
CX-397: A Novel Antithrombotic Recombinant Hirudin Analog
14
CX-397: A Novel Antithrombotic Recombinant Hirudin Analog Yasuhiko Komatsu, Yoshifumi Inoue, Masashi Sasaki, Satoru Misawa, Haruo Takaku, and Hideya Hayashi Pharmaceuticals and Biotechnology Laboratory, Japan Energy Corporation, Saitama, Japan Key Words: Antithrombotic drug—Hirudin—Thrombin inhibitor—Throm- bosis model. INTRODUCTION Among a variety of potential molecular targets of antithrombotic drugs, thrombin might be one of the most promising ones because this enzyme plays crucial roles in throm- bus formation. The coagulation cascade culminates by processing prothrombin to throm- bin, the latter of which digests fibrinogen to fibrin to begin clot formation. Thrombin also activates some upstream components of the coagulation cascade (i.e., factors V and VIII), which in turn further activate the cascade autocatalytically. In addition, since thrombin ac- tivates platelet aggregation, thrombin inhibition is one of the best ways to prevent throm- bus formation (4). Hirudin is a group of antithrombotic peptides consisting of 64 – 66 amino acids and was originally isolated from the salivary gland of the medicinal leech, Hirudo medicinalis (13,18). Hirudin tightly binds to thrombin and strongly inhibits its en- zymatic activity with reported inhibition constants of less than 1 pM. Recent develop- ments in recombinant DNA technology enabled us to produce a large amount of hirudins to be used in clinical applications. In fact, two recombinant hirudins (lepirudin and desiru- din) have already been approved. However, most of the results from clinical trials of re- combinant hirudins were less satisfactory than we had expected from animal studies (1,5,15,20). CX-397 is a novel recombinant hirudin analog having the hybrid structure of the N- and C-terminal halves of hirudin variants 1 and 3, respectively (12). This compound in- hibits thrombin more strongly than both parent hirudins (12), elicits antithrombotic effects in various animal models without serious bleeding problems (11,16,19), and is now in phase II clinical trials in Japan. In this paper, we summarize in vitro and in vivo properties 47 Cardiovascular Drug Reviews Vol. 18, No. 1, pp. 47–60 © 2000 Neva Press, Branford, Connecticut Address correspondence and reprint requests to: Dr. Hideya Hayashi, Pharmaceuticals and Biotechnology Laboratory, Japan Energy Corporation, 3-17-35 Niizo-Minami, Toda-shi, Saitama 335-8502, Japan. Fax: +81 (48) 443-1605. E-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of CX-397: A Novel Antithrombotic Recombinant Hirudin Analog

CX-397:A Novel Antithrombotic Recombinant Hirudin Analog
Yasuhiko Komatsu, Yoshifumi Inoue, Masashi Sasaki,Satoru Misawa, Haruo Takaku, and Hideya Hayashi
Pharmaceuticals and Biotechnology Laboratory,
Japan Energy Corporation, Saitama, Japan
Key Words: Antithrombotic drug—Hirudin—Thrombin inhibitor—Throm-bosis model.
INTRODUCTION
Among a variety of potential molecular targets of antithrombotic drugs, thrombinmight be one of the most promising ones because this enzyme plays crucial roles in throm-bus formation. The coagulation cascade culminates by processing prothrombin to throm-bin, the latter of which digests fibrinogen to fibrin to begin clot formation. Thrombin alsoactivates some upstream components of the coagulation cascade (i.e., factors V and VIII),which in turn further activate the cascade autocatalytically. In addition, since thrombin ac-tivates platelet aggregation, thrombin inhibition is one of the best ways to prevent throm-bus formation (4). Hirudin is a group of antithrombotic peptides consisting of 64 – 66amino acids and was originally isolated from the salivary gland of the medicinal leech,Hirudo medicinalis (13,18). Hirudin tightly binds to thrombin and strongly inhibits its en-zymatic activity with reported inhibition constants of less than 1 pM. Recent develop-ments in recombinant DNA technology enabled us to produce a large amount of hirudinsto be used in clinical applications. In fact, two recombinant hirudins (lepirudin and desiru-din) have already been approved. However, most of the results from clinical trials of re-combinant hirudins were less satisfactory than we had expected from animal studies(1,5,15,20).
CX-397 is a novel recombinant hirudin analog having the hybrid structure of the N-and C-terminal halves of hirudin variants 1 and 3, respectively (12). This compound in-hibits thrombin more strongly than both parent hirudins (12), elicits antithrombotic effectsin various animal models without serious bleeding problems (11,16,19), and is now inphase II clinical trials in Japan. In this paper, we summarize in vitro and in vivo properties
47
Cardiovascular Drug ReviewsVol. 18, No. 1, pp. 47–60© 2000 Neva Press, Branford, Connecticut
Address correspondence and reprint requests to: Dr. Hideya Hayashi, Pharmaceuticals and BiotechnologyLaboratory, Japan Energy Corporation, 3-17-35 Niizo-Minami, Toda-shi, Saitama 335-8502, Japan.Fax: +81 (48) 443-1605. E-mail: [email protected]

of this hirudin analog in comparative context with other antithrombotic drugs, includingheparin, argatroban, and recombinant hirudin variant 1 (rHV1).
DESIGN AND PRODUCTION OF CX-397
The amino acid sequences of CX-397 and its two parent hirudins, rHV1 and recom-binant hirudin variant 3 (rHV3), are depicted in Fig. 1. As shown in this figure, the N- andC-terminal halves of CX-397 were derived from rHV1 and rHV3, respectively. DNA en-coding the phoA signal–CX-397 (sequence is shown in Fig. 2A) was chemically synthe-sized and incorporated into the high-level expression vector pMT1, which was constructedby replacing the tac promoter of pMK2 (6) with the trp promoter (Fig. 2B). The nucle-otide sequence of the gene was optimized by using the codons employed in highly ex-pressed Escherichia coli genes (7). The resultant plasmid pCX397 was used to transformE. coli strain JM109 (14). For use as the master cell bank, the E. coli strain JM109 har-boring pCX397 was selected, expanded, and subdivided into 40% glycerol stocks(–80°C). For routine production of CX-397, a working cell bank was produced by ex-panding one vial of the master cell bank to make other aliquots of glycerol stocks.
Production of CX-397 was performed by using a 1-m3 jar fermenter. Firstly, a tube ofworking cell bank was cultured in a 100-mL volume in a 500-mL flask. The bacteria werethen transferred into 30-L jar fermenter and finally into the 1-m3 fermenter (culturevolume was 600 L). After 24–26 h, the CX-397 produced was purified by hydrophobic in-teraction, gel filtration, and anion exchange chromatographies. One batch of the pro-
duction run yielded about 1.5–2.5 � 109 ATU (about 100 g) of CX-397, whose specific ac-tivity was 15,500–18,800 ATU/mg.
ENZYME KINETIC STUDY
Comparison with rHV1, rHV3, and CX-397R
Enzyme kinetic study (12) revealed that the inhibition constant (Ki) of CX-397 against
human �-thrombin (0.0433 pM) was smaller than that of rHV1 (0.148 pM) or rHV3(0.0591 pM). The opposite combination analog (CX-397R; Fig. 1), whose N- and C-ter-minal halves were derived from rHV3 and rHV1, respectively, was the weakest amongthem (Ki = 0.294 pM). These results indicate that both the N-terminal half of rHV1 andthe C-terminal half of rHV3 are more beneficial than the respective opposite halves for in-hibiting thrombin. Therefore CX-397 is chimera of rHV1 and rHV3 and has a strongerthrombin inhibition ability than either parent molecule.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
48 Y. KOMATSU ET AL.
FIG. 1. Amino acid sequences of CX-397, rHV1, rHV3, and CX-397R expressed by the one-letter code. The in-hibition constants (Ki) of these peptides against human a-thrombin are shown next to their respective sequences(12). The amino acids that are different between rHV1 and rHV3 appear in bold type. Hyphens denote no corre-sponding amino acid.

Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 49
FIG. 2. Design of the synthetic E. coli alkaline phosphatase signal peptide-CX-397 gene (A) and schematic rep-resentation of CX-397 secretion vector, pCX397 (B). The synthetic gene was designed from amino acid se-quences of HV1 and HV3. Ptrp and rrnB T1T2 denote the tryptophan promoter and rrnB ribosomal terminators,respectively. Ampr, ampicillin-resistantce; phoA signal, alkaline phosphatase signal peptide gene; pUCori, repli-cation origin of pUC plasmids.

PHARMACOLOGY
In Vitro Anticoagulation Activities
Effects on various serine proteases
To elucidate the specificity of protease inhibition by CX-397, we examined the effectsof CX-397 and other protease inhibitors on various serine proteases by using syntheticfluorogenic substrates (11). The serine proteases tested were human plasma thrombin,bovine pancreas trypsin, human plasma factor Xa, human plasma plasmin, porcine pan-creatic kallikrein, and human plasma kallikrein. CX-397 inhibited thrombin but had no ef-fects on the other serine proteases, suggesting that this compound is a highly specific in-hibitor of thrombin.
Effect on coagulation times in vitro
CX-397 doubled prothrombin time (PT), activated partial thromboplastin time (APTT),
and thrombin time (TT) in rat citrated plasma at 0.88, 0.39, and 0.029 �M, respectively
(1 �M corresponds to about 100 antithrombin units (ATU)�mL). The effect of rHV1 oncoagulation times was not different from that of CX-397 (11). Although argatroban
doubled the PT and APTT at almost the same concentrations (2.1 �M for PT; 3.7 �M forAPTT) as did CX-397, heparin doubled the APTT at about a 10-fold lower concentrationthan that required for PT (4.1 U�mL for PT; 0.32 U�mL for APTT).
Effect on platelet aggregation
CX-397 inhibited the platelet aggregation induced by thrombin but did not inhibit thatinduced by collagen or ADP, suggesting again its high specificity for thrombin. The effectof rHV1 on the platelet aggregation was not different from that of CX-397 (11).
Effect on clot-bound thrombin
Clot-bound thrombin (CBT) has been thought to serve as a source of local activethrombin that plays an important role as a factor to potentiate the growth of a clot. It hasbeen postulated that CBT is protected from intrinsic inhibitors such as antithrombin (AT).This protection is thought to be a cause of the relative ineffectiveness of heparin comparedwith direct thrombin inhibitors (e.g., hirudins, argatroban) in preventing fibrin accretiononto the injury site in vivo (for review see refs. 2 and 21).
The effects of CX-397, argatroban, and heparin on CBT activities were determinedwith plasma fibrinogen used as the substrate of thrombin (Fig. 3). In this study, the plasma
clot was prepared from 100 �L of human citrated plasma by the “vacuum suction” methoddescribed here. A round-tailored filter paper (Advantec TOYO No. 2) was placed in eachwell of a 96-well multititer plate, whose bottom consisted of a membrane filter to allowvacuum filtration of the contents of each well (Milititer GV, Millipore Inc., USA). A
100-�L aliquot of human plasma was introduced into each well, and the clotting was ini-
tiated by the addition of 100 �L of thromboplastin C (Baxter Diagnostics, Switzerland) atroom temperature. Five minutes later, the plate was transferred into an incubator set at37°C and incubated for 1 h. Then the plate was set on the suction apparatus, another cir-
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
50 Y. KOMATSU ET AL.
cular filter paper was put on top of the clot, and the liquid in the clot was aspirated offacross the membrane filter. To completely wash off the constituents not bound to the clot,a solution comprising 0.1 M NaCl, 0.05 M tris-HCl [pH 7.8], and 0.1% polyethylene-glycol 6000 (Tris-buffered saline containing polyethyleneglycol; TBS-PEG) was added toeach well and aspirated off repeatedly for about 25 min. The resulting clot was shrunkenbetween the two filter papers to become a film with a disklike shape. This film was usedas the source of CBT after having been picked up together with the filter papers from thewells.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 51
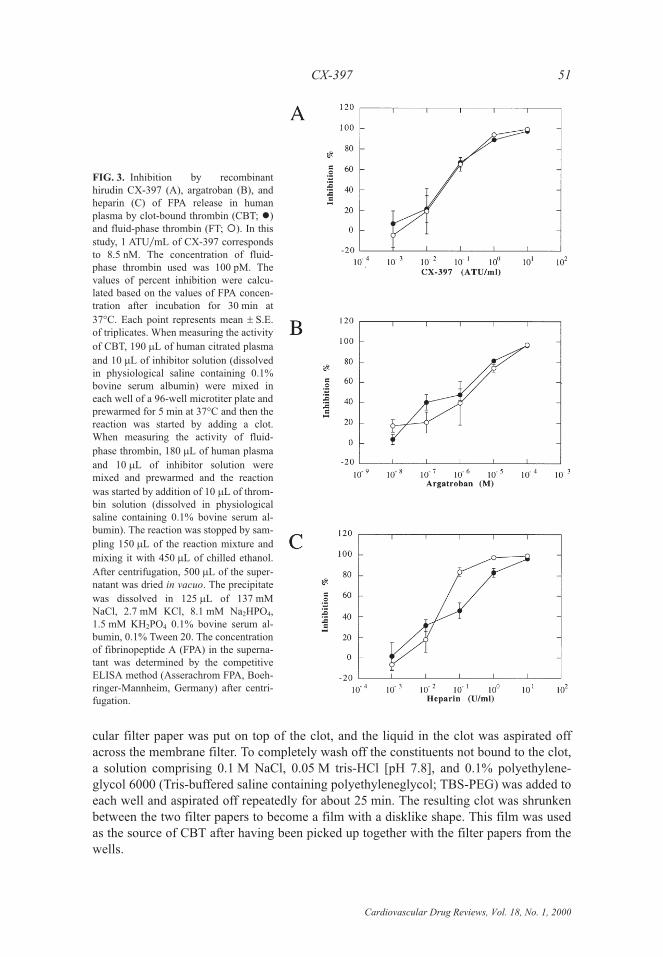
FIG. 3. Inhibition by recombinanthirudin CX-397 (A), argatroban (B), andheparin (C) of FPA release in humanplasma by clot-bound thrombin (CBT; �)and fluid-phase thrombin (FT; �). In thisstudy, 1 ATU�mL of CX-397 correspondsto 8.5 nM. The concentration of fluid-phase thrombin used was 100 pM. Thevalues of percent inhibition were calcu-lated based on the values of FPA concen-tration after incubation for 30 min at
37°C. Each point represents mean � S.E.of triplicates. When measuring the activity
of CBT, 190 �L of human citrated plasma
and 10 �L of inhibitor solution (dissolvedin physiological saline containing 0.1%bovine serum albumin) were mixed ineach well of a 96-well microtiter plate andprewarmed for 5 min at 37°C and then thereaction was started by adding a clot.When measuring the activity of fluid-
phase thrombin, 180 �L of human plasma
and 10 �L of inhibitor solution weremixed and prewarmed and the reaction
was started by addition of 10 �L of throm-bin solution (dissolved in physiologicalsaline containing 0.1% bovine serum al-bumin). The reaction was stopped by sam-
pling 150 �L of the reaction mixture and
mixing it with 450 �L of chilled ethanol.
After centrifugation, 500 �L of the super-natant was dried in vacuo. The precipitate
was dissolved in 125 �L of 137 mMNaCl, 2.7 mM KCl, 8.1 mM Na2HPO4,1.5 mM KH2PO4 0.1% bovine serum al-bumin, 0.1% Tween 20. The concentrationof fibrinopeptide A (FPA) in the superna-tant was determined by the competitiveELISA method (Asserachrom FPA, Boeh-ringer-Mannheim, Germany) after centri-fugation.

As shown in Fig. 3A, CX-397 inhibited CBT activity at almost the same concentrationas that required for fluid-phase thrombin (FT), as reported for rHV1. Argatroban, a spe-cific thrombin active site inhibitor (8,17), also inhibited CBT at the same concentration asthat needed for FT (Fig. 3B). In contrast, an approximately 4-fold higher concentration ofheparin was required to cause 50% inhibition of CBT than to inhibit FT to the same degree(Fig. 3C). These results indicate that CX-397 is more beneficial than heparin to treatthrombotic diseases in which CBT plays crucial roles, (e.g., reocclusion after PTCA, un-stable angina, etc.).
In contrast to the results mentioned above, the large protection of CBT from inhibitionby CX-397 was observed when a low-molecular–weight fluorogenic or chromogenic sub-strate was used instead of plasma fibrinogen (unpublished observation; ref. 3). However,such a result may be an artifact derived from the use of nonnatural low-molecular–weightsubstrates of thrombin, which do not interact with the anion-binding exosite on thethrombin molecule.
Antithrombotic Effects In Animal Thrombosis Models
Antithrombotic effects of CX-397 were evaluated in various animal thrombosismodels. The results are briefly described here and summarized at the end of this section.
Stasis and thrombin-induced venous thrombosis model in rats (11)
Under anesthesia with urethane, the vena cava of male Sprague–Dawley rats was ex-posed, and the test drugs were injected intravenously into a femoral vein. Five minuteslater, bovine thrombin (50 U�kg) was injected via the same route. Fifteen seconds after theinjection of thrombin, the vena cava was ligated at two sites 2 cm apart from each other.Ten minutes later, the systemic blood was withdrawn from the heart, and after another5 min, the vena cava was excised at the two ligation sites. The thrombi were picked fromthe lumen of the vein segment, dried on filter paper at room temperature overnight, andweighed. As shown in Fig. 4A, CX-397 and other antithrombotic drugs tested (rHV1,argatroban, and heparin) inhibited the thrombus formation dose-dependently, where theminimum effective doses of both hirudins were the same. By comparing the minimum ef-fective doses and bleeding risks (three-fold prolongation of template bleeding time), theratio of (risk dose)�(effective dose) or the R�E ratio can be calculated to be 12.5, 8.13,5.25, and 11.3 for CX-397, rHV1, argatroban, and heparin, respectively, indicating that thesafety margin of CX-397 in this venous thrombosis model is wider than that of argatrobanbut almost the same as that of the other two drugs, including heparin.
Glass surface-activated arterio-venous shunt thrombosis model in rats (11)
The shunt tube (25-cm length; inner and outer diameters of 0.6 and 0.9 mm, respec-tively) was made of polyethylene with a silk thread attached to its inner surface at one endand a glass tube connected to it near its other end. Under anesthesia with urethane, thejugular vein and the contralateral carotid artery of male Sprague–Dawley rats were ex-posed and the silk-thread side of the shunt tubing was inserted into the jugular vein and li-gated. Three minutes after the administration of test drugs into a tail vein, the glass-tubeside of the shunt tubing was inserted into the carotid artery; then, 5 min after drug admin-istration, blood was allowed to flow into the shunt tubing. After 5 min of perfusion, the
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
52 Y. KOMATSU ET AL.

silk-thread side was removed and the tubing was checked for occlusion. The silk threadwas then pulled out of the tube, gently washed in 3.8% sodium citrate, dried at room tem-perature overnight, and weighed. Fig. 4B shows the inhibitory effects of CX-397, rHV1,and heparin in this model. All three compounds inhibited the thrombus accretion on thesilk thread and dose-dependently decreased the occlusion rate. CX-397 was effective at1000 ATU�kg ( p < 0.01), whereas rHV1 was not effective at the same dose, suggestingthat CX-397 tended to be more effective than rHV1 in this thrombosis model. The R�Eratio was calculated in this model to be > 12.5, 4.07, and 11.3 for CX-397, rHV1, andheparin, respectively, suggesting that the safety margin of CX-397 in this arterio-venousthrombosis model is wider than that of rHV1. However, it is not clear that CX-397 is morebeneficial than heparin.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 53
FIG. 4. Effects of CX-397 andother antithrombotic drugs onthrombus formation in threedifferent types of thrombosismodels in rats. (A) Stasis- andthrombin-induced venous throm-bosis model. The thrombusweight was plotted against thedose of each drug. Argatrobanwas administered as a solutionin physiological saline con-taining 0.5 N HCl whereHCl-containing saline was usedas the vehicle control. (B)Glass surface-activated arte-rio-venous shunt thrombosismodel. The thrombus weightwas plotted against the dose ofeach drug. The number besideeach bar denotes the ratio of thenumber of no-occlusion casesto total cases. (C) Ferric chlor-ide-induced arterial thrombosismodel. The time to occlusionwas plotted against the dose ofeach drug. Argatroban was ad-ministered as a solution inphysiological saline containing0.1% HCl, where HCl-con-taining saline was administeredas the vehicle control. Eachcolumn represents the mean
� S.E. *p < 0.05, **p < 0.01 vs.control. From ref. 11 withpermission.

Ferric chloride-induced arterial thrombosis model in rats (11)
Under anesthesia with urethane, the right carotid artery of male Sprague–Dawley ratswas exposed. The surface temperature of the blood vessel was continuously monitored bya thermistor probe. Five min after the administration of test drugs into a femoral vein,
a piece of rectangular filter paper (3 � 3 mm) was placed on the carotid artery 5 mm
upstream from the thermistor probe and loaded with 10 �L of physiological salinecontaining 5% ferric chloride and 12.5% glycerol to induce arterial thrombosis. The timepoint of the rapid fall of the surface temperature was considered to be the time ofocclusion. As shown in Fig. 4C, CX-397 and the other antithrombotic drugs (i.e.,rHV1, argatroban, heparin) were effective in this ferric chloride-induced arterial throm-bosis model. When monitored by the prolongation of the time to occlusion, theminimum effective doses of CX-397 and rHV1 were 10,000 ( p < 0.01) and 20,000( p < 0.01) ATU�kg, respectively, suggesting that CX-397 was more potent than rHV1 inthis model.
The R�E rations was calculated in this model to be 1.25, 0.407, 0.525, and > 0.904 forCX-397, rHV1, argatroban, and heparin, respectively, suggesting that the safety margin ofCX-397 in this arterial thrombosis model is wider than that of both rHV1 and argatrobanbut comparable to that of heparin.
Electrolytically-induced arterial thrombosis model in dogs (16)
Under anesthesia with pentobarbital, thrombus formation was induced in the left cir-cumflex (LCX) coronary artery of male beagle dogs by an electrolytic injury. The electro-
lytic injury was induced by the application of a 100-�A continuous anodal direct currentthrough the stimulation electrode that had been inserted through the wall of the LCXartery. The blood flow was continuously monitored by an electromagnetic flow probe. Atthe end of the experiment, the intracoronary residual thrombus was removed and weighed.Drugs were administered as a bolus into the femoral vein (10 min before induction)followed by continuous drug infusion. In this canine model, CX-397 (15,000 ATU�kgbolus + 15,000 ATU�kg�h infusion; 30,000 ATU�kg bolus + 30,000 ATU�kg�h infusion)dose-dependently prevented thrombus formation, whereas heparin was not effective at thedose of 80 U�kg bolus + 60 U�kg�h infusion, which promoted the same degree of prolon-gation of template bleeding time as that caused by the highest dose of CX-397.
Photochemically-induced arterial thrombosis model in rats (19)
Under anesthesia with pentobarbital, a part of the femoral artery of male Wistar ratswas separated, and a pulsed Doppler flow probe was placed on the artery. Green light
(� = 540 nm) irradiation was done with an optic fiber positioned about 5 mm above asegment of the femoral artery proximal to the flow probe. Under irradiation, a photosensi-tized dye, rosebengal, was injected (10 mg�kg). Formation of an occlusive thrombus wasindicated by complete cessation of blood flow. In this photochemically-induced arterialthrombosis model, CX-397 dose-dependently prolonged the time to occlusion (9360 to37440 ATU�kg�h), but heparin (450 U�kg) was not effective. In template bleeding experi-ments, 37,440 ATU�kg�h of CX-397 prolonged the bleeding time only 3-fold, whereas450 U�kg of heparin prolonged it more than 6-fold, suggesting that the safety margin ofCX-397 is wider than that of heparin.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
54 Y. KOMATSU ET AL.

Disseminated intravascular coagulation (DIC) model in rats
Under anesthesia with urethane, tissue thromboplastin (10 mg�kg�h; Thromborel S,Behringwerke AG) and test drugs were simultaneously infused into right and left femoralveins, respectively, of male Sprague–Dawley rats. After a 3-h infusion, whole blood wascollected, and various coagulation parameters were then measured (i.e., PT, APTT,fibrinogen contents, platelet counts, and FDP). Both kidneys were dissected, fixed withformaldehyde, and subjected to histological observation of glomerular deposition of fibrinafter PTAH staining. In this DIC model in rats, CX-397 dose-dependently prevented allsymptoms of DIC (100 to 3000 ATU�kg�h) without any bleeding complication. Heparinand rHV1 were also effective, but the former significantly prolonged APTT at a thera-peutic dose, and the latter was less effective in the prevention of the decrease in theplatelet counts than CX-397.
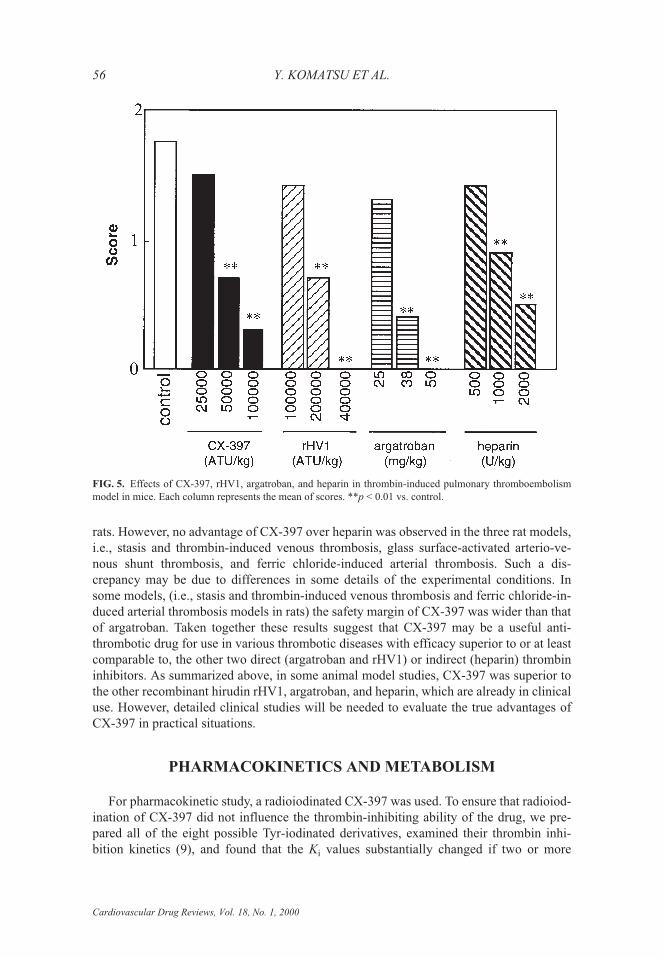
Thrombin-induced pulmonary thromboembolism model in mice
Five minutes after the administration of test drugs into a tail vein of conscious maleICR mice, bovine thrombin (2500 U�kg) was slowly (over a period of 3 sec) injected intothe contralateral vein. The behavior of the mice was observed for 20 min, and was clas-sified into the following 3 categories corresponding to scores 0, 1, and 2: score 0, no ces-sation of body-righting reflex; score 1, body-righting reflex ceased but no death within 20minutes; and score 2, death within 20 min. The score of each group was averaged andused as the index of antithrombotic action of test drugs. As shown in Fig. 5, CX-397dose-dependently inhibited the pulmonary thromboembolism in mice and the efficacy ofCX-397 was 3 to 4-fold stronger than that of rHV1. Heparin and argatroban were also ef-fective in this model, as shown in Fig. 5. When mice were pretreated with an injection ofaffinity-purified rabbit anti-rat antithrombin antibody (20 mg�kg, i.v.), the effects ofheparin were abolished, reflecting the necessity of antithrombin for the antithromboticaction of heparin. However, CX-397 was effective even in the presence of anti-antithrom-bin antibody in blood (unpublished observation), suggesting that CX-397 might be usefulin cases of severe thrombosis where patients’ antithrombin has been consumed.
Summary of antithrombotlc action of CX-397 in animal models
As mentioned above, CX-397 elicited antithrombotic action in all of the thrombosismodels examined. In four of these models (i.e., glass surface-activated arterio-venousshunt thrombosis, ferric chloride-induced arterial thrombosis, thromboplastin-inducedDIC models in rats (on platelet counts), and thrombin-induced pulmonary thromboembol-ism model in mice) the efficacy of CX-397 was greater than that of rHV1. The inhibitionconstant of CX-397 (0.0433 pM) was smaller than that of rHV1 (0.148 pM), but such adifference in Ki values might be meaningful only at very low concentrations of thrombin.So it is possible that some processes in which very low concentrations of thrombin play animportant role are involved in these four thrombosis models. Since no differences betweenCX-397 and rHV1 were detected in the other in vitro coagulation tests, differences insome unknown activity other than thrombin inhibition may be responsible for these in vivo
differences. With respect to heparin, CX-397 was more beneficial than heparin with lowerbleeding risks in two arterial thrombosis models, i.e., the electrolytically-induced arterialthrombosis model in dogs and the photochemically-induced arterial thrombosis model in
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 55
rats. However, no advantage of CX-397 over heparin was observed in the three rat models,i.e., stasis and thrombin-induced venous thrombosis, glass surface-activated arterio-ve-nous shunt thrombosis, and ferric chloride-induced arterial thrombosis. Such a dis-crepancy may be due to differences in some details of the experimental conditions. Insome models, (i.e., stasis and thrombin-induced venous thrombosis and ferric chloride-in-duced arterial thrombosis models in rats) the safety margin of CX-397 was wider than thatof argatroban. Taken together these results suggest that CX-397 may be a useful anti-thrombotic drug for use in various thrombotic diseases with efficacy superior to or at leastcomparable to, the other two direct (argatroban and rHV1) or indirect (heparin) thrombininhibitors. As summarized above, in some animal model studies, CX-397 was superior tothe other recombinant hirudin rHV1, argatroban, and heparin, which are already in clinicaluse. However, detailed clinical studies will be needed to evaluate the true advantages ofCX-397 in practical situations.
PHARMACOKINETICS AND METABOLISM
For pharmacokinetic study, a radioiodinated CX-397 was used. To ensure that radioiod-ination of CX-397 did not influence the thrombin-inhibiting ability of the drug, we pre-pared all of the eight possible Tyr-iodinated derivatives, examined their thrombin inhi-bition kinetics (9), and found that the Ki values substantially changed if two or more
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
56 Y. KOMATSU ET AL.
FIG. 5. Effects of CX-397, rHV1, argatroban, and heparin in thrombin-induced pulmonary thromboembolismmodel in mice. Each column represents the mean of scores. **p < 0.01 vs. control.

iodine were introduced into the CX-397 molecule. So a monoradioiodinated CX-397 (atTyr-3) was used for the pharmacokinetic studies. When intravenously administered to rats,CX-397 was distributed to the extracellular compartment of body fluid and disappearedaccording to the kinetics described by the open two-compartment model. The elimination
half-life of -phase was found to be around 20–30 min, and the main organ of eliminationwas the kidney (unpublished observations). In humans, almost all of the CX-397 adminis-tered was eliminated in the urine as nonmetabolized CX-397, but no nonmetabolized formcould be detected in rat urine (10). The rat urinary metabolites were a mixture of C-ter-minal–truncated CX-397 (10). Details of the pharmacokinetic study using radioiodinatedCX-397 will be reported elsewhere. Here the results of a continuous infusion study in ratsis described. Under anesthesia with urethane, CX-397 dissolved in saline was infused into
a tail vein of male Sprague–Dawley rats (625, 2500, 10,000, and 40,000 ATU�kg�h;
4 mL�kg�h). During and after the termination of the infusion, blood was collected fromthe abdominal aorta under anticoagulation with 0.38% sodium citrate, and plasma wasprepared by centrifugation. The plasma concentration of CX-397 was determined by asandwich ELISA method, and PT and APTT were also measured. As shown in Fig. 6A,the concentration of CX-397 rose within 30 min, and reached a plateau and after the termi-nation of the infusion, it immediately decreased. In accordance with the plasma CX-397concentration, PT and APTT were dose-dependently prolonged (Fig. 6B and 6C). Asshown in Fig. 6D, the plasma concentration of CX-397 was linearly dependent on the in-fusion rate at any time point during the infusion. These results indicate that the plasmaconcentration of CX-397 can be easily controlled by changing its infusion rate.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 57
FIG. 6. Plasma concentration (A), APTT (B), and PT (C) during and after intravenous administration ofCX-397 in rats. The relationships between plasma concentrations of CX-397 and its infusion rates are double-ex-ponentially plotted (D) based on the data in FIG. 6A during its infusion. The infusion times of CX-397 were 5(�), 10 (�), 30 (�), and 60 min (�). The plasma concentration was determined by a sandwich ELISA method.
Each point represents the mean � S.E. of 5 determinations. The data points in D were fitted by linear linescrossing the origin ( y = ax ) by a least squares method.

TOXICOLOGY
In the evaluations of both acute (9,700,000 ATU�kg in rats and dogs) and chronic(48,500, 162,000, and 485,000 ATU�kg in rats; 49,100, 164,000, and 491,000 ATU�kg indogs; daily administration for 4 weeks followed by 4 weeks of observation) toxicity ofCX-397 administered intravenously, neither death nor toxic symptoms other than occa-sional bleeding were observed. CX-397 had no mutagenic effects and did not affect fer-tility of rats.
PHASE I CLINICAL TRIAL
CX-397 (12,500, 25,000, 50,000, or 100,000 ATU�body) was intravenously infused for3 h into 22 healthy male volunteers. Maximal plasma concentration (Cmax) and area underthe plasma concentration curve (AUC) were elevated almost proportional to the doses.
Plasma half-lives were about 15 min for the �-phase and about 1.5 h for the -phase atany dose examined. About 75% to 95% of the CX-397 was excreted in the urine within24 h. APTT was dose-dependently prolonged after CX-397 administration. No subjectiveor objective symptoms and no changes in clinical test parameters were observed at anydose of CX-397. Anti-CX-397 and anti-ECP (E. coli-derived proteins) antibody titers re-mained under the levels that cause clinical problems. When CX-397 was administeredevery 3 days, the Cmax and AUC values for each day were almost the same as those in thesingle-day study, suggesting no accumulation. No symptoms derived from repeated appli-cation were detected. These results suggest that CX-397 obeys linear pharmacokineticswithout accumulation, has a remarkable safety profile and elicits dose-dependent anti-thrombin effects in humans.
SUMMARY
CX-397 is a recombinant hirudin analog that has the hybrid sequence of rHV1 andrHV3 (i.e., N- and C-terminal halves of rHV1 and rHV3, respectively). CX-397 inhibitsthe enzymatic activity of thrombin more strongly than its parent hirudins, rHV1 andrHV3. CX-397 does not inhibit serine proteases other than thrombin, and it prevents plate-let aggregation induced by thrombin but not that by collagen or ADP, indicating it has avery high specificity for thrombin. CX-397 was effective in all thrombosis models ex-amined, including stasis and thrombin-induced venous thrombosis model in rats, glasssurface-activated arterio-venous shunt thrombosis model in rats, ferric chloride-inducedarterial thrombosis model in rats, electrolytically-induced arterial thrombosis model indogs, photochemically-induced arterial thrombosis model in rats, DIC model in rats, andthrombin-induced pulmonary thromboembolism model in mice. In these models, data onthe effectiveness and the hemorragic risks of CX-397 and the other antithrombotic drugs(i.e., rHV1, argatroban, heparin) indicate that CX-397 is more beneficial than, or at leasthas benefits comparable to those of directly or indirectly acting thrombin inhibitors.Future clinical studies may elucidate whether CX-397 is truly more beneficial than theother antithrombotic drugs. CX-397 obeys linear pharmacokinetics, both in experimental
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
58 Y. KOMATSU ET AL.

animals and humans, and is eliminated mainly in the urine without accumulation. No se-rious toxicity was observed in either animal or phase I clinical studies. A phase II clinicaltrial is now in progress in Japan.
Acknowledgments: We are grateful to the many collaborators in Japan Energy Corporation andKaken Pharmaceutical Co. involved in the research and development of CX-397. We also thank Dr.Mitsuyoshi Nakashima for helpful advice for performing the phase I clinical study.
REFERENCES
1. Antman EM. Hirudin in acute myocardial infarction. Safety report from the Thrombolysis and Thrombin In-hibition in Myocardial Infarction (TIMI) 9A Trial. Circulation 1994;90:1624–1630.
2. Becker DL, Fredenburgh JC, Stafford AR, Weitz JI. Molecular basis for the resistance of fibrin-boundthrombin to inactivation by heparin�serpin complexes. Adv Exp Med Biol 1997;425:55–66.
3. Berry CN, Girardot C, Lecoffre C, Lunven C. Effects of the synthetic thrombin inhibitor argatroban onfibrin- or clot-incorporated thrombin: Comparison with heparin and recombinant hirudin. Thromb Haemost
1994;72:381–386.4. Fenton JW II, Ofosu FA, Brezniak DV, Hassouna HI. Thrombin and antithrombotics. Semin Thromb Hemost
1998;24:87–91.5. GUSTO IIa Investigators. Randomized trial of intravenous heparin versus recombinant hirudin for acute
coronary syndromes. Circulation 1994;90:1631–1637.6. Hibino T, Misawa S, Wakiyama M, et al. High-level expression of porcine muscle adenylate kinase in
Escherichia coli: Effects of the copy number of the gene and the translational initiation signals. J Biotechnol
1994;32:139–148.7. Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the
respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for theE. coli translational system. J Mol Biol 1981;151:389–409.
8. Kikumoto R, Tamao Y, Tezuka T, et al. Selective inhibition of thrombin by (2R,4R )-4-methyl-1-[N2-[(3-methyl-1,2,3,4-tetrahydro-8-quinolinyl) sulfonyl]-1-arginyl)]-2-piperidinecarboxylic acid. Biochemistry
1984;23:85–90.9. Komatsu Y, Hayashi H. Reevaluating the effects of tyrosine iodination of recombinant hirudin on its
thrombin inhibition kinetics. Thromb Res 1997;87:343–352.10. Komatsu Y, Hayashi H. Characterization of C-terminal–truncated urinary metabolites of a recombinant
hirudin in rats. Peptides 1999;20:1401–1409.11. Komatsu Y, Inoue Y, Goto Y, Fukazawa T, Hayashi H. Pharmacological effects of a novel recombinant hiru-
din, CX-397, in vivo and in vitro: comparison with recombinant hirudin variant-1, heparin, and argatroban.Thromb Haemost 1999;81:250–255.
12. Komatsu Y, Misawa S, Sukesada A, Ohba Y, Hayashi H. CX-397, a novel recombinant hirudin analoghaving a hybrid sequence of hirudin variants-1 and -3. Biochem Biophys Res Commun 1993;196:773–779.
13. Markwardt F. Pharmacology of hirudin: One hundred years after the first report of the anticoagulant agent inmedicinal leeches. Biomed Biochim Acta 1985;44:1007–1013.
14. Misawa S, Furuya H, Matsuda H, Abe S, Hayashi H. High level expression and secretion of biologicallyactive leech hirudin variant 1 (HVI) by Escherichia coli. In: Furusaki S, Endo I, Matsuno R, eds. Biochemi-
cal Engineering for 2001. Tokyo: Springer-Verlag, 1992;62–64.15. Neuhaus KL, von Essen R, Tebbe U, et al. Safety observations from the pilot phase of the randomized
r-Hirudin for Improvement of Thrombolysis (HIT-III) study. A study of the Arbeitsgemeinschaft LeitenderKardiologischer Krankenhausärzte (ALKK). Circulation 1994;90:1638–1642.
16. Ohyama T, Hori T, Monike M, et al. Anti-thrombotic effects of CX-397, a recombinant hirudin analog, in acanine model of coronary artery thrombosis. Thromb Haemost 1998;79:423–430.
17. Okamoto S, Hijikata A, Kikumoto R, et al. Potent inhibition of thrombin by the newly synthesized argininederivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion. Biochem
Biophys Res Commun 1981;101:440–446.18. Scharf M, Engels J, Tripier D. Primary structures of new “iso-hirudin.” FEBS Lett 1989;255:105–110.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
CX-397 59

19. Takiguchi Y, Asai F, Wada K, Hayashi H, Nakashima M. Antithrombotic effect of a novel recombinant hiru-din analogue, CX-397, in a rat arterial thrombosis model. Br J Pharmacol 1995;116:3056–3060.
20. von Essen R, Zeymer U, Tebbe U, et al. HBW 023 (recombinant hirudin) for the acceleration of thromboly-sis and prevention of coronary reocclusion in acute myocardial infarction: Results of a dose-finding study(HIT-II) by the Arbeitsgemeinschaft Leitender Kardiologischer Krankenhausärzte. Coron Artery Dis
1998;9:265–272.21. Weitz JI. Activation of blood coagulation by plaque rupture: Mechanisms and prevention. Am J Cardiol
1995;75:18B–22B.
Cardiovascular Drug Reviews, Vol. 18, No. 1, 2000
60 Y. KOMATSU ET AL.




















