
Development of an electrochemical 90Sr– 90Y generator for separation of 90Y suitable for targeted...
9
Development of an electrochemical 90 Sr– 90 Y generator for separation of 90 Y suitable for targeted therapy Rubel Chakravarty a , Usha Pandey a , Remani B. Manolkar a , Ashutosh Dash a , Meera Venkatesh a, ⁎ , M.R. Ambikalmajan Pillai b a Radiopharmaceuticals Division, Bhabha Atomic Research Centre, Mumbai-400085, India b Industrial Applications and Chemistry Section, Division of Physical and Chemical Sciences, IAEA, A 1400 Vienna, Austria Received 31 August 2007; received in revised form 22 October 2007; accepted 24 October 2007 Abstract 90 Y of high specific activity and very high radionuclidic purity (N99.998%) is essential for targeted therapy. Since the current methods used for the preparation of 90 Y from 90 Sr are not adaptable for use in a central/hospital radiopharmacy, a simple 90 Sr– 90 Y generator system based on electrochemical separation technique was developed. Methods: Two-cycle electrolysis procedure was developed for separation of 90 Y from 90 Sr in nitrate solution. The first electrolysis was performed for 90 min in 90 Sr(NO 3 ) 2 feed solution at pH 2–3 at a potential of −2.5V with 100–200 mA current using platinum electrodes. The second electrolysis was performed for 45 min in 3 mM HNO 3 at a potential of −2.5V with 100 mA current. In this step, the cathode from the first electrolysis containing 90 Y was used as anode along with a fresh circular platinum electrode as cathode. The 90 Y deposited on the circular cathode after the second electrolysis was dissolved in acetate buffer to obtain 90 Y acetate, suitable for radiolabeling. Results: N96% recovery of 90 Y could be achieved in each cycle, with an overall decay corrected yield of N90%. The recovered 90 Y had high radionuclidic purity with barely 30.2±15.2 kBq (817±411 nCi) of 90 Sr per 37 GBq (1 Ci) of 90 Y (0.817±0.411 ppm). Consistent and repeated separation could be demonstrated using up to 1.85 GBq (50 mCi) of 90 Sr. The generator was named “Kamadhenu,” the eternally milk- yielding Indian mythological cow. Conclusions: A technique suitable for adaptation at central radiopharmacies for obtaining therapeutic quantities of pure 90 Y has been developed. © 2008 Elsevier Inc. All rights reserved. Keywords: 90 Sr– 90 Y generator; Electrochemical separation; Radionuclidic purity; 90 Sr contamination; Targeted therapy; Kamadhenu 1. Introduction Radiopharmaceuticals based on 90 Y are widely used for radiolabeling various targeting molecules for the treatment of cancer as well as in radiation synoviorthesis [1–10]. The broad interest for use of 90 Y in therapeutic nuclear medicine is due to its suitable nuclear characteristics (T 1/2 64.1 h, β − max 2.28 MeV, no gamma emission) and favorable chemistry. The availability of 90 Y with very low levels of 90 Sr contamination is essential for therapeutic applications, as 90 Sr localizes in the skeleton and, owing to its long half-life (28.9 years), has a very low maximum permissible body burden of 74 kBq (2 μCi) [11]. Particulate-based radiopharmaceuticals used for treatment of liver carcinoma as well as radiosynoviorthesis can be prepared by using low specific activity 90 Y produced by neutron activation of 89 Y in a nuclear reactor. However, 90 Y of very high (near theoretical) specific activity is required for the preparation of labeled antibodies and peptides used for targeted therapy. A radionuclide generator system based on the secular equilibrium of 90 Sr decaying to 90 Y is a convenient method for the preparation of high specific activity 90 Y. Although a variety of separation technologies have been developed [12–19], the feasibility of installation of a generator system for operation in a nuclear medicine department is still an unrealistic proposition. Most of the current separation techniques involve multiple steps such as solvent extraction, ion exchange or extraction chromatogra- phy either alone or in combination. Currently, 90 Y is separated by industrial manufacturers and supplied as a Available online at www.sciencedirect.com Nuclear Medicine and Biology 35 (2008) 245 – 253 www.elsevier.com/locate/nucmedbio ⁎ Corresponding author. Fax: +91 22 25505151. E-mail address: [email protected] (M. Venkatesh). 0969-8051/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.nucmedbio.2007.10.009
Transcript of Development of an electrochemical 90Sr– 90Y generator for separation of 90Y suitable for targeted...

Available online at www.sciencedirect.com
gy 35 (2008) 245–253www.elsevier.com/locate/nucmedbio
Nuclear Medicine and Biolo
Development of an electrochemical 90Sr–90Y generatorfor separation of 90Y suitable for targeted therapy
Rubel Chakravartya, Usha Pandeya, Remani B. Manolkara, Ashutosh Dasha,Meera Venkatesha,⁎, M.R. Ambikalmajan Pillaib
aRadiopharmaceuticals Division, Bhabha Atomic Research Centre, Mumbai-400085, IndiabIndustrial Applications and Chemistry Section, Division of Physical and Chemical Sciences, IAEA, A 1400 Vienna, Austria
Received 31 August 2007; received in revised form 22 October 2007; accepted 24 October 2007
Abstract
90Y of high specific activity and very high radionuclidic purity (N99.998%) is essential for targeted therapy. Since the current methods usedfor the preparation of 90Y from 90Sr are not adaptable for use in a central/hospital radiopharmacy, a simple 90Sr–90Y generator system basedon electrochemical separation technique was developed.Methods: Two-cycle electrolysis procedure was developed for separation of 90Y from 90Sr in nitrate solution. The first electrolysis wasperformed for 90 min in 90Sr(NO3)2 feed solution at pH 2–3 at a potential of −2.5V with 100–200 mA current using platinum electrodes.The second electrolysis was performed for 45 min in 3 mM HNO3 at a potential of −2.5V with 100 mA current. In this step, the cathodefrom the first electrolysis containing 90Y was used as anode along with a fresh circular platinum electrode as cathode. The 90Y depositedon the circular cathode after the second electrolysis was dissolved in acetate buffer to obtain 90Y acetate, suitable for radiolabeling.Results: N96% recovery of 90Y could be achieved in each cycle, with an overall decay corrected yield of N90%. The recovered 90Y had highradionuclidic purity with barely 30.2±15.2 kBq (817±411 nCi) of 90Sr per 37 GBq (1 Ci) of 90Y (0.817±0.411 ppm). Consistent and repeatedseparation could be demonstrated using up to 1.85 GBq (50 mCi) of 90Sr. The generator was named “Kamadhenu,” the eternally milk-yielding Indian mythological cow.Conclusions: A technique suitable for adaptation at central radiopharmacies for obtaining therapeutic quantities of pure 90Y hasbeen developed.© 2008 Elsevier Inc. All rights reserved.
Keywords: 90Sr–90Y generator; Electrochemical separation; Radionuclidic purity; 90Sr contamination; Targeted therapy; Kamadhenu
1. Introduction
Radiopharmaceuticals based on 90Y are widely used forradiolabeling various targeting molecules for the treatment ofcancer as well as in radiation synoviorthesis [1–10]. Thebroad interest for use of 90Y in therapeutic nuclear medicine isdue to its suitable nuclear characteristics (T1/2 64.1 h, β
−max2.28MeV, no gamma emission) and favorable chemistry. Theavailability of 90Y with very low levels of 90Sr contaminationis essential for therapeutic applications, as 90Sr localizes inthe skeleton and, owing to its long half-life (28.9 years), has avery low maximum permissible body burden of 74 kBq (2μCi) [11]. Particulate-based radiopharmaceuticals used for
⁎ Corresponding author. Fax: +91 22 25505151.E-mail address: [email protected] (M. Venkatesh).
0969-8051/$ – see front matter © 2008 Elsevier Inc. All rights reserved.doi:10.1016/j.nucmedbio.2007.10.009
treatment of liver carcinoma as well as radiosynoviorthesiscan be prepared by using low specific activity 90Y producedby neutron activation of 89Y in a nuclear reactor. However,90Yof very high (near theoretical) specific activity is requiredfor the preparation of labeled antibodies and peptides used fortargeted therapy. A radionuclide generator system based onthe secular equilibrium of 90Sr decaying to 90Y is aconvenient method for the preparation of high specificactivity 90Y. Although a variety of separation technologieshave been developed [12–19], the feasibility of installation ofa generator system for operation in a nuclear medicinedepartment is still an unrealistic proposition. Most of thecurrent separation techniques involve multiple steps such assolvent extraction, ion exchange or extraction chromatogra-phy either alone or in combination. Currently, 90Y isseparated by industrial manufacturers and supplied as a

246 R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
radiochemical in inorganic form to the radiopharmacies. Thecost of 90Y is high and the availability is limited due to thetransportation logistics. Hence, the benefits of targetedtherapy using 90Y radiopharmaceuticals are availablecurrently to a small number of patients in few of thedeveloped countries.
Availability of a 90Sr–90Y radionuclide generator, whichwould be similar to the widely used 99mTc generator andcould be set up in a nuclear medicine department or in acentral radiopharmacy, is highly desirable. Althoughattempts have been reported over the last several years,this goal has not yet been accomplished [12–19]. Thedevelopment of an adsorption type generator similar tothe widely used 99Mo/99mTc column generator is notpracticable because of the possible degradation of thecolumn matrix by the intense β−radiations, which wouldreduce the useful life of the generator, which is expected tobe very long since 90Sr has a long half-life. In addition, thepossibility of unacceptable levels of 90Sr breakthroughand the lack of proper analytical methods for estimation of90Sr in the eluted 90Y are issues which need to be resolved.Finally, the production of long-lived wastes that requirecareful handling and storage is encountered with the use ofsuch generators. In addition, the security of 90Sr in nuclearpharmacies and the potential hazards in case of its misusein public domain have to be also kept in perspective. Since90Sr is a highly toxic radionuclide, it is very essential that the90Sr inventory is strictly maintained. Hence, it is suggestedthat 90Sr should be handled in a well-established, controlledlaboratory by trained personnel with strict inventorymaintenance. Facilities where the access to 90Sr is controlledby electronic devices and barriers would ensure limitedaccess only to the authorized personnel. Although all general“central nuclear pharmacy” facilities may not be appro-priately equipped for such work, some of the big facilitiesthat function under high security complying with all rulesand regulations would be able to handle this generator,taking care of all concerns regarding the safe use of 90Sr. Ourgroup had earlier developed a 90Sr–90Y generator systembased on a chelate-loaded, supported liquid membraneand had scaled up this system to 7.4 GBq (200 mCi) sothat it can be operated in a central radiopharmacy. Thissystem is currently under evaluation at the Bhabha AtomicResearch Centre [20–22].
Reischl et al. [23] demonstrated the separation of pure 86Y,the positron emission tomography (PET) radionuclide, fromirradiated 86Sr by a simple electrochemical method. Yoo et al.[24] demonstrated the use of this electrochemical separationtechnique for the preparation of 86Y for patient specificdosimetry studies prior to the administration of 90Y-basedradiopharmaceuticals. In both cases, the investigators wereable to isolate pure 86Y in usable quantities [N3.7 GBq (N100mCi)] frommilligramquantities (50–150mg) of enriched 86Sr.This method appeared simple, with minimum processingsteps, low waste generation and ability to recover the unused86Sr enriched target, and above all, the radionuclidic purity of
the resultant 86Y was acceptable for clinical work. This systemdeveloped is amenable for automation and can be operated in anuclear medicine department based PET radiochemistrylaboratory. Our strategy was to extend the use of theelectrochemical separation system for the development of a90Sr–90Y generator system. We report here the successfuldemonstration of the electrochemical method for separation of90Y from 90Sr with acceptable radionuclidic purity.
2. Materials
90Sr in equilibriumwith 90Y in 2MHNO3was available inthe Radiopharmaceuticals Division of the Bhabha AtomicResearch Centre. 85+89Sr nitrate product is regularly preparedfor commercial supply by ourDivision for use as a radiotracer.Nitric acid, ammonium hydroxide, acetone, ethyl alcohol andother reagents and chemicals were analytical grade procuredfrom SD Fine Chemicals, Mumbai, or BDH, India. Platinummetal plates of high purity (provided with material testingcertificate) were procured from M/s Hindustan Platinum,Mumbai, India. Paper chromatography strips were purchasedfrom M/s Whatman, Kent, UK.
Gamma activity of 85+89Sr was assayed using a NaI (Tl)scintillation counter (500–700 keV). 90Yand 90Sr activity whenpresent in MBq (mCi) levels were measured in an ionizationchamber. A NaI (Tl) scintillation counter was used in mostcounting experiments to measure the Bremmstrahlung radia-tions (50–500 keV) of 90Yand 90Sr when present in kBq (μCi)levels. A liquid scintillation counter (Model: Tricarb 2100TR,Packard Instrument, Minnesota, USA) was used for the finalmeasurement of Bq levels of 90Sr activity in 90Y. An HPGemultichannel analyzer, coaxial photon detector system(ORTEC, Oakridge, TN, USA) with a 0.5 keV resolution and1.8 keV to 2MeVrangewas used for identification of 85+89Sr, inthe presence of 90Y.
A potentiostat unit with 100 V compliance, a maximumcurrent of 2 Awith 1.2-nA current resolution, N1013-Ω inputimpedance,b5-pF capacitance and 10 μHz to 1 MHz built-inanalyzer for impedance was used for electrochemical studies.The apparatus for the electrochemical studies was preparedbased on the reports of Reischl et al. [23] and Yoo et al. [24].
3. Experimental
3.1. Electrochemical setup
The electrochemical separation process involved twoelectrolysis cycles — the first cycle for separation and thesecond cycle for purification of 90Y. A schematic diagram ofthe electrochemical cell is shown in Fig. 1. For initialelectrolysis, the anode and cathode used were of high purityplatinum plates and a saturated calomel electrode (SCE) wasused as the reference electrode. The electrochemical cellconsisted of an open end quartz cylinder of 50 ml capacityfitted with an acrylic cap. The electrodes were fabricated byspot welding platinum plates of size 75 (l)×10 (b)×0.5 mm

Fig. 1. Schematic diagram of the electrolysis cell used for 90Sr–90Y generator.
247R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
(thickness) to a platinum rod. The electrodes were fitted5 mm apart on the acrylic cap, which, along with theelectrodes, fitted on the mouth of the quartz cylinder.The acrylic cap was used to vertically raise and lower theelectrodes. The platinum electrodes were adjusted parallel toeach other and then connected to the power supply usingsmall screws which were embedded into the acrylic cap andtouched the rod. A provision for passing gas through anacrylic tube, which dipped into the electrolysis solution, wasgiven. A small outlet (∼2.5 mm) was provided in the acryliccap for venting the gases.
3.2. Separation of 90Y by electrolysis
Initially, to optimize the voltage to be applied for selectivedeposition of 90Y, the effect of applied potential on depositionwas studied by measuring the percentage deposition of 90Yas a function of voltage when electrochemical deposition wasperformed at pH 2.5–3.0 at room temperature. The pH of theelectrolyte is critical for successful electrochemical deposi-tion. Reischl et al. [23] had reported near-complete deposition(97±2%) of 90Yat pH 2.5–3.0, and we had adapted the samefor our experiments. However, in order to check if there arepH changes during electrolysis and also to ensure that thispH range is the most optimum, we carried out one set of
experiments to estimate the 90Y deposition as a function ofpH. Further experiments were carried out at the optimalpotential and pH 2.5–3.0.
3.2.1. Step 1: initial electrolysis90Sr was obtained as 90Sr(NO3)2 in 2 M HNO3. Of this
solution, 1.85 GBq (50 mCi) was placed in a 50 ml beakerand 37 MBq (1 mCi) of 85+89Sr as Sr(NO3)2 was added astracer to track the deposition of Sr in the electrode. The90Sr–90Y equilibrium solution was evaporated to drynessand dissolved in 30 ml of 0.001 MHNO3, which resulted in aclear solution. This 90Sr solution was then transferred to thequartz electrochemical cell. The pH was adjusted to 2.5–3.0,if necessary, by dropwise addition of 3% ammoniumhydroxide. The electrolysis cell was then covered with theacrylic cap together with the two platinum electrodes. Theelectrodes were adjusted to be fully immersed in solutionfacing each other. Argon gas was gently bubbled through thesolution for 10–15 min prior to applying current and theelectrolysis was performed potentiostatically at −2.5 V withrespect to SCE for 90 min.
3.2.2. Step 2: second electrolysis (purification step)At the end of initial electrolysis, the acrylic cap along with
the electrodes was removed from the cell while maintaining

248 R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
the current. After switching off the current, the cathodeplate was removed from the acrylic cell and washed with10 ml of acetone in order to remove any free 90Sr adsorbed onthe surface of the electrode. After washing, the electrode wasfitted to another acrylic cap into which a cylindrical platinumelectrode of dimension 2.5 (ϕ)×75(l)×0.5 mm (thickness)was attached. A fresh quartz cell was filled with 30 ml of0.003M nitric acid, the pH of which was carefully adjusted to2.5–3. The acrylic cap containing the electrodes was fitted tothe quartz cell, and Argon gas was bubbled through thesolution for 10–15 min. The cylindrical electrode wasconnected as the cathode, and the original platinum platecontaining 90Y was connected as the anode. Electrolysis wasperformed for 45 min at a constant potential of −2.5 V and100 mA current, while Argon gas was continuously bubbledthrough the solution. During this process, 90Y was leachedfrom the platinum plate and was deposited onto thecylindrical platinum electrode. At the end of electrolysis,the acrylic cap was removed from the solution whilemaintaining the voltage. The cylindrical platinum electrodewith the deposited 90Y was washed with 10 ml of acetone,followed by leaching of 90Y with 500 μl of acetate buffer atpH 4.75 into a separate glass tube.
After use, the electrodes were washed with 3 M HNO3
and made ready for subsequent experiments. The feedsolution in the first electrolytic cell was also preservedsince it retains most of the 90Sr activity loaded in the cell.This solution was then allowed to stand for growth of90Y and was further used for the next set of electrolysis afteradjusting the pH to 2.5–3.0 for obtaining 90Y. The activity ofthe electrolyte in the second cell was monitored, since itwas expected to contain traces of 90Sr and 90Y. It was thencarefully transferred into a waste container. The acetonesolutions obtained from washing the electrodes werealso pooled and added to this solution, which was theonly radioactive waste generated in this process. Over aperiod of time, the above experiments were repeated severaltimes with 1.85 GBq (50 mCi) of 90Sr, using the same feedsolution in order to ascertain the consistently acceptableperformance of the generator. The pH of the solution waschecked and adjusted, if needed, prior to each run.
3.3. Simulated study for recovery of 90Y from Sr carrieradded solution, equivalent to ∼1 Ci of 90Sr
The effects of macroscopic amounts of strontium on theelectrochemical separation of 90Y was investigated byconducting the experiments by using a Sr–Y mixtureprepared with inactive Sr and Y carrier and spiking it withan equilibrium mixture of 90Sr–90Y containing 3.7 MBq(100 μCi) of 90Sr as well as 3.7 MBq (100 μCi) of 85+89Sr.The simulated solution was prepared by dissolving 22.5 mgof anhydrous strontium carbonate and 2.32 μg of yttriumoxide in 2 M HNO3. Separation of 90Y by electrolysis wasas per the method outlined above. The activity content ofthe recovered 90Y was measured to see the electrochemicaldeposition yield.
3.4. Quality control of 90Y
A number of methods have been described in literaturefor determination of 90Sr contamination in the 90Y elutedfrom generators. For instance, a classical method availablein literature describes the separation of 90Sr and 90Y underalkaline conditions on a filter paper [25]. However, theutility of this method for quantifying very low amounts of90Sr in 90Y is not yet proven and needs to be fullyvalidated before it can be used. Some authors haverecently described a nondestructive assay method fordetermination of 90Y and 90Sr [26]. This method is usedto quantitate 90Y based on counting of a very smallfraction (34×10−6) of positron branching taking place in90Y using an HPGe detector for several hours. However,this method would be impractical for regular use in aproduction laboratory or a radiopharmacy, wherein it isessential that the radiopharmaceutical is prepared anddispatched in a span of a few hours. These authors havealso mentioned that this procedure requires sophisticatedequipment which would be difficult to find in aradiopharmacy setup. In our case, we have estimated thepurity of 90Y by measurement of the half-life of 90Y aswell as estimation of 90Sr levels. 90Sr levels wereestimated either by measuring 85+89Sr activity used astracers, using HPGe-based gamma ray spectroscopy andpaper electrophoresis or by counting the 90Sr present inthe sample after allowing the 90Y to decay for a very longperiod (∼11.5 half lives of 90Y). Further, the novel, user-friendly extraction paper chromatography (EPC) techniquethat has been recently developed in our laboratory forquality control of 90Y obtained from 90Sr–90Y generatorsystems [27] was also used to measure 90Sr in the sample.The technical details of this technique are brieflydescribed below.
3.5. Extraction paper chromatography
The procedure is based on the selective retention of 90Yby 2-ethyl hexyl-2-ethyl hexyl phosphonic acid, the chelateimpregnated at the point of spotting of the paper chromato-graphy strip (Whatman 12×1 cm). A 5 μl acetate test solutioncontaining 0.185 MBq (5 μCi) of 90Y was applied on thepaper which was developed in 0.9% saline. The paper stripwas dried, cut into 1 cm segments, placed in scintillationvials which contained 10 ml of cocktail and counted byliquid scintillation. The 90Sr activity present at the solventfront was then compared to the total applied activity to obtainthe radionuclidic impurity levels.
Estimation of 90Sr amounts in the 90Y separated was alsomade by paper electrophoresis as reported earlier [21]. Thiswas performed on a 30 cm long Whatman No. 1 chroma-tography paper using 0.03 M NaCl containing 0.15 g/L ofsodium citrate as electrolyte. The 90Y solution was applied atthe centre of the paper, and a potential of 500 V was appliedacross the paper for 2 h. The paper was dried, cut into 1 cmpieces and counted in NaI (Tl) scintillation counter.
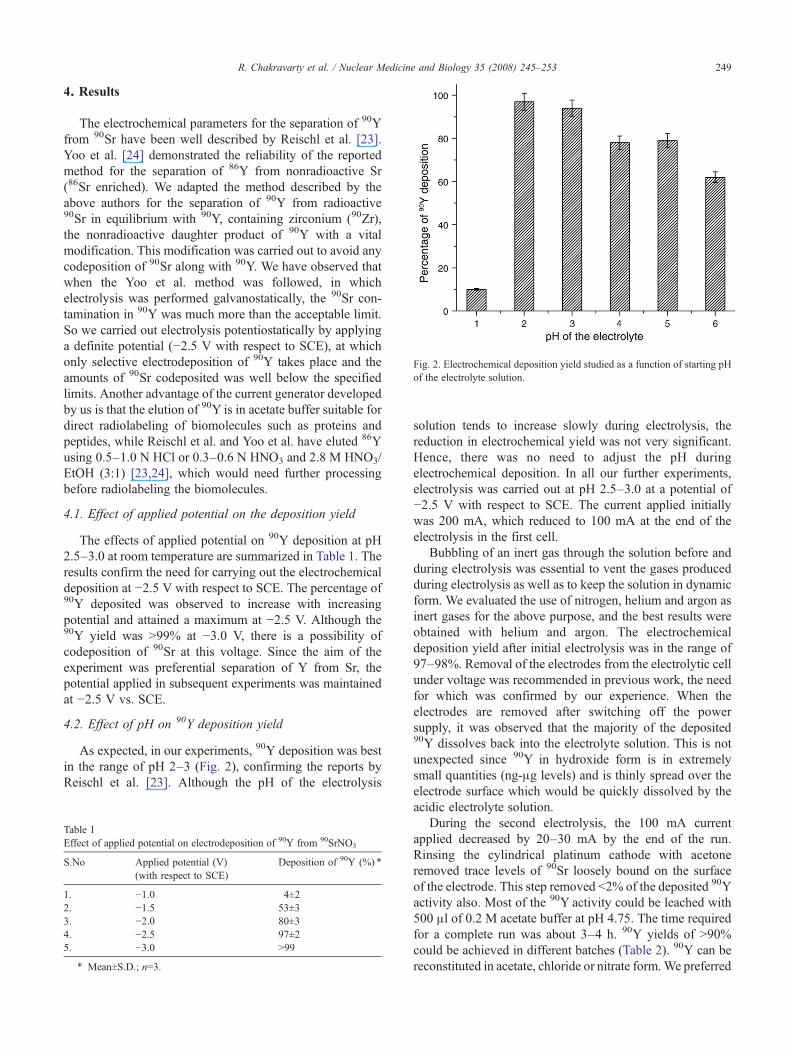
ig. 2. Electrochemical deposition yield studied as a function of starting pHf the electrolyte solution.
249R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
4. Results
The electrochemical parameters for the separation of 90Yfrom 90Sr have been well described by Reischl et al. [23].Yoo et al. [24] demonstrated the reliability of the reportedmethod for the separation of 86Y from nonradioactive Sr(86Sr enriched). We adapted the method described by theabove authors for the separation of 90Y from radioactive90Sr in equilibrium with 90Y, containing zirconium (90Zr),the nonradioactive daughter product of 90Y with a vitalmodification. This modification was carried out to avoid anycodeposition of 90Sr along with 90Y. We have observed thatwhen the Yoo et al. method was followed, in whichelectrolysis was performed galvanostatically, the 90Sr con-tamination in 90Y was much more than the acceptable limit.So we carried out electrolysis potentiostatically by applyinga definite potential (−2.5 V with respect to SCE), at whichonly selective electrodeposition of 90Y takes place and theamounts of 90Sr codeposited was well below the specifiedlimits. Another advantage of the current generator developedby us is that the elution of 90Y is in acetate buffer suitable fordirect radiolabeling of biomolecules such as proteins andpeptides, while Reischl et al. and Yoo et al. have eluted 86Yusing 0.5–1.0 N HCl or 0.3–0.6 N HNO3 and 2.8 M HNO3/EtOH (3:1) [23,24], which would need further processingbefore radiolabeling the biomolecules.
4.1. Effect of applied potential on the deposition yield
The effects of applied potential on 90Y deposition at pH2.5–3.0 at room temperature are summarized in Table 1. Theresults confirm the need for carrying out the electrochemicaldeposition at −2.5 V with respect to SCE. The percentage of90Y deposited was observed to increase with increasingpotential and attained a maximum at −2.5 V. Although the90Y yield was N99% at −3.0 V, there is a possibility ofcodeposition of 90Sr at this voltage. Since the aim of theexperiment was preferential separation of Y from Sr, thepotential applied in subsequent experiments was maintainedat −2.5 V vs. SCE.
4.2. Effect of pH on 90Y deposition yield
As expected, in our experiments, 90Y deposition was bestin the range of pH 2–3 (Fig. 2), confirming the reports byReischl et al. [23]. Although the pH of the electrolysis
Table 1Effect of applied potential on electrodeposition of 90Y from 90SrNO3
S.No Applied potential (V)(with respect to SCE)
Deposition of 90Y (%) ⁎
1. −1.0 4±22. −1.5 53±33. −2.0 80±34. −2.5 97±25. −3.0 N99
⁎ Mean±S.D.; n=3.
Fo
solution tends to increase slowly during electrolysis, thereduction in electrochemical yield was not very significant.Hence, there was no need to adjust the pH duringelectrochemical deposition. In all our further experiments,electrolysis was carried out at pH 2.5–3.0 at a potential of−2.5 V with respect to SCE. The current applied initiallywas 200 mA, which reduced to 100 mA at the end of theelectrolysis in the first cell.
Bubbling of an inert gas through the solution before andduring electrolysis was essential to vent the gases producedduring electrolysis as well as to keep the solution in dynamicform. We evaluated the use of nitrogen, helium and argon asinert gases for the above purpose, and the best results wereobtained with helium and argon. The electrochemicaldeposition yield after initial electrolysis was in the range of97–98%. Removal of the electrodes from the electrolytic cellunder voltage was recommended in previous work, the needfor which was confirmed by our experience. When theelectrodes are removed after switching off the powersupply, it was observed that the majority of the deposited90Y dissolves back into the electrolyte solution. This is notunexpected since 90Y in hydroxide form is in extremelysmall quantities (ng-μg levels) and is thinly spread over theelectrode surface which would be quickly dissolved by theacidic electrolyte solution.
During the second electrolysis, the 100 mA currentapplied decreased by 20–30 mA by the end of the run.Rinsing the cylindrical platinum cathode with acetoneremoved trace levels of 90Sr loosely bound on the surfaceof the electrode. This step removed b2% of the deposited 90Yactivity also. Most of the 90Y activity could be leached with500 μl of 0.2 M acetate buffer at pH 4.75. The time requiredfor a complete run was about 3–4 h. 90Y yields of N90%could be achieved in different batches (Table 2). 90Y can bereconstituted in acetate, chloride or nitrate form.We preferred

Table 2Electrolytic yield of 90Y, with 1.85 GBq (50 mCi) of 90Sr(NO3)2 as feedsolution
Batchno.
90Sr inelectrolyte[GBq(mCi)] a
90Ygrowthperiod(days)
Expected 90Yactivity [GBq(mCi)]
90Yrecovered[GBq(mCi)]
% 90Yrecovered
1 1.85 (50) 15 1.810 (48.93) 1.683 (45.5) 93.02 1.848 (49.95) 09 1.668 (45.09) 1.572 (42.5) 94.33 1.847 (49.92) 33 1.842 (49.80) 1.687 (45.6) 91.64 1.843 (49.81) 16 1.812 (48.97) 1.691 (45.7) 93.35 1.841 (49.76) 13 1.776 (48.01) 1.687 (45.6) 95.06 1.840 (49.72) 20 1.827 (49.37) 1.706 (46.1) 93.4
a Sr activity was calculated at each time point by correcting for decayover the span of time.
Fig. 4. Paper electrophoresis pattern of the recovered 90Y.
250 R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
to use 0.2 M acetate buffer at pH 4.75, as the solution can bedirectly used for radiolabeling studies. By appropriatelyadjusting the volume of the solution used for finaldissolution, it was possible to obtain 90Y at very highradioactivity concentrations. Further, the total liquid wastegenerated in one typical operation contained only 27 μCi(∼1 MBq) of 90Sr. Table 2 summarizes the results of thestudies to evaluate the performance of the generator overa length of time, and it was seen that consistent levels of90Y (93–95%) could be recovered from the single 90Sr feedrepeatedly. The recovery of 90Y when the feed was simulatedto represent 37 GBq (1 Ci) of 90Sr was also found to be asgood as at lower strengths of 90Sr. On analysis of the 90Yproduct, no peak corresponding to 85+89Sr was observed inthe HPGe detector confirming the purity of the product.Yields of 97–98% of 90Y deposition were observed after theinitial electrolysis step as well as the second electrolysis step.The higher yield after the second step observed in this case isperhaps due to the larger amounts of Y deposited on theelectrode, wherein the loss during the acetone washing step
Fig. 3. Radioactive decay pattern of 90Y prepared by the electrochemicalseparation method studied for ~11 half lives.
is expected to be a smaller percentage of the totalY deposited.
4.3. Sr breakthrough studies
90Sr breakthrough was studied by several means,including following the half-life of the 90Y using an LiquidSuntillation Counter. The decay pattern confirmed with the64.1 h half-life of 90Y. The absence of any deviation at thelower end of the straight line decay curve confirmed thatthe 90Y fraction is pure and contains negligible quantitiesof 90Sr. The decay was followed for nearly 700 h (∼11.5half-lives of 90Y) (Fig. 3).
In the initial experiments carried with 90Sr–90Y spikedwith 85+89Sr, the 90Y isolated was checked in an HPGedetector coupled to a 4K analyzer to evaluate the photonpeaks due to the gamma rays corresponding to 85+89Srgamma activity. The spectrum did not show the presence of
Fig. 5. Extraction paper chromatography pattern of recovered 90Y.

251R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
such peaks, thereby indirectly confirming the absence of 90Srin the recovered 90Y. In the paper electrophoresis, 90Sr and90Y were expected to move towards the cathode and anode,respectively [21]. While 90Y moved towards the anode asexpected, our studies did not show any detectable quantitiesof 90Sr in the recovered 90Y (Fig. 4).
4.4. Extraction paper chromatography
EPC pattern of the 90Y solution isolated by this novelmethod is given in Fig. 5. Estimation of the amount of90Sr by this method gave the value as ∼30.2±15.2 kBq(817±411 nCi) of 90Sr per 37 GBq (1Ci) of 90Y(0.817±0.411 ppm) for different batches. The 90Sr levelsobserved is very low and within acceptable limits.
5. Discussion
Targeted therapy using β−particle emitting radionuclidesis expected to increase substantially in the coming years. Thesale of therapeutic radiopharmaceuticals in the United Statesis projected to be about US $1.7 billion per annum by 2012[28]. Targeted therapy requires the availability of highspecific activity therapeutic radionuclides, and the choice islimited. Among the therapeutic radionuclides, generator-produced 188Re and 90Y offer the highest possible specificactivity [29]. However, the availability of 188W, the parentradionuclide of 188Re is limited because it can be producedonly in 2–3 of the available high flux research reactors in thewhole world [30]. There is an unlimited potential availabilityfor 90Y, as 90Sr is one of the major fission products andthe annual world production of 90Sr in the nuclear reactorsamounts to several hundred megacuries. Currently, spentreactor fuel is reprocessed in 5–6 countries, and thesecountries have the capability to develop the process chemistryrequired for large-scale isolation of 90Sr from fission productwaste. There is an industrial level facility for recovery of 90Sr(and other fission products) at Mayak, Russia [30]. A facilityfor the recovery of 55500 GBq (1500 Ci) of pure strontiumwas set up at the Pacific Northwest National Laboratory,Washington, DC, USA, and the process chemistry used in thatplant is documented [31]. At the Bhabha Atomic ResearchCentre, separation of 90Sr from the high level PUREX wasteusable for 90Sr–90Y generator has been developed [32]. Thisprocess could be scaled up to industrial level production.
90Sr–90Y is one of the typical examples of a secularequilibrium with a very long-lived parent (T1/2 28.8 y) anda short-lived daughter (T1/2 64.1 h). By adapting suitableseparation techniques, a few thousand Curies of usablehigh purity 90Y can be isolated from a 37 GBq (1 Ci)stock of 90Sr. A generator of 1 Ci 90Sr capacity can yield16.65–18.5 GBq (450–500 mCi) of 90Y twice a week.Supplementing the activity by adding about 10% (3.7GBq or 100 mCi) of 90Sr once in every 4–5 years will beadequate to keep the 90Y supply constant. Despite thepossibility of the abundant availability of 90Y, currently,
its cost is very high (US $15–20 per mCi of 90Y ascompared to b$1 per mCi of 131I). The decay loss duringtransportation and custom clearance at the port ofdestination further escalates the real cost. A generatorsystem adaptable in a central/hospital-based radiophar-macy will substantially reduce the cost and increase theavailability of 90Y-based radiopharmaceuticals for cancertherapy. However, controlled access, safe-keeping andstrict inventory maintenance of 90Sr have to be ensured inorder to prevent its misuse in public domain. A reliablegenerator system yielding 90Y in large quantities and with90Sr below acceptable limits is an urgent need felt by themedical community. The International Atomic EnergyAgency has hence addressed the above by initiating aCoordinated Research Project (CRP) on “Development ofGenerator Technologies for Therapeutic Radionuclides,” inwhich the Bhabha Atomic Research Centre is a partici-pant. Our efforts under the scope of the CRP are todevelop a workable generator system that can be operatedin a central/hospital radiopharmacy and to develop areliable quality control technique to estimate the radio-nuclidic purity of 90Y.
Considering the commercial importance of 90Y, a numberof internationally valid patents have been issued [33–36]. Areview of the patents reveal that the technologies are notmatured enough to be used on a regular basis. For example,Bray and Wester [33] used “purified” hydroxy ethylidenediphosphoric acid to remove the 90Y into the organic layer,followed by several steps of scrubbing, stripping, andpurification by ion exchange chromatography. This methodis further modified using 90Sr-specific resins, and a newpatent has been issued in 2006 [34]. The method iscumbersome, requiring the manipulation of large quantitiesof 90Sr activity in small volumes in several steps. Themethods described in the above patents also use a largeamount of reagents such as acids, ion exchangers, etc.,thereby giving the possibility of introduction of metallicimpurities. The generation of radioactive waste is also veryhigh in this technique. In the patent by Horwitz and Hines[35], precipitation of 90Sr and separation of the crystalline90SrNO3 by centrifugation followed by filtering is done toremove the bulk quantity of 90Sr. This is followed bypurification of 90Y by ion-specific resin, but this proceduremay not be suitable for the preparation of therapeuticallyuseful 90Y. The process also results in the production of highactive 90Sr waste, both solid and liquid, as well as the loss of90Sr. The patent by Betenekov et al. [36] describes a methodfor purification of 90Sr followed by extraction of 90Y by ionexchange resin. This method based on ion exchange usingthermoxide type of adsorbents could possibly be used as anindustrial scale process but is not expected to be superior tothe other reported methods.
The use of the electrochemical system in a cyclotron-based radiopharmacy for production of 86Y by Reischl et al.[23] and Yoo et al. [24] suggested to us that a similar system,if developed for 90Y separation from 90Sr, can also be

252 R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
operated in a central/hospital-based radiopharmacy, withadditional facilities such as a dedicated room containinga glove box with shielding, radiation detectors, liquidscintillation counter and radiation monitors. The electro-chemical method is not new but was reported more than a halfcentury earlier by Lange et al. [37] and by Hamaguchi et al[38]. However, for unknown reasons, this has not been usedfor separation of 90Y from 90Sr. The early description of thismethod by the renowned radiochemist, Strassman [37],makes it a nonpatentable invention. In the present studies, ouraim was to demonstrate the feasibility of production of highpurity 90Y from 90Sr by electrolysis.
Currently, we used up to 1.85 GBq (50 mCi) of 90Sr, andscaling up of the generator to a minimum of 37 GBq (1 Ci) of90Sr is a major goal. In the experiment with a simulated solutioncontaining 22.5 mg of Sr equivalent to about 37 GBq (1 Ci) of90Sr, we obtained similar separation and decontaminationfactors. Our further efforts will be to set up adequate facilitiesthat can handle Curie quantities of activity and standardize theseparation method. Further elaborate work is required todevelop this into a usable generator system. Potential metalcontamination of the 90Y product is not estimated, which isnecessary to ensure its use in radiolabeling studies. However, weare confident to be able to address all the issues to obtain Curiequantities of 90Y suitable for high specific activity labelingstudies. This method offers several advantages, which includesthe use of feed solution for electrolysis without furthermodification, except pH adjustment. There is insignificant lossof 90Sr activity except by natural decay. The electrodes used arereusable after proper cleaning. Minimal amounts of chemicalsare used for the whole process, and hence there is very littlepossibility of additional introduction of metal contamination.One of the metal contaminations possible is Zr, the decayproduct of 90Y. However, we expect the same will be removedby electrodeposition along with 90Y in the first few electrolysis,and repeat electrolysis will have acceptable levels of zirconium.The waste generated is expected to contain only trace levels of90Sr. The waste generated can be monitored, classified anddisposed as per regulatory requirements, and there is hardly anysolid waste coming out of the entire process.
Several modifications can be incorporated into this newmethod developed by us. These include scaling up, improvingthe electrolysis setup, working out better electrochemicalparameters in addition to the use of extremely high purityreagents to avoidmetal contamination, removal of accumulatedZr from the 90Sr feed solution before its first use, incorporatinga final step of 90Y purification with 90Sr-specific resin (ifneeded) and automation of the entire process. It is our goal tocarry out all those developments to improve this into an easilyadaptable system. The method developed, if set up in a centralradiopharmacy, will significantly reduce the cost of targetedtherapy. The method could find further applications such asrecovery of 90Sr from waste solution and its purification formaking it as a radiopharmaceutical raw material. Suitablemodification of the electrochemical parameters needs to bedeveloped depending on the specific application.
6. Summary and conclusions
An electrochemical separation procedure has beensuccessfully demonstrated for separation of 90Y from 90Srhaving high radiochemical yield and purity. The method haspractical application and can be further developed to suit awell-established/regulated central radiopharmacy. Theoperational cost of such a generator will be very less andcould provide a permanent supply of 90Y suitable fortherapeutic application. The method developed is superior toall the techniques thus far described in the literature andused by the commercial manufacturers for making 90Y. Webelieve that this electrochemical generator technology can beperfected and has the potential to be widely used resulting insubstantial benefit to the patients. Unlike other radionuclidegenerators used in the nuclear medicine departments, a90Sr–90Y generator will not need frequent replacement,since very little of the radioactivity of 90Sr is lost by decay. A10% supplementation of the 90Sr source every 4–5 years willensure near constant output from the generator. The authorswish to name this 90Sr–90Y generator as “90Y Kamadhenu,”based on the mythical cow “Kamadhenu,” which yieldsunlimited supply of milk.
Acknowledgments
This work was performed as part of the InternationalAtomic Energy Agency (IAEA) CRP on “Development ofGenerator Technologies for Therapeutic Radionuclides.” Dr.Josef Comor of the Institute of Nuclear Physics, Serbia, isacknowledged for bringing to the attention of one of theauthors (M.R.A.P.) the work done by Dr. H.J. Machulla'sgroup on 86Y separation from cyclotron-irradiated 86Sr,which forms the basis of this work. The authors are thankfulto Mr. Pramod Sherkhane and Dr. S.P. Kale, NuclearAgriculture and Biotechnology Division, Bhabha AtomicResearch Centre (BARC), for providing access to their liquidscintillation counting facility. Thanks are also due to Prof. A.Q. Contractor, Head, Department of Chemistry, IndianInstitute of Technology, Mumbai, India, and Prof. S.K.Sengupta, Department of Chemistry, Banaras Hindu Uni-versity, Varanasi, for their valuable suggestions during thecourse of the work. The authors wish to acknowledge thesupport of Dr. V. Venugopal, Director, Radiochemistry andIsotope Group, BARC. Thanks are due to Dr. M. Haji Saeid,Head, Industrial Application and Chemistry Section, and Dr.N. Ramamoorthy, Director, Division of Physical andChemical Sciences, IAEA, for their interest in this work.
References
[1] Goldenberg DM. The role of radiolabeled antibodies in the treatment ofnon-Hodgkin’s lymphoma: the coming of the age of radioimmu-notherapy. Crit Rev Oncol Hematol 2001;39:195–201.
[2] Grillo-Lopez AJ. Zevalin: the first radioimmunotherapy approved forthe treatment of lymphoma. Expert Rev Anticancer Ther 2002;2:485–93.

253R. Chakravarty et al. / Nuclear Medicine and Biology 35 (2008) 245–253
[3] Davies AJ. Radioimmunotherapy for B-cell lymphoma: Y-90 ibritu-momab tiuxetan and I-131 tositumomab. Oncogene 2007;26:3614–28.
[4] Witzig TE, Molina A, Gordon LI, Emmanouilides C, Schilder RJ,Flinn IW, et al. Long-term responses in patients with recurring orrefractory B-cell non-Hodgkin lymphoma treated with yttrium 90ibritumomab tiuxetan. Cancer 2007;109:1804–10.
[5] Paganelli G, Bartolomei M, Grana C, Ferrari M, Rocca P, Chinol M.Radioimmunotherapy of brain tumour. Neurol Res 2006;28:518–22.
[6] Ferrari M, Cremonesi M, Bartolomei M, Bodei L, Chinol M, FiorenzaM, et al. Dosimetric model for locoregional treatment of brain tumourswith 90Y conjugates: clinical applications with 90Y-DOTATOC. J NuclMed 2006;47:105–12.
[7] Breeman WA, Kwekkeboom DJ, de Blois E, de Jong M, Visser TJ,Krenning EP. Radiolabelled regulatory peptides for imaging andtherapy. Anticancer Agents Med Chem 2007;7:345–7.
[8] Kwekkeboom DJ, Mueller-Brand J, Paganelli G, Anthony LB,Pauwel S, Kvols LK, et al. Overview of results of peptide receptorradionuclide therapy with three radiolabelled somatostatin analogs.J Nucl Med 2005;46(Suppl 1):62S–6S.
[9] Gulec SA, Mesoloras G, Dezarn WA, McNeillie P, Kennedy AS.Safety and efficacy of Y-90 microsphere treatment in patients withprimary and metastatic liver cancer: the tumor selectivity of thetreatment as a function of tumor to liver flow ratio. J Transl Med2007;5:15.
[10] Wong CY, Savin M, Sherpa KM, Qing F, Campbell J, Gates VL, et al.Regional yttrium-90 microsphere treatment of surgically unresectableand chemotherapy-refractory metastatic liver carcinoma. CancerBiother Radiopharm 2006;21(4):305–13.
[11] Maximum permissible body burden and maximum permissibleconcentrations of radionuclides in air and water for occupationalexposure. National Bureau of Standards Handbook. 69, U.S.Department of Commerce, AFP 160-67;1959. p. 38.
[12] Chinol M, Hnatowich DJ. Generator produced yttrium-90 for radio-immunotherapy. J Nucl Med 1987;28:1465–70.
[13] Salutsky ML, Kirby HW. Preparation and half-life of carrier-freeyttrium-90. Anal Chem 1955;27:567–9.
[14] Skraba WJ, Arino H, Kramer HH. A new 90Sr/90Y radioisotopegenerator. Int J Appl Radiat Isot 1978;29:91–6.
[15] Wike JS, Guyer CE, Ramey DW, Phillips BP. Chemistry forcommercial scale production of yttrium-90 for medical research.Appl Radiat Isot 1990;41:861–5.
[16] Lee TW, Ting G. Study on the separation of carrier free Yttrium-90from Sr-90. Isotopenpraxis 1991;27:269–73.
[17] Dietz LM, Horwitz EP. Improved chemistry for production of yttrium-90 for medical applications. Appl Radiat Isot 1992;43:1093–101.
[18] Hsieh BT, Ting G, Hsieh HT, Shen LH. Preparation of carrier-freeyttrium-90 for medical applications by solvent extraction chromato-graphy. Appl Radiat Isot 1993;44:1473–80.
[19] Dash A, Bhattacharya PK. Preparation of a 90Sr-90Y generator usingzirconium antimonate. Appl Radiat Isot 1994;45:415–7.
[20] Achuthan PV, Dhami PS, Kannan R, Gopalakrishnan V, RamanujamA. Separation of carrier-free 90Y from high level waste by extractionchromatographic technique using 2-ethyl hexyl, 2-ethyl hexylphosphonic acid (KSM-17). Sep Sci Technol 2000;35:261–70.
[21] Venkatesh M, Pandey U, Dhami PS, Kannan R, Achuthan PV, ChitnisRR, et al. Complexation studies with 90Y from a novel 90Sr/90Ygenerator. Radiochim Acta 2001;89:413–7.
[22] Dhami PS, Naik PW, Pandey U, Dudwadkar NL, Kannan R,Achuthan PV, et al. Studies on the development of a two-stage SLMsystem for the separation of carrier-free 90Y using KSM-17 andCMPO as carriers. Sep Sci Technol 2007;42:1107–21.
[23] Reischl G, Rosch F, Machulla HJ. Electrochemical separation andpurification of yttrium-86. Radiochim Acta 2002;90:225–8.
[24] Yoo J, Tang L, Perkins TA, Rowland DJ, Laforest R, Lewis JS, et al.Preparation of high specific activity 86Y using a small medicalcyclotron. Nucl Med Biol 2005;32:891–7.
[25] Friedlander G, Kennedy JW, Macias ES, Miller JM. Nuclear andradiochemistry. 3rd Ed.; 1981. p. 296.
[26] Selwyn RG, Nickles RJ, Thomadsen BR, De Werd LA, Micka JA. Anew internal pair production branching ratio of 90Y: the development ofa non-destructive assay for 90Y and 90Sr. Appl Radiat Isot 2007;65:318–27.
[27] Pandey U, Dhami PS, Jagesia P, Venkatesh M, Pillai MRA. A novelextraction paper chromatography (EPC) technique for the radio-nuclidic purity evaluation of 90Y for clinical use. Anal Chem(in press).
[28] The world market for therapeutic radiopharmaceuticals. Biotech ReportNo. 230. http://www.biotechsystems.com/reports/230/default.asp.
[29] Pillai MRA, Chakraborty S, Das T, Venkatesh M, Ramamoorthy N.Production logistics of 177Lu for radionuclide therapy. Appl Radiat Isot2003;59:109–18.
[30] Report of first research coordination of the CRP on “development ofgenerator technologies for therapeutic radionuclides”. IAEA; 2005. p.3-18.
[31] Wester DW, Steele RT, Rinehart DE, Des Chane JR, Carson KJ, RapkoBM, et al. Large-scale purification of 90Sr from nuclear waste materialsfor production of 90Y, a therapeutic medical radioisotope. Appl RadiatIsot 2003;59:35–41.
[32] Ramanujam A, Dhami PS, Chitnis RR, Achuthan PV, Kannan R,Gopalakrishnan V, et al. Separation of strontium-90 from PUREX highlevel waste and development of 90Sr/90Y generator. BARC report2000/E/009; 2000.
[33] Bray LA, Wester DW. Method of separation of yttrium-90 fromstrontium-90, US Patent 5,512,256,1996
[34] Bray LA. Method for separating and purifying Yttrium-90 fromStrontium-90. WO 2006/025975. World Intellectual Property Organi-zation; 2006.
[35] Horwitz EP, Hines JJ. A method for isolating and purifying 90Y from90Sr in multi-curie Quantities. WO 01/80251. World IntellectualProperty Organization; 2001.
[36] Betenekov ND, Sharygin LM, Brown RW. Sr-90/Y-90 radionuclidegenerator for production of high quality Y-90 solution. WO/2006/046966. World Intellectual Property Organization; 2006.
[37] Lange G, Herrmann G, Strassmann F. Preparation of strontium-90 freeyttrium-90 by electrolysis. J Inorg Nucl Chem 1957;4:146–54.
[38] Hamaguchi H, Ikeda N, Kawashima T. Isolation of carrier free yttriumfrom the radioactive strontium-yttrium system by electrochemicaldeposition. Bunseki Kagaku 1958;7:243–6.




















