1 hour at this temperature and then cooled
to 40–45 °C. The excess amount of ester was removed under reduced pressure
to give 4,4,4-trifluoro-N-(4-methoxyphenyl)-3-oxobutanamide (37) as a dark
residue. This amide 37 was cyclized without isolation. To this residue, 76%
sulfuric acid (3.0 mL) was added and the mixture was heated to 90 °C for 12
hours, cooled at room temperature, then water (100 mL) was added with
stirring and a beige precipitate was formed, which was filtered by suction,

EXPERIMENTAL PART 173
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
washed with water until neutral and dried at 40 °C under reduced pressure to
give the corresponding compound 38. The yield was 1.402 g (71 %), beige
prisms, mp 253–254 °C (ethanol).
Rf = 0.72 (chloroform/aceton 3:7).
IR (KBr): 3432 (m), 2982 (w), 2847 (w), 1672 (s), 1621 (m), 1505 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.80 (s, 3 H, CH3O), 6.98 (s, 1 H, 3-H),
7.05 (d, J = 2.4 Hz, 1 H, 5-H), 7.35 (dd, J = 9.1 + 2.5 Hz, 1 H, 7-H), 7.40 (d, J =
9.1 Hz, 1 H, 8-H), 2.24 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 56.0 (CH3O), 106.3 (Aromatic-C), 113.9
(Aromatic-C), 118.2 (Aromatic-C), 121.1 (Aromatic-C), 134.7 (Aromatic-C),
136.1 (4-C), 154.9 (6-C), 159.9 (lactam-C=O).
MS (API-ESI neg): m/z (%) = 243 (12, M + 1), 242 (100, M).
Anal. for C11H8F3NO2 (243.19).
Calcd: C 54.33, H 3.32, N 5.76
Found: C 54.18, H 3.10, N 5.68
N-(4-methoxyphenyl)-N-methylformamide (40)
A mixture of p-anisidin (1: 29.90 g, 243.10 mmol), trimethyl orthoformate (39:
40.0 mL, 32.00 g, 301.88 mmol) and concentrated sulfuric acid (1.0 mL) was
slowly heated under stirring to 103–105 °C and the formed methanol was
distilled over a short vigreux column. During 2.5 hours the bath temperature
was increased to 170–185 °C until no more methanol was formed, then the
mixture was kept at this temperature for further 60 minutes, and cooled to 50
°C. Distillation under reduced pressure afforded N-(4-methoxyphenyl)-N-
methylformamide (40). The yield was 28.156 g (70 %), yellow oil, bp 165–170
°C/14–21 mbar, lit. bp 86–171 °C/0.2–20 torr. [Ex-17, ex-18, ex-19, ex-20].
Rf = 0.79 (chloroform/aceton 3:7).
1H-NMR (300 MHz, [D3]CDCl3): = 3.26 (s, 3 H, NCH3), 3.81 (s, 3 H, CH3O),
6.92 (d, J = 8.9 Hz, 2 H, PhH), 7.12 (d, J = 9.0 Hz, 2 H, PhH), 8.33 (s, 1 H,
CHO).
C9H11NO2 (165.19).

EXPERIMENTAL PART 174
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Methoxy-N-methylbenzenamine or 4-Methoxy-
N-methylaniline (41)
A mixture of N-(4-methoxyphenyl)-N-methylformamide (40: 17.00 g, 103.0
mmol) and 10 % hydrochloric acid (53.0 mL) was stirred and heated under
reflux for 90 minutes. Then the mixture was cooled to room temperature,
neutralized and satured with potassium carbonate. The amine separated as
colorless oil, was then extracted with diethyl ether (3 x 40 mL), the ether phase
was washed with water (50 mL) and dried over potassium carbonate. The
solvent was removed under reduced pressure and the orange product distilled
under reduced pressure to afford 4-methoxy-N-methylbenzenamine (41). The
yield was 10.445 g (74 %), pale yellow oil, bp 120–125°C/17-18 mbar, lit. bp
45–136 °C/ 0.1–19 torr. [Ex-21, ex-22, ex-23, ex-24, ex-25, ex-26, ex-27, ex-
28, ex-29, ex-30].
Rf = 0.83 (chloroform/aceton 3:7).
1H-NMR (300 MHz, [D6] DMSO): = 2.62 (d, J = 5.3 Hz, 3 H, NCH3), 3.62 (s, 3
H, CH3O), 5.14 (d, J = 8.9 Hz, 1 H, NH), 6.49 (d, J = 8.9 Hz, 2 H, PhH), 6.72 (d,
J = 8.9 Hz, 2 H, PhH).
C8H11NO (137.18).
4-Hydroxy-6-methoxy-1-methylquinolin-2(1H)-one (42)
A mixture of 4-methoxy-N-methylbenzenamine (41: 16.193 g, 118.20 mmol)
and dry malonic acid (2: 23.320 g, 224.23 mmol) in phosphoryl chloride (23.50
mL), was well stirred with thermometer, and heated to 95 °C for 90 minutes.
The crude product was cooled to 20 °C and poured onto ice/water (200 mL).
The solution was filtered and the precipitate dissolved in aqueous sodium
hydroxide (450 mL, 1 M) under warming (60 °C), then filtered from the
insoluble precipitate. The filtrate was acidified with concentrated hydrochloric
acid to pH = 1-2, and the product was filtered by suction, washed with water
and dried at 40 °C under reduced pressure to give 4-Hydroxy-6-methoxy-1-
methylquinolin-2(1H)-one (42).

EXPERIMENTAL PART 175
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The yield was 11.384 g (47 %), rose prisms, mp 247-250 °C (methanol), lit. mp
278-293 °C [ex-31].
Rf = 0.38 (chloroform/aceton 3:7).
IR (KBr): 3445 (s), 2961 (w), 2934 (w), 1643 (w), 1617 (m), 1583 (m), 1583 (w),
1524 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.52 (s, 3 H, NCH3), 3.81 (s, 3 H, CH3O),
5.93 (s, 1 H, 3-H), 7.25 (dd, J = 9.1 + 2.9 Hz, 1 H, 7-H), 7.33 (d, J = 2.9 Hz, 1
H, 5-H), 7.43 (d, J = 9.2 Hz, 1 H, 8-H), 11.46 (s, 1 H, OH).
C11H10NO2 (205.22).
4-Chloro-6-methoxy-1-methylquinolin-2(1H)-one (43)
Methode A
A solution of 4-hydroxy-6-methoxy-1-methlylquinolin-2(1H)-one (42: 2.390 g,
11.66 mmol) in phosphoroxychloride (14.0 mL) was reacted for 8 hours under
reflux and worked up using the procedure described for 5, to give 4-chloro-6-
methoxy-1-methylquinolin-2(1H)-one (43). The yield was 2.127 g (82 %), brown
prisms, mp 158-161 °C (methanol), lit. mp 161–163 °C [ex-32].
Rf = 0.84 (chloroform/aceton 3:7).
Methode B
A mixture of 4-chloro-6-methoxyquinolin-2(1H)-one (6: 1.00 g, 4.77 mmol),
iodomethane (0.70 mL, 1.60 g, 11.26 mmol), and dry sodium carbonate (1.30
g, 11.95 mmol) in dry dimethylformamide (35.0 mL) was heated to 120 °C for
25 minutes. The solution was cooled to 20 °C and poured into ice/water (50
mL). The obtained solid was filtered by suction, washed with water and dried at
40 °C under reduced pressure to afford the corresponding compound 43. The
yield was 0.861 g (81 %), brown prisms, mp 157–160 °C (methanol), lit. mp
161–163 °C [ex-32].
Rf = 0.80 (chloroform/aceton 3:7).
IR (ATR-measurement): 3437 (m), 3078 (w), 1642 (s), 1579 (w), 1564 (m), 1503
(m) cm-1.

EXPERIMENTAL PART 176
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (300 MHz, [D6]DMSO): = 3.61 (s, 3 H, NCH3), 3.86 (s, 3 H, CH3O),
6.96 (s, 1 H, 3-H), 7.35 (d, J = 2.7 Hz, 1 H, 5-H), 7.39 (dd, J = 9.1 + 2.9 Hz, 1
H, 7-H), 7.59 (d, J = 9.1 Hz, 1 H, 8-H).
C11H10ClNO2 (223.66).
6-Methoxy-1-methyl-2-oxo-1,2-dihydroquinoline-4-carbonitrile
(44)
Method A
A mixture of 4-chloro-6-methoxy-1-methylquinolin-2(1H)-one (43: 0.300 g,
1.34 mmol), sodium p-toluenesulfinate (0.500 g, 2.81 mmol) and potassium
cyanide (0.220 g, 3.38 mmol) in dimethylformamide (8.0 mL) was heated to 120
°C for 28 hours and worked up according to the procedure described for 7, to
give 6-methoxy-1-methyl-2-oxo-1,2-dihydroquinoline-4-carbonitrile (44). The
yield was 0.241 g (84 %), black-green prisms.
Rf = 0.83 (chloroform/aceton 3:7).
Method B
A mixture of 6-methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (9: 100 mg,
0.50 mmol), sodium carbonate (200 mg, 1.88 mmol) and iodomethane (0.10
mL, 0.23 g, 1.60 mmol) in dry dimethylformamide (5.0 mL) was heated to 90 °C
for 15 minutes. The solution was cooled at room temperature and poured on
ice / water (5 mL). The resulting precipitate was filtered by suction, washed
with water and dried at 40 °C under reduced pressure to give 6-methoxy-1-
methyl-2-oxo-1,2-dihydroquinoline-4-carbonitrile (44). The yield was 88 mg (82
%), brown prisms, mp 233–236 °C (ethanol).
Rf = 0.87 (chloroform/aceton 3:7).
IR (KBr): 3032 (w), 2944 (w), 2841(w), 2229 (w), 1644 (s), 1592 (m), 1573 (m),
1559 (w), 1541(w), 1505 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.64 (s, 3 H, NCH3), 3,83 (s, 3 H, CH3O),
7.36 (d, J = 2.9 Hz, 1 H, 5-H), 7.44 (dd, J = 9.3 + 2.7 Hz, 1 H, 7-H), 7.57 (d, J =
9.3 Hz, 1 H, 8-H), 8.68 (s, 1 H, 3-H).

EXPERIMENTAL PART 177
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
13C-NMR (300 MHz,[D6]DMSO): = 30.5 (CH3), 56.1 (CH3O), 106.2 (Aromatic-
C), 111.6 (Aromatic-C), 116.5 (Aromatic-C), 117.4 (Aromatic-C), 119.6
(Aromatic-C), 123.8 (Aromatic-C), 136.1 (Aromatic-C), 148.2 (3-C), 155.1 (6-C),
158.2 (lactam-C=O).
Anal. for C12H10N2O2 (214.23 ).
Calcd: C 67.28, H 4.71, N 13.08 O 14.94
Found: C 66.39, H 4.49, N 13.08 O 15.25
6-Methoxy-1-methyl-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile (45)
A mixture of 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7: 0.422
g, 1.87 mmol), iodomethane (0.30 mL, 0.684 g, 4.82 mmol), and dry sodium
carbonate (0.588 g, 5.55 mmol) in dry dimethylformamide (17.0 mL), was
heated to 120 °C for 25 minutes, and worked up as described for 43 method B,
to afford the corresponding compound 45. The yield was 0.347 g (65 %), red
prisms, mp 219–223 °C (ethanol).
Rf = 0.94 (chloroform/aceton 3:7).
IR (KBr): 3459-3438 (w, br), 3026 (w), 2978 (w), 2942(w), 2238 (w), 1657 (s),
1586 (w), 1554 (m), 1499 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.70 (s, 3 H, NCH3), 3.91 ( s, 3 H, CH3O),
7.21 (d, J = 2.3 Hz, 1 H, 5-H), 761 (dd, J = 9.4 + 2.5 Hz, 1 H, 7-H), 7.76 (d, J =
9.4 Hz, 1 H, 8-H).
13C-NMR (300 MHz,[D6]DMSO): = 31.5 (CH3), 56.1 (CH3O), 107.29 (Aromatic-
C), 112.5 (Aromatic-C), 113.3 (Aromatic-C), 114.6 (Aromatic-C), 117.3 (CN),
118.87 (CN), 125.8 (Aromatic-C), 128.5 (Aromatic-C), 136.5 (Aromatic-C),
155.9 (6-C), 156.7 (lactam-C=O).
MS (ESI pos): m/z (%) = 501 (58, [2M + Na]+), 278 (11, [M + K]
+), 262 (100, [M + Na]
+), 240
(47, [M + H]+).
Anal. for C13H9N3O2 ( 239.24 ).
Calcd: C 65.27, H 3.79, N 17.56 O 13.38
Found: C 65.10, H 3.59, N 17.17 O 13.59

EXPERIMENTAL PART 178
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Ethyl (3,4-Dicyano-6-methoxy-2-oxoquinolin-1(2H)-yl)-acetate
(46)
A mixture of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7:
0.500 g, 2.22 mmol), ethyl bromoacetate (0.6 mL, 0.906 g, 5.43 mmol) and dry
sodium carbonate (0.711 g, 6.71 mmol) in dry dimethylformamide (15.0 mL)
was heated to 90 °C for 15 minutes, and worked up as described for 43
(method B), to give ethyl (3,4-dicyano-6-methoxy-2-oxoquinolin-1(2H)-yl)-
acetate (46). The yield was 0.579 g (84 %), yellow prisms, mp 216–219 °C
(ethanol).
Rf = 0.90 (chloroform/aceton 3:7).
IR (KBr): 3143 (w), 3108 (w), 3002 (w), 2980 (w), 2942 (w), 2230 (w), 1746 (s),
1656 (s), 1557 (s), 1503 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.22 (t, J = 7.0 Hz, 3 H, CH3), 3.92 (s, 3 H,
CH3O), 4.17 (q, J = 7.0 Hz, 2 H, CH2), 5.20 (s, 2 H, NCH2), 7.26 (d, J = 2.7 Hz, 1
H, 5-H), 77.60 (dd, J = 9.4 + 2.8 Hz, 1 H, 7-H), 7.73 (d, J = 9.5 Hz, 1 H, 8-H).
13C-NMR (360 MHz, [D6]DMSO): = 14.5 (CH3), 45.6 (NCH2), 56.4 (CH3O), 62.1
(OCH2), 108.5 (aromatic-C), 112.0 (aromatic-C), 113.2 (aromatic-C), 114.2
(aromatic-C), 117.4 (aromatic-C), 118.6 (CN), 125.9 (aromatic-C), 129.8
(aromatic-C), 135.9 (4-C), 156.2 (6-C), 156.6 (lactam-C=O), 167.6 (ester-C=O).
MS (ESI pos): m/z (%) = 305 (51, [M + K]+), 334 (100,[ M + Na]
+), 312 (62, [M + H]
+).
Anal. for C16H13N3O4 ( 311.30 ).
Calcd: C 61.73, H 4.21, N 13.50
Found: C 61.48, H 4.06, N 13.34
2-Benzyloxy-6-methoxyquinoline-3,4-dicarbonitrile (48)
A mixture of 6-methoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (7: 0.90
g, 4.00 mmol), potassium carbonate (0.745 g, 5.40 mmol), and benzylchloride
(47: 0.80 mL, 0.880 g, 6.75 mmol) in dry dimethylformamide (65.0 mL) was
heated slowly to 80 °C, the mixture was kept at this temperature for 3 hours

EXPERIMENTAL PART 179
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
until tlc showed no more starting material.
Then, the temperature was raised to 110 °C for 2 hours. The solution was
filterated and the solvent was removed under reduced pressure. A thick brown
oil was obtained, which crystallized after standing over night. The purification
was performed by dry flash chromatography using toluene-dichloromethane
(3:1) as eluent to form 2-benzyloxy-6-methoxyquinoline-3,4-dicarbonitrile (48).
The yield was 0.768 g (61 %), yellow prisms, mp 318–323 °C
(toluene/dichloromethane 3:1).
Rf = 0.84 (chloroform/aceton 3:7).
IR (KBr): 3435 (s), 2924 (w), 2231 (w), 1620 (w), 1584 (m), 1505 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.98 (s, 3 H, CH3O), 5.61 (s, 2 H, CH2),
7.30-7.55 (m, 5 H, Ph-H), 7.58 (d, J = 1.6 Hz, 1 H, 5-H), 7.70 (dd, J = 9.3 + 2.8
Hz, 1 H, 7-H), 7.96 (d, J = 9.2 Hz, 1 H, 8-H).
C19H13N3O2 (315.33).
3,4-Dichloro-6-methoxy-1-methylquinolin-2(1H)-one (49)
A mixture of 3,4-dichloro-6-methoxyquinolin-2(1H)-one (13: 0.945 g, 3.87
mmol), iodomethane (0.70 mL, 1.60 g, 11.24 mmol), and dry sodium carbonate
(1.025 g, 9.68 mmol) in dry dimethylformamide (20.0 mL) was heated to 120 °C
for 25 minutes, and worked up as described for 43 (method B), to afford the
corresponding compound 49. The yield was 0.798 g (80 %), light yellow prisms,
mp 170–179 °C (methanol).
Rf = 0.86 (chloroform/aceton 3:7).
IR (ATR-measurement): 2998 (w), 2943 (w), 2838 (w), 1644 (s), 1585 (w), 1555
(m), 1494 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.67 (s, 3 H, NCH3), 3.85 (s, 3 H, CH3O),
7.31 (d, J = 2.7 Hz, 1 H, 5-H), 7.36 (dd, J = 9.2 + 2.7 Hz, 1 H, 7-H), 7.57 (d, J =
9.2 Hz, 1 H, 8-H).
13C-NMR (300 MHz, [D6]DMSO): = 31.5 (CH3), 56.1 (CH3O), 107.7 (aromatic-
C), 117.7 (aromatic-C), 118.8 (aromatic-C), 121.1 (aromatic-C), 126.1
(aromatic-C), 132.5 (aromatic-C), 139.8 (4-C), 155.5 (6-C), 156.2 (lactam-C=O).

EXPERIMENTAL PART 180
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Anal. For C11H9Cl2NO2 (258.11)
Calcd. : C 51.91 H 3.51 N 5.43
Found : C 51.06 H 3.37 N 5.41
4-Chloro-6-methoxy-7-nitroquinolin-2(1H)-one (50)
A solution of 4-chloro-6-methoxyquinolin-2(1H)-one (6: 4.00 g, 19.10 mmol) in
glacial acetic acid (35.0 mL) was treated with concentrated nitric acid (3.50 mL,
83.69 mmol) and with sodium nitrite (1.00 g, 14.50 mmol) to start a strongly
exothermic reaction. The starting material was dissolved, followed immediately
by precipitation of the product. The mixture was heated to 110 °C for 3 hours,
the solution was cooled at room temperature and poured into ice/water (150
mL). The solution was filtered by suction, washed with water and dried at 40
°C under reduced pressure to give 4-chloro-6-methoxy-7-nitroquinolin-2(1H)-
one (50). The yield was 3.991 g (82 %), yellow prisms, mp 333-336 °C
IR (KBr): 3464-3432 (m, br), 3060 (w), 2982 (w), 2949 (w), 2850 (w), 1702 (s),
1659 (s), 1628 (m), 1590 (w), 1534 (s), 1499 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.90 (s, 3 H, CH3O), 6.95 (s, 1 H, 3-H), 7.56
(s, 1 H, 5-H), 7.73 (s, 1 H, 8-H), 12.32 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 57.9 (CH3O), 108.8 (aromatic-C), 119.2
(aromatic-C), 119.5 (aromatic-C), 126.6 (aromatic-C), 133.9 (aromatic-C), 135.4
(7-C), 138.2 (4-C), 146.3 (6-C), 159.4 (lactam-C=O) .
C10H7ClN2O4 (254.63)
Ethyl (p-Methoxyphenyl)acetate (53a)
A solution of p-methoxyphenylacetic acid (52a: 35.60 g, 214.45 mmol), ethanol
(25.0 mL) and concentrated sulfuric acid (0.65 mL, 1.20 g, 12.24 mmol) in
chloroform (72.0 mL) was heated under reflux at the reversed water separator
for 5 hours. When no further water was separated, the mixture was cooled to
room temperature, and then the chloroform solution was washed with water

EXPERIMENTAL PART 181
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
(50 mL), saturated with NaHCO3-solution (50 mL), and again washed with
water (50 mL). The solvent was removed under reduced pressure and a clear
red oil was formed, which was purified by vacuum distillation to furnish ethyl
(p-methoxyphenyl)acetate (53a). The yield was 31.750 g (76 %), colorless oil,
b.p 140–155 °C/34 mbar, lit. bp 100–158.5 °C / 0.3–20 torr.[ex-33, ex-34, ex-
35, ex-36, ex-37, ex-38, ex-39, ex-40].
Rf = 0.90 (chloroform/aceton 3:7).
IR (liquid film, ATR-measurement): 2981 (w), 2960 (w), 2937 (w), 2837 (w),
1736 (s), 1613 (m), 1513 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.17 (t, J = 7.1 Hz, 3 H, CH3), 3.57 (s, 2 H,
CH2), 3.73 (s, 3 H, CH3O), 4.05 (q, J = 7.1 Hz, 2 H, OCH2), 6.87 (d, J = 8.7 Hz, 2
HBB´ Ar-H), 7.17 (d, J = 8.7 Hz, 2 HAA´ Ar-H).
C11H14O3 (194.23).
Ethyl (4-Chlorophenyl)acetate (53b)
A solution of 4-chlorophenylacetic acid (52b: 31.20 g, 183.0 mmol), ethanol
(24.0 mL) and concentrated sulfuric acid (0.60 mL, 1.104 g, 11.26 mmol) in
chloroform (70.0 mL) was reacted and worked up as described for 53a, to form
ethyl (4-Chlorophenyl)acetate (53b). The yield was 27.346 g (75 %), colorless
oil, b.p 135–140 °C/30 mbar, lit. bp 78–260 °C [ex-36b, ex-37c, ex-40a, ex-41,
ex-42, ex-43, ex-44].
Rf = 0.86 (chloroform/aceton 3:7).
IR (liquid film): 2982 (m), 1737 (s), 1493 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.17 (t, J = 7.1 Hz, 3 H, CH3), 3.67 (s, 2 H,
CH2), 4.07 (q, J = 7.1 Hz, 2 H, OCH2), 7.29 (d, J = 8.6 Hz, 2 HAA´ Ar-H), 7.37(d,
J = 8.5 Hz, 2 HBB´ Ar-H).
C10H11ClO2 (198.65).

EXPERIMENTAL PART 182
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Ethyl (4-Nitrophenyl)acetate (53c)
A solution of 4-nitrophenylacetic acid (52c: 35.60 g, 196.68 mmol), ethanol
(23.0 mL, 397.13 mmol) and concentrated sulfuric acid (0.60 mL, 1.104 g,
11.26 mmol) in chloroform (66.0 mL) was refluxed at the reversed water
separator for 5 hours. When no further water was separated, the mixture was
cooled to room temperature, which got quickly solid in contact with air. The
solid was digested with water (100 mL) under stirring, the solution was filtered
by suction, and the obtained solid was mixed with saturated NaHCO3-solution
(100 mL), filtered by suction and again mixed with water (100 mL), filtered by
suction and dried at 40 °C under reduced pressure. The solid was purified by
vacuum distillation, the product crystallized quickly in contact with air, to give
ethyl (4-nitrophenyl)acetate (53c). The yield was 35.892 g (87 %), yellowish oil,
bp. 200 °C/39mbar, lit. bp 125–197 °C / 0.1–20 torr. [ex-45].
Yellowish prisms, mp 63–65 °C, lit. mp 55.5–66 °C [ex-45a, ex-46, ex-47, ex-
48, ex-49, ex-50].
Rf = 0.90 (chloroform/aceton 3:7).
IR (KBr): 3441 (w), 3118 (w), 3058 (w), 2985 (m), 2962 (w), 2939 (w), 2907 (w),
2872 (w), 2850 (w), 2452 (w), 2337 (w), 1943 (w), 1730 (s), 1688 (w), 1604 (m),
1513 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.18 (t, J = 7.1 Hz, 3 H, CH3), 3.87 (s, 2 H,
CH2), 4.09 (q, J = 7.1 Hz, 2 H, OCH2), 7.56 (d, J = 8.8 Hz, 2 HAA´ Ar-H), 8.18 (d,
J = 8.8 Hz, 2 HBB´ Ar-H).
C10H11NO4 (209.20).
Ethyl (3-Chlorophenyl)acetate (53d)
A solution of 3-chlorophenyl-acetic acid (52d: 33.20 g, 195.0 mmol), ethanol
(24.0 ml) and concentrated sulfuric acid (0.60 mL, 1.104 g, 11.26 mmol) in
chloroform (70.0 mL) was reacted at the reversed water separator for 5 hours
and worked up as described for 53a, to give ethyl (3-chlorophenyl)acetate

EXPERIMENTAL PART 183
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
(53d). The yield was 21.682 g (56 %), colorless oil, b.p 140–145 °C/33–39
mbar, lit. bp 95-148 °C / 0.3–26 torr. [ex-41a, ex-50a, ex-51, ex-52].
Rf = 0.92 (chloroform/aceton 3:7).
IR (liquid film): 3065 (w), 2982 (m), 2938 (w), 2907 (w), 1736 (s), 1598 (w), 1574
(w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.18 (t, J = 7.1 Hz, 3 H, CH3), 3.70 (s, 2 H,
CH2), 4.08 (q, J = 7.1 Hz, 2 H, OCH2), 7.22 (dd, J = 6.4 + 2.0 Hz, 1 H, 5-H),
7.24 (t, J = 1.3 Hz, 1 H, 2-H), 7.32 (d, J = 5.2 Hz, 1 H, 4-H), 7.36 (d, J = 5.8 Hz,
1 H, 6-H).
C10H11ClO2 (198.65).
Diethyl (4-Methoxyphenyl)malonate (55a)
To a mixture of sodium hydride 60 % in mineral oil (15.00 g, 625.0 mmol)
and00000000000 diethyl carbonate (57.5 g, 59.0 mL, 487.28 mmol) in dry
tetrahydrofuran (115.0 mL), was added dropwise a solution of ethyl (4-
methoxyphenyl)acetate (53a: 18.30 g, 94.33 mmol) in dry tetrahydrofuran
(38.0 mL). The mixture was refluxed for 87 hours, cooled to room temperature,
and the solution was neutralized with a saturated aqueous solution of
ammonium chloride (150 mL). The organic phase was separated and then the
aqueous phase was extracted with diethylether (2 x 150 mL). The combined
organic phases were washed with saturated NaHCO3 solution (150 mL) and
brine (150 mL), and dried over sodium sulfate. After evaporation of the solvent
under reduced pressure, the residue was purified by vacuum distillation to give
diethyl (4-methoxyphenyl) malonate (55a). The yield was 17.368 g (69 %), clear
yellow oil, bp 190–206 °C/36.5 mbar, lit. bp 136–198 °C / 0.05–13 torr [ex-53,
ex-54, ex-55, ex-56, ex-57].
Rf = 0.89 (chloroform/aceton 3:7).
IR (liquid film): 2982 (w), 2937 (w), 2908 (w), 2839 (w), 1752 (w), 1733 (s), 1613
(w), 1514 (s) cm-1.

EXPERIMENTAL PART 184
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (300 MHz, [D6]DMSO): = 1.16 (t, J = 7.1 Hz, 6 H, 2 x CH3), 3.74 (s, 3
H, CH3O), 4.07-4.18 (m, 4 H, 2 x CH2), 4.86 (s, 1 H, CH), 6.92 (d, J = 8.7 Hz, 2
HBB´ Ar-H), 7.31 (d, J = 8.7 Hz 2 HAA´ Ar-H).
C14H18O5 (226.30).
Diethyl (4-Chlorophenyl)malonate (55b)
To a mixture of sodium hydride 60 % in mineral oil (9.80 g, 408.33 mmol) and
diethyl carbonate (114.73 g, 118.0 mL, 972.30 mmol) in dry tetrahydrofuran
(300.0 mL), was added drop wise a solution of ethyl (4-chlorophenyl)acetate
(53b: 38.60 g, 194.50 mmol) in dry tetrahydrofuran (100 mL). The mixture was
refluxed for 2 hours, and worked up as described for 55a, to give diethyl (4-
chlorophenyl)malonate (55b). The yield was 43.695 g (83 %), colorless oil, bp
170–180 °C /27 mbar, lit. bp 116–180 °C /0.05-25 torr. [ex-41a, ex-43b, ex-
44b, ex-54b, ex-55c, ex-56b, ex-57c, ex-58, ex-59, ex-60].
Rf = 0.92 (chloroform/aceton 3:7).
IR (liquid film): 2983 (w), 2935 (w), 2872 (w), 1900 (w), 1736 (s), 1597 (w), 1493
(s) cm-1.
1H-NMR (500 MHz, [D6]DMSO): = 1.17 (t, J = 7.1 Hz, 6 H, 2 x CH3), 4.10-4.17
(m, 4 H, 2 x OCH2), 5.03 (s, 1 H, CH), 7.40 (d, J = 9.1 Hz, 2 HAA´ Ar-H), 7.43 (d,
J = 9.0 Hz, 2 HBB´ Ar-H).
C13H15ClO4 (270.72).
Diethyl (3-Chlorophenyl)malonate (55d)
To a mixture of sodium hydride 60 % in mineral oil (6.00 g, 250 mmol) and
diethyl carbonate (58.20 g, 60.0 mL, 493.22 mmol) in dry tetrahydrofuran
(180.0 mL) was added dropwise a solution of ethyl (3-chlorophenyl)acetate
(53d: 19.30 g, 97.23 mmol) in dry tetrahydrofuran (60.0 mL).

EXPERIMENTAL PART 185
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The mixture was refluxed for 7 hours, and worked up as described for 55a, to
give diethyl (3-chlorophenyl)malonate (55d). The yield was 12.597 g (48 %),
colorless oil, bp 178–184 °C/22.7 mbar, lit. bp 145 –178 °C / 1.2–17 torr. [ex-
41a, ex-60b].
Rf = 0.88 (chloroform/aceton 3:7).
IR (liquid film): 2983 (m), 2937 (w), 1736 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.17 (t, J = 7.1 Hz, 6 H, 2 x CH3), 4.07-4.23
(m, 4 H, 2 x CH2), 4.86 (s, 1 H, CH), 7.36-7.40 (m, 3 H, Ar-H), 7.48 (s, 1 H, Ar-
H).
C13H15ClO4 (270.72).
4-Hydroxy-6-methoxy-3-(4-methoxyphenyl)quinolin-2(1H)-one
(57a)
A solution of p-anisidin (1: 5.30 g, 43.10 mmol) and diethyl (p-methoxyphenyl)
malonate (55a: 11.460 g, 50.71 mmol) in diphenyl ether (49.0 mL) was refluxed
at about 250–300 °C for 3 hours in the metal bath. During this time, ethanol
(3.90 mL, 67.0 mmol) was liberated. The solution was cooled to 20 °C, diluted
with cyclohexane (25.0 mL) and filtered by suction. The solid was dissolved in
warm aqueous sodium hydroxide (250.0 mL, 1 M), and filtered. To the alkaline
filtrate, concentrated hydrochloric acid was added until pH = 1–2 was reached,
the precipitate filtered by suction, washed with water and dried at 40 °C under
reduced pressure, which afforded 4-hydroxy-6-methoxy-3-(4-methoxyphenyl)-
quinolin-2(1H)-one (57a). The yield was 12.588 g (98 %), colorless prisms, mp
325 –328 °C (ethanol).
Rf = 0.79 (chloroform/aceton 3:7).
IR (ATR-measurement): 3435 (m), 3126 (w), 3054 (w), 3003 (w), 2958 (m), 2838
(w), 1647 (w), 1611 (s), 1587 (w), 1515 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.78 (s, 3 H, 6-CH3O), 3.79 (s, 3 H, Ar-
CH3O), 6.96 (d, J = 8.8 Hz, 2 HBB´ Ar-H), 7.14 (dd, J = 8.9 + 2.7 Hz, 1 H, 7-H),
7.23 (d, J = 8.9 Hz, 1 H, 8-H), 7.30 (d, J = 8.8 Hz, 2 HAA´ Ar-H),

EXPERIMENTAL PART 186
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
7.44 (d, J = 2.6 Hz, 1 H, 5-H), 10.00 (s, 1 H, NH), 11.33 (s, 1 H, OH).
Anal. for C17H15NO4 (297.31).
Calcd: C 68.68, H 5.09, N 4.71, O 21.64
Found: C 68.31 H 4.86, N 4.72, O 21.54
3-(4-Chlorophenyl)-4-hydroxy-6-methoxyquinolin-2(1H)-one
(57b)
A solution of p-anisidin (1: 1.680 g, 13.66 mmol) and diethyl p-chlorophenyl
malonate (55b: 3.70 g, 14.66 mmol) in diphenyl ether (16.0 mL) of was
refluxed for 3 hours in the metal bath. During this time, ethanol (1.40 mL,
24.04 mmol) was liberated. The reaction was worked up as described for 57a,
to give 3-(4-chlorophenyl)-4-hydroxy-6-methoxyquinolin-2(1H)-one (57b). The
yield was 3.274 g (80 %), colorless prisms, mp 347–355 °C (ethanol).
Rf = 0.68 (chloroform/aceton 3:7).
IR (KBr): 3122 (w), 3055 (w), 2999 (w), 2949 (s), 2899 (w), 2829 (w), 2741 (w),
2623 (w), 1896 (w), 1648 (s), 1607 (s), 1585 (s), 1564 (w), 1510 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.79 (s, 3 H, CH3O), 7.17 (dd, J = 8.9 + 2.7
Hz, 1 H, 7-H), 7.24 (d, J = 8.9 Hz, 1 H, 8-H), 7.39 (d, J = 8.8 Hz, 2 HAA´ Ar-H),
7.44 (d, J = 2.6 Hz, 1 H, 5-H), 7.45 (d, J = 8.8 Hz, 2 HBB´ Ar-H), 10.20 (s, 1 H,
1-NH), 11.42 (s, 1 H, OH).
Anal. for C16H12ClNO3 (301.73).
Calcd: C 63.69, H 4.01, N 4.64
Found: C 63.56, H 3.83, N 4.63
3-(3-Chlorophenyl)-4-hydroxy-6-methoxyquinolin-2(1H)-one
(57d)
A solution of p-anisidin (1: 5.273 g, 42.87 mmol) and diethyl m-chlorophenyl
malonate (55d: 11.597 g, 42.87 mmol) in diphenyl ether (50.0 mL) was refluxed

EXPERIMENTAL PART 187
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
for 3 hours in the metal bath. During this time, ethanol (3.60 mL, 61.83 mmol)
was liberated. The reaction was worked up as described for 57a, to give 3-(3-
chlorophenyl)-4-hydroxy-6-methoxyquinolin-2(1H)-one (57d). The yield was
9.413 g (73 %), colorless prisms, mp 322–325 °C (ethanol).
Rf = 0.69 (chloroform/aceton 3:7).
IR (KBr): 3127 (w), 3059 (w), 2993 (w), 2964 (s), 2936 (w), 2831 (w), 1644 (w),
1607 (s), 1587 (w), 1506 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.79 (s, 3 H, CH3O), 7.18 (dd, J = 8.9 + 2.4
Hz, 1 H, 7-H), 7.25 (d, J = 8.9 Hz, 1 H, 8-H), 7.37 (s, 1 H, Ar-H), 7.34–7.42 (m,
3 H, Ar-H), 7.45 (d, J = 2.0 Hz, 1 H, 5-H), 10.30 (s, 1 H, NH), 11.44 (s, 1 H,
OH).
Anal. for C16H12ClNO3 (301.73).
Calcd: C 63.69, H 4.01, N 4.64
Found: C 63.67, H 3.78, N 4.63
4-Hydroxy-6-methoxy-3-phenylquinolin-2(1H)-one (57e)
A solution of p-anisidin (1: 10.014 g, 81.41 mmol), and diethyl phenylmalonate
(19.344 g, 81.96 mmol) in diphenyl ether (50.0 mL) was refluxed for 3 hours in
the metal bath. During this time, ethanol (6.0 mL, 103.03 mmol) was liberated.
The reaction was worked up as described for 57a, to give 4-hydroxy-6-
methoxy-3-
phenylquinolin-2(1H)-one (57e). The yield was 21.089 g (97 %), colorless
prisms, mp 324–327 °C (ethanol), lit. mp 319–326 °C [ex-61].
Rf = 0.71 (chloroform/aceton 3:7).
IR (in KBr): 3430 - 3295 (m, br), 3126 (w), 3058 (w), 2998 (w), 2959 (w), 2832
(w), 1648 (s), 1611 (s), 1509 (m) cm-1.
1H-NMR (360 MHz, [D6]DMSO): = 3.80 (s, 3 H, CH3O), 7.16 (dd, J = 8.9 + 2.7
Hz, 1 H, 7-H), 7.24 (d, J = 8.9 Hz, 1 H, 8-H), 7.28–7.41 (m, 5 H, Ar- H), 7.46
(d, J = 2.6 Hz, 1 H, 5-H), 10.03 (s, 1 H, NH), 11.37 (s, 1 H, OH).
C16H13NO3 (267.29).

EXPERIMENTAL PART 188
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Hydroxy-6-methoxy-3-methylquinolin-2(1H)-one (57f)
A solution of p-anisidin (1: 10.00 g, 81.30 mmol) and diethylmethyl malonate
(55f: 14.0 mL, 81.30 mmol) in diphenyl ether (50.0 mL), was refluxed for 3
hours in a metal bath. During this time, ethanol (6.90 mL, 118.50 mmol) was
liberated. The reaction was worked up as described for 57a, to give 4-hydroxy-
6-methoxy-3-methylquinolin-2(1H)-one (57f). The yield was 15.715 g (94 %),
colorless prisms, mp 234–236 °C (ethanol).
Rf = 0.35 (chloroform/aceton 3:7).
IR (ATR-measurement): 3431 (s), 3290 (w), 3242 (w), 2961 (w), 2926 (w), 2834
(w), 1647 (s), 1612 (s), 1566 (w), 1509 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.99 (s, 3 H, CH3), 3.78 (s, 3 H, CH3O),
7.08 (dd, J = 8.9 + 2.7 Hz, 1 H, 7-H), 7.19 (d, J = 8.9 Hz, 1 H, 8-H), 7.34 (d, J =
2.7 Hz, 1 H, 5-H), 10.03 (s, 1 H, NH), 11.22 (s, 1 H, OH).
C11H11NO3 (205.22).
Calcd: C 64.38, H 5.40, N 6.83
Found: C 64.32, H 5.26, N 6.85
3-Ethyl-4-hydroxy-6-methoxyquinolin-2(1H)-one (57g)
A solution of p-anisidin (1: 12.50 g, 101.63 mmol) and diethylmethyl malonate
(55g: 18.0 mL, 101.11 mmol) in diphenyl ether (60.0 mL), was refluxed for 3
hours in a metal bath and worked up as described for 57a, to give 3-ethyl-4-
hydroxy-6-methoxyquinolin-2(1H)-one (57g). The yield was 9.50 g (43 %),
colorless prisms, mp 160–162 °C (ethanol), lit. mp 172 °C [ex-61a].
Rf = 0.59 (chloroform/aceton 3:7).
IR (ATR-measurement): 3368 (w), 3100-2835 (m, br), 1647 (m), 1606 (s), 1558
(m), 1508 (s) cm-1.

EXPERIMENTAL PART 189
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (300 MHz, [D6]DMSO): = 1.01 (t, J = 7.3 Hz, 1 H, CH3), 2.57 (q, J =
7.3 Hz, 2 H, CH2), 3.78 (s, 3 H, CH3O), 7.09 (dd, J = 8.9 + 2.7 Hz, 1 H, 7-H),
7.21 (d, J = 8.9 Hz, 1 H, 8-H), 7.39 (d, J = 2.7 Hz, 1 H, 5-H), 11.27 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 13.7 (CH3), 16.9 (CH2), 55.9 (CH3O), 104.7
(10-C), 113.8 (3-C), 116.4 (8-C), 116.8 (5-C), 119.3 (7-C), 132.2 (9-C), 154.3 (6-
C), 156.9 (4-C), 163.3 (lactam-C=O).
C12H13NO3 (219.24).
2,4-Dichloro-6-methoxy-3-(4-methoxyphenyl)quinoline (58a)
A solution of 4-hydroxy-6-methoxy-3-(4-methoxyphenyl)quinolin-2(1H)-one
(57a: 11.10 g, 37.37 mmol) in phosphoroxychloride (45.0 mL) was brought to
reaction under reflux for 12 hours, and worked up as described for 5, to give
2,4-dichloro-6-methoxy-3-(4-methoxyphenyl)quinoline (58a). The yield was
9.771 g (79 %), colorless prisms, mp 169–170 °C (ethanol).
Rf = 0.91 (chloroform/aceton 3:7).
IR (ATR-measurement): 3439 (s), 3059 (w), 3008 (w), 2969 (w), 2934 (w), 2837
(w), 2359 (w), 2041 (w), 1954 (w), 1898 (w), 1617 (s), 1565 (m), 1513 (s), 1490
(s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 (s, 3 H, 6-CH3O), 3.95 (s, 3 H, Ar-
CH3O), 7.08 (d, J = 8.7 Hz, 2 HBB´ Ar-H), 7.32 (d, J = 8.6 Hz, 2 HAA´ Ar-H), 7.44
(d, J = 2.6 Hz, 1 H, 5-H), 7.56 (dd, J = 9.2 + 2.7 Hz, 1 H, 7-H), 7.97 (d, J = 9.2
Hz, 1 H, 8-H).
Anal. For C17H13Cl2NO2 (334.20 ).
Calcd. : C 61.10, H 3.92, N 4.19, O 9.57
Found : C 61.08, H 3.72, N 4.17, O 9.80
2,4-Dichloro-3-(4-chlorophenyl)-6-methoxyquinoline (58b)
A solution of 3-(4-chlorophenyl)-4-hydroxy-6-methoxyquinolin-2(1H)-one (57b:
13.380 g, 44.38 mmol) in phosphoroxychloride (70.0 mL) was refluxed for 8

EXPERIMENTAL PART 190
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
hours, and worked up as described for 5, to give 2,4-dichloro-3-(4-
chlorophenyl)-6-methoxyquinoline (58b). The yield was 12.910 g (86 %),
colorless prisms, mp 167–170 °C (ethanol).
Rf = 0.94 (chloroform/aceton 3:7).
IR (KBr): 3503 (w), 3002 (w), 2978 (w), 2944 (w), 2913 (w), 2612 (w), 2435 (w),
2359 (w), 2042 (w), 1920 (w), 1788 (w), 1687 (w), 1619 (s), 1567 (m), 1492 (s)
cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.96 (s, 3 H, CH3O), 7.45 (d, J = 1.8 Hz, 1
H, 5-H), 7.47 (d, J = 8.2 Hz, 2 HBB´ Ar-H), 7.59 (d, J = 9,2 + 2.0 Hz, 1 H, 7-H),
7.63 (d, J = 8.2 Hz, 2 HAA´ Ar-H), 7.99 (d, J = 9.2 Hz, 1 H, 8-H).
Anal. For C16H10Cl3NO (338.62 ).
Calcd. : C 56.75, H 2.98, N 4.14
Found : C 56.70, H 2.77, N 4.14
2,4-Dichloro-3-(3-chlorophenyl)-6-methoxyquinoline (58d)
A solution of 4-hydroxy-3-(3-chlorophenyl)-6-methoxyquinolin-2(1H)-one (57d:
7.00 g, 23.22 mmol) in phosphoroxychloride (30.0 mL) was reacted under
reflux for 12 hours, and worked up as described for 5, to give 2,4-dichloro-3-
(3-chlorophenyl)-6-methoxyquinoline (58d). The yield was 7.597 g (97 %), beige
prisms, mp 215–217 °C (ethanol).
Rf = 0.95 (chloroform/aceton 3:7).
IR (KBr): 3073 (w), 2965 (w), 2943 (w), 2911 (w), 2845 (w), 1618 (w), 1599 (s),
1567 (m), 1492 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.97 (s, 3 H, CH3O), 7.41-7.42 (m, 1 H, Ar-
H), 7.48 (d, J = 2.6 Hz, 1 H, 5-H), 7.58-7.60 (m, 3 H, Ar-H), 7.62 (dd, J = 9.2 +
2.6 Hz, 1 H, 8-H), 8.02 (d, J = 9.2 Hz, 1 H, 8-H).
Anal. For C16H10Cl3NO (338.62 ).
Calcd. : C 56.75 H 2.98 N 4.14
Found : C 56.46 H 2.66 N 4.09

EXPERIMENTAL PART 191
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2,4-Dichloro-6-methoxy-3-phenylquinoline (58e)
A solution of 4-hydroxy-6-methoxy-3-phenylquinolin-2(1H)-one (57e: 18.50 g,
69.30 mmol) in phosphoroxychloride (80.0 mL) was brought to reaction under
reflux for 8 hours and worked up as described for 5, to give 2,4-dichloro-6-
methoxy-3-phenylquinoline (58e). The yield was 20.409 g (97 %), colorless
prisms, mp 220–222 °C (ethanol).
Rf = 0.86 (chloroform/aceton 3:7).
IR (KBr): 3438 (m), 3027 (w), 2959 (w), 2939 (w), 2908 (w), 1620 (m), 1572 (m)
cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.97 (s, 3 H, CH3O), 7.39-7.40 (m, 1 H, 7-
H), 7.42 (d, J = 1.8 Hz, 1 H, 5-H), 7.48-7.62 (m, 5 H, PhH), 8.02 (d, J = 9.2 Hz,
1 H, 8-H).
Anal. For C16H11Cl2NO (304.18 ).
Calcd. : C 63.18, H 3.65, N 4.60
Found : C 63.05, H 3.44, N 4.58
2,4-Dichloro-6-methoxy-3-methyl-quinoline (58f)
A solution of 4-hydroxy-6-methoxy-3-methylquinolin-2(1H)-one (57f: 9.50 g,
46.34 mmol) in phosphoroxychloride (54.0 mL) was brought to reaction under
reflux for 24 hours, and worked up as described for 5, to give 2,4-dichloro-6-
methoxy-3-methylquinoline (58f). The product was pure enough for further
use. The yield was 10.278 g (92 %), light yellow prisms, mp 149–151 °C
(ethanol).
Rf = 0.93 (chloroform/aceton 3:7).
IR (KBr): 3459 (s), 1622 (m), 1572 (w), 1560 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 2.60 (s, 3 H, CH3), 3.95 (s, 3 H, CH3O),
7.40 (d, J = 2.7 Hz, 1 H, 5-H), 7.49 (dd, J = 9.2 + 2.8 Hz, 1 H, 7-H), 7.91 (d, J =
9.1 Hz, 1 H, 8-H).

EXPERIMENTAL PART 192
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Anal. For C11H9Cl2NO (242.11 ).
Calcd. : C 54.57, H 3.75, N 5.79
Found : C 54.36, H 3.55, N 5.69
2,4-Dichloro-3-ethyl-6-methoxyquinoline (58g)
A solution of 3-ethyl-4-hydroxy-6-methoxyquinolin-2(1H)-one (57g: 3.150 g,
14.38 mmol) in phosphoroxychloride (17.0 mL) was brought to reaction under
reflux for 24 hours and worked up as described for 5, to give 2,4-dichloro-3-
ethyl-6-methoxyquinoline (58g).
The yield was 3.090 g (84 %), colorless prisms, mp 115–118 °C (ethanol).
Rf = 0.91 (chloroform/aceton 3:7).
IR (ATR-measurement): 3038 (w), 2971 (w), 2836 (w), 2913 (w), 2873 (w), 2359
(w), 2340 (w), 1619 (m), 1568 (m), 1557 (m), 1493 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.20 (t, J = 7.4 Hz, 3 H, CH3), 3.00 (q, J =
7.2 Hz, 2 H, CH2), 3.94 (s, 3 H, CH3O), 7.35 (s, 1 H, 5-H), 7.47 (d, J = 8.8 Hz, 7-
H), 7.86 (d, J = 8.8 Hz, 1 H, 8-H).
13C-NMR (300 MHz, [D6]DMSO): = 12.8 (CH3), 25.3 (CH3O), 56.2 (CH2), 102.6
(aromatic-C), 123.7 (aromatic-C), 126.9 (aromatic-C), 130.5 (aromatic-C), 133.6
(aromatic-C), 140.6 (4-C), 141.8 (9-C), 147.8 (2-C), 159.3 (6-C) .
Anal. for C12H11Cl2NO ( 256.13 ).
Calcd: C 56.27, H 4.33, N 5.47
Found: C 56.10, H 4.13, N 5.43
Chloro-6-methoxy-3-(4-methoxyphenyl)quinolin-2(1H)-one (59a)
A solution of 2,4-dichloro-6-methoxy-3-(4-methoxyphenyl)quinoline (58a: 7.40
g, 22.16 mmol), and 70 % methanesulfonic acid (8.0 mL) in n-butanol (55.0
mL) was heated under reflux for 48 hours and worked up following the method
described for 6, to give 4-chloro-6-methoxy-3-(4-methoxyphenyl)quinolin-

EXPERIMENTAL PART 193
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2(1H)-one (59a). The yield was 6.467 g (93 %), beige prisms, mp 275–277 °C
(ethanol).
Rf = 0.71 (chloroform/aceton 3:7).
IR (ATR-measurement): 3435 (m), 3179 (m), 3092 (w), 3046 (w), 3008 (w), 2951
(w), 2929 (w), 2832 (w), 1649 (s), 1622 (w), 1609 (w), 1595 (w), 1572 (w), 1513
(m), 1495 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.81 (s, 3 H, 6-CH3O), 3.83 (s, 3 H, Ar-
CH3O), 7.01 (d, J = 8.7 Hz, 2 HAA Ar-H), 7.27-7.36 (m, 3 H, 5-H, 7-H, 8-H, and
2 H, HBB´ Ar-H), 12.09 (s, 1 H, NH).
Anal. for C17H14ClNO3 (315.76).
Calcd: C 64.67, H 4.47, N 4.44
Found: C 64.48, H 4.27, N 4.42
4-Chloro-3-(4-chlorophenyl)-6-methoxyquinolin-2(1H)-one (59b)
A solution of 2,4-dichloro-3-(4-chlorophenyl)-6-methoxyquinoline (58b: 6.00 g,
17.73 mmol), and 70 % methanesulfonic acid (6.0 mL) in n-butanol (60.0 mL)
was heated under reflux for 45 hours and worked up following the method
described for 6, to give 4-chloro-3-(4-chlorophenyl)-6-methoxyquinolin-2(1H)-
one (59b). The yield was 5.330 g (94 %) beige prisms, mp 296–298 °C (ethanol).
Rf = 0.79 (chloroform/aceton 3:7).
IR (KBr): 3427 (m), 2957 (w), 2922 (w), 2848 (m), 2766 (w), 1666 (s), 1624 (w),
1593 (m), 1494 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 (s, 3 H, CH3O), 7.30-7.35 (m. 3 H, 5-
H, 7-H and 8–H), 7.49 (d, J = 7.7 Hz, 2 HAA´ Ar-H), 7.52 (d, J = 7.8 Hz, 2 HBB´
Ar-H), 12.18 (s, 1 H, NH).
Anal. for C16H11Cl2NO2 (320.18).
Calcd: C 60.02, H 3.46, N 4.37
Found: C 60.23, H 3.39, N 4.36

EXPERIMENTAL PART 194
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Chloro-3-(3-chlorophenyl)-6-methoxyquinolin-2(1H)-one (59d)
A solution of 2,4-dichloro-3-(3-chlorophenyl)-6-methoxyquinoline (58d: 6.00 g,
17.73 mmol), and 70 % methanesulfonic acid (3.0 mL) in n-butanol (50.0 mL)
was heated under reflux for 45 hours, and worked up following the method
described for 6, to give 4-chloro-3-(3-chlorphenyl)-6-methoxyquinolin-2(1H)-
one (59d). The compound obtained was pure enough for further use. The yield
was 5.100 g (90 %), beige prisms, mp 285–288 °C (ethanol).
Rf = 0.78 (chloroform/aceton 3:7).
IR (KBr): 2832 (w), 1658 (s), 1623 (m), 1500 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 (s, 3 H, CH3O), 7.32-7.33 (m, 3 H, Ar-
H), 7.37 (d, J = 8.6 Hz, 1 H, 8-H), 7.44 (s, 1 H, Ar-H), 7.43-7.50 (m, 2 H, 5-H,
and 7-H ), 12.21 (s, 1 H, NH).
Anal. for C16H11Cl2NO2 (320.18).
Calcd: C 60.02, H 3.46, N 4.37
Found: C 59.90, H 3.31, N 4.35
4-Chloro-6-methoxy-3-phenylquinolin-2(H)-one (59e)
A solution of 2,4-dichloro-6-methoxy-3-phenylquinoline (58e: 7.00 g, 23.03
mmol) and 70 % methanesulfonic acid (11.50 mL) in n-butanol (92.0 mL) was
heated to 110 °C for 30 hours and worked up following the method described
for 6, to afford 4-chloro-6-methoxy-3-phenylquinolin-2(H)-one (59e). The yield
was 5.860 g (89 %), colorless prisms, mp 278–281 °C (ethanol).
Rf = 0.82 (chloroform/aceton 3:7).
IR (ATR-measurement): 3523 (w), 3437 (m), 3251 (w), 3192 (s), 3087 (w), 3055
(w), 3005 (w), 2953 (w), 2926 (w), 1640 (s), 1595 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.38 (s, 3 H, CH3O), 7.29-7.48 (m, 3 H, 5-
H, 7-H 8-H, and 5-H, Ph-H), 12.14 (s, 1 H, NH).
Anal. for C16H12ClNO2 (385.73).
Calcd: C 67.26, H 4.23, N 4.90
Found: C 67.25, H 3.99, N 4.90

EXPERIMENTAL PART 195
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Chloro-6-methoxy-3-methylquinolin-2(1H)-one (59f)
A solution of 2,4-dichloro-6-methoxy-3-methylquinoline (58f: 8.50 g, 35.12
mmol) and 70 % methanesulfonic acid (7.0 mL) in ethanol (80.0 mL) was
heated to reflux for 48 hours and worked up following the method described for
6, to afford 4-chloro-6-methoxy-3-methylquinolin-2(1H)-one (59f).
The yield was 7.189 g (92 %), light yellow prisms, mp 253–256 °C (ethanol).
Rf = 0.66 (chloroform/aceton 3:7).
IR (KBr): 3431 (w), 2982 (w), 2955 (w), 2925 (w), 2818 (w), 2767 (w), 2733 (w),
1649 (s), 1625 (w), 1600 (w), 1566 (w), 1501 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 2.22 (s, 3 H, CH3), 3.81 (s, 3 H, CH3O),
7.18-7.23 (m, 2 H, 5-H and 7-H), 7.30 (d, J = 8.7 Hz, 1 H, 8-H), 11.95 (s, 1 H,
NH).
Anal. for C11H10ClNO2 (223.66).
Calcd: C 59.07, H 4.51, N 6.26
Found: C 58.95, H 4.32, N 6.17
4-Chloro-3-ethyl-6-methoxyquinolin-2(1H)-one (59g)
A solution of 2,4-dichloro-3-ethyl-6-methoxyquinoline (58g: 2.150 g, 8.40
mmol) and 70 % methanesulfonic acid (2.0 mL) in n-butanol (20.0 mL) was
heated to reflux for 48 hours and worked up following the method described
for 6, to afford 4-chloro-3-ethyl-6-methoxyquinolin-2(1H)-one (59g). The
compound was pure without recrystallization. The yield was 1.589 g (80 %),
colorless prisms, mp 215–218 °C (ethanol).
Rf = 0.79 (chloroform/aceton 3:7).
IR (ATR-measurement): 3007 (w), 2968 (w), 2957 (w), 2930 (w), 2867 (w), 2722
(w), 1651 (s), 1620 (w), 1597 (s), 1562 (w), 1498 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.07 (t, J = 7.4 Hz, 3 H, CH3), 2.72 (q, J =
7.4 Hz, 2 H, CH2), 3.81 (s, 3 H, CH3O), 7.19 (dd, J = 8.7 + 2.8 Hz, 1 H, 7-H),
7.22 (d, J = 2.3 Hz, 1 H, 5-H), 7.28 (d, J = 8.7 Hz, 1 H, 8-H), 11.95 (s, 1 H, NH).

EXPERIMENTAL PART 196
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
13C-NMR (300 MHz, [D6]DMSO): = 12.5 (CH3), 21.9 (CH2), 55.9 (CH3O), 106.5
(aromatic-C), 117.3 (aromatic-C), 118.7 (aromatic-C), 120.2 (aromatic-C), 131.8
(aromatic-C), 134.2 (aromatic-C), 139.8 (4-C), 155.0 (6-C), 160.9 (lactam-C=O).
Anal. for C12H12ClNO2 (237.69).
Calcd: C 60.64, H 5.09, N 5.89
Found: C 60.43, H 4.89, N 5.82
6-Methoxy-3-(4-methoxyphenyl)-2-oxo-1,2-dihydroquinoline-4-
carbonitrile (60a)
A mixture of 4-chloro-6-methoxy-3-(4-methoxyphenyl)quinolin-2(1H)-one (59a:
1.60 g, 5.11 mmol), sodium p-toluenesulfinate (0.910 g, 5.10 mmol), and dry
potassium cyanide (0.995 g, 15.31 mmol) in dry dimethylformamide (30.0 mL)
was heated to 120 °C for 72 hours, with vigorous stirring, and worked up
following the method described for 7, to give 6-methoxy-3-(4-methoxyphenyl)-
2-oxo-1,2-dihydroquinoline-4-carbonitrile (60a). The yield was 1.495 g (96 %),
yellow prisms, mp 275–278 °C (acetone).
Rf = 0.79 (chloroform/aceton 3:7).
IR (KBr): 3432 (m), 3148 (w), 2839 (m), 2229 (w), 1658 (s), 1624 (m), 1606 (s),
15748 (w), 1515 (w), 1501 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.84 (s, 3 H, Ar-CH3O), 3.85 (s, 3 H, 6-
CH3O), 7.06 (d, J = 8.7 Hz, 2 HBB´ Ar-H), 7.16 (d, J = 2.4 Hz, 1 H, 5-H), 7.32
(dd, J = 9.0 + 2.4 Hz, 1 H, 7-H), 7.38 (d, J = 9.0 Hz, 1 H, 8-H), 7.58 (d, J = 8.6
Hz, 2 HAA´ Ar-H), 12.41 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): =55.7 (C-6-CH3O), 55.9 (aryl-CH3O), 106.6
(aromatic-C), 113.8 (2 x aromatic-CAA´), 115.6 (aromatic-C), 1176 (aromatic-C),
118.3 (CN), 121.2 (aromatic-C),125.7 (aromatic-C),132.1 (2 x aromatic-
CBB´),132.9 (aromatic-C), 140.8 (aromatic-C), 155.4 (6-C), 159.4 (C-aryl-CH3O),
160.7 (lactam-C=O).
MS (ESI neg): m/z (%) = 306 (21, M + 1), 305 (100, M).

EXPERIMENTAL PART 197
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Anal. for C18H14N2O3 (306.32).
Calcd: C 70.58, H 4.61, N 9.15
Found: C 70.00, H 4.40, N 9.01
The elemental analysis showed a small deviation of 0.58 %.for carbon
component due to the fact that the compound 60a was not well soluble in
almost all organic solvent.
3-(4-Chlorophenyl)-6-methoxy-2-oxo-1,2-dihydroquinoline-4-
carbonitrile (60b)
A mixture of 4-chloro-3-(4-chlorophenyl)-6-methoxyquinolin-2(1H)-one (59a:
1.60 g, 5.10 mmol), sodium p-toluenesulfinate (0.890 g, 5.0 mmol), and dry
potassium cyanide (0.813 g, 12.51 mmol) in dry dimethylformamide (30.0 mL)
was heated to 120 °C for 63 hours and the solution was worked up following
the method described for 7, to give 3-(4-chlorophenyl)-6-methoxy-2-oxo-1,2-
dihydroquinoline-4-carbonitrile (60b). The yield was 1.523 g (98 %), yellow
prisms, mp 332–333.5 °C (acetone).
Rf = 0.45 (petrolether/EtOAc 1:3).
IR (ATR-measurement): 3434 (m), 2997 (w), 2916 (w), 2857 (w), 2764 (w), 2231
(w), 1670 (s), 1623 (w), 1607 (w), 1595 (w), 1503 (w), 1495 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.78 (s, 3 H, CH3O), 7.15 (d, J = 2.1 Hz, 1
H, 5-H), 7.35 (dd, J = 9.1 + 2.3 Hz, 1 H, 7-H), 7.41 (d, J = 9.0 Hz, 1 H, 8-H),
7.58 (d, J = 8.6 Hz, 2 HAA´ Ar-H), 7.63 (d, J = 8.6 Hz, 2 HBB´ Ar-H), 12.50 (s, 1
H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 56.1 (CH3O), 106.7 (10-C), 115.2 (5-C),
117.4 (4-C), 117.8 (7-C), 119.5 (CN), 121.8 (8-C), 130.8 (2 x aromatic-CBB´),
132.4 (2 x aromatic-CAA´), 132.6 (9-C), 133.4 (aryl-C), 134.7 (aryl-C), 139.9 (3-
C), 155.5 (6-C),
159.1 (lactam-C=O).
MS (ESI neg): m/z (%) = 311 (35, M + 1), 309 (100, M).
Anal. for C17H11ClN2O2 (310.75).

EXPERIMENTAL PART 198
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The elemental analysis of compound 60b could not be obtained because
compound 60b was not at all soluble in almost all organic solvent.
3-(3-Chlorophenyl)-6-methoxy-2-oxo-1,2-dihydroquinoline-4-
carbonitrile (60d)
A mixture of 4-chloro-3-(3-chlorophenyl)-6-methoxyquinolin-2(1H)-one (59d:
2.50 g, 7.81 mmol), sodium p-toluenesulfinate (1.60 g, 8.98 mmol) and dry
potassium cyanide (1.70 g, 26.15 mmol) in dry dimethylformamide (50.0 mL)
was heated to 130 °C for 43 hours with vigorous stirring and the solution was
worked up following the method described for 7, to give 3-(3-chlorophenyl)-6-
methoxy-2-oxo-1,2-dihydroquinoline-4-carbonitrile (60d). The yield was 2.347
g (97 %), green prisms, mp 328–330 °C (acetone).
Rf = 0.76 (chloroform/aceton 3:7).
IR (ATR-measurement): 2823 (w), 2230 (w), 1661 (w), 1623 (m), 1501 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.86 (s, 3 H, CH3O), 7.18 (d, J = 2.0 Hz, 1
H, 5-H), 7.37 (dd, J = 9.1 + 2.3 Hz, 1 H, 7-H), 7.41 (d, J = 9.0 Hz, 1 H, 8-H),
7.57 (m, 3 H, Ar-H), 7.70 (s, 1 H, Ar-H), 12.54 (s, 1 H, NH).
Anal. for C17H11ClN2O2 (310.74).
Calcd: C 65.71, H 3.57, N 9.01
Found: C 65.30, H 3.45, N 8.86
6-Methoxy-2-oxo-3-phenyl-1,2-dihydroquinoline-4-carbonitrile
(60e)
A mixture of 4-chloro-6-methoxy-3-phenylquinolin-2(1H)-one (59e: 3.00 g,
10.51 mmol), sodium p-toluenesulfinate (1.870 g, 10.51 mmol) and dry
potassium cyanide (2.00 g, 30.77 mmol) in dry dimethylformamide (63.0 mL)
was heated at 120 °C for 45 hours with vigorous stirring and the solution was

EXPERIMENTAL PART 199
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
worked up following the method described for 7, to give 6-methoxy-2-oxo-3-
phenyl-1,2-dihydroquinoline-4-carbonitrile (60e). The yield was 2.775 g (96 %),
yellow prisms, mp 263–265 °C (acetone).
Rf = 0.85 (chloroform/aceton 3:7).
IR (KBr): 3449 (m), 2971 (w), 2912 (w), 2849 (w), 2823 (w), 2763 (w), 2236 (w),
1667 (s), 1624 (w), 1602 (w), 1495 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.82 (s, 3 H, CH3O), 7.11 (d, J = 2.5 Hz, 1
H, 5-H), 7.30 (dd, J = 9.0 + 2.6 Hz, 1 H, 7-H), 7.35 (d, J = 9.0 Hz, 1 H, 8-H),
7.48-7.5 (m, 3 H, PhH), 7.59 (dd, J = 7.5 + 2.4 Hz, 2 H, PhH), 12.44 (s, 1 H,
NH).
13C-NMR (360 MHz, [D6]DMSO): = 55.9 (CH3), 106.6 (aromatic-C), 115.2
(aromatic-C), 117.7 (aromatic-C), 119.2 (CN), 121.5 (aromatic-C), 128.4 (PhC),
129.8 (2 x aromatic-CAA´), 130.4 (2 x aromatic-CBB´), 133.2(9-C), 133.7 (PhC),
141.1 (3-C), 155.4 (6-C), 159.3 (lactam-C=O).
MS (APCI pos): m/z (%) = 277 (100, M).
MS (APCI neg): m/z (%) = 276 (19, M + 1), 275 (100, M).
Anal. for C17H12N2O2 (276.30).
Calcd: C 73.90, H 4.38, N 10.14
Found: C 73.62, H 4.11, N 9.92
2-Chloro-6-methoxy-3-phenylquinoline-4-carbonitrile (61)
A solution of 6-methoxy-2-oxo-3-phenyl-1,2-dihydroquinoline-4-carbonitrile
(60e: 1.373 g, 5.0 mmol) in phosphoroxychloride (6.0 mL) was refluxed at 110
°C for 12 hours. The solution was cooled to 50 °C and the excess amount of
phosphoroxychloride was removed under reduced pressure. The residue was
poured onto ice/water (100 mL), filtered by suction and washed with water to
afford 2-chloro-6-methoxy-3-phenylquinoline-4-carbonitrile (61). The yield was
1.390 g (95 %), yellowish prisms, mp 242–245 °C (ethanol).
IR (KBr): 3439 (s), 2959 (w), 2236 (w), 1618 (m), 1561 (w), 1497 (m) cm-1.

EXPERIMENTAL PART 200
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1H-NMR (300 MHz, [D6]DMSO): = 4.00 (s, 3 H, CH3O), 7.36 (d, J = 2.7 Hz, 1
H, 5-H), 7.59 (s, 5 H, PhH), 7.68 (dd, J = 9.3 + 2.7 Hz, 1 H, 7-H), 8.11 (d, J =
9.3 Hz, 1 H, 8-H).
C17H11ClN2O (294.74).
Ethyl3–[(4-methoxyphenyl)amino]-
2-[(4-methoxyphenyl)carbamoyl]-3-oxopropanoate (64)
A mixture of p-anisidine (1: 0.615 g, 5.0 mmol) and triethyl
methanetricarboxylate (62: 2.320 g, 10.0 mmol) in bromobenzene (50.0 mL)
was heated under reflux to 155 °C for 6 hours. The solvent was evaporated
under reduced pressure. The crude product was digested with diethyl ether (25
mL), filtered by suction and washed with water to give ethyl 3–[(4-
methoxyphenyl)amino]-2-[(4-methoxyphenyl)carbamoyl]-3-oxopropanoate (64).
The yield was 1.570 g (88 %), grey prisms, mp 168–170 °C (ethanol).
Rf = 0.93 (chloroform/aceton 3:7).
IR (ATR-measurement): 3262 (m), 1747 (m), 1671 (s), 1609 (w), 1553 (w), 1533
(m), 1511 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.21 (t, J = 7.1 Hz, 3 H, CH3), 3.72 (s, 2 x 3
H, 2 x CH3O), 4.16 (q, J = 7.1 Hz, 2 H, OCH2), 4.65 (s, 1 H, CH), 6.90 (d, J = 8.9
Hz, 2 x 2 H, Ar-H), 7.48 (d, J = 8.9 Hz, 2 x 2 H, Ar-H), 10.12 (s, 2 x 1 H, 2
x NH).
C20H22N2O6 (386.41).

EXPERIMENTAL PART 201
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Ethyl 4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (67)
To a solution of sodium ethoxide in ethanol, prepared from sodium (3.450 g,
0.15 mol) in anhydrous ethanol (112.50 mL), a mixture of diethyl malonate
(66: 23.0 mL, 24.15 g, 0.15 mol ) and methyl anthranilate (65: 19.50 mL,
22.65 g, 0.15 mol) was added. The mixture was heated to 140–150 °C for 5
hours and the excess amount of ethanol (116.60 mL) was removed by
distillation using a short column. The resulting yellow solid material was
heated to 140–150 °C for 10 hours, the solid was cooled at ambient
temperature and triturated with diethyl ether (150.0 mL).
The solution was filtered by suction and washed with diethyl ether, the product
got dry immediately. Then it was dissolved in water (500 mL) at 50 °C, filtered
from insoluble parts and the filtrate was acidified with hydrochloric acid (2 M).
The light yellow precipitate was filtered by suction, washed several times with
water and dried at 40 °C under reduced pressure to give ethyl 4-hydroxy-2-
oxo-1,2-dihydroquinoline-3-carboxylate (67). The yield was 26.795 g (77 %),
mp 205–207 °C (methanol), lit. mp 194–304 °C [ex-62–64].
Rf = 0.57 (chloroform/aceton 3:7).
IR (ATR-measurement): 2926 (w), 2960 (w), 2910 (w), 1669 (s), 1638 (w), 1627
(s), 1609 (s), 1558 (w), 1494 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.31 (t, J = 7.1 Hz, 3 H, CH3), 4.34 (q, J =
7.1 Hz, 2 H, CH2), 7.22 (t, J = 6.6 Hz, 1 H, 6-H), 7.28 (d, J = 8.3 Hz, 1 H, 8-H),
7.62 (t, J = 7.7 Hz, 1 H, 7-H), 7.94 (d, J = 8.0 Hz, 1 H, 5-H), 11.50 (s, 1 H, NH),
13.40 (s, 1 H, OH).
C12H11NO4 (233.23).
Ethyl 2,4-dichloroquinoline-3-carboxylate (68)
A mixture of ethyl 4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carboxylate (67:
3.00 g, 12.87 mmol), in phosphoroxychloride (20.0 mL) and triethylamine (2.0
mL, 1.460 g, 14.46 mmol) was heated to 60 °C under stirring for 2 hours.

EXPERIMENTAL PART 202
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Then the mixture was poured onto crushed ice (200 mL), the obtained mixture
of yellow oil and precipitate was stirred until all solidified. The mixture was
brought to pH = 5–6 with concentrated sodium hydroxide solution, the
precipitate was filtered by suction and washed with water and dried at 40 °C
under reduced pressure, which afforded ethyl 2,4-dichloroquinoline-3-
carboxylate (68). The yield was 3.402 g (98 %), light pillow needles, mp 119–
121 °C (methanol), lit. mp 85–104 °C [ex-62b, ex-65, ex-66].
Rf = 0.93 (chloroform/aceton 3:7).
IR (ATR-measurement): 2986 (w), 1728 (s), 1613 (w), 1560 (m), 1481 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.37 (t, J = 7.1 Hz, 3 H, CH3), 4.50 (q, J =
7.1 Hz, 2 H, CH2), 7.89 (t, J = 7.9 Hz, 1 H, 6-H), 8.03 (t, J = 7.6 Hz, 1 H, 7-H),
8.10 (d, J = 8.3 Hz, 1 H, 8-H), 8.28 (d, J = 8.3 Hz, 1 H, 5-H).
C12H9Cl2NO2 (270.12).
4-Hydroxyquinolin-2(1H)-one (69)
Method A
A solution of ethyl 2,4-dichloroquinoline-3-carboxylate (68: 1.350 g, 5.0 mmol)
and 70 % methanesulfonic acid (3.1 equiv., 1.480 g, 1.00 mL, 15.42 mmol) in
ethanol ( 15.0 mL) was heated under reflux for 48 hours. The mixture was
cooled to 20 °C and poured onto ice/water (50 mL), brought to pH = 4–6 with
sodium hydroxide (2 M), filtered by suction, washed with water and dried at 40
°C under reduced pressure, to give 4-hydroxyquinolin-2(1H)-one (69). The yield
was 0.966 g (77 %), colorless prisms, mp 344–346 °C (ethanol).
Rf = 0.22 (chloroform/aceton 3:7).
Method B
A mixture of ethyl 4-chloro-2-oxo-1,2-dihydroquinoline-3-carboxylate (70: 0.60
g, 2.40 mmol), sodium p-toluenesulfinate (0.892 g, 5.01 mmol), and dry
potassium cyanide (0.387 g, 5.96 mmol) in dry dimethylformamide (15.0 mL)
was heated to 120 °C for 28 hours and the solution was worked up following

EXPERIMENTAL PART 203
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
the method described for 7, to form 4-Hydroxyquinolin-2(1H)-one (69). The
yield was 0.372 g (97 %), dark green prisms, mp > 350 °C (ethanol), lit. mp
250–360 °C [ex-3c, ex-3d, ex-67, ex-68, ex-69, ex-70, ex-71, ex-72, ex-73, ex-
74].
Rf = 0.22 (chloroform/aceton 3:7).
IR (ATR-measurement): 3093–2560 (m, br), 1653 (s), 1632 (s), 1611 (w), 1593
(s), 1505 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 5.72 (s, 1 H, 3-H), 7.15 (t, J = 7.6 Hz, 1 H,
6-H), 7.25 (d, J = 8.2 Hz, 1 H, 5-H), 7.48 (d, J = 7.7 Hz, 1 H, 7-H), 7.76 (d, J =
7.9 Hz, 1 H, 8-H), 11.18 (s, 1H, NH), 11.29 (s, 1 H, OH).
C9H7NO2 (161.16).
Ethyl 4-chloro-2-oxo-1,2-dihydroquinoline-3-carboxylate (70)
A solution of ethyl 2,4-dichloroquinoline-3-carboxylate (68: 15.00 g, 55.56
mmol) and 70 % methanesulfonic acid (1.54 equiv., 8.140 g, 5.50 mL, 84.80
mmol) in ethanol ( 90.0 mL) was heated under reflux for 48 hours. The mixture
was cooled to 20 °C and poured into ice/water (250 mL), brought to pH = 4–6
with sodium hydroxide (2 M), filtered by suction, washed with water and dried
at 40 °C under reduced pressure, to give ethyl 4-chloro-2-oxo-1,2-
dihydroquinoline-3-carboxylate (70). The compound was pure for further steps.
The yield was 9.39 g (67 %), colorless prisms, mp 198–200 °C, lit. mp 194–203
°C [ex-65, ex-75, ex-76].
Rf = 0.87 (chloroform/aceton 3:7).
IR (ATR-measurement): 3156 (w), 3101 (w), 2984 (w), 2886 (w), 2843 (w), 1736
(s), 1649 (s), 1619 (w), 1596 (m), 1566 (w), 1497 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.31 (t, J = 7.1 Hz, 3 H, CH3), 4.34 (q, J =
7.1 Hz, 2 H, CH2), 7.33–7.38 (m, 1 H, 7-H), 7.41 (dd, J = 8.1 + 1.0 Hz, 1 H, 8-
H), 7.66–7.72 (m, 1 H, 6-H), 7.90 (dd, J = 8.1 + 0.9 Hz, 1 H, 5-H), 12.50 (s, 1 H,
NH).
C12H10ClNO3 (251.67).

EXPERIMENTAL PART 204
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Ethyl [4-chloro-3-(4-chlorophenyl)-6-methoxy-2-oxoquinolin-
1(2H)-yl]acetate (72)
A mixture of 4-chloro-3-(4-chlorophenyl)-6-methoxyquinolin-2(1H)-one (59b:
1.00 g, 3.13 mmol), ethyl bromoacetate (1.310 g, 7.84 mmol) and dry sodium
carbonate (0.831 g, 7.84 mmol) in dry dimethylformamide (25.0 mL) was
heated to 120–140°C for 25 minutes. The solution was cooled to room
temperature and poured into ice/water (25 mL). The obtained solid was filtered
by suction, washed with water and dried at 40 °C under reduced pressure to
afford ethyl [4-chloro-3-(4-chlorophenyl)-6-methoxy-2-oxoquinolin-1(2H)-
yl]acetate (72). The yield was 1.199 g (95 %), colorless prisms, mp 122–124 °C
(ethanol).
Rf = 0.74 (chloroform/aceton 3:7).
IR (ATR-measurement): 2980 (w), 2838 (w), 1737 (m), 1638 (s), 1596 (w), 1570
(m), 1501 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.21 (t, J = 7.1 Hz, 3 H, CH3), 3.87 (s, 3 H,
CH3O), 4.16 (q, J = 7.1 Hz, 2 H, CH2 ), 5.14 (s, 2 H, NCH2), 7.36 (dd, J = 9.3 +
2.8
Hz, 1 H, 7-H), 7.38 (d, J = 8.6 Hz, 2 H, Ar-H), 7.49 (d, J = 2.8 Hz, 1 H, 5-H),
7.53 (d, J = 8.6 Hz, 2 H, Ar-H), 7.56 (d, J = 9.0 Hz, 1 H, 8-H).
C20H17Cl2NO4 (406.27).
4-Hydroxy-6,7-dimethoxyquinolin-2(1H)-one (74)
A mixture of 3,4-dimethoxyanilin (73: 6.12 g , 40.0 mmol) and dry malonic acid
(2: 4.38 g, 42.12 mmol) in phosphoryloxychloride (4.0 mL) was heated in an
open flask to 50 °C for 2 hours, then the temperature was raised slowly up to
90 °C and kept there for 30 minutes. The obtained residue was dissolved in
aqueous sodium hydroxide (150 mL, 0.5 M) under warming to 60 °C and the
solution was filtered. The alkaline filtrate was acidified with concentrated
hydrochloric acid to pH = 1–2.

EXPERIMENTAL PART 205
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The formed precipitate was filtered by suction, washed with water and dried at
40 °C under reduced pressure and afforded 4-hydroxy-6,7-dimethoxyquinolin-
2(1H)-one (74). The yield was 7.417 g (84 %), pale green prisms, mp 346–349
°C (ethanol), lit. mp > 320 °C [ex-76, ex-78, ex-79].
Rf = 0.08 (chloroform/aceton 3:7).
IR (KBr): 3429 (s), 2999 (w), 2941 (w), 2834 (w), 2796 (w), 1682 (m), 1642 (w),
1617 (s), 1518 (m) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.77 and 3.79 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 5.64 (s, 1 H, 3-H), 6.82 (s, 1 H, 8-H), 7.16 (s, 1 H, 5-H), 11.06 (s, 1 H
NH).
C11H11NO4 (221.21).
2,4-Dichloro-6,7-dimethoxyquinoline (75)
A solution of 4-hydroxy-6,7-dimethoxyquinolone(74: 7.828 g, 35.42 mmol) in
phosphoryl chloride (42.0 mL) was heated under reflux to 110 °C for 8 hours.
The solution was cooled to 50 °C and the excess amount of phosphoryl chloride
was removed under reduced pressure. The residue was poured onto ice/water
(400 mL), and brought to pH = 4–6 with aqueous sodium hydroxide (5 M).
The resulting precipitate was filtered by suction and washed with water to
afford 2,4-dichloro-6,7-dimethoxyquinoline (75). The yield was 7.640 g (84 %)
greyish prisms, mp 174–176 °C (ethanol), lit. mp 160–161 °C [ex-77, ex-80].
Rf = 0.88 (chloroform/aceton 3:7).
IR (KBr): 3435 (s), 3008 (w), 985 (w), 1622 (m), 1571 (m), 1505 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.95 and 3.96 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 7.35 (s, 1 H, 3-H), 7.42 (s, 1 H, 8-H), 7.72 (s, 1 H, 5-H).
C11H9Cl2NO2 (258.11).

EXPERIMENTAL PART 206
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
4-Chloro-6,7-dimethoxyquinolin-2(1H)-dione (76)
A solution of 2,4-dichloro-6,7-dimethoxyquinoline (75: 4.00 g, 15.50 mmol),
and 70 % methanesulfonic acid (4.0 mL) in n-butanol (50.0 mL) was heated
under reflux for 28 hours. The solution was cooled to room temperature and
poured onto ice/water (25 mL), brought to pH = 4–6 with sodium hydroxide (2
M), filtered by suction, washed with water and dried at 40 °C under reduced
pressure to give 4-chloro-6,7-dimethoxy-quinolon-2(1H)-one (76). The yield was
3.573 g (96 %), beige prisms, mp 272–274 °C (ethanol), lit. mp 258–259 °C [ex-
77].
Rf = 0.38 (chloroform/aceton 3:7).
IR (KBr): 3418 (m), 3261 (w), 3019 (w), 2931 (w), 2853 (w), 1656 (s), 1624 (m),
1606 (w), 1513 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.83 and 3.84 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O ), 6.60 (s, 1 H, 3-H), 6.91 (s, 1 H, 8-H), 7.17 (s, 1 H, 5-H), 11.84 (s, 1 H,
NH).
C11H10ClNO3 (239.66).
3,3-Dichloro-6,7-dimethoxyquinoline-2,4(1H,3H)-dione (77)
A suspension of 4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one (74: 10.940 g,
49.50 mmol) in dioxane (198.0 mL) was warmed to 40–50 °C and then, under
vigorous stirring, keeping the temperature between 50–60 °C, sulfuryl chloride
(10.0 mL) was added dropwise. As soon as the temperature reached 60 °C, the
reaction was stopped and cooled to 20 °C. The mixture was filtered and the
filtrate was poured onto ice/water (500 mL) under stirring, filtered by suction,
washed with water and dried at 40 °C under reduced pressure to give 3,3-
dichloro-6,7-dimethoxyquinoline-2,4(1H,3H)-dione (77). The yield was 11.751 g
(82 %), green-yellow prisms, mp 229–231 °C (ethanol).
Rf = 0.87 (chloroform/aceton 3:7).

EXPERIMENTAL PART 207
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
IR (KBr): 3430 (m), 3245 (m), 3009 (w), 2939 (w), 2849 (w), 1717 (s),
1709 (s), 1673 (s), 1612 (s), 1515 (s), 1515 (s), 1503 (w) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.80 and 3.86 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 6.68 (s, 1 H, 8-H), 7.25 (s, 1 H, 5-H), 11.29 (s, 1 H, NH).
Anal. for C11H9Cl2NO4 (209.10).
Calcd. C 45.54, H 3.13, N 4.83
Found C 45.49, H 2.96, N 4.72
3-Chloro-4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one (78)
To a solution of 3,3-dichloro-6,7-dimethoxyquinoline-2,4(1H,3H)-dione (77:
5.50 g, 26.32 mmol) in ethanol (46.0 mL) and acetic acid (23.0 mL), zinc-dust
(2.00 g, 30.58 mmol) was added in small portions and the solution was kept to
boiling. The yellow solution got decolorized (from yellow to grey-greenish),
which indicated the end of reaction. The solution was cooled to 20 °C and
filtered from zinc and salts. To the filtrate, ice/water (300 mL) was added.
The formed colorless precipitate was filtered by suction, washed with water and
dried at 40 °C under reduced pressure to form 3-chloro-4-hydroxy-6,7-
dimethoxyquinolin-2(1H)-one (78). The yield was 4.215 g (87 %), beige prisms,
mp 289–291 °C (ethanol).
Rf = 0.29 (chloroform/aceton 3:7).
IR (KBr): 3438 (m), 1525 (w), 2965 (m), 2828 (w), 1641 (s), 1620 (s), 1605 (w),
1514 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.79 and 3.80 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 6.83 (s, 1 H, 8-H), 7.29 (s, 1 H, 5-H), 11.58 (s, 1 H, NH).
Anal. for C11H10ClNO4 (255.66).
Calcd. C 51.68, H 3.94, N 5.48
Found C 51.88, H 3.84, N 5.40

EXPERIMENTAL PART 208
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
2,3,4-Trichloro-6,7-dimethoxyquinoline (79)
A solution of 3-chloro-4-hydroxy-6,7-dimethoxyquinolin-2(1H)-one (78: 3.50 g,
13.70 mmol) in phosphoryl chloride (17.0 mL) was brought to reaction under
reflux for 8 hours and worked up following the method described for 5, to form
2,3,4-trichloro-6,7-dimethoxyquinoline (79). The yield was 3.938 g (92 %),
colorless prisms, mp 187–189 °C (ethanol).
Rf = 0.90 (chloroform/aceton 3:7).
IR (KBr): 3434 (s), 3010 (w), 1620 (m), 1549 (w), 1505 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.96 and 3.98 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 7.33 (s, 1 H, 8-H), 7.44 (s, 1 H, 5-H).
Anal. for C11H8Cl3NO2 (292.55).
Calcd. C 45.16, H 2.76, N 4.79
Found C 44.92, H 2.59, N 4.59
3,4-Dichloro-6,7-dimethoxyquinolin-2(1H)-one (80)
A solution of 2,3,4-trichloro-6,7-dimethoxyquinoline (79: 3.158 g, 10.80 mmol)
in 70 % methanesulfonic acid (3.0 mL) and n-butanol (30.0 mL) was heated to
110 °C for 24 hours and worked up as described for 6, to form 3,4-dichloro-
6,7-dimethoxyquinolin-2(1H)-one (80). The yield was 2.159 g (73 %), colorless
prisms, mp 307–309 °C (ethanol).
Rf = 0.64 (chloroform/aceton 3:7).
IR (KBr): 3436 (m), 3003 (w), 2962 (w), 2900 (w), 2835 (w), 2793 (w), 1671 (s),
1626 (w), 1603 (w), 1511 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.84 (s, 2 x 3 H, 6-CH3O and 7-CH3O), 6.92
(s, 1 H, 8-H), 7.19 (s, 1 H, 5-H), 12.37 (s, 1 H, NH).
Anal. for C11H9Cl2NO3 (274.11).
Calcd. C 48.20, H 3.31, N 5.11
Found C48.32, H 3.18, N 5.00

EXPERIMENTAL PART 209
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
6,7-Dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile
(82)
Pathway A
A mixture of 4-chloro-6,7-dimethoxyquinolin-2(1H)-one (76: 1.760 g, 7.35
mmol), sodium p-toluenesulfinate (5.233 g, 29.40 mmol) and potassium
cyanide (2.40 g, 36.75 mmol) in dry dimethylformamide (44.0 mL) was heated
to 130 °C for 86 hours and worked up as described for 7, to give 6,7-
dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (82). The yield was
1.068 g (57 %), red-black prisms, mp 349–350 °C (acetone).
Rf = 0.66 (chloroform/aceton 3:7).
Pathway B
A mixture of 3,4-dichloro-6,7-dimethoxyquinolin-2(1H)-one (80: 1.270 g, 4.64
mmol), sodium p-toluenesulfinate (1.750 g, 9.73 mmol) and potassium cyanide
(1.230 g, 18.92 mmol) in dry dimethylformamide (40.0 mL) was heated to 130
°C for 5 hours and worked up as described for 7, to give 6,7-dimethoxy-2-oxo-
1,2-dihydroquinoline-3,4-dicarbonitrile (82). The yield was 0.944 g (80 %), red
prisms, mp 348 °C (acetone).
Rf = 0.64 (chloroform/aceton 3:7).
IR (KBr): 3437 (w), 2975 (w), 2946 (w), 2900 (w), 2850 (w), 2784 (w), 2700 (w),
2230 (w), 1911 (w), 1665 (s), 1619 (m), 1597 (w), 1516 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.88 and 3.90 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 6.88 (s, 1 H, 8-H), 6.98 (s, 1 H, 5-H), 12.94 (s, 1 H, NH).
13C-NMR (300 MHz, [D6]DMSO): = 56.4 (6-CH3O), 56.8 (7-CH3O), 98.4
(aromatic-C), 105.3 (aromatic-C), 113.5 (aromatic-C), 114.9 (CN), 138.3
(aromatic-C), 147.5 (6-C), 156.8 (7-C), 157.6 (lactam-C=O).
MS (API-ES neg): m/z (%) = 255 (15, M + 1), 254 (100, M).
Anal. for C13H9N3O3 (255.23).
Calcd. C 61.18, H 3.55, N 16.46
Found C 60.96, H 3.43, N 15.90

EXPERIMENTAL PART 210
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
6,7-Dimethoxy-1-methyl-2-oxo-1,2-dihydroquinoline-3,4-
dicarbonitrile (83)
A mixture of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (82:
0.170 g, 0.67 mmol), sodium carbonate (0.25 g, 2.36 mmol), and iodomethane
(0.19 g, 1.34 mmol) in dry dimethylformamide (5.0 mL) was heated to 90–100
°C for 15 minutes and worked up as described for 45, to form 6,7-dimethoxy-
1-methyl-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (83). The yield was
0.157 g (88 %), red prisms, mp 329–333 °C (ethanol).
Rf = 0.78 (chloroform/aceton 3:7).
IR (ATR-measurement): 3055 (w), 2987 (w), 2953 (w), 1119 (w), 1730 (w), 1637
(s), 1545 (m), 1513 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 3.75 and 3.92 (2 s, 2 x 3 H, 6-CH3O and 7-
CH3O), 4.06 (s, 3 H, CH3), 7.14 (s, 1 H, 8-H), 7.15 (s, 1 H, 5-H).
Anal. for C14H11N3O3 (269.26).
Calcd. C 62.45, H 4.12, N 15.61
Found C 61.94, H 3.77, N 15.11
Ethyl [3,4-Dicyano-6,7-dimethoxy-2-oxoquinolin-1(2H)-
yl]acetate (84)
Method A
A mixture of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile (82:
1.54 g, 6.0 mmol), ethyl bromoacetate (1.50 mL, 2.30 g, 13.56 mmol) and dry
sodium carbonate (1.272 g, 12.0 mmol) in dry dimethylformamide (25.0 mL)
was heated to 80 °C for 15 minutes, The mixture was cooled to room
temperature and poured onto ice/water (100 mL). The obtained solid was
filtered by suction, washed with water and dried at 40 °C under reduced
pressure. TLC analysis showed two compounds: ethyl [3,4-dicyano-6,7-
dimethoxy-2-oxoquinolin-1(2H)-yl]acetate (84) and ethyl [(3,4-dicyano-6,7-
dimethoxyquinolin-2-yl)oxy]acetate (85).

EXPERIMENTAL PART 211
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
The yield of the mixture was 1.852 g (90 %). The 2 products were separated by
recrystallization from toluene/acetone (9:1), to give as first crop compound 85
and as second crop compound 84, respectively.
The yield of compound 84 was 1.153 g (56 %), yellow prisms, mp 256–259 °C
(toluene/acetone 9:1).
Rf = 0.53 (petrolether/EtOAc 1:3).
Method B
To a solution of 6,7-dimethoxy-2-oxo-1,2-dihydroquinoline-3,4-dicarbonitrile
(82: 0.50 g, 1.96 mmol) in dry tetrahydrofurane (15.0 mL), was added dropwise
a solution of lithium diisopropylamide (1.8 M in tetrahydrofurane/
heptane/ethylbenzene, 3.0 mL). The resulting mixture was stirred at 0 °C for 1
hour, then ethyl bromoacetate (1.50 mL, 2.30 g, 13.56 mmol) was added. The
mixture was stirred for 30 minutes to 0 °C and at room temperature for 15
hours. The solution was poured onto ice/water (10 mL), filtered by suction,
washed with water and dried at 40 °C under reduced pressure, to give ethyl
(3,4-dicyano-6,7-dimethoxy-2-oxo-1,2-dihydroquinolin-1-yl)acetate (84). The
yield was 0.434 g (65 %), yellow-gold prisms, mp 251–253 °C (ethanol).
IR (ATR-measurement): 3027 (w), 2976 (w), 2226 (w), 1732 (s), 1659 (s), 1618
(m), 1542 (m), 1519 (s) cm-1.
Rf = 0.50 (petrolether/EtOAc 1:3).
1H-NMR (300 MHz, [D6]DMSO): = 1.23 (t, J = 7.1 Hz, 3 H, CH3), 3.93 and
3.99 (2 s, 2 x 3 H, 6-CH3O and 7-CH3O), 4.19 (q, J = 7.1 Hz, 2 H, CH2), 5.26 (s,
2 H, N-CH2), 7.13 (s, 1 H, 8-H), 7.17 (s, 1 H, 5-H).
13C-NMR (300 MHz, [D6]DMSO): = 14.5 (CH3), 46.0 (N-CH2), 56.5 (6-CH3O),
57.6 (7-CH3O), 61.9 (CH2), 99.2 (aromatic-C), 106.0 (aromatic-C), 107.0
(aromatic-C), 110.9 (aromatic-C), 113.5 (CN), 114.7 (CN), 128.4 (aromatic-C),
138.7 (4-C), 147.4 (6-C), 157.3 (7-C), 157.4 (lactam-C=O), 167.6 (ester-C=O).
Anal. for C17H15N3O5 (341.33).
Calcd. C 59.82, H 4.43, N 12.31
Found C 59.81, H 4.41, N 12.09

EXPERIMENTAL PART 212
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Ethyl [(3,4-Dicyano-6,7-dimethoxyquinolin-2-yl)oxy]acetate (85)
This compound was obtained as solid during the work-up of compound 84,
and recystallized from ethanol. The yield was 0.602 g (29 %), yellow prisms, mp
187-189 °C (ethanol).
Rf = 0.92 (chloroform/aceton 3:7).
IR (ATR-measurement): 2983 (w), 2938 (w), 2227 (w), 1761 (m), 1616 (w), 1571
(m), 1504 (s) cm-1.
1H-NMR (300 MHz, [D6]DMSO): = 1.21 (t, J = 7.0 Hz, 3 H, CH3), 3.99 and
4.00 (2 s, 2 x 3 H, 6-CH3O and 7-CH3O), 4.19 (q, J = 7.1 Hz, 2 H, CH2), 5.22 (s,
2 H, N-CH2), 7.24 (s, 1 H, 8-H), 7.27 (s, 1 H, 5-H).
C17H15N3O5 (341.33).

EXPERIMENTAL PART 213
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
EXPERIMENTAL PART
FOR
CHAPTER 4

EXPERIMENTAL PART 214
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
ELECTRONIC SPECTRA
UV-Vis spectra
The absorption measurements were carried out using 1 cm polystyrene PS cell
(1.50 mL, diameter = 1.0 cm, Brand, Germany) for all experiments if not
specified otherwise. UV/Visible spectra were recorded using a Shimadzu
UV/Visible scanning spectrophotometer UV-2101 PC; concentration: 10-4
mol/L.
Fluorescence data
Standard excitation and emission spectra were recorded in the same
polystyrene cells using a Perkin-Elmer LS50B luminescence spectrometer.
Fluorescence quantum yield ( ) is the fraction of molecules that emit a photon
after direct excitation by a source. Quantum yields were calculated from the
area (integration) of the uncorrected emission spectra by comparison to the
area of fluorescein at pH 13 (exc = 493, em = 512 nm, = 0.9 and at pH 3.3
(abs = 437 nm, em = 512 nm, = 0.3) [ex-81] or, in the case of absorption
maxima below 400 nm, with the known quantum yield of 6,7-dimethoxy-4-
trifluoromethylcarbostyril in DMSO (exc = 368, em = 438 nm, = 0.41) and
water (exc = 365, em = 428 nm, [ex-82].

EXPERIMENTAL PART 215
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Fluorescence quantum yield was calculated using the equation:
Sc
S
refc
ref
refA
SA
refS
Where the meaning of the letters is:
Subscript S: data of the substance.
Subscript ref: data of the reference substance.
: quantum yield.
A: absorbance data at UV/Vis maxima.
ε: excitation coefficient.
C: molar concentration (mol/L).
n: refractive index.
Excitation and emission maxima of the standard should not be too far from the
investigated substances. Absorption in the 1.0 cm cell was usually about
0.005-0.010. If fluorescence standards were measured in solvents which were
different to the sample, the following correction factors for the calculated
quantum yield were applied:
2
newn
refn
f

EXPERIMENTAL PART 216
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Preparation of the stock solutions and measurement
Purity of the compounds was confirmed by elemental analyses.
For each sample, about 1-2 mg of the dye was dissolved in exactly the volume
of dimethylsulfoxide to result in a 1x10-2 M stock solution.
Recording of spectra
For UV/visible measurement, 10 L of each stock solution were diluted to 1.0
mL in the corresponding solvent; the concentration was then 1x10-4 M.
For excitation and emission spectra, the UV/visible solution were diluted 100
times again resulting in a 1x10-6 M solution.
General slit width: 5 nm; in cases of strong fluorescence 3 nm.
For solvent not compatible with polystyrene, 1.0 cm optical glass cells (3.0 mL)
were used.
standard in: refr. index n f (CH3CN) f (H2O) f (DMSO)
CH3CN 1.344 1.000 0.984 1.224
DMSO 1.487 0.817 0.804 1.000
CH3OH 1.329 1.023 1.006 1.252
H2O 1.333 1.017 1.000 1.244
CH2Cl2 1.490 0.814 0.800 0.996

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 217
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES OF THEORETICAL PART
FOR
CHAPTERS 1, 2 AND 3

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 218
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES OF THEORETICAL PART FOR
CHAPTERS 1, 2 AND 3
[1] O. S. Park, B. S. Jang, Arch. Pharm. Res. 1995, 18, 277–281.
[2] D. R. Buckle, B. C. C. Cantello, H. Smith, B. A. Spicer, J. Med. Chem.
1975, 18, 726–732.
[3] E. Ziegler, T. Kappe, Angew. Chem. 1964, 3, 754–754.
[4] E. Ziegler, T. Kappe, R. Salvador, Monatsh. Chem. 1963, 94, 453–459.
[5] Bayer AG., British Patent 721, 171, 1954. Chem. Abstr. 1956, 2685e.
[6] Bayer AG., (U. Hörlein Inv), British Patent 733, 123, 1955. Chem. Abstr.
1956, 5010799f.
[7] C. F. Neville, M. F. Grundon, V. N. Ramchandran, J. Reisch, J. Chem.
Soc., Perkin Trans. 1 1991, 259–262.
[8] P. Roschger, W. Fiala, W. Stadlbauer, J. Heterocycl. Chem. 1992, 29,
225-231.
[9] V. Dollé, E. Fan, C. H. Nguyen, A.-M. Aubertin, A. Kirn, M. L. Andreola,
G. Jamieson, L. Tarrago-Litvak, E. Bisagni, J. Med. Chem. 1995, 38,
4679-4686.
[10] M. Cobet, M. Lucker, Phytochemistry 1971, 10, 1031.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 219
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[11] a) K. C. Majumdar, P. P. Mukhopadhyay, Synthesis, 2003, 97, b) R. L.
Yong, K. Hyuk, S. K. Wha, R. M. Kyung, K. Youngsoo, H. L. Seung,
Synthesis, 2001, 1851.
[12] Tschitschibabin, Ber. Dtsch. Chem. Ges.., 1923, 59, 1883; Ger. Pat., 406,
208; Chem. Zentr., 1925, I, 1536; Friedl Fortschr D. Teerfarbenfabrik. 14,
515.
[13] W. T. Gao, W. D. Hou, M. R. Zheng, L. J. Tang, Synth. Commun. 2010,
40, 732-738.
[14] W. Steinschifter, W. Fiala, W. Stadlbauer, J. Heterocycl. Chem., 1994,
31, 1647-52.
[15] a) P. Roschger, W. Stadlbauer, Liebigs Ann. Chem. 1990, 821; ibid., 401;
b) W. Fiala, W. Stadlbauer, J. Prakt. Chem., 1993, 335, 128.
[16] W. Stadlbauer, El-S. Badawey, G. Hojas, P. Roschger, T. Kappe,
Molecules, 2001, 6, 338-352.
[17] a) E. Ziegler, H. Junek, E. Nölken, K. Gelfert, R. Salvador, Monatsh.
Chem. 1961, 92, 814-819; b) E. Ziegler, H. G. Forraita, T. Kappe,
Monatsh. Chem. 1967, 98, 322-328.
[18] A. B. Avhale, H. Prokopcová, J. Šefčovičová, W. Steinschifter, A. E.
Täubl, G. Uray, W. Stadlbauer. Eur. J. Org. Chem., 2008, 563-71.
[19] G. C. Enoua, G. Uray, W. Stadlbauer, Proceedings of ECSOC-13, The
Thirteenth International Electronic Conference on Synthetic Organic
Chemistry, http://www.usc.es/congresos/ecsoc/13/index.htm,
November 1-30, 2009; J. A. Seijas, Shu-Kun Lin, M. P. Vázquez Tato
(Eds). CD-ROM, edition ISBN 3-906980-23-5, Published in 2009 by
MDPI, Basel, Switzerland.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 220
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[20] a) J. P. Michael, Nat. Prod. Rep., 2007, 24, 223; b) J. P. Michael, Nat.
Prod. Rep., 2005, 22, 627; c) J. P. Michael, Nat. Prod. Rep., 2004, 21,
650; d) H.-O. Kalinowski, S. Berger, S. Braun, 13C-NMR Spektroskopie,
1984, Georg Thieme Verlag Stuttgart-New York.
[21] W. Stadlbauer, A. B. Avhale, N. S. Badgujar, G. Uray, J. Heterocycl.
Chem., 2009, 46, 415-420.
[22] a) M. L. Belli, G. Illuminati, G. Marino, Tetrahedron, 1963, 19, 345; b) H.
Wohjan, Arch. Pharm., 1936, 274, 83-106; c) M. Henye, Ber. Dtsch.
Chem. Ges., 1836, 69, 1566; d) H. Rupe, A. Gassmann, Helv. Chem.
Acta. 1939, 22, 1241.
[23] W. Stadlbauer, H. Prokopcová, J. Šefčovičová, A. E. Täubl, Proceedings of
ECSOC-7, The Seventh International Electronic Conference on Synthetic
Organic Chemistry 2003; J. A. Seijas, D.-C. Ji, S.-K . Lin (Eds). CD-ROM,
edition ISBN 3-906980-13-8, MDPI, Basel (Switzerland) 2003.
[24] a) A. Hassner, C. Stumer, Tetrahedron Organic Chemistry Series Vol. 11:
Organic Syntheses Based on Name Reactions and Unnamed Reactions
(Eds.: J. E. Baldwin, P. D. Magnus), Pergamon, Elsevier Science Ltd.,
Oxford, 1994; b) H. Krauch, W. Kunz, E. Nonnenmacher, Reaktionen der
Chemie, Dr. Alfred Hüthig Verlag, Heidelberg, 1976.
[25] A. Miyashita, Y. Suziki, K. Ohta, T. Higashino, Heterocycles, 1994, 39,
345-55.
[26] a) C. Liangzhen, L. Xin, T. Xiaochun, S. Dong, Synth. Commun., 2004,
34, 1215-1221; b) J. X. Wu, B. Beck, R. X. Ren, Tretrahedron Lett., 2002,
43, 387-389; c) S. Takao, O. Kaxutoshi, J. Chem. Soc., Perkin Trans 1,
1999, 16, 2323-2326.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 221
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[27] Manfred Hesse, Herbert Meier, Bernd Zeek, Spektroskopische Methoden
in der Organischen Chemie, (3., überarbeitete Auflage; 199 Abbildungen,
94 Tabellen), 1987, Georg Thieme Verlag Stuttgart-New york.
[28] M. Takayama, J. Mass Spectrom. Soc. Jpn. 1995, 43, 177-188.
[29] a) C. O. Kappe, T. Kappe, Progress Heterocycl. Chem., 1996, 8, 1-13; b)
These results were obtained at LIPHA (Lyonnaise Industrielle
Pharmaceutique), Lyon, France.
[30] E. Malle, W. Stadlbauer, G. Ostermann, B. Hofmann, H. J. Leis, G. M.
Kostner, Eur. J. Med. Chem., 1990, 25, 137-142.
[31] R. Laschober, W. Stadlbauer, Liebigs Ann. Chem. 1990, 1083.
[32] S. Kitamura, K. Hashizume, T. Iida, E. Miyashitsa, K. Shirata, H. Kase,
J. Antibiot., 1986, 39, 1160.
[33] a) W. Neuenhaus, H. Budzikiewicz, H. Korth, G. Pulverer, Naturforsch.,
1979, 34b, 313; b) H. Budzikiewicz, U. Schaller, H. Korth, G. Pulverer,
Monatsh. Chem., 1979, 110, 974.
[34] a) W. Stadlbauer, R. Laschober, H. Lutschounig, G. Schindler, T. Kappe,
Monatsh. Chem., 1992, 123, 617-636; b) E. Ziegler, T. Kappe, Monatsh.
Chem., 1964, 94, 447-452; c) E. Ziegler, R. Salvador, T. Kappe, Monatsh.
Chem., 1962, 93, 1376-1381.
[35] a) E. Ziegler, T. Kappe, Monatsh. Chem., 1963, 94, 447; b) C. Fournier, J.
Decombe, 1967, Bull. Soc. Chem. Fr., 1967: 3367, 1967, C. R. Acad. Sci.
Paris, Ser. C. 265: 1169.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 222
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[36] a) T. Witoszynsky, PhD thesis, 1972, University of Graz/Austria, p. 58; b)
F. A. Lakhvich, V. A. Kozinetes, D. B. Rubinov, A. A. Akhrem, Zh. Org.
Khim., 1987, 23, 2626.
[37] V. H. Dang, PhD thesis, 2006, University of Graz/Austria.
[38] K. J. Hwang, Arch. Pharm. Res. 2000, 23, 31–34.
[39] A. M. Mcleod, S. Grimwood, C. Barton, L. Bristow, K. L. Saywell, G. R.
Marshall, R. G. Ball, J. Med. Chem. 1995, 38, 2239–2243.
[40] R. W. Carling, P. D. Leeson, K. W. Moore, J. D. Smith, C. R. Moyes, I. M.
Mawer, S. Thomas, T. Chan, R. Baker, A. C. Foster, S. Grimwood, J. A.
Kemp, G. R. Marshall, M. D. Tricklebank, K. L. Saywell, J. Med. Chem.
1993, 36, 3397–3408.
[41] R. W. Carling, P. D. Leeson, K. W. Moore, C. R. Moyes, M. Duncton, M. L.
Hudson, R. baker, A. C. Foster, S. Grimwood, J. A. Kemp, G. R.
Marshall, M. D. Tricklebank, K. L. Saywell, J. Med. Chem. 1997, 40,
754–765.
[42] B. S. Meldrum, Excitatory Amino Acid Antagonists, Blackwell Scientific:
Oxford, 1991.
[43] S. X. Cai, Z. L. Zhou, J. C. Huang, E. R. Whittemore, Z. O. Egbuwoku, J.
E. Hawkinson, R. M. Woodward, E. Weber, J. F. W. Keana, J. Med. Chem.
1996, 39, 4682–4686.
[44] W. Seidenfadden, D. Pawellek, in Houben-Weyl, Methoden der Org.
Chem., 4th Ed (E. Müller, ed), 1971, 10, 849.
[45] D. R. Buckle, B. C. C. Cantello, H. Smith, B. A. Spicer, J. Med. Chem.
1975, 18, 726.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 223
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[46] N. Dodia, A. Shah, Indian J. Heterocycl. Chem., 2000, 10, 69-70.
[47] W. Stadlbauer, Monatsh. Chem., 1986, 117, 1305.
[48] W. Stadlbauer, W. Fiala, M. Fischer, G. Hojas, J. Heterocycl. Chem.
2000, 37, 1253-1256.
[49] W. Stadlbauer, A. E. Täubl, H. V. Dang, C. Reidlinger, K. Zangger, J.
Heterocycl. Chem., 2006, 23, 117-125.
[50] A. E. Täubl, W. Stadlbauer, Heterocycl. Chem., 1997, 34, 989.
[51] a) S. Gabriel, Ber., 1918, 51, 1501; b) G. Lang, Dissertation, University of
Graz/Austria, 1972.
[52] V. T. Dang, Dissertation, University of Graz/Austria, 1996.
[53] T. Kappe, E. Lender, E. Ziegler, Monatsh. Chem.,1968, 99, 990-4.
[54] a) T. Kappe, E. Lender, E. Ziegler, Monatsh. Chem., 1968, 99, 2157-66;
b) T. Kappe, M. Hariri, E. Pongratz, Monatsh. Chem., 1981, 112, 1211-
1219.
[55] K. Tabacovic, I. Tabacovic, M. Trkovnik, N. Trinajstic, Liebigs Ann.
Chem., 1983, 1901.
[56] a) H.-J. Knops, L. Born, Tetrahedron Lett., 1983, 24, 2973-2976; b) L. J.
Bellamy, R. E. Rogasch, Spectrocim. Acta., 1960, 16, 30; c) A.R.
Katritzky, R.A. Jones, J. Chem. Soc., 1960, 2947.
[57] R. M. Sylverstein, G. C. Bassler, Identifcation Spectrométrique des
Composés Organiques, Masson et Cie, Gauthier-Villars, Paris-1968.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 224
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[58] M-K. Jeon, K. Kim, Tetrahedron Lett., 2000, 41, 1943-1945.
[59] a) P. Lin, J. Jiang, Tetrahedron Lett., 2000, 56, 3635-3671; b) J. T.
Welch, Tetrahedron Lett. 1987, 43, 3123-3197.
[60] a) N. S. Badgujar, M. Pazicky, P. Traar, A. Terec, G. Uray, W. Stadlbauer,
Eur. J. Org. Chem. 2006, 2715–2722; b) G. Uray, K.-H. Niederreiter, F.
Belaj, W. M. F. Fabian, Helv. Chim. Acta 1999, 82, 1408–1417; c) W. M.
F. Fabian, K. S. Niederreiter, G. Uray, W. Stadlbauer, J. Mol. Struct.
1999, 477, 209–220; d) G. A. Strohmeier, W. M. F. Fabian, G. Uray,
Helv. Chim. Acta., 2004, 87, 215–226.
[61] a) L. Limpach, Ber. Dtsch. Chem. Ges. 1931, 64, 970–971; b) W. Werner,
Tetrahedron, 1969, 25, 255.
[62] J. W. Schulenberg, S. Archer, Org. React. 1965, 14, 1.
[63] a) K. Selvakumar, K. Rangareddy, J. F. Harrod, Can. J. Chem. 2004, 82,
1244; b) N. S. Wilson, C. R. M. Sarko, G. P. Roth, Tetrahedron Lett.
2002, 43, 581.
[64] a) S. Sasatani, T. Miyazaki, K. Maruoka, H. Yamamoto, Tetrahedron Lett.
1983, 24, 4711; b) H. Agui, T. Mitani, M. Nakashita, T. J. Nakagome,
Heterocycl. Chem. 1971, 8, 357.
[65] a) J. A. F. Gardner, R. Y. Moir, C. B. Purves, Can. J. Research 1948,
26B, 681; b) R. Y. Moir, C. B. Purves, Can. J. Research, 1948, 26B, 694;
c) H. J. Burton, Chem. Soc. 1932, 546.
[66] a) L-F. Tietze, T. Eicher, Reaktionen und Synthesen im organisch-
chemischen Praktikum, 2nd ed., G. Thieme, Stuttgart, New York, 1991; b)
R. M. Roberts, P. J. Vogt, J. Am. Chem. Soc. 1956, 78, 4778.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 225
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[67] a) C. Battram, S. Charlton, B. Cuenoud, M. Dowling, R. Fairhurst, D.
Farr, J. Fozard, J. Leighton-Davies, Ch. Lewis, L. McEvoy, R. Turner, A.
Trifilieff, J. Pharmacol. Exp. Ther. 2006, 317, 762–770; b) M. Singh, A.
Chandra, B. Singh, R. Singh, Tetrahedron Lett. 2007, 48, 5987–5990.
[68] a) N. Shirode, K. Kulkarni, V. Gumaste, A. Deshmukh, Arkivoc 2005, 1,
53–64; b) Ch. Beadle, J. Boot, N. Camp, N. Dezutter, J. Findlay, L.
Hayhurst, J. Masters, R. Penariola, M. Walter, Bioorg. Med. Chem. Lett.
2005, 15, 4432–4437.
[69] a) G. Muscia, M. Bollini, A. Bruno, S. Asís, J. Chil. Chem. Soc. 2006, 51,
859–863; b) G. Moore, C. Papamicaël, V. Levacher, J. Bourguignon, G.
Dupas, Tetrahedron 2004, 60, 4197–4204; c) Ch. Mitsos, A. Zografos, O.
Igglessi-Markopoulou, J. Org. Chem. 2003, 68, 4567–4569.
[70] a) P. Renhowe, S. Pecchi, C. Shafer, T. Machajewski, E. Jazan, C. Taylor,
W. Antonios-McCrea, C. McBride, K. Frazier, M. Wiesmann, G. Lapointe,
P. Feucht, R. Warne, C. Heise, D. Menezes, K. Aardalen, H. Ye, M. He, V.
Le, J. Vora, J. Cansen, M. Wernette-Hammond, A. Harris, J. Med. Chem.
2009, 52, 278–292.
[71] a) J. Kuethe, A. Wong, C. Qu, J. Smitrovich, I. Davies, D. Hughes, J. Org.
Chem. 2005, 70, 2555–2567; b) J. Kuethe, A. Wong, I. Davies, Org. Lett.
2003, 5, 3975–3978.
[72] a) M. Fraley, W. Hoffman, K. Arrington, R. Hungate, G. Hartman, R.
McFall, K. Coll, K. Rickert, K. Thomas, G. McGaughey, Curr. Med. Chem.
2004, 11, 709–719; b) M. Fraley, S. Hambaugh, R. Hungate, Patent
WO/2001/028993.
[73] J. J. Kulagowski, R. Baker, N. R. Curtis, P. D. Leeson, I. M. Mawer, A. M.
Moseley, M. P. Ridgill, M. Rowley, I. Stansfield, A. C. Foster, S.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 226
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Grimwood, R. G. Hill, J. A. Kemp, G. R. Marshall, K. L. Saywell, M. D.
Tricklebank, J. Med. Chem. 1994, 37, 1402–1405.
[74] A. D. Chapman, N. Duermueller, B. L. Harrison, B. M. Baron, N. Parvez,
B. S. Meldrum, Eur. J. Pharmacol. 1995, 274, 83–88.
[75] M. Rowley, J. J. Kulagowski, A. P. Watt, D. Rathbone, G. I. Stevenson, R.
W. Carling, R. Baker, G. R. Marshall, J. A. Kemp, A. C. Foster, S.
Grimwood, R. Hargreaves, C. Hurley, K. L. Saywell, M. D. Tricklebank, P.
D. Leeson, J. Med. Chem. 1997, 40, 4053–4068.
[76] K. J. Hwang Arch. Pharm. Res. 2000, 23, 31–34.
[77] A. M. Mcleod, S. Grimwood, C. Barton, L. Bristow, K. L. Saywell, G. R.
Marshall, R. G. Ball, J. Med. Chem. 1995, 38, 2239–2243.
[78] R. W. Carling, P. D. Leeson, K. W. Moore, J. D. Smith, C. R. Moyes, I. M.
Mawer, S. Thomas, T. Chan, R. Baker, A. C. Foster, S. Grimwood, J. A.
Kemp, G. R. Marshall, M. D. Tricklebank, K. L. Saywell, J. Med. Chem.
1993, 36, 3397–3408.
[79] R. W. Carling, P. D. Leeson, K. W. Moore, C. R. Moyes, M. Duncton, M. L.
Hudson, R. Baker, A. C. Foster, S. Grimwood, J. H. A. Kemp, G. R.
Marshall, M. D. Tricklebank, K. L. Saywell, J. Med. Chem. 1997, 40,
754–765.
[80] B. S. Meldrum, Excitatory Amino Acid Antagonists; Blackwell Scientific:
Oxford, 1991.
[81] S. X. Cai, Z. L. Zhou, J. C. Huang, E. R. Whittemore, Z. O. Egbuwoku, J.
E. Hawkinson, R. M. Woodward, E. Weber, J. F. W. Keana, J. Med. Chem.
1996, 39, 4682–4686.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 227
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[82] H. Hayashi, I. Miwa, S. Ichikawa, N. Yoda, I. Miki, A. Ishii, M. Kono, T.
Yasuzawa, F. J. Suzuki, Med. Chem. 1993, 36, 617–626.
[83] R. J. DeVita, D. D. Hollings, M. T. Goulet, M. J. Wyvratt, M. H. Fisher,
J.-L. Lo, Y. T. Yang, K. Cheng, R. G. Smith, Bioorg. Med. Chem. Lett.
1999, 9, 2615–2620.
[84] J. H. M. Lange, C. P. Verveer, S. J. M. Osnabrug, G. M. Visser,
Tetrahedron Lett., 2001, 42, 1367-1369.
[85] R. Chênevert, M. Desjardins, Can. J. Chem. 1994, 72, 2312.
[86] a) T. Kappe, W. Stadlbauer, Molecules, 1997, 1, 255-263; b) T. Kappe,
Tetrahedron Lett., 1968, 5327.
[87] a) E. Zielgler, Österr. Chem. Ztg. 1958, 59, 155-159; b) Chimia 1970, 24,
62-64; c) E. Ziegler, H. Sterk, Monatsh. Chem., 1968, 99, 1958-1961.
[88] T. Kappe, Il Farmaco, 1999, 54, 309-315.
[89] T. Kappe, S. Ajili, W. Stadlbauer, J. Heterocycl. Chem., 1988, 25, 463-
468.
[90] A. Kutyrev, T. Kappe, J. Heterocycl. Chem., 1997, 34, 969-972.
[91]] A. Kutyrev, T. Kappe, J. Hetercyocl. Chem., 1999, 36, 237-240.
[92] J. Jampilek, R. Musiol, M. Pesko, K. Kralova, M. Vejsova, J. Carroll, A.
Coffey, J. Finster, D. Tabak, H. Niedbala, V. Kozik, J. Polanski, J. Csollei,
J. Dohnal, Molecules, 2009, 14, 1145-1159.
[93] I. V. Ukrainets, L. V. Sidorenko, E. N. Svechnikova, O. V. Shishkin,
Chem. Heterocycl. Compd. 2007, 43, 1275-1279.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 228
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[94] S. Jönsson, G. Andersson, T. Fex, T. Fristedt, G. Hedlund, K. Jansson, L.
Abramo, I. Fritzson, O. Pekarski, A. Runström, H. Sandin, I. Thuvesson,
A. Björk, J. Med. Chem., 2004, 47, 2075.
[95] K. Tsuji, G. W. Spears, K. Nakamura, T. Tojo, N. Seki, A. Sugiyama, M.
Matsuo, Bioorg. Med. Chem. Lett., 2002, 12, 85.
[96] X. Collin, J. M. Robert, M. Duflos, G. Wielgosz, G. Le Baut, C. Robin-
Dubigeon, N. Grimaud, F. Lang, and J. Y. Petit, J. Pharm. Pharmacol.,
2001, 53, 417.
[97] T. Kappe, C. Nuebling, K.–O. Westphalen, U. Kardorff, W. Deyn, M.
Gerber, and H. Walter, Ger. Pat. 1993, 4138820.
http://ep.espacenet.com.
[98] D. S. Kalinowski, P. C. Sharpe, P. V. Bernhardt, D. R. Richardson, J.
Med. Chem., 2007, 50, 6212.
[99] K. Husain, M. Abid, A. Azam, Eur. J. Med. Chem., 2007, 42, 1300.
[100] C. Biot, B. Pradines, M. H. Sergeant, J. Gut, P. J. Rosenthal, K. Chibale,
Bioorg. Med. Chem. Lett., 2007, 17, 6434.
[101] T. Varadinova, D. Kovala-Demertzi, M. Rupelieva, M. Demertzis, P.
Genova, Acta Virol., 2001, 45, 87.
[102] M. J. Mackenzie, D. Saltman, H. Hirte, J. Low, C. Johnson, G. Pond, M.
J. Moore, Invest. New Drugs, 2007, 25, 553.
[103] P. Jütten, W. Schumann, A. Härtl, H. M. Dahse, U. Gräfe, J. Med. Chem.,
2007, 50, 3661.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 229
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[104] D. S. Kalinowski, Y. Yu, P. C. Sharpe, M. Islam, Y. T. Liao, D. B. Lovejoy,
N. Kumar, P. V. Bernhardt, D. R. Richardson, J. Med. Chem., 2007, 50,
3716.
[105] A. C. Caires, Anticancer Agents Med. Chem., 2007, 7, 484.
[106] C. Kirilmis, M. Koca, A. Cukurovali, M. Ahmedzade, C. Kazaz, Molecules,
2005, 10, 1399.
[107] M. Koca, M. Ahmedzade, A. Cukurovali, C. Kazaz, Molecules, 2005, 10,
747.
[109] B. A. Wilson, R. Venkatraman, C. Whitaker, Q. Tillison, Int. J. Environ.
Res. Public Health, 2005, 2, 170.
[110] H. Elo, Z. Naturforsch, C: Biosci., 2007, 498.
[111] G. Turan-Zitouni, J. A. Fehrentz, P. Chevallet, J. Martinez, Z. A.
Kaplancikli, A. Ozdemir, M. Arslanyolu, M. T. Yildiz, Arch. Pharm.
(Weinheim), 2007, 340, 310.
[112] I. Kizilcikli, Y. D. Kurt, B. Akkurt, A. Y. Genel, S. Birteksöz, G. Otük, B.
Ulküseven, Folia Microbiol. (Praha), 2007, 52, 15.
[113] T. Rosu, A. Gulea, A. Nicolae, R. Georgescu, Molecules, 2007, 12, 782.
[114] M. F. Grundon, N. J. McCorkindale, N. M. Rodger, J. Chem. Soc. 1955,
4284.
[115] A. Khattab, PhD Thesis, Monofeia University, Egypt, 1990, pp 31 ff.
[116] W. Stadlbauer, S. Prattes, W. Fiala, J. Heterocycl. Chem., 1998, 35, 627-
636.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 230
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[117] T. Kappe, G. Baxevanidis, E Ziegler, Monatsh. Chem., 1971, 102, 1392.
[118] P. E. Hansen, S. Bolvig, T. Kappe, J. Chem. Soc., Perkin Trans., 1995, 2,
1901.
[119] I. V. Ukrainets, O. V. Gorokhova, L. V. Sidorenko, Chem. Heterocycl.
Compd. 2005, 41, 1019-1021.
[120] a) N. Shobana, P. Yeshosda, P. Shanmugam, Tetrahedron 1989, 45, 757–
762; b) V. Nadaraj, S. T. Selvi, R. Sasi, ARKIVOC 2006, 10, 82–89.
[121] a) K. Arya, M. Agarwal, Bioorg. Med. Chem. Lett. 2007, 17, 86-93; b) D.
C. Dittmer, Q. Li, D. V. Avilov, J. Org. Chem. 2005, 70, 4682-4686; c)
B. Bhudevi, P. V. Ramana, A. Mudiraj, A. R. Reddy, Indian J. Chem.
2009, 48B, 255-260.
[122] a) S.-J. Park, J.-C. Lee, K.-I. Lee, Bull. Korean Chem.Soc. 2007, 28,
1203-1205; b) J. Mpilek, R. Musiol, M. Pesko, K. Kralova, M. Vejsova,
J. Carroll, A. Coffey, J. Finster, D. Tabak, H. Niedbala, V. Kozik, J.
Polanski, J. Csollei, J. Dohnal, Molecules 2009, 14, 1145-1159.
[123] a) D. Sicker, A. Rabe, A. Zakrzewski, G. Mann, J. Prakt.Chem. (Leipziz)
1987, 329, 1063-70; b) E. Ziegler, R. Wolf, Th. Kappe, Monatsh. Chem.
1965, 96, 418-22; c) St. V. Niementowski, Ber. Dtsch. Chem. Ges.
1908, 40, 4285-94.
[124] a) F. Arndt, L. Ergener, O. Kutlu, Ber. Dtsch. Chem. Ges. 1953, 86, 951-
7; b) C. Schopf, G. Koepke, B. Kowald, F. Schulde, D. Wunderlich, Ber.
Dtsch. Chem. Ges. 1956, 89, 2877-86; c) R. T. Coutts, D. G. Wibberley,
J. Chem. Soc. 1963, 4610-12.
[125] a) I. V. Ukrainets, L. V. Sidorenko, O. V. Gorokhova, S. V. Slobodzyan,
Chem. Heterocycl. Compd. 2006, 42, 882-885; b) G. Koller, Ber. Dtsch.
Chem. Ges. 1927, 60B, 1108-11; c) C. L. Arcus, R. E. Marks, M. M.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 231
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Coombs, J. Chem. Soc. 1957, 4064-9.
[126] a) A. E. Chichibabin, Zhur. Russ. Fiz.-Khim. Obshch. 1924, 55, 7-18; b)
Cassella Farbwerke Mainkur A.-G. 1963, GB Pat. 916103; Chem. Abstr.
1963, 59, 48294.
[127] a) J. Ficini, A. Krief, Tetrahedron Lett. 1968, 8, 947-51; b) A. Meyer, P.
Heimann, Compt. Rend. 1936, 203, 264-6; c) M. C. Pirrung, F. Blume,
J. Org. Chem. 1999, 64, 3642-3649; d) S. Yu. Yunusov, G. P. Sidyakin,
Doklady Akademii Nauk USSR 1953, 12, 22-4.
[128] a) R. F. C. Brown, J. J. Hobbs, G. K. Hughes, E. Ritchie, Aust. J. Chem.
1954, 7, 348-77; b) E. H. Huntress, J. Bornstein, J. Am. Chem. Soc.
1949, 71, 745-6.
[129] a) E. K. Kainmüller, E. P. Olle, W. J. Bannwarth, J. Chem. Soc., Chem.
Commun. 2005, 5459; b) M. Turel, M. Cajlakovic, E. Austin, J. Dakin, G.
Uray, A. Lobnik, Sens. Actuators B 2008, 131, 247.
[130] a) R. A. Kramer, R. Flehr, M. U. Kumke, W. J. Bannwarth, Helv. Chem.
Acta, 2009, 92, 1933-1943; b) J. Kido, Y. Iizumi, Appl. Phys. Lett., 1998,
73, 2721; c) A. Niko, S. Tasch, F. meghdadi, C. Brandstatter, G. Leising,
J. Appl. Phys. 1997, 82, 4177; d) S. A. Van Slyke, C. H. Chen, C. W.
Tang, Appl. Phys. Lett. 1996, 69, 2160; e) S. Miyata, H. S. Nalwa, Eds.
Orgganic Electroluminescent Materials and Devices, Gordon and Breach:
Amsterdam, 1997.
[131] D. Doube, M. Blouin, C. Brideau, C. Chan, C. Desmarais, D. Ethier, J. P.
Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, P. Tagari, R. N.
Young, Bioorg. Med. Chem. Lett. 1998, 8, 1255.
[132] M. Kidwai, K. R. Bhushan, P. Sapra, R. K. Saxena, R. Gupta, Bioorg.
Med. Chem. 2000, 8, 69.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 1, 2 NAD 3 232
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[133] I. K. Moissev, M. N. Yemtsova, P. L. Trakhtenberg, D. A. Kulikowa, I.
Pskobkina, G. N. Neshchadim, N. V. Ostapchuk, Khim. Farm. Zh. 1998,
22, 1448.
[134] J. C. Craig, P. E. Person, J. Med. Chem. 1971, 14, 1221.
[135] D. Narsinh, S. Anamik, Ind. J. Pharm. Sci. 2001, 63, 211.
[136] R. D. Dillard, D. E. Pavey, D. N. Benslay, J. Med. Chem. 1973, 16, 251.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 233
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES OF THEORETICAL PART
FOR
CHAPTER 4

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 234
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES OF THEORETICAL PART
FOR
CHAPTER 4
[1] a) R. Raue, in: Ullmannns Encyclopedia of Industrial Chemistry, vol. A15,
5th ed. (Eds.: B. Elvers, S. Hawkins, G. Schulz), VCH Verlagsgesellschaft,
Weinheim, 1990, 155–157; b) B. B. Snavely, in: Organic Molecular
Photophysics, Vol. 1 (Ed.: J. B. Birks); Wiley, Chichester, 1973, chapter
5.
[2] a) R. P. Haughland, in: Handbook of Fluorescence Probes and Research
Chemicals (Ed.: K. D. Larison); Molecular Probes Inc., Eugene, USA,
1992, b) O. S. Wolfbeis, in: Molecular Spectrosopy: Methods and
Applications (Ed.: S. G. Schulman); John Wiley & Sons, New York, 1985,
chapter 3.
[3] a) H. Gold , in: Environmental quality and safety, Supplement Volume IV:
The Chemistry of Fluorescent Whitening Agents (Eds.: R. Anliker, G.
Müller, R. Raab, R. Zinkernagel), Georg Thieme, Stuttgart, 1975, 25–46;
c) J. Kido, Y. Lizumi, Appl. Phys. Lett. 1998, 73, 2721–2723.
[4] a) A. Niko, S. Tasch, F. Meghdadi, C. Brandstätter, G. Leising, J. Appl.
Phys. 1997, 82, 4177–4182; b) A. E. Siegrist, H. Hefti, H. R. Meyer, E.
Schmidt, Rev. Prog. Coloration 1987, 17, 39–55.
[5] a) Organic Luminescent Materials (Eds.: B. M. Krasovitskii, B. M. Bolotin),
Wiley-VCH, Weinheim, 1988; b) R. M. Christie, Rev. Prog. Coloration

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 235
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
1993, 23, 1–18; c) N. A. Kuznetsova, O. L. Kaliya, Russ. Chem. Rev.
1992, 61, 683–696.
[6] a) O. A. Ponomarev, E. R. Vasina, V. G. Mitina, A. A. Sukhorukov, Russ.
J. Phys. Chem. 1990, 64, 518–521; b) R. M. Christie, C.-H. Lui, Dyes
Pigm. 1999, 42, 85–93; c) C. Eggeling, R. R. J. Widengren, C. A. M.
Seidel, Anal. Chem. 1998, 70, 2651–2659.
[7] a) J. B. Gallivan, Molecular Photochem. 1970, 2, 191–211; b) G. Piszczek,
B. P. Maliwal, I. Gryczynski, D. Jonathan, J. R. Lakowicz, J. Fluoresc.
2001, 11, 101–107.
[8] M. S. Schiedel, C. A. Briehn, P. Bäuerle, Angew. Chem., Int. Ed. 2001, 40,
4677-4680.
[9] R. S. Becker, S. Chakravorti, C. A. Gartner, M. de Graca Miguel, J. Chem.
Soc., Faraday Trans. 1993, 89, 1007.
[10] U. Brackmann, Lambdachromew Laser Dyes, Lambda Physik GmbH,
Göttingen, 1986.
[11] B. B. Snavely, in: J.B. Birks (Ed.), Organic Molecular Photophysics, 1,
John Wiley and Sons, London, 1973 Chapter 5.
[12] C. V. Shank, A. Dienes, A. M. Trozzolo, J. A. Myer, Appl. Phys. Lett. 1970,
16, 405.
[13] G. A. Reynolds, K. H. Drexhage, Opt. Commun. 1975, 13, 222.
[14] A. N. Fletcher, D. E. Bliss, J. M. Kauffman, Opt. Commun. 1983, 47, 57.
[15] H. H. Grunhagen, H. T. Witt, Z. Naturforsch. 1970, 25b, 373.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 236
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[16] R. P. Haughland, in K. D. Larison (Ed.), Handbook of Fluorescent Probes
and Research Chemicals, Molecular Probes Inc, Eugene, USA, 1992.
[17] R. T. Williams, J. Roy. Inst. Chem. 1959, 83, 611.
[18] O. A. Ponomarev, E. R. Vasina, A. O. Doromenko, V. G. Mitina, Khim. Fiz
1989, 8, 854.
[19] O. A. Ponomarev, E. R. Vasina, V. G. Mitina, A. A. Sukhorukov, Zh. Fiz.
Khim. 1990, 64, 974.
[20] O. A. Ponomarev, E. R. Vasina, S. N. Yarmolenko, V. G. Mitina, N. S.
Pivnenko, Zh. Obshch. Khim. 1990, 60, 1161.
[21] O. A. Ponomarev, Yu. F. Pedash, O. V. Prezhdo, Zh. Fiz. Khim 1991, 65,
1846.
[22] R. Nakagaki, N. Kitamura, I. Aoyama, H. Ohtsubo, J. Photochem.
Photobiol. 1994, A80, 113.
[23] S. G. Schulman, A. C. Capomacchia, B. Tussey, Photochem. Photobiol.
1971, 14, 733.
[24] O. S. Wolfbeis, E. Lippert, Z. Naturforsch. 1978, 33a, 238.
[25] O. A. Ponomarev, E. P. Vasina, A. O. Doroshenko, V. G. Mitina, Zh.
Obshch. Khim. 1989, 59, 2072.
[26] G. Saroja, N. B. Sankaran, A. Samanta, Chem. Phys. Lett. 1996, 249,
392.
[27] a) R. Nakagaki, N. Kitamura, I. Aoyama, H. Ohtsubo, J. Photochem.
Photobiol. 1994, A80, 113–119; b) G. Saroja, N. B. Sankaran, A.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 237
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
Samanta, Chem. Phys. Lett. 1996, 249, 392–398.
[28] a) O. A. Ponomarev, Yu. F. Pedash, O. V. Prezhdo, Zh. Fiz. Khim. 1991,
65, 1846–1850; b) J. Chen, P. R. Selvin, J. Photochem. Photobiol. A 2000,
135, 27–32.
[29] a) M. Li, P. R. Selvin, J. Am. Chem. Soc. 1995, 117, 8132–8138; b) C.
Marzano, A. Chilin, A. Guiotto, F. Baccichetti, F. Carlassare, F. Bordin,
Farmaco 2000, 55, 650–658.
[30] a) O. A. Ponomarev, E. R. Vasina, S. N. Yarmolenko, V. G. Mitina, N. S.
Pivnenko, Zh. Obshch. Khim. 1990, 60, 1161–1170; J. Gen. Chem. USSR,
Transl. Plenum Pub. Corp. 1990, 60, 1035–1042; b) E. K. Kainmüller, E.
P. Olle, W. Bannwarth, Chem. Commun. 2005, 43, 5459–5461.
[31] A. B. Ahvale, H. Prokopcová, J. Šefčovičová, W. Steinschifter, A. E. Täubl,
G. Uray, W. Stadlbauer, Eur. J. Org. Chem. 2008, 563–571.
[32] a) F. A. Bennett, D. J. Barlow, A. N. O. Dodoo, R. C. Hider, A. B. Lansley,
M. J. Lawrence, C. Marriott, S. S. Bansal, Anal. Biochem. 1999, 270, 15–
23; b) C. Charitos, G. Kokotos, C. Tzougraki, J. Heterocycl. Chem. 2001,
38, 153–158.
[33] a) J. E. T. Corrie, V. Munasinghe, N. Ranjit, J. Heterocycl. Chem. 2000,
37, 1447–1454; b) A. P. Esteves, L. M. Rodrigues, M. E. Silva, S. Gupta,
A. M. F. Oliveira-Campos, O. Machalicky, A. J. Mendonca, Tetrahedron
2005, 61, 8625–8632; c) J. Fernandez-Carneado, M. J. Kogan, S. Castel,
E. Giralt, Angew. Chem. Int. Ed. 2004, 43, 1811–1814; d) X. Luo, X.
Naiyun, L. Cheng, D. Huang, Dyes Pigm. 2001, 51, 153–159.
[34] a) M. C. Rideout, A. C. Raymond, A. B. Burgin Jr, Nucleic Acids Res.
2004, 32, 4657–4664.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 238
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[35] a) P. D. Edwards, R. C. Mauger, K. M. Cottrell, F. X. Morris, K. K. Pine, M.
A. Sylvester, C. W. Scott, S. T. Furlong, Bioorg. Med. Chem. Lett. 2000,
10, 2291–2294; b) C. A. M. Seidel, A. Schulz, M. H. M. Sauer, J. Phys.
Chem. 1996, 100, 5541–5553.
[36] a) A. Adronov, S. L. Gilat, J. M. Frechet, K. Ohta, F. V. R. Neuwahl, G. R.
Fleming, J. Am. Chem. Soc. 2000, 122, 1175–1185; b) K. H.
Shaughnessy, P. Kim, J. F. Hartwig, J. Am. Chem. Soc. 1999, 121, 2123–
2132.
[37] a) G. A. Strohmeier, W. M. F. Fabian, G. Uray, Helv. Chim. Acta 2004, 87,
215–226; b) G. Uray, K.-H. Niederreiter, F. Belaj, W. M. F. Fabian, Helv.
Chim. Acta 1999, 82, 1408–1417; c) W. M. F. Fabian, K. S. Niederreiter,
G. Uray, W. Stadlbauer, J. Mol. Struct. 1999, 477, 209–220.
[38] N. S. Badgujar, M. Pazicky, P. Traar, A. Terec, G. Uray, W. Stadlbauer,
Eur. J. Org. Chem. 2006, 2715–2722.
[39] Uray, Sallegger, unpublished results.
[40] R. A. Kramer, R. Flehr, M. Laya, M. U. Kumke, W. Bannwarth, Helv.
Chim. Acta 2009, 92, 1933–1943.
[41] a) M. Parsons, B. Vojnovic, S. Ameer-Beg, Biochem. Soc. 2004, 431; b) Y.
Sako, S. Minoguchi T. Yanagida, Nat. Cell Biol. 2000, 2, 168; c) S.
Brasselet, E. J. G. Peterman, A. Miyawaki, W. E. Moerner, J. Phys. Chem.
2000, 104, 3676.
[42] a) A. Cha, G. E. Snyder, P. R. Selvin F. Bezanilla, Nature (London) 1999,
402, 809; b) Y. Suzuki, T. Yasunaga, R. Ohkra, T. Wakabayashi, K.
Sutoh, Nature (London) 1998, 396, 380.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 239
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[43] a) I. Rasnik, S. Myong, W. Cheng, T. M. Lohman T. Ha, J. Mol. Biol. 2004,
336, 395; b) W. Shen, M. F. Bruist, S. D. Goodman, N. C. Seeman,
Angew. Chem., Int. Ed. 2004, 43, 4750.
[44] a) D. A. Hiller, J. M. Fogg, A. M. Martin, J. M. Beechem, N. O. Reich, J. J.
Perona, Biochemistry 2003, 42, 14375; b) A. I. Dragan, J. Klass, C. Read,
M. E. A. Churchill, C. Crane-Robinson, P. L. Privalov, J. Mol. Biol. 2003,
331, 795.
[45] P. J. Rothwell, S. Berger, O. Kensch, S. Felekyan, M. Antonik, B. M.
Wçhrl, T. Restle, R. S. Goody, C. A. M. Seidel, Proc. Natl. Acad. Sci. U.S.A.
2003, 100, 1655.
[46] a) M. K. Nahas, T. J. Wilson, S. Hohng, K. Jarvie, D. M. J. Lilley, T. Ha,
Nat. Struct. Mol. Biol. 2004, 11, 1107; b) Z. Xie, N. Srividya, T. R. Sosnick,
T. Pan, N. F. Scherer, Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 534.
[47] H. D. Kim, G. U. Nienhau, T. Ha, J. W. Orr, J. R. Williamson S. Chu, Proc.
Natl. Acad. Sci. U.S.A. 2002, 99, 4284.
[48] H-K Lee, H. Cao, T. M. Rana, J. Ccomb. Chem., 2005, 7, 279-284.
[49] G. Uray, A. M. Kelterer, J. Hashim, T. N. Glasnov, C. O. Kappe, W. M. F.
Fabian, J. Mol. Struct. 2009, 929, 85-96.
[49] G. Jones II, W. R. Jackson, Chem. Phys. Lett. 1980, 72, 391.
[50] G. Jones II, in Dye Laser Principles with Applications, ed. F. J. Duarte, L.
W. Hillman, Academic Press, New York, 1990.
[51] G. Jones-II, W. R. Jackson, S. kanoktanaporn, A. M. Halpern, Opt.
Comm., 1980, 23, 315.

REFERENCES OF THEORETICAL PART FOR CHAPTERS 4 240
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[52] G. Jones II, W. R. Jackson, C. Y. Choi, W. R. Bergmark, J. Phys. Chem.,
1985, 89, 294.
[53] K. Rechthaler, G. Köhler, Chem. Phys. 1994, 189, 99.
[54] G. Jones-II, J. A. Jimenez, J. Photochem. Photobiol. B, 2001, 65, 5.
[55] W. Rettig, A. Klock, Can. J. Chem., 1986, 63, 1649.
[56] M. El-Kemary, W. Rettig, Phys. Chem. Chem. Phys. 2003, 5, 5221-5228.
[57] R. S. Moog, W. W. Davis, S. G. osttrowski, G. L. Wilson, Chem. Phys. Lett.,
1999, 299, 265.

REFERENCES FOR EXPERIMENTAL PART 241
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES FOR EXPERIMENTAL
PART

REFERENCES FOR EXPERIMENTAL PART 242
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
REFERENCES FOR EXPERIMENTAL PART
[ex-1] a) R. M. Christie, C. H. Lui, Dyes Pigm. 1999, 42, 85-93; b) C.
Eggeling, R. R. J. Widengren, C. A. M. Seidel, Anal. Chem. 1998, 70,
2651–2659.
[ex-2] L. M. Harwood, Aldrichim. Acta 1985, 18, 25.
[ex-3] a) E. Ziegler, H. G. Forraita, T. Kappe, Monatsh. Chem. 1967, 98, 324–
28; b) B. Berinzaghi, A. Muruzabal, R. Labriola, V. Deulofeu, J. Org.
Chem. 1945, 10, 181–3; c) N. Shobana, P. Yeshosda, P. Shanmugam,
Tetrahedron 1989, 45, 757–762; d) V. Nadaraj, S. T. Selvi, R. Sasi,
ARKIVOC 2006, 10, 82–89.
[ex-4] a) V. I. Akhmedzhanova, I. A. Yunosov, S. Yu, Chem. Nat. Compd.
1976, 12, 282-288; Khim. Prirod. Soedin. 1976, 12, 320-328; b) A.
Omori, N. Sonoda, S. Tsutsumi, Bull. Chem. Soc. Jpn. 1970, 43, 1135-
1138; c) W. T. Gao, W. D. Hou, M. R. Zheng, L. J. Tang, Synth.
Commun. 2010, 40, 732–738.
[ex-5] a) W. Kutney, I. W. J. Still, Can. J. Chem. 1980, 58, 1233-8; b) E. A.
Pauw, Rec. Trav. Chim. Pays-Bas 1936, 55, 215–26; c) W. R. Vaughan,
J. Am. Chem. Soc. 1946, 68, 324–5; d) G. Barnikow, J. Prakt. Chem.
1966, 34, 251-61.
[ex-6] a) A. V. Prosyanik, Z. Y. Zorin, E. L. Solov'ev, J. Org. Chem. USSR
1985, 1353–360; Zhur. Org. Khim. 1985, 21, 1485–1493; b) A. K. Sen,
P. Sengupta, J. Indian Chem. Soc. 1969, 46, 857–859.
[ex-7] a) E. Castellaneta, Gazz. Chim. Ital. 1895, 25, 538-539; b) A. K. Sen,
K. Mitra, J. Indian Chem. Soc. 1965, 42, 851–854; c) H. Nishino, K.
Ishida, H. Hashimoto, K. Kurosawa, Synthesis 1996, 888–896.

REFERENCES FOR EXPERIMENTAL PART 243
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-8] M. Sechi, M. P. Delussu, N. Pala, U. Azzena, R. Dallocchio, A. Dessi, A.
Cosseddu, N. Neamati, Molecules 2008, 13, 2442–2461.
[ex-9] G. A. Prakash, S. P. Rajendran, Heterocycl. Commun. 2001, 7, 353–358.
[ex-10] G. H. Patel, C. M. Mehta, J. Indian Chem. Soc. 1973, 50, 215–16.
[ex-11] N. Dodia, A. Shah, Indian J. Heterocycl. Chem. 2000, 10, 69–70.
[ex-12] H. Kato, J. Sakaguchi, M. Aoyama, T. Izumi, PCT Int. Appl. Hokuriku
Seiyaku EP 1104764A1 Co. LTD 2000, WO 2000009506; Chem. Abstr.
2000, 132, 180573.
[ex-13] a) K. Faber, H. Steininger, T. Kappe, J. Heterocycl. Chem. 1985, 22,
1081–5; b) A. Knierzinger, O. S. Wolfbeis, J. Heterocycl. Chem. 1980,
17, 225–229.
[ex-14] B. Berinzaghi, A. Muruzabal, R. Labriola, V. Deulofeu, J. Org. Chem.
1945, 10, 181–3.
[ex-15] a) R. Hardman, M.W. Partridge, J. Chem. Soc. 1958, 614–20; b) N. S.
Narasimhan, R. S. Mali, Tetrahedron 1974, 30, 4153, 4154, 4156.
[ex-16] J. Moon-Kook, K. Kyongtae, Tetrahedron Lett. 2000, 41, 1943–1946.
[ex-17] a) F. Dallacker, F. E. Eschelbach, Liebigs Ann. Chem. 1965, 689, 171–
178; b) V. Vosatka, A. Capek, Z. Budesinsky, Collect. Czech. Chem.
Commun. 1977, 42, 3186–91.
[ex-18] a) M. Sekiya, S. Takayama, K. Ito, J. Suzuki, K. Suzuki, Y. Terao,
Chem. Pharm. Bull. 1972, 20, 2661, 2665, 2667; b) M. Sekiya, O.
Matsuda, J. Suzuki, M. Tomie, Sekiya Chem. Pharm. Bull. 1973, 21,
372, 376; c) Z. B. Kiro, B. I. Stepanov, J. Org. Chem. USSR, 1971, 7,
2279; Zhur. Org. Khim. 1971, 7, 2196–8.

REFERENCES FOR EXPERIMENTAL PART 244
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-19] a) V. Vosatka, A. Capek, Z. Budesinsky, Coll. Czech. Chem. Commun.
1977, 42, 3186–3191; b) M. Sekiya, M. Tomie, N. J. Leonard, J. Nelson,
J. Org. Chem. 1968, 33, 318–322.
[ex-20] a) Y. Ogata, K. Tagaki, K. Mizuno, J. Org. Chem. 1982, 47, 3684–3687;
b) J. Deutsch, H. J. Nicolas, Synth. Commun. 1993, 23, 1561–1568; c)
O. Meth-Cohn, D. L. Taylor, Tetrahedron 1995, 51, 12869–12882.
[ex-21] a) E. Froehlich, E. Wedekind, Ber. Dtsch. Chem. Ges. 1907, 40, 1010;
b) W. Koenig, G. A. Becker, J. Prak. Chem. [2], 1912, 85, 353-85; c) M.
Julia, J. Bagot, Bull. Soc. Chim. France 1964, 8, 1924–1938.
[ex-22] a) A. M. Hjort, E. J. de Beer, J. S. Bruck, W. S. Ide, Pharm. Exp. Therap.
1935, 55, 152, 153, 156; b) F. Benington, R. D. Morin, L. C. Clark Jr.,
J. Org. Chem. 1958, 23, 19, 22.
[ex-23] a) K. Horiki, Heterocycles 1976, 5, 203–6; b) M. Sekiya, K. Ito, Chem.
Pharm. Bull. 1967, 15, 1339, 1340, 1342.
[ex-24] a) M. Julia, J. Lenzi, Bull. Soc. Chim. France 1962, 1051, 1054; b) J. W.
Rakshys Jr., Inorg. Chem. 1970, 9, 1521.
[ex-25] a) M. Sekiya, K. Ito, Chem. Pharm. Bull. 1966, 14, 1007–9; b) I.
Bacaloglu, K. Nadolski, R. Bacaloglu, D. Martin, J. Prakt. Chem. 1971,
313, 839–848; c) J. R. Crochet, J. R. Blanton, Synthesis 1974, 55.
[ex-26] a) W. R. Abrams, R. G. Kallen, J. Am. Chem. Soc. 1976, 98, 7777–7789;
b) B. L. Fox, R. J. Doll, J. Org. Chem. 1973, 38, 1136–1140; c) A. H.
Beckett, R. W. Daisley, J. Walker, Tetrahedron 1968, 24, 6093–6109.
[ex-27] V. K. Sharma, A. Kumar, K. P. Agarwal, Indian J. Chem. 1983, 22,
1153.
[ex-28] a) S. Padmanabhan, N. L. Reddy, J. G. Durant, Synth. Commun. 1997,
27, 691–700; b) H. Suhr, Liebigs Anna. Chem. 1965, 689, 109–17.

REFERENCES FOR EXPERIMENTAL PART 245
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-29] a) E. Spath, O. Brunner, Ber. Dtsch. Chem. Ges. 1925, 58B, 518–23.
[ex-30] N. L. Owen, C. Sflomos, J. Chem. Soc. Perkin Trans. 1. 1979, 4, 943–9.
[ex-31] a) C. M. Pirrung, F. Blume, J. Org. Chem. 1999, 64, 3642–3649; b) J. A.
Lamberton, J. R. Price, Aust. J. Chem. 1953, 6, 173-9; c) V. I. Frolowa,
A. D. Kuzovkov, P. N. Kibalchich, J. Gen. Chem. USSR 1964, 34, 3542.
[ex-32] H. Jamshed, N. T. Glasnov, M. J. Kremsner, C. O. Kappe, J. Org. Chem.
2006, 71, 1707–1710.
[ex-33] a) T. F. Dankova, T. N. Bokova, N. A. Preobrazhenskii, A. E.
Petrushchenko, I. A. Il'shtein, N. I. Shvetsov, Zhur. Obsh. Khim. 1951,
21, 787- 800; b) J. C. Cain, J. L. Simonsen, C. Smith, Proceedings J.
Chem. Soc. 1913, 29, 172; c) J. C. Cain, J. L. Simonsen, C. Smith, J.
Chem. Soc. 1913, 103, 1035–9.
[ex-34] a) L. Novak, M. Borovicka, M. Protiva, Coll. Czech. Chem. Commun.
1962, 27, 1261–72; b) R.P. Quelet, J. Gavarret, Compt. Rend. 1950,
230, 394–6; c) R. P. Quelet, J. Gavarret 1951, 987235, Fr. Pat; Chem.
Abstr. 1956, 50, 40586.
[ex-35] a) H. Sadeghian, N. Attaran, Z. Jafari, M. R. Saberi, M. Pordel, M. M.
Riazi, Bioorg. Med. Chem. 2009, 17, 2327–2335; b) R. L. Huang, K. T.
Lee, J. Chem. Soc. 1955, 4229–32.
[ex-36] a) J. Dutta, R. N. Biswas, J. Indian Chem. Soc. 1963, 40, 629–637; b) F.
Leonard, A. Wajngurt, M. Klein, H. Meyer, J. Med. Chem. 1963, 6, 539-
541; c) H. C. Brown, C. J. Kim, J. Am. Chem. Soc. 1968, 90, 2082–
2096.
[ex-37] a) P. J. Smith, A. N. Bourns, Can. J. Chem. 1974, 52, 749 - 760; b) B.
M. Bhawal, S. P. Khanapure, E. R. Biehl, Synthesis 1991, 112–114;

REFERENCES FOR EXPERIMENTAL PART 246
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
c) S. Kiyooka, Y. Ueda, K. Suzuki, Bull. Chem. Soc. Jpn. 1980, 53,
1656–1660.
[ex-38] a) K. Kindler, W. Metzendorf, D. Kwok, Ber. Dtsch. Chem. Ges. 1943,
76, 308, 317; b) R. P. Quelet, J. Gavarret, Bull. Soc. Chim. France 1950,
1075, 1078.
[ex-39] a) P. R. Carter, D. H. Hey, J. Chem. Soc. 1948, 147–9; b) K. Kindler,
Liebigs Ann. Chem. 1927, 452, 90–120; Arch. Pharm. (Weinheim,
Germany) 1929, 544; c) W. Wieniawski, Acta Pol. Pharma. 1962, 19,
285–92.
[ex-40] a) J. G. Watkinson W. Watson, B. L. Yates, J. Chem. Soc. 1963, 5437–
5444; b) Y. Izawa, H. Tomioka, M. Kutsuna, Y. Toyama, Bull. Chem.
Soc. Jpn. 1979, 52, 3465–6; c) E. M. Arnett, G. B. Klingensmith, J. Am.
Chem. Soc. 1965, 87, 1032–1037.
[ex-41] a) M. Carissimi, I. Grasso, E. Grumelli, E. Milla, F. Ravenna, Farmaco
Ed. Sci. 1962, 17, 390-413; b) M. Carissimi, F. Ravenna, (Maggioni &
C. S. p. A.), 1963, Fr. Pat. M1687; Chem. Abstr. 1963, 59, 48427.
[ex-42] a) R. Walther, A. Wetzlich, J. Prakt. Chem. [2], 1900, 61, 169-198; b) T.
Curtius, J. Prakt. Chem. [2], 1914, 89, 500, 512; c) T. Curtius, J. Prakt.
Chem., [2], 1975, 91, 7; d) J. Hooz, J. N. Bridson, J. G. Calzada, H. C.
Brown, M. M. Midland, A. B. Levy, J. Org. Chem. 1973, 38, 2574–6.
[ex-43] a) Y. Yukawa, Y. Tsuno, M. Sawada, Bull. Chem. Soc. Jpn. 1966, 39,
2274–86.
[ex-44] a) V. N. Sokolova, O. Yu. Magidson, Chem. Heterocycl. Compd. 1968, 4,
385; Khim. Geterotsikl. Soedin. 1968, 3, 519–23; b) R. Chenevert, M.
Desjardins, Can. J. Chem. 1994, 72, 2312–2317.
[ex-45] a) C. S. Rondestvedt Jr., A. H. Filbey, J. Org. Chem. 1954, 19, 119–28;
b) W. Borsche, Ber. Dtsch. Chem. Ges. 1909, 42, 3600.

REFERENCES FOR EXPERIMENTAL PART 247
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-46] a) P. Yao, X. Zhai, D. Liu, B. H. Qi, H. L. Tan, Y. C. Jin, P. Gong, Arch.
Pharm. (Weinheim, Germany), 2010, 343, 17–23; b) G. W. hielcke, E. I.
Becker, J. Org. Chem. 1950, 15, 1241–5.
[ex-47] a) A. Pathak, P.M. Mishra, Asian J. Chem. 2007, 19, 988–994, b) J. R.
Knowles, R. O. C. Norman, J. Chem. Soc. 1961, 2938–47; c) O.
Masanori, N. Yoshikazu, K. Masayuki, M. Toshihiko, Bull. Chem. Soc.
Jpn 1985, 58, 3383–4.
[ex-48] J. F. J. Dippy, J. E. Page, J. Soc. Chem. Industry 1936, 55, 190–2T.
[ex-49] a) H. P. Ward, E. F. Jenkins, J. Org. Chem. 1945, 10, 371–3; b) E.
Ferber, H. Bendix, Ber.Dtsch. Chem. Ges. 1939, 72B, 839-848; c) H. A.
Selling, Tetrahedron, 1975, 31, 2387–2390; d) J. F. Fauvarque, A.
Jutand, J. Organomet. Chem. 1979, 177, 273–281.
[ex-50] a) J. G. Watkinson, W. Watson, B. L. Yates, J. Chem. Soc. 1963, 5437-
5444; b) P M. Pailer, P. Bergthaller, Monatsh. Chem., 1968, 99, 103–
111; c) G. G. Smith, K. K. Lum, J. A. Kirby, J. Posposil, J. Org. Chem.
1969, 34, 2090; d) P. Kristian, S. Kovac, O. Hritzova, Coll. Czech.
Chem. Commun. 1969, 34, 563–571.
[ex-51] a) W. R. Boon, H. C. Carrington, N. Greenhalgh, C. H. Vasey, J. Chem.
Soc. 1954, 3263–72, b) B. Marcot, J. C. Dore, S. Munavalli, C. Viel,
Rev. Roum. Chim. 1975, 20, 91–106; c) R. M. Claramunt, R. Granados,
M. C. Repolles, Anal. Quim. 1975, 71, 206.
[ex-52] a) R. O. C. Norman, P. D. Ralph, J. Chem. Soc. 1963, 5431, 5436; b) R.
C. Cambie, S. A. Coulson, L. G. Mackay, S. J. Janssen, P. S. Rutledge,
P. D. Woodgate, J. Organomet. Chem. 1991, 3, 409, 385–409.
[ex-53] a) G. Darzens, A. Levy, Compt. Rend. 1935, 200, 469–71; b) J. C.
Bardhan, Chem. Ind. (London, U. K.), 1940, 369; c) J. B. Niederl, US.
Pat. 1942, 2297911; Chem. Abstr. 1943, 37, 8511.

REFERENCES FOR EXPERIMENTAL PART 248
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-54] a) F. Leonard (Geigy Chemical Corp.), US. Pat. Appl. 1964, 3125583;
Chem. Abstr. 1964, 60, 75223; b) S. M. M. Elshafie, Chimia 1982, 36,
343–5; c) A. Citterio, R. Santi, T. Fiorani, S. Strologo, J. Org. Chem.
1989, 54, 2703–2712.
[ex-55] a) W. M. Lauer, L. I. Hansen, J. Am. Chem. Soc. 1939, 61, 3039–41; b)
J. B. Niederl, R. T. Roth, A. A. Plentl, J. Am. Chem. Soc. 1937, 59,
1901–3; c) G. Zvilichovsky, U. Fotadar, Org. Prep. Proced. Int. 1974, 6,
5–9.
[ex-56] a) B. Cavalleri, E. Bellasio, A. Wajngurt, M. Vigevani, E. Testa,
Farmaco Ed. Sci. 1969, 24, 451–70; b) F. Leonard, A. Wajngurt, M.
Klein, H. Meyer, J. Med. Chem. 1963, 6, 539, 540.
[ex-57] a) H. Schubert, H. Zaschke, J. Prakt. Chem. 1970, 312, 494–506; b) A.
J. De Gee, J. W. Verhoeven, I. P. Dirkx, T. J. De Boer, Tetrahedron
1969, 25, 3407–3412; c) A. South Jr., R. J. Ouellette, J. Am. Chem.
Soc. 1968, 90, 7064–72; d) Geigy Chem., US. Pat., 1960, 3125583;
Chem. Abstr. 1964, 60, 13192.
[ex-58] V. N. Sokolova, O. Yu. Magidson, Chem., Heterocycl. Compd. 1968, 4,
385; Khim. Geterotsikl. Soedin. 1968, 3, 519–23.
[ex-59] P. C. U. Kuhlmann, Ger. Pat. 2362331; Chem. Abstr., 1974, 81, 91567.
[ex-60] F. M. Beringer, P. S. Forgione, Tetrahedron 1963, 19, 739–748.
[ex-61] a) W. Stadlbauer, T. Kappe, Monatsh. Chem., 1985, 116, 1005–11; b)
H. Nishimura, Y. Nagai, T. Suziki, T. Sawayama, Yakugaku Zasshi,
1970, 90, 818–828.
[ex-62] a) J. Reisch, R. A. Salehi-Artimani, A. Bathe, M. Mueller, Monatsh.
Chem., 1988, 119, 781–792; b) M. F. Grundon, N. J. McCorkindale, M.
N. Rodger, J. Chem. Soc. 1955, 4284–90.

REFERENCES FOR EXPERIMENTAL PART 249
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-63] a) I. V. Ukrainets, L. V. Sidorenko, O. V. Gorokhova, S. V. Slobodzyan,
Chem. Heterocycl. Compd. 2006, 42, 882–885; Khim. Geterotsikl.
Soedin. 2006, 42, 1022–1025; b) J. Jampilek, R. Musiol, M. Pesko, K.
Kralova, M. Vejsova, J. Carroll, A. Coffey, J. Finster, D. Tabak, H.
Niedbala, V. Kozik, J. Polanski, J. Csollei, J. Dohnal, K. S. Zentiva,
Molecules 2009, 14, 1145–1159.
[ex-64] a) M. Rowley, P. D. Leeson, G. I. Stevenson, A. M. Moseley, I. Stansfield,
I. Sanderson, L. Robinson, R. Baker, J. A. Baker, J. Med. Chem. 1993,
36, 3386–3396; b) A. Detsi, V. Bardakos, J. Markopoulos, O. Igglessi-
Markopoulou, J. Chem. Soc., Perkin Trans. 1, 1996, 24, 2909–2914.
[ex-65] a) I. V. Ukrainets, S. G. Taran, O. V. Gorokhova, N. A. Marusenko, S. N.
Kovalenko, A. V. Turov, N. I. Filimonova, S. M. Ivkov, Chem. Heterocycl.
Compd. 1995, 31, 167–175; Khim. Geterotsikl. Soedin. 1995, 2, 195–
203; b) W. Stadlbauer, S. Prattes, W. Fiala, J. Heterocycl. Chem. 1998,
35, 627–636.
[ex-66] a) I. V. Ukrainets, N. L. Bereznyakova, A. A. Davidenko, S. V.
Slobodzian, Chem. Heterocycl. Compd. 2009, 45, 952–956; Khim.
Geterotsikl. Soedin. 2008, 45, 1198–1203.
[ex-67] a) K. Arya, M. Agarwal, Bioorg. Med. Chem. Lett. 2007, 17, 86-93; b) D.
C. Dittmer, Q. Li, D. V. Avilov, J. Org. Chem. 2005, 70, 4682-4686; c)
B. Bhudevi, P. V. Ramana, A. Mudiraj, A. R. Reddy, Indian J. Chem.
2009, 48B, 255-260.
[ex-68] a) S-J. Park, J-C. Lee, K-I. Lee, Bull. Korean Chem.Soc. 2007, 28, 1203-
1205; b) J. Mpilek, R. Musiol, M. Pesko, K. Kralova, M. Vejsova, j.
Carroll, A. Coffey, J. Finster, D. Tabak, H. Niedbala, V. Kozik, J.
Polanski, J. Csollei, J. Dohnal, Molecules 2009, 14, 1145-1159.
[ex-69] a) D. Sicker, A. Rabe, A. Zakrzewski, G. Mann, J. Prakt.Chem. (Leipzig)
1987, 329, 1063-70; b) E. Ziegler, R. Wolf, Th. Kappe, Monatsh. Chem.
1965, 96, 418-22;

REFERENCES FOR EXPERIMENTAL PART 250
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
c) St. V. Niementowski, Ber. Dtsch. Chem. Ges. 1908, 40, 4285-94.
[ex-70] a) F. Arndt, L. Ergener, O. Kutlu, Ber. Dtsch. Chem. Ges. 1953, 86,
951-7; b) C. Schopf, G. Koepke, B. Kowald, F. Schulde, D. Wunderlich,
Ber. Dtsch. Chem. Ges. 1956, 89, 2877-86; c) R. T. Coutts, D. G.
Wibberley, J. Chem. Soc. 1963, 4610-12.
[ex-71] a) I. V. Ukrainets, L. V. Sidorenko, O. V. Gorokhova, S. V. Slobodzyan,.
Chem. Heterocycl. Compd. 2006, 42, 882-885; b) G. Koller, Ber. Dtsch.
Chem. Ges. 1927, 60B, 1108-11; c) C. L. Arcus, R. E. Marks, M. M.
Coombs, J. Chem. Soc. 1957, 4064-9.
[ex-72] a) A. E. Chichibabin, Zhur. Russ. Fiz.-Khim. Obshch. 1924, 55, 7-18; b)
Cassella Farbwerke Mainkur A.-G. 1963, GB Pat. 916103; Chem.
Abstr. 1963, 59, 48294.
[ex-73] a) J. Ficini, A. Krief, Tetrahedron Lett. 1968, 8, 947-51; b) A. Meyer, P.
Heimann, Compt. Rend. 1936, 203, 264-6; c) M. C. Pirrung, F. Blume,
J. Org. Chem. 1999, 64, 3642-3649; d) S. Yu. Yunusov, G. P. Sidyakin,
Doklady Akademii Nauk USSR 1953, 12, 22-4.
[ex-74] a) R. F. C. Brown, J. J. Hobbs, G. K. Hughes, E. Ritchie, Aust. J. Chem.
1954, 7, 348-77; b) E. H. Huntress, J. Bornstein, J. Am. Chem. Soc.
1949, 71, 745-6.
[ex-75] I. V. Ukrainets, L. V. Sidorenko, O. V. Gorokhova, S. V. Slobodzyan,
Chem. Heterocycl. Compd. 2006, 42, 882–885; Khim. Geterotsikl.
Soedin. 2006, 42, 1022 –1025.
[ex-76] M. F. Grundon, N. J. McCorkindale, J. Chem. Soc. 1957, 2177–85.
[ex-77] A. B. Ahvale, H. Prokopcova, J. Sefcovicova, W. Steinschifter, A. E.
Taubl, G. Uray, W. Stadlbauer, Eur. J. Org. Chem. 2008, 563–571.

REFERENCES FOR EXPERIMENTAL PART 251
PhD-Thesis-Karl-Franzens Universiy of Graz/Austria-2010 ENOUA Guy Crépin
[ex-78] F .A. L. Anet, P. T. Gilham, P. Gow, G. K. Hughes, E. Ritchie, J. Sci.
Res., A: 1952, 5, 412–19.
[ex-79] M. O. Abe, D. A. H. Taylor, Phytochemistry (Elsevier) 1971, 10, 1167–
1169.
[ex-80] N. S. Narasimhan, R. S. Mali, A. M. Gokhale, Indian J. Chem. B, 1979,
18, 115.
[ex-81] M. M. Martin and L. Lindqvist, J. Luminescence 1975, 10, 381–390.
[ex-82] N. S. Badgujar, M. Pazicky, P. Traar, A. Terec, G. Uray, W. Stadlbauer,
Eur. J. Org. Chem. 2006, 2715–2722.




















