
Compositional variability and air-sea flux of ethane and propane in the plume of a large, marine...
12
ORIGINAL Compositional variability and air-sea flux of ethane and propane in the plume of a large, marine seep field near Coal Oil Point, CA Susan Mau & Monica B. Heintz & Franklin S. Kinnaman & David L. Valentine Received: 5 February 2009 / Accepted: 14 January 2010 / Published online: 20 February 2010 # The Author(s) 2010. This article is published with open access at Springerlink.com Abstract Large quantities of methane (C1), ethane (C2), and propane (C3) emanate from shallow marine seeps near Coal Oil Point (COP), California. Concentrations of these gases were analyzed in the surface water down-current of the seep field over a 15-month period. The variable proportions of C1, C2, and C3 analyzed in gas bubbles emitted from 16 distinct seeps in the COP field encompass much of the variability found in the surface waters down- current. However, waters with disproportionate levels of C1 suggest the presence of additional C1 sources. Based on three spatial surveys, covering areas up to 280 km 2 , C2 and C3 air-sea fluxes were estimated to be in the order of 3.7 and 1.4 μmol day −1 m −2 , respectively. Only 0.6% of C2 and 0.5% of C3 in the dissolved plume originating from the COP seep field are transferred to the atmosphere in the study area, with the fate of the remainder uncertain. Introduction Although ethane (C2) and propane (C3) are less abundant than methane (C1) in the atmosphere, the higher reactivity of both gases, and the formation of more complex intermediates when oxidized make C2 and C3 important to tropospheric chemistry (Rudolph 1995; Saito et al. 2000). Most important, both gases are precursors for tropospheric ozone, a principle component of urban smog (Singh and Zimmerman 1992). Studies of nonmethane hydrocarbons (NMHCs) in- cluding C2 and C3 in the marine atmosphere and in surface seawater indicated that C2 and C3 are in part derived from oceanic sources (Saito et al. 2000). However, a database of dissolved C2 and C3 hydrocarbons in the surface water of the ocean compiled by Plass-Duelmer et al. (1995) implied that oceanic sources of C2 and C3 are small (0.16 and 0.1 Tg year −1 for C2 and C3, respectively), and play a minor role in the global budgets of NMHCs (15.5 Tg year −1 C2 as reported in Rudolph 1995, and 15–20 Tg year −1 C3 as stated in Singh and Zimmerman 1992). In contrast to concentrations found in the open ocean, concentrations of saturated hydrocarbons increase substantially in the proximity of natural oil seeps and anthropogenic sources of oil pollution (Plass- Duelmer et al. 1995). One site where gas and oil are naturally escaping the sediment and dissolving in the overlying water column is the seep field located near Coal Oil Point (COP), Santa Barbara, California. It is one of the world’ s largest and best studied seep regions, consisting of more than 1,000 seafloor seeps in water depths of 5 to 70 m (Fischer 1977; Hornafius et al. 1999). Seeps are comprised of multiple vents in the form of mm-wide discrete openings from which gas bubbles rise (Fischer 1977; Kinnaman et al. 2010, this issue). They are located above the fractured crests of three anticlines that contain oil in the Miocene-age Monterey Formation (Fischer 1977). Emanating seep gas is composed mostly of C1, C2, C3, and CO 2 along with trace gases of heavier hydrocarbons and H 2 S. More precisely, gas samples S. Mau : M. B. Heintz : F. S. Kinnaman : D. L. Valentine Department of Earth Science and Marine Science Institute, University of California, Santa Barbara, CA 93106, USA Present Address: S. Mau (*) Max Planck Institute for Marine Microbiology, Celsiusstr. 1, 28359 Bremen, Germany e-mail: [email protected] Geo-Mar Lett (2010) 30:367–378 DOI 10.1007/s00367-010-0185-z
Transcript of Compositional variability and air-sea flux of ethane and propane in the plume of a large, marine...

ORIGINAL
Compositional variability and air-sea flux of ethaneand propane in the plume of a large, marine seep field nearCoal Oil Point, CA
Susan Mau & Monica B. Heintz &
Franklin S. Kinnaman & David L. Valentine
Received: 5 February 2009 /Accepted: 14 January 2010 /Published online: 20 February 2010# The Author(s) 2010. This article is published with open access at Springerlink.com
Abstract Large quantities of methane (C1), ethane (C2),and propane (C3) emanate from shallow marine seeps nearCoal Oil Point (COP), California. Concentrations of thesegases were analyzed in the surface water down-current ofthe seep field over a 15-month period. The variableproportions of C1, C2, and C3 analyzed in gas bubblesemitted from 16 distinct seeps in the COP field encompassmuch of the variability found in the surface waters down-current. However, waters with disproportionate levels ofC1 suggest the presence of additional C1 sources. Based onthree spatial surveys, covering areas up to 280 km2, C2 andC3 air-sea fluxes were estimated to be in the order of 3.7and 1.4 μmol day−1 m−2, respectively. Only 0.6% of C2 and0.5% of C3 in the dissolved plume originating from theCOP seep field are transferred to the atmosphere in thestudy area, with the fate of the remainder uncertain.
Introduction
Although ethane (C2) and propane (C3) are less abundantthan methane (C1) in the atmosphere, the higher reactivityof both gases, and the formation of more complexintermediates when oxidized make C2 and C3 important
to tropospheric chemistry (Rudolph 1995; Saito et al.2000). Most important, both gases are precursors fortropospheric ozone, a principle component of urban smog(Singh and Zimmerman 1992).
Studies of nonmethane hydrocarbons (NMHCs) in-cluding C2 and C3 in the marine atmosphere and insurface seawater indicated that C2 and C3 are in partderived from oceanic sources (Saito et al. 2000). However,a database of dissolved C2 and C3 hydrocarbons in thesurface water of the ocean compiled by Plass-Duelmeret al. (1995) implied that oceanic sources of C2 and C3are small (0.16 and 0.1 Tg year−1 for C2 and C3,respectively), and play a minor role in the global budgetsof NMHCs (15.5 Tg year−1 C2 as reported in Rudolph1995, and 15–20 Tg year−1 C3 as stated in Singh andZimmerman 1992). In contrast to concentrations found inthe open ocean, concentrations of saturated hydrocarbonsincrease substantially in the proximity of natural oilseeps and anthropogenic sources of oil pollution (Plass-Duelmer et al. 1995).
One site where gas and oil are naturally escaping thesediment and dissolving in the overlying water columnis the seep field located near Coal Oil Point (COP),Santa Barbara, California. It is one of the world’slargest and best studied seep regions, consisting ofmore than 1,000 seafloor seeps in water depths of 5 to70 m (Fischer 1977; Hornafius et al. 1999). Seeps arecomprised of multiple vents in the form of mm-widediscrete openings from which gas bubbles rise (Fischer1977; Kinnaman et al. 2010, this issue). They are locatedabove the fractured crests of three anticlines that containoil in the Miocene-age Monterey Formation (Fischer1977). Emanating seep gas is composed mostly of C1,C2, C3, and CO2 along with trace gases of heavierhydrocarbons and H2S. More precisely, gas samples
S. Mau :M. B. Heintz : F. S. Kinnaman :D. L. ValentineDepartment of Earth Science and Marine Science Institute,University of California,Santa Barbara, CA 93106, USA
Present Address:S. Mau (*)Max Planck Institute for Marine Microbiology,Celsiusstr. 1,28359 Bremen, Germanye-mail: [email protected]
Geo-Mar Lett (2010) 30:367–378DOI 10.1007/s00367-010-0185-z
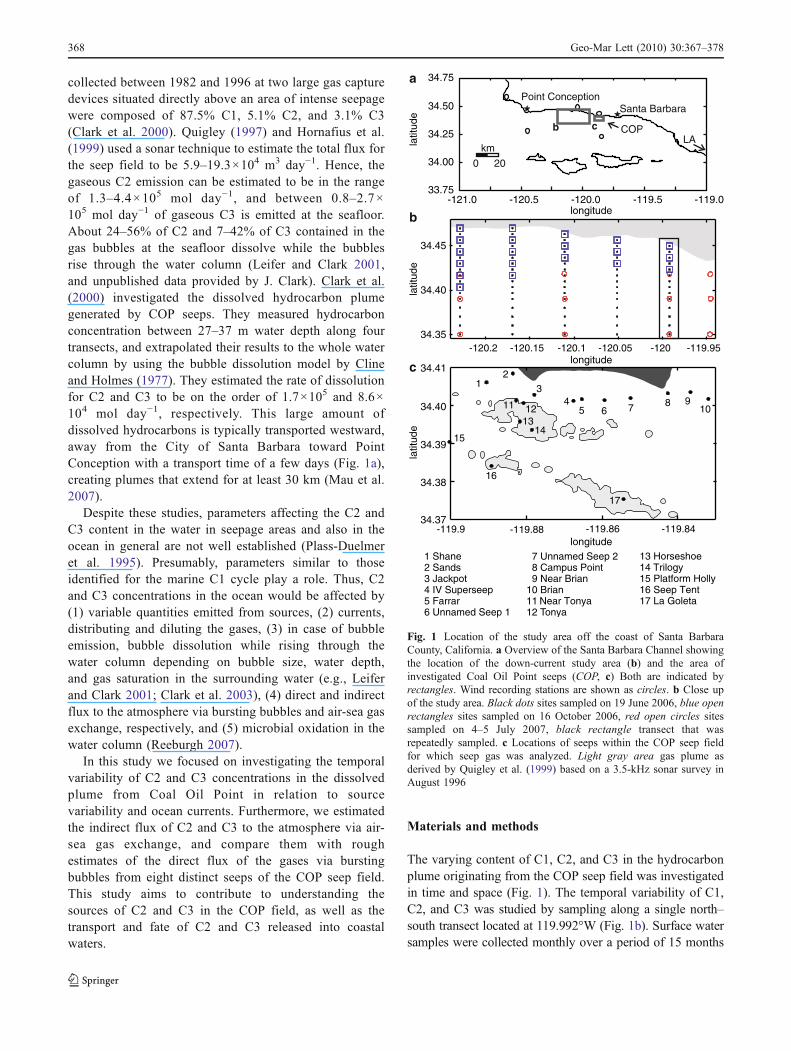
collected between 1982 and 1996 at two large gas capturedevices situated directly above an area of intense seepagewere composed of 87.5% C1, 5.1% C2, and 3.1% C3(Clark et al. 2000). Quigley (1997) and Hornafius et al.(1999) used a sonar technique to estimate the total flux forthe seep field to be 5.9–19.3×104 m3 day−1. Hence, thegaseous C2 emission can be estimated to be in the rangeof 1.3–4.4×105 mol day−1, and between 0.8–2.7×105 mol day−1 of gaseous C3 is emitted at the seafloor.About 24–56% of C2 and 7–42% of C3 contained in thegas bubbles at the seafloor dissolve while the bubblesrise through the water column (Leifer and Clark 2001,and unpublished data provided by J. Clark). Clark et al.(2000) investigated the dissolved hydrocarbon plumegenerated by COP seeps. They measured hydrocarbonconcentration between 27–37 m water depth along fourtransects, and extrapolated their results to the whole watercolumn by using the bubble dissolution model by Clineand Holmes (1977). They estimated the rate of dissolutionfor C2 and C3 to be on the order of 1.7×105 and 8.6×104 mol day−1, respectively. This large amount ofdissolved hydrocarbons is typically transported westward,away from the City of Santa Barbara toward PointConception with a transport time of a few days (Fig. 1a),creating plumes that extend for at least 30 km (Mau et al.2007).
Despite these studies, parameters affecting the C2 andC3 content in the water in seepage areas and also in theocean in general are not well established (Plass-Duelmeret al. 1995). Presumably, parameters similar to thoseidentified for the marine C1 cycle play a role. Thus, C2and C3 concentrations in the ocean would be affected by(1) variable quantities emitted from sources, (2) currents,distributing and diluting the gases, (3) in case of bubbleemission, bubble dissolution while rising through thewater column depending on bubble size, water depth,and gas saturation in the surrounding water (e.g., Leiferand Clark 2001; Clark et al. 2003), (4) direct and indirectflux to the atmosphere via bursting bubbles and air-sea gasexchange, respectively, and (5) microbial oxidation in thewater column (Reeburgh 2007).
In this study we focused on investigating the temporalvariability of C2 and C3 concentrations in the dissolvedplume from Coal Oil Point in relation to sourcevariability and ocean currents. Furthermore, we estimatedthe indirect flux of C2 and C3 to the atmosphere via air-sea gas exchange, and compare them with roughestimates of the direct flux of the gases via burstingbubbles from eight distinct seeps of the COP seep field.This study aims to contribute to understanding thesources of C2 and C3 in the COP field, as well as thetransport and fate of C2 and C3 released into coastalwaters.
Materials and methods
The varying content of C1, C2, and C3 in the hydrocarbonplume originating from the COP seep field was investigatedin time and space (Fig. 1). The temporal variability of C1,C2, and C3 was studied by sampling along a single north–south transect located at 119.992°W (Fig. 1b). Surface watersamples were collected monthly over a period of 15 months
-120.2 -120.15 -120.1 -120.05 -120 -119.95
34.45
34.40
34.35
latit
ude
longitude
108
513
43
17
911
2
16
1
12
14
6 7
34.41
34.40
34.39
34.38
34.37-119.9 -119.88 -119.86 -119.84
longitude
latit
ude
b
c
-121.0 -120.5 -120.0 -119.5 -119.033.75
34.00
34.25
34.50
34.75
longitude
latit
ude
Santa BarbaraPoint Conception
LACOP
0 20
km
b c
a
15
1 Shane2 Sands3 Jackpot4 IV Superseep5 Farrar6 Unnamed Seep 1
7 Unnamed Seep 28 Campus Point9 Near Brian
10 Brian11 Near Tonya12 Tonya
13 Horseshoe14 Trilogy15 Platform Holly16 Seep Tent17 La Goleta
Fig. 1 Location of the study area off the coast of Santa BarbaraCounty, California. a Overview of the Santa Barbara Channel showingthe location of the down-current study area (b) and the area ofinvestigated Coal Oil Point seeps (COP, c) Both are indicated byrectangles. Wind recording stations are shown as circles. b Close upof the study area. Black dots sites sampled on 19 June 2006, blue openrectangles sites sampled on 16 October 2006, red open circles sitessampled on 4–5 July 2007, black rectangle transect that wasrepeatedly sampled. c Locations of seeps within the COP seep fieldfor which seep gas was analyzed. Light gray area gas plume asderived by Quigley et al. (1999) based on a 3.5-kHz sonar survey inAugust 1996
368 Geo-Mar Lett (2010) 30:367–378
from May to November 2006, March to May 2007, and inJuly 2007 (Table 1). The transect is located 7 km west ofthe seep field, and perpendicular to the prevailing westwardcurrents. Spatial variability was explored by grid samplingcovering areas of 280, 89, and 197 km2 on 19 June 2006,16 October 2006, and 4–5 July 2007, respectively (Fig. 1b,Table 1). The sampling grid differs slightly between surveydays, but is always in the same region, down-current of theseep field. The most extensive survey was conducted on 19June 2006, sampling 79 stations along five north–southtransects. This grid matches the C1 samples from Mau et al.(2007). The area with the highest hydrocarbon concen-trations observed in June 2006 was re-sampled in October2006. In July 2007, the area was extended to the east, i.e.,toward the seep field. However, as samples were takenfrom aboard the RV Atlantis during the SEEPS 07 cruise(Studies on the Ecology and Evolution of PetroleumSeeps), near-shore sampling was limited.
Water samples for hydrocarbon analysis were collectedusing either a submersible pump or CTD-rosette. The latterwas applied for sampling aboard the RV Atlantis on 4–5July 2007, whereas the pump was used on all other dates.Samples were collected from the upper 10 m of the watercolumn; Table 1 lists the dates and sampled water depths.All sample bottles (125 ml, crimp top) were flushed withtwo volumes of water, and filled completely to eliminatebubbles. The bottles were immediately capped with butylrubber stoppers and crimp sealed. Duplicate water sampleswere always collected. A 10-ml headspace was introducedinto each bottle on the day of sampling as described byValentine et al. (2001). After headspace injection, sampleswere stored at 4°C, and analyzed within 1 month after
equilibration. Two aliquots of the headspace were eachanalyzed for C1, C2, and C3 using a gas chromatographequipped with a flame ionization detector (Kinnaman et al.2007). Replicate analyses of samples yield a precision of±2% for samples with C1 concentration ≥50 nmol L−1, and±5% for samples with C1 concentration <50 nmol L−1. ForC2 the precision is ±5% for samples with concentration≥3 nmol L−1, and ±10% for samples with concentration<3 nmol L−1; for C3 the precision is ±5% for samples withconcentration ≥1.5 nmol L−1, and ±10% for samples withconcentration <1.5 nmol L−1. C1–C3 concentrations ofduplicate water samples are within an error of 10%.
In addition to water sampling, seep gas was sampled at16 seep sites within the COP seep field, either at theseafloor or at the sea surface above these seeps (Fig. 1c,Table 2). The sampling method was similar to thatdescribed by Kinnaman et al. (2010, this issue), except forsamples from Seep Tent, La Goleta Seep, and PlatformHolly. Surface bubble samples were collected by eithersnorkeling in the bubble plume, or hanging off the side of asmall boat. Samples of gas from the reservoir underlyingPlatform Holly and from the Seep Tent were collected instainless steel canisters at an onshore processing facility(Ellwood Processing Facility, Venoco Co.), prior to treat-ment. Samples from La Goleta Seep were collected duringthe SEEPS 07 cruise aboard the RV Atlantis using aninverted funnel and container modified to enable manipu-lation by the DSV ALVIN, and to preserve the sampleduring submersible surfacing. All gas samples wereanalyzed in the laboratory as previously described by Duffyet al. (2007). Error associated with the concentrationmeasurements is ±4%.
Table 1 Surface water sampling dates, locations, number of stations, and water depths (see also Fig. 1b)
Date No. of transects Start and end location in ºN Stations Water depth (m)
Surveys along transect
16 May 2006 1 34.31–34.43 16 0.5
19 Jun. 2006 1 34.35–34.45 14 0.5, 1.5, 2.5
27 Jul. 2006 1 34.36–34.45 13 0.5, 1.5, 2.5
17 Aug. 2006 1 34.42–34.44 5 0.5, 1.5, 2.5
25 Sep. 2006 1 34.42–34.44 10 0.5
16 Oct. 2006 1 34.37–34.45 3 0.5
7 Nov. 2006 1 34.42–34.44 4 0.5
6 Mar. 2007 1 34.38–34.44 2 0.5
10 Apr. 2007 1 34.38–34.44 3 1–10 (ev. 1 m)
15 May 2007 1 34.35–34.45 3 0.5, 5, 10
4–5 Jul. 2007 1 34.35–34.42 3 5, 10
Grid surveys
19 Jun. 2006 5 34.35–34.47 79 0.5
16 Oct. 2006 5 34.35–34.47 20 0.5
4–5 Jul. 2007 4 34.35–34.42 12 5
Geo-Mar Lett (2010) 30:367–378 369
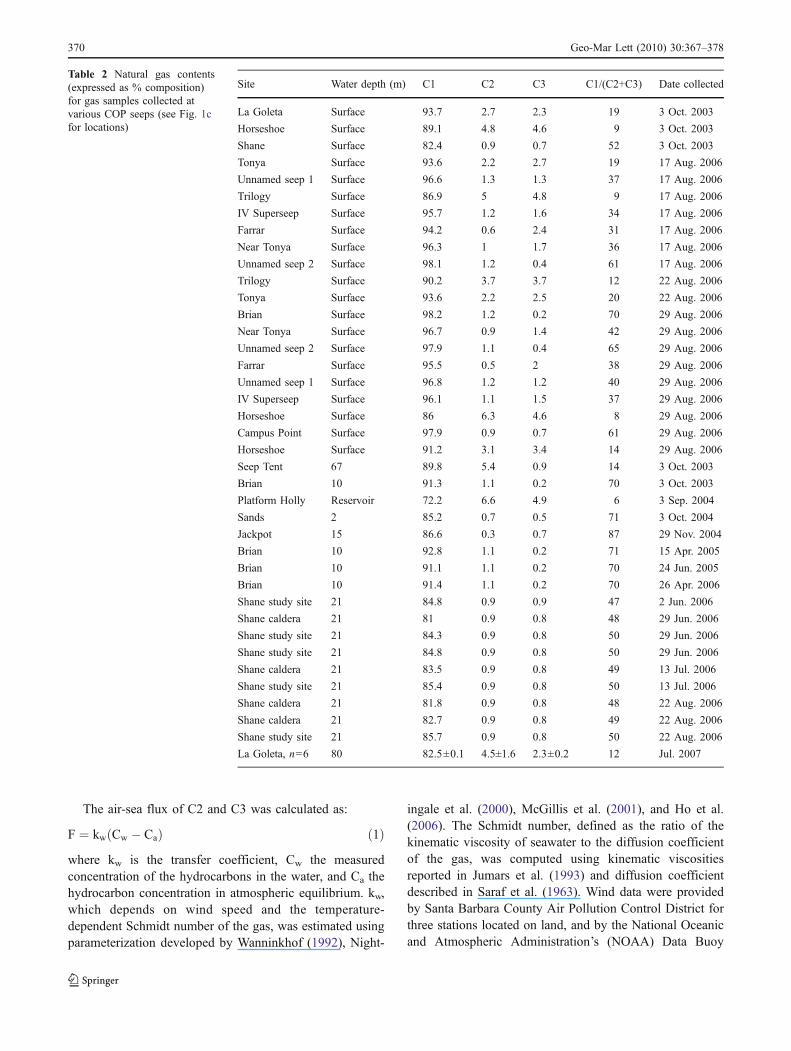
The air-sea flux of C2 and C3 was calculated as:
F ¼ kw Cw � Cað Þ ð1Þwhere kw is the transfer coefficient, Cw the measuredconcentration of the hydrocarbons in the water, and Ca thehydrocarbon concentration in atmospheric equilibrium. kw,which depends on wind speed and the temperature-dependent Schmidt number of the gas, was estimated usingparameterization developed by Wanninkhof (1992), Night-
ingale et al. (2000), McGillis et al. (2001), and Ho et al.(2006). The Schmidt number, defined as the ratio of thekinematic viscosity of seawater to the diffusion coefficientof the gas, was computed using kinematic viscositiesreported in Jumars et al. (1993) and diffusion coefficientdescribed in Saraf et al. (1963). Wind data were providedby Santa Barbara County Air Pollution Control District forthree stations located on land, and by the National Oceanicand Atmospheric Administration’s (NOAA) Data Buoy
Site Water depth (m) C1 C2 C3 C1/(C2+C3) Date collected
La Goleta Surface 93.7 2.7 2.3 19 3 Oct. 2003
Horseshoe Surface 89.1 4.8 4.6 9 3 Oct. 2003
Shane Surface 82.4 0.9 0.7 52 3 Oct. 2003
Tonya Surface 93.6 2.2 2.7 19 17 Aug. 2006
Unnamed seep 1 Surface 96.6 1.3 1.3 37 17 Aug. 2006
Trilogy Surface 86.9 5 4.8 9 17 Aug. 2006
IV Superseep Surface 95.7 1.2 1.6 34 17 Aug. 2006
Farrar Surface 94.2 0.6 2.4 31 17 Aug. 2006
Near Tonya Surface 96.3 1 1.7 36 17 Aug. 2006
Unnamed seep 2 Surface 98.1 1.2 0.4 61 17 Aug. 2006
Trilogy Surface 90.2 3.7 3.7 12 22 Aug. 2006
Tonya Surface 93.6 2.2 2.5 20 22 Aug. 2006
Brian Surface 98.2 1.2 0.2 70 29 Aug. 2006
Near Tonya Surface 96.7 0.9 1.4 42 29 Aug. 2006
Unnamed seep 2 Surface 97.9 1.1 0.4 65 29 Aug. 2006
Farrar Surface 95.5 0.5 2 38 29 Aug. 2006
Unnamed seep 1 Surface 96.8 1.2 1.2 40 29 Aug. 2006
IV Superseep Surface 96.1 1.1 1.5 37 29 Aug. 2006
Horseshoe Surface 86 6.3 4.6 8 29 Aug. 2006
Campus Point Surface 97.9 0.9 0.7 61 29 Aug. 2006
Horseshoe Surface 91.2 3.1 3.4 14 29 Aug. 2006
Seep Tent 67 89.8 5.4 0.9 14 3 Oct. 2003
Brian 10 91.3 1.1 0.2 70 3 Oct. 2003
Platform Holly Reservoir 72.2 6.6 4.9 6 3 Sep. 2004
Sands 2 85.2 0.7 0.5 71 3 Oct. 2004
Jackpot 15 86.6 0.3 0.7 87 29 Nov. 2004
Brian 10 92.8 1.1 0.2 71 15 Apr. 2005
Brian 10 91.1 1.1 0.2 70 24 Jun. 2005
Brian 10 91.4 1.1 0.2 70 26 Apr. 2006
Shane study site 21 84.8 0.9 0.9 47 2 Jun. 2006
Shane caldera 21 81 0.9 0.8 48 29 Jun. 2006
Shane study site 21 84.3 0.9 0.8 50 29 Jun. 2006
Shane study site 21 84.8 0.9 0.8 50 29 Jun. 2006
Shane caldera 21 83.5 0.9 0.8 49 13 Jul. 2006
Shane study site 21 85.4 0.9 0.8 50 13 Jul. 2006
Shane caldera 21 81.8 0.9 0.8 48 22 Aug. 2006
Shane caldera 21 82.7 0.9 0.8 49 22 Aug. 2006
Shane study site 21 85.7 0.9 0.8 50 22 Aug. 2006
La Goleta, n=6 80 82.5±0.1 4.5±1.6 2.3±0.2 12 Jul. 2007
Table 2 Natural gas contents(expressed as % composition)for gas samples collected atvarious COP seeps (see Fig. 1cfor locations)
370 Geo-Mar Lett (2010) 30:367–378
Center for two stations offshore (Fig. 1a). Ca was derivedusing Bunsen solubilities given by Mishnina et al. (1961)for C2 and Yano et al. (1974) for C3, and measured oceantemperatures and salinities.
Results
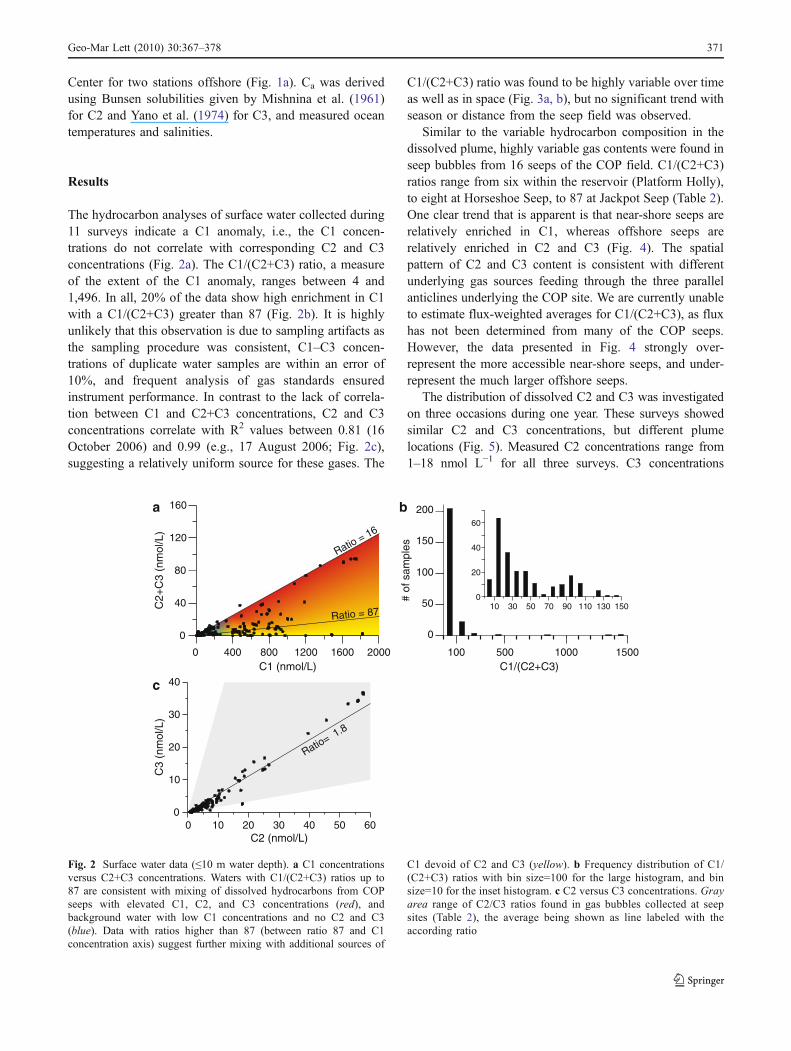
The hydrocarbon analyses of surface water collected during11 surveys indicate a C1 anomaly, i.e., the C1 concen-trations do not correlate with corresponding C2 and C3concentrations (Fig. 2a). The C1/(C2+C3) ratio, a measureof the extent of the C1 anomaly, ranges between 4 and1,496. In all, 20% of the data show high enrichment in C1with a C1/(C2+C3) greater than 87 (Fig. 2b). It is highlyunlikely that this observation is due to sampling artifacts asthe sampling procedure was consistent, C1–C3 concen-trations of duplicate water samples are within an error of10%, and frequent analysis of gas standards ensuredinstrument performance. In contrast to the lack of correla-tion between C1 and C2+C3 concentrations, C2 and C3concentrations correlate with R2 values between 0.81 (16October 2006) and 0.99 (e.g., 17 August 2006; Fig. 2c),suggesting a relatively uniform source for these gases. The
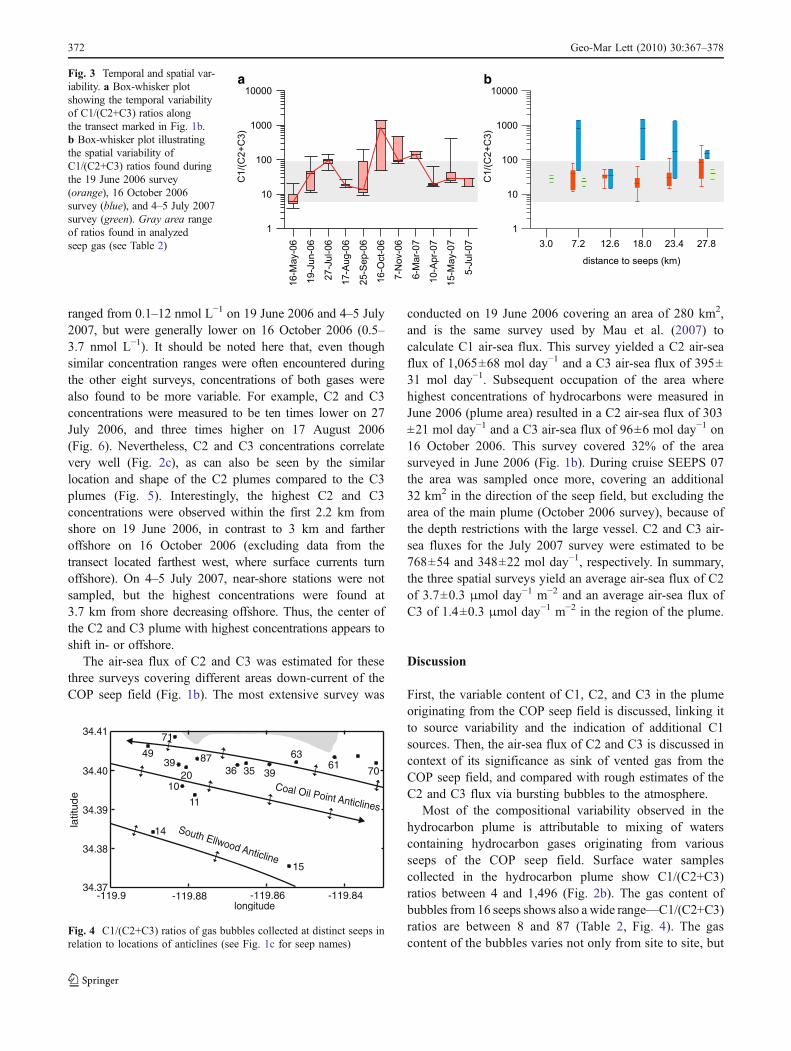
C1/(C2+C3) ratio was found to be highly variable over timeas well as in space (Fig. 3a, b), but no significant trend withseason or distance from the seep field was observed.
Similar to the variable hydrocarbon composition in thedissolved plume, highly variable gas contents were found inseep bubbles from 16 seeps of the COP field. C1/(C2+C3)ratios range from six within the reservoir (Platform Holly),to eight at Horseshoe Seep, to 87 at Jackpot Seep (Table 2).One clear trend that is apparent is that near-shore seeps arerelatively enriched in C1, whereas offshore seeps arerelatively enriched in C2 and C3 (Fig. 4). The spatialpattern of C2 and C3 content is consistent with differentunderlying gas sources feeding through the three parallelanticlines underlying the COP site. We are currently unableto estimate flux-weighted averages for C1/(C2+C3), as fluxhas not been determined from many of the COP seeps.However, the data presented in Fig. 4 strongly over-represent the more accessible near-shore seeps, and under-represent the much larger offshore seeps.
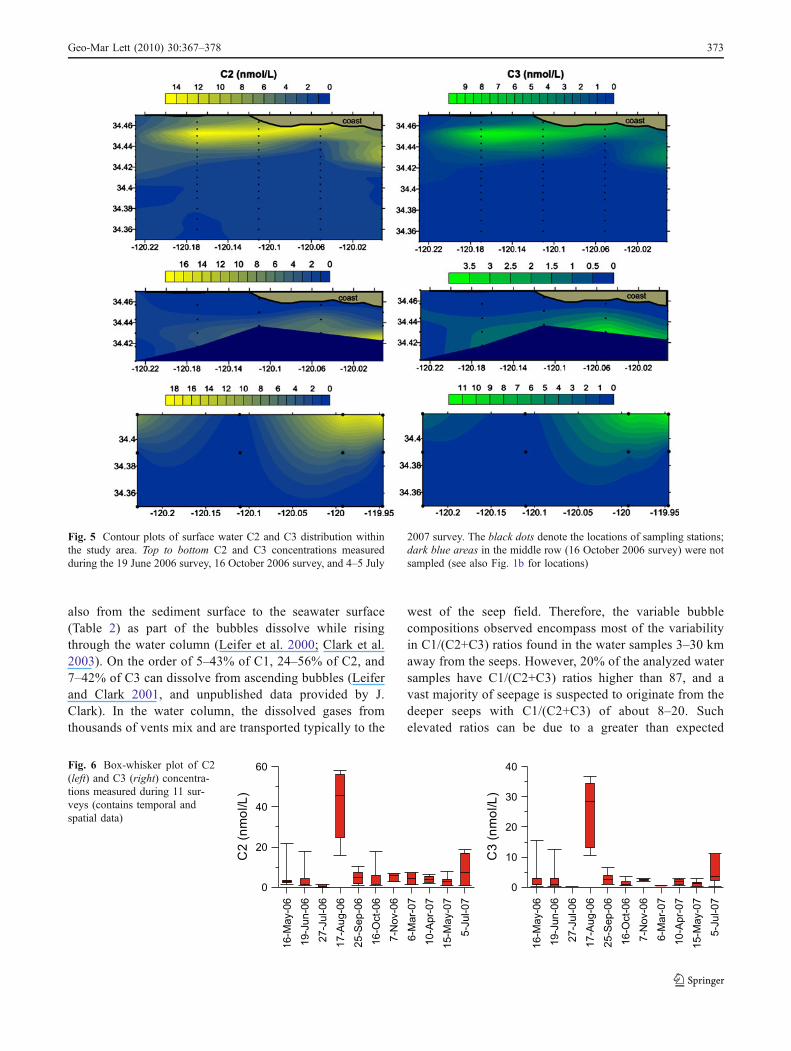
The distribution of dissolved C2 and C3 was investigatedon three occasions during one year. These surveys showedsimilar C2 and C3 concentrations, but different plumelocations (Fig. 5). Measured C2 concentrations range from1–18 nmol L−1 for all three surveys. C3 concentrations
0 400 800 1200 1600 2000
0
40
80
120
160a
Ratio = 87
C1 (nmol/L)
C2+
C3
(nm
ol/L
)
0 10 20 30 40 50 60C2 (nmol/L)
0
10
20
30
40
C3
(nm
ol/L
)
Ratio=1.8
c
# of
sam
ples
200
150
100
50
0
C1/(C2+C3)
b
Ratio = 16
100 500 1000 1500
60
40
20
010 30 50 70 90 110 130 150
Fig. 2 Surface water data (≤10 m water depth). a C1 concentrationsversus C2+C3 concentrations. Waters with C1/(C2+C3) ratios up to87 are consistent with mixing of dissolved hydrocarbons from COPseeps with elevated C1, C2, and C3 concentrations (red), andbackground water with low C1 concentrations and no C2 and C3(blue). Data with ratios higher than 87 (between ratio 87 and C1concentration axis) suggest further mixing with additional sources of
C1 devoid of C2 and C3 (yellow). b Frequency distribution of C1/(C2+C3) ratios with bin size=100 for the large histogram, and binsize=10 for the inset histogram. c C2 versus C3 concentrations. Grayarea range of C2/C3 ratios found in gas bubbles collected at seepsites (Table 2), the average being shown as line labeled with theaccording ratio
Geo-Mar Lett (2010) 30:367–378 371
ranged from 0.1–12 nmol L−1 on 19 June 2006 and 4–5 July2007, but were generally lower on 16 October 2006 (0.5–3.7 nmol L−1). It should be noted here that, even thoughsimilar concentration ranges were often encountered duringthe other eight surveys, concentrations of both gases werealso found to be more variable. For example, C2 and C3concentrations were measured to be ten times lower on 27July 2006, and three times higher on 17 August 2006(Fig. 6). Nevertheless, C2 and C3 concentrations correlatevery well (Fig. 2c), as can also be seen by the similarlocation and shape of the C2 plumes compared to the C3plumes (Fig. 5). Interestingly, the highest C2 and C3concentrations were observed within the first 2.2 km fromshore on 19 June 2006, in contrast to 3 km and fartheroffshore on 16 October 2006 (excluding data from thetransect located farthest west, where surface currents turnoffshore). On 4–5 July 2007, near-shore stations were notsampled, but the highest concentrations were found at3.7 km from shore decreasing offshore. Thus, the center ofthe C2 and C3 plume with highest concentrations appears toshift in- or offshore.
The air-sea flux of C2 and C3 was estimated for thesethree surveys covering different areas down-current of theCOP seep field (Fig. 1b). The most extensive survey was
conducted on 19 June 2006 covering an area of 280 km2,and is the same survey used by Mau et al. (2007) tocalculate C1 air-sea flux. This survey yielded a C2 air-seaflux of 1,065±68 mol day−1 and a C3 air-sea flux of 395±31 mol day−1. Subsequent occupation of the area wherehighest concentrations of hydrocarbons were measured inJune 2006 (plume area) resulted in a C2 air-sea flux of 303±21 mol day−1 and a C3 air-sea flux of 96±6 mol day−1 on16 October 2006. This survey covered 32% of the areasurveyed in June 2006 (Fig. 1b). During cruise SEEPS 07the area was sampled once more, covering an additional32 km2 in the direction of the seep field, but excluding thearea of the main plume (October 2006 survey), because ofthe depth restrictions with the large vessel. C2 and C3 air-sea fluxes for the July 2007 survey were estimated to be768±54 and 348±22 mol day−1, respectively. In summary,the three spatial surveys yield an average air-sea flux of C2of 3.7±0.3 μmol day−1 m−2 and an average air-sea flux ofC3 of 1.4±0.3 μmol day−1 m−2 in the region of the plume.
Discussion
First, the variable content of C1, C2, and C3 in the plumeoriginating from the COP seep field is discussed, linking itto source variability and the indication of additional C1sources. Then, the air-sea flux of C2 and C3 is discussed incontext of its significance as sink of vented gas from theCOP seep field, and compared with rough estimates of theC2 and C3 flux via bursting bubbles to the atmosphere.
Most of the compositional variability observed in thehydrocarbon plume is attributable to mixing of waterscontaining hydrocarbon gases originating from variousseeps of the COP seep field. Surface water samplescollected in the hydrocarbon plume show C1/(C2+C3)ratios between 4 and 1,496 (Fig. 2b). The gas content ofbubbles from 16 seeps shows also a wide range—C1/(C2+C3)ratios are between 8 and 87 (Table 2, Fig. 4). The gascontent of the bubbles varies not only from site to site, but
a bFig. 3 Temporal and spatial var-iability. a Box-whisker plotshowing the temporal variabilityof C1/(C2+C3) ratios alongthe transect marked in Fig. 1b.b Box-whisker plot illustratingthe spatial variability ofC1/(C2+C3) ratios found duringthe 19 June 2006 survey(orange), 16 October 2006survey (blue), and 4–5 July 2007survey (green). Gray area rangeof ratios found in analyzedseep gas (see Table 2)
7061
35
10
3687
15
39
71
14
49
20
11
39
63
34.41
34.40
34.39
34.38
34.37-119.9 -119.88 -119.86 -119.84
longitude
latit
ud
e
South Ellwood Anticline
Coal Oil Point Anticlines
Fig. 4 C1/(C2+C3) ratios of gas bubbles collected at distinct seeps inrelation to locations of anticlines (see Fig. 1c for seep names)
372 Geo-Mar Lett (2010) 30:367–378
also from the sediment surface to the seawater surface(Table 2) as part of the bubbles dissolve while risingthrough the water column (Leifer et al. 2000; Clark et al.2003). On the order of 5–43% of C1, 24–56% of C2, and7–42% of C3 can dissolve from ascending bubbles (Leiferand Clark 2001, and unpublished data provided by J.Clark). In the water column, the dissolved gases fromthousands of vents mix and are transported typically to the
west of the seep field. Therefore, the variable bubblecompositions observed encompass most of the variabilityin C1/(C2+C3) ratios found in the water samples 3–30 kmaway from the seeps. However, 20% of the analyzed watersamples have C1/(C2+C3) ratios higher than 87, and avast majority of seepage is suspected to originate from thedeeper seeps with C1/(C2+C3) of about 8–20. Suchelevated ratios can be due to a greater than expected
Fig. 5 Contour plots of surface water C2 and C3 distribution withinthe study area. Top to bottom C2 and C3 concentrations measuredduring the 19 June 2006 survey, 16 October 2006 survey, and 4–5 July
2007 survey. The black dots denote the locations of sampling stations;dark blue areas in the middle row (16 October 2006 survey) were notsampled (see also Fig. 1b for locations)
Fig. 6 Box-whisker plot of C2(left) and C3 (right) concentra-tions measured during 11 sur-veys (contains temporal andspatial data)
Geo-Mar Lett (2010) 30:367–378 373

amount of C1-rich seepage or a decrease of C2 and C3concentrations due to microbial oxidation.
We suggest that uncategorized seeps venting mainly C1increase C1/(C2+C3) ratios in many water samples,particularly in the samples with ratios beyond 87 (20% ofall samples). High C1 concentrations with no or low C2 andC3 (i.e., high C1/(C2+C3) ratios) were observed on, forexample, 10 April 2007 (Fig. 7). Water samples from 7–10 m water depths had elevated C1 concentrations butsimilar C2 and C3 concentrations as the water above. Wespeculate that additional C1 venting from as yet unidenti-fied seeps causes the observed C1 anomaly. Only a smallfraction of the gases emitted at the various seeps have beenanalyzed so far; thus, it is possible that there are seepsemitting primarily C1. Table 2 shows bubble compositionsfrom 16 of the better known seeps, but the COP seep fieldconsist of more than those (Fischer 1977) with the actualnumber of seeps still remaining uncertain. Thus, wepropose a three-component mixture as suggested byFig. 2a. The end members are (1) background water withlow C1 concentrations and no C2 and C3, (2) dissolvedCOP gas with elevated C1, C2, and C3 concentrations, and(3) additional water sources of high C1 concentrations butwithout C2 and C3.
Anomalous C1/(C2+C3) ratios can also be explained byother potential mechanisms. Preferential oxidation of C2and C3 by microbes could lower the concentrations withincreasing distance from the seep field, i.e., increase theC1/(C2+C3) ratio with distance. A continuous increase ofthe C1/(C2+C3) ratio with distance from the seep field wasnot observed (Fig. 3b). It should be noted that to ourknowledge no C2 or C3 oxidation rate measurements arepublished, and it is speculative if C2 and C3 arepreferentially removed by microbes. However, our assump-
tion is supported by Kinnaman et al. (2007), who reportedthat C3 and C4 are preferentially oxidized when sedimentsamples collected at COP seep sites were incubated with agas mixture of C1, C2, C3, and C4. Assuming that similarpreferences occur in the water column, we would expect toobserve a change in the C2/C3 ratio should microbialoxidation be an important process. However, C2/C3 ratiosfound in the down-current plume are relatively constant,suggesting that either there is no preferred microbialoxidation of C3 in the water column or that the process isslow and becomes more apparent farther away from theseeps. Furthermore, the C2/C3 ratios found in the bubblescollected at 16 seep sites range between 0.3 and 6.0, and94% of our data from the down-current plume fall in thatrange (Fig. 2c). Therefore, it appears that microbialoxidation of C2 and C3 is not significant in the investigatedsurface plume. Other potential sources such as the sewageoutfall located several km east of the seeps, and freshwaterinputs seem unlikely in the context of the environment.
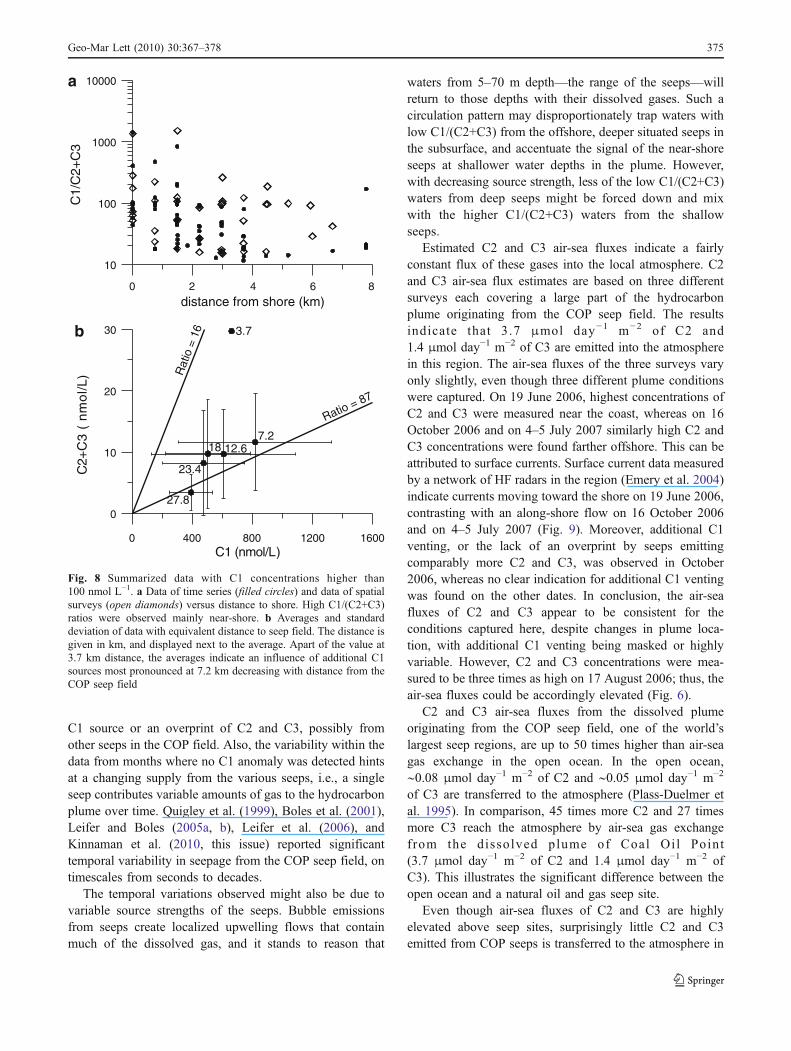
Based on our favored hypothesis of additional venting ofC1, we considered the data with high C1 concentrations(>100 nmol L−1) in more detail in order to find hints as tosource location and consistency of emission. Both thetemporal data collected along a transect ∼7 km from theseep field, and the spatial data collected along six transectslocated 3–30 km from the seeps indicate that samples withhigh C1 concentrations, and no or very low C2 and C3concentrations (high C1/(C2+C3) ratios) were mainly foundclose to the shore (Fig. 8a). In addition, the spatial data areconsistent with a source located either in the COP seep fieldor between the seep field and the transect located 7 km tothe west (Fig. 8b). Thus, the source location appears toreside near-shore and within the region ∼7 km west of theCOP seep field. Interestingly, this area includes much of anabandoned oil field. Elevated C1 concentrations comparedto C2 and C3 concentrations also suggest a longer gasmigration from the deep gas reservoir to the seafloor(Schoell 1983).
The temporal variability of the hydrocarbon compositionobserved in the down-current plume indicates inconsistentseep emissions or variable mixing. Surface currents do notcorrelate with C1/(C2+C3) ratios (Fig. 9)—that is, surfacecurrents do not indicate that water coming first through thearea where C1 seeps seem to be located becomes over-printed by gases from seeps emitting relatively more C2and C3, or vice versa. Hence, the temporal variability maybe due to changing gas sources or variable emissionstrengths. C1/(C2+C3) ratios higher than 87, as derivedfrom data collected on 16 October 2006, 7 November 2006,and 6 March 2007, suggest emission of additional C1 froma source near Coal Oil Point. In contrast, C1/(C2+C3) ratioslower than 87, as found in samples collected on all otherdates, indicate either a diminished activity of the additional
0.1 1 10 100 1000concentration (nmol/L)
10
8
6
4
2
0
de
pth
(m
)
C1
C2
C3
Fig. 7 Depth profiles of C1, C2, and C3 concentrations collected on10 April 2007 at 34.437°N, 119.992°W. Note that concentrations aredisplayed on a logarithmic scale
374 Geo-Mar Lett (2010) 30:367–378
C1 source or an overprint of C2 and C3, possibly fromother seeps in the COP field. Also, the variability within thedata from months where no C1 anomaly was detected hintsat a changing supply from the various seeps, i.e., a singleseep contributes variable amounts of gas to the hydrocarbonplume over time. Quigley et al. (1999), Boles et al. (2001),Leifer and Boles (2005a, b), Leifer et al. (2006), andKinnaman et al. (2010, this issue) reported significanttemporal variability in seepage from the COP seep field, ontimescales from seconds to decades.
The temporal variations observed might also be due tovariable source strengths of the seeps. Bubble emissionsfrom seeps create localized upwelling flows that containmuch of the dissolved gas, and it stands to reason that
waters from 5–70 m depth—the range of the seeps—willreturn to those depths with their dissolved gases. Such acirculation pattern may disproportionately trap waters withlow C1/(C2+C3) from the offshore, deeper situated seeps inthe subsurface, and accentuate the signal of the near-shoreseeps at shallower water depths in the plume. However,with decreasing source strength, less of the low C1/(C2+C3)waters from deep seeps might be forced down and mixwith the higher C1/(C2+C3) waters from the shallowseeps.
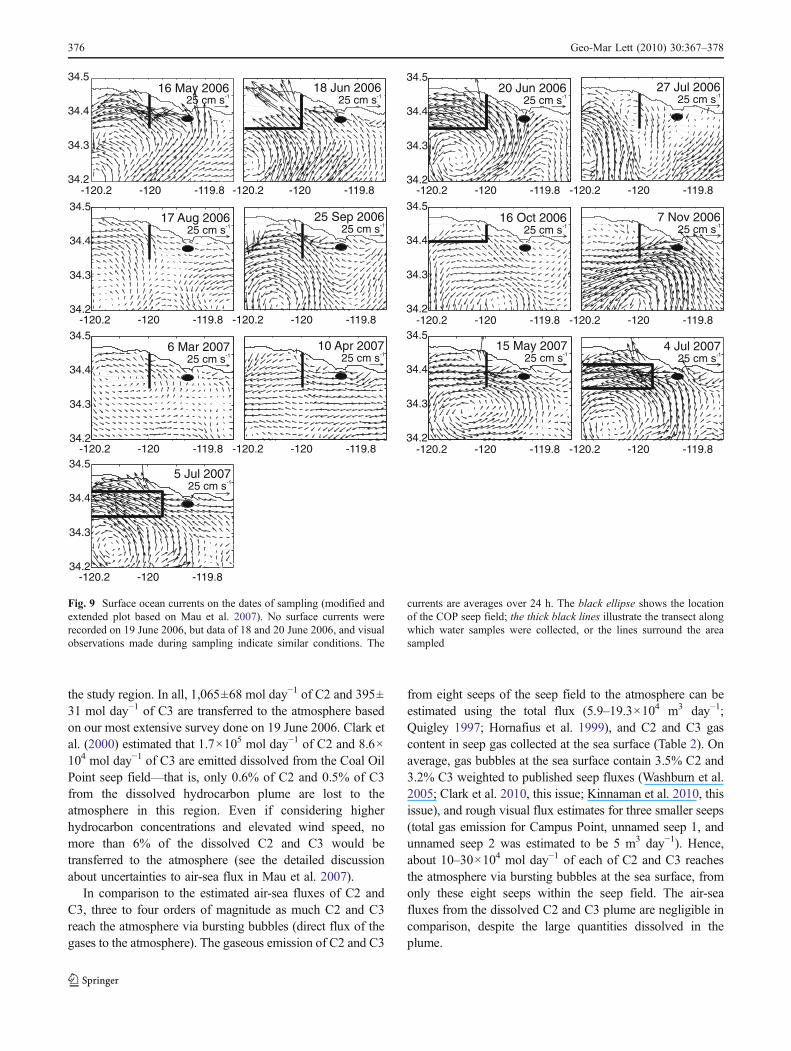
Estimated C2 and C3 air-sea fluxes indicate a fairlyconstant flux of these gases into the local atmosphere. C2and C3 air-sea flux estimates are based on three differentsurveys each covering a large part of the hydrocarbonplume originating from the COP seep field. The resultsindicate that 3.7 μmol day−1 m−2 of C2 and1.4 μmol day−1 m−2 of C3 are emitted into the atmospherein this region. The air-sea fluxes of the three surveys varyonly slightly, even though three different plume conditionswere captured. On 19 June 2006, highest concentrations ofC2 and C3 were measured near the coast, whereas on 16October 2006 and on 4–5 July 2007 similarly high C2 andC3 concentrations were found farther offshore. This can beattributed to surface currents. Surface current data measuredby a network of HF radars in the region (Emery et al. 2004)indicate currents moving toward the shore on 19 June 2006,contrasting with an along-shore flow on 16 October 2006and on 4–5 July 2007 (Fig. 9). Moreover, additional C1venting, or the lack of an overprint by seeps emittingcomparably more C2 and C3, was observed in October2006, whereas no clear indication for additional C1 ventingwas found on the other dates. In conclusion, the air-seafluxes of C2 and C3 appear to be consistent for theconditions captured here, despite changes in plume loca-tion, with additional C1 venting being masked or highlyvariable. However, C2 and C3 concentrations were mea-sured to be three times as high on 17 August 2006; thus, theair-sea fluxes could be accordingly elevated (Fig. 6).
C2 and C3 air-sea fluxes from the dissolved plumeoriginating from the COP seep field, one of the world’slargest seep regions, are up to 50 times higher than air-seagas exchange in the open ocean. In the open ocean,∼0.08 μmol day−1 m−2 of C2 and ∼0.05 μmol day−1 m−2
of C3 are transferred to the atmosphere (Plass-Duelmer etal. 1995). In comparison, 45 times more C2 and 27 timesmore C3 reach the atmosphere by air-sea gas exchangefrom the dissolved plume of Coal Oi l Point(3.7 μmol day−1 m−2 of C2 and 1.4 μmol day−1 m−2 ofC3). This illustrates the significant difference between theopen ocean and a natural oil and gas seep site.
Even though air-sea fluxes of C2 and C3 are highlyelevated above seep sites, surprisingly little C2 and C3emitted from COP seeps is transferred to the atmosphere in
0 2 4 6 8
distance from shore (km)
10
100
1000
10000C
1/C
2+C
3a
0 400 800 1200 1600C1 (nmol/L)
0
10
20
30
C2+
C3
(n
mo
l/L)
3.7
7.212.618
23.4
27.8
Ratio = 87
Rat
io=
16b
Fig. 8 Summarized data with C1 concentrations higher than100 nmol L−1. a Data of time series (filled circles) and data of spatialsurveys (open diamonds) versus distance to shore. High C1/(C2+C3)ratios were observed mainly near-shore. b Averages and standarddeviation of data with equivalent distance to seep field. The distance isgiven in km, and displayed next to the average. Apart of the value at3.7 km distance, the averages indicate an influence of additional C1sources most pronounced at 7.2 km decreasing with distance from theCOP seep field
Geo-Mar Lett (2010) 30:367–378 375
the study region. In all, 1,065±68 mol day−1 of C2 and 395±31 mol day−1 of C3 are transferred to the atmosphere basedon our most extensive survey done on 19 June 2006. Clark etal. (2000) estimated that 1.7×105 mol day−1 of C2 and 8.6×104 mol day−1 of C3 are emitted dissolved from the Coal OilPoint seep field—that is, only 0.6% of C2 and 0.5% of C3from the dissolved hydrocarbon plume are lost to theatmosphere in this region. Even if considering higherhydrocarbon concentrations and elevated wind speed, nomore than 6% of the dissolved C2 and C3 would betransferred to the atmosphere (see the detailed discussionabout uncertainties to air-sea flux in Mau et al. 2007).
In comparison to the estimated air-sea fluxes of C2 andC3, three to four orders of magnitude as much C2 and C3reach the atmosphere via bursting bubbles (direct flux of thegases to the atmosphere). The gaseous emission of C2 and C3
from eight seeps of the seep field to the atmosphere can beestimated using the total flux (5.9–19.3×104 m3 day−1;Quigley 1997; Hornafius et al. 1999), and C2 and C3 gascontent in seep gas collected at the sea surface (Table 2). Onaverage, gas bubbles at the sea surface contain 3.5% C2 and3.2% C3 weighted to published seep fluxes (Washburn et al.2005; Clark et al. 2010, this issue; Kinnaman et al. 2010, thisissue), and rough visual flux estimates for three smaller seeps(total gas emission for Campus Point, unnamed seep 1, andunnamed seep 2 was estimated to be 5 m3 day−1). Hence,about 10–30×104 mol day−1 of each of C2 and C3 reachesthe atmosphere via bursting bubbles at the sea surface, fromonly these eight seeps within the seep field. The air-seafluxes from the dissolved C2 and C3 plume are negligible incomparison, despite the large quantities dissolved in theplume.
400400
500500
600600
400
500
600
4 Jul 200725 cm s-1
-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
600600
5 Jul 200725 cm s-1
-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
20 Jun 200625 cm s-1
-120.2 -120 -119.8
400400
500500
27 Jul 200625 cm s-1
-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
17 Aug 200625 cm s-1
-120.2 -120 -119.8
400400
500500
25 Sep 200625 cm s-1
-120.2 -120 -119.8
400400
500500
7 Nov 200625 cm s-1
400400
500500
16 May 200625 cm s-1
-120.2 -120 -119.834.2
34.3
34.4
34.5
-120 -119.8400400
500500
18 Jun 200625 cm s-1
-120.2
-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
16 Oct 200625 cm s-1
-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
600600
6 Mar 200725 cm s-1
-120.2 -120 -119.8
10 Apr 200725 cm s-1
-120.2 -120 -119.8-120.2 -120 -119.834.2
34.3
34.4
34.5
400400
500500
600600
15 May 200725 cm s-1
Fig. 9 Surface ocean currents on the dates of sampling (modified andextended plot based on Mau et al. 2007). No surface currents wererecorded on 19 June 2006, but data of 18 and 20 June 2006, and visualobservations made during sampling indicate similar conditions. The
currents are averages over 24 h. The black ellipse shows the locationof the COP seep field; the thick black lines illustrate the transect alongwhich water samples were collected, or the lines surround the areasampled
376 Geo-Mar Lett (2010) 30:367–378

About an equivalent amount of C2 and C3 as transferredto the atmosphere via bursting bubbles remains dissolved inthe ocean. Of this dissolved quantity, only a small fractionundergoes air-sea gas exchange. Most likely, the remainingC2 and C3 dissolved in the water is transported offshorealong isopycnal surfaces along with C1 (Cynar andYayanos 1992); the ultimate fate is not known, butoxidation by microbes is likely.
Acknowledgements We are indebted to Shane Anderson, DavidFarrar, and Christoph Pierre for captaining the boats for watersampling. We are also grateful to the crew and scientific researchparty of the research vessel Atlantis on a cruise sponsored by theNational Science Foundation in 2007. Justin Reed, Victor Kim, andHaibing Ding helped with concentration analysis. This work waspossible by National Science Foundation CAREER grant OCE-0447395 and Department of Energy DE-NT005667. Susan Mau isfunded through a Marie Curie Outgoing International Fellowship(MOIF-CT-2006-021604) of the European Community.
Open Access This article is distributed under the terms of theCreative Commons Attribution Noncommercial License which per-mits any noncommercial use, distribution, and reproduction in anymedium, provided the original author(s) and source are credited.
References
Boles JR, Clark JF, Leifer I, Washburn L (2001) Temporal variation innatural methane seep rate due to tides, Coal Oil Point area,California. J Geophys Res 106:27077–27086
Clark J, Washburn L, Hornafius JS, Luyendyk BP (2000) Dissolvedhydrocarbon flux from natural marine seeps to the southernCalifornia Bight. J Geophys Res 105:11509–11522
Clark JF, Leifer I, Washburn L, Luyendyk BP (2003) Compositionalchanges in natural gas bubble plumes: observations from the CoalOil Point marine hydrocarbon seep field. In: Woodside JM,Garrison RE, Moore JC, Kvenvolden KA (eds) Contr 7th Int ConfGas in Marine Sediments, 7–11 October 2002, Baku, Azerbaijan.Geo-Mar Lett SI 23(3/4):187–193. doi:10.1007/s00367-003-0137-y
Clark JF, Washburn L, Schwager Emery K (2010) Variability of gascomposition and flux intensity in natural marine hydrocarbonseeps. In: Bohrmann G, Jørgensen BB (eds) Proc 9th Int ConfGas in Marine Sediments, 15–19 September 2008, Bremen. Geo-Mar Lett SI 30 (in press). doi:10.1007/s00367-009-0167-1
Cline JD, Holmes ML (1977) Submarine seepage of natural gas inNorton Sound, Alaska. Science 198:1149–1153
Cynar FJ, Yayanos AA (1992) The distribution of methane in theupper waters of the Southern California Bight. J Geophys Res97:11269–11285
Duffy M, Kinnaman F, Valentine DL, Keller E, Clark JF (2007)Gaseous emission rates from natural petroleum seeps in theUpper Ojai Valley, California. Environ Geosci 14:197–207
Emery BM, Washburn L, Harlan JA (2004) Evaluating radial currentmeasurements from CODAR high-frequency radars with mooredcurrent meters. J Atmos Ocean Technol 21:1259–1271
Fischer PJ (1977) Natural gas and oil seeps, Santa Barbara Basin,California. In: Fischer PJ (ed) California offshore gas, oil, and tarseeps. Staff Report, State of California, State Lands Commission,Sacramento, pp 1–62
Ho DT, Law CS, Smith MJ, Schlosser P, Harvey M, Hill P (2006)Measurements of air-sea gas exchange at high wind speeds in the
Southern Ocean: implications for global parameterizations. Geo-phys Res Lett 33:L16611. doi:10.1029/2006GL026817
Hornafius JS, Quigley DC, Luyendyk BP (1999) The world’s mostspectacular marine hydrocarbon seeps (Coal Oil Point, SantaBarbara Channel, California): quantification of emission. JGeophys Res 104:20703–20711
Jumars PA, Deming JW, Hill PS, Karp-Boss L, Yager PL, Dade WB(1993) Physical constraints on marine osmotrophy in an optimalforaging context. Mar Microb Food Webs 7:121–159
Kinnaman FS, Valentine DL, Tyler SC (2007) Carbon and hydrogenisotope fractionation associated with the aerobic microbialoxidation of methane, ethane, propane and butane. GeochimCosmochim Acta 71:271–283
Kinnaman FS, Kimball JB, Busso L, Birgel D, Ding H, Hinrichs K-U, Valentine DL (2010) Gas flux and carbonate occurrence at ashallow seep of thermogenic natural gas. In: Bohrmann G,Jørgensen BB (eds) Proc 9th Int Conf Gas in Marine Sedi-ments, 15–19 September 2008, Bremen. Geo-Mar Lett SI 30(in press)
Leifer I, Boles JR (2005a) Measurements of marine hydrocarbon seepflow through fractured rock and unconsolidated sediment. MarPetrol Geol 22:551–568
Leifer I, Boles JR (2005b) Turbine tent measurements of marinehydrocarbon seeps on subhourly timescales. J Geophys Res 110:C01006. doi:10.1029/2003JC002207
Leifer I, Clark J (2001) Modeling trace gases in hydrocarbon seepbubbles. Application to marine hydrocarbon seeps in the SantaBarbara Channel. Russ Geol Geophys 43:613–621
Leifer I, Clark JF, Chen RF (2000) Modifications of the localenvironment by natural marine hydrocarbon seeps. GeophysRes Lett 27:3711–3714
Leifer I, Luyendyk BP, Boles J, Clark J (2006) Natural marine seepageblowout: contribution to atmospheric methane. Glob BiogeochemCycles 20:GB3008, 10.1029/2005GB002668
Mau S, Valentine DL, Clark JF, Reed J, Camilli R, Washburn L (2007)Dissolved methane distributions and air-sea flux in the plume ofa massive seep field, Coal Oil Point, California. Geophys ResLett 34:L22603. doi:10.1029/2007GL031344
McGillis WR, Edson JB, Ware JD, Dacey JWH, Hare JE, Fairall CW,Wanninkhof R (2001) Carbon dioxide flux techniques performedduring GasEx-98. Mar Chem 75:267–280
Mishnina TA, Avdeeva OI, Bozhovskaya TK (1961) Materialy Vses.Nauchn-Issled Geol Inst 46:93–110
Nightingale PD, Malin G, Law CS, Watson AJ, Liss PS, LiddicoatMI, Boutin J, Upstill-Goddard RC (2000) In situ evaluation ofair-sea gas exchange parameterizations using novel conserva-tive and volatile tracers. Global Biogeochem Cycles 14:373–387
Plass-Duelmer C, Koppmann R, Ratte M, Rudolph J (1995) Lightnonmethane hydrocarbons in seawater. Global BiogeochemCycles 9:79–100
Quigley DC (1997) Quantifying spatial and temporal variations in thedistribution of natural marine hydrocarbon seeps in the SantaBarbara Channel, California. MSc Thesis, Department ofGeological Sciences, Santa Barbara, CA, pp 95
Quigley DC, Hornafius JS, Luyendyk BP, Francis RD, Clark J,Washburn L (1999) Decrease in natural marine hydrocarbonseepage near Coal Oil Point, California, associated with offshoreoil production. Geology 27:1047–1050
Reeburgh WS (2007) Oceanic methane biogeochemistry. Chem Rev107:486–513
Rudolph J (1995) The tropospheric distribution and budget of ethane.J Geophys Res Atmospheres 100:11369–11381
Saito T, Yokouchi Y, Kawamura K (2000) Distribution of C2-C6
hydrocarbons over the western North Pacific and eastern IndianOcean. Atmos Environ 34:4373–4381
Geo-Mar Lett (2010) 30:367–378 377

Saraf DN, Witherspoon PA, Cohen LH (1963) Diffusion coefficientsof hydrocarbons in water: method for measuring. Science142:955–956
Schoell M (1983) Genetic characterization of natural gases. AAPGBull 67:2225–2238
Singh H, Zimmerman P (1992) Atmospheric distribution and sourcesof nonmethane hydrocarbons. In: Nriagu J (ed) Gaseouspollutants: characterization and cycling. Wiley, New York,pp 177–235
Valentine DL, Blanton DC, Reeburgh WS, Kastner M (2001) Watercolumn methane oxidation adjacent to an area of active hydrate
dissociation, Eel River Basin. Geochim Cosmochim Acta65:2633–2640
Wanninkhof R (1992) Relationship between wind speed and gasexchange over the ocean. J Geophys Res 97:7373–7382
Washburn L, Clark JF, Kyriakidis P (2005) The spatial scales,distribution, and intensity of natural marine hydrocarbonseeps near Coal Oil Point, California. Mar Petrol Geol 22:569–578
Yano T, Suetaka T, Umehara T, Horiuchi A (1974) Solubilities ofmethane, ethylene, and propane in aqueous electrolyte solutions.Kagaku Kogaku 38:320–323
378 Geo-Mar Lett (2010) 30:367–378




















