
NOW suit requires sports equity - CSU Chico Digital Collections
Upload
universityCategory
view
1download
0
Cytokine Activation of Nuclear Factor �B in VascularSmooth Muscle Cells Requires Signaling Endosomes
Containing Nox1 and ClC-3Francis J. Miller Jr,* Mohammed Filali,* Gina J. Huss, Bojana Stanic, Ali Chamseddine,
Thomas J. Barna, Fred S. Lamb
Abstract—Reactive oxygen species (ROS) are mediators of intracellular signals for myriad normal and pathologic cellularevents, including differentiation, hypertrophy, proliferation, and apoptosis. NADPH oxidases are important sources ofROS that are present in diverse tissues throughout the body and activate many redox-sensitive signal transduction andgene expression pathways. To avoid toxicity and provide specificity of signaling, ROS production and metabolismnecessitate tight regulation that likely includes subcellular compartmentalization. However, the constituent elements ofNADPH oxidase-dependent cell signaling are not known. To address this issue, we examined cytokine generation ofROS and subsequent activation of the transcription factor nuclear factor �B in vascular smooth muscle cells (SMCs).Tumor necrosis factor-� and interleukin (IL)-1� stimulation of SMCs resulted in diphenylene iodonium-sensitive ROSproduction within intracellular vesicles. Nox1 and p22phox, integral membrane subunits of NADPH oxidase, coimmu-noprecipitated with early endosomal markers in SMCs. ClC-3, an anion transporter that is primarily found inintracellular vesicles, also colocalized with Nox1 in early endosomes and was necessary for tumor necrosis factor-� andinterleukin-1� generation of ROS. Cytokine activation of nuclear factor �B in SMCs required both Nox1 and ClC-3.We conclude that in response to tumor necrosis factor-� and interleukin-1�, NADPH oxidase generates ROS withinearly endosomes and that Nox1 cannot produce sufficient ROS for cell signaling in the absence of ClC-3. These databest support a model whereby ClC-3 is required for charge neutralization of the electron flow generated by Nox1 acrossthe membrane of signaling endosomes. (Circ Res. 2007;101:000-000.)
Key Words: smooth muscle cells � NAPDH oxidase � cell signaling � ion channels
In response to diverse extracellular stimuli, intracellularsignaling is dependent on generation of reactive oxygen
species (ROS) by NADPH oxidase. The catalytic core ofNADPH oxidase consists of a membrane-bound flavocyto-chrome b558 composed of a Nox (NADPH oxidase) (re-viewed elsewhere1) subunit and p22phox. The prototypicalmodel of NAPDH oxidase is found in phagosomes, where theorientation and biochemical properties of this heterodimerobligate reduction of oxygen to superoxide on the side of themembrane opposite from where NADPH and the cytosolicsubunits of the oxidase bind.2 Based on this orientation,activation of NADPH oxidase in nonphagocytes shouldgenerate superoxide into the extracellular space or, followingendocytosis, into intracellular vesicles. We hypothesized thatextracellular stimuli activating the Nox1-based NADPH ox-idase would produce superoxide in endocytotic vesicles.
The phagocyte NADPH oxidase is electrogenic, movingelectrons from cytoplasmic NADPH through the enzyme intothe phagosome to reduce oxygen to superoxide. Without
charge compensation, this electron flux rapidly depolarizesthe membrane (the voltage in the cytoplasm becomes positiverelative to the phagosome) and inactivates the oxidase.3 It isexpected that activation of NADPH oxidase in nonphago-cytes is subject to a similar requirement for charge neutral-ization. However, this mechanism has never been described.
With 9 members, the CLC family is the largest identifiedfamily of mammalian Cl� channels and its members functionin a surprisingly diverse set of biological roles, as revealed byhuman genetic diseases and murine knockout phenotypes.4
Fractionation studies aimed at determining ClC-3 functionhave localized it to the membrane of intracellular compart-ments such as endosomes and synaptic vesicles, where it hasbeen proposed to facilitate acidification.5–8 The identificationof ClC-4 and ClC-5, close structural relatives of ClC-3, aschloride-proton antiporters rather than anion channels hasplaced this role in question.9,10 We have previously demon-strated that ClC-3 is required for a normal oxidative burst inneutrophils.11 Based on these observations, we hypothesized
Original received March 6, 2007; revision received July 3, 2007; accepted July 24, 2007.From the Departments of Medicine (F.J.M., B.S., A.C.)1 and Pediatrics (M.F., G.J.H., T.J.B., F.S.L.), University of Iowa, Iowa City.*Both authors contributed equally to this work.Correspondence Francis J. Miller Jr, Department of Internal Medicine, 200 Hawkins Dr, Rm E314-4 GH, Iowa City, IA 52242. E-mail
[email protected]© 2007 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.107.151076
1
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on A
ugust 20, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on A
ugust 20, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on A
ugust 20, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on A
ugust 20, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
by guest on A
ugust 20, 2016http://circres.ahajournals.org/
Dow
nloaded from
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

that ClC-3 functions as a chloride–proton exchanger and isrequired for charge neutralization of the electron flow gener-ated by NADPH oxidase in endosomes of nonphagocytes.
Our data show that in response to inflammatory cytokines,a Nox1-based NADPH oxidase generates ROS within intra-cellular vesicles of smooth muscle cells (SMCs). Nox1 andClC-3 colocalize to early endosomes, and both are necessaryfor tumor necrosis factor (TNF)-� and interleukin (IL)-1�
generation of ROS and subsequent activation of the transcrip-tion factor nuclear factor (NF)-�B. We propose that onreceptor activation, a signaling endosome forms and compart-mentalizes intracellular ROS production. ClC-3 providescharge neutralization of the electron flow generated by Nox1.
Materials and Methods
Cultured CellsStudies were performed in human aortic SMCs (Clonetics Corp, SanDiego, Calif) and SMCs isolated from aorta of mice lacking theClcn3 gene encoding ClC-3 chloride channels12 and wild-typelittermate controls. Cells were isolated as previously described13 andgrown in 10% FCS in DMEM containing 1% basal medium Eagle,1% glutamine, 1% penicillin/streptomycin, 1% Eagle’s minimalessential medium 100� nonessential amino acids, and 2% HEPES.Studies were performed on 3 independent isolations of ClC-3–nullSMCs at passages 4 to 9 and 70% to 90% confluence.
Detection of Endosomal ROSThe detection of endosomal generation of ROS was performed usingthe fluorogenic reagent OxyBURST Green H2HFF–BSA14 (Molec-ular Probes, Invitrogen). This reagent consists of BSA covalentlylinked to the oxidant-sensitive dihydro-2�,4,5,6,7,7�-hexafluorofluo-rescein. Early endosomes develop an acidic internal pH of �6.0,which facilitates dissociation of the ligand from its receptor. Alimitation in the use of fluorophores to assess endosomal processesis the effect of pH on fluorescence intensity for many of thesecompounds. For example, as pH decreases, there is a significantreduction in fluorescence of oxidized 5(6)-carboxyfluorescein.15 Incontrast, and essential to the interpretation of our data, there is nochange in fluorescence intensity of oxidized H2HFF-BSA over thepH range of 4.5 to 7.5.15 Texas red–BSA (TXR-BSA) or –dextranconjugates were used as controls for endocytosis. Single-color cellfluorescence was analyzed by confocal microscopy or by flowcytometry. For details, see the expanded Materials and Methodssection in the online data supplement at http://circres.ahajournals.org.
Cytochemical Assessment of NADPHOxidase ActivityCells were grown to 80% confluence, and intracellular localizationof NADPH-derived ROS was detected using a method based oncerium cytochemistry,16 as detailed in the online data supplement.
AntibodiesTwo rabbit polyclonal antibodies were raised against ClC-3 andaffinity-purified. ClC-3a1 to the injected N-terminal peptides TYD-DFHTIDWVREKC and CKDRERHRRINSKKKES (Research Ge-netics) and ClC-3a2 to a coinjected keyhole limpet hemocyanin(KLH)- and BSA-conjugated N-terminal peptide of an alternativelyspliced form of ClC-3 (GenBank accession no. AF347689, PY-DGGGDSIPLRELHKRGTHYTMTNC; Bio-Synthesis). Additionalantibodies and their sources included Nox1, p22phox, and GAPDH(all from Santa Cruz Biotechnology), early endosome antigen (EEA)1(BD Transduction Laboratory), �-actin (Sigma), Rab5 (Abcam), andp65 (Imgenex).
ImmunostainingCells were grown on chamber slides, fixed in 2% paraformaldehydefor 15 minutes, and washed with PBS–Tween. Following blocking,SMCs were incubated with primary antibodies for ClC-3a2 (1:50)and p22phox (1:25) at room temperature for 2 hours. Secondaryantibodies consisted of goat anti-rabbit Alexa 488 (1:200) anddonkey anti-goat–biotin (1:500). Cells were then treated withstreptavidin–Alexa fluor 568 and TO-PRO-3. Images were scannedsequentially and merged using the Bio-Rad LaserSharp software.
ImmunoprecipitationAntibody was cross-linked to protein G–Sepharose, and affinitychromatography of SMC extracts was performed as detailed in theonline data supplement. Following elution, the collected fractionswere subjected to SDS-PAGE followed by immunoblotting usingspecified antibodies.
NADPH Oxidase ActivitySMC lysates were immunoprecipitated with anti-Rab5 antibody, andsuperoxide levels were measured in PBS–lucigenin (5 �mol/L) by anFB12 luminometer. Samples were allowed 2 minutes of darkadaptation following the addition of NADPH (10 �mol/L), with orwithout diphenylene iodonium (DPI) (10 �mol/L) and relative lightunits per second normalized to milligrams of protein used in theimmunoprecipitation.
Biotin–Streptavidin Pull-Down AssayThe NF-�B pull-down assay was performed as described in onlinedata supplement. Briefly, double-stranded oligonucleotides contain-ing the consensus sequence for NF-�B binding, and containing biotinon the 5�-nucleotide, were incubated with SMC lysate 4 hours afterIL-1�. Following Western blotting, membranes were probed withanti-p65 (Santa Cruz Biotechnology).
NF-�B Promoter ActivityNF-�B–mediated transcriptional induction was measured by infec-tion of SMCs with replication-deficient adenovirus containing theluciferase reporter gene driven by NF-�B transcriptional activa-tion.17 Luciferase activity (relative light units) was measured inreporter lysis buffer according to the protocol of the manufacturer(Promega Inc) and normalized to protein concentration. AdV12Rac1and AdN17 Rac1 were obtained from the University of Iowa VectorCore, and the AdNox1 sense and antisense were from KathyGriendling (Emory University, Atlanta, Ga); these have been char-acterized previously.18
Statistical AnalysisResults are expressed as means�SEM. Statistical comparisons wereperformed by Student’s 2-tailed t tests or ANOVA with the Tukeymultiple comparison posttest as appropriate. A value of P�0.05 wasconsidered significant.
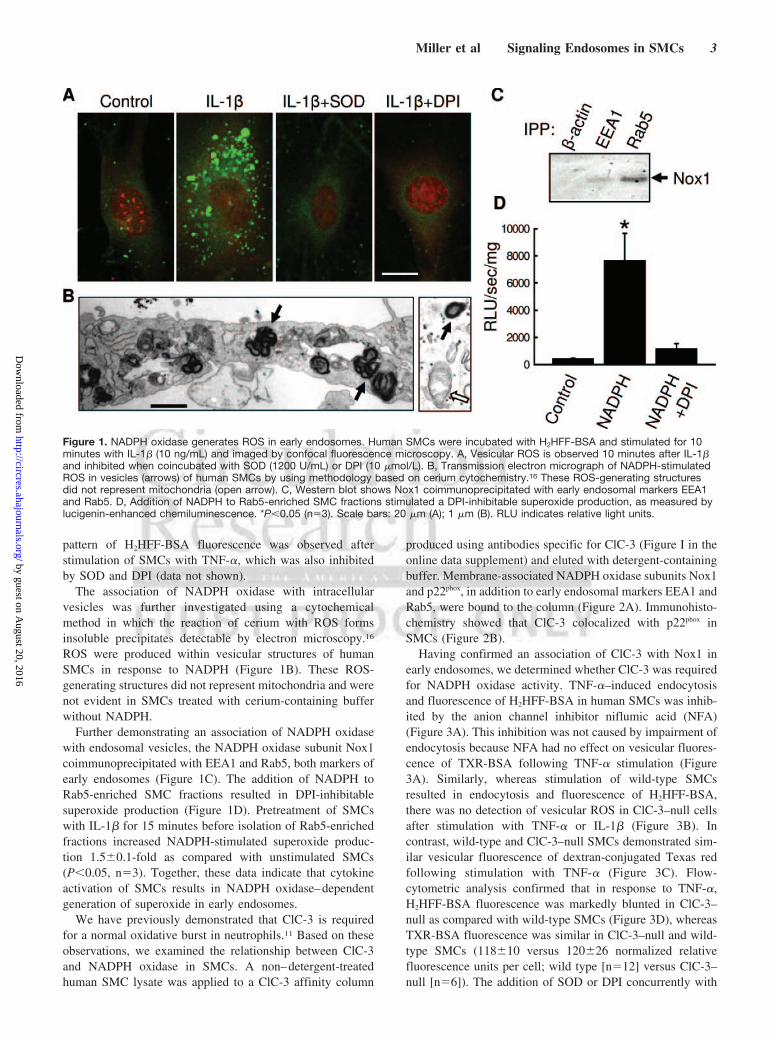
ResultsWe assessed generation of ROS using a membrane imperme-able oxidant-sensitive fluorescent probe (albumin-conjugated2�,4,5,6,7,7�-hexafluorofluorescein [H2HFF-BSA]). Intracel-lular punctate fluorescence thus indicates endocytosis of thefluorophore and production of ROS. In the presence ofH2HFF-BSA, IL-1� resulted within minutes in the appear-ance of numerous fluorescent vesicles in human SMCs(Figure 1A). Inhibition of fluorescence by superoxide dis-mutase (SOD) protein, which, like H2HFF-BSA, is endocy-tosed following stimulation with IL-1�, confirmed that thefluorescence resulted from generation of superoxide in theendosome. Inhibition of fluorescence by DPI suggestedNADPH oxidase as an enzymatic source of ROS. A similar
2 Circulation Research September 28, 2007
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
pattern of H2HFF-BSA fluorescence was observed afterstimulation of SMCs with TNF-�, which was also inhibitedby SOD and DPI (data not shown).
The association of NADPH oxidase with intracellularvesicles was further investigated using a cytochemicalmethod in which the reaction of cerium with ROS formsinsoluble precipitates detectable by electron microscopy.16
ROS were produced within vesicular structures of humanSMCs in response to NADPH (Figure 1B). These ROS-generating structures did not represent mitochondria and werenot evident in SMCs treated with cerium-containing bufferwithout NADPH.
Further demonstrating an association of NADPH oxidasewith endosomal vesicles, the NADPH oxidase subunit Nox1coimmunoprecipitated with EEA1 and Rab5, both markers ofearly endosomes (Figure 1C). The addition of NADPH toRab5-enriched SMC fractions resulted in DPI-inhibitablesuperoxide production (Figure 1D). Pretreatment of SMCswith IL-1� for 15 minutes before isolation of Rab5-enrichedfractions increased NADPH-stimulated superoxide produc-tion 1.5�0.1-fold as compared with unstimulated SMCs(P�0.05, n�3). Together, these data indicate that cytokineactivation of SMCs results in NADPH oxidase–dependentgeneration of superoxide in early endosomes.
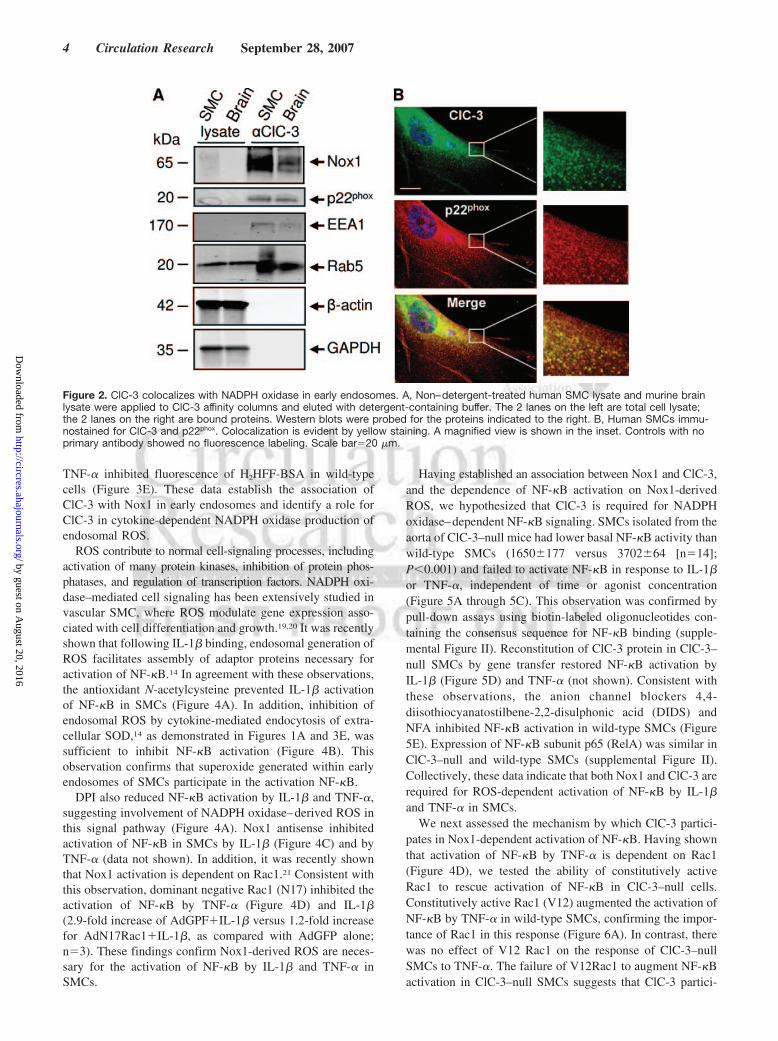
We have previously demonstrated that ClC-3 is requiredfor a normal oxidative burst in neutrophils.11 Based on theseobservations, we examined the relationship between ClC-3and NADPH oxidase in SMCs. A non–detergent-treatedhuman SMC lysate was applied to a ClC-3 affinity column
produced using antibodies specific for ClC-3 (Figure I in theonline data supplement) and eluted with detergent-containingbuffer. Membrane-associated NADPH oxidase subunits Nox1and p22phox, in addition to early endosomal markers EEA1 andRab5, were bound to the column (Figure 2A). Immunohisto-chemistry showed that ClC-3 colocalized with p22phox inSMCs (Figure 2B).
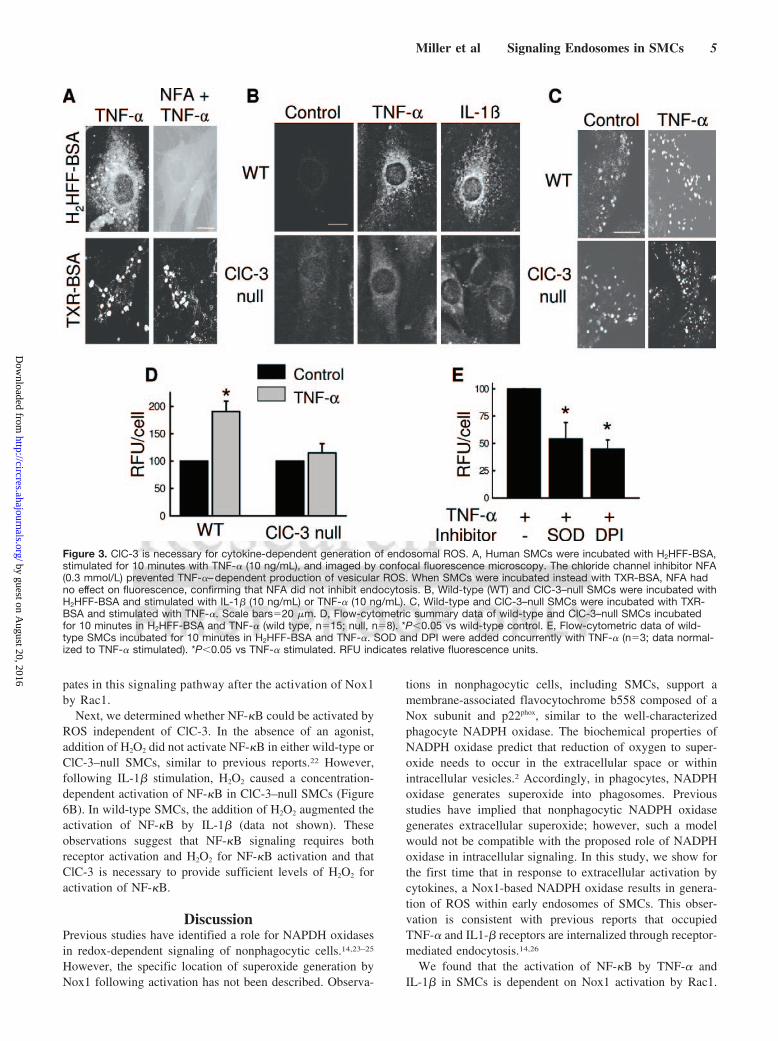
Having confirmed an association of ClC-3 with Nox1 inearly endosomes, we determined whether ClC-3 was requiredfor NADPH oxidase activity. TNF-�–induced endocytosisand fluorescence of H2HFF-BSA in human SMCs was inhib-ited by the anion channel inhibitor niflumic acid (NFA)(Figure 3A). This inhibition was not caused by impairment ofendocytosis because NFA had no effect on vesicular fluores-cence of TXR-BSA following TNF-� stimulation (Figure3A). Similarly, whereas stimulation of wild-type SMCsresulted in endocytosis and fluorescence of H2HFF-BSA,there was no detection of vesicular ROS in ClC-3–null cellsafter stimulation with TNF-� or IL-1� (Figure 3B). Incontrast, wild-type and ClC-3–null SMCs demonstrated sim-ilar vesicular fluorescence of dextran-conjugated Texas redfollowing stimulation with TNF-� (Figure 3C). Flow-cytometric analysis confirmed that in response to TNF-�,H2HFF-BSA fluorescence was markedly blunted in ClC-3–null as compared with wild-type SMCs (Figure 3D), whereasTXR-BSA fluorescence was similar in ClC-3–null and wild-type SMCs (118�10 versus 120�26 normalized relativefluorescence units per cell; wild type [n�12] versus ClC-3–null [n�6]). The addition of SOD or DPI concurrently with
Figure 1. NADPH oxidase generates ROS in early endosomes. Human SMCs were incubated with H2HFF-BSA and stimulated for 10minutes with IL-1� (10 ng/mL) and imaged by confocal fluorescence microscopy. A, Vesicular ROS is observed 10 minutes after IL-1�and inhibited when coincubated with SOD (1200 U/mL) or DPI (10 �mol/L). B, Transmission electron micrograph of NADPH-stimulatedROS in vesicles (arrows) of human SMCs by using methodology based on cerium cytochemistry.16 These ROS-generating structuresdid not represent mitochondria (open arrow). C, Western blot shows Nox1 coimmunoprecipitated with early endosomal markers EEA1and Rab5. D, Addition of NADPH to Rab5-enriched SMC fractions stimulated a DPI-inhibitable superoxide production, as measured bylucigenin-enhanced chemiluminescence. *P�0.05 (n�3). Scale bars: 20 �m (A); 1 �m (B). RLU indicates relative light units.
Miller et al Signaling Endosomes in SMCs 3
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
TNF-� inhibited fluorescence of H2HFF-BSA in wild-typecells (Figure 3E). These data establish the association ofClC-3 with Nox1 in early endosomes and identify a role forClC-3 in cytokine-dependent NADPH oxidase production ofendosomal ROS.
ROS contribute to normal cell-signaling processes, includingactivation of many protein kinases, inhibition of protein phos-phatases, and regulation of transcription factors. NADPH oxi-dase–mediated cell signaling has been extensively studied invascular SMC, where ROS modulate gene expression asso-ciated with cell differentiation and growth.19,20 It was recentlyshown that following IL-1� binding, endosomal generation ofROS facilitates assembly of adaptor proteins necessary foractivation of NF-�B.14 In agreement with these observations,the antioxidant N-acetylcysteine prevented IL-1� activationof NF-�B in SMCs (Figure 4A). In addition, inhibition ofendosomal ROS by cytokine-mediated endocytosis of extra-cellular SOD,14 as demonstrated in Figures 1A and 3E, wassufficient to inhibit NF-�B activation (Figure 4B). Thisobservation confirms that superoxide generated within earlyendosomes of SMCs participate in the activation NF-�B.
DPI also reduced NF-�B activation by IL-1� and TNF-�,suggesting involvement of NADPH oxidase–derived ROS inthis signal pathway (Figure 4A). Nox1 antisense inhibitedactivation of NF-�B in SMCs by IL-1� (Figure 4C) and byTNF-� (data not shown). In addition, it was recently shownthat Nox1 activation is dependent on Rac1.21 Consistent withthis observation, dominant negative Rac1 (N17) inhibited theactivation of NF-�B by TNF-� (Figure 4D) and IL-1�(2.9-fold increase of AdGPF�IL-1� versus 1.2-fold increasefor AdN17Rac1�IL-1�, as compared with AdGFP alone;n�3). These findings confirm Nox1-derived ROS are neces-sary for the activation of NF-�B by IL-1� and TNF-� inSMCs.
Having established an association between Nox1 and ClC-3,and the dependence of NF-�B activation on Nox1-derivedROS, we hypothesized that ClC-3 is required for NADPHoxidase–dependent NF-�B signaling. SMCs isolated from theaorta of ClC-3–null mice had lower basal NF-�B activity thanwild-type SMCs (1650�177 versus 3702�64 [n�14];P�0.001) and failed to activate NF-�B in response to IL-1�
or TNF-�, independent of time or agonist concentration(Figure 5A through 5C). This observation was confirmed bypull-down assays using biotin-labeled oligonucleotides con-taining the consensus sequence for NF-�B binding (supple-mental Figure II). Reconstitution of ClC-3 protein in ClC-3–null SMCs by gene transfer restored NF-�B activation byIL-1� (Figure 5D) and TNF-� (not shown). Consistent withthese observations, the anion channel blockers 4,4-diisothiocyanatostilbene-2,2-disulphonic acid (DIDS) andNFA inhibited NF-�B activation in wild-type SMCs (Figure5E). Expression of NF-�B subunit p65 (RelA) was similar inClC-3–null and wild-type SMCs (supplemental Figure II).Collectively, these data indicate that both Nox1 and ClC-3 arerequired for ROS-dependent activation of NF-�B by IL-1�
and TNF-� in SMCs.We next assessed the mechanism by which ClC-3 partici-
pates in Nox1-dependent activation of NF-�B. Having shownthat activation of NF-�B by TNF-� is dependent on Rac1(Figure 4D), we tested the ability of constitutively activeRac1 to rescue activation of NF-�B in ClC-3–null cells.Constitutively active Rac1 (V12) augmented the activation ofNF-�B by TNF-� in wild-type SMCs, confirming the impor-tance of Rac1 in this response (Figure 6A). In contrast, therewas no effect of V12 Rac1 on the response of ClC-3–nullSMCs to TNF-�. The failure of V12Rac1 to augment NF-�Bactivation in ClC-3–null SMCs suggests that ClC-3 partici-
Figure 2. ClC-3 colocalizes with NADPH oxidase in early endosomes. A, Non–detergent-treated human SMC lysate and murine brainlysate were applied to ClC-3 affinity columns and eluted with detergent-containing buffer. The 2 lanes on the left are total cell lysate;the 2 lanes on the right are bound proteins. Western blots were probed for the proteins indicated to the right. B, Human SMCs immu-nostained for ClC-3 and p22phox. Colocalization is evident by yellow staining. A magnified view is shown in the inset. Controls with noprimary antibody showed no fluorescence labeling. Scale bar�20 �m.
4 Circulation Research September 28, 2007
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from
pates in this signaling pathway after the activation of Nox1by Rac1.
Next, we determined whether NF-�B could be activated byROS independent of ClC-3. In the absence of an agonist,addition of H2O2 did not activate NF-�B in either wild-type orClC-3–null SMCs, similar to previous reports.22 However,following IL-1� stimulation, H2O2 caused a concentration-dependent activation of NF-�B in ClC-3–null SMCs (Figure6B). In wild-type SMCs, the addition of H2O2 augmented theactivation of NF-�B by IL-1� (data not shown). Theseobservations suggest that NF-�B signaling requires bothreceptor activation and H2O2 for NF-�B activation and thatClC-3 is necessary to provide sufficient levels of H2O2 foractivation of NF-�B.
DiscussionPrevious studies have identified a role for NAPDH oxidasesin redox-dependent signaling of nonphagocytic cells.14,23–25
However, the specific location of superoxide generation byNox1 following activation has not been described. Observa-
tions in nonphagocytic cells, including SMCs, support amembrane-associated flavocytochrome b558 composed of aNox subunit and p22phox, similar to the well-characterizedphagocyte NADPH oxidase. The biochemical properties ofNADPH oxidase predict that reduction of oxygen to super-oxide needs to occur in the extracellular space or withinintracellular vesicles.2 Accordingly, in phagocytes, NADPHoxidase generates superoxide into phagosomes. Previousstudies have implied that nonphagocytic NADPH oxidasegenerates extracellular superoxide; however, such a modelwould not be compatible with the proposed role of NADPHoxidase in intracellular signaling. In this study, we show forthe first time that in response to extracellular activation bycytokines, a Nox1-based NADPH oxidase results in genera-tion of ROS within early endosomes of SMCs. This obser-vation is consistent with previous reports that occupiedTNF-� and IL1-� receptors are internalized through receptor-mediated endocytosis.14,26
We found that the activation of NF-�B by TNF-� andIL-1� in SMCs is dependent on Nox1 activation by Rac1.
Figure 3. ClC-3 is necessary for cytokine-dependent generation of endosomal ROS. A, Human SMCs were incubated with H2HFF-BSA,stimulated for 10 minutes with TNF-� (10 ng/mL), and imaged by confocal fluorescence microscopy. The chloride channel inhibitor NFA(0.3 mmol/L) prevented TNF-�–dependent production of vesicular ROS. When SMCs were incubated instead with TXR-BSA, NFA hadno effect on fluorescence, confirming that NFA did not inhibit endocytosis. B, Wild-type (WT) and ClC-3–null SMCs were incubated withH2HFF-BSA and stimulated with IL-1� (10 ng/mL) or TNF-� (10 ng/mL). C, Wild-type and ClC-3–null SMCs were incubated with TXR-BSA and stimulated with TNF-�. Scale bars�20 �m. D, Flow-cytometric summary data of wild-type and ClC-3–null SMCs incubatedfor 10 minutes in H2HFF-BSA and TNF-� (wild type, n�15; null, n�8). *P�0.05 vs wild-type control. E, Flow-cytometric data of wild-type SMCs incubated for 10 minutes in H2HFF-BSA and TNF-�. SOD and DPI were added concurrently with TNF-� (n�3; data normal-ized to TNF-� stimulated). *P�0.05 vs TNF-� stimulated. RFU indicates relative fluorescence units.
Miller et al Signaling Endosomes in SMCs 5
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

Interestingly, neither the addition of constitutively activeRac1 nor H2O2 was sufficient to activate NF-�B in SMCsbecause they also required the addition of TNF-� or IL-1�.Together, our data support the formation of signaling endo-somes27 in SMCs on receptor binding, with subsequentgeneration of Nox1-derived ROS into the newly formedendosomes and assembly of adaptor proteins, both of whichare necessary for activation of NF-�B.14
We also found that ClC-3 is colocalized to the earlyendosome with Nox1 and p22phox in SMCs and is necessaryfor the production of ROS and activation of NF-�B bycytokines. To produce superoxide, Nox1 moves electronsfrom intracellular NADPH across flavin adenine dinucleotideand 2 hemes to reduce oxygen within the endosome. Withoutcharge compensation, this electron flux rapidly depolarizesthe plasma membrane of neutrophils and inactivates theoxidase.3 Our data support a model whereby ClC-3 functionsas a chloride–proton exchanger, necessary for charge neutral-ization of electron flow generated by Nox1 (Figure 7). Theother members of the ClC family that are most homologous toClC-3 have been shown to function as chloride–protonexchangers.9,10
The ClC family of chloride channels includes 9 members,with ClC-3 being the most abundant in SMCs.28 Although theClC-3 protein is highly conserved and ubiquitously ex-
pressed, its precise function is controversial. ClC-3 was firstproposed as a swelling-activated Cl� channel in plasma mem-branes critical for volume regulation29; however, in recentstudies, a primary intracellular localization has been proposedwhere ClC-3 participates in charge neutralization of thevacuolar ATPase (V-ATPase) during acidification of intra-cellular synaptic vesicles,5 insulin granules,6 and lysosomes.7
The complex phenotypic abnormalities of ClC-3–deficientmice denote a fundamental role for ClC-3 in cellularfunction.5,12
The ClC-3, -4, and -5 branch of the ClC family has beenhypothesized to provide shunt conductance in membranes ofintracellular organelles, permitting intraluminal acidificationby the V-ATPase.4 This hypothesis is supported by impairedendosomal chloride accumulation and acidification in ClC-3–deficient mice.8 However, if ClC-3 functions as a Cl�/H�
antiporter as previously proposed,9,10 it may be relatively illsuited for direct charge compensation of the V-ATPase.ClC-4 and ClC-5 currents rectify very strongly in the outwarddirection, preferentially conducting Cl� to the cytoplasmfrom the extracellular space. Endocytosis of this type ofchannel situates the extracellular face within the endosome,whereby strong outward rectification favors the movement ofCl� out of, rather than into endosomes. Furthermore, forClC-3 to function as a H�/Cl� antiporter and move current in
Figure 4. NF-�B activation by IL-1� and TNF-� is Nox1 dependent. NF-�B activation was measured by luciferase reporter assay. Lucif-erase activity was measured 6 hours after IL-1� (10 ng/mL) or TNF-� (10 ng/mL) treatment. A, In response to IL-1� and TNF-�, NF-�Bactivation in wild-type SMCs was inhibited by DPI (6 �mol/L) and by the antioxidant N-acetylcysteine (NAC) (30 mmol/L). B, NF-�Bactivation was inhibited whether SOD and TNF-� were washed from SMCs after 30 minutes or at the completion of the 6-hour incuba-tion. C, Adenoviral-mediated expression of antisense to Nox1 inhibited NF-�B activation by IL-1� in SMCs. D, Expression of dominantnegative Rac1 (AdN17Rac1) inhibited activation of NF-�B by TNF-�. AdGFP served as a vector control. Results are averages�SEM of3 to 5 experiments. For each experiment, values are normalized to the control value. *P�0.05 vs control, �P�0.05 vs AdGFP plusIL-1� or TNF-�.
6 Circulation Research September 28, 2007
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

the direction required to directly charge neutralize theV-ATPase (Cl� in, H� out of the endosome), each cyclewould require the removal of a proton from the endosome.Although the net charge movement of ClC-3 acting in thisdirection could still provide the required shunt current, thisprocess would be energetically unfavorable, as the V-ATPasewould need to transport twice the number of protons into theendosome to compensate for the simultaneous outward H�
flux via ClC-3. Direct compensation by ClC-3 only worksefficiently if it is acting as a simple anion channel conductingCl� into endosomes. The presence of NADPH oxidase in theendosome actually removes voltage constraints on vesicular
acidification. ClC-3 is well suited to neutralize the movementof negative current into endosomes. The oxidase generates arapid depolarization of 100 mV across the plasma mem-brane of neutrophils, and this effect may be even larger acrossphagosomal membranes.30 Therefore, the highly positiveintravesicular voltages required to activate ClC-3 may existacross the endosomal membrane on activation of the NADPHoxidase. Furthermore, a net negative intravesicular potentialwould preclude the need to provide direct charge neutraliza-tion for the V-ATPase, because movement of protons into thelumen would be highly energetically favored. In this way,ClC-3 may provide indirect charge neutralization for the
Figure 5. NF-�B activation by IL-1� and TNF-� requires ClC-3. NF-�B activation was measured by luciferase reporter assay. TNF-�activation of NF-�B was markedly reduced in ClC-3–null SMCs independent of time (A) or concentration (B). C and D, IL-1� activationof NF-�B was reduced in ClC-3–null SMCs (C) and rescued by adenoviral-mediated expression of ClC-3 (D). E, IL-1� activation ofNF-�B was inhibited by the chloride channel blockers NFA (0.3 mmol/L) and DIDS (0.3 mmol/L) in wild-type (WT) SMCs. Luciferaseactivity was measured 6 hours after IL-1� (10 ng/mL) or TNF-� (10 ng/mL) treatment. Results are averages�SEM of 3 to 5 experi-ments. For each experiment, values are normalized to the control value. *P�0.05 vs control, �P�0.05 vs IL-1� alone.
Figure 6. Role of ClC-3 in NF-�B activation. A, Constitutively active Rac1 (AdV12Rac1) augmented the activation of NF-�B by TNF-�in wild-type (WT) SMCs but not ClC-3–null SMCs in response to TNF-�. B, In the absence of IL-1�, addition of H2O2 did not activateNF-�B in ClC-3–null SMCs. However, following IL-1� stimulation, H2O2 caused a concentration-dependent activation of NF-�B in ClC-3–null SMCs. Results are averages�SEM of 3 to 5 experiments. *P�0.05 vs control, �P�0.05 vs wild-type plus TNF-�.
Miller et al Signaling Endosomes in SMCs 7
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

V-ATPase by facilitating the activity of the NADPH oxidase.Distinct from these theoretical issues, our data indicate thatactivation of V-ATPase is not necessary for ROS signaling,because inhibition of the protein with bafilomycin had noeffect on activation of NF-�B by cytokines (supplementalFigure IV).
The inability of ClC-3–null SMCs to make endosomalROS and activate NF-�B in response to TNF-� and IL-1�suggests that redox-mediated signaling events may be regu-lated by modulation of ClC-3 activity. In support of this,when ClC-3 was overexpressed in wild-type SMCs, NF-�Bactivation markedly increased (AdGFP plus IL-1�, 7.2�0.6-fold; AdClC-3 plus IL-1�, 83.3�5.3-fold compared withunstimulated control [n�3]; P�0.05). This suggests thatClC-3 is not simply required for Nox1 activation, but ratherthe level of ClC-3 activity may regulate ROS production.Several mechanisms of ClC-3 regulation have been proposed,including its phosphorylation at a unique serine residue byprotein kinase C29 and Ca2�/camodulin-dependent kinaseII.13,31 The neutrophil NADPH oxidase (Nox2) is also depen-dant on ClC-3 for intracellular ROS production. ClC-3colocalizes with the oxidase in secretory vesicles and isupregulated to phagosomes following uptake of opsonizedzymosan.11 Therefore, regulation of this anion exchanger mayrepresent a common mechanism for control of NADPHoxidase activity following activation.
The molecular mechanism by which endosomal ROSsubsequently activates redox-dependent signals is not known.Superoxide has been shown to traverse both the plasma32 andmitochondrial33 membranes via anion channels to exert cyto-plasmic effects. However, the pKa of the conjugate acid ofsuperoxide, hydrogen superoxide (HO2
.) (also known ashydroperoxyl radical) is 4.88.34 Therefore, the acidic envi-ronment of endosomes favors formation of HO2
. from super-oxide, which will react with a second proton to form H2O2.Both HO2
. and H2O2 are uncharged and can diffuse to thecytosol to activate redox-dependent signal molecules. It is
also possible that localized release of superoxide and H2O2
generate distinct and independent cytoplasmic signals.There are multiple homologs of the Nox enzymes within
cells.1 We have focused our evaluation on Nox1 because ofprevious reports identifying agonist-dependent activation ofNox1 and a subsequent role for Nox1-derived ROS in SMCsignaling.18,20,25 We cannot exclude the possibility that activ-ity of the other Nox homologs also are dependent on ClC-3 orrelated proteins. Similarly, it is not known whether othermethods of Nox1 activation in SMCs involve ClC-3–depen-dent endosomal generation of ROS. These will be importantareas of future research. Preliminary data from our laboratoryindicate that our findings are not unique to vascular SMCs.
We describe NADPH oxidase–dependent generation ofROS in early endosomes and identify ClC-3 as a criticalcomponent and a novel intermediate in redox-dependentcontrol of gene expression. Furthermore, ClC-3 may be anew target in the regulation of redox-dependent geneexpression involved in vascular disease,35 cancer,36,37 andinflammation.11
AcknowledgmentsWe thank associates of the University of Iowa Roy J. and Lucille A.Carver College of Medicine Central Microscopy Research Facility,the Flow Cytometry Facility, and the Gene Transfer Vector CoreFacility of the University of Iowa Center for Gene Therapy of CysticFibrosis and Other Genetic Diseases, which is supported by NationalInstitute of Diabetes and Digestive and Kidney Diseases/NIH grantP30 DK 54759.
Sources of FundingThis work was supported by NIH grants HL062483 (to F.S.L.,F.J.M.) and HL081750 (to F.J.M.) and by the American HeartAssociation (F.S.L.).
DisclosuresNone.
Figure 7. Proposed function of Nox1 and ClC-3 in the signaling endosome. Binding of IL-1� or TNF-� to the cell membrane initiatesendocytosis and formation of an early endosome (EEA1 and Rab5), which also contains NADPH oxidase subunits Nox1 and p22phox, inaddition to ClC-3. Nox1 is electrogenic, moving electrons from intracellular NADPH through a redox chain within the enzyme into theendosome to reduce oxygen to superoxide. ClC-3 functions as a chloride–proton exchanger, required for charge neutralization of theelectron flow generated by Nox1. The ROS generated by Nox1 result in NF-�B activation. Both ClC-3 and Nox1 are necessary for gen-eration of endosomal ROS and subsequent NF-�B activation by IL-1� or TNF-� in SMCs. In addition, redox-independent pathways notshown also contribute to the activation of NF-�B.
8 Circulation Research September 28, 2007
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

References1. Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev
Immunol. 2004;4:181–189.2. Vignais PV. The superoxide-generating NADPH oxidase: structural
aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459.
3. DeCoursey TE, Morgan D, Cherny VV. The voltage dependence ofNADPH oxidase reveals why phagocytes need proton channels. Nature.2003;422:531–534.
4. Jentsch TJ, Neagoe I, Scheel O. CLC chloride channels and transporters.Curr Opin Neurobiol. 2005;15:319–325.
5. Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, ZdebikAA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ.Disruption of ClC-3, a chloride channel expressed on synaptic vesicles,leads to a loss of the hippocampus. Neuron. 2001;29:185–196.
6. Barg S, Huang P, Eliasson L, Nelson DJ, Obermuller S, Rorsman P,Thevenod F, Renstrom E. Priming of insulin granules for exocytosis bygranular Cl(-) uptake and acidification. J Cell Sci. 2001;114:2145–2154.
7. Li X, Wang T, Zhao Z, Weinman SA. The ClC-3 chloride channelpromotes acidification of lysosomes in CHO- K1 and Huh-7 cells. Am JPhysiol Cell Physiol. 2002;282:C1483–C1491.
8. Hara-Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S, VerkmanAS. ClC-3 chloride channels facilitate endosomal acidification andchloride accumulation. J Biol Chem. 2005;280:1241–1247.
9. Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent elec-trogenic chloride/proton exchange by endosomal CLC proteins. Nature.2005;436:424–427.
10. Picollo A, Pusch M. Chloride/proton antiporter activity of mammalianCLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423.
11. Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS. Anionchannels, including ClC-3, are required for normal neutrophil oxidativefunction, phagocytosis, and transendothelial migration. J Biol Chem.2006;281:12277–12288.
12. Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC,Nehrke K, Williamson RA, Lamb FS. Altered GABAergic functionaccompanies hippocampal degeneration in mice lacking ClC-3voltage-gated chloride channels. Brain Res. 2002;958:227–250.
13. Robinson NC, Huang P, Kaetzel MA, Lamb FS, Nelson DJ. Identificationof an N-terminal amino acid of the CLC-3 chloride channel critical inphosphorylation-dependent activation of a CaMKII-activated chloridecurrent. J Physiol. 2004;556:353–368.
14. Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T,Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptorcomplexes. Mol Cell Biol. 2006;26:140–154.
15. Chen CS. Phorbol ester induces elevated oxidative activity and alkal-ization in a subset of lysosomes. BMC Cell Biol. 2002;3:21.
16. Odani K, Kobayashi T, Ogawa Y, Yoshida S, Seguchi H. ML-7 inhibitsexocytosis of superoxide-producing intracellular compartments in humanneutrophils stimulated with phorbol myristate acetate in a myosin lightchain kinase-independent manner. Histochem Cell Biol. 2003;119:363–370.
17. Sanlioglu S, Williams CM, Samavati L, Butler NS, Wang G, McCray PBJr, Ritchie TC, Hunninghake GW, Zandi E, Engelhardt JF. Lipopolysac-charide induces Rac1-dependent reactive oxygen species formation andcoordinates tumor necrosis factor-alpha secretion through IKK regulationof NF-kappa B. J Biol Chem. 2001;276:30188–30198.
18. Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL,Lambeth JD, Griendling KK. Novel gp91 phox homologues in vascularsmooth muscle cells. nox1 mediates angiotensin II-induced superoxide
formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894.
19. Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)Hoxidase-derived reactive oxygen species as mediators of angiotensin IIsignaling. Antioxid Redox Signal. 2002;4:899–914.
20. Gorlach A, Kietzmann T, Hess J. Redox signaling through NADPHoxidases: involvement in vascular proliferation and coagulation. Ann N YAcad Sci. 2002;973:505–507.
21. Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactiveoxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–17726.
22. Hoare GS, Marczin N, Chester AH, Yacoub MH. Role of oxidant stressin cytokine-induced activation of NF-kappaB in human aortic smoothmuscle cells. Am J Physiol. 1999;277:H1975–H1984.
23. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angioten-sin II stimulates NADH and NADPH oxidase activity in cultured vascularsmooth muscle cells. Circ Res. 1994;74:1141–1148.
24. Bedard K, Krause KH. The NOX family of ROS-generating NADPHoxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313.
25. Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features,expression, and regulation. Am J Physiol Regul Integr Comp Physiol.2003;285:R277–R297.
26. Mosselmans R, Hepburn A, Dumont JE, Fiers W, Galand P. Endocyticpathway of recombinant murine tumor necrosis factor in L-929 cells.J Immunol. 1988;141:3096–3100.
27. Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of ret-rograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625.
28. Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC.Expression of CLCN voltage-gated chloride channel genes in humanblood vessels. J Mol Cell Cardiol. 1999;31:657–666.
29. Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identi-fication of a volume-regulated chloride channel. Nature. 1997;390:417–421.
30. Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z,Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is thesignaling component of the macrophage migration inhibitoryfactor-CD74 receptor complex. Immunity. 2006;25:595–606.
31. Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaezel MA, Nelson DJ.Regulation of human ClC-3 channels by multifunctional Ca2�/calmod-ulin dependent protein kinase. J Biol Chem. 2001;276:20092–20100.
32. Lynch RE, Fridovich I. Permeation of the erythrocyte stroma bysuperoxide radical. J Biol Chem. 1978;253:4697–4699.
33. Weissmann N, Ebert N, Ahrens M, Ghofrani HA, Schermuly RT, HanzeJ, Fink L, Rose F, Conzen J, Seeger W, Grimminger F. Effects ofmitochondrial inhibitors and uncouplers on hypoxic vasoconstriction inrabbit lungs. Am J Respir Cell Mol Biol. 2003;29:721–732.
34. Behar D, Czapski G, Rabini J, Dorfman LM, Schwartz HA. The aciddissociation constant and decay kinetics of perhydroxyl radical. J PhysChem. 1970;74:3209–3214.
35. Hanna IR, Hilenski LL, Dikalova A, Taniyama Y, Dikalov S, Lyle A,Quinn MT, Lassegue B, Griendling KK. Functional association of nox1with p22phox in vascular smooth muscle cells. Free Radic Biol Med.2004;37:1542–1549.
36. Mitsushita J, Lambeth JD, Kamata T. The superoxide-generating oxidaseNox1 is functionally required for Ras oncogene transformation. CancerRes. 2004;64:3580–3585.
37. Lemonnier L, Shuba Y, Crepin A, Roudbaraki M, Slomianny C, MauroyB, Nilius B, Prevarskaya N, Skryma R. Bcl-2-dependent modulation ofswelling-activated Cl- current and ClC-3 expression in human prostatecancer epithelial cells. Cancer Res. 2004;64:4841–4848.
Miller et al Signaling Endosomes in SMCs 9
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

J. Barna and Fred S. LambFrancis J. Miller, Jr, Mohammed Filali, Gina J. Huss, Bojana Stanic, Ali Chamseddine, Thomas
Signaling Endosomes Containing Nox1 and ClC-3B in Vascular Smooth Muscle Cells RequiresκCytokine Activation of Nuclear Factor
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2007 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
published online August 2, 2007;Circ Res.
http://circres.ahajournals.org/content/early/2007/08/02/CIRCRESAHA.107.151076.citationWorld Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2007/08/02/CIRCRESAHA.107.151076.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on August 20, 2016
http://circres.ahajournals.org/D
ownloaded from

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
1
Expanded Materials and Methods.
Detection of endosomal ROS. The detection of endosomal generation of ROS was performed
using the fluorogenic reagent OxyBURST Green H2HFF-BSA 1 (Molecular Probes, O-
13291). This reagent consists of bovine serum albumin covalently linked to the oxidant
sensitive dihydro-2’,4,5,6,7,7’-hexafluorofluorescein. Cells were grown on chamber slides,
serum-deprived (0.5% serum) overnight, then incubated in 50 µg/ml H2HFF-BSA for 5
minutes with or without SOD (1200 units/ml), DPI (10 µmol/L), or NFA (300 µmol/L) at
37°C, and then stimulated with IL-1β or TNF-α. After 10 minutes, cells were fixed in 4%
paraformaldehyde. Nuclear counterstaining was performed with TO-PRO-3 (Molecular
Probes) for 5 minutes in some samples. Cells were cover-slide mounted with SlowFade Gold
antifade reagent (Molecular Probes) and imaged by fluorescence confocal microscopy. In
some experiments, H2HFF-BSA was replaced with BSA-conjugated to Texas Red (TXR-
BSA, 50 µg/ml, Molecular Probes), or dextran-conjugated to Texas Red (50 µg/ml,
Molecular Probes).
Flow Cytometric Analysis. SMC were rinsed in phenol red free DMEM and single color
fluorescent labeling performed by incubating cells with H2HFF-BSA (50 µg/ml) or TXR-
BSA (50 µg/ml) in phenol red free DMEM with or without TNF-α (10 ng/ml). After 10
minutes, cells were rinsed three times in ice cold PBS, collected by trypsinization,
centrifuged at 400g for 5 minutes at 4° C, resuspended in ice cold PBS, filtered, and kept on
ice in the dark for immediate analysis. Flow cytometric data were collected on a Becton
Dickinson FACS DiVa (BD Biosciences, San Jose, CA, USA) flow cytometer and analyzed
with the FlowJo software package (Tree Star, Inc., Ashland, OR). Dot plots of forward
scatter pulse area (FSC-A) vs. side scatter pulse area (SSC-A) and forward scatter pulse area

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
2
(FSC-A) vs. forward scatter pulse width (FSC-W) were generated and events gated to
exclude cell debris and aggregates. Using UV excitation and the Hoechst channel (424/44
BP filter) as a constant, autofluorescence was subtracted from the H2HFF channel to improve
signal to noise ratio. H2HFF was excited by an argon laser at 488 nm and detected with a
530/30 bandpass filter; whereas TXR was excited with a dye laser at 600 nm and detected
with a 630/22 bandpass filter. For each sample, 30,000 gated cells were analyzed. Using a
histogram of pulse area fluorescence intensity, a gate (R1) was created which contained the
upper 5% of unstimulated SMC. TNF-α stimulated samples were examined with identical
gating parameters and the frequency of cells in R1 of the stimulated sample compared to the
frequency of cells in R1 of the unstimulated sample.
Cytochemical assessment of NADPH oxidase activity. To detect intracellular localization of
NADPH-derived ROS, we used a method based on cerium cytochemistry 2. Human aortic
SMC were grown to 80% confluence and fixed with 0.1% glutaraldehyde in buffer (4° C, pH
7.4) containing 20 mmol/L HEPES, 135 mmol/L NaCl, 5 mmol/L KCL, and 5 mmol/L
glucose for 10 minutes. Cells were washed and incubated with HEPES buffer containing 20
mmol/L tricine, 1 mmol/L CeCl3, 1 mmol/L NADPH, 20 µmol/L flavin adenine dinucleotide,
and 1 mmol/L NaN3. NADPH was omitted in some samples. After 30 minutes at 37° C,
cells were washed with HEPES buffer and treated with 2% glutaraldehyde in HEPES buffer
for 10 minutes. Cells were washed with 0.1 mol/L cacodylate buffer (pH 7.4), fixed on ice
with 1% osmium tetroxide in cacodylate buffer for 30 minutes, and then washed with
cacodylate buffer before dehydration by graded ethanol. The cells were embedded in epoxy
resin, and ultrathin sections cut at 95 nm and imaged with an Hitachi 7000 transmission
electron microscope operated at a voltage of 75 kV.

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
3
Immunoaffinity column. The antibody ClC-3a2 was cross-linked to protein G-sepharose
using dimethy-pimelidate and 0.2 mol/L ethanolamine, pH 8.0. Samples were homogenized
in lysis buffer (150 mmol/L NaCl, 50 mmol/L HEPES, 20 mmol/L EDTA, 10% glycerol),
protease cocktail (Roche, 1 tablet/0.05 L) and pre-cleared with protein G-sepharose to
remove non-specific binding. Cleared lysate was loaded onto a ClC-3 immunoaffinity
column and incubated overnight at 4° C. The column was then washed three times with wash
buffer (10 mmol/L Tris-HCl, 140 mmol/L NaCl, 0.02% NaN3, 0.5% Triton X-100, 0.5% NP-
40, pH 8.0 and three times with PBS in order to remove unbound proteins. Column-bound
proteins were eluted with 50 mmol/L triethanolamine, 150 mmol/L NaCl, 1% Triton-X100,
pH 11.5, and then neutralized with 1 mol/L Tris-HCl, pH 7.0. Prior to electrophoresis, the
protein in the samples were precipitated with 10% TCA, 0.1% Deoxycholate-Na+, washed
with 80% acetone and resuspended in alkaline sample buffer. The column was regenerated
with 0.1 mol/L acetic acid and 0.2 mol/L NaCl, pH 2.61.
Immunoprecipitation. Human SMC were transferred to a glass grinder and homogenized in a
buffer containing 0.25 mol/L sucrose, 10 mmol/L ethanolamine, 1 mmol/L EDTA, 1 mmol/L
PMSF and 100 ug/ml aprotinin at pH 7.6 and cleared by centrifugation. The supernatant was
concentrated 10-fold on Amicon filters (YM50) and proteins quantified by Micro-BCA. The
resulting fraction was pre-cleared with 50 µl of washed activated protein-G-sepharose slurry
(Sigma). Protein (2.5 mg) was incubated with antibody (1 µg) for 4 hrs at 4° C on a rotary
shaker. Protein-G-sepharose was added for 45 min. The tubes were placed on ice for 10
minutes to allow separation before a 15 second pulse centrifugation at maximum speed to
collect the immunoprecipitated beads. The pellets were washed with homogenization buffer
and then PBS. After drying the pellets, the beads were resuspended by vigorous vortexing
with 50 µl of the protein-sample buffer then boiled for 15 minutes at 85°C, and centrifuged

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
4
for 5 minutes. The eluted samples were resolved on pre-cast Criterion gels (Bio-Rad) and
transferred to a Nitrocellulose membrane for 3 hours on a TRANS-BLOT SD semi-dry
transfer cell at 450 mA (Bio-Rad).
Biotin-streptavidin pull down assay. Double stranded oligonucleotides containing the
consensus sequence for NF-κB binding (5’-AGTTGAGGGGACTTTCCCAGGC-3’), and
containing biotin on the 5'-nucleotide, were incubated with SMC lysate. Cells were grown to
70% confluence and treated with IL-1β (10 ng/ml) for 4 hrs. Two hours before harvesting,
NAC (30 mmol/L) or DPI (10 µmol/L) was added to the cell media. SMC were washed with
PBS and lysed using Promega lysis buffer. After centrifugation at 12,400 rpm for 5 min, the
supernatant containing both soluble cytoplasmic and nuclear protein was quantified by
microBCA kit (Pierce). 500 pmol of double stranded NF-�B oligos were mixed with 500 µg
of protein and diluted into a final binding buffer (20 mmol/L HEPES, 5% glycerol, 100
mmol/L KCl, 5 mmol/L MgCl2 and 2 mmol/L PMSF, 0.5 ml volume, pH 8.0) for 45 min at
4°C. The DNA/protein complexes were pulled-down by adding 0.1 ml of streptavidin-
conjugated agarose beads mixed for 30 min at 4°C then left standing on ice 2 min before
centrifugation. The complexes were washed with the binding buffer and heated to 80°C for 5
min to allow the release of proteins from the streptavidin beads. Following Western blotting,
membranes were probed with anti-p65 (Santa Cruz, CA).
Adenoviral-mediated gene transfer. Adenovirus was mixed with the cationic polymer poly-L-
lysine (250 molecules/virus particle) 3 and added to SMC (70% confluence, 10-100 MOI) in
DMEM containing 2% fetal calf serum (FCS). After 16-18 hours, the media was replaced
with DMEM containing 5% FCS. Experiments were performed in SMC ~48 hours after
infection unless otherwise indicated.

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
5
Supplemental Figures
Supplemental Figure 1. Characterization of ClC-3 antibodies. a, human SMC lysates
were immunoprecipitated with three different antibodies to ClC-3: ClC-3b (#ACL-001,
Alomone Labs, Israel) directed against the C-terminus; ClC-3a1 directed against N-terminal
sequence common to all isoforms of ClC-3; and ClC-3a2 directed against N-terminal
sequence when transcription is driven by an alternative Clcn3 promoter. Immunoprecipitates
were stained with Coomassie Blue. In the lower panel, Western blotting with the ClC-3b
antibody identifies the immunoprecipitated protein as ClC-3. b, Wildtype and ClC-3 null
SMC lysate were immunoprecipitated with ClC-3a2 and probed with the ClC-3b antibody.
ClC-3 was not detected in SMC isolated from ClC-3 null mice. c, Immunostaining of
wildtype and ClC-3 null cultured SMC by anti-ClC-3a2.

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
6
Supplemental Figure 2. Expression of NF-κB subunit p65. a, Protein expression of the
NF-κB subunit p65 (RelA) was similar in wildtype and ClC-3 null SMC, and not altered by
treatment with IL-1β (10 ng/ml), TNF-α (10 ng/ml), or overexpression of ClC-3. b, Western
blot after biotin-streptavidin pulldown of p65 in IL-1β treated SMC confirm induction of NF-
κB activity by IL-1β, reduced activity of NF-κB in ClC-3 null SMC, and the inhibitory effect
of DPI (10 µmol/L) and NAC (30 mmol/L) on NF-κB activation in wildtype cells.
a b
IL-1β + + +-DPI
NAC--
-- -
-++
wt
ClC-3 null
IL-1β + + +-DPI
NAC--
-- -
-++
wt
ClC-3 null
β-ac
ti n
wt42k
42k
65k
65k
+
AdGFPAdClC-3
TNF-αIL-1β - + -
--
+
ClC-3 null
ClC-3 null
wt
+ + -
-
--+
-
-
-
-+
-+-+
+β-
acti n
wt42k
42k
65k
65k
+
AdGFPAdClC-3
TNF-αIL-1β - + -
--
+
ClC-3 null
ClC-3 null
wt
+ + -
-
--+
-
-
-
-+
-+-+
+

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
7
AdBg
lIIAd
Nox1
AdNox
1AS
Nox1
Rab5
IPP Rab5
AdBg
lIIAd
Nox1
AdNox
1AS
Nox1
Rab5
AdBg
lIIAd
Nox1
AdNox
1AS
Nox1
Rab5
IPP Rab5
Supplemental Figure 3. Characterization of Nox1 antibody. HEK293 cells were grown
to 70% confluence and the growth media replaced with serum free DMEM. Cells were
incubated with adenovirus (MOI 25) expressing sense Nox1 (AdNox1), antisense Nox1
(AdNox1AS), or no transgene (AdBglII). After 18 hours, the media was replaced with
DMEM containing 2% FBS for an additional 80 hours. Cells were homogenized in lysis
buffer, centrifuged to clear supernatant, immunoprecipitated with anti-Rab5, and then blotted
for Nox1 and Rab5.

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
8
02468
10
TNF-αBafilomycin
-- -
+++
Rel
ativ
e N
F-κB
Exp
ress
ion
* *
02468
10
TNF-αBafilomycin
-- -
+++
Rel
ativ
e N
F-κB
Exp
ress
ion
* *
Supplemental Figure 4. V-ATPase and NF-κB activation. Bafilomycin (0.1 mM), a
specific inhibitor of the V-type ATPase, failed to inhibit TNF-α induction of NF-κB in SMC,
suggesting that V-ATPase activity is not critical for cytokine mediated ROS signaling of NF-
κB (n=3, * p<0.05 vs. untreated).

Online Supplement Miller et al. Cytokine Activation of NF-κB in Vascular…
9
References
1. Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi
B, Engelhardt JF. Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to
endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140-154.
2. Odani K, Kobayashi T, Ogawa Y, Yoshida S, Seguchi H. ML-7 inhibits exocytosis of
superoxide-producing intracellular compartments in human neutrophils stimulated with
phorbol myristate acetate in a myosin light chain kinase-independent manner. Histochem
Cell Biol. 2003;119:363-370.
3. Fasbender A, Zabner J, Chillon M, Moninger TO, Puga AP, Davidson BL, Welsh MJ.
Complexes of adenovirus with polycationic polymers and cationic lipids increase the
efficiency of gene transfer in vitro and in vivo Journal of Biological Chemistry.
1997;272:6479-6489.
Copyright © 2022 FDOKUMEN














![Activation of HydA ΔEFG Requires a Preformed [4Fe4S] Cluster](https://static.fdokumen.com/doc/165x107/63164e0f0c69af6c1c0050c7/activation-of-hyda-defg-requires-a-preformed-4fe4s-cluster.jpg)





