
Connexin40 Messenger Ribonucleic Acid Is Positively Regulated by Thyroid Hormone (TH) Acting in...
31
Connexin40 mRNA is positively regulated by thyroid hormone acting in cardiac atria via the thyroid hormone receptor Norma A. S. Almeida 1 , Aline Cordeiro 1 , Danielle S. Machado 1 , Luana L. Souza 1 , Tânia M. Ortiga-Carvalho 1 , Antonio C. Campos-de-Carvalho 2 , Fredric E. Wondisford 3 , Carmen C. Pazos- Moura 1 . 1- Laboratório de Endocrinologia Molecular and 2- Laboratório de Cardiologia Celular e Molecular - Instituto de Biofísica Carlos Chagas Filho –Federal University of Rio de Janeiro, Rio de Janeiro, Brazil. 3- Metabolism Division, Department of Pediatrics, Medicine and Physiology Johns Hopkins Medical Institutes, Baltimore, USA. Address all correspondence and requests for reprints to: Carmen C Pazos-Moura, Laboratório de Endocrinologia Molecular, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21949-900, Brazil. E-mail: [email protected] Keywords: Connexin40, Connexin43, Atrial conduction, TH receptor, Hypothyroidism, Hyperthyroidism Supported by: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) Disclosure statement: The authors have nothing to disclose. Endocrinology. First published ahead of print September 11, 2008 as doi:10.1210/en.2008-0451 Copyright (C) 2008 by The Endocrine Society
Transcript of Connexin40 Messenger Ribonucleic Acid Is Positively Regulated by Thyroid Hormone (TH) Acting in...

Connexin40 mRNA is positively regulated by thyroid hormone acting in cardiac atria via
the thyroid hormone receptor
Norma A. S. Almeida1, Aline Cordeiro1, Danielle S. Machado1, Luana L. Souza1, Tânia M.
Ortiga-Carvalho1, Antonio C. Campos-de-Carvalho2, Fredric E. Wondisford3, Carmen C. Pazos-
Moura1.
1- Laboratório de Endocrinologia Molecular and 2- Laboratório de Cardiologia Celular e
Molecular - Instituto de Biofísica Carlos Chagas Filho –Federal University of Rio de Janeiro, Rio
de Janeiro, Brazil.
3- Metabolism Division, Department of Pediatrics, Medicine and Physiology Johns Hopkins
Medical Institutes, Baltimore, USA.
Address all correspondence and requests for reprints to:
Carmen C Pazos-Moura, Laboratório de Endocrinologia Molecular, Instituto de Biofísica Carlos
Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro 21949-900, Brazil. E-mail:
Keywords: Connexin40, Connexin43, Atrial conduction, TH receptor, Hypothyroidism,
Hyperthyroidism
Supported by: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq),
Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ),
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)
Disclosure statement: The authors have nothing to disclose.
Endocrinology. First published ahead of print September 11, 2008 as doi:10.1210/en.2008-0451
Copyright (C) 2008 by The Endocrine Society

Abstract
Thyroid hormone (TH) regulates many cardiac genes via nuclear thyroid receptors (TR), and
hyperthyroidism is frequently associated with atrial fibrillation. Electrical activity propagation in
myocardium depends on transfer of current at gap junctions, and Connexins (Cx) 40 and 43 are
the predominant junction proteins. In mice, Cx40, the main Connexin involved in atrial
conduction, is restricted to the atria and fibers of the conduction system, which also express
Cx43. We studied cardiac expression of Cx40 and Cx43 in conjunction with electrocardiogram
studies in mice overexpressing the dominant negative mutant TRβ Δ337T exclusively in
cardiomyocytes (MHC-mt). These mice develop the cardiac hypothyroid phenotype in the
presence of normal serum TH. Expression was also examined in wild-type mice rendered hypo-
or hyperthyroid by pharmacological treatment. Atrial Cx40 mRNA and protein levels were
decreased (85 and 55%, respectively p<0.001) in MHC-mt mice. Atrial and ventricular Cx43
mRNA levels were not significantly changed. Hypo- and hyperthyroid animals showed a 25%
decrease and 40% increase, respectively, in Cx40 mRNA abundance. However, MHC-mt mice
presented very low Cx40 mRNA expression regardless of whether they were made hypo-or
hyperthyroid. Atrial depolarization velocity, as represented by P wave duration in ECGs of
unanesthetized mice, was extremely reduced in MHC-mt mice, and to a lesser extent also in
hypothyroid mice (90% and 30% increase in P wave duration). In contrast, this measure was
increased in hyperthyroid mice (19% decrease in P wave duration). Therefore, this study reveals
for the first time that Connexin40 mRNA is up-regulated by TH acting in cardiac atria via the TH
receptor and that this may be one of the mechanisms contributing to atrial conduction alterations
in thyroid dysfunctions.

Introduction
Thyroid hormones (TH) have important inotropic and chronotropic effects on the heart
via modulation of its contractile and electrical activity. Decreased heart rate has been described in
hypothyroid patients, and sinus tachycardia is a common manifestation of hyperthyroidism that
also may lead to atrial fibrillation (1). TH are able to regulate cardiac excitation and electrical
conduction by changing the expression of ionic channels and transporters (1). Even though some
non-genomic TH effects on ionic currents have been demonstrated (2), most TH actions are
mediated by nuclear TH receptors (TR) that modulate the transcription rate of cardiac genes (1).
HCN2 mRNA in atria and ventricles and HCN4 in ventricles are major components of cardiac
pacemaker currents and are up-regulated by TH (3). The transient outward potassium current, Ito,
which is dependent on transcripts of genes coding for the ion-channel subunits KV 4.2 and KV
4.3, is markedly responsive to TH levels (4,5). Also, KV 1.5 ion channel mRNA, which
contributes to transient outward and delayed rectifier currents, is decreased by hypothyroidism
and increased by hyperthyroidism (5-7). Conversely, L-type calcium channel mRNA is down-
regulated by TH, resulting in lower inward voltage-dependent Ca+2 currents in cardiomyocytes
(5,6).
Most studies of these interactions have been performed in vivo, in situations of hypo- or
hyperthyroidism accompanied by TH extra-cardiac effects that result in hemodynamic changes or
alterations in the activity of other heart regulators, which, by themselves, can modify cardiac gene
expression and function (1). Therefore, in vivo, it is difficult to define the direct or indirect effects
of TH in the heart. We developed a transgenic mouse with cardiac-specific expression of the
mutant TH receptor β (Δ337T) driven by the α-myosin heavy chain (MHC) promoter, which is a
potent and specific cardiomyocyte promoter (8). Δ337T TRβ is a natural mutant described in
thyroid hormone resistance syndrome patients and corresponds to a point mutation in the ligand-
binding domain that renders TRβ incapable of binding T3, while preserving the ability to bind

DNA, form dimers and interact with some co-regulators (9). The Δ337T TRβ mutant exerts a
potent dominant negative effect on the function of normal TRs. The overexpression of Δ337T
TRβ in cardiomyocytes resulted in disruption of normal TR signaling pathways leading to the
hypothyroid cardiac phenotype in the presence of normal TH levels in the circulation (8),
excluding indirect effects due to systemic hypothyroidism. Therefore, these mice are a unique
model to identify direct TR-mediated effects on heart gene regulation.
Several TH receptor targets have been identified in the ventricle (1,3,5); however, less is
known about TH receptor action in atria, in spite of evidence for a very important role for TH,
especially in atrial electrical function as revealed by electrophysiological studies (10). We have
previously reported that mice harboring Δ337T TRβ only in the heart (MHC-mt) exhibited
alterations in their electrocardiograms suggestive of slow electrical conduction velocity during
the depolarization and repolarization phases of the action potential (8). Delayed repolarization
may be explained by the well-known effects of TH on the gene expression of K+ and L-type Ca+2
channels (3,5). Also, many genes involved with depolarization of the sinoatrial and
atrioventricular nodes such as L-type Ca+2 channels and HCN are targets of TH receptor gene
regulation (3,5). However, the longer duration of cardiac depolarization present in contracting
myocytes of MHC-mt mice still remains unexplained, since Na+ channel mRNA, primarily
involved in the depolarization process of these cells, has been shown to be unresponsive to TH
levels (5,11). Myocardial electrical conduction depends on intercellular transfer of current at gap
junctions, and Connexin (Cx) 40 and 43 are the main structural junction proteins in the heart. Six
subunits of Connexin bind noncovalently to form a hemichannel, or connexon. Two connexons,
one from each adjacent cell, form a hydrophilic pore that allows ions to pass from one cell to the
next. Therefore, Connexins are responsible for electrical coupling of cardiac cells (12,13). In the
mouse heart, Cx40 is restricted to the atria and atrial-ventricular conduction system, which also
express Cx43 (14,15). Cx40 and 43 have unique functions as revealed by studies in knockout

mice that demonstrated a major role for Cx40 in electrical coupling in atrium cells, while Cx43
was more important for ventricle conduction (16,17,18). There are a few controversial reports on
regulation of ventricular Cx43 mRNA by TH (5,19,20); however, it is unknown whether TH
regulate atrial Connexin expression. Therefore, considering the importance of TH for electrical
conduction in the heart, we used the MHC-mt mouse model to evaluate direct TR-mediated TH
actions on atrial and ventricular expression of Cx40 and Cx43 in conjunction with
electrocardiogram studies. The same experiments were also conducted in wild-type mice rendered
hypothyroid or hyperthyroid, in order to compare systemic hypo- and hyperthyroidism with the
cardiac selective hypothyroidism present in MHC-mt mice.
Material and Methods
Animal treatment
MHC-mt mice were generated as reported previously (8). Animals were maintained
under light/dark cycles of 12:12 h (lights at 07:00 h), weaned after 21 days, and fed chow and
water ad libitum. Southern blotting was performed to identify transgenic mice containing a
specific 2.4-kb BglII fragment using a human TR-β1 cDNA probe. Experiments were conducted
in adult transgenic mice and their wild-type littermates.
Hypothyroidism was induced by feeding animals a diet containing 0.15% 5-propyl-2-
thiouracil (PTU) for 28 days. Mice were rendered hyperthyroid by daily subcutaneous injections
of T3 (50μg/100g B.W.) for 15 days.
In all experiments, mice were sacrificed by CO2 inhalation, hearts were excised, atria and
ventricles were separated, immediately frozen in liquid nitrogen and stored at -70°C prior to
extraction of total RNA or protein. Serum was obtained from trunk blood and kept frozen at -
20°C for measurements of total T3 and TSH. All procedures were performed in accordance with

the care and use guidelines established by the Fund for the Replacement of Animals in Medical
Experiments Guide, and were approved by our Institutional Committee on Animal Care and Use.
Hormone Assays
Serum T3 concentrations were determined using a coated tube radioimmunoassay (RIA)
from ICN Pharmaceuticals (Costa Mesa, CA, USA) according to the manufacturer’s instructions.
Serum TSH was measured in 100 μL serum samples in duplicate determination by a specific
mouse TSH RIA using reagents acquired from the National Hormone and Peptide Program
(Torrance, CA), as detailed earlier (21). The minimum assay detection limit was 30ng/mL and
intra-assay variation was 6%.
Electrocardiogram recording
Mice were anesthetized (ketamine/xilazine 50/5 mg per kg, i.p.) and two steel electrodes
were implanted subcutaneously near the apex and base of the heart. The electrodes were
exteriorized through the back of the neck and terminated in a custom-made connector. Data
acquisition was performed two days after the surgical procedure, and was conducted while
unanesthetized and unrestricted animals rested quietly in individual cages (22). Records were
always started 20 minutes after linking the connector, through flexible cables, to the data
acquisition system (3A9 amplifier, Tektronix/TL-2 A/D Interface, Axon Instruments).
Electrocardiogram (ECG) recordings were obtained from each animal for 15 minutes and all
recording was performed during the same period of the day.
RNA extraction and semi-quantitative RT-PCR
Total RNA was extracted using TRIZOL™ reagent (Invitrogen), treated with RQ1
RNase-free DNase (Promega). First-strand cDNA was synthesized from 100 ng of total atrium

RNA or 200 ng of total ventricle RNA, using 50 U of reverse transcriptase (Superscript II RT;
Invitrogen). Two μL of reverse transcription mix was used in each PCR reaction. The PCR
conditions consisted of 25 cycles for Cx40 in atrium, 26 cycles for Cx43 in atrium, and 21 cycles
for Cx43 in ventricles, and each amplification was performed using the following parameters:
94°C for 60 s, 58°C for 60 s, and 72°C for 90 s, using 0.2 U of Taq DNA polymerase (Biotools).
The sequences of the forward and reverse primers were, respectively:
5’GCACACTGTGCGCATGCAGG3’ and 5’CTGCTGGCCTTACTAAGGCG3’ for Cx40,
5’ATCCAGTGGTACATCTATGG3’ and 5’CTGCTGGCTCTGCTGGAAGG3’ for Cx43 and
5’CCCCAATGTGTCCGTCGTGGAT3’ and 5’TGGAGGCCATGTAGGCCATGAGG3’ for
GAPDH, the internal control that was run within the same tube containing the primers for each
Connexin. Bands were resolved in a 1.5% agarose gel, and densitometric analysis was performed
by using Kodak Digital Science 1D 3.5. Densitometric values of the PCR product for each
Connexin were normalized to the GAPDH band.
Ribonuclease Protection Assay
α-MHC and β-MHC gene expression was evaluated by RPA with the use of specific 32P-
labeled 3’ β-MHC riboprobe, as previously described (8). This probe was obtained by reverse
transcription-polymerase chain reaction of mouse heart mRNA with the use of primers targeted to
the 3’ coding sequence and untranslated region of β-MHC. Because α-MHC and β-MHC
mRNAs are identical in their 3´coding sequence and because the riboprobes diverge only in the
3´-untranslated sequence, both isoforms of MHC mRNA were detected by the riboprobe. A
cyclophilin riboprobe was used to control for RNA quality and quantity. Forty micrograms of
total ventricular RNA was hybridized with these probes for 16 h at 42°C. The RNA-probe
mixture was then digested with RNase A (250 U/μL)/ T1 (1000 U/μL) at 37°C for 30 minutes.
Analysis of protected fragments was made in 5% polyacrylamide/8 M urea gels, which were

dried on filter paper and exposed to Kodak XAR-5 film in a cassette with an intensifying screen
at -70°C. The mRNA levels were normalized to the levels of cyclophilin mRNA.
Western blotting analysis
Atrium and ventricle tissues were excised and homogenized in an ultra-turrax T25 basic
(IKA®), in lyses buffer [50 mM Hepes, 1 mM MgCl2, 10 mM EDTA and 1% Triton X with the
protease inhibitor cocktail Complete (Roche®)], pH 6.4. Thirty micrograms of total protein per
sample was resolved by SDS-PAGE on a 12.5% gel and transferred onto a polyvinylidene
difluoride (PVDF) membrane (Westran®). Membrane was blocked with 5% nonfat dry milk
(Molico®) and incubated overnight at room temperature with anti-Cx40 antibody (Zymed® -
1:125 dilution) or anti-Cx43 (Zymed® - 1:1000 dilution), and with the internal control anti-
cyclophilin B antibody (Affinity Bioreagents® - 1:3000 dilution). Membranes were then washed
and incubated with peroxidase labeled anti-rabbit IgG antibody (Amersham® - 1:10000 dilution)
for 3 h at room temperature. All blots were then washed and incubated with a luminogen
detection reagent (ECL - Amersham®) to further exposure on autoradiograph film (Kodak®). The
protein bands were evaluated by densitometry using the software Kodak 1D 3.5. The membranes
were stained with Rouge Ponceau to evaluate the relative amounts of transferred proteins.
Statistical analysis
Data are reported as mean ± SEM. Student’s t test or one-way analysis of variance
followed by Student-Newman-Keuls multiple comparisons test was used for comparisons
between different groups (GraphPad Prism, GraphPad Software). Differences were considered to
be significant at p< 0.05.

Results
Expression of Connexin40 and 43 in Cardiac Atria of MHC-mt mice
Cardiac-specific overexpression of the mutant TRβ Δ337T, a dominant negative mutant
incapable of binding T3, resulted in specific and important reduction in atrial Connexin40
expression. As depicted in Figure 1A, in MHC-mt mice atria, Cx40 mRNA abundance was only
15% of that present in wild-type mice (p< 0.001). Semi-quantitative immunoblotting was
performed and confirmed very low protein expression of Cx40 in the atria of MHC-mt mice.
Atrial Cx40 protein was reduced by approximately 55% (p< 0.001). In addition, the weight of the
atria from MHC-mt mice was increased by approximately 56% compared to wild-type littermates
(0.51 ± 0.02 vs. 0.32 ± 0.01 mg/g body weight).
Expression of Connexin 43 in Cardiac Atria and Ventricles of MHC-mt mice
Atrial Cx43 mRNA abundance in MHC-mt mice was similar to wild-type mice (Figure
2A), while the protein content of Cx43 was slightly reduced in MHC-mt mice (19%, p< 0.05)
(Figure 2C).
Similar to the atria, ventricular Cx43 mRNA abundance (Figure 3A and B) as well as
protein content (Figure 3C and D) was not altered in MHC-mt mice, even though those mice have
a pattern of α-and β-MHC mRNA expression consistent with a hypothyroid phenotype in the
heart (8): an increase in expression of β MHC mRNA (approximately 10 times) and decrease in α
MHC mRNA (to approximately half the values of wt) (Figure 3E, F).

Hypothyroidism and hyperthyroidism in wild-type mice regulate Connexin40 mRNA
expression
Since there is no information available on whether systemic hypo- and hyperthyroidism
may change Cx40 mRNA expression, we studied expression in pharmacologically-induced hypo-
and hyperthyroid wild-type mice.
As expected, treatment with the antithyroid drug PTU resulted in low serum T3 and highly
elevated serum TSH (> 500 ng/mL). T3-treated animals (hyperthyroid group) exhibited
suppressed TSH (< 30 ng/mL) and a 3-fold increase in serum T3 (90 ± 10 vs. 341± 18 ng/mL).
Hypothyroid wild-type mice exhibited an approximately 25% reduction in atrial Cx40 mRNA
levels (p< 0.05 vs. euthyroid controls), while hyperthyroid wild-type mice showed a 40% increase
(p< 0.05 vs. euthyroid controls) (Figure 4, A and B). However, at the level of protein expression,
there were no significant changes in Cx40 (Figure 4, C and D).
MHC-mt atrial Cx40 expression does not respond to induction of systemic hypo-or
hyperthyroidism
Atrial Cx40 mRNA expression of MHC-mt mice was studied in hypo-and hyperthyroid
situations in order to evaluate the response to TH. As shown in Figure 5, atrial Cx40 mRNA of
MHC-mt mice was significantly reduced independent of the thyroid state of the animals. Both
MHC-mt mice rendered hypothyroid by PTU treatment (TSH > 500 ng/mL) or hyperthyroid by
T3 injections (serum T3: 102 ± 18 vs. 388 ± 23 ng/mL) showed a similar degree of reduction in
expression of Cx40 mRNA comparable to that observed in euthyroid MHC-mt mice. Therefore,
the disruption of TR signaling pathways caused by mutant TR rendered the animals resistant to
the effects of TH on Cx40 mRNA regulation.

Prolonged atrial conduction in MHC-mt mice
In order to evaluate surface electrical conduction, electrocardiograms (ECG) were
performed in conscious mice (Figure 6). By avoiding the use of anesthesia, the cardiac autonomic
tonus was preserved. As depicted in Table 1, MHC-mt mice exhibited reduced heart rate
(approximately 30%), a widened P wave (90% higher duration than WT) and prolonged PR
interval (18%). The P wave represents atrial depolarization, while the PR interval, measured from
the onset of atrial depolarization (P) to the onset of ventricular depolarization (Q), is used as a
rough approximation of atrial-ventricular conduction time. Therefore, MHC-mt mice have very
prolonged atrial conduction.
The QRS complex duration showed no statistical differences in relation to wild-type
littermates, reflecting a normal velocity of ventricular depolarization. However, the QT interval
was significantly increased (80% longer), and the T wave duration was more than doubled,
indicating a slower ventricular repolarization, which is in agreement with the cardiac hypothyroid
phenotype.
The same pattern of ECG alterations was observed in MHC-mt mice irrespective of
whether the mice had been made hypo- or hyperthyroid, confirming their resistance to TH action
in the heart (Table 1).
Similar to Cx40 mRNA expression, ECG measurements of hypothyroid mice showed
qualitative changes similar to those of MHC-mt mice, although there are quantitative differences
(Table 2). Compared to euthyroid wild-types, hypothyroid mice had an increased duration of P
wave, PR interval (approximately 30% and 25%, respectively), T wave and QT interval (around
45% and 33%, respectively) (p< 0.05). In contrast, wild-type hyperthyroid mice presented
decreased P wave durations and PR intervals (approximately by 19% and 22%, respectively) and
decreased T wave durations and QT intervals (approximately by 19% and 9.5%, respectively, p<
0.05). The heart rate was not significantly altered in hypothyroid mice but was increased in
hyperthyroid animals. Interestingly, bradycardia was present in the same hypothyroid mice under

anesthesia (WT: 323 ± 14; WT+PTU: 234 ± 10; WT+ T3: 531 ± 17 bpm, p< 0.05). QRS showed
no statistically significant differences between groups.
Because our previous study had shown that all ECG intervals, including the QRS
complex, were significantly increased in MHC-mt mice, and those experiments were conducted
in anesthetized mice, we performed ECG measurements in the same mice with or without
anesthesia. As depicted in Table 3, anesthesia changed the values of all ECG parameters in wild-
type and MHC-mt mice; however, the differences between wild-type and MHC-mt were similar
in conscious and anesthetized mice except for the QRS complex, which was significantly
prolonged only when ECG was performed in MHC-mt mice under anesthesia.
Discussion
The results of the present study strongly suggest, for the first time, that atrial Connexin40
mRNA expression is up-regulated by TH acting in the heart through the TH receptor (TR).
Both α1- and β1 TH receptors (TR) are present in adult mouse heart; however, TRα 1 is
more abundant (23). In the atria, both TR were identified in working myocardium cells and in
specialized cells of the conduction system, including the sinus node. In the ventricles, TR β1 has
been shown to be restricted to the peripheral conduction system, while TR α1 was detected in
myocardium cells and the peripheral conduction system (24).
According to current concepts, TR binds to DNA in the presence and absence of ligand,
showing ligand-dependent and independent activation. In positive regulation in the absence of
ligand binding, TR remains bound to co-repressors that reduce gene transcription below the basal
level; binding of T3 to receptor relieves this repression and causes additional activation by
recruiting co-activators. However, the Δ337T mutation causes TR β to be unable to bind T3, and
gene repression occurs. In addition, this mutant has been shown to have a strong dominant

negative effect on normal receptor function (25). In contrast to patients with TH resistance
syndrome who express the mutant TRβ from the endogenous locus, in our mouse model, the
mutant TRβ expression was driven by a highly active promoter in the heart, the α-MHC.
Therefore, a greater amount of mutant TR was expressed in cardiomyocytes than normal TR,
resulting in a strong negative effect on the normal functions of both TR α and β.
We had previously demonstrated that hearts of MHC-mt mice developed a hypothyroid
phenotype at the level of both gene expression and function (8,26), even in the presence of
normal circulating concentrations of TH. These animals presented important contractile systolic
and diastolic dysfunction observed in isolated hearts (8), although in vivo, they exhibited normal
cardiac performance (8, 26), in a clear adaptation to the intrinsic cardiac impairment. At the gene
level, a reduced ratio of α to -βMHC mRNA (due mainly to a massive increase in βMHC
mRNA) is consistent with cardiac hypothyroidism, and the mice showed no response to treatment
with TH, revealing a resistance to TH action in the heart (8, 26). In the present paper, the same
pattern of MHC isoform expression was observed (Figure 3, F), indicating that the level of
transgene expression was similar to that observed in our previous studies (8,26). We had shown
that the phenotype of MHC-mt mice was specifically related to the expression of mutant TR since
it was not observed in mice with cardiac overexpression of wild-type mutant TRβ, driven by the
same αMHC promoter (8). Unfortunately, this mouse line was not propagated and we were not
able to use them in the present paper.
MHC-mt mice presented a profound reduction in atrial Cx40 mRNA abundance. As
mentioned before, the cardiac-specific overexpression of dominant negative TRβ disrupts TR-
signaling pathways and affects the expression of genes known to be directly regulated by TH
receptor at the promoter level, such as α-MHC (27). Therefore, gene transcription of atrial Cx40
is regulated by a direct cardiac mechanism that is TR-dependent. In addition, TH long-term
deficiency or excess was able to decrease or increase, respectively, Cx40 mRNA abundance,

suggesting that TH action in the heart, via TR, regulates gene transcription of atrial Cx40.
However, the precise mechanism still remains to be clarified. TR may bind directly to the Cx40
gene promoter or act indirectly via regulation of, or interaction with, transcription factors that
have known binding sites in the Connexin promoter, or a combination of both of these
mechanisms. In a recent report also employing the MHC-mt mice, cross-talk between TR and
PPARα pathways in the control of specific cardiac genes was observed (28), suggesting that, in
our model, several mechanisms may contribute to a final change in Cx40 mRNA expression.
In contrast, the expression of Connexin43, the other predominant atrial Connexin, seems
not to be regulated by TR-mediated TH actions; however, there was a reduction at the protein
level, which means that MHC-mt mice may have alterations in post-transcriptional Cx43
expression. In the ventricles, no alterations of Cx43 were induced by the mutant expression.
Chamber-specific effects of TH had been reported before, such as the up-regulation of voltage
gated potassium channel, Kv.5 that occurs only in the ventricle and not in the atrium (29). Still, to
our knowledge, no previous studies have addressed the regulation of Cx43 by TH in atrial
myocardium cells. Regarding Cx43 in the ventricle, there have only been a few in vivo studies,
and they did not observe significant changes in Cx43 mRNA in response to TH (5,19). However,
the results of an in vitro study employing neonatal rat cardiomyocytes in culture indicated that
TH up-regulates Cx43 protein levels, and suggests increased coupling among T3-treated cells
(20).
The functional importance of the selective change in atrium Cx40 relies on the
fundamental role of this junctional protein in atrial electrical conduction. In normal mice, Cx40 is
restricted to atrium cells and the specialized conduction system in atrium and ventricle. Previous
studies had demonstrated that P wave and PR interval duration were increased in Cx40 knockout
mice (16,17), while mice heterozygous for a null mutation in the gene encoding Cx43, expressing
50% of the normal amount of Cx43 in the myocardium, exhibited normal atrial conduction (18).
Thus, Cx40 is the main connexin responsible for electrical coupling between atrial cells.

Therefore, the extremely low Cx40 expression in atrial cardiomyocytes of MHC-mt mice is likely
an important factor explaining the prolonged P wave duration, which is most likely due to a
reduced velocity of atrial conduction. In addition, atrial enlargement is also a potential contributor
to this increase in intra-atrial conduction time. Alterations in Na+ currents seem to be unlikely
because previous studies on atrial myocytes of hypo-or hyperthyroid animals do not support
genomic TH effects, since they revealed no alterations in Na+ channel mRNA expression or in
Na+ current densities (5,11,30). However, there is evidence of non-genomic effects of TH
inducing direct changes in the activity of Na + channels, facilitating their opening (31,32). This
may explain the fact that the net inward Na+ current in atria of hyperthyroid rats was more
sensitive to pilsicainide, a Na+-channel blocker (30). Since MHC-mt mice exhibit no changes in
serum TH, non-genomic effects are not likely to be related to the alterations observed in atrial
conduction.
Systemic hypo- and hyperthyroidism in wild-type animals induced prolonged or
shortened P wave duration. However, these changes were of lower magnitude than in MHC-mt
mice; changes in Cx40mRNA expression were insufficient to significantly change protein levels.
These differences in magnitude of response are probably related to particularities of the mouse
models. Different degrees in severity of thyroid dysfunction may be one explanation. Indeed,
MHC mutant mice have a congenital defect and thereby a more profound and prolonged state of
cardiac hypothyroidism. In addition, altered TH levels can elicit systemic responses that
compensate for primary cardiac alterations in gene expression. Therefore, it remains to be seen
whether Cx40 expression or activity may have a role in atrial conduction disturbances present in
hypo-and hyperthyroidism.
MHC-mt mice also have important bradycardia, indicating alterations in the pacemaker
activity of the sinoatrial node, which results from diminished expression of genes known to be
regulated by TH including HCN channels and Ca+2 channels (3,5,23). However, it is possible that

reduced Cx40 has some contribution, since Cx40 deletion in mice also impaired sinoatrial node
automaticity, resulting in slowing pacemaker discharge (16,33,34).
Clinical manifestations of atrial conduction disturbances in hypothyroid patients have
been reported, although they are not common (35,36,37), while atrial arrhythmias are more
prevalent in hyperthyroid patients (37). There are studies in humans associating higher expression
of Cx40 with a higher incidence of atrial fibrillation (38-40). Whether alterations of Cx40
expression or activity may have a role in the occurrence of atrial fibrillation of hyperthyroid
patients remains to be elucidated.
In conclusion, the present study shows that impairment of TR-mediated TH effects due to
expression of a TR mutant in atrial cardiac myocytes resulted in reduced atrial conduction
velocity in association with reduced expression of atrial gap junction protein, Connexin40. This is
the first report revealing that Connexin40 mRNA is up-regulated in cardiac atria by a TR-
mediated TH action. The findings of the present study highlight a very important role of TH
receptors in atrial electrical conduction.

References
1. Klein I, Ojamaa K 2001 Thyroid hormone and the cardiovascular system. N Engl J Med
334:501-509.
2. Davis PJ, Davis FB 2002 Nongenomic actions of thyroid hormone on the heart. Thyroid
12:459-466.
3. Gloss B; Trost SU, Bluhm WF, Swanson EA, Clark R, Winkfein R, Janzen KM, Giles
W, Chassende O, Samarut J, Dillmann WH 2001 Cardiac ion channel expression and
contractile function in mice with deletion of thyroid hormone receptor α or β. Endocrinology
142:544-550.
4. Shimoni Y, Fiset C, Clark RB, Dixon JE, Mc Kinnon D, Giles WR 1997 Thyroid
hormone regulates postnatal expression of transient K+ channel isoforms in rat ventricle. J
Physiol (London) 500:65-73.
5. Le Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G,
Lande G, Siavoshian S, Baró I, Pond A, Nerbonne JM, Léger JJ, Escande D, Charpentier
F 2003 Microarray analysis reveals complex remodeling of cardiac ion channel expression with
altered thyroid status. Circ Res 92:234-242.
6. Watanabe H, Ma M, Washizuka T, Komura S, Yoshida T, hosaka Y, Hatada K,
Chinushi M, Yamamoto T, Watanabe K, Aizawa Y 2003 Thyroid hormone regulates mRNA
expression and currents of ion channels in rat atrium. Biochem Biophys Res Commum 308:439-
444.
7. Ma ML, Watanabe K, Watanabe H, Hosaka Y, Komura S, Aizawa Y, Yamamoto T
2003 Different gene expression of potassium channels by thyroid hormone and an antithyroid
drug between the atrium and ventricle of rats. Jpn Heart J 44(Pt1):101-110.
8. Pazos-Moura CC, Abel ED, Boers ME, Moura E, Hampton TG, Wang J, Morgan JP,
Wondisford FE 2000 Cardiac dysfunction caused by myocardium-specific expression of a
mutant thyroid hormone receptor. Circ Res 86:700-706.
9. Usala SJ, Menke JB, Watson TL, Wondisford FE, Weintraub BD,
Bérard J, Bradley WE, Ono S, Mueller OT, Bercu BB 1991 A
homozygous deletion in the c-erbA thyroid hormone receptor gene in a
patient with generalized thyroid hormone resistance: isolation and
characterization of the mutant receptor. Mol Endocrinol 5:327-335.
10. Hu Y, Penelope Jones SV, Dillmann WH 2005 Effects of hyperthyroidism on delayed
rectifier K+ currents in left and right murine atria. Am J Physiol Heart Circ Physiol 289:H1448-
1455.
11. Sunagawa M, Yamakawa M, Shimabukuro M, Higa N, Takasu N, Kosugi T 2005
Electrophysiologic characteristics of atrial myocytes in levo-thyroxine-treated rats. Thyroid
15:3- 11.

12. Kumar NM, Gilula NB 1996 The gap junction communication channel. Cell 84:381-388.
13. Hervé JC 2004 The Connexins. Biochimica et Biophysica Acta 1662:1-2.
14. Gourdie RG, Green CR, Severs NJ 1991 Gap junction distribution in adult mammalian
myocardium revealed by an anti-peptide antibody and laser scanning confocal microscopy. J
Cell Sci 99(Pt1):41-55.
15. Delorme B, Dahl E, Jarry-Guichard T, Marics I, Briand JP, Willecke K, Gros D,
Théveniau-Ruissy M 1995 Developmental regulation of Connexin40 expression in mouse
heart correlates with the differentiation of the conduction system. Developmental Dynamics
204:358-371.
16. Kirchhoff S, Nelles E, Hagendorff A, Kruger O, Traub O, Willecke K 1998 Reduced
cardiac conduction velocity and predisposition to arrhythmias in Connexin40-deficient mice.
Curr Biol 8:299-302.
17. Simon AM, Goodenough DA, Paul DL 1998 Mice lacking Connexin40 have cardiac
conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr
Biol 8:295-298.
18. Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME,
Saffitz JE 1998 Disparate effects of deficient expression of Connexin43 on atrial and
ventricular conduction: evidence for chamber-specific molecular determinants of conduction.
Circulation 97:686-691.
19. Stock A, Sies H 2000 Thyroid hormone receptors bind to an element in the Connexin43
promoter. Biol Chem 381:973-979.
20. Tribulova N, Shneyvays V, Mamedova LK, Moshel S, Zinman T, Shainberg A,
Manoach M, Weismann P, Kostin S 2004 Enhanced connexin-43 and α-sarcomeric actin
expression in cultured heart myocytes exposed to triiodo-L-thyronine. J Mol Hist 35:463-470.
21. Oliveira KJ, Ortiga-Carvalho TM, Cabanelas A, Veiga MA, Aoki K, Ohki-Hamazaki
H, Wada K, Wada E, Pazos-Moura CC 2006. Disruption of neuromedin B receptor gene
results in dysregulation of the pituitary-thyroid axis. J Mol Endocrinol 36:73-80.
22. Pereira-Junior PP, Chaves EA, Costa-E-Sousa RH, Masuda MO, Carvalho AC,
Nascimento JH 2006 Cardiac autonomic dysfunction in rats chronically treated with anabolic
steroid. Eur J Appl Physiol 96:487-494.
23. Swanson EA, Gloss B, Belke DD, Kaneshige M, Cheng SY, Dillmann WH 2003 Cardiac
expression and function of thyroid hormone receptor β and its PV mutant. Endocrinology
144:4820-4825.
24. Stoykov I, Zandieh-Doulabi B, Moorman AF, Christoffels V, Wiersinga WM, Bakker
O 2006 Expression pattern and ontogenesis of thyroid hormone receptor isoforms in the mouse
heart. J Endocrinol 189:231-245.

25. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, Cohen RN,
Wondisford FE 2001 An unliganded thyroid hormone receptor causes severe neurological
dysfunction. Proc Natl Acad Sci (USA) 98:3998-4003.
26. Ortiga Carvalho TM, Hashimoto K, Pazos-Moura CC, Geenen D, Cohen R, Land RM,
Wondisford FE 2004 Thyroid hormone resistance in the heart: role of the thyroid hormone
receptor β isoform. Endocrinology 145:1625-1633.
27. Gustafson TA, Markham BE, Bahl JJ, Morkin E 1987 Thyroid hormone regulates
expression of a transfected alpha-myosin heavy-chain fusion gene in fetal heart cells. Proc Natl
Acad Sci (USA) 84:3122-3126.
28. Buroker NE, Young ME, Wei C, Serikawa K, Ge M, Ning XH, Portman MA 2007 The
dominant negative thyroid hormone receptor beta-mutant {Delta}337T alters PPAR{alpha}
signaling in heart. Am J Physiol Endocrinol Metab 292:E453-E460.
29. Ojamaa K, Sabet A, Kenessey A, Shenoy R, Klein I 1999 Regulation of rat cardiac Kv1.5
gene expression by thyroid hormone is rapid and chamber specific. Endocrinology 140:3170-
3176.
30. Yamakawa M, Sunagawa M, Shimabukuro M, Higa N, Takasu N, Kosugi T 2005
Effect of sodium channel blocker, pilsicainide hydrochloride, on net inward current of atrial
myocytes in thyroid hormone toxicosis rat. Thyroid 15:7.
31. Dudley SC Jr, Baumgarten CM, 1993 Bursting of cardiac sodium channels after
acute exposure to 3,5,3-triiodo-L-thyronine. Circ Res 73:301–313.
32. Wang YG, Dedkova EN, Fiening JP, Ojamaa K, Blatter LA, Lipsius SL 2003
Acute exposure to thyroid hormone increases Na+ current and intracellular Ca2+ in cat
atrial myocytes. J Physiol 548.4:491-499.
33. Hagendorff A, Schumacher B, Kirchhoff S, Lüderitz B, Willecke, K 1999 Conduction
disturbances and increased atrial vulnerability in Connexin40-deficient mice analyzed by
transesophageal stimulation. Circulation 99:1508-1515.
34. Bagwe S, Berenfeld O, Vaidya D, Morley GE, Jalife J 2005 Altered right atrial
excitation and propagation in Connexin40 knockout mice. Circulation 112:2245-2253.
35. Lindgren M, Lantz B 1998 Bradyarrhythmia profile and associated diseases in
1,265 patients with cardiac pacing. Pacing Clin Electrophysiol. 11(12):2207-15.
36. Mangiardi L, Gaita F, Brun S, Presbitero P, Nademanee K, Singh BN 1986
Atrioventricular block complicating amiodarone-induced hypothyroidism in a patient
with pre-excitation and rate-dependent bilateral bundle branch block. J Am Coll
Cardiol.Jan 7(1):180-4.

37. Klein I, Ojamaa K 2000 The cardiovascular system in hypothyroidism.
In:Braverman LE, Utiger RD,eds. Werner&Ingbar’s TheThyroid:A Fundamental and
ClinicalText, edit.8. Philadelphia:Lippincott Williams&Wilkins;777–782
38.Dupont E, Ko Y, Rothery S, Coppen SR, Baghai M, Haw M, Severs NJ 2001 The gap-
junctional protein connexin40 is elevated in patients susceptible to postoperative atrial
fibrillation. Circulation 103:842-849.
39. Firouzi M, Ramanna H, Kok B, Jongsma HJ, Koeleman BP, Doevendans PA,
Groenewegen WA, Hauer RN 2004 Association of human connexin40 gene polymorphisms
with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ Res 95:e29-e33.
40. Wetzel U, Boldt A, Lauschke J, Weigl J, Schirdewahn P, Dorszewski A, Doll N,
Hindricks G, Dhein S, Kottkamp H 2005 Expression of connexins 40 and 43 in human left
atrium in atrial fibrillation of different etiologies. Heart 91:166-170.

Figure Legends
Fig 1. Atrial expression of Connexin40 (Cx40) mRNA (A,B) and protein (C,D) in MHC-mt
mice (cardiac-specific expression of dominant negative Δ337T thyroid hormone receptorβ) and
wild-type littermates (WT). A- Representative gel of semiquantitative RT-PCR analysis of
Cx40 and GAPDH (internal control) mRNA (750 bp and 300 bp, respectively); RT-
amplification without reverse transcriptase showed no nonspecific amplification of genomic
DNA. B- Relative expression of Cx40 mRNA (normalized to GAPDH) in MHC-mt relative to
WT values; densitometric values of the WT band from each experiment were set to 1. C-
Representative autoradiograph of atrial Connexin40 (Cx40- 40 KDa) and cyclophilin (19 KDa -
internal control) analyzed by western blotting and D- Respective analysis by densitometry. Data
are means ± SEM of 2-4 independent experiments, representing 6-9 animals per group; * p<
0.001 vs. WT.
Fig 2. Expression of atrial Connexin43 (Cx43) mRNA (A,B) and protein (C,D) in MHC-mt
mice (cardiac-specific expression of dominant negative Δ337T thyroid hormone receptorβ) and
wild-type littermates (WT). A- Representative gel of semiquantitative RT-PCR analysis of
Cx43 mRNA (593 bp) and GAPDH mRNA (300 bp), used as an internal control; RT-
amplification without reverse transcriptase showed no unspecific amplification from genomic
DNA. B- Relative expression of Cx43 mRNA (normalized by GAPDH) in MHC-mt relative to
WT values; densitometric values of WT of each experiment were set to 1. C– Representative
autoradiograph of atrial Cx43 (43 KDa) and cyclophilin (19KDa - internal control) analyzed by
western blotting and D- Respective analysis by densitometry. Data are mean ± SEM of 2-4
independent experiments, representing 6-8 animals per group; * p< 0.005 vs. WT.
Fig 3. Ventricular expression of Connexin43 (Cx43) mRNA (A,B) and protein (C,D), and
myosin heavy chain (MHC) mRNA in mice with cardiac-specific expression of dominant
negative Δ337T thyroid hormone receptorβ (MHC-mt) and wild-type littermates (WT). A-
representative gel of semiquantitative RT-PCR analysis of Cx43 mRNA (593 bp) and GAPDH
mRNA (300 bp), used as an internal control; RT-amplification without reverse transcriptase
showed no nonspecific amplification from genomic DNA. B- Relative expression of Cx43
mRNA (normalized to GAPDH) in MHC-mt relative to WT values; densitometric values of WT
of each experiment were set to 1. C– Representative autoradiograph of atrial Cx43 (43 KDa)
and cyclophilin (19 KDa - internal control) analyzed by western blotting and D- respective
analysis by densitometry. E- α and βMHC isoform mRNA expression analyzed by RNAse
Protection Assay in ventricles of MHC-mt and WT mice. F - Densitometry analysis of MHC

isoform mRNA (normalized to cyclophilin) in MHC-mt relative to WT. * p< 0.001 vs. WT.
Data are means ± SEM of 2-4 independent experiments, representing 6-8 animals per group.
Fig 4. Atrial Cx40 mRNA expression in hypo (WT+PTU)- and hyperthyroid (WT+T3) wild-
type mice A- Representative gel of semiquantitative RT-PCR analysis of Cx40 mRNA (750 bp)
and GAPDH mRNA (300 bp), used as an internal control; RT-amplification without reverse
transcriptase showed no unspecific amplification from genomic DNA. B- Relative expression of
Cx40 mRNA (normalized by GAPDH) to WT values; densitometric of WT of each experiment
were set to 1. C- Representative autoradiograph of atrial Connexin40 (Cx40- 40 KDa) and
cyclophilin (19 KDa - internal control) analyzed by western blotting and D- Respective analysis
by densitometry. Data are representative of two independent experiments; values are mean ±
SEM and correspond to 5-11 animals per group; * p< 0.05 or less vs. WT.
Fig 5. Atrial Connexin40 (Cx40) expression in euthyroid (MHC-WT), hypo- (MHC-mt+PTU)
and hyperthyroid (MHC-mt+T3) mice with cardiac specific expression of dominant negative
Δ337T thyroid hormone receptorβ (MHC-mt) and wild-type littermates (WT). A-
Representative gel of semiquantitative RT-PCR analysis of Cx40 and GAPDH (internal control)
mRNA (750 and 300 bp, respectively); RT-amplification without reverse transcriptase showed
no unspecific amplification from genomic DNA. B- Relative expression of Cx40 mRNA
(normalized to GAPDH) in MHC-mt relative to WT values; densitometric values of WT of each
respective experiment were set to 1. Data correspond to two independent experiments and
values are represented as mean ± SEM; n= 5-6 animals per group; * p< 0.001 vs. WT.
Fig 6. Typical electrocardiogram (ECG) recording from a conscious wild-type littermate and
MHC-mt mouse (cardiac specific expression of dominant negative Δ337T thyroid hormone
receptorβ). ECG intervals were prolonged in MHC-mt mice. See Table 1. Note the longer P
wave duration.




Figure 1
MH
C-m
t
WT
RT
-
MW
GAPDH
Cx40
A
Cx40
Cyclo
C
MH
C-m
t
WT
MH
C-m
t
WT
WT
WT MHC-mt
Cx4
0/G
AP
DH
mR
NA
rela
tive
to W
T
0.0
0.2
0.4
0.6
0.8
1.0
*
B
0.0
0.2
0.4
0.6
0.8
1.0
*
WT MHC-mt
Cx4
0/C
yclo
rela
tive
to W
T
D

Figure 2
WT
RT
-
MW
MH
C-m
t
GAPDHCx43
A
Cx43
Cyclo
MH
C-m
t
WT
MH
C-m
t
WT
WT
MH
C-m
t
C
WT MHC-mt
Cx4
3/G
AP
DH
mR
NA
rela
tive
to W
T
0.0
0.2
0.4
0.6
0.8
1.0
1.2
B
0.0
0.2
0.4
0.6
0.8
1.0
1.2
*
Cx4
3/C
yclo
rela
tive
to W
T
WT MHC-mt
D
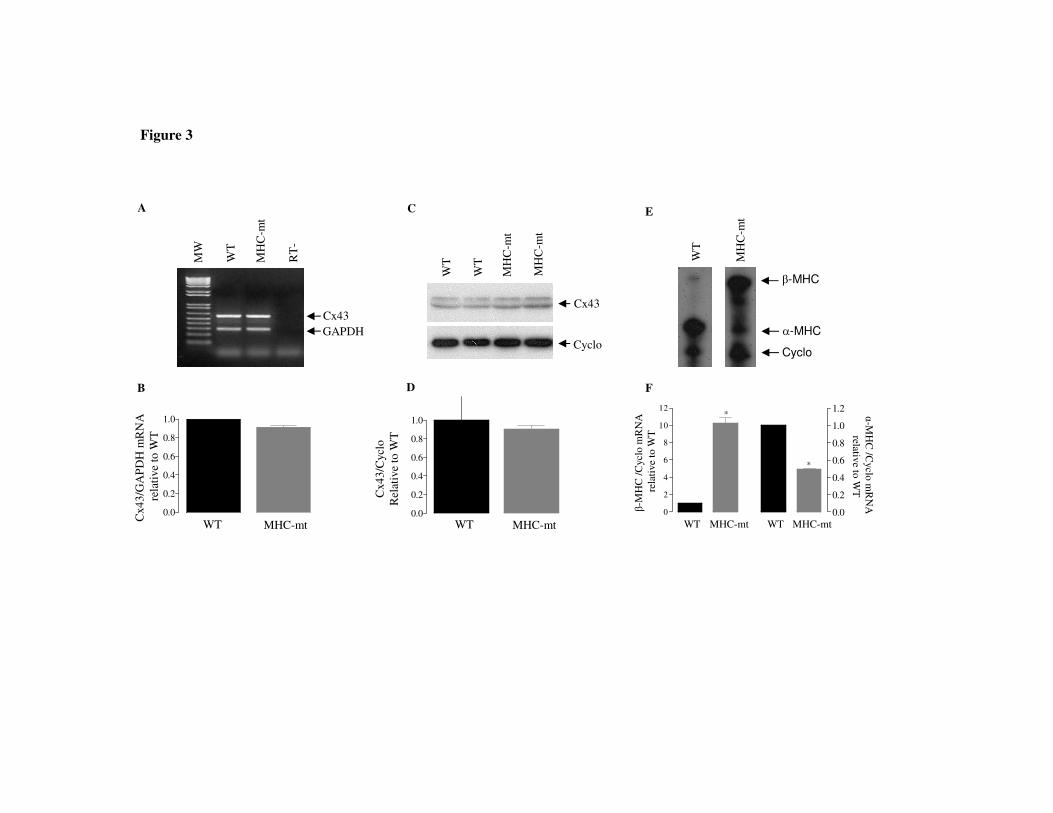
Figure 3
GAPDHCx43
WT
RT
-
MW
MH
C-m
t
A
Cx43
Cyclo
MH
C-m
t
WT
MH
C-m
t
WT
C
0.0
0.2
0.4
0.6
0.8
1.0
WT MHC-mt
Cx4
3/G
AP
DH
mR
NA
rela
tive
to W
T
B
0.0
0.2
0.4
0.6
0.8
1.0
Cx4
3/C
yclo
Rel
ativ
eto
WT
WT MHC-mt
D
Cyclo
β-MHC
α-MHC
WT
MH
C-m
tE
0
2
4
6
8
10
12
0.0
0.2
0.4
0.6
0.8
1.0
1.2*
*
β-M
HC
/Cyc
lom
RN
Are
lati
veto
WT
α-MH
C /C
yclom
RN
Arelative
to WT
F
WT MHC-mt WT MHC-mt

Figure 4
WT+PTUWT WT+T3
Cx4
0/G
AP
DH
mR
NA
rela
tive
to W
T
0.0
0.4
0.8
1.2
1.6
*
*
WT
+ P
TU
WT
WT
+T
3
RT
-
MW
GAPDH
Cx40
A
B
0.0
0.2
0.4
0.6
0.8
1.0
1.2
WT+PTUWT WT+T3
Cx4
0/C
yclo
rela
tive
to W
T
D
Cx40
Cyclo
C
WT
+ P
TU
WT
WT
+T
3

Figure 5
MW
WT
MH
C-m
t
MH
C-m
t+P
TU
MH
C-m
t+T
3
RT
-
GAPDH
Cx40
A
WT MHC-mt MHC-mt+PTU
MHC-mt+T3
Cx4
0/G
AP
DH
mR
NA
rela
tive
to W
T
0.0
0.2
0.4
0.6
0.8
1.0
**
*
B

50 ms
0,2 mV
50 ms
0,2 mV
WT
MHC-mt
A
B
10 ms
19 ms
P
P
Figure 6




















