
Synthesis of helical carbon nanofibres and its application in hydrogen desorption

Analytical Biochemistry 427 (2012) 202–210
Contents lists available at SciVerse ScienceDirect
Analytical Biochemistry
journal homepage: www.elsevier .com/locate /yabio
Mitochondrial single nucleotide polymorphism genotyping by matrix-assistedlaser desorption/ionization time-of-flight mass spectrometry using cleavablebiotinylated dideoxynucleotides
Chunmei Qiu a,b, Shiv Kumar a,b, Jia Guo a,c, Jiesheng Lu d, Shundi Shi a,b, Sergey M. Kalachikov a,b,James J. Russo a,b, Ali B. Naini d, Eric A. Schon e, Jingyue Ju a,b,⇑a Columbia Genome Center, Columbia University College of Physicians and Surgeons, New York, NY 10032, USAb Department of Chemical Engineering and Pharmacology, Columbia University, New York, NY 10027, USAc Department of Chemistry, Columbia University, New York, NY 10027, USAd Department of Clinical Pathology and Cell Biology, Columbia University College of Physicians and Surgeons, New York, NY 10032, USAe Department of Genetics and Development, Columbia University College of Physicians and Surgeons, New York, NY 10032, USA
a r t i c l e i n f o
Article history:Received 3 March 2012Received in revised form 19 April 2012Accepted 2 May 2012Available online 10 May 2012
Keywords:Cleavable biotinylated dideoxynucleotidesMALDI–TOF MSGenotypingSingle nucleotide polymorphismSingle base extensionSolid phase captureMitochondrial DNA
0003-2697/$ - see front matter � 2012 Elsevier Inc. Ahttp://dx.doi.org/10.1016/j.ab.2012.05.001
⇑ Corresponding author at: Columbia Genome CCollege of Physicians and Surgeons, New York, NY 19330.
E-mail address: [email protected] (J. Ju).1 Abbreviations used: rRNA, ribosomal RNA; tRNA,
chondrial DNA; PCR–RFLP, polymerase chain reactfragment length polymorphism; SNP, single nucleotidMS, matrix-assisted laser desorption/ionization time-SBE, single base extension; SPC, solid phase capture; TCphine; MERRF, myoclonic epilepsy with ragged red fibetype; PT, point mutation; MSBE, multiplex single basewashing; EB, ethidium bromide.
a b s t r a c t
Characterization of mitochondrial DNA (mtDNA) single nucleotide polymorphisms (SNPs) and mutationsis crucial for disease diagnosis, which requires accurate and sensitive detection methods and quantificationdue to mitochondrial heteroplasmy. We report here the characterization of mutations for myoclonic epi-lepsy with ragged red fibers syndrome using chemically cleavable biotinylated dideoxynucleotides and amass spectrometry (MS)-based solid phase capture (SPC) single base extension (SBE) assay. The methodeffectively eliminates unextended primers and primer dimers, and the presence of cleavable linkersbetween the base and biotin allows efficient desalting and release of the DNA products from solid phasefor MS analysis. This approach is capable of high multiplexing, and the use of different length linkers foreach of the purines and each of the pyrimidines permits better discrimination of the four bases by MS. Bothhomoplasmic and heteroplasmic genotypes were accurately determined on different mtDNA samples. Thespecificity of the method for mtDNA detection was validated by using mitochondrial DNA-negative cells.The sensitivity of the approach permitted detection of less than 5% mtDNA heteroplasmy levels. This indi-cates that the SPC–SBE approach based on chemically cleavable biotinylated dideoxynucleotides and MSenables rapid, accurate, and sensitive genotyping of mtDNA and has broad applications for genetic analysis.
� 2012 Elsevier Inc. All rights reserved.
The human mitochondrial genome is a double-stranded, circularDNA molecule of 16,569 bp that encodes 13 structural proteins, 2ribosomal RNAs (rRNAs),1 and 22 transfer RNAs (tRNAs) [1]. Themitochondrial DNA (mtDNA) is maternally inherited, has a high copynumber per cell, and appears as a mosaic of mutant and wild-type gen-omes (heteroplasmy) [2]. More than 200 pathogenic point mutationsin mitochondrial tRNA and rRNA genes as well as protein coding re-gions have been reported, and the hypervariable region of mtDNA is
ll rights reserved.
enter, Columbia University0032, USA. Fax: +1 212 851
transfer RNA; mtDNA, mito-ion followed by restrictione polymorphism; MALDI–TOFof-flight mass spectrometry;EP, tris(2-carboxyethyl)phos-
rs; WT, wild type; MT, mutantextension; B/W, binding and
also highly polymorphic [3,4]. These features demand precise inter-pretation of mtDNA mutation data for disease treatment, populationassociation studies, and forensic investigations [5–9].
Several technologies have been developed for mtDNA genotyp-ing, including the MitoChip [10], PCR–RFLP (polymerase chain reac-tion followed by restriction fragment length polymorphism)analysis [11], TaqMan real-time genotyping assay [12], and directSanger sequencing [10,13], yet each has some drawbacks [14,15].Most are time-consuming and fail to detect low heteroplasmy levels(<10%), PCR–RFLP and TaqMan assays are labor-intensive whendetecting multiplex single nucleotide polymorphisms (SNPs), andfalse positives often exist due to the limitations of the detectionmechanisms. Next-generation sequencing [14–16] is still too costly,inefficient, and error prone for routine targeted SNP genotyping andlow-level heteroplasmy detection. Thus, a more rapid, accurate, sen-sitive, and cost-effective method for SNP genotyping of mtDNA isneeded.
Matrix-assisted laser desorption/ionization time-of-flight massspectrometry (MALDI–TOF MS) allows rapid and accurate sample

Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210 203
measurement and provides high resolution and sensitivity. It hasbeen used for SNP genotyping coupled with hybridization [17],strand invasion-dependent cleavage of oligonucleotides [18], andsingle base extension (SBE) [19–23]; the latter has emerged as avery powerful method for multiplex SNP detection. In single baseextension, SBE primers designed to anneal adjacent to targetedSNP sites are extended by dideoxynucleotides that are comple-mentary to the nucleotides at the polymorphic positions. Thenucleotides can be identified by measuring the molecular weightof primer extension products. It is critical to stringently purifyDNA primer extension products from excess primers in the reac-tion mixture for high-fold multiplex SNP analysis and unambigu-ous MS-based detection. Therefore, in previous work, weincorporated a solid phase capture step after single base extension(SPC–SBE) reactions using biotinylated dideoxynucleotides, withisolation of SBE products on streptavidin-coated beads [23–26].However, the release of biotinylated DNA products from streptavi-din-coated surfaces requires high temperature formamide treat-ment or other harsh conditions to break the biotin–streptavidinbond and the subsequent requirement for extra ethanol precipita-tion steps, which are tedious and can result in sample loss. Further-more, the relatively small mass differences between eachdideoxynucleotide reduce the effective peak resolution, posingchallenges for low-level heteroplasmy detection due to potentialpeak overlaps. To address these problems, we have developed a no-vel set of chemically cleavable biotinylated dideoxynucleotides,ddNTPs-N3-biotin (ddATP-N3-biotin, ddGTP-N3-biotin, ddCTP-N3-biotin, and ddUTP-N3-biotin; depicted in Fig. 1), whose synthesisis described in detail in our accompanying article [27]. In thesemodified ddNTPs, the biotin moiety is attached to the 5 positionof C and U and to the 7 position of A and G via a cleavable azidolinker. The azido linker between the biotin and the nucleotide iscleavable with high efficiency by treatment with tris(2-carboxy-ethyl)phosphine (TCEP) under DNA-compatible aqueous condi-tions. In addition, the length of the linker is adjusted to increasethe mass differences among these nucleotide analogs for high-res-olution MS measurement. These modifications of the ddNTPs haveled to improved sensitivity, accuracy, and efficiency of SPCsequencing [27] and multiplex genotyping reported here.
We validated this SPC–SBE approach using cleavable biotinyla-ted ddNTPs in testing mtDNA samples for MERRF (myoclonic epi-lepsy with ragged red fibers) syndrome. MERRF syndrome, likemany other mitochondrial diseases [28,29], is a maternally inher-ited multisystemic disorder. Among the most commonly used pointmutations for clinical diagnosis of MERRF syndrome are m.8344A>G(A8344G) [30], m.8356T>C (T8356C) [31], and m.8363G>A(G8363A) [32] in the mitochondrial MT-TK gene encoding tRNALys
and m.3243A>G (A3243G) [33] and m.3255G>A (G3255A) [34] inthe mitochondrial MT-TL1 gene encoding tRNALeu. A8344G is pres-ent in more than 80% of MERRF-affected individuals [28], whereasA3243G and G3255A are mutations shared with MELAS (myopathy,encephalopathy, lactic acidosis, and stroke) syndrome [13,34,35].We have developed a 5-plex genotyping assay to simultaneouslyidentify variants at all five of these sites in mtDNA samples. Wedemonstrate that the SPC–SBE approach using cleavable biotinyla-ted ddNTPs could quantify heteroplasmy levels and detect as lowas 5% heteroplasmy. This enables rapid and accurate identificationof mtDNA SNPs at high sensitivity and specificity, offering great po-tential for mitochondrial disease diagnosis.
Materials and methods
DNA samples were extracted from homoplasmic 8344 wild-type (8344WT), homoplasmic 8344 mutant-type (8344MT), andhomoplasmic 3255 mutant-type (3255MT) cybrids, a mitochon-
drial DNA-negative cell line (rho0), a MERRF patient with the8344 point mutation (8344PT), and an anonymous healthy donor(8344WT-2). The cleavable dideoxynucleotides were synthesizedaccording to the procedures described in the accompanying article[27].
PCR amplification
The PCR primers were initially designed with Primer3 (http://www.frodo.wi.mit.edu/primer3) but further adjusted after a BLASTsearch in order to avoid nonspecific amplification of nuclear DNA.A uniplex PCR was performed to amplify region 1 containing threeof the SNP sites (A8344G, T8356C, and G8363A), whereas a 2-plexPCR was used to amplify regions 1 and 2 simultaneously, the latterof which covers the other two SNP sites (A3243G and G3255A). Theprimers were 50-GACCGGGGGTATACTACGGT-30 (region 1, for-ward), 50-GGAGGTAGGTGGTAGTTTGTG-30 (region 1, reverse), and50-TTAGTATTATACCCACACCCACCCAAG-30 (region 2, forward),50-ATTAGAATGGGTACAATGAGGAGT-30 (region 2, reverse) (pur-chased from Integrated DNA Technologies, Coralville, IA, USA, orEurofins MWG Operon, Huntsville, AL, USA). Therefore, the DNAfragments that are amplified from the 2-plex PCR include all fiveof these frequently mutated sites used for the clinical diagnosisof MERRF syndrome. The uniplex PCR was carried out in a 30-llPCR cocktail mixture containing 100 ng of mtDNA, 10 nmol ofdNTPs, 8 pmol each of forward and reverse primers (region 1, for-ward and reverse), 0.5 U of JumpStart REDTaq DNA Polymerase(Sigma–Aldrich, St. Louis, MO, USA), and the corresponding 1�polymerase reaction buffer. The 2-plex PCR consisted of 100 ngof mtDNA, 12 nmol of dNTPs, 7 pmol each of forward and reverseprimers, 0.5 U of JumpStart REDTaq DNA Polymerase, and 1� poly-merase reaction buffer in a total volume of 30 ll. Amplificationwas performed using the following PCR profile: incubation at94 �C for 2 min, followed by 25 cycles of 94 �C for 20 s, 58 �C for30 s, 72 �C for 30 s, and a final extension at 72 �C for 3 min. AfterPCR, 1 ll of PCR product from each sample was run on a 2100 Bio-analyzer (Agilent Technologies, Santa Clara, CA, USA) to visualizethe product quality. To remove excess primers and dNTPs, PCRproducts were subsequently treated with ExoSAP-IT (USB/Affyme-trix, Cleveland, OH, USA), with incubation at 37 �C for 15 min andenzyme deactivation at 80 �C for 15 min.
Relative quantification of mtDNA in the samples
The relative amounts of mtDNA in the homoplasmic samples,8344MT and 8344WT, were quantified by TaqMan real-time PCR.Region 1, which is specific to the mitochondrial genome, was cho-sen to compare the relative quantity of mtDNA between these twosamples. Here 10-fold serial dilutions from 10 to 10–5 ng/ll werecarried out on both 8344MT and 8344WT samples before real-timePCR. The real-time PCR cocktail consisted of 1 ll of diluted DNAsample, 4 pmol each of forward and reverse primers, and 10 ll ofdiluted SYBR Green mixture (Life Technologies, San Diego, CA,USA) in a total volume of 20 ll. The reaction was performed underthe following conditions: incubation at 50 �C for 2 min and 95 �Cfor 2 min, followed by 40 cycles of 95 �C for 15 s and 60 �C for30 s. Threshold cycle (Ct) values for each dilution of both sampleswere collected, and the graph of Ct values against concentrationswas plotted to determine the relative amount of mtDNA in samples8344MT and 8344WT. Based on the quantification results, the8344MT and 8344WT samples were mixed in a series of ratios(2.5, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, and 97.5% mutant type)for heteroplasmy quantitation and sensitivity analysis, targetingthe SNP at position A8344G.
Taking into account the potential for preferential PCR amplifica-tion, purified homoplasmic PCR products instead of the original
HN
N
O
H2N N
OOPO-
OOP
O-
OOP-O
O-
O
O
NH
HN5
O NH
HN
O
OS
NHHN
O
O O ON3
OO N
H
HN
ON3
OONH
OS
NHHN
O
N
NH2
O N
OOPO-
OOP
O-
OOP-O
O-
O
HN
O
O N
O
O
NH
HN5
O NH
HN
O
OS
NHHN
O
O O O
N3
OPO-
OOP
O-
OOP-O
O-
O
N
N N
NH2
O
OO N
H
HN
ON3
OONH
OS
NHHN
O
OPO-
OOP
O-
OOP-O
O-
O
ddATP-N3-biotin
ddGTP-N3-biotin
ddCTP-N3-biotin
ddUTP-N3-biotin
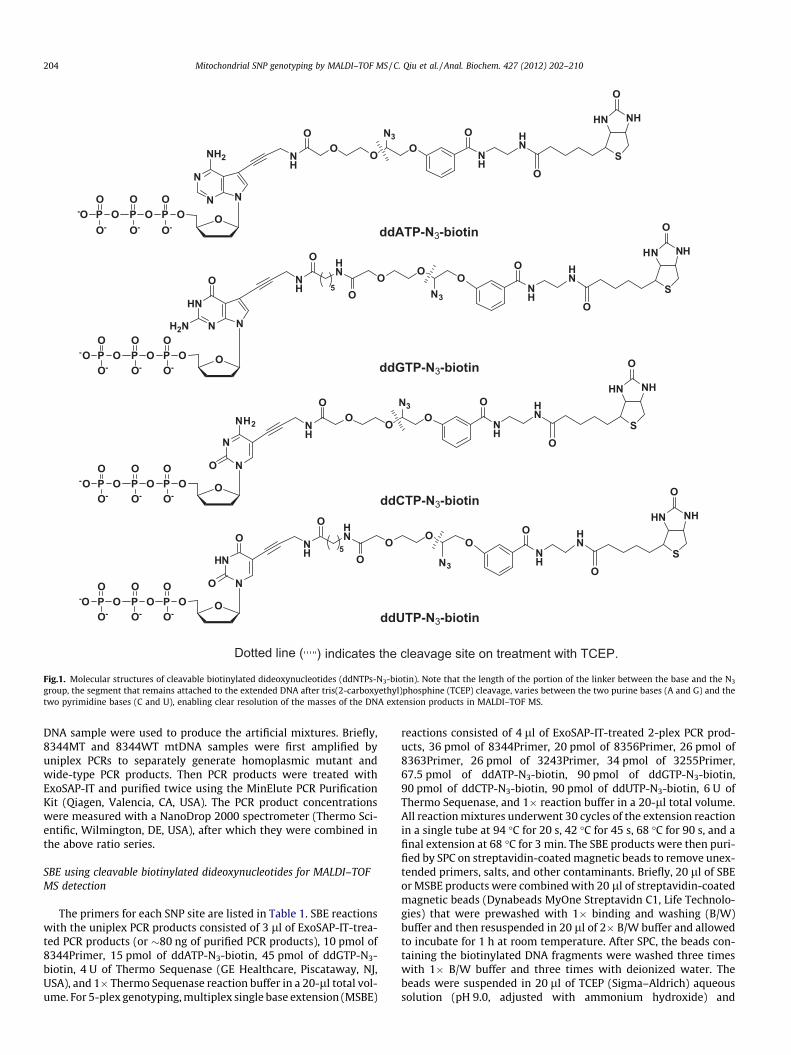
Dotted line ( ) indicates the cleavage site on treatment with TCEP.
Fig.1. Molecular structures of cleavable biotinylated dideoxynucleotides (ddNTPs-N3-biotin). Note that the length of the portion of the linker between the base and the N3
group, the segment that remains attached to the extended DNA after tris(2-carboxyethyl)phosphine (TCEP) cleavage, varies between the two purine bases (A and G) and thetwo pyrimidine bases (C and U), enabling clear resolution of the masses of the DNA extension products in MALDI–TOF MS.
204 Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210
DNA sample were used to produce the artificial mixtures. Briefly,8344MT and 8344WT mtDNA samples were first amplified byuniplex PCRs to separately generate homoplasmic mutant andwide-type PCR products. Then PCR products were treated withExoSAP-IT and purified twice using the MinElute PCR PurificationKit (Qiagen, Valencia, CA, USA). The PCR product concentrationswere measured with a NanoDrop 2000 spectrometer (Thermo Sci-entific, Wilmington, DE, USA), after which they were combined inthe above ratio series.
SBE using cleavable biotinylated dideoxynucleotides for MALDI–TOFMS detection
The primers for each SNP site are listed in Table 1. SBE reactionswith the uniplex PCR products consisted of 3 ll of ExoSAP-IT-trea-ted PCR products (or �80 ng of purified PCR products), 10 pmol of8344Primer, 15 pmol of ddATP-N3-biotin, 45 pmol of ddGTP-N3-biotin, 4 U of Thermo Sequenase (GE Healthcare, Piscataway, NJ,USA), and 1� Thermo Sequenase reaction buffer in a 20-ll total vol-ume. For 5-plex genotyping, multiplex single base extension (MSBE)
reactions consisted of 4 ll of ExoSAP-IT-treated 2-plex PCR prod-ucts, 36 pmol of 8344Primer, 20 pmol of 8356Primer, 26 pmol of8363Primer, 26 pmol of 3243Primer, 34 pmol of 3255Primer,67.5 pmol of ddATP-N3-biotin, 90 pmol of ddGTP-N3-biotin,90 pmol of ddCTP-N3-biotin, 90 pmol of ddUTP-N3-biotin, 6 U ofThermo Sequenase, and 1� reaction buffer in a 20-ll total volume.All reaction mixtures underwent 30 cycles of the extension reactionin a single tube at 94 �C for 20 s, 42 �C for 45 s, 68 �C for 90 s, and afinal extension at 68 �C for 3 min. The SBE products were then puri-fied by SPC on streptavidin-coated magnetic beads to remove unex-tended primers, salts, and other contaminants. Briefly, 20 ll of SBEor MSBE products were combined with 20 ll of streptavidin-coatedmagnetic beads (Dynabeads MyOne Streptavidn C1, Life Technolo-gies) that were prewashed with 1� binding and washing (B/W)buffer and then resuspended in 20 ll of 2� B/W buffer and allowedto incubate for 1 h at room temperature. After SPC, the beads con-taining the biotinylated DNA fragments were washed three timeswith 1� B/W buffer and three times with deionized water. Thebeads were suspended in 20 ll of TCEP (Sigma–Aldrich) aqueoussolution (pH 9.0, adjusted with ammonium hydroxide) and

Table 1SNP sites and corresponding SBE primers to generate DNA extension products.
Note: Reverse SBE primers were used for sites 8356, 8363, and 3255. The masses of the extension products (in bold black type) obtained after incorporation of the modifiedddNTPs shown in Fig. 1 refer to wild type; those in bold red type refer to the mutant type as shown in Fig. 4. (For interpretation of the references to color in this table note, thereader is referred to the Web version of this article.)
Fig.2. SPC–SBE approach for multiplex genotyping by MALDI–TOF MS usingcleavable biotinylated dideoxynucleotides. DNA fragments that contain targetSNP positions are generated by uniplex or multiplex PCR and serve as templates forSBE reactions. A set of SBE primers that are adjacent to SNP sites are used togenerate SBE products, which are then isolated from the reaction mixturecontaining unextended primers, salts, and other contaminants by streptavidin-coated magnetic beads. SBE products are then cleaved from the beads with TCEP inpreparation for MALDI–TOF MS analysis, leaving the biotin moiety still bound to thebead surface.
Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210 205
incubated at 65 �C for 15 min to cleave the biotin. The supernatantcontaining SBE fragments was desalted with a ZipTip (Millipore,Billerica, MA, USA) and characterized with a Voyager DE MALDI–TOF mass spectrometer (Applied Biosystems, Foster City, CA, USA).The entire process is shown in Fig. 2.
Direct Sanger DNA sequencing and PCR–RFLP assay
The Sanger sequencing reaction mixture consisted of 0.5 ll ofBigDye Terminator 3.1 cycle sequencing mix (Applied Biosystems),1� buffer 3.1, and 5 pmol of forward (or reverse) PCR primer. Thereaction was carried out under the following conditions: 25 cyclesof 94 �C for 20 s, 55 �C for 45 s, 68 �C for 90 s, and a final extensionat 68 �C for 3 min. After cleanup of sequencing products by sodiumacetate/ethanol precipitation and solubilization in formamide, theywere separated and analyzed on an ABI 3730xl DNA Analyzer(Applied Biosystems).
The primers used in the PCR–RFLP assay were 50-TTAAGTTAAA-GATTAAGAGG-30 (forward) and 50-TATTTTTAGTTGGGTGATGAGGAA-30 (reverse), where the 30 end of the forward primer is mis-matched to locus 8343 in the mitochondrial genome, generatinga restriction site for HaeIII if position 8344 is mutated from A toG. The PCR mixture consisted of 100 ng of mtDNA, 10 nmol ofdNTPs, 10 pmol each of the forward and reverse primers, 0.5 U ofJumpStart REDTaq DNA polymerase, and the corresponding 1�polymerase reaction buffer. The amplification was carried out with30 cycles of 94 �C for 1 min, 50 �C for 1 min, and 72 �C for 1 min.Then 20 ll of PCR product was treated by adding 10 U of HaeIII(New England Biolabs, Ipswich, MA, USA), 4 ll of 10� NEBuffer 4,and 15 ll of H2O at 37 �C. The product size was checked by separa-tion in a 15% polyacrylamide gel (Bio-Rad, Hercules, CA, USA).
Results
A8344G uniplex genotyping
A8344G is one of the most frequent mutations found in MERRFsyndrome. Genotyping by the SBE–SPC approach using cleavablebiotinylated dideoxynucleotides was first validated for this siteon six different samples: homoplasmic wild-type 8344WT, homo-plasmic mutant-type 8344MT and homoplasmic mutant-type3255MT cybrids, patient sample 8344PT, anonymous healthy sam-ple 8344WT-2, and a mitochondrial DNA-negative cell line (rho0).As shown in Fig. 3, the nucleotide at position 8344 was identifiedcorrectly for each sample, and no false-positive peak appears forthe rho0 DNA sample. The molecular weight of the 8344 SBEprimer is 4648 Da; therefore, the peak at 5098 m/z correspondsto an extension product with ddATP-N3-biotin (WT) after cleavage,whereas the peak at 5227 m/z corresponds to an extensionproduct with ddGTP-N3-biotin (MT) after cleavage. The resultsfrom Figs. 3A and 3B are indicative of the homoplasmic wild-type
form (A) at locus 8344, Fig. 3C shows the homoplasmic mutant-type form (G) at locus 8344, Fig. 3D containing the 3255 mutationshows the wild-type form at locus 8344, and Fig. 3E shows the het-eroplasmic mutant-type form (A/G), in good concordance with theRFLP assay. The specificity for detecting mtDNA polymorphismswas validated by using DNA from rho0 cells. No amplificationproducts were detected in the rho0 cell by PCR, and no peaks weredetected in the mass spectrum, as indicated in Fig. 3F.
MERRF 5-plex genotyping
Because multiple SNP sites are routinely screened in the clinicaldiagnosis for the MERRF syndrome, a 5-plex genotyping assay

Fig.3. MS detection of nucleotide variation at mtDNA locus 8344 from 8344 wild-type sample (A), anonymous healthy subject (B), 8344 mutant sample (C), 3255 mutantsample wild-type at position 8344 (D), patient sample (E), and mitochondrial DNA-negative cells (F). Samples A, C, and E are cybrid cell lines. Letters in red refer to the mutanttype. (For interpretation of the reference to color in this figure legend, the reader is referred to the Web version of this article.)
206 Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210
using the cleavable biotinylated dideoxynucleotides was devel-oped. As shown in Fig. 4, nucleotides at these five sites were iden-tified unambiguously for all of the samples, whereas the negativecontrol using rho0 cells further confirms the specificity of the assay.The molecular weights of primer peaks and their potential exten-sion products are listed in Table 1. To ensure sufficient mass differ-ences between successive primers, reverse SBE primers that annealto the complementary strand were selected for SNP sites G3255A,T8356C, and G8363A. Therefore, in the mass spectrum for the mu-tant type, transitions from C to U at locus 3255, from A to G at locus8356, and from C to U at locus 8363 were expected. As shown inFig. 4, in accordance with the uniplex genotyping results, only8344MT and PT have A to G mutations at locus 8344. Althoughlacking the mutation at position 8344, 3255MT has a C to T muta-tion at position 3255 (Fig. 4D), consistent with previous results. Nofalse positives were detected on the rho0 DNA sample (Fig. 4F).According to Table 1 and the mass spectrometric results (Fig. 4),samples 8344MT, PT, and 3255MT were diagnosed as MERRF syn-drome, whereas 8344 WT and WT-2 were determined to be clear ofthe disease, consistent with known information for these samples.
Quantitative analysis of mutations for mitochondrial sample
In contrast to heterozygous mutations in nuclear DNA, mutatedand wild-type forms of mtDNA rarely occur at 1:1 ratios. Instead,heteroplasmy can occur at any ratio between 0:100 and 100:0.Therefore, for better understanding of the mitochondrial disease,quantitative analysis of the mtDNA mutation is of particular
interest. Taking A8344G as an example, the capability of perform-ing quantitative analysis for mtDNA heteroplasmy was evaluated.Before mixing homoplasmic WT and MT samples to create differ-ent ratios of heteroplasmy, real-time PCR was first performed toestimate the relative amount of mtDNA in these samples. Theresults indicated that the 8344MT and 8344WT samples containedthe same amount of mtDNA (data not shown). Nonetheless,considering possible preferential amplification of wild-type andmutated forms, purified homoplasmic PCR products were quanti-fied and combined to generate another set of artificial mixturesfor quantitative analysis. Regardless of whether the DNA wasmixed before or after PCR, the results indicated that the PT samplecontains approximately 80 to 90% heteroplasmy at locus 8344.Quantitative results for the post-PCR mixing experiment are shownin Fig. 5. The graph was generated from the normalized signalintensities of MT and WT peaks in the mass spectra. By comparingthe mass spectrum of the patient sample (right-most bars) withthe resulting standard curve, it was suggested that its hetero-plasmy falls within the range of 80 to 90%, consistent with theinformation provided for this patient (86%).
Sensitivity of SPC–SBE MALDI–TOF MS for detecting low-heteroplasmymtDNA
For early detection, it is necessary to discriminate a very smallamount of mutated mtDNA in a majority of wild-type back-ground. Taking locus 8344 as an example, the sensitivity of thisassay was validated. With sample mixing based on two types of

Fig.4. MS results of 5-plex SNP genotyping for MERRF syndrome: (A) 8344 wild-type sample; (B) anonymous healthy donor; (C) 8344 mutant sample; (D) 3255 mutantsample; (E) patient sample; (F) mitochondrial DNA-negative cell line. Red labels refer to the mutated forms. Samples A, C, and E are cybrid cell lines. (For interpretation of thereference to color in this figure legend, the reader is referred to the Web version of this article.)
Fig.5. Quantitative analysis of position 8344 heteroplasmy in mtDNA based on themass spectral peak intensity of various ratio mixtures of purified PCR products.
Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210 207
information (real-time PCR and measurement of PCR product con-centration), 5% and occasionally 2.5% heteroplasmy was correctlyidentified, as shown in Fig. 6A and B. Similarly, as low as 5% andless consistently 2.5% wild-type DNA in a mutant background wasdetected. This further confirms the accuracy of the assay. For com-parison, Sanger sequencing and PCR–RFLP were also performed.As seen in Fig. 6C, neither 5% of mutant mtDNA (panel a) nor nor-mal mtDNA (panel b) was detected by Sanger sequencing analysis.In a PCR–RFLP assay, the enzyme HaeIII cut wild-type PCR prod-ucts into two fragments: 69 and 57 bp. The use of mismatchedprimer in the PCR enables the production of an extra restrictionsite for HaeIII if A is mutated to G at locus 8344. Thus, the 69-bp fragment is further digested into two segments: 48 and21 bp. As shown in Fig 6D, this ethidium bromide (EB)-stainedPCR–RFLP assay also failed to detect 5% heteroplasmy (lane 4).Thus, the MS-based SPC–SBE approach using cleavable biotinyla-ted ddNTPs apparently outperformed these two alternativeapproaches in terms of sensitivity.

Fig.6. Detection of low-level heteroplasmy at locus 8344 for sensitivity testing. (A) Detection of 5 and 2.5% heteroplasmy, based on original DNA sample mixing, by MALDI–TOF MS. (B) Detection of 2 and 5% heteroplasmy, based on purified PCR product mixing, by MALDI–TOF MS. Data in panels A and B demonstrate the reproducible detection of5% heteroplasmy. Red labels refer to the mutated forms. (C) Sanger sequencing analysis for 5 and 95% mixtures, which is not able to detect the heteroplasmy. Peak colorsrepresent A (green), C (blue), G (black) and T (red). (D) PCR–RFLP analysis for wild-type (lane 2), mutant-type (lane 3), 5% heteroplasmy (lane 4), and anonymous patient (lane5) samples. Low-molecular-weight (LMW) markers were used for sizing (lane 1).
208 Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210

Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210 209
Discussion
Identifying mtDNA SNPs rapidly and precisely in a cost-effectiveway is crucial for disease diagnosis and prognosis. Expanding onour previously established SPC–SBE method, we further introduceda set of chemically cleavable biotinylated dideoxynucleotides intothe SBE reactions, which enabled highly effective isolation andpurification of the SBE products and achieved higher resolutionthrough larger mass differences between the nucleotides and theircorresponding SBE products. We validated this SPC–SBE approachusing cleavable biotinylated dideoxynucleotides by identifyingmtDNA SNPs related to MERRF syndrome and further evaluatedits accuracy and sensitivity for heteroplasmy detection and quanti-fication. A uniplex assay for identifying the A8344G polymorphicsite, the most common point mutation in MERRF syndrome, wasfirst evaluated, the results of which were consistent with knowninformation. No false positives or false negatives were detected.To meet the needs of rapid and efficient clinical diagnosis, a 5-plexgenotyping assay was then developed. The results indicated thatthis method was able to precisely detect five mtDNA SNPs simulta-neously. Compared with other genotyping methods, the SPC–SBEmethod is faster and more efficient; multiple assays wereperformed in a single tube, and the work flow from sample prepa-ration to SNP identification could be accomplished in a single day,which is suitable for clinical diagnosis. We tested only 5-plex geno-typing in this study, but we previously reported up to 30-plexmutation detection with PCR products representing the p53 geneusing a prior version of this assay [25], and 40- and 50-plex multi-plexing has been achieved for two cytochrome P450 genes [26].
Although mitochondria have much higher copy numbers, theDNA extract is a mixture of mitochondrial genomes and the six or-ders of magnitude larger nuclear genome. Therefore, specificity formtDNA detection is essential. We evaluated the specificity of thisSPC–SBE approach by analyzing a mitochondrial DNA-negative cellline (rho0). No SBE products were detected by MS with either theA8344G uniplex assay (Fig. 3F) or the 5-plex genotyping assay(Fig. 4F).
In the mitochondrial genome, DNA mutations often coexist withwild-type DNA, and the proportion of mutant mtDNA will drift intime and space following cell division [36]. Different levels of het-eroplasmy are strongly associated with different clinical manifes-tations, and the disease phenotype becomes evident whenmutated forms of the mtDNA exceed a threshold level [37]. Hence,it is critical to monitor and quantify heteroplasmy, not only for dis-ease diagnosis but also for early prevention and therapy develop-ment. The capability of MALDI–TOF MS to detect the intrinsicmass of molecules and its high sensitivity make simultaneousSNP identification and quantification possible. Importantly, thelarge mass differences between our new modified nucleotides re-sult in sufficient resolution between wild-type and mutated SBEproduct peaks, clear of interference from the remaining salt peaksaround the major peak, thereby ensuring high enough resolution todetect low-level heteroplasmy. We successfully estimated the het-eroplasmy level in a patient sample after generating a quantitativecalibration curve based on mixing of homoplasmic WT and MTsamples. This SPC–SBE method using the cleavable biotinylateddideoxynucleotides was able to identify as low as 5% hetero-plasmy. As shown in Figs. 6A and 6B, in both artificial mixturemethods, low amounts of heteroplasmy were detected in a pre-dominant background of either mutant or wild-type mtDNA; San-ger sequencing and conventional EB-stained PCR–RFLP failed todiscriminate the minor allele (Figs. 6C and 6D) at this level. Be-cause mtDNA mutations might accumulate with time, the highsensitivity and accuracy of low heteroplasmy level detection willbe very helpful for routine health checking aimed at disease
prevention. Although the pathogenic threshold is usually above70% [36], dominant mutations also exist. For example, it hasbeen reported that the pathogenic threshold of a particularmitochondrial disorder was only 4 to 8% [38], in which case lowheteroplasmy detection becomes critical. Radiolabeled RFLP orreal-time PCR assays might meet the need; nonetheless, the MS-based SPC–SBE approach using cleavable biotinylated ddNTPs out-performs them in terms of simplicity, lower cost, reduced labor,and the potential for higher multiplexing. Other investigators haveused the iPLEX assay in the Sequenom MassArray system tocharacterize mitochondrial SNPs for population studies [39] anddetection of heteroplasmy [22]. This system, which also uses theMS-based SBE strategy with unmodified nucleotides, has up to40-fold SNP multiplexing ability and is able to call mitochondrialheteroplasmy to approximately 5%, but it is quite expensive andcannot be used readily for extensive parameter testing. Jiang andcoworkers [40] used electrospray ionization Fourier transformion cyclotron resonance MS to size PCR products surroundingmitochondrial SNPs, essentially a scanning approach that assessesbase composition for the amplimer and may or may not reveal theprecise mutation. Our approach reported here effectively elimi-nates unextended primers and primer dimers; the presence ofcleavable linkers between the base and biotin allows very efficientdesalting and release of the DNA products from solid phase for MSanalysis. This approach is capable of high multiplexing [26], andthe use of different length linkers for each of the purines and eachof the pyrimidines permits better discrimination of the four basesthan other MS-based SBE systems.
In conclusion, by using cleavable biotinylated nucleotides, theSPC–SBE approach coupled with MALDI–TOF MS enables rapid,accurate, and sensitive analysis of mtDNA SNPs and different levelsof heteroplasmy. Due to its sensitivity and the ability to quantifyalleles over a fairly wide range, it will allow screening of mutationsin pools of 10 or more individuals. Moreover, in the accompanyingarticle [27], we have demonstrated the advantages of these cleav-able biotinylated nucleotides for SPC MS-based sequencing, whichhas great advantage in screening for insertions and deletions com-pared with fluorescence-based Sanger sequencing.
Acknowledgment
This work was supported by National Institutes of Health (NIH)Grants R01NS060762 and R01HG004774.
References
[1] S. DiMauro, C.T. Moraes, Mitochondrial encephalomyopathies, Arch. Neurol. 50(1993) 1197–1208.
[2] D.C. Wallace, Mitochondria as chi, Genetics 179 (2008) 727–735.[3] L.C. Wong, Pathogenic mitochondrial DNA mutations in protein-coding genes,
Muscle Nerve 36 (2007) 279–293.[4] MITOMAP: a human mitochondrial genome database, 2010, <http://
www.mitomap.org>.[5] D.C. Wallace, Mitochondrial disease in man and mouse, Science 283 (1999)
1482–1486.[6] E.A. Schon, S. DiMauro, M. Hirano, R. Gilkerson, Therapeutic prospects for
mitochondrial disease, Trends Mol. Med. 16 (2010) 268–276.[7] D.C. Wallace, Mitochondrial DNA variation in human evolution and disease,
Gene 238 (1999) 211–230.[8] S. Köhnemann, H. Pfeiffer, Application of mtDNA SNP analysis in forensic
casework, Forensic Sci. Int. Genet. 5 (2011) 216–221.[9] B. Budowle, M.W. Allard, M.R. Wilson, R. Chakraborty, Forensics and
mitochondrial DNA: applications, debates, and foundations, Annu. Rev.Genomics Hum. Genet. 4 (2003) 119–141.
[10] A. Hartmann, M. Thieme, L.K. Nanduri, T. Stempfl, C. Moehle, T. Kivisild, P.J.Oefner, Validation of microarray-based resequencing of 93 worldwidemitochondrial genomes, Hum. Mutat. 30 (2009) 115–122.
[11] I.J. Holt, A.E. Harding, R.K. Petry, J.A. Morgan-Hughes, A new mitochondrialdisease associated with mitochondrial DNA heteroplasmy, Am. J. Hum. Genet.46 (1990) 428–433.

210 Mitochondrial SNP genotyping by MALDI–TOF MS / C. Qiu et al. / Anal. Biochem. 427 (2012) 202–210
[12] R.K. Bai, L.J. Wong, Detection and quantification of heteroplasmic mutantmitochondrial DNA by real-time amplification refractory mutation systemquantitative PCR analysis: a single-step approach, Clin. Chem. 50 (2004) 996–1001.
[13] B.O. Choi, J.H. Hwang, E.M. Cho, E.H. Jeong, Y.S. Hyun, H.J. Jeon, K.M. Seong, N.S.Cho, K.W. Chung, Mutational analysis of whole mitochondrial DNA in patientswith MELAS and MERRF diseases, Exp. Mol. Med. 42 (2010) 446–455.
[14] S. Tang, T. Huang, Characterization of mitochondrial DNA heteroplasmy usinga parallel sequencing system, BioTechniques 48 (2010) 287–296.
[15] M. Li, A. Schönberg, M. Schaefer, R. Schroeder, I. Nasidze, M. Stoneking,Detecting heteroplasmy from high throughput sequencing of complete humanmitochondrial DNA genomes, Am. J. Hum. Genet. 87 (2010) 237–249.
[16] Y. He, J. Wu, D.C. Dressman, C. Iacobuzio-Donahue, S.D. Markowitz, V.E.Velculescu, L.A. Diaz Jr., K.W. Kinzler, B. Volgelstein, N. Papadopoulos,Heteroplasmic mitochondrial DNA mutations in normal and tumor cells,Nature 464 (2010) 610–614.
[17] J. Stoerker, J.D. Mayo, C.N. Tetzlaff, D.A. Sarracino, I. Schwope, C. Richert, Rapidgenotyping by MALDI-monitored nuclease selection from probe libraries, Nat.Biotechnol. 18 (2000) 1213–1216.
[18] V. Lyamichev, A.L. Mast, J.G. Hall, J.R. Prudent, M.W. Kaise, T. Takova, R.W.Kwiatkowski, T.J. Sander, M. de Arruda, D.A. Arco, B.P. Neri, M.A. Brow,Polymorphism identification and quantitative detection of genomic DNA byinvasive cleavage of oligonucleotide probe, Nat. Biotechnol. 17 (1999)292–296.
[19] K. Tang, D.J. Fu, D. Julien, A. Braun, C.R. Cantor, H. Köster, Chip-basedgenotyping by mass spectrometry, Proc. Natl. Acad. Sci. USA 96 (1999)10016–10020.
[20] J. Li, J.M. Butler, Y. Tan, H. Lin, S. Royer, L. Ohler, T.A. Shaler, J.M. Hunter, D.J.Pollart, J.A. Monforte, C.H. Becker, Single nucleotide polymorphismdetermination using primer extension and time-of-flight mass spectrometry,Electrophoresis 20 (1999) 1258–1265.
[21] P. Ross, L. Hall, I.P. Smirnov, L. Haff, High level multiplex genotyping byMALDI–TOF mass spectrometry, Nat. Biotechnol. 16 (1998) 1347–1351.
[22] F.A. Xiu-Cheng, H.S. Garritsen, S.E. Tarhouny, M. Morris, S. Hahn, W. Holzgreve,X.Y. Zhong, A rapid and accurate approach to identify single nucleotidepolymorphisms of mitochondrial DNA using MALDI–TOF mass spectrometry,Clin. Chem. Lab. Med. 46 (2008) 299–305.
[23] S. Kim, H.D. Ruparel, C. Gilliam, J. Ju, Digital genotyping using molecularaffinity and mass spectrometry, Nat. Rev. Genet. 4 (2003) 1001–1008.
[24] S. Kim, J.R. Edwards, L. Deng, W. Chung, J. Ju, Solid phase capturabledideoxynucleotides for multiplex genotyping using mass spectrometry,Nucleic Acids Res. 30 (2002) e85.
[25] S. Kim, M.E. Ulz, T. Nguyen, C.M. Li, T. Sato, B. Tycko, J. Ju, Thirtyfold multiplexgenotyping of the p53 gene using solid phase capturable dideoxynucleotidesand mass spectrometry, Genomics 83 (2004) 924–931.
[26] A. Misra, J.Y. Hong, S. Kim, Multiplex genotyping of cytochrome p450 single-nucleotide polymorphisms by use of MALDI–TOF mass spectrometry, Clin.Chem. 52 (2007) 933–939.
[27] C. Qiu, S. Kumar, J. Guo, L. Yu, W. Guo, S. Shi, J.J. Russo, J. Ju, Design andsynthesis of cleavable biotinylated dideoxynucleotides for DNA sequencing bymatrix-assisted laser desorption/ionization time-of-flight mass spectrometry,Anal. Biochem. (2012). http://dx.doi.org/10.1016/i.ab.2012.04.021.
[28] S. DiMauro, M. Hirano, MERRF, in: R.A. Pagon, T.C. Bird, C.R. Dolan, K. Stephens(Eds.), GeneReviews, University of Washington, Seattle, 1993–2003.
[29] F. Pallotti, A. Baracca, E. Hernandez-Rose, W.F. Walker, G. Solaini, G. Lenaz, G.V.Melzi D’Eril, S. DiMauro, E.A. Schon, M.M. Davidson, Biochemical analysis ofrespiratory function in cybrid cell lines harbouring mitochondrial DNAmutations, Biochem. J. 384 (2004) 287–293.
[30] J.P. Masucci, M. Davidson, Y. Koga, E.A. Schon, M.P. King, In vitro analysis ofmutations causing myoclonus epilepsy with ragged-red fibers in themitochondrial tRNALys gene: two genotypes produce similar phenotypes,Mol. Cell. Biol. 384 (1995) 2872–2881.
[31] G. Silvestri, C.T. Moraes, S. Shanske, S.J. Oh, S. DiMauro, A new mtDNAmutation in the tRNALys gene associated with myoclonic epilepsy and ragged-red fibers (MERRF), Am. J. Hum. Genet. 51 (1992) 1213–1217.
[32] F.M. Santorelli, S.C. Mak, M. El-Schahawi, C. Casali, S. Shanske, T.Z. Baram, R.E.Madrid, S. DiMauro, Maternally inherited cardiomyopathy and hearing lossassociated with a novel mutation in the mitochondrial tRNALys gene (G8363A),Am. J. Hum. Genet. 58 (1996) 933–993.
[33] G.M. Fabrizi, E. Cardaioli, G.S. Grieco, T. Cavallaro, A. Malandrini, L. Manneschi,M.T. Dotti, A. Federico, G. Guazzi, The A to G transition at nt 3243 of themitochondrial tRNALeu(UUR) may cause an MERRF syndrome, J. Neurol.Neurosurg. Psychiatry 61 (1996) 47–51.
[34] Y. Nishigaki, S. Tadesse, E. Bonilla, D. Shungu, S. Hersh, B.J. Keats, C.I. Berlin,M.F. Goldberg, J. Vockley, S. DiMauro, M. Hirano, A novel mitochondrialtRNALeu(UUR) mutation in a patient with features of MERRF and Kearns–Sayresyndrome, Neuromuscul. Disord. 13 (2003) 334–340.
[35] Y. Campos, M.A. Martin, G. Lorenzo, M. Aparicio, A. Cabello, J. Arenas, SporadicMERRF/MELAS overlap syndrome associated with the 3243 tRNALeu(UUR)
mutation of mitochondrial DNA, Muscle Nerve 19 (1996) 187–190.[36] E.A. Schon, S. DiMauro, M. Hirano, R.W. Gilkerson, Therapeutic prospects for
mitochondrial disease, Trends. Mol. Med. 16 (2010) 268–276.[37] R. Rossignol, B. Faustin, C. Rocher, M. Malgat, J.P. Mazat, T. Letellier,
Mitochondrial threshold effects, Biochem. J. 370 (2003) 751–762.[38] S. Sacconi, L. Salviati, Y. Nishigaki, W.F. Walker, E. Hernandez-Rosa, E.
Trevisson, S. Delplace, C. Desnuelle, S. Shanske, M. Hirano, E.A. Schon, E.Bonilla, D.C. De Vivo, S. DiMauro, M.M. Davidson, A functionally dominantmitochondrial DNA mutation, Hum. Mol. Genet. 17 (2008) 1814–1820.
[39] M. Cerezo, V. Cerny, A. Carracedo, A. Salas, Applications of MALDI–TOF MS tolarge-scale human mtDNA population-based studies, Electrophoresis 30(2009) 3665–3673.
[40] Y. Jiang, T.A. Hall, S.A. Hofstadler, R.K. Naviaux, Mitochondrial DNA mutationdetection by electrospray mass spectrometry, Clin. Chem. 53 (2007) 195–203.
Copyright © 2022 FDOKUMEN




















