
Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve...
13
2012;72:3196-3206. Published OnlineFirst May 2, 2012. Cancer Res Aparna Rao, Jennifer L. Taylor, Nina Chi-Sabins, et al. Therapeutic T cells the Tumor Microenvironment to Improve Recruitment of Combination Therapy with HSP90 Inhibitor 17-DMAG Reconditions Updated version 10.1158/0008-5472.CAN-12-0538 doi: Access the most recent version of this article at: Material Supplementary http://cancerres.aacrjournals.org/content/suppl/2012/04/25/0008-5472.CAN-12-0538.DC1.html Access the most recent supplemental material at: Cited Articles http://cancerres.aacrjournals.org/content/72/13/3196.full.html#ref-list-1 This article cites by 49 articles, 22 of which you can access for free at: Citing articles http://cancerres.aacrjournals.org/content/72/13/3196.full.html#related-urls This article has been cited by 1 HighWire-hosted articles. Access the articles at: E-mail alerts related to this article or journal. Sign up to receive free email-alerts Subscriptions Reprints and . [email protected] To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at Permissions . [email protected] To request permission to re-use all or part of this article, contact the AACR Publications Department at on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538 on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538 on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538
Transcript of Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve...

2012;72:3196-3206. Published OnlineFirst May 2, 2012.Cancer Res Aparna Rao, Jennifer L. Taylor, Nina Chi-Sabins, et al. Therapeutic T cellsthe Tumor Microenvironment to Improve Recruitment of Combination Therapy with HSP90 Inhibitor 17-DMAG Reconditions
Updated version
10.1158/0008-5472.CAN-12-0538doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2012/04/25/0008-5472.CAN-12-0538.DC1.html
Access the most recent supplemental material at:
Cited Articles
http://cancerres.aacrjournals.org/content/72/13/3196.full.html#ref-list-1
This article cites by 49 articles, 22 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/72/13/3196.full.html#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

Microenvironment and Immunology
Combination Therapy with HSP90 Inhibitor 17-DMAGReconditions the Tumor Microenvironment to ImproveRecruitment of Therapeutic T cells
Aparna Rao1, Jennifer L. Taylor2, Nina Chi-Sabins1, Mayumi Kawabe2, William E. Gooding3,4, andWalter J. Storkus1,2,4
AbstractIneffective recognition of tumor cells by CD8þ T cells is a limitation of cancer immunotherapy. Therefore,
treatment regimens that coordinately promote enhanced antitumor CD8þ T-cell activation, delivery, and targetcell recognition should yield greater clinical benefit. Using an MCA205 sarcoma model, we show that in vitrotreatment of tumor cells with the HSP90 inhibitor 17-DMAG results in the transient (proteasome-dependent)degradation of theHSP90 client protein EphA2 and the subsequent increased recognition of tumor cells by Type-1anti-EphA2 CD8þ T cells. In vivo administration of 17-DMAG to tumor-bearing mice led to slowed tumor growth,enhanced/prolonged recognition of tumor cells by anti-EphA2 CD8þ T cells, reduced levels of myeloid-derivedsuppressor cells and regulatory T cells in the tumor microenvironment, and activation of tumor-associatedvascular endothelial cells in association with elevated levels of Type-1 tumor-infiltrating lymphocytes. Whencombined with EphA2-specific active vaccination or the adoptive transfer of EphA2-specific CD8þ T cells,17-DMAG cotreatment yielded a superior tumor therapeutic regimen that was capable of rendering animalsfree of disease. Taken together, our findings indicate that 17-DMAG functions as an immune adjuvant in thecontext of vaccines targeting EphA2. Cancer Res; 72(13); 3196–206. �2012 AACR.
IntroductionReceptor tyrosine kinases (RTK) are an extended family of
cell surface proteins (1) that bind growth factors and hormonesand play important roles in cell survival, growth, migration,and differentiation (2). In neoplastic/cancerous tissues, RTKoverexpression, mutation, and/or constitutive activation mayresult in uncontrolled proliferation and increased malignantphenotype (3).
EphA2 is an RTK that facilitates intercellular interactions viabinding to its ligands ephrin-A1, -A3, -A4, and -A5 expressed ona proximal, opposing cell surface (3). EphA2 is expressedprimarily in cells of epithelial origin in a broad range of adulttissues including lung, spleen, and kidney. In addition, EphA2 isexpressed by activated endothelial cells and is associated withtissue neovascularization in adults (4–6). Numerous studieshave described EphA2 overexpression in a variety of tumorsincluding melanoma, renal cell carcinoma, and colon carcino-
ma, where the degree of overexpression of this RTK has beenlinked to poor prognosis and increased metastatic potential(7–9). As a consequence, EphA2 has become an attractivetarget for therapeutic intervention in patients with solidtumors (10).
Currently, there are several EphA2-centric therapeutic strat-egies contemplated for translation into clinical trials, includingantibody-based strategies that antagonize the binding ofEphA2 to its ligands or which block EphA2-mediated signaltransduction (11–15). Such approaches would inherentlynegate intrinsic EphA2-associated protumor effects and pro-vide a degree of (at least transient) therapeutic efficacy that isindependent of the host immune system. However, becauseEphA2 protein levels are stabilized in tumor cells by HSP90(16, 17), a more therapeutically desirable situation wouldoccur if one were to drive EphA2 degradation via the protea-some, enhancing the likelihood for enhanced MHC class Ipresentation of derivative peptide epitopes and improvedrecognition of tumor cells by EphA2-specific CD8þ T cells(18). Because low-to-moderate avidity EphA2-specific CD8þ
T cells have been detected in the peripheral blood of patientswith renal cell carcinoma or prostate carcinoma (19, 20), levelsof circulating CD8þ T cells could also be amplified by vacci-nation for improved immune targeting of EphA2þ tumor cellsin vivo. We report that in vivo administration of the HSP90inhibitor 17-DMAG enhances EphA2þ tumor cell recognitionby specific CD8þ T cells for a period of several days, whileconcomitantly serving as: (i) a restrictor of myeloid-derivedsuppressor cells (MDSC) and regulatory T cells (Treg) and (ii)
Authors' Affiliations: Departments of 1Immunology, 2Dermatology, and3Biostatistics, University of Pittsburgh School of Medicine; and 4Universityof Pittsburgh Cancer Institute, Pittsburgh, Pennsylvania
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author:Walter J. Storkus, University of Pittsburgh Schoolof Medicine, W1041.2 Biomedical Sciences Tower, 200 Lothrop Street,Pittsburgh, PA 15213. Phone: 412-648-9981; Fax 412-383-5857; E-mail:[email protected]
doi: 10.1158/0008-5472.CAN-12-0538
�2012 American Association for Cancer Research.
CancerResearch
Cancer Res; 72(13) July 1, 20123196
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

an activator/normalizer of the blood vasculature in the TME.When applied in the context of active immunization or adop-tive CD8þ T-cell therapy, 17-DMAG co-administration led toenriched frequencies of tumor-infiltrating Type-1 (anti-EphA2)CD8þ T cells and coordinately improved treatment outcomes.
Materials and MethodsMiceSix- to 10-week-old female C57BL/6 (H-2b), and male and
female B6;129S6-Epha2tm1Jrui (EphA2�/�; H-2b) mice were pur-chased from The Jackson Laboratory and maintained in thepathogen-free animal facility in the Biomedical Sciences Towerat the University of Pittsburgh. All animal work was done inaccordance with a protocol approved by the InstitutionalAnimal Care and Use Committee (IACUC).
Tumor cell lines and tumor establishmentThe EphA2þMCA205 sarcoma andEphA2neg B16melanoma
(H-2b) cell lines were purchased from the American TypeCulture Collection. Cell lines were cultured in complete media[CM; RPMI-1640 supplemented with 100 units/mL penicillin,100 mg/mL streptomycin, 10 mmol/L L-glutamine, and 10%heat-inactivated FBS (all from Life Technologies)] ina humidified incubator at 37�C and 5% CO2. All cell lineswere negative for known mouse pathogens, including myco-plasma. Tumors were established by injection of 5 � 105
MCA205 or 1 � 105 B16 tumor cells s.c. into the right flankof syngeneic mice, with tumor size (in mm2) assessed every 3to 4 days thereafter. Mice were sacrificedwhen tumors becameulcerated or they reached a size of 400 mm2, in accordancewith IACUC guidelines.
17-DMAG–based therapyHSP90 inhibitor 17-DMAG (NSC 707545) was obtained
under a material transfer agreement from the Division ofCancer Treatment and Diagnosis at the National CancerInstitute (Bethesda, MD). For in vivo use, tumor-bearing micewere orally administered 17-DMAG or distilled water in a totalvolume of 50 mL on a daily basis (for up to 10 consecutive days),beginning approximately 18 days after tumor inoculation,when tumors were �100 mm2 in area.
Isolation of tumor, tumor-draining lymph node, andspleen cellsSingle-cell suspensions were obtained from mechanically
disrupted spleen and tumor-draining lymph node, and fromenzymatically digested tumors, as previously described (21).
Western blotMCA205 cell lines were grown to 80% to 90% confluence and
then incubated with 17-DMAG (10–1,000 nmol/L) in CM for 24to 48 hours. To assess the impact of proteasome function andendosomal acidification on EphA2 protein degradation pro-moted by 17-DMAG, MG-132 (50 mmol/L; Peptides Interna-tional), and chloroquine (50 mmol/L; Sigma-Aldrich), respec-tively, were added to cells for 3 hours. After washing in PBS,cells were cultured in the presence of 17-DMAG (500 nmol/L)
for an additional 24 hours. Harvested cells were then incubatedwith lysis buffer, and cell-free lysates were resolved by SDS-PAGE before electro-transfer onto polyvinylidene difluoridemembranes as previously described (17), before probing withpolyclonal anti-EphA2 antibody and horseradish peroxidase-conjugated goat anti-rabbit antibody reagents (both fromSanta Cruz Biotechnology). Probed proteins were visualizedby the Western Lighting Chemiluminescence Detection Kit(Perkin-Elmer) and exposed to X-Omat film (Eastman Kodak)for 5 to 7 minutes.
Immunization of EphA2�/� mice to generate EphA2-specific CD8þ T effector cells
EphA2�/�mice that are not tolerant to "self" EphA2 proteinwere vaccinatedwith syngeneic dendritic cells (DC; transducedwith recombinant adenovirus encoding mIL-12p70 as previ-ously described to generate DC.IL12; ref. 22) alone or DC.IL12pulsedwith syntheticmEphA2671–679 (FSHHNIIRL; H-2D
b classI-presented; ref. 23) and mEphA2682–689 (VVSKYKPM; H-2Kb
class I-presented; ref. 23) peptides on a weekly basis in theright flank. After 4 vaccinations, CD8þ splenic T cells (MACS-selected; Miltenyi Biotec) were analyzed for specific reactivityusing CD107 cytotoxicity and IFN-g ELISA assays.
CD107 cytotoxicity assayCD8þ T cells were cocultured with MCA205 tumor cells
(either derived from culture or single-cell suspensions ofexcised tumors) for 6 hours in the presence of anti-CD107antibody (BD Biosciences). Monensin (Sigma-Aldrich) wasadded to the culture to prevent the re-internalization ofexocytosed CD107 after the first hour of incubation (finalconcentration ¼ 10 mmol/L). Cultures were allowed to incu-bate at 37�C for an additional 5 hours before cell harvest andassessment of T-cell surface CD107 expression asmonitored byflow cytometry.
IFN-g analysesFor tumor recognition assays, splenic CD8þ T cells were
coculturedwith freshly irradiated (100Gy at room temperaturefrom a 137Cs irradiator (Gammacell40, Atomic Energy ofCanada Limited, at a dose rate of 0.87 Gy/min) tumor cellsfor 48 hours, after which, cell-free supernatants were harvestedand assessed for mIFN-g concentrations using a specific ELISA(BD Biosciences). The data are reported as mean � SD ofquadruplicate determinations. In some assays, where indicat-ed, bulk tumor-infiltrating lymphocytes (TIL)/splenocyteswere restimulated in vitro with irradiated (100 Gy) MCA205cells for 5 days at a T-cell-to-tumor cell ratio of 10:1 in CMsupplemented with 20 units/mL of recombinant human inter-leukin-2 (IL-2; PeproTech). Recovered T cells were then cul-tured inCMalone,with syngenicDCs alone, orDCs pulsedwithEphA2 peptides at a 10:1 T-cell-to-DC ratio. In additionalassays, CD8þ TIL from B16 tumor lesions or CD8þ T cellsfrom the spleens of vaccinated EphA2�/� mice were culturedwith flow-sorted CD31þ vascular endothelial cells (VEC) iso-lated from enzymatically digested B16 tumors or tumor-unin-volved kidneys harvested from untreated or treated animals,as previously described (24). T cells stimulated with 5 mg/mL
Combination Immunotherapy Using 17-DMAG
www.aacrjournals.org Cancer Res; 72(13) July 1, 2012 3197
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

anti-CD3 (eBioscience) served as a positive stimulation con-trol. In some assays, as indicated, 1mg/well (final concentrationof 5 mg/mL) of anti-Kb/-Db mAb or isotype control mAb (BDBiosciences) were added to assess the MHC class I–restrictednature of target cell recognition by T cells. For intracellularIFN-g staining, T cells were assessed after a 6 hours cultureusing an intracellular cytokine staining kit (BD Biosciences),with stained cells screened using an LSR II flow cytometer(Beckman Coulter) and data analyzed using FlowJo software(Tree Star, Inc.). Levels of IFN-g in culture supernatants werequantified by specific ELISA.
Immunofluorescence staining and imagingTumor tissue was processed and sectioned as previously
reported (24), followed by immunofluorescence staining andmicroscopy. The following primary antibodies were used forstaining sections: rat anti-mouse CD31 (BD Biosciences), rab-bit anti-mouse EphA2 (Santa Cruz Biotechnology), rat anti-mouse VCAM-1, goat anti-mouse CXCL10 (R&D Systems). Thefollowing secondary antibodies were used: donkey anti-ratAlexa Fluor 488 (Molecular Probes), donkey anti-goat Cy3(Jackson ImmunoResearch), donkey anti-rat Cy3 (JacksonImmunoResearch), goat anti-rat Fab1 fragment Cy3 (JacksonImmunoResearch), and goat anti-rat Alexa Fluor 488 (Molec-ular Probes). Terminal deoxynucleotidyl transferase–mediateddUTP nick end labeling (TUNEL) staining for detection ofapoptotic cells was done using a cell death detection kit (RocheDiagnostics) per the manufacturer's instructions. All tissuesections were briefly incubated with 40,6-diamidino-2-pheny-lindole (DAPI; Sigma-Aldrich) and thenmounted. Images werecaptured using an Olympus Provis microscope (OlympusAmerica). Isotype control and specific antibody images weretaken using the same level of exposure on the channel settings.MetaMorph (Molecular Devices) software was used for cellquantification.
Flow cytometryBefore all cell stainings, Fc receptors were blocked with an
anti-CD16/CD32 antibody (Becton Dickinson). Single-cell sus-pensionswere stained using the following fluorescently labeledantibodies: APC- or FITC-conjugated anti-CD4 and -CD8(eBioscience), FITC-conjugated anti-Gr-1, PE-conjugatedanti-CD25, and FITC-conjugated anti-CD11c (all Becton Dick-inson); FITC-conjugated anti-Class Kb/Db, anti-Class I-Ab, anti-CD107a and anti-CD107b, PE-conjugated anti-IFN-g (alleBioscience) and APC-conjugated anti-CD11b and anti-Foxp3(eBioscience); or matched, fluorochrome-labeled isotype con-trol monoclonal antibody (mAb). For Foxp3 intracellular stain-ing, CD4þ T cells were surface stained as described earlier andthen further processed using an APC-conjugated anti-mouse/rat Foxp3 Staining Kit (eBioscience) according to the manu-facturer's instructions. Fluorescently stained cells wereassessed using an LSR II flow cytometer (Beckman Coulter),with data analyzed using FlowJo software (Tree Star, Inc.).
Statistical analysisComparisons between groups were done using a 2-tailed
Student t test or 1-way ANOVA with post hoc analysis, as
indicated. All data were analyzed using SigmaStat software,version 3.5 (Systat Software). Differences with a P < 0.05 wereconsidered as significant.
Results17-DMAGaffects tumorRTK expression and viability in adose-dependent manner
17-DMAG is an HSP90 inhibitor currently being evaluated inphase I/II clinical trials (25–29). In preliminary in vitro studies,we determined that treatment of tumor cells with 17-DMAGresulted in their loss of EphA2 protein expression, with a cleardrug dose dependency (Fig. 1A). Expression of alternate tumorRTKs and known HSP90 client proteins, such as erbB2/Her2and VEGFR2 (16, 17, 30), and p53 (31), was also inhibited by 17-DMAG treatment in a dose-dependent manner (data notshown). 17-DMAG–induced loss of EphA2 protein expressionin MCA205 sarcoma cells was dependent on the proteolyticactivity of the proteasome andwas not related to the enzymaticaction of endosomes/lysosomes. Hence, addition of the pro-teasomal inhibitor MG-132 to cultures prevented tumor cellEphA2 degradation induced by 17-DMAG treatment, whereasaddition of the lysosomal inhibitor chloroquine to cultures hadno discernable effect on 17-DMAG–associated EphA2 degra-dation (Fig. 1B). Treatment of MCA205 cell cultures with 17-DMAG did not modulate the expression of MHC class Imolecules on the tumor cell surface (Fig. 1C) or tumor cellviability/apoptotic frequency (data not shown). Notably,EphA2-specific CD8þ T cells developed from EphA2�/� mice(Supplementary Fig. S1) showed increased in vitro recognitionof EphA2þ MCA205 (but not EphA2neg B16) tumor cells pre-treated with 17-DMAG (Fig. 1D; Supplementary Fig. S1).
17-DMAG promotes sarcoma regression in associationwith the altered immunophenotype of the MCA205 TME
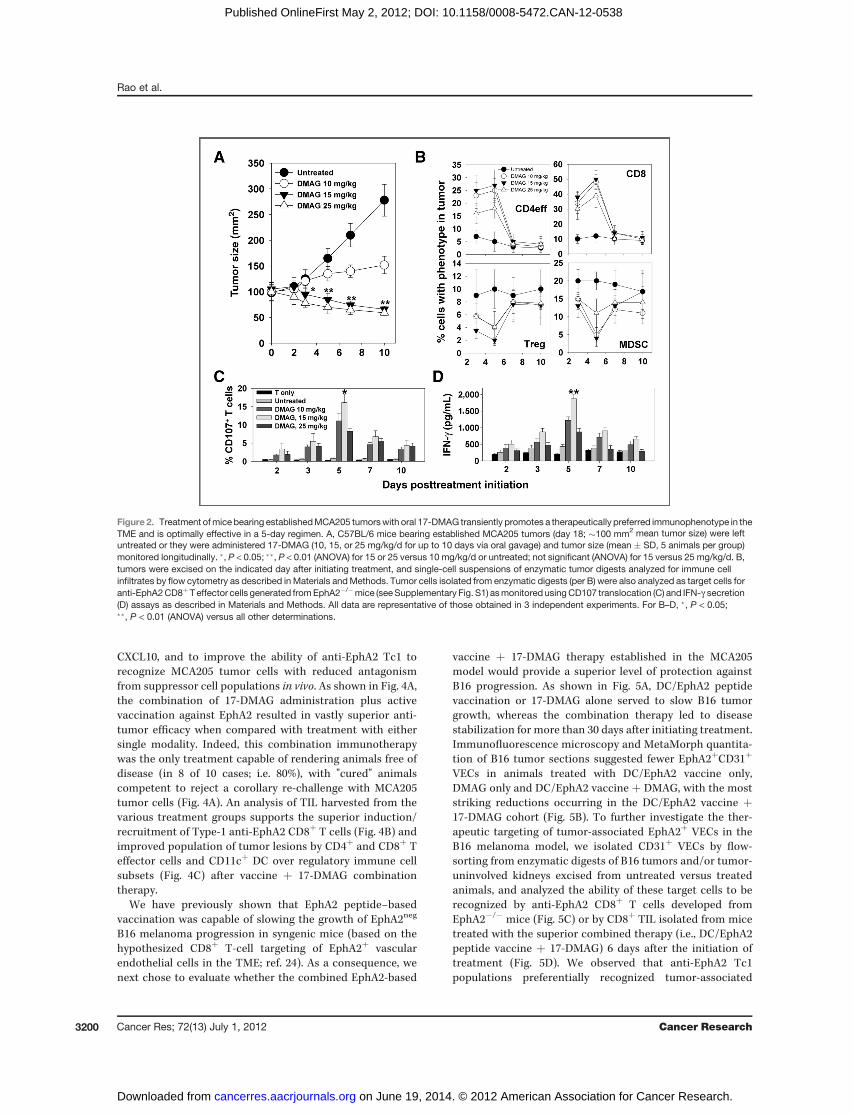
To determine how 17-DMAG would affect the growth andimmunophenotype of well-established (�100 mm2, 18-day-old) tumors, the HSP90 inhibitor was administered orally atdoses of 10, 15, and 25 mg/kg once a day for 2, 3, 5, 7, or 10consecutive days. As shown in Fig. 2A, untreated tumorsdisplayed rapidly progressive growth, whereas tumors in ani-mals treated with 17-DMAG at 10 mg/kg grew more slowly.Tumors in mice treated with 17-DMAG doses �15 mg/kgregressed during the 10 days of active drug administration.To analyze the immunophenotype of the TME, treated animalswere sacrificed 1 day after the last dose of drug, with enzy-matically digested tumors analyzed for immune cell infiltratesand the ability of the freshly isolated tumor cells to be recog-nized by EphA2-specific CD8þT cells in vitro.We observed thatall doses of 17-DMAG were capable of transiently (maximal onday 5 postinitiation of treatment) increasing the level of tumor-infiltrating CD4þ (Foxp3neg; CD4eff) and CD8þ T effector cells,while reducing the levels of tumor-associated cells bearing aCD11bþGr1þ MDSC or CD4þFoxp3þ Treg suppressor cellphenotype (Fig. 2B). Interestingly, in vivo–treated tumor cellswere better recognized by anti-EphA2 CD8þ T cells, particu-larly after 5 days of treatment with 15 mg/kg of 17-DMAG,based on both the CD107 translocation and IFN-g production
Rao et al.
Cancer Res; 72(13) July 1, 2012 Cancer Research3198
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

assays (Fig. 2C and D). Notably, treatment of animals for morethan 5 consecutive days with 17-DMAG resulted in the gradualerosion of this optimal day 5 Type-1 immunophenotype in theTME. On the basis these results, all subsequent experimentsused a standard 17-DMAG treatment regimen (i.e., 15 mg/kgprovided orally for 5 days).
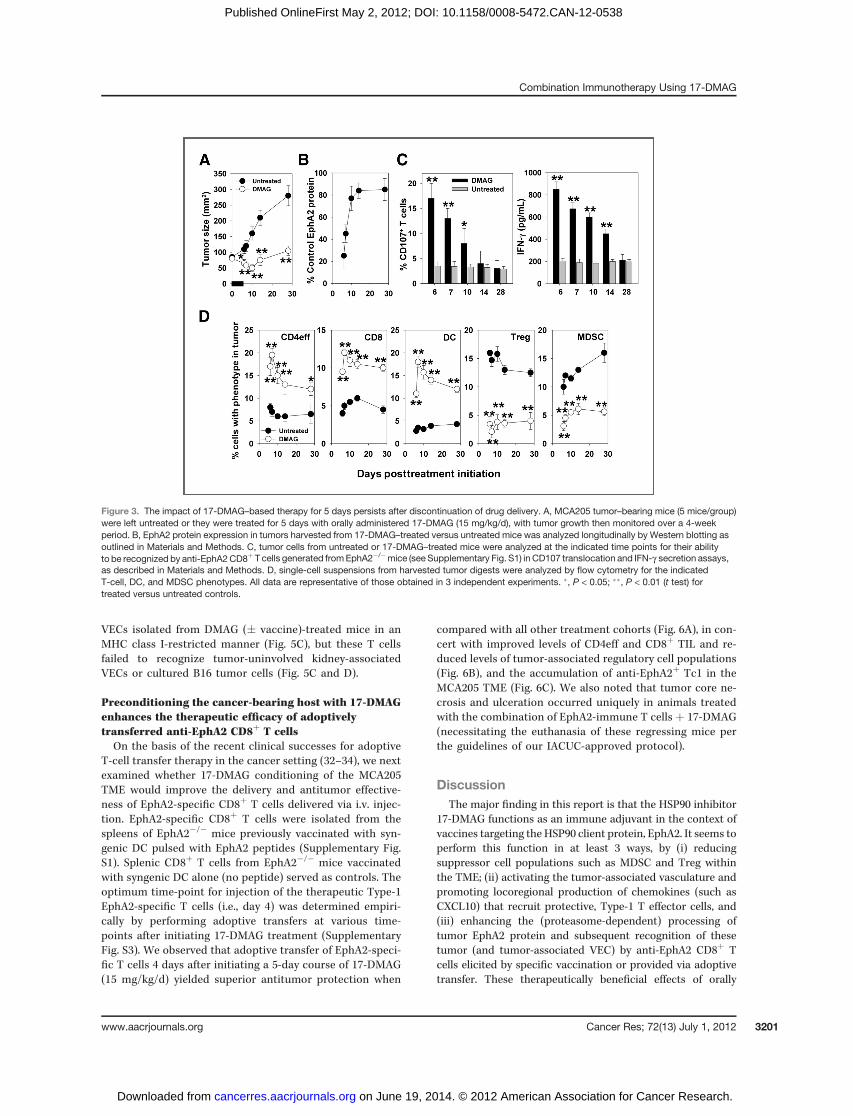
The beneficial effects of 17-DMAGadministration persisteven after discontinuation of therapy on day 5To evaluate the durability of 17-DMAG–associated immu-
nomodulation in vivo, MCA205 tumor-bearing mice weretreated with 15 mg/kg of 17-DMAG for 5 days and thenfollowed for up to 28 days. As shown in Fig. 3A, treatmentwith 17-DMAG promoted tumor regression through day 10 (5days after drug discontinuation), after which time slow tumorgrowth was observed through day 28. Tumor expression ofEphA2 protein in vivo was precipitously reduced during thedrug treatment window and only began to return to controllevels 10 to 15 days after the discontinuation of drug (Fig. 3B).The ability of anti-EphA2 CD8þ T cells to recognize in vivo–treated tumor cells remained significantly elevated throughday 10 to 14 after treatment initiation (Fig. 3C) and thepredominance of CD4þ and CD8þT effector cells (and CD11cþ
DC) over regulatory (MDSC and Treg) cells within the treatedTME persisted through day 28 in these experiments (Fig. 3D).We also observed that 17-DMAG–treated tumors displayed aprolonged, increase in expression of both VCAM-1 and theCXCR3 ligand chemokine CXCL10 in situ, even after discon-tinuation of this monotherapy (Supplementary Fig. S2). Fur-thermore, TUNEL staining of tumor sections showed increasedfrequencies of apoptotic cell death within the TME of 17-DMAG–treated versus untreated MCA205 lesions at all timepoints through day 28 (Supplementary Fig. S2).
Combination vaccination þ 17-DMAG immunotherapyyields superior antitumor efficacy
Given 17-DMAG's ability to promote the enhanced rec-ognition of treated tumor cells by anti-EphA2 CD8þ T-cell invivo, and a protective immunophenotype within the TME, wehypothesized that a combination therapy based on activevaccination against EphA2 protein along with 17-DMAGadministration would provide superior efficacy againstEphA2þ tumors. In such a paradigm, vaccine-induced,anti-EphA2 CD8þ T cells would be recruited into the TMEbased on the ability of 17-DMAG to activate tumor (VCAM-1þ) endothelial cells, to increase locoregional production of
Figure 1. 17-DMAG promotes proteasome-dependent EphA2 protein degradation in MCA205 sarcoma cells and the enhanced recognition oftumor cells by anti-EphA2 CD8þ T cells in vitro. A, MCA205 tumor cells were treated with various doses of 17-DMAG for 24 hours in vitro and thenlysed, with EphA2 and control b-actin protein expression subsequently monitored by Western blotting as described in Materials and Methods. NC,negative control lysate from EphA2neg B16 melanoma cells. B, proteasome inhibitor (MG-132), but not lysosome inhibitor chloroquine (CLQ), blocks17-DMAG (500 nmol/L)-induced degradation of EphA2 protein in MCA205 tumor cells. C, treatment of MCA205 cells with 17-DMAG at the indicateddoses for 24 hours (or 48 hours, data not shown) did not affect MHC class I expression on tumor cells. D, 17-DMAG–treated EphA2þ MCA205cells were better recognized versus control, untreated tumor cells by anti-EphA2 CD8þ T cells (developed from EphA2�/� mice, per SupplementaryFig. S1 and Materials and Methods) in CD107 translocation assays as described in Materials and Methods. All data are representative of thoseobtained in 3 independent experiments.
Combination Immunotherapy Using 17-DMAG
www.aacrjournals.org Cancer Res; 72(13) July 1, 2012 3199
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538
CXCL10, and to improve the ability of anti-EphA2 Tc1 torecognize MCA205 tumor cells with reduced antagonismfrom suppressor cell populations in vivo. As shown in Fig. 4A,the combination of 17-DMAG administration plus activevaccination against EphA2 resulted in vastly superior anti-tumor efficacy when compared with treatment with eithersingle modality. Indeed, this combination immunotherapywas the only treatment capable of rendering animals free ofdisease (in 8 of 10 cases; i.e. 80%), with "cured" animalscompetent to reject a corollary re-challenge with MCA205tumor cells (Fig. 4A). An analysis of TIL harvested from thevarious treatment groups supports the superior induction/recruitment of Type-1 anti-EphA2 CD8þ T cells (Fig. 4B) andimproved population of tumor lesions by CD4þ and CD8þ Teffector cells and CD11cþ DC over regulatory immune cellsubsets (Fig. 4C) after vaccine þ 17-DMAG combinationtherapy.
We have previously shown that EphA2 peptide–basedvaccination was capable of slowing the growth of EphA2neg
B16 melanoma progression in syngenic mice (based on thehypothesized CD8þ T-cell targeting of EphA2þ vascularendothelial cells in the TME; ref. 24). As a consequence, wenext chose to evaluate whether the combined EphA2-based
vaccine þ 17-DMAG therapy established in the MCA205model would provide a superior level of protection againstB16 progression. As shown in Fig. 5A, DC/EphA2 peptidevaccination or 17-DMAG alone served to slow B16 tumorgrowth, whereas the combination therapy led to diseasestabilization for more than 30 days after initiating treatment.Immunofluorescence microscopy and MetaMorph quantita-tion of B16 tumor sections suggested fewer EphA2þCD31þ
VECs in animals treated with DC/EphA2 vaccine only,DMAG only and DC/EphA2 vaccine þ DMAG, with the moststriking reductions occurring in the DC/EphA2 vaccine þ17-DMAG cohort (Fig. 5B). To further investigate the ther-apeutic targeting of tumor-associated EphA2þ VECs in theB16 melanoma model, we isolated CD31þ VECs by flow-sorting from enzymatic digests of B16 tumors and/or tumor-uninvolved kidneys excised from untreated versus treatedanimals, and analyzed the ability of these target cells to berecognized by anti-EphA2 CD8þ T cells developed fromEphA2�/� mice (Fig. 5C) or by CD8þ TIL isolated from micetreated with the superior combined therapy (i.e., DC/EphA2peptide vaccine þ 17-DMAG) 6 days after the initiation oftreatment (Fig. 5D). We observed that anti-EphA2 Tc1populations preferentially recognized tumor-associated
Figure 2. Treatment ofmice bearing establishedMCA205 tumorswith oral 17-DMAG transiently promotes a therapeutically preferred immunophenotype in theTME and is optimally effective in a 5-day regimen. A, C57BL/6 mice bearing established MCA205 tumors (day 18; �100 mm2 mean tumor size) were leftuntreated or they were administered 17-DMAG (10, 15, or 25 mg/kg/d for up to 10 days via oral gavage) and tumor size (mean � SD, 5 animals per group)monitored longitudinally. �, P < 0.05; ��, P < 0.01 (ANOVA) for 15 or 25 versus 10 mg/kg/d or untreated; not significant (ANOVA) for 15 versus 25 mg/kg/d. B,tumors were excised on the indicated day after initiating treatment, and single-cell suspensions of enzymatic tumor digests analyzed for immune cellinfiltrates by flow cytometry as described inMaterials andMethods. Tumor cells isolated from enzymatic digests (per B) were also analyzed as target cells foranti-EphA2CD8þTeffector cells generated fromEphA2�/�mice (seeSupplementary Fig. S1) asmonitored usingCD107 translocation (C) and IFN-g secretion(D) assays as described in Materials and Methods. All data are representative of those obtained in 3 independent experiments. For B–D, �, P < 0.05;��, P < 0.01 (ANOVA) versus all other determinations.
Rao et al.
Cancer Res; 72(13) July 1, 2012 Cancer Research3200
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538
VECs isolated from DMAG (� vaccine)-treated mice in anMHC class I-restricted manner (Fig. 5C), but these T cellsfailed to recognize tumor-uninvolved kidney-associatedVECs or cultured B16 tumor cells (Fig. 5C and D).
Preconditioning the cancer-bearing host with 17-DMAGenhances the therapeutic efficacy of adoptivelytransferred anti-EphA2 CD8þ T cellsOn the basis of the recent clinical successes for adoptive
T-cell transfer therapy in the cancer setting (32–34), we nextexamined whether 17-DMAG conditioning of the MCA205TME would improve the delivery and antitumor effective-ness of EphA2-specific CD8þ T cells delivered via i.v. injec-tion. EphA2-specific CD8þ T cells were isolated from thespleens of EphA2�/� mice previously vaccinated with syn-genic DC pulsed with EphA2 peptides (Supplementary Fig.S1). Splenic CD8þ T cells from EphA2�/� mice vaccinatedwith syngenic DC alone (no peptide) served as controls. Theoptimum time-point for injection of the therapeutic Type-1EphA2-specific T cells (i.e., day 4) was determined empiri-cally by performing adoptive transfers at various time-points after initiating 17-DMAG treatment (SupplementaryFig. S3). We observed that adoptive transfer of EphA2-speci-fic T cells 4 days after initiating a 5-day course of 17-DMAG(15 mg/kg/d) yielded superior antitumor protection when
compared with all other treatment cohorts (Fig. 6A), in con-cert with improved levels of CD4eff and CD8þ TIL and re-duced levels of tumor-associated regulatory cell populations(Fig. 6B), and the accumulation of anti-EphA2þ Tc1 in theMCA205 TME (Fig. 6C). We also noted that tumor core ne-crosis and ulceration occurred uniquely in animals treatedwith the combination of EphA2-immune T cells þ 17-DMAG(necessitating the euthanasia of these regressing mice perthe guidelines of our IACUC-approved protocol).
DiscussionThe major finding in this report is that the HSP90 inhibitor
17-DMAG functions as an immune adjuvant in the context ofvaccines targeting the HSP90 client protein, EphA2. It seems toperform this function in at least 3 ways, by (i) reducingsuppressor cell populations such as MDSC and Treg withinthe TME; (ii) activating the tumor-associated vasculature andpromoting locoregional production of chemokines (such asCXCL10) that recruit protective, Type-1 T effector cells, and(iii) enhancing the (proteasome-dependent) processing oftumor EphA2 protein and subsequent recognition of thesetumor (and tumor-associated VEC) by anti-EphA2 CD8þ Tcells elicited by specific vaccination or provided via adoptivetransfer. These therapeutically beneficial effects of orally
Figure 3. The impact of 17-DMAG–based therapy for 5 days persists after discontinuation of drug delivery. A, MCA205 tumor–bearing mice (5 mice/group)were left untreated or they were treated for 5 days with orally administered 17-DMAG (15 mg/kg/d), with tumor growth then monitored over a 4-weekperiod. B, EphA2 protein expression in tumors harvested from 17-DMAG–treated versus untreated mice was analyzed longitudinally by Western blotting asoutlined in Materials and Methods. C, tumor cells from untreated or 17-DMAG–treated mice were analyzed at the indicated time points for their abilityto be recognized by anti-EphA2CD8þ T cells generated fromEphA2�/�mice (see Supplementary Fig. S1) in CD107 translocation and IFN-g secretion assays,as described in Materials and Methods. D, single-cell suspensions from harvested tumor digests were analyzed by flow cytometry for the indicatedT-cell, DC, and MDSC phenotypes. All data are representative of those obtained in 3 independent experiments. �, P < 0.05; ��, P < 0.01 (t test) fortreated versus untreated controls.
Combination Immunotherapy Using 17-DMAG
www.aacrjournals.org Cancer Res; 72(13) July 1, 2012 3201
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

administered 17-DMAGoccurred rapidly, weremaximal by day5 of drug provision, and were sustained for a prolonged periodof 1 to 3 weeks (depending on the specific index), as long astreatment with the HSP90 inhibitor was discontinued after a 5-day course. Prolonged application of 17-DMAG for >5 daysseems to result in the erosion of its potent adjuvant-likequalities by as early as day 7 in chronic treatment protocols.Why such immunologic silencing occurs upon extended 17-DMAG administration is currently unclear. However, previousstudies have suggested the potential attenuating effects ofhigh-dose, long-term dosing of 17-DMAG on the immunesystem (7, 35, 36). We plan to intensively investigate themechanism(s) underlying the deleterious effects of more"chronic" 17-DMAG administration in future studies.
The capacity of this combination immunotherapy to targetboth EphA2þ tumor cells and/or VEC in the TME has impor-tant translational ramifications because EphA2þ cancer cellshave been reported to be more migratory (greater metastaticpotential; refs. 37–39) and the immune regulation of tumor-
associated blood vessels reduces concerns for the immuno-phenotypic status of the tumor cell population (i.e., variation inMHC and antigen expression by heterogeneous populations oftumor cells in the TME). The ability of this treatment strategyto facilitate immune targeting of stromal cells provides thepossibility of effectively treating MHC I- or antigen-loss (asmodeled by EphA2neg B16) tumor variants.
Importantly, 17-DMAG administration combined witheither active vaccination to induce anti-EphA2 Tc1 in vivo(Figs. 4A and 5A) or the adoptive transfer of anti-EphA2CD8þTcells (Fig. 6A) proved therapeutically superior to any singlecomponent modality. Both combination protocols resulted inthe rapid regression of well-established (�day 18 tumors), witha high rate of complete responses in the vaccine setting.Evidence of a protective memory CD8þ T-cell response wasevident, given the rejection of a subsequent tumor rechallengein thesemice. The only distinguishing clinical variable betweenthe 2 immunotherapy approaches was the core necrosisobserved only for tumors treated with the adoptive
Figure 4. 17-DMAG administration improves the immunogenicity and antitumor efficacy of an EphA2 peptide–based vaccine in the MCA205 tumor model. A,C57BL/6 mice bearing established EphA2þ MCA205 sarcomas (s.c. right flank) remained untreated or they were treated with DC-based vaccines (s.c., leftflank on days 0 and 7 of the treatment regimen) that contained or lacked EphA2 peptide epitopes, alone or in combination with 17-DMAG (15 mg/kg/don the first 5 days of the treatment regimen by oral gavage). Tumor size (mean � SD) is reported in mm2. All the mice in the DC/EphA2 þ 17-DMAG–treatedgroup rendered tumor-free (80%) were rechallenged (s.c., right flank) with MCA205 tumor cells on day 30 of the experiment (as indicated by arrowwith R inset) and monitored through day 60 after treatment initiation. B, CD8þ TILs recovered from tumors on day 14 after treatment initiation were assessedfor their ability to recognize syngenic control DCs pulsed with no peptide or DC pulsed with the EphA2671–679 þ EphA2682–689 peptides. After 48-hourincubation, cell-free supernatants were analyzed for IFN-g content by ELISA. Response to DC (no peptide) was <50 pg/mL in all instances. C, single-cellsuspensions of enzymatically digested day 14 (posttreatment initiation) tumors were analyzed by flow cytometry for the indicated T-cell, DC, and MDSCphenotypes as described in Materials and Methods. Each filled circle represents data from an individual animal in a given control or treatment cohort,with themeanof data indicatedbyagraybar for eachcohort. All data are representative of thoseobtained in 3 independent experiments. �,P<0.05; ��,P<0.01(ANOVA) versus all other cohorts.
Rao et al.
Cancer Res; 72(13) July 1, 2012 Cancer Research3202
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

immunotherapy (AIT) approach. The simplest explanations forthis biologic difference would reflect: (i) the comparativenumbers of specific CD8þ T cells infiltrating tumors at earlytime points (i.e., presumed to be greater in the AIT protocol),(ii) the higher functional avidity of the anti-EphA2 Tc1 gener-ated from the EphA2�/� versus wild-type (self-tolerant) miceallowing these T effector cells to more efficiently recognizetumor cells or VEC expressing modest levels of MHC I-EphA2peptide complexes on their cell surfaces in vivo, or (iii) possiblevariance in the polyfunctionality of anti-EphA2 T cells in thesetreatment cohorts. We are investigating each of these intrigu-ing possibilities in on-going experiments.HSP90 inhibitors, such as 17-DMAG (alvespimycin), have
been investigated in multiple phase I/II clinical trials overthe past several years. These drugs exhibited variable anti-tumor efficacy and toxicity when administered as singleagents (26, 27, 40–42). In a phase I study of 17-DMAGadministered i.v. to patients with advanced solid tumors,objective clinical responses (including 1 complete response)based on RECIST criteria were reported in a minority ofpatients with kidney or prostate carcinoma, melanoma or
chondrosarcoma (26). Like many chemotherapeutic agents,HSP90 inhibitors fail to exert durable anticancer efficacybased on intrinsic disease resistance or the development ofacquired resistance among treated populations of cancercells (43–45). In the case of 17-DMAG, such acquired resis-tance could be because of the ability of this agent toupregulate expression of the cytoprotective HSP70 and/orBcl-2 molecules (46, 47). Such clinical limitations reinforcethe need to evolve more effective combinational therapeuticstrategies.
Our data suggest that sustained therapeutic benefits can beobtained by combining a short (5 day) course of 17-DMAGtreatment along with an immunotherapy promoting the CD8þ
T-cell targeting of EphA2þ cells in the TME. Given the clinicalexperience suggesting onlymoderate efficacy for single-modal-ity HSP90 inhibitors, aswell as for antigen-based vaccination inthe cancer setting (26, 27, 48–50), combination protocolspredicated on these individual treatment modalities would beanticipated to provide superior clinical benefits to patients.Although our modeling has been based on combined vaccine/AIT þ 17-DMAG approaches focusing on disease-associated
Figure 5. 17-DMAG improves the antitumor efficacy of an EphA2 peptide–based vaccine in the EphA2neg B16melanomamodel based on immune targeting ofEphA2þ VEC. A, C57BL/6 mice bearing established s.c. B16 melanomas (right flank) were left untreated or treated as outlined, with tumor size (mean� SD)reported in mm2 followed for up to 30 days. ��, P < 0.001 (ANOVA) versus all other cohorts. B, day 14 (posttreatment initiation) tumors were harvestedand tissue sections analyzed by immunofluorescence microscopy and MetaMorph quantitation for coexpression of CD31 (i.e., VEC) and EphA2 proteins asdescribed in Materials and Methods. �, P < 0.05; ��, P < 0.01 (ANOVA) versus all other cohorts. Anti-EphA2 CD8þ T cells isolated from the spleens of immuneEphA2�/� mice (C; as outlined in Supplementary Fig. S1), or TIL from B16 tumor–bearing animals treated with combined DC/EphA2 peptide vaccination þ17-DMAG (D; per Supplementary Fig. 5A) were analyzed for reactivity against flow-sorted CD31þ VECs isolated from the tumors of B16-bearinganimals left untreated or treated for 6 days with DC/EphA2 vaccine only, 17-DMAG or DC/EphA2 vaccine þ 17-DMAG. CD31þ kidney VECs were alsoflow-sorted from animals treated for 6 days with DC/EphA2 vaccine þ 17-DMAG to discern autoimmunity of T cells against tumor-uninvolved VECs. C, theMHC class I–restricted nature of VEC recognition by CD8þ T cells was assessed by inclusion of anti-class I or isotype control mAb per culture well, asdescribed inMaterials andMethods. C andD, �,P < 0.05; ��,P <0.01 (t test) versus control antibody treatment or untreated controls, respectively. All data arerepresentative of those obtained in 3 independent experiments. HPF, high-power field.
Combination Immunotherapy Using 17-DMAG
www.aacrjournals.org Cancer Res; 72(13) July 1, 2012 3203
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

EphA2 protein, one could also clearly envision similar thera-peutic protocols predicated on the immune targeting of one ormore alternateHSP90 client proteins that are commonly (over)expressed by tumor cells or tumor-associated stromal cells,such as beclin 1, cyclin B, EGFR, HER2/neu, IGF1-R, PDGFR,PIM-1, STAT3, survivin, TGFbR, VEGFR1, VEGFR2, amongmany others (16, 30).
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: A. Rao, M. Kawabe, W.J. StorkusDevelopment of methodology: A. Rao, J.L. Taylor, N. Chi-Sabins, M. KawabeAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): A. RaoAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): A. Rao, W.E. Gooding, W.J. Storkus
Writing, review, and/or revision of the manuscript: A. Rao, J.L. Taylor, W.J.StorkusStudy supervision: J.L. Taylor, W.J. Storkus
AcknowledgmentsThe authors thank Drs. Devin B. Lowe, Robert L. Ferris, Theresa L.
Whiteside, Soldano Ferrone, Robert J. Binder, and Jeffrey L. Brodsky for theircareful review and useful discussions provided during the preparation of themanuscript.
Grant SupportThis work was supported by NIH grants P01 CA100327 and R01 CA114071
(W.J. Storkus). This project used the UPCI Vector Core Facility and wassupported in part by the University of Pittsburgh CCSG award P30 CA047904.
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisementin accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received February 13, 2012; revised April 4, 2012; accepted April 12, 2012;published OnlineFirst May 2, 2012.
Figure 6. 17-DMAG improves the antitumor efficacy of adoptively transferred anti-EphA2 CD8þ T cells in a combination therapy. A, C57BL/6 micebearing established s.c. MCA205 sarcomas (right flank) were left untreated or they were treated with 17-DMAG (15 mg/kg/d provided orally on the first5 days of the treatment regimen) � adoptive transfer (i.v. tail vein on day 4 of the treatment regimen) of 5 � 106 CD8þ T cells isolated from EphA2�/�
mice previously vaccinated with syngenic DCs (control T cells) or DCs loaded with the EphA2671–679 þ EphA2682–689 peptides. Tumor size wasmonitored longitudinally and is reported (mean � SD) in mm2 from 5 mice per group. All animals treated with combined anti-EphA2 (immune) T cell þDMAG therapy required euthanasia because of core necrosis on day 14 after treatment initiation. B, day 14 untreated or treated tumors underwentenzymatic digestion, with single cells analyzed by flow cytometry for the indicated T-cell, DC, and MDSC phenotypes as described in Materials andMethods. Each filled circle represents data from an individual animal/cohort with the data mean indicated by the gray bar. C, CD8þ TILs harvestedfrom day 14 (posttreatment initiation) tumors were analyzed for IFN-g secretion in response to EphA2 peptide–pulsed syngenic (control) DCs by ELISAas outlined in Materials and Methods. �, P < 0.05; ��, P < 0.01 (ANOVA) versus all other cohorts. All data are representative of those obtained in 3independent experiments.
Rao et al.
Cancer Res; 72(13) July 1, 2012 Cancer Research3204
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

References1. Robinson DR,Wu YM, Lin SF. The protein tyrosine kinase family of the
human genome. Oncogene 2000;19:5548–57.2. Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signaling as a
target for cancer intervention strategies. Endocr Relat Cancer2001;8:161–73.
3. Bache KG, Slagsvold T, Stenmark H. Defective down-regulation ofreceptor tyrosine kinases in cancer. EMBO J 2004;23:2707–12.
4. Surawska H,Ma PC, Salgia R. The role of Ephrins and eph receptors incancer. Cytokine Growth Factor Rev 2004;15:419–33.
5. ChengN,BrantleyDM, LiuH, LinQ, EnriquezM,GaleN, et al. Blockadeof EphA receptor tyrosine kinase activation inhibits vascular endothe-lial cell growth factor-induced angiogenesis. Mol Cancer Res2002;1:2–11.
6. Cheng N, Brantley DM, Chen J. The ephrins and Eph receptors inangiogenesis. Cytokine Growth Factor Rev 2002;13:75–85.
7. Herrem CJ, Tatsumi T, Olson KS, Shirai K, Finke JH, Bukowski RM,et al. Expression of EphA2 is prognostic of disease-free interval andoverall survival in surgically treated patients with renal cell carcinoma.Clin Cancer Res 2005;11:226–31.
8. Kinch MS, Moore MB, Harpole DH Jr. Predictive value of the EphA2receptor tyrosine kinase in lung cancer recurrence and survival. ClinCancer Res 2003;9:613–8.
9. Kinch MS, Carles-Kinch K. Overexpression and functional alterationsof the EphA2 tyrosine kinase in cancer. Clin Exp Metastasis 2003;20:59–68.
10. Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand insolid tumors: function and therapeutic targeting. Mol Cancer Res2008;6:1795–806.
11. Yamaguchi S, Tatsumi T, Takehara T, Sakamori R, Uemura A, Mizush-ima T, et al. Immunotherapy of murine colon cancer using receptortyrosine kinase EphA2-derived peptide-pulsed dendritic cell vaccines.Cancer 2007;110:1469–77.
12. Ansuini H, Meola A, Gunes Z, Paradisi V, Pezzanera M, Acali S,et al. Anti-EphA2 antibodies with distinct in vitro properties haveequal in vivo efficacy in pancreatic cancer. J Oncol 2009;2009:951917.
13. Brantley DM, Cheng N, Thompson EJ, Lin Q, Brekken RA, Thorpe PE,et al. Soluble EphA receptors inhibit tumor angiogenesis and progres-sion in vivo. Oncogene 2002;21:7011–26.
14. Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritch-ard S, et al. Antiangiogenic and antitumor efficacy of EphA2 receptorantagonist. Cancer Res 2004;64:910–9.
15. Landen CN Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT,Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivousing neutral liposomal small interfering RNA delivery. Cancer Res2005;65:6910–8.
16. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. NatRev Cancer 2005;5:761–72.
17. Kawabe M, Mandic M, Taylor JL, Vasquez CA, Wesa AK, NeckersLM, et al. Heat shock protein 90 inhibitor 17-dimethylaminoethy-lamino-17-demethoxygeldanamycin enhances EphA2þ tumor cellrecognition by specific CD8þ T cells. Cancer Res 2009;69:6995–7003.
18. Wesa AK, Herrem CJ, Mandic M, Taylor JL, Vasquez C, Kawabe M,et al. Enhancement in specific CD8þ T cell recognition of EphA2þ
tumors in vitro and in vivo after treatment with ligand agonists. JImmunol 2008;181:7721–7.
19. Tatsumi T, Herrem CJ, Olson WC, Finke JH, Bukowski RM, Kinch MS,et al. Disease stage variation in CD4þ and CD8þ T-cell reactivity to thereceptor tyrosine kinase EphA2 in patients with renal cell carcinoma.Cancer Res 2003;63:4481–9.
20. Alves PM, Faure O, Graff-Dubois S, Gross DA, Cornet S, Chouaib S,et al. EphA2 as target of anticancer immunotherapy: identification ofHLA-A�0201-restricted epitopes. Cancer Res 2003;63:8476–80.
21. Pardee AD, McCurry D, Alber S, Hu P, Epstein AL, Storkus WJ. AtherapeuticOX40agonist dynamically alters dendritic, endothelial, andT cell subsets within the established tumor microenvironment. CancerRes 2010;70:9041–52.
22. Komita H, Zhao X, Katakam AK, Kumar P, Kawabe M, Okada H,et al. Conditional interleukin-12 gene therapy promotes safeand effective antitumor immunity. Cancer Gene Ther 2009;16:883–91.
23. HatanoM,KuwashimaN, Tatsumi T, Dusak JE,Nishimura F, Reilly KM,et al. Vaccination with EphA2-derived T cell epitopes promotes immu-nity against both EphA2-expressing and EphA2-negative tumors. JTransl Med 2004;2:40.
24. Zhao X, Bose A, Komita H, Taylor JL, Kawabe M, Chi N, et al.Intratumoral IL-12 gene therapy results in the crosspriming of Tc1cells reactive against tumor-associated stromal antigens. Mol Ther2011;19:805–14.
25. Egorin MJ, Lagattuta TF, Hamburger DR, Covey JM, White KD,Musser SM, et al. Pharmacokinetics, tissue distribution, and metab-olism of 17-(dimethylaminoethylamino)-17-demethoxygeldanamy-cin (NSC 707545) in CD2F1 mice and Fischer 344 rats. CancerChemother Pharmacol 2002;49:7–19.
26. Pacey S, Wilson RH, Walton M, Eatock MM, Hardcastle A, ZetterlundA, et al. A phase I study of the heat shock protein 90 inhibitoralvespimycin (17-DMAG) given intravenously to patients withadvanced solid tumors. Clin Cancer Res 2011;17:1561–70.
27. Ramanathan RK, Egorin MJ, Erlichman C, Remick SC, RamalingamSS, Naret C, et al. Phase I pharmacokinetic and pharmacodynamicstudy of 17-dimethylaminoethylamino-17-demethoxygeldanamycin,an inhibitor of heat shock protein 90, in patients with advanced solidtumors. J Clin Oncol 2010;28:1520–6.
28. Lancet JE, Gojo I, Burton M, Quinn M, Tighe SM, Kersey K, et al.Phase I study of the heat shock protein 90 inhibitor alvespimycin(KOS-1022, 17-DMAG) administered intravenously twice weeklyto patients with acute myeloid leukemia. Leukemia 2010;24:699–705.
29. Kummar S, Gutierrez ME, Gardner ER, Chen X, Figg WD, Zajac-KayeM, et al. Phase I trial of 17-dimethylaminoethylamino-17-demethox-ygeldanamycin (17-DMAG), a heat shock protein inhibitor, adminis-tered twice weekly in patients with advanced malignancies. Eur JCancer 2010;46:340–7.
30. Arslan MA, Kutuk O, Basaga H. Protein kinases as drug targets incancer. Curr Cancer Drug Targets 2006;6:623–34.
31. Fukumoto R, Kiang JG. Geldanamycin analog 17-DMAG limits apo-ptosis in human peripheral blood cells by inhibition of p53 activationand its interaction with heat shock protein 90 kDa after ionizingradiation. Radiat Res 2011;176:333–45.
32. RosenbergSA,RestifoNP,Yang JC,MorganRA,DudleyME.Adoptivecell transfer: a clinical path to effective cancer immunotherapy.NatRevCancer 2008;8:299–308.
33. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF,et al. Sipuleucel-T immunotherapy for castration-resistant prostatecancer. N Eng J Med 2010;363:411–22.
34. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, SherryRM, et al. Cancer regression in patients after transfer of geneticallyengineered lymphocytes. Science 2006;314:126–9.
35. Kiang JG, Smith JT, Agravante NG. Geldanamycin analog 17-DMAGinhibits iNOS and caspases in gamma-irradiated human T cells. RadiatRes 2009;172:321–30.
36. Castro JE, Prada CE, Loria O, Kamal A, Chen L, Burrows FJ, et al.ZAP-70 is a novel conditional heat shock protein 90 (Hsp90) client:inhibition of Hsp90 leads to ZAP-70 degradation, apoptosis, andimpaired signaling in chronic lymphocytic leukemia. Blood 2005;106:2506–12.
37. Bae J, Mitsiades C, Tai YT, Bertheau R, ShammasM, Batchu RB, et al.Phenotypic and functional effects of heat shockprotein 90 inhibitionondendritic cell. J Immunol 2007;178:7730–7.
38. ZantekND, AzimiM, Fedor-ChaikenM,WangB, Brackenbury R, KinchMS. E-cadherin regulates the function of the EphA2 receptor tyrosinekinase. Cell Growth Diff 1999;10:629–38.
39. Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2overexpression causes tumorigenesis of mammary epithelial cells.Cancer Res 2001;61:2301–6.
Combination Immunotherapy Using 17-DMAG
www.aacrjournals.org Cancer Res; 72(13) July 1, 2012 3205
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

40. Lu X, Xiao L,Wang L, Ruden DM. Hsp90 inhibitors and drug resistancein cancer: the potential benefits of combination therapies of Hsp90inhibitors and other anti-cancer drugs. Biochem Pharmacol 2012;83:995–1004.
41. Gartner EM, Silverman P, SimonM, Flaherty L, Abrams J, Ivy P, et al. Aphase II study of 17-allylamino-17-demethoxygeldanamycin in meta-static or locally advanced, unresectable breast cancer. Breast CancerRes Treat 2012;131:933–7.
42. Weigel BJ, Blaney SM, Reid JM, Safgren SL, Bagatell R, Kersey J, et al.A phase I study of 17-allylaminogeldanamycin in relapsed/refractorypediatric patients with solid tumors: a Children's Oncology Groupstudy. Clin Cancer Res 2007;13:1789–93.
43. Zajac M, Gomez G, Benitez J, Martínez-Delgado B. Molecular signa-ture of response and potential pathways related to resistance to theHSP90 inhibitor, 17AAG, in breast cancer. BMC Med Genomics2010;3:44.
44. Gaspar N, Sharp SY, Pacey S, Jones C, Walton M, Vassal G, et al.Acquired resistance to 17-allylamino-17-demethoxygeldanamycin(17-AAG, tanespimycin) in glioblastoma cells. Cancer Res 2009;69:1966–75.
45. McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allyla-mino-demethoxygeldanamycin through a glutathione-mediatedmechanism. Cancer Res 2006;66:10967–75.
46. Pittet JF, Lu LN, Geiser T, Lee H, Matthay MA, Welch WJ. Stresspreconditioning attenuates oxidative injury to the alveolar epithe-lium of the lung following haemorrhage in rats. J Physiol 2001;538:583–91.
47. Kiang JG, Agravante NG, Smith JT, Bowman PD. 17-DMAG increasesBcl-2 and inhibits hemorrhage-induced increases in iNOS activation,caspase-3 activity and TNF-a. Cell Biosci 2011;1:21.
48. Bear AS,CruzCR, Foster AE. T cells as vehicles for cancer vaccination.J Biomed Biotech 2011;2011:417403.
49. Joniau S, Abrahamsson PA, Bellmunt J, Figdor C, Hamdy F, VerhagenP, et al. Current vaccination strategies for prostate cancer. Eur Urol2012;61:290–306.
50. Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, et al. Aphase I/II clinical trial investigating the adverse and therapeutic effectsof a postoperative autologous dendritic cell tumor vaccine in patientswith malignant glioma. J Clin Neurosci 2011;18:1048–54.
Rao et al.
Cancer Res; 72(13) July 1, 2012 Cancer Research3206
on June 19, 2014. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 2, 2012; DOI: 10.1158/0008-5472.CAN-12-0538

Correction
Correction: Combination Therapy with HSP90Inhibitor 17-DMAG Reconditions the TumorMicroenvironment to Improve Recruitment ofTherapeutic T Cells
In this article (Cancer Res. 2012;72:3196–206), which appeared in the July 1, 2012issue of Cancer Research (1), the NIH grant P50 CA121973 was missing from theGrant Support section of this article. The authors regret this error.
Reference1. Rao A, Taylor JL, Chi-Sabins N, KawabeM, GoodingWE, StorkusWJ. Combination therapy with
HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment oftherapeutic T cells. Cancer Res 2012;72:3196–206.
Published OnlineFirst October 9, 2012.doi: 10.1158/0008-5472.CAN-12-3249�2012 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 5431




















