
Compositional analysis of a polymer-induced liquid-precursor (PILP) amorphous CaCO3 phase
10
Compositional analysis of a polymer-induced liquid-precursor (PILP) amorphous CaCO 3 phase Lijun Dai, Elliot P. Douglas, Laurie B. Gower * Department of Materials Science and Engineering, University of Florida, Gainesville, FL 32611, USA Received 15 March 2007; received in revised form 19 September 2007 Available online 4 December 2007 Abstract Amorphous calcium carbonate (ACC) has been of keen interest in the biomimetics field because of recent evidence which suggests it plays an important role in biomineralization. In this report, an in vitro model system is used to examine the composition of an amorphous phase generated by polyanionic process-directing agents, such as the sodium salt of polyaspartic acid (Pasp), which is considered a simple mimic to the proteins associated with calcific biominerals. This additive leads to the formation of a highly hydrated, amorphous mineral precursor to calcium carbonate (CaCO 3 ), referred to as a polymer-induced liquid-precursor (PILP) phase. The precursor phase was col- lected by centrifugation, and the quantity of precursor phase and the water content were determined. It was found that Pasp promotes and stabilizes the amorphous precursor, which has a composition that steadily changes with time as the polymer and water are excluded. Elemental analysis was used to investigate the role of the polymer in influencing the calcium/carbonate ratio, the water content, and the amount of precursor phase. Raman and ATR–FTIR spectroscopy were used to compare the compositions of the precursor phases gen- erated with different polymeric concentrations. The role of Pasp in generating and stabilizing the ACC precursor phase is discussed. Ó 2007 Elsevier B.V. All rights reserved. PACS: 81.10.Dn; 61.46.Bc; 64.70.Ja Keywords: Biomaterials; Biopolymers; Crystal growth; Colloids; Nano-clusters; Phases and equilibria; Hydration 1. Introduction Amorphous calcium carbonate (ACC) has been of increasing interest in biomimetic and biomineralization studies due to recent discoveries that reveal its important role in morphological control in biological systems [1–9]. For instance, ACC is found to be a precursor phase for aragonite in mollusk larval shell formation [10]. In sea urchin larval species and the adult spine, ACC is the tran- sient precursor phase of calcite [8]. To study the formation mechanisms of the ACC phase in vitro, different additives such as metal ions, phosphonate, polymers, and different reaction conditions have been explored to synthesize ACC [11–23]. Biogenic and synthetic ACC can both exist in stable and transient forms [4–6,8]. The underlying mechanisms of how ACC is generated and stabilized under various conditions are still being debated, especially as to the role(s) of associ- ated macromolecules. It has been found that proteins from stable biogenic ACC are rich in the amino acid residues glutamic acid and glutamine, while proteins from transient biogenic ACC are rich in aspartic acid [5]. In addition, it is proposed that magnesium and phosphate ions cooperate with the associated proteins to stabilize ACC [5]. However, it is still not understood how one macromolecule could sta- bilize around 150 000 molecules of calcium carbonate [5]. In the stabilization of ACC, water is also proposed to play an important role in preventing the reorganization of ACC into the anhydrous form of calcium carbonate [5]. 0022-3093/$ - see front matter Ó 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.jnoncrysol.2007.10.022 * Corresponding author. Tel.: +1 352 846 3337; fax: +1 352 846 3355. E-mail address: [email protected]fl.edu (L.B. Gower). www.elsevier.com/locate/jnoncrysol Available online at www.sciencedirect.com Journal of Non-Crystalline Solids 354 (2008) 1845–1854
-
Upload
independent -
Category
Documents
-
view
9 -
download
0
Transcript of Compositional analysis of a polymer-induced liquid-precursor (PILP) amorphous CaCO3 phase

Available online at www.sciencedirect.com
www.elsevier.com/locate/jnoncrysol
Journal of Non-Crystalline Solids 354 (2008) 1845–1854
Compositional analysis of a polymer-induced liquid-precursor(PILP) amorphous CaCO3 phase
Lijun Dai, Elliot P. Douglas, Laurie B. Gower *
Department of Materials Science and Engineering, University of Florida, Gainesville, FL 32611, USA
Received 15 March 2007; received in revised form 19 September 2007Available online 4 December 2007
Abstract
Amorphous calcium carbonate (ACC) has been of keen interest in the biomimetics field because of recent evidence which suggests itplays an important role in biomineralization. In this report, an in vitro model system is used to examine the composition of an amorphousphase generated by polyanionic process-directing agents, such as the sodium salt of polyaspartic acid (Pasp), which is considered a simplemimic to the proteins associated with calcific biominerals. This additive leads to the formation of a highly hydrated, amorphous mineralprecursor to calcium carbonate (CaCO3), referred to as a polymer-induced liquid-precursor (PILP) phase. The precursor phase was col-lected by centrifugation, and the quantity of precursor phase and the water content were determined. It was found that Pasp promotesand stabilizes the amorphous precursor, which has a composition that steadily changes with time as the polymer and water are excluded.Elemental analysis was used to investigate the role of the polymer in influencing the calcium/carbonate ratio, the water content, and theamount of precursor phase. Raman and ATR–FTIR spectroscopy were used to compare the compositions of the precursor phases gen-erated with different polymeric concentrations. The role of Pasp in generating and stabilizing the ACC precursor phase is discussed.� 2007 Elsevier B.V. All rights reserved.
PACS: 81.10.Dn; 61.46.Bc; 64.70.Ja
Keywords: Biomaterials; Biopolymers; Crystal growth; Colloids; Nano-clusters; Phases and equilibria; Hydration
1. Introduction
Amorphous calcium carbonate (ACC) has been ofincreasing interest in biomimetic and biomineralizationstudies due to recent discoveries that reveal its importantrole in morphological control in biological systems [1–9].For instance, ACC is found to be a precursor phase foraragonite in mollusk larval shell formation [10]. In seaurchin larval species and the adult spine, ACC is the tran-sient precursor phase of calcite [8]. To study the formationmechanisms of the ACC phase in vitro, different additivessuch as metal ions, phosphonate, polymers, and different
0022-3093/$ - see front matter � 2007 Elsevier B.V. All rights reserved.
doi:10.1016/j.jnoncrysol.2007.10.022
* Corresponding author. Tel.: +1 352 846 3337; fax: +1 352 846 3355.E-mail address: [email protected] (L.B. Gower).
reaction conditions have been explored to synthesizeACC [11–23].
Biogenic and synthetic ACC can both exist in stable andtransient forms [4–6,8]. The underlying mechanisms of howACC is generated and stabilized under various conditionsare still being debated, especially as to the role(s) of associ-ated macromolecules. It has been found that proteins fromstable biogenic ACC are rich in the amino acid residuesglutamic acid and glutamine, while proteins from transientbiogenic ACC are rich in aspartic acid [5]. In addition, it isproposed that magnesium and phosphate ions cooperatewith the associated proteins to stabilize ACC [5]. However,it is still not understood how one macromolecule could sta-bilize around 150000 molecules of calcium carbonate [5].In the stabilization of ACC, water is also proposed to playan important role in preventing the reorganization of ACCinto the anhydrous form of calcium carbonate [5].

H20(v) + NH3(v) + CO2(v)
(NH3)2CO3(s)
Pasp(aq) + Ca2+(aq) + CO3
2-(aq)
PILP droplets
Fig. 1. Schematic illustration of the experimental setup. A 150 mL glassbeaker containing crushed ammonium carbonate powder was placed in a500 mL glass beaker containing 250 mL of a 20 mM CaCl2 solution with avariable amount of polyaspartate (Pasp). The top of the big glass beakerwas then sealed with parafilm to make a vapor diffusion chamber for thedecomposition products of the ammonium carbonate. The PILP dropletswere collected by centrifugation when the solution became cloudy.
1846 L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854
Spectroscopic methods such as Infrared and Ramanspectroscopy have been widely used to investigate hydratedand anhydrous phases of calcium carbonate [4–6,8,24,25].Water is weakly active in Raman spectroscopy, which istherefore an advantageous technique for investigatinghydrated phases.
Here we present a detailed study of an amorphousCaCO3 phase generated with and without Pasp, with focuson the compositional variation throughout the process. Inparticular, this study is focused on a polymer-inducedliquid-precursor (PILP) amorphous phase, which we haveproposed may play a fundamental role in the morphogen-esis of biominerals. In this PILP process, liquid–liquidphase separation occurs within the crystallizing solutioncontaining micromolar amounts of Pasp, with the minorphase consisting of droplets of a highly hydrated amor-phous precursor to the CaCO3 mineral. The fluidic charac-ter of the colloidal droplets allows them to coalesce into avariety of non-equilibrium morphologies, many of whichbear a striking resemblance to the features found in calcificbiominerals [26–31].
Evidence of a liquid-like ACC phase has also beenreported by Xu et al. [19] (under conditions of high super-saturation, with and without a polymeric process-directingagent), who describe the ‘wetting’ characteristics of ACCparticles which coalesce into films on favorably chargedsurfaces. Faatz et al. [16,17] have also generated ACC par-ticles without polymer (under basic conditions to hydrolyzea carbonate releasing compound), and suggest that par-tially coalesced particles are evidence of the liquid-likecharacter of the early stage ACC. Dense liquid phases,metastable with respect to the solid phase, have also beenobserved in some protein crystallizations that proceedthrough a two-step nucleation process (first-quasidropletsof a densified disordered intermediate form, which are thenfollowed by the nucleation of ordered crystalline embryoswithin these droplets) and a phase diagram has been rigor-ously determined for the L–L phase transformation of amodel protein, lysozyme [32–36].
Wohlrab et al. [37] have identified a PILP type processin amino acids, whereby a combination of oppositelycharged polymers and amino acids (e.g. glutamic acid withPEI, or L-lysine and L-histidine with polyacrylic acid) led todroplets of a liquid-phase precursor which could be stabi-lized for months in the presence of ethanol, enabling adetailed characterization of the role of the polymer in theprocess. The chemical shifts in 1H NMR of the glutamicacid in the PILP phase, the zeta potential of PILP droplets,as well as FTIR data and pH dependence data, all indicatea strong interaction between the polymers and the aminoacids within or on the surface of the induced PILP droplets.Of particular relevance here is the observation that, uponcrystallization, the amount of polymer was greatly reduced.
In the work reported herein, Raman and infrared spec-troscopy was used to investigate the role of Pasp in gener-ation and stabilization of the amorphous CaCO3 PILPphase. The vapor diffusion technique that we commonly
use to form the PILP droplets [26–31] was scaled up to gen-erate sufficient phase for compositional analysis. A calciumchloride solution containing the polymer additive wasgradually raised in supersaturation with carbonate bydecomposition of ammonium carbonate into NH3 andCO2 vapor. The cloudy precipitate was collected by centri-fugation at various stages of the reaction, and character-ized by vibrational spectroscopy and elemental analysis.Part of the gelatinous pellet was additionally filtered toremove loosely bound water. The time-dependent variationof composition of this polymer-induced ACC phase wasthen determined, as described below.
2. Experimental procedure
2.1. Materials
Polyaspartate Æ sodium salt (Mw = 6000 g/mol) waspurchased from Sigma. The CaCl2 Æ 2H2O (99.99+%) and(NH4)2CO3 powders (A.C.S. reagent) were obtained fromAldrich. Double deionized water was used for all solutions.
2.2. Generation of precursor phase
A scaled up version of the vapor diffusion method [38,39]was used to accumulate quantities of PILP phase sufficientfor compositional analysis. The experimental setup isillustrated in Fig. 1. The (NH4)2CO3 powder which wascontained in a 150 mL glass beaker diffused into 250 mL
0
5
10
15
20
25
30
35
40
0 1 2 3 4 5 6 7 8
20ug/ml Pasp, decant,washed and dried mineral
100ug/ml Pasp, decant,washed and dried mineral
20ug/ml Pasp, directly driedPILP phase
100ug/ml Pasp, directlydried PILP phase
Time (Hour)
Pas
p w
t% in
col
lect
ed P
ILP
pha
se
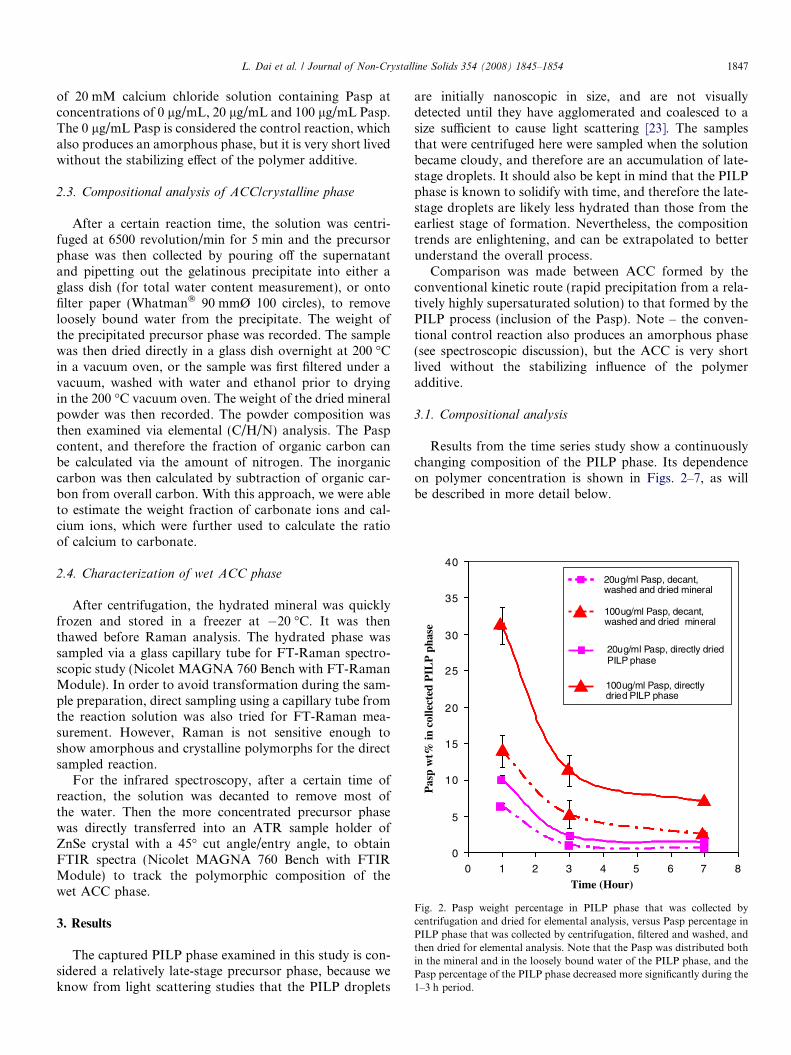
Fig. 2. Pasp weight percentage in PILP phase that was collected bycentrifugation and dried for elemental analysis, versus Pasp percentage inPILP phase that was collected by centrifugation, filtered and washed, andthen dried for elemental analysis. Note that the Pasp was distributed bothin the mineral and in the loosely bound water of the PILP phase, and thePasp percentage of the PILP phase decreased more significantly during the1–3 h period.
L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854 1847
of 20 mM calcium chloride solution containing Pasp atconcentrations of 0 lg/mL, 20 lg/mL and 100 lg/mL Pasp.The 0 lg/mL Pasp is considered the control reaction, whichalso produces an amorphous phase, but it is very short livedwithout the stabilizing effect of the polymer additive.
2.3. Compositional analysis of ACC/crystalline phase
After a certain reaction time, the solution was centri-fuged at 6500 revolution/min for 5 min and the precursorphase was then collected by pouring off the supernatantand pipetting out the gelatinous precipitate into either aglass dish (for total water content measurement), or ontofilter paper (Whatman� 90 mmØ 100 circles), to removeloosely bound water from the precipitate. The weight ofthe precipitated precursor phase was recorded. The samplewas then dried directly in a glass dish overnight at 200 �Cin a vacuum oven, or the sample was first filtered under avacuum, washed with water and ethanol prior to dryingin the 200 �C vacuum oven. The weight of the dried mineralpowder was then recorded. The powder composition wasthen examined via elemental (C/H/N) analysis. The Paspcontent, and therefore the fraction of organic carbon canbe calculated via the amount of nitrogen. The inorganiccarbon was then calculated by subtraction of organic car-bon from overall carbon. With this approach, we were ableto estimate the weight fraction of carbonate ions and cal-cium ions, which were further used to calculate the ratioof calcium to carbonate.
2.4. Characterization of wet ACC phase
After centrifugation, the hydrated mineral was quicklyfrozen and stored in a freezer at �20 �C. It was thenthawed before Raman analysis. The hydrated phase wassampled via a glass capillary tube for FT-Raman spectro-scopic study (Nicolet MAGNA 760 Bench with FT-RamanModule). In order to avoid transformation during the sam-ple preparation, direct sampling using a capillary tube fromthe reaction solution was also tried for FT-Raman mea-surement. However, Raman is not sensitive enough toshow amorphous and crystalline polymorphs for the directsampled reaction.
For the infrared spectroscopy, after a certain time ofreaction, the solution was decanted to remove most ofthe water. Then the more concentrated precursor phasewas directly transferred into an ATR sample holder ofZnSe crystal with a 45� cut angle/entry angle, to obtainFTIR spectra (Nicolet MAGNA 760 Bench with FTIRModule) to track the polymorphic composition of thewet ACC phase.
3. Results
The captured PILP phase examined in this study is con-sidered a relatively late-stage precursor phase, because weknow from light scattering studies that the PILP droplets
are initially nanoscopic in size, and are not visuallydetected until they have agglomerated and coalesced to asize sufficient to cause light scattering [23]. The samplesthat were centrifuged here were sampled when the solutionbecame cloudy, and therefore are an accumulation of late-stage droplets. It should also be kept in mind that the PILPphase is known to solidify with time, and therefore the late-stage droplets are likely less hydrated than those from theearliest stage of formation. Nevertheless, the compositiontrends are enlightening, and can be extrapolated to betterunderstand the overall process.
Comparison was made between ACC formed by theconventional kinetic route (rapid precipitation from a rela-tively highly supersaturated solution) to that formed by thePILP process (inclusion of the Pasp). Note – the conven-tional control reaction also produces an amorphous phase(see spectroscopic discussion), but the ACC is very shortlived without the stabilizing influence of the polymeradditive.
3.1. Compositional analysis
Results from the time series study show a continuouslychanging composition of the PILP phase. Its dependenceon polymer concentration is shown in Figs. 2–7, as willbe described in more detail below.
0
2
4
6
8
10
12
14
16
0 1 2 3 4 5 6 7 8
0 μg/ml Pasp
20 μg/ml Pasp
100 μg/ml Pasp
Time (Hour)
Wei
ght
rati
o of
Wat
er/d
ried
min
eral
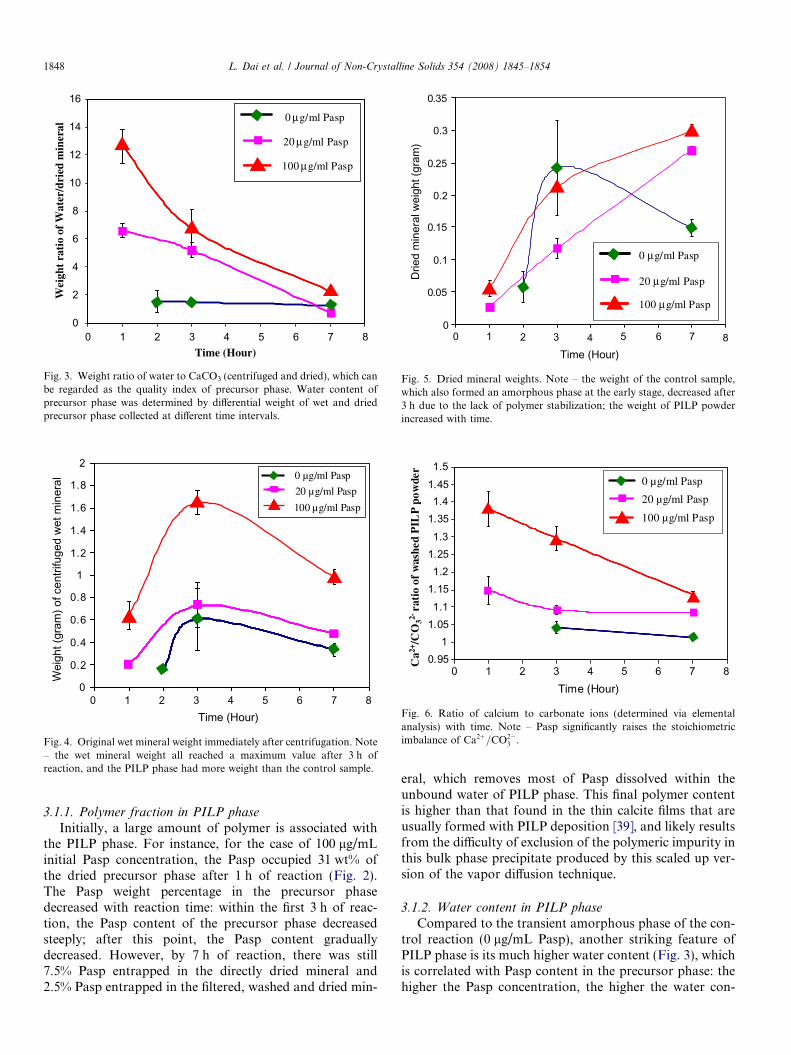
Fig. 3. Weight ratio of water to CaCO3 (centrifuged and dried), which canbe regarded as the quality index of precursor phase. Water content ofprecursor phase was determined by differential weight of wet and driedprecursor phase collected at different time intervals.
Wei
ght (
gram
) of c
entri
fuge
d w
et m
iner
al
Time (Hour)
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
2
0 1 2 3 4 5 6 7 8
0 µg/ml Pasp
20 µg/ml Pasp
100 µg/ml Pasp
Fig. 4. Original wet mineral weight immediately after centrifugation. Note– the wet mineral weight all reached a maximum value after 3 h ofreaction, and the PILP phase had more weight than the control sample.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0 1 4 5 6 7 8Time (Hour)
Drie
d m
iner
al w
eigh
t (gr
am)
0 g/ml Pasp
20 g/ml Pasp
100 g/ml Pasp
32
Fig. 5. Dried mineral weights. Note – the weight of the control sample,which also formed an amorphous phase at the early stage, decreased after3 h due to the lack of polymer stabilization; the weight of PILP powderincreased with time.
Time (Hour)
0.951
1.051.1
1.151.2
1.251.3
1.351.4
1.451.5
0 1 2 3 4 5 6 7 8
Ca2+
/CO
32- r
atio
of
was
hed
PIL
P p
owde
r
0 µg/ml Pasp
20 µg/ml Pasp
100 µg/ml Pasp
Fig. 6. Ratio of calcium to carbonate ions (determined via elementalanalysis) with time. Note – Pasp significantly raises the stoichiometricimbalance of Ca2þ=CO2�
3 .
1848 L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854
3.1.1. Polymer fraction in PILP phase
Initially, a large amount of polymer is associated withthe PILP phase. For instance, for the case of 100 lg/mLinitial Pasp concentration, the Pasp occupied 31 wt% ofthe dried precursor phase after 1 h of reaction (Fig. 2).The Pasp weight percentage in the precursor phasedecreased with reaction time: within the first 3 h of reac-tion, the Pasp content of the precursor phase decreasedsteeply; after this point, the Pasp content graduallydecreased. However, by 7 h of reaction, there was still7.5% Pasp entrapped in the directly dried mineral and2.5% Pasp entrapped in the filtered, washed and dried min-
eral, which removes most of Pasp dissolved within theunbound water of PILP phase. This final polymer contentis higher than that found in the thin calcite films that areusually formed with PILP deposition [39], and likely resultsfrom the difficulty of exclusion of the polymeric impurity inthis bulk phase precipitate produced by this scaled up ver-sion of the vapor diffusion technique.
3.1.2. Water content in PILP phase
Compared to the transient amorphous phase of the con-trol reaction (0 lg/mL Pasp), another striking feature ofPILP phase is its much higher water content (Fig. 3), whichis correlated with Pasp content in the precursor phase: thehigher the Pasp concentration, the higher the water con-
0.2
Frac
tion
of P
asp
Time (Hour)
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
0 1 2 3 4 5 6 7 8 9 10
20µg/ml Pasp; decant, washed and dried PILP100µg/ml Pasp; decant, washed and dried PILP20µg/ml Pasp; directly dried PILP100µg/ml Pasp; directly dried PILP
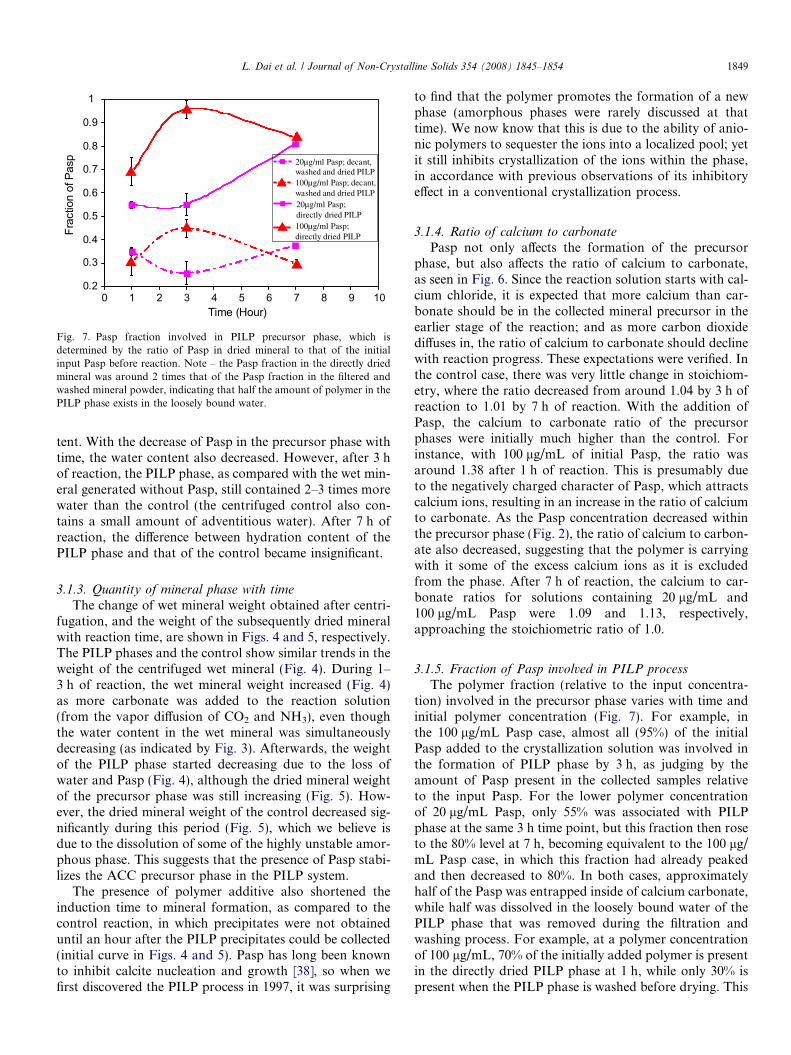
Fig. 7. Pasp fraction involved in PILP precursor phase, which isdetermined by the ratio of Pasp in dried mineral to that of the initialinput Pasp before reaction. Note – the Pasp fraction in the directly driedmineral was around 2 times that of the Pasp fraction in the filtered andwashed mineral powder, indicating that half the amount of polymer in thePILP phase exists in the loosely bound water.
L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854 1849
tent. With the decrease of Pasp in the precursor phase withtime, the water content also decreased. However, after 3 hof reaction, the PILP phase, as compared with the wet min-eral generated without Pasp, still contained 2–3 times morewater than the control (the centrifuged control also con-tains a small amount of adventitious water). After 7 h ofreaction, the difference between hydration content of thePILP phase and that of the control became insignificant.
3.1.3. Quantity of mineral phase with time
The change of wet mineral weight obtained after centri-fugation, and the weight of the subsequently dried mineralwith reaction time, are shown in Figs. 4 and 5, respectively.The PILP phases and the control show similar trends in theweight of the centrifuged wet mineral (Fig. 4). During 1–3 h of reaction, the wet mineral weight increased (Fig. 4)as more carbonate was added to the reaction solution(from the vapor diffusion of CO2 and NH3), even thoughthe water content in the wet mineral was simultaneouslydecreasing (as indicated by Fig. 3). Afterwards, the weightof the PILP phase started decreasing due to the loss ofwater and Pasp (Fig. 4), although the dried mineral weightof the precursor phase was still increasing (Fig. 5). How-ever, the dried mineral weight of the control decreased sig-nificantly during this period (Fig. 5), which we believe isdue to the dissolution of some of the highly unstable amor-phous phase. This suggests that the presence of Pasp stabi-lizes the ACC precursor phase in the PILP system.
The presence of polymer additive also shortened theinduction time to mineral formation, as compared to thecontrol reaction, in which precipitates were not obtaineduntil an hour after the PILP precipitates could be collected(initial curve in Figs. 4 and 5). Pasp has long been knownto inhibit calcite nucleation and growth [38], so when wefirst discovered the PILP process in 1997, it was surprising
to find that the polymer promotes the formation of a newphase (amorphous phases were rarely discussed at thattime). We now know that this is due to the ability of anio-nic polymers to sequester the ions into a localized pool; yetit still inhibits crystallization of the ions within the phase,in accordance with previous observations of its inhibitoryeffect in a conventional crystallization process.
3.1.4. Ratio of calcium to carbonate
Pasp not only affects the formation of the precursorphase, but also affects the ratio of calcium to carbonate,as seen in Fig. 6. Since the reaction solution starts with cal-cium chloride, it is expected that more calcium than car-bonate should be in the collected mineral precursor in theearlier stage of the reaction; and as more carbon dioxidediffuses in, the ratio of calcium to carbonate should declinewith reaction progress. These expectations were verified. Inthe control case, there was very little change in stoichiom-etry, where the ratio decreased from around 1.04 by 3 h ofreaction to 1.01 by 7 h of reaction. With the addition ofPasp, the calcium to carbonate ratio of the precursorphases were initially much higher than the control. Forinstance, with 100 lg/mL of initial Pasp, the ratio wasaround 1.38 after 1 h of reaction. This is presumably dueto the negatively charged character of Pasp, which attractscalcium ions, resulting in an increase in the ratio of calciumto carbonate. As the Pasp concentration decreased withinthe precursor phase (Fig. 2), the ratio of calcium to carbon-ate also decreased, suggesting that the polymer is carryingwith it some of the excess calcium ions as it is excludedfrom the phase. After 7 h of reaction, the calcium to car-bonate ratios for solutions containing 20 lg/mL and100 lg/mL Pasp were 1.09 and 1.13, respectively,approaching the stoichiometric ratio of 1.0.
3.1.5. Fraction of Pasp involved in PILP process
The polymer fraction (relative to the input concentra-tion) involved in the precursor phase varies with time andinitial polymer concentration (Fig. 7). For example, inthe 100 lg/mL Pasp case, almost all (95%) of the initialPasp added to the crystallization solution was involved inthe formation of PILP phase by 3 h, as judging by theamount of Pasp present in the collected samples relativeto the input Pasp. For the lower polymer concentrationof 20 lg/mL Pasp, only 55% was associated with PILPphase at the same 3 h time point, but this fraction then roseto the 80% level at 7 h, becoming equivalent to the 100 lg/mL Pasp case, in which this fraction had already peakedand then decreased to 80%. In both cases, approximatelyhalf of the Pasp was entrapped inside of calcium carbonate,while half was dissolved in the loosely bound water of thePILP phase that was removed during the filtration andwashing process. For example, at a polymer concentrationof 100 lg/mL, 70% of the initially added polymer is presentin the directly dried PILP phase at 1 h, while only 30% ispresent when the PILP phase is washed before drying. This

1090
712
280
156
1440 1750
Free H2O
C
C
C
C
C C
5.01090
712
280
156
1440 17500.00.51.01.52.02.53.03.54.04.5
0 500 1000 1500 2000 2500 3000 3500 4000
1090
712
280
156
1440 1750
Inte
nsity
Inte
nsity
Inte
nsity
Free H2O
C
C
C
C
C C
1090
283
1h3h
Free H2O
CC
1h3h
Free H2O
CC
2801090
2850 2880
CC
Free H2O
1h
3h
Raman shift (cm-1)
Raman shift (cm-1)
Raman shift (cm-1)
0 500 1000 1500 2000 2500 3000 3500 4000
0 500 1000 1500 2000 2500 3000 3500 4000
CC
Free H2O
1h
3h
A
B
C
Fig. 8. Raman spectroscopic analysis of centrifuged wet mineral. Com-parison of control sample (no polymer), 20 lg/mL Pasp, 100 lg/mL Pasp.Peaks labeled C refer to calcite. The spurious peak at 2850/2880 cm�1 isfrom the polyethylene parafilm used to seal the bottom of the capillarytube that holds the centrifuged wet mineral. (A) Control precipitate after3 h of reaction. (Typically, the major peak of ACC also shows at1090 cm�1, which is rather broad; but the peak here is sharp, indicatingvery little ACC phase is present. Also, note the free water peak located at3000–3500 cm�1 is small. (B) Addition of 20 lg/mL Pasp, and (C) 100 lg/mL Pasp. In both cases, the bottom spectrum corresponds to samplecollected after 1 h of reaction; the top spectrum to sample collected after3 h of reaction. Note the free water peak is significantly enhanced byaddition of Pasp.
1850 L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854
implies that the remaining 40% of the polymer is looselyheld and can easily be removed.
Although the weight fraction of Pasp in the dried pre-cursor mineral was decreasing dramatically during the 1–3 h of reaction period (Fig. 2), more PILP phase was beingformed throughout the reaction (Fig. 5). This indicates thatthe Pasp either continuously produces PILP phase, or, as itis excluded from the precursor back into the solution, it hasthe capability to initiate more PILP precipitates (as long asthe calcium concentration is sufficient). We believe the lat-ter is the case because nearly all (95%) of the polymer con-centration that was added into the reaction was found inthe early stage precipitate of the 100 lg/mL case (Fig. 7).
3.1.6. The effects of calcium to carbonate ratio on driedmineral weight
The control reaction leads to a calcium to carbonateratio of 1.04 by 3 h of reaction, which decreased to 1.01by 7 h of reaction. The slight non-stoichiometric ratioand/or the metastability of the amorphous phase may bea reason why the mineral weight decreased after 3 h ofreaction for the control. In contrast, when polymer is pres-ent the amount of mineral present continues to increasethroughout the reaction, even though the calcium to car-bonate ratio is even further from stoichiometry. Appar-ently the polymer plays a role in not only the formationof the PILP phase, but also in stabilizing the amorphousmineral that forms, although the mechanism of this stabil-ization has not yet been determined.
3.2. Spectroscopic analysis of mineral evolution
3.2.1. Raman study of centrifuged wet mineral
Without the presence of Pasp, calcite was the majorproduct formed (Fig. 8(A)). With 20 lg/mL Pasp, theACC phase was stabilized and no crystalline peak showedup after 1 h of reaction; after 3 h of reaction, a small frac-tion of calcite was generated (Fig. 8(B)). The area of waterpeak (located at 3000–3500 cm�1) relative to crystalline cal-cite increased remarkably with the increase of Pasp(Fig. 8(A)–(C)).
3.2.2. FTIR study of precursor evolution
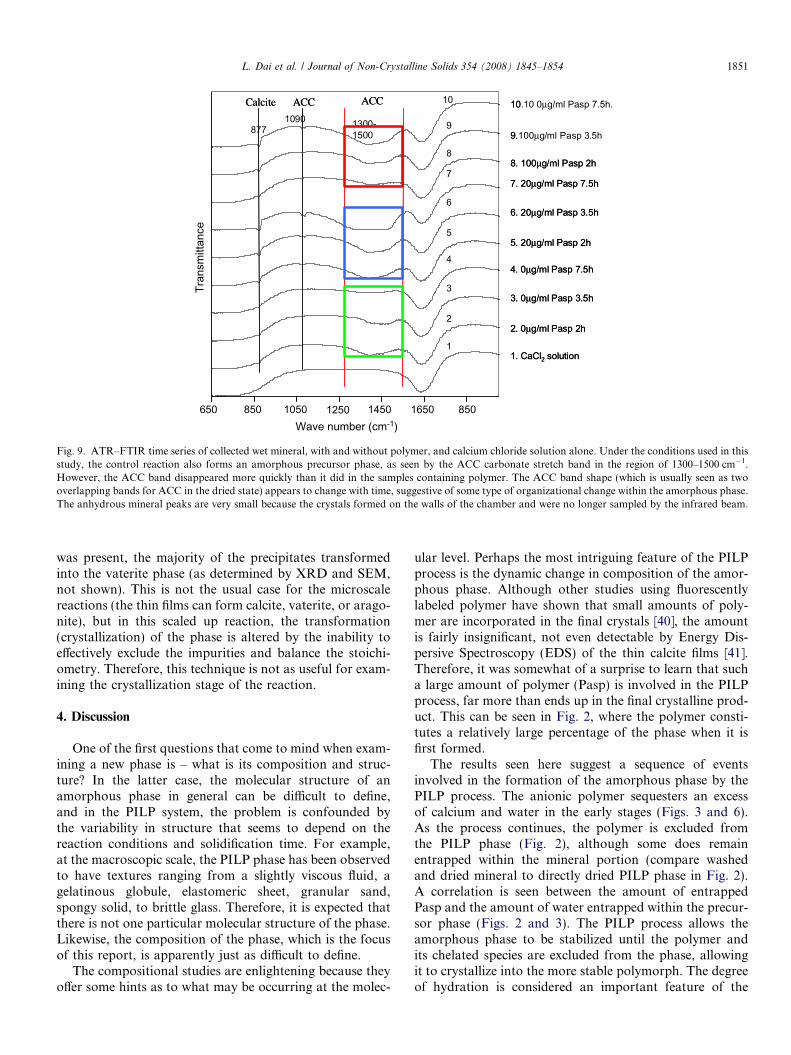
ATR–FTIR was used to examine the polymorphs of thedecanted wet mineral. Fig. 9 shows the FTIR results. Peakslocated at 1090 cm�1 and 1300–1500 cm�1 belong to theamorphous phase, and the peak located at 877 cm�1
belongs to calcite [4–6]. In the control reaction (withoutPasp), measurements at 2 h of reaction showed that themineral did pass through an ACC stage; but the ACCphase was relatively unstable and after 7.5 h of reaction,there was essentially no ACC phase in the reaction solu-tion. The mineral was visually observed to precipitate ontothe wall or bottom of the glass beaker at around 3 h, andwas not in the beam path, therefore a peak representativeof the crystalline mineral peak is not observed once theamorphous phase begins crystallizing, and only the disap-
pearance of the amorphous peak is evidence of this change.For the solutions containing Pasp, the band at the range of1300–1500 cm�1 became broader and deeper with time,indicating an increasing amount of amorphous phase wasbeing generated. Therefore, the polymer stabilizes theamorphous phase, allowing more to be accumulated withtime. The calcite peak is quite small because the ACCphase is dominant (whose FTIR peaks are seen at1090 cm�1 and 1300–1500 cm�1). Although some calcite
2. 0μg/ml Pasp 2h
1. CaCl2 solution
3. 0μg/ml Pasp 3.5h
4. 0μg/ml Pasp 7.5h
5. 20μg/ml Pasp 2h
6. 20μg/ml Pasp 3.5h
7. 20μg/ml Pasp 7.5h
8. 100μg/ml Pasp 2h
9.
10
2
1
3
4
5
6
7
8
9
10
8771090 1300-
1500
2. 0μg/ml Pasp 2h
1. CaCl2 solution
3. 0μg/ml Pasp 3.5h
4. 0μg/ml Pasp 7.5h
5. 20μg/ml Pasp 2h
6. 20μg/ml Pasp 3.5h
7. 20μg/ml Pasp 7.5h
8. 100μg/ml Pasp 2h
9.100μg/ml Pasp 3.5h
10.10 0μg/ml Pasp 7.5h.
2
1
3
4
5
6
7
8
9
10
8771090 1300-
1500
ACC
Tran
smitt
ance
ACCCalcite
650 850 1050 1250 1450 1650 850
ACCACCCalcite
Wave number (cm-1)
Fig. 9. ATR–FTIR time series of collected wet mineral, with and without polymer, and calcium chloride solution alone. Under the conditions used in thisstudy, the control reaction also forms an amorphous precursor phase, as seen by the ACC carbonate stretch band in the region of 1300–1500 cm�1.However, the ACC band disappeared more quickly than it did in the samples containing polymer. The ACC band shape (which is usually seen as twooverlapping bands for ACC in the dried state) appears to change with time, suggestive of some type of organizational change within the amorphous phase.The anhydrous mineral peaks are very small because the crystals formed on the walls of the chamber and were no longer sampled by the infrared beam.
L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854 1851
was present, the majority of the precipitates transformedinto the vaterite phase (as determined by XRD and SEM,not shown). This is not the usual case for the microscalereactions (the thin films can form calcite, vaterite, or arago-nite), but in this scaled up reaction, the transformation(crystallization) of the phase is altered by the inability toeffectively exclude the impurities and balance the stoichi-ometry. Therefore, this technique is not as useful for exam-ining the crystallization stage of the reaction.
4. Discussion
One of the first questions that come to mind when exam-ining a new phase is – what is its composition and struc-ture? In the latter case, the molecular structure of anamorphous phase in general can be difficult to define,and in the PILP system, the problem is confounded bythe variability in structure that seems to depend on thereaction conditions and solidification time. For example,at the macroscopic scale, the PILP phase has been observedto have textures ranging from a slightly viscous fluid, agelatinous globule, elastomeric sheet, granular sand,spongy solid, to brittle glass. Therefore, it is expected thatthere is not one particular molecular structure of the phase.Likewise, the composition of the phase, which is the focusof this report, is apparently just as difficult to define.
The compositional studies are enlightening because theyoffer some hints as to what may be occurring at the molec-
ular level. Perhaps the most intriguing feature of the PILPprocess is the dynamic change in composition of the amor-phous phase. Although other studies using fluorescentlylabeled polymer have shown that small amounts of poly-mer are incorporated in the final crystals [40], the amountis fairly insignificant, not even detectable by Energy Dis-persive Spectroscopy (EDS) of the thin calcite films [41].Therefore, it was somewhat of a surprise to learn that sucha large amount of polymer (Pasp) is involved in the PILPprocess, far more than ends up in the final crystalline prod-uct. This can be seen in Fig. 2, where the polymer consti-tutes a relatively large percentage of the phase when it isfirst formed.
The results seen here suggest a sequence of eventsinvolved in the formation of the amorphous phase by thePILP process. The anionic polymer sequesters an excessof calcium and water in the early stages (Figs. 3 and 6).As the process continues, the polymer is excluded fromthe PILP phase (Fig. 2), although some does remainentrapped within the mineral portion (compare washedand dried mineral to directly dried PILP phase in Fig. 2).A correlation is seen between the amount of entrappedPasp and the amount of water entrapped within the precur-sor phase (Figs. 2 and 3). The PILP process allows theamorphous phase to be stabilized until the polymer andits chelated species are excluded from the phase, allowingit to crystallize into the more stable polymorph. The degreeof hydration is considered an important feature of the

1852 L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854
PILP process because fluidic colloids can coalesce, andtherefore be dynamically molded and shaped into a varietyof non-equilibrium morphologies (many of which dupli-cate the features found in biogenic minerals [26,28–30,39,42,43]).
The concentration of the polymer also affects the rate ofprecursor formation, where the higher the polymer concen-tration, the quicker the precursor phase is formed (Figs. 4and 5). The polymer even promotes the formation of theamorphous phase faster than mineral formation in the con-trol reaction (without polymer). The polymer induces andstabilizes the precursor phase with an excess of calciumas the phase is first formed. The non-stoichiometric balancethen diminishes with time as polymer is excluded, evidentlytaking along with it the excess calcium and associatedhydration waters as it leaves the phase. The FTIR dataindicates that an amorphous precursor exists for both thecontrol reaction and the polymer containing reaction, butthe ACC compositions differ, and this may affect the stabil-ity of the amorphous phase.
In a somewhat analogous system, Wang et al. [44,45]have determined the mechanism of fiber formation inBaSO4 fiber bundles which occurs through an amorphousprecursor mechanism in the presence of polyacrylate(PAA–Na+). They use analytical ultracentrifugation(AUC) and time-dependent pH/conductivity measurementsto show that the first formed species are PAA–Ba2+ com-plexes and aggregates. These complexes then slowly releasethe Ba2+ ions because of the competition of RCOO� withthe SO2�
4 ions. The surrounding supersaturation is thus low-ered such that mineralization proceeds primarily throughthe amorphous nanoparticles that are produced throughion-complexation with the polymer. The ensuing crystalli-zation is described as a mesoscale transformation of theprecursor particles (into fibrous assemblies) because thenucleation and growth processes have already ended (asdetermined by pH measurements, which show that thesupersaturation has become constant).
As is the case with PAA, Pasp is known to be a potentchelator of calcium ions due to the carboxylate groups,which are fully deprotonated at a pH of 7 [46]. Therefore,in an analogous fashion to the BaSO4 system of Wang et al.[44,45], the Pasp likewise initially localizes a high concen-tration of the calcium ions into Pasp–Ca2+ complexes,and then the Ca2+ ions gradually get released because ofthe competition of RCOO� groups with the CO2�
3 ions.Indeed, the specificity of the polymer seems relatively un-important with respect to the PILP process, as we haveseen that the PILP products formed using polyacrylate inplace of polyaspartate (of similar molecular weight) arequite similar (both form droplets and mineral films). Aswas the case in Wang’s system, cloudiness is not observedwith only the addition of calcium, presumably due to thelow molecular weight of the polymer additive. Cloudinessonly appears upon slow addition of the counterion(carbonate in our system), at which point the Pasp–Ca2+
complexes have accumulated sufficient counterion to form
the amorphous droplets. In our prior studies, the earlystage droplets in solution have been observed by dynamiclight scattering to be around 100 nm in diameter [27],which grow by aggregation and coalescence to a size of sev-eral microns. Smaller nanoparticles have been observed byAFM in the colloidal texture of CaCO3 films, which isdependent on the functionality of the surface [28]. Smoothfilms form by coalescence of the droplets if a substrate isplaced within a reasonably small diffusional distance fromwhere the droplets are formed (such as a couple millime-ters) so that the nanoparticles retain the fluidic characteris-tics long enough to coalesce into films. On occasion, wehave also observed fiber bundles that appear identical tothe BaSO4 system of Wang et al. [44,45], but this is notthe usual case (unpublished results), probably because weare not using the limiting supersaturation conditions thatWang uses to create a mesoscopic transformation process.
Faatz et al. [16,17] have put forth a ‘hypothetical’ phasediagram for ACC formation to suggest that the liquid–liquid phase separation occurs via binodal decompositioninto a calcium carbonate poor and rich phase. TheirACC system is somewhat simpler than ours because poly-mer additive was not used; instead the ACC was generatedunder basic conditions (to hydrolyze a carbonate releasingcompound), and they suggest that partially coalesced par-ticles are evidence of the liquid-like character of the earlystage ACC. It is not clear how their ACC particles are sta-bilized in their system (since one does not usually find suchlong-lived ACC at this moderate supersaturation; perhapsthis is due to the alkyl by-products). The liquid-like charac-ter appears to be more limited in their system (as judgingfrom the data provided), which may suggest that polymerenhances the hydration characteristics of the amorphousphase (in agreement with our results). However, it is notclear if their basic reaction proceeds via a calcium hydrox-ide gel (since the pH is not provided), which might impartsome coalescence characteristics to the products. There-fore, comparison between these two systems is difficult interms of deciphering the role of the polymer on ACC for-mation/stabilization. Given the non-homogeneity of thecrystallizing solution due to the sequestering ability of thepolymer, it cannot be said with certainty what type ofphase transformation (i.e., binodal or spinodal) is occur-ring within these localized regions of high supersaturation.As described by Wang et al. [44], who find that differentcomplexes are formed depending on the solution concen-trations of the Ba2+ and polymer, the polymer-directedmineralization process is complicated because it appearsto be a kinetically driven complex-formation process ratherthan a thermodynamically driven one.
It should be noted that the kinetics of the formation andsolidification process for this scaled up reaction are likelydifferent as compared to the colloidal, nanoscale reactionsthat we usually use to mimic biomineral features. The dif-fusional distances required for polymer exclusion may bedifficult to surmount in the bulk gelatinous phase. Never-theless, the dynamic composition trends are likely to be

L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854 1853
qualitatively similar, and can provide insight into themolecular mechanisms. In other words, when the PILPdroplets are at the nano to micron size scale, it should befar easier to drive off the polymer and water as it tries toreach the more thermodynamically stable state. This fitswith our observations that the PILP phase seems to rapidlysolidify when it deposits on a substrate (such as a glassslide), yet large accumulated droplets remain in the fluidicstate much longer [39].
Although there is not a singular composition of thePILP phase, there are good reasons for calling PILP a‘phase’ (and not simply a chelation complex) because thereare distinct phase boundaries which separate the PILPdroplets from the surrounding crystallizing solution oncethe carbonate counterion has been incorporated. In fact,it is these phase boundaries that delineate the morphologyof the final crystal because when the precursor transforms(solidifies and crystallizes), the crystals retain the shape ofthe precursor phase. An amorphous phase can transforminto the more stable crystalline state by two pathways,(1) dissolution and re-precipitation, or (2) solid-state trans-formation. Both are observed in the PILP system, but it isthe latter case that is more desirable because it provides avaluable mechanism for controlling crystal morphology.Although ion mobility is not provided by high temperaturein this aqueous based system (as is the case for most solid-state transformations), in the PILP system, the excess watermay provide the freedom of motion for ordering of theions, particularly once the chelating agent is removed,therefore providing a means for a pseudo-solid-state trans-formation. Because the overall shape is retained, the pro-cess is commonly referred to as a pseudomorphictransformation. This pseudomorphic transformation isnot seen for all ACC phases (most ACC dissolves andrecrystallizes), and may result from the stabilizing influenceof the polymer. The advantage of such a process is that avariety of non-equilibrium morphologies can be generatedbecause the final crystals retain the shape of the precursorphase. For example, this is highlighted by the formation ofmineral fibers by a solution-precursor-solid (SPS) mecha-nism, which constrains the mineral to one-dimensionalgrowth from a PILP ‘flux’ droplet [29,31]. Likewise, thephase boundaries are thought to play a critical role in theintrafibrillar mineralization of collagen to form bone-likenanostructures, in which we have proposed the mechanismutilizes capillary forces applied to the phase boundaries ofthe PILP phase [31,42].
One of the most striking features of the PILP process isthe evolution of the composition of the amorphous phase.In other words, the ACC composition is continuouslychanging and there is no single ACC phase for which thecomposition can be measured and compared to the singu-lar ACC phases reported in the literature [4–6,8,16]. Forexample, Weiner and coworkers [47] distinguish betweena transient anhydrous ACC form and a metastablehydrated ACC with a composition of CaCO3–H2O. It isnot clear how the anhydrous ACC phase which has little
to no water can reorganize into the crystalline structure.In contrast, we find that water aids in the crystallizationof the ACC phase prepared by the PILP process. Forexample, air-dried samples crystallize much more slowlythan hydrated samples remaining in the crystallizing solu-tion (unpublished results), presumably because the hydra-tion waters provide more mobility for organization of theions. Given that these biogenic amorphous phases are asso-ciated with highly charged proteins containing high levelsof aspartate residues (and magnesium, which enhancesthe PILP process), it seems reasonable to speculate that ifit were possible to capture the early stage of the reactionin the biological systems, one might find that the amor-phous phase passes through a dynamical compositionpathway, as seen here.
Weiner and coworkers have also shown through X-rayabsorption spectroscopy that even for biogenic ACC ofthe same chemical composition, the local order is differentfor different ACC samples, which appears to influence thepolymorph of the final crystalline biomineral [48]. Thestructure of the amorphous phase was not examined inour system, but one can imagine that with this changingcomposition the structure will also vary with time, and thiscould markedly influence the transformation (crystalliza-tion) stage of the process, such as the role of polymericmatrices in templating the crystal phase and orientation.These compositional studies show the early stages of thetransformation process, where the phase solidifies as thepolymer and water are excluded. The later stage of thetransformation pathway (i.e., solidification and crystalliza-tion) was not examined in this study because of the kineticvariance of the scaled up reaction. Future reports will delveinto this important stage of the process.
5. Conclusions
The PILP process results in the formation of an amor-phous phase which is induced and stabilized by a polymericadditive. Ultimately, most of the polymer is excluded fromthe final mineral phase, so the polymer can perhaps be con-sidered as a catalyst, one that speeds up the ‘nucleation’ ofthe amorphous phase by sequestering a concentrated ionpool, but also slows down the transformation stage ofthe reaction by inhibiting organization and crystallization.The stabilization of the amorphous phase is importantbecause it provides time for the phase to be molded andshaped into a variety of non-equilibrium morphologies,which then are ‘locked in’ as the phase solidifies and under-goes a pseudomorphic transformation. The stoichiometicbalance also changes throughout the process, and the ratioof Ca2+ to CO2�
3 may influence the subsequent crystalliza-tion rate, as well as the resulting polymorph.
Overall, the composition, and therefore structure, of theamorphous phase is continuously changing throughout themineralization process, making it an extremely difficult sys-tem to characterize, but an extremely useful system formanipulating crystal morphologies.

1854 L. Dai et al. / Journal of Non-Crystalline Solids 354 (2008) 1845–1854
Acknowledgment
Financial support provided by the National ScienceFoundation (Grant DMR-0094209) is gratefullyacknowledged.
References
[1] S. Raz, P.C. Hamilton, F.H. Wilt, S. Weiner, L. Addadi, Adv. Funct.Mater. 13 (2003) 480.
[2] E. Beniash, J. Aizenberg, L. Addadi, S. Weiner, Proc. Roy. Soc.Lond. B – Biol. Sci. 264 (1997) 461.
[3] J. Aizenberg, G. Lambert, L. Addadi, S. Weiner, Adv. Mater. 8(1996) 222.
[4] S. Weiner, Y. Levi-Kalisman, S. Raz, L. Addadi, Connect. TissueRes. 44 (2003) 214.
[5] L. Addadi, S. Raz, S. Weiner, Adv. Mater. 15 (2003) 959.[6] J. Aizenberg, G. Lambert, S. Weiner, L. Addadi, J. Am. Chem. Soc.
124 (2002) 32.[7] J. Aizenberg, S. Weiner, L. Addadi, Connect. Tissue Res. 44 (2003)
20.[8] Y. Politi, T. Arad, E. Klein, S. Weiner, L. Addadi, Science 306 (2004)
1161.[9] S. Weiner, L. Addadi, Geochim. Cosmochim. Acta 66 (2002) A827.
[10] I.M. Weiss, N. Tuross, L. Addadi, S. Weiner, J. Exp. Zool. 293 (2002)478.
[11] J. Donners, B.R. Heywood, E.W. Meijer, R.J.M. Nolte, C. Roman,A. Schenning, N. Sommerdijk, Chem. Commun. (2000) 1937.
[12] Y. Kojima, A. Kawanobe, T. Yasue, Y. Arai, Nippon Seram. Kyo.Gak. – J. Ceram. Soc. Jpn. 102 (1994) 1128.
[13] X.R. Xu, J.T. Han, K. Cho, Chem. Mater. 16 (2004) 1740.[14] L.B. Gower, D.J. Odom, J. Cryst. Growth 210 (2000) 719.[15] S. Raz, S. Weiner, L. Addadi, Adv. Mater. 12 (2000) 38.[16] M. Faatz, F. Grohn, G. Wegner, Adv. Mater. 16 (2004) 996.[17] M. Faatz, F. Grohn, G. Wegner, Mater. Sci. Eng. C – Biomim.
Supramol. Syst. 25 (2005) 153.[18] B. Guillemet, M. Faatz, F. Grohn, G. Wegner, Y. Gnanou, Langmuir
22 (2006) 1875.[19] X. Xu, J.T. Han, K. Cho, Langmuir 21 (2005) 4801.[20] A.W. Xu, Q. Yu, W.F. Dong, M. Antonietti, H. Colfen, Adv. Mater.
17 (2005) 2217.[21] H. Colfen, Curr. Opin. Colloid Interf. Sci. 8 (2003) 23.[22] E. Loste, R.J. Park, J. Warren, F.C. Meldrum, Adv. Funct. Mater. 14
(2004) 1211.
[23] I. Sethman, A. Putnis, O. Grassman, P. Lobmann, Am. Mineral. 90(2005) 1213.
[24] M.M. Tlili, M. Ben Amor, C. Gabrielli, S. Joiret, G. Maurin, P.Rousseau, J. Raman Spectrosc. 33 (2002) 10.
[25] G. Falini, S. Fermani, M. Gazzano, A. Ripamonti, J. Mater. Chem. 8(1998) 1061.
[26] X. Cheng, L.B. Gower, Biotechnol. Prog. 22 (1) (2005) 141.[27] E. DiMasi, T. Liu, M.J. Olszta, L.B. Gower, in: K.H. Sandhage, S.
Yang, T. Douglas, A.R. Parker, E. DiMa si (Eds.), Biol. Bio-InspiredMater. Devices, Warrendale, PA, 2005, p. K10.6.1.
[28] Y.-Y. Kim, E.P. Doulgas, L.B. Gower, Langmuir 23 (9) (2007) 4862.[29] M. Olszta, S. Gajjeraman, M. Kaufman, L. Gower, Chem. Mater. 16
(12) (2004) 2355.[30] M.J. Olszta, E.P. Douglas, L.B. Gower, Calcified Tissue Int. 72 (2003)
583.[31] M.J. Olszta, D.J. Odom, E.P. Douglas, L.B. Gower, Connect. Tissue
Res. 44 (2003) 326.[32] D.N. Petsev, X. Wu, O. Galkin, P.G. Vekilov, J. Phys. Chem. B 107
(2003) 3921.[33] M. Shah, O. Galkin, P.G. Vekilov, J. Chem. Phys. 121 (15) (2004)
7505.[34] L.F. Filobelo, O. Galkin, P.G. Vekilov, J. Chem. Phys. 123 (2005).[35] W.C. Pan, A.B. Kolomeisky, P.G. Vekilov, J. Chem. Phys. 122
(2005).[36] D. Kashchiev, P.G. Vekilov, A.B. Kolomeisky, J. Chem. Phys. 122
(2005).[37] S. Wohlrab, H. Colfen, M. Antonietti, Angew. Chem. – Int. Ed. 44
(2005) 4087.[38] L. Addadi, J. Moradiam, E. Shay, N.G. Maroudas, S. Weiner, Proc.
Natl. Acad. Sci. USA 84 (1987) 2732.[39] L.B. Gower, D.J. Odom, J. Cryst. Growth 210 (4) (2000) 719.[40] L. Dai, X. Cheng, L.B. Gower, Cryst. Growth Design (in
preparation)..[41] L.A. Gower, D.A. Tirrell, J. Cryst. Growth 191 (1–2) (1998) 153.[42] M.J. Olszta, X. Cheng, S.S. Jee, R. Kumar, Y.-Y. Kim, M.J.
Kaufman, E.P. Douglas, L.B. Gower, Materials Science & Engineer-ing – Reports, 2007, doi:10.1016/j.mser.2007.05.001 .
[43] F.F. Amos, D.M. Sharbaugh, D.R. Talham, L.B. Gower, M. Fricke,D. Volkmer, Langmuir 23 (4) (2007) 1988.
[44] T. Wang, H. Colfen, Langmuir 22 (2006) 8975.[45] T. Wang, A. Reinecke, H. Colfen, Langmuir 22 (2006) 8986.[46] Y.-T. Wu, C. Grant, Langmuir 18 (2002) 6813.[47] S. Weiner, I. Sagi, L. Addadi, Science 309 (2005) 1027.[48] Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi, I. Sagi, Adv. Funct.
Mater. 12 (1) (2002) 43.














![[hal-00256385, v1] Modelling improvisatory and compositional ...](https://static.fdokumen.com/doc/165x107/6324da46cedd78c2b50c4d83/hal-00256385-v1-modelling-improvisatory-and-compositional-.jpg)





