Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers
7
Please cite this article in press as: Westwood JA, et al. Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.leukres.2014.05.010 ARTICLE IN PRESS G Model LR-5175; No. of Pages 7 Leukemia Research xxx (2014) xxx–xxx Contents lists available at ScienceDirect Leukemia Research journa l h om epage: www.elsevier.com/locate/leukres Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers Jennifer A. Westwood a , Geoffrey M. Matthews a,b,1 , Jake Shortt a,b,1 , David Faulkner c , Hollie J. Pegram a , Connie P.M. Duong a , Marta Chesi d , P. Leif Bergsagel d , Leslie L. Sharp e , Richard D. Huhn f , Phillip K. Darcy a,g,h , Ricky W. Johnstone a,b , Michael H. Kershaw a,g,h,∗ a Division of Cancer Research, Peter MacCallum Cancer Centre, St Andrew’s Place, East Melbourne 3002, Australia b Cancer Therapeutics Program, Gene Regulation Laboratory, Peter MacCallum Cancer Centre, St Andrew’s Place, East Melbourne 3002, Australia c Department of Pathology, Peter MacCallum Cancer Centre, East Melbourne 3002, Australia d Comprehensive Cancer Center, Mayo Clinic Arizona, Scottsdale, AZ 85259, USA e Oncology Research Unit, Pfizer Inc., San Diego, CA 92121, USA f Pfizer Corporation, New London CT 06320, USA g Sir Peter MacCallum Department of Oncology, University of Melbourne, Parkville 3010, Australia h Department of Immunology, Monash University, Prahran 3181, Australia a r t i c l e i n f o Article history: Received 20 February 2014 Received in revised form 16 May 2014 Accepted 19 May 2014 Available online xxx Keywords: Multiple myeloma Lymphoma E-Myc Vk*myc Anti-CD137 Anti-CD40 a b s t r a c t In order to stimulate antigen presentation and T cell activity against cancer, we treated three different tumor models in mice with the monoclonal antibodies anti-CD40 plus anti-CD137 (BiMab). In a sub- cutaneous transplantable MC38 colon cancer model, there was significant enhancement in the survival of mice following BiMab treatment. Anti-CD40 has shown considerable success against lymphoma in previous studies by other investigators, and we also showed in this study that, in a model of E-Myc lymphoma, there was a statistically significant enhancement of survival of mice following BiMab treat- ment. Following the success of the BiMab treatment in the previous two models, we wished to determine if it would be successful in a mouse model of multiple myeloma. Firstly, we tested a transplantable model of disease in which multiple myeloma cells derived from Vk*MYC mice were injected intravenously. A minor proportion of anti-CD137 and BiMab treated mice experienced prolongation of life beyond 250 days. Then we tested the therapy in a spontaneously occurring multiple myeloma model, in Vk*MYC transgenic mice. The majority of mice treated survived longer than control mice, although statistical significance was not demonstrated. © 2014 Elsevier Ltd. All rights reserved. 1. Introduction We wished to determine if the combined delivery of two mon- oclonal murine antibodies, anti-CD40 and anti-CD137 (hereafter called BiMab), could impact on hematological malignancies. There is evidence that both antibodies with the addition of anti-DR5 can impact on a variety of subcutaneous tumors [1]. However, to sim- plify translation of this approach, we wished to see if reducing this to just the two antibodies in BiMab could be effectual. CD40 is expressed on antigen presenting cells and leads to their activation, ∗ Corresponding author at: Sir Peter MacCallum Department of Oncology, Univer- sity of Melbourne, Parkville 3010, Australia. Tel.: +61 3 9656 1177; fax: +61 3 9656 1411. E-mail address: [email protected] (M.H. Kershaw). 1 These authors contributed equally to this work. generation of CTL and eradication of BCL 1 lymphoma in mice [2]. Agonist antibodies specific for CD40 have been demonstrated to be effective against malignancies in mice and in the clinic [3]. CD137 (4-1BB) is expressed on activated T cells, and ligation leads to T cell proliferation and activation. Anti-CD137 is an agonistic anti- body and has been shown to inhibit tumor growth in a model of multiple myeloma in mice [4]. We decided to use these two ther- apies in combination to target hematological malignancies with elevated expression of the oncogene MYC, which is over expressed in a large proportion of tumors [5]. Deregulated expression of MYC induces B-cell neoplasm development when expression of the MYC locus is driven by the immunoglobulin promoters [6]. We utilized two different transplantable hematological cancers, E-Myc [7,8] B-cell lymphoma and Vk*MYC [9] multiple myeloma, each derived from transgenic mice, injected intravenously to test the combina- tion therapy. We also treated spontaneously generated cancers in Vk*MYC transgenic mice. http://dx.doi.org/10.1016/j.leukres.2014.05.010 0145-2126/© 2014 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Combination anti-CD137 and anti-CD40 antibody therapy in murine myc-driven hematological cancers

L
Cm
JHRa
b
c
d
e
f
g
h
a
ARRAA
KMLEVAA
1
ociipte
sT
h0
ARTICLE IN PRESSG ModelR-5175; No. of Pages 7
Leukemia Research xxx (2014) xxx–xxx
Contents lists available at ScienceDirect
Leukemia Research
journa l h om epage: www.elsev ier .com/ locate / leukres
ombination anti-CD137 and anti-CD40 antibody therapy in murineyc-driven hematological cancers
ennifer A. Westwooda, Geoffrey M. Matthewsa,b,1, Jake Shortta,b,1, David Faulknerc,ollie J. Pegrama, Connie P.M. Duonga, Marta Chesid, P. Leif Bergsageld, Leslie L. Sharpe,ichard D. Huhnf, Phillip K. Darcya,g,h, Ricky W. Johnstonea,b, Michael H. Kershawa,g,h,∗
Division of Cancer Research, Peter MacCallum Cancer Centre, St Andrew’s Place, East Melbourne 3002, AustraliaCancer Therapeutics Program, Gene Regulation Laboratory, Peter MacCallum Cancer Centre, St Andrew’s Place, East Melbourne 3002, AustraliaDepartment of Pathology, Peter MacCallum Cancer Centre, East Melbourne 3002, AustraliaComprehensive Cancer Center, Mayo Clinic Arizona, Scottsdale, AZ 85259, USAOncology Research Unit, Pfizer Inc., San Diego, CA 92121, USAPfizer Corporation, New London CT 06320, USASir Peter MacCallum Department of Oncology, University of Melbourne, Parkville 3010, AustraliaDepartment of Immunology, Monash University, Prahran 3181, Australia
r t i c l e i n f o
rticle history:eceived 20 February 2014eceived in revised form 16 May 2014ccepted 19 May 2014vailable online xxx
eywords:ultiple myeloma
ymphoma
a b s t r a c t
In order to stimulate antigen presentation and T cell activity against cancer, we treated three differenttumor models in mice with the monoclonal antibodies anti-CD40 plus anti-CD137 (BiMab). In a sub-cutaneous transplantable MC38 colon cancer model, there was significant enhancement in the survivalof mice following BiMab treatment. Anti-CD40 has shown considerable success against lymphoma inprevious studies by other investigators, and we also showed in this study that, in a model of E�-Myclymphoma, there was a statistically significant enhancement of survival of mice following BiMab treat-ment. Following the success of the BiMab treatment in the previous two models, we wished to determineif it would be successful in a mouse model of multiple myeloma. Firstly, we tested a transplantable model
�-Myck*mycnti-CD137nti-CD40
of disease in which multiple myeloma cells derived from Vk*MYC mice were injected intravenously. Aminor proportion of anti-CD137 and BiMab treated mice experienced prolongation of life beyond 250days. Then we tested the therapy in a spontaneously occurring multiple myeloma model, in Vk*MYCtransgenic mice. The majority of mice treated survived longer than control mice, although statisticalsignificance was not demonstrated.
. Introduction
We wished to determine if the combined delivery of two mon-clonal murine antibodies, anti-CD40 and anti-CD137 (hereafteralled BiMab), could impact on hematological malignancies. Theres evidence that both antibodies with the addition of anti-DR5 canmpact on a variety of subcutaneous tumors [1]. However, to sim-
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
lify translation of this approach, we wished to see if reducing thiso just the two antibodies in BiMab could be effectual. CD40 isxpressed on antigen presenting cells and leads to their activation,
∗ Corresponding author at: Sir Peter MacCallum Department of Oncology, Univer-ity of Melbourne, Parkville 3010, Australia.el.: +61 3 9656 1177; fax: +61 3 9656 1411.
E-mail address: [email protected] (M.H. Kershaw).1 These authors contributed equally to this work.
ttp://dx.doi.org/10.1016/j.leukres.2014.05.010145-2126/© 2014 Elsevier Ltd. All rights reserved.
© 2014 Elsevier Ltd. All rights reserved.
generation of CTL and eradication of BCL1 lymphoma in mice [2].Agonist antibodies specific for CD40 have been demonstrated to beeffective against malignancies in mice and in the clinic [3]. CD137(4-1BB) is expressed on activated T cells, and ligation leads to Tcell proliferation and activation. Anti-CD137 is an agonistic anti-body and has been shown to inhibit tumor growth in a model ofmultiple myeloma in mice [4]. We decided to use these two ther-apies in combination to target hematological malignancies withelevated expression of the oncogene MYC, which is over expressedin a large proportion of tumors [5]. Deregulated expression of MYCinduces B-cell neoplasm development when expression of the MYClocus is driven by the immunoglobulin promoters [6]. We utilizedtwo different transplantable hematological cancers, E�-Myc [7,8]
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
B-cell lymphoma and Vk*MYC [9] multiple myeloma, each derivedfrom transgenic mice, injected intravenously to test the combina-tion therapy. We also treated spontaneously generated cancers inVk*MYC transgenic mice.
IN PRESSG ModelL
2 mia Research xxx (2014) xxx–xxx
mlipocdtecb
2
2
VnTsi
2
RIFTawcs
bof(awm
gasd
cmdts
2
htfidfiacfegwm(1(m
Ici
0
20
40
60
80
100
0 20 40 60 80 100Days since tumor injected
Perc
ent s
urvi
val
MAC4CD40 itCD40 ipCD137CD40 it + CD137CD40 ip + CD137 P=0.0204
P=0.0039
A
BMAC4α
ααα
α
ααααα
αα
α
αCD40 itCD40 ipCD137CD40 it + CD137CD40 ip + CD137
0
20
40
60
80
100
120
140
160
10 15 20 25 302
T um
or s
ize
(mm
)Days since tumor injected
**
*
***** *
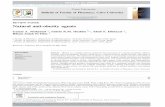
Fig. 1. BiMab is effective against a subcutaneous model of MC38. C57BL6 miceinjected SC with 1 × 106 MC38 tumor cells were treated with either 100 �g �CD40
2.4. Flow cytometry
CD40-expressing cells in mice were identified by flow cytometry. ThreeC57BL/6.ptprca (WEHI) (CD45.1+) mice were injected intravenously with either
0
20
40
60
80
100
0 10 20 30 40 50Days since tumor injected
Perc
ent s
urvi
val
MAC4100CD40CD13750CD40 + CD137100CD40 + CD137200CD40 + CD137400CD40 + CD137
P=0.0437
P=0.0104
ARTICLER-5175; No. of Pages 7
J.A. Westwood et al. / Leuke
The Vk*MYC multiple myeloma model of disease in mice [9]imics human multiple myeloma by generating a clinically simi-
ar disease in mice: indolent course of disease, with organ damagencluding renal dysfunction, bone disease and anemia, and elevatedaraprotein levels in the blood. This disease results from the MYCncogene activation, under the kappa light chain (Vk) promoter 1,onditionally expressed in maturing B cells expressing adenosineeaminase. In the E�-Myc lymphoma model, on the other hand,he MYC gene is driven by the Ig heavy chain enhancer and causesnhanced proliferation of precursor and maturing B lymphoid cells,ausing enlarged lymph nodes, spleen, and thymus, thoracic verte-ral column tumors, and disseminated lymphoma.
. Materials and methods
.1. Cell line
The mouse (C57BL/6) MC38 colon carcinoma tumor cell line (ATCC, Manassas,A, USA) was maintained in complete medium consisting of RPMI (Gibco, Life Tech-ologies, Grand Island, NY) with 10% heat-inactivated fetal calf serum (FCS; MultiSer,hermo Trace, Melbourne) and additives (2 mM glutamine (Gibco)), 100 �g/mltreptomycin (Sigma–Aldrich, St Louis, MO) and 100 U/ml penicillin (Sigma–Aldrich)n a humidified incubator at 37 ◦C with 5% CO2.
.2. Mouse tumor models
Wild type female C57BL/6 mice were purchased from either Australianesources Centre (Canning Vale, WA, Australia) or from the Walter and Eliza Hall
nstitute of Medical Research (WEHI, Bundoora, VIC, Australia), at age 8–12 weeks.or the MC38 mouse model, 1 × 106/100 �l cells were injected subcutaneously (SC).reatment started on day 10, when tumors averaged 33 mm2 in area, after randomssignment and then allocation of mice to equal size range treatment groups. Tumorsere measured (longest and shortest dimensions) every 2–3 days and tumor area
alculated. Mice were culled when tumors reached 200 mm2 in size or at the firstigns of stress.
The Vk*MYC and E�-Myc tumor models were established as systemic diseasey intravenous injection. For the transplantable Vk*MYC model, 3 × 105 cells/200 �lf Vk*MYC clone 4929 (derived by GM and JS) were injected intravenously (IV) intoemale C57BL/6 mice. On days 37 and 56 mice were monitored for serum paraproteinM spike) levels, detected by serum protein electrophoresis (SPEP) [10]. On day 63fter tumor injection, mice were randomly assigned into groups for treatment. Miceere again monitored for serum M spike levels just prior to being culled due toorbidity. All mice were necropsied on death.
For the Vk*MYC spontaneous tumor model, male and female Vk*MYC trans-enic mice (52–56 weeks old) were monitored for serum M spike levels, randomlyssigned to treatment groups and treated. Mice were again monitored for serum Mpike levels just prior to being culled due to morbidity. All mice were necropsied oneath.
For the E�-Myc model, C57BL/6 female mice were injected IV with 2 × 105
ells/200 �l of E�-Myc clone 4242 [8,11] (derived from serial passage in C57BL/6ice of pooled cells from axial, brachial, inguinal and mesenteric lymph nodes). On
ay 7 after tumor cells were injected, mice were randomly assigned to groups andreatment was started. The number of myeloma and lymphoma cells injected wasufficient to ensure reliable engraftment of tumor cells in mice [10,12–14].
.3. Treatment
Mouse anti-CD40 antibody (�CD40) clone FGK-45 (supernatant generated in-ouse from the hybridoma kindly provided by A. Rolink, University of Basel [2])reatment consisted of either intratumoral (IT) injections with 100 �g/100 �l for therst dose followed by 50–100 �g/100 �l IT injections for 3 successive doses at 2–3ay intervals thereafter, or intraperitoneal (IP) injections with 100 �g/200 �l for therst dose followed by 50 �g/100 �l IP injections for the 3 successive doses. Mousenti-CD137 antibody (�CD137) (supernatant generated in-house from hybridomalone 3H3 [15]) treatment consisted of IP injections of 100 �g/200 �l, every 3–4 daysor a total of 3–6 injections. A combination of both antibodies (BiMab) was deliv-red to some groups at the same dose as the monotherapies. MAC4 (supernatantenerated in-house) an isotype control antibody for both monoclonal antibodies,as given to control groups at the doses of 200 �g/200 �l IP. The spontaneousodel of Vk*MYC received 3 courses of either BiMab or isotype control antibody
MAC4), injected IP at the dose of 100 �g for each antibody on days 0, 4, 7, 10, 13,6 (first course), 63, 66, 70, 74, 77, 81 (second course), 154, 158, 162, 165, 168, 172third course), that is with 2–2.5 months between courses. Disease progression was
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
onitored by measuring M spike levels. Mice were culled at the first signs of stress.For experiments depicted in Fig. 1, mice were treated with either 100 �g �CD40
T or IP either as a monotherapy on day 10 and then 50 �g on days 13, 15 and 17, orombined with 100 �g �CD137 IP (injected on days 10, 13 and 17). For experimentsn Fig. 2, �CD40 was injected IP at different doses as shown on days 7, 10, 13 and
IT or IP either as a monotherapy on day 10 and then 50 �g on days 13, 15 and 17, orcombined with 100 �g �CD137 IP (injected on days 10, 13 and 17). A, survival andB, tumor growth (n = 5 mice/group). *P < 0.05, **P < 0.005.
16 after tumor injection, and �CD137 was injected IP on days 7, 10, 13, 16, 19 and22. In Fig. 3A, mice were treated on days 63, 66, and 70 after tumor injection with100 �g �CD40, and/or on days 63, 66, 70, 74, 77 and 81 with 100 �g �CD137 orMAC4 (n = 5 mice/group). In Fig. 3B, mice were treated on days 42, 45, and 48 aftertumor injection with 100 �g �CD40, and/or on days 42, 45, 48, 52, 55, 59 with 100 �g�CD137 or MAC4.
Ethics statement: This study was carried out in strict accordance with the rec-ommendations of the Victorian Bureau of Animal Welfare, Department of PrimaryIndustries, and the National Health and Medical Research Council’s Australian codeof practice for the care and use of animals for scientific purposes. The protocol wasapproved by the Peter MacCallum Cancer Centre Animal Experimentation EthicsCommittee under Permit number E396. All efforts were made to minimize suffering.
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
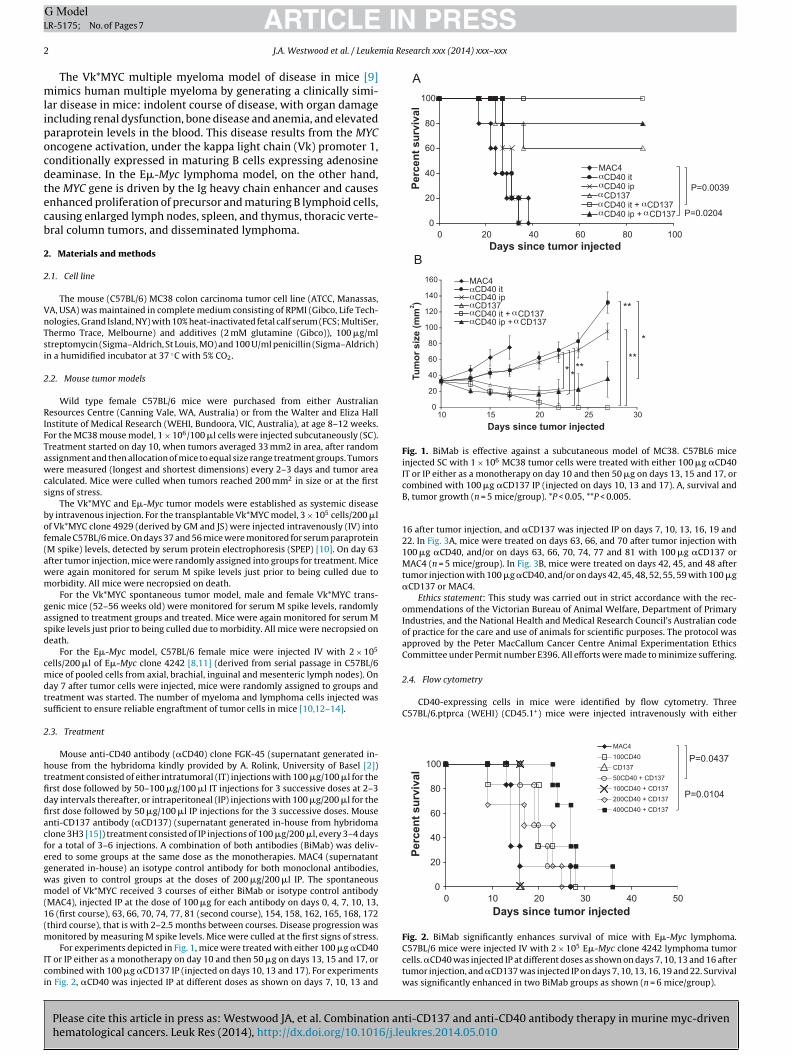
Fig. 2. BiMab significantly enhances survival of mice with E�-Myc lymphoma.C57BL/6 mice were injected IV with 2 × 105 E�-Myc clone 4242 lymphoma tumorcells. �CD40 was injected IP at different doses as shown on days 7, 10, 13 and 16 aftertumor injection, and �CD137 was injected IP on days 7, 10, 13, 16, 19 and 22. Survivalwas significantly enhanced in two BiMab groups as shown (n = 6 mice/group).
ARTICLE IN PRESSG ModelLR-5175; No. of Pages 7
J.A. Westwood et al. / Leukemia Research xxx (2014) xxx–xxx 3
MAC4CD40
αα
α
CD137CD40 + �CD137
BPe
rce
nt
su
rviv
al
*
0
20
40
60
80
100
0 50 100 150 200 250 300 350 400 450 500
A
0 50 100 150 200 250 300 350 400 450 500 550
0
20
40
60
80
100
Days since tumor injected
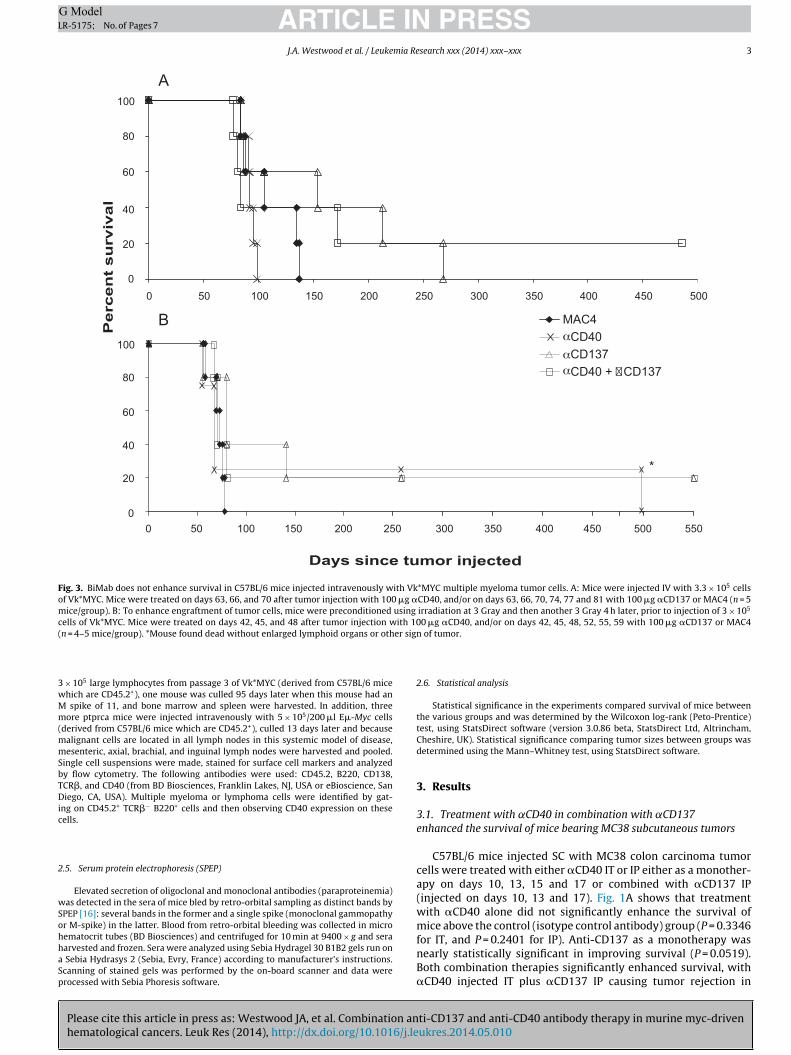
Fig. 3. BiMab does not enhance survival in C57BL/6 mice injected intravenously with Vk*MYC multiple myeloma tumor cells. A: Mice were injected IV with 3.3 × 105 cellsof Vk*MYC. Mice were treated on days 63, 66, and 70 after tumor injection with 100 �g �CD40, and/or on days 63, 66, 70, 74, 77 and 81 with 100 �g �CD137 or MAC4 (n = 5mice/group). B: To enhance engraftment of tumor cells, mice were preconditioned using irradiation at 3 Gray and then another 3 Gray 4 h later, prior to injection of 3 × 105
cells of Vk*MYC. Mice were treated on days 42, 45, and 48 after tumor injection with 100 �g �CD40, and/or on days 42, 45, 48, 52, 55, 59 with 100 �g �CD137 or MAC4( er sign
3wMm(mmSbTDic
2
wSohhaSp
n = 4–5 mice/group). *Mouse found dead without enlarged lymphoid organs or oth
× 105 large lymphocytes from passage 3 of Vk*MYC (derived from C57BL/6 micehich are CD45.2+), one mouse was culled 95 days later when this mouse had an
spike of 11, and bone marrow and spleen were harvested. In addition, threeore ptprca mice were injected intravenously with 5 × 105/200 �l E�-Myc cells
derived from C57BL/6 mice which are CD45.2+), culled 13 days later and becausealignant cells are located in all lymph nodes in this systemic model of disease,esenteric, axial, brachial, and inguinal lymph nodes were harvested and pooled.
ingle cell suspensions were made, stained for surface cell markers and analyzedy flow cytometry. The following antibodies were used: CD45.2, B220, CD138,CR�, and CD40 (from BD Biosciences, Franklin Lakes, NJ, USA or eBioscience, Saniego, CA, USA). Multiple myeloma or lymphoma cells were identified by gat-
ng on CD45.2+ TCR�− B220+ cells and then observing CD40 expression on theseells.
.5. Serum protein electrophoresis (SPEP)
Elevated secretion of oligoclonal and monoclonal antibodies (paraproteinemia)as detected in the sera of mice bled by retro-orbital sampling as distinct bands by
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
PEP [16]: several bands in the former and a single spike (monoclonal gammopathyr M-spike) in the latter. Blood from retro-orbital bleeding was collected in microematocrit tubes (BD Biosciences) and centrifuged for 10 min at 9400 × g and seraarvested and frozen. Sera were analyzed using Sebia Hydragel 30 B1B2 gels run on
Sebia Hydrasys 2 (Sebia, Evry, France) according to manufacturer’s instructions.canning of stained gels was performed by the on-board scanner and data wererocessed with Sebia Phoresis software.
of tumor.
2.6. Statistical analysis
Statistical significance in the experiments compared survival of mice betweenthe various groups and was determined by the Wilcoxon log-rank (Peto-Prentice)test, using StatsDirect software (version 3.0.86 beta, StatsDirect Ltd, Altrincham,Cheshire, UK). Statistical significance comparing tumor sizes between groups wasdetermined using the Mann–Whitney test, using StatsDirect software.
3. Results
3.1. Treatment with ˛CD40 in combination with ˛CD137enhanced the survival of mice bearing MC38 subcutaneous tumors
C57BL/6 mice injected SC with MC38 colon carcinoma tumorcells were treated with either �CD40 IT or IP either as a monother-apy on days 10, 13, 15 and 17 or combined with �CD137 IP(injected on days 10, 13 and 17). Fig. 1A shows that treatmentwith �CD40 alone did not significantly enhance the survival ofmice above the control (isotype control antibody) group (P = 0.3346
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
for IT, and P = 0.2401 for IP). Anti-CD137 as a monotherapy wasnearly statistically significant in improving survival (P = 0.0519).Both combination therapies significantly enhanced survival, with�CD40 injected IT plus �CD137 IP causing tumor rejection in
ARTICLE IN PRESSG ModelLR-5175; No. of Pages 7
4 J.A. Westwood et al. / Leukemia Research xxx (2014) xxx–xxx
F ith trt rbitalF ment
1t(
3e
iilmE11sB�rwg
3m
Vt8sttolr(aa(m
injected IP at the dose of 100 �g for each antibody as detailed inMethods. Three mice were found dead in the BiMab group (2 onday 7 and one on day 52) after initiation of therapy. Their M spikelevels taken two weeks prior to treatment were predominantly
0
20
40
60
80
100
0 50 100 150 200 250 300 350 400 450
Days since treatment started
Perc
ent s
urvi
val
MAC4BiMab
**
Fig. 5. BiMab enhances survival of the majority of Vk*MYC transgenic mice. Vk*MYCtransgenic mice were treated with 3 courses of either BiMab or isotype control
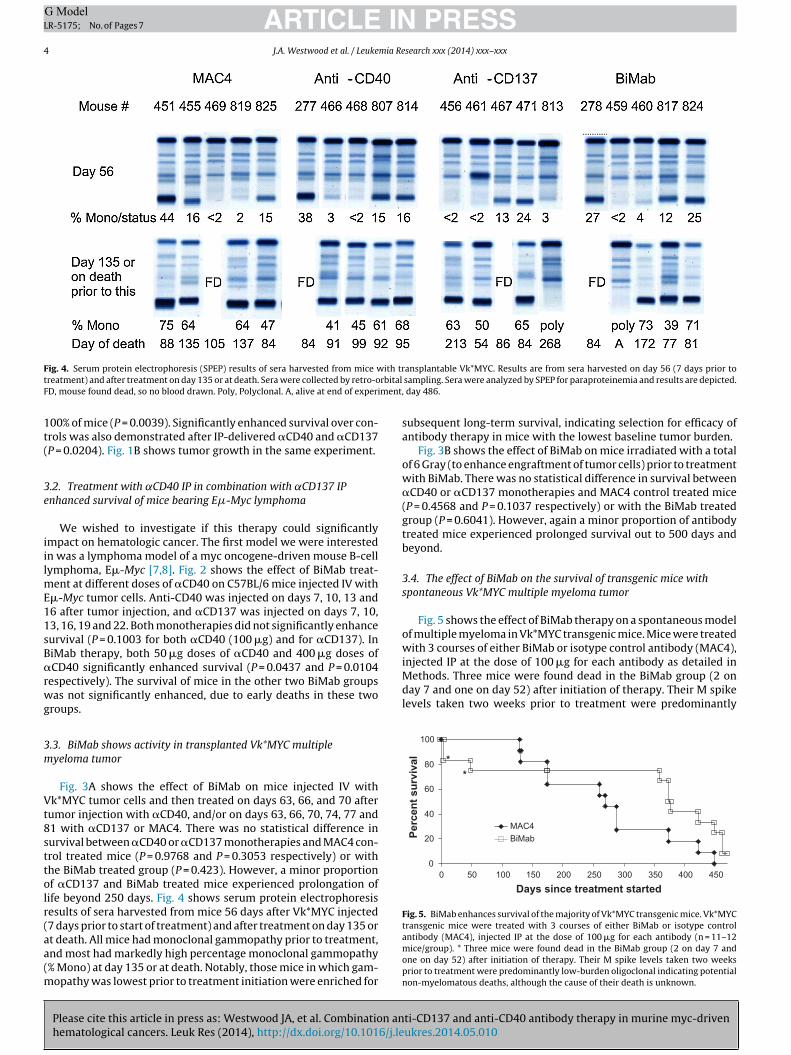
ig. 4. Serum protein electrophoresis (SPEP) results of sera harvested from mice wreatment) and after treatment on day 135 or at death. Sera were collected by retro-oD, mouse found dead, so no blood drawn. Poly, Polyclonal. A, alive at end of experi
00% of mice (P = 0.0039). Significantly enhanced survival over con-rols was also demonstrated after IP-delivered �CD40 and �CD137P = 0.0204). Fig. 1B shows tumor growth in the same experiment.
.2. Treatment with ˛CD40 IP in combination with ˛CD137 IPnhanced survival of mice bearing E�-Myc lymphoma
We wished to investigate if this therapy could significantlympact on hematologic cancer. The first model we were interestedn was a lymphoma model of a myc oncogene-driven mouse B-cellymphoma, E�-Myc [7,8]. Fig. 2 shows the effect of BiMab treat-
ent at different doses of �CD40 on C57BL/6 mice injected IV with�-Myc tumor cells. Anti-CD40 was injected on days 7, 10, 13 and6 after tumor injection, and �CD137 was injected on days 7, 10,3, 16, 19 and 22. Both monotherapies did not significantly enhanceurvival (P = 0.1003 for both �CD40 (100 �g) and for �CD137). IniMab therapy, both 50 �g doses of �CD40 and 400 �g doses ofCD40 significantly enhanced survival (P = 0.0437 and P = 0.0104
espectively). The survival of mice in the other two BiMab groupsas not significantly enhanced, due to early deaths in these two
roups.
.3. BiMab shows activity in transplanted Vk*MYC multipleyeloma tumor
Fig. 3A shows the effect of BiMab on mice injected IV withk*MYC tumor cells and then treated on days 63, 66, and 70 after
umor injection with �CD40, and/or on days 63, 66, 70, 74, 77 and1 with �CD137 or MAC4. There was no statistical difference inurvival between �CD40 or �CD137 monotherapies and MAC4 con-rol treated mice (P = 0.9768 and P = 0.3053 respectively) or withhe BiMab treated group (P = 0.423). However, a minor proportionf �CD137 and BiMab treated mice experienced prolongation ofife beyond 250 days. Fig. 4 shows serum protein electrophoresisesults of sera harvested from mice 56 days after Vk*MYC injected7 days prior to start of treatment) and after treatment on day 135 or
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
t death. All mice had monoclonal gammopathy prior to treatment,nd most had markedly high percentage monoclonal gammopathy% Mono) at day 135 or at death. Notably, those mice in which gam-
opathy was lowest prior to treatment initiation were enriched for
ansplantable Vk*MYC. Results are from sera harvested on day 56 (7 days prior to sampling. Sera were analyzed by SPEP for paraproteinemia and results are depicted., day 486.
subsequent long-term survival, indicating selection for efficacy ofantibody therapy in mice with the lowest baseline tumor burden.
Fig. 3B shows the effect of BiMab on mice irradiated with a totalof 6 Gray (to enhance engraftment of tumor cells) prior to treatmentwith BiMab. There was no statistical difference in survival between�CD40 or �CD137 monotherapies and MAC4 control treated mice(P = 0.4568 and P = 0.1037 respectively) or with the BiMab treatedgroup (P = 0.6041). However, again a minor proportion of antibodytreated mice experienced prolonged survival out to 500 days andbeyond.
3.4. The effect of BiMab on the survival of transgenic mice withspontaneous Vk*MYC multiple myeloma tumor
Fig. 5 shows the effect of BiMab therapy on a spontaneous modelof multiple myeloma in Vk*MYC transgenic mice. Mice were treatedwith 3 courses of either BiMab or isotype control antibody (MAC4),
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
antibody (MAC4), injected IP at the dose of 100 �g for each antibody (n = 11–12mice/group). * Three mice were found dead in the BiMab group (2 on day 7 andone on day 52) after initiation of therapy. Their M spike levels taken two weeksprior to treatment were predominantly low-burden oligoclonal indicating potentialnon-myelomatous deaths, although the cause of their death is unknown.
ARTICLE IN PRESSG ModelLR-5175; No. of Pages 7
J.A. Westwood et al. / Leukemia Research xxx (2014) xxx–xxx 5
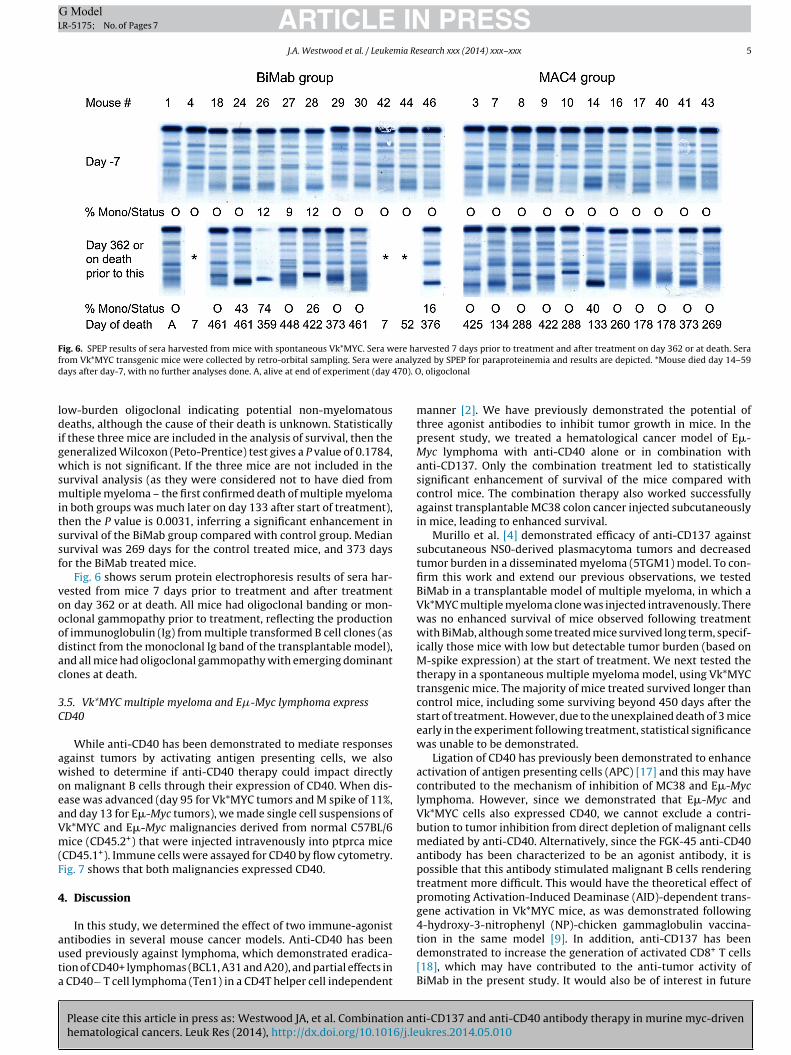
Fig. 6. SPEP results of sera harvested from mice with spontaneous Vk*MYC. Sera were harvested 7 days prior to treatment and after treatment on day 362 or at death. Seraf analyd 70). O
ldigwsmitssf
vooodac
3C
awoeaVm(F
4
auta
rom Vk*MYC transgenic mice were collected by retro-orbital sampling. Sera wereays after day-7, with no further analyses done. A, alive at end of experiment (day 4
ow-burden oligoclonal indicating potential non-myelomatouseaths, although the cause of their death is unknown. Statistically
f these three mice are included in the analysis of survival, then theeneralized Wilcoxon (Peto-Prentice) test gives a P value of 0.1784,hich is not significant. If the three mice are not included in the
urvival analysis (as they were considered not to have died fromultiple myeloma – the first confirmed death of multiple myeloma
n both groups was much later on day 133 after start of treatment),hen the P value is 0.0031, inferring a significant enhancement inurvival of the BiMab group compared with control group. Medianurvival was 269 days for the control treated mice, and 373 daysor the BiMab treated mice.
Fig. 6 shows serum protein electrophoresis results of sera har-ested from mice 7 days prior to treatment and after treatmentn day 362 or at death. All mice had oligoclonal banding or mon-clonal gammopathy prior to treatment, reflecting the productionf immunoglobulin (Ig) from multiple transformed B cell clones (asistinct from the monoclonal Ig band of the transplantable model),nd all mice had oligoclonal gammopathy with emerging dominantlones at death.
.5. Vk*MYC multiple myeloma and E�-Myc lymphoma expressD40
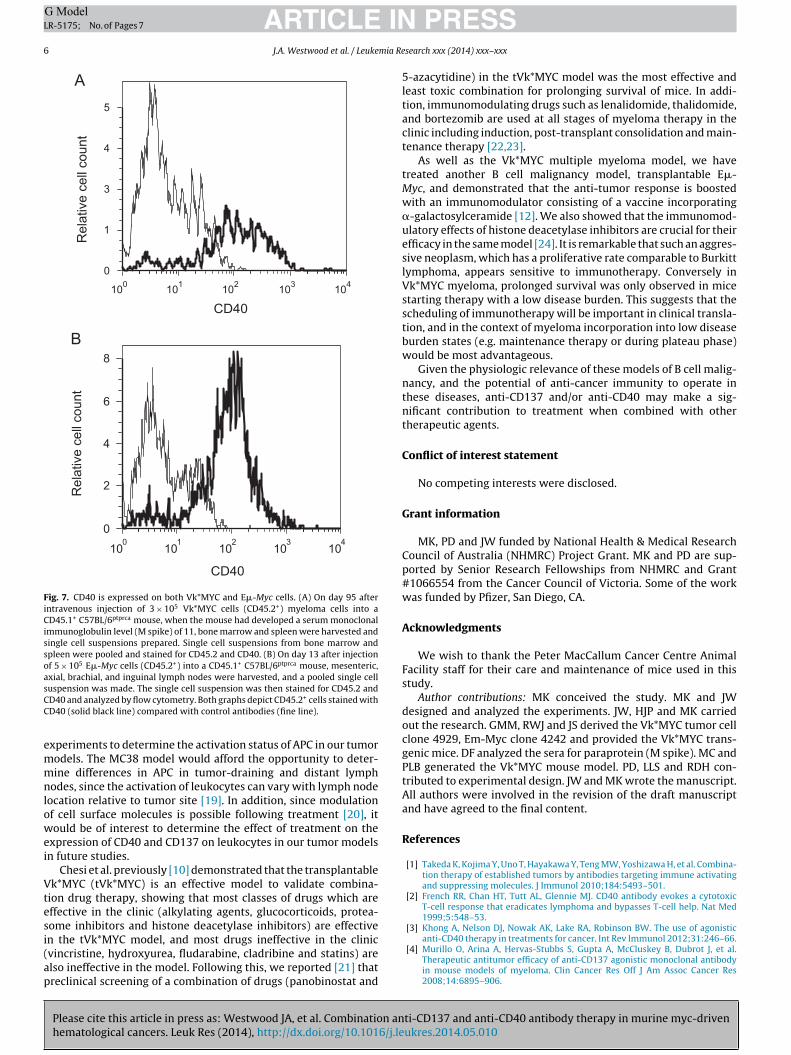
While anti-CD40 has been demonstrated to mediate responsesgainst tumors by activating antigen presenting cells, we alsoished to determine if anti-CD40 therapy could impact directly
n malignant B cells through their expression of CD40. When dis-ase was advanced (day 95 for Vk*MYC tumors and M spike of 11%,nd day 13 for E�-Myc tumors), we made single cell suspensions ofk*MYC and E�-Myc malignancies derived from normal C57BL/6ice (CD45.2+) that were injected intravenously into ptprca mice
CD45.1+). Immune cells were assayed for CD40 by flow cytometry.ig. 7 shows that both malignancies expressed CD40.
. Discussion
In this study, we determined the effect of two immune-agonist
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
ntibodies in several mouse cancer models. Anti-CD40 has beensed previously against lymphoma, which demonstrated eradica-ion of CD40+ lymphomas (BCL1, A31 and A20), and partial effects in
CD40− T cell lymphoma (Ten1) in a CD4T helper cell independent
zed by SPEP for paraproteinemia and results are depicted. *Mouse died day 14–59, oligoclonal
manner [2]. We have previously demonstrated the potential ofthree agonist antibodies to inhibit tumor growth in mice. In thepresent study, we treated a hematological cancer model of E�-Myc lymphoma with anti-CD40 alone or in combination withanti-CD137. Only the combination treatment led to statisticallysignificant enhancement of survival of the mice compared withcontrol mice. The combination therapy also worked successfullyagainst transplantable MC38 colon cancer injected subcutaneouslyin mice, leading to enhanced survival.
Murillo et al. [4] demonstrated efficacy of anti-CD137 againstsubcutaneous NS0-derived plasmacytoma tumors and decreasedtumor burden in a disseminated myeloma (5TGM1) model. To con-firm this work and extend our previous observations, we testedBiMab in a transplantable model of multiple myeloma, in which aVk*MYC multiple myeloma clone was injected intravenously. Therewas no enhanced survival of mice observed following treatmentwith BiMab, although some treated mice survived long term, specif-ically those mice with low but detectable tumor burden (based onM-spike expression) at the start of treatment. We next tested thetherapy in a spontaneous multiple myeloma model, using Vk*MYCtransgenic mice. The majority of mice treated survived longer thancontrol mice, including some surviving beyond 450 days after thestart of treatment. However, due to the unexplained death of 3 miceearly in the experiment following treatment, statistical significancewas unable to be demonstrated.
Ligation of CD40 has previously been demonstrated to enhanceactivation of antigen presenting cells (APC) [17] and this may havecontributed to the mechanism of inhibition of MC38 and E�-Myclymphoma. However, since we demonstrated that E�-Myc andVk*MYC cells also expressed CD40, we cannot exclude a contri-bution to tumor inhibition from direct depletion of malignant cellsmediated by anti-CD40. Alternatively, since the FGK-45 anti-CD40antibody has been characterized to be an agonist antibody, it ispossible that this antibody stimulated malignant B cells renderingtreatment more difficult. This would have the theoretical effect ofpromoting Activation-Induced Deaminase (AID)-dependent trans-gene activation in Vk*MYC mice, as was demonstrated following4-hydroxy-3-nitrophenyl (NP)-chicken gammaglobulin vaccina-
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
tion in the same model [9]. In addition, anti-CD137 has beendemonstrated to increase the generation of activated CD8+ T cells[18], which may have contributed to the anti-tumor activity ofBiMab in the present study. It would also be of interest in future
ARTICLE ING ModelLR-5175; No. of Pages 7
6 J.A. Westwood et al. / Leukemia Re
Rel
ativ
e ce
ll co
unt
100 101 102 103 1040
1
3
4
5
CD40
CD40
100
10 1
10 2
10 3
10 4
0
2
4
6
8
Rel
ativ
e ce
ll co
unt
A
B
Fig. 7. CD40 is expressed on both Vk*MYC and E�-Myc cells. (A) On day 95 afterintravenous injection of 3 × 105 Vk*MYC cells (CD45.2+) myeloma cells into aCD45.1+ C57BL/6ptprca mouse, when the mouse had developed a serum monoclonalimmunoglobulin level (M spike) of 11, bone marrow and spleen were harvested andsingle cell suspensions prepared. Single cell suspensions from bone marrow andspleen were pooled and stained for CD45.2 and CD40. (B) On day 13 after injectionof 5 × 105 E�-Myc cells (CD45.2+) into a CD45.1+ C57BL/6ptprca mouse, mesenteric,axial, brachial, and inguinal lymph nodes were harvested, and a pooled single cellsCC
emmnlowei
Vtesi(ap
uspension was made. The single cell suspension was then stained for CD45.2 andD40 and analyzed by flow cytometry. Both graphs depict CD45.2+ cells stained withD40 (solid black line) compared with control antibodies (fine line).
xperiments to determine the activation status of APC in our tumorodels. The MC38 model would afford the opportunity to deter-ine differences in APC in tumor-draining and distant lymph
odes, since the activation of leukocytes can vary with lymph nodeocation relative to tumor site [19]. In addition, since modulationf cell surface molecules is possible following treatment [20], itould be of interest to determine the effect of treatment on the
xpression of CD40 and CD137 on leukocytes in our tumor modelsn future studies.
Chesi et al. previously [10] demonstrated that the transplantablek*MYC (tVk*MYC) is an effective model to validate combina-
ion drug therapy, showing that most classes of drugs which areffective in the clinic (alkylating agents, glucocorticoids, protea-ome inhibitors and histone deacetylase inhibitors) are effective
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
n the tVk*MYC model, and most drugs ineffective in the clinicvincristine, hydroxyurea, fludarabine, cladribine and statins) arelso ineffective in the model. Following this, we reported [21] thatreclinical screening of a combination of drugs (panobinostat and
PRESSsearch xxx (2014) xxx–xxx
5-azacytidine) in the tVk*MYC model was the most effective andleast toxic combination for prolonging survival of mice. In addi-tion, immunomodulating drugs such as lenalidomide, thalidomide,and bortezomib are used at all stages of myeloma therapy in theclinic including induction, post-transplant consolidation and main-tenance therapy [22,23].
As well as the Vk*MYC multiple myeloma model, we havetreated another B cell malignancy model, transplantable E�-Myc, and demonstrated that the anti-tumor response is boostedwith an immunomodulator consisting of a vaccine incorporating�-galactosylceramide [12]. We also showed that the immunomod-ulatory effects of histone deacetylase inhibitors are crucial for theirefficacy in the same model [24]. It is remarkable that such an aggres-sive neoplasm, which has a proliferative rate comparable to Burkittlymphoma, appears sensitive to immunotherapy. Conversely inVk*MYC myeloma, prolonged survival was only observed in micestarting therapy with a low disease burden. This suggests that thescheduling of immunotherapy will be important in clinical transla-tion, and in the context of myeloma incorporation into low diseaseburden states (e.g. maintenance therapy or during plateau phase)would be most advantageous.
Given the physiologic relevance of these models of B cell malig-nancy, and the potential of anti-cancer immunity to operate inthese diseases, anti-CD137 and/or anti-CD40 may make a sig-nificant contribution to treatment when combined with othertherapeutic agents.
Conflict of interest statement
No competing interests were disclosed.
Grant information
MK, PD and JW funded by National Health & Medical ResearchCouncil of Australia (NHMRC) Project Grant. MK and PD are sup-ported by Senior Research Fellowships from NHMRC and Grant#1066554 from the Cancer Council of Victoria. Some of the workwas funded by Pfizer, San Diego, CA.
Acknowledgments
We wish to thank the Peter MacCallum Cancer Centre AnimalFacility staff for their care and maintenance of mice used in thisstudy.
Author contributions: MK conceived the study. MK and JWdesigned and analyzed the experiments. JW, HJP and MK carriedout the research. GMM, RWJ and JS derived the Vk*MYC tumor cellclone 4929, Em-Myc clone 4242 and provided the Vk*MYC trans-genic mice. DF analyzed the sera for paraprotein (M spike). MC andPLB generated the Vk*MYC mouse model. PD, LLS and RDH con-tributed to experimental design. JW and MK wrote the manuscript.All authors were involved in the revision of the draft manuscriptand have agreed to the final content.
References
[1] Takeda K, Kojima Y, Uno T, Hayakawa Y, Teng MW, Yoshizawa H, et al. Combina-tion therapy of established tumors by antibodies targeting immune activatingand suppressing molecules. J Immunol 2010;184:5493–501.
[2] French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxicT-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med1999;5:548–53.
[3] Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BW. The use of agonistic
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
anti-CD40 therapy in treatments for cancer. Int Rev Immunol 2012;31:246–66.[4] Murillo O, Arina A, Hervas-Stubbs S, Gupta A, McCluskey B, Dubrot J, et al.
Therapeutic antitumor efficacy of anti-CD137 agonistic monoclonal antibodyin mouse models of myeloma. Clin Cancer Res Off J Am Assoc Cancer Res2008;14:6895–906.

ING ModelL
mia Re
[
[
[
[
[
[
[
[
[
[
[
[
[
[
ARTICLER-5175; No. of Pages 7
J.A. Westwood et al. / Leuke
[5] Klapproth K, Wirth T. Advances in the understanding of MYC-induced lym-phomagenesis. Brit J Haematol 2010;149:484–97.
[6] Wiman KG, Clarkson B, Hayday AC, Saito H, Tonegawa S, Hayward WS. Activa-tion of a translocated c-myc gene: role of structural alterations in the upstreamregion. Proc Natl Acad Sci USA 1984;81:6798–802.
[7] Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al.The c-myc oncogene driven by immunoglobulin enhancers induces lymphoidmalignancy in transgenic mice. Nature 1985;318:533–8.
[8] Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. TheE mu-myc transgenic mouse. A model for high-incidence spontaneous lym-phoma and leukemia of early B cells. J Exp Med 1988;167:353–71.
[9] Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in aconditional mouse model of post-germinal center malignancies. Cancer Cell2008;13:167–80.
10] Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, et al. Drugresponse in a genetically engineered mouse model of multiple myeloma ispredictive of clinical efficacy. Blood 2012;120:376–85.
11] Shortt J, Martin BP, Newbold A, Hannan KM, Devlin JR, Baker AJ, et al. Combinedinhibition of PI3K-related DNA damage response kinases and mTORC1 inducesapoptosis in MYC-driven B-cell lymphomas. Blood 2013;121:2964–74.
12] Mattarollo SR, West AC, Steegh K, Duret H, Paget C, Martin B, et al. NKT celladjuvant-based tumor vaccine for treatment of myc oncogene-driven mouseB-cell lymphoma. Blood 2012;120:3019–29.
13] Newbold A, Salmon JM, Martin BP, Stanley K, Johnstone RW. The roleof p21 and p27 in HDACi-mediated tumor cell death and cell cycle
Please cite this article in press as: Westwood JA, et al. Combination anhematological cancers. Leuk Res (2014), http://dx.doi.org/10.1016/j.le
arrest in the Emu-myc model of B-cell lymphoma. Oncogene 2013,http://dx.doi.org/10.1038/onc.2013.482 [Epub ahead of print].
14] Devlin JR, Hannan KM, Ng PY, Bywater MJ, Shortt J, Cullinane C, et al. AKT sig-nalling is required for ribosomal RNA synthesis and progression of Emu-MycB-cell lymphoma in vivo. FEBS J 2013;280:5307–16.
[
PRESSsearch xxx (2014) xxx–xxx 7
15] Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, et al.Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicateestablished tumors. Nat Med 1997;3:682–5.
16] Longsworth LG, Shedlovsky T, Macinnes DA. Electrophoretic patterns of normaland pathological human blood serum and plasma. J Exp Med 1939;70:399–413.
17] Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, AlberG. Ligation of CD40 on dendritic cells triggers production of high levels ofinterleukin-12 and enhances T cell stimulatory capacity: T-T help via APC acti-vation. J Exp Med 1996;184:747–52.
18] Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, et al. Eradicationof established tumors in mice by a combination antibody-based therapy. NatMed 2006;12:693–8.
19] Vuletic A, Jurisic V, Jovanic I, Milovanovic Z, Nikolic S, Konjevic G. Distributionof several activating and inhibitory receptors on CD3(−)CD56(+) NK cells inregional lymph nodes of melanoma patients. J Surg Res 2013;183:860–8.
20] Jurisic V, Srdic-Rajic T, Konjevic G, Bogdanovic G, Colic M. TNF-alpha inducedapoptosis is accompanied with rapid CD30 and slower CD45 shedding fromK-562 cells. J Membr Biol 2011;239:115–22.
21] Matthews GM, Lefebure M, Doyle MA, Shortt J, Ellul J, Chesi M, et al. Pre-clinical screening of histone deacetylase inhibitors combined with ABT-737,rhTRAIL/MD5-1 or 5-azacytidine using syngeneic Vk*MYC multiple myeloma.Cell Death Disease 2013;4:e798.
22] Ludwig H, Miguel JS, Dimopoulos MA, Palumbo A, Garcia Sanz R, Powles R, et al.International Myeloma Working Group recommendations for global myelomacare. Leukemia 2013;28:981–92.
23] Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: immuno-
ti-CD137 and anti-CD40 antibody therapy in murine myc-drivenukres.2014.05.010
logical, molecular and epigenetic targets in cancer therapy. Oncogene2013;32:4191–202.
24] West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, et al.An intact immune system is required for the anticancer activities of histonedeacetylase inhibitors. Cancer Res 2013;73:7265–76.