
Direct measurements of interfacial interactions between pectin and κ-casein and implications for...
13
Direct measurements of interfacial interactions between pectin and j-casein and implications for the stabilisation of calcium-free casein micelle mimics A. Cucheval a,c , M.A. Al-Ghobashy a , Y. Hemar a,b , D. Otter c , M.A.K. Williams a,b, * a Institute of Fundamental Sciences, Massey University, Manawatu, New Zealand b The MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand c Fonterra Research Centre, Dairy Farm Road, Private Bag 11 029, Palmerston North, New Zealand article info Article history: Received 11 May 2009 Accepted 22 June 2009 Available online 27 June 2009 Keywords: j-Casein Pectin SPR DWS j-Casein coated beads Interfacial interactions abstract Using Surface Plasmon Resonance (SPR) it has been shown that the fine structure of the anionic polysac- charide pectin strongly influences its interfacial interaction with a j-casein layer coated onto a gold sur- face (via a dextran linker) in the pH range 3.5–6.8, with the highest SPR signal being observed for pectin with the lowest charge density tested (a degree of methylesterification (DM) around 90%). Furthermore, the Brownian motions of j-casein coated polystyrene beads (used to provide calcium- free ‘model casein micelles’) were studied in pectin solutions using Diffusing Wave Spectroscopy (DWS) and microscopy, and were compared with measurements made on naked beads. At every pH value studied (with the exception of 3.5), bridging of the protein-covered probe particles was observed for pec- tins of both DM 28 and DM 78. However, no aggregated complexes were found in these model casein micelle systems when pectin of an unusually high DM was used (90%). It was hypothesised that having a limited number of binding regions of spatially limited extent max- imises the number of chains binding to the protein layer (as found with the SPR measurement), encour- ages the formation of loops and trains, and additionally limits the potential for destabilisation via bridging. Ó 2009 Elsevier Inc. All rights reserved. 1. Introduction While the detailed internal structure of native casein micelles is still a source of controversy to some extent [1], it is well known that j-casein molecules form an entropy-rich brush at their sur- face, ensuring the steric stabilisation of these bio-colloids [2]. The destabilisation of this steric barrier initiates aggregation and depending on the phase volume can cause precipitation or the for- mation of colloidal-networks consisting primarily of aggregated micelles. Pectin, an anionic polysaccharide extracted from plants is commonly used as a ‘stabilizer’ in acid milk drinks where it inhibits this precipitation; and as a ‘thickener’ in other dairy des- serts, where it has been assumed to form a gel in the serum aque- ous phase [3]. Pectin is composed of three pectic polysaccharides [4] homoga- lacturonan (HG), rhamnogalacturonan I (RGI) and rhamnogalactu- ronan II (RGII). HG is a linear polymer of (1-4)-linked a-D- galacturonic acid and its methylesterified counterpart. The ratio of methylesterified galacturonic acid units to the total galacturonic acid content is given by the degree of methylesterification (DM) and determines how the polymer interacts with other molecules. RGI has a backbone consisting of the repeating disaccharide rham- nose–galacturonic acid and carries glycan side chains. RGII has a backbone of (1-4)-linked a-D-galacturonic acid and has many con- served complex sugar side chains. Typical extraction processes modify the in vivo fine structure of pectin and most commercially available samples are routinely found to consist of primarily linear homogalacturonan chains (around 90%). There is good evidence that in acidified milk systems pectin de- rives its functionality by absorbing onto casein micelles via electro- static interactions at pH values less than around 5.3 [5], preventing the flocculation that would otherwise be initiated by the pH-in- duced collapse of the j-casein layer [6]. In such a scenario, the polysaccharide adsorbs onto the casein micelle via its charged re- gions, while the other parts of the molecule protrude into solution as loop and tails [7]. Knowing more about the details of this inter- action might allow pectins with particular DMs and charge distri- butions to be identified as offering maximal functionality. In commercial systems, high methoxyl (HM) pectins have empirically been shown to stabilize acid milk drinks more effi- ciently than those of low DM [8,9]. However, the ability of pectin 0021-9797/$ - see front matter Ó 2009 Elsevier Inc. All rights reserved. doi:10.1016/j.jcis.2009.06.052 Abbreviations: DWS, diffusive wave spectroscopy; SPR, surface plasmon reso- nance; DM, degree of methylesterification. * Corresponding author. Address: Institute of Fundamental Sciences, Massey University, Private Bag 11222, Palmerston North, Manawatu 4442, New Zealand. Fax: +64 3505682. E-mail address: [email protected] (M.A.K. Williams). Journal of Colloid and Interface Science 338 (2009) 450–462 Contents lists available at ScienceDirect Journal of Colloid and Interface Science www.elsevier.com/locate/jcis
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Direct measurements of interfacial interactions between pectin and κ-casein and implications for...

Journal of Colloid and Interface Science 338 (2009) 450–462
Contents lists available at ScienceDirect
Journal of Colloid and Interface Science
www.elsevier .com/locate / jc is
Direct measurements of interfacial interactions between pectin and j-caseinand implications for the stabilisation of calcium-free casein micelle mimics
A. Cucheval a,c, M.A. Al-Ghobashy a, Y. Hemar a,b, D. Otter c, M.A.K. Williams a,b,*
a Institute of Fundamental Sciences, Massey University, Manawatu, New Zealandb The MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealandc Fonterra Research Centre, Dairy Farm Road, Private Bag 11 029, Palmerston North, New Zealand
a r t i c l e i n f o
Article history:Received 11 May 2009Accepted 22 June 2009Available online 27 June 2009
Keywords:j-CaseinPectinSPRDWSj-Casein coated beadsInterfacial interactions
0021-9797/$ - see front matter � 2009 Elsevier Inc. Adoi:10.1016/j.jcis.2009.06.052
Abbreviations: DWS, diffusive wave spectroscopynance; DM, degree of methylesterification.
* Corresponding author. Address: Institute of FuUniversity, Private Bag 11222, Palmerston North, MaFax: +64 3505682.
E-mail address: [email protected] (M.A.K.
a b s t r a c t
Using Surface Plasmon Resonance (SPR) it has been shown that the fine structure of the anionic polysac-charide pectin strongly influences its interfacial interaction with a j-casein layer coated onto a gold sur-face (via a dextran linker) in the pH range 3.5–6.8, with the highest SPR signal being observed for pectinwith the lowest charge density tested (a degree of methylesterification (DM) around 90%).
Furthermore, the Brownian motions of j-casein coated polystyrene beads (used to provide calcium-free ‘model casein micelles’) were studied in pectin solutions using Diffusing Wave Spectroscopy(DWS) and microscopy, and were compared with measurements made on naked beads. At every pH valuestudied (with the exception of 3.5), bridging of the protein-covered probe particles was observed for pec-tins of both DM 28 and DM 78. However, no aggregated complexes were found in these model caseinmicelle systems when pectin of an unusually high DM was used (90%).
It was hypothesised that having a limited number of binding regions of spatially limited extent max-imises the number of chains binding to the protein layer (as found with the SPR measurement), encour-ages the formation of loops and trains, and additionally limits the potential for destabilisation viabridging.
� 2009 Elsevier Inc. All rights reserved.
1. Introduction
While the detailed internal structure of native casein micelles isstill a source of controversy to some extent [1], it is well knownthat j-casein molecules form an entropy-rich brush at their sur-face, ensuring the steric stabilisation of these bio-colloids [2]. Thedestabilisation of this steric barrier initiates aggregation anddepending on the phase volume can cause precipitation or the for-mation of colloidal-networks consisting primarily of aggregatedmicelles. Pectin, an anionic polysaccharide extracted from plantsis commonly used as a ‘stabilizer’ in acid milk drinks where itinhibits this precipitation; and as a ‘thickener’ in other dairy des-serts, where it has been assumed to form a gel in the serum aque-ous phase [3].
Pectin is composed of three pectic polysaccharides [4] homoga-lacturonan (HG), rhamnogalacturonan I (RGI) and rhamnogalactu-ronan II (RGII). HG is a linear polymer of (1-4)-linked a-D-
ll rights reserved.
; SPR, surface plasmon reso-
ndamental Sciences, Masseynawatu 4442, New Zealand.
Williams).
galacturonic acid and its methylesterified counterpart. The ratioof methylesterified galacturonic acid units to the total galacturonicacid content is given by the degree of methylesterification (DM)and determines how the polymer interacts with other molecules.RGI has a backbone consisting of the repeating disaccharide rham-nose–galacturonic acid and carries glycan side chains. RGII has abackbone of (1-4)-linked a-D-galacturonic acid and has many con-served complex sugar side chains. Typical extraction processesmodify the in vivo fine structure of pectin and most commerciallyavailable samples are routinely found to consist of primarily linearhomogalacturonan chains (around 90%).
There is good evidence that in acidified milk systems pectin de-rives its functionality by absorbing onto casein micelles via electro-static interactions at pH values less than around 5.3 [5], preventingthe flocculation that would otherwise be initiated by the pH-in-duced collapse of the j-casein layer [6]. In such a scenario, thepolysaccharide adsorbs onto the casein micelle via its charged re-gions, while the other parts of the molecule protrude into solutionas loop and tails [7]. Knowing more about the details of this inter-action might allow pectins with particular DMs and charge distri-butions to be identified as offering maximal functionality.
In commercial systems, high methoxyl (HM) pectins haveempirically been shown to stabilize acid milk drinks more effi-ciently than those of low DM [8,9]. However, the ability of pectin

Table 1Characteristics of pectin samples used in this study.
DM/% Mw/g mol�1 Origin and treatment
DM 90 90 �31 000 Sigma Aldrich, St. Louis, USADM 78 78 30 000–100 000 Fluka Biochemica, SwitzerlandDM 28 28 30 000–100 000 Homemade by alkali deesterification with
DM 78 Fluka Biochemica, Switzerland asstarting material
A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 451
to stabilize a casein micelle dispersion is clearly strongly depen-dant on its molecular availability, which can be severely compro-mised by its binding with calcium [10]. Indeed, it has beenrecently shown that (i) the serum calcium liberated in milk sys-tems as they are acidified [11] is sufficient to gel low DM pectinsand (ii) that simply the presence of low DM pectin in samples ofmilk at natural pH (6.8) modifies the calcium mineral balance. Asa result susceptible pectin fine structures can then be trapped bycalcium and are thus not able to interact with caseins [10]. In otherwords, the interaction between pectin and the calcium in the ser-um forbids the direct assessment of the effect of pectin fine struc-ture on the direct interaction of pectin with casein micelles. One ofthe pathways available to study the interaction in absence of cal-cium is to use sodium caseinate [3,9] instead of native micelles.Pereyra et al. [9] compared the efficiency of LM and HM pectin instabilizing an acidified sodium caseinate dispersion and concludedthat, at low pH, higher DM pectins were more effective. However,sodium caseinate exists as small self-assembled protein particlesof casein aggregates in equilibrium with free casein molecules[12,13] and is not found in a form akin to the native casein micelleswith a j-casein layer at its surface.
In this work, the direct interfacial interaction of pectin with j-casein has been investigated in the absence of calcium. Firstly, theeffect of the pectin fine structure on the amount of binding onto aj-casein layer was studied by Surface Plasmon Resonance (SPR) asa function of pH. j-Casein was covalently attached to the gold sur-face of an SPR chip by a standard coupling procedure and changesin the resonance angle were monitored while pectin solutions wereflowed over the immobilized j-casein. Three different pectins withdistinct degrees of methylesterification (DM 90, 78, 28) weretested. Secondly, we then investigated the interaction of pectinwith j-casein in a system that more closely mimics the casein mi-celle, but without calcium present. More specifically, j-caseincoated polystyrene particles were used as ‘synthetic casein mi-celles’ which were mixed into pectin solutions at low phase vol-umes. The mean square displacements of the j-casein coatedparticles were obtained with Diffusing Wave Spectroscopy (DWS)and compared to those found with the naked beads. Additionally,the systems were visualised by microscopy and the results ob-tained were interpreted within a coherent framework incorporat-ing the results from the SPR study.
2. Materials and methods
2.1. Materials
2.1.1. Control of surface chemistry2.1.1.1. Covalent immobilization of j-casein onto an SPR sensorchip. j-Casein (Sigma, St. Louis, USA) was covalently immobilizedon the surface of one cell of a CM5 sensor chip by a standard aminecoupling procedure (Pharmacia Biosensor, 1994). Such chips con-tain two cells which are pre-coated by carboxylmethyl dextran.The immobilization reaction involved an amide group of j-casein,which can be from the N-terminal amino acid or from the sidechains of the amino acids lysine, glutamine, aspartame or arginine.The final step of the coupling procedure involved the use of etha-nolamine to block any untreated charged groups on the dextranlinker.
Prior to covalent immobilization, a pH scouting exercise (car-ried out in acetate buffer 10 mM pH 4, 4.5, 5 and 5.5) determinedpH 4 as the optimum pH for the immobilization procedure. Thedextran surface was first activated with N-hydroxysuccinimide(NHS, Biacore AB) and N-ethyl-N0-(dimethylaminopropyl)-carbodi-imide (EDC, Biacore AB) and then j-casein was flowed over thecell. The binding level was 3000 RU. The second cell incorporatedon the chip was used as a control, and was activated with the same
procedure (NHS and EDC) but subsequently blocked with ethanol-amine (Biacore AB). The binding response was calculated by thesubtraction of the signal obtained from the sample cell minus thatfrom blocked control cell, in order to correct the response for non-specific binding. The running buffer used for the immobilizationprocedure was HBS buffer (10 mM HEPES buffer at pH 7.4 contain-ing 150 mM NaCl, 3 mM EDTA, and 0.005% of the nonionic surfac-tant polyoxyethylenesorbitan (P20)) (Biacore AB).
Once immobilized on the sensor chip the state of the bound j-casein was qualitatively compared to that of micellar j-casein byinvestigating the action of the enzyme chymosin (a j-casein cleav-ing enzyme). Chymosin (at a concentration of 84 Units, in HEPESbuffer pH 6.8 10 mM) was flowed, at 25 �C, over the CM5 chip cov-ered with j-casein, twice for 540 s at a 1 ll/min, and changes inthe baseline were recorded. The availability of the immobilizedj-casein to chymosin provided some insight into the potential ef-fect of the covalent immobilization procedure on the state of theprotein itself.
2.1.1.2. Passive absorption of j-casein onto the surface of beads. Poly-styrene beads of diameter 465 nm (2.62% w/v stock solutions) pur-chased from Polyscience Inc. (Warrington, PA) were used withoutmodification or with a coating of j-casein (Sigma Aldrich, Ger-many) produced by passive absorption. More specifically, to createj-casein coated particles, 1.5 mg of j-casein was added to 1 mL ofthe mother beads solution in the range 3.5–5 mg/m2 needed forfull surface coverage [14,15]. The efficiency of the coating wastested by particle size measurements with DWS. The diameter ofthe treated particles increased, by �20 nm consistent with previ-ous observations.
2.1.2. Sample preparation and interaction studyPectins with different fine structures were used in this study,
the degree of methylesterification, molecular weight and originof which are reported in Table 1. The lack of a quoted range inthe molecular weight of the DM 90 sample should not be takento imply it is monodisperse, simply that the only information cur-rently available is a viscosity average molecular weight; it shouldbe noted that pectin samples so highly methylesterified are cur-rently rare.
For the sample studied by SPR, 0.1% w/w pectin solutions wereprepared by stirring overnight at room temperature to insure gooddissolution, in 10 mM acetate buffer, at pH 3.5, 4.5 or 5.3 and addi-tionally in HEPES buffer (Sigma, USA) at pH 6.8. The running bufferfor the SPR analysis was 10 mM HEPES, pH 7.4 (Sigma, USA). Allbuffers were filtered and degassed. The temperature was main-tained at 25 �C. A large range of pectin concentration (0.001–0.5%w/w) was tested at pH 4.5 with a high DM pectin (which accordingto the literature should present the most favourable conditions forthe interaction) in order to determine the most suitable pectin con-centration to investigate the effect of pH and pectin fine structure.A pectin concentration of 0.1% w/w was judged optimal in therange tested and was used to study the interaction of pectin withj-casein using SPR. The experiment was performed with the ana-lyte first being injected (60 s, 10 ll/min), followed by a dissociationphase (30 s) in running buffer and finally a regeneration step

452 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
(120 s) with glycine pH 2.5 (10 mM). Each experiment was re-peated three times, and the mean value recorded (the standarddeviation of which was always less than 5%).
For the sample studied by DWS, 1% or 1.5% w/w mother pectinsolution was prepared by stirring overnight at room temperatureto ensure complete dissolution. The pH values of the mother solu-tions were then adjusted using NaOH (1 and 0.1 M) and HCl (1 and0.1 M). Appropriate amounts of pectin and j-casein coated or bareparticles were subsequently mixed at �700 rpm so that the finalconcentration achieved was 0.8% w/w of polystyrene beads and0.4% w/w pectin. The pH was then checked and the sample storedat 20 �C for 30 min before the measurements were taken to allowthe temperature to equilibrate.
2.2. Methods
2.2.1. SPRThe SPR analyses were done on a Biacore X100 (Biacore AB,
Uppsala, Sweden) with a CM5 sensor chip (Biacore AB, Uppsala,Sweden) which has a gold surface coated with carboxyl methyldextran. The interaction of j-casein with pectin was investigatedby first immobilizing covalently j-casein (the ligand) on the sensorchip, as described in Section 2.1.1.1. A pectin sample (the analyte)was then flowed over the j-casein coated surface in a controlledmanner. Any change in the surface properties resulting from theinteraction was detected in the reflected light intensity by the pho-todetector and expressed in resonance units (RU). The change inthe resonance angle versus time (the so-called Sensorgram) givesinsight into the interaction of the ligand and analyte, and can beseparated into an association and dissociation phase. The surfaceis regenerated after each interaction analysis with an appropriatesolution (glycine, pH 2.5) which does not affect the immobilized li-gand but strips off all the absorbed species.
2.2.2. DWSDiffusing Wave Spectroscopy (DWS) is a light scattering method
which can be applied to turbid samples. Each photon goingthrough the sample encounters multiple scattering events, suchthat the photons path can be considered as a random walk. In thisstudy, the light is scattered by bare (or j-casein coated) beads. Theautocorrelation function of the scattered light owing to the motionof the scatters can be calculated from the temporal intensityfluctuation:
gð1ÞðtÞ �1b
IðtÞIð0ÞihhIi2
� 1
!1=2
ð1Þ
where b is a constant characteristic of the optical set-up, I(t) theintensity of the detected light at the time t. DWS has been carriedout in transmission mode which allows writing the temporal auto-correlation function as follows [16]:
g1ðtÞ
¼
L=l�þ4=3z0=l�þ2=3 sinh z0
l�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞiq� �
þ23
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞicosh z0l�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞiq� �s( )
1þ 8t3s
� �sinh L
l�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞiq� �
þ43
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞicosh Ll�
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffik2
0hDr2ðsÞiq� �s
ð2Þ
where l* is the transport mean path, z0 the penetration depth (con-sidered equal to l* in these experiments), L is the thickness of thesample (4 mm), k0 = 2pn/k the wave vector of the light and hDr2(s)iis the mean square displacement (MSD) of the particle. Hence, whenl* is known, the experimentally determined correlation function canbe turned into a plot of MSD versus lag time, by inverting Eq. (2)
with a zero-crossing routine. l* can be calculated by fitting the auto-correlation for a reference sample of known viscosity (water) withthe viscous Stokes–Einstein substitution [16]. Subsequently l* forfuture samples is obtained by scaling the value obtained for water,based on the change in transmitted intensity when the sample isintroduced, compared to the water experiment. It is known thatfor non-absorbing slabs of thickness L, the transmitted intensity isdirectly proportional to ð5l�=3LÞ=ð1þ 4l�=3LÞ, so that by measuringthe change in transmittance, the change in l� can be calculated.Theexperimental set-up has been fully described elsewhere [17,18].Briefly, a laser light source with a wavelength of 633 nm (35*mWHe Ne Melles Griot laser) diffused through the sample, containedin a plastic cuvette of 10 mm width, 50 mm height and 4 mm pathlength. The transmitted scattered light was collected using a singleoptical fibre (P1-3223-PC-5, Thorlabs Inc., Germany) and was de-tected with a photomultiplier tube module (Hamamatsu HC120-08). The auto-correlation analysis was performed using a Malvern7132 correlator. Tests were run for 3 min to ensure low noise inten-sity autocorrelation functions. The comparison of the motion of thej-casein coated particles and the bare particles motion in pectinsolutions provided information on interactions between biopoly-meric components. Indeed, while the Brownian motion of the parti-cles reflects the rheological properties of the surrounding systemfor both the bare and coated beads; it is also affected for the coatedbeads by the interaction between the j-casein coating and the pec-tic polymers.
2.2.3. MicroscopyPhotomicrographs of the j-casein coated and bare beads in
water or pectin solution were obtained in brightfield using a Leicamicroscope (Leica Microsystems AG, Wetzler, Germany) using a60� water-immersion objective.
3. Results and discussion
3.1. Interaction of j-casein with pectin studied by SPR
3.1.1. State of the immobilized j-casein on the sensor chip surface3.1.1.1. Comparison with the conformation on the casein micellesurface. j-casein was covalently bound to the carboxylmethyl dex-tran coating of the gold surface of the sensor chip as previously de-scribed. The covalent binding involved at least one amide group ofthe j-casein from the N-terminal or an amino acid side chain.Being mindful that covalent binding may not be without repercus-sions on the orientation of the protein on the surface, and thus itsinteraction with pectin, we aimed to qualitatively compare thestate of j-casein immobilized on the SPR chip to the same mole-cule as it is manifest on the surface of a casein micelle. Are the ori-entation and the conformation of the protein similar? Looking atthe amino acid sequence for j-casein, the amino acids with amidegroups in their side chains (glutamine, lysine, aspartame, and argi-nine) are mostly grouped in the first 100 amino acids but there arealso a few in the region, close to the C terminus, which constitutethe ‘‘hairy” portion sticking out from the casein micelle. To havemore insight into the effect of covalent bonding on the state andconformation of j-casein, we studied the ability of chymosin tohydrolyse the covalently immobilized j-casein. Chymosin was flo-wed, in two successive injections; at pH 6.8 over the j-caseincoated and reference cells. The baseline levels were recorded be-fore the first chymosin injection and after the end of each injection.
Fig. 1 shows the resulting sensorgram and the resonance angle(RU) versus time for the signal from the j-casein cell, once the ref-erence cell data have been subtracted. Section A shows the initialbaseline; Section B the binding and subsequent action of the en-zyme, and C the baseline after washing off the enzyme; in Sections

Fig. 1. Effect of chymosin on j-casein immobilized on the SPR chip. Injection of chymosin (84 units) at 1 ll/min during 540 s. Section A shows the initial baseline; section Bthe binding and subsequent action of the enzyme, and C the baseline after washing off the enzyme; in Sections D and E the injection of the enzyme and subsequent washingcycle is repeated.
A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 453
D and E the injection of the enzyme and subsequent washing cycleis repeated. The baseline level can be observed to decrease aftereach chymosin injection as is highlighted in the inset. The enzymeis thus able to interact at least with part of the covalently immobi-lized protein and to hydrolyse it, reducing the density of polymerin the vicinity of the surface and hence its refractive index. The partof the j-casein available for hydrolysis was found to be importantenough that after the injection of chymosin and regeneration of thesurface, the refractive index of the surface had changed. The de-crease of the baseline level was found to be more pronounced fol-lowing the first injection compared with the second. It seems then,that almost all ‘available’ j-casein was hydrolysed during the firstinjection. In conclusion, this accessibility of the bound ligand tochymosin is an indication that conformation and orientation ofthe j-casein on the sensor chip is not significantly different fromthat found on the surface of the casein micelle.
3.1.1.2. Effect of pH on the immobilized j-casein conformation. As theinteraction between j-casein and pectin was studied as a functionof pH, the effect of pH on the immobilized ligand itself was firstinvestigated. As reported in the introduction, in milk j-caseinforms a hairy brush layer on the casein micelle surface. Indeed atpH 6.8, it has been reported that the protein protrudes into solu-tion by around 12 nm [19]; with this size reported to be pH sensi-tive (rationalised by differing charge states expected fromexamining the pKa of the constituent amino acids). Between pH6.7 and 5.8, a slight size decrease in the average diameter of around10 nm has previously been observed by light scattering measure-ments [2,20], while an acidification of the system to below pH 5 in-duces the collapse of the j-casein as charges are removed from thechain and its hydrophobic character dominates [6]. One might ex-pect then changes of a similar type in the covalently immobilizedj-casein with pH, and the extent of these was investigated as aprelude to studying the effects of the addition of pectin.
The effect of pH on the immobilized j-casein conformation wasinvestigated by flowing buffer (10 mM) with different pH values(3.5, 4.5, 5.3 and 6.8) over both the j-casein coated and reference
cells. Changes in the refractive index of the surface close to the sen-sor chip were recorded via monitoring the resonance angle withSPR. Fig. 2 shows illustrative sensorgrams and the response (RU)as the resonance signal recorded for the j-casein coated cell aftersubtraction of the reference cell data, versus pH. A positive re-sponse was recorded for every pH change; from the running bufferat pH 7.4, to each pH in the range tested: 3.5–6.8. The response in-creases as the pH decreases although clearly not linearly: the valuemeasured for pH values of 6.8 and 5.3 are in the same range,whereas further acidification of the j-casein coated surface to pHvalues of 4.5 and 3.5 induced significant increases in the recordedresponse. We thus observed a net effect of pH on the refractive in-dex of the molecular layer covalently linked to the gold surfacethat tentatively might be interpreted as the result of the collapseof the j-casein layer, indeed akin to that which causes micelledestabilisation in milk.
However, it should be considered that the polymer layer in-cludes the carboxymethyl dextran coating on the gold surface ofthe CM5 chip which has been used for the coupling reaction inaddition the j-casein covalently bound by reaction of its aminegroup to the dextran carboxyl group. The effect of pH on the reso-nance angle for proteins covalently bonded onto carboxymethyldextran coated CM5 sensor chips has previously been investigatedfor dihydrofolate reductase [21], myoglobin (Mb) and for cyto-chrome c, concanavalin A, and poly-L-lysine [22]. Sota et al. [21],by comparison of their SPR data to circular dichroism spectra, con-cluded that the changes in the SPR signal corresponded to confor-mational changes of the reductase during acid denaturation. Onthe other hand, Paynter et al. [22] did not observe concurrentchanges in dichroism spectra accompanying SPR manifest changes,and suggested that the SPR data might be explained by electro-static interactions between the immobilized biomolecules andthe carboxylmethyl dextran. Indeed, higher SPR signals were re-corded when the immobilized ligand and the carboxylmethyl dex-tran were of opposite sign. However, in our experiments withimmobilized j-casein the highest SPR signal was obtained at pH3.5 where j-casein and carboxylmethyl dextran are both net neg-

Fig. 2. The change in the j-casein state (response in RU) versus pH.
454 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
atively charged. The simplest explanation would seem to be thatchanges in the SPR signal versus pH obtained for covalently immo-bilized j-casein reflects modifications in the conformation of theligand itself. (It should again be emphasised that the final step ofthe coupling procedure we employed involved the use of ethanol-amine in an attempt to block any untreated charged groups on thedextran linker.) As the pH decreases below pH 5.3, the j-caseinlayer coils closer to the gold surface increasing the density andthe local refractive index. As the pH moves below 4.5, the SPR sig-nal increases significantly as the j-casein layer collapses further.This interpretation is consistent with what is known about thepH sensitivity of j-casein as part of a casein micelle; as the pH islowered, the j-casein brush collapses on the surface of the micelle.Furthermore, this provides further evidence, in addition to the chy-mosin sensitivity, that the behaviour of the immobilised j-caseinlayer used should not be too far removed from that of the bio-assembled casein micelle coating.
3.1.2. Effect of pectin fine structure on the interactionAs the pH dependant conformation of the ligand is clearly re-
flected in the SPR signal, as described above, care must be takento take this into account when interpreting the results of experi-ments carried out to investigate the interaction of pectin and j-casein. It has been clearly shown that in acidified milk systemspectin absorbs onto casein micelles by electrostatic interactions[5,6]. However, the effect of the amount and distribution of nega-tive charges on the polysaccharide backbone on the interactionwith the j-casein layer on the surface of the casein micelle is stillunclear. (Recently such considerations have been addressedregarding the interaction of pectin with b-lactoglobulin [23].)While high methoxyl pectins are generally reported to be moreefficient in stabilizing acid milk drinks [8] when calcium is present(in serum and additionally leaking from casein micelles), calciumsensitive pectins are trapped preventing a direct assessment ofthe interaction with the casein protein itself [10]. Here, the direct
interaction of the main casein on the surface of the micelle, j-case-in, and the polysaccharide, was tested, in absence of calcium.Although it might be argued that this is not as directly relevantfor milk, it does help in the fundamental understanding of theinteraction, particularly in relation to the pectin fine structure.The investigation was carried out using three pectin fine struc-tures: common low (DM 28), and high methoxy pectins (DM 78);and a more unusually high DM sample (DM 90). These were flowedover a j-casein coated surface in turn and changes in the refractiveindex of the surface were recorded versus pH by measuring theresonance angle by SPR as described previously herein.
Fig. 3 shows the SPR signal at a given pH obtained by subtract-ing the response recorded when just the buffer was used (Section3.1.1) from the one recorded with the pectin analytes present. Pec-tin samples dissolved in buffers (0.1% w/w) at different pH values(6.8, 5.3, 4.5, and 3.5) were flowed over the cell at a low flow rate(10 ll/min) for 60 s, as previously described in the experimentalsection, and the SPR signal (taken 10 s before the end of the injec-tion) was taken as the binding response.
At pH 6.8, a small positive SPR response was recorded for everypectin fine structure. This indicates that the presence of the poly-saccharide changes the refractive index of the layer on the goldsurface, which is most simply explained by the interaction of pec-tin with the j-casein immobilized on the sensor chip and by thebinding of some of the pectin molecules. At this pH, the net chargefor both molecules is negative but at around one pH unit higherthan the isoelectric point of j-casein (5.9), around 10% of thecharges on the protein are still positive and pectin seems to be ableto bind j-casein through these positive patches. It should be notedhowever that it has been clearly shown that at this pH, pectin doesnot adsorb onto casein micelles [5]. This divergence at pH 6.8 be-tween the interaction with j-casein coupled onto a gold surfaceor on the outer of a casein micelle could be explained by the lowerpI (4.6) of the micelle, resulting from the other proteins present inthe interior of the assembled biocolloid.

Fig. 3. Effect of pectin structure on pectin-j casein interfacial interaction versuspH: (j) DM 78, (h) DM 90, (N) DM 28. Relative response (RU) obtained bysubtraction of the response obtained at the same pH for j-casein alone (Fig. 2).
a
b
Fig. 4. Temporal autocorrelation function measured by DWS for (a) bare polysty-rene beads and (b) j-coated beads, in 10 mM buffer at different pHs (j) 3.5, (h) 4.6,
A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 455
At pH 6.8, pectins presented a higher binding response in theorder DM 90 > DM 78 > DM 28. Higher SPR values are recordedfor pectins with more methylesterified backbones e.g. those lessnegatively charged. By SPR, the variation of refractive index ofthe layer next to the surface is followed; it is not the strength ofthe interaction per-se but the amount of polysaccharide bindingon the j-casein coated surface (and in detail the effect of this inter-action on the protein conformation) and hence density of materialclose to the surface which is being probed. At pH 6.8, a pectin mol-ecule with a higher DM has less charged binding sites that caninteract with the relatively small number of positive patches onj-casein, thus leaving more available sites for other chains to bindto. In contrast, in the sample of lower DM, each individual mole-cule can bind more patches on the protein, essentially saturatingthe surface at a fewer number of pectin molecules bound. Whenthe pH is decreased to 5.3, the SPR signal for all three pectins de-creases; slightly for the HM pectins and in a more pronouncedmanner for the LM pectin. At this pH, the net charge of the j-caseinis positive, while pectin is still highly negatively charged. Underthese conditions, there are more available binding sites on the pro-tein so that one pectin molecule is more likely to bind to multiplesites, restricting the number of chains that might interact with thej-casein layer. This pH decrease has the greatest effect on thebinding of the low DM pectin, which with a large number of neg-ative blocks on the pectin backbone are most likely to ‘lie down’on the surface, essentially blocking many positives sites on the pro-tein and limiting the number of adsorbed chains.
As the pH is reduced further to 4.5, the binding level becomeseven more dependent on the pectin DM. Higher SPR responsesare registered for pectin with higher DM and the signal for lowmethoxyl pectin is weak. At this pH, the interaction is likely tobe the strongest, as more of 90% of the charges are of opposite signbetween the two biopolymers. More positive sites are available onthe protein to bind pectin than at pH 5.3 and around the same per-centage of negative charges are present on the pectin backbone. Itmakes sense then that the difference between the amount of poly-saccharide chains containing differing amounts of potential bind-ing sites for the same number of binding sites on the protein willbe more pronounced. The weak signal observed with LM pectinlikely results from a strong interaction of the polysaccharide withthe j-casein on multiple sites of its backbone driving each mole-cule to ‘lay flat’ on the j-casein layer with limited formation of
loops and tails; and limiting available sites for further moleculesto bind.
Finally at pH 3.5, the binding level is still influenced by the pec-tin structure. The SPR signal is similar for DM 78 but an importantdecrease is observed for the DM 90 and DM 28. By decreasing thepH by one unit further, it is the amount of binding sites on thepolysaccharide that is primarily affected (as the pKa of the carbox-ylic group is traversed, while the number of charges on the proteinchanges only slightly). At this pH, considerably less than 50% of theunmethylesterified acid galacturonic residues will be charged andpresumably for the DM 90 sample, this only leaves a few percent ofthe galacturonic acid residues charged which may not be in runs ofsufficiently charged patches to take part in binding. For the DM 78sample, the binding response is similar to that at pH 4.5, showingthat this polysaccharide still has enough charged patches to bindsuccessfully. In contrast, a negative signal is observed for the lowmethoxyl pectin (DM 28). This change in the SPR signal whenthe pectin is flowed over the protein layer, despite its negative nat-ure, can only be explained by interaction between the polymer andthe j-casein, as the effect of pH on the protein is eliminated by tak-ing it as a reference. Its decreasing nature indicates that the bio-polymer layer next to the gold surface is less dense when thelow methoxyl pectin is present than it is for the protein on itsown, suggesting that its presence lessens the collapse of the pro-tein at low pHs, as measured in the control experiments describedin Section 3.1.1.
While, to date, the effect of pectin DM on the efficiency of thepolysaccharide to stabilize acidified casein micelle systems has
(d) 5.3, (s) 6.8.

456 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
mostly been studied with high (DM 60–80) and low methoxyl pec-tin, our results indicate that ‘unusually high’ DM pectin should beof particular interest in stabilisation, especially at pH valuesaround 4.5 where the highest pectin binding is observed with thisstructure. The relatively small percentage of charge on such ahighly methylesterified structure implies that for potential bindingsites to the protein to be formed small charged regions suffice. Theeffectiveness of this fine pectin structure provides hints about thesize of such ‘epitopes’ needed on the pectin backbone for the inter-action. Indeed, even if we consider the pKa of the unmethylesteri-fied groups to be approximately that of galacturonic acidmonomers so that more than 90% of the unmethylesterified resi-dues are charged, then for the average degree of polymerisationof the DM 90 sample used herein, each chain would have around13 occurrences of single negatively charged galacturonic acid res-idues, one occurrence of a site with two consecutive charged resi-dues, and a negligible probability of any runs of three consecutivecharges. The effective ‘epitope’ on the pectin backbone is thuslikely to consist of �2 charged residues next to each other.
3.2. Interaction of ‘‘model casein micelles” with pectin
Having elucidated the effect of pectin fine structure on the bind-ing of pectin to a j-casein layer at different pH values in the ab-sence of calcium, the interaction was investigated on anintermediate model system: a ‘synthetic casein micelle’ withoutcalcium. It might be argued that this model is far from a nativecasein micelle; however it is an interesting tool to get betterunderstanding of the role of the interfacial interaction. The Brown-
Fig. 5. Scatterer mean square displacements as a function of lag time for j-casein coated aj-casein coated beads, (h) bare beads.
ian motion of these models ‘synthetic casein micelles’ was re-corded by DWS in pectin solutions, and by comparison with theirmotion in water, the interfacial interaction between the surround-ing polymer matrices and the j-casein coated beads was studied.The state of these model synthetic micelles was also followed bylight microscopy. Specifically, calcium-free model casein micelleswere synthesised by surface modification of polystryrene particles,as described in the experimental section. j-caseins were bound byphysisorption to the bead surface, mainly through their hydropho-bic residues leaving the hydrophilic residues exposed in the solu-tion [19]. The coating by j-casein led to an increase of 20 nm indiameter (measured by DWS) which agrees well with measure-ment reported previously [14,15]. This layer thickness suggeststhat the j-casein must form loops and tails on the bead surface[15]. In similar experiments to those carried out herein with pro-tein layers, the action of chymosin on j-casein coated particleshas been reported previously and was found to be similar to its ac-tion on the casein micelle, highlighting that the conformation andorientation of j-casein in these model systems may not be too dis-similar to when it forms part of the native casein micelle [14,24].Furthermore, it has been recently shown by the comparison ofthe motion of casein micelles with j-casein coated beads in acidmilk systems that such beads can be excellent casein micelle mod-els [25].
3.2.1. j-Casein coated probes versus naked particles in waterThe effect of the j-casein coating on the Brownian motion of
polystyrene beads was first investigated in water. These prelimin-ary experiments serve as controls in the study of the effect of pec-
nd bare polystyrene beads in water, calculated form the data presented in Fig. 4. (j)

a
b
Fig. 6. Temporal autocorrelation functions measured by DWS for (a) bare beads and(b) j-coated beads in 0.4% HM pectin solution, at different pH values (j) 3.5, (h)4.6, (d) 5.3, (s) 6.8.
Fig. 7. Micrographs showing the system j-casein coated polystyrene beads and D
A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 457
tin on the motion of coated and naked particles. The behaviour ofbare and j-casein coated beads (0.8% w/w) in a low concentrationbuffer (10 mM) were analysed versus pH. DWS was used to recordthe evolution of the autocorrelation function of the transmittedlight scattered by the coated and uncoated beads, respectively.Fig. 4 shows the autocorrelation functions, g1(s) as a function ofthe time lag s, as measured by DWS at different pHs. For everypH, they decrease to zero at long times for both the bare and thej-casein coated beads, characteristic of a diffusive motion in aNewtonian fluid. The correlation functions of the bare beads atall pHs between 3.5 and 6.8 (Fig. 4a) are similar which was ex-pected as the size of the scatterers is constant and there is no sig-nificant change in viscosity of water with pH.
However, for the j-casein coated beads (Fig. 4b), the autocorre-lation function was not measurable for every pH. Indeed at pH 4.6,the system with j-casein coated beads in water forms macroscopicprecipitates, and an analysis of this bulk phase separated and het-erogeneous system with DWS is not relevant. The difference ofbehaviour for the coated and bare beads at pH 4.6 could be ex-plained by the fact that the layer of j-casein on the beads doesnot only change the size of the particles but also gives the surfacea more hydrophobic character when compared with the nakedbeads. Subsequently when the pH drops and the j-casein layer col-lapses (as seen in the SPR experiment) aggregation is induced; thesame phenomenology as observed with casein micelles them-selves. For the other pH values, where the polymer layer is still ableto sterically stabilize the particles (Fig. 4b), the autocorrelationfunction was observed to move slightly to higher correlation timewhen compared with the naked beads, corresponding well withthe change in size induced by the coating.
Subsequently, the mean square displacements of the scattererswere obtained from the autocorrelation functions by inversion ofEq. (2), with a zero-crossing routine as previously described inthe experimental section and elsewhere [16]. Fig. 5 shows the cor-responding mean square displacements (MSD) for bare and coatedparticles as a function of time lag s, as measured by DWS at pH: 3.5(A), 4.6 (B), 5.3 (C) and 6.8 (D) derived from the data shown in
M 78 pectin (1% w/w) at different pH values: (a) 3.5, (b) 4.6, (c) 5.3, (d) 6.8.

Fig. 8. Scatterer mean square displacements as a function of lag time for j-caseincoated and bare polystyrene beads in HM pectin (0.4%), calculated form the datapresented in Fig. 6. (j) j-casein coated beads, (h) bare beads.
458 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
Fig. 4. The slope of the MSD versus time lag follows the samepower law dependency with a slope close to 1, characteristic to dif-fusive motion in a viscous medium, as expected. The fact that thej-casein coated particles move slightly less than the bare ones inthe same time at each pH and that the intensities observed for bothtypes of particles were similar, is again evident, consistent with thesize increase of the coated scatterers.
3.2.2. j-Casein coated probes versus naked particles in HM pectinsolutions
The motion of the same bare and coated particles was probedand compared in HM pectin solution in order to investigate theinteraction of pectin with synthetic ‘model casein micelles’ (with-out the complication of calcium). While in the system with barebeads the dynamics of the scatterers reflects the viscoelastic prop-erties of the medium: i.e. the pectin solution (as in conventionalmicrorheology), in systems containing j-casein coated particles,the displacement of the probes can also reflect interactions be-tween the particle surface and the surrounding pectin polymers.
Systems containing 0.4% w/w high methoxypectin (DM 78) and0.8% w/w j-casein coated, or, bare particles were adjusted to dif-ferent pHs between 6.8 and 3.5 and DWS was used to record thefluctuation of the transmitted intensity scattered by the coatedor uncoated beads. Fig. 6 shows the autocorrelation functions,g1(s), as a function of the time lag s, measured by DWS at differentpHs. For the bare beads system (Fig. 6a), the g1(s) values decreaseto zero at long times and are extremely similar for every pH, char-acteristic of Newtonian fluids of similar viscosity. This is expectedas the size of these scatterers does not change with pH, and pH-in-duced changes in the viscosity for high DM pectin solutions arelimited (owing to the low concentration and limited number ofionizable groups on the polymer). No pectin-induced depletionflocculation was observed either in the DWS experiments or bymicroscopy, over long time scales (>12 h).
Interestingly, j-casein coated beads in pectin solution (Fig. 6b)did not show any bulk precipitation at any pH, including 4.6;whereas in water, bulk phase separation had been evident (Section3.2.1). It was however evident from the autocorrelation functionsof the j-casein coated beads in pectin solution that the functionalforms had changed in comparison to the experiments with barebeads, especially at pHs 4.6, 5.3, and 6.8. This change in the shapeof the autocorrelation function could be explained by the localaggregation of the scatterers themselves, which fits well with thehigher scattered intensity observed for the j-casein coated beadsystem compared with the naked one for these three pHs.
This hypothesis was confirmed by microscopy of these systemsin a large range of pectin concentration (Fig. 7) (small aggregates ofj-casein-coated particles can be visualised at all pH values exam-ined with the exception of 3.5). This limited local aggregation is theresult of the presence of pectin in the system: it was not observedat these pH values for the same j-casein coated beads in water,nor, importantly, did it occur for bare beads in pectin solution:both the protein coating and the polysaccharide solution are re-quired. As such, the bridging of the j-casein coated micelles bythe polysaccharide would seem to offer a natural explanation forthe observed phenomena.
The MSDs obtained by inversion of the correlation functionswere analysed. Fig. 8 shows the MSD versus time lag for the barebeads and their j-casein coated counterparts in 0.4% pectin solu-tion for different pH values. At pH 3.5 (Fig. 8a), the MSD for thej-casein coated and bare particles are almost identical, as expectedwhere no aggregates were visualised by microscopy. Although pec-tin and j-casein do carry opposite charges at this pH, the negativecharge on the pectin backbone is low. Even though SPR measure-ments did show (Section 3.1.2) that at this pH, some pectin wasbinding on the j-casein (when immobilized on a gold surface via
a dextran linker rather than a bead surface) it is possible that therarity of charged binding regions on the high DM polymers atlow pH values make bridging less likely and stabilisation a morecommon outcome. At higher pHs (Fig. 8b–d) different slopes at

A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 459
high frequency and subtle but distinct differences in the shapes ofthe MSD are observed, with a change of slope at time lag around0.1 ms, for the j-casein coated scatterers. Once again it is impor-tant to emphasise that this change of slope has not been observed
a
Fig. 9. Scatterer mean square displacements as a function of lag time for j-casein coatedat different pH. (j) j-casein coated beads, (h) bare beads.
in any systems investigated without the presence of pectin, or inpectin solution with bare beads. Such MSD shape changes havepreviously been observed however in acidified skimmed milk sys-tems (10% w/w solid content) with high methoxypectin (0.1% w/
b
and bare polystyrene beads in 0.4% w/w pectin solution a: HM pectin, b: LM pectin,

460 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
w), and was suggested to provide a signature of the bridging of thecasein micelles by pectin [10].
Further, hints of such a sigmoidal shapes have also been foundpreviously in studies of colloidal gels embedded in polymer solu-tions of varying visco-elasticity [26]. We hypothesise then that
Fig. 10. Scatterer mean square displacements as a function of lag time for j-casein coatej-casein coated beads, (h) bare beads. Right hand side: micrographs showing the syste
the change of slope and shape in the MSD reflects the same phe-nomena previously observed with casein micelles: model j-caseincoated micelles are bridged by pectin and the aggregates observedby microscopy are complexes of the synthetic micelles, sterically
d and bare polystyrene beads in 0.4% w/w DM 90 pectin solution at different pH. (j)m j-casein beads.
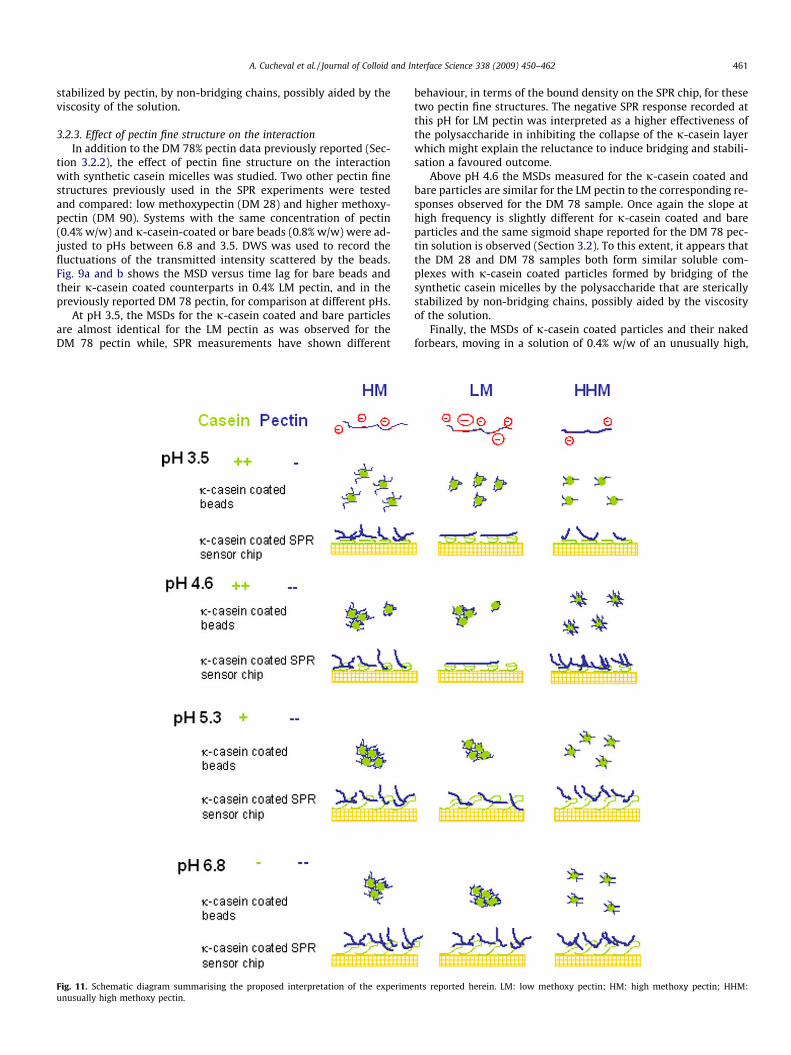
A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462 461
stabilized by pectin, by non-bridging chains, possibly aided by theviscosity of the solution.
3.2.3. Effect of pectin fine structure on the interactionIn addition to the DM 78% pectin data previously reported (Sec-
tion 3.2.2), the effect of pectin fine structure on the interactionwith synthetic casein micelles was studied. Two other pectin finestructures previously used in the SPR experiments were testedand compared: low methoxypectin (DM 28) and higher methoxy-pectin (DM 90). Systems with the same concentration of pectin(0.4% w/w) and j-casein-coated or bare beads (0.8% w/w) were ad-justed to pHs between 6.8 and 3.5. DWS was used to record thefluctuations of the transmitted intensity scattered by the beads.Fig. 9a and b shows the MSD versus time lag for bare beads andtheir j-casein coated counterparts in 0.4% LM pectin, and in thepreviously reported DM 78 pectin, for comparison at different pHs.
At pH 3.5, the MSDs for the j-casein coated and bare particlesare almost identical for the LM pectin as was observed for theDM 78 pectin while, SPR measurements have shown different
Fig. 11. Schematic diagram summarising the proposed interpretation of the experimeunusually high methoxy pectin.
behaviour, in terms of the bound density on the SPR chip, for thesetwo pectin fine structures. The negative SPR response recorded atthis pH for LM pectin was interpreted as a higher effectiveness ofthe polysaccharide in inhibiting the collapse of the j-casein layerwhich might explain the reluctance to induce bridging and stabili-sation a favoured outcome.
Above pH 4.6 the MSDs measured for the j-casein coated andbare particles are similar for the LM pectin to the corresponding re-sponses observed for the DM 78 sample. Once again the slope athigh frequency is slightly different for j-casein coated and bareparticles and the same sigmoid shape reported for the DM 78 pec-tin solution is observed (Section 3.2). To this extent, it appears thatthe DM 28 and DM 78 samples both form similar soluble com-plexes with j-casein coated particles formed by bridging of thesynthetic casein micelles by the polysaccharide that are stericallystabilized by non-bridging chains, possibly aided by the viscosityof the solution.
Finally, the MSDs of j-casein coated particles and their nakedforbears, moving in a solution of 0.4% w/w of an unusually high,
nts reported herein. LM: low methoxy pectin; HM: high methoxy pectin; HHM:

462 A. Cucheval et al. / Journal of Colloid and Interface Science 338 (2009) 450–462
DM 90, pectin sample are shown in Fig. 10 as well as the micro-graphs for the coated particles, once more as a function of pH. Instark contrast to the behaviour observed for the other fine struc-tures, here the MSDs measured for the j-casein coated particlesare almost identical to those of their naked counterparts, for everypH. In solutions of this pectin the j-casein coated particles did notaggregate in the pH range tested, which was confirmed withmicroscopy (Fig. 10).
While pectin with a DM of 90% is only ever going to be weaklynegatively charged regardless of the pH, the SPR measurementscarried out herein conclusively showed that (i) DM 90 does bindonto a j-casein immobilized layer at every pH tested and (ii) owingto its sparing attachment to multiple sites on the protein, the ac-tual amount of bound polymer is higher than for the other finestructures. The effectiveness of DM 90 in stabilizing the micellemimics over the pH range tested, while simultaneously avoidingnot only macroscopic precipitation, but also local bridging-medi-ated aggregation, reinforces the hypothesis that ideally stabilisingfine structures would be those containing the minimal amount ofbinding regions per chain.
4. Conclusions
Utilising model casein micelles (in order to avoid the complica-tions of calcium binding) LM and HM pectin samples have beenshown to produce similar phenomenology when added to the sys-tem at different pHs (6.8–3.5). This includes (i) the stabilisation ofj-casein coated particles at pH 3.5. This is consistent either (forHM samples) with the binding observed in SPR coupled with a lackof multiple binding patches on each chain owing to the low charge,or with the stabilisation of the protein collapse also seen in SPR atlow DM. (ii) The formation of bridged soluble complexes at higherpH values, confirmed by microscopy, and reminiscent in their DWSsignature of the findings of previous work on bridged micelles inacid milk gels.
Additionally it has been shown that unusually HM pectins (DM90) do not exhibit the formation of such synthetic casein micelle-pectin complexes over a considerably broader pH range, insteadstabilising the dispersion to aggregation. This is corroborated bythe SPR study that clearly shows the largest polymer densitybound to the j-casein coated SPR chip for this fine structure; sup-porting the hypothesis that having a minimal number of binding
regions per polymer chain is beneficial; both allowing the proteinbinding sites to be used more efficiently in harvesting the solutionfor more stabilising chains, and ensuring that where multiple sitesexist large loops, trains and tails can be formed. Fig. 11 shows aschematic diagram summarising the proposed interpretation ofthe experiments reported herein.
Acknowledgments
The authors acknowledge financial support from Fonterra andFRST for a PhD scholarship for A Cucheval and would also like tothank Assoc Prof Ashton Partridge for providing access to the Bia-core instrument.
References
[1] D.S. Horne, Curr. Opin. Colloid Interface Sci. 11 (2006) 148.[2] C.G. deKruif, E.B. Zhulina, Colloids Surf. B 117 (1996) 151.[3] L. Matia-Merino, K. Lau, E. Dickinson, Food Hydrocolloids 18 (2004) 271.[4] W.G.T. Williats, P. Knox, J.D. Mikkelsen, Trends Food Sci. Technol. 17 (2006) 97.[5] A. Maroziene, C.G. de Kruif, Food Hydrocolloids 14 (2000) 391.[6] R. Tuinier, C. Rolin, C.G. de Kruif, Biomacromolecules 3 (2002) 632.[7] R.H. Tromp, C.G. De Kruif, M. van Eijk, C. Rolin, Food Hydrocolloids 18 (2004)
565.[8] J.R. Liu, A. Nakamura, M. Corredig, J. Agric. Food Chem. 54 (2006) 6241.[9] R. Pereyra, K.A. Schmidt, L. Wicker, J. Agric. Food chem. 45 (1997) 3448.
[10] A. Cucheval, R.R. Vincent, Y. Hemar, D. Otter, M.A.K. Williams, Colloid Polym.Sci. 287 (2009) 695.
[11] F.M. Harte, C. Montes, M. Adams, J. Dairy Sci. 90 (2007) 2705.[12] K. Creamer, G.P. Berry, J. Dairy Res. 42 (1975) 169.[13] B. Chu, Z. Zhou, G.W. Wu, H.M. Farrell, J. Colloid Interface Sci. 170 (1995) 102.[14] S.G. Anema, Int. Dairy J. 7 (1997) 553.[15] J. Leaver, D.V. Brooksbank, D.S. Horne, J. Colloid Interface Sci. 162 (1994) 463.[16] D.A. Weitz, D.J. Pine, W. Brown, in: W Brown (Ed.), Dynamic Light Scattering,
Oxford University Press, Oxford, 1993, p. 652.[17] Y. Hemar, D.N. Pinder, Biomacromolecules 7 (2006) 674.[18] M.A.K. Williams, R.R. Vincent, D.N. Pinder, Y. Hemar, J. Non-Newtonian Fluid
Mech. 149 (2008) 63.[19] D.G. Dalgleish, C. Halt, J. Colloid Interface Sci. 123 (1988) 80.[20] M. Alexander, D.G. Dalgleish, Colloids Surf. B 38 (2004) 83.[21] H. Sota, Y. Hasegawa, M. Iwakura, Anal. Chem. 70 (1998) 2019.[22] S. Paynter, D.A. Russell, D.A. Anal, Biochem. 309 (2002) 85.[23] B.L.H.M. Sperber, H.A. Schols, M.A. C Stuart, W. Norde, A.G.J. Voragen, Food
Hydrocolloids 23 (2009) 765.[24] J. Leaver, in: A.J. Schwarz, C.I. Contescu (Eds.), Surfaces of Nanoparticles and
Porous Materials, CRC Press, Boca Raton, Fl, 1999.[25] A. Cucheval, R.R. Vincent, Y. Hemar, D. Otter, M.A.K. Williams, Langmuir
(2009), in press.[26] E.E. Pashkovski, J.G. Masters, A. Mehreteab, Langmuir 19 (2003) 3589.




















