
Presence and utility of intrinsically disordered regions in kinases

Conformational Selection and Folding-upon-binding ofIntrinsically Disordered Protein CP12 RegulatePhotosynthetic Enzymes Assembly*□S
Received for publication, February 8, 2012, and in revised form, April 5, 2012 Published, JBC Papers in Press, April 18, 2012, DOI 10.1074/jbc.M112.350355
Simona Fermani‡1, Xavier Trivelli§¶�1, Francesca Sparla**1, Anton Thumiger**, Matteo Calvaresi‡, Lucia Marri**,Giuseppe Falini‡, Francesco Zerbetto‡, and Paolo Trost**2
From the ‡Department of Chemistry “G. Ciamician,” University of Bologna, 40126 Bologna, Italy, §University Lille Nord de France,59000 Lille, France, ¶Universite des Sciences et Technologies de Lille, Unite de Glycobiologie Structurale et Fonctionelle, 59655Villeneuve d’Ascq, France, �CNRS, UMR 8576, 59655 Villeneuve d’Ascq, France, and **Department of Experimental EvolutionaryBiology, University of Bologna, 40126 Bologna, Italy
Background: In the dark CP12 is oxidized and regulates photosynthetic GAPDH.Results: The disordered C terminus of oxidized CP12 gets ordered when bound to GAPDH.Conclusion:Transient complexes between GAPDH and selected conformations of CP12 evolve into a stable binary complex inwhich CP12 blocks GAPDH catalytic sites.Significance:Disordered proteins can bind structured partners through a synergistic combination of conformational selectionand folding-upon-binding.
Carbon assimilation in plants is regulated by the reduction ofspecific protein disulfides by light and their re-oxidation in thedark. The redox switch CP12 is an intrinsically disordered pro-tein that can form two disulfide bridges. In the dark oxidizedCP12 forms an inactive supramolecular complex with glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) and phosphoribu-lokinase, two enzymes of the carbon assimilation cycle. Here weshow that binding of CP12 to GAPDH, the first step of ternarycomplex formation, follows an integrated mechanism that com-bines conformational selection with induced folding steps. Ini-tially, a CP12 conformation characterized by a circular structuralmotif including the C-terminal disulfide is selected by GAPDH.Subsequently, the induced folding of the flexible C-terminal tail ofCP12 in the active site of GAPDH stabilizes the binary complex.Formation of several hydrogen bonds compensates the entropiccost of CP12 fixation and terminates the interaction mechanismthat contributes to carbon assimilation control.
In oxygen phototrophs a complex regulatory system basedon thioredoxins ensures that carbon fixation is active duringthe day and nil during the night (1). In the light, chloroplastthioredoxins are reduced by photosystem I via ferredoxin and
ferredoxin:thioredoxin reductase (2) and impose a reducedstate to their targets (protein dithiols). In the dark, re-oxidationof protein dithiols to disulfides is allowed (1).Besides other proteins and regulatory systems, coordinated
light/dark regulation involves the participation of (i) glyceral-dehyde-3-phosphate dehydrogenase (GAPDH), which cata-lyzes the only reducing step of the Calvin-Benson cycle of car-bon assimilation (3), (ii) phosphoribulokinase, which consumesa third of the ATP required for CO2 fixation into sugars, and(iii) the scaffold protein CP12, an ubiquitous regulatory proteinof oxygenic photosynthetic organisms that contains, with fewexceptions, two cleavable disulfide bridges (4–9).GAPDH of land plants includes two plastidic isoforms
named A4 and A2B2 (3). The former is a stable, constitutivelyactive tetramer, whereas the latter can form A4B4 and A8B8oligomers in the presence of NAD(H) (3, 5, 9). Thioredoxin fspecifically reduced a disulfide bridge in the C-terminal exten-sion (CTE)2 of subunits B, thereby relieving the inhibition ofenzyme activity caused by oxidized CTE (10). The CTE ishomologous to the C-terminal half of CP12 (4), andA4-GAPDH may bind CP12 in a similar way, as A2B2-GAPDHdoes bind its own CTE (11). In land plants only, the autono-mous regulation ofAB-GAPDHco-exists with the regulation ofA4-GAPDH by CP12 (3, 5), ubiquitous in oxygen phototrophs.
The three proteins A4-GAPDH, phosphoribulokinase, andCP12 form a supramolecular complex where the activity ofboth GAPDH and phosphoribulokinase is inhibited (4–6, 12).The CP12-assembled complex, stored in the dark in chloro-plasts, is rapidly dissociated by the onset of light (13). Becauseboth GAPDH and phosphoribulokinase activities are inhibitedin the complex but are fully recovered upon dissociation (10),
* This work was supported by the Italian Ministry of University and Research(MiUR, projects FIRB and PRIN 2008). This work was also supported by TresGrand Equipement de Recherche (TGE) Resonance Magnetique Nuclear(RMN) a Tres Haut Champs (THC) Fr3050.
□S This article contains supplemental Tables S1–S3, Figs. S1–S7, and Video S1.The atomic coordinates and structure factors (codes 3QV1 and 3RVD) have been
deposited in the Protein Data Bank, Research Collaboratory for Structural Bioin-formatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
NMR structural models and chemical shift assignments have been deposited inthe Protein Data Bank and Biological Magnetic Resonance Data Bank underaccession codes 2LJ9 and 17926, respectively.
1 These authors equally contributed to the work.2 To whom correspondence should be addressed: Dept. of Experimental Evolu-
tionary Biology, University of Bologna, Via Irnerio 42, 40126 Bologna, Italy. Tel.:39-051-2091329; Fax: 39-051-242576; E-mail: [email protected].
2 The abbreviations used are: CTE, C-terminal extension; IDP, intrinsically dis-ordered protein; MR, molecular replacement; HSQC, heteronuclear singlequantum correlation; MD, molecular dynamics; PCA, principal componentanalysis.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 287, NO. 25, pp. 21372–21383, June 15, 2012© 2012 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
21372 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from
http://www.jbc.org/content/suppl/2012/04/18/M112.350355.DC1.html Supplemental Material can be found at:

CP12 could effectively contribute to themodulation of the Cal-vin-Benson cycle under the natural variable light/dark regime(13). The thioredoxin-dependent CP12may, therefore, work asa light-sensitive redox switch in chloroplasts and was recentlyshown by antisense technology to be required for normalgrowth and development in transgenic tobacco plants (14).Moreover, in the cyanobacterium Synechococcus elongatusPCC7942, growth of CP12-knock out mutants was inhibitedunder light/dark cycle but not in continuous light (7). Interest-ingly, CP12 is also coded by the ultrasmall genome of cya-nophages infecting marine cyanobacteria (e.g. Synechococcus),where CP12-inhibition of the Calvin-Benson cycle may favorNADPHproduction (essential for phage replication) by the oxi-dative pentose phosphate pathway (15).Differently from what was recently demonstrated by the
x-ray structure of A4-GAPDH-CP124 complex from S. elonga-tus (16), Arabidopsis thaliana GAPDH (isoform A4) can bindtwo CP12 monomers per GAPDH tetramer (17). Formation ofthe GAPDH-CP12 binary complex requires, as a seeminglycompulsory condition, that NAD or NADH are bound to thedehydrogenase (12) and that C-terminal cysteines of CP12 areoxidized into a disulfide (4, 17, 18).Only after formation of the binary CP12-GAPDH complex,
interaction with dimeric phosphoribulokinase leads to stabili-zation of an inactive ternary complex made by two GAPDHtetramers and two phosphoribulokinase dimers linkedtogether by four CP12 monomers (17). The stability of theternary complex is controlled by thioredoxins, which canreduce CP12 disulfides (10). Also, nucleotides (NADP(H),ATP) and the GAPDH substrate 1,3-bisphosphoglyceratecan dissociate the ternary complex leading to enzyme reac-tivation (4, 6, 7, 12).CP12 is a small protein of about 80 amino acids. It is intrin-
sically disordered both in the green alga Chlamydomonas rein-hardtii (6) and in the higher plant A. thaliana (17, 8) and pre-dicted disordered in other phototrophs (e.g. Synechococcus) (8).Intrinsically disordered proteins (IDPs) constitute a significantfraction of eukaryotic proteomes, including plants (19, 20), andoften play a role as scaffolds in the assembly of supramolecularcomplexes (21, 22). At odds with most IDPs, an uncommonproperty of most CP12 is the presence of four conserved cys-teines able to form two consecutive disulfide bridges (4, 6, 8,17). Although formation of disulfides increases the overalldegree of order in terms of �-helix content, even fully oxidizedCP12 appears largely devoid of secondary structures in circulardichroism spectra (6, 8).Intrinsically disordered proteins (such as CP12) may bind to
structured partners (like GAPDH) in different ways (23, 24). Ina model based on conformational selection (25), associationwith a structured partner occurs when a single conformation ofthe disordered protein, appreciably populated in solution, isselected by the target and stabilized into the complex. In such acase, the structure of the IDP in the complex corresponds toone of the several accessible conformational states that arepresent in the free energy landscape (26, 27). Alternatively, thefinal structure of the IDP in the complex is only reachedthrough amultistep folding process that takes place upon bind-ing (23). The structure of the IDP in the complex often becomes
markedly different from native unbound states (28–30). Theinduced folding can be initiated by the recognition of pre-formed structural elements in the IDP (Molecular RecognitionFeatures (31)) by the structured partner (32). Although exam-ples of both types of mechanisms are known (27–30), theircombinationmay also be active in real systems (23). A synergis-tic model has been proposed to reconcile both hypotheses (24).With the aim of unraveling the interaction mechanism
between the intrinsically disordered proteinCP12 and its struc-tured partner GAPDH, here we describe theNMR structures ofthe C-terminal region of free-oxidizedCP12 in solution and thecrystal structure of the GAPDH-CP12 binary complex fromA. thaliana at 2.0Å. Its formation is the first crucial step towardthe assembly of the GAPDH-CP12-phosphoribulokinase ter-nary complex.Molecular interactions between CP12 and GAPDH in the
Arabidopsis complex were found very similar to those observedin the Synechococcus complex (16), indicative of a high degreeof conservation in this regulatory system (4). On the otherhand, although CP12 bound to A4-GAPDH(NAD) (this work)was found in the same cleft occupied by CTE in A2B2-GAPDH-(NADP) (11), the two structures diverged completely in termsof folding and molecular interactions.Leveraging on the NMR structural ensemble of the C-termi-
nal part of CP12, the crystal structure of the binary complex,and molecular dynamics (MD) simulations, we develop amodel of binding that starts from the intrinsically disorderedstructure of CP12. The interaction model between CP12 andGAPDH, proposed in the following, presents features con-sistent with a synergistic mechanism including both confor-mational selection and induced folding steps. Additionalcomplexity is added by the redox-regulation of the system.
EXPERIMENTAL PROCEDURES
Protein Preparations—Recombinant CP12 (isoform 2, TAIRdata base: At3g62410) and A4-GAPDH (TAIR data base:At3g26650) from A. thaliana were purified as described (12).After His-tag removal (12), CP12 was treated overnight with 20mM oxidized DTT to ensure disulfide formation. Purified CP12and A4-GAPDH were quantified by UV-absorbance (12) andstored at �20 °C in 25 mM potassium phosphate, pH 7.5.
For crystallization trials, preformed binary complex (10mg�ml�1) was prepared by mixing A4-GAPDH and CP12 in a1:2 ratio plus 1 mM NAD in 25 mM potassium phosphate, pH7.5. For NMR experiments, labeled CP12 was prepared in M9medium containing 1 g�liter�1 15NH4Cl with or without 5g�liter�1 D-[13C6]glucose (17). The His tag was not removed.Crystallization, Data Collection, and Processing—GAPDH-
CP12 complex crystals were produced both by crystallization ofthe preformed complex in solution and by soaking crystals ofrecombinant A4-GAPDH (33) in a CP12 solution. The vapordiffusionmethodwas used to screen several crystallization con-ditions at 277 or 293 K. A drop of 2-�l complex solution plus 2�l of reservoir solution was equilibrated against 750 �l of res-ervoir solution. The best diffracting crystals were obtained inhanging drop at 277Kwith 15% (w/v) PEG 4000, 0.1 MMES, pH6.5, 0.6 M NaCl, 1 mM NAD as reservoir solution.
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21373
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

For soaking experiments, crystals of recombinant A4-GAPDH(33) were transferred in a 5-�l drop containing 2.4 M ammoniumsulfate, 0.1 M sodium citrate, 1 mM NAD, and 3.125 mg�ml�1
CP12. The drops were equilibrated against 750 �l of 2.4 M
ammonium sulfate, 0.1 M sodium citrate for 4 days at 293 K.Data from co-crystallized and soaked complex crystals were
collected at European Synchrotron Radiation Facility (Grenoble)at beam lines ID14-3 and ID14-1, respectively at 100 K. Co-crys-tallized crystals diffracted to a maximum resolution of 1.98 Å,and data were processed with IMOSFLM (34), POINTLESS,and SCALA (35). Soaked crystals diffracted to a maximum res-olution of 2.7 Å, and data were processed with DENZO andscaled with SCALEPACK (36). Space groups, unit cell parame-ters, and diffraction data statistics for both structures arereported in Table 1.Structure Determination and Refinement—Both structures
were solved by molecular replacement (MR) by the programAMORE (37) using the structure of A. thaliana A4-GAPDH(33) as the search model. The MR procedures gave unequivo-cally two and three solutions for co-crystallized and soakedcomplexes, respectively. Refinement was performed with REF-MAC 5.2.005 (38) (co-crystallization) or CNS 1.3 (39) (soaking)and manual rebuilding with Coot (40).CP12, NAD, and sulfate ions were inserted after few refine-
ment cycles in the electron density regions not occupied byGAPDH chains. In the final stages of refinement and modelbuilding, water molecules were added. Stereochemical qualityof the models was checked with PROCHECK (41). Ramachan-dran plots show that 99% (co-crystallization) and 97.8% (soak-ing) of residues lie in the most favored plus additional allowedregions.Only 0.5%of residues are in disallowed regions for bothstructures. Refinement statistics are reported in Table 1.NMR Analysis—NMR samples (non labeled or uniformly
15N- or 13C,15N-labeledCP12) contained about 1mMprotein inNMR buffer (25 mM potassium phosphate, 95% H2O, 5% D2O,0.02% NaN3) at pH 7.0. The binary complex was made in vitrobymixing 0.2mMU-15N-labeledCP12with 0.5mMA4-GAPDHand 2.7 mM NAD in NMR buffer. NMR experiments wererecorded at 20 °C on Bruker Avance 400, Avance 600, andAvanceII 800 spectrometers and on an Agilent vnmrs 800spectrometer.Backbone resonance assignments were obtained from a
series of standard heteronuclear experiments. NMR distancerestraints were extracted from three-dimensional 1H,15NNOESY-HSQC and two-dimensional NOESY with 60- and150-ms mixing times.NMRdata were processed and analyzed usingNMRPipe (42)
and NMRView (43). All proton dimensions were referenced to1 mM trimethylsilyl propionate signal, and 13C and 15N dimen-sions were referenced indirectly using the 1H/X frequencyratios of the zero point. Homonuclear NOE assignment wasmade manually. The resulting distance restraints, 40 backbonedihedral predictions from TALOS� (44), and 2 hydrogenbonds extracted from a long-range three-dimensional BEST-TROSY-HNCO (45) were used as input for CNS (39). The 20structures with the lowest total energies were selected. Refine-ment statistics are reported in Table 2. The stereochemicalquality of the models was checked with PROCHECK (41). Res-
idues inmost favored regions and in additional allowed regionsrepresent 81.7 and 18.3%, respectively.Molecular Dynamics—All-atom molecular dynamics simu-
lations were carried out with AMBER (46). AMBER 99 forcefield parameters (47) were employed. Water molecules weredescribed by the TIP3P model. An equilibration protocol con-sisting of three individual steps was applied resulting in anunconstrained well tempered NPT ensemble (isothermal-iso-baric ensemble, moles (N), pressure (P), and temperature (T)are conserved).TheCartesian coordinates of CP12 are taken from the crystal
structure. The structure is relaxed (minimized with SANDERusing steepest descent algorithm) to remove the constraintimposed by the binding with A4-GAPDH. The explicit netcharge of CP12 is neutralized adding Na� counterions at posi-tions of high negative electric potential, then the protein is sol-vatedwith explicit water. An 8Å radius buffer of TIP3Pwater isput around CP12 in each direction. To allow the water box torelax before running molecular dynamics, about 5000 steps ofsteepest descent minimization were performed with SANDER.In the first 1000 steps CP12 was restrained to its original posi-tion using a force constant of 5 kcal�mol�1�A�2, then the con-straint is released to relax the entire system. Particle MeshEwald summation was used throughout (cutoff radius of 10 Åfor the direct space sum). H-atoms were frozen with theSHAKE algorithm (48), and a time step of 2 fs was applied in allequilibration runs. After successfully minimization, the systemwas heated from 0 to 300 K. To avoid wild fluctuations in thesolute, a weak restraint (5 kcal�mol�1�A�2) on CP12 atoms wasapplied in an NVT ensemble (canonical ensemble, moles (N),volume (V), and temperature (T) are conserved) and tempera-ture coupling according to Langevin for 100 ps. Constant pres-sure was then applied, so that the density of water could relax.Temperature was 300 K. Safe removal of the restraints on CP12was possible. A further 400 ps of equilibration inNPT ensembleresembling laboratory conditions were carried out. Finally aproduction run of 50 ns was performed. Snapshot structureswere saved into individual trajectory files every 25,000 timesteps, i.e. every 2 ps of molecular dynamics.
RESULTS
GAPDH-boundCP12Contains aCircular StructuralMotif—A. thaliana CP12 (isoform 2) is an intrinsically disordered pro-tein with little secondary structure also in the oxidized statewhere two intramolecular disulfide bridges are present (17, 8).In line with the disorder, all attempts to grow crystals of CP12failed. A different crystallographic approach exploited theinteraction of CP12 with GAPDH. It is reasoned that CP12might undergo a disorder-to-order transition upon binding to astructured protein. GAPDH-assisted crystallization of CP12was attempted both by setting-up co-crystallization trials withpreformed GAPDH-CP12 complex in solution and by soakingA4-GAPDH(NAD) crystals within a solution of oxidized CP12.The crystal structure of recombinant GAPDH (isoform A4), incomplexwithNAD,was recently solved at 2.6Å resolution (33).Both approaches, co-crystallization and soaking, producedx-ray diffracting crystals amenable to structural studies. Twostructures of theGAPDH-CP12 binary complexwere refined at
Disorder to Order Transitions in CP12 Interacting with GAPDH
21374 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

2.0 Å (co-crystallized complex) and 2.7 Å (soaked complex)(Table 1). The following description of the GAPDH-CP12binary complex is mostly based on the higher resolution dataproduced by co-crystallization. The conclusions are fully sup-ported also by the data obtained from soaked crystals.In the co-crystallized complex structure, the asymmetric
unit contained one tetramer and one dimer of GAPDH thatgenerated a second tetramer by symmetry. For simplicity, thedifferent subunits in the model (all A subunits with identicalsequence) have different labels (chains ABCD, Fig. 1a; andchains-EF, not shown). Additional electron density was associ-ated to NAD molecules (one bound to each GAPDH subunit)and to CP12 (one for each pair of GAPDH subunits; supple-mental Fig. S1). Depending on the cleft, only the last 19–21residues of CP12 could be modeled (chains G, H, and I), indi-cating that the N-terminal region of CP12 is largely disorderedwithin the crystal. This conclusion is consistent with theGAPDH-CP12 complex structure from S. elongatuswhere only23C-terminal residueswere placed in the electron densitymap,but the intactness of CP12 in the crystals was confirmed bymass spectroscopy (16).Two CP12 molecules were bound to each tetramer of
GAPDH. CP12 occupied an eccentric position within the cleft(Fig. 1b) roughly similar to the position of CTE in the structureof spinach A2B2-GAPDH complexed with NADP (11). Thenegatively charged C-terminal part of CP12 fits in a positivelycharged binding site of GAPDH, whose surface potentialmainly depends on Arg-77 and Arg-191 belonging to two dif-ferent subunits (Fig. 1c). Although GAPDH clefts containedtwo symmetrical binding sites, only one was occupied by CP12,consistent with the stoichiometry of the binary complex insolution (17). Half-occupation of CP12 binding sites was alsoobserved in GAPDH crystals soaked with excess CP12 (supple-mental Fig. S2). In both co-crystallized and CP12-soaked com-
plexes the two CP12s were either on the same side of thetetramer (e.g. symmetric to axis-P) or on opposite sides (e.g.symmetric to axis-Q, supplemental Fig. S2), suggesting that thetype of CP12 occupationwas influenced by the crystallographicenvironment.Differently, in the structure of the binary complex from
S. elongatus both symmetrical positions of the GAPDH cleftwere occupied by CP12, forming an hetero-octamer in whichfour CP12 molecules are bound to tetrameric GAPDH (16).The full occupation of GAPDH clefts by CP12 in the cyanobac-terial complex was probably favored by the presence of smallamino acids at the CP12-CP12 interface (Ala-67, Ala-68, Leu-71). In Arabidopsis CP12, the replacement of these residues bymore bulky side chains (Thr-70, Asn-71, Arg-74)might preventdouble occupation of theGAPDHcleft. Beside this difference inthe binary complex stoichiometry, the overall complex struc-tures of Synechococcus and Arabidopsis are quite similar, andtheir superposition gave root mean square deviation valuesranging between 0.8 and 1.1 Å over about 1400 C� atoms ofCP12 and GAPDH and between 0.8 and 1.3 Å over 19–22 C�
atoms of CP12 alone. As previously observed (16), the overallstructure of theCTE in spinachA2B2-GAPDH(NADP) (11)wasinstead completely different and not superimposable withCP12 in either Synechococcus or Arabidopsis complexes.
Apart from the initial (Asp-58–Pro-59) and the final C-ter-minal residues (Asn-78) of CP12, the backbones of all otherresidues were unambiguously positioned and displayed thesame structure (Fig. 1, d and e). The influence of CP12 bindingon the overall tetrameric structure ofGAPDHwas limited (sup-plemental Fig. S3a). The model of the C-terminal fragment ofCP12 includes an eight-residue amphipathic �-helix-C (for theC terminus, from Pro-59 to Asp-66) followed by a seven-resi-due loop-C (Asn-67–Cys-73) and by a five-residue C-terminaltail (C-tail, Arg-74–Asn-78) (Fig. 1, d and e). Loop-C consists oftwo consecutive�-turns (Asn-67–Thr-70 andThr-70–Cys-73)stabilized by hydrogen bonds (Fig. 1f). Cys-73 establishes adisulfide bridge with Cys-64 (belonging to �-helix-C). The�-helix-C, loop-C, and the disulfide bridge constitute the cir-cular structural motif of CP12. The C-tail has no apparent sec-ondary structure.Side-chain-mediated interconnections between different
portions of CP12 reinforce the structure (Fig. 1g). The �-he-lix-C is capped by the side chain of Asn-67, the first residue ofloop-C (supplemental Table S1a). The first two residues of theC-tail (Arg-74 and Thr-75) are hydrogen-bonded with loop-C.The last three residues of the C-tail are completely free frominteractions with other residues of CP12 (Fig. 1g).C-tail of CP12 Blocks GAPDH Catalytic Sites—Interactions
between GAPDH and the two portions of CP12 entail mainlyH-bonds. Their number differs depending on the portion ofCP12 considered. Similarly to other chains, the well resolvedchain G of co-crystallized CP12 makes 13 short-range interac-tions with GAPDH.There are four interactions between the circular structural
motif and GAPDH. The �-helix-C (CP12-chain G) binds toGAPDH (chain D) via a single hydrogen bond with Arg-191.Glu-69 (in the first �-turn of loop-C) and Arg-77 (GAPDH)form a salt bridge (Fig. 2a). Arg-191 and Arg-77 have a role in
TABLE 1Data collection and refinement statistics for A4-GAPDH/CP12 complexstructuresOne crystal was used for each structure. Values in parentheses are for highest res-olution shell.
Co-crystallizedcomplex Soaked complex
Data collectionSpace group C2221 I222Cell dimensionsa, b, c (Å) 142.7, 246.0, 139.0 153.2, 188.8, 312.2�, �, � (°) 90.0, 90.0, 90.0 90.0, 90.0, 90.0
Resolution (Å) 34.2 -1.98 (2.08-1.98) 95.3-2.70 (2.80-2.70)Rsym 0.139 (0.716) 0.098 (0.587)I/�I 5.9 (1.3) 16.7 (2.1)Completeness (%) 99.2 (98.2) 96.6 (84.0)Redundancy 3.3 (3.2) 11.3 (7.9)
RefinementResolution (Å) 34.2-2.0 94.6-2.7No. reflections 153,928 118,509Rwork/Rfree 0.225/0.272 0.247/0.319No. atomsProtein 15,827 26,538Ligand/ion 299 580Water 191 556
B-FactorsProtein 32.4 63.9Ligand/ion 25.7 63.4Water 21.2 43.4
Root mean square deviationsBond lengths (Å) 0.016 0.009Bond angles (°) 1.74 1.40
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21375
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

the positioning of CP12 within GAPDH deep clefts (Fig. 1b).The second �-turn of loop-C forms two additional H-bondswith GAPDH (Fig. 2a); one of them, which involves the sidechains of Glu-72 of CP12 and Thr-33 of GAPDH (chain A), isthe only link between CP12 and the distal GAPDH subunit ofthe cleft (Fig. 2a). Moreover, the carboxyl group of Glu-72approaches to the adenine ribose hydroxyl groups of NAD boundto the distal subunit (distance Glu-72.OE2-NAD335.O2B 3.5 Å).All other interactions between CP12 and GAPDH involve thevicinal subunit (GAPDH-chain D for CP12-chain G; supple-mental Table S1b).There are nine H-bonds between the terminal C-tail of CP12
(chain G) and GAPDH. Two of them are fully conserved in allnine CP12 chains of either co-crystallized and CP12-soakedcomplexes; the main-chain carbonyl of CP12-Arg-74 isH-bonded with the side chain of GAPDH-His-190, and theside-chain hydroxyl of CP12-Tyr-76 is H-bonded with NAD(Fig. 2b, supplemental Table S1b). Residues Tyr-76 and Glu-72represent two of the six highly conserved residues of the CP12
C-terminal region from oxygenic photosynthetic organisms.Both amino acids seem to have a specific function in complexformation in presence of NAD (16). Tyr-76 contributes tocofactor recognition by interacting with its backbone phos-phate group; at the same time Glu-72 prevents NADP bindingto GAPDH by steric hindrance and electrostatic repulsionbetween its carboxylic group and the 2�-phosphate of the pyri-dine nucleotide. The competition between Glu-72 and the2�-phosphate of NADP(H) explains why the GAPDH-CP12complex does form in the presence of NAD(H) but notNADP(H) (12). Other interactions involving the C-tail vary inthe different CP12 chains and always engage GAPDH residuesof the catalytic sites (Fig. 2, b and c, and supplemental TableS1b).Each GAPDH subunit contains one Ps and one Pi site (Fig.
2c). When A4-GAPDH crystals were grown in the presence ofammonium sulfate, each P-site hosted a sulfate ion, mimickingthe binding of the phosphate groups of the substrate (33). In theco-crystallized complex (crystals grown in the absence of
FIGURE 1. The structure of CP12 bound to GAPDH. a, two CP12 molecules (purple, chains G and H) are bound to GAPDH (chains ABCD; co-crystalliza-tion). Each CP12 is inserted in a cleft between two GAPDH subunits. b, shown is the top view of the cleft formed by GAPDH chain A and D. Four arginines,two per GAPDH subunit, delimit the cleft. CP12 chain G interacts with Arg-191 and Arg-77 of GAPDH-chain D. Arg-191 and Arg-77 of GAPDH chain Adelimit a symmetrical binding site for CP12. c, shown is the same view as in panel -b, with colors representing the electrostatic surface potentialcalculated by GRASP (54). The potential of CP12 (mesh) is mainly negative (red), and the GAPDH left is largely positive (blue). d, shown is the amino acidsequence of the portion of CP12 detected in the crystals. e, shown is superimposition of 3 and 6 CP12 chains from co-crystallization and soaking. The�-helix-C (red), loop-C (gray), and the disulfide (S atoms of Cys-64 and Cys-73 in yellow) form the circular structural motif. The last five residues are inextended conformation and form the C-tail (blue). f, loop-C is made of two consecutive �-turns. The backbone of CP12-chain G plus T70 side chain areshown. Each �-turn (Asn-67–Thr-70 and Thr-70 –Cys-73) is stabilized by two H-bonds. These H-bonds, found in most CP12 chains, were shorter than 3.1Å, except for N67.O-T70.N, which was 3.5 Å. g, the circular structure is reinforced by several intramolecular H-bonds. Only side chains involved ininteractions are shown. Although the first two residues of the C-tail are H-bonded with the circular structural motif, the last three are completely free tointeract with GAPDH. Interactions are listed in supplemental Table S1.
Disorder to Order Transitions in CP12 Interacting with GAPDH
21376 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

ammonium sulfate), these pockets are entirely occupied byCP12 in two of fourGAPDHsubunits (GAPDHchainsC andD;Fig. 2c; see also Fig. 1, a and b). The backbone of CP12 inter-acted with the side chains of the charged residues forming thePs sites (Asp-181, Arg-231, Arg-195) andwithNAD via the sidechain of Tyr-76 (Fig. 2c). The last two residues of CP12 formeda variety of interactionswith hydroxylated residues of the Pi site(Thr-150, Thr-208; Fig. 2c) and in some chains also with cata-lytic Cys-149 (supplemental Table S1b). The interactions ofCP12 within P-sites engage the same residues that stabilize sul-fates in CP12-free GAPDH (33).Occupation of P-sites by CP12 was also observed in the
binary complex obtained by soaking (supplemental Fig. S3b).Because these crystals were grown in the presence of ammo-nium sulfate, this finding demonstrates that CP12 can competewith and displace co-crystallized sulfate ions. P-sites of theopposite subunits that are not involved in CP12 binding werenormally occupied by sulfate ions (supplemental Fig. S3b).Major interactions between CP12 and GAPDH were con-
served in both Arabidopsis and Synechococcus complexes (16)(supplemental Table S2). CP12 residues Glu-72 and Tyr-76(Arabidopsis numbering) are engaged in the most relevantinteractions and conserved in both species. Arg-74 in Arabi-dopsis CP12 is replaced by leucine in Synechococcus (supple-mental Fig. S4a), but the main chain carbonyl group of bothresidues is hydrogen-bonded to a homologous GAPDH histi-dine (His-190 in Arabidopsis, His-195 in Synechococcus; sup-plemental Table S2). Also the position of CP12 and GAPDHinteracting partners, including the coenzyme, is similar in bothstructures (supplemental Fig. S4b). Although the side chain ori-entation of Arg-191 in Arabidopsis GAPDH diverged from thehomologous Arg196 in Synechococcus, the correspondingH-bond with CP12 glutamate involves main chain atoms and isfully conserved (supplemental Table S2).NMR Confirms CP12-GAPDH Interaction in Solution—The
NMR spectrum (1H,15N HSQC) of CP12 in solution showedlimited dispersion of signals. The chemical shifts of many back-
bone amide protons fell in the random coil region (8–8.5 ppm,Fig. 3a). The backbone amide protons of only 44 over 74 non-proline residues gave detectable peaks. All missing signalsbelonged to the central part of the protein, between Val-10 andAla-48. A short nine-residue N-terminal peptide (Ala-1–Asp-9) and a long 30-residue C-terminal peptide (Arg-49–Asn-78) were assigned (Fig. 3b).In NMR experiments conducted with 15N-labeled CP12 and
unlabeled GAPDH(NAD), all CP12 signals from residuesAsp-55 to Asn-78 disappeared. They include all resolved resi-dues in binary complex crystals (Fig. 3, a and b, and Fig. 1d).This result strongly suggests direct interactions or close prox-imity of this portion of CP12 with GAPDH, which is largeenough (145 kDa) to broaden the interacting CP12 signalsbelow the detection limit. Residues more distant from the Cterminus of CP12, e.g. Ser-46, retained sufficient mobility andwere still detectable. The results are consistent with what isexpected if the entire C-terminal portion of CP12 is inserted inthe deep cleft delimited by two GAPDH subunits. No new sig-nals from the central part of CP12 appeared when the binarycomplex was formed.Free CP12 Contains Circular Structural Motif—The struc-
ture of the C-terminal part of oxidized CP12 was modeled onthe basis of NMR data (Table 2; Fig. 4, a and b). Twenty lowenergy models were selected. They all contain the �-helix-Cencompassing residues Pro-59 toAsp-66, helix-capped byAsn-67. This portion of CP12 is superimposable in all NMR andx-ray structures (Fig. 4d).Solution heteronuclear 15NNOE experiments indicated that
the residues of CP12 circular motif have slow dynamics in thebinary complex (Fig. 4c). In most NMR models residues ofloop-C (Asn-67–Cys-73) have torsion angles typical of�-turns,and the loop-C adopted a helicoidal structure formed by twoconsecutive �-turns (Fig. 4e). However, the position of the loopwith respect to the �-helix-C can vary in the different models(Fig. 4f).
FIGURE 2. The interactions of CP12 with GAPDH. a, the circular structural motif of CP12 (�-helix-C, red), loop-C (gray), and disulfide (S-atoms, yellow)is weakly bound to GAPDH. The CP12-chain G (schematic and sticks) lies in the GAPDH cavity formed by chains A (yellow) and D (white, surface and sticks).Only side chains involved in interactions (dashes) are shown and labeled when visible. The �-helix-C is bound to GAPDH-chain D through a singleH-bond between the side chains of Glu-61 (CP12) and Arg-191 (GAPDH). Three more interactions fix loop-C. None of these interactions are completelyconserved in other CP12 chains (supplemental Table S1b). b, most of the interactions between CP12 and GAPDH involve the C-tail. CP12-chain G makes9 H-bonds with GAPDH chain D. The two H-bonds, R74.O(CP12)-H190.NE2(GAPDH) and Y76.OH(CP12)-NAD.O1A, are fully conserved in all CP12 chains.Other interactions are more variable but often involve the GAPDH residues forming the substrate-phosphates binding sites (Ps-site, Asp-181, Arg-195,Arg-231; Pi-site, Thr-150, Thr-208). c, the C-tail of CP12 blocks the GAPDH catalytic site. In the structure of A4-GAPDH (33) two sulfates ions deriving fromthe precipitation medium occupy the Ps and Pi sites of each GAPDH subunit. A superimposition of this structure with the structure of the binary complexGAPDH-CP12 shows that the C-tail of bound CP12 occupies both P-sites. The figure illustrates how the C-tail of the GAPDH-CP12 complex would clashwith the sulfates bound to A4-GAPDH (33).
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21377
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

The last five residues forming the C-tail of CP12 in solutionare predicted dynamic (TALOS�, Fig. 4a) and display low val-ues of heteronuclear 15N NOE (Fig. 4c) and limited homo-
nuclear 1H,1H NOE signals (Fig. 4b). These features indicatehigh structural dynamism.The ensemble of 20 CP12 models obtained by NMR was
structurally clustered. The two major structural clusters (N1and N2) including 11 and 4 models, respectively, were neatlyseparated and represented alternative structures of CP12 C ter-minus in solution. The nine crystallographic CP12 models(three from co-crystallization, six from soaking) formedanother homogeneous cluster (C1) thatmapped closely to clus-ter N1 in the principal-component analysis (Fig. 5a). In clusterN1, both the �-helix-C and the first �-turn of loop-C (i.e. fromPro-59 to Thr-70) were nicely superimposed with crystallo-graphic CP12 structures. FromThr-70 to Asn-78, the structureof clusters N1 and C1 diverged completely (Fig. 5b). Except forthe �-helix-C, all other NMR clusters diverged from crystallo-graphic cluster C1 for the entire CP12 C-terminal structure.Exploring Region of Cluster C1 by Molecular Dynamics—
Starting from CP12 crystallographic structure, we carried outMD simulations of the last 21 residues of CP12 in explicit waterfor 50 ns to explore the conformational space around clusterC1. Projection of the trajectory in the conformational land-scape shows that MDmodels occupy a wide region of the map,overlapping with both crystallographic cluster C1 and NMRclusterN1 (Fig. 5a). TheB-factors from theMD trajectory (sup-plemental Fig. S5a) are in agreement with NMR data and iden-tify a region of 15 residues with slow dynamics (Pro-9–Arg-74)that corresponds to the circular structural motif (�-helix-C/loop-C/disulfide). The C-tail is highly dynamical, also inaccordance with the NMR data (Fig. 4, a, b, and c).In longer simulations in implicit water for 100 ns, the trajec-
tories provide further atomistic details (supplemental VideoS1). In the Pro-59/Asp-66 region, �-helix-C temporarily breaksdown in two 310 helices due to the conformational restraint ofthe Cys-64 residue, which is involved in the sulfur-bridge (sup-plemental Fig. S5). The conformation of the remaining part ofCP12 was mostly turn, as in the NMR models. Asn-71–Cys-73andArg-74-Tyr-76 tend to form short 310 helices before or afterthe disulfide or, alternatively, to give a short-lived �-helix. Thedisulfide limits the dynamics of Cys-73 and hinders the forma-tion of a well defined structure. In 1 of 20 NMRmodels, a short�-helix was found in this region (supplemental Fig. S5d). Pop-ulations of theH-bonds in percentages are given in supplemen-tal Table S3. The top ranking hydrogen bonds stabilize the�-helix-C and �-turns/310-helices of loop-C. No stableH-bonds involve residues of the C-tail.MD and NMR Concur—Both NMR and MD indicated that
the last 20 amino acids of free, oxidized CP12 can be dividedinto two regions. The first region is characterized by slowdynamics and corresponds to the circular structural motif(�-helix-C/loop-C/disulfide). The second region is a fasterdynamics region that corresponds to the C-tail.The “slow” circular motif (Pro-59–Cys-73) of CP12 popu-
lates twominima (Fig. 6, a–d, and supplemental Fig. S5). In thefree energy surface obtained from MD simulations, the whitedots represent the crystallographic structure. The most popu-latedMDconformation, representing the absoluteminimum, issimilar to the conformation of the circular structural motifobserved in the crystals (cluster C1) and in cluster N1 of NMR
FIGURE 3. NMR analysis of oxidized CP12 in solution. a, superimposition of1H,15N HSQC spectra of oxidized 15N-labeled CP12 either alone (black spots) orwith unlabeled GAPDH (red spots), both samples containing 2.7 mM NAD.Oxidized CP12 showed limited dispersion of signals, and many residues gaveno detectable resonances. Assignment of the C-terminal portion of CP12(Arg-49 –Asn-78) was complete. In the presence of unlabeled GAPDH (red spots),all signals belonging to the C terminus of CP12 (from Asp-55 to Asn-78; bluelabels) disappeared, indicating direct interaction with or close proximity toGAPDH (145 kDa). Assigned residues that do not belong to the C terminus ofCP12 were little affected by GAPDH (red labels). s.c., side chain; Rs.c., arginine sidechain. b, in the primary structure of CP12, assigned residues are underlined. Thecentral part of the protein was almost completely NMR silent. Blue residues gavesignals that disappeared under conditions of complex formation with GAPDH.Signals of red residues were little or not affected by GAPDH.
TABLE 2NMR and refinement statistics for CP12 structuresPairwise root mean square deviation was calculated among 20 refined structures.
CP12
NMR distance and dihedral constraintsDistance constraintsTotal NOE 165Intra-residue 69Inter-residueSequential (�i � j� � 1) 66Medium range (�i � j� � 4) 29Long range (�i � j� � 5) 1Intermolecular 0
Hydrogen bonds 2Total dihedral angle restraints 40
� 20� 20
Structure statisticsViolations (mean and S.D.)Distance constraints (Å) 0.54 � 0.59Dihedral angle constraints (°) 1.08 � 1.06
Deviations from idealized geometryBond lengths (Å) 0.0060 � 0.00091Bond angles (°) 0.8429 � 0.0524Impropers (°) 0.8814 � 0.1057
Average pairwise root mean squaredeviation (Å)
Residues: 59–66/59–70/59–73
Heavy 0.855/1.284/1.966Backbone 0.216/0.754/1.286
Disorder to Order Transitions in CP12 Interacting with GAPDH
21378 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

FIGURE 5. Relationships between NMR, crystal, and MD structures of CP12. a, shown is clusterization by principal component analysis (PCA). Data projectedon the PCA eigenvectors of the NMR models are characterized by the smallest eigenvalues, PCA1 and PCA2. The 20 NMR models (red) formed 5 clusters (N1 toN5). The 9 CP12 crystal chains (blue) formed cluster C1. Gray empty circles represent MD models. Superimposition of all models of each cluster is shown inconventional color code (�-helix-C, red; loop-C, gray; C-tail, blue). The circular structural motif (�-helix-C/loop-C/disulfide) adopts a different conformation ineach NMR cluster. In cluster N1, this conformation is the closest to the one of crystal cluster C1. The C-tail is disordered in all NMR models and only in crystal-lographic models adopted a univocal extended conformation. b, shown is the structure of CP12 chain G (purple) in the cleft formed by GAPDH chain A (yellow)and GAPDH chain D (white) of the co-crystallized complex. A representative CP12 model (blue) belonging to NMR cluster N1 was superimposed on CP12 crystalstructure (superimposition performed on the Pro-59 –Thr-70 segment, i.e. �-helix-C plus first �-turn of loop-C). At the level of the C� (CA) of residue Thr-70 thetwo CP12 molecules diverge. We propose that after the interaction between GAPDH and CP12 in cluster-N1 conformation, the second �-turn of CP12 (blue) flipstoward the crystal conformation (purple) where Glu-72 can form two H-bonds with GAPDH (supplemental Table S1b). Such major conformational changewould trigger the slipping of the flexible C-tail along the groove that fixes CP12 in GAPDH catalytic sites.
FIGURE 4. The structural ensemble of the C-terminal portion of oxidized CP12 in solution. a, TALOS� analysis of CP12 C terminus is shown. The torsionangles of residues Ser-57–Arg-74 were predicted (green bars). This region corresponds to the circular structural motif of crystallized binary complexes. The lastfour residues of the C-tail (Thr-75–Asn-78) were predicted to be dynamic (blue). b, 165 homonuclear (n) NOE signals were recorded and classified as intraresi-due; i � j, red, sequential; �i � j� � 1 (gray), medium range; 1 � �i � j� �5 (blue); long range, �i � j� �4 (purple). The majority of NOE signals involve residues of the�-helix-C (Leu-60 –Asn-67). c, heteronuclear (he) NOE values demonstrate the relative rigidity of the circular structural motif (Leu-60 –Cys-73) in contrast withthe C-tail (Arg-74 –Asn-78). d, superimposition of 20 models of CP12 C terminus (Asp-58 –Asn-78) was calculated from NMR data. The color code is the same asin Fig. 1: �-helix-C (red), loop-C (gray), C-tail (blue). Disulfides are in stick representation with sulfur atoms in yellow. The figure also includes the crystallographicstructure of CP12 chain G (purple). The structural alignment was based on the �-helix-C (Pro-59 –Asp-66), present in all 20 NMR models and crystallographicCP12. e, the aligned loops-C of the 20 NMR showed a helicoidal structure formed by two �-turns (Asn-67–Thr-70; Thr-70 –Cys-73). The position of the C� ofcrucial residues is indicated. Crystallized loop-C could not be aligned with NMR loops-C. f, loop-C-based alignment of the 20 NMR models shows that the angleof insertion of loop-C with �-helix-C varies substantially in the different models.
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21379
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from
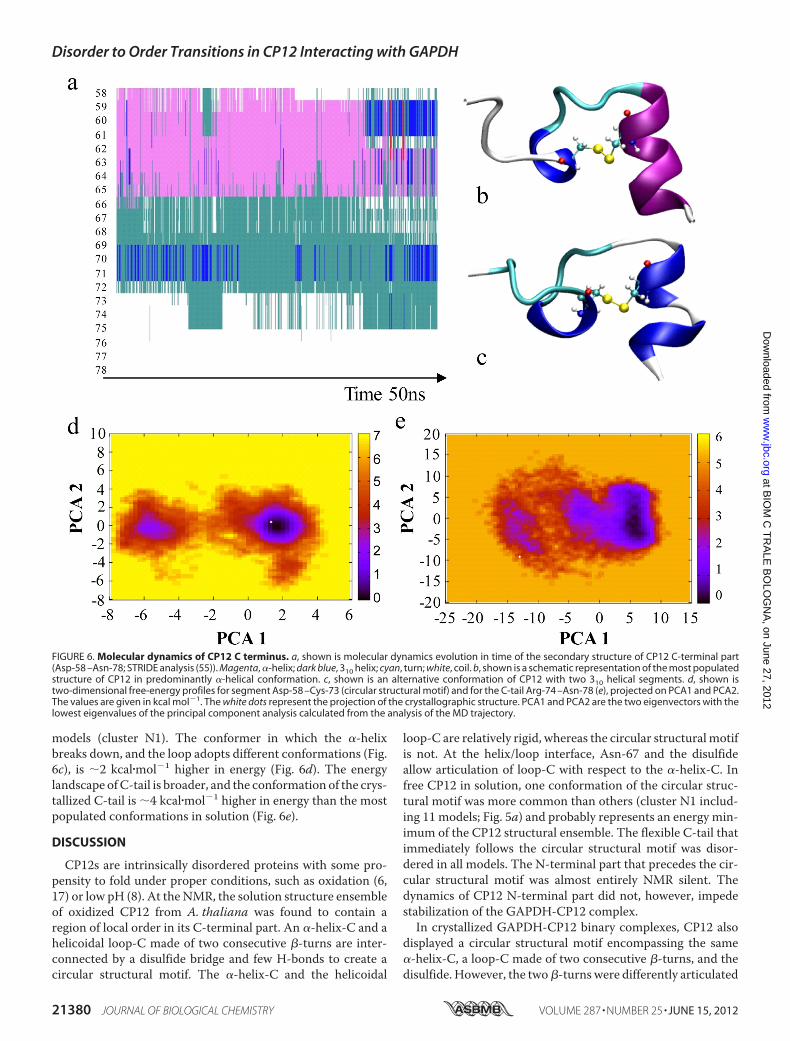
models (cluster N1). The conformer in which the �-helixbreaks down, and the loop adopts different conformations (Fig.6c), is 2 kcal�mol�1 higher in energy (Fig. 6d). The energylandscape ofC-tail is broader, and the conformation of the crys-tallized C-tail is 4 kcal�mol�1 higher in energy than the mostpopulated conformations in solution (Fig. 6e).
DISCUSSION
CP12s are intrinsically disordered proteins with some pro-pensity to fold under proper conditions, such as oxidation (6,17) or low pH (8). At the NMR, the solution structure ensembleof oxidized CP12 from A. thaliana was found to contain aregion of local order in its C-terminal part. An �-helix-C and ahelicoidal loop-C made of two consecutive �-turns are inter-connected by a disulfide bridge and few H-bonds to create acircular structural motif. The �-helix-C and the helicoidal
loop-C are relatively rigid, whereas the circular structural motifis not. At the helix/loop interface, Asn-67 and the disulfideallow articulation of loop-C with respect to the �-helix-C. Infree CP12 in solution, one conformation of the circular struc-tural motif was more common than others (cluster N1 includ-ing 11 models; Fig. 5a) and probably represents an energy min-imum of the CP12 structural ensemble. The flexible C-tail thatimmediately follows the circular structural motif was disor-dered in all models. The N-terminal part that precedes the cir-cular structural motif was almost entirely NMR silent. Thedynamics of CP12 N-terminal part did not, however, impedestabilization of the GAPDH-CP12 complex.In crystallized GAPDH-CP12 binary complexes, CP12 also
displayed a circular structural motif encompassing the same�-helix-C, a loop-C made of two consecutive �-turns, and thedisulfide. However, the two�-turns were differently articulated
FIGURE 6. Molecular dynamics of CP12 C terminus. a, shown is molecular dynamics evolution in time of the secondary structure of CP12 C-terminal part(Asp-58 –Asn-78; STRIDE analysis (55)). Magenta, �-helix; dark blue, 310 helix; cyan, turn; white, coil. b, shown is a schematic representation of the most populatedstructure of CP12 in predominantly �-helical conformation. c, shown is an alternative conformation of CP12 with two 310 helical segments. d, shown istwo-dimensional free-energy profiles for segment Asp-58 –Cys-73 (circular structural motif) and for the C-tail Arg-74 –Asn-78 (e), projected on PCA1 and PCA2.The values are given in kcal mol�1. The white dots represent the projection of the crystallographic structure. PCA1 and PCA2 are the two eigenvectors with thelowest eigenvalues of the principal component analysis calculated from the analysis of the MD trajectory.
Disorder to Order Transitions in CP12 Interacting with GAPDH
21380 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

at the level of Thr-70 (Fig. 5b), the residue that ends the first�-turn and initiates the second. The crystallized loop-C (Fig. 1f)was, therefore, not helicoidal as that of CP12 solution struc-tures (Fig. 4e). Despite this difference, the connection betweenloop-C and�-helix-C (via the joint residue Asn-67) is similar inboth the crystallized binary complex and in cluster N1 of CP12solution structures (Fig. 5b). All other NMR alternative struc-tures are different in this respect. The extended conformationof the C-tail embedded in the GAPDH was never found insolution.The circular structural motif �-helix-C/loop-C/disulfide is
likely to play a major role in the process of binding, in agree-ment with PONDRVL-XT (49) analysis of CP12 (supplementalFig. S6). Short ordered sequences within large unstructuredregions are often involved in binding interactions (49). Thesemolecular recognition features (31) often include amphipathic�-helices (23) and fold either before or after binding (50).PONDR VL-XT predicts a short segment of nine residues cor-responding to amphipatic �-helix-C as the only ordered ele-ment of the entire CP12, strongly suggesting a role in binding.The disorder of the rest of CP12 in solution is experimentallysupported also by its ability to diffuse into GAPDH crystals,allowing formation of binary complexes by soaking.We predict that an encounter complex forms by interaction
of GAPDH with oxidized CP12 in cluster-N1 conformation(one of the possible conformations of the NMR landscape; Fig.5, a and b). Although subject to rapidmovements that can evenbreak the �-helix-C, the circular structural motif of cluster N1is the most stable conformation of the MD landscape (Fig. 6, aand b). In the encounter complex, the �-helix-C and the first�-turn of loop-C lodge in aGAPDHcavity delimited by positiveside chains (Arg-77, Arg-191; Fig. 1, b and c). Few (and variable)interactions stabilize this binding; often one single interactionlinking the �-helix-C to GAPDH (Arg-191) and an ionic bondbetween the side chains of Glu-69 (CP12) and Arg-77(GAPDH). The essential role of these two GAPDH arginines inCP12 binding was proved in the orthologous Chlamydomonassystem (51, 52) and confirmed by the crystallographic structureof the binary complex from S. elongatus (16). Therefore, theGAPDH-CP12 encounter complex is the result of a prelimi-nary, albeit essential, conformational selection performed byGAPDH on the CP12 structural ensemble. Our data do notsupport the recently proposed model of CP12 folding uponbinding to GAPDH (16), although induced folding steps areindeed involved in the stabilization of the encounter complex.Evolution of the encounter complex into the final stable
structure of the crystallized binary complex implies a majorrearrangement of the rest of CP12, whereas the general foldingof the enzyme is not influenced. The possibility of CP12 Glu-72to form H-bonds with different GAPDH residues induces thesecond�-turn to flip in theGAPDHcavity (Fig. 5b), thus adopt-ing the conformation observed in the crystallized binarycomplex.The flip of the second�-turn starts the zippering process that
drives the C-tail to fit into the GAPDH groove that leads to thesubstrate-phosphate binding sites (Fig. 2c). The crystal struc-ture of the binary complex suggests that the energy of CP12binding to GAPDH (enthalpic term) must crucially depend on
the C-tail, which is anchored to GAPDH by up to 12 H-bonds(supplemental Table S1b). In this view, the inability of reducedCP12 to bindGAPDH(12) is simply a consequence of the desta-bilization of this structural motif in the absence of the disulfidebridge.Without the initial recognition of the circular structuralmotif, the binding of the C-tail is hindered by its dynamism.Although the capability of reduced CP12 to bind GAPDH wasreported in C. reinhardtii (53), we could not confirm the sametype of binding in the Arabidopsis system.3
The previously measured negative entropic term of 5kcal�mol�1 at room temperature for the CP12-GAPDH inter-action (17) can be attributed to the stiffening of the C-tails inthe GAPDH-CP12 complex. The enthalpic counterpart is rep-resented by the great number of short-distance interactionsthat are formed between theC-tail of CP12 andGAPDH,whichcan supply the energy necessary for the binding (�15kcal�mol�1; Ref. 17). We propose that the interaction of CP12with GAPDH follows a synergistic mechanism (24) that com-bines conformational selection, triggered by CP12 oxidation,and folding upon binding.Conformational selection is important for the initial recog-
nition event when GAPDH samples the CP12 conformationalensemble and fishes out the oxidized molecules with the pre-formed circular structural motif in apt conformation. Foldingupon binding occurs only in a second step. The flip of the sec-ond �-turn helps the flexible C-tail of the CP12 molecule rec-ognized by GAPDH to slip into the catalytic site where itassumes an ordered, extended conformation. The MD showthat in solution this extended conformation of the C-tail is notusual. The calculations show that for the Arg-74–Asn-78 seg-ment the energy cost is 4 kcal/mol for folding upon binding(Fig. 6e). The process allows going from the more stable con-formation of the tail in water to that of the bound state. Picto-rially, oxidation baits the fishing rod of CP12 with the circularstructuralmotif�-helix-C/loop-C/disulfide, andupon catchingits GAPDH-prey, the terminal part of the rod stiffens.
Acknowledgments—We thank the European Synchrotron RadiationFacility (Grenoble, France) for access to ID14 beam lines. We thankEwen Lescop (ICSN, UPR CNRS 2301, Gif sur Yvette, France),Nathalie Sibille, Guy Lippens, Isabelle Landrieu (UGSF,UMRCNRS/Universite Lille1 8576, Villeneuve d’Ascq, France), Pierre Gans,Adrien Favier, Bernard Brutscher (IBS, UMR CNRS/CEA/UniversiteGrenoble1 5075, Grenoble, France) for helpful discussions. The 600-and 800-MHz NMR facilities used in this study were funded by theRégion Nord-Pas de Calais (France), FEDER, Ministère de la Recher-che, the CNRS, the Université Lille1-Science et Technologies, and theInstitut Pasteur de Lille.
REFERENCES1. Buchanan, B. B., and Balmer, Y. (2005) Redox regulation. A broadening
horizon. Annu. Rev. Plant Biol. 56, 187–2202. Dai, S., Friemann, R., Glauser, D. A., Bourquin, F., Manieri, W., Schür-
mann, P., and Eklund, H. (2007) Structural snapshots along the reactionpathway of ferredoxin-thioredoxin reductase. Nature 448, 92–96
3. Trost, P., Fermani, S., Marri, L., Zaffagnini, M., Falini, G., Scagliarini, S.,
3 L. Marri, unpublished information.
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21381
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

Pupillo, P., and Sparla, F. (2006) Thioredoxin-dependent regulation ofphotosynthetic glyceraldehyde-3-phosphate dehydrogenase. Autono-mous vs. CP12-dependent mechanisms. Photosynth. Res. 89, 263–275
4. Wedel, N., and Soll, J. (1998) Evolutionary conserved light regulation ofCalvin cycle activity by NAPDH-mediated reversible phosphoribuloki-nase/CP12/glyceraldehyde-3-phosphate-dehydrogenase complex disso-ciation. Proc. Natl. Acad. Sci. U.S.A. 95, 9699–9704
5. Scheibe, R., Wedel, N., Vetter, S., Emmerlich, V., and Sauermann, S. M.(2002) Co-existence of two regulatory NADP-glyceraldehyde 3-phos-phate dehydrogenase complexes in higher plant chloroplasts. Eur.J. Biochem. 269, 5617–5624
6. Graciet, E., Gans, P.,Wedel, N., Lebreton, S., Camadro, J.M., andGontero,B. (2003) The small protein CP12. A protein linker for supramolecularcomplex assembly. Biochemistry 42, 8163–8170
7. Tamoi,M.,Miyazaki, T., Fukamizo, T., and Shigeoka, S. (2005) TheCalvincycle in cyanobacteria is regulated by CP12 via NAD(H)/NADP(H) ratiounder light/dark conditions. Plant J. 42, 504–513
8. Marri, L., Pesaresi, A., Valerio, C., Lamba, D., Pupillo, P., Trost, P., andSparla, F. (2010) In vitro characterization of Arabidopsis CP12 isoformsreveals common biochemical and molecular properties. J. Plant Physiol.167, 939–950
9. Howard, T. P., Lloyd, J. C., and Raines, C. A. (2011) Interspecies variationin the oligomeric states of the higher plant Calvin cycle enzymes glyceral-dehyde-3-phosphate dehydrogenase and phosphoribulokinase. J. Exp.Bot. 62, 3799–3805
10. Marri, L., Zaffagnini, M., Collin, V., Issakidis-Bourguet, E., Lemaire, S. D.,Pupillo, P., Sparla, F., Miginiac-Maslow, M., and Trost, P. (2009) Promptand easy activation by specific thioredoxins of Calvin cycle enzymes ofArabidopsis thaliana associated in the GAPDH-CP12/PRK supramolecu-lar complex.Mol. Plant 2, 259–269
11. Fermani, S., Sparla, F., Falini, G., Martelli, P. L., Casadio, R., Pupillo, P.,Ripamonti, A., and Trost, P. (2007) Molecular mechanism of thioredoxinregulation in photosynthetic A2B2-glyceraldehyde-3-phosphate dehy-drogenase. Proc. Natl. Acad. Sci. U.S.A. 104, 11109–11114
12. Marri, L., Trost, P., Pupillo, P., and Sparla, F. (2005) Reconstitution andproperties of the recombinant glyceraldehyde-3-phosphate dehydroge-nase/CP12/phosphoribulokinase supramolecular complex of Arabidop-sis. Plant Physiol. 139, 1433–1443
13. Howard, T. P., Metodiev, M., Lloyd, J. C., and Raines, C. A. (2008) Thiore-doxin-mediated reversible dissociation of a stromalmultiprotein complexin response to changes in light availability. Proc. Natl. Acad. Sci. U.S.A.105, 4056–4061
14. Howard, T. P., Fryer, M. J., Singh, P., Metodiev, M., Lytovchenko, A.,Obata, T., Fernie, A. R., Kruger, N. J., Quick,W. P., Lloyd, J. C., and Raines,C. A. (2011) Antisense suppression of the small chloroplast protein CP12in tobacco alters carbon partitioning and severely restricts growth. PlantPhysiol. 157, 620–631
15. Thompson, L. R., Zeng, Q., Kelly, L., Huang, K. H., Singer, A. U., Stubbe, J.,and Chisholm, S. W. (2011) Phage auxiliary metabolic genes and the redi-rection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci.U.S.A. 108, E757–E764
16. Matsumura, H., Kai, A., Maeda, T., Tamoi, M., Satoh, A., Tamura, H.,Hirose, M., Ogawa, T., Kizu, N., Wadano, A., Inoue, T., and Shigeoka, S.(2011) Structure basis for the regulation of glyceraldehyde-3-phosphatedehydrogenase activity via the intrinsically disordered protein CP12.Structure 19, 1846–1854
17. Marri, L., Trost, P., Trivelli, X., Gonnelli, L., Pupillo, P., and Sparla, F.(2008) Spontaneous assembly of photosynthetic supramolecular com-plexes as mediated by the intrinsically unstructured protein CP12. J. Biol.Chem. 283, 1831–1838
18. Lebreton, S., Andreescu, S., Graciet, E., andGontero, B. (2006)Mapping ofthe interaction site of CP12 with glyceraldehyde-3-phosphate dehydro-genase from Chlamydomonas reinhardtii. Functional consequences forglyceraldehyde-3-phosphate dehydrogenase. FEBS J. 273, 3358–3369
19. Ward, J. J., Sodhi, J. S.,McGuffin, L. J., Buxton, B. F., and Jones, D. T. (2004)Prediction and functional analysis of native disorder in proteins from thethree kingdoms of life. J. Mol. Biol. 26, 635–645
20. Sun, X., Jones, W. T., Harvey, D., Edwards, P. J., Pascal, S. M., Kirk, C.,
Considine, T., Sheerin, D. J., Rakonjac, J., Oldfield, C. J., Xue, B., Dunker,A. K., and Uversky, V. N. (2010) N-terminal domains of DELLA proteinsare intrinsically unstructured in the absence of interaction with GID1/gibberellic acid receptors. J. Biol. Chem. 285, 11557–11571
21. Cortese, M. S., Uversky, V. N., and Dunker, A. K. (2008) Intrinsic disorderin scaffold proteins. Getting more from less. Prog. Biophys. Mol. Biol. 98,85–106
22. Tompa, P., and Fuxreiter, M. (2008) Fuzzy complexes. Polymorphism andstructural disorder in protein-protein interactions. Trends Biochem. Sci.33, 2–8
23. Wright, P. E., and Dyson, H. J. (2009) Linking folding and binding, Curr.Opin. Struct. Biol. 19, 31–38
24. Espinoza-Fonseca L.M. (2009) Reconciling bindingmechanisms of intrin-sically disordered proteins. Biochem. Biophys. Res. Commun. 382,479–482
25. Boehr, D. D., Nussinov, R., and Wright, P. E. (2009) The role of dynamicconformational ensembles in biomolecular recognition. Nat. Chem. Biol.5, 789–796
26. Dyson, H. J., and Wright, P. E. (2005) Intrinsically unstructured proteinsand their functions, Nat. Rev. Mol. Cell Biol. 6, 197–208
27. Kjaergaard, M., Teilum, K., and Poulsen, F. M. (2010) Conformationalselection in the molten globule state of the nuclear coactivator bindingdomain of CBP. Proc. Natl. Acad. Sci. U.S.A. 107, 12535–12540
28. Sugase, K., Dyson, H. J., and Wright P. E. (2007) Mechanism of coupledfolding and binding of an intrinsically disordered protein. Nature 447,1021–1025
29. Wang, T., Darwin, K. H., and Li, H. (2010) Binding-induced folding ofprokaryotic ubiquitin-like protein on the Mycobacterium proteasomalATPase targets substrates for degradation. Nat. Struct. Mol. Biol. 17,1352–1357
30. Lee, C.W.,Martinez-Yamout,M. A., Dyson, H. J., andWright, P. E. (2010)Structure of the p53 transactivation domain in complex with the nuclearreceptor coactivator binding domain of CREB-binding protein. Biochem-istry 49, 9964–9971
31. Mohan, A., Oldfield, C. J., Radivojac, P., Vacic, V., Cortese, M. S., Dunker,A. K., andUversky, V.N. (2006)Analysis ofmolecular recognition features(MoRFs), J. Mol. Biol. 362, 1043–1059
32. Fuxreiter, M., Simon, I., Friedrich, P., and Tompa, P. (2004) Preformedstructural elements feature in partner recognition by intrinsically unstruc-tured proteins. J. Mol. Biol. 338, 1015–1026
33. Fermani, S., Sparla, F., Marri, L., Thumiger, A., Pupillo, P., Falini, G., andTrost, P. (2010) The crystal structure of photosynthetic glyceraldehyde-3-phosphate dehydrogenase (isoform A4) from Arabidopsis thaliana incomplex with NAD. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun.66, 621–626
34. Leslie, A. G. W. (1992) Recent changes to the MOSFLM package for pro-cessing film and image plate data. Joint CCP4 � ESF-EAMCB Newsletteron Protein Crystallography 26, 27–33
35. Evans, P. (2006) Scaling and assessment of data quality.ActaCrystallogr. DBiol. Crystallogr. 62, 72–82
36. Otwinowsky, Z., and Minor, W. (1997) Processing of X-ray diffractiondata collected in oscillation mode.Methods Enzymol. 276, 307–326
37. Navaza, J. (1994) AMoRe. An automated package for molecular replace-ment. Acta Crystallogr. A 50, 157–163
38. Vagin, A. A., Steiner, R. A., Lebedev, A. A., Potterton, L., McNicholas, S.,Long, F., and Murshudov, G. N. (2004) REFMAC5 dictionary: organiza-tion of prior chemical knowledge and guidelines for its use. Acta Crystal-logr. D Biol. Crystallogr. 60, 2184–2195
39. Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P.,Grosse-Kunstleve, R.W., Jiang, J. S., Kuszewski, J., Nilges,M., Pannu,N. S.,Read, R. J., Rice, L. M., Simonson, T., andWarren, G. L. (1998) Crystallog-raphy and NMR system (CNS). A new software system for macromolec-ular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54,905–921
40. Emsley, P., and Cowtan, K. (2004) Coot. Model-building tools for molec-ular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132
41. Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M.(1993) PROCHECK: a program to check the stereochemical quality of
Disorder to Order Transitions in CP12 Interacting with GAPDH
21382 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 287 • NUMBER 25 • JUNE 15, 2012
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

protein structures. J. Appl. Crystallogr. 26, 283–29142. Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A.
(1995) NMRPipe. A multidimensional spectral processing system basedon UNIX pipes. J. Biomol. NMR 6, 277–293
43. Johnson, B. A., and Blevins, R. A. (1994) NMRView. A computer programfor the visualization and analysis ofNMRdata. J. Biomol. NMR 4, 603–614
44. Shen, Y., Delaglio, F., Cornilescu, G., and Bax, A. (2009) TALOS�. Ahybrid method for predicting protein backbone torsion angles fromNMRchemical shifts. J. Biomol. NMR 44, 213–223
45. Favier, A., and Brutscher, B. (2011) Recovering lost magnetization. Polar-ization enhancement in biomolecular NMR. J. Biomol. NMR 49, 9–15
46. Case, D. A., Cheatham, T. E., 3rd, Darden, T., Gohlke, H., Luo, R., Merz,K. M., Jr., Onufriev, A., Simmerling, C., Wang, B., andWoods, R. J. (2005)The Amber biomolecular simulation programs. J. Comput. Chem. 26,1668–1688
47. Cornell,W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K.M., Ferguson,D.M., Spellmeyer, D. C., Fox, T., Caldwell, J.W., andKollman, P. A. (1995)A second generation force field for the simulation of proteins, nucleicacids, and organic molecules. J. Am. Chem. Soc. 117, 5179–5197
48. van Gunsteren, W. F., and Berendsen, H. J. C. (1997) Algorithms for mac-romolecular dynamics and constraint dynamics. Mol. Physiol. 34,1311–1327
49. Garner, E., Romero, P., Dunker, A. K., Brown, C., andObradovic, Z. (1999)
Predicting binding regions within disordered proteins. Genome Inform.Ser. Workshop Genome Inform. 10, 41–50
50. Oldfield, C. J., Cheng, Y., Cortese, M. S., Romero, P., Uversky, V. N., andDunker, A. K. (2005) Coupled folding and binding with �-helix-formingmolecular recognition elements. Biochemistry 44, 12454–12470
51. Graciet, E., Mulliert, G., Lebreton, S., and Gontero, B. (2004) Involvementof two positively charged residues of Chlamydomonas reinhardtii glycer-aldehyde-3-phosphate dehydrogenase in the assembly process of a bi-enzyme complex involved in CO2 assimilation. Eur. J. Biochem. 271,4737–4744
52. Erales, J., Mekhalfi, M., Woudstra, M., and Gontero, B. (2011) Molecularmechanism of NADPH-glyceraldehyde-3-phosphate dehydrogenase reg-ulation through the C terminus of CP12 in Chlamydomonas reinhardtii.Biochemistry 50, 2881–2888
53. Erales, J., Lignon, S., and Gontero, B. (2009) CP12 from Chlamydomonasreinhardtii, a permanent specific “chaperone-like” protein of glyceralde-hydes-3-phosphate dehydrogenase. J. Biol. Chem. 284, 12735–12744
54. Nicholls, A., Sharp, K. A., and Honig, B. (1991) Protein folding and asso-ciation. Insights from the interfacial and thermodynamic properties ofhydrocarbons. Proteins Struct. Funct. Genet. 11, 281–296
55. Frishman, D., and Argos, P. (1995) Knowledge-based protein secondarystructure assignment. Proteins 23, 566–579
Disorder to Order Transitions in CP12 Interacting with GAPDH
JUNE 15, 2012 • VOLUME 287 • NUMBER 25 JOURNAL OF BIOLOGICAL CHEMISTRY 21383
at BIO
M C
TR
ALE
BO
LOG
NA
, on June 27, 2012w
ww
.jbc.orgD
ownloaded from

1
Conformational selection and folding-upon-binding of an intrinsically
disordered protein regulate photosynthetic enzymes assembly
Simona Fermani, Xavier Trivelli, Francesca Sparla, Anton Thumiger, Matteo Calvaresi,
Lucia Marri, Giuseppe Falini, Francesco Zerbetto, Paolo Trost
Supplementary Figure S1 – CP12 electron density map.
2[Fo-Fc] electron density map around CP12 molecule (chain G) in co-crystallized
binary complex, at 1σ cut-off. The CP12 is represented in sticks, GAPDH in ribbon.
Atom color codes: carbon, green; oxygen, red; nitrogen, blue; sulphur, yellow.

2
Supplementary Figure S2 - Asymmetric units and position of CP12 molecules
in binary complexes obtained by co-crystallization and by soaking.
Panels a and d show, in cartoon representation, the asymmetric units of crystals of
GAPDH/CP12 binary complex obtained by either co-crystallization (left) or soaking
(right). In co-crystallization, the A.U. contains 6 chains of GAPDH (A to F, one
tetramer and one dimer, different colours) and 3 chains of CP12 (G to I, green). In
soaked crystals, the asymmetric unit contains 10 GAPDH chains (A to H, and OQ; two
tetramers and one dimer, different colours) and 6 CP12 chains (I,J,M,N,L,K, green).
Panels b and c (co-crystallization data), and, e, f and g (soaking data), schematically
show the binding of CP12 to GAPDH in the different binary complexes. Two CP12
molecules are bound to each GAPDH tetramer, one in each deep cleft that contains
two binding sites for CP12. The location of CP12 in one of the clefts does not appear
to be related to the position of CP12 in the other cleft. In the schematic drawing of the
A
BC
D
G
H
F
EC
D
I
F
EF’
E’
I
I’
AGD
B
HFC
EI
a
b
c
A
BC
D
I
J
E
FG
H
N
M
Q
O
K
O
’
’
O
K
L L
’
K’
L
A
B
C
DI
J
EF
GH
M
N
O
QL
K
e
f
g
d
Co-crystallization Soaking
P-axis
R-axis
Q-axis

3
binary complex, the dashed circles represent the four possible positions that can be
occupied by the α-helices-C of CP12. The green colour indicates the actual occupation.
Co-crystallization:
(b) The GAPDH tetramer ABCD binds CP12-chains G and H. In the reported view,
CP12-chain-G occupies the position in front of GAPDH-chain-A, and CP12-chain-H
occupies the position behind GAPDH-chain-B. Symmetry axis are shown.
(c) The GAPDH dimer EF together with CP12-chain-I generates by symmetry a second
binary complex in which the two CP12 are located behind GAPDH-chains-E and -E’.
The type of CP12 occupation in the EE’FF’ symmetry-generated tetramer is therefore
different from that of the independent tetramer ABCD.
Soaking:
(e) The ABCD tetramer binds CP12-chain-I in front of GAPDH-chain-A and CP12-
chain-J in front of GAPDH-chain-C.
(f) In the tetramer EFGH the two CP12 occupy different positions: CP12-chain-N is in
front of GAPDH-chain-E, but CP12-chain-M is behind GAPDH-chain-F.
(g) The GAPDH dimer OQ is associated with two CP12 molecules (K,L) showing 50%
occupation (light green). In the symmetry-generated binary complex (OO’QQ’), all
four binding sites are thus statistically 50% occupied by CP12 molecules. This
interpretation is consistent with the experimental data that A4-GAPDH binds two CP12
molecules (Marri et al.,(2008) J. Biol. Chem. 283, 1831-1838).

4
Supplementary Table S1- Intra-CP12 and CP12-GAPDH molecular
interactions.
a) CP12 intramolecular interactions. Distance cut-off of 3.1 Å. Residues highlighted
in red belong to helix-C, in gray to loop-C and in blue to the C-tail. The most
conserved interactions in the 9 CP12 chains are highlighted yellow.
CP12 d (Å)
Residue Atom Residue Atom Co-cryst. complex Soaked complex
G H I I J K L M N
Ser57 OG Leu60 N 2.8
Asp58 O Leu60 N 3.1
Asp58 O Glu61 N 2.9
Asp58 O Glu62 N 3.0 3.1
Asp58 OD1 Glu61 OE2 3.0
Asp58 OD2 Leu60 N 2.8 3.1
Asp58 OD2 Glu61 N 2.6
Pro59 O Glu61 N 2.9 2.6
Pro59 O Glu62 N 2.6 2.8
Pro59 O Tyr63 N 2.7 3.1 2.9
Leu60 O Glu62 N 3.1 2.8 3.0
Leu60 O Tyr63 N 3.1 2.5 2.8
Leu60 O Cys64 N 3.0 3.1 2.8 3.1 2.9 2.8 3.1 2.9 3.0
Glu61 O Cys64 N 3.1
Glu62 O Lys65 N 3.0 2.9 3.0 3.1
Glu62 O Asp66 OD2 2.6
Glu62 OE1/2 Lys65 NZ 2.3
Tyr63 O Lys65 N 2.9 2.7
Tyr63 O Asp66 N 3.1 3.1 3.0 2.7
Tyr63 O Asp66 OD1 2.8
Tyr63 O Asn67 N 2.9 2.9 3.1 2.9 2.9
Tyr63 O Asn67 ND2 2.8 2.4 3.1 2.6
Cys64 O Asp66 N 2.9 2.8 2.8
Cys64 O Asn67 N 3.0 2.8 2.9
Cys64 O Pro68 N 3.1 2.9 3.1
Asn67 O Glu69 N 2.9 3.0
Asn67 O Thr70 N 3.0 3.1
Asn67 O Thr70 OG1 2.9 3.0 3.0 2.9 2.6 2.8 2.9 3.0
Pro68 O Thr70 N 3.1
Pro68 O Arg74 N 3.1
Glu69 N Glu69 OE1/2 2.7 2.8 2.9 2.4 3.1 2.4 2.4 3.1
Glu69 O Arg74 NE 3.1 3.0
Thr70 O Glu72 N 3.1 3.0 3.1
Thr70 O Cys73 N 3.1 3.0 2.9 3.1 2.9 3.0 2.6
Thr70 OG1 Cys73 SG 2.9 2.9 3.1 3.0 2.7
Thr70 O Arg74 N 3.0 3.1 3.1 3.0 3.0 3.0
Asn71 O Cys73 N 3.0
Asn71 OD1 Arg74 NH1 3.1 3.0 3.0
Asn71 OD1 Arg74 NH2 2.6 2.9 2.6 2.7 2.5
Cys73 O Thr75 N 3.1 3.0 3.1
Cys73 O Thr75 OG1 2.8 2.6 2.9 3.0 2.6 2.6 2.5
Tyr76 O Asn78 N 3.0

5
b) CP12-GAPDH interactions. Distance cut-off 3.1 Å. CP12 residues highlighted in
red belong to helix-C, in yellow to loop-C and in blue to the C-tail. The most
conserved interactions are highlighted yellow. C = GAPDH chain. d = distance
in Å.
Co-cryst. complex Soaked complex CP12 GAPDH CP12-G CP12-H CP12-I CP12-I CP12-J CP12-K CP12-L CP12-M CP12-N
Residue Atom Residue Atom C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
C d
(Å)
Asp55 O Gly60A O Q’ 3.0
Ser57 O Lys38 N Q’ 2.8
Asp58 N Lys38 NZ E 3.1
Asp58 OD1 Lys38 NZ B 3.1
Asp58 OD2 Glu61 OE2 E’ 2.5
Leu60 N Gly34 O B 3.1
Glu61 OE1 Arg191 NH2
Glu61 OE2 Arg191 NH1 D 3.1
Glu61 OE1/2 Arg191 NE D 2.7 G 2.7
Glu62 OE2 Arg77 NH1
Tyr63 OH Arg77 NH1/2 C 2.7 F 2.9
Asn67 OD1 Arg77 NH2 D 2.8 H 2.9
Glu69 OE1 Arg77 NH2 D 2.7 D 2.7
Glu69 OE2 Arg77 NH2 O 2.8
Glu69 OE2 Arg77 NH1 O 2.8 Q 3.1
Glu69 OE1 NAD335 N7A H 3.1
Glu69 OE1 NAD335 N6A H 3.1
Asn71 O Arg183 NH1 Q 3.1
Glu72 O Arg191 N D 2.9 F 2.9 D 3.1 O 3.1 H 2.7
Glu72 OE1 Thr33 OG1 A 2.7 Q’ 3.1
Glu72 OE1/2 Ser188 OG C 3.1 Q 2.5 H 2.5
Glu72 OE1 NAD335 O2B A 2.9 F 3.1
Arg74 NH1 Arg183 NH2 O’ 2.9 E 3.0
Arg74 NH2 Arg183 NH2 E 2.8
Arg74 NH2 Gly180 O D 3.1
Arg74 NE Gly180 O C 3.0
Arg74 O His190 NE2 D 2.7 C 2.5 F 2.6 D 2.7 B 3.0 O 2.6 G 2.9 H 2.5
Arg74 O Asp181 OD1/2 O 2.4 G 2.6
Thr75 N Asp181 OD1/2 O 2.7 G 3.1
Thr75 OG1 Arg191 NH1 D 3.0 O 3.0 G 2.8
Thr75 OG1 Arg191 NE F 3.0
Tyr76 N Arg195 NH1 D 2.9 B 2.9
Tyr76 N Asp181 OD1 D 2.9 C 2.9 F 3.0 G 2.5 H 2.6
Tyr76 N Asp181 OD2 O 2.6 H 2.8
Tyr76 O Arg195 NH1 D 2.4 B 2.9
Tyr76 O Arg195 NH2 D 2.8 F 2.7 B 3.1 O 2.7 G 2.8
Tyr76 O Asp181 OD1 D 3.1 G 3.1
Tyr76 O Arg231 NH2 D 2.9 C 3.0
Tyr76 OH NAD335 O1A D 2.9 C 2.9 F 3.1 D 2.7 B 3.0 O 2.7 G 2.8 H 2.4
Tyr76 OH NAD335 PA H 3.1
Tyr76 OH NAD335 O3 H 2.8
Asp77 OD1 Thr208 O B 3.1
Asp77 OD1 Thr208 OG1 O 3.0
Asp77 OD1 Gly209 N D 2.9
Asn78 O Arg231 NH2 D 3.0 B 2.7

6
Asn78 O Arg231 NE B 3.1
Asn78 O Thr179 OG1 B 2.9
Asn78 O Thr150 OG1 D 2.4
Asn78 O Ser148 OG O 2.8
Asn78 O Ser207 O G 3.1
Asn78 O Thr208 OG1 O 2.8
Asn78 OXT Thr208 OG1 D 2.3
Asn78 OD1 Cys149 SG D 3.0 B 3.0 G 2.9
Asn78 OD1 His176 NE2 G 3.1
Asn78 ND2 Cys149 SG D 2.4 B 2.9
Asn78 ND2 Thr150 N D 2.7
Asn78 ND2 Thr150 OG1 D 2.8
Asn78 ND2 Arg231 NE O 2.8
Asn78 ND2 Arg231 NH2 G 2.6

7
a) b)
Supplementary Figure S3 – Effects of CP12 binding on GAPDH.
a) C-alpha superimposition of the A4-GAPDH (chains A, B, C, D, cyan) structure
from co-crystallized binary complex with the A4-GAPDH structure (magenta;
Fermani et al., (2010) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun.
66, 621-626). The CP12 molecules (chains G,H) belonging to the binary
complex are show in purple as cartoon representation. The superimposition
indicates that the interaction between the complex partners does not affect the
structure of GAPDH.
b) Partial displacing of sulphate ions by CP12 in soaked binary complexes. The
figure shows the binding sites for sulphate ions and CP12 (chain I) in the cleft
formed by GAPDH-chain-A and of GAPDH-chain-D in soaked binary complex.
The sulphate and the side chains of GAPDH residues forming the P-sites are
represented in ball-and –sticks, CP12 (purple) and GAPDH (chain A cyan, and
chain D blue) in cartoon. All P-sites were occupied by sulphate ions before the
soaking process (Fermani et al. (2010) Acta Crystallogr. Sect. F Struct. Biol.
Cryst. Commun. 66, 621-626). In this complex, CP12-chain-I occupies Ps- and
Pi-sites of GAPDH-chain-D, thereby displacing the sulphates from both P-sites.
On the contrary, both Ps- and Pi-sites of GAPDH-chain-A are normally occupied
by sulphate ions (SO4336 and SO4337, respectively).

8
Supplementary Table S2 – Correspondence between major CP12-GAPDH
interactions in Arabidopsis thaliana and Synechococcus elongatus complex
structures
Selected interactions between CP12 and GAPDH in the Arabidopsis thaliana (At)
binary complex (left side) were observed in at least two over three CP12 chains of the
co-crystallized complex (see Supplementary Tab. 1). Corresponding interactions
between CP12 and GAPDH in the complex from Synechococcus elongatus (Se, right side)(Matsumura et al. (2011) Structure 19, 1846–1854) were selected on the basis
of sequence and structural alignment (see Supplementary Fig. S4).
At-CP12 At-GAPDH Se-GAPDH Se-GAPDH
E72 O R191 N E69 O R196 N
R74 O H190 NE2 L71 O H195 NE2
Y76 N D181 OD1 Y73 N D187 OD1
Y76 O R195 NH2 Y73 O R200 NH2
Y76 O R231 NH2 Y73 O R236 NH2
Y76 OH NAD335 O1A Y73 OH NAD340 O1A

9
a)
64 73
| |
Q9LZP9_ARATH GSDPLEEYCKDNPETNECRTYDN
Q6BBK3_SYNE7 TEPFFGDYCSENPDAAECLIYDD
| |
61 70
b)
Supplementary Figure S4 – Sequence and structural alignment between from
Arabidopsis and Synecochoccus CP12-GAPDH binary complexes.
a) Sequence alignment of the co-crystallized portion of CP12 from Arabidopsis thaliana (Q9LZP9_ARATH) and Synechococcus elongatus (Q6BBK3_SYNE7).
Cysteines are highlighted in yellow and conserved residues in gray. The
residues involved in the most relevant interactions with GAPDH are red.
b) Superimposition of CP12-GAPDH complexes from Arabidopsis thaliana (At) and
Synechococcus elongatus (Se)(Matsumura et al. (2011) Structure 19, 1846–1854). The C trace of At-CP12 (chain G from co-crystallized complex,
magenta) and Se-CP12 (chain H from Cu-complex, cyan) are represented as
ribbons. The side chains of At-CP12 residues involved in most relevant
interactions with GAPDH (i.e. observed at least in two over three CP12 chains in the co-crystallized complex), are represented in sticks and superimposed to the
corresponding residues of Se-CP12. The GAPDH residues involved in these
interactions are also represented in sticks, gray for At-GAPDH and lightpink for
Se-GAPDH.

10
Supplementary Figure S5 - MD calculations.
a) B-factor profile for CP12 residues Pro59-Asn78 calculated by ptraj program
from 50 ns simulation.
α-helix-C loop-C C-tail
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24
α-h
elix-C
Lo
op
-CC
-ta
il
b c d
a
e f
d

11
b,c) Evolution in time of the secondary structure of Asp58-Asn78 CP12 (STRIDE
analysis) for trajectory of the dynamics in explicit solvent (b), and in implicit solvent
(c).
d) STRIDE analysis of the 20 NMR model structures.
Color code for panels b,c,d: magenta: α-helix; dark blue: 3-10 helix; cyan: turn;
white: coil.
The site-specific propensity for secondary structure formation was determined via the
STRIDE program (Frishman and Argos, (1995) Proteins 23, 566-579). The STRIDE
program consists of a knowledge-based algorithm that uses hydrogen-bond energy
and statistically derived backbone torsional angle information to determine the
secondary structure assignments in maximal agreement with crystallographers
designations.
e,f) Two-dimensional free-energy profiles for segments Asp58-Cys73 in explicit
solvent (e) and in implicit solvent (f), as a function of PCA1 and PCA2. The values are
given in kcal mol–1. The energy landscape of the protein is visualized by means of
free-energy functions, which are projected as contour lines onto a two-dimensional
space formed by the PCA1/PCA2 axes. These coordinates are derived from a principal
component analysis (Garcia, (1992) Phys. Rev. Lett. 66, 2696). The free-energy
change associated with the passage between two different states of a system in
thermodynamic equilibrium is given by ΔG = -RT (ln p1/p2). Here, R is the ideal gas
constant, T is the absolute temperature, and pi is the probability of finding the system
in state i. The two-dimensional space defined by the PCA1 and PCA2 axes has been
divided into a grid and the free energy has been calculated for each bin of the grid on
the basis of the previous equation. The whole set of G values was shifted in such a
way that the lowest value of the free-energy surface corresponds to zero. Thus, the
reported ΔG values represent the transfer free energies with respect to the bin that
has been set to zero. To obtain the pi values the trajectory at ambient temperature
was projected onto the PCA1/PCA2 space and pi corresponds to the number of times
the trajectory ‘visits’ a given bin.

12
Supplementary Table S3 - Calculated CP12 intra-molecular interactions
lifetime.
Lifetime obtained from analysis of the trajectory file using ‘ptraj’ package (an AMBER
module). It is estimated the percentage for the existence of every H-bond during the
whole simulation (an ‘active’ hydrogen bond requires a distance between acceptor and
donor shorter than 3.5 A, and a bond angle larger than 120°). We denote this quantity as the ‘lifetime’ of a given H-bond, expressed as a percentage of existence on
the whole trajectory.
OTHR70/NCYS73 90.78% OGLU62/NLYS65 44.22% OASN67/OG1THR70 18.47% OD1ASP77/NEARG74 8.42% OASP77/NH1ARG74 7.18%
OD1ASN67/NGLU69 86.60% OACE57/NGLU61 42.53% OGLU61/NCYS64 15.96% OE2GLU62/NZLYS65 8.42% OASP77/ND2ASN71 6.88%
OTHR70/NARG74 73.33% OTYR63/NASP66 39.08% OGLU69/NH1ARG74 15.84% OE1GLU62/NZLYS65 8.29% OASN78/NH1ARG74 6.79%
OASP58/NGLU62 64.14% OPRO68/NTHR70 38.26% OCYS64/NASN67 14.29% OD2ASP77/NASN78 8.26% OD2ASP77/NEARG74 6.58%
OTYR63/NASN67 63.72% OACE57/NLEU60 37.64% OGLU69/NH2ARG74 12.23% OD2ASP77/NH2ARG74 8.08% OGLU69/ND2ASN67 6.02%
OLEU60/NCYS64 61.51% OPRO59/NGLU62 30.97% OD1ASP77/NASN78 10.10% OE1GLU72/ND2ASN21 7.86% OE1GLU62/NZLYS65 5.74%
OGLU62/NASP66 60.15% OLEU60/NTYR63 26.07% OTYR76/NH1ARG74 10.01% OASN67/NTHR70 7.70% OE2GLU62/NZLYS65 5.39%
OPRO59/NTYR63 52.67% OPRO59/NCYS64 21.64% OE2GLU72/ND2ASN71 9.86% OE1GLU62/NZLYS65 7.66% OG1THR70/NCYS73 5.29%
OASP58/NGLU61 52.46% OASN71/NARG74 21.42% OD1ASP77/NH2ARG74 9.19% OG1THR70/NGLU72 7.56%
OGLU61/NLYS65 44.70% OGLU69/NEARG74 21.26% OE2GLU62/NZLYS65 8.47% OARG74/NASP77 7.43%

13
Supplementary Figure S6 - PONDR VL-XT analysis of CP12.
The disorder predictor PONDR VL-XT (Garner et al., (1999) Genome Informatics, 10,
41-50) predicts that CP12 is 88% disordered (Average Prediction Score: 0.6890). A
stretch of 8 amino acids, from Ser57 to Lys65 (marked in bold), is the only part of the
78-residues protein which is predicted to be ordered, based on a PONDR score lower
than 0.5. This predicted ordered stretch nicely matches the alpha-helix-C which, in
both NMR models and in crystallized CP12, starts with Pro59 and ends with Asp66.
Prediction was performed on the sequence of CP12 (isoform 2) of Arabidopsis thaliana
at www.pondr.com. Short ordered segments within large disordered regions predicted
by PONDR are often involved in interactions (Garner et al., (1999) Genome
Informatics, 10, 41-50). Alpha-helix-C is involved in the formation of the encounter
complex between CP12 and GAPDH. In this sense, alpha-helix-C acts as a Molecular
Recognition Feature (Mohan et al., (2006) J. Mol. Biol. 362, 1043–1059).
SDPLEEYCKDNPETNECRTYDN
α-helix-C loop-C C-tail
Copyright © 2022 FDOKUMEN













![Intrinsically radiolabelled [(59)Fe]-SPIONs for dual MRI/radionuclide detection](https://static.fdokumen.com/doc/165x107/6335c40d379741109e00c5c6/intrinsically-radiolabelled-59fe-spions-for-dual-mriradionuclide-detection.jpg)






