
Consequences of genetic manipulations of gonadotrophins and gonadotrophin receptors in mice
8
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
Transcript of Consequences of genetic manipulations of gonadotrophins and gonadotrophin receptors in mice

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Annales d’Endocrinologie 71 (2010) 170–176
Journées Klotz 2010
Consequences of genetic manipulations of gonadotrophins andgonadotrophin receptors in mice
Conséquences des manipulations génétiques des gonadotrophines et des récepteurs desgonadotrophines chez la souris
H. Peltoketo a, A. Rivero-Müller b, P. Ahtiainen b, M. Poutanen b,c, I. Huhtaniemi a,b,∗a Department of Surgery and Cancer, Imperial College London, IRDB Building, Hammersmith Campus, Du Cane Road, London W12 0NN, United Kingdom
b Department of Physiology, University of Turku, Kiinamyllynkatu 10, 20520 Turku, Finlandc The Turku Centre for Disease Models, University of Turku, Kiinamyllynkatu 10, 20520 Turku, Finland
Available online 2 April 2010
Presented by Jacques Young
Résumé
Nous avons produit au cours de ces dernières années différents modèles de souris génétiquement modifiées (transgénique [TG], knockout[KO] et knockin [KI]) afin d’étudier les fonctions normales ou aberrantes des gonadotrophines et de leurs récepteurs. Dans cette revue, nousrésumons nos constatations les plus récentes dans ces modèles animaux. Le premier point correspond à la cascade des phénotypes extragonadiquesengendrée par l’hyperstimulation ovarienne chez les souris TG surexprimant la sous-unité � de l’hCG humain et présentant des taux sériques élevésd’hormone lutéinisante (LH)/hCG bioactive. Des concentrations sériques très élevées de progestérone plutôt que d’estrogènes sont responsablesde l’induction de prolactinomes hypophysaires et d’une ascension consécutive des taux de prolactine (PRL). À côté de l’estradiolémie normaleavec progestéronémie élevée, les concentrations élevées de prolactine induisent un développement lobulo-alvéolaire de la glande mammaire avecfinalement la formation des tumeurs malignes négatives pour les récepteurs aux estrogènes et à la progestérone. Un autre modèle de souris TGexprimant un mutant constitutionnellement actif de récepteur de la FSH (FSHR) se présente avec un phénotype nettement ovarien caractérisé par undéveloppement folliculaire avancé, une déplétion, des follicules hémorragiques, des tératomes et une infertilité. Un troisième modèle de souris TGcoexprimant des mutants déficitaires aussi bien dans le domaine de liaison que de signalisation du LHCGR sur un fond génétique KO pour le gène dumême récepteur, fournit des arguments convaincants en faveur du fait que la complémentarité fonctionnelle à travers une homo-di/oligomérisationest un mécanisme physiologique d’activation des récepteurs couplés aux protéines G (GPCR) de classe A. Considérés dans leur ensemble, lesmodèles de souris génétiquement modifiées sont de puissants outils pour élucider les fonctions normales et pathologiques des gonadotrophines etde leurs récepteurs.© 2010 Elsevier Masson SAS. Tous droits réservés.
Mots clés : Gonadotrophines ; Récepteurs des gonadotrophines ; Hormone lutéinisante (LH) ; Gonadotrophine chorionique humaine (hCG) ; Hormone stimulant lafolliculogénèse (FSH) ; Souris transgénique (TG) ; Souris knock-out (KO) ; Ovaires ; Testicules ; Hypophyse ; Lutéome ; Prolactinome ; Récepteurs couplés auxprotéines G (GPCR) ; Dimérisation
Abstract
We have produced over the years several genetically modified mouse models (transgenic [TG], knockout [KO] and knockin [KI]) for thestudy of normal and aberrant functions of gonadotrophins and their receptors. We summarise in the present review some of our recent findingson these animal models. One is the cascade of extragonadal phenotypes triggered by ovarian hyperstimulation in TG mice overexpressing thehuman choriongonadotrophin (hCG) β-subunit and presenting with elevated levels of serum luteinising hormone (LH)/hCG bioactivity. Massivelyelevated levels of serum progesterone, rather than oestrogens, are responsible for the induction of pituitary prolactinomas and the subsequently
∗ Corresponding author.E-mail address: [email protected] (I. Huhtaniemi).
0003-4266/$ – see front matter © 2010 Elsevier Masson SAS. All rights reserved.doi:10.1016/j.ando.2010.02.022

Author's personal copy
H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176 171
elevated prolactin (PRL) levels. Along with normal oestradiol and elevated progesterone levels, the increased concentration of PRL induceslobuloalveolar development of the mammary gland, with ultimate formation of oestrogen and progesterone receptor-negative malignant tumours.Another TG mouse model expressing a constitutively activating mutant form of the follicle-stimulating hormone receptor (FSHR) presents witha strong ovarian phenotype inducing advanced follicular development and depletion, haemorrhagic follicles, teratomas and infertility. A third TGmouse model, coexpressing binding- and signalling-deficient mutants of LHCGR in the KO background for the same receptor (R) gene providedconvincing evidence that functional complementation through homo-di/oligomerisation is a physiologically relevant mode of activation of classA G protein-coupled receptors (GPCR). Taken together, genetically modified mouse models provide powerful tools for the elucidation of normaland pathological functions of gonadotrophins and their R.© 2010 Elsevier Masson SAS. All rights reserved.
Keywords: Gonadotrophins; Gonadotrophin receptors; Luteinising hormone (LH); Human chorionic gonadotrophin (hCG); Follicle-stimulating hormone (FSH);Transgenic (TG) mice; Knockout (KO) Mice; Ovary; Testis; Pituitary; Luteoma; Prolactinoma; G protein-coupled receptor (GPCR) dimerisation
1. Introduction
The hypothalamic-pituitary-gonadal (HPG) axis forms thebackbone of the endocrine regulation of reproductive func-tions. The key hormones and receptors (R) functional alongthis axis are the hypothalamic decapeptide gonadotrophin-releasing hormone (GnRH), its pituitary R GnRHR, the twopituitary gonadotrophins, follicle-stimulating hormone (FSH),luteinising hormone (LH) and their cognate gonadal R, follicle-stimulating hormone receptor (FSHR) and LHCGR respectively.Although numerous physiological and pathophysiological stud-ies have delineated the functions of GnRH, FSH, LH and theirR in great detail, we have recently learned many interesting andimportant new details about their functions from human muta-tions and genetically modified mice [1,2]. Human mutations ofnearly all hormones and R of the HPG axis have been described,and knockout (KO) and transgenic animal models have beengenerated for the respective genes. Mutations in genes encodinghormones invariably lead to their inactivation, whereas those inR genes can either inactivate the R protein or lead into its consti-tutive ligand-independent activation, both of them evoking theirspecific phenotypes.
Our laboratory has contributed to some of the geneticallymodified animal models of aberrant gonadotrophin function,including the LHCGR KO [3], a constitutively activatingmutation of FSHR [4], constitutive overexpression of α- andβ-subunits of hCG [5,6], and a mouse model demonstrat-ing homo-di/oligomeric LHCGR activation through functionalcomplementation [7].
2. Tumorigenesis in human choriongonadotrophinoverexpressing transgenic mice
To find out phenotypic effects of enhanced LH/hCG action,we produced two TG mouse models, one expressing under thehuman ubiquitin C promoter the human glycoprotein hormonecommonα-subunit (Cα) minigene [5], the other expressing underthe same promoter cDNA of the hCG β-subunit [6]. The femalehCGβ TG mice had about 40-fold elevation of bioactive LH/CGlevels in serum, due to dimerisation of the TG hCG� producedin the pituitary gland with endogenously produced C� sub-unit. The double-TG mice (Cα/hCGβ) presented ubiquitous TGexpression of both subunits and consequently brought about a1000-fold elevation in circulating bioactive LH/CG levels. The
phenotypes of the two mouse models were rather similar, prob-ably because maximal LH/hCG effect was achieved already inthe single hCGβ-TG mice with the moderate LH/hCG elevation.
It was interesting to assess to what extent we could repli-cate in the mice the phenotypes of human activating mutationsin LHCGR, i.e. early-onset precocious puberty in males and noapparent phenotype in females [2]. Conspicuously, the findingson the TG mice were quite different, with no advancement ofpuberty in males and very strong ovarian and extraovarian phe-notypes in females. The phenotypes of these mice have beenreported in details elsewhere [5,6,8–10].
The female hCGβ-TG mice developed precocious puberty,as monitored by advancement of the age of vaginal opening byabout 7 days. Adult mice developed massive obesity weighingabout twice as much as their non-TG littermates at 4 monthsof age [6]. The mice were infertile with signs of dioestrus andpseudopregnancy in the vaginal epithelium. Ovarian histologyin adult age revealed massive luteinisation with multiple luteo-mas and haemorrhagic cysts, and oocytes were often trappedinside the masses of luteoma tissue, reminding the human syn-drome of luteinised unruptured follicles (LUF). Uterine weightof adult hCGβ-TG mice did not differ from WT controls butshowed signs of strong progesterone action. The ovaries pro-duce peripubertally elevated levels of oestradiol, but later onthey were similar to WT controls, apparently because of thestrong luteinisation and paucity of nonluteinised granulose cells.In adult age, the main ovarian steroid product was progesterone,reaching levels of > 1 �mol/l, and also serum testosterone wasabout 5-fold elevated (about 5 nmol/l).
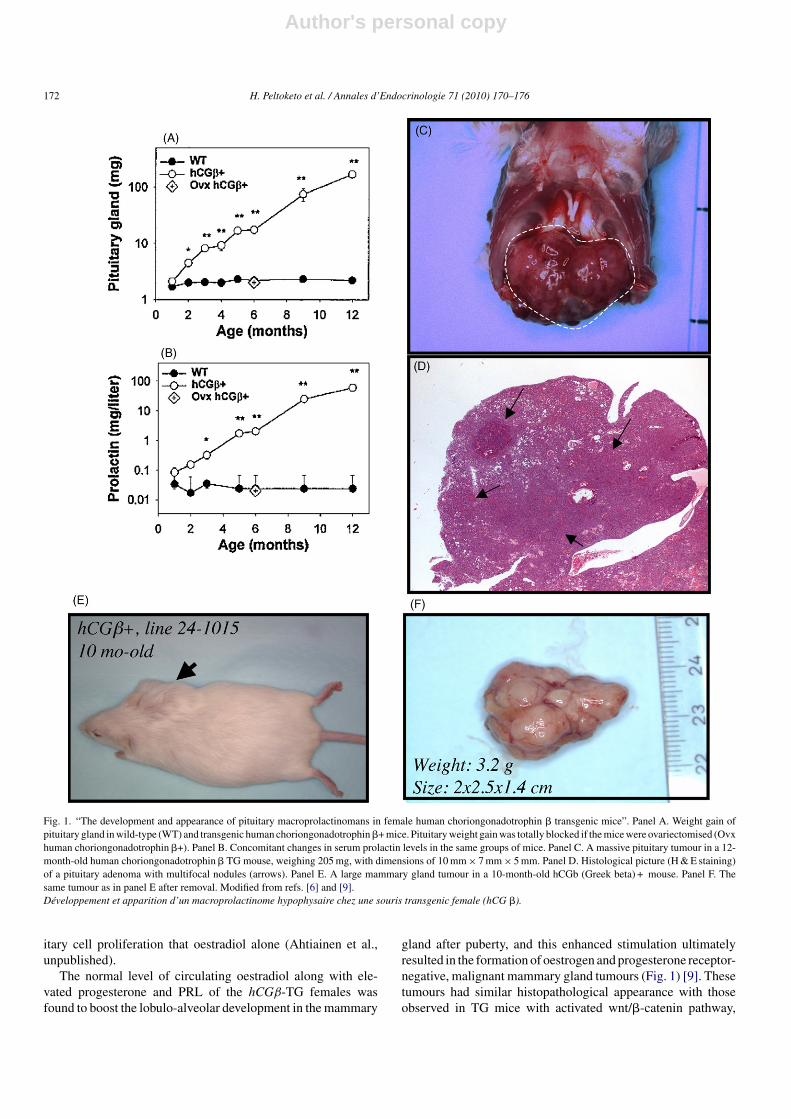
The mice developed gradually after puberty massive PRL-secreting pituitary adenomas (Fig. 1). The finding was surprisingin the absence of increased oestradiol levels, known to pro-mote the formation of prolactinomas. We therefore examinedin greater detail whether progesterone, produced in high con-centrations by ovaries of these mice, could be involved in theprolactinoma formation (Ahtiainen et al., unpublished). Indeed,we found several lines of evidence for direct involvement ofovarian steroidogenesis, most likely the combination of nor-mal oestradiol production and massively elevated progesterone,in the induction of the tumours. This was demonstrated forinstance by the total lack of pituitary tumorigenesis in gonadec-tomised mice despite the continuously elevated LH/hCG levels.Cell culture studies showed that a combination of oestradiolplus progesterone was more effective in stimulating pitu-
Author's personal copy
172 H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176
Fig. 1. “The development and appearance of pituitary macroprolactinomans in female human choriongonadotrophin � transgenic mice”. Panel A. Weight gain ofpituitary gland in wild-type (WT) and transgenic human choriongonadotrophin �+ mice. Pituitary weight gain was totally blocked if the mice were ovariectomised (Ovxhuman choriongonadotrophin �+). Panel B. Concomitant changes in serum prolactin levels in the same groups of mice. Panel C. A massive pituitary tumour in a 12-month-old human choriongonadotrophin � TG mouse, weighing 205 mg, with dimensions of 10 mm × 7 mm × 5 mm. Panel D. Histological picture (H & E staining)of a pituitary adenoma with multifocal nodules (arrows). Panel E. A large mammary gland tumour in a 10-month-old hCGb (Greek beta) + mouse. Panel F. Thesame tumour as in panel E after removal. Modified from refs. [6] and [9].Développement et apparition d’un macroprolactinome hypophysaire chez une souris transgenic female (hCG �).
itary cell proliferation that oestradiol alone (Ahtiainen et al.,unpublished).
The normal level of circulating oestradiol along with ele-vated progesterone and PRL of the hCGβ-TG females wasfound to boost the lobulo-alveolar development in the mammary
gland after puberty, and this enhanced stimulation ultimatelyresulted in the formation of oestrogen and progesterone receptor-negative, malignant mammary gland tumours (Fig. 1) [9]. Thesetumours had similar histopathological appearance with thoseobserved in TG mice with activated wnt/�-catenin pathway,

Author's personal copy
H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176 173
showing increased expression of β-catenin [11], in compli-ance with human breast tumours. Transdifferentiation wasalso observed in the TG mammary tumours, accompanied byabnormal expression of the Wnt genes in the tumorous andnontumorous mammary gland tissue. Specifically, increasedexpression of Wnt5b and up-regulation of Wnt7b and −5b werefound in the hCGβ-TG mammary glands at the age of 3 months.These responses of the wnt cascade appeared to occur indepen-dently of the changes in ovarian steroidogenesis. Thus, our TGmodel revealed a novel link between enhanced hCG action andupregulation of wnt signaling in the mammary gland, resulting in�-catenin-stabilizing mammary tumorigenesis. The novel find-ing of hCG up-regulating wnt7b and wnt5b could contribute topregnancy-induced breast cancer in humans.
3. Ovarian dysfunction associated with enhancedfollicle-stimulating hormone action
We and others have generated several genetically modifiedmouse models to phenocopy consequences of enhanced FSH
action, by expressing exogenous FSH or constitutively activeFSHR in mice (Table 1). Typically, increased FSH level and basalR activity lead to biphasic effects on ovarian function. Modestincrease in FSH concentration [12] and expression of a consti-tutively active mFshrD580H [4] bring about improved fecundityin the form of increased litter size, while more progressiveand stronger influences eventually lead to infertility [4,12,13].The ovaries of the former mice, characteristically, contain morecorpora lutea (CL) than their WT littermates, demonstratingincreased recruitment of growing follicles and ovulation. ThemFshrD580H expressing ovaries present with significantly higherproportion of Ki67-positive small follicles than those in WTanimals, indicating that the transgene expression increases pro-liferation of granulosa cells and, thus, growth of primary follicles[4]. In addition to increasing the proliferation of granulosacells, FSH can decrease apoptosis of granulosa cells by inter-acting with other signaling pathways such as Activin/Smad3[14] and P13K/Akt [15], and the excessive hormone/R activa-tion may therefore also enhance the number of surviving largefollicles.
Table 1Gain-of-function mouse models for follicle-stimulating hormone pathway.
Model Abbreviationa Description Female fertility Major ovarianabnormalities
Reference
TG (FSH�) hFSHβ TG line with FSHβ minigene Fertile No obvious abnormalities [26]
TG (Mt1-CGAMt1-FSH�)Low serumconcentration
MT-hFSH Cross-breeds of Mt1promoter-CGA and Mt1promoter – FSHβ TG lines
Fertile No obvious abnormalities [13]
TG (Mt1-CgaMt1-FSH�)High serumconcentration
MT-hFSH Cross-breeds ofMt1-promoter-CGA andMt1-promoter – FSHβ TGlines
Infertile Haemorrhagic cysts,elevated E2 and Pproduction, disruptedoestrous cycle, no CL
[13]
TG (rInsl2-CGA-rIns2-FSH�)< 22 w
TG-FSH TG line with Ins2-promoter-CGA-Ins2-promoter – FSHβ
Fertile, increased littersize
Increased in size and totalCL count
[12]
TG (rInsl2-CGA;rIns2-FSH�) > 23 w
TG-FSH TG line with Ins2-promoter-CGA-Ins2-promoter – FSHβ
Premature infertility Increased in size and totalCL count
[12]
TG (rAbp-FSHR), TG(rAbp-FSHRD567G),Hpg-TG(rAbp-FSHRD567G)
TG-FSHRwt,TG-FSHR*D567G
TG lines withAbp-promoter-FSHR andAbp-promoter-FSHRD567Ginwt and hpg background
Fertile No obvious abnormalities [27,28]
FVB/N-TG(AMH-FSHR)
mFshr-WT TG line withAMH-promoter-Fshr
Fertile No obvious abnormalities [4]
FVB/N-TG(AMH-FSHRD580H):low expression, strongCAM
mFshrD580H TG line withAMH-promoter-FshrD580H
Fertile, increased littersize
No obvious abnormalities [4]
FVB/N-TG(AMH-FSHRD580H)High expression, strongCAM
mFshrD580H TG line withAMH-promoter-FshrD580H
Infertile Haemorrhagic, disruptedoestrous cycle, mildlyelevated E2 production,accelerated loss offollicles, occasionalteratomas and LUFs
[4]
FVB/N-Tg(AMH-FSHRD580Y)Moderate CAM
mFshrD580Y TG line withAMH-promoter-FshrD580Y
Fertile Haemorrhagic [4]
B6; 129-FshrD580Y
Moderate CAMmFshrD580Y-KI KI line with FshrD580Y Fertile Haemorrhagic [4]
CAM: constitutively active mutant; E2: estradiol; P: progesterone; CL: corpora lutea; LUF: luteinised unovulated follicle; w: week; KI: knockin; TG: transgenic.a Abbreviation of the mouse line as used in the original article.

Author's personal copy
174 H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176
Robust activation of the FSH signalling, on the other hand,has grave consequences on function of the ovary, leading eventu-ally to infertility. The ultimate example is the methallothioneinpromoter-drive MT-hFSH� mouse line producing supraphysi-ological levels of serum FSH [13]. The females are infertilewith disrupted estrous cycle, presenting with severely hemor-rhagic and cystic ovaries, and missing late-stage follicles andCL. Less vigorous FSH action, though, can also harm ovar-ian function; even heterozygous KI mice for a milder form ofconstitutively activated FSHR, mFshrD580Y, develop multiplehemorrhagic ovarian follicles (Table 1) [4]. Furthermore, theprogressively rising serum FSH concentration in the MT-hFSH�mice leads to premature infertility that may be due to accel-erated loss of small follicles and recruitment of poor-qualityfollicles, leading to embryo-fetal resorption [12]. Finally, theTG-mFshrD580H, expressed at similar level to the endogenousFshr in WT littermates, causes several structural and functionalabnormalities in ovaries with increasing severity upon aging [4].Of note, FSHRD580H is active without ligand stimulation, andthus, the affected mice escape the effects of negative feedbackregulation of the pituitary.
Characteristically, the most affected TG-mFshrD580H femalesare infertile, but despite disturbed oestrus cycles, follicles fromthe primordial to antral stage, as well as corpora lutea, are presentin their ovaries [4]. This suggests on-going follicular matu-ration up to ovulation and luteinisation, but occasional LUFsare perceived indicating unsuccessful ovulations. Simultane-ously, the production of oestradiol is modestly but significantlyincreased, while the concentration of serum FSH is suppressed.The hemorrhagic follicles with red blood cells leaking fromthe theca cell layer and infiltrating the granulosa cell layer arealso common in all our models with constitutively active FSHR(Table 1) [4]. Hence, the recruitment of poor-quality folliclesand/or angiogenetic effects of the constitutively active R resem-ble observations made in patients with ovarian hyperstimulationsyndrome (OHSS).
In addition to the pseudopregnancy-type acyclicity and occa-sional unsuccessful ovulations of the TG-mFshrD580H females,their fertility is compromised by the accelerated loss of smallfollicles, primordial and primary in particular. The folliclesshow increased granulosa cell proliferation, and thus, enhancedrecruitment of them to the grow pathway [4]. Moreover, a sub-group of the mFshrD580H mice develop teratomas associatedwith almost complete lack of follicles in the affected ovary.Most of the tumours observed are mature benign cysts, but infew cases, foci of immature tissue suggestive of teratocarci-noma or chorioncarcinoma have been detected. In addition tothe mFshrD580H females, TG-hCG��+ mice develop ovarianteratomas (Rulli et al., unpublished). Thus, high gonadotrophinstimulation of granulosa cells may interfere with oocyte matura-tion and subsequently induce parthenogenetic oocyte activation.
4. Functional complementation in vivo of binding andsignalling deficient LHCGR mutants
Hormone (FSH, LH or hCG) binding to gonadotrophinreceptors results in the activation of intracellular signalling cas-
cades via G-protein coupling to specific intracellular regionsof the R. The cyclic AMP/protein kinase A cascade is themost important second messenger in the activation of LHCGRand FSHR. Gonadotrophin receptors belong to the large familyof G protein-coupled receptors (GPCR), which are responsi-ble for transmitting signals to all senses (vision, odour, touch),neurotransmitters and a number of hormones. All GPCRs con-stitute structurally from a seven transmembrane core, which isresponsible for signal transmission [16]. A large extracellularligand-binding domain (about half of size of the R molecule) isa typical feature of the glycoprotein hormone (LH/CG, FSH andTSH) R [2].
Ligand-induced activation is the norm for almost all GPCRswith a few exceptions (see below) and a few orphan R for whichno ligand has yet been identified. Whether the GPCRs func-tion as a single-hormone/single R monomer or as R complexes(dimer/oligomer), especially in the case of the large class A ofGPCRs, has been a highly debated topic. Although the phe-nomenon of GPCR di/oligomerisation in cell line experimentsin vitro has been known for over 30 years, its importance hasonly been demonstrated recently on obligatory heterodimericGPCRs such as GABAB, taste (T1R1-3) metabotropic gluta-mate (mGluR) and calcium-sensing R, where only heterodimersare involved in signal transduction [17,18]. The main ques-tions, however, have remained unanswered, i.e. whether the Rare activated in physiological conditions in vivo as monomersor di/oligomers, and whether the two possible modes activatesimilar or dissimilar signal transduction cascades?
In order to assess the significance of LHCGR functionas homodimers in vivo, we took advantage of the comple-mentary molecular models of intermolecular cooperation ofgonadotrophin receptors, shown to be functional in vitro by thegroup of Ji [19–21]. Using LHCGR complementary mutants(one deficient of ligand binding and the other one of signalling),it was possible to partially rescue the cAMP response to LH/hCGstimulation in cell cultures, when both mutants were coexpressedin the same cell, while the mutants were completely inactivewhen expressed alone.
Using the same principle, similar mutations were introducedto bacterial artificial chromosome (BAC) clones that contain theentire mouse genomic LHCGR gene, including the regulatoryregions (promoter, introns, and UTRs) to assure correct spatio-temporal expression in a physiologically meaningful fashion.Two TG mouse lines were created using the modified BACs [7].As expected, the TG mice expressed LHCGR mutants mainlyin gonads, as their WT counterparts, and each line harbour-ing either a binding-deficient LHCGR or a signalling-deficientLHCGR were crossed with the previously produced LHCGR KO(LuRKO) mice [3], to obtain mice without endogenous LHCGRbut expressing either one of the inactive LHCGR mutants. Thesingle LHCGR mutant/LuRKO mice, as LuRKO mice with theLHCGR deletion alone, were hypogonadal and infertile, withdefective postnatal development of gonads and genital struc-tures, due to totally missing LH-stimulated gonadal sex steroidproduction [3]. However, when both mutant R were coexpressedin LuRKO mice, gonadal development and spermatogenesiswere completely rescued [7], as demonstrated by their normal

Author's personal copy
H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176 175
Fig. 2. Gonadal and genital phenotypes of (A) LHCGR knockout (luteinizing hormone receptor knockout [LuRKO]) mice. B. Transgenic mice expressing a ligandbinding-deficient mutant of LHCGR in the LuRKO background. C. Transgenic mice expressing a signalling-deficient mutant of LHCGR in the LuRKO background.D. Transgenic mice coexpressing the ligand-binding and signalling deficient LHCGR mutants in the LuRKO background. E. WT controls. Modified from ref. [7].Phénotypes gonadiques et génitaux de (A) souris LHCGR knock-out (luteinizing hormone receptor knockout [LuRKO]). B. Souris transgéniques exprimant un mutantde LHCGR déficitaire dans le domaine de liaison sur un fond génétique (LuRKO). C. Souris transgénique exprimant un mutant LHCGR déficitaire dans le domaine designalisation sur un fond génétique LuRKO. D. Souris transgénique coexprimant un mutant LHCGR déficitaire à la fois dans le domaine de liaison et de signalisationsur un fond génétique LuRKO. E. Témoins normaux (d’après la référence 7).
size of testes and extragonadal sex organs (Fig. 2) and normalfertility. The extent of the rescuing could not only be observedat macroscopic level but also at molecular level, where LH-dependent gene expression was nearly fully recovered in doubleTG but not in single TG mice [7]. The only explanation of thisfinding is that the two differently inactivated R molecules, whencoexpressed in the same cells, can form functional dimers whereone defective molecule capable of hormone binding can activateanother R mutant that is only able to transmit the signal. To ourknowledge, this is the first demonstration of functional relevanceof GPCR homodimerisation in physiological conditions.
5. Conclusions
The ability to modify large DNA molecules [22,23], to manip-ulate the genome [24] and to create genetically modified animalmodels, provides extremely valuable tools for the characterisa-tion of complex biological phenomena at the physiological level.Indeed, almost 130 years ago Charles R. Darwin envisaged thatthe understanding of these processes requires experimental invivo models: “. . . physiology cannot possibly progress exceptby means of experiments on living animals. . .” [25].
Increased LH/hCG activity in female mice has serious func-tional consequences, including ovarian hyperstimulation withluteinisation and massive overproduction of progesterone, in theface of normal oestrogen production. The ovarian stimulationhas multiple indirect extragonadal consequences, including the
development of pituitary prolactinomas and malignant mam-mary gland tumours. If extrapolated to humans, and takenthe importance of endogenous progesterone and syntheticprogestins in female reproductive functions and their pharma-cotherapy, it is relevant to revisit the potential role of thesehormones in the origin and growth of human prolactinomas andmammary tumours.
Overproduction of FSH and gain-of-function mutations ofthe Fshr have several functional consequences in female mice.While modest increase in FSH action temporarily boosts thefecundity of the mice, more vigorous actions lead to an arrayof abnormalities in ovarian function. A similar array of pheno-types, such as OHSS, premature ovarian failure, formation ofhemorrhagic cysts, abnormal hormone balance of the pituitary-gonadal axis and teratomas, may also be possible in humansexpressing constitutively activating mutations of FSHR, not yetdescribed.
The molecules of gonadotrophin receptors have been demon-strated to be much more flexible than previously thought, beingable to generate multiple interactions with G proteins, resultingin multiple signalling cascades, and possibly even with neigh-bouring R through homo- and hetero-di/oligomerization. Thereasons for such flexibility might involve the previously men-tioned functions (transport, coupling, cooperativity, etc), butalso signal amplification at the membrane level maintaining Rspecificity and, in the case of hetero-di/oligomerisation, phar-macological diversity. Yet, the most important finding of our

Author's personal copy
176 H. Peltoketo et al. / Annales d’Endocrinologie 71 (2010) 170–176
study was that intermolecular cooperation was able to rescueLHCGR function in the absence of WT R, suggesting that theseinteractions may also occur in physiological conditions in livingorganisms. We expect that also other GPCRs will show similarmolecular cooperation, either to generate biased signals or thefull response.
Conflicts of interest
None.
References
[1] Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility path-ways. Nature Cell Biol 2002;4 Suppl:s41–9.
[2] Themmen APN, Huhtaniemi IT. Mutations of gonadotropins andgonadotropin receptors: elucidating the physiology and pathophysiologyof pituitary-gonadal function. Endocr Rev 2000;21:551–83.
[3] Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal butarrested postnatal sexual development of luteinizing hormone receptorknockout (LuRKO) mice. Mol Endocrinol 2001;15:172–83.
[4] Peltoketo H, Strauss L, Karjalainen R, Xhang M, Stamp G, Segaloff D,et al. Female mice expressing constitutively active mutants of FSH recep-tor with a phenotype of premature follicle depletion and estrogen access.Endocrinology 2010 [Epub ahead of print].
[5] Rulli SB, Ahtiainen P, Makela S, Toppari J, Poutanen M, HuhtaniemiI. Elevated steroidogenesis, defective reproductive organs, and infertilityin transgenic male mice overexpressing human chorionic gonadotropin.Endocrinology 2003;144:4980–90.
[6] Rulli SB, Kuorelahti A, Karaer O, Pelliniemi LJ, Poutanen M, HuhtaniemiI. Reproductive disturbances, pituitary lactotrope adenomas, and mammarygland tumors in transgenic female mice producing high levels of humanchorionic gonadotropin. Endocrinology 2002;143:4084–95.
[7] Rivero-Müller A, Chou Y-Y, Ji I, Lajic S, Hanyaloglu AC, Jonas K,et al. Rescue of defective G-protein-coupled receptor function in vivoby intermolecular co-operation. Proc Natl Acad Sci U S A 2010;107:2319–24.
[8] Ahtiainen P, Rulli SB, Shariatmadari R, Pelliniemi LJ, Toppari J, Pouta-nen M, et al. Fetal but not adult Leydig cells are susceptible to adenomaformation in response to persistently high hCG level: a study on hCGoverexpressing transgenic mice. Oncogene 2005;24:7301–9.
[9] Kuorelahti A, Rulli S, Huhtaniemi I, Poutanen M. Human chorionicgonadotropin (hCG) up-regulates wnt5b and wnt7b in the mammarygland, and hCGbeta transgenic female mice present with mammary Glandtumors exhibiting characteristics of the Wnt/beta-catenin pathway activa-tion. Endocrinology 2007;148:3694–703.
[10] Yarram SJ, Perry MJ, Christopher TJ, Westby K, Brown NL, Lammi-nen T, et al. Luteinizing hormone receptor knockout (LuRKO) miceand transgenic human chorionic gonadotropin (hCG)-overexpressingmice (hCG alphabeta+) have bone phenotypes. Endocrinology 2003;144:3555–64.
[11] Miyoshi K, Rosner A, Nozawa M, Byrd C, Morgan F, Landesman-BollagE, et al. Activation of different Wnt/beta-catenin signaling componentsin mammary epithelium induces transdifferentiation and the formation ofpilar tumors. Oncogene 2002;21(36):5548–56.
[12] McTavish KJ, Jimenez M, Walters KA, Spaliviero J, Groome NP, ThemmenAP, et al. Rising follicle-stimulating hormone levels with age acceleratefemale reproductive failure. Endocrinology 2007;148:4432–9.
[13] Kumar TR, Palapattu G, Wang P, Woodruff TK, Boime I, Byrne MC, etal. Transgenic models to study gonadotropin function: the role of follicle-stimulating hormone in gonadal growth and tumorigenesis. Mol Endocrinol1999;13:851–65.
[14] Tomic D, Miller KP, Kenny HA, Woodruff TK, Hoyer P, Flaws JA. Ovarianfollicle development requires Smad3. Mol Endocrinol 2004;18:2224–40.
[15] Hunzicker-Dunn M, Maizels ET. FSH signalling pathways in immaturegranulosa cells that regulate target gene expression: branching out fromprotein kinase A. Cell Signal 2006;18:1351–9.
[16] Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, etal. The G protein-coupled receptor repertoires of human and mouse. ProcNatl Acad Sci U S A 2003;100:4903–8.
[17] Pin JP, Kniazeff J, Binet V, Liu J, Maurel D, Galvez T, et al. Activationmechanism of the heterodimeric GABA(B) receptor. Biochem Pharmacol2004;68:1565–72.
[18] Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al.GABA(B)-receptor subtypes assemble into functional heteromeric com-plexes. Nature 1998;396:683–7.
[19] Lee C, Ji IJ, Ji TH. Use of defined-function mutants to access receptor-receptor interactions. Methods 2002;27:318–23.
[20] Lee C, Ji I, Ryu K, Song Y, Conn PM, Ji TH. Two defective heterozygousluteinizing hormone receptors can rescue hormone action. J Biol Chemi2002;277:15795–800.
[21] Ji I, Lee C, Jeoung M, Koo Y, Sievert GA, Ji TH. Trans-activation ofmutant follicle-stimulating hormone receptors selectively generates onlyone of two hormone signals. Mol Endocrinol 2004;18:968–78.
[22] Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic forDNA engineering using recombination in Escherichia coli. Nature Genet1998;20:123–8.
[23] Rivero-Muller A, Lajic S, Huhtaniemi I. Assisted large fragment insertionby Red/ET-recombination (ALFIRE) – an alternative and enhanced methodfor large fragment recombineering. Nucl Acid Res 2007;35:e78.
[24] Thomas KR, Folger KR, Capecchi MR. High frequency targeting of genesto specific sites in the mammalian genome. Cell 1986;44:419–28.
[25] Darwin C. Mr. Darwin on vivisection. Nature 1881;23:583.[26] Kumar TR, Fairchild-Huntress V, Low MJ. Gonadotrope-specific expres-
sion of the human follicle-stimulating hormone beta-subunit gene inpituitaries of transgenic mice. Mol Endocrinol 1992;6:81–90.
[27] Haywood M, Tymchenko N, Spaliviero J, Koch A, Jimenez M, GromollJ, et al. An activated human follicle-stimulating hormone (FSH) recep-tor stimulates FSH-like activity in gonadotropin-deficient transgenic mice.Mol Endocrinol 2002;16:2582–91.
[28] Allan CM, Lim P, Robson M, Spaliviero J, Handelsman DJ. Transgenicmutant D567G but not wild-type human FSH receptor overexpression pro-vides FSH-independent and promiscuous glycoprotein hormone Sertoli cellsignaling. Am J Physiol 2009;296:E1022–8.




















