
Crystal structures of triosephosphate isomerase from methicillin resistant Staphylococcus aureus...
13
Research paper Crystal structures of triosephosphate isomerase from methicillin resistant Staphylococcus aureus MRSA252 provide structural insights into novel modes of ligand binding and unique conformations of catalytic loop Somnath Mukherjee 1 , Amlan Roychowdhury, Debajyoti Dutta, Amit Kumar Das * Department of Biotechnology, Indian Institute of Technology, Kharagpur 721302, West Bengal, India article info Article history: Received 4 January 2012 Accepted 4 July 2012 Available online 17 July 2012 Keywords: Triosephosphate isomerase Crystal structures Conformational flexibility “almost closed” conformation “swung in” “swung out” abstract Staphylococcus aureus is one of the most dreaded pathogens worldwide and emergence of notorious antibiotic resistant strains have further exacerbated the present scenario. The glycolytic enzyme, tri- osephosphate isomerase (TIM) is one of the cell envelope proteins of the coccus and is involved in biofilm formation. It also plays an instrumental role in adherence and invasion of the bacteria into the host cell. To structurally characterize this important enzyme and analyze it’s interaction with different inhibitors, substrate and transition state analogues, the present article describes several crystal structures of SaTIM alone and in complex with different ligands: glycerol-3-phosphate (G3P), glycerol-2-phosphate (G2P), 3- phosphoglyceric acid (3PG) and 2-phosphoglyceric acid (2PG). Unique conformations of the catalytic loop 6 (L6) has been observed in the different complexes. It is found to be in “almost closed” confor- mation in both subunits of the structure complexed to G3P. However L6 adopts the open conformation in presence of G2P and 2PG. The preference of the conformation of the catalytic loop can be correlated with the position of the phosphate group in the ligand. Novel modes of binding have been observed for G2P and 3PG for the very first time. The triose moiety is oriented away from the catalytic residues and occupies an entirely different position in some subunits. A completely new binding site for phosphate has also been identified in the complex with 2PG which differs substantially from the conventional phosphate binding site of the ligand in the crystal structures of TIM determined so far. Ó 2012 Elsevier Masson SAS. All rights reserved. 1. Introduction Glycolysis is the biochemical pathway by which a glucose molecule is converted to pyruvate with the generation of two molecules of ATP that meets the principal energy demand of living cells. Triosephosphate isomerase (TIM, EC: 5.3.1.1) is a ubiquitous glycolytic enzyme catalyzing the isomerization of dihydroxyace- tone phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP) through the intermediate formation of cis-enediolate in fifth step of the glycolytic pathway. The uphill direction from DHAP to GAP is essential for optimal throughput during glycolysis. The high cata- lytic capacity of TIM is a diffusion controlled process and according to Knowles, the enzyme appears to have arrived at the end of its evolutionary development as a catalyst [1]. The catalytic reaction involves several steps of intermolecular protonation and deproto- nation between the enzyme and substrate by the active participa- tion of the catalytic glutamate, histidine (Fig. 1) and a catalytic loop of ten residues. This loop acts as a rigid lid and rests on flange residues that function as hinges. The catalytic mechanism in TIM is believed to be the result of stronger bonding of the enzyme to its transition state than to the initial Michaelis complex [2]. Various schemes for the reaction mechanism have been postulated [3] and kinetic isotope effects and isotope “washout” experiments suggests that the reaction proceeds through a planar cis-enediolate inter- mediate [4] stabilized by the catalytic loop. The energetic landscape of the mechanistic pathway is characterized by several steps of competitive timescales [5]. However, despite numerous experi- mental evidences [4,6e10] and theoretical studies [11,12], the Abbreviations: SaTIM, triosephosphate isomerase from Staphylococcus aureus; PfTIM, triosephosphate isomerase from Plasmodium falciparum; BsTIM, tri- osephosphate isomerase from Bacillus stearothermophilus; TbTIM, triosephosphate isomerase from Trypanosoma brucei; r.m.s.d.C a , root mean square deviation of C a ; G3P, glycerol-3-phosphate; G2P, glycerol-2-phosphate; 3PG, 3-phosphoglyceric acid; 2PG, 2-phosphoglyceric acid; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde-3-phosphate. * Corresponding author. Tel.: þ91 3222 283756; fax: þ91 3222 278707/255303. E-mail address: [email protected] (A.K. Das). 1 Present Address: Department of Biochemistry and Molecular Biology, Univer- sity of Chicago, Chicago, IL 60637, USA. Contents lists available at SciVerse ScienceDirect Biochimie journal homepage: www.elsevier.com/locate/biochi 0300-9084/$ e see front matter Ó 2012 Elsevier Masson SAS. All rights reserved. http://dx.doi.org/10.1016/j.biochi.2012.07.001 Biochimie 94 (2012) 2532e2544
Transcript of Crystal structures of triosephosphate isomerase from methicillin resistant Staphylococcus aureus...

at SciVerse ScienceDirect
Biochimie 94 (2012) 2532e2544
Contents lists available
Biochimie
journal homepage: www.elsevier .com/locate/b iochi
Research paper
Crystal structures of triosephosphate isomerase from methicillin resistantStaphylococcus aureus MRSA252 provide structural insights into novel modesof ligand binding and unique conformations of catalytic loop
Somnath Mukherjee 1, Amlan Roychowdhury, Debajyoti Dutta, Amit Kumar Das*
Department of Biotechnology, Indian Institute of Technology, Kharagpur 721302, West Bengal, India
a r t i c l e i n f o
Article history:Received 4 January 2012Accepted 4 July 2012Available online 17 July 2012
Keywords:Triosephosphate isomeraseCrystal structuresConformational flexibility“almost closed” conformation“swung in”“swung out”
Abbreviations: SaTIM, triosephosphate isomerasePfTIM, triosephosphate isomerase from Plasmodiosephosphate isomerase from Bacillus stearothermophisomerase from Trypanosoma brucei; r.m.s.d.Ca, root mG3P, glycerol-3-phosphate; G2P, glycerol-2-phosphacid; 2PG, 2-phosphoglyceric acid; DHAP, dihydroglyceraldehyde-3-phosphate.* Corresponding author. Tel.: þ91 3222 283756; fax
E-mail address: [email protected] (A.K. Da1 Present Address: Department of Biochemistry an
sity of Chicago, Chicago, IL 60637, USA.
0300-9084/$ e see front matter � 2012 Elsevier Mashttp://dx.doi.org/10.1016/j.biochi.2012.07.001
a b s t r a c t
Staphylococcus aureus is one of the most dreaded pathogens worldwide and emergence of notoriousantibiotic resistant strains have further exacerbated the present scenario. The glycolytic enzyme, tri-osephosphate isomerase (TIM) is one of the cell envelope proteins of the coccus and is involved in biofilmformation. It also plays an instrumental role in adherence and invasion of the bacteria into the host cell.To structurally characterize this important enzyme and analyze it’s interaction with different inhibitors,substrate and transition state analogues, the present article describes several crystal structures of SaTIMalone and in complex with different ligands: glycerol-3-phosphate (G3P), glycerol-2-phosphate (G2P), 3-phosphoglyceric acid (3PG) and 2-phosphoglyceric acid (2PG). Unique conformations of the catalyticloop 6 (L6) has been observed in the different complexes. It is found to be in “almost closed” confor-mation in both subunits of the structure complexed to G3P. However L6 adopts the open conformation inpresence of G2P and 2PG. The preference of the conformation of the catalytic loop can be correlated withthe position of the phosphate group in the ligand. Novel modes of binding have been observed for G2Pand 3PG for the very first time. The triose moiety is oriented away from the catalytic residues andoccupies an entirely different position in some subunits. A completely new binding site for phosphatehas also been identified in the complex with 2PG which differs substantially from the conventionalphosphate binding site of the ligand in the crystal structures of TIM determined so far.
� 2012 Elsevier Masson SAS. All rights reserved.
1. Introduction
Glycolysis is the biochemical pathway by which a glucosemolecule is converted to pyruvate with the generation of twomolecules of ATP that meets the principal energy demand of livingcells. Triosephosphate isomerase (TIM, EC: 5.3.1.1) is a ubiquitousglycolytic enzyme catalyzing the isomerization of dihydroxyace-tone phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP)
from Staphylococcus aureus;um falciparum; BsTIM, tri-ilus; TbTIM, triosephosphateean square deviation of Ca;
ate; 3PG, 3-phosphoglycericxyacetone phosphate; GAP,
: þ91 3222 278707/255303.s).d Molecular Biology, Univer-
son SAS. All rights reserved.
through the intermediate formation of cis-enediolate in fifth step ofthe glycolytic pathway. The uphill direction from DHAP to GAP isessential for optimal throughput during glycolysis. The high cata-lytic capacity of TIM is a diffusion controlled process and accordingto Knowles, the enzyme appears to have arrived at the end of itsevolutionary development as a catalyst [1]. The catalytic reactioninvolves several steps of intermolecular protonation and deproto-nation between the enzyme and substrate by the active participa-tion of the catalytic glutamate, histidine (Fig. 1) and a catalytic loopof ten residues. This loop acts as a rigid lid and rests on flangeresidues that function as hinges. The catalytic mechanism in TIM isbelieved to be the result of stronger bonding of the enzyme to itstransition state than to the initial Michaelis complex [2]. Variousschemes for the reaction mechanism have been postulated [3] andkinetic isotope effects and isotope “washout” experiments suggeststhat the reaction proceeds through a planar cis-enediolate inter-mediate [4] stabilized by the catalytic loop. The energetic landscapeof the mechanistic pathway is characterized by several steps ofcompetitive timescales [5]. However, despite numerous experi-mental evidences [4,6e10] and theoretical studies [11,12], the

Fig. 1. Probable catalytic mechanism of TIM: Various schemes for the isomerization of DHAP to GAP have been postulated. The first and the last step are unambiguous but there arethree alternative proposals for the formation of the intermediate (Path A, B and C). The enediol and enediolate are respectively abbreviated as EDL and EDT. The cis-EDL-1 and EDL-2are formed by intramolecular proton transfer between the EDT-1 and His95 (Path A) or catalytic Glu167 (Path C) respectively. Path B depicts an intramolecular proton transfer fromC1 of EDT-1 to C2 of EDT-2. EDL-1, EDT-2 and EDL-2 are mutually interconvertible by series of intramolecular proton transfers [3]. TIM abstracts the pro-R hydrogen from C1 of DHAPand stereospecifically introduces the proton at C2 (R-configuration) of GAP.
S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2533
precise chemical mechanism of the multistep reaction catalyzed byTIM is not fully elucidated and is still under extensive investigation(Fig. 1). Dynamics of loop 6 in the catalytic mechanism have alsoreceived considerable attention. NMR studies on the motion of thecatalytic loop suggest that loop movement and product release areprobably concerted and is perhaps the rate limiting step in thecatalysis [13,14]. The data from temperature jump relaxationstudies of the loop motion is in conformity of a model where rate ofthe loop opening is dependent on the nature of the ligands [15].Interactions among the different loops of the enzyme in coordi-nating motions and catalytic activity have also been studied. Theseresults suggest that interactions between loop 6 and loop 7 arenecessary to provide the proper chemical context for the enzymaticreaction to occur and these interactions play a significant role inmodulating the chemical dynamics near the active site [16].Nevertheless, the participation of loop 6 residues to allow ligandbinding, in properly positioning the catalytic base with suitablegeometry to protect the reaction intermediate in its closed state,and finally its release in open state are still not well understood[17]. This also varies among species and hence requires extensiveinvestigation.
Staphylococcus aureus is one of the most dreaded, opportunistic,nosocomial human pathogens responsible for minor skin infectionsto life-threatening diseases such as meningitis, pneumonia, oste-omyelitis, endocarditis, septic arthritis, toxic shock syndrome andsepticaemia [18]. Amidst vast technical and medical advancements,it still continues to wreak havoc worldwide and remains one of theleading causes for morbidity and mortality. The resistance of this
“golden staph” to all prevalent frontline antimicrobials hasincreased the menace. In fact the notorious methicillin resistantstrain of S. aureus (MRSA) has already become an endemic in thelast decade. Recent reports on the recalcitrance of this bacterium tomodern glycopeptide antibiotics like vancomycin have increasedthe concern to tackle the vancomycin resistant and vancomycinintermediate S. aureus (VRSA and VISA) [19,20].
The methicillin resistant S. aureus MRSA252 possesses a singletriosephosphate iosmerase (SaTIM: SAR0830) comprised of 253amino acids. SaTIM is one of the components of the cell envelopeproteins from this pathogen [21] and is involved in the biofilmformation. TIM is identified as one of the five genes that is upre-gulated during biofilm formation, which may be due to oxygenlimitation in the deeper layers of the biofilm [22]. It is evident thatduring biofilm formation, the upregulation of glycolytic enzymesare stimulated by many factors other than oxygen limitation in thecell. This reflects their putative role not only in complex microbialpopulations but also in reduced antimicrobial susceptibility.Proteins localized on the surfaces of microbes have diverse inter-actions with other cells and molecules. So during the process ofinfection, these proteins can be instrumental for adherence to andinvasion into the host [21,23,24]. Usually, glycolytic enzymes act inthe cytoplasm of microbes, but accumulating evidence indicatesthat some glycolytic enzyme proteins, such as GAPDH, enolase, andTIM, are present on the cell surface, where they may have multiplefunctions [23,25]. The presence of TIM on the surface of the path-ogen has also been confirmed by proteomics analysis [21], agglu-tination test and scanning immunoelectron microscopy [26]. TIM

S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442534
has been identified as a candidate adhesion molecule for theinteraction between the coccus and the fungal pathogen Crypto-coccus neoformans [27]. TIM on Paracoccidioides brasiliensis inter-acts with laminin and fibronectin. Thus, TIM is important inadherence to and invasion into the host cells, indicating that TIMmay be a virulence factor for the pathogen [28]. TIM of Schistosomamansoni, a causative agent of parasitic infection, is a protectiveantigen in mice, suggesting that it might be a potential vaccinecandidate [29]. Extensive analysis of the interactions between TIMand several other bioreactive host proteins like human fibrinogen,fibronectin, thrombin and plaminogen reveal that SaTIM is a plas-minogen binding protein like enolase and GAPDH [30]. GAPDH andenolase in S. aureus promote the invasion of the pathogen into thehost tissue by initiating proteolysis through plasminogen binding.However, SaTIM is involved in the inhibition of fibrinolysis byarresting the conversion of plasminogen to plasmin. All thesefindings suggest a novel role of the enzyme on the surface ofS. aureus in addition to glycolysis.
As glycolytic enzymes are responsible for the production of ATPin the cells, they are necessary for the pathogen’s sustenance andare generally lucrative targets for inhibitor design. The structure ofGAPDH1 from MRSA252 has already been determined [31]. Struc-tural characterization of an enzyme like SaTIM which has severalother important roles in pathogenesis in addition to glycolysis isthus imperative. A scrupulous analysis of interactions of the aminoacid residues of SaTIM with different inhibitors and transition stateanalogues at the atomic level is an absolute necessity. Moreover, itis evident from the Protein Data Bank [32] that out of 120 structuresof TIM submitted so far, eukaryotic TIM (87.2%) has been the subjectof most extensive researchwhile the enzyme from only a handful ofprokaryotic species (w10%) has been structurally characterized.This gives us ample opportunities to explore different structuralaspects of SaTIM.
To elucidate all the structural aspects of the enzyme includingconformational flexibility of the catalytic loop, modes of bindingof different substrate surrogates, inhibitors and transition stateanalogues, the present article describes five different crystalstructures of wild type SaTIM alone and in complex with variousphosphate containing ligands like glycerol-3-phosphate (G3P),glycerol-2-phosphate (G2P), 3-phosphoglyceric acid (3PG) and 2-phosphoglyceric acid (2PG) (Fig. 2). These ligands are either
Fig. 2. Ligands of TIM: Structures of various ligands used in the atomic resolutioncomplexes of SaTIM (a) G3P: Glycerol-3-phosphate (b) G2P: Glycerol-2-phosphate (c)3PG: 3-phosphoglyceric acid (d) 2PG: 2-phosphoglyceric acid. The atoms arenumbered following the numbering in the coordinate files.
structurally similar to the physiological substrate, GAP, or areinhibitors or transition state analogues. G3P is analogous to GAP;the only difference is in the oxidation state of C1. 2PG and 3PGhave an additional negative charge at the extra oxygen atom atC1. Unlike GAP, the phosphate group in G2P and 2PG are attachedto C2. Earlier structural studies of TIM-ligand complexes suggestthat suitable substitution at C2 of 3PG could be potentiallyinteresting as a lead for drug design [17]. The crystal structuresof SaTIM complexed with these different ligands show how theinteractions of carboxy, phosphate and hydroxyl groups ofthe ligands with the protein are altered due to change in theoxidation state of C1 and disposition of the phosphate group. Thedifferent conformations of loop 6 observed as a result of ligandbinding in these different structures have also been analyzed.This is the first study of TIM to report unique modes of binding ofG2P and 3PG. A completely new binding site for phosphatehas also been identified in the complex with 2PG for the firsttime.
2. Materials and methods
2.1. Materials
G3P, G2P, 2PG and 3PG were purchased from Sigma Aldrich,USA. Other reagents and chemicals used for protein purificationand crystallization were of analytical grade.
2.2. Crystallization, data collection and structure determination
Cloning, over-expression, purification and crystallization ofSaTIM have been described [33]. The respective complexes of SaTIMwere obtained by incubating the purified protein with 20 mM ofG3P, G2P, 2PG and 3PG. Crystals of SaTIM and its complexes grewfrom 1.6 M trisodium citrate dihydrate, pH 6.5 after 3 days at 298 Kby hanging drop vapour diffusion method at a concentration of80 mg/ml. The protein also crystallized from 2.4 M sodium malo-nate, pH 7.0 but diffracted to a nominal resolution of 2.4 Å incomparison to the crystals from trisodium citrate which diffractedto 1.9 Å. So, all the discussions in this article will be limited to thestructural information obtained from crystals from trisodiumcitrate.
All the diffraction data were collected at our home sourceequipped with a Rigaku R Axis IVþþ detector with Cu Ka X-rays(1.5418 Å) generated by a Rigaku Micromax HF007 Microfocusrotating anode X-ray generator. The crystals were cryoprotected[34] with mother liqour and flash cooled in liquid nitrogen streamat 100 K. Data were processed with XDS [35] and scaled usingSCALA [36] in CCP4 suite [37]. Data collection statistics are detailedin Table 1A. The structure of SaTIM was determined by molecularreplacement using MOLREP [38] within CCP4 package with tri-osephosphate isomerase from Bacillus stearothermophilus (BsTIM,PDB entry: 2BTM) [39] as the starting model. Matthews coefficientsuggests the presence of two molecules in the asymmetric unit. Apromising solution with a homodimeric structure was obtainedonly in the space group P43212 which is identical to that obtainedfrom AutoRickshaw [40]. The initial model obtained was subjectedto cycles of rigid body refinement to generate the 2FoeFc and FoeFcmap followed by consecutive cycles of TLS and restrained refine-ment in Refmac5 [41] until a map with interpretable electrondensity was obtained. Cycles of restrained refinement alternatedwith manual model building in COOT [42] to improve the quality ofthe model. Noncrystallographic restraints (NCS) were imposed onparts of the protein molecules that do not structurally differ widelyamong the two subunits (r.m.s.d.Ca < 1.0 Å). This was ultimatelyfollowed bymodelling the solvent molecules. After the preliminary

Table 1Summary of (A) Data collection and (B) Refinement statistics.
(A) SaTIM SaTIM-G3P SaTIM-G2P SaTIM-2PG SaTIM-3PG
Data collectionSpace group P43212Cell Parameters (a, b, c) (Å) 79.5, 79.5, 175.0 81.1, 81.1, 174.6 79.0, 79.0, 175.0 79.0, 79.0, 175.1 80.6, 80.6, 174.0Resolution (Å) 72.37e1.90 (2.00e1.90) 29.83e2.15 (2.26e2.15) 29.63e2.50 (2.63e2.50) 29.32e2.07 (2.18e2.07) 29.56e2.25 (2.37e2.25)Completeness (%) 99.5 (97.9) 98.7 (91.7) 99.5 (99.0) 99.3 (96.9) 99.6 (98.7)Redundancy 13.50 (12.10) 13.00 (11.20) 14.0 (13.9) 13.7 (11.4) 5.8 (5.7)I/s(I) 29.5 (5.5) 19.5 (4.8) 27.4 (5.3) 22.1 (5.3) 15.1 (4.1)Rmerge (%)a 5.8 (43.3) 10.2 (43.9) 8.6 (52.4) 8.2 (45.4) 8.1 (46.6)Rmeas (%)b 6.2 (53.7) 10.6 (46.0) 8.9 (54.4) 8.5 (47.6) 8.9 (51.3)Rpim (%)c 1.7 (15.2) 2.9 (13.4) 2.4 (14.5) 2.3 (13.7) 3.7 (21.1)PDB code 3M9Y 3UWU 3UWZ 3UWV 3UWW
(B) SaTIM SaTIM-G3P SaTIM-G2P SaTIM-2PG SaTIM-3PG
RefinementResolution (Å) 20.0e1.90 20.0e2.15 20.0e2.50 20.0e2.07 20.0e2.25No. of reflections 44,825 32,120 18,891 32,582 26 625Rwork (%)d 15.9 18.1 18.9 18.5 20.3Rfree (%)d 20.5 22.3 23.1 22.2 24.6No. of atomsProtein 3847 3835 3833 3846 3809Ligands 13 33 20 22 30Ions 2 e e 3 1Water 481 373 147 323 211Average B factors (Å2)Protein 31.2 28.9 40.2 29.6 35.1Citrate 43.7 43.0 e e e
G3P e 28.9 e e e
G2P e e 52.4 e e
2PG e e e 58.2 e
3PG e e e e 52.4Sodium 26.9 e e e 34.9DTT e e e e 34.9Water 30.4 25.4 25.9 29.6 22.7R.m.s. deviationsBond Angle (�) 1.842 1.089 0.978 1.125 1.067Bond Length (Å) 0.025 0.009 0.008 0.010 0.008Ramachandran PlotMost favoured, additionally allowed, generously allowed (%) 97.0, 2.4, 0.6 93.9, 5.0, 0.7 93.4, 5.7, 0.5 93.5, 4.9, 0.9 94.4, 3.8, 1.1
a Rmerge ¼ P
hkl
P
ijIiðhklÞ � hIðhklÞij=P
hkl
P
iIiðhklÞ.
b Rmeas ¼ P
hkl
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiN=ðN � 1Þp P
ijIiðhklÞ � hIðhklÞij=P
hkl
P
iIiðhklÞ.
c Rp:i:m: ¼ P
hkl
ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffi1=ðN � 1Þp P
ijIiðhklÞ � hIðhklÞij=P
hkl
P
iIiðhklÞ.
d Rwork is crystallographic R factor. Rfree is calculated based on 5% of total reflections excluded from refinement.
S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2535
refinement of the polypeptide chain, the ligands were placedunambiguously according to the difference electron maps calcu-lated. The coordinates of citric acid was taken from the Refmaclibrary. Each peak contoured at 3s in the FoeFc map was identifiedas water molecule, taking into consideration their geometricconstraints as well. The progress of the refinement was monitoredby a steady decrease and convergence of Rwork and Rfree values [43].The stereochemical quality of the model was validated usingPROCHECK [44].
The structures of SaTIM_G3P, SaTIM_G2P, SaTIM_2PG and SaT-IM_3PGwere determined by difference Fourier maps phased on theatomic coordinates of SaTIM. Model building was done in COOTandrefinement in Refmac5 in a manner similar to the protein withoutligands. Atomic coordinates and the corresponding restraints(crystallographic information file) of the ligands used in refinementwere obtained from PRODRG [45]. All of them were fitted by COOTinto the generated difference electron density maps. Overallrefinement statistics are enlisted in Table 1B.
Structural homologues to SaTIM in PDB were searched by theBLASTP server [46]. Structure superpositions were done using theLSQKAB [47], interface analysis and accessible surface area calcu-lations were done using PISA [48]. Figs. 1 and 2 are generated byChemDraw Ultra Version 6.0* [65] and Figs. 3e8 are generated by
PyMOL [49]. Simulated annealing FoeFc omit maps were generatedusing CNS [50].
3. Results
3.1. Overall structure
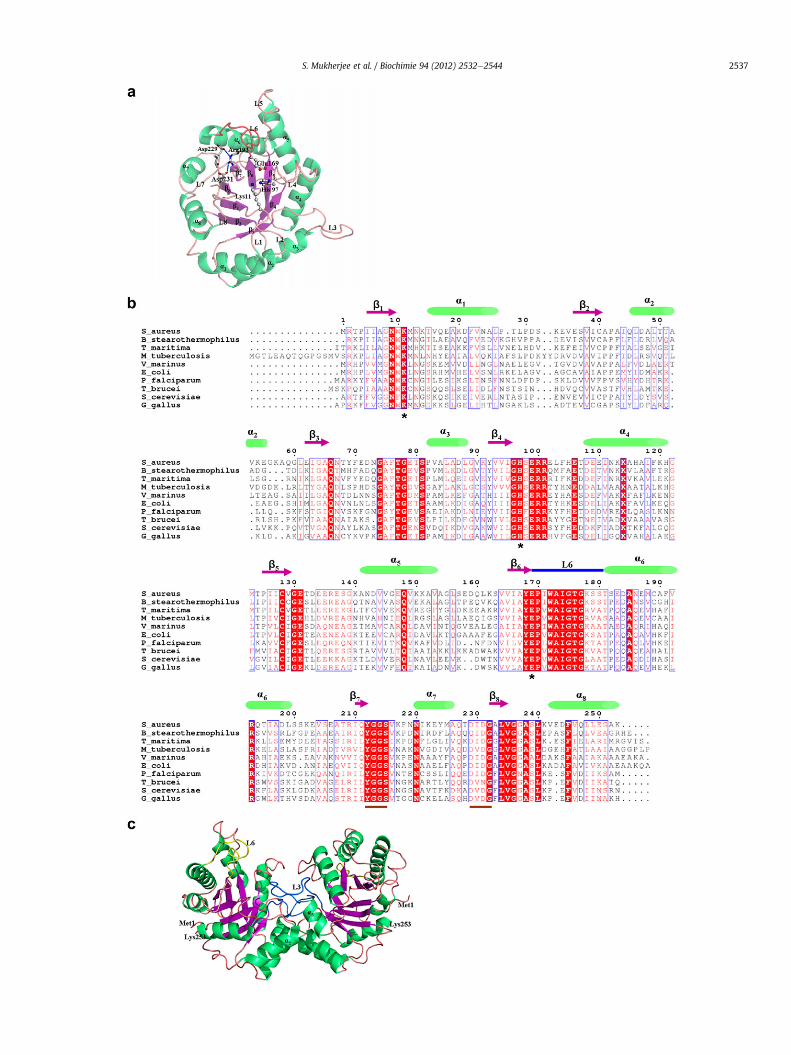
SaTIM crystallizes in P43212 space group consisting of twomolecules, A and B in the asymmetric unit related by a non-crystallographic two fold axis of symmetry. The two subunits in theasymmetric unit are identical with an r.m.s.d.Ca of 0.19 Å. Eachsubunit is composed of 253 amino acid residues and adoptsa classical alpha/beta barrel structure. Most of the residues in theprotein are solvent exposed and the solvent accessible area of eachsubunit is 11,586 Å2. The overall folding of the SaTIM monomer, (b/a)8, is well conserved in other structures of TIM as well. There areeight beta strands which form the inner core of the barrel sur-rounded by eight alpha helices on the outer layer (Fig. 3a). The coreof the beta barrel is predominantly lined by hydrophobic residueslike Ile6, Val38, Ile39, Val165 and Ile210. The secondary structuralelements are interconnected by loops of varying lengths. Thestrands, helices and the loops connecting the strands to the helicesof the TIM barrel are numbered 1e8 and the loops connecting the

S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442536
helices to the strands are designated as LA-LG. The central betabarrel consists of eight parallel beta strands comprising of theresidues 5e9 (b1), 36e40(b2), 62e65 (b3), 92e95 (b4),125e129 (b5),165e168 (b6), 210e212 (b7), 232e235 (b8) and the outer eighthelices comprise of the residues 15e26 (a1), 45e55 (a2), 82e88(a3), 108e121 (a4), 141e153 (a5), 181e199 (a6), 220e226 (a7),242e253 (a8). The catalytically active loop, L6, showing the “hinge-bend” motion in the enzyme upon ligand binding, comprises resi-dues 169e180. This loop is found in both “open” and “closed”conformations in the different structures of SaTIM that will bediscussed in detail in the later sections of this article. The flexiblenature of this long unstructured loop is evident from its highertemperature factor values and poorly defined electron densitymaps for some residues. The average B factor for the residues of L6in subunit A of 3M9Y (SaTIM crystallized from 1.6 M trisodiumcitrate) is 54.6 Å2 while that of the other residues is 33.0 Å2. Thecatalytically active residues, Lys11, His97 and Glu169 are conserved(Fig. 3b) and reside in flexible L1, L4 and L6 respectively. All of theseresidues are flexible, Glu169 being the most among them. Animportant salt bridge between NE of conserved Arg193 and OD1 ofAsp229 of the 229DIDG232 motif exists in SaTIM. OD2 of the secondaspartic acid (Asp231) of the DIDG motif is also hydrogen bondedwith the NH2 group of the conserved Arg193 (Fig. 3a). Althoughthese salt bridges are distant from both the active site and thedimeric interface, the interaction between the higly conservedarginine located in a unique helix of a connecting network and thefirst aspartic acid residue of the well conserved “DX(D/N)G” motif(Fig. 3b) has an important role in the folding, stability and catalysisof the enzyme [51].
Superimposition of subunit A of SaTIM with that of nearesthomologous structure, BsTIM (PDB code: 2BTM) gives an r.m.s.d.Ca
of 0.77 Å. Insignificant differences are observed in the helices andstrands while the variations in L6 (169e180) are noticeable. Inunliganded SaTIM, L6 was found to be in an open conformationwhile the same adopts a closed conformation in 2BTM. Minutedifference in orientations of more ordered alpha helices and betastrands are due to insertions and deletions. All the structures ofSaTIM presented in this paper are within the range of expectedstereochemical parameters (Table 1B).
3.2. Intersubunit interaction
TIM is always biologically active as a dimer. The interfacebetween the two subunits is extensively stabilized by a number ofhydrogen bonds and salt bridges among the different polar residuesresiding in L1, L2, L3 and L4 (Table S1) where the maximumstabilizing contribution is observed from the L3 residues. Theprotruding loop L3 of one subunit docks into a deep pocket of theother subunit (Fig. 3c) formed by the loops L1 and L4 in which twoof the three key residues Lys11 and His97 are located. The dimericinterface of one subunit is close to the catalytic site of the other.Residues of a2 and a3 also have some additional stabilizinghydrophobic and hydrophilic interaction that contribute substan-tially to the process of dimerization. The overall surface area buriedin dimeric association is 1615 Å2 per subunit which constitutes 14%of the total solvent accessible surface area of the individualmonomers.
3.3. Proteineligand interaction
3.3.1. SaTIM-G3P complexG3P is positioned in the active site pocket of the protein formed
by the catalytically active residues Lys11, His97 and Glu169 in bothsubunits in an identical orientation. One end of the substrateanalogue (C1-C2) that corresponds to the reactive end of the
substrate (DHAP or GAP) is involved in a number of polar interac-tions with the catalytically active residues while the phosphategroup positioned at the other terminus is primarily involved ina number of water mediated interactions (Fig. 4). The importantpolar interactions of the ligand with the protein atoms are enlistedin Table 2A. OE1 and OE2 of Glu169 are at distances of 2.7 and 3.6 Åfrom O1 of G3P while the same are at distances of 2.5 and3.4 Å fromO2 of the ligand in both subunits. NE2 of His97 is 2.9 and2.8 Å away from O1 and O2 of G3P in subunit A whereas the cor-responding distances in the other subunit are 3.0 and 2.9 Årespectively. Existence of a strong hydrogen bond between the NE2of His97 and the oxygen atoms of the substrate analogue ispresumably relevant for activation of the proton transfer during thecourse of catalysis. The proximity of ND2 of Asn9 to O1 of G3P (2.9 Åin both subunits) is also observed. Almost identical distancesbetween the interacting atoms of the protein and the oxygen atomsat the reactive end of the ligand is in conformity with the suggestedcis eneediol transition state [52]. Among the catalytically activeresidues, only Lys11 is involved in direct interaction with thebridging oxygen atom (O1P) of the phosphate group at 3.4 and 3.5 Åin subunits A and B respectively. This interaction is important asthis is thought to be one of the determinants of substrate specificityfor TIM [53]. Fig. 4 highlights the refined 2FoeFc map of the ligandwith some important active site interactions in the subunit A. Apartfrom being stabilized by a number of water mediated interactions,the phosphate group of G3P is also stabilized by direct hydrogenbonded interactions with the protein atoms. Nitrogen of thepeptide backbone of Ser215 is at a distance of 2.8 and 2.9 Å awayfrom O4P of phosphate in subunits A and B respectively. Directinteractions between the oxygen atoms (O2P and O3P) of thephosphate with the nitrogen atoms of the polypeptide chain of L8residues (Gly236, Gly237) hold the phosphate group in place. Themost important interaction is however the hydrogen bond betweenthe NH atom of the peptide backbone of Gly175 and the O4P ofphosphate group. O4P of G3P is positioned at a distance of 2.8 and2.9 Å from the NH atom of Gly175 in subunits A and B respectively.This interaction primarily responsible for placing the flexiblecatalytic loop, L6, in close proximity to the ligand, is considered tobe the fingerprint of the closed conformation of the catalytic loop inalmost all the ligand bound structures of TIM [53]. Five structuralwaters (351, 356, 385, 386 and 389 in subunit A and 272, 304, 307,308, 317 and 339 in subunit B) bound to the phosphate group ofG3P are observed. These waters are fully conserved in the crystalstructure of unliganded SaTIM (3M9Y) as well. However, the watermolecules in the active site cavity that were involved in directhydrogen bonding with the catalytic residues in the unboundstructure of SaTIM (255, 276, 493 in subunit A and 303, 332 insubunit B) are expelled from the pocket upon ligand binding.
3.3.2. SaTIM-G2P complexG2P binds to the active site of the enzyme in both subunits.
Fig. 5a shows the fit of the ligand in subunit A into the unbiasedFoeFc omit map. The density of the ligand is comparatively better insubunit A. In subunit A, the reactive end of the ligand is positionedtowards the catalytic residues and is stabilized by a number ofdirect or water mediated hydrogen bonding interactions withthem. O31 of G2P is hydrogen bondedwith NZ of catalytically activeLys11 at a distance of 2.9 Å. Unlike O1 of G3P in subunit A, O31 ofG2P is not directly hydrogen bonded to the NE2 of His97 but isengaged in hydrogen bonding to the catalytic histidinemediated bywater 280. Although bound in an orientation similar to G3P insubunit A of SaTIM_G3P, G2P is further away from the active sitepocket. The phosphorus atom of the phosphate group of G2P inchain A of SaTIM_G2P is 1.2 Å from the corresponding phosphorusatom of G3P in chain A of SaTIM_G3P. Apart from a number of
S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2537

Fig. 4. G3P binding in SaTIM_G3P: Stereoview of refined 2FoeFc electron density map ofG3P in subunit A of SaTIM_G3P. The map is contoured at 1.0s. Some of the polarinteractions stabilizing the ligand in the active site pocket are highlighted in green. Themost important interaction between NH group of Gly175 and O4P of G3P responsiblefor the closed conformation of L6 is highlighted. The interaction between Lys11 andbridging oxygen atom (O1P) is one of the principal determinants of substrate speci-ficity for TIM. (For interpretation of the references to colour in this figure legend, thereader is referred to the web version of this article.)
S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442538
hydrogen bonding interactions with the conserved water mole-cules (255, 275 and 296), the phosphate group is extensivelyengaged in direct hydrogen bonding interactions with polar sidechains of Ser215, and the main chain nitrogen atom of Gly236 andGly237 of L8.
Although no significant differences in the conformation of thepolypeptide fold are noticed between the two subunits of theSaTIM_G2P complex (r.m.s.d.Ca¼ 0.07 Å2), G2P binds to the subunitB in a completely inverted orientation (Fig. 5b) which is observed inTIM for the first time. Although the phosphate group resides ina similar position as observed in the subunit A, the triose moiety iscompletely flipped out in the opposite direction. Presenting the L6residues of subunit B in a manner identical to subunit A (as shownin Fig. 5a), the inverted orientation of G2P in subunit B can easily bevisualized (Sup. Fig. 1). The distance measured between the C2atoms of G2P in the two subunits on superimposition is 4.3 Å(Fig. 5c). Although higher temperature factors (67.7 Å2) of theligand bound in this manner account for its higher flexibility ascompared to the one in subunit A (37.0 Å2), nevertheless it isstabilized by a number of polar interactions. The conserved stabi-lizing interactions of the phosphate group with the L8 residues andSer215 of L7 remain unperturbed. As the other end of the ligandresides in a different position, some of the hydrogen bondinginteractions with active site residues like His97 and structuralwater molecules are compromised. Hydrogen bonding between NZof Lys11 and O31 of G2P is however retained. The loss of theconserved interactions is duly compensated by some new interac-tions arising from conserved water molecules (Table 2B). O3P ofG2P in subunit B is stabilized by hydrogen bonding with structuralwater (314 in B chain) that is a member of the conserved watercluster network in the vicinity of the active site cavity.
Fig. 3. Structural features of SaTIM: (a) Cartoon representation of a single subunit of SaTIM whis highlighted in red. Important salt bridges of conserved Arg193 with Asp229 and Asp2alignment of amino acid residues of SaTIM (3M9Y) with TIM from Bacillus stearothermopmarinus (1AW1), Escherichia coli (1TRE), Plasmodium falciparum (1O5X), Trypanosoma bruceiThe alignment file has been formatted by ESPRIPT [64]. The secondary structural elements ocatalytically active Lys11, His97 and Glu169. The gaps are introduced to optimize the alignmeat the C terminus of the TIM barrel are underlined. (c) Spatial organization of the two mocrystallographic axis of symmetry. Most of the intersubunit interactions are formed by the Lhydrophilic interactions that contribute substantially to the process of dimerization. The cafigure legend, the reader is referred to the web version of this article.)
The most notable difference between SaTIM_G3P and SaT-IM_G2P complex is the orientation of flexible loop L6. In G2P boundcomplex, L6 adopts the open conformation in both subunits. Onsuperposition of the respective subunits of the two complexes, Ca ofGly175 in subunit A of SaTIM_G2P is at a distance of 7.0 Å away fromthat in subunit A of SaTIM_G3P (Fig. 5c). The stabilizing interactionsbetween the oxygen atoms of phosphate group of the ligand andthe peptide backbone of the L6 residues are lost. All the residuescan be mapped into the electron density in subunit A while nodensity could be obtained for Gly175, Thr176 and Gly177 in subunitB. Temperature factors of the other L6 residues are comparable inboth subunits.
3.3.3. SaTIM_3PG complexLike G3P and G2P, 3PG is also bound to the active site cavity in
both subunits of SaTIM. The active site of the enzyme is largeenough to accommodate the extra oxygen atom of the ligandwithout significant perturbation of the active site geometry asobserved from low r.m.s.d.Ca values when superimposed with thecorresponding subunit of SaTIM_G3P complex. The phosphategroup in both subunits share similar stabilizing interactions withthe peptide backbone of the conserved L8 residues as in othercomplexes. Apart from direct interactions with the nitrogen atomof the polypeptide chain, the phosphate group extensively interactswith a number of water molecules surrounding the active sitepocket. Most of these are structurally conserved in almost all thestructures of the complexes of SaTIM discussed here.
The ligand in subunit B occupies a position identical to that ofG3P in SaTIM_G3P complex. Fig. 6a justifies the correct positioningof the ligand in (R)-configuration into the unbiased FoeFc peakabove 6s. The carboxylate end of the ligand is positioned towardsthe catalytically active residues and is stabilized by hydrogenbonding with NE2 of His97, ND2 of Asn9, O of Leu234 and N ofVal235. The important interaction of the phosphate atom and thenitrogen of Gly175 that holds the L6 in closed conformation isretained in contrast to SaTIM_G2P where the loop adopts the openconformation.
The binding of 3PG to subunit A is significantly different.Although the phosphate group resides in the same position, thecarboxylate end is oriented away from the binding pocket. Fittingthe ligand in the generated unbiased FoeFc omit map in an orien-tation identical to the subunit B, resulted in generation of someunresolved negative density over the C1-C3 segment. However,fitting the ligand with the carboxylate end away from the active siteresidues solved the problem (Fig. 6b). The binding of 3PG in thissubunit is very similar to the binding of G2P in the subunit B ofSaTIM_G2P complex. The carboxylate oxygen atoms stabilize theligand through a number of polar interactions. Similar to O3 of G2P,O1 of 3PG is hydrogen bonded to NZ of catalytically active Lys11 ata distance of 2.9 Å. The other carboxylate oxygen, O2, is ata distance of 2.8 Å from the N atom of Gly237 and is also hydrogenbonded toWat320 andWat356 of the subunit A (Fig. 6b). O3 of 3PGis also hydrogen bonded to the peptide nitrogen of Gly237 andsome conserved water molecules. The position of O1, O2 and O3
ich shows the overall (b/a)8 fold. The catalytic loop L6 (169e180) in open conformation31of conserved 229DIDG232 motif are shown. (b) Structure based multiple sequencehilus (2BTM), Thermotoga maritima (1B9B), Mycobacterium tuberculosis (3GVG), Vibrio(4TIM), Saccharomyces cerevisiae (1YPI) and Gallus gallus (1TPH) using CLUSTALW [63].f SaTIM and the catalytic loop L6 are highlighted. Asterisks (*) indicate fully conservednt and are indicated by dots. The well conserved “YGGS” and “DX(D/N)G”motifs locatedlecules of SaTIM in asymmetric unit. The two subunits are related by a two fold non-3 residues. Residues of a2 and a3 also have some additional stabilizing hydrophobic andtalytic loop L6 is coloured yellow. (For interpretation of the references to colour in this

Fig. 5. G2P binding in SaTIM_G2P: Unbiased simulated annealing FoeFc omit maps(contoured at 3.5s) of G2P in (a) subunit A and (b) subunit B in SaTIM_G2P. C1 of G2P ispointed towards the catalytic residues Lys11, His96 and Glu169 in subunit A. The ligandis stabilized by a number of direct and water mediated interactions with the active siteresidues, some of which are highlighted in blue in (a). In subunit B (b), G2P resides ina completely inverted orientation with the triose group flipped away from the catalyticresidues. Some interactions stabilizing the ligand in this new orientation are high-lighted in purple. Conserved structural water molecules lying in the vicinity of theactive site pocket are shown as non-bonded spheres in (a) and (b). L6 is in “open”configuration in both subunits. The unbiased omit map was calculated from the refinedstructure after removal of the ligand. (c) Superposition of G2P in A (green) and B(violet) subunit of SaTIM_G2P. The ligand is shown in ball and stick. The distancebetween the corresponding C2 atom is 4.3 Å. L6, coloured lime green is in the openconformation in both subunits in SaTIM_G2P while the same, coloured blue, adoptsclosed conformation in SaTIM_G3P. On superposition, Ca of Gly175 (shown in sticks) inSaTIM_G2P is 7.0 Å from that in SaTIM_G3P. (For interpretation of the references tocolour in this figure legend, the reader is referred to the web version of this article.)
S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2539
atoms from 3PG in subunit B are occupied by Wat317, 316 and 360respectively. Although there is a paucity of number of hydrogenbonding interactions of the triose moiety in the subunit A ascompared to the subunit B, the new interactions that develop dueto this particular orientation of the ligand are sufficient to stabilizethe ligand as observed from comparable refined temperaturefactors of 3PG in both subunits. In contrast to subunit B, L6 is foundto be in the open conformation. Thus, the conserved stabilizinginteractions of the ligand with L6 residues are completely lost.Different conformation of the catalytic loop and altered binding ofthe ligand is very clear upon superposition of the two subunits inSaTIM_3PG (Fig. 6c). This is the only complex of SaTIM discussedherewhere the catalytic loop adopts two different conformations inthe two different subunits.
3.3.4. TIM_2PG complex2PG is an inhibitor of TIM which is similar in structure to 3PG
but differs in the position of the phosphate. Like all the otherligands discussed so far (G3P, G2P and 3PG), 2PG to the active sitecavity in both subunits of SaTIM although in a completely differentmanner. 2PG binds to both subunits in almost similar orientationalthough subtle differences can not be ruled out. Only the (R)-isomer could be modelled into the FoeFc omit density map (Fig. 7a).The active site interactions are summarized in Table 3. The orien-tation and position of the bound ligand is unique compared to theall the other ligand bound structures of TIM structurally charac-terized so far. A completely new phosphate ion binding site of theligand has been identified. The phosphorus atom of 2PG in subunitA of SaTIM_2PG complex is 3.0, 3.4 and 2.9 Å, away from thephosphorus atom of G3P, 3PG and G2P of the correspondingsubunit in the respective complexes (Fig. 7b). The phosphate grouphere is stabilized by polar interactions with NZ of catalytic Lys11while the oxygen atom (O3) attached to C3 is hydrogen bonded toNZ group of Lys217. This hydrogen bonding interaction is weaker insubunit B in comparison to subunit A. The carboxylate oxygenatoms of the ligand are extensively stabilized by the peptidebackbone of Gly236 and Gly237. Due to its unique orientation, thehydrogen bonding interactions of the carboxylate oxygen atoms ofthe ligand with the polar groups of the catalytic residues arecompletely lost. Compared to the other ligands, this inhibitor is notso deep seated in the active site cavity which can be accounted forits comparatively higher temperature factor. The catalytic loop L6 isfound in open conformation in both subunits with a low r.m.s.d.Ca
of 0.1 Å. This is concordant with the fact that the stabilizing inter-action between the phosphate oxygen atom and the peptidebackbone of Gly175 is completely disrupted upon ligand binding inthis new orientation.
4. Discussion
The present work describes different crystal structures of SaTIMand its complexes with various substrate analogues and inhibitorswith a focus to delineate the differential and interesting modes ofligand binding along with conformational variation of the catalyticloop L6. In all the structures, the protein crystallizes in same spacegroup with two molecules in the asymmetric unit. Extensiveresearch on the enzymatic activity of the engineered monomericvariants of TIM claims a complete dependency of catalytic effi-ciency of the enzyme on its dimeric state [54e56]. This is also truefor SaTIM (data not shown). Although DHAP is the physiologicalsubstrate of TIM, we were not able to trap the Michaelis complex ofthe enzyme with the substrate. Due to high catalytic efficiency ofthe enzyme, the crystals started dissolving instantly upon soakingwith the substrate. Optimizing the substrate concentration, soakingtime and carrying out the soaking experiments at 4 �C did not help

Fig. 6. 3PG binding in SaTIM_3PG: Stereoview of simulated annealing FoeFc omit maps of 3PG in (a) subunit B and (b) subunit A in SaTIM_3PG. In (a), the carboxylate end of theligand is stabilized by the catalytic residues. Some of the stabilizing interactions are highlighted in violet. The “closed” conformation of L6 is stabilized by the hydrogen bondinginteractions between the NH group of Gly175 and O4P of 3PG. Glu169 is in the “swung-out” (c1w 60�) conformation and is involved in a water mediated interactionwith O3 of 3PG.In (b), the carboxylate end of the ligand is oriented away from the active site residues and is only stabilized by Lys11 and Gly237 residues of the protein. The position of carboxylategroup of 3PG in subunit B are here occupied by water molecules some of which are labelled. L6 is in the “open conformation” in subunit A as the stabilizing interactions of the loopresidues with the ligand are completely lost. C2 in 2PG in both subunits adopt a (R) configuration. The electron density maps were computed using the weighted Fo and Fccoefficients after exclusion of the ligand from the refined model and contoured at 3.5s. (c) Superposition of subunit A (green) and B (magenta). L6 adopts two different orientationsin the two subunits. The active site residues and L6 are highlighted. The carboxylate group of 3PG is subunit A is oriented away from the active site pocket. (For interpretation of thereferences to colour in this figure legend, the reader is referred to the web version of this article.)
S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442540
either. Mutation of the catalytically active residues may have beena solution to the problem but we are more prompted to analyze theinteractions of the active site residues with the different substrateanalogues and inhibitors. This will provide a pool of informationabout the substrate binding along with conformational variation ofthe catalytic loop L6. In all the structures of unliganded andliganded SaTIM discussed in this article, interestingmodes of ligandbinding along with different conformation of L6 residues have beenobserved. Each of these issues will be addressed and analyzed.
4.1. Conformation of catalytic loop
Conformational flexibility is a common feature of enzymes andis often used to control chemical reactivity or to deliver a signal fromonemacromolecule to another. Such a common element is an activesite loop, wherein an open form can facilitate ligand binding andrelease while the closed form prepares, controls, or protects thereaction intermediates [15]. Conformational flexibility of the cata-lytic loop in TIM has been the subject of intensive research. Inter-esting loop movement has been extensively correlated with thecatalytic mechanism of this enzyme [6e10,13]. The loop is usuallyfound to be present in the open conformation in the absence of theligands and closes upon ligand binding to avoid the deleteriouseffect of hydrolysis [1]. However, in some organisms like Plasmo-dium falciparum, L6 has been reported to be predominantly presentin open conformation in presence of some specific ligands [57].
Nevertheless, closed conformation of the loop has also beenobserved in PfTIM [53].In addition, the trypanosomal enzymeexhibits an “almost closed” conformation which can switch to“completely closed” conformation in presence of a phosphatecontaining ligand [58]. In our case, the conformations of L6 vary notonly with ligands but marked differences in the conformation arealso observed between the different subunits upon ligand binding.Although crystallized from a high concentration of trisodiumcitrate, the unliganded structure of SaTIM exhibits open loopconformation in spite of the bound citrate ion. This is howeverjustifiable from the fact the citrate is much bigger in size than thephosphate containing ligands used. So it resides in a completelydifferent position and the interactions of citrate with the L6 resi-dues that are primarily responsible for the closed conformation ofthe loop are absent in the unliganded structure. The conformationof L6 in both subunits of SaTIM-G3P complex is closed as expectedin the presence of the ligand. However, careful structural super-imposition of the L6 residues of the subunit A of SaTIM_G3P withthe corresponding residues of subunit 2 of trypanosomal TIM (PDBentry: 5TIM, 6TIM) shows that the catalytic loop in SaTIM_G3Padopts the “almost closed” conformation instead of completely“closed” conformation. Ca of Gly175 of SaTIM_G3P (subunit A) isat a distance of 0.9 Å from the corresponding atom of Gly173(subunit 2) of 5TIM where the catalytic loop is in the “almostclosed” conformation but at a distance of 1.5 Å from the same in6TIM where the catalytic loop is in the completely “closed”

Fig. 7. 2PG binding in SaTIM_2PG: (a) Stereoviewof simulated annealing FoeFc omitmaps(contoured at 3.5s) of 2PG. Some polar interactions with the amino acid residues andstructurally conserved water molecules are highlighted. The phosphate group occupiesa completely new position. L6 is in the “open” conformation in both subunits. (b)Superimposed view of 2PG (olive) in subunit A of SaTIM_2PG with G3P (grey), G2P(green) and 3PG (blue) of SaTIM_G3P, SaTIM_G2P and SaTIM_3PG respectively. The Ca
atoms of subunit A of the different complexes were superposed using the programLSQKAB. The phosphorus atomof 2PG is 3.0, 3.4 and 2.9 Å away from that in G3P, 3PG andG2P respectively. The ligands are shown in sticks. (For interpretation of the references tocolour in this figure legend, the reader is referred to the web version of this article.)
Fig. 8. Superposition of L6: Superposition of the L6 residues of SaTIM_G3P (subunit A,coloured red) with the corresponding residues of 5TIM (subunit 2: “almost closed”conformation, coloured magenta), 5TIM (subunit 1: “open” conformation, colouredgreen) and 6TIM (subunit 2: “closed” conformation, coloured cyan). L6 adopts an“almost closed” conformation in both subunits of SaTIM_G3P. Ca of Gly175 (red) is only0.9 Å away from the corresponding atom of Gly173 in subunit 2 (magenta) of 5TIM butat a distance of 1.5 Å from the same in subunit 2 (cyan) of 6TIM. (For interpretation ofthe references to colour in this figure legend, the reader is referred to the web versionof this article.)
Table 2Polar interactions between protein and ligand atoms in (A) SaTIM_G3P and (B)SaTIM_G2P (B subunit). The maximum distance cutoff value is 3.6 Å
(A) SaTIM_G3P
A subunit B subunit
G3P Protein atom Distance (Å) G3P Protein atom Distance (Å)
O1 His97[NE2] 3.0 O1 His97[NE2] 3.0O1 Glu169[OE1] 3.6 O1 Glu169[OE1] 3.6O1 Glu169[OE2] 2.7 O1 Glu169[OE2] 2.7O1 Asn9[ND2] 2.9 O1 Asn9[ND2] 2.9O2 His97[NE2] 2.9 O2 His97[NE2] 2.9O2 Glu169[OE1] 2.5 O2 Glu169[OE1] 2.5O2 Glu169[OE2] 3.4 O2 Glu169[OE2] 3.4O2 Lys11[NZ] 3.4 O2 Lys11[NZ] 3.3O1P Lys11[NZ] 3.4 O1P Lys11[NZ] 3.5O1P Gly236[N] 3.6 O1P Gly236[N] 3.6O1P Wat386[O] 3.4 O4P Wat317 3.5O4P Gly175[N] 2.8 O4P Gly175[N] 2.9O4P Ser215[N] 2.7 O2P Ser215[N] 2.7O2P Gly236[N] 2.8 O2P Gly236[N] 2.8O2P Wat385[O] 2.6 O2P Wat304[O] 2.6O2P Wat389[O] 2.6 O3P Wat308[O] 2.6O3P Gly237[N] 2.9 O3P Gly237[N] 2.8O3P Wat356[O] 2.5 O3P Wat339[O] 2.6O3P Wat386[O] 2.7 O3P Wat317[O] 2.8
(B) SaTIM_G2P (B subunit)
G2P Protein Distance (Å)
O11 Lys217[NZ] 2.8O31 Lys11[NZ] 2.5O31 Wat319[O] 3.2O1P Gly237[N] 3.0O2P Wat317[O] 2.6O2P Wat305[O] 2.5O3P Ser215[OG] 3.2O3P Wat314[O] 3.3
S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2541
conformation (Fig. 8). The loop is in the open conformation insubunit 1 of 5TIM.
A unique binding of G2P has been observed in one of thesubunits of the SaTIM_G2P. Although the phosphate ion occupiesa position similar to subunit A, the orientation of the triose group asobserved in subunit B has never been observed before. The onlyother G2P bound structure of TIM is the W168F mutant from P.falciparum (1WOA) where the loop is reported to be in the openconformation and disordered due to the mutation [59]. Role of thetryptophan residue in stabilizing the loop in closed conformationhas already beenwell established. This residue (Trp172) is howeverpresent in SaTIM. In spite of its presence, the reason behind the
Table 3Proteineligand interactions in SaTIM_2PG in A and B subunits. All the polar inter-actions between 2.2 and 3.6 Å are listed.
A subunit B subunit
2PG Protein atom Distance (Å) 2PG Protein atom Distance (Å)
O1 Gly236[N] 3.1 O1 Wat258[O] 2.9O1 Wat342[O] 2.5 O2 Wat258[O] 3.5O2 Wat333[O] 2.7 O2 Wat277[O] 2.9O2 Wat360[O] 3.4 O2 Wat335[O] 2.5O3 Gly237[N] 3.1 O2 Gly236[N] 3.1O3 Wat315[O] 3.0 O3 Lys217[NZ] 3.6O3 Wat354[O] 3.0 O3 Wat336[O] 2.5O3P Lys11[NZ] 2.4 O1P Lys11[NZ] 3.5O3P Wat360[O] 2.4 O1P Gly237[N] 3.6O4P Lys11[NZ] 3.6 O1P Wat313[O] 3.6O4P Wat318[O] 3.3 O2P Lys11[NZ] 2.9
O2P Wat298[O] 3.5O2P Wat313[O] 2.7O3P Lys11[NZ] 2.7

S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442542
adoption of disordered and open conformation of L6 residues inSaTIM_G2P remains unclear. To analyze the plausible cause, all theinteractions of the residues with the crystallographic symmetrymates were analyzed. No such stabilizing crystal contacts wereobserved. The only other way to unambiguously ascertain thereason behind the open conformation of the catalytic loop in thisparticular ligand bound structure is to subject each of the residuesto site directed mutagenesis and to structurally characterize theinteractions responsible for this particular conformation.
Among all the liganded structures of SaTIM discussed in thisarticle, only in SaTIM_3PG, does L6 adopt two different conforma-tions in the two subunits. This is particularly interesting. The openconformation of the loop has also been observed in structures ofPfTIM bound to 3PG which is attributed to the S96F mutation.Phe96 is unique to PfTIM. A careful comparison with the trypano-somal enzyme shows that extensive clashes between Phe96 andIle172 in the closed conformation may be anticipated to force theloop in open conformation [57]. However, the correspondingresidue in SaTIM is Ser98 and suffers no unfavourable steric inter-action with any of the residues in closed conformation. Concomi-tant with the dual conformation of L6, the conserved 212YGGS215
motif (Fig. 3b) also exhibits significant change in the two subunits.In subunit A,4 and j of Gly214 are 142.0 and 87 0.6� while the samein the subunit B are �82.4 and �175.7� respectively. The side chainof Ser215 also adopts two different conformations, the conforma-tion in subunit B stabilizing the closed form of L6 by forming stronghydrogen bonds with the residues at its tip. The other residues ofthe motif also contribute sufficiently to the stabilization of theclosed conformation of the catalytic loop. Detailed analysis of thecrystal contacts failed to provide any convincing reason behind thedisposition of L6 residues in different conformations in thedifferent subunits. Nevertheless, to rationalize this dual confor-mation of the catalytic loop, extensive comparison with previouslysolved TIM structures [32] has been carried out. Supported by bulkof available structural details, this particular characteristic of thecatalytic loop can possibly be correlated with the persistentconformational heterogeneity of the enzyme as also observed inrabbit muscle isomerase [60]. A completely new mode of bindinghas been observed for the inhibitor 2PG. The phosphate groupresiding in an entirely different position fails to stabilize the loop inthe closed conformation. The ligand in its unique orientation hasbeen superimposed and compared with all the high resolutionstructure of PfTIM complexed with 2PG (PDB entry: 1O5X) andTbTIM_2PG complex (PDB entry: 4TIM). However, the position ofthe bound inhibitor in SaTIM_2PG is entirely different from thatobserved in TbTIM [61] and both the “LIGO” and “LIGC” forms inPfTIM [17]. On careful comparison with all the twenty four struc-tures of TIM complexed with transition state analogue 2-phosphoglycolic acid (2PGA) determined so far, the position of2PG in SaTIM is observed to be significantly different from all ofthem. The carboxylate group occupies the position of the phos-phate binding site. The carboxylate anion is smaller in size than thephosphate group. So it’s not unlikely that the phosphate bindingsite has sufficient space to room smaller carboxylate group. What ismore interesting is that the phosphate groupwas accommodated inthe site which was occupied by the carboxylate group in 3PG. Asmentioned earlier, compared to other ligands, the ligand was not sodeep seated within the active site pocket. The loose binding of theligand gave sufficient space for the phosphate group to be accom-modated in the carboxylate binding site. Moreover, in both subunitsL6 adopts open conformation providing a more spacious active sitein which the carboxylate group of the ligand is free to reside ina different position. By comparing the conformations of the L6residues in all the liganded structures of SaTIM, it can perhaps bereasonably inferred that the open conformation of the catalytic
loop has a preference for the phosphate group attached to C2 atomin contrast to the phosphate at C3 which is more preferred forclosed loop conformation.
4.2. Flexibility of the active site and structural adaptation of theenzyme upon ligand binding
The active site of triosephosphate isomerase is inherently flex-ible. Different types of phosphate and phosphonate containingligands like inhibitors, substrates and transition state analogues areobserved to be bound in the active site cavity with altered orien-tations and differential modes of binding. Most of the active siteresidues display a single conformation. These include the catalyti-cally active residues Lys11 and His97. In contrast, other residues inthe active site are extremely flexible depending upon the natureand position of the ligand. Apart from the conformations of theresidues of the catalytic loop which have been extensively eluci-dated in the preceding section, the most notable conformationalvariation is observed in case of Glu169. This residue in particularoccupies one of the two discrete conformations: “swung in” or“swung out” dependent on the nature of the ligand bound to theactive site. In three of the four liganded structures, Glu169 is in thecatalytically active (swung in, c1 w �60�) conformation. However,it is in the “swung out” (c1 w 60�) conformation in the structurecomplexed to 3PG (Fig. 6a). Although catalytically inactive, thisconformation is particularly stabilized by some polar interactionswith the Ser98 and O3 of 3PG via a water mediated interaction. Theapparent reason behind the adoption of this “swung out” confor-mation in spite of the ligand in subunit B oriented similarly to thatin G3P complex is inexplicable. In SaTIM_G3P, the carboxylate sidechain is strongly hydrogen bonded with the hydroxyl groups of theligand. Some of the hydrogen bonding interactions of the ligandwith the carboxylate side chain from Glu169 and NE2 of His95 aresubstantially strong and are termed as “low barrier hydrogenbonds” (LBHB) by Cleland [2]. They have energies of formation inthe range of 10e20 kcal mol�1. This energy is thought to facilitatethe catalysis by lowering the free energy of the transition state. It isbelieved that the catalytic glutamate and the histidine are the mostprobable residues facilitating the two proton transfers respectivelyin the catalytic mechanism of TIM [3,62]. The activation barrier ofthese proton transfer steps are postulated to be significantly low-ered by these LBHB highlighting their importance in the catalysis ofthis almost “evolutionary perfect enzyme”.
5. Conclusion
Crystal structures of SaTIM alone and in complex with differentphosphate containing ligands provide interesting insights intodifferential modes of ligand binding and conformational flexibilityof the catalytic loop. L6 has been found to be “almost closed”conformation in both subunits of SaTIM-G3P while it is in the openconformation in the subunits of complexes with G2P and 2PG. Inthe complex with 3PG, it is in the open conformation in subunit Abut closed in subunit B. Preference of the conformation of thecatalytic loop can be correlated with the position of the phosphategroup in the ligand. Open conformation of the catalytic loop hasa preference for the phosphate group attached to C2 atom incontrast to the phosphate at C3 which is more preferred for closedloop conformation. Unique modes of binding have also beenobserved for G2P and 3PG for the very first time. The triose moietyis oriented away from the catalytic residues and occupies anentirely different position in some subunits. A completely newbinding site for phosphate has also been identified in the complexwith 2PG which differs substantially from the conventional

S. Mukherjee et al. / Biochimie 94 (2012) 2532e2544 2543
phosphate binding site of the ligand in the crystal structures of TIMdetermined so far.
5.1. Protein data bank accession numbers
Coordinates and structure factor amplitudes have beensubmitted in the protein data bank under the accession codes:3M9Y (SaTIM crystallized from 1.6 M trisodium citrate dihydrate),3UWY (SaTIM crystallized from 2.4 M sodium malonate), 3UWU(SaTIM_G3P), 3UWZ (SaTIM_G2P), 3UWV (SaTIM_2PG), 3UWW(SaTIM_3PG).
Acknowledgements
The authors like to acknowledge Department of Biotechnology,Government of India for financial assistance and setting up thecrystallographic facility at IIT Kharagpur. SM and ARC thank Councilof Scientific and Industrial research and University GrantsCommission, Government of India for individual fellowship. Theauthors gratefully acknowledge Central Research Facility of IITKharagpur. The authors thank Dr. Baisakhee Saha, Department ofBiotechnology, IIT Kharagpur for helpful suggestions. The genomicDNA of MRSA252 was a kind gift from Dr. H.G. Wiker, University ofBergen, Norway.
Appendix A. Supplementary information
Supplementary data related to this article can be found online athttp://dx.doi.org/10.1016/j.biochi.2012.07.001.
References
[1] J.R. Knowles, Enzyme catalysis: not different, just better, Nature 350 (1991)121e124.
[2] W.W. Cleland, M.M. Kreevoy, Low barrier hydrogen bonds and enzymiccatalysis, Science 264 (1994) 1887e1890.
[3] Q. Cui, M. Karplus, Triosephosphate isomerase: a theoretical comparison ofalternative pathways, J. Am. Chem. Soc. 123 (2001) 2284e2290.
[4] W.J. Albery, J.R. Knowles, Free-energy profile of the reaction catalyzed bytriosephosphate isomerase, Biochemistry 15 (1976) 5627e5631.
[5] S.C. Blacklow, R.T. Raines, W.A. Lim, P.D. Zamore, J.R. Knowles,Triosephosphate isomerase catalysis is diffusion controlled. Appendix:analysis of triose phosphate equilibria in aqueous solution by 31P NMR,Biochemistry 27 (1988) 1158e1167.
[6] R.C. Davenport, P.A. Bash, B.A. Seaton, M. Karplus, G.A. Petsko, D. Ringe,Structure of the triosephosphate isomerase-phosphoglycolohydroxamatecomplex: an analogue of the intermediate on the reaction pathway,Biochemistry 30 (1991) 5821e5826.
[7] T.K. Harris, R.N. Cole, F.I. Comer, A.S. Mildvan, Proton transfer in the mecha-nism of triosephosphate isomerase, Biochemistry 37 (1998) 16828e16838.
[8] J.M. Herlihy, S.G. Maister, W.J. Albery, J.R. Knowles, Energetics oftriosephosphate isomerase: the fate of the 1(R)-3H label of tritiateddihydroxyacetone phsophate in the isomerase reaction, Biochemistry 15(1976) 5601e5607.
[9] E.A. Komives, L.C. Chang, E. Lolis, R.F. Tilton, G.A. Petsko, J.R. Knowles,Electrophilic catalysis in triosephosphate isomerase: the role of histidine-95,Biochemistry 30 (1991) 3011e3019.
[10] E. Lolis, G.A. Petsko, Crystallographic analysis of the complex betweentriosephosphate isomerase and 2-phosphoglycolate at 2.5 Å resolution:implications for catalysis, Biochemistry 29 (1990) 6619e6625.
[11] J. Aqvist, M. Fothergill, Computer simulation of the triosephosphate isomerasecatalyzed reaction, J. Biol. Chem. 271 (1996) 10010e10016.
[12] P.A. Bash, M.J. Field, R.C. Davenport, G.A. Petsko, D. Ringe, M. Karplus,Computer simulation and analysis of the reaction pathway of triosephosphateisomerase, Biochemistry 30 (1991) 5826e5832.
[13] S. Rozovsky, A.E. McDermott, The time scale of the catalytic loop motion intriosephosphate isomerase, J. Mol. Biol. 310 (2001) 259e270.
[14] S. Rozovsky, G. Jogl, L. Tong, A.E. McDermott, Solution-state NMRinvestigations of triosephosphate isomerase active site loop motion: ligandrelease in relation to active site loop dynamics, J. Mol. Biol. 310 (2001)271e280.
[15] R. Desamero, S. Rozovsky, N. Zhadin, A. McDermott, R. Callender, Active siteloop motion in triosephosphate isomerase: T-jump relaxation spectroscopy ofthermal activation, Biochemistry 42 (2003) 2941e2951.
[16] Y. Wang, R.B. Berlow, J.P. Loria, Role of loop-loop interactions in coordinatingmotions and enzymatic function in triosephosphate isomerase, Biochemistry48 (2009) 4548e4556.
[17] S. Parthasarathy, K. Eaazhisai, H. Balaram, P. Balaram, M.R. Murthy, Structureof Plasmodium falciparum triose-phosphate isomerase-2-phosphoglyceratecomplex at 1.1 Å resolution, J. Biol. Chem. 278 (2003) 52461e52470.
[18] G.L. Archer, Staphylococcus aureus: a well-armed pathogen, Clin. Infect. Dis.26 (1998) 1179e1181.
[19] K. Hiramatsu, H. Hanaki, T. Ino, K. Yabuta, T. Oguri, F.C. Tenover, Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycinsusceptibility, J. Antimicrob. Chemother. 40 (1997) 135e136.
[20] F. Menichetti, Current and emerging serious gram-positive infections, Clin.Microbiol. Infect. 11 (2005) 22e28.
[21] C.L. Gatlin, R. Pieper, S.T. Huang, E. Mongodin, E. Gebregeorgis, P.P. Parmar,D.J. Clark, H. Alami, L. Papazisi, R.D. Fleischmann, S.R. Gill, S.N. Peterson,Proteomic profiling of cell envelope-associated proteins from Staphylococcusaureus, Proteomics 6 (2006) 1530e1549.
[22] P. Becker, W. Hufnagle, G. Peters, M. Herrmann, Detection of differential geneexpression in biofilm-forming versus planktonic populations of Staphylo-coccus aureus using micro-representational-difference analysis, Appl. Environ.Microbiol. 67 (2001) 2958e2965.
[23] S. Bergmann, S. Hammerschmidt, Fibrinolysis and host response in bacterialinfections, Thromb. Haemost. 98 (2007) 512e520.
[24] N. Harraghy, M. Hussain, A. Haggar, T. Chavakis, B. Sinha, M. Herrmann,J.I. Flock, The adhesive and immunomodulating properties of the multifunc-tional Staphylococcus aureus protein Eap, Microbiology 149 (2003)2701e2707.
[25] V. Pancholi, G.S. Chhatwal, Housekeeping enzymes as virulence factors forpathogens, Int. J. Med. Microbiol. 293 (2003) 391e401.
[26] M. Yamaguchi, R. Ikeda, M. Nishimura, S. Kawamoto, Localization by scanningimmunoelectron microscopy of triosephosphate isomerase, the moleculesresponsible for contact-mediated killing of Cryptococcus, on the surface ofStaphylococcus, Microbiol. Immunol. 54 (2010) 368e370.
[27] H. Furuya, R. Ikeda, Interaction of triosephosphate isomerase from the cellsurface of Staphylococcus aureus and alpha-(1/3)-mannooligosaccharidesderived from glucuronoxylomannan of Cryptococcus neoformans, Microbi-ology 155 (2009) 2707e2713.
[28] L.A. Pereira, S.N. Bao, M.S. Barbosa, J.L. da Silva, M.S. Felipe, J.M. de Santana,M.J. Mendes-Giannini, C.M. de Almeida Soares, Analysis of the Para-coccidioides brasiliensis triosephosphate isomerase suggests the potential foradhesin function, FEMS Yeast Res. 7 (2007) 1381e1388.
[29] A. Ribeiro de Jesus, I. Araújo, O. Bacellar, A. Magalhães, E. Pearce, D. Harn,M. Strand, E.M. Carvalho, Human immune responses to Schistosoma mansonivaccine candidate antigens, Infect. Immun. 68 (2000) 2797e2803.
[30] H. Furuya, R. Ikeda, Interaction of triosephosphate isomerase from Staph-ylococcus aureus with plasminogen, Microbiol. Immunol. 55 (2011)855e862.
[31] S. Mukherjee, D. Dutta, B. Saha, A.K. Das, Crystal structure of glyceraldehyde-3-phosphate dehydrogenase 1 from methicillin-resistant Staphylococcusaureus MRSA252 provides novel insights into substrate binding and catalyticmechanism, J. Mol. Biol. 401 (2010) 949e968.
[32] F.C. Bernstein, T.F. Koetzle, G.J. Williams, E.F. Meyer Jr., M.D. Brice, J.R. Rodgers,O. KennarD, T. Shimanouchi, M. Tasumi, The protein data bank. A computer-based archival file for macromolecular structures, Eur. J. Biochem. 80 (1977)319e324.
[33] S. Mukherjee, D. Dutta, B. Saha, A.K. Das, Expression, purification, crystalli-zation and preliminary X-ray diffraction studies of triosephosphate isomerasefrom methicillin-resistant Staphylococcus aureus (MRSA252), Acta Cryst. F65(2009) 398e401.
[34] T.Y. Teng, Mounting of crystals for macromolecular crystallography in a free-standing thin film, J. Appl. Cryst. 23 (1990) 387e391.
[35] W. Kabsch, Automatic processing of rotation diffraction data from crystals ofinitially unknown symmetry and cell constants, J. Appl. Cryst. 26 (1993)795e800.
[36] P.R. Evans, Scaling and assessment of data quality, Acta Cryst. D62 (2005)72e82.
[37] Collaborative Computational Project, Number 4, the CCP4 suite: programs forprotein crystallography, Acta Cryst. D50 (1994) 760e763.
[38] A. Vagin, A. Teplyakov, MOLREP: an automated program for molecularreplacement, J. Appl. Cryst. 30 (1997) 1022e1025.
[39] M. Alvarez, J. Wouters, D. Maes, V. Mainfroid, F. Rentier-Delrue, L. Wyns,E. Depiereux, J.A. Martial, Lys13 plays a crucial role in the functional adap-tation of the thermophilic triose-phosphate isomerase from Bacillus stear-othermophilus to high temperatures, J. Biol. Chem. 274 (1999) 19181e19187.
[40] S. Panjikar, V. Parthasarathy, V.S. Lamzin, M.S. Weiss, P.A. Tucker, Auto-Rickshaw: an automated crystal structure determination platform as an effi-cient tool for the validation of an X-ray diffraction experiment, Acta Cryst. D61(2005) 449e457.
[41] G.N. Murshudov, A.A. Vagin, E.J. Dodson, Refinement of macromolecularstructures by the maximum-likelihood method, Acta Cryst. D53 (1997)240e255.
[42] P. Emsley, K. Cowtan, Coot: model-building tools for molecular graphics, ActaCryst. D60 (2004) 2126e2132.
[43] A.T. Brünger, Free R value: a novel statistical quantity for assessing theaccuracy of crystal structures, Nature 355 (1992) 472e475.

S. Mukherjee et al. / Biochimie 94 (2012) 2532e25442544
[44] R.A. Laskowski, M.W. MacArthur, D.S. Moss, J.M. Thornton, PROCHECK:a program to check the stereochemical quality of protein structures, J. Appl.Cryst. 26 (1993) 283e291.
[45] A.W. Schüttelkopf, D.M. van Aalten, PRODRG: a tool for high-throughputcrystallography of protein-ligand complexes, Acta Cryst. D60 (2004)1355e1363.
[46] S.F. Altschul, L.M. Thomas, A.S. Alejandro, J. Zhang, Z. Zhang, W. Miller,D.J. Lipman, Gapped BLAST and PSI-BLAST: a new generation of proteindatabase search programs, Nucleic Acids Res. 25 (1997) 3389e3402.
[47] W. Kabsch, A solution for the best rotation to relate two sets of vectors, ActaCryst. A32 (1976) 922e923.
[48] E. Krissinel, K. Henrick, Inference of macromolecular assemblies from crys-talline state, J. Mol. Biol. 372 (2007) 774e797.
[49] W.L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific LLC,San Carlos, CA, 2002, Available at: www.pymol.org.
[50] A.T. Brünger, P.D. Adams, L.M. Rice, New applications of simulated annealingin X-ray crystallography and solution NMR, Structure 5 (1997) 325e336.
[51] I. Kursula, S. Partanen, A.M. Lambeir, R.K. Wierenga, The importance of theconserved Arg191-Asp227 salt bridge of triosephosphate isomerase forfolding, stability, and catalysis, FEBS Lett. 518 (2002) 39e42.
[52] T. Alber, D.W. Banner, A.C. Bloomer, G.A. Petsko, D. Phillips, P.S. Rivers, I.A.Wilson,On the three-dimensional structure and catalyticmechanism of triose phosphateisomerase, Philos. Trans. R. Soc. Lond. B. Biol. Sci. 293 (1981) 159e171.
[53] S. Parthasarathy, G. Ravindra, H. Balaram, P. Balaram, M.R. Murthy, Structureof the Plasmodium falciparum triosephosphate isomerase-phosphoglycolatecomplex in two crystal forms: characterization of catalytic loop open andclosed conformations in the ligand-bound state, Biochemistry 41 (2002)13178e13188.
[54] T.V. Borchert, R. Abagyan, R. Jaenicke, R.K. Wierenga, Design, creation, andcharacterization of a stable, monomeric triosephosphate isomerase, Proc. Natl.Acad. Sci. U. S.A.. 91 (1994) 1515e1518.
[55] T.V. Borchert, J.P. Zeelen, W. Schliebs, M. Callens, W. Minke, R. Jaenicke,R.K. Wierenga, An interface point-mutation variant of triosephosphateisomerase is compactly folded and monomeric at low protein concentrations,FEBS Lett. 367 (1995) 315e318.
[56] M. Salin, E.G. Kapetaniou, M. Vaismaa, M. Lajunen, M. Casteleijn, P. Neubauer,L. Salmon, R.K. Wierenga, Crystallographic binding studies with an engineeredmonomeric variant of triosephosphate isomerase, Acta Cryst. D66 (2010)934e944.
[57] S. Parthasarathy, H. Balaram, P. Balaram, M.R. Murthy, Structures of Plasmo-dium falciparum triosephosphate isomerase complexed to substrateanalogues: observation of the catalytic loop in the open conformation in theligand-bound state, Acta Cryst. D58 (2002) 1992e2000.
[58] R.K. Wierenga, M.E. Noble, G. Vriend, S. Nauche, W.G. Hol, Refined 1.83 Åstructure of trypanosomal triosephosphate isomerase crystallized in thepresence of 2.4 M ammonium sulphate. A comparison with the structure ofthe trypanosomal triosephosphate isomerase-glycerol-3-phosphate complex,J. Mol. Biol. 220 (1991) 995e1015.
[59] K. Eaazhisai, H. Balaram, P. Balaram, M.R. Murthy, Structures of unligandedand inhibitor complexes of W168F, a Loop6 hinge mutant of Plasmodiumfalciparum triosephosphate isomerase: observation of an intermediate posi-tion of loop6, J. Mol. Biol. 343 (2004) 671e684.
[60] R. Aparicio, S.T. Ferreira, I. Polikarpov, Closed conformation of the active siteloop of rabbit muscle triosephosphate isomerase in the absence of substrate:evidence of conformational heterogeneity, J. Mol. Biol. 334 (2003)1023e1041.
[61] M.E. Noble, C.L. Verlinde, H. Groendijk, K.H. Kalk, R.K. Wierenga, W.G. Hol,Crystallographic and molecular modeling studies on trypanosomal tri-osephosphate isomerase: a critical assessment of the predicted and observedstructures of the complex with 2-phosphoglycerate, J. Med. Chem. 34 (1991)2709e2718.
[62] W.J. Albery, J.R. Knowles, Deuterium and tritium exchange in enzyme kinetics,Biochemistry 15 (1976) 5588e5600.
[63] J.D. Thompson, D.G. Higgins, T.J. Gibson, CLUSTAL W: improving the sensi-tivity of progressive multiple sequence alignment through sequenceweighting, position-specific gap penalties and weight matrix choice, NucleicAcids Res. 22 (1994) 4673e4680.
[64] P. Gouet, E. Courcelle, D.I. Stuart, F. Metoz, ESPript: multiple sequence align-ments in Postscript, Bioinformatics 15 (1999) 305e308.
[65] http://www.cambridgesoft.com.




















