
Discovery and characterization of pseudocyclic cystine-knot α-amylase inhibitors with high...
26
Accepted Article This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/febs.12939 This article is protected by copyright. All rights reserved. Discovery and Characterization of Pseudocyclic Cystine-Knot α-Amylase Inhibitors with High Resistance to Heat and Proteolytic Degradation Phuong Quoc Thuc Nguyen, Shujing Wang, Akshita Kumar, Li Jian Yap, Thuy Thanh Luu, Julien Lescar, and James P Tam* School of Biological Sciences, Nanyang Technological University, Singapore 637551 Correspondence: James P Tam, School of Biological Sciences, Nanyang Technological University, Singapore; Fax: 65- 6515 1632; Email: [email protected] Running Title: Pseudocyclic Cystine-Knot α-Amylase Inhibitors Abbreviations AAI, Amaranth α-amylase inhibitor; TMA, Tenebrio molitor α-amylase; CK, cystine-knot; MALDI-TOF MS, matrix-assisted laser desorption/ionisation-time of flight mass spectrometry; HPLC, high performance liquid chromatography; UPLC, ultra performance liquid chromatography; RACE, rapid amplification of cDNA ends Keywords: wrightide; pseudocyclics, cystine knot; α-amylase inhibitors, cis proline Article type : Original Article Abstract Obesity and type-2 diabetes are chronic metabolic diseases that could be benefited by the use of α- amylase inhibitors to manage starch intake. The pseudocyclics, wrightides Wr-AI1 to Wr-AI3, isolated from an Apocynaceae plant show promising potentials for further development as orally active α-amylase inhibitors. These linear peptides retain the stability known for cystine knot peptides in harsh treatment. They are resistant to treatment by heat, endopeptidase or exopeptidase, characteristics of cyclic cystine knot peptides. Our NMR and crystallography analysis
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Discovery and characterization of pseudocyclic cystine-knot α-amylase inhibitors with high...

Acc
epte
d A
rtic
le
This article has been accepted for publication and undergone full peer review but has not been
through the copyediting, typesetting, pagination and proofreading process, which may lead to
differences between this version and the Version of Record. Please cite this article as doi:
10.1111/febs.12939
This article is protected by copyright. All rights reserved.
Discovery and Characterization of Pseudocyclic Cystine-Knot α-Amylase Inhibitors
with High Resistance to Heat and Proteolytic Degradation
Phuong Quoc Thuc Nguyen, Shujing Wang, Akshita Kumar, Li Jian Yap, Thuy Thanh Luu, Julien
Lescar, and James P Tam*
School of Biological Sciences, Nanyang Technological University, Singapore 637551
Correspondence: James P Tam, School of Biological Sciences, Nanyang Technological University,
Singapore; Fax: 65- 6515 1632; Email: [email protected]
Running Title: Pseudocyclic Cystine-Knot α-Amylase Inhibitors
Abbreviations
AAI, Amaranth α-amylase inhibitor; TMA, Tenebrio molitor α-amylase; CK, cystine-knot; MALDI-TOF
MS, matrix-assisted laser desorption/ionisation-time of flight mass spectrometry; HPLC, high
performance liquid chromatography; UPLC, ultra performance liquid chromatography; RACE, rapid
amplification of cDNA ends
Keywords: wrightide; pseudocyclics, cystine knot; α-amylase inhibitors, cis proline
Article type : Original Article
Abstract
Obesity and type-2 diabetes are chronic metabolic diseases that could be benefited by the use of α-
amylase inhibitors to manage starch intake. The pseudocyclics, wrightides Wr-AI1 to Wr-AI3,
isolated from an Apocynaceae plant show promising potentials for further development as orally
active α-amylase inhibitors. These linear peptides retain the stability known for cystine knot
peptides in harsh treatment. They are resistant to treatment by heat, endopeptidase or
exopeptidase, characteristics of cyclic cystine knot peptides. Our NMR and crystallography analysis

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
also showed that wrightides, currently the smallest proteinaceous α -amylase inhibitors reported,
contain the backbone-twisting cis proline which is preceded by a non-aromatic residue rather than
a conventional aromatic residue. Modeled structure and molecular dynamics study of Wr-AI1 in
complex with yellow meal worm α-amylase suggested that despite similar structure and cystine
knot fold, members of knottin-type α-amylase inhibitors may bind to insect α-amylase via a
different set of interactions. Finally, we showed that the precursors of pseudocyclic cystine knot α-
amylase inhibitors and their biosynthesis in plants follow secretory protein synthesis pathway.
Together, our work provides insights for the use of the pseudocyclic α-amylase inhibitors as useful
leads for developing orally active peptidyl bioactives as well as an alternative scaffold to cyclic
peptides for engineering metabolic-stable human α-amylase inhibitors.
Database
The nucleotide sequences for Wr-AI1 to Wr-AI3 genes have been deposited in the GenBank database
under GenBank accession numbers KF679826, KF679827, KF679828, respectively. The Wr-AI1
solution structure solved of 10 ensembles with the lowest target function is available in Protein Data Bank
(PDB) with accession code 2MAU. The coordinates of Wr-AI1 crystal structure is available in PDB
database with accession code 4BFH.
Introduction
Small disulfide-rich, proteinaceous bioactives are prominently found in toxins, hormones, growth factors,
and protease inhibitors [1]. Many contain a cystine-knot (CK) motif, with a three-disulfide knotted
structure which is formed by two disulfide bonds, together with the connecting backbones, forming an
embedded ring through which the third bond penetrates [2]. Of particular interest in drug development is
the knottin family of CK peptides containing 25 to 45 amino acid residues, and often, possessing protease
inhibitory functions from which the name was derived [3]. Knottins form compact and defined structures
with extensive internal H-bonding, endowing them with resistance to proteolytic degradation by
endopeptidases and denaturation by heat or chemicals as shown by numerous studies including those
using sequencing experiments to determine their primary structures. Certain CK peptides of the knottin
family have further evolved as macrocycles such as cyclotides, harboring cyclic cystine-knot (CCK) with
no termini, a feature that has further bestowed them resistance to degradation by exopeptidases [4].
Cyclotides, generally consisting of 28-37 amino acids, are known be ultra-stable to proteolytic and heat
degradation, and possess robust quality comparable to small-molecule drug candidates. All these features
bode well for developing orally-active peptidyl bioactives.
In a program to identify potentially orally-active peptidyl bioactives to treat metabolic diseases such as
diabetes, we have initiated mass spectroscopy (MS) profiling to identify cysteine-rich peptidyl α-amylase
inhibitors in traditional medicines. Plants and microorganisms produce a diverse group of proteinaceous
α-amylase inhibitors that function in defense pathways. These inhibitors vary greatly in structure and size,
ranging from small peptides (3 kDa), such as Amaranth α-amylase inhibitor (AAI) [5], to large proteins
such as α-AI1, a 23-kDa α-amylase inhibitor from kidney bean (Phaseolus vulgaris) [6]. They are
structurally classified into seven groups: knottin-type, -thionin-like, CM-proteins, Kunitz-type,
thaumatin-like, legume-lectin-like and microbial inhibitors [7]. These classes of α-amylase inhibitors have
attracted attention as tools in agriculture and for anti-diabetic management.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
The smallest proteinaceous α-amylase inhibitor known to date, the 3-kDa AAI, is currently the only
member reported for the knottin-type group. AAI comprises 32 amino acid residues harboring a CK core.
This inhibitor specifically inhibits the yellow mealworm Tenebrio molitor α-amylase (TMA), but is
inactive against human and bovine α-amylases [5]. Although detailed structural study of the inhibition
mechanism of AAI on TMA has been reported, little is known about its knottin-type homologs as well as
their genetic precursors.
Here, we report on the discovery and characterization of a group of linear knottins with characteristics
and potential use in drug development comparable to those of CCK peptides. Using a combination of
proteomic and genomic methods, we identified three AAI-like α-amylase inhibitors, wrightide-amylase-
inhibitors Wr-AI1 to Wr-AI3 from the medicinal plant Wrightia religiosa (Apocynaceae family). We
showed that they are resistant not only to heat treatment and endopeptidase degradation, but also to
exopeptidase. The structure of Wr-AI1 was analyzed in both solution and crystal form by NMR and X-ray
crystallography (to 1.25 Å resolution), respectively. Modeling the Wr-AI1 – TMA complex using
docking and molecular dynamics suggests that α-amylase inhibition by knottins occurs via an overall-
shape-fitting mechanism rather than through a particular set of polar or ionic interaction in the TMA
active site pocket. We also showed that the precursors of knottin-type α-amylase inhibitors contain a
three-domain structure common for CK peptides. Taken together, our study provides new insights into the
sequence, structure and biosynthesis of CK α-amylase inhibitors which could be used as stable scaffold in
engineering human α-amylase inhibitors.
Results
Isolation of α-amylase inhibitors from W. religiosa
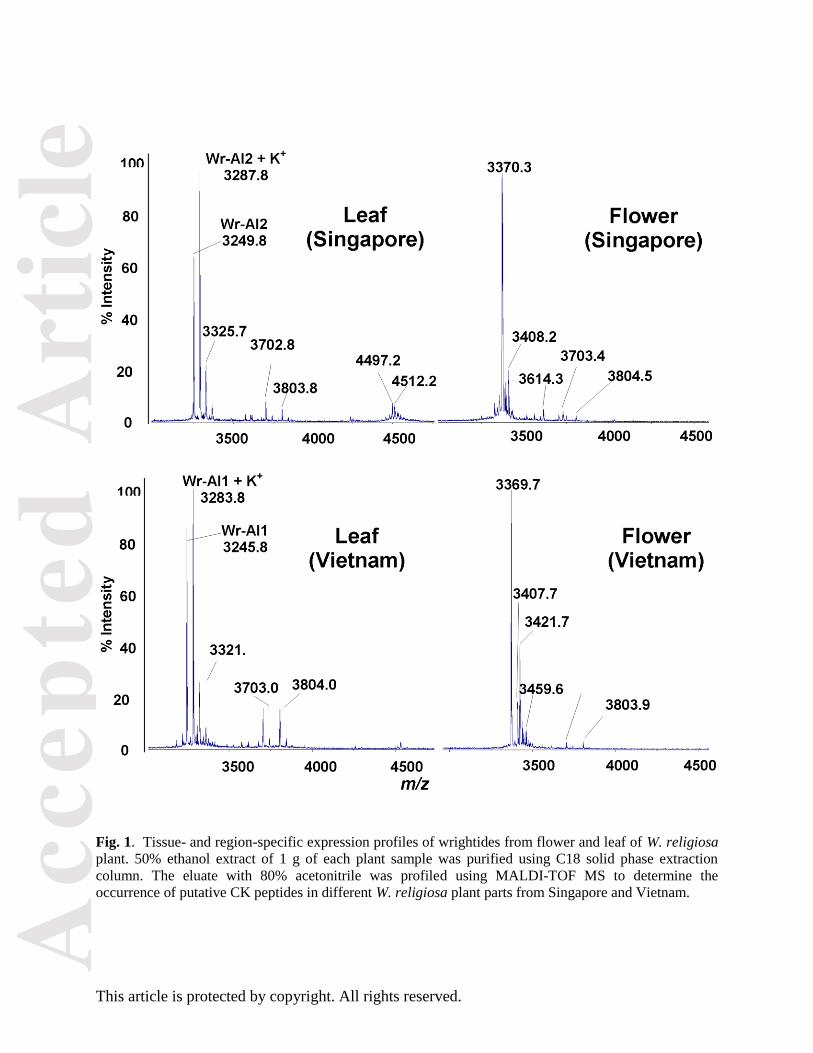
Our preliminary MS profiling of crude extract of W. religiosa leaves and flowers revealed strong positive
signals in the mass range of 3–5 kDa, indicative of cysteine-rich peptides (Fig. 1). Thus, we performed
extraction of the putative cysteine-rich peptides from fresh W. religiosa leaves from Vietnam and
Singapore in 50% ethanol and purified them through several rounds of reversed-phase and strong cation-
exchange high-performance liquid chromatography (HPLC). The most abundant peptides from Vietnam
and Singapore leaves were named wrightide-amylase-inhibitors Wr-AI1 and Wr-AI2, respectively. Each
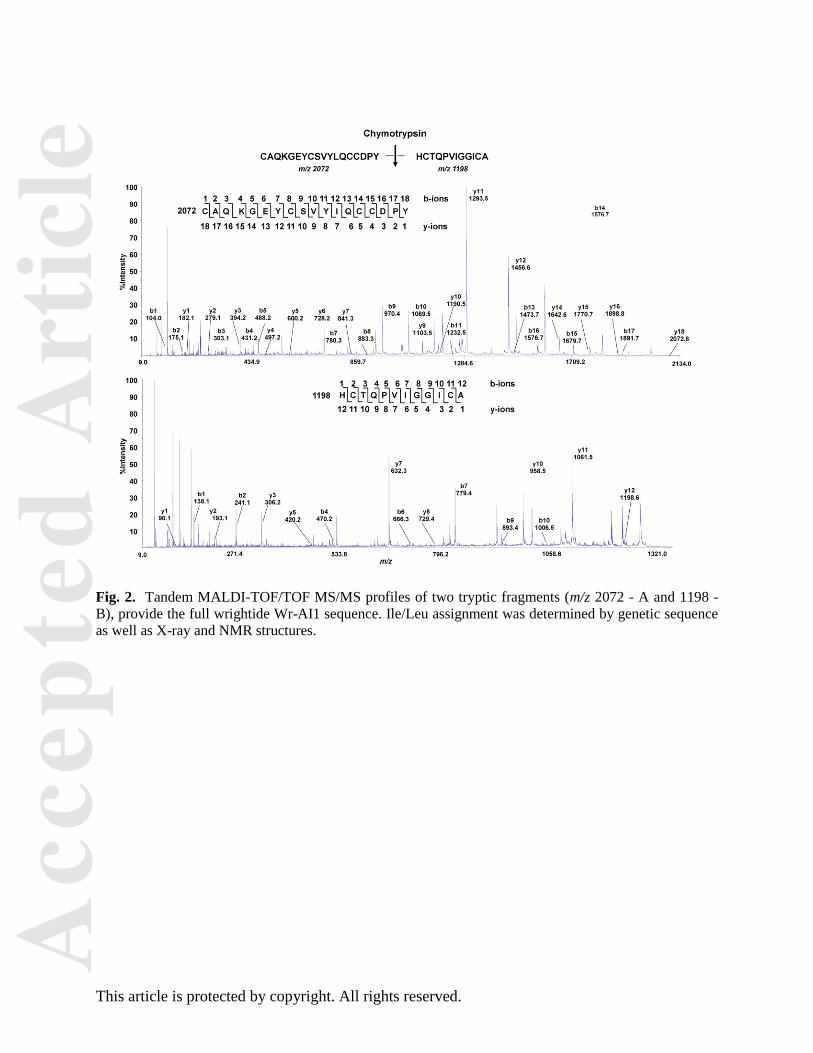
purified wrightide was fully reduced by dithiothreitol and then digested with trypsin and chymotrypsin.
The resulting fragments were sequenced by tandem mass spectrometry and their sequences deduced by
analyzing b- and y-ions (Fig. 2). By genetic analysis, we also obtained the sequence of wrightide Wr-AI3,
which could not be detected in MS profile.
Wrightides Wr-AI1 to Wr-AI3, all 30 amino acid residues in length, contain 6 cysteine, 3 glycine and 2
proline residues. Together, these three residues account for >35% of the sequences. Wrightides share high
sequence homology among each other (93-96%), differing by one or two residues (Fig. 3), and high
sequence homology with AAI (48%).
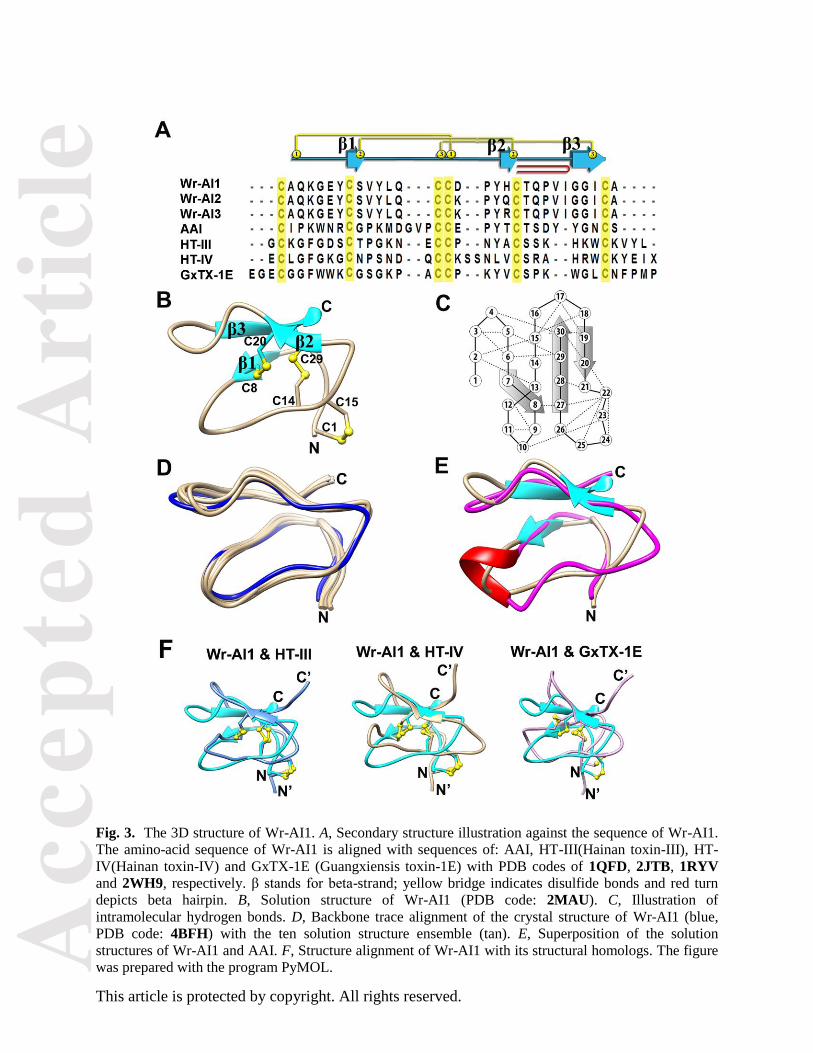
Solution structure of Wr-AI1 by 1H NMR
Using the distance, dihedral angle, and hydrogen bond restraints derived from 1H NMR experiments, the
solution structures of Wr-AI1 showed that it adopts similar CK scaffold as does AAI with the same three

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
disulfide linkages as Cys I-IV, Cys II-V, and Cys III-VI, where Cys III-VI is the penetrating disulfide
bond (Fig. 3A & 3B). The structure contains three short β-strands: Tyr7-Cys8, His19-Cys20, and Gly27-
Ala30, where His19-Ala30 forms a β-hairpin (Fig. 3C). The β-strands are connected by four β-turns, two
pointing towards the N- and C-terminal ends on one side of the molecule, and the other two to the
opposite side. This compact fold is also stabilized by an abundance of intra-molecular hydrogen bonds
(Fig. 3C) as reported in other cysteine-rich peptides such as plant denfensin PhD1 and cyclotide kalata B5
[8, 9]. Moreover, the structure is devoid of N- or C-terminal tails that would extend away from the CK
core stabilized by three disulfide bonds. Around 30% of the amide proton signals remained in the one
dimensional (1D) spectra after 18 h H/D exchange in D2O. These amide protons are identified as
hydrogen bond donors based on the structures. Wr-AI1 contains two prolines, whose conformations were
identified as cis Pro17 and trans Pro23. The proline cis and trans conformations were confirmed by the
observation of NOE crosspeaks HN
Asp16 - H
Pro17 and HN
Gln22 - HPro23, respectively, since the H
strips of
these four residues were not identified from the noise region of H2O around 4.7 ppm.
Structure of Wr-AI1 by X-ray crystallography
Wr-AI1 showed a high propensity to form fiber-like precipitates at neutral pH. Crystals of Wr-AI1
suitable for X-ray crystallography were successfully obtained after 1-day incubation and diffracted to a
1.25 Å resolution at a synchrotron beamline. The complete peptide chain comprising 30 residues was
unambiguously traced (Movie S1), clearly confirming the disulfide connectivity of Cys I-IV, Cys II-V,
and Cys III-VI as determined by NMR experiment. In addition, the crystal structure of Wr-AI1 agreed
with its solution structure ensemble (Fig. 3D) with an average RMSD value of 0.93 Å for backbone
atoms. Minor differences were observed mainly in the loop region and side chain orientations, including
the disulfide bonds. This could be due to the flexibility in solution of the loop region and side chains. This
observation is consistent with the overlapping of the chemical shifts of Hβ
Cys which created ambiguities in
clearly defining the orientation of disulfide bonds by NMR.
A systematic search for homologous structures deposited in the PDB using the DALI server
(http://www.ebi.ac.uk/) returned four homologous structures with a Z factor greater than 3.0 (Fig. 3A).
The structure of Wr-AI1 is most similar to that of AAI (PDB code: 1QFD in its free form and 1CLV as a
complex with the α-amylase from the yellow meal worm): a superposition of 29 α-carbon atoms returns
an RMSD of 1.10 Å with a strict conservation observed for the inhibitor core and disulfide bridges.
Variations between the two structures are confined to the two turns connecting the individual inhibitor
strands that come in contact with the α-amylase upon complex formation. The structure of Wr-AI1 also
resembles those of several spider toxins (Fig. 3F): the Hainan Toxin III and IV (PDB codes 2JTB and
1RYV, respectively), which are neuronal sodium channel inhibitors comprising 33 and 35 amino-acids
[10], and the GXTX-1E high affinity tarantula toxin (PDB code: 2WH9), which is a potassium channel
inhibitor [11].
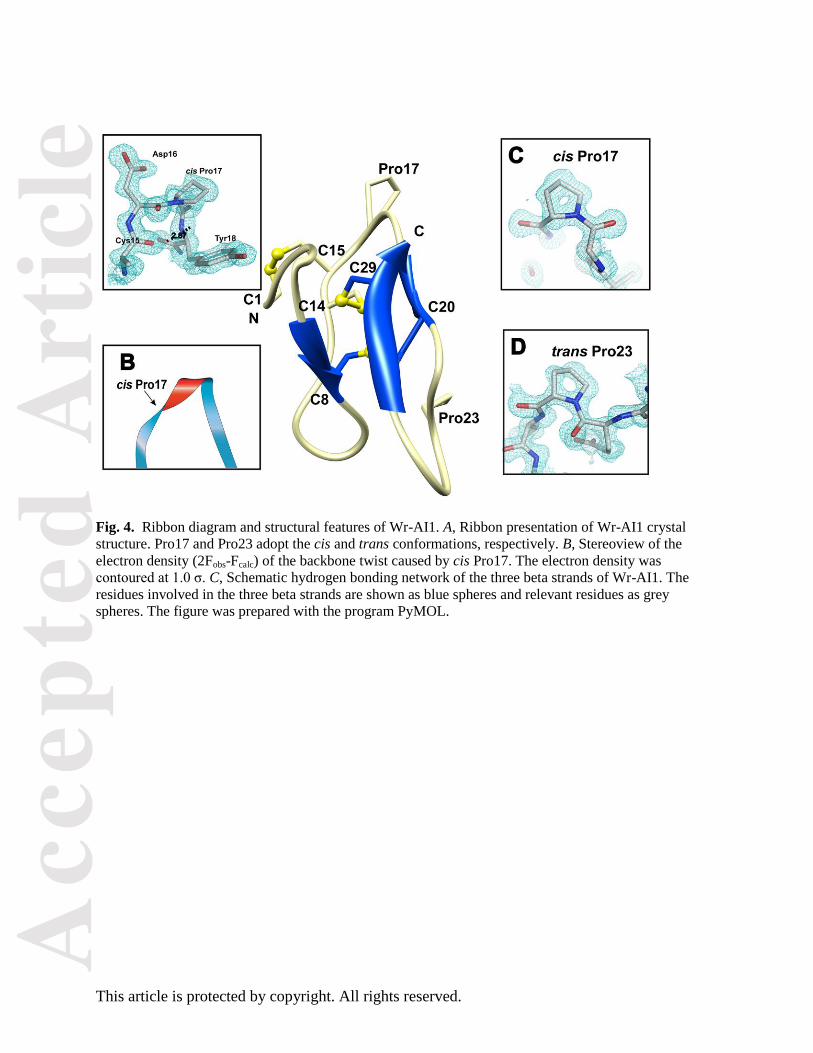
Further analysis of the crystal structure unambiguously established the two proline residues as cis Pro17
and trans Pro23 (Fig. 4). In the structure of Wr-AI1, the cis peptide bond between Asp16 and Pro17
causes a local backbone twist (Mobius-like structure similar to Mobius cyclotides). This energetically
unfavorable twist is partly stabilized by a strong hydrogen bond between main-chain atoms of Cys15 and
Tyr18 (Table 3 shows the list of intramolecular hydrogen bonds). Previous studies showed that the cis
conformation occurs at higher frequency in X-Pro peptides where X is an aromatic residue [12]. This high
occurrence is explained by the interaction between the aromatic side chain and the proline residue which
gives rise to ring-current-induced shifts for the cis conformers but not the trans conformers in NMR

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
experiments. Here we observed a parallel stacking between the phenol ring of Tyr18 and pyrrolidine ring
of Pro17. Significant shifts from the average chemical shifts of Hδ (2.39 with reference average value
3.63) and Hγ (0.71 with reference average value 2.02) were also observed based on the NMR assignments.
The distance between the centers of Pro17 pyrrolidine ring and Tyr18 phenol ring is 4.9 Å. Thus, the
interaction between the aromatic side chain and the pyrrolidone ring could contribute to the stabilization
of Pro17 cis form.
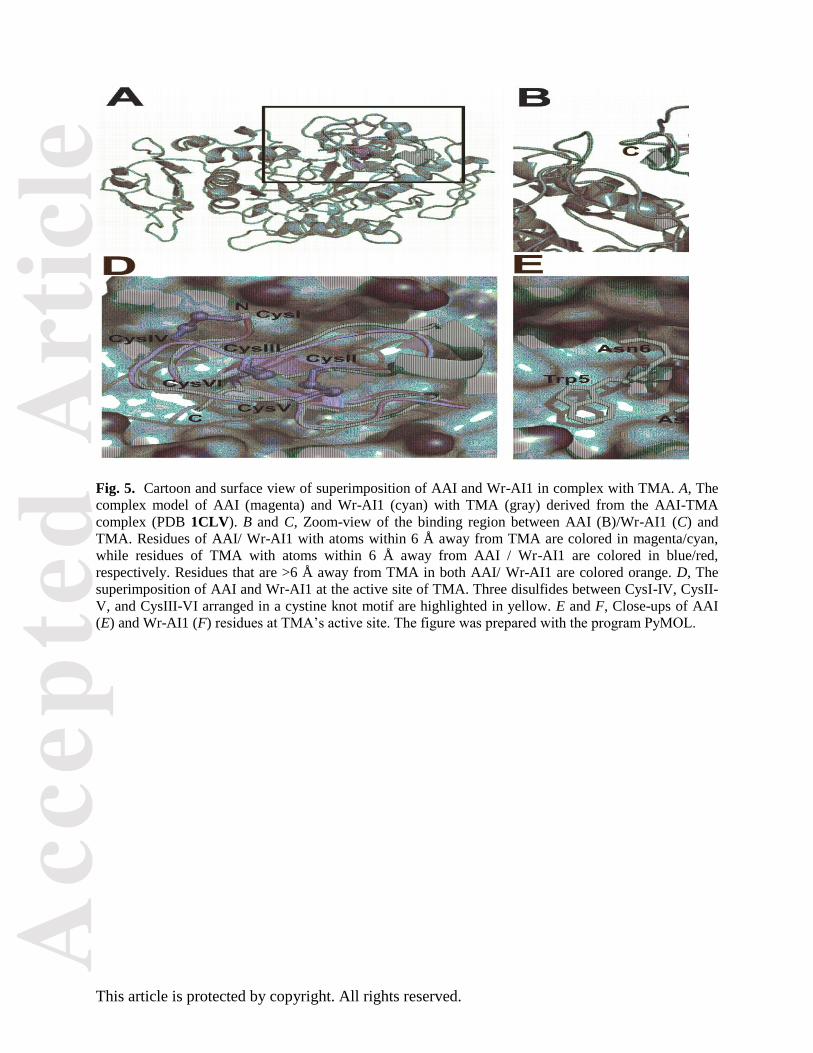
Modeled complex between Wr-AI1 and TMA
The complex between TMA and AAI was previously characterized by Pereira and coworkers using X-
ray crystalloraphy [13]. In this structure, AAI inserts into a V-shaped crevice located at the interface of
TMA domains A and B that forms the active site accommodating the carbohydrate residues. A total of 18
residues of AAI are in contact (distance <4.0 Å) with 24 residues of TMA. Among them, several residues
of the AAI inhibitor occupy or block the entrance to the six carbohydrate-binding subsites at the TMA’s
active site cleft. These residues include Lys4-Arg7, Met12, Tyr27, and Tyr28, all of which are located at
the hydrophilic accessible surface of the inhibitor molecule. Particularly, Arg7 forms a salt bridge with
the catalytic residue Asp287 from TMA. This residue is also involved in a water-mediated hydrogen-
bonding network with two other catalytic residues Glu222 and Asp185 from TMA. To better understand
how Wr-AI1 can inhibit the amylase activity of TMA, we built an atomic model for their interaction using
the AAI-TMA complex as a template (Fig. 5) and assuming an overall conservation of the molecular
orientation of the inhibitors in the TMA active site pocket. With the exception of Gly27 (Wr-AI1
numbering), no residue located at the interface with the enzyme is strictly conserved between Wr-AI1 and
AAI. Upon complex formation the buried surface area is 1831 Å2 which is comparable to the AAI-TMA
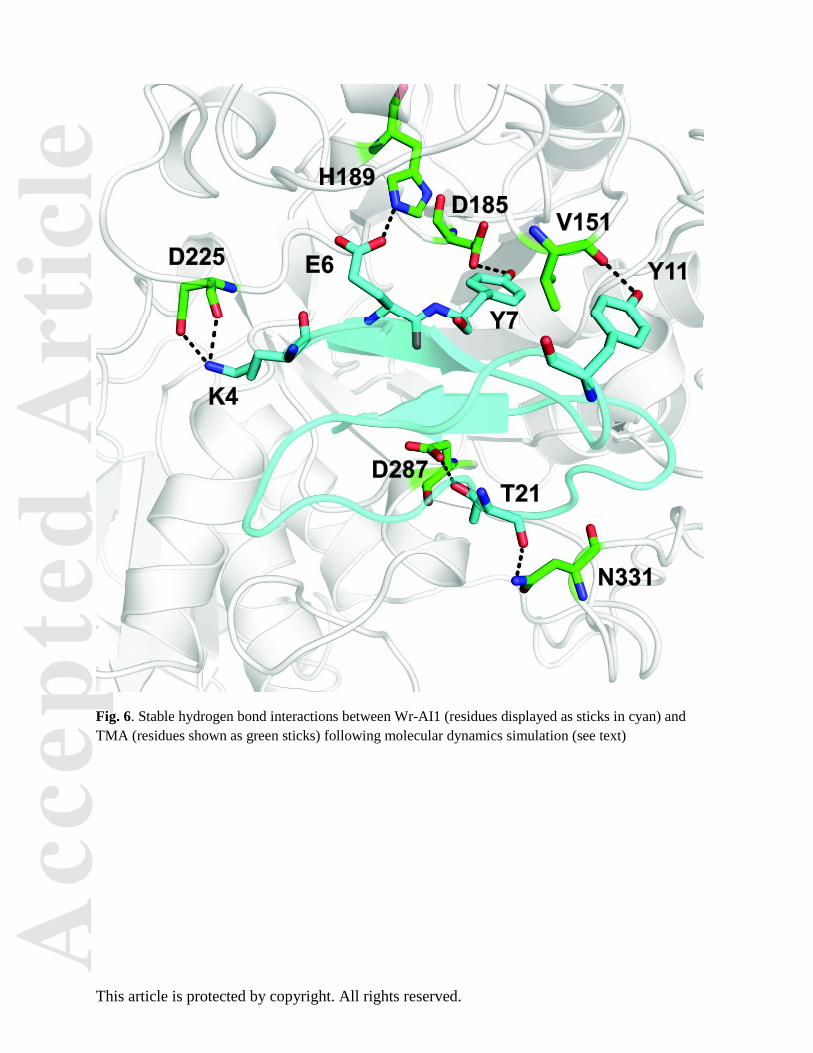
complex (2085 Å2) [13]. The network of interactions that stabilizes the Wr-AI1 complex is detailed in
Table 3 and Fig. 6 (Movie S2). Residues Lys4, Glu6, Tyr7 and Thr21 from Wr-AI1 provide hydrogen
bonds with several negatively charged residues from TMA (Fig. 6). This set of H-bonds are preserved in
>95% of all configurations sampled along the last 250 ns of the molecular dynamics simulation. The total
ΔG is -40.56 (1.07) kcal/mol for this comlex. An analysis of the various components contributing to
molecular complex stabilization gives van der Waals: -88.880.93, electrostatics: 138.444.47 and
nonpolar solvation -77.493.79 kcal/mol. This analysis suggests that the enthalpic contribution of the
association between TMA and Wr-AI1 is mainly driven by van der Waals interactions with a smaller
contribution of electrostatic interaction.
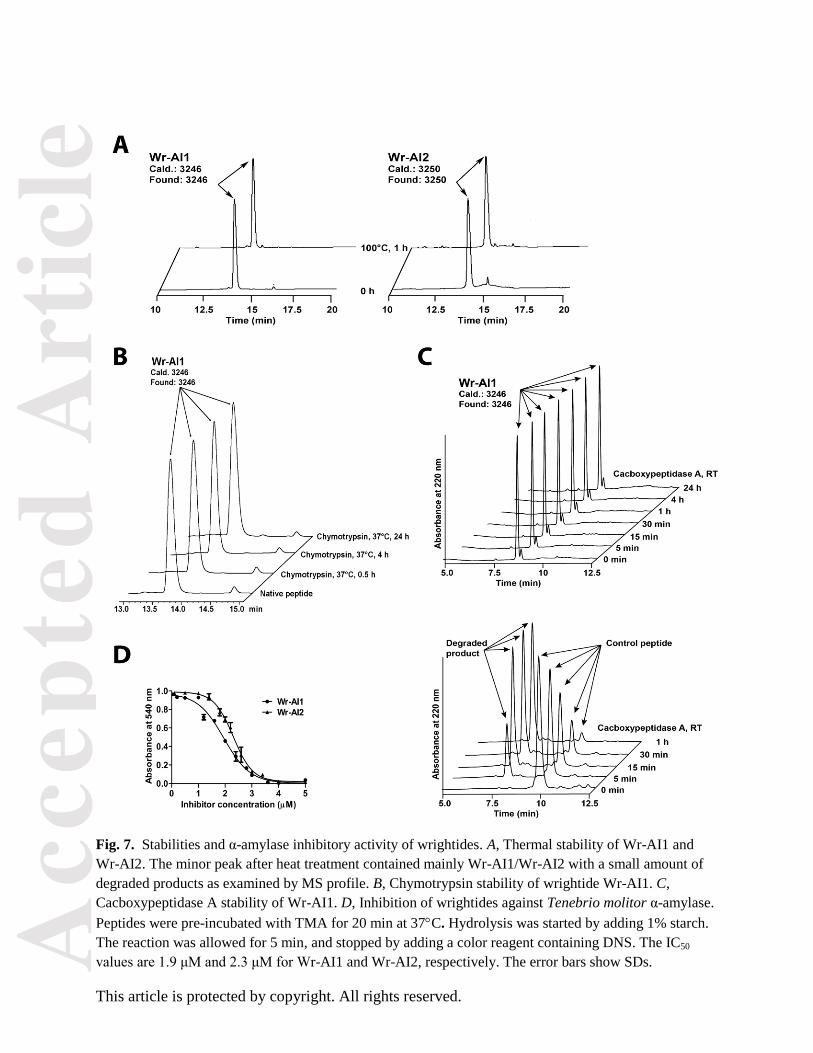
Heat and proteolytic stability
To determine whether wrightides would survive degradation in boiling or protease treatment, parameters
important for administering decoctions in traditional medicines, Wr-AI1 was heated at 100C for 1 h or
incubated with chymotrypsin or cacboxypeptidase A for 4 h. More than 95% of Wr-AI1 remaining intact
was observed at the same retention time in the ultra performance liquid chromatography (UPLC) profiles
after heat treatment (Fig. 7A). The MS profiles of corresponding peaks showed that both peaks contained
native Wr-AI1 (m/z 3246) with a small amount of degraded products.
To show that the cystine-knot structure is important for its proteolytic stability, Wr-AI1 was fully reduced
by dithiothreitol and served as the control in chymotrypsin stability assay. A 9-amino-acid linear peptide
was used as the control in cacboxypeptidase A assay. The control peptides were almost completely
hydrolyzed after a 4-hour incubation with chymotrypsin or 1-hour treatment with cacboxypeptidase A at
37C. Under similar conditions, the native peptide Wr-AI1 was resistant to protease degradation with

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
>95% of peptides remaining intact (Fig. 7B and 7C). Our results provide strong evidence for the stability
of wrightides against thermal, endopeptidase and exopeptidase treatments.
α-amylase inhibitory activity
We performed inhibition assays with TMA and α-amylases from human saliva, porcine pancreas, and
fungus (Aspergillus oryzae) using the Bernfeld method [14]. Preliminary results showed that both
wrightides Wr-AI1 and Wr-AI2 exhibited inhibitory activities against TMA in a dose-dependent manner
with IC50 of 1.9 and 2.3 μM, respectively (Fig. 7D). Similar to AAI, Wr-AI1 and Wr-AI2 did not inhibit
α-amylases from fungus or mammals at concentrations up to 100 μM.
Biological activity of amylase inhibitors
Cytotoxic, hemolytic, and antibacterial activities of Wr-AI1 and Wr-AI2 were tested. In our experiments,
wrightides did not show appreciable toxic, hemolytic or antibacterial activity at concentrations up to 100
μM.
Cloning of wrightide-encoding genes
Using 3 and 5 RACE (rapid amplification of cDNA ends) PCRs, we obtained the Wr-AI2 full-length
gene from RNA extract from a Singapore plant. Subsequently we used two primers derived from the 5
and 3 untranslated regions of Wr-AI2 clone and successfully amplified DNA sequences of Wr-AI1, Wr-
AI2, and a novel wrightide, Wr-AI3, which was not found at protein level.
Fig. 8 shows the deduced 87-amino-acid precursors of Wr-AI1 to Wr-AI3 and their alignment with
previously characterized CK trypsin inhibitor and -conotoxin precursors. In general, wrightide
precursors contain a 21-residue ER signal sequence followed by a pro-domain of 36 residues and a 30-
residue wrightide domain at the C-terminus. Comparison between RACE and DNA PCRs showed that
wrightide genes contain a phase-one intron in the middle of ER signal. The signal sequence and pro-
domain of wrightide precursors are almost identical except for three residue differences in the pro-domain
and several silent mutations at the gene level as highlighted in Fig. 8A.
Discussion
In this paper, we used proteomic, genomic, and structural methods to characterize the 30-residue knottin-
type α-amylase inhibitors, Wr-AI1 to Wr-AI3, from W. religiosa of the Apocynaceae family. Since the
discovery of AAI in 1994, the 32-residue AAI has remained the only representative of the knottin group
that exhibits α-amylase inhibitory activity [5]. The discovery of wrightides thus extends the list of the
family of knottin α-amylase inhibitors. With two fewer amino acids than AAI, the wrightide family
represents the smallest proteinaceous α-amylase inhibitors reported. Interestingly, these wrightides are
resistant to both heat denaturation and proteolytic degradation, including exopeptidase treatment. Thus,
wrightides take on the favorable stability features of cyclic CK peptides such as cyclotides. In wrightides,
their N- and C-termini are protected by disulfide bonds at the ultimate or penultimate residues. Our

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
structural analysis showed that this arrangement enables the termini to loop back to the peptide chain via
disulfide bonds like a “pseudocyclics”, particularly at the N-terminus of wrightides. These pseudocylic
cystine-knot (PCK) peptides, with or without one extra residue flanking at the disulfide-looping terminus,
would likely escape the degradation by exopeptidases
The backbone-twisting cis proline in PCK inhibitors
The presence of cis proline in naturally occurring cysteine-rich peptides generally causes a twist in the
peptide backbone. This was used as a benchmark to classify cyclotides into Möbius (with cis Pro) and the
bracelet (without cis Pro) subfamilies [4]. In this work, we found that PCK wrightide Wr-AI1 also
contains one backbone-twisting cis proline. Also in our unreported work, we found PCK peptides in three
other Apocynaceae plants, each of which containing 3-4 proline residues and four determined by NMR
spectroscopy to have 1-2 cis proline residues. Together, these results suggested that the occurrence of cis
proline bond could be underestimated in proline-rich cysteine-rich peptides.
Surveys of protein databases revealed approximately 35% and 6–8% cis proline in small polypeptides and
native proteins, respectively [15]. The percentage of cis proline amide bond increases to as high as 12–
16% when proline is preceded by an aromatic residue in protein primary sequences. The steric repulsion
of the pyrrolidine rings of a proline residue with the two neighboring Cα atoms generally renders the cis
configuration energetically less favorable than the trans configuration. In peptides with cis proline
preceded by an aromatic residue, clustering of the aromatic side chain and the pyrrolidine ring provides
stability to the sterically constrained cis proline, which is manifested in part by the selective ring-current-
induced shifts of proline Hα and H
β in NMR spectroscopy [12]. Our analysis of the Wr-AI1 structure
demonstrated the occurrence of one cis proline in X-Pro amide bonds, where X is a non-aromatic residue.
The backbone twist caused by this cis proline is likely stabilized by the hydrogen bond between
neighboring residues Cys15 and Tyr18 rather than direct stacking of the preceding aromatic side chain
and proline residue. Thus, the study of local interactions that stabilize the cis Pro residues in Wr-AI1 and
other PCK α-amylase inhibitors may reveal diverse mechanisms of cis Pro formation in cis proline-rich
peptides.
Shape-fitting inhibition mechanism between PCK α-amylase inhibitors and TMA
A model of the interaction between Wr-AI1 and TMA was constructed assuming an overall conservation
of molecular orientation in the TMA’s active site pocket compared to AAI. Interestingly, despite the lack
of sequence conservation between both peptide inhibitors, several side chains that project from the
surface of the two inhibitors are placed in similar positions in the active-site crevice of TMA and small
movements would allow them to form equivalent contacts with the enzyme. Molecular dynamics study of
the modeled complex suggests that Wr-AI1 binds to TMA’s active-site depression via an interaction
network built largely on nonpolar interactions and completely lacking the critical salt bridge observed for
AAI (Table 3). A crystal structure for the TMA-Wr-AI1 complex is needed to confirm this hypothesis.
Wrightides follow the biosynthesis pathway for secretory proteins
Our genetic analysis showed that wrightide precursors consist of an endoplasmic reticulum signal domain
with a phase-one intron, a pro-domain, and a single wrightide domain at its C-terminus. The gene

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
organization, starting with a signal peptide, provides hints of the biosynthesis pathway of wrightides
which are gene-encoded and ER-targeted following the conventional pathway for secretory proteins (Fig.
8B) as suggested for many cysteine-rich peptides [16]. The signal peptide is generally removed by SPase
I from the precursor to release the pro-peptide. A single cleavage between the 36-residue pro-domain and
the 30-residue functional domain subsequently produces the native wrightide. These characteristics
distinguish them as ribosomally synthesized peptides from smaller peptides of 5–12 residues that are
synthesized by non-ribosomal multi-enzyme complexes [17, 18].
The three-domain precursor structure is commonly found in many CK peptides, both cyclic and linear.
Examples for such cyclic plant CK peptide precursors include cyclotides from Rubiaceae, Violaceae, and
Solanaceae families [17, 19] and squash trypsin inhibitors from M. cochinchinensis [20] whereas selected
examples for linear plant CK peptide precursors are acyclic cyclotides from Violaceae, Rubiaceae, and
Poaceae families [21-23] and towel gourd trypsin inhibitors [24]. This precursor organization is also used
by animals such as cone snails to encode ion channel blockers ω-conotoxins [25] and δ-conotoxins [26]. It
should be noted that within the plant kingdom, diverse structures are used to organize CK peptide
precursors. One example is precursors with multiple repeats of cyclic or linear CK peptides such as those
reported for cyclotides from Rubiaceae and Violaceae families [17, 19] as well as TIPTOP squash trypsin
inhibitors [20]. Another example is chimeric precursors that encode both CK peptides and other types of
proteins, such as cliotide precursors encoding both cyclotides and legume albumin PA1a in C. ternatea
plant [27, 28]. Given such diversity of precursor organization even within a CK peptide family,
understanding about the genetic sequences of each CK peptide family, here PCK α-amylase inhibitors, is
thus beneficial for their applications in crop protection and also provides insights into their biosynthesis.
Knottin-type α-amylase inhibitors with applications in engineering peptidyl bioactives
The CK structure has been employed in nature as scaffolds of a variety of unrelated protein families
found in microbes, animals, and plants. Particularly, α-amylase inhibitors adopting CK fold are small,
extraordinarily stable against heat, endopeptidase as well as exopeptidase degradation, and highly tolerant
to sequence variation [29]. Thus, small CK peptides such as wrightides with MW 3-5 kDa contain
appealing features as potential peptide therapeutics [30, 31]. First, the small size renders wrightides more
accessible to chemical synthesis [32, 33]. Second, the CK peptides in general are highly tolerant to
sequence variations and the spacing of the half cystines, allowing α-amylase inhibitors to potentially serve
as a scaffold for protein engineering to attain new functions, such as in the successful grafting of the
bradykinin-antagonist peptides DALK or DAK onto cyclotide kalata B1 scaffold [34].
A potential application of considerable interest to drug development is the engineering of wrightides to be
orally active mammalian α-amylase inhibitors in treating obesity and type-2 diabetes mellitus (T2DM).
Literature precedents showed that extended hydrophobic interactions could be important for AAI
inhibition on mammalian α-amylases [35]. Human salivary α-amylase (HSA, PDB code: 3DHP) and
TMA share high sequence homology (65%) and structural homology (Z-score of 57.1 with 468 equivalent
residues at rmsd of 1 Å, by DALI server). Superimposing HSA:Wr-AI1 complex with TMA:Wr-AI1
complex reveals four additional loops present in HSA at the interface of the active site, including loops
Asn53-Phe55, Asn137-Gly146, Gly304-Ala310, and Trp344-Val358. The conformational flexibility of
these loops might be responsible for the low affinity binding of Wr-AI1 to HSA. Our docking
experiments suggested that it is possible that careful incorporation of aromatic and positively charged
residues into wrightide templates could improve their contact with the negatively charged enzyme active
sites to render wrightides active against mammalian α-amylases. In this regard, our work showing the

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
interaction of Wr-AI1 and TMA provides new insights for a structure-guided approach to design
potentially useful orally active α-amylase inhibitors for managing T2DM and obesity.
Experimental procedures
Isolation of α-amylase inhibitors
800 g of W. religiosa leaves was homogenized and extracted twice in 50% (v/v) ethanol. After
centrifugation (7000 rpm, 10 min), the supernatant was partitioned with dichloromethane. The aqueous
upper layer was concentrated, filtered, and loaded onto a C18 flash column (Grace Davison, US). Elution
was done with increasing concentrations of ethanol. The presence of cysteine-rich peptides in all fractions
was monitored by matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-
TOF MS). To purify individual peptides, several dimensions of strong cation exchange and reversed-
phase HPLC were employed.
De novo sequencing with MALDI-TOF MS/MS
About 40 μg of each purified peptide was dissolved in 50 mM ammonium bicarbonate buffer (pH 7.8)
containing 50 mM dithiothreitol and incubated at 37C for 2 h. Digestion with endoproteinase Glu-C,
trypsin or chymotrypsin was carried out at room temperature for 5 min and subsequently sequenced by
MALDI-TOF MS/MS as previously described [27]. Isobaric residues were assigned based on gene
sequences for Wr-AI1 and Wr-AI2 and confirmed based on X-ray or NMR structure for Wr-AI1. The
peptide Wr-AI3 was only determined at gene level.
Solution structure determination using NMR spectroscopy
The NMR sample was prepared by dissolving lyophilized Wr-AI1 into 95% H2O/5% D2O or 99.9% D2O
directly (~1 mM protein and pH/pD 3.3). All NMR experiments were carried out on a Bruker 600 MHz
NMR spectrometer equipped with a cryogenic probe. Two dimensional (2D) total correlation
spectroscopy (TOCSY) and nuclear Overhauser spectroscopy (NOESY) experiments were performed
with mixing times 80 ms and 200 ms, respectively [36]. The 2D data were acquired at 298 K. Water
suppression was achieved using modified WATERGATE pulse sequences [37]. The NMR spectra were
processed with NMRPipe software [38]. The amides involved in hydrogen bonding were identified by the
hydrogen-deuterium exchange 1D 1H experiment [39].
Sequence specific assignments were achieved based on the 2D TOCSY and NOESY, and NOEs were
assigned from the 2D NOESY using the software NMRspy
(http://yangdw.science.nus.edu.sg/Software&Scripts/NMRspy/index.htm). The chemical shifts are
deposited in BioMagResBank (accession number: 18983). Distance restraints were derived from the peak
intensities of the assigned NOEs. Dihedral angles φ were obtained from 3JHN-Hα coupling constants
measured from the 1D 1H spectrum. Hydrogen bond restraints were incorporated based on the observation
of amide protons in the 1D 1H spectra recorded after resuspending the lyophilized Wr-AI1 in D2O for up
to 18 h at 25C.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Structure was calculated with simulated annealing approach with CYANA 2.0 [40]. Distance restraints
are divided into three classes: 1.8<d<3.4 Å (strong NOEs), 1.8<d<4.2 Å (medium NOEs) and 1.8<d<5.5
Å (weak NOEs). Disulfide bond restraints of 2.0<d (Sγi, S
γj)<2.1 Å, 3.0<d (C
βi, S
γj)<3.1 Å and 3.0<d (S
γi,
Cβ
j)<3.1 Å were employed for structure calculation. During the structure calculation, hydrogen bond
restraints of 1.8-2.2 Å for the NH-O distance and 2.2-3.2 Å for the N-O distance were applied on nine
identified hydrogen bonds according to the slowly exchanging amide protons. Φ angles were constrained
to the range of -150° to -90° for 3JHN-Hα>8 Hz. Structures were displayed and analyzed using software
PyMOL and program PROCHECK-NMR, respectively [41]. The experimental and structural statistics are
summarized in Table 1.
Crystal structure determination
Using sitting drop vapor diffusion method, the native crystals were obtained from the mixture of 1 µl of
Wr-AI1 solution (4.8 mg/ml) and 1 µl of precipitant solution (3.6 M sodium formate, 10% glycerol) after
one-day incubation at 16C. The crystals were stabilized in the precipitant solution supplemented with
40% (v/v) glycerol and flash-frozen in liquid nitrogen. Diffraction intensities to 1.25 Å resolution were
collected at 100 K at the Swiss Light Source Beamline PXIII using a Dectris Pilatus 6M detector.
Integration, scaling and merging of intensities were carried out using programs XDS [42] and SCALA
[43] from the CCP4 suite [44]. Data collection statistics are summarized in Table 2.
The structure was determined by molecular replacement using the program PHASER [45]. The search
probe was the structure of AAI (PDB code 1CLV [13]). The program Arp-Warp [46] was used for chain
tracing and map improvement and the resulting model was corrected manually (Table 2). The
Ramachandran plot calculated using PROCHECK [47] revealed that 87% of the residues were in the most
favoured region and 13% of the residues in additional allowed region.
Molecular docking and molecular dynamics study of the Wr-AI1-TMA complex
To investigate the stability of the TMA:Wr-AI1 complex obtained from docking (which was initially
obtained by simply superimposing Wr-AI1 onto AAI in the TMA-AAI crystal structure), we performed
three molecular dynamics simulation of 500 ns each, using ACEMD [48] and an all atom ff12SB force
field parameters. Hydrogen atoms were added to this initial complex using the Xleap module of AMBER
[49]. The system was solvated with TIP3P water molecules to form a box with at least 10 Å separating
the solute atoms and the edge of the box. A total of 92 Na+ and 70 Cl
- ions, corresponding to a salt
concentration of 150 mM were added to the system by replacing water molecules in random position.
Before the dynamic simulations, the solvated system was relieved of any unfavourable interactions by
subjecting it to 100 steps of energy minimization. Harmonic restraints during the equilibration were
placed on C 1 kcal.mol-1
.Å2 to the energy minimized co-ordinates. The system was heated
up to 300 K in steps of 100 K, followed by the gradual removal of the positional restraints and a 10 ns
unrestrained equilibration at 300 K. Analysis of the resulting trajectories revealed that the simulated
complex reached stability after 50 ns, with RMSD less than 1.8 Å. The first 10 ns of simulation were
performed in NPT, the production run of 500 ns was performed in NVT. The simulation temperature of
300 K was set using Langevin dynamics, with a collision frequency of 0.1 ps-1
. The pressure was
maintained at 1 atm using weak coupling with a pressure relaxation time of 1 ps. During the simulation,
all long range electrostatic interactions were treated with particle mesh Ewald methods [50] using a real
space cut-off distance of 9 Å. Bonds involving hydrogen atoms were constrained using the M-SHAKE
algorithm [51]. A time step of 4 fs with hydrogen mass repartitioning was used and coordinates were

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
saved every 100 ps. Hydrogen bond analysis was performed using the PTRAJ module in AMBER for the
last 250 ns of the stabilised trajectory using a distance cut off of 3.5 Å. Binding energy analysis based on
MM/GBSA [52] was performed on the simulated trajectory to calculate the free energy of binding of Wr-
AI1 peptide to TMA. For binding energy calculation, a total of 100 structures were extracted at regular
intervals from the last 250 ns of the trajectory. A salt concentration of 150 mM and Born implicit solvent
model of 2 (igb=2) [53] was used. The binding surface area was calculated using NACCESS software
[54]. Simulation trajectories were visualized in VMD [55] and figures were generated using PyMol.
Heat stability test
Purified Wr-AI1 was heated in boiling water for 1 h and then subjected to UPLC. The peptide Wr-AI1
without heat treatment was used as a control. Collected peaks from UPLC were monitored by MALDI-
TOF MS.
Proteolytic stability test
Purified Wr-AI1 was incubated with or without chymotrypsin (at a final peptide:enzyme ratio of 10:1
mol/mol) in 20 mM ammonium bicarbonate (pH = 7.8) at 37C for 4 h. Purified Wr-AI1 completely
reduced with 50 mM dithiothreitol (2 h, 37C) was treated in the same way and used as a control. Treated
samples or controls were subjected to UPLC and the collected peaks were monitored by MALDI-TOF
MS.
In cacboxypeptidase A stability assay, Wr-AI1 was incubated with or without enzyme (at final
peptide:enzyme ratio of 40:1 mol/mol) in 50 mM Tris-HCl, 1 M NaCl (pH 7.5) at room temperature for
up to 24 h. A linear 9-residue peptide was used as a control. Degradation products were monitored by
UPLC and MALDI-TOF MS.
Assay for α-amylase activity
Alpha-amylase was isolated from yellow mealworm, larvae of Tenebrio molitor, following the procedure
described previously [56]. Assays for α-amylase were carried out in 96-well plates following Bernfeld
method [14]. Enzyme TMA with or without treatment with peptides (20 min, 37C) was incubated with
1% starch (in 20 mM sodium phosphate buffer, pH 6.7) for 5 min. Color reagent (3,5-dinitrosalicilic acid
and sodium potassium tartrate, Sigma) [57] was dispensed into each well and color developed for 20 min
at 100C. Absorbance at 540 nm was read to determine the α-amylase activity. Similar inhibition
experiments were performed for human salivary, porcine pancreatic and Aspergillus oryzae α-amylases
(Sigma).
Hemolysis assay
Fresh type AB blood was donated by a healthy volunteer. The hemolysis assay was performed as
described elsewhere [27].

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Cytotoxicity assay
Cytotoxicity of the purified wrightides was tested using PrestoBlue™ Cell Viability Reagent (Invitrogen).
African green monkey kidney (Vero) cells seeded onto 96-well plate were incubated with Wr-AI1 and
Wr-AI2 at 1 to 100 μM for 24 h at 37C. After incubation, the wells were dispensed with Presto Blue
reagent and left at 37C for 2 h. The fluorescence was subsequently read as instructed by the
manufacturer. 1% Triton X-100 solution was used as positive control.
Antibacterial assay
The antibacterial activity of wrightides was assessed using radial diffusion assay as described previously
[58] on Gram negative Escherichia coli (FDA strain Seattle 1946) and Gram positive Staphylococcus
aureus bacteria. D4R, an in-house peptide dendrimer with potent antibacterial activity, was used as a
positive control. The experiments were done in duplicate.
Cloning of α-amylase inhibitor genes
Total RNA extraction was performed using PureLinkTM
Mini RNA purification kit (Invitrogen) with
additional 3% of 2-mercaptoethanol and 4% of polyvinylpyrrolidone added to the lysis buffer. Total RNA
extract of Singapore W. religiosa leaves was subsequently converted to 3' and 5' RACE cDNA libraries
using 3' RACE System for Rapid Amplification of cDNA Ends (Invitrogen) and SMARTer™ RACE
cDNA Amplification Kit (Clontech), respectively. 3' RACE PCR products using the degenerate primer
targeting the sequence CAQKGE (5'-TGTGCTCArAArGGnGA-3') were gel-purified, cloned into
pGEM®-T Easy Vector (Promega) and sequenced. A reverse primer based on the newly obtained partial
sequence was designed to reveal the remaining encoding gene in 5' RACE PCR. To determine the DNA
sequences of wrightide genes, we performed PCR on W. religiosa DNA extract using two primers:
Wr2speF (5'-TAGGCGCAAACAACATGGCT-AAGC-3') and Wr2speR (5'-CCACATAGCTCG-
TAGAACAAGCTTACAG-3'). The endoplasmic reticulum signal peptides were predicted using SignalP
3.0 (http://www.cbs.dtu.dk/services/-SignalP-3.0/).
Acknowledgments
We thank Giang K.T. Nguyen, Teo C. H. and Lam Y. S. for technical assistance in this project. This
research was supported in part by the Competitive Research Grant by National Research Foundation in
Singapore (NRF-CRP8-2011-05)
Author contribution
PQTN performed or was involved in all the experiments presented in this paper except NMR experiment;
SW performed NMR spectroscopy experiment and sequence calculation; AK analyzed MD results and
enzyme alignment; LJY performed X-ray crystallization experiment; TTL contributed to peptide and
enzyme extraction; JL analyzed X-ray crystallography data, built the structure and modeled the complex;
JPT analyzed the data. PQTN, JL, and JPT contributed mainly to manuscript preparation. All the authors
discussed the results and contributed to the writing of the manuscript.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
References
1. Cheek, S., Krishna, S. S. & Grishin, N. V. (2006) Structural classification of small, disulfide-rich protein domains, J Mol Biol. 359, 215-37. 2. Pallaghy, P. K., Nielsen, K. J., Craik, D. J. & Norton, R. S. (1994) A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides, Protein Sci. 3, 1833-9. 3. Le Nguyen, D., Heitz, A., Chiche, L., Castro, B., Boigegrain, R. A., Favel, A. & Coletti-Previero, M. A. (1990) Molecular recognition between serine proteases and new bioactive microproteins with a knotted structure, Biochimie. 72, 431-435. 4. Craik, D. J., Daly, N. L., Bond, T. & Waine, C. (1999) Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif, J Mol Biol. 294, 1327-36. 5. Chagolla-Lopez, A., Blanco-Labran, A. & Patthy, A. (1994) A Novel a-Amylase Inhibitor from Amaranth (Amaranthus hypocondriacus) Seeds, J Biol Chem. 269, 23675-23680. 6. Le Berre-Anton, V., Bompard-Gilles, C., Payan, F. & Rouge, P. (1997) Characterization and functional properties of the alpha-amylase inhibitor (alpha-AI) from kidney bean (Phaseolus vulgaris) seeds, Biochim Biophys Acta. 14, 31-40. 7. Svensson, B., Fukuda, K., Nielsen, P. K. & Bønsager, B. C. (2004) Proteinaceous alpha-amylase inhibitors, Biochim Biophys Acta (BBA) - Proteins & Proteomics. 1696, 145-156. 8. Plan, M. R., Rosengren, K. J., Sando, L., Daly, N. L. & Craik, D. J. (2010) Structural and biochemical characteristics of the cyclotide kalata B5 from Oldenlandia affinis, Peptide Science. 94, 647-658. 9. Janssen, B. J. C., Schirra, H. J., Lay, F. T., Anderson, M. A. & Craik, D. J. (2003) Structure of Petunia hybrida Defensin 1, a Novel Plant Defensin with Five Disulfide Bonds†, Biochemistry. 42, 8214-8222. 10. Li, D., Xiao, Y., Xu, X., Xiong, X., Lu, S., Liu, Z., Zhu, Q., Wang, M., Gu, X. & Liang, S. (2004) Structure-activity relationships of hainantoxin-iv and structure determination of active and inactive sodium channel blockers, J Biol Chem. 279, 37734-37740. 11. Lee, S., Milescu, M., Jung, H. H., Lee, J. Y., Bae, C. H., Lee, C. W., Kim, H. H., Swartz, K. J. & Kim, J. I. (2010) Solution structure of GxTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in Kv2.1 potassium channels, Biochemistry. 49, 5134-5142. 12. Wu, W. J. & Raleigh, D. P. (1998) Local control of peptide conformation: stabilization of cis proline peptide bonds by aromatic proline interactions, Biopolymers. 45, 381-94. 13. Pereira, P. J., Lozanov, V., Patthy, A., Huber, R., Bode, W., Pongor, S. & Strobl, S. (1999) Specific inhibition of insect alpha-amylases: yellow meal worm alpha-amylase in complex with the amaranth alpha-amylase inhibitor at 2.0 A resolution, Structure. 7, 1079-88. 14. Bernfeld, P. (1955) Amylases a and b, Methods Enz. 1, 149-158. 15. Milner-White, E. J., Bell, L. H. & Maccallum, P. H. (1992) Pyrrolidine ring puckering in cis and trans-proline residues in proteins and polypeptides. Different puckers are favoured in certain situations, J Mol Biol. 228, 725-34. 16. Mergaert, P., Nikovics, K., Kelemen, Z., Maunoury, N., Vaubert, D., Kondorosi, A. & Kondorosi, E. (2003) A novel family in Medicago truncatula consisting of more than 300 nodule-specific genes coding for small, secreted polypeptides with conserved cysteine motifs, Plant Physiol. 132, 161-173. 17. Jennings, J, W., C, W., D, C. & M., A. (2001 Sep 11) Biosynthesis and insecticidal properties of plant cyclotides: the cyclic knotted proteins from Oldenlandia affinis., Proc Natl Acad Sci USA. 98, 10614-9. 18. Marahiel, M. A. (2009) Working outside the protein-synthesis rules: insights into non-ribosomal peptide synthesis, Journal of Peptide Science. 15, 799-807. 19. Dutton, J. L., Renda, R. F., Waine, C., Clark, R. J., Daly, N. L., Jennings, C. V., Anderson, M. A. & Craik, D. J. (2004) Conserved structural and sequence elements implicated in the processing of gene-encoded circular proteins, J Biol Chem. 279, 46858-67.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
20. Mylne, J. S., Chan, L. Y., Chanson, A. H., Daly, N. L., Schaefer, H., Bailey, T. L., Nguyencong, P., Cascales, L. & Craik, D. J. (2012) Cyclic peptides arising by evolutionary parallelism via asparaginyl-endopeptidase-mediated biosynthesis, Plant Cell. 24, 2765-78. 21. Nguyen, G. K., Zhang, S., Wang, W., Wong, C. T., Nguyen, N. T. & Tam, J. P. (2011) Discovery of a linear cyclotide from the bracelet subfamily and its disulfide mapping by top-down mass spectrometry, J Biol Chem. 286, 44833-44. 22. Ireland, D. C., Colgrave, M. L. & Craik, D. J. (2006) A novel suite of cyclotides from Viola odorata: sequence variation and the implications for structure, function and stability, Biochem J. 400, 1–12. 23. Nguyen, G. K., Lian, Y., Pang, E. W., Nguyen, P. Q., Tran, T. D. & Tam, J. P. (2013) Discovery of linear cyclotides in monocot plant Panicum laxum of Poaceae family provides new insights into evolution and distribution of cyclotides in plants, J Biol Chem. 288, 3370-80. 24. Ling, M. H., Qi, H. Y. & Chi, C. W. (1993) Protein, cDNA, and genomic DNA sequences of the towel gourd trypsin inhibitor. A squash family inhibitor, J Biol Chem. 268, 810-4. 25. Colledge, C. J., Hunsperger, J. P., Imperial, J. S. & Hillyard, D. R. (1992) Precursor structure of omega-conotoxin GVIA determined from a cDNA clone, Toxicon. 30, 1111-6. 26. Woodward, S. R., Cruz, L. J., Olivera, B. M. & Hillyard, D. R. (1990) Constant and hypervariable regions in conotoxin propeptides, The EMBO journal. 9, 1015-20. 27. Nguyen, G. K. T., Zhang, S., Nguyen, N. T. K., Nguyen, P. Q. T., Chiu, M. S., Hardjojo, A. & Tam, J. P. (2011) Discovery and characterization of novel cyclotides originated from chimeric precursors consisting of albumin-1 chain a and cyclotide domains in the Fabaceae family, J Biol Chem. 286, 24275-24287. 28. Poth, A. G., Colgrave, M. L., Lyons, R. E., Daly, N. L. & Craik, D. J. (2011) Discovery of an unusual biosynthetic origin for circular proteins in legumes, Proc Natl Acad Sci USA. 108, 10127-32. 29. Norton, R. S. & Pallaghy, P. K. (1998) The cystine knot structure of ion channel toxins and related polypeptides, Toxicon. 36, 1573-83. 30. Tam, J. P. & Lu, Y.-A. (1997) Synthesis of large cyclic cystine-knot peptide by orthogonal coupling strategy using unprotected peptide precursor, Tetrahedron Letters. 38, 5599-5602. 31. Taichi, M., Hemu, X., Qiu, Y. & Tam, J. P. (2013) A thioethylalkylamido (TEA) thioester surrogate in the synthesis of a cyclic peptide via a tandem acyl shift, Organic Letters. 15, 2620-2623. 32. Wong, C. T. T., Taichi, M., Nishio, H., Nishiuchi, Y. & Tam, J. P. (2011) Optimal oxidative folding of the novel antimicrobial cyclotide from Hedyotis biflora requires high alcohol concentrations, Biochem. 50, 7275-7283. 33. Tam, J. P., Lu, Y.-A. & Yu, Q. (1999) Thia Zip Reaction for Synthesis of Large Cyclic Peptides: Mechanisms and Applications†, J Am Chem Soc. 121, 4316-4324. 34. Wong, C. T. T., Rowlands, D. K., Wong, C.-H., Lo, T. W. C., Nguyen, G. K. T., Li, H.-Y. & Tam, J. P. (2012) Orally active peptidic bradykinin b1 receptor antagonists engineered from a cyclotide scaffold for inflammatory pain treatment, Angew Chem Int Ed. 51, 5620-5624. 35. Pereira, P. J. B., Lozanov, V., Patthy, A., Huber, R., Bode, W., Pongor, S. & Strobl, S. (1999) Specific inhibition of insect alpha-amylases: yellow meal worm alpha-amylase in complex with the Amaranth alpha-amylase inhibitor at 2.0 Å resolution, Structure (London, England : 1993). 7, 1079-1088. 36. Kumar, A., Ernst, R. R. & Wuthrich, K. (1980) A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules, Biochem Biophys Res Commun. 95, 1-6. 37. Piotto, M., Saudek, V. & Sklenar, V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions, J Biomol NMR. 2, 661-5. 38. Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J. & Bax, A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J Biomol NMR. 6, 277-93. 39. Saether, O., Craik, D. J., Campbell, I. D., Sletten, K., Juul, J. & Norman, D. G. (1995) Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata B1, Biochem. 34, 4147-58. 40. Guntert, P., Mumenthaler, C. & Wuthrich, K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA, J Mol Biol. 273, 283-98.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
41. Laskowski, R. A., Rullmannn, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR, J Biomol NMR. 8, 477-86. 42. Kabsch, W. (2001) Integration, scaling, space-group assignment and post refinement in International Tables for Crystallography Volume F: Crystallography of biological macromolecules (Rossmann, M. G. & Arnold, E., eds) pp. 218-225, Springer Netherlands. 43. Evans, P. (2006) Scaling and assessment of data quality, Acta Crystallogr Sect D. 62, 72-82. 44. Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E. B., Leslie, A. G., McCoy, A., McNicholas, S. J., Murshudov, G. N., Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A. & Wilson, K. S. (2011) Overview of the CCP4 suite and current developments, Acta Crystallogr D Biol Crystallogr. 67, 235-42. 45. McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007) Phaser crystallographic software, J Appl Crystallogr. 40, 658-674. 46. Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7, Nat Protocols. 3, 1171-1179. 47. Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures, J Appl Crystallogr. 26, 283-291. 48. Harvey, M. J., Giupponi, G. & De Fabritiis, G. (2009) ACEMD: Accelerating biomolecular dynamics in the microsecond time scale, J Chem Theory Comput. 5, 1632-1639. 49. Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) Comparison of simple potential functions for simulating liquid water, J Chem Phys. 79, 926-935. 50. Darden, T., York, D. & Pedersen, L. (1993) Particle mesh Ewald - an N.Log(N) method for ewald sums in large systems, J Chem Phys. 98, 10089-10092. 51. Krautler, V., Van Gunsteren, W. F. & Hunenberger, P. H. (2001) A fast SHAKE: Algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations, J Comput Chem. 22, 501-508. 52. Bashford, D. & Case, D. A. (2000) Generalized born models of macromolecular solvation effects, Annu Rev Phys Chem. 51, 129-152. 53. Onufriev, A., Bashford, D. & Case, D. A. (2000) Modification of the generalized Born model suitable for macromolecules, J Phys Chem B. 104, 3712-3720. 54. Hubbard, S. J. & Thornton, J. M. (1993) NACCESS, Department of Biochemistry and Molecular Biology, University College, London 55. Humphrey, W., Dalke, A. & Schulten, K. (1996) VMD: Visual molecular dynamics, J Mol Graph Model. 14, 33-38. 56. Strobl, S., Gomis-Rüth, F.-X., Maskos, K., Frank, G., Huber, R. & Glockshuber, R. (1997) The α-amylase from the yellow meal worm: complete primary structure, crystallization and preliminary X-ray analysis, FEBS Letters. 409, 109-114. 57. Miller, G. L. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar, Analytical Chem. 31, 426-429. 58. Lehrer, R. I., Rosenman, M., Harwig, S. S. S. L., Jackson, R. & Eisenhauer, P. (1991) Ultrasensitive assays for endogenous antimicrobial polypeptides, J Immunol Methods. 137, 167-173.
Supporting information
Movie S1. Stick representation of Wr-AI1 with 2Fo-Fc map overlaid. The complete chain was
unambiguously traced and contains three disulfide bonds in a cystine-knot motif.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Movie S2. Movie of MD simulation trajectory of TMA bound to Wr-AI1. The TMA protein (white) and
Wr-AI1 (blue) are shown as "cartoon". Interacting residues from TMA (Asp225, His189, Asp185,
Val151, and Asp287) and Wr-AI1 (Lys4, Glu6, Tyr7, Tyr11 and Thr21) are shown as sticks. See also
Table 3. For clarity purpose, hydrogen atoms are not shown.
Table 1. NMR experimental and structural statistics of Wr-AI1
NOE constraints 681
Intra-residue(|i-j|=0) 332
Sequential (|i-j|=1) 193
Medium-range (1<|i-j|<5) 36
Long-range (|i-j|≥5) 120
Dihedral angle restraints 10
Hydrogen bonds 7
PROCHECK-NMR Ramachandran plot (%)
Most favored region 70.9
Additionally allowed region 28.7
Generously allowed region 0.4
Disallowed region 0
Average maximum violations per structure
Distance (Å) 0.02±0.002
Van der waals (Å) 3.7±0.4
Torsion angles (°) 0.25±0.11
CYANA target function value (Å2) 1.35±0.15
Average RMSD to mean structure (Å)
All back bone atoms (1-30) 0.38±0.08
All heavy atoms (1-30) 0.85±0.12
Table 2. Crystallography data collection and refinement statistics of Wr-AI1
Statistics of data collection
Space group
Cell parametersa P212121
a,b,c (Å) a=16.19, b=29.13, c=47.70
α,β,γ (o) α =β=γ=90
Resolution range (Å) 29.1 – 1.25 (1.28-1.25)
Observed reflections 21,115 (2,864)
Unique reflections 6,362 (856)
Completeness (%) 95.4 (90.3)

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Multiplicity 3.3 (3.3)
Rmergeb 0.029 (0.071)
Mean I/σ(I) 23.2 (11.9)
Statistics of refinement
Resolution range (Å) 24.88 – 1.25 (1.28-1.25)
Number of reflections used for refinement 6,017 (410)
Number of reflections for Rfree calculation (5%) 317 (21)
Rfactor (%)#, Rfree (%)
* 15.8, 15.9
Number of non hydrogen atoms 223
Water molecules 41
Mean B-factor (Å2)
Protein whole chain 5.4
Water 32.9
r.m.s. deviations from ideality
Bond lengths (Å) 0.020
Bond angles (°) 1.85
Ramachandran plot (%)
Most favoured regions 87
Additional allowed regions 13
G-factor$ 0.07
aThe numbers in parentheses refer to the last (highest) resolution shell.
bRmerge = ΣhΣi|Ihi-<Ih>|/Σh,i Ihi, where Ihi is the i
th observation of the reflection h, while <Ih> is its mean
intensity. #Rfactor = Σh||Fobs(h)| - |Fcalc(h)|| / Σ |Fobs(h)|
*Rfree was calculated with 5% of reflections excluded from the whole refinement procedure.
$G factor is the overall measure of structure quality from PROCHECK.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
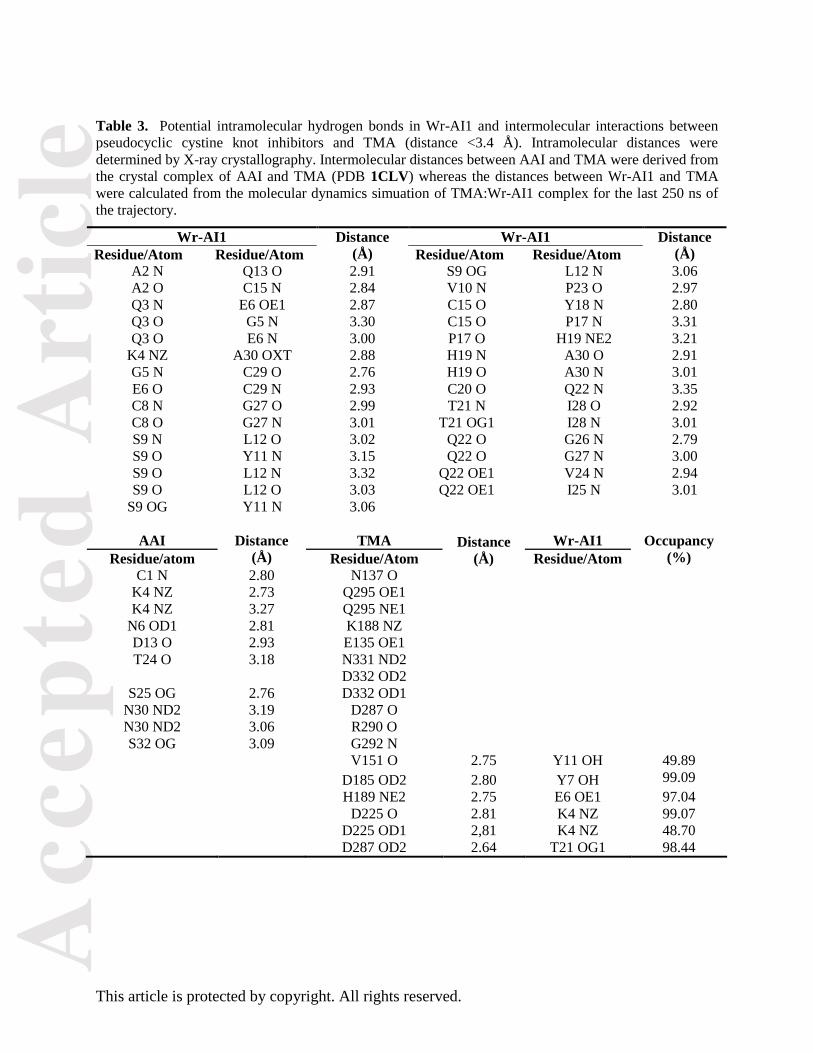
Table 3. Potential intramolecular hydrogen bonds in Wr-AI1 and intermolecular interactions between
pseudocyclic cystine knot inhibitors and TMA (distance <3.4 Å). Intramolecular distances were
determined by X-ray crystallography. Intermolecular distances between AAI and TMA were derived from
the crystal complex of AAI and TMA (PDB 1CLV) whereas the distances between Wr-AI1 and TMA
were calculated from the molecular dynamics simuation of TMA:Wr-AI1 complex for the last 250 ns of
the trajectory.
Wr-AI1 Distance
(Å)
Wr-AI1 Distance
(Å) Residue/Atom Residue/Atom Residue/Atom Residue/Atom
A2 N Q13 O 2.91 S9 OG L12 N 3.06
A2 O C15 N 2.84 V10 N P23 O 2.97
Q3 N E6 OE1 2.87 C15 O Y18 N 2.80
Q3 O G5 N 3.30 C15 O P17 N 3.31
Q3 O E6 N 3.00 P17 O H19 NE2 3.21
K4 NZ A30 OXT 2.88 H19 N A30 O 2.91
G5 N C29 O 2.76 H19 O A30 N 3.01
E6 O C29 N 2.93 C20 O Q22 N 3.35
C8 N G27 O 2.99 T21 N I28 O 2.92
C8 O G27 N 3.01 T21 OG1 I28 N 3.01
S9 N L12 O 3.02 Q22 O G26 N 2.79
S9 O Y11 N 3.15 Q22 O G27 N 3.00
S9 O L12 N 3.32 Q22 OE1 V24 N 2.94
S9 O L12 O 3.03 Q22 OE1 I25 N 3.01
S9 OG Y11 N 3.06
AAI Distance
(Å)
TMA Distance
(Å)
Wr-AI1 Occupancy
(%) Residue/atom Residue/Atom Residue/Atom
C1 N 2.80 N137 O
K4 NZ 2.73 Q295 OE1
K4 NZ 3.27 Q295 NE1
N6 OD1 2.81 K188 NZ
D13 O 2.93 E135 OE1
T24 O 3.18 N331 ND2
D332 OD2
S25 OG 2.76 D332 OD1
N30 ND2 3.19 D287 O
N30 ND2 3.06 R290 O
S32 OG 3.09 G292 N
V151 O 2.75 Y11 OH 49.89
D185 OD2 2.80 Y7 OH 99.09
H189 NE2 2.75 E6 OE1 97.04
D225 O 2.81 K4 NZ 99.07
D225 OD1 2,81 K4 NZ 48.70
D287 OD2 2.64 T21 OG1 98.44
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 1. Tissue- and region-specific expression profiles of wrightides from flower and leaf of W. religiosa
plant. 50% ethanol extract of 1 g of each plant sample was purified using C18 solid phase extraction
column. The eluate with 80% acetonitrile was profiled using MALDI-TOF MS to determine the
occurrence of putative CK peptides in different W. religiosa plant parts from Singapore and Vietnam.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 2. Tandem MALDI-TOF/TOF MS/MS profiles of two tryptic fragments (m/z 2072 - A and 1198 -
B), provide the full wrightide Wr-AI1 sequence. Ile/Leu assignment was determined by genetic sequence
as well as X-ray and NMR structures.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 3. The 3D structure of Wr-AI1. A, Secondary structure illustration against the sequence of Wr-AI1.
The amino-acid sequence of Wr-AI1 is aligned with sequences of: AAI, HT-III(Hainan toxin-III), HT-
IV(Hainan toxin-IV) and GxTX-1E (Guangxiensis toxin-1E) with PDB codes of 1QFD, 2JTB, 1RYV
and 2WH9, respectively. β stands for beta-strand; yellow bridge indicates disulfide bonds and red turn
depicts beta hairpin. B, Solution structure of Wr-AI1 (PDB code: 2MAU). C, Illustration of
intramolecular hydrogen bonds. D, Backbone trace alignment of the crystal structure of Wr-AI1 (blue,
PDB code: 4BFH) with the ten solution structure ensemble (tan). E, Superposition of the solution
structures of Wr-AI1 and AAI. F, Structure alignment of Wr-AI1 with its structural homologs. The figure
was prepared with the program PyMOL.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 4. Ribbon diagram and structural features of Wr-AI1. A, Ribbon presentation of Wr-AI1 crystal
structure. Pro17 and Pro23 adopt the cis and trans conformations, respectively. B, Stereoview of the
electron density (2Fobs-Fcalc) of the backbone twist caused by cis Pro17. The electron density was
contoured at 1.0 σ. C, Schematic hydrogen bonding network of the three beta strands of Wr-AI1. The
residues involved in the three beta strands are shown as blue spheres and relevant residues as grey
spheres. The figure was prepared with the program PyMOL.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 5. Cartoon and surface view of superimposition of AAI and Wr-AI1 in complex with TMA. A, The
complex model of AAI (magenta) and Wr-AI1 (cyan) with TMA (gray) derived from the AAI-TMA
complex (PDB 1CLV). B and C, Zoom-view of the binding region between AAI (B)/Wr-AI1 (C) and
TMA. Residues of AAI/ Wr-AI1 with atoms within 6 Å away from TMA are colored in magenta/cyan,
while residues of TMA with atoms within 6 Å away from AAI / Wr-AI1 are colored in blue/red,
respectively. Residues that are >6 Å away from TMA in both AAI/ Wr-AI1 are colored orange. D, The
superimposition of AAI and Wr-AI1 at the active site of TMA. Three disulfides between CysI-IV, CysII-
V, and CysIII-VI arranged in a cystine knot motif are highlighted in yellow. E and F, Close-ups of AAI
(E) and Wr-AI1 (F) residues at TMA’s active site. The figure was prepared with the program PyMOL.
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 6. Stable hydrogen bond interactions between Wr-AI1 (residues displayed as sticks in cyan) and
TMA (residues shown as green sticks) following molecular dynamics simulation (see text)
Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 7. Stabilities and α-amylase inhibitory activity of wrightides. A, Thermal stability of Wr-AI1 and
Wr-AI2. The minor peak after heat treatment contained mainly Wr-AI1/Wr-AI2 with a small amount of
degraded products as examined by MS profile. B, Chymotrypsin stability of wrightide Wr-AI1. C,
Cacboxypeptidase A stability of Wr-AI1. D, Inhibition of wrightides against Tenebrio molitor α-amylase.
Peptides were pre-incubated with TMA for 20 min at 37C. Hydrolysis was started by adding 1% starch.
The reaction was allowed for 5 min, and stopped by adding a color reagent containing DNS. The IC50
values are 1.9 μM and 2.3 μM for Wr-AI1 and Wr-AI2, respectively. The error bars show SDs.

Acc
epte
d A
rtic
le
This article is protected by copyright. All rights reserved.
Fig. 8. Precursor structure of wrightides Wr-AI. A, Alignment of wrightide precursors with precursors of
other CK peptides including -conotoxin SO-3 and towel gourd trypsin inhibitor TGTI-II. The
endoplasmic reticulum signal peptide was assigned using SignalP 3.0 for -conotoxin SO-3 while this
domain is not recognized by SignalP 3.0 for TGTI-II. B, Secretory protein synthesis pathway for
wrightides.




















