
Deletion of the Activated Protein-1 Transcription Factor JunD Induces Oxidative Stress and...
55
Volpe, Thomas F. Lüscher and Francesco Cosentino Tadeusz Malinski, Giovanni G. Camici, Christian M. Matter, Fatima Mechta-Grigoriou, Massimo Coppolino, Enrico Perna, Pavani Mocharla, Alexander Akhmedov, Ruslan Kubant, Lucia Rohrer, Francesco Paneni, Elena Osto, Sarah Costantino, Bogdan Mateescu, Sylvie Briand, Giuseppe Age-Related Endothelial Dysfunction Deletion of the AP-1 Transcription Factor JunD Induces Oxidative Stress and Accelerates Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2013 American Heart Association, Inc. All rights reserved. is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231 Circulation published online February 14, 2013; Circulation. http://circ.ahajournals.org/content/early/2013/02/14/CIRCULATIONAHA.112.000826 World Wide Web at: The online version of this article, along with updated information and services, is located on the http://circ.ahajournals.org/content/suppl/2013/02/14/CIRCULATIONAHA.112.000826.DC1.html Data Supplement (unedited) at: http://circ.ahajournals.org//subscriptions/ is online at: Circulation Information about subscribing to Subscriptions: http://www.lww.com/reprints Information about reprints can be found online at: Reprints: document. Permissions and Rights Question and Answer available in the Permissions in the middle column of the Web page under Services. Further information about this process is Once the online version of the published article for which permission is being requested is located, click Request can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office. Circulation Requests for permissions to reproduce figures, tables, or portions of articles originally published in Permissions: by guest on February 18, 2013 http://circ.ahajournals.org/ Downloaded from
Transcript of Deletion of the Activated Protein-1 Transcription Factor JunD Induces Oxidative Stress and...

Volpe, Thomas F. Lüscher and Francesco CosentinoTadeusz Malinski, Giovanni G. Camici, Christian M. Matter, Fatima Mechta-Grigoriou, MassimoCoppolino, Enrico Perna, Pavani Mocharla, Alexander Akhmedov, Ruslan Kubant, Lucia Rohrer,
Francesco Paneni, Elena Osto, Sarah Costantino, Bogdan Mateescu, Sylvie Briand, GiuseppeAge-Related Endothelial Dysfunction
Deletion of the AP-1 Transcription Factor JunD Induces Oxidative Stress and Accelerates
Print ISSN: 0009-7322. Online ISSN: 1524-4539 Copyright © 2013 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation published online February 14, 2013;Circulation.
http://circ.ahajournals.org/content/early/2013/02/14/CIRCULATIONAHA.112.000826World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circ.ahajournals.org/content/suppl/2013/02/14/CIRCULATIONAHA.112.000826.DC1.htmlData Supplement (unedited) at:
http://circ.ahajournals.org//subscriptions/
is online at: Circulation Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer available in the
Permissions in the middle column of the Web page under Services. Further information about this process isOnce the online version of the published article for which permission is being requested is located, click Request
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the Editorial Office.Circulation Requests for permissions to reproduce figures, tables, or portions of articles originally published inPermissions:
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
1
Deletion of the AP-1 Transcription Factor JunD Induces Oxidative Stress and
Accelerates Age-Related Endothelial Dysfunction
Running title: Paneni et al.; JunD in vascular disease
Francesco Paneni, MD1,2*; Elena Osto, MD, PhD1,3*; Sarah Costantino, PhD1,4; Bogdan Mateescu, PhD5; Sylvie Briand, PhD1,3 ; Giuseppe Coppolino, MD1; Enrico Perna, MD6; Pavani
Mocharla, MSc1,3; Alexander Akhmedov, PhD1,3; Ruslan Kubant, MD7; Lucia Rohrer, PhD8; Tadeusz Malinski, PhD7; Giovanni G. Camici, PhD1,3; Christian M. Matter, MD1,3; Fatima
Mechta-Grigoriou, PhD5; Massimo Volpe, MD2,6; Thomas F. Lüscher MD1,3; Francesco Cosentino, MD, PhD1,3,6
1Cardiology and Cardiovascular Research, Institute of Physiology and University Hospital, Zürich, Switzerland; 2IRCCS Neuromed, Pozzilli, Italy; 3Zürich Center for Integrative Human
Physiology (ZIHP), University of Zurich, Switzerland; 4Dept of Experimental Medicine, Section of Pharmacology, Second University of Study of Naples, Italy; 5Stress and Cancer Laboratory,
Institut Curie, Inserm U830, Paris, France; 6Cardiology, Dept of Clinical and Molecular Medicine, “Sapienza” University, Rome, Italy; 7Dept of Biochemistry, Ohio University, Athens,
Ohio; 8Institute of Clinical Chemistry, University Hospital, Zürich, Switzerland *The authors contributed equally to this work
Address for Correspondence:
Francesco Cosentino, MD, PhD
Cardiology & Cardiovascular Research
Institute of Physiology
University of Zurich-Irchel
Winterthurerstrasse, 190
CH-8057 Zurich, Switzerland
Tel: 41-44-635 6470
Fax: 41-44-635 6827
E-mail: [email protected]
Journal Subject Code: [95] Endothelium/vascular type/nitric oxide
Francesco Cosentino, MD, PhD1,3,6
1Cardiology and Cardiovascular Research, Institute of Physiology and University Hospital, Zürich, Swwitzerland; 2IRCCS Neuromed, Pozzilli,, Italy; 3Zürich Center for Integrative Human
PhPhysysysioioiollologygygy ((ZZZIHPHPHP),), University of Zurich, Switzzzerere laand; 4Dept of ExEE peeriririmmmental Medicine, Sectionofoof PPPhah rmacaccooologogogy,y,y SSececcononnd d d UnUnivivivererersisitytyy oof f StStududy y oof NNapplelees,s,s, IIItat lyyy;;; 555StStreresssss aandd CCCananancecec r r LaLabobob rararatototoryry,
Institut CCururieii ,, InInsssermrmrm UUU8383830,0, PPPaararisss, FFrannnccee; 6CaCaCardiioiollologgygy, DeDeepttt oof ff CCCliininiccacalll anandd d MoMoMolell cucuculaalarr MeMeMedid cine, “SSapiiiennnza””” UUUniivveverrsrsitty,y RRRomomee, Ittaalyy; 7DeDeDeptpt ooof f f BiBiBiocccheemimistryy, OOhOhioio UUnivvverrsrsititty,, Athhheens,
OhOhhioioio;; 88IInInsststitituuutee e ofof CCClilinininicacacal l l ChChhememmisisisttry,y,y, UUUniiiveveersrsititity y y HoHoospspspititalalal,, , ZüZüürriichchh,, SwSwSwitittzezezerrlananndd d*T*Thehe aututhoh rs cconontrtribibututed eeququalallyly tto o tht isis wworork k
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
2
Abstract:
Background—Reactive oxygen species (ROS) are major determinants of vascular aging. JunD, a
member of the activated protein 1 (AP-1) family of transcription factors, is emerging as a major
gatekeeper against oxidative stress. However, its contribution to ROS homeostasis in the
vasculature remains unknown.
Methods and Results—Endothelium-dependent vasorelaxation was impaired in young and old
JunD-/- mice (6 and 22 month old) as compared with age-matched wild-type (WT). JunD-/- mice
displayed an age-independent decline of endothelial NO release and eNOS activity as well as
increased mitochondrial superoxide formation and peroxynitrite levels. Furthermore, vascular
expression and activity of free radical scavengers manganese and extracellular superoxide
dismutase as well as aldehyde dehydrogenase 2 were reduced while NADPH oxidase subunits
p47phox, Nox2 and Nox4 were upregulated. These redox changes were associated with premature
vascular aging as shown by reduced teIomerase activity, increased -gal positive cells,
upregulation of senescence markers p16INK4a and p53 as well as mitochondrial disruption.
Interestingly, old WT mice showed a reduction of JunD expression and transcriptional activity
due to promoter hypermethylation and binding with tumor suppressor menin, respectively. By
contrast, JunD overexpression blunted age-induced endothelial dysfunction. In human endothelial
cells, JunD knockdown exerted a similar impairment of O2-/NO balance which was prevented by
concomitant NADPH inhibition. In parallel, JunD expression was reduced in monocytes from old
vs. young healthy subjects and correlated with mRNA levels of scavenging and oxidant enzymes.
Conclusions—JunD provides protection in aging-induced endothelial dysfunction and may
represent a novel target to prevent ROS-driven vascular aging.
Key words: aging, endothelial function, oxidative stress, vascular biology
p p y ,,
expression and activity of free radical scavengers manganese and extracellular susuupepeperorooxixixidedede
dismutase as well as aldehyde dehydrogenase 2 were reduced while NADPH oxidase subunits
p4p447p7p7phhohox, NNNoxoxox2 anand d NoNox4x4 wwerre e upuprer gugulaatet d. TThehessese redoxox chc angegeess weerere aassocciaatet d wiwithh pprematur
vavav scccular aging ass ssshowwwnnn byy rreededucedded teeeIooomerrrasse aaccttiivitytyy, , , ininincrcreeasseddd dd -ggalal pooosiittivi e cccelllll s,,,
upupreregugugulalalatititioonon oof f f sesesenenenescscscenenencecec mmarararkekekerrsrs pp16166INKINKINK4a4a4 aaandndnd ppp535353 aaass s wewewellllll aas mimimitototochchchononondrdriaiaiall l dididissrsrupuptititiononon..
nterestinglg y,y,y oooldldld WWWTT T mimim cecee ssshohoh weweedd aa rereeduduductctctioioonn n ofofo JJJunununD DD eexexprprpresesessisiionnn aaandndnd tttrrrff aananscscriririptpttioioionanaal activityy
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
3
Introduction
The prevalence of cardiovascular disease is markedly age-dependent and a major cause of
myocardial infarction and death among elderly patients. A key alteration of aging arteries is the
development of endothelial dysfunction which is associated with a reduction of the
bioavailability of nitric oxide (NO).1-3 The primary mechanism of aging-associated endothelial
dysfunction is oxidative stress, a state in which generation of reactive oxygen species (ROS)
exceeds the capacity of cellular antioxidant defense systems4. Of note, superoxide anion (O2-)
inactivates endothelium-derived NO.5-7 From interaction of O2- with NO, the highly reactive
peroxynitrite (ONOO-) is formed.5 As a consequence, vasodilation as well as the antithrombotic
and anti-inflammatory effects mediated by the endothelium are impaired resulting in vascular
damage.8, 9 Although the accumulation of ROS generation is the most widely accepted cause of
vascular aging4,10, a thorough understanding of the molecular mechanisms of altered redox
signaling in endothelial cells remains to be accomplished.
Activator protein-1 (AP-1) is a collection of dimeric complexes made by different
members of three families of DNA binding proteins: Jun, Fos and ATF/CREB.11-13 These
members assemble to form AP-1 transcription factors whose activity is strongly influenced by
their specific components as well as their cellular environment.12 JunD, the most recent gene of
the Jun family, regulates cell growth and survival and protects against oxidative stress by
modulating genes involved in antioxidant defense and ROS production. Along these lines, JunD-
/- mice exhibit shortened life span and increased incidence of aggressive cancers.14-16 Recent
evidence demonstrated an accumulation of ROS in JunD-/- murine embryonic fibroblasts which
was reduced by treatment with ascorbate.17 Gene expression profiling of JunD-/- cells showed
downregulation of several free radical scavenging enzymes associated with an increase in
and anti-inflammatory effects mediated by the endothelium are impaired resultingngg iin nn vavavascscscululularara
damage.8, 9 Although the accumulation of ROS generation is the most widely accepted cause of
vaascscculululaarar aaagigigingng4,1100, , a a thorough understanding of thththe mmolecular meechchc annisissmmms of altered redox
iiignnnala ing in endnddooto hhehelililiala cccelelellslsls rreremamaaininns s ttoto be aacacccommpmpllishhheedd.
AcActititivavvatotorrr prprprototeeinn-n-11 (A(AAP-P-P 1)1)1) isisis aa ccololollelelectctioioion n ofoff dimimimererericicic cococommpmplelelexexex sss mmamaddede bbby y y dididiffffferrrennnt t
members of tthrhrhreeeee fffamamamilili ieii s ofofof DDNANANA bbbindndndininng g g prprprototo eieieinsnsn ::: JuJun,n,n FFFososo aaandndn AAATFTFTF/C/C/CREREREB.B.B 111111-131313 TTTheh se
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
4
expression of ROS-producing NADPH oxidase.17 By contrast, overexpression of JunD blunting
redox signaling abolished ROS production and apoptosis.17, 18
In this study, we tested the hypothesis that JunD may be a critical regulator of vascular
homeostasis by investigating its role in ROS-driven vascular aging.
Methods
A detailed description of the methods used in this study is provided in Supplemental Material.
Animals
Young (6 month old) and old (22 month old) JunD-/- and wild-type (WT) littermates male mice
were obtained from Institut Curie (Paris, France). Control and knockout mice shared an identical
genetic background because both mice lines are of the same C57BL6/129Sv background. Due to
male sterility, the JunD deficient colony was maintained through the breeding of heterozygous
animals.14 Genotyping was performed by PCR on tail samples, as previously described.14 Mice
were housed in temperature-controlled cages (20°C to 22°C), fed ad libitum and maintained on a
12/12-hour light/dark cycle. All animal experiments were approved by the local institutional
animal care committee.
Tissue harvesting and organ chamber experiments
On the day of the experiment, mice were anesthetized through the intraperitoneal administration
of 50 mg/kg sodium pentobarbital and then were euthanized. The chest and abdomen were
opened with a medial sternotomy. The aorta was cleaned from adhering connective tissue under
a dissection microscope and immediately used for organ chamber experiments.
Measurements of mitochondrial O2-, NO and ONOO- from mouse aorta
Superoxide anion was measured in mitochondria isolated from mouse aorta by using ESR-
were obtained from Institut Curie (Paris, France). Control and knockout mice shahaareedd d ananan iiidededentntn icical
genetic background because both mice lines are of the same C57BL6/129Sv background. Due to
mamalelele sssteteteriririlililitytyty,, thhe ee JJuJunD deficient colony was maaaiininttaained throughh tttheh bbrerereeede ing of heterozygous
annimmmals.14 Gennotottypppinnggg wawawasss pepeperrfrfororrmmemedd bbyy PCRCRCR onnn ttaail sssaamampppleeses, aaas pprerevivv oouuslsllyy y ddedescsccririibebebedd.d 14144 MMMicicce e
weweererer hhhououseseed d d inin ttememmpepeeraratutut rre-c-ccononntrtrtrololollleledd d cacacagegeges (2(2(20°0°°CC tooo 222°2°2 CCC),, fefef ddd adadad llibibbitttumumm aandndn mmmaaaintnttaiinenen dd d ononn a
12/12-hour lligigighththt/d/ddararark k cycycyclle.e.e AAAllll anananimimmalall eexpxpxperererimimimenenentststs wwererereee apapapprprprovovoveddd bbby y y thththe e e lolol cacacal l l ininnstststititi utu ional
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
5
spectroscopy analysis, as previously reported.19 In situ measurements of NO and ONOO- were
carried out with electrochemical nanosensors.20, 21
Isolation of mitochondrial and cytosolic fraction
Mitochondria were isolated by centrifugation, as previously reported.19
Mitochondrial swelling assay
Forty μg of isolated mitochondria from mouse aortas in swelling buffer (250 mmol/L sucrose, 10
mmol/L MOPS, 5 μmol/L EGTA, 2 mmol/L MgCl2, 5 mmol/L KH2PO4, 5 mmol/L pyruvate, 5
mmol/L malate) were incubated with 150 μmol/L of calcium chloride (CaCl2) in a final volume
of 200 μL in a 96-well plate for 20 minutes. Absorbance was read every 30 seconds at 520 nm.22
Mitochondrial DNA damage detection
Assessment of mitochondrial DNA damage was performed by using quantitative PCR-based
amplification of a large fragment of mitochondrial DNA, as previously described23.
Western blotting
Frozen samples of aortas were pulverized and dissolved in lysis buffer (120 mmol/L sodium
chloride, 50 mmol/L Tris, 20 mmol/L sodium fluoride, 1 mmol/L benzamidine, 1 mmol/L DTT,
1 mmol/L EDTA, 6 mmol/L EGTA, 15 mmol/L sodium pyrophosphate, 0.8 μg/mL leupeptin, 30
mmol/L p-nitrophenyl phosphate, 0.1 mmol/L PMSF, and 1% NP-40)v for immunoblotting.
Detection of membrane translocation of NADPH subunit p47phox
This method is reported in Supplemental Material.
MnSOD and ecSOD activity
MnSOD and ecSOD activity was measured in isolated mitochondria and aortic lysates,
respectively, by using a commercially available kit (OxiSelectTM Superoxide Dismutase
Activity Assay, Cell Biolabs, Inc. CA, USA).
Mitochondrial DNA damage detection
Assessment of mitochondrial DNA damage was performed by using quantitative PCR-based
ammplplplififificicicatatatioioionn off aaa llaarge fragment of mitochondrriaiaial DDNA, as prevvioioouslylyy dddese cribed23.
WeWeWeststern blotttitiinnng
FrFrozozozenenen ssamammplplpleses ooof f f aoaorrtrtaass wwererre e pupupulvlvlvererizizzededed aaanndnd didid sssssoololveeed dd ininin llysysysisisi bbufufuffefef rr r (11120200 mmmmomomol/l/l/LLL sosooddidiumumm
chloride, 50 mmmmomom l/l/l/LLL TrTrTrisii , 202020 mmmmomom l/l//L LL sososodididiumumum ffflululuororrididde,e 111 mmmmomomol/l/l/L LL bebebenznznzamamamidididinine,e,e, 111 mmmmomm l/L DTT,
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
6
NADPH and xanthine oxidase activity
Commercially available kits were used to assess NADPH and xanthine oxidase activity in aortic
lysates of JunD-/- and WT mice (abcam®, USA).
Aldehyde dehydrogenase 2 activity
Aldehyde dehydrogenase 2 (ALDH2) activity was determined in mitochondria isolated from
mouse aorta of JunD-/- and WT mice, according to the manufacturer recommendations (ALDH2
activity assay kit, abcam®, USA).
Co-immunoprecipitation
This method is reported in Supplemental Material.
Immunofluorescence
Thoracic aorta segments from all mice were embedded in optimum cutting temperature and
stored at -80°C. Five-micrometer-thick slices were then cut, blocked with BSA1% for 1 h, and
incubated overnight at 4°C with anti-3 nitrotyrosine (Upstate Biotechonology, USA), CD31 (BD
Biosciences, CA, USA), p47phox, Nox2, Nox4, p53, p16INK4a and JunD antibodies (Santa Cruz
Biotechnology, Nunningen, Switzerland). Fluorescence-labeled secondary antibodies (Alexa
Fluor 546 and 488, Molecular Probes, USA) were applied for 1 h. Successively, slides were
incubated with 4,6 diamidino-2-phenylindole hydrochloride (DAPI) solution (Vector
Laboratories, CA, USA) for 10 minutes and analyzed by fluorescence microscopy (Olympus).
Quantification of immunofluorescence was performed by counting stained cells on total cells.
JunD promoter methylation in mouse aorta
For each analyzed genomic DNA sample, 30 ng of genomic DNA was digested with methylation
sensitive enzyme (Ms) or with methylation dependent enzyme (Md) and also with both
methylation sensitive and dependent enzymes (Msd) in 15μl total volume including 5X digestion
mmunofluorescence
Thoracic aorta segments from all mice were embedded in optimum cutting temperature and
ttororrededed aaattt -8-8-80°0°0°C. FFFiivive-micrometer-thick slices weeererr tthen cut, blockckkede wiwiwiththth BSA1% for 1 h, and
nncuuubab ted overernininighhht atat 444°C°C°C wwwititithh ananantititi-3-3 nnnitroottyyrrosiinneee (UUUppspstataatete BBiiototecechohohononooloogygygy,, USUSSA)A)A), CDCDCD313131 (BBBD
BiBiioososcicicienene cecees,s,s, CCAA,A, UUUSASAA),),, pp47477phphphoxoxox, , NoNoN x2x2x2,, NoNoNox4x4x4,, pp5p53,3,3 ppp16166INKINK444a aanndnd JJJununu DDD anantititiboboodidieseses ((Saaantntta a CCCruuzuz
Biotechnologoggy,y,y NNNunununniniingngngennn, , , SwSwSwitititzezez rlrlrlanannd)d)d .. FlFlFluououorererescscscenenencecee-l-llabababelele ededed sssecccononondadadaryryry aaantnttibibibodododieieiesss (Alexa
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
7
buffer overnight at 37°C.
Nuclear extracts and JunD binding activity
Nuclear protein and junD binding activity were assessed by using commercially available kits
(Active Motif, Rixensart, Belgium).
In vivo knockdown of JunD
In vivo knockdown of JunD was performed by injecting a predesigned siRNA specifically
targeting JunD (Santa Cruz Biotechnology, Nunningen, Switzerland) together with a cationic
transfection reagent (JetPEI, Polyplus Transfection), as previously reported.19
JunD overexpression in mice
Old male mice (22 months) were injected i.v either with 40μg of cloning vector (Origene,
PCMV6-Kan/Neo, PCMV6KN) or a predesigned mouse JunD cDNA clone (Origene,
MC200652) together with a cationic transfection reagent (JetPEI, Polyplus Transfection),
according to the manufacturer’s instructions.
Telomerase activity
The catalytic activity of telomerase was assessed by quantitative PCR, as described previously.24
Beta-galactosidase staining
See Supplemental Material for a description of this method.
Cell culture
Human aortic endothelial cells (HAECs, passages 5 to 8) were cultured in EGM-2 containing 2%
FCS for 12 h and then transfected with JunD, p47phox, Nox2, Nox4 as well as scrambled
siRNAs. After 72 hours cells were harvested for immunoblotting, ESR measurements and
assessment of NADPH activity.
Real-time PCR
Old male mice (22 months) were injected i.v either with 40μg of cloning vector ((O(Oririr ggeenenene, ,
PCMV6-Kan/Neo, PCMV6KN) or a predesigned mouse JunD cDNA clone (Origene,
MCMCC202020060606525252) ) ) tttogegeethththeer with a cationic transfection n n rereagent (JetPEI,I,, PPollypypypllulus Transfection),
acccooordr ing to tthehe mmmannufuu acacactutturereer’r’r’s s ininnststtruructctiionsss.
TeTeelololomememerarasesese aactctiiiviitity y y
The catalyticc acacactitivivivitytyty ooof f f teelololomemem rararaseses wwwasasas aaassssssesesessesesedd d bybyby qqquauaantntntititi atata iviviveee PCCCR,R,R, aaas s s dededescsccririribebebedd d prprp eviously.242424
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
8
All PCR experiments were performed using the SYBR Green JumpStart kit (Sigma Aldrich,
USA). A detailed description of this methods including primer sequences is reported in
Supplemental Material.
Measurements of O2-, and NO by ESR spectroscopy
O2– and NO levels in intact cells was assessed by electron spin resonance (ESR) spectroscopy
analysis as previously described25
Small interfering RNA transfection
HAECs were transfected at 70–80% confluence using the Lipofectamine reagent (Invitrogen,
Carlsbad, CA, USA) for 4 h at 37°C in EBM-2. Commercially available human JunD, p47phox,
Nox2 and Nox4 siRNAs were purchased from Santa Cruz (Santa Cruz Biotechnology,
Nunningen, Switzerland). As a control, a predesigned scrambled siRNA was used (Microsynth,
5’ UAC ACA CUC UCG UCU CU dTdT 3').
Subjects
Twelve young (24±3 years) and fourteen old (65±6 years) male healthy volunteers were
consecutively enrolled at the Blood Donation Service of the University Hospital Zürich. All of
the subjects were non smokers, normotensive and free of overt cardiovascular disease, as
determined by health history questionnaire and physical examination. All participants signed an
informed consent.
Isolation of peripheral blood monocytes
Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by density gradient
centrifugation over Ficoll-Paque. Blood monocytes were recovered by adherence to 100-mm
plastic tissue culture plates (Griener, Frinckenhausen, Germany) in the presence of 10% fetal
calf serum for 1h at 37°C in 5% CO2/air. Non-adherent cells were removed from monocyte
Nox2 and Nox4 siRNAs were purchased from Santa Cruz (Santa Cruz Biotechnnoolologoggyy,y,
Nunningen, Switzerland). As a control, a predesigned scrambled siRNA was used (Microsynth,
5’ UUUACACAC AAACACACA CCUCUCUC UCG UCU CU dTdT 3').
SSSubbjbjects
TwTwwelelelveveve yyyououungnng ((22424±3±3± yyyeeaarsrs) anana ddd fofofouruurteteeenenn oooldldld (((6665±5±±66 yeyeeararrs)s)s) mmalalalee hehehealalalththyyy vvovoluluuntntnteeeerrsrs wwwererre
consecutivellyyy enenenroroolllllededd aaat ththhee e BlBlBlooooood d DoDoDonananatitit ononon SSSererrvvvicicce e e ofoff tthehehe UUUnininivevev rsrsrsititity y y HoHoHospsps itittalalal ZZZürürüricici h. All of
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
9
monolayers by washing with phosphate buffered saline (PBS). Isolated monocytes were cultured
in RPMI medium supplemented with 10% FBS. Finally, cells were collected for real-time PCR.
Statistical analysis
All data are presented as means ± SEM. Statistical comparisons were made by using the Student
t test for unpaired data and 1-way ANOVA, followed by Bonferroni’s post-hoc test, when
appropriate. Spearman's correlation analysis was used to assess correlation between variables.
Probability values less than 0.05 were considered statistically significant. All analyses were
performed with GraphPad Prism Software (version 5.0).
Results
Genetic deletion of JunD accelerates aging-induced endothelial dysfunction
To investigate the role of JunD in vascular aging, we examined endothelium-dependent
responses in 6 and 22 month old JunD-/- and WT mice. Young JunD-/- mice showed an
impairment of endothelium-dependent relaxation to acetylcholine (10-9-10-6 mol/L) which was
similar to the one observed in old WT (Figure 1A), suggesting a premature endothelial aging in
animals lacking the AP-1 transcription factor JunD. Moreover, old JunD-/- exhibited a more
pronounced impairment of vasorelaxation compared with age-matched WT mice (Figure 1A).
Addition of the free radical scavenger polyethylene glycol-superoxide dismutase (PEG-SOD,
150 U/mL) restored acetylcholine-induced relaxation not only in old WT but also in JunD-/-
irrespective of age (Figure 1B). Endothelium-independent relaxation to sodium nitroprusside
(10-10-10-5 mol/L) was similar in all groups (Figure 1C). These findings indicate a link between
JunD deletion, ROS generation, and endothelial dysfunction.
Mitochondrial derangement and oxidative stress in JunD-/-mice
Results
Genetic deletion of JunD accelerates aging-induced endothelial dysfunction
Too iiinvnvnveesestititigagagatetet thehehe rrolo e of JunD in vascular aginggg,, wwwe examined enenndoththheleleliuium-dependent
eespppono ses in 66 aandndnd 22222 momomontntnth h h oloold d JJuJunDnDn -/-- anddd WWWT mimim cee.. YYoYoununung g JJuJunnDnD-/--/-/ mmiicece sshhohoweweed dd anan
mmpapapairirirmemem ntntnt oooff enennddooththeeliiuiumm-deded pepependndndenenttt rrelelelaxaxaxatttioioion n tooo tt acccetettylylylcccholololininne (1(1(100-999-11100-6-66 mmmololo /L/LL))) whwhhiccch h wwwasss
imilar to thee ooonenen ooobsbsb erere vevv d d d ininn ooldldld WWWT T (((FiFiFigugugurerere 11AAA),)) sssugugggegegestststinining g g a a a prprrememematata ururureee enenndododothththeleleliai l aging in
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
10
Superoxide anion (O2-) was measured by ESR spectroscopy in mitochondria isolated from mouse
aorta to directly assess the oxidative stress burden in the presence or absence of JunD.
Mitochondrial O2- levels were elevated in young animals lacking JunD as in old WT littermates
(Figure 2A). O2- production further increased in 22 month old JunD-/- mice (Figure 2A). To
investigate the impact of JunD deletion on mitochondrial function, mitochondria isolated from
mouse aortas were challenged with calcium overload (150 mol/L CaCl2) and the rate of
mitochondrial swelling was determined by light scattering.
A stable absorbance at 520 nm throughout the 20-minute time course was observed in
mitochondria from young WT. By contrast, mitochondria from young JunD-/- displayed a
significant decrease in absorbance following calcium overload (Figure 2B, Supplemental
Figure 1). Interestingly, young JunD-/- and old WT littermates showed a comparable
mitochondrial disruption (Figure 2B). A similar pattern of mitochondrial DNA fragmentation
and increased caspase 3 activity was observed (Figure 2C, Supplemental Figure 2).
In agreement with increased mitochondrial O2- generation, NO release from single aortic
endothelial cells was markedly impaired in both young and old JunD-/- mice as compared with
age-matched WT littermates (Figure 2D). Blunted NO availability was associated with an early
reduction of both activating eNOS Ser-1177 phosphorylation and total protein in JunD-/- mice
(Figure 2E, Supplemental Figure 3). Furthermore, peroxynitrite (ONOO-) levels and 3-
nitrotyrosine immunostaining were also elevated in young JunD-/- to an even higher level than
aged control animals (Figure 2F and G).
Aging downregulates JunD expression
In agreement with the effects of JunD deletion on endothelium-dependent relaxation,
immunofluorescence analysis of the mouse aorta revealed that JunD expression is mostly
ignificant decrease in absorbance following calcium overload (Figure 2B, Supppplplememmenenentatatal ll
Figure 1). Interestingly, young JunD-/- and old WT littermates showed a comparable
mimitotoochchchoonondrdrdriiaial l didisrsrsruupuption (Figure 2B). A similarrr pppaatttern of mitocchohoh nddriririaalal DNA fragmentation
anndd d ini creased d cacaspsppassee 33 acaccttitivivivittyty wwwasasas oobsbsservveeddd (Fiiigguureee 222C,C,, SSupuppplplememenennttaal l FiFiFigugureree 222).).
InIn aaagrgrgreeeememementnt wwwititth h inincrcrc eaeaasesesed d d mimiitooochchchonono drdrdriaiaal OOO22-- gggeneneneeeratatatioioi n,n, NNNOO reeeleleasasase e frfromomom sinnnglglle e aoaoortrttici
endothelial ceceelllll sss wawawass s mamamarkkkedededlyll imimimpapaairi ededed iiin n n boboboththh yyyouououngngn aaandndnd oooldldd JuJuJ nDnDnD //-/- mmmiciciceee asasas cccomomompapapared with
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
11
confined to the vascular endothelium (Figure 3A). To strengthen the link between AP-1
transcription factor JunD and age-dependent, ROS-mediated endothelial dysfunction, its protein
expression was assessed in aortic lysates obtained from young and old WT mice. Interestingly,
old mice showed a downregulation of JunD expression compared with young animals (Figure
3B). This finding was confirmed by immunofluorescence analysis showing reduced JunD-
specific staining (Figure 3C). As expected, JunD-/- mice did not show any protein signal (Figure
3C).
Then, we investigated whether JunD downregulation was induced at the transcriptional
level by epigenetic changes.
Quantitative analysis of JunD promoter methylation, an important repressor of gene
transcription, showed a significant increase in methylated CpG dinucleotides in old as compared
with young mice (Figure 3D).
Together with blunted expression, we found that JunD transcriptional activity is reduced
in aged vessels (Figure 3E). The tumor suppressor menin, a critical modulator of JunD activity,
co-precipitated with JunD only in the aorta of old mice leading to blunted JunD phosphorylation
(Figure 3F, Supplemental Figure 4).
To further elucidate the relevance of aging-induced JunD downregulation, silencing of
JunD was performed by injecting i.v. JunD siRNA together with a cationic transfection reagent.
A predesigned scrambled siRNA, used as a negative control, did not affect JunD expression
(Supplemental Figure 5A). Interestingly, as observed for JunD-/- mice, siRNA-mediated
knockdown of JunD exerted a significant impairment of acetylcholine-induced vasorelaxation
both in young and old WT mice (Supplemental Figure 5B).
Restoration of JunD expression improves vascular function in old mice
Quantitative analysis of JunD promoter methylation, an important represssssorr off f gegegenenene
ranscription, showed a significant increase in methylated CpG dinucleotides in old as compared
wiwiththh yyyoououngngng mmmice ee (((FFFigure 3D).
Togethherer wwwitthhh blbll nununtetetedd d exexprprpreesessiionn, wwwe foundndnd thahaattt JJuunnDDD trtrannnsccririipptp iioonanaal l acactitiivivivitytyt iiis rrereduuducceed tt
nn aaagegegedd d vevesssssseleelss (((FiFiigugug rrre 33EE). TThehehe tttumuumooror sssupupupprprpresesessosoor r memeenininin,n,n, aa cccririr ttiicacaall l mommoddudulalaatototor rr ofoff JJJuuunDDD aacactitivvvitytyty,
co-precipitateteed d d wiwiwiththth JJunununD onononlyll iiin n n ththhe e aoaoaortrtrta aa ofofof oooldldd mmmicici e leleleadadadinining g g tototo bbblllfff unununtetetedd d JuJuJunDnDD ppphohohospspsphorylationnn
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
12
Since JunD deletion is associated with early endothelial dysfunction we determined whether
restoration of its expression exerts protective effects against age-induced, ROS-dependent
endothelial dysfunction. In vivo overexpression of JunD was performed by injecting i.v. a
predesigned cDNA clone. This approach resulted in a significant overexpression of JunD in the
aorta of old mice whereas injection of cloning vector did not exert any significant effect
(Supplemental Figure 6). Interestingly, JunD overexpression improved acetylcholine-dependent
relaxation compared with vector-treated mice (Figure 3G). This finding indicates that JunD is
critically involved in age-dependent endothelial dysfunction.
JunD affects the balance between ROS-scavenging and -generating enzymes
Expression and activity of MnSOD, ecSOD as well as ALDH2 was decreased in the aorta of
JunD-/- mice as compared with age-matched littermates (Figure 4 A-C). Similar findings were
observed for glutathione peroxidase-1 (GPX-1) and xanthine oxidase (Supplemental Figure 7).
By contrast, NADPH oxidase subunits p47phox, Nox2 and Nox4 were already upregulated in
young JunD-/- and further increased with aging (Figure 5A-C, Supplemental Figure 8).
Upregulation of NADPH isoforms was mainly confined to the vascular endothelium, as assessed
by immunofluorescence (Figure 5A-C). These findings were supported by a significant increase
of NADPH activity as well as p47phox membrane translocation in JunD-/- mice as compared
with age-matched littermates (Figure 5D, Supplemental Figure 9). Consistently, endothelial
superoxide anion generation was elevated in young JunD-/- and further increased in old mice
(Supplemental Figure 10).
To further investigate the link between JunD and NADPH in the vascular endothelium,
selective dowregulation of JunD was achieved by using siRNA technology. Interestingly, pre-
treatment with the NADPH inhibitor apocynin blunted impairment of acetylcholine-induced
Expression and activity of MnSOD, ecSOD as well as ALDH2 was decreased inn tthehee aaoroortatata ooof f f
JunD-/- mice as compared with age-matched littermates (Figure 4 A-C). Similar findings were
obbseseservrvvededed fffororo ggluutatatatththione peroxidase-1 (GPX-1) aaandndd xanthine oxidddasa e (((SuSuSupplemental Figure 7).
BBy ccontrast, NNADADADPHPHPH ooxixixidadadasesese susububuunninitsss ppp47ppphooox, NoNoN x222 aaandndd NNNoxox44 wewereree aalrlreaeaadydydy uuprppregegegululu atttedeed iiin n
yoyoununu ggg JuJunDnDD-/--/--/- anannd fufurtrttheheer r ininccrcreaeaasesesed d d wiwiiththh aaaggigingngg ((FFFiggugurrre 555AA-A-C,C,C, SSSupupupplplp eemmenentatatal l FiFiF gugugurrre 888)...
Upregulationn ooof f f NANANADPDPDPH HH isissofofofororrmsmsm wwwasss mamamainininlylyly cconononfifiinenn d d d tototo tthehehe vvvasaa ccculululararar eeendndndotothehehelilil umumum,,, as assesseddd
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
13
relaxation in the aortic rings of JunD siRNA-treated young and old mice as well as in old WT
animals (Figure 5E).
JunD deletion accelerates ROS-induced vascular aging
Since JunD deletion is associated with an early burst of oxidative stress we investigated markers
of vascular senescence in age-matched WT and JunD-/-. Telomerase activity was blunted in
young JunD-/- as compared with age-matched WT mouse aorta and no further impairment was
observed upon aging. This finding indicated a vascular senescence phenotype in young animals
lacking JunD (Figure 6A). -galactosidase staining further supported the early occurrence of
vascular aging in JunD-/- mice (Figure 6B). Accordingly, the expression of aging markers such
as tumor suppressor p53 and cyclin dependent kinase inhibitor p16INK4a was increased in these
mice (Figure 6 C-D, Supplemental Figure 11).
Gene silencing of JunD in human endothelial cells
To translate the findings observed in JunD-/- mice to the human endothelium, we downregulated
JunD in human aortic endothelial cells using siRNA technology. Selective JunD siRNA
markedly reduced its expression, while scrambled siRNA did not exert any effect (Figure 7A).
In line with our findings in the mouse, knockdown of JunD elicited a marked upregulation of
NADPH isoforms and increased enzyme activity (Figure 7A-B, Supplemental Figure 12).
These findings were associated with O2- overproduction and blunted NO availability, as assessed
by ESR spectroscopy (Figure 7C-D). Of interest, the NO/O2- balance was restored either by
NADPH inhibitor apocynin or concomitant knockdown of the NADPH oxidase subunits
p47phox and Nox2, indicating that this enzyme critically regulates ROS generation in the
absence of JunD (Figure 7C-D, Supplemental Figure 13). By contrast, Nox4 downregulation
did not exert any inhibitory effect on superoxide anion generation (Supplemental Figure 13). In
as tumor suppressor p53 and cyclin dependent kinase inhibitor p16INK4a was incrreeeasesesedd d ininin ttthehehesese
mice (Figure 6 C-D, Supplemental Figure 11).
GeGennene sssilililenenncicicinnng ooofff JuJ nD in human endotheliall cecc lllls
TTo tttrar nslate thehe fffinnndidiinnngsss obobobseseserrvrvededd iinn n JJJunnnD-/--/-/- mmmiceee ttoo thehehe hhumummanan eenndndotothehh lliumumm,, wewe dddowowownnnreegegulululattteed
JuunDnDnD iiinn n huhuumamam n n aaoaorrtrticic enndndoto hheheliliialalal ccceleellslss usususininingg g sisisiRNRNRNAAA teeechchhnononolooogygyg .. SeSeSelelel ccctivvvee JuJuJunDnDnD sssiRiRRNANAA
markedly redducucucededed iiitstst eeexpxpx reeessssssioii n,n,n, wwwhihih leee sscrcrcraaambmbmblelel d d d sisiiRNRNRNA A A dididid dd nononot t exexexererert t ananany y y efefffefefectctct (((FiFiFigug re 7A).
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
14
agreement with increased ROS generation, PEG-SOD blunted O2- and preserved NO availability
following silencing of JunD (Figure 7C-D). Moreover, we also confirmed that JunD is required
for eNOS expression and activation in human endothelial cells. Indeed, eNOS protein expression
and Ser1177 activating phosphorylation were significantly reduced by JunD knockdown (Figure
7E).
Age-dependent downregulation of JunD in old healthy subjects
To exploit the role of JunD gene in human vascular aging we assessed JunD mRNA expression
in peripheral monocytes obtained from young and old human subjects free of overt
cardiovascular disease and risk factors. Interestingly, JunD protein and gene expression were
significantly reduced in old compared with young individuals (Figure 8A-B). Moreover, mRNA
levels significantly correlated with gene expression of scavenger MnSOD, ecSOD (Figure 8C-
D) as well as NADPH p47phox and Nox2 subunits (Figure 8E-F). These results suggest that
downregulation of JunD may represent an important mechanism of increased oxidative stress
burden in human aging.
Discussion
JunD is emerging as a major gatekeeper against oxidative stress15, but its contribution to ROS
homeostasis in the vasculature remains unknown. The present study demonstrates for the first
time that genetic deletion of JunD is associated with premature endothelial dysfunction and
vascular aging via ROS generation. Several lines of evidence support our conclusions. In
contrast to young WT, age-matched mice lacking JunD showed an early impairment of
endothelium-dependent relaxation which was restored by ROS scavenging enzyme PEG-SOD. A
marked increase of mitochondrial O2- paralleled by reduced endothelial NO bioavailability and
ignificantly reduced in old compared with young individuals (Figure 8A-B). MMoMorerereovvverrer,, mRmRmRNNA
evels significantly correlated with gene expression of scavenger MnSOD, ecSOD (Figure 8C-
DD) ) asaas www lelellll asaa NADADADPHP p47phox and Nox2 subunininitsts (Figure 8E-FF).)) Thehehessese results suggest that
ddodowwnwnregulationon ooff f JuunDnDnD mmmayayay rrrepeprereressesenntt aaan immmppporttaannnt mmmeecechahaanniismsmm oof f inincccreeaaseseed d d oxoxidididatatativivee e stststreressss
buurdrdrdenenen iinn huhuhummamannn agagagining.g.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
15
elevated ONOO- concentrations, were already found in the aorta of young JunD-/- mice. JunD
deletion was associated with downregulation of free radical scavengers as well as increased
expression and activity of ROS-generating NADPH oxidase. JunD expression was reduced in old
as compared with young WT mice, implicating that the protective role of this transcription factor
is diminished with aging. Interestingly, overexpression of JunD rescued age-induced endothelial
dysfunction. Oxidative stress burden in JunD-/- mice led to premature vascular senescence. Last
but not least, JunD was downregulated in peripheral monocytes of old healthy subjects and
correlated with scavenging and oxidant enzymes, suggesting a potential translation of our
findings to the clinical context.
The AP-1 transcription factor JunD modulates different target genes involved in
proliferation, growth, and survival13, 26, 27. Gerald and colleagues previously reported an
increased ROS generation in immortalized JunD-/- cells.17 By contrast, redox signaling involved
in tumor angiogenesis was blunted by JunD overexpression. Notably, JunD-/- mice display
reduced lifespan.14-16 In the present study, double immunostaining with the endothelial marker
CD31 showed that JunD is expressed in the vascular endothelium. Hence, we investigated
whether its genetic deletion was associated with a premature oxidative phenotype in the
vasculature.
JunD-/- mice displayed a ROS-dependent impairment of endothelium-dependent
relaxation already at young age. Indeed, the free radical scavenger SOD restored endothelial
function. In line with these findings, O2- generation, peroxynitrite levels and 3-nitrotyrosine
immunostaining were also prematurely elevated while NO availability was blunted in young
mice carrying the genetic disruption of JunD. These mice also showed increased DNA
fragmentation, swelling of the mitochondria and caspase 3 activation, markers of apoptosis. The
The AP-1 transcription factor JunD modulates different target genes invoolvllvedede iin nn
proliferation, growth, and survival13, 26, 27. Gerald and colleagues previously reported an
nncrcrreaeaeasseseddd ROROROS gegegenneneration in immortalized JunDDD-/-/-/- ccells.17 By contntntrar stt,, rereredod x signaling involved
nn tuumumor angioogegeenneesisiiss s wwawasss blblbluununteteddd bbby y JuJuunDD ovvereeexxppreesssssioionnn. NNotottabbblyly,dd JuJuJ nDnDD-/--/--/- mimicecece dddisisi plplp aayay
eedududucececedd d lilifefefespsspanan..1444-1616 IIIn n ththee prprpresessenenent t t ststuududy,y,y dddououublblb ee immmmmumunononosststaaainininininng g wiwiw ththh ttthehe eeendndndotothehehellialall mmmararkkkerr r
CD31 showeed d d thththatatat JJJunununD DD isss eexpxx rereresssss ededed iinn n thththe e e vavavascscs ulululararr eendndndotototheheheliliiumumum. HeHeHencncnce,e,e, wwwe e inininvevevestststigigigated
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
16
increase in oxidative stress was associated with impaired balance between ROS-scavenging and
producing enzymes. Expression and activity of ecSOD, MnSOD, GPX1 and xanthine oxidase
were significantly reduced in knock-out mice, suggesting that JunD is essential for their
transcription. In addition, the mitochondrial reductase ALDH2 was almost abolished in young
JunD-/- as compared with age-matched littermates. This scavenging enzyme has been recently
reported to reduce ischemia-reperfusion injury and to protect against cardiac arrhythmias.28-30
We show that JunD deletion is affecting ALDH2 expression and activity in mouse aorta and its
downregulation may contribute to the abnormal vascular redox state. Our findings are in line
with the established role of AP-1 in the activation of several genes encoding a set of detoxifying
defensive proteins. Indeed, antioxidant response elements (ARE) have been identified in the
promoters of scavenging enzymes.31 By contrast, we found that p47phox, Nox2 and Nox4, key
subunits of ROS-generating NADPH oxidase were upregulated in the endothelium of young
JunD-/- mice. Notably, the NADPH inhibitor apocynin was able to improve acetylcholine-
induced vasorelaxation in the aorta of WT mice with downregulation of JunD induced by siRNA
technology. In this regard, we have recently reported that in vivo delivery of siRNA with a
cationic reagent targets the vascular endothelium.19 As observed for JunD-/- mice, silencing of
JunD elicited a similar impairment of vasorelaxation. Transient knockdown of the transcription
factor allowed us to investigate the effects of gene downregulation more specifically than in the
setting of genetic deletion, increasing the physiological relevance of our findings. On the other
hand, we also showed that overexpression of JunD blunted endothelial dysfunction in old mice.
This latter experiment strongly suggests that modulation of JunD expression plays a role in ROS-
mediated vascular dysfunction.
The oxidant-rich milieu observed in young JunD-/- mice was associated with increased
defensive proteins. Indeed, antioxidant response elements (ARE) have been identnttififieiei dd d ininin ttthehehe
promoters of scavenging enzymes.31 By contrast, we found that p47phox, Nox2 and Nox4, key
uubububunininitststs ooof f f ROROR S-S--ggegennerating NADPH oxidase weweererr upregulated innn thee eeenndndothelium of young
JuJunDnDD-/- mice. NoNoNotaaabllyy,, thehehe NNNADADADPHPHH iinnhhiibbitorr aaapoccycynninn n wwawas s ababblele too imimppproovove e acaccetetylylylchchholollinini ee-e-
nndududucececedd d vavaasososorerelalaaxaaatitionon iiin n ththeee aoaoortrtrta a a ofof WWWTT T mmimicecece wwwitthth dddowowwnnrnregeggululu atata ioioion n n ooof JJununnD D D inini dududucccedd d byyy ssiRiRRNNANA
echnology. InInn ttthihih ss s rerer gagagardrr ,, wewewe hhhavavave e rerer cececentntntlylyly rrrepepepooortrtrtededed tthahaat tt ininin vvvivivivooo dedeelililiveveveryryry ooof f sisisiRNRNRNA A A wiww th a
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
17
NO breakdown, as indicated by elevated ONOO- and protein nitrosylation. However, we also
found a decreased expression of eNOS in the aorta of these mice which may contribute to the
reduced NO bioavailability. Previous evidence reported that AP-1 is required for eNOS
transcription.32 Interestingly, our findings suggest that AP-1 complexes lacking JunD may not be
able to warrant eNOS gene expression. JunD inactivation also blunted eNOS Ser1177
phosphorylation, in line with the notion that ROS suppress enzyme activity.33 In addition we
showed that oxidative stress in JunD-/- was associated with early features of vascular aging,
including reduced telomerase activity, increased -gal staining as well as upregulation of the
senescence markers p53 and p16INK4a. Importantly, the extent of vascular senescence observed in
young JunD-/- mice was similar as in old WT mice, suggesting that JunD deletion accelerates
ROS-driven vascular aging. The importance of premature senescence in this setting is
strengthened by the notion that cardiovascular disease, in particular myocardial infarction and
stroke, exhibit a strong age-dependency.34
To test JunD role in the human endothelium, JunD siRNA-mediated knockdown was
performed in HAECs. Silencing of JunD increased NADPH activity and ROS generation,
whereas concomitant treatment with apocynin or knockdown of p47phox and Nox2 subunits
prevented oxidative stress, confirming that JunD is a critical modulator of NADPH activity also
in human endothelial cells. By contrast, gene silencing of Nox4 did not abolish superoxide
production in line with the observation that this subunit is involved in H2O2 generation.35
Although Nox4 is upregulated in aging35 and Nox4-derived H2O2 has been recently linked to
DNA damage, mitochondrial dysfunction and senescence phenotypes36-38, other evidence suggest
a protective role against vascular ischemic or inflammatory stress.39, 40 Whether the observed
Nox4 upregulation represents a futile compensatory mechanism or contributes to vascular aging
young JunD-/- mice was similar as in old WT mice, suggesting that JunD deletiononn aacccc elelelerreratatatesese
ROS-driven vascular aging. The importance of premature senescence in this setting is f
ttrereengngngthththenennededed bby y thththee notion that cardiovascular dddisisiseaase, in particulularaa mmyoyoyoccardial infarction and
ttroookke, exhibit t a a sttroronngg aaagegee-d-d-deepepenenndededencnccyy.3434
ToTo ttteseest t JuJuunnDD rrololee e inin ttthehehe hhhumumumanann eendndndootothehehelilil umumum,, JuJuunDnDnD ssiRRRNANANA-m-m-mededdiaaateted dd knknnocockdkdkdowwwn wawasss
performed inn HHHAEAEAECsCsC .. SiSS leencncncinini g g g ofofo JJJununnDDD ininincrcrcreaeaeaseseed d d NANAN DPDPDPH H H acacctititivivv tytyty aaandndnd RRROSOSO gggenenenerereratata ion,
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
18
in JunD-/- mice remains unclear.
Another important finding of the present study is that JunD expression was
downregulated in old as compared with young WT animals. Since oxidative stress contributes to
aging, we speculate that JunD is crucial in maintaining a redox balance in the vasculature, while
its deletion is associated with premature ROS-mediated endothelial dysfunction. This conclusion
is supported by our previous work showing that normalization of ROS prevents age-related
vascular dysfunction.41 In our effort to elucidate the molecular basis of age-dependent JunD
downregulation, we investigated the epigenetic modulation of JunD expression at the
transcriptional level. Quantitative analysis of JunD promoter methylation showed a significant
hypermethylation of CpG dinucleotides in old as compared with young mice. Indeed, DNA
methylation is an important repressor of gene transcription in mammals.42 Beside epigenetic
changes, we found that JunD transcriptional activity was reduced in aortas of aged vs. young
mice. In this regard, it was recently shown that the tumor suppressor menin binds JunD leading
to inhibition of its activity.43 Accordingly, in our experimental setting, menin co-precipitates
with JunD only in aged but not in young vessels and this interaction inhibits JunD activating
phosphorylation. Hence, transcriptional and posttranslational modification may explain the
decrease of JunD expression and activity in the vasculature of old mice.
Of note, JunD gene expression was downregulated in peripheral monocytes of old
healthy subjects and significantly correlated with ROS-scavenging and generating enzymes.
Although we did not confirm the age-dependent decline of endothelial function in our cohort,
several studies have reported a strong correlation between age and endothelial dysfunction.44-46
In conclusion, the present work demonstrates for the first time that JunD is critically
involved in age-induced oxidative stress in the endothelium and controls vascular senescence.
hypermethylation of CpG dinucleotides in old as compared with young mice. Indnddeeeedd,d DDDNANANA
methylation is an important repressor of gene transcription in mammals.42 Beside epigenetic
chhananangegeges,s, wwwe ee fffoununnddd tht at JunD transcriptional actiiivvivittyy was reduced d ini aaororortatatas of aged vs. young
mmiccece. In this reregagaarrdd, ititit wwwasass rrecececeentntlylyly sshhoowwwn tthahaat thhee ttumomomorr susuuppppprreeesssor r mmmenninin n bibibindndsss JuJuunDnDnD llleeaeadidid nngng
oo iiinhnhnhibibibititioioonn n ofof iittts aactctiiiviitity.y.433 AAAccccccorororddidingngglylyy,, ininin oooururu eexpxpxpeererimimmenenentaaalll ssetete tititingngn ,,, mmemeniniin nn cococ -p-p-prerereciipppittatatetesss
with JunD onnlylyly iin n n agagagededed bbututut nnnotoo iiin n n yoyooununng gg vevevesssssselelels s ananand d d ththisisis iiintntnterereracacactitit ononn iiinnnhihihibibibitststs JJunununD D D acacactit vating
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
19
The strength of our study is the consistent observation of JunD as a protector of endothelial
homeostasis in different experimental settings including knockout mice, human endothelial cells
and healthy individuals.
In perspective, these findings may provide the rationale to pharmacologically modulate
JunD expression for the prevention of cardiovascular disease.
Funding Sources: This study was supported by grants from the Swiss Heart Foundation,
Fondazione Roma, Italy (to F.C.) and the Swiss National Research Foundation to T.F.L (3100-
06811802/1). F.P was the recipient of a PhD program in Experimental Medicine at the
University of Rome “Sapienza” and B.M. of a post-doctoral fellowship from INSERM and the
French "Association pour la Recherche sur le cancer (ARC)".
Conflict of Interest Disclosures: None
References: 1. Sawabe M. Vascular aging: From molecular mechanism to clinical significance. Geriatr Gerontol Int. 2010;10 Suppl 1:S213-220. 2. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012;110:1109-1124. 3. Cosentino F, Francia P, Camici GG, Pelicci PG, Luscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66shc protein. Arterioscler Thromb Vasc Biol. 2008;28:622-628. 4.Chen K, Keaney JF, Jr. Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep. 2012;14:476-483.
5. van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731-1744. 6. Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial nitric oxide synthase activity. Cardiovasc Res. 1999;43:274-278.
y p p p
French "Association pour la Recherche sur le cancer (ARC)".
Conflict of Interest Disclosures: None
RReffeferences:
1.. SSSawawwababee M.M.M VVaasascucuc lalaar agaga iningg:g: FFFrororom mm mmomolllecececuuulararar mmmececchahaaninismsmsm to oo clclc iininicicicalala sssigggninififificacaancnce.ee. GeGeerriaatatrr Gerorontnt lol IIntn . 2020100;1;100 Suppppl 1:1:S2S21313-2200. .
22 DDaiai DDFF RRababininovovititchch PPSS UUngngvavariri ZZ MMititocochohondndririaa anandd cacardrdioiovavascsculularar aagigingng CiCircrc RReses
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
20
7. Toda N. Age-related changes in endothelial function and blood flow regulation. Pharmacol Ther. 2012;133:159-176. 8. Epstein SE, Lassance-Soares RM, Faber JE, Burnett MS. Effects of aging on the collateral circulation, and therapeutic implications. Circulation. 2012;125:3211-3219. 9. Camici GG, Sudano I, Noll G, Tanner FC, Luscher TF. Molecular pathways of aging and hypertension. Curr Opin Nephrol Hypertens. 2009;18:134-137. 10. Labunskyy VM, Gladyshev VN. Role of reactive oxygen species-mediated signaling in aging. Antioxid Redox Signal. 2012; doi:10.1089/ars.2012.4891. 11. Hai T, Curran T. Cross-family dimerization of transcription factors fos/jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991;88:3720-3724. 12. Persengiev SP, Green MR. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis. 2003;8:225-228. 13. Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian jun proteins: Redundancy and specificity. Oncogene. 2001;20:2378-2389. 14. Thepot D, Weitzman JB, Barra J, Segretain D, Stinnakre MG, Babinet C, Yaniv M. Targeted disruption of the murine jund gene results in multiple defects in male reproductive function. Development. 2000;127:143-153. 15. Laurent G, Solari F, Mateescu B, Karaca M, Castel J, Bourachot B, Magnan C, Billaud M, Mechta-Grigoriou F. Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab. 2008;7:113-124. 16. Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, Richardson M, Rigaill G, Parrini MC, Lucchesi C, Bellanger D, Stern MH, Dubois T, Sastre-Garau X, Delattre O, Vincent-Salomon A, Mechta-Grigoriou F. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211-230.
17. Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, Pouyssegur J, Yaniv M, Mechta-Grigoriou F. Jund reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781-794. 18. Zhou H, Gao J, Lu ZY, Lu L, Dai W, Xu M. Role of c-fos/jund in protecting stress-induced cell death. Cell Prolif. 2007;40:431-444. 19. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Luscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66shc suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012; 111:278-29. 20. Erdei N, Toth A, Pasztor ET, Papp Z, Edes I, Koller A, Bagi Z. High-fat diet-induced
13. Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian jun proteins: Redduuundadad ncnccy y y ananand dd pecificity. Oncogene. 2001;20:2378-2389.
14. Theppot D, , WeW itzman JB, Barra J, Segretain D, Stinnakre MG, Babinet C, Yaniv M. Targeteddiisrsrrupupuptititiononn ooofff thee mmumurine jund gene results in muuultltltippple defects in mamam le rrrepepepror ductive function. DeDeDeveveelol pmennttt... 202000000 ;1;127277:1:1143434 -1-1535353..
15155. LLaL urent G,G SSolollari FF, Maatteeeesccu u B,B,B KKKaaaracaaa MMM, CCCaasastetelll J,J,J, BBBoouuraachchhot BB, MaMaMagngnnanan CCC, BBiBillllaauud M,M,M MeMeechchchtatata-G-Griririgoggoririoouou FF. . OxOxOxididattivivive e stststrereressss cccononontrtrtriibibutututeses too o agaggininngg g bybyby eeetttttt nhnhhananancicic nnng pppananncrcrreaeae titiic c c anngggioogogenennesessisi and d ininsulilin n signgnalining.g. Cellll MMetetabab. 202 08;7;7:1:11313-1-1242 .
1616 ToToulullelecc AA GGereralaldd DD DDesespopouyuy GG BoBoururacachohott BB CCarardodonn MM LLefeforortt SS RRicichahardrdsosonn MM RRigigaiaillll
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
21
reduction in nitric oxide-dependent arteriolar dilation in rats: Role of xanthine oxidase-derived superoxide anion. Am J Physiol Heart Circ Physiol. 2006;291:H2107-2115. 21. Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676-678. 22. Borillo GA, Mason M, Quijada P, Volkers M, Cottage C, McGregor M, Din S, Fischer K, Gude N, Avitabile D, Barlow S, Alvarez R, Truffa S, Whittaker R, Glassy MS, Gustafsson AB, Miyamoto S, Glembotski CC, Gottlieb RA, Brown JH, Sussman MA. Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circ Res. 2010;106:1265-1274. 23. Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtdna damage and repair using quantitative pcr. Methods Mol Biol. 2002;197:159-176. 24. Kajstura J, Bai Y, Cappetta D, Kim J, Arranto C, Sanada F, D'Amario D, Matsuda A, Bardelli S, Ferreira-Martins J, Hosoda T, Leri A, Rota M, Loscalzo J, Anversa P. Tracking chromatid segregation to identify human cardiac stem cells that regenerate extensively the infarcted myocardium. Circ Res. 2012;111:894-906. 25. Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, Doerries C, Horvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: Restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163-173. 26. Hernandez JM, Floyd DH, Weilbaecher KN, Green PL, Boris-Lawrie K. Multiple facets of jund gene expression are atypical among ap-1 family members. Oncogene. 2008;27:4757-4767. 27. Jochum W, Passegue E, Wagner EF. Ap-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401-2412. 28. Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, Munzel T, Burkle A, Bachschmid MM, Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280-289. 29. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493-1495. 30. Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, Mochly-Rosen D, Levi R. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation. 2010;122:771-781. 31. Venugopal R, Jaiswal AK. Nrf2 and nrf1 in association with jun proteins regulate antioxidant
nfarcted myocardium. Circ Res. 2012;111:894-906.
25. Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, DDoeoeerrrrrrieiesss CCC, rHorvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant tress impairs in vivo reendothelialization capacity y of endothelial progegenitor cells from patients
wiwiththh tttypypypee 222 didid abbeteteteeses mellitus: Restoration by theee pepeperroxisome proliliifef raatototorr-r-activated receptor-gagaammmmma agonnniiist t roroosisis glglitittazaza ononone.e. CiCiCircrcrcululatatioionn.. 20200707;1;1166:163633-1-1173737 .
26266. HHeH rnandedez z JMMM, Flloyoyyd DDHH,, , WeW ilillbabab ececcher KKKN, GrGrG eeeen n n PLPLPL,, BBBoriiis--Lawwwriiee KKK. MMulttipiplele ffaaccetss oof uundndnd gggenene ee exexexprpresesssiiionon aareree aatyypipip cacaalll amamamononng gg apapap-1-1 fffamammililly y mememembmbmbererrs.s.s OnOnOncococ gggennene. 20202008080 ;2;2;277:7:47775777-4-476767677.
27. Jochum WWW,, , PaPaPassssssegegegueueu EEE, , , WaWaWagngngnererr EF.F.F AAAp-p-p 111 ininin mmmououo seee dddevevevelelelopopopmemementntnt aaandndnd tttumumumorororigigigenenenese is.OnOncocogegenene 20200101;2;20:0:24240101 2-2414122
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
22
response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145-3156. 32. Srinivasan S, Hatley ME, Bolick DT, Palmer LA, Edelstein D, Brownlee M, Hedrick CC. Hyperglycaemia-induced superoxide production decreases enos expression via ap-1 activation in aortic endothelial cells. Diabetologia. 2004;47:1727-1734. 33. Ladurner A, Schmitt CA, Schachner D, Atanasov AG, Werner ER, Dirsch VM, Heiss EH. Ascorbate stimulates endothelial nitric oxide synthase enzyme activity by rapid modulation of its phosphorylation status. Free Radic Biol Med. 2012;52:2082-2090. 34. Kovacic JC, Moreno P, Nabel EG, Hachinski V, Fuster V. Cellular senescence, vascular disease, and aging: Part 2 of a 2-part review: Clinical vascular disease in the elderly. Circulation. 2011;123:1900-1910.
35. Touyz RM, Montezano AC. Vascular nox4: A multifarious NADPH oxidase. Circ Res. 2012;110:1159-1161. 36. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Al Ghuzlan A, Bidart JM, Schlumberger M, Dupuy C. Ros-generating NADPH oxidase nox4 is a critical mediator in oncogenic h-ras-induced DNA damage and subsequent senescence. Oncogene. 2012;31:1117-1129. 37. Lener B, Koziel R, Pircher H, Hutter E, Greussing R, Herndler-Brandstetter D, Hermann M, Unterluggauer H, Jansen-Durr P. The NADPH oxidase nox4 restricts the replicative lifespan of human endothelial cells. Biochem J. 2009;423:363-374. 38. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253-1264. 39. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, Shibata R, Sato K, Walsh K, Keaney JF, Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731-740. 40. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012;110:1217-1225. 41. Francia P, Schiavoni M, Gatti CD, Bachschmid M, Savoia C, Volpe M, Luscher TF, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;108:254-254. 42. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145-2156.
36. Weyemi U, Lagente-Chevallier O, Boufraqech M, Prenois F, Courtin F, Caiilllllouuu BB, , TaTaTalblblbototo MDardalhon M, Al Ghuzlan A, Bidart JM, Schlumberger M, Dupuy C. Ros-generatatinininggg NANANADPDPDPHHHoxidase nox4 is a critical mediator in oncogenic h-ras-induced DNA damage and subsequent enescence. OnO cogegene. 2012;31:1117-1129.
37377. LLeLener B,B, KKKozozzieieiel l R,R,, PPPiririrchchc erer HHH,, HuHutttterer EE,,, GrGreue sssssiing g R,R,R, HHHere ndnddllelerr-r-BrBrananndsd teettttererer DDD, , HeHermrmrmanannnn n M,M UUntteterluggauer r HH,H JJJanansssenn-n-DuDuDurrrrrr PP. ThThThe e NANADPDPPHHH oxxxidddaseee nnonoxx4x4 rresestricictsts ttthehee rrepepeplililicacatititivevee lllifififessspapannn ooof huhuhummaman endooththeliiaialll celllls.. Biiooochhhem m JJ... 22000009;4232323:36633--3374744. . JJJ
38. AgAgo T,T, KKuru odo a a J,J, PPaiinn J, FFu u CC, Li H,H SaSadodoshshimima J.J UUprpregegulu attioion n ofof noxox4 4 byby hhypperertropophihic ctimuli prommotototesese aaapopopoptptp osoo isss aaandndn mmmititococo hohohondndndriririalalal dddysysy fufufuncncn titiiononon iin nn cacacardrdr iaaac c c mymymyocococytyty eseses... CiCiCircrcrc RRes.
20201010;1;10606:1:1252533-12126464
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
23
43. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both mll and jund but has opposite effects on transcription. Nature. 2012;482:542-546. 44. Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of no availability and oxidative stress in humans. Hypertension. 2001;38:274-279. 45. Wray DW, Nishiyama SK, Harris RA, Zhao J, McDaniel J, Fjeldstad AS, Witman MA, Ives SJ, Barrett-O'Keefe Z, Richardson RS. Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension. 2012;59:818-824. 46. Ghiadoni L, Faita F, Salvetti M, Cordiano C, Biggi A, Puato M, Di Monaco A, De Siati L, Volpe M, Ambrosio G, Gemignani V, Muiesan ML, Taddei S, Lanza GA, Cosentino F. Assessment of flow-mediated dilation reproducibility: A nationwide multicenter study. J Hypertens. 2012;30:1399-1405.
Figure Legends:
Figure 1. (A) Age-dependent changes of endothelium-dependent relaxation in aortic rings from
WT and JunD / aortas. Line graphs show concentration–response curves to acetylcholine (Ach).
(B) Effect of free-radical scavenger polyethylene glycol-superoxide dismutase (PEG-SOD) on
response to Ach. (C) Endothelium-independent relaxation to the NO donor sodium nitroprusside
(SNP) across the experimental groups. Results are presented as mean ± SEM, n= 5-7 per group,
*P<0.05. NE, norepinephrine.
Figure 2. (A) ESR spectroscopy analysis of mitochondrial superoxide anion (O2-) generation;
(B) line graphs represent time-course of light absorbance decrease in the absence (red line) and
in the presence (blue line) of calcium overload (CaCl2, 150�mol/L); (C) assessment of
mitochondrial DNA integrity by real-time PCR in mitochondria isolated from mouse aorta of
young and old WT or JunD-/- mice. (D) Electrochemical analysis of nitric oxide (NO)
concentrations in single endothelial cells from aortic rings of young and old WT or JunD-/- mice.
Figure Legends:
Figure 1. (A) Age-dependent changes of endothelium-dependent relaxation in aortic rings from
WTWTWT aaandnd JuJunDnDnD //// aaorortaas.s LLinne grgrapaphsh sshoow w concncenennttrrationon–rrese ponsnsseee cuurvrvees to accete yly chhollinne (Ach)
BBB) EEffect of freeee-rrraddiiccaaal scaaavvevengerer ppooolyyyethyylyleene ggllycool-l-l-sususupepeeroxxxiddde diissmmmuttatassse (PEPEEGGG-SOSOSOD)D) oonnn
eespsponononsesese tttooo AAcAch.h.h (((CCC) ) ) EnEnEndodod thheleleliuiuiumm-m-inindedeepepependndndenenenttt rererelalalaxaxax tititiononon tttooo ttthhehe NNNO OO dododonononor r sosoodididiumumum nnitititrororoprprprususussisisideded
SNP)) acrosssss ttthehehe eeexpxpperere imimimenenentatatal grgrouououppsps. ReReResusuultlttsss aarare e prprpresesennenteted dd asasas mmeaeaannn ±±± SESESEMM,M, nnn== 5-5-5 7 77 peper grg oup,p
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
24
(E) Representative Western blots of activating eNOS Ser1177 phosphorylation and eNOS
expression. (F) Endothelial peroxynitrite (ONOO-) levels across the experimental groups. (G)
Immunofluorescence showing 3-nitrotyrosine (3-NT) staining in aortic cross-sections obtained
from young and old WT or JunD-/- mice. Nuclei stained with DAPI (blue). Bar graphs indicate
quantification of 3-NT stained cells on total cells. Results are presented as mean ± SEM; n=4-5
per group. eNOS, endothelial nitric oxide synthase.
Figure 3. (A) Representative fluorescence microscopy images and relative quantification
showing preferential expression of JunD in the vascular endothelium, as indicated by co-staining
with the endothelial marker CD31 (merged image). Quantification of immunofluorescence was
performed by counting double stained cells (JunD+ and CD31+) on total cells. Nuclei stained
with DAPI (blue). (B) Representative Western blot and densitometric quantification of JunD
protein expression from aortic lysates of young and old WT mice. (C) Immunofluorescence
showing JunD staining in aortic cross-sections obtained from young and old WT or JunD-/- mice.
Nuclei stained with DAPI (blue). Fluorescence microscopy images as well as relative
quantification indicates a significant downregulation of JunD expression with aging. As
expected, JunD signal was absent in aortas from JunD-/- mice. (D) Quantitative analysis of JunD
promoter methylation in mouse aorta of young and old WT mice. UM, unmethylated; IM,
intermediately methylated; HM, hypermethylated CpG dinucleotides. (E) Bar graphs indicate
decline of JunD transcriptional activity with aging. (F) Representative Western blots showing the
interaction of JunD with menin and subsequent JunD dephosphorylation in old vs. young mice.
IB, immunoblotting; IP, immunoprecipitation. (G) Line graphs show cumulative concentration
response curves to acetylcholine-dependent relaxation in aortic rings of old WT mice receiving
with the endothelial marker CD31 (merged image). Quantification of immunofluuooreeesccenenencecece wwwasa
performed by counting double stained cells (JunD+ and CD31+) on total cells. Nuclei stained
wiwiththh DDDAPAPAPII I (b(b(blue)e)e).. ((BB) Representative Western bbbllolott and densitommetete ric c quququana tification of JunD ff
prottteie n expresssisiiononon ffrorom m aoaoortrtrticicic llyysasasatetetes ofoff youuunnng andndnd olddd WWTT T mimim cce. ((CC))) Immmmumumunnonoflfluouuorerer scsccenenncece
hhowowowininingg g JuJuunDnDnD ssttataiinniningg iinin aaororrtitic cc crcrcrosooss-s-sesesectctctioioionsss oobbbtaaiineeed d frfrfromomom yyyoououngngng aanndnd ooldldd WWWT T ororor uJuunDnDD-/--/- mmmiicice
Nuclei stainededd wwwititth h h DADADAPIPP (((blblblueueue).).) FFlululuorrresesescececennncecec mmmiciccrororoscscopopopy yy imimimagagagesess aaasss wewewellllll aaas s rererelalalatitiiveveve
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
25
vector (pCMV, filled circle) or JunD cDNA clone (open circles). Results are presented as mean
± SEM; n=4-8 per group. ND, not detectable; NE, norepinephrine.
Figure 4. Protein expression and activity of (A) MnSOD, (B) ecSOD and (C) ALDH2 from
aortic lysates of young and old WT or JunD-/- mice. Results are presented as mean ± SEM; n=4
per group. MnSOD, manganese superoxide dismutase; ecSOD, extracellular superoxide
dismutase; ALDH2, aldehyde dehydrogenase 2.
Figure 5. Western blot and immunofluorescence showing expression of NADPH subunits (A)
p47phox (B) Nox2 and (C) Nox4 in the 4 experimental groups. Nuclei stained with DAPI (blue,
magnification 10X). For each panel, bar graphs show densitometric analysis of Western blot. (D)
NADPH activity in aortic lysates from young and old WT or JunD-/- mice. (E) Maximal
relaxation to acetylcholine (Ach, 10-6M) before and after incubation with the NADPH inhibitor
apocynin (100μM) assessed in aortic rings from young and old WT mice treated with scrambled
or JunD siRNA. Results are presented as mean ± SEM; n=4-6 per group, *p<0.05.
Figure 6. (A) Bar graphs show telomerase activity in young and old WT or JunD-/- mice. (B)
Aortic cross-sections showing -gal positive cells in the 4 experimental groups (magnification
10X). (C-D) Representative Western blots and immunofluorescence showing expression of the
senescence markers p53 and p16INK4a in age-matched WT and JunD-/- mice. Nuclei stained with
DAPI (blue, magnification 10X). For each panel, bar graphs show densitometric analysis of
Western blot. Results are presented as mean ± SEM; n=4-5 per group, *p<0.05.
p47phox (B) Nox2 and (C) Nox4 in the 4 experimental groups. Nuclei stained wiwiiththh DDAPAPAPI II (b(b(blulue,,
magnification 10X). For each panel, bar graphs show densitometric analysis of Western blot. (D)
NANAADPDPDPHHH acacactititivvvityyy iiinnn aortic lysates from young annnddd ooold WT or JunDnDD-/- mmmicicice.e (E) Maximal
eelaaaxax tion to acaceteetyylchchholoo ininineee (A(A(Acchch,, 101010-6-6M)M)M) beffforrre annnddd afffteteterr ininncuuubabattioonon wwiti hhh ththhee NANANADPDPDPHH innnhihhibibib tooor
apapoococynynyninin (((1010100μ0μM)M)M) aassssesesssesed inini aaaororortititicc ririingnggs ss ffrfromomom yyyououungngg aaandndnd ooldldld WWWTTT mimimiceee ttrereeatatatedede wwwiitith sscscrarar mmbmblleed d
or JunD siRNNNA.A.A. RRResesesululltstss arerere ppprereesesesentnttedee aaas s mememeananan ±± SSSEMEMEM;; n=n=n 4-4-4 6 6 6 pepeper r grgrgrouououp,p,p, *p<p<p 0.0.0 050505..
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

DOI: 10.1161/CIRCULATIONAHA.112.000826
26
Figure 7. (A) Representative Western blots and densitometric quantification of NADPH subunits
following JunD knockdown in human aortic endothelial cells. Scrambled siRNA (scr.siRNA)
was used as a control. (B) Bar graphs show NADPH oxidase activity in the presence or in the
absence of JunD silencing. (C-D) ESR spectroscopy analysis of superoxide anion (O2-) and nitric
oxide (NO) production in HAECs in the presence or in the absence of JunD knockdown,
NADPH inhibitor apocynin and PEG-SOD. (E) Representative Western blots and densitometric
quantification of eNOS and activating eNOS Ser1177 phosphorylation after JunD knockdown.
Results are presented as mean ± SEM; n=6-9 per group. eNOS, endothelial nitric oxide synthase.
Figure 8. (A) Box plots show JunD mRNA and (B) representative Western blot with
quantification of JunD protein expression in peripheral blood monocytes from young and old
healthy subjects. (C-F) Gene expression of MnSOD, ecSOD, p47phox and Nox2 and relative
correlations with JunD transcript. Results are presented as mean ± SEM; n=12-14 per group.
TBP, TATA binding protein; MnSOD, manganese superoxide dismutase; ecSOD, extracellular
superoxide dismutase.
Figure 8. (A) Box plots show JunD mRNA and (B) representative Western blot t wwiwiththt
quantification of JunD protein expression in peripheral blood monocytes from young and old
heealalalthththyy y susuubjbjbjeecctss.. (((CCC-F) Gene expression of MnSSSODODOD, ecSOD, p477phpp oxoxx aaanndn Nox2 and relative
coorrrrele ations wwitithh h JJuunDnDnD tttrararansnnscrcrcriipipt.t. RRResesuulultts arrre ppressseenntedd d asas mmmeeaeann ±± SESEM;M;M nn==1=122-2-14144 pperere gggrorooupupp.
TBTBBP,P,P, TTTATATAAA bbibinnddininng g ppproototeie n;n;; MMMnnnSOSOSOD,D,, mmmananangagaanenen ssse ssupuppererroxoxoxididdee e ddiismmmututu aaasee;e; eecScSc ODODOD, , eeextrtrracccelelluluulaaar r
uperoxide ddisissmumum tatataseses ..
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 1 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 2 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 3 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 4 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 5 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 6 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 7 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

Figure 8 by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

SUPPLEMENTAL MATERIAL for
Deletion of the AP-1 Transcription Factor JunD
Induces Oxidative Stress and Accelerates Age-Related
Endothelial Dysfunction
Francesco Paneni MD1,2*, Elena Osto MD, PhD1,3*, Sarah Costantino PhD1,4,
Bogdan Mateescu PhD5, Sylvie Briand, PhD1,3 Giuseppe Coppolino MD1, Enrico Perna
MD6, Pavani Mocharla MSc1,3, Alexander Akhmedov, PhD1,3, Ruslan Kubant MD7,
Lucia Rohrer, PhD8, Tadeusz Malinski PhD7, Giovanni G. Camici PhD1,3, Christian M.
Matter MD1,3, Fatima Mechta-Grigoriou PhD5, Massimo Volpe MD2,6,
Thomas F. Lüscher MD1,3, Francesco Cosentino MD,PhD1,6
1Cardiology and Cardiovascular Research, Institute of Physiology and University Hospital,
Zürich, Switzerland; 2IRCCS Neuromed, Pozzilli, Italy; 3Zürich Center for Integrative
Human Physiology (ZIHP), University of Zurich, Switzerland; 4Department of Experimental
Medicine, Section of Pharmacology, Second University of Study of Naples, Italy; 5Stress
and Cancer Laboratory, Institut Curie, Inserm U830, Paris, France; 6Cardiology,
Department of Clinical and Molecular Medicine, “Sapienza” University, Rome, Italy;
7Department of Biochemistry, Ohio University, Athens, Ohio; 8Institute of Clinical
Chemistry, University Hospital, Zürich, Switzerland
*The authors contributed equally to this work Address for Correspondence: Francesco Cosentino, MD, PhD Cardiology & Cardiovascular Research Institute of Physiology
University of Zurich-Irchel Winterthurerstrasse, 190 CH-8057 Zurich, Switzerland Tel: 41-44-635 6470 Fax: 41-44-635 6827 Email: [email protected]
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

2
Supplemental Methods
Tissue Harvesting
On the day of the experiment, mice were anesthetized through the intraperitoneal
administration of 50 mg/kg sodium pentobarbital and then euthanized. The chest and
abdomen were opened with a medial sternotomy. The entire aorta from the heart to the
iliac bifurcation was excised and placed immediately in cold modified Krebs-Ringer
bicarbonate solution (pH 7.4, 37°C, 95% O2; 5% CO2) of the following composition
(mmol/L): NaCl (118.6), KCl (4.7), CaCl2 (2.5), KH2PO4 (1.2), MgSO4 (1.2), NaHCO3
(25.1), glucose (11.1), and calcium EDTA (0.026). The aorta was cleaned from adhering
connective tissue under a dissection microscope and immediately used for organ chamber
experiments or snap-frozen in liquid nitrogen and stored at -80°C.
Organ chamber experiments
For endothelial function experiments, aortas were cut into 2-3 mm rings which were
connected to an isometric force transducer (Multi-Myograph 610M, Danish Myo
Technology, Denmark), suspended in an organ chamber filled with 5 mL Krebs-Ringer
bicarbonate solution (37°C, pH 7.4), and bubbled with 95% O2, 5% CO2. Isometric tension
was recorded continuously. After a 30 minute equilibration period, rings were gradually
stretched to the optimal point of their length–tension curve as determined by the
contraction in response to potassium chloride (100 mmol/L). Concentration–response
curves were obtained in a cumulative fashion.1 Several rings cut from the same artery
were studied in parallel. Responses to acetylcholine (Ach, 10-9 to 10-6 mol/L, Sigma
Aldrich) in the presence or the absence of polyethylene glycol-superoxide dismutase
(PEG-SOD, 150 U/mL, Sigma Aldrich) and apocynin (100 µM, Sigma Aldrich) were
recorded during submaximal contraction to norepinephrine (NE, 10-6 mol/L). PEG-SOD
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

3
and apocynin were added to the organ chamber 30 and 60 minutes before the assessment
of endothelium-dependent relaxation to Ach, respectively. The nitric oxide (NO) donor
sodium nitroprusside (SNP, 10-10 to 10-5 mol/L, Sigma Aldrich) was added to test
endothelium-independent relaxation. Relaxations were expressed as percentages of pre-
contraction to norepinephrine.
Measurements of mitochondrial O2- by ESR spectroscopy
O2- generation in mitochondria isolated from mouse aorta was assessed by electron spin
resonance (ESR) spectroscopy analysis using the spin trap 1-hydroxy-3-methoxycarbonyl-
2,2,5,5-tetramethyl-pyrrolidine (CMH). Twenty five µg of isolated mitochondria were
resuspended in Krebs Hepes Buffer (KHB, PH 7.35) containing desferoxamine (25µM) and
DETC (5µM). O2- production was determined by following the oxidation of CP-H to
paramagnetic 3-carboxy-proxyl (CP). Signals were quantified by measuring the total
amplitude after correction of baseline and subtraction of background signals.
Measurements of endothelial NO and ONOO- from mouse aorta
In situ measurements of NO and ONOO- were carried out with three electrochemical
nanosensors combined into one working unit with a total diameter of 2.0 - 2.5 µm. Their
design was based on previously developed and well-characterized chemically modified
carbon-fiber technology.2, 3 Amperometry was performed with a computer-based Gamry
VFP600 multichannel potentiostat. A current at the peak potential characteristic for NO
(0.65 V) oxidation and ONOO- (-0.40 V) reduction was directly proportional to the local
concentrations of these compounds in the immediate vicinity of the sensor. Linear
calibration curves (current vs. concentration) were constructed for each sensor from 10
nmol/L to 2 mol/L before and after measurements with aliquots of NO and ONOO-
standard solutions, respectively. At a constant distance of the sensors from the surface of
the endothelial cell, the reproducibility of measurements is high (5-12%). The consumption
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

4
of redox species by nanosensors depends on the area of the electrode (< 0.12 µm2) and
the duration time of electrolysis (~5-10 s). For the amperometric measurements used, it
varied between 0.04-0.1% of the NO and ONOO- peak concentration. This value is
negligible compared with the experimental error. The position of nanosensors (x,y, z
coordinates) versus the endothelial cell was established with the help of a computer
controlled micromanipulator. In order to establish a constant distance from cells, the
module of sensors was lowered until it reached the surface of the cell membrane. After
that, the sensors were slowly raised 4 + 1 µm (z coordinates) from the surface of cells. The
sensors were then moved horizontally (x, y coordinates) and positioned above a surface of
randomly chosen single endothelial cells in an aortic ring. Acetylcholine (Ach), were then
injected with a nanoinjector that was also positioned by a computer controlled-
micromanipulator.
Isolation of mitochondrial and cytosolic fraction
Thoracic aortas were suspended in the mitochondrial buffer containing 10mM MOPS (pH
7.2), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 10 µg/mL leupeptin, 10 µg/mL aprotinin,
and 0.25 M sucrose, and gently homogenized with a Dounce homogenizer (30 strokes).
The homogenate was centrifuged at 750g for 10 minutes at 4°C to remove nuclei and
unbroken cells, and the supernatant was centrifuged at 10,000g for 15 minutes. The
resultant mitochondrial pellet was resuspended in 40 µL of standard lysis buffer (see
Western blot for details) while the supernatant was collected as the cytosolic fraction.
Mitochondrial DNA damage detection
Briefly, each 30 ng of total DNA was used in two PCR reactions, one amplifying a short
117-bp product of mtDNA and the other amplifying a long fragment of 10-kb product of
mtDNA. The amount of PCR products from the 10-kb amplification reflects the quality of
the template mtDNA, whereas the 117-bp amplification serves as an internal control for
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

5
template concentration.4 Primer sequences are shown in Supplemental Table 1. PCR
reaction was run with the following conditions: 10-kb PCR, denaturation at 94°C for 15
seconds, annealing, and extension at 68°C for 12 minutes for 20 amplifying cycles,
followed by a final 10 minutes extension;117-bp PCR, 94°C for 30 seconds, 60°C for 45
seconds, 72°C for 45 seconds for 20 cycles, and followed by final extension at 72°C for 10
minutes. mtDNA integrity was determined by calculating the ratios of amount of 10 kb
products to that of 117-bp products.
Western blotting
Frozen samples of aorta and HAECs were lysed for immunoblotting (120 mmol/L sodium
chloride, 50 mmol/L Tris, 20 mmol/L sodium fluoride, 1 mmol/L benzamidine, 1 mmol/L
DTT, 1 mmol/L EDTA, 6 mmol/L EGTA, 15 mmol/L sodium pyrophosphate, 0.8 µg/mL
leupeptin, 30 mmol/L p-nitrophenyl phosphate, 0.1 mmol/L PMSF, and 1% NP-40) for
immunoblotting. Cell debris were removed by centrifugation (12 000 g) for 10 minutes at
4°C. The samples (20 µg) were added with 5X Laemmli’s SDS-PAGE sample buffer (0.35
mol/L Tris-Cl, pH 6.8, 15% SDS, 56.5% glycerol, 0.0075% bromophenol blue), followed by
heating at 99°C for 5 minutes, and then subjected to 10% SDS-PAGE gel for
electrophoresis. Proteins were then transferred onto Immobilon-P filter papers (Millipore
AG, Bedford, MA) with a semidry transfer unit (Hoefer Scientific, San Francisco, CA). The
membranes were then blocked with 5% milk in TBS-Tween buffer (0.1% Tween 20; pH
7.5) for 1 hour at room temperature and incubated with JunD, eNOS, GpX1, p47phox,
Nox2, Nox4, ALDH2, p16INK4a and p53 antibodies (Santa Cruz Biotechnology, Nunningen,
Switzerland); phospho eNOS Ser-1177 (Transduction Laboratories, Lexington, USA);
MnSOD and ecSOD (Upstate Biotechonology, USA). Anti-rabbit and anti-mouse
secondary antibodies were bought from GE Healthcare (Buckinghamshire, United
Kingdom). The immunoreactive bands were detected by an enhanced chemiluminescence
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

6
system (Millipore, Billerica, USA). Related signals were quantified using a Scion image
software (Scion Corp, Frederick, USA).
Detection of membrane translocation of NADPH subunit p47phox
Segments of thoracic aortas were homogenized in a lysis buffer [50 mmol/L HEPES (pH
7.4), 50 mmol/L NaCl, 1 mmol/L MgCl2, 2 mmol/L EDTA, 1 mmol/L PMSF, 10 mg/ml
leupeptin, 1 mmol/L NaVO4, and 0.1% Triton X-100] for 5 minutes on ice. Aortic lysates
were centrifuged at 15 000 rpm at 4°C for 15 minutes. After the supernatant had been
collected as the cytosol fraction, the pellet was resuspended in 1% Triton X-100 in the lysis
buffer and centrifuged at 15 000 rpm and 4°C for 15 minutes. The supernatant was then
collected as the membrane fraction. Equal amounts (25 μg) of protein from each fraction
were subjected to SDS-polyacrylamide gel electrophoresis followed by immunoblotting
with anti-p47phox antibody (Santa Cruz Biotechnology, Nunningen, Switzerland).
Co-immunoprecipitation
Segments of mouse aorta were lysed in ice-cold lysis buffer containing 50 mmol/L Tris-
HCl, pH 7.4, 100 mmol/L NaF, 15 mmol/L Na4P2O7, 1 mmol/L Na3VO4, 1% Triton X-100,
and 1 mmol/L phenylmethylsulfonyl fluoride. Lysates were centrifuged at 10,000 g to
remove insoluble material. For immunoprecipitation, precleared lysates were incubated
with anti-menin1 antibody overnight at 4°C. Lysates were precleared by incubation with 50
μL of protein A/G-agarose for 2 h at 4°C with rocking. Agarose beads were pelleted by
centrifugation at 1000 g. Immunoprecipitated proteins were eluted from the beads by
boiling for 5 min in SDS sample buffer and immunoblotted with anti-JunD antibody (1:1000
dilution). Bound antibody was visualized using an enhanced chemiluminescence system
(Millipore, Billerica, USA) after incubation of the blot with peroxidase-conjugated
secondary antibody for 1 h.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

7
JunD promoter methylation in mouse aorta
For each analyzed genomic DNA sample, 30 ng of genomic DNA was digested with
methylation sensitive enzyme (Ms) or with methylation dependent enzyme (Md) and also
with both methylation sensitive and dependent enzymes (Msd) in 15µl total volume
including 5X digestion buffer overnight at 37°C. In parallel, 30 ng of each genomic DNA
sample was mock-treated (Mo) under identical conditions with the exception that water
was substituted for digestion enzyme. Following digestion, enzyme activity was heat
inactivated at 65°C for 20 min. A total of 6 ng digested DNA was analyzed by quantitative
real-time PCR. A predesigned primer for mouse JunD promoter was obtained from SA
Biosciences (Frederick, MD, USA). Standard quantitative PCR cycling conditions were
used with a ‘hot’ plate read of 72°C for 1 min. The melt curve of each amplicon was
calculated within a temperature gradient from 60 to 95°C at 1°C increments with a 15 s
hold time for each read. The cycle number at which the Ms and Md digested sample
crossed the threshold was subtracted from the cycle number at which the Mo-treated
sample crossed the threshold to determine the ΔCt of the locus. Since Ms/Md digests only
DNA including purine-5mC, thereby decreasing the amplifiable copies of loci containing
DNA methylation and increasing the Ct relative to the mock-treated sample, increasing
ΔCt values reflect increasing levels of local DNA methylation.
Nuclear extracts and JunD binding activity
Nuclear protein was obtained by using a nuclear extraction kit (Active Motif, Rixensart,
Belgium). Aorta segments were lysed in hypotonic buffer for 15 min before centrifugation,
isolated nuclei were resuspended in a hypertonic buffer, and nuclear protein was extracted
by incubation on a rotator for 30 min. Nuclear and cytosolic fractions were collected after
centrifugation. The DNA binding reaction was carried out with 5 μg of nuclear protein in a
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

8
96-well plate coated with consensus sequences for JunD for 1 h at room temperature.
After washing, JunD antibody (Active Motif) was added and incubated for 1 h, followed by
incubation with a horseradish peroxidase-conjugated secondary antibody. Finally, JunD
DNA binding was assessed spectrophotometrically at 450 nm.
In vivo knockdown of JunD
In vivo knockdown of JunD was performed by injecting a predesigned siRNA specifically
targeting JunD (Santa Cruz, USA). A scrambled-siRNA was used as a negative control
(Microsynth, 5’-UAC ACA CUC UCG UCU CU dTdT-3'). Amount of JunD siRNA was
selected based on dose optimization studies (data not shown). The siRNA mix at the final
dose of 1.6 mg/kg was incubated with the in vivo-jetPEI delivery reagent (Polyplus
Transfection) for 15 min at room temperature and injected i.v in a final volume of 100 L,
as previously reported.5 Successful knockdown of JunD was assessed by Western blot in
aortic lysates. We have previously characterized the distribution of siRNA injected together
with the vivo-jetPEI delivery reagent. Labelling of siRNA sequences with a fluorescent tag
showed its exclusive distribution to the endothelium within the vessel wall.5
JunD overexpression in mice
Old male mice (22 months) were injected i.v either with 40µg of cloning vector (Origene,
PCMV6-Kan/Neo, PCMV6KN) or a predesigned mouse JunD cDNA clone (Origene,
MC200652) together with a cationic transfection reagent (JetPEI, Polyplus Transfection),
according to the manufacturer’s instructions (http://www.polyplus-transfection.com).
I.v. injections were repeated every other day for 1 week and mice harvested for organ
chamber experiments, as described in the methods section. Time course studies showed
that JunD overexpression was already evident after 24 hours and injections were repeated
to warrant a stable overexpression before ex-vivo assessment of endothelial function.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

9
Telomerase activity
The catalytic activity of telomerase was assessed by quantitative PCR, as described
previously.6 Segments of mouse aorta were homogenized in CHAPS buffer and
centrifuged at 4°C. Two different protein concentrations, 0.5 μg and 1 μg, were employed
to document the specificity of the assay. Lysates were incubated in a solution containing
reverse transcriptase reaction mix and Taq polymerase (TRAPEZE RT Telomerase
Detection Kit, Chemicon) at 30°C for 30 minutes. Telomerase positive cells were used as
positive control while CHAPS buffer in the absence of protein lysates was used as
negative control. PCR cycling conditions were as follows: 95°C for 2.0 minutes; 40 cycles
of 94°C for 15 seconds; and 59°C for 60 seconds. Data were collected at the 59°C stage
of each cycle.
Beta-galactosidase staining
Senescence-associated β-galactosidase (SA-β-gal) activity is detectable using the artificial
substrate 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-gal) and allows the identification
of senescent cells in vivo. The ability to induce SA-β-gal activity is a manifestation of
residual lysosomal activity at suboptimal pH (pH 6). It becomes detectable in the course of
senescence because of the increased lysosomal content in senescent cells. Thoracic
aorta segments from all the experimental groups were embedded in optimum cutting
temperature and stained for β-galactosidase activity. Briefly, aortas were fixed in Fixative
Solution (25% glutaraldehyde in PBS/MgCl2) for 1h and incubated at 37°C for 6h with fresh
Staining Solution (0.6 M potassium ferricyanide, 0.6M potassium ferrocyanide, 1 M MgCl2,
10% Igepal, 10% sodium deoxycholate, 40 mg/ml X-gal solution, PBS). Staining was
evident in 6 hours. SA-β-gal positive cells were visualized under an optical microscope
(Olympus, 10x magnification).
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

10
Cell culture
Human aortic endothelial cells (HAECs, passages 5 to 8) were purchased from Clonetics
(Allschwil, Switzerland) and grown in fibronectin-coated 75cm2 flasks in optimized
endothelial growth medium-2 (EGM-2, Clonetics, Walkersville, USA) supplemented with
10% FCS. Cells were detached by using Tripsin/EDTA for 90 seconds and reseeded in
fibronectin-coated 3 cm-cell culture dishes or 12 multiwell plates. For experiments, HAECs
were cultured in EGM-2 containing 2% FCS for 12 h and then transfected with JunD,
p47phox and scrambled siRNAs. After 72 hours cells were harvested for immunoblotting,
ESR measurements and assessment of NADPH activity.
Real-time PCR
Total RNA was extracted from human peripheral blood monocytes using TRIzol Reagent
(Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendations. Conversion
of total cellular RNA to cDNA was carried out with Moloney murine leukemia virus reverse
transcriptase and random hexamer primers (Amersham Biosciences, Piscataway, NJ) in a
final volume of 33 μl using 1 μg of RNA. Real time PCR was performed in an MX3000P
PCR cycler (Stratagene, Amsterdam, The Netherlands) by using the SYBR Green
JumpStart kit (Sigma). Each reaction (25 μl) contained 2 μl cDNA, 10 pmol of each primer,
0.25 μl of internal reference dye and 12.5 μl of JumpStart Taq ReadyMix (containing
buffer, dNTPs, stabilizers, SYBR Green, Taq polymerase and JumpStart Taq antibody).
Primers for JunD, MnSOD, ecSOD, p47phox, Nox2, Nox4, 117-pb and 10-kb were
synthesized by Microsynth and relative sequences are shown in Supplemental Table1.
The amplification program consisted of 1 cycle at 95 °C for 10 min, followed by 40 cycles
with a denaturing phase at 95 °C for 30 s, an annealing phase at 56 °C for 30 s, and an
elongation phase at 72 °C for 30 s. A melting curve analysis was performed after
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

11
amplification to verify the accuracy of the amplicon, and PCR products were analyzed on
an ethidium bromide stained 1% agarose gel. Data were normalised to results obtained
with primers specific for human TBP and mouse GAPDH.
Measurement of O2- and NO in HAECs
O2– generation in intact cells was assessed by electron spin resonance (ESR)
spectroscopy analysis using the spin trap 1-hydroxy-3-methoxycarbonyl-2,2,5,5-
tetramethyl-pyrrolidine (CMH). O2- production was determined by following the oxidation of
CP-H to paramagnetic 3-carboxy-proxyl (CP).7 Endothelial NO release was examined by
ESR spectroscopy analysis with the use of the spin-trap colloid Fe(DETC)2. Briefly, cells
were washed and resuspended in 900 µL of Krebs-HEPES buffer (37°C), 300 µL of colloid
Fe(DETC)2 (final concentration 285 µmol/L) was added to each sample and incubated at
37°C for 60 minutes. Samples were scraped on ice, cells were collected from each sample
and tubes were centrifuged at 4°C at 5000 rpm for 10 minutes. Cells were resuspended
and aspirated into 1 mL syringes which were frozen immediately in liquid nitrogen.5 All
ESR spectra relative to O2–, NO and ONOO- measurements were recorded with the use of
NOX-E.5-ESR spectrometer (Bruker, Bremen, Germany). Signals were quantified by
measuring the total amplitude after correction of baseline and subtraction of background
signals.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

12
Primer Sequence
Human
JunD forward 5’ - ATC GAC ATC GAC ACG CAG GAG - 3’
JunD reverse 5’ - CTC CGT GTT CTG ACT CTT CAG G - 3’
MnSOD forward 5’ - TAG GGC TGA GGT TTG TCC AG - 3’
MnSOD reverse 5’ - GGA GAA GTA CCA GGA GGC GT - 3’
ecSOD forward 5’ - CGA GTC AGA GTT GGG CTC C - 3’
ecSOD reverse 5’ - CTC TCT TGG AGG AGC TGG AA - 3’
ALDH2 forward 5’- CCT CTC CAG TGG ACG GAT T - 3’
ALDH2 reverse 5’ - CGA GGT CTT CTG CAA CCA G - 3’
p47phox forward 5’ - GAC AGG TCC TGC CAT TTC AC - 3’
p47phox reverse 5’ - CAG TCA TGG GGG ACA CTT T - 3’
Nox2 forward 5’ - TCA CTT CCT CCA CCA AAA CC - 3’
Nox2 reverse 5’ - GGG ATT GGG CAT TCC TTT AT - 3’
Nox4 forward 5’ - CTT CCG TTG GTT TGC AGA TT - 3’
Nox4 reverse 5’ - TGG GTC CAC AAC AGA AAA CA - 3’
TBP forward 5’ - CGT GGC TCT CTT ATC CTC ATG - 3’
TBP reverse 5’ - GCC CGA ACC GCC GAA TAT A - 3’
Mouse
10-kb forward 5’ - GCCAGCCTGACCCATAGCCATAATAT-3’
10-kb reverse 5’ - GAGAGATTTTATGGGTGTAATGCGG - 3’
117-pb forward 5’ - CCCAGCTACTACCATCATTCAAGT - 3’
117-pb reverse 5’ - GATGGTTTGGGAGATTGGTTGATG - 3’
GAPDH forward 5’ - TGC CCC CAT GTT TGT GAT G - 3’
GAPDH reverse 5’ - TGT GGT CAT GAG CCC TTC C - 3’
Supplemental Table 1. Primers used for real-time PCR
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

13
Supplemental Figures
Supplemental Figure 1. Bar graphs showing percentage decrease of 520-nm absorbance
at 20 minutes in calcium-treated mitochondria isolated from aorta of young and old WT or
JunD-/- mice. is shown. Results are presented as mean ± SEM; n=4 per group.
Supplemental Figure 2. Caspase 3 activity in aortic lysates of young and old WT or
JunD-/- mice. Results are presented as mean ± SEM; n=4 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

14
Supplemental Figure 3. Densitometric quantification of activating eNOS Ser1177
phosphorylation and eNOS expression in the 4 experimental groups. Results are
presented as mean ± SEM; n=4 per group.
Supplemental Figure 4. Densitometric quantification of activating JunD phosphorylation
in young and old WT mice. Results are presented as mean ± SEM; n=4 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from
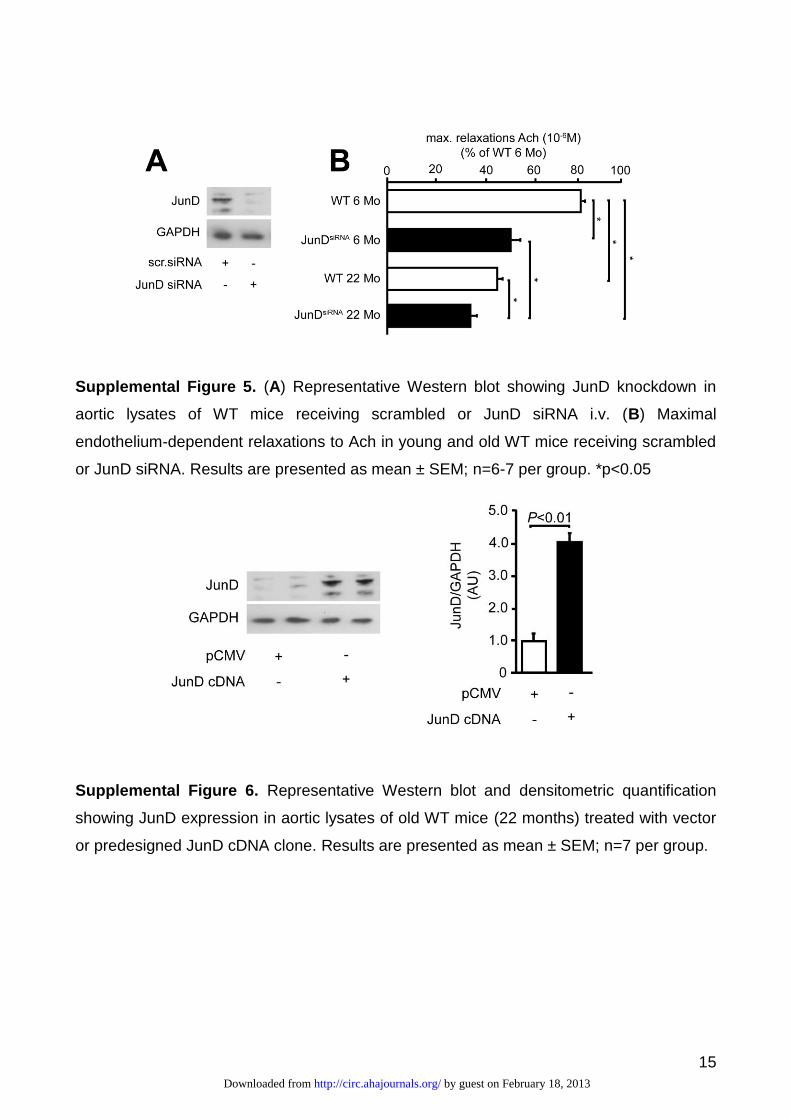
15
Supplemental Figure 5. (A) Representative Western blot showing JunD knockdown in
aortic lysates of WT mice receiving scrambled or JunD siRNA i.v. (B) Maximal
endothelium-dependent relaxations to Ach in young and old WT mice receiving scrambled
or JunD siRNA. Results are presented as mean ± SEM; n=6-7 per group. *p<0.05
Supplemental Figure 6. Representative Western blot and densitometric quantification
showing JunD expression in aortic lysates of old WT mice (22 months) treated with vector
or predesigned JunD cDNA clone. Results are presented as mean ± SEM; n=7 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

16
Supplemental Figure 7. (A) Representative Western blot and densitometric quantification
of GPX1 in age-matched WT and JunD-/- mice. (B) Xanthine oxidase activity in aortic
lysates from the 4 experimental groups. Results are presented as mean ± SEM; n=4 per
group. GPX1, glutathione peroxidase 1.
Supplemental Figure 8. Quantification of immunofluorescence for the NADPH subunits
p47phox, Nox2 and Nox4. Analysis was performed by counting positive cells on total cells.
Results are presented as mean ± SEM; n=3-4 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

17
Supplemental Figure 9. Representative Western blot and densitometric quantification
showing membrane translocation of the NADPH subunit p47phox in aortic lysates of age
matched WT and JunD-/- mice. Results are presented as mean ± SEM; n=4 per group.
Supplemental Figure 10. Electrochemical analysis of superoxide anion (O2-) in single
endothelial cells from aortic rings of young and old WT or JunD-/- mice. Results are
presented as mean ± SEM; n=4 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

18
Supplemental Figure 11. Quantification of immunofluorescence for the aging markers
p53 and p16INK4a. Analysis was performed by counting positive cells on total cells. Results
are presented as mean ± SEM; n=3-4 per group.
Supplemental Figure 12. Densitometric quantification of p47phox, Nox2 and Nox4
expression following JunD silencing in HAECs. Results are presented as mean ± SEM;
n=4-5 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

19
Supplemental Figure 13. (A-C) siRNA-mediated knockdown of p47phox, Nox2 and Nox4
in HAECs. (D) ESR analysis showing superoxide anion generation following JunD
knockdown with or without concomitant silencing of NADPH isoforms. Results are
presented as mean ± SEM; n=5 per group.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from

20
Supplemental References 1. Osto E, Matter CM, Kouroedov A, Malinski T, Bachschmid M, Camici GG, Kilic U,
Stallmach T, Boren J, Iliceto S, Luscher TF, Cosentino F. c-Jun N-terminal kinase 2
deficiency protects against hypercholesterolemia-induced endothelial dysfunction
and oxidative stress. Circulation. 2008;118:2073-2080.
2. Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a
porphyrinic-based microsensor. Nature. 1992;358:676-678.
3. Erdei N, Toth A, Pasztor ET, Papp Z, Edes I, Koller A, Bagi Z. High-fat diet-induced
reduction in nitric oxide-dependent arteriolar dilation in rats: role of xanthine
oxidase-derived superoxide anion. Am J Physiol Heart Circ Physiol.
2006;291:H2107-2115.
4. Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and
repair using quantitative PCR. Methods Mol Biol. 2002;197:159-176.
5. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Luscher TF,
Cosentino F. Gene Silencing of the Mitochondrial Adaptor p66Shc Suppresses
Vascular Hyperglycemic Memory in Diabetes. Circ Res. 2012;111:278-89
6. Kajstura J, Bai Y, Cappetta D, Kim J, Arranto C, Sanada F, D'Amario D, Matsuda A,
Bardelli S, Ferreira-Martins J, Hosoda T, Leri A, Rota M, Loscalzo J, Anversa P.
Tracking chromatid segregation to identify human cardiac stem cells that
regenerate extensively the infarcted myocardium. Circ Res. 2012;111:894-906.
7. Sorrentino SA, Bahlmann FH, Besler C, Muller M, Schulz S, Kirchhoff N, Doerries
C, Horvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser
U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial
progenitor cells from patients with type 2 diabetes mellitus: restoration by the
peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation.
2007;116:163-173.
by guest on February 18, 2013http://circ.ahajournals.org/Downloaded from




















